Vantobra 170 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Vantobra 170 mg roztok k rozprašování
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna ampulka s jednou dávkou o objemu 1,7 ml obsahuje tobramycinum 170 mg. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Roztok k rozprašování.
Čirý až nažloutlý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Vantobra je indikován k léčbě chronických plicních infekcí vyvolaných Pseudomonas aeruginosa při cystické fibróze (CF) u pacientů ve věku 6 let a starších.
Je nutné přihlížet k oficiálním doporučením, týkajícím se správného použití antibakteriálních látek.
4.2 Dávkování a způsob podání
Dávkování
Dávka přípravku Vantobra je stejná pro všechny pacienty ve schváleném věkovém rozpětí bez ohledu na věk nebo tělesnou hmotnost. Doporučená dávka je jedna ampulka (170 mg/1,7 ml) podávaná dvakrát denně (tj. celková denní dávka jsou 2 ampulky) po dobu 28 dnů. Interval mezi 2 dávkami se má co nejvíce blížit 12 hodinám a nemá být kratší než 6 hodin.
Přípravek Vantobra se používá ve střídavých cyklech po 28 dnech. Je nutné dodržovat cyklus 28 dnů aktivní terapie (období s léčbou) a 28 dnů bez terapie (období bez léčby).
Vynechání dávky
V případě, že si pacient zapomene vzít dávku a do další dávky zbývá nejméně 6 hodin, je třeba aby inhaloval dávku co nejdříve. Pokud do další plánované dávky zbývá méně než 6 hodin, pacient má počkat na další dávku a neinhalovat více, aby vynechanou dávku nahradil.
Trvání léčby
Léčba má pokračovat v cyklech tak dlouho, dokud se lékař domnívá, že má pacient z léčby klinický prospěch s tím, že vezme v úvahu skutečnost, že údaje o bezpečnosti přípravku Vantobra při dlouhodobé léčbě nejsou k dispozici. Pokud je zřejmé, že dochází ke klinickému zhoršení stavu funkce plic, je zapotřebí zvážit přídatnou nebo alternativní léčbu pseudomonádové infekce. Viz také informace o klinickém přínosu a snášenlivosti v bodech 4.4, 4.8 a 5.1.
Zvláštní populace
Starší pacienti (>65 let)
U této populace neexistují dostatečné údaje na podporu doporučení pro úpravu dávky, nebo proti ní.
Porucha funkce ledvin
U této populace existují žádné údaje na podporu doporučení pro úpravu dávky přípravku Vantobra, nebo proti ní. Viz také informace o nefrotoxicitě v bodě 4.4 a informace o vylučování v bodě 5.2.
Porucha funkce jater
U pacientů s poruchou funkce jater nebyly prováděny žádné studie. Účinek poruchy funkce jater na expozici tobramycinu se neočekává, protože se tobramycin nemetabolizuje.
Pacienti po transplantaci orgánů
O použití inhalovaného tobramycinu u pacientů po transplantaci orgánů neexistují adekvátní údaje. Pro pacienty po transplantaci orgánů nelze vydat žádná doporučení pro úpravu dávky, nebo proti ní.
Pediatrická populace
Neexistuje žádné relevantní použití přípravku Vantobra u dětí mladších 6 let.
Způsob podání Inhalační podání.
Přípravek Vantobra se podává inhalací pomocí ruční soupravy inhalátoru Tolero, který je součástí balení. Podrobné pokyny k použití viz bod 6.6.
Přípravek Vantobra se nesmí podávat žádnou jinou cestou ani se nesmí použít žádné jiné zařízení, než které je součástí balení. Použití alternativního netestovaného inhalačního systému může změnit ukládání léčivé látky v plicích. Následkem toho se může změnit účinnost a bezpečnost přípravku.
Jestliže pacienti užívají několik inhalačních léčivých přípravků a podstupují hrudní fyzioterapii, doporučuje se, aby byl přípravek Vantobra použit jako poslední.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití Ototoxicita
U parenterálně podávaných aminoglykosidů byla hlášena ototoxicita, která se projevuje jako sluchová toxicita (ztráta sluchu) a vestibulární toxicita. Vestibulární toxicita se může projevovat vertigem, ataxií nebo závratěmi. Tinitus může být varovným příznakem ototoxicity, a proto nástup tohoto příznaku je důvodem k opatrnosti.
U parenterálně podávaných aminoglykosidů byla hlášena sluchová toxicita diagnostikovaná na základě subjektivního vnímání ztráty sluchu nebo audiometrického vyšetření a lze ji rovněž předpokládat při inhalační cestě podání. Ztráta sluchu se objevovala v otevřených klinických studiích a postmarketingovém sledování u některých pacientů s dlouhodobým předchozím či konkomitantním používáním intravenózně podávaných aminoglykosidů v anamnéze. Je nutné, aby lékaři vzali v úvahu aminoglykosidy jako potenciální příčinu vestibulární a kochleární toxicity a během léčby přípravkem Vantobra provedli příslušná vyšetření sluchových funkcí.
U pacientů s predispozicí k riziku vzhledem k předchozí dlouhodobé systémové léčbě aminoglykosidy může být nezbytné zvážit audiologické vyšetření před zahájením léčby přípravkem Vantobra. Pokud bude pacient udávat tinitus nebo ztrátu sluchu během léčby aminoglykosidy, je nutné, aby lékař zvážil doporučení k audiologickému vyšetření.
Nefrotoxicita
Nefrotoxicita je spojována s parenterální léčbou aminoglykosidy. V klinických hodnoceních s inhalovaným tobramycinem a přípravkem Vantobra však nefrotoxicita prokázána nebyla. Při předepisování přípravku Vantobra pacientům se známou renální dysfunkcí či s podezřením na ni je zapotřebí postupovat s opatrností. Podle současné klinické praxe je třeba vyšetřit základní funkce ledvin. Hladiny urey a kreatininu je zapotřebí vyšetřit znovu po každých 6 úplných cyklech léčby přípravkem Vantobra (180 dnů léčby nebulizovaným aminoglykosidem).
