Temodal 2,5 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Temodal 5 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje temozolomidum 5 mg.
Pomocná látka se známým účinkem:
Jedna tvrdá tobolka obsahuje 132,8 mg bezvodé laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka (tobolka).
Tvrdé tobolky mají neprůsvitné bílé tělo, neprůsvitné zelené víčko a jsou potištěny černým inkoustem. Na víčku je vytištěn nápis „Temodal“. Na těle tobolky je vytištěn nápis „5 mg“, logo společnosti Schering-Plough a dva proužky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Temodal je určen k léčbě:
- dospělých pacientů s nově diagnostikovaným multiformním glioblastomem souběžně s radioterapií (RT) a následně jako monoterapeutická léčba.
- dětí ve věku od tří let, dospívajících a dospělých pacientů s maligním gliomem, jako je multiformní glioblastom nebo anaplastický astrocytom, u nichž se po standardní léčbě projevují recidivy nebo progrese.
4.2 Dávkování a způsob podání
Temodal smí být předepisován výhradně lékařem se zkušenostmi s onkologickou léčbou mozkových
nádorů.
Může se podávat antiemetická léčba (viz bod 4.4).
Dávkování
Dospělí _pacienti s nově diagnostikovaným multiformním glioblastomem
Temodal je podáván v kombinaci s fokální radioterapií (fáze souběžné léčby) s následnými až 6 cykly monoterapie temozolomidem (TMZ) (monoterapeutická fáze).
Fáze souběžné léčby
TMZ se podává perorálně v dávce 75 mg/m2 denně po dobu 42 dnů souběžně s fokální radioterapií (60 Gy podáno ve 30 frakcích). Snižování dávek se nedoporučuje, ale každý týden by se mělo rozhodnout o pozdržení nebo přerušení podávání TMZ podle kritérií hematologické a nehematologické toxicity. Podávání TMZ může pokračovat po celou dobu 42 dnů souběžné fáze (až do 49 dnů), jestliže jsou splněny všechny následující podmínky:
- absolutní počet neutrofilů (ANC) > 1,5 x 109/l
- počet trombocytů > 100 x 109/l
- všeobecná kritéria toxicity (CTC - common toxicity criteria) nehematologické toxicity < stupeň 1 (s výjimkou alopecie, nevolnosti a zvracení).
Během léčby by se měl každý týden vyšetřit celkový krevní obraz. Podávání TMZ by mělo být během fáze souběžné léčby dočasně přerušeno nebo trvale ukončeno podle kritérií hematologické a nehematologické toxicity, jak je uvedeno v Tabulce 1.
|
Tabulka 1. Přerušení nebo ukončení podávání TMZ během souběžné léčby radioterapií a TMZ | ||
|
Toxicita |
TMZ přerušení3 |
TMZ ukončení |
|
Absolutní počet neutrofilů |
> 0,5 a <1,5 x 109/l |
< 0,5 x 109/l |
|
Počet trombocytů |
> 10 a < 100 x 109/l |
<10 x 109/l |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 2 |
CTC stupeň 3 nebo 4 |
a: Léčba se souběžně podávaným TMZ může pokračovat, jestliže jsou splněny všechny následující podmínky: absolutní počet neutrofilů > 1,5 x 109/l; počet trombocytů > 100 x 109/l; CTC nehematologická toxicita < stupeň 1 (s výjimkou alopecie, nausey, zvracení).
Monoterapeutická fáze
Čtyři týdny po ukončené souběžné fázi TMZ + RT se TMZ podává až v 6 cyklech monoterapeutické léčby. Dávka Cyklu 1 (monoterapie) je 150 mg/m2 jednou denně po dobu 5 dnů, následovaná 23 dny bez léčby. Na začátku Cyklu 2 je dávka zvýšena na 200 mg/m2, jestliže CTC nehematologická toxicita pro Cyklus 1 je stupeň < 2 (s výjimkou alopecie, nausey a zvracení), absolutní počet neutrofilů (ANC) je > 1,5 x 109/l a počet trombocytů je > 100 x 109/l. Jestliže není dávka zvýšena v Cyklu 2, nemělo by být zvýšení prováděno v následných cyklech. Je-li jednou zvýšena, zůstává dávka 200 mg/m2 na den pro prvních 5 dnů každého následujícího cyklu s výjimkou vzniku toxicity. Snížení dávky a ukončení podávání v průběhu monoterapeutické fáze by se mělo provádět podle Tabulek 2 a 3.
V průběhu léčby by měl být ve dni 22 (21 dnů po první dávce TMZ) vyšetřen celkový krevní obraz. Dávka by měla být redukována nebo podávání přípravku ukončeno podle Tabulky 3.
|
Tabulka 2. Úrovně dávky TMZ pro monoterapeutickou léčbu | ||
|
Úroveň dávky |
Dávka TMZ (mg/m2/den) |
Poznámky |
|
-1 |
100 |
Redukce kvůli předchozí toxicitě |
|
0 |
150 |
Dávka během Cyklu 1 |
|
1 |
200 |
Dávka během Cyklů 2-6 při absenci toxicity |
|
Tabulka 3. Snížení dávky nebo ukončení podávání TMZ během monoterapeutické léčby | ||
|
Toxicita |
Redukce TMZ o 1 úroveň dávkya |
Ukončení TMZ |
|
Absolutní počet neutrofilů |
<1,0 x 109/l |
Viz poznámka pod čarou b |
|
Počet trombocytů |
< 50 x 109/l |
Viz poznámka pod čarou b |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 3 |
CTC stupeň 4b |
a: Úrovně dávky TMZ jsou uvedeny v Tabulce 2. b: TMZ se ukončí jestliže:
• výsledkem úrovně dávky -1 (100 mg/m2) je stále neakceptovatelná toxicita
• jestliže se po redukci dávky znovu objeví stejný stupeň 3 nehematologické toxicity (s výjimkou alopecie, nausey, zvracení).
Dospělí_pacienti a děti ve věku 3 let nebo starší s recidivujícím nebo _progresivním maligním gliomem:
Léčebný cyklus zahrnuje 28 dnů. U pacientů, kteří dosud nebyli léčeni chemoterapií, se TMZ podává perorálně v dávce 200 mg/m2 jedenkrát denně po dobu prvních 5 dnů s následným přerušením léčby po dobu 23 dnů (celkem 28 dnů). U nemocných, kteří již dříve prodělali chemoterapii, činí zahajovací dávka 150 mg/m2 jedenkrát denně, která se pak ve druhém cyklu zvyšuje na 200 mg/m2 jedenkrát denně po dobu 5 dnů, pokud nejsou přítomny známky hematotoxicity (viz bod 4.4).
Zvláštní _ populace
Pediatrická populace
U dětí ve věku 3 let nebo starších by se měl TMZ používat pouze pro léčbu recidivujícího nebo progresivního maligního gliomu. Zkušenosti u těchto dětí jsou velmi omezené (viz body 4.4 a 5.1). Bezpečnost a účinnost TMZ u dětí ve věku do 3 let nebyla stanovena. Nejsou dostupné žádné údaje.
Pacienti s poruchou funkce jater či ledvin
Farmakokinetika TMZ je u pacientů s normální jaterní funkcí a s mírnou nebo středně těžkou jaterní dysfunkcí srovnatelná. Údaje o podání TMZ u pacientů s těžkou jaterní dysfunkcí (Třída C podle Childovy klasifikace) ani s dysfunkcí ledvin nejsou k dispozici. Vzhledem k uvedeným farmakokinetickým vlastnostem TMZ je však pravděpodobné, že ani u nemocných s těžkou jaterní dysfunkcí nebo dysfunkcí ledvin kteréhokoli stupně není snižování jeho dávek nutné. Při podávání TMZ těmto nemocným je však nutná zvýšená opatrnost.
Starší pacienti
Z populační analýzy farmakokinetiky u pacientů ve věku 19-78 let vyplývá, že clearance TMZ není ovlivňována věkem. Nicméně se zdá, že starší pacienti (ve věku > 70 let) mají vyšší riziko vzniku neutropenie a trombocytopenie (viz bod 4.4).
Způsob podání
Temodal tvrdé tobolky by měl podáván nalačno.
Tobolky se musejí polykat vcelku a zapíjet sklenicí vody a nesmějí se otevírat nebo kousat.
Pokud se po podání dávky dostaví zvracení, neměla by být další dávka podána tentýž den.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Hypersenzitivita na dakarbazin (DTIC).
Těžká myelosuprese (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Oportunní infekce a reaktivace infekcí
Během léčby pomocí TMZ byly pozorovány oportunní infekce (jako je pneumonie způsobená Pneumocystis jirovecii) a reaktivace infekcí (jako jsou HBV, CMV) (viz bod 4.8).
Pneumonie způsobená Pneumocystis _ jirovecii
U pacientů, kteří dostávali souběžně TMZ a RT v pilotní studii s prodlouženým harmonogramem 42 dnů, bylo zjištěno zvýšené riziko vzniku pneumonie způsobené Pneumocystis jirovecii (PCP). Proto je nutná profylaxe PCP u všech pacientů, kteří dostávají souběžně TMZ a RT po dobu 42 denního režimu (maximálně 49 dnů) bez ohledu na počet lymfocytů. Jestliže se objeví lymfopenie, je třeba pokračovat v profylaxi, dokud se neobnoví stupeň lymfopenie < 1.
Výskyt PCP může být vyšší, když je TMZ podáván během delšího dávkovacího režimu. Nicméně všichni pacienti, kteří dostávají TMZ, zvláště pacienti dostávající steroidy, by měli být pečlivě sledováni pro vznik PCP, bez ohledu na režim. U pacientů užívajících TMZ, zvláště v kombinaci s dexamethasonem nebo jinými steroidy, byly hlášeny případy fatálního respiračního selhání.
HBV
Byla hlášena hepatitida způsobená reaktivací viru hepatitidy B (HBV), v některých případech vedoucí ke smrti. Před zahájením léčby pacientů s pozitivní sérologií na hepatitidu B (včetně těch s aktivním onemocněním) je nezbytná konzultace s odborníky na onemocnění jater. Během léčby musí být pacienti náležitě monitorováni a vedeni.
Hepatotoxicita
U pacientů léčených TMZ bylo hlášeno poškození jater, včetně smrtelného selhání jater (viz bod 4.8). Před zahájením léčby je nutno provést testy jaterních funkcí. Pokud jsou výsledky abnormální, musí lékař před zahájením léčby temozolomidem vyhodnotit poměr přínosů a rizik, včetně potenciálu ke smrtelnému selhání jater. U pacientů se 42denním léčebným cyklem se musí testy jaterních funkcí opakovat uprostřed tohoto cyklu. Pro všechny pacienty platí, že jaterní funkce je nutno zkontrolovat po každém léčebném cyklu. U pacientů s výraznými abnormalitami jaterních funkcí musí lékař vyhodnotit poměr přínosů a rizik vyplývajících z pokračování v léčbě. Jaterní toxicita se může objevit několik nebo i více týdnů po poslední léčbě temozolomidem.
Malignity
Byly též velmi vzácně hlášeny případy myelodysplastického syndromu a sekundárních malignit, včetně myeloidní leukémie (viz bod 4.8).
Antiemetická léčba
Nausea a zvracení jsou velmi často spojené s TMZ.
Před nebo po podání TMZ se může podávat antiemetická léčba.
Antiemetická profylaxe se doporučuje před počáteční dávkou souběžné fáze a velmi doporučuje během monoterapeutické fáze.
Pacienti s recidivujícím nebo _progresivním maligním gliomem
Antiemetika může být třeba podat pacientům, u nichž v předchozích léčebných cyklech došlo k těžšímu zvracení (stupeň 3 nebo 4).
Laboratorní parametry
U pacientů léčených TMZ může dojít k myelosupresi, včetně dlouhotrvající pancytopenie, která může vést k aplastické anémii, jež v některých případech skončila fatálně. V některých případech souběžná expozice léčivým přípravkům spojených s výskytem aplastické anémie, zahrnující karbamazepin, fenytoin a sulfamethoxazol/trimethoprim, hodnocení komplikuje. Před zahájením podávání léku by měly být splněny následující laboratorní parametry: ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Kompletní krevní obraz je třeba vyšetřit ve 22. dni (21 dnů po podání 1. dávky) nebo v průběhu 48 hodin po tomto dni, a pak v týdenních intervalech do té doby, dokud není ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Jestliže v průběhu kteréhokoli cyklu poklesne ANC na < 1,0 x 109/l nebo počet trombocytů na < 50 x 109/l, měla by být v následném cyklu dávka o jeden stupeň snížena (viz bod 4.2). Jednotlivé stupně dávky činí 100 mg/m2, 150 mg/m2 a 200 mg/m2. Nejnižší doporučená dávka je 100 mg/m2.
Pediatrická populace
S podáváním TMZ dětem ve věku nižším než 3 roky nejsou dosud žádné klinické zkušenosti. U starších dětí a dospívajících jsou zkušenosti velmi omezené (viz body 4.2 a 5.1).
Starší pacienti (ve věku >70 let)
U starších pacientů je ve srovnání s mladšími osobami pravděpodobně vyšší riziko neutropenie a trombocytopenie. Vzhledem k tomu je při podávání TMZ ve vyšším věku nutná zvýšená opatrnost.
Pacienti mužského pohlaví
Muži léčení TMZ by měli být upozorněni, aby se do 6 měsíců od užití poslední dávky nestali otci a aby se před zahájením léčby informovali o možnosti kryokonzervace spermatu (viz bod 4.6).
Laktóza
Tento léčivý přípravek obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo glukózo-galaktózovou malabsorpcí by proto neměli tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Podávání TMZ spolu s ranitidinem nemělo v oddělené studii fáze I za následek žádné změny rozsahu absorpce temozolomidu nebo expozice jeho aktivnímu metabolitu monomethyl- triazenoimidazol-karboxamidu (MTIC).
Podávání TMZ spolu s jídlem vedlo k poklesu Cmax o 33 % a zmenšení plochy pod křivkou koncentrace (AUC) o 9 %.
Protože nelze vyloučit, že změna Cmax může být klinicky významná, neměl by se Temodal podávat současně s jídlem.
Výsl edky analýzy populační farmakokinetiky ve II. fázi klinických studií prokazují, že nebyla clearance TMZ ovlivněna současným podáním dexamethasonu, prochlorperazinu, fenytoinu, karbamazepinu, ondansetronu, antagonistů H2 receptorů nebo fenobarbitalu. Současné podávání kyseliny valproové bylo spojeno s malým, nicméně statisticky významným poklesem clearance TMZ.
Dosud nebyly provedeny žádné studie, které by hodnotily účinek TMZ na metabolismus nebo eliminaci jiných léků. Nicméně vzhledem k tomu, že TMZ neprochází přeměnou v játrech a na plazmatické bílkoviny se váže jen v malé míře, je málo pravděpodobné, že by ovlivňoval farmakokinetiku jiných léčivých přípravků (viz bod 5.2).
V kombinaci s jinými myelosupresivními látkami může TMZ zvýšit pravděpodobnost myelosuprese. Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání léku těhotným ženám nejsou k dispozici. V preklinických studiích u potkanů a králíků, kteří TMZ dostávali v dávce 150 mg/m2, byly prokázány teratogenní a/nebo toxické účinky na plod (viz bod 5.3). Temodal by neměl být podáván těhotným ženám. Pokud je přece jen nutno zvážit použití během těhotenství, je třeba pacientku seznámit s potenciálním rizikem pro plod.
Kojení
Není známo, zda se TMZ vylučuje do mateřského mléka; kojení by se proto mělo po dobu léčby TMZ přerušit.
Ženy ve fertilním věku.
Ženám ve fertilním věku je třeba doporučit používání účinné antikoncepce k zabránění otěhotnění v průběhu užívání TMZ.
Mužská plodnost
TMZ může mít genotoxické účinky. Proto se mužům léčeným temozolomidem nedoporučuje, aby se do 6 měsíců od užití poslední dávky stali otci, a naopak se doporučuje, vzhledem k možnosti ireverzibilní neplodnosti po léčbě TMZ, ještě před jejím zahájením konzultovat s lékařem možnost kryokonzervace předem odebraných spermií.
4.7 Účinky na schopnost řídit a obsluhovat stroje
TMZ má malý vliv na schopnost řídit nebo obsluhovat stroje v důsledku únavy a ospalosti (viz bod 4.8.).
4.8 Nežádoucí účinky
Zkušenosti z klinických hodnocení
U pacientů léčených TMZ ať už užívaným v kombinaci s RT nebo ve formě monoterapie následující po RT v případě nově diagnostikovaného multiformního glioblastomu, nebo ve formě monoterapie pacientů s recidivujícím nebo progresivním gliomem, byly velmi často hlášené nežádoucí účinky podobné: nausea, zvracení, zácpa, anorexie, bolest hlavy a únava. Křeče byly hlášeny velmi často u
nově diagnostikovaných pacientů s multiformním glioblastomem léčených monoterapií, vyrážka byla hlášena velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených TMZ v kombinaci s RT nebo také ve formě monoterapie a často u pacientů s recidivujícím gliomem. Většina hematologických nežádoucích účinků byla hlášena u obou indikací často nebo velmi často (Tabulky 4 a 5); četnost laboratorních nálezů stupně 3 a 4 je uvedena za každou tabulkou.
V tabulkách jsou nežádoucí účinky klasifikovány podle orgánových systémů a četnosti. Zařazení do skupin četností se provádí podle následujícího pravidla: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Nově diagnostikovaný multiformní glioblastom
Tabulka 4 ukazuje nežádoucí účinky, které se objevily při léčbě u pacientů s nově diagnostikovaným multiformním glioblastomem během souběžné a monoterapeutické fáze léčby.
|
Tabulka 4: Nežádoucí účinky, které se objevily během souběžné a monoterapeutické léčby u pacientů s nově diagnostikovaným multiformním glioblastomem | ||
|
Třída orgánového systému |
TMZ + souběžná RT n=288* |
TMZ monoterapie n=224 |
|
Infekce a infestace | ||
|
Časté: |
Infekce, Herpes simplex, infekce rány, faryngitida, orální kandidóza |
Infekce, orální kandidóza |
|
Méně časté: |
Herpes simplex, herpes zoster, symptomy podobné chřipce | |
|
Poruchy krve a lymfatického systému | ||
|
Časté: |
Neutropenie, trombocytopenie, lymfopenie, leukopenie |
Febrilní neutropenie, trombocytopenie, anémie, leukopenie |
|
Méně časté: |
Febrilní neutropenie, anémie |
Lymfopenie, petechie |
|
Endokrinní poruchy | ||
|
Méně časté: |
Cushingoid |
Cushingoid |
|
Poruchy metabolismu a výživy | ||
|
Velmi časté: | ||
|
Časté: |
Hyperglykémie, pokles hmotnosti |
Pokles hmotnosti |
|
Méně časté: |
Hypokalémie, zvýšení alkalické fosfatázy, zvýšení hmotnosti |
Hyperglykémie, zvýšení hmotnosti |
|
Psychiatrické poruchy | ||
|
Časté: |
Úzkost, emoční labilita, nespavost | |
|
Méně časté: |
Neklid, apatie, poruchy chování, deprese, halucinace |
Halucinace, amnézie |
|
Poruchy nervového systému | ||
|
Velmi časté: |
Konvulze, bolest hlavy | |
|
Časté: |
Konvulze, porucha vědomí, somnolence, afázie, porucha rovnováhy, závrať, zmatenost, porucha paměti, porucha koncentrace, neuropatie, parestézie, porucha řeči, třes |
Hemiparéza, afázie, porucha rovnováhy, somnolence, zmatenost, závrať, porucha paměti, porucha koncentrace, dysfázie, neurologická porucha (NOS), neuropatie, periferní neuropatie, parestézie, porucha řeči, třes |
|
Méně časté: |
Status epilepticus, extrapyramidální porucha, hemiparéza, ataxie, poruchy kognice, dysfázie, abnormální chůze, hyperestézie, hypestézie, neurologická porucha (NOS), periferní neuropatie |
Hemiplegie, ataxie, abnormální koordinace, abnormální chůze, hyperestézie, smyslová porucha |
|
Poruchy oka | ||
|
Časté: |
Rozmazané vidění |
Defekt zorného pole, rozmazané vidění, diplopie |
|
Méně časté: |
Hemianopie, zmenšená ostrost vidění, poruchy zraku, defekt zorného pole, bolest očí |
Zmenšená ostrost vidění, bolest očí, suché oči |
|
Poruchy ucha a labyrintu | ||
|
Časté: |
Porucha sluchu |
Porucha sluchu, tinitus |
|
Méně časté: |
Otitis media, tinitus, hyperakuze, bolest ucha |
Hluchota, závrať, bolest ucha |
|
Srdeční poruchy | ||
|
Méně časté: |
Palpitace | |
|
Cévní poruchy | ||
|
Časté: |
Hemoragie, edém, edém nohy |
Hemoragie, hluboká žilní trombóza, edém nohy |
|
Méně časté: |
Cerebrální hemoragie, hypertenze |
Plicní embolie, edém, periferní edém |
|
Respirační, hrudní a mediastinální poruchy | ||
|
Časté: | ||
|
Méně časté: |
Pneumonie, infekce horních cest dýchacích, nosní kongesce |
Pneumonie, sinusitida, infekce horních cest dýchacích, bronchitida |
|
Gastrointestinální poruchy | ||
|
Velmi časté: |
Zácpa, nausea, zvracení |
Zácpa, nausea, zvracení |
|
Časté: |
Stomatitida, průjem, bolest břicha, dyspepsie, dysfagie | |
|
Méně časté: |
Abdominální distenze, fekální inkontinence, gastrointestinální porucha (NOS), gastroenteritida, hemeroidy | |
|
Poruchy kůže a podkožní tkáně | ||
|
Velmi časté: |
Vyrážka, alopecie |
Vyrážka, alopecie |
|
Časté: |
Dermatitida, suchá kůže, erytém, svědění |
Suchá kůže, svědění |
|
Méně časté: |
Exfoliace kůže, fotosenzitivní reakce, abnormální pigmentace |
Erytém, abnormální pigmentace, zvýšené pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Časté: |
Svalová slabost, artralgie |
Svalová slabost, artralgie, muskuloskeletální bolest, myalgie |
|
Méně časté: |
Myopatie, bolest v zádech, muskuloskeletální bolest, myalgie |
Myopatie, bolest v zádech |
|
Poruchy ledvin a močových cest | ||
|
Časté: |
Časté močení, močová inkontinence |
Močová inkontinence |
|
Méně časté: | ||
|
Poruchy reprodukčního systému a prsu | ||
|
Méně časté: |
Impotence |
Vaginální hemoragie, menoragie, amenorea, vaginitida, bolest prsou |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Velmi časté: |
Únava |
Únava |
|
Časté: |
Alergická reakce, horečka, radiační poškození, edém obličeje, bolest, změna chuti |
Alergická reakce, horečka, radiační poškození, bolest, změna chuti |
|
Méně časté: |
Astenie, zrudnutí, návaly horka, zhoršení stavu, rigory, změna barvy jazyka, parosmie, žízeň |
Astenie, edém obličeje, bolest, zhoršení stavu, rigory, zubní potíže |
|
Vyšetření | ||
|
Časté: |
Zvýšené ALT |
Zvýšené ALT |
|
Méně časté: |
Zvýšené jaterní enzymy, zvýšené gama GT, zvýšené AST | |
*Pacient který byl randomizován pouze do větve RT, obdržel Temodal + RT.
Laboratorní výsledky
Byla zjištěna myelosuprese (neutropenie a trombocytopenie), která je známou toxicitou limitující dávku většiny cytotoxických látek včetně TMZ. Když byly zkombinovány abnormality laboratorních testů a nežádoucí účinky v souběžných a monoterapeutických fázích léčby, byly pozorovány u 8 % pacientů abnormality neutrofilů stupeň 3 nebo stupeň 4, včetně neutropenických případů. Stupeň 3 nebo stupeň 4 abnormalit trombocytů, včetně trombocytopenických případů, byl pozorován u 14 % pacientů, kteří dostávali TMZ.
Recidivující nebo progresivní maligní gliom
Nejčastěji se vyskytujícími nežádoucími účinky ve vztahu k léčbě byly v provedených klinických studiích gastrointestinální poruchy, především nausea (43 %) a zvracení (36 %). Tyto reakce dosahovaly obvykle pouze stupně 1 nebo 2 (0 - 5 epizod zvracení během 24 hodin) a buď spontánně odezněly, nebo byly snadno léčitelné standardní antiemetickou léčbou. Incidence těžké nausey a zvracení činila 4 %.
