Vargatef 100 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Vargatef 100 mg měkké tobolky Vargatef 150 mg měkké tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tobolka obsahuje nintedanibum 100 mg (ve formě nintedanibi esilas). Jedna tobolka obsahuje nintedanibum 150 mg (ve formě nintedanibi esilas).
Pomocné látky se známým účinkem:
Jedna tobolka obsahuje 1,2 mg sójového lecithinu. Jedna tobolka obsahuje 1,8 mg sójového lecithinu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Měkká tobolka.
Neprůhledné, oválné měkké želatinové tobolky broskvové barvy s černým potiskem na jedné straně -symbolem společnosti Boehringer Ingelheim a „100“.
Neprůhledné, oválné měkké želatinové tobolky hnědé barvy s černým potiskem na jedné straně -symbolem společnosti Boehringer Ingelheim a „150“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Vargatef je v kombinaci s docetaxelem indikován k léčbě dospělých pacientů s lokálně pokročilým metastazujícím nebo lokálně rekurentním nemalobuněčným karcinomem plic (NSCLC), histologicky prokázaným adenokarcinomem, po chemoterapii první linie.
4.2 Dávkování a způsob podání
Léčbu přípravkem Vargatef musí zahajovat a vést lékař, který má zkušenosti s použitím protinádorové léčby.
Dávkování
Doporučená dávka nintedanibu je 200 mg dvakrát denně podaných s odstupem přibližně 12 hodin v den 2 až 21 standardního 21denního cyklu léčby docetaxelem.
Přípravek Vargatef nelze užít v týž den, kdy je podána chemoterapie docetaxelem (= den 1).
Jestliže dojde k vynechání dávky nintedanibu, je jeho podávání třeba obnovit podáním doporučené dávky v následující plánovaný termín. Vynechanou dávku nelze nahradit zvýšením jednotlivé denní dávky nintedanibu nad doporučenou dávku. Doporučená maximální denní dávka 400 mg nesmí být překročena.
Pacienti mohou pokračovat v léčbě nintedanibem po ukončení docetaxelu tak dlouho, dokud lze pozorovat přínos léčby, nebo do doby, kdy se objeví nepřijatelná toxicita.
Dávkování, způsoby podávání a změny dávek docetaxelu, prosím, vyhledejte v příslušných informacích o docetaxelu.
Úpravy dávky
Jako úvodní opatření k zvládání nežádoucích účinků (viz tabulky 1 a 2) je třeba léčbu nintedanibem přerušit do doby, než je příslušný nežádoucí účinek zvládnut do té míry, že lze pokračovat v léčbě (stupeň 1 nebo výchozí stav).
V léčbě lze pokračovat podáváním nižších dávek nintedanibu. Doporučuje se dávku upravovat postupně po 100 mg denně (tedy snížení o 50 mg na jednu dávku), v závislosti na bezpečnosti a snášenlivosti každého pacienta, jak je uvedeno v tabulce 1 a tabulce 2.
V případě, že nežádoucí účinek/účinky přetrvávají, tedy pokud pacient netoleruje dávku 100 mg dvakrát denně, léčbu přípravkem Vargatefje třeba trvale ukončit. V případě specifických zvýšení hladin aspartátaminotransferázy (AST)/ alaninaminotransferázy (ALT) na > 3x horní hranice normy (upper limit normal, ULN) společně se zvýšením celkového bilirubinu na > 2x ULN a alkalické fosfatázy (ALKP) < 2x ULN (viz tabulka 2) je třeba léčbu přípravkem Vargatef přerušit. Pokud není zjištěna jiná příčina, je třeba léčbu přípravkem Vargatef trvale ukončit (viz rovněž bod 4.4).
Tabulka 1: Doporučené úpravy dávky přípravku Vargatef (nintedanib) v případě výskytu průjmu,
zvracení a jiných nehematologických nebo hematologických nežádoucích účinků
|
Nežádoucí účinek dle CTCAE* |
Úprava dávky |
|
Průjem stupně >2 po dobu delší než 7 po sobě následujících dnů, přestože byla podána protiprůjmová léčba NEBO Průjem stupně > 3, přestože byla podána protiprůjmová léčba. |
Po přerušení léčby a zlepšení na stupeň 1 nebo výchozí stav, snížení dávky z 200 mg dvakrát denně na 150 mg dvakrát denně a - jestliže je 2. snížení dávky považováno za nutné - ze 150 mg dvakrát denně na 100 mg dvakrát denně. |
|
Zvracení stupně > 2 A/NEBO Nauzea stupně > 3 přestože byla podávána antiemetická léčba | |
|
Ostatní nehematologické nebo hematologické nežádoucí účinky stupně > 3 |
CTCAE: obecná terminologická kritéria nežádoucích příhod (Common Terminology Criteria for Adverse Events)
Tabulka 2: Doporučené úpravy dávky přípravku Vargatef (nintedanib) v případě zvýšených
hladin AST a/nebo ALT a bilirubinu
|
Zvýšení hladin AST/ALT a bilirubinu |
Úprava dávky |
|
Zvýšení hladin AST a/nebo ALT na > 2,5x ULN současně se zvýšením celkového bilirubinu na > 1,5x ULN NEBO Zvýšení hladin AST a/nebo ALT na > 5x ULN |
Po přerušení léčby a srovnání hodnot transamináz na < 2,5x ULN současně s normálním bilirubinem, snížení dávky z 200 mg dvakrát denně na 150 mg dvakrát denně a - jestliže je 2. snížení dávky považováno za nutné - ze 150 mg dvakrát denně na 100 mg dvakrát denně. |
|
Zvýšení hladin AST a/nebo ALT na > 3x ULN současně s nárůstem celkového bilirubinu na > 2x ULN a ALKP < 2x ULN |
Pokud není zjištěna jiná příčina, je třeba léčbu přípravkem Vargatef trvale ukončit. |
Zvláštní _ populace
Pediatrická populace
Bezpečnost a účinnost přípravku Vargatef u dětí ve věku 0-18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Starší pacienti (> 65 let)
U starších pacientů nebyly zjištěny rozdíly v bezpečnosti a účinnosti.
V pivotní studii 1199.13 bylo 85 pacientů (12,9 % pacientů s histologicky prokázaným adenokarcinomem) ve věku > 70 let (medián věku: 72 let, rozpětí: 70-80 let) (viz bod 5.1).
Na základě věku pacienta není třeba upravovat úvodní dávku (viz bod 5.2).
Rasa a tělesná hmotnost
Na základě populačních analýz farmakokinetiky (PK) není třeba a priori upravovat dávku přípravku Vargatef (viz bod 5.2). U pacientů černé a afroamerické rasy jsou k dispozici jen omezené údaje o bezpečnosti.
Porucha funkce ledvin
Ledvinami je vyloučeno méně než 1 % jednorázové dávky nintedanibu (viz bod 5.2). U pacientů s mírnou nebo středně těžkou poruchou funkce ledvin není třeba upravovat úvodní dávku. Bezpečnost, účinnost a farmakokinetika nintedanibu nebyla u pacientů s těžkou poruchou funkce ledvin (clearance kreatininu < 30 ml/min) studována.
Porucha funkce jater
Nintedanib je vylučován převážně žlučí a stolicí (> 90 %). U pacientů s poruchou funkce jater se expozice zvýšila (Child Pugh A, Child Pugh B, viz bod 5.2). Na základě klinických údajů není u pacientů s lehkou poruchou funkce jater třeba upravovat úvodní dávku (Child Pugh A; viz bod 4.4). Bezpečnost a účinnost nintedanibu nebyla studována u pacientů s poruchou funkce jater klasifikovanou jako Child Pugh B a C. Léčba pacientů se středně těžkou (Child Pugh B) nebo těžkou (Child Pugh C) poruchou funkce jater přípravkem Vargatef se nedoporučuje (viz bod 5.2).
Způsob podání
Tobolky přípravku Vargatef musí být užívány perorálně, pokud možno s jídlem, spolknuty celé s vodou, nesmějí se žvýkat ani drtit.
4.3 Kontraindikace
Hypersenzitivita na nintedanib, arašídy nebo sóju nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Gastrointestinální poruchy
Průjem byl nejčastěji hlášeným gastrointestinálním nežádoucím účinkem, přičemž se objevoval v úzké časové souvislosti s podáním docetaxelu (viz bod 4.8). V klinické studii LUME-Lung 1 (viz bod 5.1) měla většina pacientů mírný až středně závažný průjem. Průjem je třeba při prvních příznacích léčit adekvátní hydratací a léčivými přípravky proti průjmu, například loperamidem, přičemž může být nutné přerušit léčbu, snížit dávku nebo ukončit léčbu přípravkem Vargatef (viz bod 4.2).
Nauzea a zvracení, většinou mírné nebo středně závažné intenzity, byly často hlášenými gastrointestinálními nežádoucími účinky (viz bod 4.8). Přestože je poskytnuta odpovídající podpůrná péče, může být nutné přerušit léčbu, snížit dávku nebo ukončit léčbu přípravkem Vargatef (viz bod 4.2). Jako podpůrnou péči při výskytu nauzey a zvracení lze použít léčivé přípravky s antiemetickými vlastnostmi, např. glukokortikoidy, antihistaminika nebo antagonisty 5-HT3 receptoru, a adekvátní hydrataci.
V případě dehydratace je třeba podávat elektrolyty a tekutiny. V případě výskytu příslušných gastrointestinálních nežádoucích příhod je třeba sledovat plazmatické hladiny elektrolytů.
Neutropenie a sepse
V porovnání s monoterapií docetaxelem byla u pacientů léčených přípravkem Vargatef v kombinaci s docetaxelem pozorována vyšší frekvence neutropenie stupně > 3 dle CTCAE.
Byly pozorovány následné komplikace, jako je sepse či febrilní neutropenie.
Během léčby, obzvláště během kombinované léčby s docetaxelem, je třeba sledovat krevní obrazy. Celkový krevní obraz pacientů, kterým je podávána léčba nintedanibem v kombinaci s docetaxelem, je třeba často sledovat na začátku každého léčebného cyklu a kolem nejnižších hladin, a dle klinické indikace po podání posledního cyklu kombinační terapie.
Funkce jater
Bezpečnost a účinnost nintedanibu nebyla studována u pacientů se středně těžkou (Child Pugh B) nebo těžkou (Child Pugh C) poruchou funkce jater. Léčba přípravkem Vargatef se proto u těchto pacientů nedoporučuje (viz bod 4.2). Vzhledem ke zvýšené expozici se u pacientů s lehkou poruchou funkce jater může zvýšit riziko nežádoucích účinků (Child Pugh A, viz body 4.2 a 5.2).
Podávání nintedanibu bylo spojeno se zvýšením hladin jaterních enzymů (ALT, AST, ALKP, gama-glutamyltransferáza) či bilirubinu, s potenciálně vyšším rizikem u pacientek. Ve většině případů byla tato zvýšení reverzibilní.
Před zahájením kombinační léčby přípravkem Vargatef s docetaxelem je třeba vyšetřit hladiny transamináz, ALKP a bilirubinu. Hodnoty je třeba sledovat na základě klinické indikace nebo periodicky v průběhu léčby, tedy na začátku každého léčebného cyklu v kombinační fázi s docetaxelem, a měsíčně, je-li po ukončení docetaxelu přípravek Vargatef nadále podáván jako monoterapie.
Jsou-li naměřena relevantní zvýšení hladin jaterních enzymů, může být nutné přerušit léčbu, snížit dávku nebo ukončit léčbu přípravkem Vargatef (viz bod 4.2). Je třeba vyšetřit jiné možné příčiny zvýšení hladin jaterních enzymů a podle potřeby provést příslušná opatření. V případě specifických změn hodnot jaterních testů (AST/ALT > 3x ULN; celkový bilirubin > 2x ULN a ALKP < 2x ULN) je léčbu přípravkem Vargatef třeba přerušit. Pokud není zjištěna jiná příčina, je třeba léčbu přípravkem Vargatef trvale ukončit (viz bod 4.2).