Sledování koncentrace tobramvcinu v séru
U pacientů se známou nebo suspektní sluchovou či ledvinovou dysfunkcí je nutné sledovat sérové koncentrace tobramycinu. Pokud dojde k výskytu ototoxicity nebo nefrotoxicity u pacienta dostávajícího přípravek Vantobra, je nutné léčbu tobramycinem přerušit, dokud sérová koncentrace nepoklesne pod 2 pg/ml.
Sérové koncentrace vyšší než 12 pg/ml jsou spojeny s toxicitou tobramycinu a léčbu je nutné přerušit, pokud koncentrace překročila tuto úroveň.
Sérová koncentrace tobramycinu má být sledována pouze pomocí validovaných metod. Odběr vzorku krve z prstu se nedoporučuje kvůli riziku kontaminace vzorku.
Bronchospasmus
Bronchospasmus se může vyskytnout při inhalaci léčivých přípravků a byl hlášen při použití inhalačního tobramycinu. Bronchospasmus je nutné léčit v souladu s běžným medicínským postupem.
První dávka přípravku Vantobra se má podat pod dohledem lékaře po podání bronchodilatancia, pokud je součástí stávajícího léčebného režimu pacienta. Je zapotřebí změřit FEVi před inhalací a po ní.
Pokud se prokáže bronchospasmus vyvolaný léčbou, lékař má pečlivě vyhodnotit, zda výhody pokračujícího používání přípravku Vantobra převažují nad riziky pro pacienta. Pokud bude podezření na alergickou reakci, je nutné přípravek Vantobra vysadit.
Neuromuskulární poruchy
Přípravek Vantobra je nutné používat s velkou opatrností u pacientů s neuromuskulárními poruchami, jako je parkinsonismus nebo jiná onemocnění vyznačující se myastenií, včetně myasthenia gravis, protože aminoglykosidy mohou zhoršovat svalovou slabost díky potenciálnímu účinku na neuromuskulární funkci podobnému působení kurare.
Hemoptýza
Inhalace rozprášených roztoků tobramycinu může vyvolat kašlací reflex. Léčba přípravkem Vantobra u pacientů s aktivní, závažnou hemoptýzou by se měla zahajovat pouze v případě, že přínos léčby převáží riziko vyvolání dalšího krvácení.
Vznik rezistence
Vývoj P. aeruginosa rezistentní na antibiotika a superinfekce ostatními patogeny představují potenciální rizika spojená s terapií antibiotiky. Vznik rezistence během inhalační terapie tobramycinem by mohl omezit možnosti léčby během akutních exacerbací; sledování je nezbytné.
Další opatření
Pacienty léčené současně parenterálními aminoglykosidy (nebo jakýmkoliv léčivem ovlivňujícím renální exkreci, například diuretika) je nutné sledovat, z klinického hlediska je vhodné vzít v úvahu riziko kumulativní toxicity. To zahrnuje i sledování sérových koncentrací tobramycinu.
U pacientů s kolonizací Burkholderia cepacia nebyla studována bezpečnost a účinnost.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí. Na základě profilu interakce pro tobramycin po intravenózním podání a podání ve formě aerosolu se souběžné a/nebo následné použití přípravku Vantobra nedoporučuje s jinými léčivými přípravky s nefrotoxickým či ototoxickým potenciálem, jako jsou:
- amfotericin B, cefalotin, cyklosporin, takrolimus, polymyxiny (riziko zvýšené nefrotoxicity);
- sloučeniny platiny (riziko zvýšené nefrotoxicity a ototoxicity);
Souběžné používání přípravku Vantobra s diuretiky (například kyselinou etakrynovou, furosemidem, močovinou nebo manitolem) se nedoporučuje. Tyto sloučeniny mohou zvýšit toxicitu aminoglykosidů změnou koncentrací antibiotika v séru a tkáni (viz bod 4.4).
Mezi další léčivé přípravky, u nichž bylo hlášeno, že zvyšují potenciální toxicitu parenterálně podávaných aminoglykosidů, patří:
- anticholinesterázy, botulotoxin (neuromoskulární účinky).
V klinických studiích pacienti používající inhalovaný tobramycin pokračovali v používání alfadornázy, bronchodilatancií, inhalačních kortikosteroidů a makrolidů. Nebyl odhalen žádný důkaz o interakcích léčivého přípravku s těmito léky.
4.6 Fertilita, těhotenství a kojení
Údaje o parenterálním podávání tobramycinu těhotným ženám jsou omezené. Adekvátní údaje o použití tobramycinu podávaného ve formě inhalací těhotným ženám nejsou k dispozici. Studie na zvířatech nenaznačují teratogenní účinek tobramycinu (viz bod 5.3). Aminoglykosidy však mohou způsobit poškození plodu (např. vrozená hluchota a nefrotoxicita), když je u těhotné ženy dosaženo vysokých systémových koncentrací. Systémová expozice po inhalaci přípravku Vantobra je velmi nízká (viz bod 5.2). Pokud se přípravek Vantobra bude používat během těhotenství nebo pokud pacientka otěhotní během používání přípravku Vantobra, je nutné ji informovat o potenciálním nebezpečí pro plod.
Přípravek Vantobra nemá být podáván během těhotenství, pokud výhody pro matku nepřeváží nad riziky pro plod či novorozence.
Kojení
Tobramycin se po systémovém podání vylučuje do lidského mateřského mléka. Množství tobramycinu vyloučeného do mateřského mléka po inhalačním podání není známo, předpokládá se však, že bude velmi nízké s ohledem na nízkou systémovou expozici. Vzhledem k potenciální ototoxicitě a nefrotoxicitě u novorozenců je zapotřebí rozhodnout, zda ukončit kojení, nebo přerušit léčbu přípravkem Vantobra s ohledem na význam léčby pro matku.
Fertilita
Ve studiích se zvířaty nebyl pozorován žádný účinek na samčí či samičí fertilitu po subkutánním podání (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vantobra nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
V kontrolovaných klinických studiích s přípravkem Vantobra byly nejčastějšími nežádoucími účinky u pacientů s cystickou fibrózou s infekcí P. aeruginosa kašel a dysfonie.