Tabulka 5 obsahuje nežádoucí účinky, které byly hlášeny v průběhu klinických studií pro recidivující nebo progresivní maligní gliom a po uvedení přípravku Temodal na trh.
|
Tabulka 5. Nežádoucí účinky u pacientů s recidivujícím nebo progresivním maligním gliomem | |
|
Infekce a infestace | |
|
Vzácné: |
Oportunní infekce včetně pneumonie způsobené PCP |
|
Poruchy krve a lymfatického systému | |
|
Velmi časté: |
Neutropenie nebo lymfopenie (stupeň 3-4) trombocytopenie (stupeň 3-4) |
|
Méně časté: |
Pancytopenie, anémie (stupeň 3-4), leukopenie |
|
Poruchy metabolismu a výživy | |
|
Velmi časté: | |
|
Časté: |
Pokles hmotnosti |
|
Poruchy nervového systému | |
|
Velmi časté: | |
|
Časté: |
Somnolence, závratě, parestézie |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté: | |
|
Gastrointestinální poruchy | |
|
Velmi časté: |
Zvracení, nausea, zácpa |
|
Časté: | |
|
Poruchy kůže a podkožní tkáně | |
|
Časté: |
Vyrážka, pruritus, alopecie, |
|
Velmi vzácné: |
Multiformní erytém, erytrodermie, urtikarie, exantém |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté: |
Únava |
|
Časté: | |
|
Velmi vzácné: |
Alergické reakce, včetně anafylaxe, angioedém |
Laboratorní výsledky
Trombocytopenie a neutropenie stupně 3 nebo 4 byly zaznamenány u 19 %, resp. 17 % pacientů léčených pro maligní gliom. K hospitalizaci a/nebo přerušení léčby TMZ vedly v 8 %, resp. 4 % případů. Myelosuprese byla předvídatelná (obvykle k ní došlo během několika prvních cyklů, s nadirem mezi 21. dnem a 28. dnem) a úprava k normě byla rychlá, obvykle v průběhu 1-2 týdnů. Kumulace myelosupresivních účinků prokázána nebyla. Přítomnost trombocytopenie může zvýšit riziko krvácení a přítomnost neutropenie nebo leukopenie může zvýšit riziko infekce.
Pohlaví
Do analýzy populační farmakokinetiky založené na zkušenostech z klinického hodnocení bylo zahrnuto 101 žen a 169 mužů, u kterých byla dostupná měření nadiru neutrofilů, a 110 žen a 174 mužů, u kterých byla dostupná měření nadiru krevních destiček. U žen bylo ve srovnání s muži v prvním cyklu terapie zaznamenáno více neutropenií stupně 4 (ANC< 0,5 x 109/l), 12 % vs 5 %, a trombocytopenií (< 20 x 109/l), 9 % vs 3 %. V souboru dat od 400 jedinců s rekurentním gliomem se v prvním cyklu terapie objevila neutropenie stupně 4 u 8 % žen vs 4 % mužů a trombocytopenie stupně 4 u 8 % žen vs 3 % mužů. Ve studii s 288 jedinci s nově diagnostikovaným multiformním glioblastomem se v prvním cyklu terapie vyskytla neutropenie stupně 4 u 3 % žen vs 0 % mužů a trombocytopenie stupně 4 u 1 % žen vs 0 % mužů.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s rekurentním gliomem mozkového kmene nebo rekurentním astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Ačkoliv jsou údaje omezené, tolerance u dětí se očekává být podobná jako u dospělých. Bezpečnost TMZ u dětí ve věku do 3 let nebyla stanovena.
Zkušenosti po uvedení na trh
Po uvedení přípravku na trh byly hlášeny další následující závažné nežádoucí účinky:
|
Tabulka 6. Souhrn příhod hlášených u temozolomidu po jeho uvedení na trh | |
|
Infekce a infestace* | |
|
Méně časté: |
infekce způsobená cytomegaloviry, reaktivace infekce způsobené virem, jako je cytomegalovirus, virus hepatitidy BŤ |
|
Poruchy krve a lymfatického systému | |
|
Velmi vzácné: |
dlouhotrvající pancytopenie, aplastická anémieŤ |
|
Novotvary benigní, maligní a blíže neurčené | |
|
Velmi vzácné: |
myelodysplastický syndrom (MDS), sekundární malignity, včetně myeloidní leukémie |
|
Endokrinní poruchy | |
|
Méně časté: |
diabetes insipidus |
|
Respirační, hrudní a mediastinální poruchy | |
|
Velmi vzácné: |
intersticiální pneumonitida/pneumonitida, plicní fibróza, respirační selhání1 |
|
Poruchy jater a žlučových cest* | |
|
Časté: |
zvýšení jaterních enzymů |
|
Méně časté: |
hyperbilirubinemie, cholestáza, hepatitida, poškození jater, selhání jater1 |
|
Poruchy kůže a podkožní tkáně | |
|
Velmi vzácné: |
toxická epidermální nekrolýza, Stevens-Johnsonův syndrom |
t Včetně smrtelných případů
Frekvence odhadované na základě relevantních klinických studií.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinicky byly u pacientů ověřeny dávky 500, 750, 1 000, a 1 250 mg/m2 (celková dávka za 5denní cyklus). Toxicita limitující dávku byla toxicita hematologická a byla pozorována u všech dávek, ale lze očekávat, že při vyšších dávkách bude závažnější. U jednoho pacienta došlo k předávkování, když užil dávku 10 000 mg (celková dávka během jednoho 5denního cyklu), nežádoucí účinky hlášené v tomto případě byly pancytopenie, pyrexie, multiorgánové selhání a smrt. Byly popsány případy, kdy pacienti doporučenou dávku užívali po dobu delší než 5 dnů (až 64 dnů) s hlášenými nežádoucími příhodami včetně suprese kostní dřeně s nebo bez rozvoje infekce, v některých případech závažné, dlouhotrvající a vedoucí ke smrti. V případě předávkování je nezbytné hematologické zhodnocení. Podle potřeby by měla být zajištěna podpůrná léčba.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: protinádorové léky - ostatní alkylační látky, ATC kód: L01A X03 Mechanismus účinku
Temozolomid je triazen, který za fyziologické hodnoty pH prochází rychlou chemickou konverzí na aktivní monometyl-triazenoimidazol-karboxamid (MTIC). Předpokládá se, že cytotoxické účinky MTIC jsou primárně důsledkem alkylace na O6 pozici guaninu, s následnou alkylací rovněž na pozici N7. Následně vznikající cytotoxické léze jsou vysvětlovány nejspíše aberantní reparací methylovaného zbytku.
Klinická účinnost a bezpečnost
Nově diagnostikovaný multiformní glioblastom
Celkem 573 pacientů bylo randomizováno do skupin dostávajících buď TMZ + RT (n=287) nebo RT samotnou (n=286). Pacienti ve větvi TMZ + RT dostávali souběžně TMZ (75 mg/m2) jednou denně od prvního až do posledního dne RT po dobu 42 dnů (maximálně 49 dnů). Pak následovala monoterapie TMZ (150 - 200 mg/m2) ve dnech 1-5 každého 28denního cyklu, až v 6 cyklech, začínající 4 týdny po ukončení RT. Pacienti v kontrolní větvi dostávali pouze RT. Během RT a souběžné TMZ terapie byla nutná profylaxe pneumonie Pneumocystis jirovecii (PCP).
TMZ byl podáván jako záchranná terapie vnásledné fázi 161 pacientům z 282 (57 %) ve větvi dostávající pouze RT a 62 pacientům z 277 (22 %) ve větvi dostávající TMZ + RT.
Poměr rizika (HR) pro celkové přežívání byl 1,59 (95 % CI pro HR=1,33 - 1 ,91) s log-rank p < 0,0001 ve prospěch TMZ větve. Odhadnutá pravděpodobnost přežívání 2 roky nebo více (26 % vs 10 %) je vyšší ve větvi RT + TMZ. Přidání souběžně podávaného TMZ k RT následované monoterapií TMZ v léčbě pacientů s nově diagnostikovaným multiformním glioblastomem prokázalo statisticky významné zlepšení v celkovém přežívání (overall survival=OS) ve srovnání s RT samotnou (Graf 1).
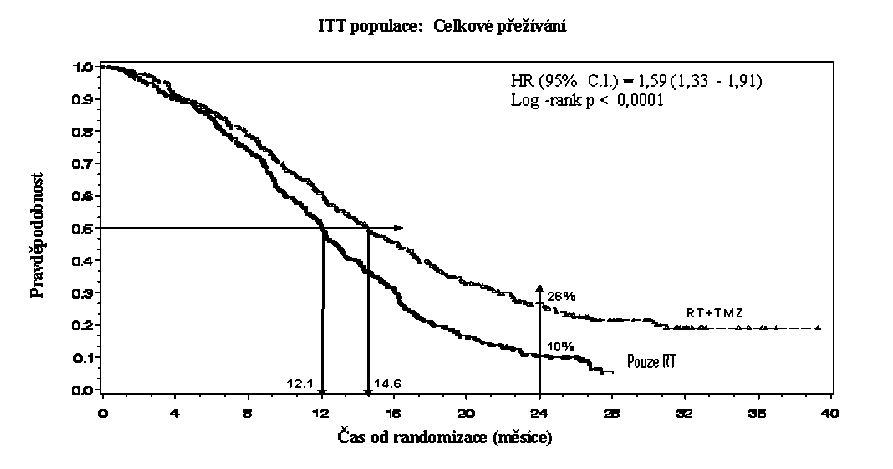
Graf 1 Kaplanovy-Meierovy křivky pro celkové přežívání (ITT populace=intent-to- treat population)
Výsledky studie nebyly konzistentní v podskupině pacientů se slabým výkonnostním statusem (WHO PS=2, n=70), kde celkové přežívání a čas do progrese byly podobné v obou větvích. Zdá se však, že u této skupiny pacientů nejsou přítomna žádná nepřijatelná rizika.
Recidivující nebo _progresivní maligní gliom
Data o klinické účinnosti u pacientů s multiformním glioblastomem (Karnofského výkonnostní status [KPS] > 70), u něhož dochází k progresi nebo recidivě po chirurgickém výkonu a RT, vycházela ze dvou klinických studií s perorálním TMZ. V prvním případě šlo o nekomparativní studii u 138 pacientů (29 % bylo předtím léčeno chemoterapií), ve druhém o randomizovanou studii kontrolovanou léčivou látkou s TMZ vs prokarbazinem u celkem 225 pacientů (67 % z nich bylo předtím léčeno chemoterapií na bázi nitrosourey). V obou studiích bylo primárním hodnotícím kritériem přežívání bez známek progrese (PFS - progression-free survival), hodnocených na základě zobrazení magnetickou rezonancí (MRI) nebo na základě zhoršení neurologických funkcí. V nekomparativní studii bylo u 19 % pacientů dosaženo 6měsíční PFS, medián přežívání bez známek progrese byl 2,1 měsíce a medián celkové doby přežívání byl 5,4 měsíce. Objektivní počet terapeutických odpovědí na základě hodnocení MRI dosahoval 8 %.
V randomizované studii kontrolované léčivou látkou bylo šestiměsíční PFS u pacientů léčených TMZ významně vyšší než u těch, kteří dostávali prokarbazin (21 % vs 8 %; chí-kvadrát p=0,008)
s mediánem 2,89, respektive 1,88 měsíce (log rank p=0,0063). Medián přežití byl 7,34 měsíce pro TMZ a 5,66 měsíce pro prokarbazin (log rank p=0,33). Po 6 měsících byl podíl přežívajících pacientů v TMZ skupině významně vyšší (60 %) než ve skupině prokarbazinové (44 %; chí-kvadrát p=0,019). U pacientů, kteří byli předtím léčeni chemoterapií, bylo dosaženo kladného efektu u těch, kteří měli KPS > 80.
Údaje o době do zhoršení neurologického statusu, jakož i o době do zhoršení výkonnostního statusu (pokles na KPS < 70 nebo pokles nejméně o 30 bodů), hovoří ve prospěch TMZ oproti prokarbazinu. Medián doby do progrese v těchto hodnotících kritériích byl u TMZ o 0,7 - 2,1 měsíce delší než u prokarbazinu (log rank p < 0,01 - 0,03).
Recidivující anaplastický astrocytom
V multicentrické prospektivní studii fáze II, v níž byly hodnoceny bezpečnost a účinnost perorálně podávaného TMZ v léčbě pacientů s anaplastickým astrocytomem při prvním relapsu, bylo zjištěno šestiměsíční PSF u 46 % léčených. Medián PSF činil 5,4 měsíce. Medián celkového přežití činil 14,6 měsíce. Procento odpovědí na léčbu, hodnocené centrálně činilo 35 % (13 % kompletních, 43 % částečných odpovědí) pro populaci intent-to-treat (ITT), n=162. U 43 pacientů došlo ke stabilizaci onemocnění. Šestiměsíční přežití bez klinické příhody bylo pro populaci intent-to-treat 44 %, přičemž medián přežití bez klinické příhody činil 4,6 měsíce, což je podobné výsledkům hodnocení přežívání bez známek progrese. Z histologického hlediska byla účinnost obdobná. Objektivní radiologicky prokázaná odpověď na léčbu nebo zachování stavu beze známek progrese velmi dobře korelovaly se zachovanou nebo zlepšenou kvalitou života.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s recidivujícím gliomem mozkového kmene nebo recidivujícím astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Tolerance TMZ byla podobná jako u dospělých.
5.2 Farmakokinetické vlastnosti
TMZ se spontánně hydrolyzuje při fyziologickém pH primárně na aktivní látku, 3-metyl-(triazen-1-yl)imidazol-4-karboxamid (MTIC). MTIC se spontánně hydrolyzuje na 5-amino-imidazol-4-karboxamid (AIC), známý meziprodukt biosyntézy purinů a nukleových kyselin a na metylhydrazin, o kterém se předpokládá, že je aktivní alkylační látkou. Předpokládá se, že cytotoxicita MTIC je primárně způsobena alkylací DNA zejména na pozicích guaninu O6 a N7' Expozice MTIC a AIC je ~ 2,4 %, respektive 23 % v poměru k AUC TMZ. In vivo byl ti/2 MTIC podobný jako pro TMZ,
1,8 hodin.
Absorpce
Po perorálním podání dospělým pacientům se TMZ rychle absorbuje a vrcholné koncentrace dosahuje již za 20 minut po podání (střední doba je mezi 0,5 a 1,5 hodiny). Po perorálním podání 14C-značeného TMZ byla průměrná fekální exkrece 14C v průběhu 7 dnů po podání dávky 0,8 %, což svědčilo o úplné absorpci.
Distribuce v organismu
TMZ vykazuje nízkou vazebnou afinitu k proteinům (10 % až 20 %) proto se neočekává, že by interagoval s látkami s vysokou vazebnou afinitou k proteinům.
PET studie u lidí a preklinické údaje ukazují, že TMZ rychle prostupuje hematoencefalickou bariérou a je přítomný v mozkomíšním moku. Průnik do mozkomíšního moku (CSF) byl potvrzen u jednoho pacienta; expozice CSF na základě AUC TMZ byla přibližně 30 % plazmatické expozice, což odpovídá údajům u zvířat.
Eliminace z organismu
Plazmatický poločas (t1/2) je přibližně 1,8 hodin. Hlavní způsob eliminace 14C je ledvinami. Po perorálním podání se objevuje přibližně 5 až 10 % dávky v nezměněné podobě v moči v průběhu 24 hodin a zbytek se vylučuje ve formě temozolomidové kyseliny, 5-aminoimidazol-4-karboxamidu (AIC) nebo neidentifikovaných polárních metabolitů.
Plazmatické koncentrace vzrůstají v závislosti na velikosti dávky. Plazmatická clearance, distribuční objem a poločas jsou na velikosti dávky nezávislé.
Zvláštní populace
V analýze populační farmakokinetiky TMZ bylo zjištěno, že plazmatická clearance TMZ nezávisí na věku, renální funkci nebo kouření. V oddělené farmakokinetické studii byly plazmatické farmakokinetické profily u pacientů s mírnou až střední jaterní dysfunkcí stejné jako profily u pacientů s normální funkcí jater.
U dětských pacientů byla AUC větší než u dospělých; nicméně maximální tolerovaná dávka (MTD) činila 1 000 mg/m2 na jeden cyklus jak u dětí, tak u dospělých.
5.3 Předklinické údaje vztahující se k bezpečnosti
U potkanů a psů byly prováděny studie toxicity s jednotlivým cyklem (5 dnů podávání léku, 23 dnů bez léčby) 3 a 6 cyklů léčby. Primární cíle toxických účinků tvořila kostní dřeň, lymforetikulární systém, testes, gastrointestinální ústrojí a při vyšších dávkách, které byly pro 60 % - 100 % testovaných potkanů a psů letální, docházelo k degeneraci retiny. Většina toxických účinků byla reverzibilní, s výjimkou nežádoucích účinků na mužský reprodukční systém a degeneraci retiny. Nicméně vzhledem k tomu, že dávky podmiňující retinální degeneraci se pohybovaly v rozmezí dávek letálních, a v klinických studiích nebyl žádný srovnatelný účinek pozorován, nepřikládá se tomuto nálezu klinická významnost.
TMZ je embryotoxická, teratogenní a genotoxická alkylační látka. TMZ má toxické účinky větší u potkanů a psů než u člověka, a klinická dávka se blíží minimální letální dávce u potkanů a psů. Za citlivé ukazatele toxicity se považují poklesy počtu leukocytů a trombocytů v závislosti na velikosti dávky. Ve studii u potkanů, v níž bylo aplikováno šest cyklů terapie, bylo zjištěno několik druhů novotvarů, včetně karcinomu mléčné žlázy, kožního keratoakanthomu a adenomu bazálních buněk, zatímco ve studiích u psů žádné nádorové ani prenádorové změny pozorovány nebyly. Zdá se, že potkani jsou vůči onkogenním účinkům TMZ mimořádně citliví, neboť první tumory se u nich objevují již po třech měsících od zahájení podávání léku. Jde o dobu krátkou dokonce i pro alkylační látky.
Výsledky aberačních testů s chromosomy Ames/salmonella a lymfocytů z lidské periferní krve prokázaly pozitivní mutagenní reakci.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky: laktóza,
koloidní bezvodý oxid křemičitý, sodná sůl karboxymethylškrobu typu A, kyselina vinná, kyselina stearová.
Obal tobolky: želatina,
oxid titaničitý (E 171), natrium-lauryl-sulfát, žlutý oxid železitý (E 172), indigokarmín (E 132),
Potisk:
šelak,
propylenglykol, čištěná voda, roztok amoniaku, hydroxid draselný, černý oxid železitý (E 172).
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Balení lahvička
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
Balení sáček
Uchovávejte při teplotě do 30°C.
6.5 Druh obalu a obsah balení
Balení lahvička
Skleněná lahvička třídy I jantarové barvy s polypropylenovými uzávěry jištěnými před dětmi, obsahujícími 5 nebo 20 tvrdých tobolek.
Papírová skládačka (krabička) obsahuje jednu lahvičku.
Balení sáček
Sáčky jsou složené z lineárního nízkohustotního polyethylenu (nejvnitřnější vrstva), aluminia a polyethylen-tereftalátu.
Jeden sáček obsahuje 1 tvrdou tobolku a je dodáván v papírové krabičce.
Papírová skládačka obsahuje 5 nebo 20 tvrdých tobolek, jednotlivě zatavených v sáčcích.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Tobolky by neměly být otevřeny. Pokud dojde k poškození celistvosti tobolky, zabraňte kontaktu jejího práškového obsahu s kůží a sliznicemi. Pokud přijde Temodal do styku s kůží nebo sliznicí, je třeba neprodleně a řádně ho smýt mýdlem a vodou.
Pacientům by mělo být doporučeno uchovávat tobolky mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/98/096/001
EU/1/98/096/002
EU/1/98/096/024
EU/1/98/096/025
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 26. ledna 1999.
Datum posledního prodloužení registrace: 26. ledna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Temodal 20 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje temozolomidum 20 mg.
Pomocná látka se známým účinkem:
Jedna tvrdá tobolka obsahuje 182,2 mg bezvodé laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka (tobolka).
Tvrdé tobolky mají neprůsvitné bílé tělo, neprůsvitné žluté víčko a jsou potištěny černým inkoustem. Na víčku je vytištěn nápis „Temodal“. Na těle tobolky je vytištěn nápis „20 mg“, logo společnosti Schering-Plough a dva proužky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Temodal je určen k léčbě:
- dospělých pacientů s nově diagnostikovaným multiformním glioblastomem souběžně s radioterapií (RT) a následně jako monoterapeutická léčba.
- dětí ve věku od tří let, dospívajících a dospělých pacientů s maligním gliomem, jako je multiformní glioblastom nebo anaplastický astrocytom, u nichž se po standardní léčbě projevují recidivy nebo progrese.
4.2 Dávkování a způsob podání
Temodal smí být předepisován výhradně lékařem se zkušenostmi s onkologickou léčbou mozkových
nádorů.
Může se podávat antiemetická léčba (viz bod 4.4).
Dávkování
Dospělí _pacienti s nově diagnostikovaným multiformním glioblastomem
Temodal je podáván v kombinaci s fokální radioterapií (fáze souběžné léčby) s následnými až 6 cykly monoterapie temozolomidem (TMZ) (monoterapeutická fáze).
Fáze souběžné léčby
TMZ se podává perorálně v dávce 75 mg/m2 denně po dobu 42 dnů souběžně s fokální radioterapií (60 Gy podáno ve 30 frakcích). Snižování dávek se nedoporučuje, ale každý týden by se mělo rozhodnout o pozdržení nebo přerušení podávání TMZ podle kritérií hematologické a nehematologické toxicity. Podávání TMZ může pokračovat po celou dobu 42 dnů souběžné fáze (až do 49 dnů), jestliže jsou splněny všechny následující podmínky:
- absolutní počet neutrofilů (ANC) > 1,5 x 109/l
- počet trombocytů > 100 x 109/l
- všeobecná kritéria toxicity (CTC - common toxicity criteria) nehematologické toxicity < stupeň 1 (s výjimkou alopecie, nevolnosti a zvracení).
Během léčby by se měl každý týden vyšetřit celkový krevní obraz. Podávání TMZ by mělo být během fáze souběžné léčby dočasně přerušeno nebo trvale ukončeno podle kritérií hematologické a nehematologické toxicity, jak je uvedeno v Tabulce 1.
|
Tabulka l.Přerušení nebo ukončení podávání TMZ během souběžné léčby radioterapií a TMZ | ||
|
Toxicita |
TMZ přerušení3 |
TMZ ukončení |
|
Absolutní počet neutrofilů |
> 0,5 a <1,5 x 109/l |
< 0,5 x 109/l |
|
Počet trombocytů |
> 10 a < 100 x 109/l |
<10 x 109/l |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 2 |
CTC stupeň 3 nebo 4 |
a: Léčba se souběžně podávaným TMZ může pokračovat, jestliže jsou splněny všechny následující podmínky: absolutní počet neutrofilů > 1,5 x 109/l; počet trombocytů > 100 x 109/l; CTC nehematologická toxicita < stupeň 1 (s výjimkou alopecie, nausey, zvracení).
Monoterapeutická fáze
Čtyři týdny po ukončené souběžné fázi TMZ + RT se TMZ podává až v 6 cyklech monoterapeutické léčby. Dávka Cyklu 1 (monoterapie) je 150 mg/m2 jednou denně po dobu 5 dnů, následovaná 23 dny bez léčby. Na začátku Cyklu 2 je dávka zvýšena na 200 mg/m2, jestliže CTC nehematologická toxicita pro Cyklus 1 je stupeň < 2 (s výjimkou alopecie, nausey a zvracení), absolutní počet neutrofilů (ANC) je > 1,5 x 109/l a počet trombocytů je > 100 x 109/l. Jestliže není dávka zvýšena v Cyklu 2, nemělo by být zvýšení prováděno v následných cyklech. Je-li jednou zvýšena, zůstává dávka 200 mg/m2 na den pro prvních 5 dnů každého následujícího cyklu s výjimkou vzniku toxicity. Snížení dávky a ukončení podávání v průběhu monoterapeutické fáze by se mělo provádět podle Tabulek 2 a 3.
V průběhu léčby by měl být ve dni 22 (21 dnů po první dávce TMZ) vyšetřen celkový krevní obraz. Dávka by měla být redukována nebo podávání přípravku ukončeno podle Tabulky 3.
|
Tabulka 2. Úrovně dávky TMZ pro monoterapeutickou léčbu | ||
|
Úroveň dávky |
Dávka TMZ (mg/m2/den) |
Poznámky |
|
-1 |
100 |
Redukce kvůli předchozí toxicitě |
|
0 |
150 |
Dávka během Cyklu 1 |
|
1 |
200 |
Dávka během Cyklů 2-6 při absenci toxicity |
|
Tabulka 3. Snížení dávky nebo ukončení podávání TMZ během monoterapeutické léčby | ||
|
Toxicita |
Redukce TMZ o 1 úroveň dávkya |
Ukončení TMZ |
|
Absolutní počet neutrofilů |
<1,0 x 109/l |
Viz poznámka pod čarou b |
|
Počet trombocytů |
< 50 x 109/l |
Viz poznámka pod čarou b |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 3 |
CTC stupeň 4b |
a: Úrovně dávky TMZ jsou uvedeny v Tabulce 2. b: TMZ se ukončí jestliže:
• výsledkem úrovně dávky -1 (100 mg/m2) je stále neakceptovatelná toxicita
• jestliže se po redukci dávky znovu objeví stejný stupeň 3 nehematologické toxicity (s výjimkou alopecie, nausey, zvracení).
Léčebný cyklus zahrnuje 28 dnů. U pacientů, kteří dosud nebyli léčeni chemoterapií, se TMZ podává perorálně v dávce 200 mg/m2 jedenkrát denně po dobu prvních 5 dnů s následným přerušením léčby po dobu 23 dnů (celkem 28 dnů). U nemocných, kteří již dříve prodělali chemoterapii, činí zahajovací dávka 150 mg/m2 jedenkrát denně, která se pak ve druhém cyklu zvyšuje na 200 mg/m2 jedenkrát denně po dobu 5 dnů, pokud nejsou přítomny známky hematotoxicity (viz bod 4.4).
