Uptravi 1200 Mikrogramů
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Uptravi 200 mikrogramů potahované tablety Uptravi 400 mikrogramů potahované tablety Uptravi 600 mikrogramů potahované tablety Uptravi 800 mikrogramů potahované tablety Uptravi 1 000 mikrogramů potahované tablety Uptravi 1 200 mikrogramů potahované tablety Uptravi 1 400 mikrogramů potahované tablety Uptravi 1 600 mikrogramů potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Uptravi 200 mikrogramů potahované tablety
Jedna potahovaná tableta obsahuje selexipagum 200 mikrogramů.
Uptravi 400 mikrogramů potahované tablety
Jedna potahovaná tableta obsahuje selexipagum 400 mikrogramů.
Uptravi 600 mikrogramů potahované tablety
Jedna potahovaná tableta obsahuje selexipagum 600 mikrogramů.
Uptravi 800 mikrogramů potahované tablety
Jedna potahovaná tableta obsahuje selexipagum 800 mikrogramů.
Uptravi 1 000 mikrogramů potahované tablety
Jedna potahovaná tableta obsahuje selexipagum 1 000 mikrogramů.
Uptravi 1 200 mikrogramů potahované tablety
Jedna potahovaná tableta obsahuje selexipagum 1 200 mikrogramů.
Uptravi 1 400 mikrogramů potahované tablety
Jedna potahovaná tableta obsahuje selexipagum 1 400 mikrogramů.
Uptravi 1 600 mikrogramů potahované tablety
Jedna potahovaná tableta obsahuje selexipagum 1 600 mikrogramů.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta
Uptravi 200 mikrogramů potahované tablety
Kulaté, světle žluté, potahované tablety na jedné straně s vyraženým “2”. Uptravi 400 mikrogramů potahované tablety
Kulaté, červené, potahované tablety na jedné straně s vyraženým “4”. Uptravi 600 mikrogramů potahované tablety
Uptravi 800 mikrogramu potahované tablety
Kulaté, zelené, potahované tablety na jedné straně s vyraženým “8”.
Uptravi 1 000 mikrogramů potahované tablety
Kulaté, oranžové, potahované tablety na jedné straně s vyraženým “10”. Uptravi 1 200 mikrogramů potahované tablety
Kulaté, tmavě fialové, potahované tablety na jedné straně s vyraženým “12”. Uptravi 1 400 mikrogramů potahované tablety
Kulaté, tmavě žluté, potahované tablety na jedné straně s vyraženým “14”.
Uptravi 1 600 mikrogramů potahované tablety
Kulaté, hnědé, potahované tablety na jedné straně s vyraženým “16”.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Uptravi je indikován k dlouhodobé léčbě plicní arteriální hypertenze (PAH) u dospělých pacientů s funkční klasifikací II—III WHO, a to buď v kombinované terapii u pacientů, u nichž není dostatečná léčba antagonistou endothelinového receptoru (ERA) a/nebo inhibitorem fosfodiesterázy typu 5 (PDE-5), nebo v monoterapii u pacientů, kteří nejsou kandidáty pro tyto terapie.
Účinnost byla prokázána u populace s PAH, včetně idiopatické a dědičné PAH, PAH spojené s poruchami pojivové tkáně a PAH spojené s upravenou prostou vrozenou srdeční vadou (viz bod 5.1).
4.2 Dávkování a způsob podání
Léčbu smí zahájit a sledovat pouze lékař se zkušenostmi s léčbou PAH.
Dávkování
Individuální titrace dávky
U každého pacienta je nutno provést vzestupnou titraci na nejvyšší individuálně tolerovanou dávku, která se může pohybovat od 200 mikrogramů podávaných dvakrát denně do 1 600 mikrogramů podávaných dvakrát denně (individualizovaná udržovací dávka).
Doporučená zahajovací dávka je 200 mikrogramů podávaných dvakrát denně s odstupem přibližně 12 hodin. Tato dávka se zvyšuje po 200 mikrogramech podávaných dvakrát denně, obvykle v týdenních intervalech. Na začátku léčby a při každém kroku vzestupné titrace se doporučuje užít první dávku večer. Během titrace dávky se mohou objevit některé nežádoucí účinky, které odrážejí mechanismus účinku přípravku Uptravi (jako je bolest hlavy, průjem, nauzea a zvracení, bolest čelisti, myalgie, bolest končetin, artralgie a zrudnutí). Jsou obvykle přechodné nebo zvládnutelné symptomatickou léčbou (viz bod 4.8). Pokud však pacient dosáhne dávky, kterou netoleruje, je nutno dávku snížit na předchozí úroveň.
U pacientů, u kterých byla vzestupná titrace omezena z jiných důvodů, než jsou nežádoucí účinky odrážející mechanismus účinku přípravku Uptravi, lze zvážit druhý pokus o pokračování ve vzestupné titraci na nejvyšší individuálně tolerovanou dávku do maximální dávky 1 600 mikrogramů dvakrát denně.
Individualizovaná udržovací dávka
Nejvyšší tolerovaná dávka dosažená během titrace se má udržovat. Pokud bude léčba časem při dané dávce hůře snášena, je nutno zvážit symptomatickou léčbu a/nebo snížení dávky na nejbližší nižší dávku.
Přerušení a ukončení léčby
Pokud se dávka vynechá, má se užít co nejdříve. Vynechaná dávka se nemá užít, pokud je další dávka plánována v průběhu přibližně 6 hodin.
Pokud se léčba vynechá na 3 nebo více dní, podávání přípravku Uptravi se má znovu zahájit s nižší dávkou a tu pak titrovat.
S náhlým ukončením podávání přípravku Uptravi u pacientů s PAH jsou omezené zkušenosti. Důkazy akutního rebound fenoménu nebyly pozorovány.
Pokud se však přijme rozhodnutí o vysazení přípravku Uptravi, má se provést postupně při současném zavádění alternativní léčby.
Starší pacienti (> 65 let)
U starších lidí není nutná žádná úprava dávkovacího režimu (viz bod 5.2). U pacientů starších 75 let jsou klinické zkušenosti omezené, proto se u této populace přípravek Uptravi má používat opatrně (viz bod 4.4).
Porucha funkce jater
Přípravek Uptravi se nemá podávat pacientům s těžkou poruchou funkce jater (Child-Pugh třída C; viz bod 4.4). U pacientů se středně těžkou poruchou funkce jater (Child-Pugh třída B) má být zahajovací dávka přípravku Uptravi 200 mikrogramů jednou denně a ta se zvyšuje v týdenních intervalech o 200 mikrogramů podávaných jednou denně do výskytu nežádoucích účinků, které odrážejí mechanismus účinku selexipagu, jež pacient nemůže tolerovat nebo je nelze lékařsky zvládnout.
U pacientů s mírnou poruchou funkce jater (Child-Pugh třída A) není úprava dávkovacího režimu nutná.
Porucha funkce ledvin
U pacientů s mírnou nebo středně těžkou poruchou ledvin není úprava dávkovacího režimu nutná.
U pacientů s těžkou poruchou funkce ledvin (odhadovaná rychlost glomerulární filtrace [eGFR]
< 30 ml/min/1,73 m2) není změna zahajovací dávky nutná; titrace dávek se u těchto pacientů má provádět s opatrností (viz bod 4.4).
Pediatrická populace (< 18 let)
Bezpečnost a účinnost přípravku Uptravi u dětí ve věku od 0 do méně než 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje. Podávání selexipagu u pediatrické populace se nedoporučuje. Studie na zvířatech ukazují na zvýšené riziko intususcepce, nicméně klinický význam těchto zjištění není znám (viz bod 5.3).
Způsob podání
Perorální podání.
Potahované tablety se užívají perorálně ráno a večer. Ke zlepšení snášenlivosti se doporučuje užívat přípravek Uptravi s jídlem a na začátku každé fáze vzestupné titrace užívat první zvýšenou dávku večer.
Tablety se nesmí dělit, drtit ani žvýkat a mají se zapíjet vodou.
Pacienty se slabým zrakem nebo nevidomé pacienty je nutno poučit, aby si během titračního období při užívání přípravku Uptravi vyžádali pomoc další osoby.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Těžká ischemická choroba srdeční nebo nestabilní angina pectoris.
• Infarkt myokardu v posledních 6 měsících.
• Dekompenzované srdeční selhání, pokud není pod pečlivým lékařským dohledem.
• Závažné arytmie.
• Cerebrovaskulární příhody (např. tranzitorní ischemická ataka, cévní mozková příhoda) v posledních 3 měsících.
• Vrozené nebo získané vady chlopní s klinicky relevantními poruchami funkce myokardu, které nesouvisí s plicní hypertenzí.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek Uptravi má vasodilatační vlastnosti, které mohou vést ke snížení krevního tlaku. Předtím, než se přípravek Uptravi předepíše, mají lékaři pečlivě uvážit, zda pacienti s určitými základními chorobami nemohou být vasodilatačními účinky nepříznivě postiženi (např. pacienti na antihypertenzní léčbě nebo pacienti s klidovou hypotenzí, hypovolémií, závažnou obstrukcí výtoku z levé komory nebo s autonomní dysfunkcí).
Hyperthyroidismus
U přípravku Uptravi byl pozorován hyperthyroidismus. Jsou-li přítomny známky nebo příznaky hyperthyroidismu, doporučují se podle klinické indikace testy funkce štítné žlázy.
Plicní venookluzivní choroba
U vasodilatačních látek (zejména prostacyklinů) byly při použití u pacientů s plicní venookluzivní chorobou hlášeny případy plicního edému. Pokud se tedy při podávání přípravku Uptravi pacientům s PAH objeví známky plicního edému, je nutno zvážit možnost plicní venookluzivní choroby. Pokud se tato domněnka potvrdí, léčbu přípravkem Uptravi je nutno ukončit.
Starší pacienti (> 65 let)
U pacientů ve věku nad 75 let jsou se selexipagem jen omezené klinické zkušenosti, proto se u této populace má přípravek Uptravi používat s opatrností (viz bod 4.2).
Porucha funkce jater
U pacientů s těžkou poruchou funkce jater (Child-Pugh třída C) nejsou se selexipagem žádné klinické zkušenosti, proto se těmto pacientům přípravek Uptravi nemá podávat. U subjektů se středně těžkou poruchou funkce jater (Child-Pugh třída B; viz bod 5.2) je expozice selexipagu a jeho aktivnímu metabolitu zvýšena. U pacientů se středně těžkou poruchou funkce jater se přípravek Uptravi má dávkovat jednou denně (viz bod 4.2).
Porucha funkce ledvin
U pacientů s těžkou poruchou funkce ledvin (eGFR < 30 ml/min/1,73 m2) se má při titraci dávek postupovat s opatrností. S přípravkem Uptravi u pacientů na dialýze nejsou žádné zkušenosti (viz bod 5.2), proto se u nich přípravek Uptravi nemá používat.
Ženy ve fertilním věku
Ženy ve fertilním věku mají během léčby selexipagem používat účinnou antikoncepci.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vliv jiných léčivých přípravků na selexipag
Selexipag se hydrolyzuje na aktivní metabolit jatemí karboxylesterázou 1 (CES1; viz bod 5.2). Selexipag a jeho aktivní metabolit podléhají oxidační metabolizaci zprostředkované CYP2C8 a CYP3A4. Glukuronidace aktivního metabolitu je katalyzována UGT1A3 a UGT2B7. Selexipag a jeho aktivní metabolit jsou substráty OATP1B1 a OATP1B3. Selexipag je slabým substrátem efluxní pumpy P-gp. Aktivní metabolit je slabým substrátem proteinu rezistence karcinomu prsu (breast cancer resistance protein - BCRP).
Farmakokinetika selexipagu a jeho aktivního metabolitu není ovlivněna warfarinem.
Inhibitory nebo induktory CYP2C8, UGT1A3 a UGT2B7
Vliv inhibitorů CYP2C8 (gemfibrozil), inhibitorů UGT1A3 a UGT2B7 (kyselina valproová, probenecid a flukonazol), induktorů CYP2C8 (rifampicin, rifapentin) nebo induktorů UGT1A3 a UGT2B7 (rifampicin) na expozici selexipagu a jeho aktivnímu metabolitu nebyl studován. Při podávání těchto léčivých přípravků současně s přípravkem Uptravi je nutná opatrnost. Potenciální farmakokinetické interakce se silnými inhibitory nebo induktory těchto enzymů nelze vyloučit.
