Translarna 125 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Translarna 125 mg granule pro perorální suspenzi
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden sáček obsahuje atalurenum 125 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Granule pro perorální suspenzi. Bílé až téměř bílé granule.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Translarna je indikován k léčbě Duchennovy svalové dystrofie vzniklé v důsledku nonsense mutace v genu pro dystrofin u chodících pacientů ve věku od 5 let (viz bod 5.1). U nechodících pacientů účinnost nebyla prokázána.
Přítomnost nonsense mutace v genu pro dystrofin má být stanovena genetickými testy (viz bod 4.4).
4.2 Dávkování a způsob podání
Léčbu přípravkem Translarna má zahajovat pouze specializovaný lékař se zkušenostmi s léčbou Duchennovy/Beckerovy svalové dystrofie.
Dávkování
Ataluren se podává perorálně každý den ve 3 dávkách.
První dávka se užívá ráno, druhá v poledne a třetí večer. Doporučené intervaly mezi dávkami jsou 6 hodin mezi ranní a polední dávkou, 6 hodin mezi polední a večerní dávkou a 12 hodin mezi večerní dávkou a první dávkou podanou následující den.
Doporučená dávka přípravku je 10 mg/kg tělesné hmotnosti ráno, 10 mg/kg tělesné hmotnosti v poledne a 20 mg/kg tělesné hmotnosti večer (pro dosažení celkové denní dávky 40 mg/kg tělesné hmotnosti).
Přípravek Translarna je dostupný v sáčcích obsahujících 125 mg, 250 mg nebo 1 000 mg atalurenu. Následující tabulka poskytuje informace, kolik sáčků o určité síle je třeba použít při přípravě doporučené dávky pro dané rozmezí tělesné hmotnosti.
|
Rozmezí tělesné hmotnosti (kg) |
Počet sáčků | |||||||||
|
Ráno |
V poledne |
Večer | ||||||||
|
125mg sáčky |
250mg sáčky |
1 000mg sáčky |
125mg sáčky |
250mg sáčky |
1 000mg sáčky |
125mg sáčky |
250mg sáčky |
1 000mg sáčky | ||
|
12 |
14 |
1 |
0 |
0 |
1 |
0 |
0 |
0 |
1 |
0 |
|
15 |
16 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
1 |
0 |
|
17 |
20 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
|
21 |
23 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
1 |
0 |
|
24 |
26 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
0 |
|
27 |
31 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
0 |
|
32 |
35 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
0 |
|
36 |
39 |
1 |
1 |
0 |
1 |
1 |
0 |
0 |
3 |
0 |
|
40 |
44 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
3 |
0 |
|
45 |
46 |
0 |
2 |
0 |
0 |
2 |
0 |
1 |
3 |
0 |
|
47 |
55 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
0 |
1 |
|
56 |
62 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
1 |
1 |
|
63 |
69 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
1 |
1 |
|
70 |
78 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
2 |
1 |
|
79 |
86 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
3 |
1 |
|
87 |
93 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
3 |
1 |
|
94 |
105 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
|
106 |
111 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
|
112 |
118 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
|
119 |
125 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
2 |
2 |
Opožděná nebo vynechaná dávka
Jestliže zpoždění při podání atalurenu činí méně než 3 hodiny po ranní či polední dávce nebo méně než 6 hodin po večerní dávce, dávka by měla být užita beze změny následujícího dávkovacího schématu. Jestliže je zpoždění delší než 3 hodiny po ranní nebo polední dávce nebo delší než 6 hodin po večerní dávce, neměla by se dávka užívat a pacient by měl pokračovat podle obvyklého dávkovacího schématu. V případě vynechání dávky by pacienti neměli dávku zdvojnásobovat ani užívat dávku navíc. Je důležité podávat správnou dávku. Zvýšení doporučené dávky může být spojeno se snížením účinnosti.
Zvláštní populace
Starší lidé
Bezpečnost a účinnost atalurenu u pacientů ve věku 65 let a starších nebyla dosud stanovena (viz bod 5.2.)
Insuficience ledvin a jater
Bezpečnost a účinnost atalurenu u pacientů s poruchou funkce ledvin a jater nebyla dosud stanovena (viz bod 4.4).
Pediatrická populace
Bezpečnost a účinnost přípravku Translarna u dětí ve věku od 6 měsíců do 5 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Translarna se užívá perorálně po smísení do formy suspenze v tekutině nebo v polotekuté stravě. Sáčky se mají otevírat až v době přípravy dávky. Celý obsah každého sáčku by se měl smísit s minimálně 30 ml tekutiny (vody, mléka, ovocného džusu) nebo 3 lžícemi polotekuté stravy (jogurtu
nebo jablečného protlaku). Připravená dávka by se před podáním měla dobře promísit. Množství tekutin nebo polotekuté stravy lze zvýšit podle preferencí pacienta. Pacienti by měli užít celou dávku.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Souběžné užívání intravenózních aminoglykosidů (viz bod 4.4 a 4.5).
4.4 Zvláštní upozornění a opatření pro použití Pacienti, kteří nenesou nonsense mutaci
Onemocnění pacientů musí být založeno na nosičství nonsense mutace v genu pro dystrofin, stanovené genetickými testy. Pacientům, kteří nonsense mutaci nenesou, nemá být ataluren podáván.
Porucha funkce ledvin a jater
Pacienti s poruchami funkce ledvin či jater by měli být pečlivě sledováni.
Změny lipidového profilu
Protože u některých pacientů v klinických studiích byly hlášeny změny lipidového profilu (zvýšené triglyceridy a cholesterol), u pacientů s DMD s nonsense mutací, kterým je podáván ataluren, se doporučuje každoročně nebo podle klinického stavu pacienta i častěji monitorovat celkový cholesterol, LDL, HDL a triglyceridy.
Hypertenze při současném užívání systémových kortikosteroidů
Protože v klinických studiích byla u některých pacientů při současném užívání systémových kortikosteroidů hlášena hypertenze, u pacientů s DMD s nonsense mutací, kterým je podáván ataluren souběžně s kortikosteroidy, se doporučuje každých 6 měsíců nebo podle klinického stavu pacienta i častěji monitorovat klidový systolický a diastolický krevní tlak.
Monitorování renálních funkcí
Protože v kontrolované studii nonsense mutace v genu pro dystrofin byl pozorován mírný vzestup průměrné hodnoty sérového kreatininu, dusíku močoviny (BUN) v krvi a cystatinu C, u pacientů s DMD s nonsense mutací, kterým je podáván ataluren, se doporučuje každých 6 až 12 měsíců nebo podle klinického stavu pacienta i častěji monitorovat hladiny sérového kreatininu, BUN a cystatinu C.
Možné interakce s jinými léčivými přípravky
Při současném podávání atalurenu s léčivými přípravky, které jsou substráty či induktory UGT1A9, inhibitory BCRP nebo substráty OAT1, OAT3 nebo OATP1B3 (viz bod 4.5), je zapotřebí opatrnosti.
Aminoglykosidy
Bylo prokázáno, že aminoglykosidy snižují čtecí aktivitu atalurenu in vitro. Bylo navíc zjištěno, že ataluren zvyšuje nefrotoxicitu intravenózních aminoglykosidů. Je nutné se vyvarovat současného podávání těchto léčivých přípravků s atalurenem (viz bod 4.3). Protože není znám mechanismus, kterým ataluren zvyšuje nefrotoxicitu intravenózních aminoglykosidů, souběžné použití jiných nefrotoxických léčivých přípravků s atalurenem se nedoporučuje. Pokud se této situaci nelze vyhnout (např. vankomycin k léčbě MRSA), doporučuje se pečlivé monitorování renálních funkcí (viz bod 4.5).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Aminoglykosidy
Ataluren by neměl být podáván současně s intravenózními aminoglykosidy, protože v klinické studii u pacientů s nmCF byly zjištěny případy snížených renálních funkcí (viz bod 4.3).
U několika pacientů s nmCF léčených atalurenem a intravenózními aminoglykosidy spolu s dalšími antibiotiky z důvodu exacerbace cystické fibrózy došlo ke zvýšení hladin sérového kreatininu. Zvýšení sérového kreatininu ve všech případech ustoupilo při ukončení podávání intravenózních aminoglykosidů bez ohledu na pokračování nebo přerušení podávání přípravku Translarna. Tato zjištění naznačují, že současné podávání přípravku Translarna a intravenózních aminoglykosidů může zvyšovat nefrotoxický účinek aminoglykosidů. Proto je-li nezbytná léčba intravenózními aminoglykosidy, měla by být léčba přípravkem Translarna přerušena a poté znovu zahájena 2 dny po ukončení podávání aminoglykosidů. Účinek současného podávání atalurenu s jinými nefrotoxickými léčivými přípravky není znám.
V některých z těchto případů může být přispívajícím faktorem dehydratace. Pacienti by měli při užívání atalurenu zachovávat dostatečnou hydrataci. Viz bod 4.4.
Účinek jiných léčivých přípravků na farmakokinetiku atalurenu
Studie in vitro ukázaly, že ataluren je substrátem UGT1A9 a proteinu BRCP (proteinu rezistence u karcinomu prsu). Při současném podávání atalurenu s léčivými přípravky, které jsou induktory UGT1A9 (např. mykofenolát mofetilu) nebo inhibitory BCRP (např. cyklosporinu), je zapotřebí opatrnosti.
Účinek atalurenu na farmakokinetiku jiných léčivých přípravků
Studie in vitro ukázaly, že ataluren je inhibitorem UGT1A9, OAT1 (transportéru organických aniontů 1), OAT3 (transportéru organických aniontů 3) a OATP1B3 (transportního polypeptidu organických aniontů). Když je ataluren podáván současně s léčivými přípravky, které jsou substráty UGT1A9, OAT1, OAT3 nebo OATP1B3 (např. oseltamivirem, acyklovirem, ciprofloxacinem, kaptoprilem, furosemidem, bumetanidem, valsartanem, pravastatinem, rosuvastatinem, atorvastatinem, pitavastatinem), je zapotřebí opatrnosti vzhledem k riziku zvýšení koncentrace těchto léčivých přípravků.
Na základě studií in vitro se očekává, že ataluren není inhibitorem ani p-gp mediovaného transportu ani metabolismu zprostředkovaného cytochromem P450. Podobně se očekává, že ataluren není in vivo induktorem izoenzymů cytochromu P450.
Současné podávání kortikosteroidů (deflazakortu, prednisonu nebo prednisolonu) s atalurenem neovlivňuje plazmatické koncentrace atalurenu. Při současném podávání atalurenu nebyla pozorována klinicky významná změna plazmatických koncentrací kortikosteroidů. Tyto údaje nenaznačují zjevnou interakci léčivo-léčivo mezi kortikosteroidy a atalurenem a není nutná úprava dávky.
Léčivé přípravky, které postihují transportér p-glykoprotein
In vitro ataluren není substrátem transportéru p-glykoprotein. Farmakokinetika atalurenu pravděpodobně není ovlivněna léčivými přípravky, které inhibují transportér p-glykoprotein.
4.6 Fertilita, těhotenství a kojení
Není k dispozici dostatek údajů o použití atalurenu u těhotných žen. Studie na zvířatech prokázaly reprodukční toxicitu pouze v dávkách, které jsou toxické pro matku (viz bod 5.3).
Jako preventivní opatření se doporučuje vyhnout se použití atalurenu během těhotenství.
Kojení
Není známo, zda se ataluren/metabolity vylučují do lidského mateřského mléka. Dostupné farmakodynamické/toxikologické údaje u zvířat prokázaly vylučování atalurenu/metabolitů do mléka (viz bod 5.3). Riziko pro kojené novorozence/kojence nelze vyloučit.
Kojení má být během léčby atalurenem přerušeno.
Fertilita
Neklinické údaje na základě standardní studie samčí a samičí toxicity u potkanů neodhalily riziko pro člověka (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Účinek atalurenu na řízení, jízdu na kole a obsluhu strojů nebyl testován. Pacienti, u kterých dochází k závratím, by měli být při řízení, jízdě na kole nebo při obsluze strojů opatrní.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
V klinických studiích u pacientů s Duchennovou svalovou dystrofií způsobenou nonsense mutací (nmDMD) byly nejčastějšími nežádoucími účinky při doporučené dávce nevolnost, zvracení a bolest hlavy. Tyto nežádoucí účinky obecně nevyžadovaly lékařský zásah a u žádného z pacientů nedošlo k přerušení léčby atalurenem z důvodu nežádoucích účinků.
Tabulkový přehled nežádoucích účinků
Nežádoucí účinky hlášené v klinické studii u převážně pediatrických pacientů s nmDMD léčených doporučenou dávkou 10 - 10 - 20 mg/kg jsou klasifikovány podle tříd orgánových systémů a frekvence MedDRA. Skupiny frekvencí jsou definovány podle následující konvence: velmi časté (> 1/10) a časté (> 1/100 až < 1/10). V každé skupině četností jsou nežádoucí účinky uvedeny v pořadí podle klesající závažnosti.
Tabulka 1: Nežádoucí účinky přípravku Translama v kontrolované studii nmDMD
|
Třída orgánových systémů |
Velmi časté |
Časté |
Frekvence neznámá |
|
Poruchy metabolismu a výživy |
Snížená chuť k jídlu |
Změna lipidového profilu (zvýšené triglyceridy a cholesterol) | |
|
Poruchy nervového systému |
Závratě | ||
|
Cévní poruchy |
Hypertenze | ||
|
Respirační, hrudní a mediastinální poruchy |
Kašel, epistaxe | ||
|
Gastrointestinální poruchy |
Bolest horní části břicha, nadýmání, průjem, břišní dyskomfort, bolest břicha, zácpa, regurgitace | ||
|
Poruchy kůže a podkožní tkáně |
Erytém | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolest končetin | ||
|
Poruchy ledvin a močových cest |
Enuréza, ledvinná cysta, polakisurie, abnormální zbarvení moči |
Změna testů renálních funkcí (zvýšený kreatinin, dusík močoviny v krvi, cystatin C) | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie, únava, snížení tělesné hmotnosti |
Popis vybraných nežádoucích účinků
Sérové lipidy
V průběhu kontrolované studie nmDMD byly průměrné celkové hladiny cholesterolu a triglyceridů ve výchozím bodě normální a zvyšovaly se, přičemž dosáhly hraničně vysokých nebo vysokých hodnot. Hodnoty se v časné fázi studie obvykle stabilizovaly a při pokračování léčby se dále nezvyšovaly.
Testy renálních funkcí
Během kontrolované studie nmDMD byl pozorován mírný vzestup průměrných hodnot sérového kreatininu, dusíku močoviny (BUN) v krvi a cystatinu C. Hodnoty se v časné fázi studie obvykle stabilizovaly a při pokračování léčby se dále nezvyšovaly.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V*.
4.9 Předávkování
U zdravých dobrovolníků, kterým byla podána jedna perorální dávka 200 mg/kg atalurenu, došlo k přechodným, málo intenzivním symptomům bolesti hlavy, nevolnosti, zvracení a průjmu. U těchto subjektů nebyly pozorovány žádné závažné nežádoucí účinky. V případě podezření na předávkování by měla být poskytnuta podpůrná lékařská péče včetně konzultace se zdravotnickým pracovníkem a pečlivého sledování klinického stavu pacienta.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: {skupina}, ATC kód: dosud nepřidělen Mechanismus účinku
Nonsense mutace v DNA má za následek předčasný stop kodon v mRNA. Tento předčasný stop kodon v mRNA způsobuje onemocnění tím, že ukončuje translaci ještě před vytvořením proteinu o plné délce. Ataluren umožňuje ribozomální čtení mRNA obsahující tento předčasný stop kodon, které vede k tvorbě proteinu o plné délce.