Zvláštní _ populace
Pediatrická populace
U dětí ve věku 3 let nebo starších by se měl TMZ používat pouze pro léčbu recidivujícího nebo progresivního maligního gliomu. Zkušenosti u těchto dětí jsou velmi omezené (viz body 4.4 a 5.1). Bezpečnost a účinnost TMZ u dětí ve věku do 3 let nebyla stanovena. Nejsou dostupné žádné údaje.
Pacienti s poruchou funkce jater či ledvin
Farmakokinetika TMZ je u pacientů s normální jaterní funkcí a s mírnou nebo středně těžkou jaterní dysfunkcí srovnatelná. Údaje o podání TMZ u pacientů s těžkou jaterní dysfunkcí (Třída C podle Childovy klasifikace) ani s dysfunkcí ledvin nejsou k dispozici. Vzhledem k uvedeným farmakokinetickým vlastnostem TMZ je však pravděpodobné, že ani u nemocných s těžkou jaterní dysfunkcí nebo dysfunkcí ledvin kteréhokoli stupně není snižování jeho dávek nutné. Při podávání TMZ těmto nemocným je však nutná zvýšená opatrnost.
Starší pacienti
Z populační analýzy farmakokinetiky u pacientů ve věku 19-78 let vyplývá, že clearance TMZ není ovlivňována věkem. Nicméně se zdá, že starší pacienti (ve věku > 70 let) mají vyšší riziko vzniku neutropenie a trombocytopenie (viz bod 4.4).
Způsob podání
Temodal tvrdé tobolky by měl podáván nalačno.
Tobolky se musejí polykat vcelku a zapíjet sklenicí vody a nesmějí se otevírat nebo kousat.
Pokud se po podání dávky dostaví zvracení, neměla by být další dávka podána tentýž den.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Hypersenzitivita na dakarbazin (DTIC).
Těžká myelosuprese (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Oportunní infekce a reaktivace infekcí
Během léčby pomocí TMZ byly pozorovány oportunní infekce (jako je pneumonie způsobená Pneumocystis jirovecii) a reaktivace infekcí (jako jsou HBV, CMV) (viz bod 4.8).
Pneumonie způsobená Pneumocystis _ jirovecii
U pacientů, kteří dostávali souběžně TMZ a RT v pilotní studii s prodlouženým harmonogramem 42 dnů, bylo zjištěno zvýšené riziko vzniku pneumonie způsobené Pneumocystis jirovecii (PCP). Proto je nutná profylaxe PCP u všech pacientů, kteří dostávají souběžně TMZ a RT po dobu 42 denního režimu (maximálně 49 dnů) bez ohledu na počet lymfocytů. Jestliže se objeví lymfopenie, je třeba pokračovat v profylaxi, dokud se neobnoví stupeň lymfopenie < 1.
Výskyt PCP může být vyšší, když je TMZ podáván během delšího dávkovacího režimu. Nicméně všichni pacienti, kteří dostávají, TMZ zvláště pacienti dostávající steroidy, by měli být pečlivě sledováni pro vznik PCP, bez ohledu na režim. U pacientů užívajících TMZ, zvláště v kombinaci s dexamethasonem nebo jinými steroidy, byly hlášeny případy fatálního respiračního selhání.
HBV
Byla hlášena hepatitida způsobená reaktivací viru hepatitidy B (HBV), v některých případech vedoucí ke smrti. Před zahájením léčby pacientů s pozitivní sérologií na hepatitidu B (včetně těch s aktivním onemocněním) je nezbytná konzultace s odborníky na onemocnění jater. Během léčby musí být pacienti náležitě monitorováni a vedeni.
Hepatotoxicita
U pacientů léčených TMZ bylo hlášeno poškození jater, včetně smrtelného selhání jater (viz bod 4.8). Před zahájením léčby je nutno provést testy jaterních funkcí. Pokud jsou výsledky abnormální, musí lékař před zahájením léčby temozolomidem vyhodnotit poměr přínosů a rizik, včetně potenciálu ke smrtelnému selhání jater. U pacientů se 42denním léčebným cyklem se musí testy jaterních funkcí opakovat uprostřed tohoto cyklu. Pro všechny pacienty platí, že jaterní funkce je nutno zkontrolovat po každém léčebném cyklu. U pacientů s výraznými abnormalitami jaterních funkcí musí lékař vyhodnotit poměr přínosů a rizik vyplývajících z pokračování v léčbě. Jaterní toxicita se může objevit několik nebo i více týdnů po poslední léčbě temozolomidem.
Malignity
Byly též velmi vzácně hlášeny případy myelodysplastického syndromu a sekundárních malignit, včetně myeloidní leukémie (viz bod 4.8).
Antiemetická léčba:
Nausea a zvracení jsou velmi často spojené s TMZ.
Před nebo po podání TMZ se může podávat antiemetická léčba.
Dospělí _pacienti s nově diagnostikovaným multiformním glioblastomem
Antiemetická profylaxe se doporučuje před počáteční dávkou souběžné fáze a velmi doporučuje během monoterapeutické fáze.
Antiemetika může být třeba podat pacientům, u nichž v předchozích léčebných cyklech došlo k těžšímu zvracení (stupeň 3 nebo 4).
Laboratorní parametry
U pacientů léčených TMZ může dojít k myelosupresi, včetně dlouhotrvající pancytopenie, která může vést k aplastické anémii, jež v některých případech skončila fatálně. V některých případech souběžná expozice léčivým přípravkům spojených s výskytem aplastické anémie, zahrnující karbamazepin, fenytoin a sulfamethoxazol/trimethoprim, hodnocení komplikuje. Před zahájením podávání léku by měly být splněny následující laboratorní parametry: ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Kompletní krevní obraz je třeba vyšetřit ve 22. dni (21 dnů po podání 1. dávky) nebo v průběhu 48 hodin po tomto dni, a pak v týdenních intervalech do té doby, dokud není ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Jestliže v průběhu kteréhokoli cyklu poklesne ANC na < 1,0 x 109/l nebo počet trombocytů na < 50 x 109/l, měla by být v následném cyklu dávka o jeden stupeň snížena (viz bod 4.2). Jednotlivé stupně dávky činí 100 mg/m2, 150 mg/m2 a 200 mg/m2. Nejnižší doporučená dávka je 100 mg/m2.
Pediatrická populace
S podáváním TMZ dětem ve věku nižším než 3 roky nejsou dosud žádné klinické zkušenosti. U starších dětí a dospívajících jsou zkušenosti velmi omezené (viz body 4.2 a 5.1).
Starší pacienti (ve věku >70 let)
U starších pacientů je ve srovnání s mladšími osobami pravděpodobně vyšší riziko neutropenie a trombocytopenie. Vzhledem k tomu je při podávání TMZ ve vyšším věku nutná zvýšená opatrnost.
Pacienti mužského pohlaví
Muži léčení TMZ by měli být upozorněni, aby se do 6 měsíců od užití poslední dávky nestali otci a aby se před zahájením léčby informovali o možnosti kryokonzervace spermatu (viz bod 4.6).
Laktóza
Tento léčivý přípravek obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo glukózo-galaktózovou malabsorpcí by proto neměli tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Podávání TMZ spolu s ranitidinem nemělo v oddělené studii fáze I za následek žádné změny rozsahu absorpce temozolomidu nebo expozice jeho aktivnímu metabolitu monomethyl- triazenoimidazol-karboxamidu (MTIC).
Podávání TMZ spolu s jídlem vedlo k poklesu Cmax o 33 % a zmenšení plochy pod křivkou koncentrace (AUC) o 9 %.
Protože nelze vyloučit, že změna Cmax může být klinicky významná, neměl by se Temodal podávat současně s jídlem.
Výsledky analýzy populační farmakokinetiky ve II. fázi klinických studií prokazují, že nebyla clearance TMZ ovlivněna současným podáním dexamethasonu, prochlorperazinu, fenytoinu, karbamazepinu, ondansetronu, antagonistů H2 receptorů nebo fenobarbitalu. Současné podávání kyseliny valproové bylo spojeno s malým, nicméně statisticky významným poklesem clearance TMZ.
Dosud nebyly provedeny žádné studie, které by hodnotily účinek TMZ na metabolismus nebo eliminaci jiných léků. Nicméně vzhledem k tomu, že TMZ neprochází přeměnou v játrech a na plazmatické bílkoviny se váže jen v malé míře, je málo pravděpodobné, že by ovlivňoval farmakokinetiku jiných léčivých přípravků (viz bod 5.2).
V kombinaci s jinými myelosupresivními látkami může TMZ zvýšit pravděpodobnost myelosuprese. Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání léku těhotným ženám nejsou k dispozici. V preklinických studiích u potkanů a králíků, kteří TMZ dostávali v dávce 150 mg/m2, byly prokázány teratogenní a/nebo toxické účinky na plod (viz bod 5.3). Temodal by neměl být podáván těhotným ženám. Pokud je přece jen nutno zvážit použití během těhotenství, je třeba pacientku seznámit s potenciálním rizikem pro plod.
Kojení
Není známo, zda se TMZ vylučuje do mateřského mléka; kojení by se proto mělo po dobu léčby TMZ přerušit.
Ženy ve fertilním věku.
Ženám ve fertilním věku je třeba doporučit používání účinné antikoncepce k zabránění otěhotnění v průběhu užívání TMZ.
Mužská plodnost
TMZ může mít genotoxické účinky. Proto se mužům léčeným temozolomidem nedoporučuje, aby se do 6 měsíců od užití poslední dávky stali otci a naopak se doporučuje, vzhledem k možnosti ireverzibilní neplodnosti po léčbě TMZ, ještě před jejím zahájením konzultovat s lékařem možnost kryokonzervace předem odebraných spermií.
4.7 Účinky na schopnost řídit a obsluhovat stroje
TMZ má malý vliv na schopnost řídit nebo obsluhovat stroje v důsledku únavy a ospalosti (viz bod 4.8.).
4.8 Nežádoucí účinky
Zkušenosti z klinických hodnocení
U pacientů léčených TMZ ať už užívaným v kombinaci s RT nebo ve formě monoterapie následující po RT v případě nově diagnostikovaného multiformního glioblastomu nebo ve formě monoterapie pacientů s recidivujícím nebo progresivním gliomem, byly velmi často hlášené nežádoucí účinky podobné: nausea, zvracení, zácpa, anorexie, bolest hlavy a únava. Křeče byly hlášeny velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených monoterapií, vyrážka byla hlášena velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených TMZ v kombinaci s RT nebo také ve formě monoterapie a často u pacientů s recidivujícím gliomem. Většina hematologických nežádoucích účinků byla hlášena u obou indikací často nebo velmi často (Tabulky 4 a 5); četnost laboratorních nálezů stupně 3 a 4 je uvedena za každou tabulkou.
V tabulkách jsou nežádoucí účinky klasifikovány podle orgánových systémů a četnosti. Zařazení do skupin četností se provádí podle následujícího pravidla: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Nově diagnostikovaný multiformní glioblastom
Tabulka 4 ukazuje nežádoucí účinky, které se objevily při léčbě u pacientů s nově diagnostikovaným multiformním glioblastomem během souběžné a monoterapeutické fáze léčby.
|
Tabulka 4: Nežádoucí účinky, které se objevily během souběžné a monoterapeutické léčby u pacientů s nově diagnostikovaným multiformním glioblastomem | ||
|
Třída orgánového systému |
TMZ + souběžná RT n=288* |
TMZ monoterapie n=224 |
|
Infekce a infestace | ||
|
Časté: |
Infekce, Herpes simplex, infekce rány, faryngitida, orální kandidóza |
Infekce, orální kandidóza |
|
Méně časté: |
Herpes simplex, herpes zoster, symptomy podobné chřipce | |
|
Poruchy krve a lymfatického systému | ||
|
Časté: |
Neutropenie, trombocytopenie, lymfopenie, leukopenie |
Febrilní neutropenie, trombocytopenie, anémie, leukopenie |
|
Méně časté: |
Febrilní neutropenie, anémie |
Lymfopenie, petechie |
|
Endokrinní poruchy | ||
|
Méně časté: |
Cushingoid |
Cushingoid |
|
Poruchy metabolismu a výživy | ||
|
Velmi časté: | ||
|
Časté: |
Hyperglykémie, pokles hmotnosti |
Pokles hmotnosti |
|
Méně časté: |
Hypokalémie, zvýšení alkalické fosfatázy, zvýšení hmotnosti |
Hyperglykémie, zvýšení hmotnosti |
|
Psychiatrické poruchy | ||
|
Časté: |
Úzkost, emoční labilita, nespavost | |
|
Méně časté: |
Neklid, apatie, poruchy chování, deprese, halucinace |
Halucinace, amnézie |
|
Poruchy nervového systému | ||
|
Velmi časté: |
Konvulze, bolest hlavy | |
|
Časté: |
Konvulze, porucha vědomí, somnolence, afázie, porucha rovnováhy, závrať, zmatenost, porucha paměti, porucha koncentrace, neuropatie, parestézie, porucha řeči, třes |
Hemiparéza, afázie, porucha rovnováhy, somnolence, zmatenost, závrať, porucha paměti, porucha koncentrace, dysfázie, neurologická porucha (NOS), neuropatie, periferní neuropatie, parestézie, porucha řeči, třes |
|
Méně časté: |
Status epilepticus, extrapyramidální porucha, hemiparéza, ataxie, poruchy kognice, dysfázie, abnormální chůze, hyperestézie, hypestézie, neurologická porucha (NOS), periferní neuropatie |
Hemiplegie, ataxie, abnormální koordinace, abnormální chůze, hyperestézie, smyslová porucha |
|
Poruchy oka | ||
|
Časté: |
Rozmazané vidění |
Defekt zorného pole, rozmazané vidění, diplopie |
|
Méně časté: |
Hemianopie, zmenšená ostrost vidění, poruchy zraku, defekt zorného pole, bolest očí |
Zmenšená ostrost vidění, bolest očí, suché oči |
|
Poruchy ucha a labyrintu | ||
|
Časté: |
Porucha sluchu |
Porucha sluchu, tinitus |
|
Méně časté: |
Otitis media, tinitus, hyperakuze, bolest ucha |
Hluchota, závrať, bolest ucha |
|
Srdeční poruchy | ||
|
Méně časté: |
Palpitace | |
|
Cévní poruchy | ||
|
Časté: |
Hemoragie, edém, edém nohy |
Hemoragie, hluboká žilní trombóza, edém nohy |
|
Méně časté: |
Cerebrální hemoragie, hypertenze |
Plicní embolie, edém, periferní edém |
|
Respirační, hrudní a mediastinální poruchy | ||
|
Časté: | ||
|
Méně časté: |
Pneumonie, infekce horních cest dýchacích, nosní kongesce |
Pneumonie, sinusitida, infekce horních cest dýchacích, bronchitida |
|
Gastrointestinální poruchy | ||
|
Velmi časté: |
Zácpa, nausea, zvracení |
Zácpa, nausea, zvracení |
|
Časté: |
Stomatitida, průjem, bolest břicha, dyspepsie, dysfagie | |
|
Méně časté: |
Abdominální distenze, fekální inkontinence, gastrointestinální porucha (NOS), gastroenteritida, hemeroidy | |
|
Poruchy kůže a podkožní tkáně | ||
|
Velmi časté: |
Vyrážka, alopecie |
Vyrážka, alopecie |
|
Časté: |
Dermatitida, suchá kůže, erytém, svědění |
Suchá kůže, svědění |
|
Méně časté: |
Exfoliace kůže, fotosenzitivní reakce, abnormální pigmentace |
Erytém, abnormální pigmentace, zvýšené pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Časté: |
Svalová slabost, artralgie |
Svalová slabost, artralgie, muskuloskeletální bolest, myalgie |
|
Méně časté: |
Myopatie, bolest v zádech, muskuloskeletální bolest, myalgie |
Myopatie, bolest v zádech |
|
Poruchy ledvin a močových cest | ||
|
Časté: |
Časté močení, močová inkontinence |
Močová inkontinence |
|
Méně časté: | ||
|
Poruchy reprodukčního systému a prsu | ||
|
Méně časté: |
Impotence |
Vaginální hemoragie, menoragie, amenorea, vaginitida, bolest prsou |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Velmi časté: |
Únava |
Únava |
|
Časté: |
Alergická reakce, horečka, radiační poškození, edém obličeje, bolest, změna chuti |
Alergická reakce, horečka, radiační poškození, bolest, změna chuti |
|
Méně časté: |
Astenie, zrudnutí, návaly horka, zhoršení stavu, rigory, změna barvy jazyka, parosmie, žízeň |
Astenie, edém obličeje, bolest, zhoršení stavu, rigory, zubní potíže |
|
Vyšetření | ||
|
Časté: |
Zvýšené ALT |
Zvýšené ALT |
|
Méně časté: |
Zvýšené jaterní enzymy, zvýšené gama GT, zvýšené AST | |
*Pacient který byl randomizován pouze do větve RT, obdržel Temodal + RT.
Laboratorní výsledky
Byla zjištěna myelosuprese (neutropenie a trombocytopenie), která je známou toxicitou limitující dávku většiny cytotoxických látek včetně TMZ.. Když byly zkombinovány abnormality laboratorních testů a nežádoucí účinky v souběžných a monoterapeutických fázích léčby, byly pozorovány u 8 % pacientů abnormality neutrofilů stupeň 3 nebo stupeň 4, včetně neutropenických případů. Stupeň 3 nebo stupeň 4 abnormalit trombocytů, včetně trombocytopenických případů, byl pozorován u 14 % pacientů, kteří dostávali TMZ.
Recidivující nebo progresivní maligní gliom
Nejčastěji se vyskytujícími nežádoucími účinky ve vztahu k léčbě byly v provedených klinických studiích gastrointestinální poruchy, především nausea (43 %) a zvracení (36 %). Tyto reakce dosahovaly obvykle pouze stupně 1 nebo 2 (0 - 5 epizod zvracení během 24 hodin) a buď spontánně odezněly, nebo byly snadno léčitelné standardní antiemetickou léčbou. Incidence těžké nausey a zvracení činila 4 %.
Tabulka 5 obsahuje nežádoucí účinky, které byly hlášeny v průběhu klinických studií pro recidivující nebo progresivní maligní gliom a po uvedení přípravku Temodal na trh.
_Tabulka 5. Nežádoucí účinky u pacientů s recidivujícím nebo progresivním maligním gliomem
Infekce a infestace
|
Vzácné: |
Oportunní infekce včetně pneumonie způsobené PCP |
|
Poruchy krve a lymfatického systému | |
|
Velmi časté: |
Neutropenie nebo lymfopenie (stupeň 3-4) trombocytopenie (stupeň 3-4) |
|
Méně časté: |
Pancytopenie, anémie (stupeň 3-4), leukopenie |
|
Poruchy metabolismu a výživy | |
|
Velmi časté: | |
|
Časté: |
Pokles hmotnosti |
|
Poruchy nervového systému | |
|
Velmi časté: | |
|
Časté: |
Somnolence, závratě, parestézie |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté: | |
|
Gastrointestinální poruchy | |
|
Velmi časté: |
Zvracení, nausea, zácpa |
|
Časté: | |
|
Poruchy kůže a podkožní tkáně | |
|
Časté: |
Vyrážka, pruritus, alopecie, |
|
Velmi vzácné: |
Multiformní erytém, erytrodermie, urtikarie, exantém |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté: |
Únava |
|
Časté: | |
|
Velmi vzácné: |
Alergické reakce, včetně anafylaxe, angioedém |
Laboratorní výsledky
Trombocytopenie a neutropenie stupně 3 nebo 4 byly zaznamenány u 19 %, resp. 17 % pacientů léčených pro maligní gliom. K hospitalizaci a/nebo přerušení léčby TMZ vedly v 8 %, resp. 4 % případů. Myelosuprese byla předvídatelná (obvykle k ní došlo během několika prvních cyklů, s nadirem mezi 21. dnem a 28. dnem) a úprava k normě byla rychlá, obvykle v průběhu 1-2 týdnů. Kumulace myelosupresivních účinků prokázána nebyla. Přítomnost trombocytopenie může zvýšit riziko krvácení a přítomnost neutropenie nebo leukopenie může zvýšit riziko infekce.
Pohlaví
Do analýzy populační farmakokinetiky založené na zkušenostech z klinického hodnocení bylo zahrnuto 101 žen a 169 mužů, u kterých byla dostupná měření nadiru neutrofilů, a 110 žen a 174 mužů, u kterých byla dostupná měření nadiru krevních destiček. U žen bylo ve srovnání s muži v prvním cyklu terapie zaznamenáno více neutropenií stupně 4 (ANC< 0,5 x 109/l), 12 % vs 5 %, a trombocytopenií (< 20 x 109/l), 9 % vs 3 %. V souboru dat od 400 jedinců s rekurentním gliomem se v prvním cyklu terapie objevila neutropenie stupně 4 u 8 % žen vs 4 % mužů a trombocytopenie stupně 4 u 8 % žen vs 3 % mužů. Ve studii s 288 jedinci s nově diagnostikovaným multiformním glioblastomem se v prvním cyklu terapie vyskytla neutropenie stupně 4 u 3 % žen vs 0 % mužů a trombocytopenie stupně 4 u 1 % žen vs 0 % mužů.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s rekurentním gliomem mozkového kmene nebo rekurentním astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Ačkoliv jsou údaje omezené, tolerance u dětí se očekává být podobná jako u dospělých. Bezpečnost TMZ u dětí ve věku do 3 let nebyla stanovena.
Zkušenosti po uvedení na trh
Po uvedení přípravku na trh byly hlášeny další následující závažné nežádoucí účinky:
|
Tabulka 6. Souhrn příhod hlášených u temozolomidu po jeho uvedení na trh | |
|
Infekce a infestace* | |
|
Méně časté: |
infekce způsobená cytomegaloviry, reaktivace infekce způsobené virem, jako je cytomegalovirus, virus hepatitidy BŤ |
|
Poruchy krve a lymfatického systému | |
|
Velmi vzácné: |
dlouhotrvající pancytopenie, aplastická anémieŤ |
|
Novotvary benigní, maligní a blíže neurčené | |
|
Velmi vzácné: |
myelodysplastický syndrom (MDS), sekundární malignity, včetně myeloidní leukémie |
|
Endokrinní poruchy | |
|
Méně časté: |
diabetes insipidus |
|
Respirační, hrudní a mediastinální poruchy | |
|
Velmi vzácné: |
intersticiální pneumonitida/pneumonitida, plicní fibróza, respirační selhání1 |
|
Poruchy jater a žlučových cest* | |
|
Časté: |
zvýšení jaterních enzymů |
|
Méně časté: |
hyperbilirubinemie, cholestáza, hepatitida, poškození jater, selhání jater1 |
|
Poruchy kůže a podkožní tkáně | |
|
Velmi vzácné: |
toxická epidermální nekrolýza, Stevens-Johnsonův syndrom |
t Včetně smrtelných případů
Frekvence odhadované na základě relevantních klinických studií.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinicky byly u pacientů ověřeny dávky 500, 750, 1 000, a 1 250 mg/m2 (celková dávka za 5-denní cyklus). Toxicita limitující dávku byla toxicita hematologická a byla pozorována u všech dávek, ale lze očekávat, že při vyšších dávkách bude závažnější. U jednoho pacienta došlo k předávkování, když užil dávku 10 000 mg (celková dávka během jednoho 5-denního cyklu), nežádoucí účinky hlášené v tomto případě byly pancytopenie, pyrexie, multiorgánové selhání a smrt. Byly popsány případy, kdy pacienti doporučenou dávku užívali po dobu delší než 5 dnů (až 64 dnů) s hlášenými nežádoucími příhodami včetně suprese kostní dřeně s nebo bez rozvoje infekce, v některých případech závažné, dlouhotrvající a vedoucí ke smrti. V případě předávkování je nezbytné hematologické zhodnocení. Podle potřeby by měla být zajištěna podpůrná léčba.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: protinádorové léky - ostatní alkylační látky, ATC kód: L01A X03 Mechanismus účinku
Temozolomid je triazen, který za fyziologické hodnoty pH prochází rychlou chemickou konverzí na aktivní monometyl-triazenoimidazol-karboxamid (MTIC). Předpokládá se, že cytotoxické účinky MTIC jsou primárně důsledkem alkylace na O6 pozici guaninu, s následnou alkylací rovněž na pozici N7. Následně vznikající cytotoxické léze jsou vysvětlovány nejspíše aberantní reparací methylovaného zbytku.
Klinická účinnost a bezpečnost
Nově diagnostikovaný multiformní glioblastom
Celkem 573 pacientů bylo randomizováno do skupin dostávajících buď TMZ + RT (n=287) nebo RT samotnou (n=286). Pacienti ve větvi TMZ + RT dostávali souběžně TMZ (75 mg/m2) jednou denně od prvního až do posledního dne RT po dobu 42 dnů (maximálně 49 dnů). Pak následovala monoterapie TMZ (150 - 200 mg/m2) ve dnech 1-5 každého 28-denního cyklu, až v 6 cyklech, začínající 4 týdny po ukončení RT. Pacienti v kontrolní větvi dostávali pouze RT. Během RT a souběžné TMZ terapie byla nutná profylaxe pneumonie Pneumocystis jirovecii (PCP).
TMZ byl podáván jako záchranná terapie vnásledné fázi 161 pacientům z 282 (57 %) ve větvi dostávající pouze RT a 62 pacientům z 277 (22 %) ve větvi dostávající TMZ + RT.