Inhibitory a induktory CYP3A4
Za přítomnosti 400/100 mg lopinaviru/ritonaviru dvakrát denně, což jsou silné inhibitory CYP3A4, se expozice selexipagu zvýšila přibližně 2krát, zatímco expozice aktivnímu metabolitu selexipagu se nezměnila. S ohledem na 37násobně vyšší potenci aktivního metabolitu není tento účinek klinicky relevantní. Jelikož silný inhibitor CYP3A4 neměl na farmakokinetiku aktivního metabolitu vliv, což naznačuje, že metabolizace prostřednictvím CYP3A4 není při eliminaci aktivního metabolitu důležitá, žádný účinek induktorů CYP3A4 na farmakokinetiku aktivního metabolitu se neočekává.
Specifické terapie PAH
V placebem kontrolované klinické studii fáze 3 u pacientů s PAH vedlo použití selexipagu v kombinaci jak s ERA, tak s inhibitorem PDE-5 k o 30 % nižší expozici aktivnímu metabolitu.
Inhibitory transportéru (lopinavir/ritonavir)
Za přítomnosti 400/100 mg lopinaviru/ritonaviru dvakrát denně, což jsou silné inhibitory OATP (OATP1B1 a OATP1B3) a P-gp, se expozice selexipagu zvýšila přibližně 2krát, zatímco expozice aktivnímu metabolitu selexipagu se nezměnila. S ohledem na to, že většina farmakologického účinku je zprostředkována aktivním metabolitem, není tento účinek klinicky relevantní.
Vliv selexipagu na jiné léčivé přípravky
Selexipag a jeho aktivní metabolit v klinicky relevantních koncentracích neinhibují enzymy cytochromu P450. Selexipag a jeho aktivní metabolit neinhibují transportní proteiny. Nepředpokládá se, že by selexipag a jeho aktivní metabolit v klinicky relevantních koncentracích indukoval enzymy cytochromu P450 v játrech a ledvinách. In vitro data naznačují, že by selexipag mohl být induktorem jak CYP3A4, tak CYP2C9 ve střevě.
Antikoagulancia nebo inhibitory agregace trombocytů
Selexipag je in vitro inhibitorem agregace trombocytů. V placebem kontrolované studii fáze 3 u pacientů s PAH nebylo u selexipagu v porovnání s placebem zjištěno zvýšení rizika krvácení, včetně situací, kdy byl selexipag podáván s antikoagulancii (jako je heparin, antikoagulancia kumarinového typu) nebo inhibitory agregace trombocytů. Ve studii u zdravých subjektů selexipag (400 mikrogramů dvakrát denně) po jedné dávce 20 mg warfarinu nezměnil expozici S-warfarinu (substrát CYP2C9) ani R-warfarinu (substrát CYP3A4). Selexipag neovlivňoval farmakodynamické účinky warfarinu na mezinárodní normalizovaný poměr.
Hormonální kontraceptiva
Specifické studie lékových interakcí s hormonálními kontraceptivy nebyly provedeny. Jelikož selexipag neměl vliv na expozici substrátu CYP3A4 R-warfarinu ani substrátu CYP2C9 S-warfarinu, snížení účinnosti hormonálních kontraceptiv se nepředpokládá.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Ženy ve fertilním věku musí během léčby selexipagem používat účinnou antikoncepci. Těhotenství
Údaje o podávání selexipagu těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky. U zvířecích druhů použitých ve studiích reprodukční toxicity vykazoval selexipag a jeho aktivní metabolit in vitro 20 až 80 krát nižší účinnost na prostacyklinový (IP) receptor ve srovnání s účinností u lidí. Proto je bezpečnostní rozpětí s ohledem na potenciální účinky na reprodukci související s IP receptorem nižší než pro potenciální účinky nesouvisející s IP receptorem (viz bod 5.3).
Podávání přípravku Uptravi se v těhotenství a u žen v reprodukčním věku, které nepoužívají antikoncepci, nedoporučuje.
Kojení
Není známo, zda se selexipag nebo jeho metabolity vylučují do lidského mateřského mléka. U potkanů se selexipag nebo jeho metabolity do mléka vylučují (viz bod 5.3). Riziko pro kojené děti nelze vyloučit. Přípravek Uptravi se během kojení nemá užívat.
Fertilita
K dispozici nejsou žádné klinické údaje. Ve studiích na potkanech selexipag vyvolával přechodné poruchy ovulačního cyklu, které neměly vliv na plodnost (viz bod 5.3). Význam tohoto zjištění u lidí není znám.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Uptravi má mírný vliv na schopnost řídit nebo obsluhovat stroje. Při zvažování pacientovy schopnosti řídit nebo obsluhovat stroje je nutné vzít v úvahu klinický stav pacienta a profil nežádoucích účinků selexipagu (jako je bolest hlavy nebo hypotenze).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nejčastěji hlášenými nežádoucími účinky jsou bolest hlavy, průjem, nauzea a zvracení, bolest čelisti, myalgie, bolest končetin, artralgie a zrudnutí. Tyto účinky jsou častější během fáze titrace dávky. Většina těchto účinků je mírné až střední intenzity.
Tabulkový seznam nežádoucích účinků
Bezpečnost selexipagu byla hodnocena v dlouhodobé, placebem kontrolované studii fáze 3, do které bylo zařazeno 1 156 pacientů se symptomatickou PAH. Střední hodnota doby trvání léčby byla 76,4 týdne (medián 70,7 týdne) u pacientů léčených selexipagem versus 71,2 týdne (medián 63,7 týdne) u pacientů léčených placebem. Expozice selexipagu trvala až 4,2 roku.
Dále jsou uvedeny nežádoucí účinky zjištěné v pivotní klinické studii. V každé skupině četnosti jsou nežádoucí účinky uvedeny podle klesající závažnosti.
|
Třída orgánových systémů |
Velmi časté (> 1/10) |
Časté (> 1/100 až < 1/10) |
Méně časté (> 1/1 000 až < 1/100) |
|
Poruchy krve a lymfatického systému |
Snížení hemoglobinu | ||
|
Endokrinní poruchy |
Hyperthyroidismus Snížení thyreostimulačního hormonu (viz bod 4.4) | ||
|
Poruchy metabolismu a výživy |
Snížení chuti k jídlu Snížení tělesné hmotnosti | ||
|
Poruchy nervového systému |
Bolest hlavy* | ||
|
Srdeční poruchy |
Sinusová tachykardie (viz bod 4.4) | ||
|
Cévní poruchy |
Zrudnutí* |
Hypotenze (viz bod 4.4) | |
|
Respirační, hrudní a mediastinální poruchy |
Nazofaryngitida (neinfekční) |
Ucpaný nos | |
|
Gastrointestinální poruchy |
Průjem* Zvracení * Nauzea* | ||
|
Poruchy kůže a podkožní tkáně |
Kopřivka Erytém | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolest čelisti* Myalgie* Artralgie* Bolest končetin* | ||
|
Celkové poruchy a reakce v místě aplikace |
Bolest |
* Viz bod Popis vybraných nežádoucích účinků.
Popis vybraných nežádoucích účinků
Farmakologické účinky spojené s titrací a udržovací léčbou
Nežádoucí účinky související s mechanismem účinku selexipagu byly pozorovány často, zejména během fáze individuální titrace dávky a jsou uvedeny v následující tabulce:
|
Nežádoucí účinky podobné účinkům prostacyklinu |
Titrace |
Udržovací léčba | ||
|
Selexipag |
Placebo |
Selexipag |
Placebo | |
|
64 % |
28 % |
40 % |
20 % | |
|
36 % |
12 % |
30 % |
13 % | |
|
29 % |
13 % |
20 % |
10 % | |
|
Bolest čelisti |
26 % |
4 % |
21 % |
4 % |
|
Myalgie |
15 % |
5 % |
9 % |
3 % |
|
Bolest končetin |
14 % |
5 % |
13 % |
6 % |
|
14 % |
4 % |
8 % |
6 % | |
|
Zrudnutí |
11 % |
4 % |
10 % |
3 % |
|
Artralgie |
7 % |
5 % |
9 % |
5 % |
Tyto účinky jsou obvykle přechodné nebo zvládnutelné symptomatickou léčbou. Léčbu kvůli těmto nežádoucím účinkům přerušilo 7,5 % pacientů léčených selexipagem. Přibližná míra výskytu závažných nežádoucích účinků byla 2,3 % ve skupině léčené selexipagem a 0,5 % ve skupině léčené placebem. V klinické praxi bylo pozorováno, že gastrointestinální příhody reagují na protiprůjmové a antiemetické přípravky a na přípravky proti nauzee a/nebo na léčivé přípravky na funkční gastrointestinální poruchy. Příhody spojené s bolestí byly často léčeny analgetiky (jako je paracetamol).
Pokles hemoglobinu
V placebem kontrolované studii fáze 3 u pacientů s PAH se střední hodnota absolutních změn hemoglobinu při pravidelných návštěvách v porovnání s výchozími hodnotami pohybovala od -0,34 do -0,02 g/dl ve skupině léčené selexipagem v porovnání s -0,05 až 0,25 g/dl ve skupině léčené placebem. Pokles hemoglobinu z výchozí koncentrace na hodnoty pod 10 g/dl byl hlášen u 8,6 % pacientů léčených selexipagem a u 5,0 % pacientů léčených placebem.
Testy funkce štítné žlázy
V placebem kontrolované studii fáze 3 u pacientů s PAH byl hyperthyroidismus hlášen u 1,6 % pacientů ve skupině léčené selexipagem v porovnání s žádným případem ve skupině léčené placebem (viz bod 4.4). Při většině návštěv bylo u skupiny léčené selexipagem pozorováno snížení (až do -0,3 MU/l z výchozího mediánu 2,5 MU/l) mediánu thyreostimulujícího hormonu. Ve skupině léčené placebem byla zřejmá malá změna mediánu hodnot. V žádné ze skupin nedošlo k významné změně trijodothyroninu či thyroxinu.
Zvýšení srdeční frekvence
V placebem kontrolované studii fáze 3 u pacientů s PAH bylo 2 až 4 hodiny po podání dávky pozorováno přechodné zvýšení střední hodnoty srdeční frekvence o 3 až 4 tepy za minutu. Vyšetření elektrokardiogramu ukázalo sinusovou tachykardii u 11,3 % pacientů ve skupině léčené selexipagem v porovnání s 8,8 % ve skupině léčené placebem (viz též body 4.4 a 5.1).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Byly hlášeny ojedinělé případy předávkování až 3 200 mikrogramy. Jediným hlášeným důsledkem byla mírná, přechodná nauzea. Při předávkování se podle potřeby musí přijmout podpůrná opatření. Dialýza je pravděpodobně neúčinná, protože selexipag a jeho aktivní metabolit jsou silně vázány na proteiny.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antikoagulancia, antitrombotika, kromě heparinu, ATC kód: B01AC27 Mechanismus účinku
Selexipag je selektivním agonistou receptoru IP, který se liší od prostacyklinu a jeho analogů. Selexipag se hydrolyzuje prostřednictvím CES1 na aktivní metabolit, který je přibližně 37násobně účinnější, než selexipag. Selexipag a aktivní metabolit jsou agonisté receptoru IP s vysokou afinitou a vysokou selektivitou k receptoru IP v porovnání s jinými prostanoidovými receptory (EPj-EP4, DP, FP a TP). Selektivita vůči receptorům EPj, EP3, FP a TP je důležitá, protože tyto receptory jsou dobře popsanými kontraktilními receptory v gastrointestinálním traktu a cévách. Selektivní neaktivita vůči receptorům EP2, EP4 a DPi je důležitá proto, že tyto receptory zprostředkovávají účinky oslabující imunitu.
Stimulace receptoru IP selexipagem a aktivním metabolitem vede k vasodilatačním a antiproliferativním a antifibrotickým účinkům. V modelu PAH na potkanech brání selexipag remodelaci srdce a plic a vyvolává proporční pokles tlaku v plicích a periferii, což naznačuje, že periferní vasodilace odráží farmakodynamické účinky na plíce. V modelu na potkanech nevyvolává selexipag in vitro desenzitizaci receptoru IP ani tachyfylaxi.
Farmakodynamické účinky
Elektrofyziologie srdce
V důkladné studii QT u zdravých dobrovolníků nevykázaly opakované dávky 800 a 1 600 mikrogramů selexipagu dvakrát denně žádný účinek na srdeční repolarizaci (interval QTc) ani vodivost (intervaly PR a QRS), přičemž na srdeční frekvenci měly mírně zrychlující účinky (na placebo a dle výchozích hodnot upravené zvýšení tepové frekvence dosáhlo 6 až 7 tepů za minutu 1,5 až 3 hodiny po podání dávky 800 mikrogramů selexipagu a 9 až 10 tepů za minutu ve stejných časech po dávce 1 600 mikrogramů selexipagu).