Krvácení
Inhibice VEGFR může být spojena se zvýšeným rizikem krvácení. V klinické studii (LUME-Lung 1; viz bod 5.1) s přípravkem Vargatef byla frekvence krvácení v obou léčebných ramenech srovnatelná (viz bod 4.8).
Mírná až středně závažná epistaxe představovala nejčastější krvácivou příhodu. Většina fatálních krvácivých příhod byla spojena s nádorem. Nevyskytly se žádné rozdíly v krvácení do dýchacích cest či fatálním krvácení a nebylo nahlášeno žádné intracerebrální krvácení.
Pacienti s nedávným plicním krvácením (> 2,5 ml červené krve), stejně jako pacienti s centrálně lokalizovanými nádory s radiologicky potvrzenou lokální invazí do velkých krevních cév nebo radiologicky prokázanými kavitujícími nebo nekrotickými nádory, byli z klinických studií vyřazeni. Proto se nedoporučuje tyto pacienty léčit přípravkem Vargatef.
Terapeutická antikoagulace
K dispozici nejsou žádné údaje pro pacienty s dědičnou predispozicí ke krvácení ani pacienty, kterým byly před zahájením léčby přípravkem Vargatef podávány plné dávky antikoagulační léčby.
U pacientů na chronické nízkodávkové terapii nízkomolekulárními hepariny nebo kyselinou acetylsalicylovou nebyla zjištěna zvýšená frekvence krvácení. Pacienti, u nichž došlo k rozvoji tromboembolických příhod během léčby a u kterých bylo nutné použít antikoagulační léčbu, mohli pokračovat v léčbě přípravkem Vargatef a neprokázala se u nich zvýšená frekvence krvácivých příhod. U pacientů, kteří souběžně užívají antikoagulační léčbu, například warfarin nebo fenprokumon, je třeba pravidelně sledovat případný výskyt změn protrombinového času, mezinárodního normalizovaného poměru (INR) a epizod klinického krvácení.
Mozkové metastázy Stabilní mozkové metastázy
U pacientů s adekvátně předléčenými mozkovými metastázami, které byly stabilní po dobu > 4 týdnů před zahájením léčby přípravkem Vargatef, nebyla zjištěna zvýšená frekvence cerebrálního krvácení.
U těchto pacientů je však třeba pečlivě sledovat případný výskyt příznaků cerebrálního krvácení.
Aktivní mozkové metastázy
Pacienti s aktivními mozkovými metastázami byli vyřazeni z klinických studií a není doporučeno je léčit přípravkem Vargatef.
Venózní tromboembolie
U pacientů léčených přípravkem Vargatef existuje zvýšené riziko venózní tromboembolie, včetně hluboké žilní trombózy. U pacientů je třeba pečlivě sledovat případný výskyt tromboembolických příhod. Léčbu přípravkem Vargatef je třeba ukončit u pacientů s život ohrožujícími venózními tromboembolickými reakcemi.
Arteriální tromboembolické příhody
Ve fázi 3 studie 1199.13 (LUME -Lung 1) byla frekvence arteriálních tromboembolických příhod v obou léčebných ramenech srovnatelná. Pacienti s nedávnou anamnézou infarktu myokardu nebo cévní mozkové příhody byli z této studie vyřazeni. U pacientů s idiopatickou plicní fibrózou (IPF) léčených monoterapií nintedanibem však byla pozorována zvýšená frekvence arteriálních tromboembolických příhod. Opatrnosti je zapotřebí při léčbě pacientů se zvýšeným kardiovaskulárním rizikem, včetně pacientů se známou koronární nemocí. U pacientů, u kterých dojde k rozvoji příznaků akutní ischemie myokardu, je třeba zvážit přerušení léčby.
Gastrointestinální perforace
V klinické studii byla frekvence gastrointestinálních perforací v léčebných ramenech srovnatelná. Na základě mechanismu účinku však pacienti léčení přípravkem Vargatef mohou mít zvýšené riziko gastrointestinálních perforací. Obzvláštní péče je zapotřebí při léčbě pacientů po předchozí operaci břicha nebo s nedávnou anamnézou perforace dutých orgánů. Léčbu přípravkem Vargatef je proto možné zahájit nejméně 4 týdny po velkém operačním výkonu. U pacientů, u kterých dojde ke gastrointestinální perforaci, je třeba léčbu přípravkem Vargatef trvale ukončit.
Komplikace při hojení ran
Na základě mechanismu účinku může nintedanib narušit hojení ran. Ve studii LUME-Lung 1 nebyla zjištěna zvýšená frekvence narušeného hojení ran. Nebyly provedeny žádné studie, které by zkoumaly specificky účinek nintedanibu na hojení ran. Léčbu přípravkem Vargatefje proto možné zahájit nebo -v případě perioperativního přerušení - obnovit pouze na základě klinického zvážení adekvátního hojení.
Vliv na QT interval
V programu klinického hodnocení nebylo u nintedanibu zjištěno žádné prodloužení QT intervalu (viz bod 5.1).
Vzhledem k tomu, že u několika jiných inhibitorů tyrosinkinázy je znám účinek na QT interval, při podávání nintedanibu pacientům, u kterých může dojít k prodloužení QTc intervalu, je třeba opatrnosti.
Alergická reakce
Je známo, že potraviny obsahující sóju způsobují u osob s alergií na sóju alergické reakce, včetně závažné anafylaxe. U pacientů se známou alergií na bílkoviny obsažené v arašídech existuje zvýšené riziko závažných reakcí na přípravky obsahující sóju.
Zvláštní populace
Expozice nintedanibu se zvyšovala lineárně s věkem pacientů, inverzně korelovala s hmotností a byla obecně vyšší u pacientů asijské rasy. To může vést ke zvýšenému riziku rozvoje vyšších hladin jaterních enzymů. Doporučuje se pečlivě sledovat pacienty s několika těmito rizikovými faktory.
Ve studii 1199.13 (LUME-Lung 1) byla u pacientů léčených nintedanibem a docetaxelem, jejichž tělesná hmotnost byla nižší než 50 kg, vyšší frekvence závažných nežádoucích účinků než u pacientů s hmotností > 50 kg; počet pacientů, jejichž tělesná hmotnost byla nižší než 50 kg, však byl nízký. Doporučuje se proto pečlivě sledovat pacienty s hmotností < 50 kg.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Interakční studie byly provedeny pouze u dospělých.
Glykoprotein P (P-gp)
Nintedanib je substrát P-gp (viz bod 5.2). Ve specifické lékové interakční studii došlo při společném podávání s potentním inhibitorem P-gp ketokonazolem ke zvýšení expozice nintedanibu 1,61krát vzhledem k AUC a 1,83krát vzhledem k Cmax. V lékové interakční studii s potentním induktorem P-gp rifampicinem došlo při společném podávání s rifampicinem v porovnání s podáváním samotného nintedanibu k poklesu expozice nintedanibu na 50,3 % vzhledem k AUC a na 60,3 % vzhledem k Cmax. Při společném podávání s nintedanibem mohou silné inhibitory P-gp (např. ketokonazol nebo erythromycin) zvýšit expozici nintedanibu. V takových případech je třeba pečlivě sledovat, zda pacienti nintedanib snášejí. Léčba nežádoucích účinků může vyžadovat přerušení léčby, snížení dávky nebo ukončení léčby přípravkem Vargatef (viz bod 4.2).
Potentní induktory P-gp (např. rifampicin, karbamazepin, fenytoin a třezalka tečkovaná) mohou snižovat expozici nintedanibu. Společné podávání s nintedanibem je třeba pečlivě zvážit.
Enzymy cytochromu (CYP)
CYP dráhy byly součástí biotransformace nintedanibu pouze v malé míře. V předklinických studiích nintedanib a jeho metabolity, volná kyselá frakce BIBF 1202 a její glukuronid BIBF 1202 glukuronid, neinhibovaly ani neindukovaly enzymy CYP (viz bod 5.2). Pravděpodobnost lékových interakcí s nintedanibem na základě CYP metabolismu je proto považována za nízkou.
Společné podávání s jinými léčivými přípravky
Společné podávání nintedanibu s docetaxelem (75 mg/m2) nezměnilo v relevantní míře farmakokinetiku ani jednoho léčivého přípravku.
Potenciál pro interakce nintedanibu s hormonálními antikoncepčními přípravky nebyl zjišťován.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku / Antikoncepce
Nintedanib může poškozovat lidský plod (viz bod 5.3). Ženy ve fertilním věku léčené přípravkem Vargatefje třeba poučit, aby se v době, kdy jsou léčeny tímto přípravkem, vyhnuly otěhotnění a aby během léčby a ještě nejméně 3 měsíce po poslední dávce přípravku Vargatef užívaly účinnou antikoncepci. Vzhledem k tomu, že nebyl zkoumán účinek nintedanibu na metabolismus a účinnost antikoncepčních přípravků, je třeba jako další formu antikoncepce používat bariérové metody, aby nedošlo k otěhotnění.
Údaje o podávání přípravku Vargatef těhotným ženám nejsou k dispozici, ale předklinické studie na zvířatech prokázaly reprodukční toxicitu této léčivé látky (viz bod 5.3). Vzhledem k tomu, že nintedanib může poškozovat také lidský plod, lze jej v těhotenství použít pouze tehdy, když klinický stav vyžaduje léčbu. Těhotenský test je třeba provést minimálně před zahájením léčby přípravkem Vargatef.
Pacientky je třeba poučit, aby v případě, že během léčby přípravkem Vargatef otěhotní, informovaly svého lékaře nebo lékárníka.
Jestliže pacientka v průběhu léčby přípravkem Vargatef otěhotní, musí být informována o potenciálním nebezpečí pro plod. Je třeba zvážit ukončení léčby přípravkem Vargatef.
Kojení
Informace o vylučování nintedanibu a jeho metabolitů do lidského mateřského mléka nejsou k dispozici.
Předklinické studie prokázaly, že se do mléka kojících samic potkanů vylučují malá množství nintedanibu a jeho metabolitů (< 0,5 % podané dávky). Riziko pro novorozence/kojence nelze vyloučit. Kojení má být během léčby přípravkem Vargatef přerušeno.
Fertilita
Předklinické zkoumání nepotvrdilo negativní vliv na mužskou fertilitu (viz bod 5.3). Údaje o potenciálních účincích nintedanibu na ženskou fertilitu ze studií s lidmi ani zvířaty nejsou k dispozici.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Vargatef má malý vliv na schopnost řídit a obsluhovat stroje. Pacienty je třeba poučit, aby byli v průběhu léčby přípravkem Vargatef při řízení a obsluze strojů opatrní.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Údaje o bezpečnosti poskytnuté v bodech níže vycházejí z globální, dvojitě zaslepené randomizované pivotní fáze 3 studie 1199.13 (LUME-Lung 1), která porovnávala léčbu nintedanibem a docetaxelem oproti placebu a docetaxelu u pacientů s lokálně pokročilým nebo metastazujícím nebo rekurentním NSCLC po léčbě první linie. Nejčastěji hlášenými nežádoucími účinky léku specifickými pro nintedanib byly průjem, zvýšené hodnoty jaterních enzymů (ALT s AST) a zvracení. Tabulka 3 poskytuje souhrn nežádoucích účinků dle třídy orgánových systémů. Informace k léčbě vybraných nežádoucích účinků jsou uvedeny v bodě 4.4. Informace o vybraných nežádoucích účincích pozorovaných ve studii LUME-Lung 1 jsou popsány níže.