Klinická zkušenost s roztoky tobramycinu k rozprašování uvádí dysfonii a tinitus u pacientů léčených tobramycinem. Epizody tinitu byly přechodné a ustoupily bez přerušení léčby tobramycinem .
Příležitostně může u pacientů s anamnézou dlouhodobého předchozího nebo souběžného užívání intravenózních aminoglykosidů dojít ke ztrátě sluchu. Parenterálně podávané aminoglykosidy bývají spojovány s hypersenzitivitou, ototoxicitou a nefrotoxicitou (viz bod 4.4).
Dlouhodobé údaje o bezpečnosti nejsou pro přípravek Vantobra k dispozici (viz také bod 5.1).
Tabulkový přehled nežádoucích účinků
Nežádoucí účinky hlášené pro roztok tobramycinu k rozprašování jsou uvedeny v tabulce 1.
Nežádoucí účinky léčivého přípravku jsou uvedeny níže podle tříd orgánových systémů MedDRA. U každé třídy orgánových systémů jsou nežádoucí účinky léčivého přípravku seřazeny podle frekvence a účinky s nejvyšší frekvencí výskytu jsou uvedeny jako první. V každé skupině frekvencí jsou nežádoucí účinky seřazeny podle klesající závažnosti. Kromě toho jsou příslušné kategorie frekvencí vyjádřeny pomocí následující konvence: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100); vzácné (> 1/10000 až < 1/1000); velmi vzácné (< 1/10000).
Tabulka 1 Nežádoucí účinky
|
Třída orgánových systémů |
Kategorie frekvence |
Nežádoucí reakce |
|
Infekce a infestace | ||
|
Vzácné | ||
|
Velmi vzácné |
Mykotická infekce Orální kandidóza | |
|
Poruchy krve a lymfatického systému | ||
|
Velmi vzácné |
Lymfadenopatie | |
|
Poruchy imunitního systému | ||
|
Velmi vzácné |
Hypersenzitivita | |
|
Poruchy metabolismu a výživy | ||
|
Vzácné | ||
|
Poruchy nervového systému | ||
|
Vzácné |
Závrať Afonie Bolest hlavy | |
|
Velmi vzácné |
Somnolence | |
|
Poruchy ucha a labyrintu | ||
|
Vzácné |
Ztráta sluchu Tinitus | |
|
Velmi vzácné |
Bolest ucha Onemocnění ucha | |
|
Cévní poruchy | ||
|
Vzácné |
Hemoptýza Epistaxe | |
|
Respirační, hrudní a mediastinální poruchy | ||
|
Méně časté |
Dysfonie Faryngitida | |
|
Vzácné |
Plicní porucha Diskomfort v oblasti hrudníku Produktivní kašel Rinitida Bronchospasmus | |
|
Velmi vzácné |
Hypoxie |
|
Hyperventilace Sinusitida | ||
|
Gastrointestinální poruchy | ||
|
Vzácné |
Zvracení Ulcerace v ústech Nauzea Dysgeuzie | |
|
Velmi vzácné |
Bolesti v břišní krajině | |
|
Poruchy kůže a podkožní tkáně | ||
|
Vzácné | ||
|
Velmi vzácné |
Kopřivka Pruritus | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Velmi vzácné | ||
|
Celkové poruchy a reakce v místě aplikace | ||
|
Vzácné |
Astenie Pyrexie Bolest Bolest hrudníku | |
|
Velmi vzácné | ||
|
Vyšetření | ||
|
Vzácné |
Funkční vyšetření (test) plic snížené |
Pediatrická populace
Mezi populacemi pediatrických a dospělých pacientů léčených přípravkem Vantobra nebyl žádný rozdíl v bezpečnostním profilu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměrů přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Inhalační podání vede k nízké systémové biologické dostupnosti tobramycinu. Příznaky předávkování aerosolem mohou zahrnovat silný chrapot.
V případě náhodného požití přípravku Vantobra je toxicita nepravděpodobná, protože se tobramycin špatně absorbuje z intaktního gastrointestinálního traktu.
V případě neúmyslného podání přípravku Vantobra intravenózní cestou se mohou objevit známky a příznaky parenterálního předávkování tobramycinem včetně závratí, tinitu, vertiga, ztráty sluchu, dechové tísně a/nebo neuromuskulární blokády a zhoršení funkce ledvin.
Akutní toxicitu je nutné léčit okamžitým vysazením přípravku Vantobra a musí se provést základní testy funkce ledvin Při sledování předávkování může být užitečné vyšetření sérových koncentrací tobramycinu. V případě jakéhokoliv předávkování je zapotřebí zvážit možnost lékové interakce se změnami eliminace přípravku Vantobra nebo jiných léčivých přípravků.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antibakteriální látky pro systémové užití, aminoglykosidová antibiotika.
ATC kód: J01GB01
Mechanismus účinku
Tobramycin je aminoglykosidové antibiotikum, které produkují bakterie Streptomyces tenebrarius. Primárně působí tak, že narušuje syntézu proteinů, což má za následek pozměněnou permeabilitu buněčné membrány, progresivní narušení buněčného obalu a případně i buněčnou smrt. Působí baktericidně ve stejné nebo mírně vyšší koncentraci, než je inhibiční koncentrace.
Hraniční hodnoty
Stanovené limity citlivosti pro parenterální podání tobramycinu jsou nevhodné při podání léčivého přípravku ve formě aerosolu. Sputum pacientů s cystickou fibrózou vykazuje inhibiční účinek na lokální biologickou aktivitu rozprašovaných aminoglykosidů. To znamená, že koncentrace ve sputu po léčbě inhalovaným tobramycinem musí být desetkrát až dvacetpětkrát vyšší než minimální inhibiční koncentrace (MIK) jak pro supresi růstu P. aeruginosa, tak kontrolu baktericidní aktivity. V kontrolovaných klinických studiích dosahovaly u 97 % pacientů léčených tobramycinem ve formě roztoku k rozprašování koncentrace ve sputu 10násobku nejvyšší MIK P. aeruginosa kultivované od pacienta a 95 % pacientů léčených tobramycinem ve formě roztoku k rozprašování dosáhlo 25násobku nejvyšší MIK.