Koagulační faktory
Ve studiích fáze 1 a 2 byl u selexipagu pozorován mírný pokles hladin von Willebrandova faktoru (vWF) v plasmě; hodnoty vWF zůstávaly nad dolním limitem normálního rozmezí.
Plicní hemodynamika
Dvojitě zaslepená, placebem kontrolovaná klinické studie fáze 2 hodnotila hemodynamické proměnné po 17 týdnech léčby u pacientů s PAH funkční klasifikace WHO II—III, kteří současně dostávali ERA a/nebo inhibitory PDE-5. Pacienti titrující selexipag na individuálně tolerovanou dávku (vzestupy po 200 mikrogramech dvakrát denně až do dávky 800 mikrogramů dvakrát denně; N = 33) dosáhli v porovnání s placebem (N = 10) statisticky významné střední hodnoty snížení plicní cévní rezistence
0 30,3 % (95% interval spolehlivosti [CI]: -44,7 %; -12,2 %; p = 0,0045) a zvýšení srdečního indexu (medián léčebného účinku) 0,41 l/min/m2 (95% CI: 0,10; 0,71).
Klinická účinnost a bezpečnost
Účinnost u pacientů s PAH
Účinky selexipagu na progresi PAH byly prokázány v multicentrické, dlouhodobé (maximální trvání expozice přibližně 4,2 roku), dvojitě zaslepené, placebem kontrolované studii fáze 3 s paralelní skupinou vedené počtem příhod (event-driven study) u 1 156 pacientů se symptomatickou (WHO FC I-IV) PAH. Pacienti byli randomizováni buď do skupiny léčené placebem (N = 582) nebo selexipagem (N = 574) dvakrát denně. Dávka se zvyšovala v týdenních intervalech po 200 mikrogramech podávaných dvakrát denně s cílem stanovit individuální udržovací dávku (200 až
1 600 mikrogramů dvakrát denně).
Primárním kritériem hodnocení použitým ve studii byla doba do výskytu první příhody morbidity nebo mortality až do ukončení léčby. Kritérium hodnocení bylo definováno jako složené, jako kombinace úmrtí (ze všech příčin) nebo hospitalizace kvůli PAH nebo progrese PAH vedoucí k potřebě transplantace plic nebo balónkové atriální septostomie nebo zahájení parenterální léčby prostanoidy nebo chronické léčby kyslíkem nebo jiných příhod progrese nemoci (pacienti s výchozí funkční klasifikací WHO II nebo III) potvrzené zkrácením (> 15 %) vzdálenosti ušlé za 6 minut (6MWD, 6-minute walk distance) v porovnání s výchozí hodnotou a zhoršením funkční klasifikace WHO nebo (pacienti s výchozí funkční klasifikací WHO III nebo IV) potvrzené zkrácením (> 15 %) vzdálenosti ušlé za 6 minut (6MWD) v porovnání s výchozí hodnotou a potřebou další specifické léčby PAH.
Všechny příhody byly potvrzeny nezávislou posudkovou komisí, jíž nebylo známo, do které léčebné skupiny je pacient zařazen.
Střední hodnota věku byla 48,1 roku (rozmezí 18 až 80 let věku), přičemž většina účastníků byla bělošské rasy (65,0 %) a ženského pohlaví (79,8 %). 17,9 % pacientů bylo ve věku > 65 a 1,1 %
> 75 let. Přibližně 1 % pacientů mělo při zařazení do studie funkční klasifikaci WHO I, 46 % mělo funkční klasifikaci WHO II, 53 % funkční klasifikaci WHO III a 1 % funkční klasifikaci WHO IV.
Nejčastější etiologií u populace zařazené do studie byla idiopatická nebo dědičná PAH (58 %), následovaná PAH v důsledku poruch pojivové tkáně (29 %), PAH spojenou s upravenou prostou vrozenou srdeční vadou (10 %) a PAH související s jinými etiologiemi (léčiva a toxiny [2 %] a HIV [1 %]).
V době zařazení do studie byla většina pacientů (80 %) léčena stabilní dávkou terapie specifické pro PAH, buď ERA (15 %) nebo inhibitorem PDE-5 (32 %) nebo jak ERA, tak inhibitorem PDE-5 (33 %).
Celkový medián trvání dvojitě zaslepené léčby byl 63,7 týdne u skupiny léčené placebem a 70,7 týdne u skupiny léčené selexipagem. 23 % pacientů léčených selexipagem dosáhlo udržovacích dávek v rozmezí 200 až 400 mikrogramů, 31 % dosáhlo dávek v rozmezí 600 až 1 000 mikrogramů a 43 % dosáhlo dávek v rozmezí 1 200 až 1 600 mikrogramů.
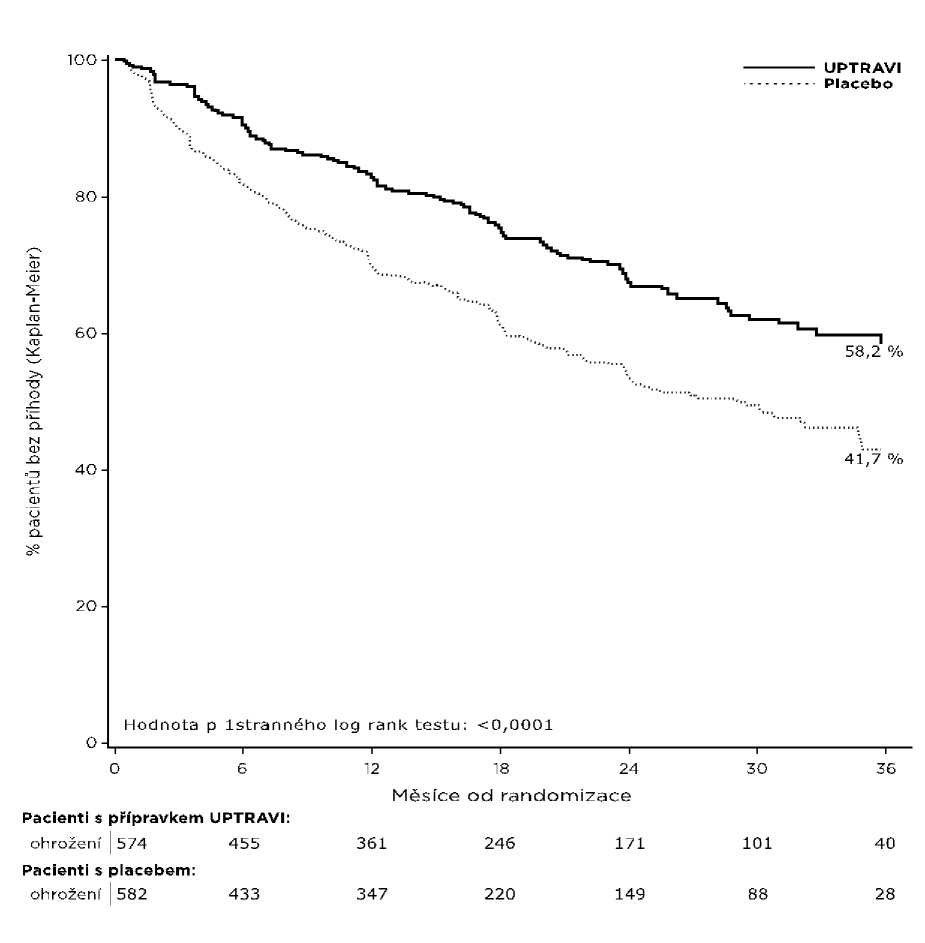
Léčba selexipagem v dávce 200 až 1 600 mikrogramů dvakrát denně vedla v porovnání s placebem ke 40% snížení (poměr rizik [HR] 0,60; 99% interval spolehlivosti [CI]: 0,46; 0,78; hodnota p jednostranného log-rank testu < 0,0001) výskytu morbiditních nebo mortalitních příhod až 7 dní po poslední dávce (Obrázek 1). Příznivý účinek selexipagu byl primárně přičítán snížení hospitalizací kvůli PAH a snížení jiných příhod progrese nemoci (Tabulka 1).
Obrázek 1 Kaplan-Meierovy odhady první příhody morbidity a mortality
|
Kritéria hodnocení & statistika |
Pacienti s příhodami |
Porovnání léčby: selexipag vs. placebo | ||||
|
Placebo (N=582) |
Selexipag (N=574) |
Absolutní snížení rizika |
Relativní snížení rizika (99% CI) |
HR (99% CI) |
Hodnota p | |
|
Příhoda morbidita-mortalita a |
58,2 % |
41,7 % |
16,5 % |
40 % (22 %; 54 %) |
0,60 (0,46; 0,78) |
< 0,0001 |
|
Hospitalizace kvůli PAH b n (%) |
109 (18,7 %) |
78 (13,6 %) |
5,1 % |
33 % (2 %; 54 %) |
0,67 (0,46; 0,98) |
0,04 |
|
Progrese nemoci b n (%) |
100 (17,2 %) |
38 (6,6 %) |
10,6 % |
64 % (41 %; 78 %) |
0,36 (0,22; 0,59) |
< 0,0001 |
|
Zahájení i.v./s.c. podávání prostanoidu nebo oxygenoterapie b c n (%) |
15 (2,6 %) |
11 (1,9 %) |
0,7 % |
32 % (-90 %; 76 %) |
0,68 (0,24; 1,90) |
0,53 |
|
Úmrtí do EOT + 7 dní d n (%) |
37 (6,4 %) |
46 (8,0 %) |
-1,7 % |
-17 % (-107 %; 34 %) |
1,17 (0,66; 2,07) |
0,77 |
|
Úmrtí do uzavření studie d n (%) |
105 (18,0 %) |
100 (17,4 %) |
0,6 % |
3 % (-39 %; 32 %) |
0,97 (0,68; 1,39) |
0,42 |
CI = interval spolehlivosti; EOT = ukončení léčby; HR = poměr rizik; i.v. = intravenózní; PAH = plicní arteriální hypertenze;
s.c. = subkutánní.
(a) % pacientů s příhodou po 36 měsících = 100 x (1 - Kaplan-Meierův odhad); poměr rizik odhadnut za pomoci Coxova proporčního modelu rizik; hodnota p nestratifikovaného jednostranného log-rank testu
(b) % pacientů s příhodou v rámci primárního kritéria hodnocení do EOT + 7 dní; poměr rizik odhadnut pomocí Aalen-Johansenovy metody; dvoustranná hodnota p pomocí Grayova testu
(c) Zahrnuje „Potřebu transplantace plic nebo septostomie atrií“ (1 pacient léčený selexipagem a 2 léčení placebem)
(d) % pacientů s příhodou do EOT + 7 dní (uzavření studie); poměr rizik odhadnut za pomoci Coxova proporčního modelu rizik; hodnota p nestratifikovaného jednostranného log-rank testu
Nárůst počtu úmrtí až do ukončení léčby + 7 dní, ale ne do uzavření studie byl dále zkoumán pomocí matematického modelování, které ukázalo, že nerovnováha v počtu úmrtí je v souladu s předpokladem neutrálního vlivu na úmrtnost z důvodu PAH a snížení nefatálních událostí.
Pozorované účinky selexipagu v porovnání s placebem na primární kritéria hodnocení byly konzistentní bez ohledu na dosaženou individualizovanou udržovací dávku, jak je prokázáno poměrem rizik u tří předem definovaných kategorií (0,60 pro 200 až 400 mikrogramů dvakrát denně; 0,53 pro 600 až 1 000 mikrogramů dvakrát denně a 0,64 pro 1 200 až 1 600 mikrogramů dvakrát denně), což je v souladu s celkovým léčebným účinkem (0,60).
Účinnost selexipagu na primární kritérium hodnocení byla konzistentní u všech podskupin určených věkem, pohlavním, rasou, etiologií, geografickou oblastí, funkční třídou WHO i při podávání v monoterapii nebo v kombinaci s ERA nebo inhibitorem PDE-5 nebo v trojkombinaci jak s ERA, tak s inhibitorem PDE-5.
Doba do úmrtí souvisejícího s PAH nebo do hospitalizace kvůli PAH byla hodnocena jako sekundární kritérium hodnocení. Riziko příhody u tohoto kritéria hodnocení bylo v porovnání s placebem u pacientů léčených selexipagem sníženo o 30 % (poměr rizik [HR] 0,70; 99% interval spolehlivosti [CI]: 0,50; 0,98; hodnota p jednostranného log-rank testu = 0,0031). Procenta pacientů s příhodou ve 36. měsíci byla 28,9 % ve skupině léčené selexipagem a 41,3 % ve skupině léčené placebem, s absolutním snížením rizika 12,4 %.