Tabulkový přehled nežádoucích účinků
Tabulka 3 shrnuje frekvence nežádoucích účinků léku, které byly nahlášeny v pivotní studii LUME-Lung 1 u pacientů s NSCLC s histologicky potvrzeným adenokarcinomem (n = 320).
K rozdělení nežádoucích účinků dle frekvence jsou použity následující termíny: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000), velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit).
V rámci každé frekvence jsou nežádoucí účinky uvedeny v pořadí dle snižující se závažnosti.
Tabulka 3: Souhrn nežádoucích účinků dle frekvenční kategorie
|
Třída orgánových systémů |
Velmi časté (> 1/10) |
Časté (> 1/100 < 1/10) |
Méně časté (> 1/1000 < 1/100) |
|
Infekce a infestace |
Febrilní neutropenie, abscesy, sepse | ||
|
Poruchy krve a lymfatického systému |
Neutropenie (včetně febrilní neutropenie) | ||
|
Poruchy metabolismu a výživy |
Snížená chuť k jídlu, elektrolytová nerovnováha |
Dehydratace | |
|
Poruchy nervového systému |
Periferní neuropatie | ||
|
Cévní poruchy |
Krvácení1'1 |
Venózní tromboembolie, hypertenze | |
|
Gastrointestinální poruchy |
Perforace1' | ||
|
Poruchy jater a žlučových cest |
Zvýšení alaninaminotransferázy, zvýšení aspartátaminotransferázy , zvýšení alkalické fosfatázy v krvi |
Hyperbilirubinemie, zvýšení gama-glutamyltransferázy | |
|
Poruchy kůže a podkožní tkáně |
Mukozitida (včetně stomatitidy), vyrážka |
X) Frekvence nebyla vyšší u pacientů léčených nintedanibem a docetaxelem
v porovnání s placebem a docetaxelem.
Popis vybraných nežádoucích účinků
Průjem se vyskytl u 43,4 % (> stupeň 3: 6,3 %) pacientů s adenokarcinomem v rameni s nintedanibem. Většina nežádoucích účinků se objevila v úzké časové souvislosti s podáním docetaxelu. U většiny pacientů průjem odezněl po přerušení léčby, nasazení léčby proti průjmu a snížení dávky nintedanibu.
Doporučená opatření a úpravy dávky v případě výskytu průjmu jsou uvedeny v bodě 4.4, respektive 4.2.
Zvýšení hladin jaterních enzymů a hyperbilirubinemie
Nežádoucí účinky související s játry se objevily u 42,8 % pacientů léčených nintedanibem. Přibližně jedna třetina těchto pacientů měla s játry spojené nežádoucí účinky stupně závažnosti > 3. U pacientů se zvýšenými jaterními testy bylo vhodným opatřením ověřené postupné snižování dávky a ukončení léčby bylo nutné pouze u 2,2 % pacientů. U většiny pacientů bylo zvýšení jaterních testů reverzibilní.
Informace ke zvláštním populacím, doporučeným opatřením a úpravám dávek v případě zvýšených hladin jaterních enzymů a bilirubinu jsou uvedeny v bodech 4.4, respektive 4.2.
Neutropenie, febrilní neutropenie a sepse
Sepse a febrilní neutropenie byly hlášeny jako následné komplikace neutropenie. Frekvence výskytu sepse (1,3 %) a febrilní neutropenie (7,5 %) byly při léčbě nintedanibem vyšší než v rameni s placebem. Je důležité, aby během léčby, obzvláště během kombinované léčby s docetaxelem, byl u pacientů sledován krevní obraz (viz bod 4.4).
Krvácení
Ačkoli je vzhledem k mechanismu účinku nintedanibu krvácení jeho očekávaným nežádoucím účinkem, výskyt krvácení byl v obou léčebných skupinách pacientů s adenokarcinomem srovnatelný (placebo: 11,1 %, nintedanib: 10,9%).
Perforace
Na základě mechanismu účinku lze u pacientů léčených nintedanibem očekávat výskyt perforací.
Podíl pacientů s gastrointestinální perforací však byl nízký.
Periferní neuropatie
Periferní neuropatie se objevuje také při léčbě docetaxelem. Periferní neuropatie byla hlášena u 16,5 % pacientů ve skupině s placebem a u 19,1 % pacientů v rameni s nintedanibem.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Pro předávkování nintedanibem neexistuje specifické antidotum ani léčba. Nejvyšší jednorázová dávka nintedanibu podaná ve studiích fáze 1 bylo 450 mg jednou denně. U 2 pacientů navíc došlo k předávkování maximální dávkou 600 mg dvakrát denně (b.i.d.) po dobu až osmi dnů. Zjištěné nežádoucí příhody byly v souladu se známým bezpečnostním profilem nintedanibu, tedy zvýšené hladiny jaterních enzymů a gastrointestinální příznaky. U obou pacientů tyto nežádoucí účinky odezněly. V případě předávkování je třeba přerušit léčbu a zahájit odpovídající obecná podpůrná opatření.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, inhibitory proteinkináz.
ATC kód: L01XE31.
Mechanismus účinku
Nintedanib je trojitý inhibitor angiokináz, který blokuje aktivitu kináz receptorů vaskulárního endoteliálního růstového faktoru (VEGFR 1-3), destičkového růstového faktoru (PDGFR a a B) a fibroblastového růstového faktoru (FGFR 1-3). Nintedanib se kompetitivně váže na vazebnou kapsu těchto receptorů pro adenosintrifosfát (ATP) a blokuje intracelulární signalizaci, která je zásadní pro proliferaci a přežití endoteliálních stejně jako perivaskulárních buněk (pericyty a buňky cévní hladké svaloviny). Dochází navíc k inhbici proteinové tyrosinkinázy podobné Fms (Flt)-3, proteinové tyrosinkinázy specifické pro lymfocyty (Lck) a proto-onkogenní proteinové tyrosinkinázy Src (Src).
Farmakodvnamické účinky
Nádorová angiogeneze je klíčovým rysem, který přispívá k růstu nádorů, jejich progresi a tvorbě metastáz, přičemž k jejímu spuštění dochází převážně uvolněním proangiogenních faktorů vylučovaných nádorovou buňkou (např. VEGF a bFGF), jejichž úkolem je zajistit, aby endoteliální a perivaskulární buňky hostitele podporovaly dodávání kyslíku a živin cévním systémem hostitele.
V předklinických modelech onemocnění nintedanib, použitý samostatně, účinně narušil tvorbu a udržování cévního systému nádoru, což vedlo k inhibici růstu a následně zastavení růstu nádoru. Obzvláště léčba xenograftů nádorů nintedanibem vedla k rychlému poklesu hustoty mikrocév v nádoru, poklesu pokrytí cév pericyty a poklesu perfuze nádoru.
Měření pomocí dynamické kontrastní magnetické rezonance (DCE-MRI) prokázala antiangiogenní účinek nintedanibu u lidí. Nebyl zřetelně závislý na dávce, avšak většina odpovědí byla zjištěna při dávce > 200 mg. Logistická regrese odhalila statisticky významnou souvislost mezi antiangiogenním účinkem a expozicí nintedanibu. Účinky na DCE-MRI byly zřetelné 24-48 hodin po užití první dávky léčivého přípravku a při kontinuální léčbě po dobu několika týdnů přetrvávaly nebo došlo i k jejich nárůstu. Nebyla zjištěna korelace mezi DCE-MRI odpovědí a následným klinicky významným snížením velikosti cílové leze, avšak DCE-MRI odpověď byla spojená se stabilizací onemocnění.
Klinická účinnost a bezpečnost
Účinnost v pivotní studii fáze 3 LUME-Lung 1
Účinnost a bezpečnost přípravku Vargatef byla zjišťována u 1314 dospělých pacientů s lokálně pokročilým, metastatickým nebo rekurentním NSCLC po jedné předchozí linii chemoterapie.
„Lokálně rekurentní“ bylo definováno jako lokální rekurence nádoru bez metastáz při vstupu do studie. Do studie bylo zařazeno 658 pacientů (50,1 %) s adenokarcinomem, 555 pacientů (42,2 %) s dlaždicobuněčným karcinomem a 101 pacientů (7,7 %) s nádory s jinou histologií.
Pacienti byli randomizováni (1:1) k podávání nintedanibu 200 mg perorálně dvakrát denně v kombinaci se 75 mg/m2 docetaxelu intravenózně každých 21 dnů (n = 655) nebo placeba perorálně dvakrát denně v kombinaci se 75 mg/m2 docetaxelu každých 21 dnů (n = 659). Randomizace byla stratifikována dle výkonnostního indexu Eastern Cooperative Oncology Group (ECOG) (0 oproti 1), předléčby bevacizumabem (ano oproti ne), mozkové metastázy (ano oproti ne) a histologie nádoru (dlaždicová nebo jiná histologie).
Charakteristiky pacientů byly mezi oběma léčebnými rameny vyvážené jak v celé populaci, tak v podskupinách dle histologie. 72,7 % pacientů v celé populaci byli muži. Většina pacientů byla jiné než asijské rasy (81,6 %), medián věku byl 60,0 let, výchozí výkonnostní index ECOG byl 0 (28,6 %) nebo 1 (71,3 %); jeden pacient měl výchozí výkonnostní index ECOG 2. 5,8 % pacientů mělo při vstupu do studie stabilní mozkové metastázy a 3,8 % bylo po předchozí léčbě bevacizumabem. Stadium onemocnění pacientů zařazených do studie LUME-Lung 1 bylo určeno v době diagnózy za použití 6. nebo 7. vydání Union Internationale Contre le Cancer (UICC) / American Joint Committee on Cancer (AJCC). V celkové populaci bylo 16,0 % pacientů ve stadiu onemocnění < IIIB/IV, 22,4 % pacientů ve stadiu onemocnění IIIB a 61,6 % pacientů ve stadiu onemocnění IV. 9,2 % pacientů vstoupilo dle výchozího hodnocení do studie s lokálně rekurentním stadiem onemocnění. V celkové populaci bylo 15,8 % pacientů ve stadiu onemocnění < IIIB/IV, 15,2 % pacientů ve stadiu onemocnění IIIB a 69,0 % pacientů ve stadiu onemocnění IV.
5,8 % pacientů s adenokarcinomem vstoupilo dle výchozího hodnocení do studie s lokálně rekurentním stadiem onemocnění.
Primárním cílovým parametrem bylo přežití bez progrese onemocnění (progression-free survival,
PFS) hodnocené nezávislou hodnotící komisí (independent review committee, IRC) u intent-to-treat (ITT) populace a testované histologicky. Celkové přežití (overall survival, OS) bylo hlavním sekundárním cílovým parametrem. Dalšími účinnostními cíli byla objektivní odpověď, kontrola onemocnění, změna velikosti nádoru a se zdravím spojená kvalita života.
Přidání nintedanibu k docetaxelu vedlo v celkové populaci ke statisticky významnému snížení rizika progrese nebo úmrtí stanoveného nezávislou posuzovací komisí o 21 % (poměr rizik (HR) 0,79; 95 % interval spolehlivosti (CI): 0,68-0,92; p = 0,0019). Tento výsledek byl potvrzen v následné analýze
PFS (HR 0,85, 95 % CI: 0,75-0,96; p = 0,0070), ve které byly použity všechny příhody zaznamenané v době závěrečné analýzy OS. Analýza celkového přežití v celkové populaci nedosáhla statistické významnosti (HR 0,94; 95% CI: 0,83-1,05).
Je třeba vzít v úvahu, že předem naplánované analýzy podle histologie prokázaly statisticky významný rozdíl v celkovém přežití mezi léčebnými rameny pouze u populace s adenokarcinomem (Tabulka 4).