Citlivost
Protože konvenční limity citlivosti pro inhalační cestu podání nejsou k dispozici, musí se při definování organizmů jako citlivých, či necitlivých na nebulizovaný tobramycin postupovat opatrně.
V klinických studiích s přípravkem TOBI se u většiny pacientů s izoláty P. aeruginosa s MIK tobramycinu < 128 pg/ml ve výchozím stavu projevilo zlepšení funkce plic po léčbě přípravkem TOBI. Pacienti s izoláty/1. aeruginosa s MIK ^ 128 pg/ml ve výchozím stavu mají nižší pravděpodobnost vzniku klinické odpovědi. Zlepšení funkce plic se však objevilo u sedmi ze 13 pacientů (54%) v placebem kontrolovaných klinických studiích, kteří poskytli izoláty s MIK ^ 128 pg/ml během používání přípravku TOBI.
Na základě údajů in vitro a/nebo zkušeností z klinických studií lze očekávat, že organismy spojované s plicními infekcemi u cystické fibrózy budou reagovat na terapii přípravkem Vantobra následujícím způsobem:
|
Citlivé |
Pseudomonas aeruginosa Haemophilus influenzae Staphylococcus aureus |
|
Necitlivé |
Burkholderia cepacia Stenotrophomonas maltophilia Alcaligenes xylosoxidans |
Léčba s dávkovým režimem 28 dnů nasazení a 28 dnů vysazení přípravku v klinických studiích prokázala malé, ale jasné zvýšení MIK tobramycinu, amikacinu a gentamicinu pro testované izoláty P. aeruginosa. Každých dalších 6 měsíců léčby vedlo k aditivnímu zvýšení, v podobném rozsahu jako bylo zvýšení pozorované po 6 měsících v kontrolovaných klinických studiích. Impermeabilita, definovaná jako celková absence citlivosti na všechny aminoglykosidy, je mechanismem rezistence
na aminoglykosidy s největší prevalencí pozorovaným u P. aeruginosa izolované u pacientů s cystickou fibrózou chronicky infikovaných. P. aeruginosa izolovaná od pacientů s cystickou fibrózou rovněž manifestovala adaptivní rezistenci na aminoglykosidy, která se mění po skončení antibiotické léčby zpět na citlivost.
Další informace
Nej sou žádné důkazy o tom, že by pacienti léčení až po 18 měsíců roztokem tobramycinu k rozprašování byli více ohroženi infekcí B. cepacia, S. maltophilia nebo A. xylosoxidans, než by se očekávalo u neléčených pacientů. Druhy Aspergillus byly ze sputa léčených pacientů získávány častěji, přesto byly klinické následky, jako je alergická bronchopulmonální aspergilóza (ABPA), hlášeny vzácně a s podobnou frekvencí jako v kontrolní skupině.
Charakteristiky aerosolu
Tabulka 2: Srovnávací údaje klinické testovací a referenční šarže:
Vantobra/ruční inhalátor Tolero1, and TOBI/PARI LC PLUS2.
|
Parametr/léčivý přípravek/zařízení |
Vantobra/Tolero |
TOBI/PARI LC PLUS |
|
Celkem podáno léčivého přípravku [mg±SD] |
96 ± 4,4 |
101 ± 8,5 |
|
Vdechnutelná dávka < 5 pm [mg±SD] |
72 ± 6,5 |
65 ± 7,1 |
|
Rychlost podávání léčivého přípravku [mg/min] |
27 ± 5,0 |
7 ± 0,9 |
|
Hmotnostní střední aerodynamický průměr [pm ± SD] |
3,8 ± 0,3 |
3,6 ± 0,4 |
|
Geometrická směrodatná odchylka ±SD |
1,5 ± 0,0 |
2,3 ± 0,2 |
|
Doba rozprašování [min] |
3,9 ± 0,6 |
15,3 ± 0,6 |
Výsledky z dechové simulace a měření kaskádovým impaktorem.
1 ve spojení s regulátorem eBase nebo eFlow rapid
2 zapojeno s kompresorem PARI Boy SX
Rychlost dávkování léčivého přípravku Vantobra nezávisí na způsobu dýchání, tj. dospělý či dítě, což je v protikladu k inhalátoru PARI LC PLUS s tryskou.
Klinická účinnost a bezpečnost
Omezené údaje z jedné kontrolované klinické studie v průběhu jednoho léčebného cyklu naznačují, že zlepšení funkce plic zůstalo zachováno nad výchozím stavem během 28denního období bez léčby.
Na základě výsledků klinického hodnocení 12012.101 vzrostlo zlepšení funkce plic FEV 1 % vůči výchozímu stavu o 8,2 ± 9,4 % u přípravku Vantobra a o 4,8 ± 9,6 % u referenční terapie v prvním léčebném cyklu, což prokazuje noninferiorní účinnost (p=0,0005). Snížení CFU, jakožto ukazatel suprese P. aeruginosa, bylo srovnatelné u přípravku Vantobra a u referenčního přípravku.
5.2 Farmakokinetické vlastnosti
Absorpce a distribuce
Očekává se, že systémová expozice tobramycinu po inhalaci přípravku Vantobra pochází primárně z inhalovaného podílu léčivého přípravku, protože se tobramycin neabsorbuje po perorálním podání ve vyhodnotitelném množství. Inhalace rozprášeného tobramycinu vede k vysokým koncentracím ve sputu a nízkým hladinám v plazmě.
Srovnávací data aerosolu naleznete v tabulce 2 v bodě 5.1.
Na konci 4týdenního dávkovacího cyklu přípravku Vantobra (170 mg/1,7 ml dvakrát denně) u pacientů s cystickou fibrózou bylo dosaženo maximálních koncentrací tobramycinu v plazmě (Cmax) 1,27 ± 0,81 pg/ml přibližně za jednu hodinu po inhalaci. Koncentrace ve sputu byly vyšší a proměnlivější při Cmax 1,951 +
2,187 pg/g. Po podání jedné dávky přípravku Vantobra 170 mg zdravým dobrovolníkům bylo dosaženo Cmax
1,1 + 0,4 pg/ml po tmax přibližně 4 hodiny.
Distribuce
Tobramycin je vázán na plazmové proteiny z méně než 10 %.