Počet pacientů, u nichž do ukončení léčby bylo první příhodou úmrtí v důsledku PAH nebo hospitalizace kvůli PAH bylo 102 (17,8 %) ve skupině léčené selexipagem a 137 (23,5 %) ve skupině léčené placebem. Úmrtí v důsledku PAH jako složka kritéria hodnocení bylo pozorováno u 16 (2,8 %) pacientů léčených selexipagem a 14 (2,4 %) léčených placebem. Hospitalizace kvůli PAH byla pozorována u 86 (15,0 %) pacientů léčených selexipagem a u 123 (21,1 %) pacientů léčených placebem. Selexipag v porovnání s placebem snižoval riziko hospitalizace kvůli PAH jako první příhody (poměr rizik 0,67; 99 % interval spolehlivosti: 0,46; 0,98; hodnota p jednostranného log-rank testu = 0,04).
Celkový počet úmrtí ze všech příčin do uzavření studie byl 100 (17,4 %) ve skupině léčené selexipagem a 105 (18,0 %) ve skupině léčené placebem (poměr rizik 0,97; 99% interval spolehlivosti: 0,68; 1,39). Počet úmrtí v důsledku PAH do uzavření studie byl 70 (12,2 %) ve skupině léčené selexipagem a 83 (14,3 %) ve skupině léčené placebem.
Symptomatická kritéria hodnocení
Jako sekundární kritérium hodnocení byla hodnocena námahová kapacita. Medián výchozí 6MWD při zařazení do studie byl 376 m (rozmezí: 90 až 482 m) u pacientů léčených selexipagem a 369 m (rozmezí: 50 až 515 m) u pacientů léčených placebem. Léčba selexipagem vedla k mediánu na placebo upraveného účinku na 6MWD měřenému při minimu (tj. přibližně 12 hodin po dávce) 12 m ve 26. týdnu (99% interval spolehlivosti: 1, 24 m; hodnota p jednostranného log-rank testu = 0,0027).
U pacientů bez souběžné léčby specifické pro PAH byl na placebo upravený léčebný účinek měřený při minimu 34 m (99% interval spolehlivosti: 10, 63 m).
Kvalita života byla hodnocena u podsouboru pacientů ve studii GRIPHON využívající dotazník Cambridge Pulmonary Hypertension Outcome Review (CAMPHOR). Od zařazení do studie do 26. týdne nebyl žádný významný účinek léčby pozorován.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Uptravi u jedné nebo více podskupin pediatrické populace v indikaci léčba plicní arteriální hypertenze (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika selexipagu a jeho aktivního metabolitu byla studována primárně u zdravých subjektů. Farmakokinetika selexipagu a aktivního metabolitu jak po jednotlivé dávce, tak po opakovaném podání byla do jednotlivé dávky 800 mikrogramů a opakovaných dávek 1 800 mikrogramů dvakrát denně úměrná dávce. Po podání opakovaných dávek se rovnovážného stavu selexipagu a aktivního metabolitu dosáhlo za 3 dny. Po podání opakovaných dávek nedošlo k akumulaci v plasmě, a to ani u mateřské sloučeniny, ani u aktivního metabolitu.
U zdravých subjektů byla mezisubjektová variabilita expozice (plocha pod křivkou za dobu dávkovacího intervalu) v rovnovážném stavu 43 % u selexipagu a 39 % u aktivního metabolitu. Variabilita expozice u jednoho subjektu byla 24 % u selexipagu a 19 % u aktivního metabolitu.
Expozice selexipagu a aktivnímu metabolitu v rovnovážném stavu u pacientů s PAH a u zdravých subjektů byly podobné. Farmakokinetika selexipagu a aktivního metabolitu u pacientů s PAH nebyla ovlivněna závažností nemoci a neměnila se v čase.
Absorpce
Selexipag se rychle absorbuje a v játrech se hydrolyzuje prostřednictvím CES1 na aktivní metabolit.
Maximálních pozorovaných plasmatických koncentrací selexipagu a jeho aktivního metabolitu se po peroráním podání dosáhne během 1 až 3 hodin, respektive 3 až 4 hodin.
Absolutní biologická dostupnost selexipagu u lidí je přibližně 49 %. To je pravděpodobně důsledkem first pass efektu selexipagu, protože plazmatické koncentrace aktivního metabolitu jsou po podání stejné perorální a intravenózní dávky podobné.
Za přítomnosti potravy byla expozice selexipagu po jednotlivé dávce 400 mikrogramů zvýšena o 10 % u osob bělošské rasy a snížena o 15 % u Japonců, zatímco expozice aktivnímu metabolitu byla snížena o 27 % (osoby bělošské rasy) a 12 % (Japonci). Více subjektů hlásilo nežádoucí účinky po podání nalačno, než po podání po jídle.
Distribuce
Selexipag a jeho aktivní metabolit se silně váží na plasmatické proteiny (přibližně 99 % celkem a stejnou měrou na albumin a alfa1-kyselý glykoprotein). Distribuční objem selexipagu v rovnovážném stavu je 11,7 litru.
Biotransformace
Selexipag prochází enzymatickou hydrolýzou acylsulfonamidu zprostředkovanou CES1, čímž vzniká aktivní metabolit. Oxidační metabolizace katalyzovaná CYP3A4 a CYP2C8 vede k tvorbě hydroxylovaných a dealkylovaných produktů. Glukuronidace aktivního metabolitu se účastní UGT1A3 a UGT2B7. S výjimkou aktivního metabolitu nepřesahuje žádný z metabolitů cirkulujících v lidské plasmě 3 % celkového materiálu souvisejícího s léčivem. Jak u zdravých subjektů, tak u pacientů s PAH je v rovnovážném stavu expozice aktivnímu metabolitu přibližně 3- až 4krát vyšší, než expozice mateřské látce.
Eliminace
Eliminace selexipagu probíhá hlavně metabolizací se střední hodnotou biologického poločasu 0,8 až 2,5 hodiny. Biologický poločas aktivního metabolitu je 6,2 až 13,5 hodiny. Celková tělesná clearance selexipagu je 17,9 l/h. Vyloučení u zdravých subjektů bylo úplné 5 dní po podání a docházelo k němu primárně stolicí (počítáno na 93 % podané dávky) v porovnání s 12 % vyloučenými močí.
Zvláštní populace
U zdravých subjektů ani u pacientů s PAH nebyl pozorován žádný klinicky relevantní vliv pohlaví, rasy, věku ani tělesné hmotnosti na farmakokinetiku selexipagu a jeho aktivního metabolitu.
Porucha funkce ledvin
U subjektů s těžkou poruchou funkce ledvin (eGFR < 30 ml/min/1,73 m2) bylo pozorováno 1,4- až 1,7násobné zvýšení expozice (maximální plasmatické koncentrace a plocha pod křivkou průběhu koncentrace v plasmě v čase) selexipagu a jeho aktivnímu metabolitu.
Porucha funkce jater
U subjektů s mírnou (Child-Pugh třída A) nebo středně těžkou (Child-Pugh třída B) poruchou funkce jater byla expozice selexipagu v porovnání se zdravými subjekty 2krát, respektive 4krát vyšší. Expozice aktivnímu metabolitu u subjektů s mírnou poruchou funkce jater zůstávala téměř nezměněna a u subjektů se středně těžkou poruchou funkce jater byla dvojnásobná. Selexipag se podával pouze 2 subjektům s těžkou (Child-Pugh třída C) poruchou funkce jater. Expozice selexipagu a jeho aktivnímu metabolitu u těchto dvou subjektů byla podobná expozici u subjektů se středně těžkou (Child-Pugh třída B) poruchou funkce jater.
Na základě modelování a simulačních údajů ze studie u subjektů s poruchou funkce jater se předpokládá, že expozice selexipagu v rovnovážném stavu u subjektů se středně těžkou poruchou funkce jater (Child-Pugh třída B) po režimu podávání jednou denně bude přibližně 2krát vyšší, než u zdravých subjektů při režimu podávání dvakrát denně. Předpokládá se, že expozice aktivnímu metabolitu v rovnovážném stavu u těchto pacientů při režimu podávání jednou denně bude podobná jako u zdravých subjektů při režimu podávání dvakrát denně. Subjekty s těžkou poruchou funkce jater (Child-Pugh třída C) vykazovaly podobnou predikovanou expozici v rovnovážném stavu jako subjekty se středně těžkou poruchou funkce jater při režimu podávání jednou denně.
5.3 Předklinické údaje vztahující se k bezpečnosti
Při studiích toxicity opakovaných dávek u hlodavců vyvolal silný pokles krevního tlaku jako důsledek přehnaného farmakologického účinku přechodné klinické projevy a snižení spotřeby krmiva a přírůstku tělesné hmotnosti. U dospělých a mladých psů byly jako hlavní cílové orgány po léčbě selexipagem zjištěna střeva a kosti/kostní dřeň. U mladých psů bylo zjištěno zpoždění uzavření femorální a/nebo tibiální epifyzální růstové destičky. Hladina, při které se nepozoruje žádný nežádoucí účinek, nebyla stanovena. U mladých psů byla sporadicky pozorována intususcepce v důsledku účinků na motilitu střev souvisejících s prostacyklinem. Rozmezí bezpečnosti upravené na potenci receptoru IP u aktivního metabolitu bylo 2násobné (založeno na celkové expozici) ve vztahu k terapeutické expozici u lidí. Toto nebylo ve studiích toxicity na myších a potkanech zjištěno. Vzhledem k druhově specifické citlivosti u psů ohledně vzniku intususcepce se toto zjištění nepovažuje za relevantní pro dospělé lidi.
Má se za to, že zvýšená osifikace kostí a související změny v kostní dřeni ve studích na psech je důsledkem aktivace receptorů EP4 u psů. Protože lidské receptory EP4 nejsou selexipagem ani jeho aktivním metabolitem aktivovány, je tento účinek druhově specifický, a proto není pro lidi relevantní.
Selexipag a aktivní metabolit nejsou na základě celkových důkazů z provedených studií genotoxicity genotoxické.
Ve dvouletých studiích karcinogenity vyvolával selexipag zvýšenou incidenci adenomů štítné žlázy u myší a adenomů Leydigových buněk u potkanů. Mechanismus je specifický pro hlodavce. Pouze u potkanů byla po dvou letech léčby zaznamenána tortuozita retinálních arteriol. Z mechanického hlediska se má za to, že je tento účinek vyvolán celoživotní vasodilatací a následnými změnami v oční hemodynamice. Další histopatologické nálezy byly u selexipagu pozorovány pouze při expozicích dostatečně přesahujících maximální expozici u lidí, což ukazuje na malou relevanci pro lidi.
Ve studii fertility provedené na potkanech bylo při expozicích 173násobně přesahujících terapeutické expozice (na základě celkové expozice) pozorováno prodloužení ovulačních cyklů vedoucí k nárůstu počtu dnů do kopulace, hladina při které se nepozoruje žádný účinek byla 30krát vyšší než při terapeutické expozici. Jinak parametry fertility ovlivněny nebyly.
Selexipag nebyl u potkanů ani králíků teratogenní (rozmezí expozice nad terapeutickou expozicí bylo 13násobné u selexipagu a 43násobné u aktivního metabolitu, založeno na celkové expozici). Bezpečnostní rozpětí s ohledem na potenciální účinky na reprodukci související s prostacyklinovým IP receptorem bylo 20 pro fertilitu a 5 u potkanů a 1 u králíků (na základě volné expozice) pro embryofetální vývoj, při adaptaci na rozdíly v potenci receptorů. Ve studii pre-/postnatálního vývoje u potkanů nevyvolával selexipag žádné účinky na reprodukční funkce matek a potomstva.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety
Mannitol (E421),
kukuřičný škrob,
částečně substituovaná hyprolosa,
hyprolosa,
magnesium-stearát.
Potah tablety
Uptravi 200 mikrogramů potahovaná tableta:
Hypromelosa,
propylenglykol,
oxid titaničitý (E171),
žlutý oxid železitý (E172),
karnaubský vosk.
Uptravi 400 mikrogramů potahovaná tableta:
Hypromelosa,
propylenglykol,
oxid titaničitý (E171),
červený oxid železitý (E172),
karnaubský vosk.
Uptravi 600 mikrogramů potahovaná tableta:
Hypromelosa,
propylenglykol,
oxid titaničitý (E171),
červený oxid železitý (E172),
černý oxid železitý (E172),
karnaubský vosk.
Uptravi 800 mikrogramů potahovaná tableta:
Hypromelosa,
propylenglykol,
oxid titaničitý (E171),
žlutý oxid železitý (E172),
černý oxid železitý (E172),
karnaubský vosk.