Jak je uvedeno v tabulce 4, přidání nintedanibu k docetaxelu vedlo v celkové populaci ke statisticky významnému snížení rizika progrese nebo úmrtí, a to o 23 % u populace s adenokarcinomem (HR 0,77; 95% CI: 0,62-0,96). U souvisejících cílů studie, jako je kontrola onemocnění a změna velikosti nádoru, došlo v souladu s tímto pozorováním k významnému zlepšení.
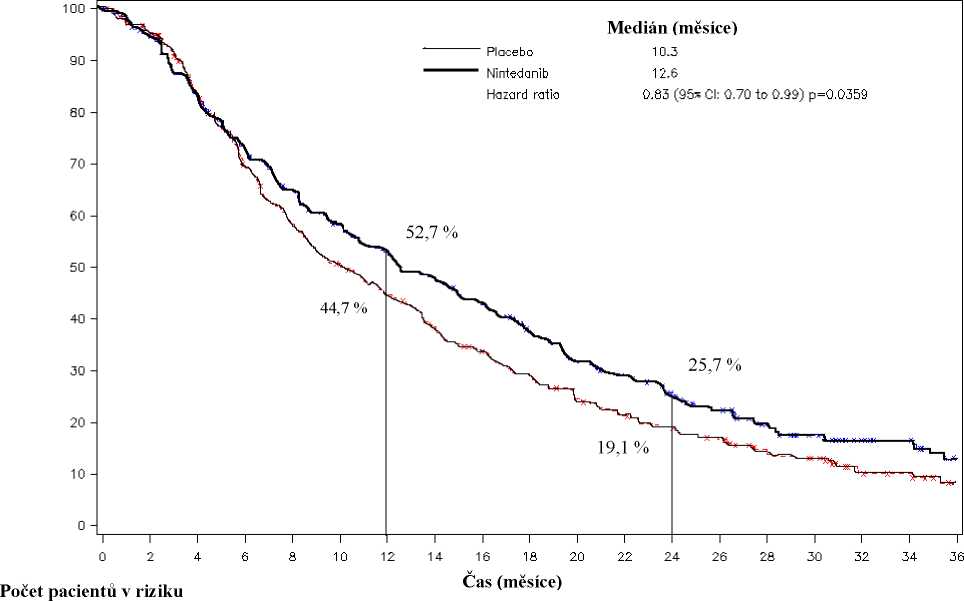
Tabulka 4: Výsledky účinnosti studie LUME-Lung 1 pro pacienty s histologicky potvrzeným adenokarcinomem
|
Vargatef + Docetaxel |
Placebo + Docetaxel | |
|
Přežití bez progrese onemocnění (PFS)* - primární analýza | ||
|
Počet pacientů |
277 |
285 |
|
Počet úmrtí nebo progresí (%) |
152 (54,9) |
180 (63,2) |
|
Medián PFS [měsíce] |
4,0 |
2,8 |
|
HR (95% CI) |
0,77 (0,62; 0,96) | |
|
Hodnota p pro stratifikovaný log-rank test** |
0,0193 | |
|
Přežití bez progrese onemocnění (PFS)*** - následná analýza | ||
|
Počet pacientů |
322 |
336 |
|
Počet úmrtí nebo progresí (%) |
255 (79,2) |
267 (79,5) |
|
Medián PFS [měsíce] |
4,2 |
2,8 |
|
HR (95% CI) |
0,84 (0,71; 1,00) | |
|
Hodnota p pro stratifikovaný log-rank test** |
0,0485 | |
|
Kontrola onemocnění [%] |
60,2 44,0 | |
|
Odds ratio (95% CI)+ |
1,93 (1,42; 2,64) | |
|
hodnota p+ |
<0,0001 | |
|
Objektivní odpověď [%] |
4,7 |
3,6 |
|
Odds ratio (95% CI)+ |
1,32 (0,61; 2,93) | |
|
hodnota p+ |
0,4770 | |
|
Zmenšení nádoru [%]° |
-7,76 |
-0,97 |
|
hodnota p° |
0,0002 | |
|
Celkové přežití (OS)*** | ||
|
Počet pacientů |
322 |
336 |
|
Počet úmrtí (%) |
259 (80,4) |
276 (82,1) |
|
Medián OS [měsíce] |
12,6 |
10,3 |
|
HR (95% CI) |
0,83 (0,70; 0,99) | |
|
Hodnota p pro stratifikovaný log-rank test* |
0,0359 | |
HR: poměr rizik; CI: interval spolehlivosti
Primární analýza PFS na základě 713 zjištěných PFS příhod dle hodnocení IRC v celkové ITT populaci (332 příhod u pacientů s adenokarcinomem).
Stratifikováno podle výchozího indexu ECOG PS (0 oproti 1), mozkových metastáz ve výchozím období (ano oproti ne) a předchozí léčby bevacizumabem (ano oproti ne).
Analýza OS a následná analýza PFS provedená poté, co se v celkové ITT populaci vyskytlo 1121 případů úmrtí (535 příhod u pacientů s adenokarcinomem).
+ Odds ratio a hodnota p byly získány z logistického regresního modelu upraveného dle výchozího indexu ECOG (0 oproti 1).
° Upravený průměr nejlepší % změny od výchozího stavu a hodnota p vycházející z ANOVA modelu upraveného dle výchozího indexu ECOG PS (0 oproti 1), mozkových metastáz ve výchozím období (ano oproti ne) a předchozí léčby bevacizumabem (ano oproti ne).
U pacientů s adenokarcinomem bylo prokázáno statisticky významné zlepšení OS ve prospěch léčby nintedanibem s docetaxelem se 17% snížením rizika úmrtí (HR 0,83, p = 0,0359) a mediánem zlepšení OS 2,3 měsíce (10,3 oproti 12,6 měsíce, Obrázek 1).
Obrázek 1: Kaplan-Meierova křivka celkového přežití u pacientů s histologicky potvrzeným adenokarcinomem dle léčebné skupiny ve studii LUME-Lung 1
Pravděpodobnost přežití (%)
Pllc.l-'- ■ ■■ ■
Nintedamb 322 302 263 230 203 ISO 163 149 131 113 96 B7 72 59 46 36 25 22 10
Předem definované hodnocení bylo provedeno u populace pacientů s adenokarcinomem, kteří vstoupili do studie s obzvláště špatnou prognózou léčby, konkrétně pacientů, u kterých došlo v průběhu nebo krátce po léčbě 1. linie, podané před vstupem do studie, k progresi. Do této populace patřili pacienti s adenokarcinomem, u kterých byla ve výchozím období identifikována progrese, a kteří vstoupili do studie méně než 9 měsíců od zahájení léčby první linie. Léčba těchto pacientů nintedanibem v kombinaci s docetaxelem snížila riziko úmrtí o 25 % v porovnání s placebem s docetaxelem (HR 0,75; 95% CI: 0,60-0,92; p = 0,0073). Medián OS se zlepšil o 3 měsíce (nintedanib:
10,9 měsíců; placebo: 7,9 měsíců). V post-hoc analýze pacientů s adenokarcinomem, kteří progredovali a vstoupili do studie > 9 měsíců od zahájení první linie léčby, nedosáhl rozdíl statistické významnosti (HR pro OS: 0,89, 95% CI: 0,66-1,19).
Podíl pacientů s adenokarcinomem ve stadiu < IIIB/IV při diagnóze byl nízký a mezi léčebnými skupinami vyvážený (placebo: 54 pacientů (16, %); nintedanib: 50 pacientů, (15,5 %)). HR pro PFS a OS byl u těchto pacientů 1,24 (95% CI: 0,68, 2,28), respektive 1,09 (95% CI: 0,70, 1,70). Soubor byl nicméně malý, nebyla přítomna žádná významná interakce a CI byl široký a zahrnoval HR pro OS celkové populace s adenokarcinomem.
Kvalita života
Léčba nintedanibem významně nezměnila dobu do zhoršení předdefinovaných příznaků kašle, dušnosti a bolesti, ale vedla k významnému zhoršení na škále příznaků průjmu. Celkový přínos léčby nintedanibem však neměl negativní vliv na kvalitu života hodnocenou pacienty.
Vliv na QT interval
QT/QTc byl měřen a analyzován ve specifické studii srovnávající monoterapii nintedanibem s monoterapií sunitinibem u pacientů s karcinomem ledvinové buňky. V této studii neprodlužovaly jednotlivé perorální dávky 200 mg nintedanibu ani opakované perorální dávky 200 mg nintedanibu podávané dvakrát denně po dobu 15 dnů QTcF interval. Nebyla však provedena žádná důkladná studie QT intervalu s nintedanibem podávaným v kombinaci s docetaxelem.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Vargatef u všech podskupin pediatrické populace u nemalobuněčného karcinomu plic (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Nintedanib dosáhl maximálních plazmatických koncentrací přibližně 2-4 hodiny po perorálním podání měkkých želatinových tobolek ve stavu sytosti (rozpětí 0,5-8 hodin). Absolutní biologická dostupnost 100 mg dávky byla u zdravých dobrovolníků 4,69 % (90 % CI: 3,615-6,078). Absorpce a biologická dostupnost jsou sníženy účinky transportérů a významným metabolismem při prvním průchodu játry. Proporcionalita dávky byla prokázána zvýšením expozice nintedanibu (rozpětí dávek 50-450 mg jednou denně a 150-300 mg dvakrát denně. Rovnovážného stavu plazmatických koncentrací bylo dosaženo nejdéle do týdne od podání dávky.
Po požití stravy se expozice nintedanibu zvýšila o přibližně 20 % v porovnání s podáním nalačno (CI: 95,3-152,5%) a absorpce byla zpožděna (medián tmax nalačno: 2,00 hod.; po jídle: 3,98 hod).
Distribuce
Nintedanib má nejméně dvoufázovou kinetiku. Po intravenózní infuzi byl zjištěn vysoký distribuční objem (Vss: 1050 l, 45,0 % gCV).
Vazba nintedanibu na bílkoviny lidské krevní plazmy in vitro byla vysoká, vázaná frakce činila 97,8 %. Za hlavní vazebnou bílkovinu je považován sérový albumin. Nintedanib je distribuován především do plazmy, přičemž poměr krev ku plazmě je 0,869.
Biotransformace
Převažující metabolickou reakcí u nintedanibu je hydrolytické štěpení esterázami, které vede k tvorbě volné kyselé frakce BIBF 1202. BIBF 1202 je následně glukuronidována UGT enzymy, především UGT 1A1, UGT 1A7, UGT 1A8 a UGT 1A10 na BIBF 1202 glukuronid.
CYP dráhy byly součástí biotransformace nintedanibu pouze v malé míře, přičemž hlavním enzymem byl CYP 3A4. V lidské studii absorpce, distribuce, metabolismu a eliminace (ADME) nebylo možné hlavní metabolit CYP dráhy v plazmě zjistit. In vitro představoval metabolismus závislý na CYP přibližně 5 % v porovnání s 25 % esterického štěpení.
Přestože je BIBF 1202 aktivní na cílových receptorech původní látky, v předklinických experimentech in vivo nebyla jeho účinnost prokázána.
Eliminace
Celková plazmatická clearance po intravenózní infůzi byla vysoká (CL: 1390 ^nl/^nin, 28,8 % gCV).
V podobě nezměněné léčivé látky bylo během 48 hodin močí vyloučeno přibližně 0,05 % perorálně podané dávky (31,5 % gCV) a přibližně 1,4 % intravenózně podané dávky (24,2 % gCV); renální clearance byla 20 ml/min (32,6 % gCV). Hlavní cestou eliminace lékové radioaktivity po perorálním podání [14C] nintedanibu bylo vylučování stolicí a žlučí (93,4 % dávky, 2,61 % gCV).