Poměr rizika (HR) pro celkové přežívání byl 1,59 (95 % CI pro HR=1,33 - 1,91) s log-rank p < 0,0001 ve prospěch TMZ větve. Odhadnutá pravděpodobnost přežívání 2 roky nebo více (26 % vs 10 %) je vyšší ve větvi RT + TMZ. Přidání souběžně podávaného TMZ k RT následované monoterapií TMZ v léčbě pacientů s nově diagnostikovaným multiformním glioblastomem prokázalo statisticky významné zlepšení v celkovém přežívání (overall survival=OS) ve srovnání s RT samotnou (Graf 1).
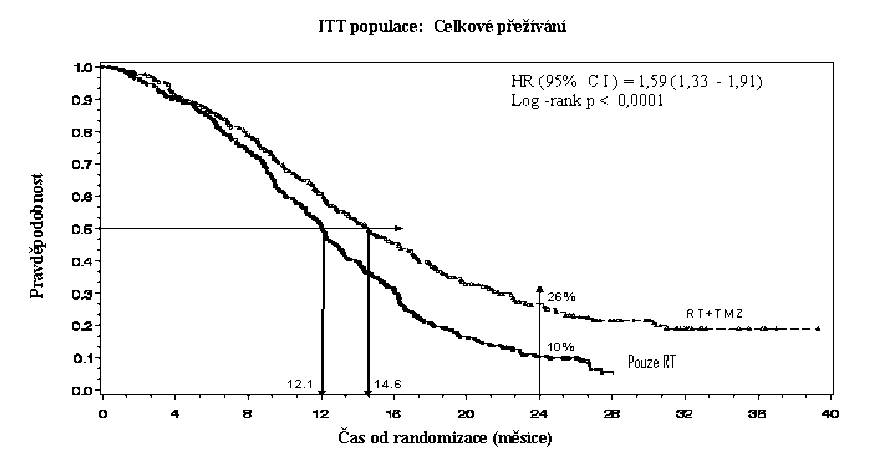
Graf 1 Kaplanovy-Meierovy křivky pro celkové přežívání (ITT populace=intent-to- treat population)
Výsledky studie nebyly konzistentní v podskupině pacientů se slabým výkonnostním statusem (WHO PS=2, n=70), kde celkové přežívání a čas do progrese byly podobné v obou větvích. Zdá se však, že u této skupiny pacientů nejsou přítomna žádná nepřijatelná rizika.
Recidivující nebo _progresivní maligní gliom
Data o klinické účinnosti u pacientů s multiformním glioblastomem (Karnofského výkonnostní status [KPS] > 70), u něhož dochází k progresi nebo recidivě po chirurgickém výkonu a RT, vycházela ze dvou klinických studií s perorálním TMZ. V prvním případě šlo o nekomparativní studii u 138 pacientů (29 % bylo předtím léčeno chemoterapií), ve druhém o randomizovanou studii kontrolovanou léčivou látkou s TMZ vs prokarbazinem u celkem 225 pacientů (67 % z nich bylo předtím léčeno chemoterapií na bázi nitrosourey). V obou studiích bylo primárním hodnotícím kritériem přežívání bez známek progrese (PFS - progression-free survival), hodnocených na základě zobrazení magnetickou rezonancí (MRI) nebo na základě zhoršení neurologických funkcí. V nekomparativní studii bylo u 19 % pacientů dosaženo 6měsíční PFS, medián přežívání bez známek progrese byl 2,1 měsíce a medián celkové doby přežívání byl 5,4 měsíce. Objektivní počet terapeutických odpovědí na základě hodnocení MRI dosahoval 8 %.
V randomizované studii kontrolované léčivou látkou bylo šestiměsíční PFS u pacientů léčených TMZ významně vyšší než u těch, kteří dostávali prokarbazin (21 % vs 8 %; chí-kvadrát p=0,008)
s mediánem 2,89, respektive 1,88 měsíce (log rank p=0,0063). Medián přežití byl 7,34 měsíce pro TMZ a 5,66 měsíce pro prokarbazin (log rank p=0,33). Po 6 měsících byl podíl přežívajících pacientů v TMZ skupině významně vyšší (60 %) než ve skupině prokarbazinové (44 %; chí-kvadrát p=0,019). U pacientů, kteří byli předtím léčeni chemoterapií, bylo dosaženo kladného efektu u těch, kteří měli KPS > 80.
Údaje o době do zhoršení neurologického statusu, jakož i o době do zhoršení výkonnostního statusu (pokles na KPS < 70 nebo pokles nejméně o 30 bodů), hovoří ve prospěch TMZ oproti prokarbazinu. Medián doby do progrese v těchto hodnotících kritériích byl u TMZ o 0,7 - 2,1 měsíce delší než u prokarbazinu (log rank p < 0,01 - 0,03).
Recidivující anaplastický astrocytom
V multicentrické prospektivní studii fáze II, v níž byly hodnoceny bezpečnost a účinnost perorálně podávaného TMZ v léčbě pacientů s anaplastickým astrocytomem při prvním relapsu, bylo zjištěno šestiměsíční PSF u 46 % léčených. Medián PSF činil 5,4 měsíce. Medián celkového přežití činil 14,6 měsíce. Procento odpovědí na léčbu, hodnocené centrálně, činilo 35 % (13 % kompletních, 43 % částečných odpovědí) pro populaci intent-to-treat (ITT), n=162. U 43 pacientů došlo ke stabilizaci onemocnění. Šestiměsíční přežití bez klinické příhody bylo pro populaci intent-to-treat 44 %, přičemž medián přežití bez klinické příhody činil 4,6 měsíce, což je podobné výsledkům hodnocení přežívání bez známek progrese. Z histologického hlediska byla účinnost obdobná. Objektivní radiologicky prokázaná odpověď na léčbu nebo zachování stavu beze známek progrese velmi dobře korelovaly se zachovanou nebo zlepšenou kvalitou života.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s recidivujícím gliomem mozkového kmene nebo recidivujícím astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Tolerance TMZ byla podobná jako u dospělých.
5.2 Farmakokinetické vlastnosti
TMZ se spontánně hydrolyzuje při fyziologickém pH primárně na aktivní látku, 3-metyl-(triazen-1-yl)imidazol-4-karboxamid (MTIC). MTIC se spontánně hydrolyzuje na 5-amino-imidazol-4-karboxamid (AIC), známý meziprodukt biosyntézy purinů a nukleových kyselin a na metylhydrazin, o kterém se předpokládá, že je aktivní alkylační látkou. Předpokládá se, že cytotoxicita MTIC je primárně způsobena alkylací DNA zejména na pozicích guaninu O6 a N7' Expozice MTIC a AIC je ~ 2,4 %, respektive 23 % v poměru k AUC TMZ. In vivo byl ti/2 MTIC podobný jako pro TMZ,
1,8 hodin.
Absorpce
Po perorálním podání dospělým pacientům se TMZ rychle absorbuje a vrcholné koncentrace dosahuje již za 20 minut po podání (střední doba je mezi 0,5 a 1,5 hodiny). Po perorálním podání 14C-značeného TMZ byla průměrná fekální exkrece 14C v průběhu 7 dnů po podání dávky 0,8 %, což svědčilo o úplné absorpci.
Distribuce v organismu
TMZ vykazuje nízkou vazebnou afinitu k proteinům (10 % až 20 %) proto se neočekává, že by interagoval s látkami s vysokou vazebnou afinitou k proteinům.
PET studie u lidí a preklinické údaje ukazují, že TMZ rychle prostupuje hematoencefalickou bariérou a je přítomný v mozkomíšním moku. Průnik do mozkomíšního moku (CSF) byl potvrzen u jednoho pacienta; expozice CSF na základě AUC TMZ byla přibližně 30 % plazmatické expozice, což odpovídá údajům u zvířat.
Eliminace z organismu
Plazmatický poločas (t1/2) je přibližně 1,8 hodiny. Hlavní způsob eliminace 14C je ledvinami. Po perorálním podání se objevuje přibližně 5 až 10 % dávky v nezměněné podobě v moči v průběhu 24 hodin a zbytek se vylučuje ve formě temozolomidové kyseliny, 5-aminoimidazol-4-karboxamidu (AIC) nebo neidentifikovaných polárních metabolitů.
Plazmatické koncentrace vzrůstají v závislosti na velikosti dávky. Plazmatická clearance, distribuční objem a poločas jsou na velikosti dávky nezávislé.
Zvláštní populace
V analýze populační farmakokinetiky TMZ bylo zjištěno, že plazmatická clearance TMZ nezávisí na věku, renální funkci nebo kouření. V oddělené farmakokinetické studii byly plazmatické farmakokinetické profily u pacientů s mírnou až střední jaterní dysfunkcí stejné jako profily u pacientů s normální funkcí jater.
U dětských pacientů byla AUC větší než u dospělých; nicméně maximální tolerovaná dávka (MTD) činila 1 000 mg/m2 na jeden cyklus jak u dětí, tak u dospělých.
5.3 Předklinické údaje vztahující se k bezpečnosti
U potkanů a psů byly prováděny studie toxicity s jednotlivým cyklem (5 dnů podávání léku, 23 dnů bez léčby) 3 a 6 cyklů léčby. Primární cíle toxických účinků tvořila kostní dřeň, lymforetikulární systém, testes, gastrointestinální ústrojí a při vyšších dávkách, které byly pro 60 % - 100 % testovaných potkanů a psů letální, docházelo k degeneraci retiny. Většina toxických účinků byla reverzibilní, s výjimkou nežádoucích účinků na mužský reprodukční systém a degeneraci retiny. Nicméně vzhledem k tomu, že dávky podmiňující retinální degeneraci se pohybovaly v rozmezí dávek letálních, a v klinických studiích nebyl žádný srovnatelný účinek pozorován, nepřikládá se tomuto nálezu klinická významnost.
TMZ je embryotoxická, teratogenní a genotoxická alkylační látka. TMZ má toxické účinky větší u potkanů a psů než u člověka, a klinická dávka se blíží minimální letální dávce u potkanů a psů. Za citlivé ukazatele toxicity se považují poklesy počtu leukocytů a trombocytů v závislosti na velikosti dávky. Ve studii u potkanů, v níž bylo aplikováno šest cyklů terapie, bylo zjištěno několik druhů
novotvarů, včetně karcinomu mléčné žlázy, kožního keratoakanthomu a adenomu bazálních buněk, zatímco ve studiích u psů žádné nádorové ani prenádorové změny pozorovány nebyly. Zdá se, že potkani jsou vůči onkogenním účinkům TMZ mimořádně citliví, neboť první tumory se u nich objevují již po třech měsících od zahájení podávání léku. Jde o dobu krátkou dokonce i pro alkylační látky.
Výsledky aberačních testů s chromosomy Ames/salmonella a lymfocytů z lidské periferní krve prokázaly pozitivní mutagenní reakci.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky: laktóza,
koloidní bezvodý oxid křemičitý, sodná sůl karboxymethylškrobu typu A, kyselina vinná, kyselina stearová.
Obal tobolky: želatina,
oxid titaničitý (E171), natrium-lauryl-sulfát, žlutý oxid železitý (E 172),
Potisk:
šelak,
propylenglykol, čištěná voda, roztok amoniaku, hydroxid draselný, černý oxid železitý (E 172).
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Balení lahvička
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
Balení sáček
Uchovávejte při teplotě do 30°C.
6.5 Druh obalu a obsah balení
Balení lahvička
Skleněná lahvička třídy I jantarové barvy s polypropylenovými uzávěry jištěnými před dětmi, obsahujícími 5 nebo 20 tvrdých tobolek.
Papírová skládačka (krabička) obsahuje jednu lahvičku.
Balení sáček
Sáčky jsou složené z lineárního nízkohustotního polyethylenu (nejvnitřnější vrstva), aluminia a polyethylen-tereftalátu.
Jeden sáček obsahuje 1 tvrdou tobolku a je dodáván v papírové krabičce.
Papírová skládačka obsahuje 5 nebo 20 tvrdých tobolek, jednotlivě zatavených v sáčcích.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Tobolky by neměly být otevřeny. Pokud dojde k poškození celistvosti tobolky, zabraňte kontaktu jejího práškového obsahu s kůží a sliznicemi. Pokud přijde Temodal do styku s kůží nebo sliznicí, je třeba neprodleně a řádně ho smýt mýdlem a vodou.
Pacientům by mělo být doporučeno uchovávat tobolky mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/98/096/003
EU/1/98/096/004
EU/1/98/096/013
EU/1/98/096/014
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 26. ledna 1999.
Datum posledního prodloužení registrace: 26. ledna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Temodal 100 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje temozolomidum 100 mg.
Pomocná látka se známým účinkem:
Jedna tvrdá tobolka obsahuje 175,7 mg bezvodé laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka (tobolka).
Tvrdé tobolky mají neprůsvitné bílé tělo, neprůsvitné růžové víčko a jsou potištěny černým inkoustem. Na víčku je vytištěn nápis „Temodal“. Na těle tobolky je vytištěn nápis „100 mg“, logo společnosti Schering-Plough a dva proužky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Temodal je určen k léčbě:
- dospělých pacientů s nově diagnostikovaným multiformním glioblastomem souběžně s radioterapií (RT) a následně jako monoterapeutická léčba.
- dětí ve věku od tří let, dospívajících a dospělých pacientů s maligním gliomem, jako je multiformní glioblastom nebo anaplastický astrocytom, u nichž se po standardní léčbě projevují recidivy nebo progrese.
4.2 Dávkování a způsob podání
Temodal smí být předepisován výhradně lékařem se zkušenostmi s onkologickou léčbou mozkových
nádorů.
Může se podávat antiemetická léčba (viz bod 4.4).
Dávkování
Dospělí _pacienti s nově diagnostikovaným multiformním glioblastomem
Temodal je podáván v kombinaci s fokální radioterapií (fáze souběžné léčby) s následnými až 6 cykly monoterapie temozolomidem (TMZ) (monoterapeutická fáze).
Fáze souběžné léčby
TMZ se podává perorálně v dávce 75 mg/m2 denně po dobu 42 dnů souběžně s fokální radioterapií (60 Gy podáno ve 30 frakcích). Snižování dávek se nedoporučuje, ale každý týden by se mělo rozhodnout o pozdržení nebo přerušení podávání TMZ podle kritérií hematologické a nehematologické toxicity. Podávání TMZ může pokračovat po celou dobu 42 dnů souběžné fáze (až do 49 dnů), jestliže jsou splněny všechny následující podmínky:
- absolutní počet neutrofilů (ANC) > 1,5 x 109/l
- počet trombocytů > 100 x 109/l
- všeobecná kritéria toxicity (CTC - common toxicity criteria) nehematologické toxicity < stupeň 1 (s výjimkou alopecie, nevolnosti a zvracení).
Během léčby by se měl každý týden vyšetřit celkový krevní obraz. Podávání TMZ by mělo být během fáze souběžné léčby dočasně přerušeno nebo trvale ukončeno podle kritérií hematologické a nehematologické toxicity, jak je uvedeno v Tabulce 1.
|
Tabulka 1. Přerušení nebo ukončení podávání TMZ během souběžné léčby radioterapií a TMZ | ||
|
Toxicita |
TMZ přerušení3 |
TMZ ukončení |
|
Absolutní počet neutrofilů |
> 0,5 a <1,5 x 109/l |
< 0,5 x 109/l |
|
Počet trombocytů |
> 10 a < 100 x 109/l |
<10 x 109/l |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 2 |
CTC stupeň 3 nebo 4 |
a: Léčba se souběžně podávaným TMZ může pokračovat, jestliže jsou splněny všechny následující podmínky: absolutní počet neutrofilů > 1,5 x 109/l; počet trombocytů > 100 x 109/l; CTC nehematologická toxicita < stupeň 1 (s výjimkou alopecie, nausey, zvracení).
Monoterapeutická fáze
Čtyři týdny po ukončené souběžné fázi TMZ + RT se TMZ podává až v 6 cyklech monoterapeutické léčby. Dávka Cyklu 1 (monoterapie) je 150 mg/m2 jednou denně po dobu 5 dnů, následovaná 23 dny bez léčby. Na začátku Cyklu 2 je dávka zvýšena na 200 mg/m2, jestliže CTC nehematologická toxicita pro Cyklus 1 je stupeň < 2 (s výjimkou alopecie, nausey a zvracení), absolutní počet neutrofilů (ANC) je > 1,5 x 109/l a počet trombocytů je > 100 x 109/l. Jestliže není dávka zvýšena v Cyklu 2, nemělo by být zvýšení prováděno v následných cyklech. Je-li jednou zvýšena, zůstává dávka 200 mg/m2 na den pro prvních 5 dnů každého následujícího cyklu s výjimkou vzniku toxicity. Snížení dávky a ukončení podávání v průběhu monoterapeutické fáze by se mělo provádět podle Tabulek 2 a 3.
V průběhu léčby by měl být ve dni 22 (21 dnů po první dávce TMZ) vyšetřen celkový krevní obraz. Dávka by měla být redukována nebo podávání přípravku ukončeno podle Tabulky 3.
|
Tabulka 2. Úrovně dávky TMZ pro monoterapeutickou léčbu | ||
|
Úroveň dávky |
Dávka TMZ (mg/m2/den) |
Poznámky |
|
-1 |
100 |
Redukce kvůli předchozí toxicitě |
|
0 |
150 |
Dávka během Cyklu 1 |
|
1 |
200 |
Dávka během Cyklů 2-6 při absenci toxicity |
|
Tabulka 3. Snížení dávky nebo ukončení podávání TMZ během monoterapeutické léčby | ||
|
Toxicita |
Redukce TMZ o 1 úroveň dávkya |
Ukončení TMZ |
|
Absolutní počet neutrofilů |
<1,0 x 109/l |
Viz poznámka pod čarou b |
|
Počet trombocytů |
< 50 x 109/l |
Viz poznámka pod čarou b |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 3 |
CTC stupeň 4b |
a: Úrovně dávky TMZ jsou uvedeny v Tabulce 2.
b: TMZ se ukončí jestliže:
• výsledkem úrovně dávky -1 (100 mg/m2) je stále neakceptovatelná toxicita
• jestliže se po redukci dávky znovu objeví stejný stupeň 3 nehematologické toxicity (s výjimkou alopecie, nausey, zvracení).
Dospělí_pacienti a děti ve věku 3 let nebo starší s recidivujícím nebo _progresivním maligním gliomem:
Léčebný cyklus zahrnuje 28 dnů. U pacientů, kteří dosud nebyli léčeni chemoterapií, se TMZ podává perorálně v dávce 200 mg/m2 jedenkrát denně po dobu prvních 5 dnů s následným přerušením léčby po dobu 23 dnů (celkem 28 dnů). U nemocných, kteří již dříve prodělali chemoterapii, činí zahajovací dávka 150 mg/m2 jedenkrát denně, která se pak ve druhém cyklu zvyšuje na 200 mg/m2 jedenkrát denně po dobu 5 dnů, pokud nejsou přítomny známky hematotoxicity (viz bod 4.4).
Zvláštní _ populace
Pediatrická populace
U dětí ve věku 3 let nebo starších by se měl TMZ používat pouze pro léčbu recidivujícího nebo progresivního maligního gliomu. Zkušenosti u těchto dětí jsou velmi omezené (viz body 4.4 a 5.1). Bezpečnost a účinnost TMZ u dětí ve věku do 3 let nebyla stanovena. Nejsou dostupné žádné údaje.
Pacienti s poruchou funkce jater či ledvin
Farmakokinetika TMZ je u pacientů s normální jaterní funkcí a s mírnou nebo středně těžkou jaterní dysfunkcí srovnatelná. Údaje o podání TMZ u pacientů s těžkou jaterní dysfunkcí (Třída C podle Childovy klasifikace) ani s dysfunkcí ledvin nejsou k dispozici. Vzhledem k uvedeným farmakokinetickým vlastnostem TMZ je však pravděpodobné, že ani u nemocných s těžkou jaterní dysfunkcí nebo dysfunkcí ledvin kteréhokoli stupně není snižování jeho dávek nutné. Při podávání TMZ těmto nemocným je však nutná zvýšená opatrnost.
Starší pacienti
Z populační analýzy farmakokinetiky u pacientů ve věku 19-78 let vyplývá, že clearance TMZ není ovlivňována věkem. Nicméně se zdá, že starší pacienti (ve věku > 70 let) mají vyšší riziko vzniku neutropenie a trombocytopenie (viz bod 4.4).
Způsob podání
Temodal tvrdé tobolky by měl podáván nalačno.
Tobolky se musejí polykat vcelku a zapíjet sklenicí vody a nesmějí se otevírat nebo kousat.
Pokud se po podání dávky dostaví zvracení, neměla by být další dávka podána tentýž den.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Hypersenzitivita na dakarbazin (DTIC).
Těžká myelosuprese (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Oportunní infekce a reaktivace infekcí
Během léčby pomocí TMZ byly pozorovány oportunní infekce (jako je pneumonie způsobená Pneumocystis jirovecii) a reaktivace infekcí (jako jsou HBV, CMV) (viz bod 4.8).
Pneumonie způsobená Pneumocystis _jirovecii
U pacientů, kteří dostávali souběžně TMZ a RT v pilotní studii s prodlouženým harmonogramem 42 dnů, bylo zjištěno zvýšené riziko vzniku pneumonie způsobené Pneumocystis jirovecii (PCP). Proto je nutná profylaxe PCP u všech pacientů, kteří dostávají souběžně TMZ a RT po dobu 42 denního režimu (maximálně 49 dnů) bez ohledu na počet lymfocytů. Jestliže se objeví lymfopenie, je třeba pokračovat v profylaxi, dokud se neobnoví stupeň lymfopenie < 1.
Výskyt PCP může být vyšší, když je TMZ podáván během delšího dávkovacího režimu. Nicméně všichni pacienti, kteří dostávají TMZ, zvláště pacienti dostávající steroidy, by měli být pečlivě sledováni pro vznik PCP, bez ohledu na režim. U pacientů užívajících TMZ, zvláště v kombinaci s dexamethasonem nebo jinými steroidy, byly hlášeny případy fatálního respiračního selhání.
HBV
Byla hlášena hepatitida způsobená reaktivací viru hepatitidy B (HBV), v některých případech vedoucí ke smrti. Před zahájením léčby pacientů s pozitivní sérologií na hepatitidu B (včetně těch s aktivním onemocněním) je nezbytná konzultace s odborníky na onemocnění jater. Během léčby musí být pacienti náležitě monitorováni a vedeni.
Hepatotoxicita
U pacientů léčených TMZ bylo hlášeno poškození jater, včetně smrtelného selhání jater (viz bod 4.8). Před zahájením léčby je nutno provést testy jaterních funkcí. Pokud jsou výsledky abnormální, musí lékař před zahájením léčby temozolomidem vyhodnotit poměr přínosů a rizik, včetně potenciálu ke smrtelnému selhání jater. U pacientů se 42denním léčebným cyklem se musí testy jaterních funkcí opakovat uprostřed tohoto cyklu. Pro všechny pacienty platí, že jaterní funkce je nutno zkontrolovat po každém léčebném cyklu. U pacientů s výraznými abnormalitami jaterních funkcí musí lékař vyhodnotit poměr přínosů a rizik vyplývajících z pokračování v léčbě. Jaterní toxicita se může objevit několik nebo i více týdnů po poslední léčbě temozolomidem.
Malignity
Byly též velmi vzácně hlášeny případy myelodysplastického syndromu a sekundárních malignit, včetně myeloidní leukémie (viz bod 4.8).
Antiemetická léčba:
Nausea a zvracení jsou velmi často spojené s TMZ.
Před nebo po podání TMZ se může podávat antiemetická léčba.
Antiemetická profylaxe se doporučuje před počáteční dávkou souběžné fáze a velmi doporučuje během monoterapeutické fáze.
Pacienti s recidivujícím nebo _progresivním maligním gliomem
Antiemetika může být třeba podat pacientům, u nichž v předchozích léčebných cyklech došlo k těžšímu zvracení (stupeň 3 nebo 4).
Laboratorní parametry
U pacientů léčených TMZ může dojít k myelosupresi, včetně dlouhotrvající pancytopenie, která může vést k aplastické anémii, jež v některých případech skončila fatálně. V některých případech souběžná expozice léčivým přípravkům spojených s výskytem aplastické anémie, zahrnující karbamazepin, fenytoin a sulfamethoxazol/trimethoprim, hodnocení komplikuje. Před zahájením podávání léku by měly být splněny následující laboratorní parametry: ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Kompletní krevní obraz je třeba vyšetřit ve 22. dni (21 dnů po podání 1. dávky) nebo v průběhu 48 hodin po tomto dni, a pak v týdenních intervalech do té doby, dokud není ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Jestliže v průběhu kteréhokoli cyklu poklesne ANC na < 1,0 x 109/l nebo počet trombocytů na < 50 x 109/l, měla by být v následném cyklu dávka o jeden stupeň snížena (viz bod 4.2). Jednotlivé stupně dávky činí 100 mg/m2, 150 mg/m2 a 200 mg/m2. Nejnižší doporučená dávka je 100 mg/m2.
Pediatrická populace
S podáváním TMZ dětem ve věku nižším než 3 roky nejsou dosud žádné klinické zkušenosti. U starších dětí a dospívajících jsou zkušenosti velmi omezené (viz body 4.2 a 5.1).
Starší pacienti (ve věku >70 let)
U starších pacientů je ve srovnání s mladšími osobami pravděpodobně vyšší riziko neutropenie a trombocytopenie. Vzhledem k tomu je při podávání TMZ ve vyšším věku nutná zvýšená opatrnost.