Farmakodynamické účinky
Neklinické experimenty in vitro s buňkami a rybími larvami s nonsense mutacemi pěstovanými v roztoku atalurenu ukázaly, že ataluren umožňuje ribozomální čtení, přičemž křivka závislosti odpovědi na koncentraci má zvonovitý tvar (tvar obráceného písmene U). Existují hypotézy, že in vivo může být křivka závislosti odpovědi na dávce také zvonovitá, údaje in vivo jsou ale příliš omezené na to, aby tuto hypotézu bylo možné potvrdit u myšího modelu DMD s nonsense mutací a u člověka.
Neklinické studie in vitro naznačují, že kontinuální expozice atalurenu může být důležitá z hlediska maximalizace aktivity a že účinky léčivé látky na ribozomální čtení kodonů předčasného ukončení vymizí krátce po vysazení atalurenu.
Klinická účinnost a bezpečnost
Bezpečnost a účinnost přípravku Translarna byla hodnocena v randomizované dvojitě zaslepené placebem kontrolované multicentrické studii Duchennovy svalové dystrofie s nonsense mutací (nmDMD) u 174 pacientů mužského pohlaví ve věku 5 až 20 let. Bylo nutné, aby všichni pacienti byli chodící, což bylo definováno jako schopnost ujít během screeningového 6minutového testu chůze (6MWT) > 75 metrů bez podpůrných pomůcek. U pacientů muselo být také zdokumentováno potvrzení přítomnosti nonsense mutace v genu pro dystrofin, zjištěné pomocí genového sekvenování. Většina pacientů (90 %) ve všech léčených skupinách byli běloši. Pacienti byli randomizováni v poměru 1:1:1 a po dobu 48 týdnů dostávali 3krát denně (ráno, v poledne, večer) ataluren nebo placebo, přičemž 57 pacientů dostávalo placebo, 57 pacientů dostávalo ataluren v dávce 10 - 10 -20 mg/kg a 60 pacientů dostávalo ataluren v dávce 20 - 20 - 40 mg/kg; studii dokončilo 173 pacientů. Primárním cílovým parametrem z hlediska účinnosti byl účinek atalurenu na chůzi, posuzovaný podle změny vzdálenosti (6MWD), kterou pacient ujde během 6minutového testu chůze (6MWT). Post hoc analýza ukázala, že u pacientů dostávajících 10 - 10 - 20 mg/kg atalurenu došlo mezi výchozím bodem a 48. týdnem k poklesu vzdálenosti 6MWD průměrně o 12,9 metrů, zatímco u pacientů dostávajících placebo došlo k poklesu této vzdálenosti průměrně o 44,1 metrů (Obr. 1). Průměrná změna vzdálenosti 6MWD mezi výchozím bodem a 48. týdnem byla tedy v rameni s atalurenem 10 -10 - 20 mg/kg o 31,3 metrů lepší než v rameni s placebem (p=0,056). Ve statistickém modelu odhadu byl průměrný rozdíl 31,7 metru (upravená hodnota p=0,0367). Mezi atalurenem v dávce 20 - 20 -40 mg/kg a placebem nebyly rozdíly. Tyto výsledky ukazují, že ataluren v dávce 10 - 10 - 20 mg/kg snižuje u pacientů s nmDMD ztrátu schopnosti chůze.
Obrázek 1. Průměrná změna vzdálenosti, kterou pacient ujde za 6 minut
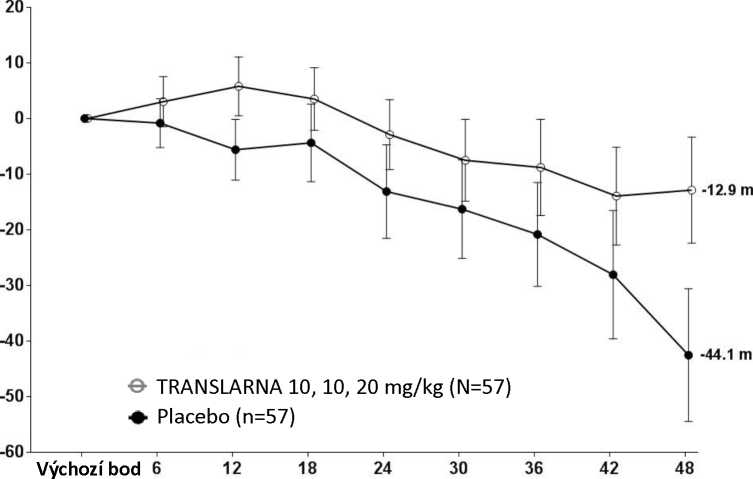
Týdny
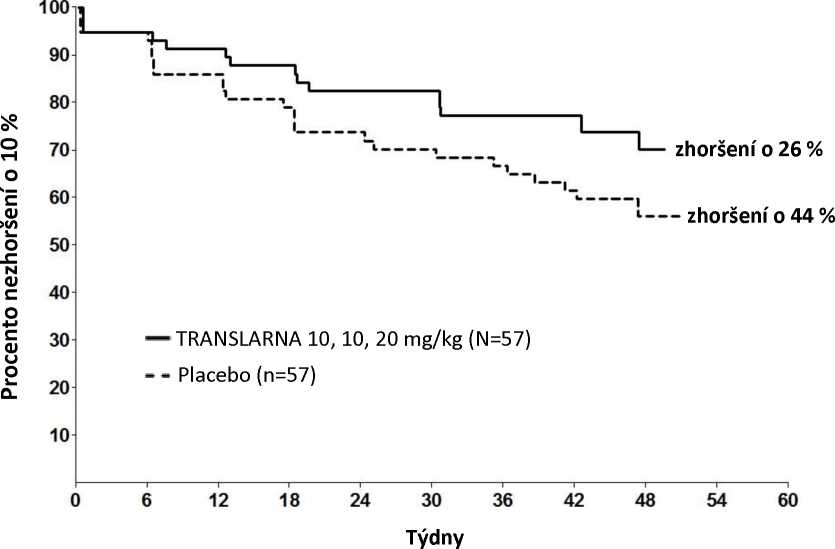
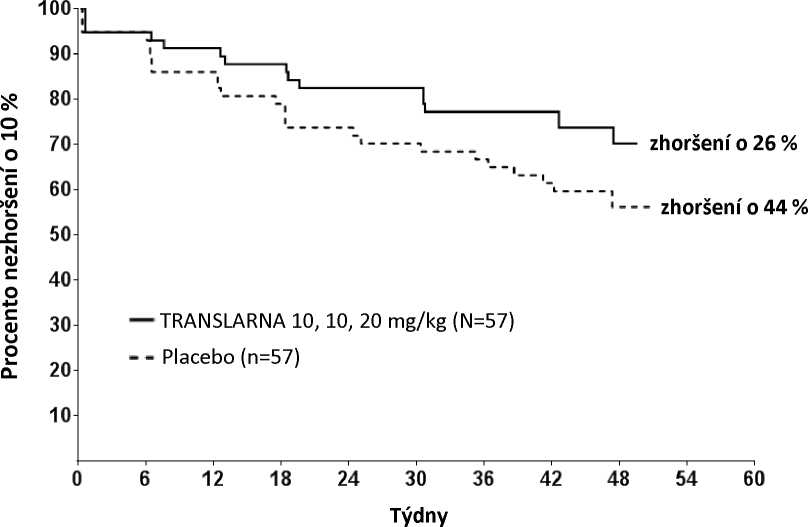
Post hoc analýza doby do trvalého zhoršení vzdálenosti 6MWD o 10 % ukázala, že v rameni s atalurenem 10 - 10 - 20 mg/kg došlo do 48. týdne k progresi u 26 % pacientů oproti 44 % pacientů ve skupině s placebem (p=0,0652) (obrázek 2). Mezi atalurenem v dávce 20 - 20 - 40 mg/kg a placebem nebyly rozdíly. Tyto výsledky ukazují, že za 48 týdnů došlo ke zhoršení vzdálenosti 6MWD u nižšího počtu pacientů dostávajících ataluren 10 - 10 - 20 mg/kg.
Obrázek 2. Kaplan-Meierova křivka doby do trvalého zhoršení vzdálenosti 6MWD o 10 %
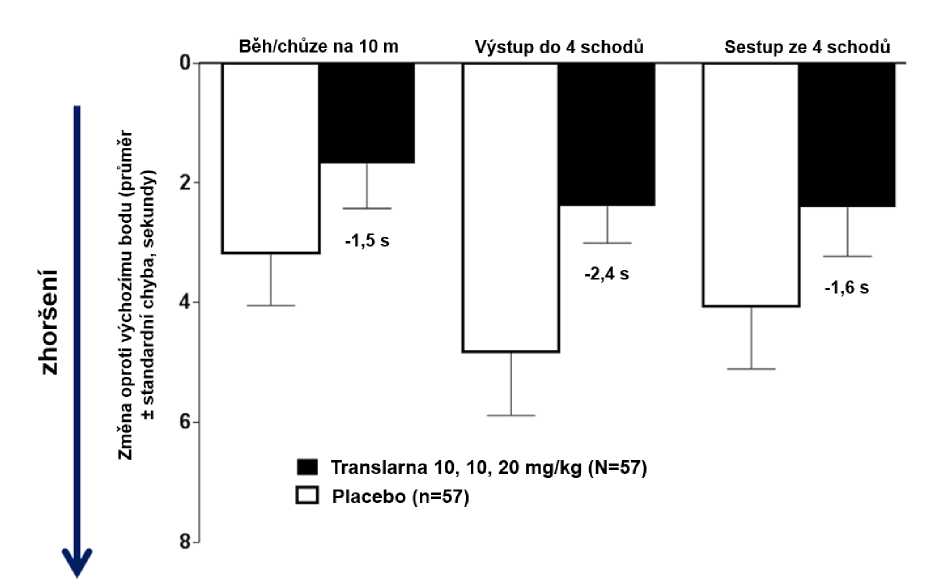
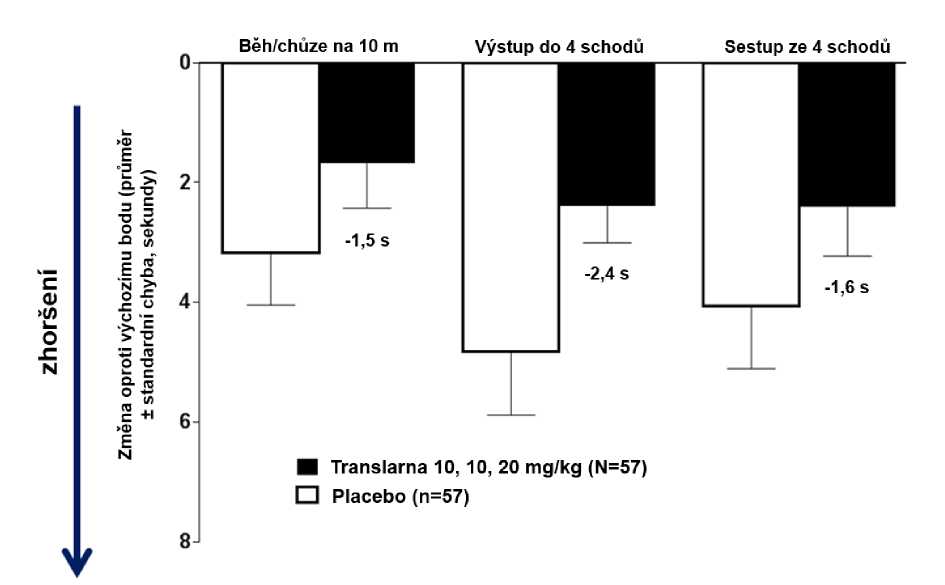
Ve funkčních testech s měřením času (TFT), testech času potřebného k uběhnutí/ujití 10 metrů, k výstupu do 4 schodů a k sestupu ze 4 schodů byl u pacientů léčených atalurenem prokázán menší nárůst času potřebného k uběhnutí/ujití 10 metrů, výstupu do 4 schodů a sestupu ze 4 schodů, což naznačuje zpomalení progrese nmDMD vůči placebu.
Průměrná změna ve funkčních testech s měřením času mezi výchozím bodem a 48. týdnem byla v rameni s atalurenem 10 - 10 - 20 mg/kg lepší než u placeba u času potřebného k uběhnutí/ujití 10 metrů (lepší o 1,5 sekundy), času potřebného k výstupu do 4 schodů (lepší o 2,4 sekundy) i času potřebného k sestupu ze 4 schodů (lepší o 1,6 sekundy), obrázek 3.
Obrázek 3. Průměrná změna ve funkčních testech s měřením času
Výsledky testu vzdálenosti 6MWD u pacientů s výchozí vzdáleností 6MWD < 350 metrů.
U pacientů s výchozí vzdáleností 6MWD < 350 metrů byla průměrná změna vzdálenosti 6MWD mezi výchozím bodem a 48. týdnem v rameni s atalurenem 10 - 10 - 20 mg/kg o 68 metrů lepší než v rameni s placebem (p=0,053).
U těchto pacientů byla průměrná změna ve funkčních testech s měřením času mezi výchozím bodem a 48. týdnem v rameni s atalurenem 10 - 10 - 20 mg/kg lepší než u placeba u času potřebného k uběhnutí/ujití 10 metrů (lepší o 3,5 sekundy), času potřebného k výstupu do 4 schodů (lepší o 6,4 sekundy) i času potřebného k sestupu ze 4 schodů (lepší o 5,0 sekundy).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s atalurenem u nmDMD u dvou podskupin pediatrické populace ve věku od narození do 28 dní a u kojenců od 28 dní do 6 měsíců v souladu s Plánem pediatrického výzkumu (PIP), ve schválené indikaci (informace o použití u dětí viz bod 4.2).
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s atalurenem u nmDMD u jedné podskupiny pediatrické populace ve věku od 6 měsíců do 5 let v souladu s Plánem pediatrického výzkumu (PIP), ve schválené indikaci (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že j sou očekávány další důkazy o jeho přínosech.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Podávání atalurenu v dávkách upravených podle tělesné hmotnosti (mg/kg) vedlo u dětí i dospívajících s nmDMD se širokým rozmezím tělesné hmotnosti k podobným hodnotám expozice v ustáleném stavu (AUC). Ačkoli je ataluren ve vodě prakticky nerozpustný, po perorálním podání ve formě suspenze se snadno vstřebává.
Obecné charakteristiky atalurenu po podání
Absorpce
Vrcholné plazmatické hladiny atalurenu se u subjektů, které dostávaly léčivý přípravek do 30 minut po jídle, udržovaly přibližně po dobu 1,5 hodiny. Biologická dostupnost atalurenu je na základě vylučování radioaktivity v moči ve studii s jednou dávkou radioaktivně značeného atalurenu odhadována na > 55 %. Plazmatické koncentrace atalurenu v ustáleném stavu se zvyšují úměrně se zvyšující se dávkou. U atalurenu v dávce mezi 10 a 50 mg/kg jsou plazmatické koncentrace v ustáleném stavu dávkově závislé a po opakovaném podání dávky nebyla pozorována akumulace.