Uptravi 1 000 mikrogramů potahovaná tableta:
Hypromelosa,
propylenglykol,
oxid titaničitý (E171),
červený oxid železitý (E172),
žlutý oxid železitý (E172),
karnaubský vosk.
Uptravi 1 200 mikrogramů potahovaná tableta:
Hypromelosa,
propylenglykol,
oxid titaničitý (E171),
černý oxid železitý (E172),
červený oxid železitý (E172),
karnaubský vosk.
Uptravi 1 400 mikrogramů potahovaná tableta:
Hypromelosa,
propylenglykol,
oxid titaničitý (E171),
žlutý oxid železitý (E172),
karnaubský vosk.
Uptravi 1 600 mikrogramů potahovaná tableta: Hypromelosa,
propylenglykol, oxid titaničitý (E171), černý oxid železitý (E172), červený oxid železitý (E172), žlutý oxid železitý (E172), karnaubský vosk.
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Blistr z polyamidu / hliníku / HDPE / PE s vloženým vysoušedlem / HDPE uzavřený hliníkovou fólií. Uptravi 200 mikrogramů potahované tablety
Papírové krabičky s 10 nebo 60 potahovanými tabletami nebo 140 potahovanými tabletami (titrační balení).
Uptravi 400 mikrogramů, 600 mikrogramů, 800 mikrogramů, 1 000 mikrogramů, 1 200 mikrogramů, 1 400 mikrogramů a 1 600 mikrogramů potahované tablety
Papírové krabičky se 60 potahovanými tabletami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky na likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Actelion Registration Ltd Chiswick Tower 13th Floor 389 Chiswick High Road London W4 4AL Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1083/001
EU/1/15/1083/002
EU/1/15/1083/003
EU/1/15/1083/004
EU/1/15/1083/005
EU/1/15/1083/006
EU/1/15/1083/007
EU/1/15/1083/008
EU/1/15/1083/009
EU/1/15/1083/010
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ /VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Actelion Pharmaceuticals Deutschland GmbH Konrad-Goldmann-Strasse 5b 79100 Freiburg Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Před uvedením přípravku Uptravi na trh v každém členském státě odsouhlasí držitel rozhodnutí o registraci Distribuční plán s národní regulační autoritou.
Distribuční plán má za cíl usnadnit identifikaci předepisujících lékařů, aby jim mohly být zpřístupňovány příslušné informace o bezpečném a účinném používání přípravku Uptravi a poskytovány prostředky pro minimalizaci rizik, zejména pokud jde o potenciální riziko chyby v medikaci. Distribuční plán má zahrnovat tři klíčové principy, které budou zahrnuty v každém systému ve všech členských státech. Jsou to:
• Identifikace a vedení seznamu všech lékařů předepisujících přípravek Uptravi;
• Distribuce souprav všem zjištěným předepisujícím lékařům, aby se minimalizovalo riziko zejména chyb v medikaci;
• Sledování obdržení soupravy předepisujícími lékaři.
Držitel rozhodnutí o registraci zajistí, že všem zdravotníkým pracovníkům, kteří hodlají přípravek Uptravi předepisovat a/nebo vydávat, bude poskytnuta souprava pro předepisujícího obsahující následující:
• Souhrn údajů o přípravku Uptravi;
• Průvodní dopis zdravotnickému pracovníkovi;
• Pokyny pro zdravotnického pracovníka k titraci dávek na laminované kartě formátu A4;
• Průvodce titrací pro pacienta;
• Příbalová informace pro pacienta.
Průvodní dopis zdravotnickému pracovníkovi má vysvětlit, že účelem těchto edukačních materiálů je snížit riziko chyb v medikaci v důsledku dostupnosti více možných sil tablet a dávek, a má poskytnout seznam obsahu soupravy pro předepisujícího.
Pokyny pro zdravotnického pracovníka k titraci dávek na laminované kartě formátu A4 jsou určeny ke snížení rizika chyb v medikaci v důsledku titrační fáze při zahájení léčby přípravkem Uptravi a má obsahovat následující klíčové prvky:
• Koncepci dávkování a titrace;
• Postup k dosažení udržovací dávky (titrační fáze);
• Očekávané nežádoucí účinky v průběhu titrační fáze a způsob jejich zvládnutí;
• Podporu a návod pro zdravotnické pracovníky k jasné komunikaci s pacientem během jeho první návštěvy, jakož i převzetí zodpovědnosti za kontaktování pacienta v průběhu titrační fáze, usnadnění komunikace mezi zdravotnickými pracovníky a pacientem (nutnost kontaktu a naplánování kontaktů prostřednictvím telefonu).
Průvodce titrací pro pacienta, který má být používán zdravotnickými pracovníky při diskusích s pacientem má obsahovat následující klíčové prvky:
• Laická jazyková verze pokynů pro zdravotnického pracovníka k titraci dávek na laminované kartě formátu A4;
• Deník, který má usnadnit užívání přípravku Uptravi a slouží jako připomínka pro pacienty (např., aby kontaktovali svého lékaře), a je místem pro záznam užívání tablet;
• Informace o bezpečném a účinném užívání prípravku Uptravi v jazyce pro laiky.
Průvodce titrací pro pacienta spolu s příbalovou informací má být poskytnuta pacientovi po předvedení. Pacienti obdrží identický průvodce titrací a příbalovou informaci ve svém titračním balení přípravku Uptravi.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Uptravi 200 mikrogramů potahované tablety selexipagum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVYCH LÁTEK
Jedna potahovaná tableta obsahuje selexipagum 200 mikrogramů
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
140 potahovaných tablet Titrační balení
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Actelion Registration Ltd Chiswick Tower 13th Floor 389 Chiswick High Road London W4 4AL Velká Británie
12 REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1083/003
EU/1/15/1083/001
EU/1/15/1083/002
EU/1/15/1083/004
EU/1/15/1083/005
EU/1/15/1083/006
EU/1/15/1083/007
EU/1/15/1083/008
EU/1/15/1083/009
EU/1/15/1083/010
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Uptravi 200 mikrogramů
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Uptravi 200 mikrogramů potahované tablety Uptravi 400 mikrogramů potahované tablety Uptravi 600 mikrogramů potahované tablety Uptravi 800 mikrogramů potahované tablety Uptravi 1 000 mikrogramů potahované tablety Uptravi 1 200 mikrogramů potahované tablety Uptravi 1 400 mikrogramů potahované tablety Uptravi 1 600 mikrogramů potahované tablety
selexipagum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje selexipagum 200 mikrogramů Jedna potahovaná tableta obsahuje selexipagum 400 mikrogramů Jedna potahovaná tableta obsahuje selexipagum 600 mikrogramů Jedna potahovaná tableta obsahuje selexipagum 800 mikrogramů Jedna potahovaná tableta obsahuje selexipagum 1 000 mikrogramů Jedna potahovaná tableta obsahuje selexipagum 1 200 mikrogramů Jedna potahovaná tableta obsahuje selexipagum 1 400 mikrogramů Jedna potahovaná tableta obsahuje selexipagum 1 600 mikrogramů
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
10 potahovaných tablet 60 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Actelion Registration Ltd Chiswick Tower 13th Floor 389 Chiswick High Road London W4 4AL Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1083/001
EU/1/15/1083/002
EU/1/15/1083/003
EU/1/15/1083/004
EU/1/15/1083/005
EU/1/15/1083/006
EU/1/15/1083/007
EU/1/15/1083/008
EU/1/15/1083/009
EU/1/15/1083/010
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Uptravi 200 mikrogramů Uptravi 400 mikrogramů Uptravi 600 mikrogramů
Uptravi 800 mikrogramů Uptravi 1 000 mikrogramů Uptravi 1 200 mikrogramů Uptravi 1 400 mikrogramů Uptravi 1 600 mikrogramů
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTRY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Uptravi 200 mikrogramů tablety Uptravi 400 mikrogramů tablety Uptravi 600 mikrogramů tablety Uptravi 800 mikrogramů tablety Uptravi 1 000 mikrogramů tablety Uptravi 1 200 mikrogramů tablety Uptravi 1 400 mikrogramů tablety Uptravi 1 600 mikrogramů tablety
selexipagum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Actelion
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Uptravi 200 mikrogramů potahované tablety Uptravi 400 mikrogramů potahované tablety Uptravi 600 mikrogramů potahované tablety Uptravi 800 mikrogramů potahované tablety Uptravi 1 000 mikrogramů potahované tablety Uptravi 1 200 mikrogramů potahované tablety Uptravi 1 400 mikrogramů potahované tablety Uptravi 1 600 mikrogramů potahované tablety Selexipagum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Uptravi a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Uptravi užívat
3. Jak se přípravek Uptravi užívá
4. Možné nežádoucí účinky
5 Jak přípravek Uptravi uchovávat
6. Obsah balení a další informace
1. Co je přípravek Uptravi a k čemu se používá
Uptravi je léčivý přípravek, který obsahuje léčivou látku selexipag.Ppůsobí na krevní cévy podobným způsobem jako přirozená látka prostacyklin, uvolňuje je a rozšiřuje.
Přípravek Uptravi se používá k dlouhodbé léčbě plicní arteriální hypertenze (PAH) u dospělých pacientů, u kterých kterých není dostatečná léčba jinými typy přípravků na léčbu PAH známými jako jsou antagonisté endothelinového receptoru a inhibitory fosfodiesterázy typu 5. Přípravek Uptravi může být také používán samostatně, pokud pacient není kandidátem pro tyto přípravky.
Lze jej používat samotný nebo s dalšími léčivými přípravky používanými k léčbě PAH jako jsou antagonisté endothelinového receptoru nebo inhibitory fosfodiesterázy typu 5.
PAH je vysoký krevní tlak v cévách, které vedou krev ze srdce do plic (plicní tepny). U lidí s PAH se tyto tepny zužují, takže srdce musí pracovat silněji, aby jimi krev přečerpalo. To může u lidí vyvolávat únavu, točení hlavy, dušnost nebo jiné příznaky.
Napodobením účinku prostacyklinu přípravek Uptravi rozšiřuje plicní tepny a snižuje jejich ztuhnutí. To srdci usnadňuje přečerpávat jimi krev. Tím ulevuje od příznaků PAH a zlepšuje průběh nemoci.
2. Čemu musíte věnovat pozornost, než začnete přípravek Uptravi užívat
- jestliže jste alergický(á) na selexipag nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- Jestliže máte problémy se srdcem, jako je:
- špatný průtok krve srdeční svalem (těžká ischemická choroba srdeční nebo nestabilní angina pectoris); příznaky mohou zahrnovat bolesti na hrudi
- infarkt myokardu v posledních 6 měsících
- slabé srdce (dekompenzované srdeční selhání), které není pod pečlivým lékařským dohledem
- silně nepravidelný tep
- porucha srdečních chlopní (vrozená nebo získaná), která způsobuje nedostatečnou práci srdce (nesouvisející s plicní hypertenzí)
Upozornění a opatření
Před užitím přípravku Uptravi se poraďte se svým lékařem nebo zdravotní sestrou, jestliže
- užíváte léky na vysoký krevní tlak
- máte nízký krevní tlak spojený s příznaky jako je točení hlavy
- u Vás nedávno došlo k velké ztrátě krve nebo tekutin jako např. při těžkém průjmu nebo zvracení
- máte problémy se štítnou žlázou
- máte závažné problémy s ledvinami nebo podstupujete dialýzu
- máte nebo jste měl(a) problémy vyplývající z nedostatečné funkce jater
Jestliže zaznamenáte některý z výše uvedených příznaků nebo se Váš stav změní, ihned to oznamte svému lékaři.
Děti a dospívající
Nepodávejte tento přípravek dětem mladším než 18 let, protože přípravek Uptravi nebyl na dětech hodnocen.
Starší pacienti
S přípravkem Uptravi u pacientů starších 75 let jsou jen omezené zkušenosti. Přípravek Uptravi se má u této věkové skupiny používat opatrně.
Další léčivé přípravky a přípravek Uptravi
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Užívání dalších léků může ovlivnit způsob, jakým přípravek Uptravi účinkuje.
Jestliže užíváte některý z následujících léků, poraďte se s lékařem, který Vás na PAH léčí, nebo jeho zdravotní sestrou:
- gemfibrozil (lék používaný ke snížení hladin tuků [lipidů] v krvi)
- kyselina valproová (lék používaný k léčbě epilepsie)
- probenecid (lék používaný k léčbě dny)
- flukonazol, rifampicin nebo rifapentin (antibiotika používaná k léčbě infekcí)
Těhotenství a kojení
Přípravek Uptravi se v těhotenství a během kojení nedoporučuje. Pokud jste žena ve věku, která může mít děti, musíte během užívání přípravku Uptravi používat účinnou antikoncepční metodu. Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Přípravek Uptravi může vyvolat nežádoucí účinky, jako j sou bolesti hlavy a nízký krevní tlak (viz bod 4), které mohou mít vliv na schopnost řídit; zhoršit schopnosti řídit mohou také příznaky Vaší nemoci.