Renální vylučování přispívalo k celkové clearance jen v malé míře (0,649 % dávky, 26,3 % gCV). Celkové množství zjištěné látky (recovery) bylo považováno za úplné (více než 90 %) do 4 dnů po podání dávky. Terminální poločas nintedanibu byl mezi 10 a 15 hod. (gCV % přibližně 50 %).
Linearita/nelinearita
Farmakokinetiku nintedanibu lze považovat za lineární vzhledem k času (tedy údaje pro podání jednotlivých dávek lze extrapolovat na opakované dávky). Při opakovaném podání docházelo k akumulaci 1,04krát u Cmax a 1,38krát u AUC. Minimální koncentrace nintedanibu zůstaly stabilní po dobu více než jednoho roku.
Další informace o lékových interakcích
Metabolismus
Vzhledem k tomu, že nintedanib, BIBF 1202 ani glukuronid BIBF 1202 v předklinických studiích neinhibovaly ani neindukovaly CYP enzymy a nintedanib nebyl v relevantní míře metabolizován CYP enzymy, nepředpokládá se, že budou existovat lékové interakce mezi nintedanibem a substráty CYP, CYP inhibitory a CYP induktory.
Transport
Nintedanib je substrát P-gp. Informace o interakčním potenciálu nintedanibu s tímto transportérem jsou uvedeny v bodě 4.5. Bylo prokázáno, že nintedanib není in vitro substrátem ani inhibitorem OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, ani MRP-2. Nintedanib rovněž nebyl substrátem BCRP. In vitro byl pozorován pouze slabý inhibiční potenciál na OCT-1, BCRP a P-gp, což je považováno za klinicky málo významné. Totéž platí pro nintedanib jako substrát OCT-1.
Farmakokinetické/farmakodynamické vztahy
V exploratorních analýzách farmakokinetiky nežádoucích příhod byla vyšší expozice nintedanibu spojena se zvýšenými hladinami jaterních enzymů, avšak ne s gastrointestinálními nežádoucími příhodami.
Analýzy vztahu mezi PK a účinností nebyly pro klinické cíle studie provedeny. Logistická regrese odhalila statisticky významnou souvislost mezi expozicí nintedanibu a odpovědí na DCE-MRI.
Populační analýza farmakokinetiky u zvláštních populací
Farmakokinetické vlastnosti nintedanibu byly podobné u zdravých dobrovolníků, onkologických pacientů a pacientů z cílové populace. Expozice nintedanibu nebyla ovlivněna pohlavím (korigováno dle tělesné hmotnosti), přítomností mírné či středně závažné poruchy funkce ledvin (odhadnuto na základě clearance kreatininu), přítomností jaterních metastáz, výkonnostním indexem ECOG, požíváním alkoholu ani genotypem P-gp.
Populační analýzy PK naznačily středně významný vliv na expozici nintedanibu v závislosti na věku, tělesné hmotnosti a rase (viz níže). Vzhledem k velkým interindividuálním rozdílům v expozici zjištěným v klinické studii LUME-Lung 1 nejsou tyto účinky považovány za klinicky významné. Doporučuje se však pečlivě sledovat pacienty s několika těmito rizikovými faktory (viz bod 4.4).
Věk
Expozice nintedanibu se zvyšovala lineárně s věkem. AUCT,ss se u 45letého pacienta snížila o 16 % (5. percentil) a zvýšila o 13 % u 76letého pacienta (95. percentil) v porovnání s pacientem s mediánem věku 62 let. Věkové rozpětí pacientů v analýze bylo 29 až 85 let; přibližně 5 % populace bylo starších 75 let.
Studie u pediatrických populací nebyly provedeny.
Tělesná hmotnost
Byl zjištěn inverzní vztah mezi tělesnou hmotností a expozicí nintedanibu. AUCT,ss se zvětšila o 25 % u 50kg pacienta (5. percentil) a snížila o 19 % u 100kg pacienta (95. percentil) v porovnání s pacientem s mediánem hmotnosti 71,5 kg.
Rasa
Oproti bělochům byla geometrická průměrná expozice nintedanibu u pacientů z Číny, Tchaj-wanu a Indie o 33 % vyšší a u pacientů z Koreje o 22 % nižší (korigováno dle tělesné hmotnosti). Vzhledem k velkým interindividuálním rozdílům v expozici nejsou tyto účinky považovány za klinicky významné. Údaje od osob černé rasy byly velmi omezené, ale ve stejném rozpětí, jako u bělochů.
Porucha funkce jater
V dedikované studii fáze I s jednorázovou dávkou a v porovnání se zdravými jedinci byla expozice nintedanibu dle Cmax a AUC 2,2krát vyšší u dobrovolníků s lehkou poruchou funkce jater (Child Pugh A; 90% CI 1,3 - 3,7 u Cmax a 1,2 - 3,8 u AUC). U dobrovolníků se středně těžkou poruchou funkce jater (Child Pugh B) byla v porovnání se zdravými dobrovolníky expozice 7,6krát vyšší dle Cmax (90% CI 4,4 - 13,2) a 8,7krát vyšší dle AUC (90% CI 5,7 - 13,1). Pacienti s těžkou poruchou funkce jater (Child Pugh C) nebyli studováni.
5.3 Předklinické údaje vztahující se k bezpečnosti
Obecná toxikologie
Studie toxicity po podání jednorázové dávky potkanům a myším naznačily nízký potenciál nintedanibu k akutní toxicitě. V toxikologických studiích s opakovaným podáváním u potkanů byly nežádoucí účinky (např. ztluštění epifyzárních štěrbin, léze na řezácích) spojené s mechanismem účinku nintedanibu (tedy inhibice VEGFR-2). Tyto změny jsou známy i u jiných VEGFR-2 inhibitorů a mohou být považovány za skupinový účinek.
Ve studiích toxicity s jinými živočichy než hlodavci byl pozorován průjem a zvracení spojené se snížením příjmu potravy a ztrátou tělesné hmotnosti.
U potkanů, psů a opic druhu Cynomolgus nebyl zjištěn nárůst hladin jaterních enzymů. Mírné zvýšení hladin jaterních enzymů, které nebylo spojeno se závažnými nežádoucími účinky, jako je průjem, bylo zjištěno pouze u opic druhu Rhesus.
Reprodukční toxicita
Studie samčí fertility a časného embryonálního vývoje do nidace u potkanů neprokázala účinky na samčí reproduktivní soustavu a samčí fertilitu.
U potkanů byly embryofetální úmrtnost a teratogenní účinky pozorovány při expozicích nižších než je expozice lidí při maximální doporučené dávce pro člověka (MRHD) 200 mg b.i.d. Při subterapeutických hladinách expozice byly rovněž zaznamenány účinky na vývoj axiálního skeletu a na vývoj velkých cév.
U králíků byly při expozici přibližně 8krát vyšší než při MRHD pozorovány účinky na embryofetální úmrtnost. Při expozici přibližně 4krát vyšší než při MRHD byly pozorovány teratogenní účinky na aortické oblouky v kombinaci s účinky na srdce a urogenitální ústrojí, a při expozici přibližně 3krát vyšší než při MRHD účinky na embryofetální vývoj axiálního skeletu.
U potkanů byly malé dávky radioaktivně značeného nintedanibu a/nebo jeho metabolitů vyloučeny do mléka (< 0,5 % podané dávky).
Studie genotoxicity nenaznačily žádný mutagenní potenciál nintedanibu.
6. FARMACEUTICKÉ ÚDAJE 6.1 Seznam pomocných látek
Obsah tobolky
střední nasycené triacylglyceroly tvrdý tuk
sójový lecithin (E322)
Obal tobolky
želatina
glycerol 85 %
oxid titaničitý (E171)
červený oxid železitý (E172)
žlutý oxid železitý (E172)
Potiskový inkoust šelak
černý oxid železitý (E172) propylenglykol (E1520)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení Al/Al blistry, každý obsahuje 10 tobolek.
Velikosti balení: 60 nebo 120 tobolek nebo vícenásobné balení se 120 (2 x 60) tobolkami (2 krabičky, každá se 60 tobolkami,zabalenými v plastové fólii). Velikost balení: 60 tobolek.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH
Binger Strasse 173
55216 Ingelheim am Rhein
Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/954/001
EU/1/14/954/002
EU/1/14/954/003
EU/1/14/954/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. listopadu 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Boehringer Ingelheim Pharma GmbH & Co. KG
Binger Strasse 173
55216 Ingelheim am Rhein
NĚMECKO
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky;
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Za účelem výzkumu vhodného bio-/nádorového markéru (včetně VEGF) k přesnější identifikaci a výběru cílové populace pacientů, u nichž by byla nejvyšší pravděpodobnost přínosnosti léčby nintedanibem, žadatel provede a předloží výsledky Výzkumného programu biomarkerů, včetně: 1. U krevních vzorků získaných ve studiích LUME-Lung 1 a LUME-Lung 2 |
Výsledky budou předkládány každoročně. |
|
bude zhodnocena genetická variabilita zárodečné linie s ohledem na angiogenní faktory, včetně VEGF a jeho receptorů. 2. Jednoramenná studie s cílem zjistit, zda lze genetické/genomické markery (samotné nebo v kombinaci s klinickými kovariáty) použít k predikci celkového přežití (OS) u NSCLC pacientů vhodných pro léčbu nintedanibem. 3. Údaje o bio-/nádorových markerech ze všech klinických studií klinického programu s nintedanibem. Žadatel zařadí sběr materiálu pro zkoumání biomarkerů a analýzy údajů o biomarkerech do studijních protokolů všech nových studií s nintedanibem v onkologii, které budou v budoucnu naplánovány, a ve kterých to bude klinicky vhodné. |
Předložení závěrečné zprávy z jednoramenné studie: Q3 2021 |
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA (100 MG)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Vargatef 100 mg měkké tobolky nintedanibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje nintedanibum 100 mg (ve formě nintedanibi esilas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sóju. Další informace jsou uvedeny v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
60 x 1 měkká tobolka 120 x 1 měkká tobolka
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/954/001
EU/1/14/954/002
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Vargatef 100 mg
KRABIČKA (100 MG - 60 TOBOLEK VE VÍCENÁSOBNÉM BALENÍ - BEZ „BLUE BOX“ INFORMACE)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Vargatef 100 mg měkké tobolky nintedanibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje nintedanibum 100 mg (ve formě nintedanibi esilas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sóju. Další informace jsou uvedeny v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
60 x 1 měkká tobolka. Část vícenásobného balení, nelze prodat samostatně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/954/003
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Vargatef 100 mg
VNĚJŠÍ OBAL (100 MG - VÍCENÁSOBNÉ BALENÍ 120 TOBOLEK - S „BLUE BOX“ INFORMACÍ)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Vargatef 100 mg měkké tobolky nintedanibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje nintedanibum 100 mg (ve formě nintedanibi esilas).
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Vícenásobné balení: 120 (2 balení po 60 x 1) měkkých tobolek.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/954/003
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Vargatef 100 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Vargatef 150 mg měkké tobolky nintedanibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje nintedanibum 150 mg (ve formě nintedanibi esilas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sóju. Další informace jsou uvedeny v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
60 x 1 měkká tobolka
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/954/004
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Vargatef 150 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR (100 MG)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Vargatef 100 mg tobolky Nintedanibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Před použitím neotvírejte.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Vargatef 150 mg tobolky Nintedanibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Před použitím neotvírejte.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Vargatef 100 mg měkké tobolky
Nintedanibum
▼ Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Vargatef a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Vargatef užívat
3. Jak se přípravek Vargatef užívá
4. Možné nežádoucí účinky
5. Jak přípravek Vargatef uchovávat
6. Obsah balení a další informace
1. Co je přípravek Vargatef a k čemu se používá
Tobolky přípravku Vargatef obsahují léčivou látku nintedanib. Nintedanib blokuje aktivitu skupiny bílkovin, které jsou zapojeny do vývoje nových krevních cév, které nádorové buňky potřebují jako zdroj výživy a kyslíku. Tím, že nintedanib zablokuje aktivitu těchto bílkovin, je schopen potlačit růst a šíření nádoru.