Distribuce
In vitro se ataluren z 99,6 % váže na lidské plazmatické proteiny a jeho vazba je nezávislá na plazmatické koncentraci. Ataluren se nedistribuuje do červených krvinek.
Biotransformace
Ataluren je metabolizován konjugací prostřednictvím enzymů UGT (uridindifosfát-glukuronyl-transferáz), převážně UGT1A9 v játrech a ve střevě.
In vivo byl jediným metabolitem detekovaným v plazmě po perorálním podání radioaktivně značeného atalurenu ataluren-O-ip-acylglukuronid; expozice tomuto metabolitu u lidí činila přibližně 8 % plazmatické hodnoty AUC pro ataluren.
Eliminace
Plazmatický poločas atalurenu se pohybuje v rozmezí 2-6 hodin a není ovlivněn ani dávkou ani opakovaným podáním. Eliminace atalurenu pravděpodobně závisí na jaterní a střevní glukuronidaci atalurenu, po níž následuje renální exkrece vzniklého glukuronidového metabolitu.
Po podání jedné perorální dávky radioaktivně značeného atalurenu došlo k vyloučení přibližně poloviny podané radioaktivní látky ve stolici a zbytek byl vyloučen močí. V moči tvoří nezměněný ataluren < 1 % a acylglukuronidový metabolit 49 % podané dávky.
Linearita/nelinearita
U atalurenu v dávce mezi 10 a 50 mg/kg jsou plazmatické koncentrace v ustáleném stavu dávkově závislé a po opakovaném podání dávky nebyla pozorována akumulace. Na základě údajů od zdravých dobrovolníků byla relativní biologická dostupnost atalurenu v ustáleném stavu přibližně o 40 % nižší než po zahajovací dávce. Odhaduje se, že relativní biologická dostupnost začíná klesat přibližně po 60 hodinách od podání první dávky. Ustáleného stavu je dosaženo po přibližně dvou týdnech podávání přípravku třikrát denně.
Charakteristika u specifických skupin subjektů nebo pacientů
Věk
Na základě údajů od subjektů ve věkovém rozmezí 5 až 57 let nemá věk zjevný vliv na plazmatickou expozici atalurenu. Úprava dávky podle věku není nutná.
Pohlaví
V klinických studiích nmDMD nebyly zkoumány ženy. V jiných populacích se účinek pohlaví na plazmatickou expozici atalurenu nicméně neprojevil.
Rasa
Farmakokinetika atalurenu pravděpodobně není významně ovlivněna polymorfismy UTG1A9 v bělošské populaci. Vzhledem k nízkému počtu jiných ras zahrnutých do klinických studií nelze odvodit ohledně účinku UTG1A9 v jiných etnických skupinách žádné závěry.
Porucha funkce ledvin nebo jater
U pacientů s poruchou funkce ledvin nebo jater nebyly s přípravkem Translarna provedeny žádné studie. Pacienti s poruchou funkce ledvin nebo jater by měli být pečlivě sledováni.
Nechodící pacienti
Ztráta schopnosti chůze nevede ke zjevným rozdílům ani u relativní biologické dostupnosti v ustáleném stavu ani u zjevné clearance. U pacientů, kteří přestanou být schopni chůze, není nutná úprava dávky.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Byl dostupný standardní balík studií reprodukční toxicity. Nebyly pozorovány účinky na mužskou ani ženskou fertilitu, nebyly ale zkoumány účinky časné léčby v dětství na fertilitu v dospělosti. U potkanů a králíků byla za přítomnosti toxicity pro matku zjištěna embryonální/fetální toxicita (např. zvýšená časná resorpce, postimplantační ztráty, snížené množství životaschopných plodů) a příznaky opožděného vývoje (zvýšené množství skeletálních variací). Expozice na úrovni dávky bez pozorovaného nežádoucího účinku (NOAEL) byla podobná (králík) nebo 4krát vyšší (potkan) než systémová expozice u člověka (10 - 10 - 20 mg/kg/den). Ukázalo se, že radioaktivně značený ataluren prochází placentou. Při testování jedné relativně nízké dávky 30 mg/kg radioaktivně značeného atalurenu u matky byla koncentrace fetální radioaktivity < 27 % koncentrace v plazmě matky. Ve studii prenatální/postnatální vývojové toxicity u potkanů byla při expozici ve výši přibližně 5násobku expozice u člověka pozorována významná toxicita pro matku i účinky na tělesnou hmotnost a vývoj chůze u potomstva. Systémová expozice u matky na úrovni dávky bez pozorovaného účinku (NOEL) u neonatální toxicity odpovídala přibližně 3násobku expozice u člověka. Při jedné relativně nízké dávce 30 mg/kg radioaktivně značeného atalurenu u matky odpovídala nejvyšší naměřená koncentrace radioaktivity v potkaním mléce 37 % koncentrace v plazmě matky. Přítomnost radioaktivity v plazmě mláďat potvrdila u mláďat absorpci z mléka.
U myší se ve studiích s opakovanou perorální dávkou při systémové expozici ekvivalentní 0,3násobku hodnoty AUC v ustáleném stavu u pacientů, kterým je podáván přípravek Translarna v ranní, polední a večerní dávce 10 - 10 - 20 mg/kg a vyšší, objevila renální toxicita (nefróza distálního nefronu).
U 26týdenního transgenního myšího modelu kancerogenity nebyly zjištěny žádné známky kancerogenity. V dvouleté studii kancerogenity u potkanů byl zjištěn jeden případ hibernomu. Při expozici mnohem vyšší než u pacientů byl navíc zjištěn zvýšený výskyt (vzácných) nádorů močového měchýře. Významný výskyt nádorů močového měchýře u člověka není považován za pravděpodobný.
Jedna ze dvou studií s opakovanou dávkou u 26týdenních potkanů, zahájená u 4-5týdenních potkanů, ukázala na dávce závislý vzestup incidence maligního hibernomu, tumoru u potkanů vzácného.
V dvouleté studii kancerogenity u potkanů byl navíc při nejvyšší dávce zjištěn jeden případ maligního hibernomu. Základní incidence tohoto typu nádoru u potkanů i u člověka je velmi nízká
a mechanismus způsobující tyto nádory ve studiích u potkanů (včetně vztahu k léčbě atalurenem) není znám. Význam pro člověka není znám.
V jednoleté studii u 10-12týdenních psů byly zjištěny nálezy na nadledvinách (lokální zánět a degenerace oblastí kůry produkujících glukokortikoidy) a mírné narušení produkce kortizolu po exogenní stimulaci adrenokortikotropním hormonem. Tyto nálezy byly pozorovány u psů při systémové expozici ekvivalentní 0,8násobku hodnoty AUC v ustáleném stavu u pacientů, kterým je podáván přípravek Translarna v ranní, polední a večerní dávce 10 - 10 - 20 mg/kg a vyšší. Ve studii distribuce u potkanů byla pozorována vysoká koncentrace atalurenu v nadledvinách.
Kromě výše zmíněných účinků bylo ve studiích s opakovanou dávkou zjištěno několik dalších méně závažných nežádoucích účinků, zvláště snížený hmotnostní přírůstek, snížený příjem potravy a zvýšená hmotnost jater bez histologické korelace a s nejasným klinickým významem. Studie u potkanů a psů také ukázaly změny lipidů v plazmě (cholesterolu a triglyceridů) naznačující změny metabolismu tuků.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Polydextrosa (E1200)
Makrogol Poloxamer Mannitol (E421)
Krospovidon
Hydroxyetylcelulóza
Umělá vanilková příchuť: maltodextrin, umělé příchutě a propylenglykol Koloidní bezvodý oxid křemičitý (E551)
Magnesium-stearát
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky
Každou připravenou dávku je nejlépe užít bezprostředně po její přípravě. Jestliže připravená dávka není zkonzumována do 24 hodin od přípravy při skladování v chladničce (2-8 °C) nebo do 3 hodin při pokojové teplotě (15-30 °C), měla by být zlikvidována.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Tepelně uzavřený laminovaný sáček z hliníkové fólie: polyethylentereftalát (odolný vůči porušení dětmi), polyethylen (zbarvení a polyesterový/fóliový spoj), hliníková fólie (ochrana proti vlhkosti), lepidlo (ze třídy polyurethanů), kopolymer etylenu a kyseliny metakrylové (těsnicí pryskyřice pro zachování neporušeného balení).
Balení 30 sáčků.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Sáčky by se měly otevírat až v době přípravy dávky. Celý obsah každého sáčku by se měl smísit s minimálně 30 ml tekutiny (vody, mléka, ovocného džusu) nebo 3 lžícemi polotekuté stravy (jogurtu
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
PTC Therapeutics International Limited 77 Sir John Rogerson’s Quay Dublin 2 Irsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/902/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 31 července 2014
Datum posledního prodloužení registrace: 28. července 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Translarna 250 mg granule pro perorální suspenzi
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden sáček obsahuje atalurenum 250 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Granule pro perorální suspenzi. Bílé až téměř bílé granule.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Translarna je indikován k léčbě Duchennovy svalové dystrofie vzniklé v důsledku nonsense mutace v genu pro dystrofin u chodících pacientů ve věku od 5 let (viz bod 5.1). U nechodících pacientů účinnost nebyla prokázána.
Přítomnost nonsense mutace v genu pro dystrofin má být stanovena genetickými testy (viz bod 4.4).
4.2 Dávkování a způsob podání
Léčbu přípravkem Translarna má zahajovat pouze specializovaný lékař se zkušenostmi s léčbou Duchennovy/Beckerovy svalové dystrofie.
Dávkování
Ataluren se podává perorálně každý den ve 3 dávkách.
První dávka se užívá ráno, druhá v poledne a třetí večer. Doporučené intervaly mezi dávkami j sou 6 hodin mezi ranní a polední dávkou, 6 hodin mezi polední a večerní dávkou a 12 hodin mezi večerní dávkou a první dávkou podanou následující den.
Doporučená dávka přípravku je 10 mg/kg tělesné hmotnosti ráno, 10 mg/kg tělesné hmotnosti v poledne a 20 mg/kg tělesné hmotnosti večer (pro dosažení celkové denní dávky 40 mg/kg tělesné hmotnosti).
Přípravek Translarna je dostupný v sáčcích obsahujících 125 mg, 250 mg nebo 1 000 mg atalurenu. Následující tabulka poskytuje informace, kolik sáčků o určité síle je třeba použít při přípravě doporučené dávky pro dané rozmezí tělesné hmotnosti.
|
Rozmezí tělesné hmotnosti (kg) |
Počet sáčků | |||||||||
|
Ráno |
V poledne |
Večer | ||||||||
|
125mg sáčky |
250mg sáčky |
1 000mg sáčky |
125mg sáčky |
250mg sáčky |
1 000mg sáčky |
125mg sáčky |
250mg sáčky |
1 000mg sáčky | ||
|
12 |
14 |
1 |
0 |
0 |
1 |
0 |
0 |
0 |
1 |
0 |
|
15 |
16 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
1 |
0 |
|
17 |
20 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
|
21 |
23 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
1 |
0 |
|
24 |
26 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
0 |
|
27 |
31 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
0 |
|
32 |
35 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
0 |
|
36 |
39 |
1 |
1 |
0 |
1 |
1 |
0 |
0 |
3 |
0 |
|
40 |
44 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
3 |
0 |
|
45 |
46 |
0 |
2 |
0 |
0 |
2 |
0 |
1 |
3 |
0 |
|
47 |
55 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
0 |
1 |
|
56 |
62 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
1 |
1 |
|
63 |
69 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
1 |
1 |
|
70 |
78 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
2 |
1 |
|
79 |
86 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
3 |
1 |
|
87 |
93 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
3 |
1 |
|
94 |
105 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
|
106 |
111 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
|
112 |
118 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
|
119 |
125 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
2 |
2 |
Opožděná nebo vynechaná dávka
Jestliže zpoždění při podání atalurenu činí méně než 3 hodiny po ranní či polední dávce nebo méně než 6 hodin po večerní dávce, dávka by měla být užita beze změny následujícího dávkovacího schématu. Jestliže je zpoždění delší než 3 hodiny po ranní nebo polední dávce nebo delší než 6 hodin po večerní dávce, neměla by se dávka užívat a pacient by měl pokračovat podle obvyklého dávkovacího schématu. V případě vynechání dávky by pacienti neměli dávku zdvojnásobovat ani užívat dávku navíc. Je důležité podávat správnou dávku. Zvýšení doporučené dávky může být spojeno se snížením účinnosti.
Zvláštní populace
Starší lidé
Bezpečnost a účinnost atalurenu u pacientů ve věku 65 let a starších nebyla dosud stanovena (viz bod 5.2.)
Insuficience ledvin a jater
Bezpečnost a účinnost atalurenu u pacientů s poruchou funkce ledvin a jater nebyla dosud stanovena (viz bod 4.4).
Pediatrická populace
Bezpečnost a účinnost přípravku Translarna u dětí ve věku od 6 měsíců do 5 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Translarna se užívá perorálně po smísení do formy suspenze v tekutině nebo v polotekuté stravě. Sáčky se mají otevírat až v době přípravy dávky. Celý obsah každého sáčku by se měl smísit s minimálně 30 ml tekutiny (vody, mléka, ovocného džusu) nebo 3 lžícemi polotekuté stravy (jogurtu
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Souběžné užívání intravenózních aminoglykosidů (viz bod 4.4 a 4.5).
4.4 Zvláštní upozornění a opatření pro použití Pacienti, kteří nenesou nonsense mutaci
Onemocnění pacientů musí být založeno na nosičství nonsense mutace v genu pro dystrofin, stanovené genetickými testy. Pacientům, kteří nonsense mutaci nenesou, nemá být ataluren podáván.
Porucha funkce ledvin a jater
Pacienti s poruchami funkce ledvin či jater by měli být pečlivě sledováni.
Změny lipidového profilu
Protože u některých pacientů v klinických studiích byly hlášeny změny lipidového profilu (zvýšené triglyceridy a cholesterol), u pacientů s DMD s nonsense mutací, kterým je podáván ataluren, se doporučuje každoročně nebo podle klinického stavu pacienta i častěji monitorovat celkový cholesterol, LDL, HDL a triglyceridy.
Hypertenze při současném užívání systémových kortikosteroidů
Protože v klinických studiích byla u některých pacientů při současném užívání systémových kortikosteroidů hlášena hypertenze, u pacientů s DMD s nonsense mutací, kterým je podáván ataluren souběžně s kortikosteroidy, se doporučuje každých 6 měsíců nebo podle klinického stavu pacienta i častěji monitorovat klidový systolický a diastolický krevní tlak.
Monitorování renálních funkcí
Protože v kontrolované studii nonsense mutace v genu pro dystrofin byl pozorován mírný vzestup průměrné hodnoty sérového kreatininu, dusíku močoviny (BUN) v krvi a cystatinu C, u pacientů s DMD s nonsense mutací, kterým je podáván ataluren, se doporučuje každých 6 až 12 měsíců nebo podle klinického stavu pacienta i častěji monitorovat hladiny sérového kreatininu, BUN a cystatinu C.
Možné interakce s jinými léčivými přípravky
Při současném podávání atalurenu s léčivými přípravky, které jsou substráty či induktory UGT1A9, inhibitory BCRP nebo substráty OAT1, OAT3 nebo OATP1B3 (viz bod 4.5), je zapotřebí opatrnosti.