3. Jak se přípravek Uptravi užívá
Přípravek Uptravi smí předepisovat pouze lékař se zkušenostmi s léčbou PAH. Vždy užívejte přípravek Uptravi přesně podle pokynů svého lékaře. Pokud si nejste jistý(á) nebo pokud máte nějaké otázky, poraďte se se svým lékařem.
Pokud máte slabý zrak nebo trpíte nějakým typem slepoty, vyžádejte si během období titrace dávek přípravku Uptravi pomoc další osoby.
Zjišťování dávky, která je pro Vás vhodná
Na začátku léčby budete užívat nejnižší dávku. Ta činí jednu 200mikrogramovou tabletu ráno a další 200mikrogramovou tabletu večer. Léčba se má zahájit večer. Váš lékař Vám dá pokyn, že máte dávku postupně zvyšovat. Tento postup se nazývá titrace. Umožní tělu si na nový lék zvyknout. Cílem titrace je dosažení nejvhodnější dávky. Tou bude nejvyšší dávka, kterou budete moci snášet, která může dosáhnout maximální dávky 1 600 mikrogramů ráno a večer.
První balení tablet, které dostanete, bude obsahovat světle žluté 200mikrogramové tablety.
Váš lékař Vám dá pokyn, abyste dávky zvyšoval(a) postupně, obvykle každý týden, nicméně interval mezi zvýšeními dávky může být delší.
Při každém kroku přidáte jednu 200mikrogramovou tabletu k ranní dávce a další 200mikrogramovou tabletu k večerní dávce. Zvýšená dávka se má poprvé užít večer. Níže uvedený diagram znázorňuje počet tablet, které se během prvních 4 kroků mají užít každé ráno a každý večer.
Jeden dávkovači krok trvá přibližně 1 týden
POČÁTEČNÍ
DÁVKA
KROK 1
Ráno: jedna 200mikrogramová tableta Večer: jedna 200mikrogramová tableta
(Celková denní dávka: 400 mikrogramů)
KROK 2
Ráno: dvě
200mikrogramové tablety Večer: dvě 200mikrogramové tablety
(Celková denní dávka: 800 mikrogramů)
KROK 3
Ráno: tři
200mikrogramové tablety Večer: tři
200mikrogramové
tablety
(Celková denní dávka:
1 200 mikrogramů)
KROK 4
Ráno: čtyři 200mikrogramové tablety Večer: čtyři 200mikrogramové tablety
(Celková denní dávka:
1 600 mikrogramů)

Pokud Vám lékař dá pokyn k dalšímu zvyšování dávky a k postoupení ke kroku 5, lze to provést užitím jedné zelené 800mikrogramové tablety a jedné světle žluté 200mikrogramové tablety ráno a jedné 800mikrogramové tablety a jedné 200mikrogramové tablety večer.
Pokud Vám lékař řekne, že máte dávku zvyšovat dále, přidáte při každém dalším kroku jednu 200mikrogramovou tabletu k ranní dávce a jednu 200mikrogramovou tabletu k večerní dávce. Zvýšená dávka se má poprvé užít večer. Maximální dávka přípravku Uptravi je 1 600 mikrogramů ráno a 1 600 mikrogramů večer. Ne každý pacient však této dávky dosáhne, protože různí pacienti vyžadují různé dávky.
Diagram níže znázorňuje počet tablet, které máte užít každé ráno a každý večer při každém kroku, počínaje krokem 5.
200mikrogramová tableta
NEJVYSSI
DÁVKA
800mikrogramová tableta (Použijte u kroků 5 až 8, aby se snížil počet tablet potřebných na dávku.)

KROK 5
Ráno: jedna 800mikrogramová a jedna
200mikrogramová tableta Večer: jedna 800mikrog rámová a jedna
200mikrogramová
tableta
(Celková denní dávka:
2 000 mikrogramů)
KROK 6
Ráno: jedna 800mikrog rámová a dvě
200mikrogramové tablety Večer: jedna 800mikrogramová a dvě
200mikrogramové
tablety
(Celková denní dávka:
2 400 mikrogramů)
KROK 7
Ráno: jedna 800mikrogramová a tři
200mikrogramové tablety Večer: jedna 800mikrogramová a tři
200mikrogramové
tablety
(Celková denní dávka:
2 800 mikrogramů)
KROK 8
Ráno: jedna 800mikrog rámová a čtyři
200mikrogramové tablety Večer: jedna 800mikrogramová a čtyři
200mikrogramové
tablety
(Celková denní dávka:
3 200 mikrogramů)
Titrační balíček rovněž obsahuje průvodce titrací, kde jsou uvedeny informace o postupu titrace, a který Vám umožní zaznamenávat si počet tablet, které budete každý den užívat.
Nezapomeňte si do titračního deníku zapsat počet tablet, které každý den užijete. Titrační kroky obvykle trvají přibližně 1 týden. Pokud Vám dá lékař pokyn, abyste každý titrační krok podloužil(a) na dobu delší než 1 týden, jsou v deníku další stránky, které Vám umožní titraci zaznamenávat. Nezapomeňte se s lékařem, který Vás na PAH léčí, nebo s jeho zdravotní sestrou, během titrace pravidelně radit.
Přechod na nižší dávku kvůli nežádoucím účinkům
Během titrace se mohou u Vás objevit nežádoucí účinky, jako je bolest hlavy, průjem, pocit na zvracení (nauzea), nevolnost (zvracení), bolest čelisti, bolest svalů, bolest nohou, bolest kloubů nebo zrudnutí v obličeji (viz bod 4). Pokud je pro Vás těžké tyto nežádoucí účinky snášet, poraďte se se svým lékařem, jak je zvládnout nebo léčit. Jsou k dispozici způsoby léčby, které mohou pomoci tyto nežádoucí účinky zmírnit. Například léky proti bolesti jako je paracetamol Vám mohou pomoci léčit bolesti a bolest hlavy.
Pokud nežádoucí účinky nelze léčit nebo pokud se postupně při dávce, kterou užíváte, nezlepšují, může Váš lékař dávku upravit snížením počtu 200mikrogramových světle žlutých tablet, které užíváte, o jednu ráno a o jednu večer. Dále uvedený diagram znázorňuje přechod na nižší dávku. K tomuto kroku přistupte jedině, pokud Vám k tomu dal pokyn Váš lékař.
KROK DOLŮ
START KROK NAHORU
KROK 6
Cílem je dosáhnout nejvyšší dávky s nežádoucími účinky, které můžete tolerovat
Krok dolů Pokud nebude možné tolerovat nežádoucí účinky
ZPĚT KE KROKU 5
KROK 1
Ráno: jedna 200mikrogramová tableta Večer: jedna 200mikrog rámová tableta
(Celková denní dávka: 400 mikrogramů)
Zvyšujte svoji dávku přibližně každý týden přidáním 200mikrogramové tablety ke své ranní a večerní dávce
Pokud jsou Vaše nežádoucí účinky po snížení dávky zvládnutelné, může Váš lékař rozhodnout, že u této dávky zůstanete. Další informace naleznete v bodě Udržovací dávka uvedeném dále.
Udržovací dávka
Nejvyšší dávka, kterou můžete během titrace snést, se stane Vaší udržovací dávkou. Udržovací dávka je dávka, kterou máte dál pravidelně užívat.
Váš lékař Vám bude jako udržovací dávku předepisovat tabletu o vhodné síle. To Vám umožní užívat místo několika tablet jednu tabletu ráno a jednu večer.
Úplný popis tablet přípravku Uptravi, včetně barev a značení, naleznete v bodě 6 této příbalové informace.
V průběhu času může Váš lékař udržovací dávku podle potřeby upravit.
Pokud Vás kdykoli při užívání stejné dávky po dlouhou dobu postihnou nežádoucí účinky, které nebudete moci snášet nebo nežádoucí účinky, které budou mít vliv na Vaše normální denní činnosti, obraťte se na svého lékaře, protože může být potřeba snížit dávku. Lékař Vám pak předepíše nižší sílu obsaženou v jedné tabletě. Nezapomeňte zlikvidovat nepoužité tablety (viz bod 5).
Přípravek Uptravi užívejte jednou ráno a jednou večer s odstupem přibližně 12 hodin.
Tablety užívejte s jídlem, protože to může vést k lepší snášenlivosti přípravku. Tablety polykejte celé a zapíjejte je sklenicí vody. Tablety nerozdělujte, nedrťte a nežvýkejte.
Jestliže jste užil(a) více přípravku Uptravi, než jste měl(a)
Jestliže jste užil(a) více tablet, než máte, požádejte lékaře o radu.
Jestliže jste zapomněl(a) přípravek Uptravi užít, dávku užijte ihned, jakmile si vzpomenete, poté pokračujte v užívání tablet v obvyklou dobu. Pokud již téměř nastal čas užít další dávku (6 hodin do další dávky), opomenutou dávku vynechte a pokračujte v užívání přípravku v obvyklou dobu. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou tabletu.
Jestliže jste přestal(a) užívat přípravek Uptravi
Náhlé ukončení léčby přípravkem Uptravi může mít za následek zhoršení příznaků. Přípravek Uptravi nepřestávejte užívat, dokud Vám k tomu nedá pokyn Váš lékař. Váš lékař Vám může poradit, abyste před úplným ukončením léčby dávku postupně snižoval(a).
Pokud z jakéhokoli důvodu přestanete užívat přípravek Uptravi po více než 3 po sobě následující dny (pokud vynecháte 3 ranní a 3 večerní dávky neboli 6 nebo více dávek v řadě), ihned se obraťte na svého lékaře, protože může být potřeba dávku upravit, aby se zabránilo nežádoucím účinkům.
Váš lékař může rozhodnout, že se léčba obnoví s nižší dávkou, která se bude postupně zvyšovat na předchozí udržovací dávku.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i přípravek Uptravi nežádoucí účinky. Nežádoucí účinky Vás mohou postihnout nejen během titračního období, kdy se dávka zvyšuje, ale i později, až budete užívat stejnou dávku po dlouhou dobu.
Pokud Vás postihne některý s těchto nežádoucích účinků: bolest hlavy, průjem, pocit na zvracení (nauzea), nevolnost (zvracení), bolest čelisti, bolest svalů, bolest nohou, bolest kloubů nebo zarudnutí v obličeji, které nemůžete snášet nebo které nelze léčit, musíte se obrátit na svého lékaře, protože dávka, kterou užíváte, může být příliš vysoká a může být potřeba ji snížit.
Velmi časté nežádoucí účinky (mohou postihnout více než 1 z 10 osob)
- Bolest hlavy
- Zarudnutí (zarudnutí obličeje)
- Pocit na zvracení a zvracení (pocit nevolnosti a nevolnost)
- Průjem
- Bolest čelisti, bolest svalů, bolest kloubů, bolest nohou
- Nazofaryngitida (ucpaný nos)
Časté nežádoucí účinky (mohou postihnout až 1 z 10 osob)
- Anémie (nízký počet červených krvinek)
- Hyperthyroidismus (nadměrně aktivní štítná žláza)
- Snížení chuti k jídlu
- Snížení tělesné hmotnosti
- Hypotenze (nízký krevní tlak)
- Bolest žaludku
- Bolest
- Změny výsledků některých krevních testů, včetně krevního obrazu nebo testů funkce štítné žlázy
- Vyrážky, včetně kopřivky, mohou vyvolat pálivé nebo píchavé pocity a zarudnutí kůže
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 osob)
- Zrychlený tep
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Uptravi uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a na blistru za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Žádné zvláštní podmínky pro likvidaci.
6. Obsah balení a další informace
Co přípravek Uptravi obsahuje
- Léčivou látkou je selexipagum.
Uptravi 200 mikrogramů potahované tablety obsahuje selexipagum 200 mikrogramů Uptravi 400 mikrogramů potahované tablety obsahuje selexipagum 400 mikrogramů Uptravi 600 mikrogramů potahované tablety obsahuje selexipagum 600 mikrogramů Uptravi 800 mikrogramů potahované tablety obsahuje selexipagum 800 mikrogramů Uptravi 1 000 mikrogramů potahované tablety obsahuje selexipagum 1 000 mikrogramů Uptravi 1 200 mikrogramů potahované tablety obsahuje selexipagum 1 200 mikrogramů Uptravi 1 400 mikrogramů potahované tablety obsahuje selexipagum 1 400 mikrogramů Uptravi 1 600 mikrogramů potahované tablety obsahuje selexipagum 1 600 mikrogramů
- Pomocnými látkami j sou:
V jádru tablety:
mannitol (E421), kukuřičný škrob, částečně substituovaná hyprolosa, hyprolosa, magnesium-stearát
V potahu tablety:
hypromelosa, propylenglykol, oxid titaničitý (E171), karnaubský vosk a oxidy železa (viz níže). Uptravi 200 mikrogramů potahované tablety obsahuje žlutý oxid železitý (E172).