Pacienti mužského pohlaví
Muži léčení TMZ by měli být upozorněni, aby se do 6 měsíců od užití poslední dávky nestali otci a aby se před zahájením léčby informovali o možnosti kryokonzervace spermatu (viz bod 4.6).
Laktóza
Tento léčivý přípravek obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo glukózo-galaktózovou malabsorpcí by proto neměli tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Podávání TMZ spolu s ranitidinem nemělo v oddělené studii fáze I za následek žádné změny rozsahu absorpce temozolomidu nebo expozice jeho aktivnímu metabolitu monomethyl- triazenoimidazol-karboxamidu (MTIC).
Podávání TMZ spolu s jídlem vedlo k poklesu Cmax o 33 % a zmenšení plochy pod křivkou koncentrace (AUC) o 9 %.
Protože nelze vyloučit, že změna Cmax může být klinicky významná, neměl by se Temodal podávat současně s jídlem.
Výsledky analýzy populační farmakokinetiky ve II. fázi klinických studií prokazují, že nebyla clearance TMZ ovlivněna současným podáním dexamethasonu, prochlorperazinu, fenytoinu, karbamazepinu, ondansetronu, antagonistů H2 receptorů nebo fenobarbitalu. Současné podávání kyseliny valproové bylo spojeno s malým, nicméně statisticky významným poklesem clearance TMZ.
Dosud nebyly provedeny žádné studie, které by hodnotily účinek TMZ na metabolismus nebo eliminaci jiných léků. Nicméně vzhledem k tomu, že TMZ neprochází přeměnou v játrech a na plazmatické bílkoviny se váže jen v malé míře, je málo pravděpodobné, že by ovlivňoval farmakokinetiku jiných léčivých přípravků (viz bod 5.2).
V kombinaci s jinými myelosupresivními látkami může TMZ zvýšit pravděpodobnost myelosuprese. Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání léku těhotným ženám nejsou k dispozici. V preklinických studiích u potkanů a králíků, kteří TMZ dostávali v dávce 150 mg/m2, byly prokázány teratogenní a/nebo toxické účinky na plod (viz bod 5.3). Temodal by neměl být podáván těhotným ženám. Pokud je přece jen nutno zvážit použití během těhotenství, je třeba pacientku seznámit s potenciálním rizikem pro plod.
Kojení
Není známo, zda se TMZ vylučuje do mateřského mléka; kojení by se proto mělo po dobu léčby TMZ přerušit.
Ženy ve fertilním věku.
Ženám ve fertilním věku je třeba doporučit používání účinné antikoncepce k zabránění otěhotnění v průběhu užívání TMZ.
Mužská plodnost
TMZ může mít genotoxické účinky. Proto se mužům léčeným temozolomidem nedoporučuje, aby se do 6 měsíců od užití poslední dávky stali otci a naopak se doporučuje, vzhledem k možnosti ireverzibilní neplodnosti po léčbě TMZ, ještě před jejím zahájením konzultovat s lékařem možnost kryokonzervace předem odebraných spermií.
4.7 Účinky na schopnost řídit a obsluhovat stroje
TMZ má malý vliv na schopnost řídit nebo obsluhovat stroje v důsledku únavy a ospalosti (viz bod 4.8.).
4.8 Nežádoucí účinky
Zkušenosti z klinických hodnocení
U pacientů léčených TMZ ať už užívaným v kombinaci s RT nebo ve formě monoterapie následující po RT v případě nově diagnostikovaného multiformního glioblastomu nebo ve formě monoterapie pacientů s recidivujícím nebo progresivním gliomem, byly velmi často hlášené nežádoucí účinky podobné: nausea, zvracení, zácpa, anorexie, bolest hlavy a únava. Křeče byly hlášeny velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených monoterapií, vyrážka byla hlášena velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených TMZ nebo v kombinaci s RT nebo také ve formě monoterapie a často u pacientů s recidivujícím gliomem. Většina hematologických nežádoucích účinků byla hlášena u obou indikací často nebo velmi často (Tabulky 4 a 5); četnost laboratorních nálezů stupně 3 a 4 je uvedena za každou tabulkou.
V tabulkách jsou nežádoucí účinky klasifikovány podle orgánových systémů a četnosti. Zařazení do skupin četností se provádí podle následujícího pravidla: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Nově diagnostikovaný multiformní glioblastom
Tabulka 4 ukazuje nežádoucí účinky, které se objevily při léčbě u pacientů s nově diagnostikovaným multiformním glioblastomem během souběžné a monoterapeutické fáze léčby.
|
Tabulka 4: Nežádoucí účinky, které se objevily během souběžné a monoterapeutické léčby u pacientů s nově diagnostikovaným multiformním glioblastomem | ||
|
Třída orgánového systému |
TMZ + souběžná RT n=288* |
TMZ monoterapie n=224 |
|
Infekce a infestace | ||
|
Časté: |
Infekce, Herpes simplex, infekce rány, faryngitida, orální kandidóza |
Infekce, orální kandidóza |
|
Méně časté: |
Herpes simplex, herpes zoster, symptomy podobné chřipce | |
|
Poruchy krve a lymfatického systému | ||
|
Časté: |
Neutropenie, trombocytopenie, lymfopenie, leukopenie |
Febrilní neutropenie, trombocytopenie, anémie, leukopenie |
|
Méně časté: |
Febrilní neutropenie, anémie |
Lymfopenie, petechie |
|
Endokrinní poruchy | ||
|
Méně časté: |
Cushingoid |
Cushingoid |
|
Poruchy metabolismu a výživy | ||
|
Velmi časté: | ||
|
Časté: |
Hyperglykémie, pokles hmotnosti |
Pokles hmotnosti |
|
Méně časté: |
Hypokalémie, zvýšení alkalické fosfatázy, zvýšení hmotnosti |
Hyperglykémie, zvýšení hmotnosti |
|
Psychiatrické poruchy | ||
|
Časté: |
Úzkost, emoční labilita, nespavost | |
|
Méně časté: |
Neklid, apatie, poruchy chování, deprese, halucinace |
Halucinace, amnézie |
|
Poruchy nervového systému | ||
|
Velmi časté: |
Konvulze, bolest hlavy | |
|
Časté: |
Konvulze, porucha vědomí, somnolence, afázie, porucha rovnováhy, závrať, zmatenost, porucha paměti, porucha koncentrace, neuropatie, parestézie, porucha řeči, třes |
Hemiparéza, afázie, porucha rovnováhy, somnolence, zmatenost, závrať, porucha paměti, porucha koncentrace, dysfázie, neurologická porucha (NOS), neuropatie, periferní neuropatie, parestézie, porucha řeči, třes |
|
Méně časté: |
Status epilepticus, extrapyramidální porucha, hemiparéza, ataxie, poruchy kognice, dysfázie, abnormální chůze, hyperestézie, hypestézie, neurologická porucha (NOS), periferní neuropatie |
Hemiplegie, ataxie, abnormální koordinace, abnormální chůze, hyperestézie, smyslová porucha |
|
Poruchy oka | ||
|
Časté: |
Rozmazané vidění |
Defekt zorného pole, rozmazané vidění, diplopie |
|
Méně časté: |
Hemianopie, zmenšená ostrost vidění, poruchy zraku, defekt zorného pole, bolest očí |
Zmenšená ostrost vidění, bolest očí, suché oči |
|
Poruchy ucha a labyrintu | ||
|
Časté: |
Porucha sluchu |
Porucha sluchu, tinitus |
|
Méně časté: |
Otitis media, tinitus, hyperakuze, bolest ucha |
Hluchota, závrať, bolest ucha |
|
Srdeční poruchy | ||
|
Méně časté: |
Palpitace | |
|
Cévní poruchy | ||
|
Časté: |
Hemoragie, edém, edém nohy |
Hemoragie, hluboká žilní trombóza, edém nohy |
|
Méně časté: |
Cerebrální hemoragie, hypertenze |
Plicní embolie, edém, periferní edém |
|
Respirační, hrudní a mediastinální poruchy | ||
|
Časté: | ||
|
Méně časté: |
Pneumonie, infekce horních cest dýchacích, nosní kongesce |
Pneumonie, sinusitida, infekce horních cest dýchacích, bronchitida |
|
Gastrointestinální poruchy | ||
|
Velmi časté: |
Zácpa, nausea, zvracení |
Zácpa, nausea, zvracení |
|
Časté: |
Stomatitida, průjem, bolest břicha, dyspepsie, dysfagie | |
|
Méně časté: |
Abdominální distenze, fekální inkontinence, gastrointestinální porucha (NOS), gastroenteritida, hemeroidy | |
|
Poruchy kůže a podkožní tkáně | ||
|
Velmi časté: |
Vyrážka, alopecie |
Vyrážka, alopecie |
|
Časté: |
Dermatitida, suchá kůže, erytém, svědění |
Suchá kůže, svědění |
|
Méně časté: |
Exfoliace kůže, fotosenzitivní reakce, abnormální pigmentace |
Erytém, abnormální pigmentace, zvýšené pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Časté: |
Svalová slabost, artralgie |
Svalová slabost, artralgie, muskuloskeletální bolest, myalgie |
|
Méně časté: |
Myopatie, bolest v zádech, muskuloskeletální bolest, myalgie |
Myopatie, bolest v zádech |
|
Poruchy ledvin a močových cest | ||
|
Časté: |
Časté močení, močová inkontinence |
Močová inkontinence |
|
Méně časté: | ||
|
Poruchy reprodukčního systému a prsu | ||
|
Méně časté: |
Impotence |
Vaginální hemoragie, menoragie, amenorea, vaginitida, bolest prsou |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Velmi časté: |
Únava |
Únava |
|
Časté: |
Alergická reakce, horečka, radiační poškození, edém obličeje, bolest, změna chuti |
Alergická reakce, horečka, radiační poškození, bolest, změna chuti |
|
Méně časté: |
Astenie, zrudnutí, návaly horka, zhoršení stavu, rigory, změna barvy jazyka, parosmie, žízeň |
Astenie, edém obličeje, bolest, zhoršení stavu, rigory, zubní potíže |
|
Vyšetření | ||
|
Časté: |
Zvýšené ALT |
Zvýšené ALT |
|
Méně časté: |
Zvýšené jaterní enzymy, zvýšené gama GT, zvýšené AST | |
*Pacient který byl randomizován pouze do větve RT, obdržel Temodal + RT.
Laboratorní výsledky
Byla zjištěna myelosuprese (neutropenie a trombocytopenie), která je známou toxicitou limitující dávku většiny cytotoxických látek včetně TMZ. Když byly zkombinovány abnormality laboratorních testů a nežádoucí účinky v souběžných a monoterapeutických fázích léčby, byly pozorovány u 8 % pacientů abnormality neutrofilů stupeň 3 nebo stupeň 4, včetně neutropenických případů. Stupeň 3 nebo stupeň 4 abnormalit trombocytů, včetně trombocytopenických případů, byl pozorován u 14 % pacientů, kteří dostávali TMZ.
Recidivující nebo progresivní maligní gliom
Nejčastěji se vyskytujícími nežádoucími účinky ve vztahu k léčbě byly v provedených klinických studiích gastrointestinální poruchy, především nausea (43 %) a zvracení (36 %). Tyto reakce dosahovaly obvykle pouze stupně 1 nebo 2 (0 - 5 epizod zvracení během 24 hodin) a buď spontánně odezněly, nebo byly snadno léčitelné standardní antiemetickou léčbou. Incidence těžké nausey a zvracení činila 4 %.
Tabulka 5 obsahuje nežádoucí účinky, které byly hlášeny v průběhu klinických studií pro recidivující nebo progresivní maligní gliom a po uvedení přípravku Temodal na trh.
|
Tabulka 5. Nežádoucí účinky u pacientů s recidivujícím nebo progresivním maligním gliomem | |
|
Infekce a infestace | |
|
Vzácné: |
Oportunní infekce včetně pneumonie způsobené PCP |
|
Poruchy krve a lymfatického systému | |
|
Velmi časté: |
Neutropenie nebo lymfopenie (stupeň 3-4) trombocytopenie (stupeň 3-4) |
|
Méně časté: |
Pancytopenie, anémie (stupeň 3-4), leukopenie |
|
Poruchy metabolismu a výživy | |
|
Velmi časté: | |
|
Časté: |
Pokles hmotnosti |
|
Poruchy nervového systému | |
|
Velmi časté: | |
|
Časté: |
Somnolence, závratě, parestézie |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté: | |
|
Gastrointestinální poruchy | |
|
Velmi časté: |
Zvracení, nausea, zácpa |
|
Časté: | |
|
Poruchy kůže a podkožní tkáně | |
|
Časté: |
Vyrážka, pruritus, alopecie, |
|
Velmi vzácné: |
Multiformní erytém, erytrodermie, urtikarie, exantém |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté: |
Únava |
|
Časté: | |
|
Velmi vzácné: |
Alergické reakce, včetně anafylaxe, angioedém |
Laboratorní výsledky
Trombocytopenie a neutropenie stupně 3 nebo 4 byly zaznamenány u 19 %, resp. 17 % pacientů léčených pro maligní gliom. K hospitalizaci a/nebo přerušení léčby TMZ vedly v 8 %, resp. 4 % případů. Myelosuprese byla předvídatelná (obvykle k ní došlo během několika prvních cyklů, s nadirem mezi 21. dnem a 28. dnem) a úprava k normě byla rychlá, obvykle v průběhu 1-2 týdnů. Kumulace myelosupresivních účinků prokázána nebyla. Přítomnost trombocytopenie může zvýšit riziko krvácení a přítomnost neutropenie nebo leukopenie může zvýšit riziko infekce.
Pohlaví
Do analýzy populační farmakokinetiky založené na zkušenostech z klinického hodnocení bylo zahrnuto 101 žen a 169 mužů, u kterých byla dostupná měření nadiru neutrofilů, a 110 žen a 174 mužů, u kterých byla dostupná měření nadiru krevních destiček. U žen bylo ve srovnání s muži v prvním cyklu terapie zaznamenáno více neutropenií stupně 4 (ANC< 0,5 x 109/l), 12 % vs 5 %, a trombocytopenií (< 20 x 109/l), 9 % vs 3 %. V souboru dat od 400 jedinců s rekurentním gliomem se v prvním cyklu terapie objevila neutropenie stupně 4 u 8 % žen vs 4 % mužů a trombocytopenie stupně 4 u 8 % žen vs 3 % mužů. Ve studii s 288 jedinci s nově diagnostikovaným multiformním glioblastomem se v prvním cyklu terapie vyskytla neutropenie stupně 4 u 3 % žen vs 0 % mužů a trombocytopenie stupně 4 u 1 % žen vs 0 % mužů.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s rekurentním gliomem mozkového kmene nebo rekurentním astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Ačkoliv jsou údaje omezené, tolerance u dětí se očekává být podobná jako u dospělých. Bezpečnost TMZ u dětí ve věku do 3 let nebyla stanovena.
Zkušenosti po uvedení na trh
Po uvedení přípravku na trh byly hlášeny další následující závažné nežádoucí účinky:
|
Tabulka 6. Souhrn příhod hlášených u temozolomidu po jeho uvedení na trh | |
|
Infekce a infestace* | |
|
Méně časté: |
infekce způsobená cytomegaloviry, reaktivace infekce způsobené virem, jako je cytomegalovirus, virus hepatitidy BŤ |
|
Poruchy krve a lymfatického systému | |
|
Velmi vzácné: |
dlouhotrvající pancytopenie, aplastická anémieŤ |
|
Novotvary benigní, maligní a blíže neurčené | |
|
Velmi vzácné: |
myelodysplastický syndrom (MDS), sekundární malignity, včetně myeloidní leukémie |
|
Endokrinní poruchy | |
|
Méně časté: |
diabetes insipidus |
|
Respirační, hrudní a mediastinální poruchy | |
|
Velmi vzácné: |
intersticiální pneumonitida/pneumonitida, plicní fibróza, respirační selhání1 |
|
Poruchy jater a žlučových cest* | |
|
Časté: |
zvýšení jaterních enzymů |
|
Méně časté: |
hyperbilirubinemie, cholestáza, hepatitida, poškození jater, selhání jater1 |
|
Poruchy kůže a podkožní tkáně | |
|
Velmi vzácné: |
toxická epidermální nekrolýza, Stevens-Johnsonův syndrom |
t Včetně smrtelných případů
Frekvence odhadované na základě relevantních klinických studií.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinicky byly u pacientů ověřeny dávky 500, 750, 1 000, a 1 250 mg/m2 (celková dávka za 5-denní cyklus). Toxicita limitující dávku byla toxicita hematologická a byla pozorována u všech dávek, ale lze očekávat, že při vyšších dávkách bude závažnější. U jednoho pacienta došlo k předávkování, když užil dávku 10 000 mg (celková dávka během jednoho 5-denního cyklu), nežádoucí účinky hlášené v tomto případě byly pancytopenie, pyrexie, multiorgánové selhání a smrt. Byly popsány případy, kdy pacienti doporučenou dávku užívali po dobu delší než 5 dnů (až 64 dnů) s hlášenými nežádoucími příhodami včetně suprese kostní dřeně s nebo bez rozvoje infekce, v některých případech závažné, dlouhotrvající a vedoucí ke smrti. V případě předávkování je nezbytné hematologické zhodnocení. Podle potřeby by měla být zajištěna podpůrná léčba.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: protinádorové léky - ostatní alkylační látky, ATC kód: L01A X03 Mechanismus účinku
Temozolomid je triazen, který za fyziologické hodnoty pH prochází rychlou chemickou konverzí na aktivní monometyl-triazenoimidazol-karboxamid (MTIC). Předpokládá se, že cytotoxické účinky MTIC jsou primárně důsledkem alkylace na O6 pozici guaninu, s následnou alkylací rovněž na pozici N7. Následně vznikající cytotoxické léze jsou vysvětlovány nejspíše aberantní reparací methylovaného zbytku.
Klinická účinnost a bezpečnost
Nově diagnostikovaný multiformní glioblastom
Celkem 573 pacientů bylo randomizováno do skupin dostávajících buď TMZ + RT (n=287) nebo RT samotnou (n=286). Pacienti ve větvi TMZ + RT dostávali souběžně TMZ (75 mg/m2) jednou denně od prvního až do posledního dne RT po dobu 42 dnů (maximálně 49 dnů). Pak následovala monoterapie TMZ (150 - 200 mg/m2) ve dnech 1-5 každého 28-denního cyklu, až v 6 cyklech, začínající 4 týdny po ukončení RT. Pacienti v kontrolní větvi dostávali pouze RT. Během RT a souběžné TMZ terapie byla nutná profylaxe pneumonie Pneumocystis jirovecii (PCP).
TMZ byl podáván jako záchranná terapie vnásledné fázi 161 pacientům z 282 (57 %) ve větvi dostávající pouze RT a 62 pacientům z 277 (22 %) ve větvi dostávající TMZ + RT.
Poměr rizika (HR) pro celkové přežívání byl 1,59 (95 % CI pro HR=1,33 - 1,91) s log-rank p < 0,0001 ve prospěch TMZ větve. Odhadnutá pravděpodobnost přežívání 2 roky nebo více (26 % vs 10 %) je vyšší ve větvi RT + TMZ. Přidání souběžně podávaného TMZ k RT následované monoterapií TMZ v léčbě pacientů s nově diagnostikovaným multiformním glioblastomem prokázalo statisticky významné zlepšení v celkovém přežívání (overall survival=OS) ve srovnání s RT samotnou (Graf 1).
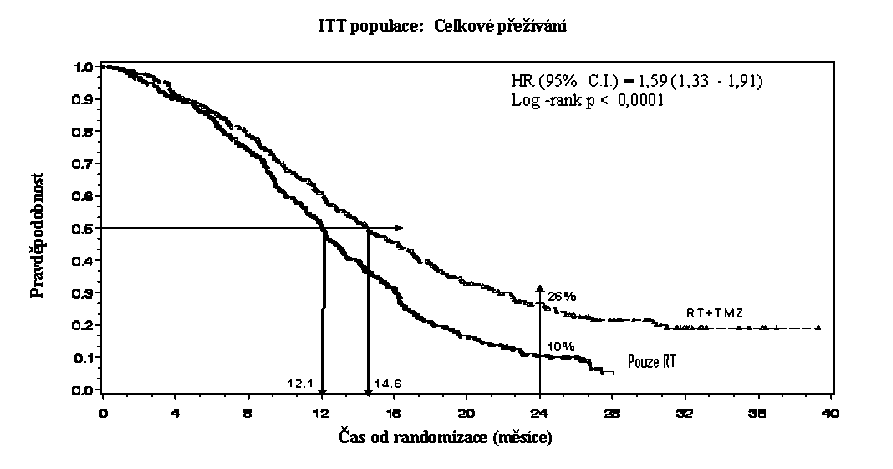
Graf 1 Kaplanovy-Meierovy křivky pro celkové přežívání (ITTpopulace=intent-to- treatpopulation)
Výsledky studie nebyly konzistentní v podskupině pacientů se slabým výkonnostním statusem (WHO PS=2, n=70), kde celkové přežívání a čas do progrese byly podobné v obou větvích. Zdá se však, že u této skupiny pacientů nejsou přítomna žádná nepřijatelná rizika.
Recidivující nebo _progresivní maligní gliom
Data o klinické účinnosti u pacientů s multiformním glioblastomem (Karnofského výkonnostní status [KPS] > 70), u něhož dochází k progresi nebo recidivě po chirurgickém výkonu a RT, vycházela ze dvou klinických studií s perorálním TMZ. V prvním případě šlo o nekomparativní studii u 138 pacientů (29 % bylo předtím léčeno chemoterapií), ve druhém o randomizovanou studii kontrolovanou léčivou látkou s TMZ vs prokarbazinem u celkem 225 pacientů (67 % z nich bylo předtím léčeno chemoterapií na bázi nitrosourey). V obou studiích bylo primárním hodnotícím kritériem přežívání bez známek progrese (PFS - progression-free survival), hodnocených na základě zobrazení magnetickou rezonancí (MRI) nebo na základě zhoršení neurologických funkcí. V nekomparativní studii bylo u 19 % pacientů dosaženo 6měsíční PFS, medián přežívání bez známek progrese byl 2,1 měsíce a medián celkové doby přežívání byl 5,4 měsíce. Objektivní počet terapeutických odpovědí na základě hodnocení MRI dosahoval 8 %.
V randomizované studii kontrolované léčivou látkou bylo šestiměsíční PFS u pacientů léčených TMZ významně vyšší než u těch, kteří dostávali prokarbazin (21 % vs 8 %; chí-kvadrát p=0,008)
s mediánem 2,89, respektive 1,88 měsíce (log rank p=0,0063). Medián přežití byl 7,34 měsíce pro TMZ a 5,66 měsíce pro prokarbazin (log rank p=0,33). Po 6 měsících byl podíl přežívajících pacientů v TMZ skupině významně vyšší (60 %) než ve skupině prokarbazinové (44 %; chí-kvadrát p=0,019). U pacientů, kteří byli předtím léčeni chemoterapií, bylo dosaženo kladného efektu u těch, kteří měli KPS > 80.
Údaje o době do zhoršení neurologického statusu, jakož i o době do zhoršení výkonnostního statusu (pokles na KPS < 70 nebo pokles nejméně o 30 bodů), hovoří ve prospěch TMZ oproti prokarbazinu. Medián doby do progrese v těchto hodnotících kritériích byl u TMZ o 0,7 - 2,1 měsíce delší než u prokarbazinu (log rank p < 0,01 - 0,03).
Recidivující anaplastický astrocytom
V multicentrické prospektivní studii fáze II, v níž byly hodnoceny bezpečnost a účinnost perorálně podávaného TMZ v léčbě pacientů s anaplastickým astrocytomem při prvním relapsu, bylo zjištěno šestiměsíční PSF u 46 % léčených. Medián PSF činil 5,4 měsíce. Medián celkového přežití činil 14,6 měsíce. Procento odpovědí na léčbu, hodnocené centrálně činilo 35 % (13 % kompletních, 43 % částečných odpovědí) pro populaci intent-to-treat (ITT), n=162. U 43 pacientů došlo ke stabilizaci onemocnění. Šestiměsíční přežití bez klinické příhody bylo pro populaci intent-to-treat 44 %, přičemž medián přežití bez klinické příhody činil 4,6 měsíce, což je podobné výsledkům hodnocení přežívání bez známek progrese. Z histologického hlediska byla účinnost obdobná. Objektivní radiologicky prokázaná odpověď na léčbu nebo zachování stavu beze známek progrese velmi dobře korelovaly se zachovanou nebo zlepšenou kvalitou života.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s recidivujícím gliomem mozkového kmene nebo recidivujícím astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Tolerance TMZ byla podobná jako u dospělých.
5.2 Farmakokinetické vlastnosti
TMZ se spontánně hydrolyzuje při fyziologickém pH primárně na aktivní látku, 3-metyl-(triazen-1-yl)imidazol-4-karboxamid (MTIC). MTIC se spontánně hydrolyzuje na 5-amino-imidazol-4-karboxamid (AIC), známý meziprodukt biosyntézy purinů a nukleových kyselin a na metylhydrazin, o kterém se předpokládá, že je aktivní alkylační látkou. Předpokládá se, že cytotoxicita MTIC je
primárně způsobena alkylací DNA zejména na pozicích guaninu O6 a N7' Expozice MTIC a AIC je ~
2,4 %, respektive 23 % v poměru k AUC TMZ. In vivo byl ti/2 MTIC podobný jako pro TMZ,
1,8 hodin.