Aminoglykosidy
Bylo prokázáno, že aminoglykosidy snižují čtecí aktivitu atalurenu in vitro. Bylo navíc zjištěno, že ataluren zvyšuje nefrotoxicitu intravenózních aminoglykosidů. Je nutné se vyvarovat současného podávání těchto léčivých přípravků s atalurenem (viz bod 4.3). Protože není znám mechanismus, kterým ataluren zvyšuje nefrotoxicitu intravenózních aminoglykosidů, souběžné použití jiných nefrotoxických léčivých přípravků s atalurenem se nedoporučuje. Pokud se této situaci nelze vyhnout (např. vankomycin k léčbě MRSA), doporučuje se pečlivé monitorování renálních funkcí (viz bod 4.5).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Aminoglykosidy
Ataluren by neměl být podáván současně s intravenózními aminoglykosidy, protože v klinické studii u pacientů s nmCF byly zjištěny případy snížených renálních funkcí (viz bod 4.3).
U několika pacientů s nmCF léčených atalurenem a intravenózními aminoglykosidy spolu s dalšími antibiotiky z důvodu exacerbace cystické fibrózy došlo ke zvýšení hladin sérového kreatininu. Zvýšení sérového kreatininu ve všech případech ustoupilo při ukončení podávání intravenózních aminoglykosidů bez ohledu na pokračování nebo přerušení podávání přípravku Translarna. Tato zjištění naznačují, že současné podávání přípravku Translarna a intravenózních aminoglykosidů může zvyšovat nefrotoxický účinek aminoglykosidů. Proto je-li nezbytná léčba intravenózními aminoglykosidy, měla by být léčba přípravkem Translarna přerušena a poté znovu zahájena 2 dny po ukončení podávání aminoglykosidů. Účinek současného podávání atalurenu s jinými nefrotoxickými léčivými přípravky není znám.
V některých z těchto případů může být přispívajícím faktorem dehydratace. Pacienti by měli při užívání atalurenu zachovávat dostatečnou hydrataci. Viz bod 4.4.
Účinek jiných léčivých přípravků na farmakokinetiku atalurenu
Studie in vitro ukázaly, že ataluren je substrátem UGT1A9 a proteinu BRCP (proteinu rezistence u karcinomu prsu). Při současném podávání atalurenu s léčivými přípravky, které jsou induktory UGT1A9 (např. mykofenolát mofetilu) nebo inhibitory BCRP (např. cyklosporinu), je zapotřebí opatrnosti.
Účinek atalurenu na farmakokinetiku jiných léčivých přípravků
Studie in vitro ukázaly, že ataluren je inhibitorem UGT1A9, OAT1 (transportéru organických aniontů 1), OAT3 (transportéru organických aniontů 3) a OATP1B3 (transportního polypeptidu organických aniontů). Když je ataluren podáván současně s léčivými přípravky, které jsou substráty UGT1A9, OAT1, OAT3 nebo OATP1B3 (např. oseltamivirem, acyklovirem, ciprofloxacinem, kaptoprilem, furosemidem, bumetanidem, valsartanem, pravastatinem, rosuvastatinem, atorvastatinem, pitavastatinem), je zapotřebí opatrnosti vzhledem k riziku zvýšení koncentrace těchto léčivých přípravků.
Na základě studií in vitro se očekává, že ataluren není inhibitorem ani p-gp mediovaného transportu ani metabolismu zprostředkovaného cytochromem P450. Podobně se očekává, že ataluren není in vivo induktorem izoenzymů cytochromu P450.
Současné podávání kortikosteroidů (deflazakortu, prednisonu nebo prednisolonu) s atalurenem neovlivňuje plazmatické koncentrace atalurenu. Při současném podávání atalurenu nebyla pozorována klinicky významná změna plazmatických koncentrací kortikosteroidů. Tyto údaje nenaznačují zjevnou interakci léčivo-léčivo mezi kortikosteroidy a atalurenem a není nutná úprava dávky.
Léčivé přípravky, které postihují transportér p-glykoprotein
In vitro ataluren není substrátem transportéru p-glykoprotein. Farmakokinetika atalurenu pravděpodobně není ovlivněna léčivými přípravky, které inhibují transportér p-glykoprotein.
4.6 Fertilita, těhotenství a kojení
Není k dispozici dostatek údajů o použití atalurenu u těhotných žen. Studie na zvířatech prokázaly reprodukční toxicitu pouze v dávkách, které jsou toxické pro matku (viz bod 5.3).
Jako preventivní opatření se doporučuje vyhnout se použití atalurenu během těhotenství.
Kojení
Není známo, zda se ataluren/metabolity vylučují do lidského mateřského mléka. Dostupné farmakodynamické/toxikologické údaje u zvířat prokázaly vylučování atalurenu/metabolitů do mléka (viz bod 5.3). Riziko pro kojené novorozence/kojence nelze vyloučit.
Kojení má být během léčby atalurenem přerušeno.
Fertilita
Neklinické údaje na základě standardní studie samčí a samičí toxicity u potkanů neodhalily riziko pro člověka (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Účinek atalurenu na řízení, jízdu na kole a obsluhu strojů nebyl testován. Pacienti, u kterých dochází k závratím, by měli být při řízení, jízdě na kole nebo při obsluze strojů opatrní.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
V klinických studiích u pacientů s Duchennovou svalovou dystrofií způsobenou nonsense mutací (nmDMD) byly nejčastějšími nežádoucími účinky při doporučené dávce nevolnost, zvracení a bolest hlavy. Tyto nežádoucí účinky obecně nevyžadovaly lékařský zásah a u žádného z pacientů nedošlo k přerušení léčby atalurenem z důvodu nežádoucích účinků.
Tabulkový přehled nežádoucích účinků
Nežádoucí účinky hlášené v klinické studii u převážně pediatrických pacientů s nmDMD léčených doporučenou dávkou 10 - 10 - 20 mg/kg jsou klasifikovány podle tříd orgánových systémů a frekvence MedDRA. Skupiny frekvencí jsou definovány podle následující konvence: velmi časté (> 1/10) a časté (> 1/100 až < 1/10). V každé skupině četností jsou nežádoucí účinky uvedeny v pořadí podle klesající závažnosti.
Tabulka 2: Nežádoucí účinky přípravku Translama v kontrolované studii nmDMD
|
Třída orgánových systémů |
Velmi časté |
Časté |
Frekvence neznámá |
|
Poruchy metabolismu a výživy |
Snížená chuť k jídlu |
Změna lipidového profilu (zvýšené triglyceridy a cholesterol) | |
|
Poruchy nervového systému |
Závratě | ||
|
Cévní poruchy |
Hypertenze | ||
|
Respirační, hrudní a mediastinální poruchy |
Kašel, epistaxe | ||
|
Gastrointestinální poruchy |
Bolest horní části břicha, nadýmání, průjem, břišní dyskomfort, bolest břicha, zácpa, regurgitace | ||
|
Poruchy kůže a podkožní tkáně |
Erytém | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolest končetin | ||
|
Poruchy ledvin a močových cest |
Enuréza, ledvinná cysta, polakisurie, abnormální zbarvení moči |
Změna testů renálních funkcí (zvýšený kreatinin, dusík močoviny v krvi, cystatin C) | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie, únava, snížení tělesné hmotnosti |
Popis vybraných nežádoucích účinků
Sérové lipidy
V průběhu kontrolované studie nmDMD byly průměrné celkové hladiny cholesterolu a triglyceridů ve výchozím bodě normální a zvyšovaly se, přičemž dosáhly hraničně vysokých nebo vysokých hodnot. Hodnoty se v časné fázi studie obvykle stabilizovaly a při pokračování léčby se dále nezvyšovaly.
Testy renálních funkcí
Během kontrolované studie nmDMD byl pozorován mírný vzestup průměrných hodnot sérového kreatininu, dusíku močoviny (BUN) v krvi a cystatinu C. Hodnoty se v časné fázi studie obvykle stabilizovaly a při pokračování léčby se dále nezvyšovaly.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V*.
4.9 Předávkování
U zdravých dobrovolníků, kterým byla podána jedna perorální dávka 200 mg/kg atalurenu, došlo k přechodným, málo intenzivním symptomům bolesti hlavy, nevolnosti, zvracení a průjmu. U těchto subjektů nebyly pozorovány žádné závažné nežádoucí účinky. V případě podezření na předávkování by měla být poskytnuta podpůrná lékařská péče včetně konzultace se zdravotnickým pracovníkem a pečlivého sledování klinického stavu pacienta.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: {skupina}, ATC kód: dosud nepřidělen Mechanismus účinku
Nonsense mutace v DNA má za následek předčasný stop kodon v mRNA. Tento předčasný stop kodon v mRNA způsobuje onemocnění tím, že ukončuje translaci ještě před vytvořením proteinu o plné délce. Ataluren umožňuje ribozomální čtení mRNA obsahující tento předčasný stop kodon, které vede k tvorbě proteinu o plné délce.
Farmakodynamické účinky
Neklinické experimenty in vitro s buňkami a rybími larvami s nonsense mutacemi pěstovanými v roztoku atalurenu ukázaly, že ataluren umožňuje ribozomální čtení, přičemž křivka závislosti odpovědi na koncentraci má zvonovitý tvar (tvar obráceného písmene U). Existují hypotézy, že in vivo může být křivka závislosti odpovědi na dávce také zvonovitá, údaje in vivo jsou ale příliš omezené na to, aby tuto hypotézu bylo možné potvrdit u myšího modelu DMD s nonsense mutací a u člověka.
Neklinické studie in vitro naznačují, že kontinuální expozice atalurenu může být důležitá z hlediska maximalizace aktivity a že účinky léčivé látky na ribozomální čtení kodonů předčasného ukončení vymizí krátce po vysazení atalurenu.
Klinická účinnost a bezpečnost
Bezpečnost a účinnost přípravku Translarna byla hodnocena v randomizované dvojitě zaslepené placebem kontrolované multicentrické studii Duchennovy svalové dystrofie s nonsense mutací (nmDMD) u 174 pacientů mužského pohlaví ve věku 5 až 20 let. Bylo nutné, aby všichni pacienti byli chodící, což bylo definováno jako schopnost ujít během screeningového 6minutového testu chůze (6MWT) > 75 metrů bez podpůrných pomůcek. U pacientů muselo být také zdokumentováno potvrzení přítomnosti nonsense mutace v genu pro dystrofin, zjištěné pomocí genového sekvenování. Většina pacientů (90 %) ve všech léčených skupinách byli běloši. Pacienti byli randomizováni v poměru 1:1:1 a po dobu 48 týdnů dostávali 3krát denně (ráno, v poledne, večer) ataluren nebo placebo, přičemž 57 pacientů dostávalo placebo, 57 pacientů dostávalo ataluren v dávce 10 - 10 -20 mg/kg a 60 pacientů dostávalo ataluren v dávce 20 - 20 - 40 mg/kg; studii dokončilo 173 pacientů. Primárním cílovým parametrem z hlediska účinnosti byl účinek atalurenu na chůzi, posuzovaný podle změny vzdálenosti (6MWD), kterou pacient ujde během 6minutového testu chůze (6MWT). Post hoc analýza ukázala, že u pacientů dostávajících 10 - 10 - 20 mg/kg atalurenu došlo mezi výchozím bodem a 48. týdnem k poklesu vzdálenosti 6MWD průměrně o 12,9 metrů, zatímco u pacientů dostávajících placebo došlo k poklesu této vzdálenosti průměrně o 44,1 metrů (Obr. 1). Průměrná změna vzdálenosti 6MWD mezi výchozím bodem a 48. týdnem byla tedy v rameni s atalurenem 10 -10 - 20 mg/kg o 31,3 metrů lepší než v rameni s placebem (p=0,056). Ve statistickém modelu odhadu byl průměrný rozdíl 31,7 metru (upravená hodnota p=0,0367). Mezi atalurenem v dávce 20 - 20 -40 mg/kg a placebem nebyly rozdíly. Tyto výsledky ukazují, že ataluren v dávce 10 - 10 - 20 mg/kg snižuje u pacientů s nmDMD ztrátu schopnosti chůze.
Obrázek 1. Průměrná změna vzdálenosti, kterou pacient ujde za 6 minut
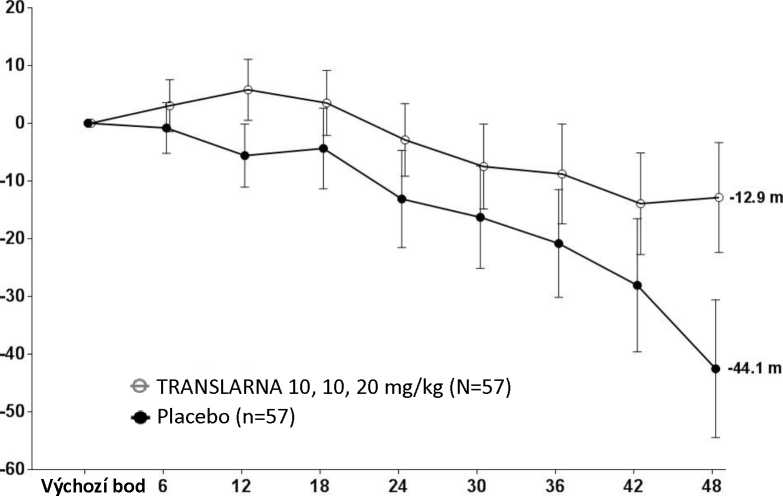
Týdny
Post hoc analýza doby do trvalého zhoršení vzdálenosti 6MWD o 10 % ukázala, že v rameni s atalurenem 10 - 10 - 20 mg/kg došlo do 48. týdne k progresi u 26 % pacientů oproti 44 % pacientů ve skupině s placebem (p=0,0652) (obrázek 2). Mezi atalurenem v dávce 20 - 20 - 40 mg/kg a placebem nebyly rozdíly. Tyto výsledky ukazují, že za 48 týdnů došlo ke zhoršení vzdálenosti 6MWD u nižšího počtu pacientů dostávajících ataluren 10 - 10 - 20 mg/kg.
Obrázek 2. Kaplan-Meierova křivka doby do trvalého zhoršení vzdálenosti 6MWD o 10 %
Ve funkčních testech s měřením času (TFT), testech času potřebného k uběhnutí/ujití 10 metrů, k výstupu do 4 schodů a k sestupu ze 4 schodů byl u pacientů léčených atalurenem prokázán menší nárůst času potřebného k uběhnutí/ujití 10 metrů, výstupu do 4 schodů a sestupu ze 4 schodů, což naznačuje zpomalení progrese nmDMD vůči placebu.
Průměrná změna ve funkčních testech s měřením času mezi výchozím bodem a 48. týdnem byla v rameni s atalurenem 10 - 10 - 20 mg/kg lepší než u placeba u času potřebného k uběhnutí/ujití 10 metrů (lepší o 1,5 sekundy), času potřebného k výstupu do 4 schodů (lepší o 2,4 sekundy) i času potřebného k sestupu ze 4 schodů (lepší o 1,6 sekundy), obrázek 3.
Obrázek 3. Průměrná změna ve funkčních testech s měřením času
Výsledky testu vzdálenosti 6MWD u pacientů s výchozí vzdáleností 6MWD < 350 metrů.