Uptravi 400 mikrogramů potahované tablety obsahuje červený oxid železitý (E172).
Uptravi 600 mikrogramů potahované tablety obsahuje červený oxid železitý a černý oxid železitý (E172).
Uptravi 800 mikrogramů potahované tablety obsahuje žlutý oxid železitý a černý oxid železitý (E172).
Uptravi 1 000 mikrogramů potahované tablety obsahuje červený oxid železitý a žlutý oxid železitý (E172).
Uptravi 1 200 mikrogramů potahované tablety obsahuje černý oxid železitý a červený oxid železitý (E172).
Uptravi 1 400 mikrogramů potahované tablety obsahuje žlutý oxid železitý (E172).
Uptravi 1 600 mikrogramů potahované tablety obsahuje černý oxid železitý, červený oxid železitý a žlutý oxid železitý (E172).
Uptravi 200 mikrogramů potahované tablety: kulaté, světle žluté, potahované tablety na jedné straně s vyraženým “2”.
Uptravi 400 mikrogramů potahované tablety: kulaté, červené, potahované tablety na jedné straně s vyraženým “4”.
Uptravi 600 mikrogramů potahované tablety: kulaté, světle fialové, potahované tablety na jedné straně s vyraženým “6”.
Uptravi 800 mikrogramů potahované tablety: kulaté, zelené, potahované tablety na jedné straně s vyraženým “8”.
potahované tablety: kulaté, oranžové, potahované tablety na jedné
Uptravi 1 000 mikrogramů
straně s vyraženým “10”.
potahované tablety: kulaté, tmavě fialové, potahované tablety na jedné
Uptravi 1 200 mikrogramů
straně s vyraženým “12”.
potahované tablety: kulaté, tmavě žluté, potahované tablety na jedné
Uptravi 1 400 mikrogramů
straně s vyraženým “14”.
potahované tablety: kulaté, hnědé, potahované tablety na jedné straně s
Uptravi 1 600 mikrogramů
vyraženým “16”.
Uptravi 200 mikrogramů potahované tablety se dodává blistrech po 10 nebo 60 tabletách nebo po 140 tabletách (titrační balíček).
Uptravi 400 mikrogramů, 600 mikrogramů, 800 mikrogramů, 1 000 mikrogramů, 1 200 mikrogramů, 1 400 mikrogramů a 1 600 mikrogramů potahované tablety se dodává v blistrech po 60 tabletách.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Actelion Registration Ltd Chiswick Tower 13th Floor 389 Chiswick High Road London W4 4AL Velká Británie Tel: +44 20 8987 3320
Výrobce
Actelion Pharmaceuticals Deutschland GmbH Konrad-Goldmann-Strasse 5b 79100 Freiburg Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Actelion Pharmaceuticals Belgium N.V. UAB ALGOL PHARMA
Tél/Tel: +32-(0)15 284 777 Tel: +370 37 40 86 81
Luxembourg/Luxemburg
Btarapna
AKBaXHM Afl Ten.: +359 2 807 50 00
Actelion Pharmaceuticals Belgium N.V. Tél/Tel: +32-(0)15 284 777
|
Česká republika Actelion Pharmaceuticals CZ, s.r.o. Tel: +420 221 968 006 |
Magyarország Actelion Pharmaceuticals Hungaria Kft. Tel: +36-1-413-3270 |
|
Danmark Actelion Danmark, Filial af Actelion Pharmaceuticals Sverige AB, Sverige Tlf: +45 3694 45 95 |
Malta Actelion Pharmaceuticals Ltd Tel: +44 208 987 3333 |
|
Deutschland Actelion Pharmaceuticals Deutschland GmbH Tel: +49 761 45 64 0 |
Nederland Actelion Pharmaceuticals Nederland B.V Tel: +31 (0)348 435950 |
|
Eesti Algol Pharma OU Tel: +372 605 6014 |
Norge Actelion Pharmaceuticals Sverige AB, Filial Norge Tlf: +47 22480370 |
|
EXlába Actelion Pharmaceuticals Ekkág A.E. Tnk: +30 210 675 25 00 |
Osterreich Actelion Pharmaceuticals Austria GmbH Tel: +43 1 505 4527 |
|
Espaňa Actelion Pharmaceuticals Espana S.L. Tel: +34 93 366 43 99 |
Polska Actelion Pharma Polska Sp. z o.o. Tel: +48 (22) 262 31 00 |
|
France Actelion Pharmaceuticals France SAS Tél: +33 1 58 62 32 32 |
Portugal Actelion Pharmaceuticals Portugal Lda. Tel: +351 21 358 6120 |
|
Hrvatska Medis Adria d.o.o. Tel: + 385 (0) 1 2303 446 |
Románia Geneva Romfarm International SRL Tel: + 40 (021) 231 3561 |
|
Ireland Actelion Pharmaceuticals UK Ltd Tel: +44 208 987 3333 |
Slovenija Medis d.o.o. Tel: +386-(0)1 589 69 00 |
|
Island Actelion Pharmaceuticals Sverige AB Sími: +46 (0)8 544 982 50 |
Slovenská republika Actelion Pharmaceuticals SK, s.r.o. Tel: +420 221 968 006 |
|
Italia Actelion Pharmaceuticals Italia S.r.l. Tel: +39 0542 64 87 40 |
Suomi/Finland Actelion Pharmaceuticals Sverige AB, Filial Finland Puh/Tel: +358 9 2510 7720 |
|
Kúnpoq Actelion Pharmaceuticals Ekkág A.E. Tnk: +30 210 675 25 00 |
Sverige Actelion Pharmaceuticals Sverige AB Tel: +46 8 544 982 50 |
United Kingdom
Latvija
Algol Pharma SIA Tel: +371 6761 9365
Actelion Pharmaceuticals UK Ltd Tel: +44 208 987 3333
Tato příbalová informace byla naposledy revidována měsíc RRRR
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Strana 1_
Uptravi potahované tablety selexipag
Průvodce titrací
Zahájení léčby přípravkem Uptravi
Před zahájením léčby si prosím přečtěte doprovodnou příbalovou informaci pro pacienty.
Strana 2 Strana 3
|
Obsah Jak máte užívat přípravek Uptravi?..........................4 Jak máte zvyšovat dávku?.........................................6 Jaké jsou kroky?........................................................8 Kdy máte přejít na nižší dávku?.............................10 Přechod na nižší dávku............................................12 |
Když se dostanete na udržovací dávku............ Jestliže zapomenete užít přípravek Uptravi...... Jestliže přestanete užívat přípravek Uptravi..... Titrační deník............................................. |
.....14 .....16 ....17 ......18 |
|
Strana 4 |
Strana 5 | |
|
Jak máte užívat přípravek Uptravi? |
Léčba přípravkem Uptravi probíhá ve dvou fázích: | |
|
Uptravi je léčivý přípravek, který se užívá každé ráno a | ||
|
večer k léčbě plicní arteriální hypertenze, rovněž |
Titrace | |
|
nazývané PAH. |
Během několika prvních týdnů se budete ve spoluráci | |
|
s Vaším lékařem snažit najít pro Vás vhodnou dávku | ||
|
Zahajovací dávka přípravku Uptravi je |
přípravku Uptravi. Váš lékař může rozhodnout, že | |
|
200 mikrogramů jednou ráno a jednou večer. |
přejdete od zahajovací dávky přípravku Uptravi na | |
|
První dávka přípravku Uptravi se má užít večer. |
vyšší dávku. Váš lékař může rozhodnout, že přejdete | |
|
Každou dávku musíte zapít sklenicí vody, přičemž je |
na nižší dávku. Tento postup se nazývá titrace. | |
|
lepší tabletu užít při jídle. |
Umožní tělu postupně si na lék zvyknout. | |
|
Udržovací fáze | ||
|
Jakmile lékař nalezne dávku, která je pro Vás vhodná, | ||
budete tuto dávku užívat pravidelně. Nazývá se udržovací dávka.
Jak máte zvyšovat dávku?
Léčbu zahájíte dávkou 200 mikrogramů ráno a večer a po dohodě s lékařem nebo zdravotní sestrou přejdete na další dávku.
Zvýšená dávka se má poprvé užít večer. Každý krok obvykle trvá přibližně 1 týden. Nalezení pro Vás vhodné dávky může trvat několik týdnů.
Cílem je dosáhnout dávky, která je pro léčbu nejvhodnější.
Tato dávka bude Vaší udržovací dávkou.
Každý pacient s PAH je jiný. Ne každý bude mít stejnou udržovací dávku.
Někteří pacienti mohou jako svou udržovací dávku užívat dávku 200 mikrogramů ráno a večer, zatímco jiní dosáhnou nejvyšší dávky 1 600 mikrogramů ráno a večer.
Další mohou dosáhnout udržovací dávku někde mezi tím. Důležité je, abyste dosáhl(a) pro Vás nejvhodnější dávky.
^ Jaké jsou to kroky?
200mlkrogramová tableta
n <Uvfcov«:i krok Irvi přibližné 1 týden
ISOOmlkrogramová tableta (Použlltc u kroků & až 8. aby se snížil počet tablet potřebných na dávku.)
200mikrogramová tableta
NEJVYSŠI
DÁVKA
POČÁTEČNÍ
DÁVKA
KROK 1 Ráno: jedna 200mlkrogramová tableta Večer: jedna 200mlkrogramová tableta (Celková denní dávka: 400 mikrogramů)
KROK 2 Ráno: dvě
200mikrog rámové tablety Večer: dvé 200mikrog rámové tablety (Celková denní dávka: 800 mikrogramů)
KROK 3 Ráno: tři
200mikr©gramové
tablety Večer: tři
200mikrogramov0 tablety (Celková denní dávka:
1200 mikrogramů)
KROK 4
Ráno: čtyř* 200mikrogramové tablety večer čtyři 200mikrogramové tablety
(Celková denní davka: 1 600 mikrogramů)
KROK 5 Ráno: jedna 800mlkrogramová a jedna
200mikrogramová tableta Večer jedna SOOmlkrogramová a jedna
200mikrogramová
tableta
(Celková denní dávka: 2 000 mikrogramů)
KROK 6 Ráno: jedna
800mikrogramova
a tři
200mlkrogramové tablety Večer jedna 800mikrogramová a tři
200mikrogramove
tablety
(Celková denní dávka: 2 800 mikrogramů)
KROK 7 Ráno: jedna 800mikrogramová a
tři 200mikrogramove
tablety Večer: jedna 800mikrogramová a tři 200mikrogramové tablety
(Celková denní dávka:
2 800 mikrogramů)
KROK 8 Ráno: jedna 800mlkrogramová a čtyři
200mikrogramové tablety Večer jedna 800mikrog rámová a čtyři
200mikrogramové
tablety
(Celková denní dávka: 3 200 mikrogramů)
(Tatúety ve skutečnosti nemalí tyto rormáry)
4 Kdy máte přejít na nižší dávku?
Jako u všech léků, mohou se u Vás při zvyšování dávky přípravku Uptravi vyskytnout nežádoucí účinky.
Pokud se u Vás vyskytnou nežádoucí účinky, sdělte to svému lékaři nebo zdravotní sestře. K dispozici je dostupná terapie, která může pomoci ulevit od těchto nežádoucích účinků.
Nejčastějšími nežádoucími účinky (mohou postihnout více než 1 z 10 osob), které se u Vás mohou při užívání přípravku Uptravi vyskytnout, jsou:
• bolest hlavy • průjem • pocit na zvracení • zvracení
• bolest čelisti • bolest svalů • bolest nohou • bolest kloubů • zarudnutí v obličeji
Úplný seznam nežádoucích účinků naleznete v příbalové informaci._
Pokud nežádoucí účinky nemůžete snášet i když se je lékař nebo zdravotní sestra pokusili léčit, mohou Vám doporučit přejít na nižsí dávku.
Pokud Vám lékař nebo zdravotní sestra doporučí přejít na nižší dávku, musíte ráno i večer užít o jednu 200mikrogramovou tabletu méně.
Přejít na nižší dávku smíte pouze po poradě s lékařem, který léčí Vaši PAH, nebo jeho zdravotní sestrou. Tento postup snižování dávky Vám pomůže najít dávku, která je pro Vás vhodná, a která se rovněž nazývá udržovací dávka.