Absorpce
Po perorálním podání dospělým pacientům se TMZ rychle absorbuje a vrcholné koncentrace dosahuje již za 20 minut po podání (střední doba je mezi 0,5 a 1,5 hodiny). Po perorálním podání 14C-značeného TMZ byla průměrná fekální exkrece 14C v průběhu 7 dnů po podání dávky 0,8 %, což svědčilo o úplné absorpci.
Distribuce v organismu
TMZ vykazuje nízkou vazebnou afinitu k proteinům (10 % až 20 %) proto se neočekává, že by interagoval s látkami s vysokou vazebnou afinitou k proteinům.
PET studie u lidí a preklinické údaje ukazují, že TMZ rychle prostupuje hematoencefalickou bariérou a je přítomný v mozkomíšním moku. Průnik do mozkomíšního moku (CSF) byl potvrzen u jednoho pacienta; expozice CSF na základě AUC TMZ byla přibližně 30 % plazmatické expozice, což odpovídá údajům u zvířat.
Eliminace z organismu
Plazmatický poločas (t1/2) je přibližně 1,8 hodiny. Hlavní způsob eliminace 14C je ledvinami. Po perorálním podání se objevuje přibližně 5 až 10 % dávky v nezměněné podobě v moči v průběhu 24 hodin a zbytek se vylučuje ve formě temozolomidové kyseliny, 5-aminoimidazol-4-karboxamidu (AIC) nebo neidentifikovaných polárních metabolitů.
Plazmatické koncentrace vzrůstají v závislosti na velikosti dávky. Plazmatická clearance, distribuční objem a poločas jsou na velikosti dávky nezávislé.
Zvláštní populace
V analýze populační farmakokinetiky TMZ bylo zjištěno, že plazmatická clearance TMZ nezávisí na věku, renální funkci nebo kouření. V oddělené farmakokinetické studii byly plazmatické farmakokinetické profily u pacientů s mírnou až střední jaterní dysfunkcí stejné jako profily u pacientů s normální funkcí jater.
U dětských pacientů byla AUC větší než u dospělých; nicméně maximální tolerovaná dávka (MTD) činila 1 000 mg/m2 na jeden cyklus jak u dětí, tak u dospělých.
5.3 Předklinické údaje vztahující se k bezpečnosti
U potkanů a psů byly prováděny studie toxicity s jednotlivým cyklem (5 dnů podávání léku, 23 dnů bez léčby) 3 a 6 cyklů léčby. Primární cíle toxických účinků tvořila kostní dřeň, lymforetikulární systém, testes, gastrointestinální ústrojí a při vyšších dávkách, které byly pro 60 % - 100 % testovaných potkanů a psů letální, docházelo k degeneraci retiny. Většina toxických účinků byla reverzibilní, s výjimkou nežádoucích účinků na mužský reprodukční systém a degeneraci retiny. Nicméně vzhledem k tomu, že dávky podmiňující retinální degeneraci se pohybovaly v rozmezí dávek letálních, a v klinických studiích nebyl žádný srovnatelný účinek pozorován, nepřikládá se tomuto nálezu klinická významnost.
TMZ je embryotoxická, teratogenní a genotoxická alkylační látka. TMZ má toxické účinky větší u potkanů a psů než u člověka, a klinická dávka se blíží minimální letální dávce u potkanů a psů. Za citlivé ukazatele toxicity se považují poklesy počtu leukocytů a trombocytů v závislosti na velikosti dávky. Ve studii u potkanů, v níž bylo aplikováno šest cyklů terapie, bylo zjištěno několik druhů novotvarů, včetně karcinomu mléčné žlázy, kožního keratoakanthomu a adenomu bazálních buněk, zatímco ve studiích u psů žádné nádorové ani prenádorové změny pozorovány nebyly. Zdá se, že potkani jsou vůči onkogenním účinkům TMZ mimořádně citliví, neboť první tumory se u nich objevují již po třech měsících od zahájení podávání léku. Jde o dobu krátkou dokonce i pro alkylační látky.
Výsledky aberačních testů s chromosomy Ames/salmonella a lymfocytů z lidské periferní krve prokázaly pozitivní mutagenní reakci.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky: laktóza,
koloidní bezvodý oxid křemičitý, sodná sůl karboxymethylškrobu typu A, kyselina vinná, kyselina stearová.
Obal tobolky: želatina,
oxid titaničitý (E171), natrium-lauryl-sulfát, červený oxid železitý (E 172),
Potisk:
šelak,
propylenglykol, čištěná voda, roztok amoniaku, hydroxid draselný, černý oxid železitý (E 172).
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Balení lahvička
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
Balení sáček
Uchovávejte při teplotě do 30°C.
6.5 Druh obalu a obsah balení
Balení lahvička
Skleněná lahvička třídy I jantarové barvy s polypropylenovými uzávěry jištěnými před dětmi, obsahujícími 5 nebo 20 tvrdých tobolek.
Papírová skládačka (krabička) obsahuje jednu lahvičku.
Balení sáček
Sáčky jsou složené z lineárního nízkohustotního polyethylenu (nejvnitřnější vrstva), aluminia a polyethylen-tereftalátu.
Jeden sáček obsahuje 1 tvrdou tobolku a je dodáván v papírové krabičce.
Papírová skládačka obsahuje 5 nebo 20 tvrdých tobolek, jednotlivě zatavených v sáčcích.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Tobolky by neměly být otevřeny. Pokud dojde k poškození celistvosti tobolky, zabraňte kontaktu jejího práškového obsahu s kůží a sliznicemi. Pokud přijde Temodal do styku s kůží nebo sliznicí, je třeba neprodleně a řádně ho smýt mýdlem a vodou.
Pacientům by mělo být doporučeno uchovávat tobolky mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/98/096/005
EU/1/98/096/006
EU/1/98/096/015
EU/1/98/096/016
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 26. ledna 1999.
Datum posledního prodloužení registrace: 26. ledna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Temodal 140 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje temozolomidum 140 mg.
Pomocná látka se známým účinkem:
Jedna tvrdá tobolka obsahuje 246 mg bezvodé laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka (tobolka).
Tvrdé tobolky mají neprůsvitné bílé tělo, modré víčko a jsou potištěny černým inkoustem. Na víčku je vytištěn nápis „Temodal“. Na těle tobolky je vytištěn nápis „140 mg“, logo společnosti Schering-Plough a dva proužky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Temodal je určen k léčbě:
- dospělých pacientů s nově diagnostikovaným multiformním glioblastomem souběžně s radioterapií (RT) a následně jako monoterapeutická léčba.
- dětí ve věku od tří let, dospívajících a dospělých pacientů s maligním gliomem, jako je multiformní glioblastom nebo anaplastický astrocytom, u nichž se po standardní léčbě projevují recidivy nebo progrese.
4.2 Dávkování a způsob podání
Temodal smí být předepisován výhradně lékařem se zkušenostmi s onkologickou léčbou mozkových
nádorů.
Může se podávat antiemetická léčba (viz bod 4.4).
Dávkování
Dospělí _pacienti s nově diagnostikovaným multiformním glioblastomem
Temodal je podáván v kombinaci s fokální radioterapií (fáze souběžné léčby) s následnými až 6 cykly monoterapie temozolomidem (TMZ) (monoterapeutická fáze).
Fáze souběžné léčby
TMZ se podává perorálně v dávce 75 mg/m2 denně po dobu 42 dnů souběžně s fokální radioterapií (60 Gy podáno ve 30 frakcích). Snižování dávek se nedoporučuje, ale každý týden by se mělo rozhodnout o pozdržení nebo přerušení podávání TMZ podle kritérií hematologické a nehematologické toxicity. Podávání TMZ může pokračovat po celou dobu 42 dnů souběžné fáze (až do 49 dnů), jestliže jsou splněny všechny následující podmínky:
- absolutní počet neutrofilů (ANC) > 1,5 x 109/l
- počet trombocytů > 100 x 109/l
- všeobecná kritéria toxicity (CTC - common toxicity criteria) nehematologické toxicity < stupeň 1 (s výjimkou alopecie, nevolnosti a zvracení).
Během léčby by se měl každý týden vyšetřit celkový krevní obraz. Podávání TMZ by mělo být během fáze souběžné léčby dočasně přerušeno nebo trvale ukončeno podle kritérií hematologické a nehematologické toxicity, jak je uvedeno v Tabulce 1.
|
Tabulka 1. Přerušení nebo ukončení podávání TMZ během souběžné léčby radioterapií a TMZ | ||
|
Toxicita |
TMZ přerušení3 |
TMZ ukončení |
|
Absolutní počet neutrofilů |
> 0,5 a <1,5 x 109/l |
< 0,5 x 109/l |
|
Počet trombocytů |
> 10 a < 100 x 109/l |
<10 x 109/l |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 2 |
CTC stupeň 3 nebo 4 |
a: Léčba se souběžně podávaným TMZ může pokračovat, jestliže jsou splněny všechny následující podmínky: absolutní počet neutrofilů > 1,5 x 109/l; počet trombocytů > 100 x 109/l; CTC nehematologická toxicita < stupeň 1 (s výjimkou alopecie, nausey, zvracení).
Monoterapeutická fáze
Čtyři týdny po ukončené souběžné fázi TMZ + RT se TMZ podává až v 6 cyklech monoterapeutické léčby. Dávka Cyklu 1 (monoterapie) je 150 mg/m2 jednou denně po dobu 5 dnů, následovaná 23 dny bez léčby. Na začátku Cyklu 2 je dávka zvýšena na 200 mg/m2, jestliže CTC nehematologická toxicita pro Cyklus 1 je stupeň < 2 (s výjimkou alopecie, nausey a zvracení), absolutní počet neutrofilů (ANC) je > 1,5 x 109/l a počet trombocytů je > 100 x 109/l. Jestliže není dávka zvýšena v Cyklu 2, nemělo by být zvýšení prováděno v následných cyklech. Je-li jednou zvýšena, zůstává dávka 200 mg/m2 na den pro prvních 5 dnů každého následujícího cyklu s výjimkou vzniku toxicity. Snížení dávky a ukončení podávání v průběhu monoterapeutické fáze by se mělo provádět podle Tabulek 2 a 3.
V průběhu léčby by měl být ve dni 22 (21 dnů po první dávce TMZ) vyšetřen celkový krevní obraz. Dávka by měla být redukována nebo podávání přípravku ukončeno podle Tabulky 3.
|
Tabulka 2. Úrovně dávky TMZ pro monoterapeutickou léčbu | ||
|
Úroveň dávky |
Dávka TMZ (mg/m2/den) |
Poznámky |
|
-1 |
100 |
Redukce kvůli předchozí toxicitě |
|
0 |
150 |
Dávka během Cyklu 1 |
|
1 |
200 |
Dávka během Cyklů 2-6 při absenci toxicity |
|
Tabulka 3. Snížení dávky nebo ukončení podávání TMZ během monoterapeutické léčby | ||
|
Toxicita |
Redukce TMZ o 1 úroveň dávkya |
Ukončení TMZ |
|
Absolutní počet neutrofilů |
<1,0 x 109/l |
Viz poznámka pod čarou b |
|
Počet trombocytů |
< 50 x 109/l |
Viz poznámka pod čarou b |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 3 |
CTC stupeň 4b |
a: Úrovně dávky TMZ jsou uvedeny v Tabulce 2.
b: TMZ se ukončí jestliže:
• výsledkem úrovně dávky -1 (100 mg/m2) je stále neakceptovatelná toxicita
• jestliže se po redukci dávky znovu objeví stejný stupeň 3 nehematologické toxicity (s výjimkou alopecie, nausey, zvracení).
Dospělí_pacienti a děti ve věku 3 let nebo starší s recidivujícím nebo _progresivním maligním gliomem:
Léčebný cyklus zahrnuje 28 dnů. U pacientů, kteří dosud nebyli léčeni chemoterapií, se TMZ podává perorálně v dávce 200 mg/m2 jedenkrát denně po dobu prvních 5 dnů s následným přerušením léčby po dobu 23 dnů (celkem 28 dnů). U nemocných, kteří již dříve prodělali chemoterapii, činí zahajovací dávka 150 mg/m2 jedenkrát denně, která se pak ve druhém cyklu zvyšuje na 200 mg/m2 jedenkrát denně po dobu 5 dnů, pokud nejsou přítomny známky hematotoxicity (viz bod 4.4).
Zvláštní _ populace
Pediatrická populace
U dětí ve věku 3 let nebo starších by se měl TMZ používat pouze pro léčbu recidivujícího nebo progresivního maligního gliomu. Zkušenosti u těchto dětí jsou velmi omezené (viz body 4.4 a 5.1). Bezpečnost a účinnost TMZ u dětí ve věku do 3 let nebyla stanovena. Nejsou dostupné žádné údaje.
Pacienti s poruchou funkce jater či ledvin
Farmakokinetika TMZ je u pacientů s normální jaterní funkcí a s mírnou nebo středně těžkou jaterní dysfunkcí srovnatelná. Údaje o podání TMZ u pacientů s těžkou jaterní dysfunkcí (Třída C podle Childovy klasifikace) ani s dysfunkcí ledvin nejsou k dispozici. Vzhledem k uvedeným farmakokinetickým vlastnostem TMZ je však pravděpodobné, že ani u nemocných s těžkou jaterní dysfunkcí nebo dysfunkcí ledvin kteréhokoli stupně není snižování jeho dávek nutné. Při podávání TMZ těmto nemocným je však nutná zvýšená opatrnost.
Starší pacienti
Z populační analýzy farmakokinetiky u pacientů ve věku 19-78 let vyplývá, že clearance TMZ není ovlivňována věkem. Nicméně se zdá, že starší pacienti (ve věku > 70 let) mají vyšší riziko vzniku neutropenie a trombocytopenie (viz bod 4.4).
Způsob podání
Temodal tvrdé tobolky by měl podáván nalačno.
Tobolky se musejí polykat vcelku a zapíjet sklenicí vody a nesmějí se otevírat nebo kousat.
Pokud se po podání dávky dostaví zvracení, neměla by být další dávka podána tentýž den.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Hypersenzitivita na dakarbazin (DTIC).
Těžká myelosuprese (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Oportunní infekce a reaktivace infekcí
Během léčby pomocí TMZ byly pozorovány oportunní infekce (jako je pneumonie způsobená Pneumocystis jirovecii) a reaktivace infekcí (jako jsou HBV, CMV) (viz bod 4.8).
Pneumonie způsobená Pneumocystis _ jirovecii
U pacientů, kteří dostávali souběžně TMZ a RT v pilotní studii s prodlouženým harmonogramem 42 dnů, bylo zjištěno zvýšené riziko vzniku pneumonie způsobené Pneumocystis jirovecii (PCP). Proto je nutná profylaxe PCP u všech pacientů, kteří dostávají souběžně TMZ a RT po dobu 42 denního režimu (maximálně 49 dnů) bez ohledu na počet lymfocytů. Jestliže se objeví lymfopenie, je třeba pokračovat v profylaxi, dokud se neobnoví stupeň lymfopenie < 1.
Výskyt PCP může být vyšší, když je TMZ podáván během delšího dávkovacího režimu. Nicméně všichni pacienti, kteří dostávají TMZ, zvláště pacienti dostávající steroidy, by měli být pečlivě sledováni pro vznik PCP, bez ohledu na režim. U pacientů užívajících TMZ, zvláště v kombinaci s dexamethasonem nebo jinými steroidy, byly hlášeny případy fatálního respiračního selhání.
HBV
Byla hlášena hepatitida způsobená reaktivací viru hepatitidy B (HBV), v některých případech vedoucí ke smrti. Před zahájením léčby pacientů s pozitivní sérologií na hepatitidu B (včetně těch s aktivním onemocněním) je nezbytná konzultace s odborníky na onemocnění jater. Během léčby musí být pacienti náležitě monitorováni a vedeni.
Hepatotoxicita
U pacientů léčených TMZ bylo hlášeno poškození jater, včetně smrtelného selhání jater (viz bod 4.8). Před zahájením léčby je nutno provést testy jaterních funkcí. Pokud jsou výsledky abnormální, musí lékař před zahájením léčby temozolomidem vyhodnotit poměr přínosů a rizik, včetně potenciálu ke smrtelnému selhání jater. U pacientů se 42denním léčebným cyklem se musí testy jaterních funkcí opakovat uprostřed tohoto cyklu. Pro všechny pacienty platí, že jaterní funkce je nutno zkontrolovat po každém léčebném cyklu. U pacientů s výraznými abnormalitami jaterních funkcí musí lékař vyhodnotit poměr přínosů a rizik vyplývajících z pokračování v léčbě. Jaterní toxicita se může objevit několik nebo i více týdnů po poslední léčbě temozolomidem.
Malignity
Byly též velmi vzácně hlášeny případy myelodysplastického syndromu a sekundárních malignit, včetně myeloidní leukémie (viz bod 4.8).
Antiemetická léčba
Nausea a zvracení jsou velmi často spojené s TMZ.
Před nebo po podání TMZ se může podávat antiemetická léčba.
Antiemetická profylaxe se doporučuje před počáteční dávkou souběžné fáze a velmi doporučuje během monoterapeutické fáze.
Pacienti s recidivujícím nebo _progresivním maligním gliomem
Antiemetika může být třeba podat pacientům, u nichž v předchozích léčebných cyklech došlo k těžšímu zvracení (stupeň 3 nebo 4).
Laboratorní parametry
U pacientů léčených TMZ může dojít k myelosupresi, včetně dlouhotrvající pancytopenie, která může vést k aplastické anémii, jež v některých případech skončila fatálně. V některých případech souběžná expozice léčivým přípravkům spojených s výskytem aplastické anémie, zahrnující karbamazepin, fenytoin a sulfamethoxazol/trimethoprim, hodnocení komplikuje. Před zahájením podávání léku by měly být splněny následující laboratorní parametry: ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Kompletní krevní obraz je třeba vyšetřit ve 22. dni (21 dnů po podání 1. dávky) nebo v průběhu 48 hodin po tomto dni, a pak v týdenních intervalech do té doby, dokud není ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Jestliže v průběhu kteréhokoli cyklu poklesne ANC na < 1,0 x 109/l nebo počet trombocytů na < 50 x 109/l, měla by být v následném cyklu dávka o jeden stupeň snížena (viz bod 4.2). Jednotlivé stupně dávky činí 100 mg/m2, 150 mg/m2 a 200 mg/m2. Nejnižší doporučená dávka je 100 mg/m2.
Pediatrická populace
S podáváním TMZ dětem ve věku nižším než 3 roky nejsou dosud žádné klinické zkušenosti. U starších dětí a dospívajících jsou zkušenosti velmi omezené (viz body 4.2 a 5.1).
Starší pacienti (ve věku >70 let)
U starších pacientů je ve srovnání s mladšími osobami pravděpodobně vyšší riziko neutropenie a trombocytopenie. Vzhledem k tomu je při podávání TMZ ve vyšším věku nutná zvýšená opatrnost.
Pacienti mužského pohlaví
Muži léčení TMZ by měli být upozorněni, aby se do 6 měsíců od užití poslední dávky nestali otci a aby se před zahájením léčby informovali o možnosti kryokonzervace spermatu (viz bod 4.6).
Laktóza
Tento léčivý přípravek obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo glukózo-galaktózovou malabsorpcí by proto neměli tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Podávání TMZ spolu s ranitidinem nemělo v oddělené studii fáze I za následek žádné změny rozsahu absorpce temozolomidu nebo expozice jeho aktivnímu metabolitu monomethyl- triazenoimidazol-karboxamidu (MTIC).
Podávání TMZ spolu s jídlem vedlo k poklesu Cmax o 33 % a zmenšení plochy pod křivkou koncentrace (AUC) o 9 %.
Protože nelze vyloučit, že změna Cmax může být klinicky významná, neměl by se Temodal podávat současně s jídlem.
Výsledky analýzy populační farmakokinetiky ve II. fázi klinických studií prokazují, že nebyla clearance TMZ ovlivněna současným podáním dexamethasonu, prochlorperazinu, fenytoinu, karbamazepinu, ondansetronu, antagonistů H2 receptorů nebo fenobarbitalu. Současné podávání kyseliny valproové bylo spojeno s malým, nicméně statisticky významným poklesem clearance TMZ.
Dosud nebyly provedeny žádné studie, které by hodnotily účinek TMZ na metabolismus nebo eliminaci jiných léků. Nicméně vzhledem k tomu, že TMZ neprochází přeměnou v játrech a na plazmatické bílkoviny se váže jen v malé míře, je málo pravděpodobné, že by ovlivňoval farmakokinetiku jiných léčivých přípravků (viz bod 5.2).
V kombinaci s jinými myelosupresivními látkami může TMZ zvýšit pravděpodobnost myelosuprese. Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání léku těhotným ženám nejsou k dispozici. V preklinických studiích u potkanů a králíků, kteří TMZ dostávali v dávce 150 mg/m2, byly prokázány teratogenní a/nebo toxické účinky na plod (viz bod 5.3). Temodal by neměl být podáván těhotným ženám. Pokud je přece jen nutno zvážit použití během těhotenství, je třeba pacientku seznámit s potenciálním rizikem pro plod.
Kojení
Není známo, zda se TMZ vylučuje do mateřského mléka; kojení by se proto mělo po dobu léčby TMZ přerušit.
Ženy ve fertilním věku.
Ženám ve fertilním věku je třeba doporučit používání účinné antikoncepce k zabránění otěhotnění v průběhu užívání TMZ.
Mužská plodnost
TMZ může mít genotoxické účinky. Proto se mužům léčeným temozolomidem nedoporučuje, aby se do 6 měsíců od užití poslední dávky stali otci a naopak se doporučuje, vzhledem k možnosti ireverzibilní neplodnosti po léčbě TMZ, ještě před jejím zahájením konzultovat s lékařem možnost kryokonzervace předem odebraných spermií.
4.7 Účinky na schopnost řídit a obsluhovat stroje
TMZ má malý vliv na schopnost řídit nebo obsluhovat stroje v důsledku únavy a ospalosti (viz bod 4.8.).
4.8 Nežádoucí účinky
Zkušenosti z klinických hodnocení
U pacientů léčených TMZ ať už užívaným v kombinaci s RT nebo ve formě monoterapie následující po RT v případě nově diagnostikovaného multiformního glioblastomu nebo ve formě monoterapie pacientů s recidivujícím nebo progresivním gliomem, byly velmi často hlášené nežádoucí účinky podobné: nausea, zvracení, zácpa, anorexie, bolest hlavy a únava. Křeče byly hlášeny velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených monoterapií, vyrážka byla hlášena velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených TMZ nebo v kombinaci s RT nebo také ve formě monoterapie a často u pacientů s recidivujícím gliomem. Většina hematologických nežádoucích účinků byla hlášena u obou indikací často nebo velmi často (Tabulky 4 a 5); četnost laboratorních nálezů stupně 3 a 4 je uvedena za každou tabulkou.