U pacientů s výchozí vzdáleností 6MWD < 350 metrů byla průměrná změna vzdálenosti 6MWD mezi výchozím bodem a 48. týdnem v rameni s atalurenem 10 - 10 - 20 mg/kg o 68 metrů lepší než v rameni s placebem (p=0,053).
U těchto pacientů byla průměrná změna ve funkčních testech s měřením času mezi výchozím bodem a 48. týdnem v rameni s atalurenem 10 - 10 - 20 mg/kg lepší než u placeba u času potřebného k uběhnutí/ujití 10 metrů (lepší o 3,5 sekundy), času potřebného k výstupu do 4 schodů (lepší o 6,4 sekundy) i času potřebného k sestupu ze 4 schodů (lepší o 5,0 sekundy).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s atalurenem u nmDMD u dvou podskupin pediatrické populace ve věku od narození do 28 dní a u kojenců od 28 dní do 6 měsíců v souladu s Plánem pediatrického výzkumu (PIP), ve schválené indikaci (informace o použití u dětí viz bod 4.2).
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s atalurenem u nmDMD u jedné podskupiny pediatrické populace ve věku od 6 měsíců do 5 let v souladu s Plánem pediatrického výzkumu (PIP), ve schválené indikaci (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že jsou očekávány další důkazy o jeho přínosech.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Podávání atalurenu v dávkách upravených podle tělesné hmotnosti (mg/kg) vedlo u dětí i dospívajících s nmDMD se širokým rozmezím tělesné hmotnosti k podobným hodnotám expozice v ustáleném stavu (AUC). Ačkoli je ataluren ve vodě prakticky nerozpustný, po perorálním podání ve formě suspenze se snadno vstřebává.
Obecné charakteristiky atalurenu po podání
Absorpce
Vrcholné plazmatické hladiny atalurenu se u subjektů, které dostávaly léčivý přípravek do 30 minut po jídle, udržovaly přibližně po dobu 1,5 hodiny. Biologická dostupnost atalurenu je na základě vylučování radioaktivity v moči ve studii s jednou dávkou radioaktivně značeného atalurenu odhadována na > 55 %. Plazmatické koncentrace atalurenu v ustáleném stavu se zvyšují úměrně se zvyšující se dávkou. U atalurenu v dávce mezi 10 a 50 mg/kg jsou plazmatické koncentrace v ustáleném stavu dávkově závislé a po opakovaném podání dávky nebyla pozorována akumulace.
Distribuce
In vitro se ataluren z 99,6 % váže na lidské plazmatické proteiny a jeho vazba je nezávislá na plazmatické koncentraci. Ataluren se nedistribuuje do červených krvinek.
Biotransformace
Ataluren je metabolizován konjugací prostřednictvím enzymů UGT (uridindifosfát-glukuronyl-transferáz), převážně UGT1A9 v játrech a ve střevě.
In vivo byl jediným metabolitem detekovaným v plazmě po perorálním podání radioaktivně značeného atalurenu ataluren-O-ip-acylglukuronid; expozice tomuto metabolitu u lidí činila přibližně 8 % plazmatické hodnoty AUC pro ataluren.
Eliminace
Plazmatický poločas atalurenu se pohybuje v rozmezí 2-6 hodin a není ovlivněn ani dávkou ani opakovaným podáním. Eliminace atalurenu pravděpodobně závisí na jaterní a střevní glukuronidaci atalurenu, po níž následuje renální exkrece vzniklého glukuronidového metabolitu.
Po podání jedné perorální dávky radioaktivně značeného atalurenu došlo k vyloučení přibližně poloviny podané radioaktivní látky ve stolici a zbytek byl vyloučen močí. V moči tvoří nezměněný ataluren < 1 % a acylglukuronidový metabolit 49 % podané dávky.
Linearita/nelinearita
U atalurenu v dávce mezi 10 a 50 mg/kg jsou plazmatické koncentrace v ustáleném stavu dávkově závislé a po opakovaném podání dávky nebyla pozorována akumulace. Na základě údajů od zdravých dobrovolníků byla relativní biologická dostupnost atalurenu v ustáleném stavu přibližně o 40 % nižší než po zahajovací dávce. Odhaduje se, že relativní biologická dostupnost začíná klesat přibližně po 60 hodinách od podání první dávky. Ustáleného stavu je dosaženo po přibližně dvou týdnech podávání přípravku třikrát denně.
Charakteristika u specifických skupin subjektů nebo pacientů
Věk
Na základě údajů od subjektů ve věkovém rozmezí 5 až 57 let nemá věk zjevný vliv na plazmatickou expozici atalurenu. Úprava dávky podle věku není nutná.
Pohlaví
V klinických studiích nmDMD nebyly zkoumány ženy. V jiných populacích se účinek pohlaví na plazmatickou expozici atalurenu nicméně neprojevil.
Rasa
Farmakokinetika atalurenu pravděpodobně není významně ovlivněna polymorfismy UTG1A9 v bělošské populaci. Vzhledem k nízkému počtu jiných ras zahrnutých do klinických studií nelze odvodit ohledně účinku UTG1A9 v jiných etnických skupinách žádné závěry.
Porucha funkce ledvin nebo jater
U pacientů s poruchou funkce ledvin nebo jater nebyly s přípravkem Translarna provedeny žádné studie. Pacienti s poruchou funkce ledvin nebo jater by měli být pečlivě sledováni.
Nechodící pacienti
Ztráta schopnosti chůze nevede ke zjevným rozdílům ani u relativní biologické dostupnosti v ustáleném stavu ani u zjevné clearance. U pacientů, kteří přestanou být schopni chůze, není nutná úprava dávky.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Byl dostupný standardní balík studií reprodukční toxicity. Nebyly pozorovány účinky na mužskou ani ženskou fertilitu, nebyly ale zkoumány účinky časné léčby v dětství na fertilitu v dospělosti. U potkanů a králíků byla za přítomnosti toxicity pro matku zjištěna embryonální/fetální toxicita (např. zvýšená časná resorpce, postimplantační ztráty, snížené množství životaschopných plodů) a příznaky opožděného vývoje (zvýšené množství skeletálních variací). Expozice na úrovni dávky bez pozorovaného nežádoucího účinku (NOAEL) byla podobná (králík) nebo 4krát vyšší (potkan) než systémová expozice u člověka (10 - 10 - 20 mg/kg/den). Ukázalo se, že radioaktivně značený ataluren prochází placentou. Při testování jedné relativně nízké dávky 30 mg/kg radioaktivně značeného atalurenu u matky byla koncentrace fetální radioaktivity < 27 % koncentrace v plazmě matky. Ve studii prenatální/postnatální vývojové toxicity u potkanů byla při expozici ve výši přibližně 5násobku expozice u člověka pozorována významná toxicita pro matku i účinky na tělesnou hmotnost a vývoj chůze u potomstva. Systémová expozice u matky na úrovni dávky bez pozorovaného účinku (NOEL) u neonatální toxicity odpovídala přibližně 3násobku expozice u člověka. Při jedné relativně nízké dávce 30 mg/kg radioaktivně značeného atalurenu u matky odpovídala nejvyšší naměřená koncentrace radioaktivity v potkaním mléce 37 % koncentrace v plazmě matky. Přítomnost radioaktivity v plazmě mláďat potvrdila u mláďat absorpci z mléka.
U myší se ve studiích s opakovanou perorální dávkou při systémové expozici ekvivalentní 0,3násobku hodnoty AUC v ustáleném stavu u pacientů, kterým je podáván přípravek Translarna v ranní, polední a večerní dávce 10 - 10 - 20 mg/kg a vyšší, objevila renální toxicita (nefróza distálního nefronu).
U 26týdenního transgenního myšího modelu kancerogenity nebyly zjištěny žádné známky kancerogenity. V dvouleté studii kancerogenity u potkanů byl zjištěn jeden případ hibernomu. Při expozici mnohem vyšší než u pacientů byl navíc zjištěn zvýšený výskyt (vzácných) nádorů močového měchýře. Významný výskyt nádorů močového měchýře u člověka není považován za pravděpodobný.
Jedna ze dvou studií s opakovanou dávkou u 26týdenních potkanů, zahájená u 4-5týdenních potkanů, ukázala na dávce závislý vzestup incidence maligního hibernomu, tumoru u potkanů vzácného.
V dvouleté studii kancerogenity u potkanů byl navíc při nejvyšší dávce zjištěn jeden případ maligního hibernomu. Základní incidence tohoto typu nádoru u potkanů i u člověka je velmi nízká
a mechanismus způsobující tyto nádory ve studiích u potkanů (včetně vztahu k léčbě atalurenem) není znám. Význam pro člověka není znám.
V jednoleté studii u 10-12týdenních psů byly zjištěny nálezy na nadledvinách (lokální zánět a degenerace oblastí kůry produkujících glukokortikoidy) a mírné narušení produkce kortizolu po exogenní stimulaci adrenokortikotropním hormonem. Tyto nálezy byly pozorovány u psů při systémové expozici ekvivalentní 0,8násobku hodnoty AUC v ustáleném stavu u pacientů, kterým je podáván přípravek Translarna v ranní, polední a večerní dávce 10 - 10 - 20 mg/kg a vyšší. Ve studii distribuce u potkanů byla pozorována vysoká koncentrace atalurenu v nadledvinách.
Kromě výše zmíněných účinků bylo ve studiích s opakovanou dávkou zjištěno několik dalších méně závažných nežádoucích účinků, zvláště snížený hmotnostní přírůstek, snížený příjem potravy a zvýšená hmotnost jater bez histologické korelace a s nejasným klinickým významem. Studie u potkanů a psů také ukázaly změny lipidů v plazmě (cholesterolu a triglyceridů) naznačující změny metabolismu tuků.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Polydextrosa (E1200)
Makrogol Poloxamer Mannitol (E421)
Krospovidon
Hydroxyetylcelulóza
Umělá vanilková příchuť: maltodextrin, umělé příchutě a propylenglykol Koloidní bezvodý oxid křemičitý (E551)
Magnesium-stearát
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky
Každou připravenou dávku je nejlépe užít bezprostředně po její přípravě. Jestliže připravená dávka není zkonzumována do 24 hodin od přípravy při skladování v chladničce (2-8 °C) nebo do 3 hodin při pokojové teplotě (15-30 °C), měla by být zlikvidována.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Tepelně uzavřený laminovaný sáček z hliníkové fólie: polyethylentereftalát (odolný vůči porušení dětmi), polyethylen (zbarvení a polyesterový/fóliový spoj), hliníková fólie (ochrana proti vlhkosti), lepidlo (ze třídy polyurethanů), kopolymer etylenu a kyseliny metakrylové (těsnicí pryskyřice pro zachování neporušeného balení).
Balení 30 sáčků.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Sáčky by se měly otevírat až v době přípravy dávky. Celý obsah každého sáčku by se měl smísit s minimálně 30 ml tekutiny (vody, mléka, ovocného džusu) nebo 3 lžícemi polotekuté stravy (jogurtu
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
PTC Therapeutics International Limited 77 Sir John Rogerson’s Quay Dublin 2 Irsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/902/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 31 července 2014
Datum posledního prodloužení registrace: 28. července 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Translarna 1 000 mg granule pro perorální suspenzi
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden sáček obsahuje atalurenum 1 000 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Granule pro perorální suspenzi. Bílé až téměř bílé granule.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Translarna je indikován k léčbě Duchennovy svalové dystrofie vzniklé v důsledku nonsense mutace v genu pro dystrofin u chodících pacientů ve věku od 5 let (viz bod 5.1). U nechodících pacientů účinnost nebyla prokázána.
Přítomnost nonsense mutace v genu pro dystrofin má být stanovena genetickými testy (viz bod 4.4).
4.2 Dávkování a způsob podání
Léčbu přípravkem Translarna má zahajovat pouze specializovaný lékař se zkušenostmi s léčbou Duchennovy/Beckerovy svalové dystrofie.
Dávkování
Ataluren se podává perorálně každý den ve 3 dávkách.
První dávka se užívá ráno, druhá v poledne a třetí večer. Doporučené intervaly mezi dávkami j sou 6 hodin mezi ranní a polední dávkou, 6 hodin mezi polední a večerní dávkou a 12 hodin mezi večerní dávkou a první dávkou podanou následující den.
Doporučená dávka přípravku je 10 mg/kg tělesné hmotnosti ráno, 10 mg/kg tělesné hmotnosti v poledne a 20 mg/kg tělesné hmotnosti večer (pro dosažení celkové denní dávky 40 mg/kg tělesné hmotnosti).
Přípravek Translarna je dostupný v sáčcích obsahujících 125 mg, 250 mg nebo 1 000 mg atalurenu. Následující tabulka poskytuje informace, kolik sáčků o určité síle je třeba použít při přípravě doporučené dávky pro dané rozmezí tělesné hmotnosti.
|
Rozmezí tělesné hmotnosti (kg) |
Počet sáčků | |||||||||
|
Ráno |
V poledne |
Večer | ||||||||
|
125mg sáčky |
250mg sáčky |
1 000mg sáčky |
125mg sáčky |
250mg sáčky |
1 000mg sáčky |
125mg sáčky |
250mg sáčky |
1 000mg sáčky | ||
|
12 |
14 |
1 |
0 |
0 |
1 |
0 |
0 |
0 |
1 |
0 |
|
15 |
16 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
1 |
0 |
|
17 |
20 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
|
21 |
23 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
1 |
0 |
|
24 |
26 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
0 |
|
27 |
31 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
0 |
|
32 |
35 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
0 |
|
36 |
39 |
1 |
1 |
0 |
1 |
1 |
0 |
0 |
3 |
0 |
|
40 |
44 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
3 |
0 |
|
45 |
46 |
0 |
2 |
0 |
0 |
2 |
0 |
1 |
3 |
0 |
|
47 |
55 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
0 |
1 |
|
56 |
62 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
1 |
1 |
|
63 |
69 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
1 |
1 |
|
70 |
78 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
2 |
1 |
|
79 |
86 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
3 |
1 |
|
87 |
93 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
3 |
1 |
|
94 |
105 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
|
106 |
111 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
|
112 |
118 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
|
119 |
125 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
2 |
2 |
Opožděná nebo vynechaná dávka
Jestliže zpoždění při podání atalurenu činí méně než 3 hodiny po ranní či polední dávce nebo méně než 6 hodin po večerní dávce, dávka by měla být užita beze změny následujícího dávkovacího schématu. Jestliže je zpoždění delší než 3 hodiny po ranní nebo polední dávce nebo delší než 6 hodin po večerní dávce, neměla by se dávka užívat a pacient by měl pokračovat podle obvyklého dávkovacího schématu. V případě vynechání dávky by pacienti neměli dávku zdvojnásobovat ani užívat dávku navíc. Je důležité podávat správnou dávku. Zvýšení doporučené dávky může být spojeno se snížením účinnosti.
Zvláštní populace
Starší lidé
Bezpečnost a účinnost atalurenu u pacientů ve věku 65 let a starších nebyla dosud stanovena (viz bod 5.2.)
Insuficience ledvin a jater
Bezpečnost a účinnost atalurenu u pacientů s poruchou funkce ledvin a jater nebyla dosud stanovena (viz bod 4.4).