Biotransformace
Tobramycin se nemetabolizuje a primárně se vylučuje do moči v nezměněném stavu.
Eliminace
Eliminace tobramycinu podávaného inhalační cestou nebyla zkoumána.
Po intravenózním podání se systémově absorbovaný tobramycin eliminuje glomerulární filtrací. Poločas eliminace tobramycinu ze séra je přibližně 2 hodiny.
Neabsorbovaný tobramycin se po inhalačním podání pravděpodobně primárně eliminuje ve vykašlávaném sputu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity, kancerogenního potenciálu a reprodukční a vývojové toxicity odhalily, že hlavním rizikem pro člověka je renální toxicita a ototoxicita. Ve studiích toxicity po opakovaném podání bylo prokázáno, že cílovými orgány toxicity jsou ledviny a vestibulární/kochleární funkce. Obecně platí, že se toxicita projevuje při vyšších systémových hladinách tobramycinu, než jsou hladiny dosažitelné inhalací doporučené klinické dávky.
S inhalačně podávaným tobramycinem nebyly prováděny žádné studie reprodukční toxicity. Subkutánní podání při dávkách 100 mg/kg/den u potkanů a maximální tolerovaná dávka 20 mg/kg/den u králíků během organogeneze nebyla teratogenní. Teratogenita nemohla být vyhodnocena při vyšších parenterálních dávkách u králíků, protože vyvolávala maternální toxicitu a potrat. Na základě dostupných údajů ze studií se zvířaty nelze vyloučit riziko toxicity (např. ototoxicity) při prenatálních hladinách expozice. Tobramycin nezhoršoval fertilitu u samců nebo samic potkanů při subkutánních dávkách až do 100 mg/kg/den.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný
Chlorid vápenatý
Síran hořečnatý
Kyselina sírová (k úpravě pH)
Hydroxid sodný (k úpravě pH)
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky v inhalátoru.
6.3 Doba použitelnosti
3 roky
Obsah jednodávkové ampulky použijte bezprostředně po otevření (viz bod 6.6).
Stabilita po otevření sáčku: 4 týdny, pokud je uchováván při teplotě do 25 °C
6.4 Zvláštní opatření pro uchovávání Uchovávejte v chladničce (2 °C - 8 °C).
Podmínky uchovávání tohoto léčivého přípravku po jeho prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Přípravek Vantobra je dodáván v polyetylenových (PE) ampulkách, které jsou baleny do uzavřeného sáčku z hliníkové fólie (8 ampulek na sáček).
Vnější krabička obsahuje:
• Jednu krabičku s léčivým přípravkem: 56 ampulek s roztokem k rozprašování v 7 sáčcích.
• Jedna krabičku s ručním inhalátorem Tolero.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Obsah jedné ampulky vyprázdněte do nádržky na lék ručního inhalátoru Tolero a podávejte inhalací, dokud v nádržce nezůstane žádný lék. Ruční inhalátor Tolero lze ovládat buď regulátorem eBase, nebo ovládací jednotkou eFlow rapid. Parametry výkonu ze studií pro charakterizaci aerosolu in vitro jsou shodné pro tyto dva regulátory a jsou uvedeny v bodě 5.1, tabulka 2.
• Rozprašování je nutné provádět v dobře větrané místnosti.
• Ruční inhalátor se musí udržovat během provozu ve vodorovné poloze.
• Pacient musí během inhalace sedět vzpřímeně. Inhalaci je zapotřebí provádět při normálním způsobu dýchání bez přerušení.
• Ruční inhalátor Tolero se musí vyčistit a dezinfikovat způsobem uvedeným v pokynech k použití zařízení.
Přípravek Vantobra je čirý až nažloutlý roztok, a je možné pozorovat určitou variabilitu zbarvení, která však neznamená ztrátu účinnosti, pokud bude přípravek uchováván podle doporučení.
Roztok přípravku Vantobra je sterilní, vodný přípravek určený pouze k jednorázovému použití. Obsah celé ampulky se musí použít ihned po otevření a jakýkoliv nespotřebovaný roztok se musí zlikvidovat, protože neobsahuje konzervační látky. Otevřená ampulka se nikdy nesmí uchovávat pro opakované použití.
Používejte nový ruční inhalátor Tolero pro každý léčebný cyklus (28 dnů léčby), inhalátor je dodáván s léčivým přípravkem.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
PARI Pharma GmbH Moosstrasse 3 D-82319 Starnberg Německo
Tel: +49 (0) 89 - 74 28 46 - 10
Fax: +49 (0) 89 - 74 28 46 - 30
E-mail: info@paripharma.com
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/932/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE 18 březen 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
PARI Pharma GmbH Lochhamer Schlag 21 82166 Graefelfing NĚMECKO
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivých přípravků je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 12 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
Vantobra 170 mg roztok k rozprašování Tobramycinum
Jedna ampulka o objemu 1,7 ml obsahuje tobramycinum 170 mg.
Pomocné látky: chlorid sodný, chlorid vápenatý, síran hořečnatý, voda na injekci, kyselina sírová a hydroxid sodný k úpravě pH.
Balení obsahuje
• jednu krabičku s: 56 ampulkami s roztokem k rozprašování v 7 sáčcích;
• jednu krabičku s ručním inhalátorem Tolero.
Před použitím si přečtěte jak příbalovou informaci přípravku Vantobra, tak pokyny k použití ručního inhalátoru Tolero.
Inhalační podání.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PARI Pharma GmbH Moosstrasse 3 D-82319 Starnberg Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/932/001
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Vantobra 170 mg
Vantobra 170 mg roztok k rozprašování Tobramycinum
Jedna ampulka o objemu 1,7 ml obsahuje tobramycinum 170 mg.
Pomocné látky: chlorid sodný, chlorid vápenatý, síran hořečnatý, voda na injekci kyselina sírová a hydroxid sodný k úpravě pH.
Balení obsahuje 56 ampulek s roztokem k rozprašování v 7 sáčcích.
Před použitím si přečtěte jak příbalovou informaci přípravku Vantobra, tak pokyny k použití ručního inhalátoru Tolero.