Tento přípravek se používá v kombinaci s jiným protinádorovým léčivým přípravkem (docetaxel) k léčbě rakoviny plic zvané nemalobuněčný karcinom plic (non-small cell lung cancer, NSCLC). Je určen pro dospělé pacienty se specifickým typem NSCLC (,,adenokarcinom“), kteří již byli léčeni jedním jiným léčivým přípravkem určeným k léčbě tohoto nádorového onemocnění, ale u nichž nádor začal znovu růst.
2. Čemu musíte věnovat pozornost, než začnete přípravek Vargatef užívat
Neužívejte přípravek Vargatef:
- jestliže jste alergický(á) na nintedanib, arašídy nebo sóju nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím tohoto přípravku se poraďte se svým lékařem nebo lékárníkem,
- jestliže jste někdy měl(a) problémy s játry; jestliže máte nebo jste měl(a) problémy s krvácením, obzvláště krvácení do plic v nedávné době
- jestliže užíváte léčivé přípravky k ředění krve (jako je warfarin, fenprokumon, heparin nebo kyselina acetylsalicylová) k prevenci tvorby krevních sraženin. Léčba přípravkem Vargatef může zvyšovat riziko krvácení
- jestliže jste v nedávné době podstoupil(a) nebo plánujete podstoupit operaci. Nintedanib může ovlivnit způsob, jak se Vám hojí rány. Léčba přípravkem Vargatef proto obvykle bude v případě, že máte být operován(a), přerušena. Lékař rozhodne o tom, kdy bude léčba tímto přípravkem obnovena
- jestliže máte rakovinu, která se rozšířila do mozku
Na základě těchto informací může lékař provést některé krevní testy, například zkontrolovat funkci Vašich jater a zjistit, jak rychle se Vaše krev sráží. Lékař s Vámi výsledky těchto testů prodiskutuje a rozhodne, zda Vám může být podán přípravek Vargatef.
Během užívání tohoto přípravku svého lékaře okamžitě informujte,
- jestliže dostanete průjem. Je důležité léčit průjem při prvních příznacích (viz bod 4)
- jestliže budete mít horečku, protože ta by mohla být příznakem febrilní neutropenie nebo sepse (viz bod 4)
- jestliže pociťujete silnou bolest v oblasti žaludku, máte horečku, zimnici, je Vám nevolno, zvracíte nebo máte napnuté břicho nebo se cítíte nafouklý(á), protože to by mohly být příznaky proděravění stěny trávicího traktu („gastrointestinální perforace“)
- jestliže pociťujete bolest v končetině, nebo ji máte oteklou, zarudlou či teplou, protože to by mohly být příznaky krevní sraženiny v jedné z Vašich žil
- jestliže máte velké krvácení
- jestliže pociťujete tlak nebo bolest na hrudi, typicky na levé straně těla, bolest v šíji, čelisti, rameni nebo paži, máte zrychlený srdeční tep, jste dušný(á), máte pocit na zvracení, zvracíte, protože to by mohly být příznaky srdečního infarktu
- jestliže se kterýkoli z nežádoucích účinků, které se u Vás případně vyskytnou (viz bod 4), stane závažným
Děti a dospívající
Tento léčivý přípravek nebyl u dětí a dospívajících studován, a nesmí být proto dětmi a dospívajícími mladšími 18 let užíván.
Další léčivé přípravky a přípravek Vargatef
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, včetně rostlinných přípravků a léků, které jste získal(a) bez lékařského předpisu.
Tento léčivý přípravek se může vzájemně ovlivňovat s některými jinými léky. Následující léky mohou zvyšovat hladiny nintedanibu, léčivé látky obsažené v přípravku Vargatef, v krvi a tak mohou zvyšovat riziko nežádoucích účinků (viz bod 4):
- Ketokonazol (používaný k léčbě plísňových infekcí)
- Erythromycin (používaný k léčbě bakteriálních infekcí)
Následující léky mohou snižovat hladiny nintedanibu v krvi, a tak mohou snižovat účinnost přípravku Vargatef:
- Rifampicin (antibiotikum používané k léčbě tuberkulózy)
- Karbamazepin, fenytoin (používané k léčbě záchvatů křečí)
- Třezalka tečkovaná (rostlinný přípravek k léčbě deprese)
Těhotenství
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Neužívejte tento přípravek během těhotenství, neboť může poškodit Vaše nenarozené dítě a způsobovat vrozené vady. Ženy, které mohou otěhotnět, musí během užívání přípravku Vargatef a nejméně 3 měsíce po ukončení léčby, používat účinnou kombinaci antikoncepčních metod, včetně bariérových metod jako druhé formy antikoncepce. Prodiskutujte pro Vás nejvhodnější způsoby antikoncepce se svým lékařem.
Okamžitě informujte svého lékaře nebo lékárníka v případě, že během léčby přípravkem Vargatef otěhotníte.
Kojení
Není známo, zda se léčivá látka vylučuje do lidského mateřského mléka a zda by mohla poškodit kojence. Ženy by proto během léčby přípravkem Vargatef neměly kojit.
Plodnost
Účinek tohoto přípravku na plodnost u lidí nebyl hodnocen.
Řízení dopravních prostředků a obsluha strojů
Jestliže se necítíte dobře, neměl(a) byste řídit ani obsluhovat stroje.
Přípravek Vargatef obsahuje sóju
Tobolky obsahují sójový lecithin. Neužívejte tento přípravek, jestliže jste alergický(á) na arašídy nebo sóju.
3. Jak se přípravek Vargatef užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Přípravek Vargatef neužívejte ve stejný den, jako chemoterapii docetaxelem.
Tobolky spolkněte celé s vodou, nežvýkejte je ani nedrťte. Doporučuje se užít tobolky s jídlem, tedy během nebo těsně před nebo po jídle.
Doporučená dávka jsou čtyři tobolky denně (to je celkem 400 mg nintedanibu denně). Neužívejte víc než tuto dávku.
Tuto denní dávku je třeba rozdělit na dvě dávky o dvou tobolkách, které užijete s odstupem přibližně 12 hodin, například dvě tobolky ráno a dvě tobolky večer. Tyto dvě dávky je třeba užít každý den v přibližně stejnou dobu. Užívání přípravku tímto způsobem zajistí, že se v těle udržuje stejné množství nintedanibu.
Snížení dávky
Jestliže doporučenou dávku 400 mg denně z důvodu nežádoucích účinků netolerujete (viz bod 4), lékař může denní dávku přípravku Vargatef snížit. Sám (sama) dávku nesnižujte ani léčbu neukončujte bez předchozí konzultace s lékařem.
Lékař může doporučenou dávku snížit na 300 mg denně (dvě 150mg tobolky). V takovém případě Vám lékař k užívání předepíše přípravek Vargatef 150 mg měkké tobolky.
V případě potřeby Vám může lékař denní dávku dále snížit na 200 mg denně (dvě 100mg tobolky). Pokud se tak stane, lékař Vám předepíše odpovídající sílu tobolek.
V obou případech užijte jednu tobolku příslušné síly s jídlem dvakrát denně s odstupem přibližně 12 hodin (například jednu tobolku ráno a jednu tobolku večer) v přibližně stejnou dobu během dne.
Jestliže lékař ukončil Vaši chemoterapii docetaxelem, je třeba, abyste pokračoval(a) v užívání přípravku Vargatef dvakrát denně.
Jestliže jste užil(a) více přípravku Vargatef, než jste měl(a)
Okamžitě kontaktujte svého lékaře nebo lékárníka.
Jestliže jste zapomněl(a) užít přípravek Vargatef
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Další dávku přípravku Vargatef užijte dle plánu v další plánovanou dobu a v dávce doporučené lékařem nebo lékárníkem.
Jestliže jste přestal(a) užívat přípravek Vargatef
Nepřerušujte užívání přípravku Vargatef bez předchozí porady s lékařem. Je důležité, abyste tento přípravek užíval(a) každý den po celou dobu, kdy Vám jej lékař předepisuje. Jestliže tento přípravek neužíváte dle doporučení svého lékaře, může se stát, že tato protinádorová léčba nebude správně fungovat.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Musíte být obzvlášť obezřetný(á) v případě, že se u Vás v průběhu léčby přípravkem Vargatef objeví následující nežádoucí účinky:
■ Průjem (velmi časté, může postihnout více než 1 pacienta z 10):
Průjem může vést ke ztrátě tekutin a důležitých solí (elektrolyty jako je sodík nebo draslík) z těla. Při prvních příznacích průjmu pijte velká množství vody a okamžitě kontaktujte svého lékaře. Po poradě s lékařem začněte co nejdříve užívat vhodnou léčbu průjmu, například loperamid.
■ Febrilní neutropenie a sepse (časté, může postihnout 1 až 10 pacientů ze 100) :
Léčba přípravkem Vargatef může vést ke snížení počtu určitého typu bílých krvinek (neutropenie), které jsou důležité pro obranu těla proti bakteriálním a plísňovým infekcím. Jako důsledek neutropenie se může objevit horečka (febrilní neutropenie) a otrava krve (sepse). Jestliže budete mít horečku, okamžitě informujte svého lékaře.
Během léčby přípravkem Vargatef bude lékař pravidelně vyšetřovat Vaše krvinky a zjišťovat, zda nemáte příznaky infekce, jako je zánět, horečka nebo únava.
Během léčby tímto přípravkem byly pozorovány následující nežádoucí účinky:
Velmi časté nežádoucí účinky (mohou postihnout více než 1 pacienta z 10)
■ Průjem - viz výše
■ Pocit bolesti, necitlivosti a/nebo brnění v prstech u rukou a u nohou (periferní neuropatie)
■ Nevolnost (pocit na zvracení)
■ Zvracení
■ Bolest břicha
■ Krvácení
■ Snížení počtu bílých krvinek (neutropenie)
■ Zánět sliznic vystýlajících trávicí trakt, včetně boláků a vřídků v ústech (mukozitida, včetně stomatitidy)
■ Vyrážka
■ Snížená chuť k jídlu
■ Elektrolytová nerovnováha
■ Zvýšené hodnoty jaterních enzymů (alaninaminotransferáza, aspartátaminotransferáza, krevní alkalická fosfatáza) v krvi, což je patrné z krevních testů
Časté nežádoucí účinky (mohou postihnout 1 až 10 pacientů ze 100)
■ Otrava krve (sepse) - viz výše
■ Febrilní neutropenie (snížení počtu bílých krvinek společně s horečkou)
■ Krevní sraženiny v žilách (žilní tromboembolie)
■ Vysoký krevní tlak (hypertenze)
■ Ztráta tekutin (dehydratace)
■ Abscesy
■ Žloutenka (hyperbilirubinemie)
■ Zvýšené hodnoty jaterních enzymů (gama-glutamyltransferázy) v krvi na základě krevních testů
Méně časté nežádoucí účinky (méně časté, mohou postihnout 1 až 10 pacientů z 1000)
■ Výskyt proděravění stěny trávicího traktu (gastrointestinální perforace)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Vargatef uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce, obalu a blistrech. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Nepoužívejte tento přípravek, pokud si všimnete, že je blistr, ve kterém jsou tobolky umístěny, otevřený nebo je tobolka poničená.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Vargatef obsahuje
- Léčivou látkou v přípravku Vargatefje nintedanibum. Jedna měkká tobolka obsahuje nintedanibum 100 mg (ve formě nintedanibi esilas).