Pediatrická populace
Bezpečnost a účinnost přípravku Translarna u dětí ve věku od 6 měsíců do 5 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Translarna se užívá perorálně po smísení do formy suspenze v tekutině nebo v polotekuté stravě. Sáčky se mají otevírat až v době přípravy dávky. Celý obsah každého sáčku by se měl smísit s minimálně 30 ml tekutiny (vody, mléka, ovocného džusu) nebo 3 lžícemi polotekuté stravy (jogurtu
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Souběžné užívání intravenózních aminoglykosidů (viz bod 4.4 a 4.5).
4.4 Zvláštní upozornění a opatření pro použití Pacienti, kteří nenesou nonsense mutaci
Onemocnění pacientů musí být založeno na nosičství nonsense mutace v genu pro dystrofin, stanovené genetickými testy. Pacientům, kteří nonsense mutaci nenesou, nemá být ataluren podáván.
Porucha funkce ledvin a jater
Pacienti s poruchami funkce ledvin či jater by měli být pečlivě sledováni.
Změny lipidového profilu
Protože u některých pacientů v klinických studiích byly hlášeny změny lipidového profilu (zvýšené triglyceridy a cholesterol), u pacientů s DMD s nonsense mutací, kterým je podáván ataluren, se doporučuje každoročně nebo podle klinického stavu pacienta i častěji monitorovat celkový cholesterol, LDL, HDL a triglyceridy.
Hypertenze při současném užívání systémových kortikosteroidů
Protože v klinických studiích byla u některých pacientů při současném užívání systémových kortikosteroidů hlášena hypertenze, u pacientů s DMD s nonsense mutací, kterým je podáván ataluren souběžně s kortikosteroidy, se doporučuje každých 6 měsíců nebo podle klinického stavu pacienta i častěji monitorovat klidový systolický a diastolický krevní tlak.
Monitorování renálních funkcí
Protože v kontrolované studii nonsense mutace v genu pro dystrofin byl pozorován mírný vzestup průměrné hodnoty sérového kreatininu, dusíku močoviny (BUN) v krvi a cystatinu C, u pacientů s DMD s nonsense mutací, kterým je podáván ataluren, se doporučuje každých 6 až 12 měsíců nebo podle klinického stavu pacienta i častěji monitorovat hladiny sérového kreatininu, BUN a cystatinu C.
Možné interakce s jinými léčivými přípravky
Při současném podávání atalurenu s léčivými přípravky, které jsou substráty či induktory UGT1A9, inhibitory BCRP nebo substráty OAT1, OAT3 nebo OATP1B3 (viz bod 4.5), je zapotřebí opatrnosti.
Aminoglykosidy
Bylo prokázáno, že aminoglykosidy snižují čtecí aktivitu atalurenu in vitro. Bylo navíc zjištěno, že ataluren zvyšuje nefrotoxicitu intravenózních aminoglykosidů. Je nutné se vyvarovat současného podávání těchto léčivých přípravků s atalurenem (viz bod 4.3). Protože není znám mechanismus, kterým ataluren zvyšuje nefrotoxicitu intravenózních aminoglykosidů, souběžné použití jiných nefrotoxických léčivých přípravků s atalurenem se nedoporučuje. Pokud se této situaci nelze vyhnout (např. vankomycin k léčbě MRSA), doporučuje se pečlivé monitorování renálních funkcí (viz bod 4.5).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Aminoglykosidy
Ataluren by neměl být podáván současně s intravenózními aminoglykosidy, protože v klinické studii u pacientů s nmCF byly zjištěny případy snížených renálních funkcí (viz bod 4.3).
U několika pacientů s nmCF léčených atalurenem a intravenózními aminoglykosidy spolu s dalšími antibiotiky z důvodu exacerbace cystické fibrózy došlo ke zvýšení hladin sérového kreatininu. Zvýšení sérového kreatininu ve všech případech ustoupilo při ukončení podávání intravenózních aminoglykosidů bez ohledu na pokračování nebo přerušení podávání přípravku Translarna. Tato zjištění naznačují, že současné podávání přípravku Translarna a intravenózních aminoglykosidů může zvyšovat nefrotoxický účinek aminoglykosidů. Proto je-li nezbytná léčba intravenózními aminoglykosidy, měla by být léčba přípravkem Translarna přerušena a poté znovu zahájena 2 dny po ukončení podávání aminoglykosidů. Účinek současného podávání atalurenu s jinými nefrotoxickými léčivými přípravky není znám.
V některých z těchto případů může být přispívajícím faktorem dehydratace. Pacienti by měli při užívání atalurenu zachovávat dostatečnou hydrataci. Viz bod 4.4.
Účinek jiných léčivých přípravků na farmakokinetiku atalurenu
Studie in vitro ukázaly, že ataluren je substrátem UGT1A9 a proteinu BRCP (proteinu rezistence u karcinomu prsu). Při současném podávání atalurenu s léčivými přípravky, které jsou induktory UGT1A9 (např. mykofenolát mofetilu) nebo inhibitory BCRP (např. cyklosporinu), je zapotřebí opatrnosti.
Účinek atalurenu na farmakokinetiku jiných léčivých přípravků
Studie in vitro ukázaly, že ataluren je inhibitorem UGT1A9, OAT1 (transportéru organických aniontů 1), OAT3 (transportéru organických aniontů 3) a OATP1B3 (transportního polypeptidu organických aniontů). Když je ataluren podáván současně s léčivými přípravky, které jsou substráty UGT1A9, OAT1, OAT3 nebo OATP1B3 (např. oseltamivirem, acyklovirem, ciprofloxacinem, kaptoprilem, furosemidem, bumetanidem, valsartanem, pravastatinem, rosuvastatinem, atorvastatinem, pitavastatinem), je zapotřebí opatrnosti vzhledem k riziku zvýšení koncentrace těchto léčivých přípravků.
Na základě studií in vitro se očekává, že ataluren není inhibitorem ani p-gp mediovaného transportu ani metabolismu zprostředkovaného cytochromem P450. Podobně se očekává, že ataluren není in vivo induktorem izoenzymů cytochromu P450.
Současné podávání kortikosteroidů (deflazakortu, prednisonu nebo prednisolonu) s atalurenem neovlivňuje plazmatické koncentrace atalurenu. Při současném podávání atalurenu nebyla pozorována klinicky významná změna plazmatických koncentrací kortikosteroidů. Tyto údaje nenaznačují zjevnou interakci léčivo-léčivo mezi kortikosteroidy a atalurenem a není nutná úprava dávky.
Léčivé přípravky, které postihují transportér p-glykoprotein
In vitro ataluren není substrátem transportéru p-glykoprotein. Farmakokinetika atalurenu pravděpodobně není ovlivněna léčivými přípravky, které inhibují transportér p-glykoprotein.
4.6 Fertilita, těhotenství a kojení
Není k dispozici dostatek údajů o použití atalurenu u těhotných žen. Studie na zvířatech prokázaly reprodukční toxicitu pouze v dávkách, které jsou toxické pro matku (viz bod 5.3).
Jako preventivní opatření se doporučuje vyhnout se použití atalurenu během těhotenství.
Kojení
Není známo, zda se ataluren/metabolity vylučují do lidského mateřského mléka. Dostupné farmakodynamické/toxikologické údaje u zvířat prokázaly vylučování atalurenu/metabolitů do mléka (viz bod 5.3). Riziko pro kojené novorozence/kojence nelze vyloučit.
Kojení má být během léčby atalurenem přerušeno.
Fertilita
Neklinické údaje na základě standardní studie samčí a samičí toxicity u potkanů neodhalily riziko pro člověka (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Účinek atalurenu na řízení, jízdu na kole a obsluhu strojů nebyl testován. Pacienti, u kterých dochází k závratím, by měli být při řízení, jízdě na kole nebo při obsluze strojů opatrní.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
V klinických studiích u pacientů s Duchennovou svalovou dystrofií způsobenou nonsense mutací (nmDMD) byly nejčastějšími nežádoucími účinky při doporučené dávce nevolnost, zvracení a bolest hlavy. Tyto nežádoucí účinky obecně nevyžadovaly lékařský zásah a u žádného z pacientů nedošlo k přerušení léčby atalurenem z důvodu nežádoucích účinků.
Tabulkový přehled nežádoucích účinků
Nežádoucí účinky hlášené v klinické studii u převážně pediatrických pacientů s nmDMD léčených doporučenou dávkou 10 - 10 - 20 mg/kg jsou klasifikovány podle tříd orgánových systémů a frekvence MedDRA. Skupiny frekvencí jsou definovány podle následující konvence: velmi časté (> 1/10) a časté (> 1/100 až < 1/10). V každé skupině četností jsou nežádoucí účinky uvedeny v pořadí podle klesající závažnosti.
Tabulka 3: Nežádoucí účinky přípravku Translama v kontrolované studii nmDMD
|
Třída orgánových systémů |
Velmi časté |
Časté |
Frekvence neznámá |
|
Poruchy metabolismu a výživy |
Snížená chuť k jídlu |
Změna lipidového profilu (zvýšené triglyceridy a cholesterol) | |
|
Poruchy nervového systému |
Závratě | ||
|
Cévní poruchy |
Hypertenze | ||
|
Respirační, hrudní a mediastinální poruchy |
Kašel, epistaxe | ||
|
Gastrointestinální poruchy |
Bolest horní části břicha, nadýmání, průjem, břišní dyskomfort, bolest břicha, zácpa, regurgitace | ||
|
Poruchy kůže a podkožní tkáně |
Erytém | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolest končetin | ||
|
Poruchy ledvin a močových cest |
Enuréza, ledvinná cysta, polakisurie, abnormální zbarvení moči |
Změna testů renálních funkcí (zvýšený kreatinin, dusík močoviny v krvi, cystatin C) | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie, únava, snížení tělesné hmotnosti |
Popis vybraných nežádoucích účinků
Sérové lipidy
V průběhu kontrolované studie nmDMD byly průměrné celkové hladiny cholesterolu a triglyceridů ve výchozím bodě normální a zvyšovaly se, přičemž dosáhly hraničně vysokých nebo vysokých hodnot. Hodnoty se v časné fázi studie obvykle stabilizovaly a při pokračování léčby se dále nezvyšovaly.
Testy renálních funkcí
Během kontrolované studie nmDMD byl pozorován mírný vzestup průměrných hodnot sérového kreatininu, dusíku močoviny (BUN) v krvi a cystatinu C. Hodnoty se v časné fázi studie obvykle stabilizovaly a při pokračování léčby se dále nezvyšovaly.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V*.
4.9 Předávkování
U zdravých dobrovolníků, kterým byla podána jedna perorální dávka 200 mg/kg atalurenu, došlo k přechodným, málo intenzivním symptomům bolesti hlavy, nevolnosti, zvracení a průjmu. U těchto subjektů nebyly pozorovány žádné závažné nežádoucí účinky. V případě podezření na předávkování by měla být poskytnuta podpůrná lékařská péče včetně konzultace se zdravotnickým pracovníkem a pečlivého sledování klinického stavu pacienta.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: {skupina}, ATC kód: dosud nepřidělen Mechanismus účinku
Nonsense mutace v DNA má za následek předčasný stop kodon v mRNA. Tento předčasný stop kodon v mRNA způsobuje onemocnění tím, že ukončuje translaci ještě před vytvořením proteinu o plné délce. Ataluren umožňuje ribozomální čtení mRNA obsahující tento předčasný stop kodon, které vede k tvorbě proteinu o plné délce.
Farmakodynamické účinky
Neklinické experimenty in vitro s buňkami a rybími larvami s nonsense mutacemi pěstovanými v roztoku atalurenu ukázaly, že ataluren umožňuje ribozomální čtení, přičemž křivka závislosti odpovědi na koncentraci má zvonovitý tvar (tvar obráceného písmene U). Existují hypotézy, že in vivo může být křivka závislosti odpovědi na dávce také zvonovitá, údaje in vivo jsou ale příliš omezené na to, aby tuto hypotézu bylo možné potvrdit u myšího modelu DMD s nonsense mutací a u člověka.
Neklinické studie in vitro naznačují, že kontinuální expozice atalurenu může být důležitá z hlediska maximalizace aktivity a že účinky léčivé látky na ribozomální čtení kodonů předčasného ukončení vymizí krátce po vysazení atalurenu.
Klinická účinnost a bezpečnost
Bezpečnost a účinnost přípravku Translarna byla hodnocena v randomizované dvojitě zaslepené placebem kontrolované multicentrické studii Duchennovy svalové dystrofie s nonsense mutací (nmDMD) u 174 pacientů mužského pohlaví ve věku 5 až 20 let. Bylo nutné, aby všichni pacienti byli chodící, což bylo definováno jako schopnost ujít během screeningového 6minutového testu chůze (6MWT) > 75 metrů bez podpůrných pomůcek. U pacientů muselo být také zdokumentováno potvrzení přítomnosti nonsense mutace v genu pro dystrofin, zjištěné pomocí genového sekvenování. Většina pacientů (90 %) ve všech léčených skupinách byli běloši. Pacienti byli randomizováni v poměru 1:1:1 a po dobu 48 týdnů dostávali 3krát denně (ráno, v poledne, večer) ataluren nebo placebo, přičemž 57 pacientů dostávalo placebo, 57 pacientů dostávalo ataluren v dávce 10 - 10 -20 mg/kg a 60 pacientů dostávalo ataluren v dávce 20 - 20 - 40 mg/kg; studii dokončilo 173 pacientů. Primárním cílovým parametrem z hlediska účinnosti byl účinek atalurenu na chůzi, posuzovaný podle změny vzdálenosti (6MWD), kterou pacient ujde během 6minutového testu chůze (6MWT). Post hoc analýza ukázala, že u pacientů dostávajících 10 - 10 - 20 mg/kg atalurenu došlo mezi výchozím bodem a 48. týdnem k poklesu vzdálenosti 6MWD průměrně o 12,9 metrů, zatímco u pacientů dostávajících placebo došlo k poklesu této vzdálenosti průměrně o 44,1 metrů (Obr. 1). Průměrná změna vzdálenosti 6MWD mezi výchozím bodem a 48. týdnem byla tedy v rameni s atalurenem 10 -10 - 20 mg/kg o 31,3 metrů lepší než v rameni s placebem (p=0,056). Ve statistickém modelu odhadu byl průměrný rozdíl 31,7 metru (upravená hodnota p=0,0367). Mezi atalurenem v dávce 20 - 20 -40 mg/kg a placebem nebyly rozdíly. Tyto výsledky ukazují, že ataluren v dávce 10 - 10 - 20 mg/kg snižuje u pacientů s nmDMD ztrátu schopnosti chůze.