Inhalační podání.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Uchovávejte v chladničce.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PARI Pharma GmbH Moosstrasse 3 D-82319 Starnberg Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/932/001
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Vantobra 170 mg
Vantobra 170 mg roztok k rozprašování Tobramycinum
Jedna ampulka o objemu 1,7 ml obsahuje tobramycinum 170 mg.
Pomocné látky: chlorid sodný, chlorid vápenatý, síran hořečnatý, voda na injekci kyselina sírová a hydroxid sodný k úpravě pH.
Obsahuje 8 ampulek.
Před použitím si přečtěte jak příbalovou informaci přípravku Vantobra, tak pokyny k použití ručního inhalátoru Tolero.
Inhalační podání.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Uchovávejte v chladničce.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PARI Pharma GmbH Moosstrasse 3 D-82319 Starnberg Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/932/001
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU AMPULKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Vantobra 170 mg roztok k rozprašování Tobramycinum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
PARI Pharma GmbH
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE
c.s.:
5. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Vantobra 170 mg roztok k rozprašování
Tobramycinum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoliv další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoliv nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek V antobra a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Vantobra používat
3. Jak se přípravek Vantobra používá
4. Možné nežádoucí účinky
5. Jak přípravek Vantobra uchovávat
6. Obsah balení a další informace
1. Co je přípravek Vantobra a k čemu se používá Co je přípravek Vantobra
Přípravek Vantobra obsahuje antibiotikum nazývané tobramycin. Patří do třídy antibiotik nazývaných aminoglykosidy.
K čemu se přípravek Vantobra používá
Přípravek Vantobra se používá u pacientů s cystickou fibrózou ve věku 6 let a starších k léčbě plicních infekcí způsobených bakterií nazývanou Pseudomonas aeruginosa.
Pseudomonas aeruginosa je bakterie, která často infikuje plíce pacientů s cystickou fibrózou v určitém období jejich života. Pokud nebude infekce řádně léčena, bude nadále poškozovat plíce a způsobovat další dýchací obtíže.
Jak přípravek Vantobra působí
Když inhalujete přípravek Vantobra, antibiotikum se může dostat přímo do plic a bojovat s bakteriemi způsobujícími infekci. Působí tak, že narušuje tvorbu bílkovin, které bakterie potřebují ke stavbě svých buněčných stěn. To bakterie poškodí a nakonec i zabije.
2. Čemu musíte věnovat pozornost, než začnete přípravek Vantobra používat Nepoužívejte přípravek Vantobra:
• jestliže jste alergický(a) (přecitlivělý(á)) na tobramycin, na jakýkoliv typ aminoglykosidových antibiotik nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Pokud je to váš případ, oznamte to svému lékaři před použitím přípravku Vantobra.
Upozornění a opatření
Poraďte se se svým lékařem, pokud jste někdy měl(a) kterékoliv z následujících onemocnění:
• potíže se sluchem (včetně ušních šelestů a závratí)
• ledvinové obtíže
• pocit tísně na hrudi
• krev ve sputu (hlenovitá hmota, kterou vykašláváte)
• svalovou slabost, která přetrvává nebo se během doby zhoršuje, příznak většinou dávaný do souvislosti s onemocněními, jako jsou myastenie (svalová slabost) nebo Parkinsonova choroba.
Pokud se Vás týká cokoliv z výše uvedeného, oznamte to svému lékaři před použitím přípravku Vantobra.
Pokud budete mít obtíže se sluchem nebo s funkcí ledvin, lékař vám může odebrat vzorky krve, aby sledoval množství přípravku Vantobra ve vašem těle.
Vdechování léků může způsobit pocit tísně na hrudi v důsledku zúžení dýchacích cest, to se může přihodit i s přípravkem Vantobra. Lékař vám může doporučit používání jiných vhodných léčivých přípravků k rozšíření dýchacích cest před použitím přípravku Vantobra.
Kmeny bakterií Pseudomonas se mohou po určité době stát rezistentními na léčbu antibiotikem. To znamená, že přípravek Vantobra nemusí po určité době tak dobře působit, jak by měl. Jestliže se toho obáváte, pohovořte si o tom se svým lékařem.
Pokud rovněž používáte tobramycin nebo jiné injekční aminoglykosidové antibiotikum, může to zvyšovat riziko nežádoucích účinků a lékař to bude podle potřeby sledovat.
Děti
Léčivý přípravek není určen k použití u dětí mladších 6 let.
Další léčivé přípravky a přípravek Vantobra
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte nebo jste v nedávné době užíval(a), a to i o lécích, které jsou dostupné bez lékařského předpisu.
Následující léčiva neužívejte, pokud používáte přípravek Vantobra:
• furosemid, diuretikum (tablety zvyšující tvorbu a vylučování moči)
• j iná léčiva s diuretickým účinkem, j ako j e močovina nebo mannitol
• jiná léčiva, která mohou poškodit ledviny nebo sluch:
o amfotericin B, cefalotin, polymyxiny (slouží k léčbě mikrobiálních infekcí), cyklosporin, takrolimus (slouží ke snížení aktivity imunitního systému). Tato léčiva mohou poškodit ledviny. o sloučeniny platiny, jako je karboplatina a cisplatina (používají se k léčbě některých forem nádorových onemocnění). Tato léčiva mohou poškodit ledviny a sluch.
Dále uvedené léky mohou zvyšovat rizika škodlivých účinků, pokud vám budou podávány během současného injekčního podávání tobramycinu nebo jiných aminoglykosidových antibiotik:
• anticholinesterázy, jako jsou neostigmin a pyridostigmin (používané k léčbě svalové slabosti) nebo botulotoxin. Tyto léky mohou způsobovat propuknutí nebo zhoršení svalové slabosti.
Pokud užíváte jeden či více shora uvedených léků, informujte o tom svého lékaře předtím, než budete přípravek Vantobra užívat.
Přípravek Vantobra nemíchejte ani neřeďte s jiným lékem ve svém ručním inhalátoru Tolero, který je dodáván společně s přípravkem Vantobra.