V tabulkách jsou nežádoucí účinky klasifikovány podle orgánových systémů a četnosti. Zařazení do skupin četností se provádí podle následujícího pravidla: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Nově diagnostikovaný multiformní glioblastom
Tabulka 4 ukazuje nežádoucí účinky, které se objevily při léčbě u pacientů s nově diagnostikovaným multiformním glioblastomem během souběžné a monoterapeutické fáze léčby.
|
Tabulka 4: Nežádoucí účinky, které se objevily během souběžné a monoterapeutické léčby u pacientů s nově diagnostikovaným multiformním glioblastomem | ||
|
Třída orgánového systému |
TMZ + souběžná RT n=288* |
TMZ monoterapie n=224 |
|
Infekce a infestace | ||
|
Časté: |
Infekce, Herpes simplex, infekce rány, faryngitida, orální kandidóza |
Infekce, orální kandidóza |
|
Méně časté: |
Herpes simplex, herpes zoster, symptomy podobné chřipce | |
|
Poruchy krve a lymfatického systému | ||
|
Časté: |
Neutropenie, trombocytopenie, lymfopenie, leukopenie |
Febrilní neutropenie, trombocytopenie, anémie, leukopenie |
|
Méně časté: |
Febrilní neutropenie, anémie |
Lymfopenie, petechie |
|
Endokrinní poruchy | ||
|
Méně časté: |
Cushingoid |
Cushingoid |
|
Poruchy metabolismu a výživy | ||
|
Velmi časté: | ||
|
Časté: |
Hyperglykémie, pokles hmotnosti |
Pokles hmotnosti |
|
Méně časté: |
Hypokalémie, zvýšení alkalické fosfatázy, zvýšení hmotnosti |
Hyperglykémie, zvýšení hmotnosti |
|
Psychiatrické poruchy | ||
|
Časté: |
Úzkost, emoční labilita, nespavost | |
|
Méně časté: |
Neklid, apatie, poruchy chování, deprese, halucinace |
Halucinace, amnézie |
|
Poruchy nervového systému | ||
|
Velmi časté: |
Konvulze, bolest hlavy | |
|
Časté: |
Konvulze, porucha vědomí, somnolence, afázie, porucha rovnováhy, závrať, zmatenost, porucha paměti, porucha koncentrace, neuropatie, parestézie, porucha řeči, třes |
Hemiparéza, afázie, porucha rovnováhy, somnolence, zmatenost, závrať, porucha paměti, porucha koncentrace, dysfázie, neurologická porucha (NOS), neuropatie, periferní neuropatie, parestézie, porucha řeči, třes |
|
Méně časté: |
Status epilepticus, extrapyramidální porucha, hemiparéza, ataxie, poruchy kognice, dysfázie, abnormální chůze, hyperestézie, hypestézie, neurologická porucha (NOS), periferní neuropatie |
Hemiplegie, ataxie, abnormální koordinace, abnormální chůze, hyperestézie, smyslová porucha |
|
Poruchy oka | ||
|
Časté: |
Rozmazané vidění |
Defekt zorného pole, rozmazané vidění, diplopie |
|
Méně časté: |
Hemianopie, zmenšená ostrost vidění, poruchy zraku, defekt zorného pole, bolest očí |
Zmenšená ostrost vidění, bolest očí, suché oči |
|
Poruchy ucha a labyrintu | ||
|
Časté: |
Porucha sluchu |
Porucha sluchu, tinitus |
|
Méně časté: |
Otitis media, tinitus, hyperakuze, bolest ucha |
Hluchota, závrať, bolest ucha |
|
Srdeční poruchy | ||
|
Méně časté: |
Palpitace | |
|
Cévní poruchy | ||
|
Časté: |
Hemoragie, edém, edém nohy |
Hemoragie, hluboká žilní trombóza, edém nohy |
|
Méně časté: |
Cerebrální hemoragie, hypertenze |
Plicní embolie, edém, periferní edém |
|
Respirační, hrudní a mediastinální poruchy | ||
|
Časté: | ||
|
Méně časté: |
Pneumonie, infekce horních cest dýchacích, nosní kongesce |
Pneumonie, sinusitida, infekce horních cest dýchacích, bronchitida |
|
Gastrointestinální poruchy | ||
|
Velmi časté: |
Zácpa, nausea, zvracení |
Zácpa, nausea, zvracení |
|
Časté: |
Stomatitida, průjem, bolest břicha, dyspepsie, dysfagie | |
|
Méně časté: |
Abdominální distenze, fekální inkontinence, gastrointestinální porucha (NOS), gastroenteritida, hemeroidy | |
|
Poruchy kůže a podkožní tkáně | ||
|
Velmi časté: |
Vyrážka, alopecie |
Vyrážka, alopecie |
|
Časté: |
Dermatitida, suchá kůže, erytém, svědění |
Suchá kůže, svědění |
|
Méně časté: |
Exfoliace kůže, fotosenzitivní reakce, abnormální pigmentace |
Erytém, abnormální pigmentace, zvýšené pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Časté: |
Svalová slabost, artralgie |
Svalová slabost, artralgie, muskuloskeletální bolest, myalgie |
|
Méně časté: |
Myopatie, bolest v zádech, muskuloskeletální bolest, myalgie |
Myopatie, bolest v zádech |
|
Poruchy ledvin a močových cest | ||
|
Časté: |
Časté močení, močová inkontinence |
Močová inkontinence |
|
Méně časté: | ||
|
Poruchy reprodukčního systému a prsu | ||
|
Méně časté: |
Impotence |
Vaginální hemoragie, menoragie, amenorea, vaginitida, bolest prsou |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Velmi časté: |
Únava |
Únava |
|
Časté: |
Alergická reakce, horečka, radiační poškození, edém obličeje, bolest, změna chuti |
Alergická reakce, horečka, radiační poškození, bolest, změna chuti |
|
Méně časté: |
Astenie, zrudnutí, návaly horka, zhoršení stavu, rigory, změna barvy jazyka, parosmie, žízeň |
Astenie, edém obličeje, bolest, zhoršení stavu, rigory, zubní potíže |
|
Vyšetření | ||
|
Časté: |
Zvýšené ALT |
Zvýšené ALT |
|
Méně časté: |
Zvýšené jaterní enzymy, zvýšené gama GT, zvýšené AST | |
*Pacient který byl randomizován pouze do větve RT, obdržel Temodal + RT.
Laboratorní výsledky
Byla zjištěna myelosuprese (neutropenie a trombocytopenie), která je známou toxicitou limitující dávku většiny cytotoxických látek včetně TMZ. Když byly zkombinovány abnormality laboratorních testů a nežádoucí účinky v souběžných a monoterapeutických fázích léčby, byly pozorovány u 8 % pacientů abnormality neutrofilů stupeň 3 nebo stupeň 4, včetně neutropenických případů. Stupeň 3 nebo stupeň 4 abnormalit trombocytů, včetně trombocytopenických případů, byl pozorován u 14 % pacientů, kteří dostávali TMZ.
Recidivující nebo progresivní maligní gliom
Nejčastěji se vyskytujícími nežádoucími účinky ve vztahu k léčbě byly v provedených klinických studiích gastrointestinální poruchy, především nausea (43 %) a zvracení (36 %). Tyto reakce dosahovaly obvykle pouze stupně 1 nebo 2 (0 - 5 epizod zvracení během 24 hodin) a buď spontánně odezněly, nebo byly snadno léčitelné standardní antiemetickou léčbou. Incidence těžké nausey a zvracení činila 4 %.
Tabulka 5 obsahuje nežádoucí účinky, které byly hlášeny v průběhu klinických studií pro recidivující nebo progresivní maligní gliom a po uvedení přípravku Temodal na trh.
|
Tabulka 5. Nežádoucí účinky u pacientů s recidivujícím nebo progresivním maligním gliomem | |
|
Infekce a infestace | |
|
Vzácné: |
Oportunní infekce včetně pneumonie způsobené PCP |
|
Poruchy krve a lymfatického systému | |
|
Velmi časté: |
Neutropenie nebo lymfopenie (stupeň 3-4) trombocytopenie (stupeň 3-4) |
|
Méně časté: |
Pancytopenie, anémie (stupeň 3-4), leukopenie |
|
Poruchy metabolismu a výživy | |
|
Velmi časté: | |
|
Časté: |
Pokles hmotnosti |
|
Poruchy nervového systému | |
|
Velmi časté: | |
|
Časté: |
Somnolence, závratě, parestézie |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté: | |
|
Gastrointestinální poruchy | |
|
Velmi časté: |
Zvracení, nausea, zácpa |
|
Časté: | |
|
Poruchy kůže a podkožní tkáně | |
|
Časté: |
Vyrážka, pruritus, alopecie, |
|
Velmi vzácné: |
Multiformní erytém, erytrodermie, urtikarie, exantém |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté: |
Únava |
|
Časté: | |
|
Velmi vzácné: |
Alergické reakce, včetně anafylaxe, angioedém |
Laboratorní výsledky
Trombocytopenie a neutropenie stupně 3 nebo 4 byly zaznamenány u 19 %, resp. 17 % pacientů léčených pro maligní gliom. K hospitalizaci a/nebo přerušení léčby TMZ vedly v 8 %, resp. 4 % případů. Myelosuprese byla předvídatelná (obvykle k ní došlo během několika prvních cyklů, s nadirem mezi 21. dnem a 28. dnem) a úprava k normě byla rychlá, obvykle v průběhu 1-2 týdnů. Kumulace myelosupresivních účinků prokázána nebyla. Přítomnost trombocytopenie může zvýšit riziko krvácení a přítomnost neutropenie nebo leukopenie může zvýšit riziko infekce.
Pohlaví
Do analýzy populační farmakokinetiky založené na zkušenostech z klinického hodnocení bylo zahrnuto 101 žen a 169 mužů, u kterých byla dostupná měření nadiru neutrofilů, a 110 žen a 174 mužů, u kterých byla dostupná měření nadiru krevních destiček. U žen bylo ve srovnání s muži v prvním cyklu terapie zaznamenáno více neutropenií stupně 4 (ANC< 0,5 x 109/l), 12 % vs 5 %, a trombocytopenií (< 20 x 109/l), 9 % vs 3 %. V souboru dat od 400 jedinců s rekurentním gliomem se v prvním cyklu terapie objevila neutropenie stupně 4 u 8 % žen vs 4 % mužů a trombocytopenie stupně 4 u 8 % žen vs 3 % mužů. Ve studii s 288 jedinci s nově diagnostikovaným multiformním glioblastomem se v prvním cyklu terapie vyskytla neutropenie stupně 4 u 3 % žen vs 0 % mužů a trombocytopenie stupně 4 u 1 % žen vs 0 % mužů.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s rekurentním gliomem mozkového kmene nebo rekurentním astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Ačkoliv jsou údaje omezené, tolerance u dětí se očekává být podobná jako u dospělých. Bezpečnost TMZ u dětí ve věku do 3 let nebyla stanovena.
Zkušenosti po uvedení na trh
Po uvedení přípravku na trh byly hlášeny další následující závažné nežádoucí účinky:
|
Tabulka 6. Souhrn příhod hlášených u temozolomidu po jeho uvedení na trh | |
|
Infekce a infestace* | |
|
Méně časté: |
infekce způsobená cytomegaloviry, reaktivace infekce způsobené virem, jako je cytomegalovirus, virus hepatitidy BŤ |
|
Poruchy krve a lymfatického systému | |
|
Velmi vzácné: |
dlouhotrvající pancytopenie, aplastická anémieŤ |
|
Novotvary benigní, maligní a blíže neurčené | |
|
Velmi vzácné: |
myelodysplastický syndrom (MDS), sekundární malignity, včetně myeloidní leukémie |
|
Endokrinní poruchy | |
|
Méně časté: |
diabetes insipidus |
|
Respirační, hrudní a mediastinální poruchy | |
|
Velmi vzácné: |
intersticiální pneumonitida/pneumonitida, plicní fibróza, respirační selhání1 |
|
Poruchy jater a žlučových cest* | |
|
Časté: |
zvýšení jaterních enzymů |
|
Méně časté: |
hyperbilirubinemie, cholestáza, hepatitida, poškození jater, selhání jater1 |
|
Poruchy kůže a podkožní tkáně | |
|
Velmi vzácné: |
toxická epidermální nekrolýza, Stevens-Johnsonův syndrom |
t Včetně smrtelných případů
Frekvence odhadované na základě relevantních klinických studií.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinicky byly u pacientů ověřeny dávky 500, 750, 1 000, a 1 250 mg/m2 (celková dávka za 5-denní cyklus). Toxicita limitující dávku byla toxicita hematologická a byla pozorována u všech dávek, ale lze očekávat, že při vyšších dávkách bude závažnější. U jednoho pacienta došlo k předávkování, když užil dávku 10 000 mg (celková dávka během jednoho 5-denního cyklu), nežádoucí účinky hlášené v tomto případě byly pancytopenie, pyrexie, multiorgánové selhání a smrt. Byly popsány případy, kdy pacienti doporučenou dávku užívali po dobu delší než 5 dnů (až 64 dnů) s hlášenými nežádoucími příhodami včetně suprese kostní dřeně s nebo bez rozvoje infekce, v některých případech závažné, dlouhotrvající a vedoucí ke smrti. V případě předávkování je nezbytné hematologické zhodnocení. Podl e potřeby by měla být zajištěna podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: protinádorové léky - ostatní alkylační látky, ATC kód: L01A X03 Mechanismus účinku
Temozolomid je triazen, který za fyziologické hodnoty pH prochází rychlou chemickou konverzí na aktivní monometyl-triazenoimidazol-karboxamid (MTIC). Předpokládá se, že cytotoxické účinky MTIC jsou primárně důsledkem alkylace na O6 pozici guaninu, s následnou alkylací rovněž na pozici N7. Následně vznikající cytotoxické léze jsou vysvětlovány nejspíše aberantní reparací methylovaného zbytku.
Klinická účinnost a bezpečnost
Nově diagnostikovaný multiformní glioblastom
Celkem 573 pacientů bylo randomizováno do skupin dostávajících buď TMZ + RT (n=287) nebo RT samotnou (n=286). Pacienti ve větvi TMZ + RT dostávali souběžně TMZ (75 mg/m2) jednou denně od prvního až do posledního dne RT po dobu 42 dnů (maximálně 49 dnů). Pak následovala monoterapie TMZ (150 - 200 mg/m2) ve dnech 1-5 každého 28-denního cyklu, až v 6 cyklech, začínající 4 týdny po ukončení RT. Pacienti v kontrolní větvi dostávali pouze RT. Během RT a souběžné TMZ terapie byla nutná profylaxe pneumonie Pneumocystis jirovecii (PCP).
TMZ byl podáván jako záchranná terapie vnásledné fázi 161 pacientům z 282 (57 %) ve větvi dostávající pouze RT a 62 pacientům z 277 (22 %) ve větvi dostávající TMZ + RT.
Poměr rizika (HR) pro celkové přežívání byl 1,59 (95 % CI pro HR=1,33 - 1,91) s log-rank p < 0,0001 ve prospěch TMZ větve. Odhadnutá pravděpodobnost přežívání 2 roky nebo více (26 % vs 10 %) je vyšší ve větvi RT + TMZ. Přidání souběžně podávaného TMZ k RT následované monoterapií TMZ v léčbě pacientů s nově diagnostikovaným multiformním glioblastomem prokázalo statisticky významné zlepšení v celkovém přežívání (overall survival=OS) ve srovnání s RT samotnou (Graf 1).
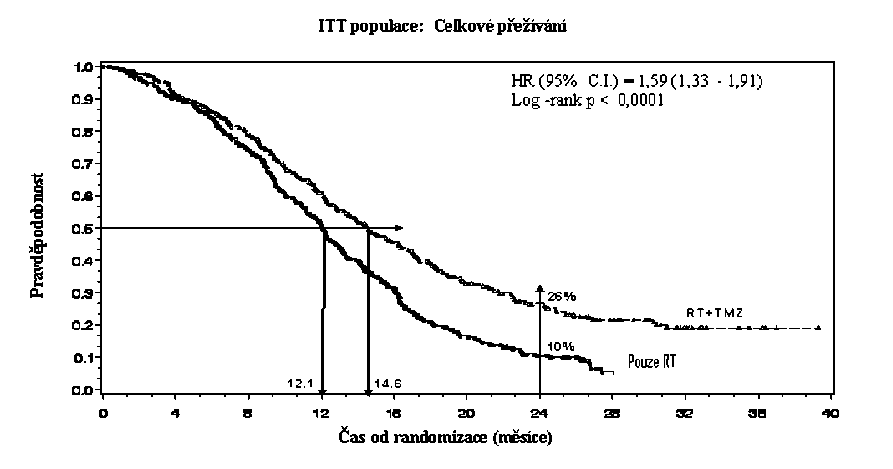
Výsledky studie nebyly konzistentní v podskupině pacientů se slabým výkonnostním statusem (WHO PS=2, n=70), kde celkové přežívání a čas do progrese byly podobné v obou větvích. Zdá se však, že u této skupiny pacientů nejsou přítomna žádná nepřijatelná rizika.
Recidivující nebo _progresivní maligní gliom
Data o klinické účinnosti u pacientů s multiformním glioblastomem (Karnofského výkonnostní status [KPS] > 70), u něhož dochází k progresi nebo recidivě po chirurgickém výkonu a RT, vycházela ze dvou klinických studií s perorálním TMZ. V prvním případě šlo o nekomparativní studii u 138 pacientů (29 % bylo předtím léčeno chemoterapií), ve druhém o randomizovanou studii kontrolovanou léčivou látkou s TMZ vs prokarbazinem u celkem 225 pacientů (67 % z nich bylo předtím léčeno chemoterapií na bázi nitrosourey). V obou studiích bylo primárním hodnotícím kritériem přežívání bez známek progrese (PFS - progression-free survival), hodnocených na základě zobrazení magnetickou rezonancí (MRI) nebo na základě zhoršení neurologických funkcí. V nekomparativní studii bylo u 19 % pacientů dosaženo 6měsíční PFS, medián přežívání bez známek progrese byl 2,1 měsíce a medián celkové doby přežívání byl 5,4 měsíce. Objektivní počet terapeutických odpovědí na základě hodnocení MRI dosahoval 8 %.
V randomizované studii kontrolované léčivou látkou bylo šestiměsíční PFS u pacientů léčených TMZ významně vyšší než u těch, kteří dostávali prokarbazin (21 % vs 8 %; chí-kvadrát p=0,008)
s mediánem 2,89, respektive 1,88 měsíce (log rank p=0,0063). Medián přežití byl 7,34 měsíce pro TMZ a 5,66 měsíce pro prokarbazin (log rank p=0,33). Po 6 měsících byl podíl přežívajících pacientů v TMZ skupině významně vyšší (60 %) než ve skupině prokarbazinové (44 %; chí-kvadrát p=0,019). U pacientů, kteří byli předtím léčeni chemoterapií, bylo dosaženo kladného efektu u těch, kteří měli KPS > 80.
Údaje o době do zhoršení neurologického statusu, jakož i o době do zhoršení výkonnostního statusu (pokles na KPS < 70 nebo pokles nejméně o 30 bodů), hovoří ve prospěch TMZ oproti prokarbazinu. Medián doby do progrese v těchto hodnotících kritériích byl u TMZ o 0,7 - 2,1 měsíce delší než u prokarbazinu (log rank p < 0,01 - 0,03).
Recidivující anaplastický astrocytom
V multicentrické prospektivní studii fáze II, v níž byly hodnoceny bezpečnost a účinnost perorálně podávaného TMZ v léčbě pacientů s anaplastickým astrocytomem při prvním relapsu, bylo zjištěno šestiměsíční PSF u 46 % léčených. Medián PSF činil 5,4 měsíce. Medián celkového přežití činil
14,6 měsíce. Procento odpovědí na léčbu, hodnocené centrálně činilo 35 % (13 % kompletních, 43 % částečných odpovědí) pro populaci intent-to-treat (ITT), n=162. U 43 pacientů došlo ke stabilizaci onemocnění. Šestiměsíční přežití bez klinické příhody bylo pro populaci intent-to-treat 44 %, přičemž medián přežití bez klinické příhody činil 4,6 měsíce, což je podobné výsledkům hodnocení přežívání bez známek progrese. Z histologického hlediska byla účinnost obdobná. Objektivní radiologicky prokázaná odpověď na léčbu nebo zachování stavu beze známek progrese velmi dobře korelovaly se zachovanou nebo zlepšenou kvalitou života.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s recidivujícím gliomem mozkového kmene nebo recidivujícím astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Tolerance TMZ byla podobná jako u dospělých.
5.2 Farmakokinetické vlastnosti
TMZ se spontánně hydrolyzuje při fyziologickém pH primárně na aktivní látku, 3-metyl-(triazen-1-yl)imidazol-4-karboxamid (MTIC). MTIC se spontánně hydrolyzuje na 5-amino-imidazol-4-
karboxamid (AIC), známý meziprodukt biosyntézy purinů a nukleových kyselin a na metylhydrazin, o kterém se předpokládá, že je aktivní alkylační látkou. Předpokládá se, že cytotoxicita MTIC je primárně způsobena alkylací DNA zejména na pozicích guaninu O6 a N7' Expozice MTIC a AIC je ~
2,4 %, respektive 23 % v poměru k AUC TMZ. In vivo byl ti/2 MTIC podobný jako pro TMZ,
1,8 hodin.
Absorpce
Po perorálním podání dospělým pacientům se TMZ rychle absorbuje a vrcholné koncentrace dosahuje již za 20 minut po podání (střední doba je mezi 0,5 a 1,5 hodiny). Po perorálním podání 14C-značeného TMZ byla průměrná fekální exkrece 14C v průběhu 7 dnů po podání dávky 0,8 %, což svědčilo o úplné absorpci.
Distribuce v organismu
TMZ vykazuje nízkou vazebnou afinitu k proteinům (10 % až 20 %) proto se neočekává, že by interagoval s látkami s vysokou vazebnou afinitou k proteinům.
PET studie u lidí a preklinické údaje ukazují, že TMZ rychle prostupuje hematoencefalickou bariérou a je přítomný v mozkomíšním moku. Průnik do mozkomíšního moku (CSF) byl potvrzen u jednoho pacienta; expozice CSF na základě AUC TMZ byla přibližně 30 % plazmatické expozice, což odpovídá údajům u zvířat.
Eliminace z organismu
Plazmatický poločas (t1/2) je přibližně 1,8 hodiny. Hlavní způsob eliminace 14C je ledvinami. Po perorálním podání se objevuje přibližně 5 až 10 % dávky v nezměněné podobě v moči v průběhu 24 hodin a zbytek se vylučuje ve formě temozolomidové kyseliny, 5-aminoimidazol-4-karboxamidu (AIC) nebo neidentifikovaných polárních metabolitů.
Plazmatické koncentrace vzrůstají v závislosti na velikosti dávky. Plazmatická clearance, distribuční objem a poločas jsou na velikosti dávky nezávislé.
Zvláštní populace
V analýze populační farmakokinetiky TMZ bylo zjištěno, že plazmatická clearance TMZ nezávisí na věku, renální funkci nebo kouření. V oddělené farmakokinetické studii byly plazmatické farmakokinetické profily u pacientů s mírnou až střední jaterní dysfunkcí stejné jako profily u pacientů s normální funkcí jater.
U dětských pacientů byla AUC větší než u dospělých; nicméně maximální tolerovaná dávka (MTD) činila 1 000 mg/m2 na jeden cyklus jak u dětí, tak u dospělých.
5.3 Předklinické údaje vztahující se k bezpečnosti
U potkanů a psů byly prováděny studie toxicity s jednotlivým cyklem (5 dnů podávání léku, 23 dnů bez léčby) 3 a 6 cyklů léčby. Primární cíle toxických účinků tvořila kostní dřeň, lymforetikulární systém, testes, gastrointestinální ústrojí a při vyšších dávkách, které byly pro 60 % - 100 % testovaných potkanů a psů letální, docházelo k degeneraci retiny. Většina toxických účinků byla reverzibilní, s výjimkou nežádoucích účinků na mužský reprodukční systém a degeneraci retiny. Nicméně vzhledem k tomu, že dávky podmiňující retinální degeneraci se pohybovaly v rozmezí dávek letálních, a v klinických studiích nebyl žádný srovnatelný účinek pozorován, nepřikládá se tomuto nálezu klinická významnost.
TMZ je embryotoxická, teratogenní a genotoxická alkylační látka. TMZ má toxické účinky větší u potkanů a psů než u člověka, a klinická dávka se blíží minimální letální dávce u potkanů a psů. Za citlivé ukazatele toxicity se považují poklesy počtu leukocytů a trombocytů v závislosti na velikosti dávky. Ve studii u potkanů, v níž bylo aplikováno šest cyklů terapie, bylo zjištěno několik druhů novotvarů, včetně karcinomu mléčné žlázy, kožního keratoakanthomu a adenomu bazálních buněk, zatímco ve studiích u psů žádné nádorové ani prenádorové změny pozorovány nebyly. Zdá se, že potkani jsou vůči onkogenním účinkům TMZ mimořádně citliví, neboť první tumory se u nich objevují již po třech měsících od zahájení podávání léku. Jde o dobu krátkou dokonce i pro alkylační látky.
Výsledky aberačních testů s chromosomy Ames/salmonella a lymfocytů z lidské periferní krve prokázaly pozitivní mutagenní reakci.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky: laktóza,
koloidní bezvodý oxid křemičitý, sodná sůl karboxymethylškrobu typu A, kyselina vinná, kyselina stearová.
Obal tobolky: želatina,
oxid titaničitý (E171), natrium-lauryl-sulfát, indigokarmín (E 132),
Potisk:
šelak,
propylenglykol, čištěná voda, roztok amoniaku, hydroxid draselný, černý oxid železitý (E 172).
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Balení lahvička
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
Balení sáček
Uchovávejte při teplotě do 30°C.
6.5 Druh obalu a obsah balení
Balení lahvička
Skleněná lahvička třídy I jantarové barvy s polypropylenovými uzávěry jištěnými před dětmi, obsahujícími 5 nebo 20 tvrdých tobolek.
Papírová skládačka (krabička) obsahuje jednu lahvičku.
Balení sáček
Sáčky jsou složené z lineárního nízkohustotního polyethylenu (nejvnitřnější vrstva), aluminia a polyethylen-tereftalátu.
Jeden sáček obsahuje 1 tvrdou tobolku a je dodáván v papírové krabičce.
Papírová skládačka obsahuje 5 nebo 20 tvrdých tobolek, jednotlivě zatavených v sáčcích.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Tobolky by neměly být otevřeny. Pokud dojde k poškození celistvosti tobolky, zabraňte kontaktu jejího práškového obsahu s kůží a sliznicemi. Pokud přijde Temodal do styku s kůží nebo sliznicí, je třeba neprodleně a řádně ho smýt mýdlem a vodou.
Pacientům by mělo být doporučeno uchovávat tobolky mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/98/096/009
EU/1/98/096/010
EU/1/98/096/017
EU/1/98/096/018
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 26. ledna 1999.
Datum posledního prodloužení registrace: 26. ledna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Temodal 180 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje temozolomidum 180 mg.
Pomocná látka se známým účinkem:
Jedna tvrdá tobolka obsahuje 316,3 mg bezvodé laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka (tobolka).
Tvrdé tobolky mají neprůsvitné bílé tělo, neprůsvitné oranžové víčko a jsou potištěny černým inkoustem. Na víčku je vytištěn nápis „Temodal“. Na těle tobolky je vytištěn nápis „180 mg“, logo společnosti Schering-Plough a dva proužky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Temodal je určen k léčbě:
- dospělých pacientů s nově diagnostikovaným multiformním glioblastomem souběžně s radioterapií (RT) a následně jako monoterapeutická léčba.
- dětí ve věku od tří let, dospívajících a dospělých pacientů s maligním gliomem, jako je multiformní glioblastom nebo anaplastický astrocytom, u nichž se po standardní léčbě projevují recidivy nebo progrese.
4.2 Dávkování a způsob podání
Temodal smí být předepisován výhradně lékařem se zkušenostmi s onkologickou léčbou mozkových
nádorů.