Obrázek 1. Průměrná změna vzdálenosti, kterou pacient ujde za 6 minut
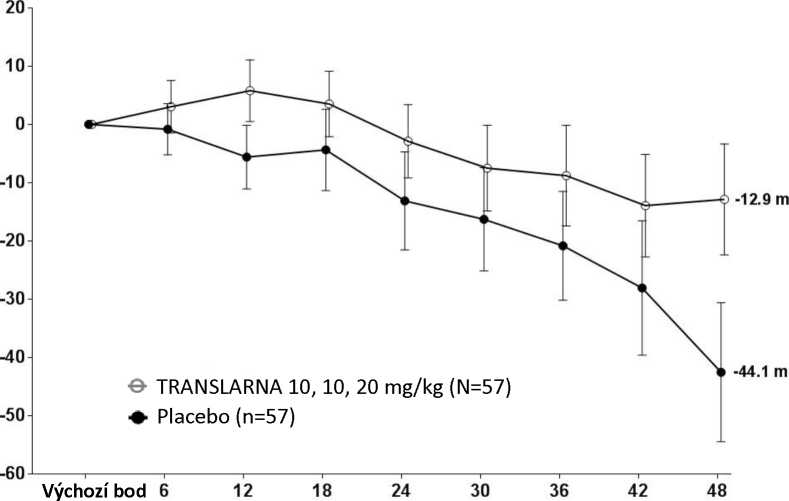
Týdny
Post hoc analýza doby do trvalého zhoršení vzdálenosti 6MWD o 10 % ukázala, že v rameni s atalurenem 10 - 10 - 20 mg/kg došlo do 48. týdne k progresi u 26 % pacientů oproti 44 % pacientů ve skupině s placebem (p=0,0652) (obrázek 2). Mezi atalurenem v dávce 20 - 20 - 40 mg/kg a placebem nebyly rozdíly. Tyto výsledky ukazují, že za 48 týdnů došlo ke zhoršení vzdálenosti 6MWD u nižšího počtu pacientů dostávajících ataluren 10 - 10 - 20 mg/kg.
Obrázek 2. Kaplan-Meierova křivka doby do trvalého zhoršení vzdálenosti 6MWD o 10 %
100 TI
90
V
s°
os
O
70
5 60
>10
O
-5 50 0)
o 40
4->
c
QJ
u 30 o
20
10
zhoršení o 26 %
■i__
zhoršení o 44 %
TRANSLARNA 10, 10, 20 mg/kg (N=57) Placebo (n=57)
12 18 24 30 36
Týdny
42
48
54
60
Ve funkčních testech s měřením času (TFT), testech času potřebného k uběhnutí/ujití 10 metrů, k výstupu do 4 schodů a k sestupu ze 4 schodů byl u pacientů léčených atalurenem prokázán menší nárůst času potřebného k uběhnutí/ujití 10 metrů, výstupu do 4 schodů a sestupu ze 4 schodů, což naznačuje zpomalení progrese nmDMD vůči placebu.
Průměrná změna ve funkčních testech s měřením času mezi výchozím bodem a 48. týdnem byla v rameni s atalurenem 10 - 10 - 20 mg/kg lepší než u placeba u času potřebného k uběhnutí/ujití 10 metrů (lepší o 1,5 sekundy), času potřebného k výstupu do 4 schodů (lepší o 2,4 sekundy) i času potřebného k sestupu ze 4 schodů (lepší o 1,6 sekundy), obrázek 3.
Obrázek 3. Průměrná změna ve funkčních testech s měřením času
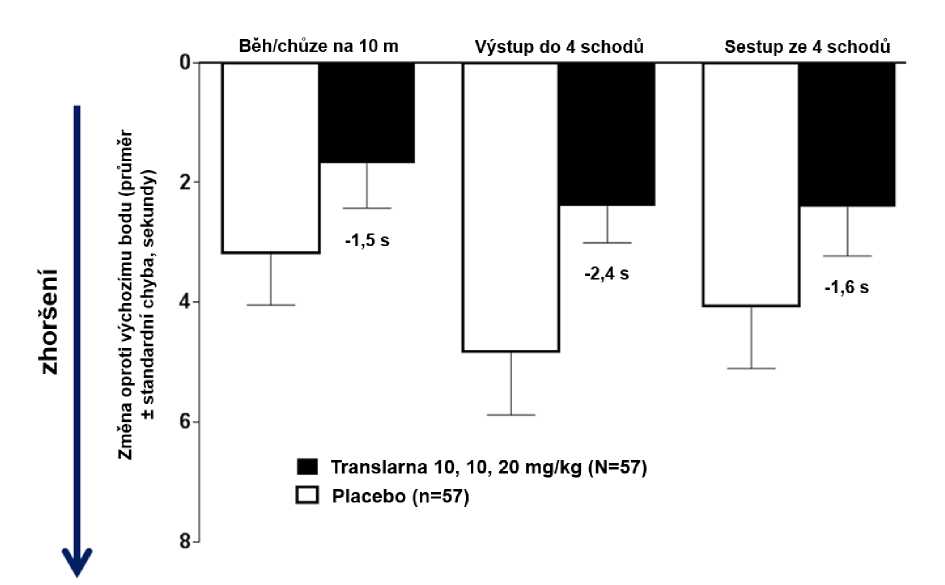
Výsledky testu vzdálenosti 6MWD u pacientů s výchozí vzdáleností 6MWD < 350 metrů
U pacientů s výchozí vzdáleností 6MWD < 350 metrů byla průměrná změna vzdálenosti 6MWD mezi výchozím bodem a 48. týdnem v rameni s atalurenem 10 - 10 - 20 mg/kg o 68 metrů lepší než v rameni s placebem (p=0,053).
U těchto pacientů byla průměrná změna ve funkčních testech s měřením času mezi výchozím bodem a 48. týdnem v rameni s atalurenem 10 - 10 - 20 mg/kg lepší než u placeba u času potřebného k uběhnutí/ujití 10 metrů (lepší o 3,5 sekundy), času potřebného k výstupu do 4 schodů (lepší o 6,4 sekundy) i času potřebného k sestupu ze 4 schodů (lepší o 5,0 sekundy).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s atalurenem u nmDMD u dvou podskupin pediatrické populace ve věku od narození do 28 dní a u kojenců od 28 dní do 6 měsíců v souladu s Plánem pediatrického výzkumu (PIP), ve schválené indikaci (informace o použití u dětí viz bod 4.2).
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s atalurenem u nmDMD u jedné podskupiny pediatrické populace ve věku od 6 měsíců do 5 let v souladu s Plánem pediatrického výzkumu (PIP), ve schválené indikaci (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že j sou očekávány další důkazy o jeho přínosech.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Podávání atalurenu v dávkách upravených podle tělesné hmotnosti (mg/kg) vedlo u dětí i dospívajících s nmDMD se širokým rozmezím tělesné hmotnosti k podobným hodnotám expozice v ustáleném stavu (AUC). Ačkoli je ataluren ve vodě prakticky nerozpustný, po perorálním podání ve formě suspenze se snadno vstřebává.
Obecné charakteristiky atalurenu po podání
Absorpce
Vrcholné plazmatické hladiny atalurenu se u subjektů, které dostávaly léčivý přípravek do 30 minut po jídle, udržovaly přibližně po dobu 1,5 hodiny. Biologická dostupnost atalurenu je na základě vylučování radioaktivity v moči ve studii s jednou dávkou radioaktivně značeného atalurenu odhadována na > 55 %. Plazmatické koncentrace atalurenu v ustáleném stavu se zvyšují úměrně se zvyšující se dávkou. U atalurenu v dávce mezi 10 a 50 mg/kg jsou plazmatické koncentrace v ustáleném stavu dávkově závislé a po opakovaném podání dávky nebyla pozorována akumulace.
Distribuce
In vitro se ataluren z 99,6 % váže na lidské plazmatické proteiny a jeho vazba je nezávislá na plazmatické koncentraci. Ataluren se nedistribuuje do červených krvinek.
Biotransformace
Ataluren je metabolizován konjugací prostřednictvím enzymů UGT (uridindifosfát-glukuronyl-transferáz), převážně UGT1A9 v játrech a ve střevě.
In vivo byl jediným metabolitem detekovaným v plazmě po perorálním podání radioaktivně značeného atalurenu ataluren-O-ip-acylglukuronid; expozice tomuto metabolitu u lidí činila přibližně 8 % plazmatické hodnoty AUC pro ataluren.
Eliminace
Plazmatický poločas atalurenu se pohybuje v rozmezí 2-6 hodin a není ovlivněn ani dávkou ani opakovaným podáním. Eliminace atalurenu pravděpodobně závisí na jaterní a střevní glukuronidaci atalurenu, po níž následuje renální exkrece vzniklého glukuronidového metabolitu.
Po podání jedné perorální dávky radioaktivně značeného atalurenu došlo k vyloučení přibližně poloviny podané radioaktivní látky ve stolici a zbytek byl vyloučen močí. V moči tvoří nezměněný ataluren < 1 % a acylglukuronidový metabolit 49 % podané dávky.
Linearita/nelinearita
U atalurenu v dávce mezi 10 a 50 mg/kg jsou plazmatické koncentrace v ustáleném stavu dávkově závislé a po opakovaném podání dávky nebyla pozorována akumulace. Na základě údajů od zdravých dobrovolníků byla relativní biologická dostupnost atalurenu v ustáleném stavu přibližně o 40 % nižší než po zahajovací dávce. Odhaduje se, že relativní biologická dostupnost začíná klesat přibližně po 60 hodinách od podání první dávky. Ustáleného stavu je dosaženo po přibližně dvou týdnech podávání přípravku třikrát denně.
Charakteristika u specifických skupin subjektů nebo pacientů
Věk
Na základě údajů od subjektů ve věkovém rozmezí 5 až 57 let nemá věk zjevný vliv na plazmatickou expozici atalurenu. Úprava dávky podle věku není nutná.
Pohlaví
V klinických studiích nmDMD nebyly zkoumány ženy. V jiných populacích se účinek pohlaví na plazmatickou expozici atalurenu nicméně neprojevil.
Rasa
Farmakokinetika atalurenu pravděpodobně není významně ovlivněna polymorfismy UTG1A9 v bělošské populaci. Vzhledem k nízkému počtu jiných ras zahrnutých do klinických studií nelze odvodit ohledně účinku UTG1A9 v jiných etnických skupinách žádné závěry.
Porucha funkce ledvin nebo jater
U pacientů s poruchou funkce ledvin nebo jater nebyly s přípravkem Translarna provedeny žádné studie. Pacienti s poruchou funkce ledvin nebo jater by měli být pečlivě sledováni.
Nechodící pacienti
Ztráta schopnosti chůze nevede ke zjevným rozdílům ani u relativní biologické dostupnosti v ustáleném stavu ani u zjevné clearance. U pacientů, kteří přestanou být schopni chůze, není nutná úprava dávky.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Byl dostupný standardní balík studií reprodukční toxicity. Nebyly pozorovány účinky na mužskou ani ženskou fertilitu, nebyly ale zkoumány účinky časné léčby v dětství na fertilitu v dospělosti. U potkanů a králíků byla za přítomnosti toxicity pro matku zjištěna embryonální/fetální toxicita (např. zvýšená časná resorpce, postimplantační ztráty, snížené množství životaschopných plodů) a příznaky opožděného vývoje (zvýšené množství skeletálních variací). Expozice na úrovni dávky bez pozorovaného nežádoucího účinku (NOAEL) byla podobná (králík) nebo 4krát vyšší (potkan) než systémová expozice u člověka (10 - 10 - 20 mg/kg/den). Ukázalo se, že radioaktivně značený ataluren prochází placentou. Při testování jedné relativně nízké dávky 30 mg/kg radioaktivně značeného atalurenu u matky byla koncentrace fetální radioaktivity < 27 % koncentrace v plazmě matky. Ve studii prenatální/postnatální vývojové toxicity u potkanů byla při expozici ve výši přibližně 5násobku expozice u člověka pozorována významná toxicita pro matku i účinky na tělesnou hmotnost a vývoj chůze u potomstva. Systémová expozice u matky na úrovni dávky bez pozorovaného účinku (NOEL) u neonatální toxicity odpovídala přibližně 3násobku expozice u člověka. Při jedné relativně nízké dávce 30 mg/kg radioaktivně značeného atalurenu u matky odpovídala nejvyšší naměřená koncentrace radioaktivity v potkaním mléce 37 % koncentrace v plazmě matky. Přítomnost radioaktivity v plazmě mláďat potvrdila u mláďat absorpci z mléka.
U myší se ve studiích s opakovanou perorální dávkou při systémové expozici ekvivalentní 0,3násobku hodnoty AUC v ustáleném stavu u pacientů, kterým je podáván přípravek Translarna v ranní, polední a večerní dávce 10 - 10 - 20 mg/kg a vyšší, objevila renální toxicita (nefróza distálního nefronu).
U 26týdenního transgenního myšího modelu kancerogenity nebyly zjištěny žádné známky kancerogenity. V dvouleté studii kancerogenity u potkanů byl zjištěn jeden případ hibernomu. Při expozici mnohem vyšší než u pacientů byl navíc zjištěn zvýšený výskyt (vzácných) nádorů močového měchýře. Významný výskyt nádorů močového měchýře u člověka není považován za pravděpodobný.
Jedna ze dvou studií s opakovanou dávkou u 26týdenních potkanů, zahájená u 4-5týdenních potkanů, ukázala na dávce závislý vzestup incidence maligního hibernomu, tumoru u potkanů vzácného.
V dvouleté studii kancerogenity u potkanů byl navíc při nejvyšší dávce zjištěn jeden případ maligního hibemomu. Základní incidence tohoto typu nádoru u potkanů i u člověka je velmi nízká
a mechanismus způsobující tyto nádory ve studiích u potkanů (včetně vztahu k léčbě atalurenem) není znám. Význam pro člověka není znám.
V jednoleté studii u 10-12týdenních psů byly zjištěny nálezy na nadledvinách (lokální zánět
a degenerace oblastí kůry produkujících glukokortikoidy) a mírné narušení produkce kortizolu po exogenní stimulaci adrenokortikotropním hormonem. Tyto nálezy byly pozorovány u psů při systémové expozici ekvivalentní 0,8násobku hodnoty AUC v ustáleném stavu u pacientů, kterým je podáván přípravek Translarna v ranní, polední a večerní dávce 10 - 10 - 20 mg/kg a vyšší. Ve studii distribuce u potkanů byla pozorována vysoká koncentrace atalurenu v nadledvinách.
Kromě výše zmíněných účinků bylo ve studiích s opakovanou dávkou zjištěno několik dalších méně závažných nežádoucích účinků, zvláště snížený hmotnostní přírůstek, snížený příjem potravy a zvýšená hmotnost jater bez histologické korelace a s nejasným klinickým významem. Studie u potkanů a psů také ukázaly změny lipidů v plazmě (cholesterolu a triglyceridů) naznačující změny metabolismu tuků.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Polydextrosa (E1200)
Makrogol Poloxamer Mannitol (E421)
Krospovidon
Hydroxyetylcelulóza
Umělá vanilková příchuť: maltodextrin, umělé příchutě a propylenglykol Koloidní bezvodý oxid křemičitý (E551)
Magnesium-stearát
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky
Každou připravenou dávku je nejlépe užít bezprostředně po její přípravě. Jestliže připravená dávka není zkonzumována do 24 hodin od přípravy při skladování v chladničce (2-8 °C) nebo do 3 hodin při pokojové teplotě (15-30 °C), měla by být zlikvidována.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Tepelně uzavřený laminovaný sáček z hliníkové fólie: polyethylentereftalát (odolný vůči porušení dětmi), polyethylen (zbarvení a polyesterový/fóliový spoj), hliníková fólie (ochrana proti vlhkosti), lepidlo (ze třídy polyurethanů), kopolymer etylenu a kyseliny metakrylové (těsnicí pryskyřice pro zachování neporušeného balení).