Pokud používáte několik různých způsobů léčby cystické fibrózy, provádějte to v následujícím pořadí:
1. bronchodilatační terapie, například salbutamol
2. fyzioterapie hrudníku
3. jiné inhalované léky
4. přípravek Vantobra
Ověřte si toto pořadí u svého lékaře.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek používat.
Není známo, zda inhalování tohoto léčivého přípravku během těhotenství způsobuje nežádoucí účinky. Pokud budete tobramycin a další aminoglykosidová antibiotika dostávat ve formě injekce, může to poškodit nenarozené dítě a způsobit hluchotu a ledvinové obtíže.
Pokud kojíte, měla byste se poradit s lékařem, než začnete tento léčivý přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Neočekává se, že přípravek Vantobra ovlivní vaši schopnost řídit motorová vozidla nebo obsluhovat stroje.
3. Jak se přípravek Vantobra používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se
svým lékařem.
Doporučená dávka přípravku jsou dvě ampulky každý den (jedna ráno a jedna večer) po 28 dnů.
• Dávka je stejná pro všechny osoby ve věku 6 let a starší.
• Inhalujte ústy celý obsah jedné ampulky ráno a druhé ampulky večer pomocí ručního inhalátoru Tolero.
• Nejlepší je, aby se interval mezi dvěma dávkami co nejvíce blížil 12 hodinám, ale tento interval musí být nejméně 6 hodin.
• Poté, co jste užíval(a) svůj přípravek po 28 dnů, následuje 28denní přestávka, během níž nebudete inhalovat žádný přípravek Vantobra. Poté zahájíte další cyklus (jak je znázorněno na obrázku).
• Je důležité, abyste dodržoval(a) užívání přípravku dvakrát denně během 28 dnů s léčbou a abyste dodržoval(a) 28denní cyklus s léčbou /28denní cyklus bez léčby.

|
S přípravkem Vantobra |
BEZ přípravku Vantobra |
|
Používejte přípravek Vantobra dvakrát denně po 28 dní |
Dalších 28 dnů přípravek Vantobra nepoužívejte |

Opakujte cyklus
Pokračujte v užívání přípravku Vantobra tímto cyklickým způsobem tak dlouho, jak Vám lékař určí.
Pokud budete mít dotazy ohledně délky používání přípravku Vantobra, obraťte se na svého lékaře nebo lékárníka.
Příprava přípravku Vantobra k inhalaci
• Přípravek Vantobra používejte pouze s ručním inhalátorem Tolero zobrazeným na následujícím obrázku, abyste měli jistotu, že inhalujete správnou dávku. Nepoužívejte ruční inhalátor Tolero pro žádný jiný lék.
• Před použitím si přečtěte pokyny k použití dodávané s ručním inhalátorem.
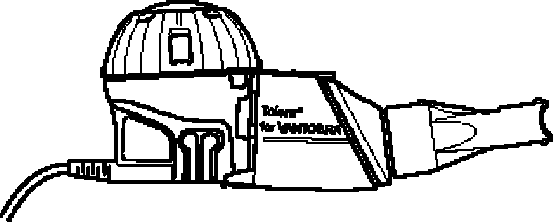
• Ujistěte se, že máte regulátor eFlowrapid nebo eBase k připojení ručního inhalátoru Tolero. Příslušný regulátor vám může předepsat ošetřující lékař nebo si jej můžete zakoupit samostatně.
• Omyjte si důkladně ruce mýdlem a vodou.
• Vyjměte jednu ampulku přípravku Vantobra ze sáčku z hliníkové folie těsně před inhalací.
• Uchovávejte zbývající přípravek v chladničce v původní krabičce.
• Položte všechny díly ručního inhalátoru Tolero na čistý, suchý papírový nebo látkový ručník. Dbejte na to, aby byl ruční inhalátor na rovném, stabilním povrchu.
• Sestavte ruční inhalátor Tolero podle obrázku v pokynech pro použití ručního inhalátoru.
• Držte ampulku ve svislé poloze a lehce na ní poklepejte předtím, než odlomíte horní část ampulky, aby nedošlo k rozlití. Vyprázdněte obsah jedné ampulky do nádobky na lék ručního inhalátoru.
• Začněte s léčbou vsedě ve vzpřímené poloze v dobře větrané místnosti. Držte ruční inhalátor ve vodorovné poloze a normálně dýchejte ústy. Nedýchejte nosem. Pokračujte v pohodlném nadechování a vydechování až do ukončení léčby. Po podání veškerého léku uslyšíte tón „léčba dokončena“.
• Pokud budete potřebovat léčbu z jakéhokoliv důvodu přerušit, stiskněte a přidržte tlačítko On/Off (zapnuto/vypnuto) na celou jednu sekundu. Chcete-li léčbu začít znovu, stiskněte a přidržte tlačítko On/Off (zapnuto/vypnuto) opět na jednu celou sekundu, aby se léčba obnovila.
• Ruční inhalátor Tolero se musí vyčistit a dezinfikovat způsobem uvedeným v pokynech k použití přístroje.
• Používejte nový ruční inhalátor Tolero pro každý léčebný cyklus (28 dnů s léčbou), inhalátor je dodáván s léčivým přípravkem.
Nepoužívejte alternativní netestovaný inhalační systém, protože může změnit množství léčiva, které se dostane do plic. To může ovlivnit účinnost a bezpečnost přípravku.
Jestliže jste použil(a) více přípravku Vantobra, než jste měl(a)
Pokud vdechnete příliš mnoho přípravku Vantobra, může se objevit chraptivý hlas. Informujte o tom co nejdříve svého lékaře. Pokud přípravek Vantobra požijete, je nepravděpodobné, že by to způsobilo závažné obtíže, protože se tobramycin ze žaludku špatně absorbuje, přesto o tom co nejdříve informujte svého lékaře.
Jestliže jste zapomněl(a) použít přípravek Vantobra
Pokud zapomenete použít přípravek Vantobra a zbývá-li do vaší příští dávky více než 6 hodin, použijte svoji dávku co nejdříve. Jinak počkejte na svoji další dávku. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat přípravek Vantobra
Nepřestávejte používat přípravek Vantobra, pokud vám to ošetřující lékaře nepřikáže, jinak byste mohl(a) ztratit dostatečnou kontrolu nad plicní infekcí a mohlo by dojít k jejímu zhoršení.