- Pomocnými látkami jsou:
Obsah tobolky: Střední nasycené triacylglyceroly, tvrdý tuk, sójový lecithin (E322)
Obal tobolky: Želatina, glycerol 85%, oxid titaničitý (E171), červený oxid železitý (E172),
žlutý oxid železitý (E172)
Potiskový inkoust: Šelak, černý oxid železitý (E172), propylenglykol (E1520)
Jak přípravek Vargatef vypadá a co obsahuje toto balení
Přípravek Vargatef 100 mg měkké tobolky jsou neprůhledné, oválné tobolky broskvové barvy s černým potiskem na jedné straně - symbolem společnosti Boehringer Ingelheim a číslicí „100“.
K dispozici jsou tři velikosti balení přípravku Vargatef 100 mg měkké tobolky:
■ Jedna krabička obsahující 60 tobolek (6 hliníkových blistrů, každý obsahuje 10 tobolek).
■ Jedna krabička obsahující 120 tobolek (12 hliníkových blistrů, každý obsahuje 10 tobolek).
■ Vícenásobné balení obsahující 120 tobolek (2 krabičky, každá obsahuje 60 tobolek, krabičky jsou spojené balicí folií).
Na trhu nemusí být všechny velikosti balení přípravku Vargatef 100 mg měkké tobolky.
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Německo
Výrobce
Boehringer Ingelheim Pharma GmbH & Co. KG
Binger Strasse 173
D-55216 Ingelheim am Rhein
Německo
|
Belgie/Belgique/Belgien SCS Boehringer Ingelheim Comm.V Tél/Tel: +32 2 773 33 11 |
Lietuva Boehringer Ingelheim RCV GmbH & Co KG Lietuvos filialas Tel: +370 37 473922 |
|
Btarapna EbopHHrep HHrenxaňM P^B rMÓX h Ko. Kr - KnoH Ebnrapna Ten: +359 2 958 79 98 |
Luxembourg/Luxemburg SCS Boehringer Ingelheim Comm.V Tél/Tel: +32 2 773 33 11 |
|
Česká republika Boehringer Ingelheim spol. s r.o. Tel: +420 234 655 111 |
Magyarország Boehringer Ingelheim RCV GmbH & Co KG Magyarországi Fióktelepe Tel: +36 1 299 8900 |
|
Danmark Boehringer Ingelheim Danmark A/S Tlf: +45 39 15 88 88 |
Malta Boehringer Ingelheim Ltd. Tel: +44 1344 424 600 |
|
Deutschland Boehringer Ingelheim Pharma GmbH & Co. KG Tel: +49 (0) 800 77 90 900 |
Nederland Boehringer Ingelheim b.v. Tel: +31 (0) 800 22 55 889 |
|
Eesti Boehringer Ingelheim RCV GmbH & Co KG Eesti filiaal Tel: +372 612 8000 |
Norge Boehringer Ingelheim Norway KS Tlf: +47 66 76 13 00 |
|
EXláSa Boehringer Ingelheim Ellas A.E. T^: +30 2 10 89 06 300 |
Osterreich Boehringer Ingelheim RCV GmbH & Co KG Tel: +43 1 80 105-0 |
|
Espaňa Boehringer Ingelheim Espana, S.A. Tel: +34 93 404 51 00 |
Polska Boehringer Ingelheim Sp. z o.o. Tel: +48 22 699 0 699 |
|
France Boehringer Ingelheim France S.A.S. Tél: +33 3 26 50 45 33 |
Portugal Boehringer Ingelheim, Unipessoal, Lda. Tel: +351 21 313 53 00 |
|
Hrvatska Boehringer Ingelheim Zagreb d.o.o. Tel: +385 1 2444 600 |
Románia Boehringer Ingelheim RCV GmbH & Co KG Viena - Sucursala Bucuresti Tel: +40 21 302 2800 |
|
Ireland Boehringer Ingelheim Ireland Ltd. Tel: +353 1 295 9620 |
Slovenija Boehringer Ingelheim RCV GmbH & Co KG Podružnica Ljubljana Tel: +386 1 586 40 00 |
Ísland
Vistor hf.
Sími: +354 535 7000
Italia
Boehringer Ingelheim Italia S.p.A.
Tel: +39 02 5355 1
Kúnpoq
Boehringer Ingelheim Ellas A.E.
T^: +30 2 10 89 06 300
Latvija
Boehringer Ingelheim RCV GmbH & Co KG
Latvijas filiale
Tel: +371 67 240 011
Slovenská republika
Boehringer Ingelheim RCV GmbH & Co KG organizačná zložka Tel: +421 2 5810 1211
Suomi/Finland
Boehringer Ingelheim Finland Ky Puh/Tel: +358 10 3102 800
Sverige
Boehringer Ingelheim AB Tel: +46 8 721 21 00
United Kingdom
Boehringer Ingelheim Ltd.
Tel: +44 1344 424 600
Tato příbalová informace byla naposledy revidována <{měsíc/RRRR}>.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http: //www .ema. europa. eu.
Příbalová informace: informace pro pacienta
Vargatef 150 mg měkké tobolky
Nintedanibum
▼ Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Vargatef a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Vargatef užívat
3. Jak se přípravek Vargatef užívá
4. Možné nežádoucí účinky
5. Jak přípravek Vargatef uchovávat
6. Obsah balení a další informace
1. Co je přípravek Vargatef a k čemu se používá
Tobolky přípravku Vargatef obsahují léčivou látku nintedanib. Nintedanib blokuje aktivitu skupiny bílkovin, které jsou zapojeny do vývoje nových krevních cév, které nádorové buňky potřebují jako zdroj výživy a kyslíku. Tím, že nintedanib zablokuje aktivitu těchto bílkovin, je schopen potlačit růst a šíření nádoru.
Tento přípravek se používá v kombinaci s jiným protinádorovým léčivým přípravkem (docetaxel) k léčbě rakoviny plic zvané nemalobuněčný karcinom plic (non-small cell lung cancer, NSCLC). Je určen pro dospělé pacienty se specifickým typem NSCLC (,,adenokarcinom“), kteří již byli léčeni jedním jiným léčivým přípravkem určeným k léčbě tohoto nádorového onemocnění, ale u nichž nádor začal znovu růst.
2. Čemu musíte věnovat pozornost, než začnete přípravek Vargatef užívat
Neužívejte přípravek Vargatef:
- jestliže jste alergický(á) na nintedanib, arašídy nebo sóju nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím tohoto přípravku se poraďte se svým lékařem nebo lékárníkem,
- jestliže jste někdy měl(a) problémy s játry; jestliže máte nebo jste měl(a) problémy s krvácením, obzvláště krvácení do plic v nedávné době
- jestliže užíváte léčivé přípravky k ředění krve (jako je warfarin, fenprokumon, heparin nebo kyselina acetylsalicylová) k prevenci tvorby krevních sraženin. Léčba přípravkem Vargatef může zvyšovat riziko krvácení
- jestliže jste v nedávné době podstoupil(a) nebo plánujete podstoupit operaci. Nintedanib může ovlivnit způsob, jak se Vám hojí rány. Léčba přípravkem Vargatef proto obvykle bude v případě, že máte být operován(a), přerušena. Lékař rozhodne o tom, kdy bude léčba tímto přípravkem obnovena
- jestliže máte rakovinu, která se rozšířila do mozku
Na základě těchto informací může lékař provést některé krevní testy, například zkontrolovat funkci Vašich jater a zjistit, jak rychle se Vaše krev sráží. Lékař s Vámi výsledky těchto testů prodiskutuje a rozhodne, zda Vám může být podán přípravek Vargatef.
Během užívání tohoto přípravku svého lékaře okamžitě informujte,
- jestliže dostanete průjem. Je důležité léčit průjem při prvních příznacích (viz bod 4)
- jestliže budete mít horečku, protože ta by mohla být příznakem febrilní neutropenie nebo sepse (viz bod 4)
- jestliže pociťujete silnou bolest v oblasti žaludku, máte horečku, zimnici, je Vám nevolno, zvracíte nebo máte napnuté břicho nebo se cítíte nafouklý(á), protože to by mohly být příznaky proděravění stěny trávicího traktu („gastrointestinální perforace“)
- jestliže pociťujete bolest v končetině, nebo ji máte oteklou, zarudlou či teplou, protože to by mohly být příznaky krevní sraženiny v jedné z Vašich žil
- jestliže máte velké krvácení
- jestliže pociťujete tlak nebo bolest na hrudi, typicky na levé straně těla, bolest v šíji, čelisti, rameni nebo paži, máte zrychlený srdeční tep, jste dušný(á), máte pocit na zvracení, zvracíte, protože to by mohly být příznaky srdečního infarktu
- jestliže se kterýkoli z nežádoucích účinků, které se u Vás případně vyskytnou (viz bod 4), stane závažným
Děti a dospívající
Tento léčivý přípravek nebyl u dětí a dospívajících studován, a nesmí být proto dětmi a dospívajícími mladšími 18 let užíván.
Další léčivé přípravky a přípravek Vargatef
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, včetně rostlinných přípravků a léků, které jste získal(a) bez lékařského předpisu.
Tento léčivý přípravek se může vzájemně ovlivňovat s některými jinými léky. Následující léky mohou zvyšovat hladiny nintedanibu, léčivé látky obsažené v přípravku Vargatef, v krvi a tak mohou zvyšovat riziko nežádoucích účinků (viz bod 4):
- Ketokonazol (používaný k léčbě plísňových infekcí)
- Erytromycin (používaný k léčbě bakteriálních infekcí)
Následující léky mohou snižovat hladiny nintedanibu v krvi, a tak mohou snižovat účinnost přípravku Vargatef:
- Rifampicin (antibiotikum používané k léčbě tuberkulózy)
- Karbamazepin, fenytoin (používané k léčbě záchvatů křečí)
- Třezalka tečkovaná (rostlinný přípravek k léčbě deprese)
Těhotenství
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Neužívejte tento přípravek během těhotenství, neboť může poškodit Vaše nenarozené dítě a způsobovat vrozené vady. Ženy, které mohou otěhotnět, musí během užívání přípravku Vargatef a nejméně 3 měsíce po ukončení léčby, používat účinnou kombinaci antikoncepčních metod, včetně bariérových metod jako druhé formy antikoncepce. Prodiskutujte pro Vás nejvhodnější způsoby antikoncepce se svým lékařem.
Okamžitě informujte svého lékaře nebo lékárníka v případě, že během léčby přípravkem Vargatef otěhotníte.
Kojení
Není známo, zda se léčivá látka vylučuje do lidského mateřského mléka a zda by mohla poškodit kojence. Ženy by proto během léčby přípravkem Vargatef neměly kojit.
Plodnost
Účinek tohoto přípravku na plodnost u lidí nebyl hodnocen.
Řízení dopravních prostředků a obsluha strojů
Jestliže se necítíte dobře, neměl(a) byste řídit ani obsluhovat stroje.
Přípravek Vargatef obsahuje sóju
Tobolky obsahují sójový lecithin. Jestliže jste alergický(á) na arašídy nebo sóju, tento přípravek neužívejte.
3. Jak se přípravek Vargatef užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Přípravek Vargatef neužívejte ve stejný den, jako chemoterapii docetaxelem.
Tobolky spolkněte celé s vodou, nežvýkejte je ani nedrťte. Doporučuje se užít tobolku s jídlem, tedy během nebo těsně před nebo po jídle.
Doporučená dávka jsou dvě tobolky denně (to je celkem 300 mg nintedanibu denně). Neužívejte víc než tuto dávku.
Tuto denní dávku je třeba rozdělit na dvě dávky o jedné tobolce, které užijete s odstupem přibližně 12 hodin, například jedna tobolka ráno a jedna tobolka večer. Tyto dvě dávky je třeba užít každý den v přibližně stejnou dobu. Užívání přípravku tímto způsobem zajistí, že se v těle udržuje stejné množství nintedanibu.