Balení 30 sáčků.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Sáčky by se měly otevírat až v době přípravy dávky. Celý obsah každého sáčku by se měl smísit s minimálně 30 ml tekutiny (vody, mléka, ovocného džusu) nebo 3 lžícemi polotekuté stravy (jogurtu nebo jablečného protlaku). Připravená dávka by se před podáním měla dobře promísit. Množství tekutin nebo polotekuté stravy lze zvýšit podle preferencí pacienta.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
PTC Therapeutics International Limited 77 Sir John Rogerson’s Quay Dublin 2 Irsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/902/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 31 července 2014
Datum posledního prodloužení registrace: 28. července 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ
POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO
PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
Název a adresa výrobce odpovědného za propouštění šarží Almac Pharma Services Ltd.
Seagoe Industrial Estate Craigavon
Co. Armagh BT63 5UA Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
Tato registrace byla schválena postupem tzv. podmíněného schválení, a proto podle čl. 14 odst. 7 nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
Dokončit multicentrickou randomizovanou dvojitě zaslepenou placebem kontrolovanou konfirmační studii zaměřenou na posouzení účinnosti a bezpečnosti atalurenu 10, 10, 20 mg/kg u pacientů s Duchennovou svalovou dystrofií způsobenou nonsense mutací (studii PTC124-GD-020-DMD). |
Předložení závěrečné zprávy: do 4. čtvrtletí 2015 |
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Translama 125 mg granule pro perorální suspenzi atalurenum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Jeden sáček obsahuje atalurenum 125 mg
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Granule pro perorální suspenzi 30 sáčků
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PTC Therapeutics International Limited 77 Sir John Rogerson’s Quay Dublin 2 Irsko
EU/1/13/902/001
13. ČÍSLO ŠARŽE
Č. šarže
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Translama 125 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Translama 125 mg granule pro perorální suspenzi atalurenum
Perorální podání
2. ZPŮSOB PODÁNÍ_
Před použitím čtěte příbalovou informaci.
3. POUŽITELNOST_
EXP
4. ČÍSLO ŠARŽE_
Č. šarže
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
125 mg
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Translama 250 mg granule pro perorální suspenzi atalurenum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Jeden sáček obsahuje atalurenum 250 mg
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Granule pro perorální suspenzi 30 sáčků
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PTC Therapeutics International Limited 77 Sir John Rogerson’s Quay Dublin 2 Irsko
EU/1/13/902/002
13. ČÍSLO ŠARŽE
Č. šarže
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Translama 250 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Translama 250 mg granule pro perorální suspenzi atalurenum
Perorální podání
2. ZPŮSOB PODÁNÍ_
Před použitím čtěte příbalovou informaci.
3. POUŽITELNOST_
EXP
4. ČÍSLO ŠARŽE_
Č. šarže
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
250 mg
6. JINÉ

Translama 1 000 mg granule pro perorální suspenzi atalurenum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Jeden sáček obsahuje atalurenum 1 000 mg
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Granule pro perorální suspenzi 30 sáčků
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PTC Therapeutics International Limited 77 Sir John Rogerson’s Quay Dublin 2 Irsko
EU/1/13/902/003
13. ČÍSLO ŠARŽE
Č. šarže
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Translama 1 000 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Translama 1 000 mg granule pro perorální suspenzi atalurenum
Perorální podání
2. ZPŮSOB PODÁNÍ_
Před použitím čtěte příbalovou informaci.
3. POUŽITELNOST_
EXP
4. ČÍSLO ŠARŽE_
Č. šarže
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 000 mg
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Translarna 125 mg granule pro perorální suspenzi Translarna 250 mg granule pro perorální suspenzi Translarna 1 000 mg granule pro perorální suspenzi
Atalurenum
'VTento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Translarna a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Translarna užívat
3. Jak se přípravek Translarna užívá
4. Možné nežádoucí účinky
5. Jak přípravek Translarna uchovávat
6. Obsah balení a další informace
1. Co je přípravek Translarna a k čemu se používá
Translarna je léčivý přípravek, který obsahuje léčivou látku ataluren.
Přípravek Translarna se používá k léčbě Duchennovy svalové dystrofie způsobené specifickým genetickým defektem, který ovlivňuje normální funkci svalů.
Přípravek Translarna se používá k léčbě pacientů ve věku od 5 let, kteří jsou schopni chůze.
Aby se potvrdilo, že tento léčivý přípravek je pro léčbu Vašeho onemocnění vhodný, před zahájením léčby přípravkem Translarna budete Vy nebo Vaše dítě svým lékařem testováni.
Jak přípravek Translarna působí?
Duchennova svalová dystrofie je způsobena genetickými změnami, které vedou k abnormalitě u svalového proteinu zvaného dystrofin, který je nutný pro správné fungování svalů. Přípravek Translarna umožňuje tvorbu funkčního dystrofinu a napomáhá správnému fungování svalů.
2. Čemu musíte věnovat pozornost, než začnete přípravek Translarna užívat Neužívejte přípravek Translarna
- jestliže jste alergický(á) na ataluren nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6),
- jestliže jste léčen určitými antibiotiky, např. gentamycinem, tobramycinem nebo streptomycinem podávanými injekčně do žíly.
Upozornění a opatření
Lékař Vám musí provést krevní test potvrzující, že Vaše onemocnění je vhodné léčit přípravkem Translarna. Jestliže máte jakékoli potíže s játry či ledvinami, lékař Vám bude pravidelně kontrolovat jaterní a ledvinné funkce.
Váš lékař u Vás bude testovat hladiny lipidů (tuků, např. cholesterolu a triglyceridů) v krvi a Vaše ledvinné funkce každých 6 až 12 měsíců. Jestliže užíváte léčivý přípravek ze skupiny kortikosteroidů, bude Váš lékař každých 6 měsíců sledovat Váš krevní tlak.
Děti a dospívající
Nepoužívejte tento léčivý přípravek u dětí mladších 5 let, protože u této skupiny pacientů nebyl testován.
Další léčivé přípravky a přípravek Translarna
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Přípravek Translarna neužívejte zejména s injekčně podávanými antibiotiky gentamycinem, tobramycinem nebo streptomycinem. Mohlo by dojít k ovlivnění Vašich ledvinných funkcí.
Informujte svého lékaře, pokud užíváte některé z následujících léčiv:
|
Léčivý přípravek |
Obvyklý důvod předepsání |
|
acyklovir |
léčba planých neštovic [varicelly] |
|
adefovir |
léčba chronické hepatitidy (žloutenky) B a/nebo HIV |
|
atorvastatin |
snižování hladin tuků v krvi |
|
benzylpenicilin |
závažné infekce |
|
bumetanid |
léčba nebo prevence městnavého srdečního selhání |
|
kaptopril |
léčba nebo prevence městnavého srdečního selhání |
|
cyklosporin |
prevence rejekce (odmítnutí) transplantovaného orgánu |
|
ciprofloxacin |
léčba infekcí |
|
famotidin |
léčba aktivního duodenálního vředu, gastroezofageální refluxní nemoci |
|
furosemid |
léčba nebo prevence městnavého srdečního selhání |
|
metotrexát |
revmatoidní artritida, psoriáza |
|
mykofenolát mofetil |
prevence rejekce (odmítnutí) transplantovaného orgánu |
|
olmesartan |
esenciální hypertenze u dospělých |
|
oseltamivir |
prevence chřipky |
|
fenobarbital |
navození spánku, prevence záchvatů |
|
pitavastatin |
snižování hladin tuků v krvi |
|
pravastatin |
snižování hladin tuků v krvi |
|
rifampicin |
léčba tuberkulózy |
|
rosuvastatin |
snižování hladin tuků v krvi |
|
sitagliptin |
diabetes typu 2 |
|
telmisartan |
léčba nebo prevence městnavého srdečního selhání |
|
valsartan |
léčba nebo prevence městnavého srdečního selhání |
Tato léčiva nebyla s přípravkem Translarna testována a Váš lékař se může rozhodnout Vás proto pečlivě sledovat.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat. Jestliže při užívání přípravku Translarna otěhotníte, poraďte se okamžitě se svým lékařem, protože během těhotenství nebo kojení se užívání přípravku Translarna nedoporučuje.
Řízení dopravních prostředků a obsluha strojů
Jestliže pociťujete závrať, neřiďte, nejezděte na kole a neobsluhujte stroje.
3. Jak se přípravek Translarna užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Přípravek Translarna je dostupný v sáčcích v následujících silách: 125 mg, 250 mg a 1 000 mg atalurenu v jednom sáčku. Váš lékař či lékárník Vám řekne, jaký počet sáčků o jaké síle máte pokaždé užívat.
Vaše dávka přípravku Translarna závisí na Vaší tělesné hmotnosti. Doporučená dávka přípravku je 10 mg/kg tělesné hmotnosti ráno, 10 mg/kg tělesné hmotnosti v poledne a 20 mg/kg tělesné hmotnosti večer (pro dosažení celkové denní dávky 40 mg/kg tělesné hmotnosti).
Léčivý přípravek se užívá perorálně v podobě směsi s tekutinou nebo polotekutou stravou.
Sáček otevírejte až v době užívání léčivého přípravku a použijte celý obsah sáčku. Granule promíchejte s 30 ml tekutiny (vody, mléka, ovocného džusu) nebo 3 lžícemi polotekuté stravy (jogurtu nebo jablečného protlaku). Připravenou dávku před užitím dobře promíchejte. Množství tekutin nebo polotekuté stravy lze zvýšit podle Vašich preferencí.
Tabulka dávkování
|
Rozmezí tělesné hmotnosti (kg) |
Počet sáčků | |||||||||
|
Ráno |
V poledne |
Večer | ||||||||
|
125mg sáčky |
250mg sáčky |
1 000mg sáčky |
125mg sáčky |
250mg sáčky |
1 000mg sáčky |
125mg sáčky |
250mg sáčky |
1 000mg sáčky | ||
|
12 |
14 |
1 |
0 |
0 |
1 |
0 |
0 |
0 |
1 |
0 |
|
15 |
16 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
1 |
0 |
|
17 |
20 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
|
21 |
23 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
1 |
0 |
|
24 |
26 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
0 |
|
27 |
31 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
0 |
|
32 |
35 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
0 |
|
36 |
39 |
1 |
1 |
0 |
1 |
1 |
0 |
0 |
3 |
0 |
|
40 |
44 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
3 |
0 |
|
45 |
46 |
0 |
2 |
0 |
0 |
2 |
0 |
1 |
3 |
0 |
|
47 |
55 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
0 |
1 |
|
56 |
62 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
1 |
1 |
|
63 |
69 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
1 |
1 |
|
70 |
78 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
2 |
1 |
|
79 |
86 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
3 |
1 |
|
87 |
93 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
3 |
1 |
|
94 |
105 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
|
106 |
111 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
|
112 |
118 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
|
119 |
125 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
2 |
2 |
Přípravek Translama užívejte perorálně 3krát denně, ráno, v poledne a večer. Mezi ranní a polední dávkou má být interval 6 hodin, mezi polední a večerní dávkou interval 6 hodin a mezi večerní dávkou a první dávkou následujícího dne interval 12 hodin. Přípravek Translama můžete např. užívat v 7 hodin ráno spolu se snídaní, ve 13 hodin spolu s obědem a opět přibližně v 19 hodin spolu s večeří.
Aby při užívání přípravku Translama nedocházelo k dehydrataci, pijte pravidelně vodu či jiné tekutiny.
Jestliže jste užil(a) více přípravku Translarna, než jste měl(a)
Jestliže užijete více přípravku Translarna, než je doporučená dávka, obraťte se na svého lékaře.
Může u Vás dojít k mírné bolesti hlavy, nevolnosti, zvracení či průjmu.
Jestliže jste zapomněl(a) užít přípravek Translarna
Jestliže se při užívání přípravku Translarna opozdíte o méně než 3 hodiny při podání ranní nebo polední dávky nebo o méně než 6 hodin při podání večerní dávky, dávku si vezměte. Nezapomeňte si příští dávku vzít včas.
Jestliže se při užívání přípravku Translarna opozdíte o více než 3 hodiny při podání ranní nebo polední dávky nebo o více než 6 hodin při podání večerní dávky, dávku neužívejte. Další dávku si nicméně vezměte včas.
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Je důležité užít správnou dávku. Jestliže užijete více, než je doporučená dávka, nemusí být přípravek Translarna u Vás tak účinný při léčbě příznaků onemocnění.
Jestliže jste přestal(a) užívat přípravek Translarna
Nepřestávejte užívat přípravek Translarna bez konzultace se svým lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Po užívání přípravku Translarna se u Vás může objevit jeden nebo více nežádoucích účinků:
Velmi časté nežádoucí účinky (mohou postihnout více než 1 z 10 osob):
- bolest hlavy,
- pocit nevolnosti,
- zvracení.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 osob):
- snížená chuť k j ídlu,
- úbytek hmotnosti,
- závratě,
- vysoký krevní tlak,
- kašel,
- krvácení z nosu,
- zácpa,
- průjem,
- nadýmání,
- regurgitace (návrat nestrávené potravy do úst bez nauzey a dávení),
- břišní dyskomfort,
- vyrážka,
- bolest horních nebo dolních končetin,
- ledvinná cysta,
- abnormálně časté močení,
- nechtěný únik moči,
- abnormální zbarvení moči,
- horečka,
- únava.
Neznámá frekvence (z dostupných údajů ji nelze určit)
- zvýšení hladin lipidů v krvi,
- zvýšení hodnot testů hodnotících funkci ledvin.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V*. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Translarna uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a sáčku. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Každou připravenou dávku užijte bezprostředně po přípravě. Jestliže připravenou dávku neužijete do 24 hodin při skladování v chladničce (2-8 °C) nebo do 3 hodin při pokojové teplotě (15-30 °C), dávku zlikvidujte.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Translarna obsahuje
Přípravek Translarna je dostupný ve třech silách s obsahem 125 mg, 250 mg a 1 000 mg léčivé látky zvané ataluren. Dalšími složkami jsou: polydextrosa (E1200), makrogol, poloxamer, manitol (E421), krospovidon, hydroxyetylcelulóza, umělá vanilková příchuť (maltodextrin, umělá barviva a propylenglykol), koloidní bezvodý oxid křemičitý (E551), magnesium-stearát.
Jak přípravek Translarna vypadá a co obsahuje toto balení
Přípravek Translarna jsou bílé až téměř bílé granule pro perorální suspenzi v sáčku.
Přípravek Translarna je dostupný v balení obsahujícím 30 sáčků.
Držitel rozhodnutí o registraci
PTC Therapeutics International Limited 77 Sir John Rogerson’s Quay Dublin 2 Irsko
Výrobce
Almac Pharma Services 22 Seagoe Industrial Estate Craigavon BT63 5QD Velká Británie
Tato příbalová informace byla naposledy revidována <{MM.RRRR}> <{měsíc RRRR}>.
Tomuto léčivému přípravku bylo uděleno tzv. podmíněné schválení. Znamená to, že informace o tomto přípravku budou přibývat.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tato příbalová informace bude podle potřeby aktualizována.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
PŘÍLOHA IV
ZÁVĚRY TÝKAJÍCÍ SE UDĚLENÍ PODMÍNĚNÉ REGISTRACE PŘEDKLÁDANÉ EVROPSKOU AGENTUROU PRO LÉČIVÉ PŘÍPRAVKY
Závěry předložené Evropskou agenturou pro léčivé přípravky:
• Podmíněná registrace přípravku
Výbor CHMP posoudil žádost a je toho názoru, že poměr přínosů a rizik je příznivý, a proto doporučuje, aby přípravku byla udělena podmíněná registrace, jak je podrobněji popsáno v Evropské veřejné zprávě o hodnocení.
63