Postupné snižovaní
KROK NAHORU
KROK DOLU
UDRŽOVACÍ DAVKA
KROK 4
KROK 5
KROK 6
ZPĚT KE
JEDINÁ TABLETA,
Jeden dávkovači krok trvá přibližní 1 týden
STEJNÁ DAVKA
200mlkrogramova tableta
r Pokud nebude moM tolerovat netadouci
Z*nit. užíval
nelldouciml uCKwy
účinky, jdit
dívky
krok zpil
BOOmlkrogramova tableta
:C tikoví denní dívki
200mlkrogramoví tableta
(Použijte u kroku 5 až 8. aby se snížil
počet tablet potřebných na dávku )
(Tablety ve skutečnosti nemají tyto rozměry.)
Kdy se dostanete na udržovací dávku
Nejvyšší dávka, kterou budete moci snášet během titrace, bude Vaší udržovací dávkou. Udržovací dávka je dávka, kterou máte dál pravidelně užívat.
Váš lékař nebo zdravotní sestra mohou jako udržovací dávku předepisovat ekvivalentní sílu v jedné tabletě. To Vám umožní užívat místo několika tablet pro jednu dávku pouze jednu tabletu ráno a jednu večer.
Například pokud během titrace byla nejvyšší tolerovaná
dávka 1 200 mikrogramů jednou ráno a jednou večer:

Jestliže zapomenete užít přípravek Uptravi
Pokud zapomenete užít dávku, užijte ji jakmile si vzpomenete, poté tablety užívejte v obvyklý čas. Pokud do další dávky zbývá 6 hodin nebo méně, zapomenutou dávku vynechte a lék dál užívejte v obvyklý čas.
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou tabletu.
Jestliže přestanete užívat přípravek Uptravi
Přípravek Uptravi nepřestávejte užívat, dokud Vám k tomu nedá pokyn Váš lékař nebo zdravotní sestra. Pokud z jakéhokoli důvodu přestanete užívat přípravek Uptravi po více než 3 po sobě následující dny (pokud vynecháte 6 dávek v řadě), obraťte se ihned na lékaře, který Vás na PAH léčí, nebo jeho zdravotní sestru, protože může být potřeba dávku upravit, aby se zamezilo vzniku nežádoucích účinků.
Váš lékař nebo zdravotní sestra může určit, abyste léčbu obnovil(a) na nižší dávce a tu postupně zvyšoval(a) na předchozí udržovací dávku._
|
Titrační deník |
Nezapomeňte se s lékařem, který Vás na PAH léčí, nebo jeho zdravotní sestrou pravidelně radit. |
|
Přečtěte si, prosím, pečlivě pokyny uvedené v příbalové informaci. |
Zapište si pokyny svého lékaře nebo zdravotní sestry: |
|
Následující stránky deníku Vám pomohou sledovat počet tablet, které máte během titrace užívat ráno a večer. | |
|
Použijte je k zápisování počtu tablet, které ráno a večer užíváte. |
Telefon a e-mail do ordinace lékaře: |
|
Každý krok obvykle trvá 1 týden, pokud Vám lékař nebo zdravotní sestra nedá jiný pokyn. Pokud u Vás titrační kroky trvají déle než 1 týden, jsou zde další |
Telefon na lékárníka: |
|
stránky deníku k jejich zapisování. |
Poznámky: |
|
Ke sledování průběhu prvních týdnů léčby (kroky 1-4), kdy užíváte pouze 200mikrogramové tablety, použijte strany 19 až 27. | |
|
^ Pokud j sou Vám předepisovány j ak | |
|
200mikrogramové, tak 800mikrogramové tablety, použijte strany 28 až 37 (kroky 5-8). |
Strana 20 Strana 21
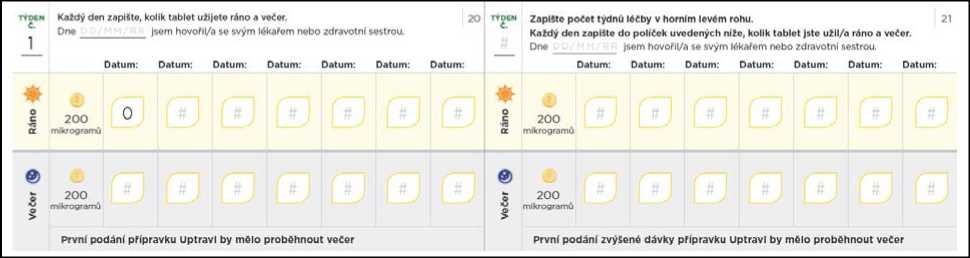
Strana 22 Strana 23
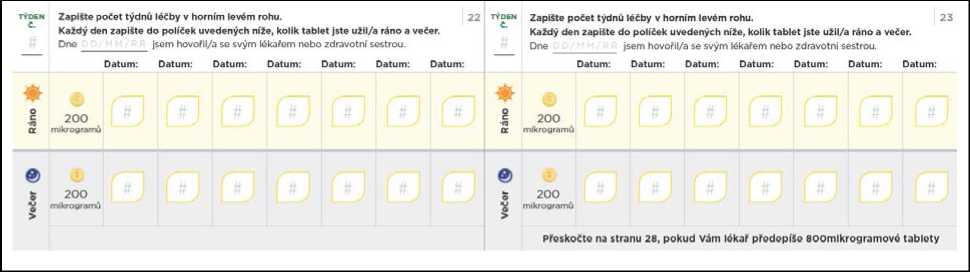
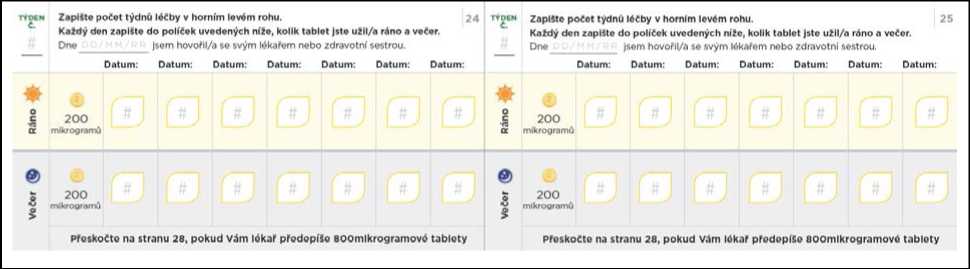
Strana 26 Strana 27
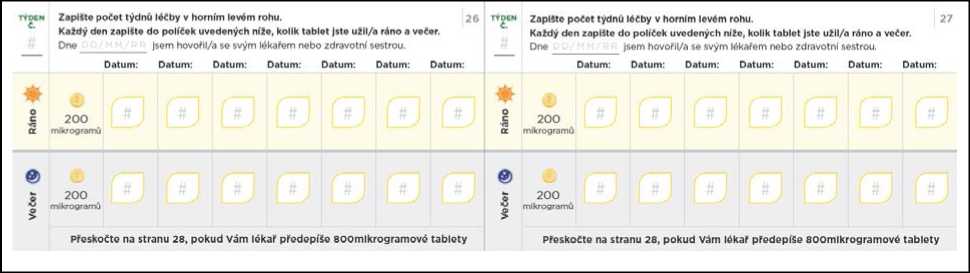
Strana 28 Strana 29
Následující stránky deníku použijte, pokud Vám lékař nebo zdravotní sestra předepíší vedle 200mikrogramových tablet i 800mikrogramové tablety.
Na stránkách deníku si zaškrtávejte, že jste k předepsanému počtu 200mikrogramových tablet každý den ráno a večer užil(a) jednu 800mikrogramovou tabletu.
■
200mikrogramová tableta
SOOmikrogramová tableta (Použijte u kroků 5 až S, aby se snížil počet tablet potřebných na dávku.)
Nezapomeňte se s lékařem, který Vás na PAH léčí, nebo jeho zdravotní sestrou pravidelně radit.
Zapište si pokyny svého lékaře nebo zdravotní sestry: Telefon a e-mail do ordinace lékaře:
Telefon na lékárníka:
Poznámky:
Strana 30
Strana 31
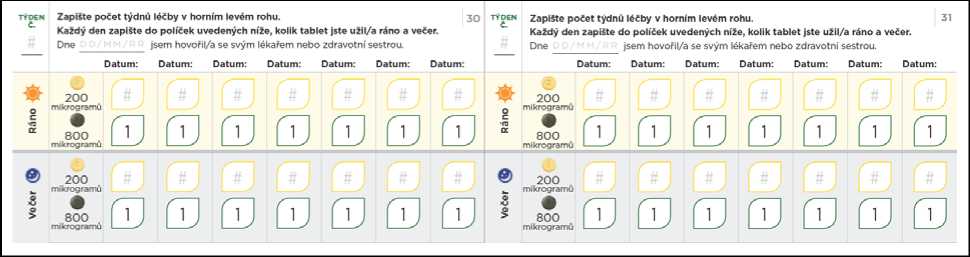
tvocn Zaplit* počet týdnů WC by v horním lovům rohu. 32 tydot Zaplito počet týdnů léčby v horním lovům rohu.
Každý don zaplito do poličok uvodoných nfío, kolik tablot jste užll/a ráno a vočor. Každý don zaplito do políček uvodoných nížo, kolik tablet Jste užll/a ráno a večer.
Dne ■ Jsem hovořil/a se svým lékařem nebo zdravotní sestrou. Dne jsem hovořil/a se svým lékařem nebo zdravotní sestrou.
|
* 0 Ě S |
1 200 mikrogramů • 800 mikrogramů |
Saturn: 1 0_ |
datum: 1 0L |
datum: 0 |
Datum: 0 |
Datum: 0 |
Datum: 10 |
Datum: 0 |
* 1 a |
1 200 mikrogramů • 800 mikrogramů |
Datum: 0 |
Datum: 1 Ój |
Datum: 1 0 |
Datum: 0 |
Datum: 0 |
Datum: 0 |
Datum: 0 |
|
® i s |
200 mikrogramů • 800 mikrogramů |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
® S $ |
200 mikrogramů • 800 mikrogramů |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
Strana 34 Strana 35
tVosm Zaplit* počot týdnů léčby v horním lovům rohu. 34 tVdsm Zaplito počet týdnů léčby v horním lovům rohu. 35
Každý den zaplito do políček uvodoných níže, kolik tablet Jste užll/a ráno a večer. Každý don zaplito do poličok uvodoných nížo, kolik tablot Jste užll/a ráno a večer.
Dne ■ ■ '''1 Jsem hovořil/a se svým lékařem nebo zdravotní sestrou. Dne Jsem hovořll/a se svým lékařem nebo zdravotní sestrou.
|
♦ O c e |
1 200 mikrogramů • 800 mikrogramů |
Datum: 10 |
Datum: 0 |
Datum: 0 |
Datum: 0 |
Datum: 0 |
Datum: JjJ |
Datum: d |
❖ © ■1 |
200 mikrogramů m 800 mikrogramů |
Datum: 0 |
Datum: 0_ |
Datum: 0 |
Datum: 0 |
Datum: 0 |
Datum: 0 |
Datum: 0 |
|
® s 5 |
200 mikrogramů • 800 mikrogramů |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
® a v ? |
200 mikrogramů • 800 mikrogramů |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
Strana 36 Strana 37
tVosm Zaplito počet týdnů léčby v horním lovům rohu. 36 tVdsm Zaplito počet týdnů léčby v horním lovům rohu. 37
' Každý den zaplito do poliček uvodoných nížo, kolik tablet Jste užil/a ráno a večer. ' Každý don zaplito do poličok uvodoných niž*, kolik tablot Jst* užil/a ráno a večer.
Dne " " Jsem hovořil/a se svým lékařem nebo zdravotní sestrou. Dne Jsem hovořll/a se svým lékařem nebo zdravotní sestrou.
Datum: Datum: Datum: Datum: Datum: Datum: Datum: Datum: Datum: Datum: Datum: Datum: Datum: Datum:
■0 200 #1 í # I (#J f#J Í#J [#J (#J ^ 200
mikrogramů „ mikrogramú
|
0 c e |
• 800 mikrogramů |
0_ |
0 |
0 |
0 |
0 |
0 |
O, |
O E ’S |
• 800 mikrogramů |
0 |
0_ |
0 |
0 |
0 |
0 |
0 |
|
® s 5 |
200 mikrogramů • 800 . mikrogramů |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
® a v s |
200 mikrogramů • 800 mikrogramů |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
Strana 38 Strana 39
|
Poznámky | |
|
Strana 40 | |
|
Actelion Pharmaceuticals Ltd. | |
46