Snížení dávky
Jestliže doporučenou dávku 300 mg denně z důvodu nežádoucích účinků netolerujete (viz bod 4), lékař může Vaši doporučenou denní dávku přípravku Vargatef snížit na 200 mg denně (dvě 100mg tobolky). V takovém případě Vám lékař k užívání předepíše přípravek Vargatef 100 mg měkké tobolky.
Užijte jednu tobolku této síly s jídlem dvakrát denně s odstupem přibližně 12 hodin (například jednu tobolku ráno a jednu tobolku večer) v přibližně stejnou dobu během dne.
Sám (sama) dávku nesnižujte ani léčbu neukončujte bez předchozí konzultace s lékařem.
Jestliže lékař ukončil Vaši chemoterapii docetaxelem, je třeba, abyste pokračoval(a) v užívání přípravku Vargatef dvakrát denně.
Jestliže jste užil(a) více přípravku Vargatef, než jste měl(a)
Okamžitě kontaktujte svého lékaře nebo lékárníka.
Jestliže jste zapomněl(a) užít přípravek Vargatef
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Další dávku přípravku Vargatef užijte dle plánu v další plánovanou dobu a v dávce doporučené lékařem nebo lékárníkem.
Jestliže jste přestal(a) užívat přípravek Vargatef
Nepřerušujte užívání přípravku Vargatef bez předchozí porady s lékařem. Je důležité, abyste tento přípravek užíval(a) každý den po celou dobu, kdy Vám jej lékař předepisuje. Jestliže tento přípravek neužíváte dle doporučení svého lékaře, může se stát, že tato protinádorová léčba nebude správně fungovat.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Musíte být obzvlášť obezřetný(á) v případě, že se u Vás v průběhu léčby přípravkem Vargatef objeví následující nežádoucí účinky:
■ Průjem (velmi časté, může postihnout více než 1 pacienta z 10):
Průjem může vést ke ztrátě tekutin a důležitých solí (elektrolyty jako je sodík nebo draslík) z těla. Při prvních příznacích průjmu pijte velká množství vody a okamžitě kontaktujte svého lékaře. Po poradě s lékařem začněte co nejdříve užívat vhodnou léčbu průjmu, například loperamid.
■ Febrilní neutropenie a sepse (časté, může postihnout 1 až 10 pacientů ze 100) :
Léčba přípravkem Vargatef může vést ke snížení počtu určitého typu bílých krvinek (neutropenie), které jsou důležité pro obranu těla proti bakteriálním a plísňovým infekcím. Jako důsledek neutropenie se může objevit horečka (febrilní neutropenie) a otrava krve (sepse). Jestliže budete mít horečku, okamžitě informujte svého lékaře.
Během léčby přípravkem Vargatef bude lékař pravidelně vyšetřovat Vaše krvinky a zjišťovat, zda nemáte příznaky infekce, jako je zánět, horečka nebo únava.
Během léčby tímto přípravkem byly pozorovány následující nežádoucí účinky:
Velmi časté nežádoucí účinky (mohou postihnout více než 1 pacienta z 10)
■ Průjem - viz výše
■ Pocit bolesti, necitlivosti a/nebo brnění v prstech u rukou a u nohou (periferní neuropatie)
■ Nevolnost (pocit na zvracení)
■ Zvracení
■ Bolest břicha
■ Krvácení
■ Snížení počtu bílých krvinek (neutropenie)
■ Zánět sliznic vystýlajících trávicí trakt, včetně boláků a vřídků v ústech (mukozitida, včetně stomatitidy)
■ Vyrážka
■ Snížená chuť k jídlu
■ Elektrolytová nerovnováha
■ Zvýšené hodnoty jaterních enzymů (alaninaminotransferáza, aspartátaminotransferáza, krevní alkalická fosfatáza) v krvi, což je patrné z krevních testů
Časté nežádoucí účinky (mohou postihnout 1 až 10 pacientů ze 100)
■ Otrava krve (sepse) - viz výše
■ Febrilní neutropenie (snížení počtu bílých krvinek společně s horečkou)
■ Krevní sraženiny v žilách (žilní tromboembolie)
■ Vysoký krevní tlak (hypertenze)
■ Ztráta tekutin (dehydratace)
■ Abscesy
■ Žloutenka (hyperbilirubinemie)
■ Zvýšené hodnoty jaterních enzymů (gama-glutamyltransferázy) v krvi na základě krevních testů
Méně časté nežádoucí účinky (méně časté, mohou postihnout 1 až 10 pacientů z 1000)
■ Výskyt proděravění stěny trávicího traktu (gastrointestinální perforace)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Vargatef uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a blistrech. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Nepoužívejte tento přípravek, pokud si všimnete, že je blistr, ve kterém jsou tobolky umístěny, otevřený nebo je tobolka poničená.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Vargatef obsahuje
- Léčivou látkou v přípravku Vargatefje nintedanibum. Jedna tobolka přípravku Vargatef 150 mg měkké tobolky obsahuje nintedanibum 150 mg (ve formě nintedanibi esilas).
- Pomocnými látkami jsou:
Obsah tobolky: Střední nasycené triacylglyceroly, tvrdý tuk, sójový lecithin (E322)
Obal tobolky: Želatina, glycerol 85%, oxid titaničitý (E171), červený oxid železitý (E172),
žlutý oxid železitý (E172)
Potiskový inkoust: Šelak, černý oxid železitý (E172), propylenglykol (E1520)
Jak přípravek Vargatef vypadá a co obsahuje toto balení
Přípravek Vargatef 150 mg měkké tobolky jsou neprůhledné, oválné tobolky hnědé barvy s černým potiskem na jedné straně - symbolem společnosti Boehringer Ingelheim a číslicí „150“.
Jedna krabička obsahuje 60 tobolek (6 hliníkových blistrů, každý obsahuje 10 tobolek).
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Německo
Výrobce
Boehringer Ingelheim Pharma GmbH & Co. KG
Binger Strasse 173
D-55216 Ingelheim am Rhein
Německo
|
Belgie/Belgique/Belgien SCS Boehringer Ingelheim Comm.V Tél/Tel: +32 2 773 33 11 |
Lietuva Boehringer Ingelheim RCV GmbH & Co KG Lietuvos filialas Tel: +370 37 473922 |
|
Btarapna EbopHHrep HHrenxaňM P^B rMÓX h Ko. Kr - KnoH Ebnrapna Ten: +359 2 958 79 98 |
Luxembourg/Luxemburg SCS Boehringer Ingelheim Comm.V Tél/Tel: +32 2 773 33 11 |
|
Česká republika Boehringer Ingelheim spol. s r.o. Tel: +420 234 655 111 |
Magyarország Boehringer Ingelheim RCV GmbH & Co KG Magyarországi Fióktelepe Tel: +36 1 299 8900 |
|
Danmark Boehringer Ingelheim Danmark A/S Tlf: +45 39 15 88 88 |
Malta Boehringer Ingelheim Ltd. Tel: +44 1344 424 600 |
|
Deutschland Boehringer Ingelheim Pharma GmbH & Co. KG Tel: +49 (0) 800 77 90 900 |
Nederland Boehringer Ingelheim b.v. Tel: +31 (0) 800 22 55 889 |
|
Eesti Boehringer Ingelheim RCV GmbH & Co KG Eesti filiaal Tel: +372 612 8000 |
Norge Boehringer Ingelheim Norway KS Tlf: +47 66 76 13 00 |
|
EXláSa Boehringer Ingelheim Ellas A.E. T^: +30 2 10 89 06 300 |
Osterreich Boehringer Ingelheim RCV GmbH & Co KG Tel: +43 1 80 105-0 |
|
Espaňa Boehringer Ingelheim Espana, S.A. Tel: +34 93 404 51 00 |
Polska Boehringer Ingelheim Sp. z o.o. Tel: +48 22 699 0 699 |
|
France Boehringer Ingelheim France S.A.S. Tél: +33 3 26 50 45 33 |
Portugal Boehringer Ingelheim, Unipessoal, Lda. Tel: +351 21 313 53 00 |
|
Hrvatska Boehringer Ingelheim Zagreb d.o.o. Tel: +385 1 2444 600 |
Románia Boehringer Ingelheim RCV GmbH & Co KG Viena - Sucursala Bucuresti Tel: +40 21 302 2800 |
|
Ireland Boehringer Ingelheim Ireland Ltd. Tel: +353 1 295 9620 |
Slovenija Boehringer Ingelheim RCV GmbH & Co KG Podružnica Ljubljana Tel: +386 1 586 40 00 |
Slovenská republika
Ísland
Vistor hf.
Sími: +354 535 7000
Italia
Boehringer Ingelheim Italia S.p.A. Tel: +39 02 5355 1
Boehringer Ingelheim RCV GmbH & Co KG organizačná zložka Tel: +421 2 5810 1211
Suomi/Finland
Boehringer Ingelheim Finland Ky Puh/Tel: +358 10 3102 800
Sverige
Boehringer Ingelheim AB Tel: +46 8 721 21 00
United Kingdom
Boehringer Ingelheim Ltd. Tel: +44 1344 424 600
Kúnpoq
Boehringer Ingelheim Ellas A.E.
T^: +30 2 10 89 06 300
Latvija
Boehringer Ingelheim RCV GmbH & Co KG
Latvijas filiale
Tel: +371 67 240 011
Tato příbalová informace byla naposledy revidována <{měsíc/RRRR}>.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY V REGISTRACI
Vědecké závěry
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizované zprávy o bezpečnosti (PSUR) pro nintedanib (onkologické indikace) dospěl výbor CHMP k těmto vědeckým závěrům:
V poslední PSUR přípravku Vargatef držitel rozhodnutí o registraci předložil kumulativní hodnocení nežádoucího účinku „Zvýšená hladina gama-glutamyltransferázy (GGT)“ V celkové populaci a u pacientů s adenokarcinomem ve studii 1199.13 byly nežádoucí příhody zvýšené hladiny GGT častější při léčbě nintedanibem a docetaxelem než docetaxelem samotným. Většina nežádoucích příhod byla stupně 1/2. Nežádoucí příhody byly zvládnutelné snížením dávky. Trvalé vysazení studijní medikace nebylo obvykle nutné. Nežádoucí příhody zvýšené hladiny GGT byly hlášeny i při léčbě monoterapií nintedanibem.
Hodnocení případů odhalilo další faktory, které pravděpodobně přispěly ke zvýšení hladiny GGT, jako např. souběžná léčba přípravky, u nichž je známo, že mohou zvyšovat hladiny jaterních enzymů a/nebo bilirubinu, a souběžná onemocnění, např. progrese nádoru do jater, žlučový kámen nebo cholestáza. Ve většině případů došlo ke zvýšení hladiny GGT společně se zvýšením alaninaminotransferázy (ALT), aspartátaminotransferázy (AST) a alkalické fosfatázy (ALKP).
Zvýšené hladiny GGT byly u většiny pacientů reverzibilní. Na základě této kumulativní analýzy zastává výbor PRAC stanovisko, že existují dostatečné důkazy k vytvoření závěru, že léčba nintedanibem (onkologické indikace) může vést ke zvýšení hladin GGT.
Na základě údajů uvedených v hodnocené PSUR tedy výbor PRAC považuje změny v informacích o léčivých přípravcích obsahujících nintedanib (onkologické indikace) za oprávněné.
Výbor CHMP souhlasí s vědeckými závěry výboru PRAC.
Zdůvodnění změny v registraci
Na základě vědeckých závěrů týkajících se nintedanibu (onkologické indikace) výbor CHMP zastává stanovisko, že poměr přínosů a rizik léčivého přípravku obsahujícího nintedanib (onkologické indikace) zůstává nezměněný, a to pod podmínkou, že v informacích o přípravku budou provedeny navrhované změny.
Výbor CHMP doporučuje změnu v registraci.
52