Máte-li jakékoliv další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Některé nežádoucí účinky mohou být závažné
• tíseň na hrudi s dýchacími obtížemi (vzácné, postihuje až 1 z 1000 osob)
• alergické reakce včetně kopřivky a svědění (velmi vzácné, postihují až 1 z 10000 osob).
Pokud se objeví kterýkoliv z výše uvedených účinků, přestaňte používat přípravek Vantobra a okamžitě o tom informujte svého lékaře.
Lidé s cystickou fibrózou mohou mít mnoho příznaků nemoci. Ty se mohou stále vyskytovat během používání přípravku Vantobra, ale neměly by být tak časté nebo horší než dříve.
Pokud se vám bude zdát, že se základní plicní onemocnění zhoršilo v době, kdy používáte přípravek Vantobra, informujte o tom neprodleně svého lékaře.
Mezi ostatní nežádoucí účinky může patřit:
Méně časté (mohou postihnout až 1 osobu ze 100)
• dušnost
• změna hlasu (chrapot)
• zhoršení kašle
• bolesti v krku
Vzácné (mohou postihnout až 1 osobu z 1000)
• laryngitida (zánět hrtanu, který může způsobit změnu hlasu, bolesti v krku a polykací obtíže)
• ztráta hlasu
• bolest hlavy, slabost
• krvácení z nosu, zvýšená nosní sekrece (rýma)
• ušní šelest (normálně přechodný), ztráta sluchu, závrať
• vykašlávání krve, větší tvorba sputa než obvykle, nepříjemný pocit na hrudi, astma, horečka
• poruchy chuti, pocit na zvracení (nauzea), vředy v ústech, zvracení, ztráta chuti k jídlu
• vyrážka
• bolest na hrudi nebo celková bolest
• zhoršení výsledků při testu funkce plic
Velmi vzácné (mohou postihnout až 1 osobu z 10000)
• kvasinkové infekce v ústech či krku, například moučnivka
• otok lymfatických žláz
• spavost
• bolesti ucha, problémy s uchem
• hyperventilace, nízké hladiny kyslíku v krvi, zánět vedlejších nosních dutin
• průjem, bolest žaludku a v okolí žaludku
• červené puchýřky, pupeny na kůži
• kopřivka, svědění
• bolest zad
• celkový pocit nevolnosti
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Vantobra uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na ampulce, sáčku nebo na krabičce za „Použitelné do“ nebo „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce. v
Uchovávejte v chladničce (2 °C - 8 °C). Pokud nebudete mít k dispozici chladničku (například když lék přepravujete), můžete uchovávat krabičku s lékem (otevřené a neotevřené sáčky) při teplotě do 25 °C až po 4 týdny. Pokud byl přípravek uchováván při pokojové teplotě delší dobu než 4 týdny, musí být rovněž zlikvidován podle místních požadavků.
Nepoužívejte tento přípravek, pokud si všimnete, že se roztok zakalil, nebo pokud jsou v něm částice.
Otevřenou ampulku nikdy neuchovávejte. Po otevření ampulku okamžitě použijte a jakýkoliv zbývající přípravek je nutné zlikvidovat.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Vantobra obsahuje
- Léčivou látkou je tobramycinum. Jedna ampulka obsahuje tobramycinum 170 mg jako jednotlivou dávku.
- Dalšími složkami (pomocnými látkami) jsou: chlorid sodný, chlorid vápenatý, síran hořečnatý, voda na injekci, kyselina sírová a hydroxid sodný k úpravě pH.
Jak přípravek Vantobra vypadá a co obsahuje toto balení
Přípravek Vantobra roztok k rozprašování je dodáván v ampulce připravený k použití.
Přípravek Vantobra je čirý až nažloutlý roztok, který se může změnit až na tmavší žlutou barvu. Tím se nezmění způsob, jak přípravek Vantobra působí, za předpokladu, že byly dodrženy pokyny k uchovávání.
Ampulky jsou baleny do sáčků. Jeden sáček obsahuje osm ampulek, což vystačí na čtyři dny léčby.
Přípravek Vantobra je k dispozici s ručním inhalátorem Tolero. Dodává se v krabičce, která obsahuje dvě vnitřní krabičky, v jedné je léčivý přípravek (56 ampulek s roztokem k rozprašování v 7 sáčcích) a v druhém je ruční inhalátor. Balení stačí na jeden cyklus léčby trvající 28 dnů.
Držitel rozhodnutí o registraci a výrobce
PARI Pharma GmbH Moosstrasse 3 D-82319 Starnberg Německo
Tel: +49 89 - 74 28 46 - 10
Fax: +49 89 - 74 28 46 30
E-mail: info@paripharma.com
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Deutschland und Osterreich
PARI Pharma GmbH Lochhamer Schlag 21 D-82166 Gráfelfing Tel.: +49 89 - 74 28 46 - 10 E-Mail: info@paripharma.com
Italia
NEUPHARMA S.r.l. Via Lorenzo Respighi 7 I-00197 Roma>
Tel: +39 0542 26540
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
Příloha IV
• Podobnost
Výbor CHMP je toho názoru, že přípravek Vantobra je podobný registrovanému léčivému přípravku (registrovaným léčivým přípravkům) pro vzácná onemocnění ve smyslu článku 3 nařízení Komise (ES) č. 847/2000, jak je podrobněji popsáno v Evropské veřejné zprávě o hodnocení.
• Odlišnost
Výbor CHMP je toho názoru, že v souladu s článkem 8 nařízení (ES) č. 141/2000 a článkem 3 nařízení Komise (ES) č. 847/2000 byla doložena tato odlišnost uvedená v čl. 8 odst. 3 uvedeného nařízení, jak je podrobněji popsáno v Evropské veřejné zprávě o hodnocení: žadatel v žádosti prokázal, že léčivý přípravek, třebaže je podobný registrovanému léčivému přípravku Tobi Podhaler, je bezpečnější, účinnější nebo jinak klinicky nadřazený (jak je definováno v článku 3 nařízení Komise (ES) č. 847/2000) pro stejnou léčebnou indikaci.
33