Může se podávat antiemetická léčba (viz bod 4.4).
Dávkování
Dospělí _pacienti s nově diagnostikovaným multiformním glioblastomem
Temodal je podáván v kombinaci s fokální radioterapií (fáze souběžné léčby) s následnými až 6 cykly monoterapie temozolomidem (TMZ) (monoterapeutická fáze).
Fáze souběžné léčby
TMZ se podává perorálně v dávce 75 mg/m2 denně po dobu 42 dnů souběžně s fokální radioterapií (60 Gy podáno ve 30 frakcích). Snižování dávek se nedoporučuje, ale každý týden by se mělo rozhodnout o pozdržení nebo přerušení podávání TMZ podle kritérií hematologické a nehematologické toxicity. Podávání TMZ může pokračovat po celou dobu 42 dnů souběžné fáze (až do 49 dnů), jestliže jsou splněny všechny následující podmínky:
- absolutní počet neutrofilů (ANC) > 1,5 x 109/l
- počet trombocytů > 100 x 109/l
- všeobecná kritéria toxicity (CTC - common toxicity criteria) nehematologické toxicity < stupeň 1 (s výjimkou alopecie, nevolnosti a zvracení).
Během léčby by se měl každý týden vyšetřit celkový krevní obraz. Podávání TMZ by mělo být během fáze souběžné léčby dočasně přerušeno nebo trvale ukončeno podle kritérií hematologické a nehematologické toxicity, jak je uvedeno v Tabulce 1.
|
Tabulka 1. Přerušení nebo ukončení podávání TMZ během souběžné léčby radioterapií a TMZ | ||
|
Toxicita |
TMZ přerušení3 |
TMZ ukončení |
|
Absolutní počet neutrofilů |
> 0,5 a <1,5 x 109/l |
< 0,5 x 109/l |
|
Počet trombocytů |
> 10 a < 100 x 109/l |
<10 x 109/l |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 2 |
CTC stupeň 3 nebo 4 |
a: Léčba se souběžně podávaným TMZ může pokračovat, jestliže jsou splněny všechny následující podmínky: absolutní počet neutrofilů > 1,5 x 109/l; počet trombocytů > 100 x 109/l; CTC nehematologická toxicita < stupeň 1 (s výjimkou alopecie, nausey, zvracení).
Monoterapeutická fáze
Čtyři týdny po ukončené souběžné fázi TMZ + RT se TMZ podává až v 6 cyklech monoterapeutické léčby. Dávka Cyklu 1 (monoterapie) je 150 mg/m2 jednou denně po dobu 5 dnů, následovaná 23 dny bez léčby. Na začátku Cyklu 2 je dávka zvýšena na 200 mg/m2, jestliže CTC nehematologická toxicita pro Cyklus 1 je stupeň < 2 (s výjimkou alopecie, nausey a zvracení), absolutní počet neutrofilů (ANC) je > 1,5 x 109/l a počet trombocytů je > 100 x 109/l. Jestliže není dávka zvýšena v Cyklu 2, nemělo by být zvýšení prováděno v následných cyklech. Je-li jednou zvýšena, zůstává dávka 200 mg/m2 na den pro prvních 5 dnů každého následujícího cyklu s výjimkou vzniku toxicity. Snížení dávky a ukončení podávání v průběhu monoterapeutické fáze by se mělo provádět podle Tabulek 2 a 3.
V průběhu léčby by měl být ve dni 22 (21 dnů po první dávce TMZ) vyšetřen celkový krevní obraz. Dávka by měla být redukována nebo podávání přípravku ukončeno podle Tabulky 3.
|
Tabulka 2. Úrovně dávky TMZ pro monoterapeutickou léčbu | ||
|
Úroveň dávky |
Dávka TMZ (mg/m2/den) |
Poznámky |
|
-1 |
100 |
Redukce kvůli předchozí toxicitě |
|
0 |
150 |
Dávka během Cyklu 1 |
|
1 |
200 |
Dávka během Cyklů 2-6 při absenci toxicity |
|
Tabulka 3. Snížení dávky nebo ukončení podávání TMZ během monoterapeutické léčby | ||
|
Toxicita |
Redukce TMZ o 1 úroveň dávkya |
Ukončení TMZ |
|
Absolutní počet neutrofilů |
<1,0 x 109/l |
Viz poznámka pod čarou b |
|
Počet trombocytů |
< 50 x 109/l |
Viz poznámka pod čarou b |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 3 |
CTC stupeň 4b |
a: Úrovně dávky TMZ jsou uvedeny v Tabulce 2.
b: TMZ se ukončí jestliže:
• výsledkem úrovně dávky -1 (100 mg/m2) je stále neakceptovatelná toxicita
• jestliže se po redukci dávky znovu objeví stejný stupeň 3 nehematologické toxicity (s výjimkou alopecie, nausey, zvracení).
Dospělí_pacienti a děti ve věku 3 let nebo starší s recidivujícím nebo _progresivním maligním gliomem:
Léčebný cyklus zahrnuje 28 dnů. U pacientů, kteří dosud nebyli léčeni chemoterapií, se TMZ podává perorálně v dávce 200 mg/m2 jedenkrát denně po dobu prvních 5 dnů s následným přerušením léčby po dobu 23 dnů (celkem 28 dnů). U nemocných, kteří již dříve prodělali chemoterapii, činí zahajovací dávka 150 mg/m2 jedenkrát denně, která se pak ve druhém cyklu zvyšuje na 200 mg/m2 jedenkrát denně po dobu 5 dnů, pokud nejsou přítomny známky hematotoxicity (viz bod 4.4).
Zvláštní _ populace
Pediatrická populace
U dětí ve věku 3 let nebo starších by se měl TMZ používat pouze pro léčbu recidivujícího nebo progresivního maligního gliomu. Zkušenosti u těchto dětí jsou velmi omezené (viz body 4.4 a 5.1). Bezpečnost a účinnost TMZ u dětí ve věku do 3 let nebyla stanovena. Nejsou dostupné žádné údaje.
Pacienti s poruchou funkce jater či ledvin
Farmakokinetika TMZ je u pacientů s normální jaterní funkcí a s mírnou nebo středně těžkou jaterní dysfunkcí srovnatelná. Údaje o podání TMZ u pacientů s těžkou jaterní dysfunkcí (Třída C podle Childovy klasifikace) ani s dysfunkcí ledvin nejsou k dispozici. Vzhledem k uvedeným farmakokinetickým vlastnostem TMZ je však pravděpodobné, že ani u nemocných s těžkou jaterní dysfunkcí nebo dysfunkcí ledvin kteréhokoli stupně není snižování jeho dávek nutné. Při podávání TMZ těmto nemocným je však nutná zvýšená opatrnost.
Starší pacienti
Z populační analýzy farmakokinetiky u pacientů ve věku 19-78 let vyplývá, že clearance TMZ není ovlivňována věkem. Nicméně se zdá, že starší pacienti (ve věku > 70 let) mají vyšší riziko vzniku neutropenie a trombocytopenie (viz bod 4.4).
Způsob podání
Temodal tvrdé tobolky by měl podáván nalačno.
Tobolky se musejí polykat vcelku a zapíjet sklenicí vody a nesmějí se otevírat nebo kousat.
Pokud se po podání dávky dostaví zvracení, neměla by být další dávka podána tentýž den.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Hypersenzitivita na dakarbazin (DTIC).
Těžká myelosuprese (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Oportunní infekce a reaktivace infekcí
Během léčby pomocí TMZ byly pozorovány oportunní infekce (jako je pneumonie způsobená Pneumocystis jirovecii) a reaktivace infekcí (jako jsou HBV, CMV) (viz bod 4.8).
Pneumonie způsobená Pneumocystis _ jirovecii
U pacientů, kteří dostávali souběžně TMZ a RT v pilotní studii s prodlouženým harmonogramem 42 dnů, bylo zjištěno zvýšené riziko vzniku pneumonie způsobené Pneumocystis jirovecii (PCP). Proto je nutná profylaxe PCP u všech pacientů, kteří dostávají souběžně TMZ a RT po dobu 42 denního režimu (maximálně 49 dnů) bez ohledu na počet lymfocytů. Jestliže se objeví lymfopenie, je třeba pokračovat v profylaxi, dokud se neobnoví stupeň lymfopenie < 1.
Výskyt PCP může být vyšší, když je TMZ podáván během delšího dávkovacího režimu. Nicméně všichni pacienti, kteří dostávají TMZ, zvláště pacienti dostávající steroidy, by měli být pečlivě sledováni pro vznik PCP, bez ohledu na režim. U pacientů užívajících TMZ, zvláště v kombinaci s dexamethasonem nebo jinými steroidy, byly hlášeny případy fatálního respiračního selhání.
HBV
Byla hlášena hepatitida způsobená reaktivací viru hepatitidy B (HBV), v některých případech vedoucí ke smrti. Před zahájením léčby pacientů s pozitivní sérologií na hepatitidu B (včetně těch s aktivním onemocněním) je nezbytná konzultace s odborníky na onemocnění jater. Během léčby musí být pacienti náležitě monitorováni a vedeni.
Hepatotoxicita
U pacientů léčených TMZ bylo hlášeno poškození jater, včetně smrtelného selhání jater (viz bod 4.8). Před zahájením léčby je nutno provést testy jaterních funkcí. Pokud jsou výsledky abnormální, musí lékař před zahájením léčby temozolomidem vyhodnotit poměr přínosů a rizik, včetně potenciálu ke smrtelnému selhání jater. U pacientů se 42denním léčebným cyklem se musí testy jaterních funkcí opakovat uprostřed tohoto cyklu. Pro všechny pacienty platí, že jaterní funkce je nutno zkontrolovat po každém léčebném cyklu. U pacientů s výraznými abnormalitami jaterních funkcí musí lékař vyhodnotit poměr přínosů a rizik vyplývajících z pokračování v léčbě. Jaterní toxicita se může objevit několik nebo i více týdnů po poslední léčbě temozolomidem.
Malignity
Byly též velmi vzácně hlášeny případy myelodysplastického syndromu a sekundárních malignit, včetně myeloidní leukémie (viz bod 4.8).
Antiemetická léčba
Nausea a zvracení jsou velmi často spojené s TMZ.
Před nebo po podání TMZ se může podávat antiemetická léčba.
Antiemetická profylaxe se doporučuje před počáteční dávkou souběžné fáze a_velmi doporučuje během monoterapeutické fáze.
Pacienti s recidivujícím nebo _progresivním maligním gliomem
Antiemetika může být třeba podat pacientům, u nichž v předchozích léčebných cyklech došlo k těžšímu zvracení (stupeň 3 nebo 4).
Laboratorní parametry
U pacientů léčených TMZ může dojít k myelosupresi, včetně dlouhotrvající pancytopenie, která může vést k aplastické anémii, jež v některých případech skončila fatálně. V některých případech souběžná expozice léčivým přípravkům spojených s výskytem aplastické anémie, zahrnující karbamazepin, fenytoin a sulfamethoxazol/trimethoprim, hodnocení komplikuje. Před zahájením podávání léku by měly být splněny následující laboratorní parametry: ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Kompletní krevní obraz je třeba vyšetřit ve 22. dni (21 dnů po podání 1. dávky) nebo v průběhu 48 hodin po tomto dni, a pak v týdenních intervalech do té doby, dokud není ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Jestliže v průběhu kteréhokoli cyklu poklesne ANC na < 1,0 x 109/l nebo počet trombocytů na < 50 x 109/l, měla by být v následném cyklu dávka o jeden stupeň snížena (viz bod 4.2). Jednotlivé stupně dávky činí 100 mg/m2, 150 mg/m2 a 200 mg/m2. Nejnižší doporučená dávka je 100 mg/m2.
Pediatrická populace
S podáváním TMZ dětem ve věku nižším než 3 roky nejsou dosud žádné klinické zkušenosti. U starších dětí a dospívajících jsou zkušenosti velmi omezené (viz body 4.2 a 5.1).
Starší pacienti (ve věku >70 let)
U starších pacientů je ve srovnání s mladšími osobami pravděpodobně vyšší riziko neutropenie a trombocytopenie. Vzhledem k tomu je při podávání TMZ ve vyšším věku nutná zvýšená opatrnost.
Pacienti mužského pohlaví
Muži léčení TMZ by měli být upozorněni, aby se do 6 měsíců od užití poslední dávky nestali otci a aby se před zahájením léčby informovali o možnosti kryokonzervace spermatu (viz bod 4.6).
Laktóza
Tento léčivý přípravek obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo glukózo-galaktózovou malabsorpcí by proto neměli tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Podávání TMZ spolu s ranitidinem nemělo v oddělené studii fáze I za následek žádné změny rozsahu absorpce temozolomidu nebo expozice jeho aktivnímu metabolitu monomethyl- triazenoimidazol-karboxamidu (MTIC).
Podávání TMZ spolu s jídlem vedlo k poklesu Cmax o 33 % a zmenšení plochy pod křivkou koncentrace (AUC) o 9 %.
Protože nelze vyloučit, že změna Cmax může být klinicky významná, neměl by se Temodal podávat současně s jídlem.
Výsledky analýzy populační farmakokinetiky ve II. fázi klinických studií prokazují, že nebyla clearance TMZ ovlivněna současným podáním dexamethasonu, prochlorperazinu, fenytoinu, karbamazepinu, ondansetronu, antagonistů H2 receptorů nebo fenobarbitalu. Současné podávání kyseliny valproové bylo spojeno s malým, nicméně statisticky významným poklesem clearance TMZ.
Dosud nebyly provedeny žádné studie, které by hodnotily účinek TMZ na metabolismus nebo eliminaci jiných léků. Nicméně vzhledem k tomu, že TMZ neprochází přeměnou v játrech a na plazmatické bílkoviny se váže jen v malé míře, je málo pravděpodobné, že by ovlivňoval farmakokinetiku jiných léčivých přípravků (viz bod 5.2).
V kombinaci s jinými myelosupresivními látkami může TMZ zvýšit pravděpodobnost myelosuprese. Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání léku těhotným ženám nejsou k dispozici. V preklinických studiích u potkanů a králíků, kteří TMZ dostávali v dávce 150 mg/m2, byly prokázány teratogenní a/nebo toxické účinky na plod (viz bod 5.3). Temodal by neměl být podáván těhotným ženám. Pokud je přece jen nutno zvážit použití během těhotenství, je třeba pacientku seznámit s potenciálním rizikem pro plod.
Kojení
Není známo, zda se TMZ vylučuje do mateřského mléka; kojení by se proto mělo po dobu léčby TMZ přerušit.
Ženy ve fertilním věku.
Ženám ve fertilním věku je třeba doporučit používání účinné antikoncepce k zabránění otěhotnění v průběhu užívání TMZ.
Mužská plodnost
TMZ může mít genotoxické účinky. Proto se mužům léčeným temozolomidem nedoporučuje, aby se do 6 měsíců od užití poslední dávky stali otci a naopak se doporučuje, vzhledem k možnosti ireverzibilní neplodnosti po léčbě TMZ, ještě před jejím zahájením konzultovat s lékařem možnost kryokonzervace předem odebraných spermií.
4.7 Účinky na schopnost řídit a obsluhovat stroje
TMZ má malý vliv na schopnost řídit nebo obsluhovat stroje v důsledku únavy a ospalosti (viz bod 4.8.).
4.8 Nežádoucí účinky
Zkušenosti z klinických hodnocení
U pacientů léčených TMZ ať už užívaným v kombinaci s RT nebo ve formě monoterapie následující po RT v případě nově diagnostikovaného multiformního glioblastomu nebo ve formě monoterapie pacientů s recidivujícím nebo progresivním gliomem, byly velmi často hlášené nežádoucí účinky podobné: nausea, zvracení, zácpa, anorexie, bolest hlavy a únava. Křeče byly hlášeny velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených monoterapií, vyrážka byla hlášena velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených TMZ v kombinaci s RT nebo také ve formě monoterapie a často u pacientů s recidivujícím gliomem. Většina hematologických nežádoucích účinků byla hlášena u obou indikací často nebo velmi často (Tabulky 4 a 5); četnost laboratorních nálezů stupně 3 a 4 je uvedena za každou tabulkou.
V tabulkách jsou nežádoucí účinky klasifikovány podle orgánových systémů a četnosti. Zařazení do skupin četností se provádí podle následujícího pravidla: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Nově diagnostikovaný multiformní glioblastom
Tabulka 4 ukazuje nežádoucí účinky, které se objevily při léčbě u pacientů s nově diagnostikovaným multiformním glioblastomem během souběžné a monoterapeutické fáze léčby.
|
Tabulka 4: Nežádoucí účinky, které se objevily během souběžné a monoterapeutické léčby u pacientů s nově diagnostikovaným multiformním glioblastomem | ||
|
Třída orgánového systému |
TMZ + souběžná RT n=288* |
TMZ monoterapie n=224 |
|
Infekce a infestace | ||
|
Časté: |
Infekce, Herpes simplex, infekce rány, faryngitida, orální kandidóza |
Infekce, orální kandidóza |
|
Méně časté: |
Herpes simplex, herpes zoster, symptomy podobné chřipce | |
|
Poruchy krve a lymfatického systému | ||
|
Časté: |
Neutropenie, trombocytopenie, lymfopenie, leukopenie |
Febrilní neutropenie, trombocytopenie, anémie, leukopenie |
|
Méně časté: |
Febrilní neutropenie, anémie |
Lymfopenie, petechie |
|
Endokrinní poruchy | ||
|
Méně časté: |
Cushingoid |
Cushingoid |
|
Poruchy metabolismu a výživy | ||
|
Velmi časté: | ||
|
Časté: |
Hyperglykémie, pokles hmotnosti |
Pokles hmotnosti |
|
Méně časté: |
Hypokalémie, zvýšení alkalické fosfatázy, zvýšení hmotnosti |
Hyperglykémie, zvýšení hmotnosti |
|
Psychiatrické poruchy | ||
|
Časté: |
Úzkost, emoční labilita, nespavost | |
|
Méně časté: |
Neklid, apatie, poruchy chování, deprese, halucinace |
Halucinace, amnézie |
|
Poruchy nervového systému | ||
|
Velmi časté: |
Konvulze, bolest hlavy | |
|
Časté: |
Konvulze, porucha vědomí, somnolence, afázie, porucha rovnováhy, závrať, zmatenost, porucha paměti, porucha koncentrace, neuropatie, parestézie, porucha řeči, třes |
Hemiparéza, afázie, porucha rovnováhy, somnolence, zmatenost, závrať, porucha paměti, porucha koncentrace, dysfázie, neurologická porucha (NOS), neuropatie, periferní neuropatie, parestézie, porucha řeči, třes |
|
Méně časté: |
Status epilepticus, extrapyramidální porucha, hemiparéza, ataxie, poruchy kognice, dysfázie, abnormální chůze, hyperestézie, hypestézie, neurologická porucha (NOS), periferní neuropatie |
Hemiplegie, ataxie, abnormální koordinace, abnormální chůze, hyperestézie, smyslová porucha |
|
Poruchy oka | ||
|
Časté: |
Rozmazané vidění |
Defekt zorného pole, rozmazané vidění, diplopie |
|
Méně časté: |
Hemianopie, zmenšená ostrost vidění, poruchy zraku, defekt zorného pole, bolest očí |
Zmenšená ostrost vidění, bolest očí, suché oči |
|
Poruchy ucha a labyrintu | ||
|
Časté: |
Porucha sluchu |
Porucha sluchu, tinitus |
|
Méně časté: |
Otitis media, tinitus, hyperakuze, bolest ucha |
Hluchota, závrať, bolest ucha |
|
Srdeční poruchy | ||
|
Méně časté: |
Palpitace | |
|
Cévní poruchy | ||
|
Časté: |
Hemoragie, edém, edém nohy |
Hemoragie, hluboká žilní trombóza, edém nohy |
|
Méně časté: |
Cerebrální hemoragie, hypertenze |
Plicní embolie, edém, periferní edém |
|
Respirační, hrudní a mediastinální poruchy | ||
|
Časté: | ||
|
Méně časté: |
Pneumonie, infekce horních cest dýchacích, nosní kongesce |
Pneumonie, sinusitida, infekce horních cest dýchacích, bronchitida |
|
Gastrointestinální poruchy | ||
|
Velmi časté: |
Zácpa, nausea, zvracení |
Zácpa, nausea, zvracení |
|
Časté: |
Stomatitida, průjem, bolest břicha, dyspepsie, dysfagie | |
|
Méně časté: |
Abdominální distenze, fekální inkontinence, gastrointestinální porucha (NOS), gastroenteritida, hemeroidy | |
|
Poruchy kůže a podkožní tkáně | ||
|
Velmi časté: |
Vyrážka, alopecie |
Vyrážka, alopecie |
|
Časté: |
Dermatitida, suchá kůže, erytém, svědění |
Suchá kůže, svědění |
|
Méně časté: |
Exfoliace kůže, fotosenzitivní reakce, abnormální pigmentace |
Erytém, abnormální pigmentace, zvýšené pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Časté: |
Svalová slabost, artralgie |
Svalová slabost, artralgie, muskuloskeletální bolest, myalgie |
|
Méně časté: |
Myopatie, bolest v zádech, muskuloskeletální bolest, myalgie |
Myopatie, bolest v zádech |
|
Poruchy ledvin a močových cest | ||
|
Časté: |
Časté močení, močová inkontinence |
Močová inkontinence |
|
Méně časté: | ||
|
Poruchy reprodukčního systému a prsu | ||
|
Méně časté: |
Impotence |
Vaginální hemoragie, menoragie, amenorea, vaginitida, bolest prsou |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Velmi časté: |
Únava |
Únava |
|
Časté: |
Alergická reakce, horečka, radiační poškození, edém obličeje, bolest, změna chuti |
Alergická reakce, horečka, radiační poškození, bolest, změna chuti |
|
Méně časté: |
Astenie, zrudnutí, návaly horka, zhoršení stavu, rigory, změna barvy jazyka, parosmie, žízeň |
Astenie, edém obličeje, bolest, zhoršení stavu, rigory, zubní potíže |
|
Vyšetření | ||
|
Časté: |
Zvýšené ALT |
Zvýšené ALT |
|
Méně časté: |
Zvýšené jaterní enzymy, zvýšené gama GT, zvýšené AST | |
*Pacient který byl randomizován pouze do větve RT, obdržel Temodal + RT.
Laboratorní výsledky
Byla zjištěna myelosuprese (neutropenie a trombocytopenie), která je známou toxicitou limitující dávku většiny cytotoxických látek včetně TMZ. Když byly zkombinovány abnormality laboratorních testů a nežádoucí účinky v souběžných a monoterapeutických fázích léčby, byly pozorovány u 8 % pacientů abnormality neutrofilů stupeň 3 nebo stupeň 4, včetně neutropenických případů. Stupeň 3 nebo stupeň 4 abnormalit trombocytů, včetně trombocytopenických případů, byl pozorován u 14 % pacientů, kteří dostávali TMZ.
Recidivující nebo progresivní maligní gliom
Nejčastěji se vyskytujícími nežádoucími účinky ve vztahu k léčbě byly v provedených klinických studiích gastrointestinální poruchy, především nausea (43 %) a zvracení (36 %). Tyto reakce dosahovaly obvykle pouze stupně 1 nebo 2 (0 - 5 epizod zvracení během 24 hodin) a buď spontánně odezněly, nebo byly snadno léčitelné standardní antiemetickou léčbou. Incidence těžké nausey a zvracení činila 4 %.
Tabulka 5 obsahuje nežádoucí účinky, které byly hlášeny v průběhu klinických studií pro recidivující nebo progresivní maligní gliom a po uvedení přípravku Temodal na trh.
|
Tabulka 5. Nežádoucí účinky u pacientů s recidivujícím nebo progresivním maligním gliomem | |
|
Infekce a infestace | |
|
Vzácné: |
Oportunní infekce včetně pneumonie způsobené PCP |
|
Poruchy krve a lymfatického systému | |
|
Velmi časté: |
Neutropenie nebo lymfopenie (stupeň 3-4) trombocytopenie (stupeň 3-4) |
|
Méně časté: |
Pancytopenie, anémie (stupeň 3-4), leukopenie |
|
Poruchy metabolismu a výživy | |
|
Velmi časté: | |
|
Časté: |
Pokles hmotnosti |
|
Poruchy nervového systému | |
|
Velmi časté: | |
|
Časté: |
Somnolence, závratě, parestézie |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté: | |
|
Gastrointestinální poruchy | |
|
Velmi časté: |
Zvracení, nausea, zácpa |
|
Časté: | |
|
Poruchy kůže a podkožní tkáně | |
|
Časté: |
Vyrážka, pruritus, alopecie, |
|
Velmi vzácné: |
Multiformní erytém, erytrodermie, urtikarie, exantém |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté: |
Únava |
|
Časté: | |
|
Velmi vzácné: |
Alergické reakce, včetně anafylaxe, angioedém |
Laboratorní výsledky
Trombocytopenie a neutropenie stupně 3 nebo 4 byly zaznamenány u 19 %, resp. 17 % pacientů léčených pro maligní gliom. K hospitalizaci a/nebo přerušení léčby TMZ vedly v 8 %, resp. 4 % případů. Myelosuprese byla předvídatelná (obvykle k ní došlo během několika prvních cyklů, s nadirem mezi 21. dnem a 28. dnem) a úprava k normě byla rychlá, obvykle v průběhu 1-2 týdnů. Kumulace myelosupresivních účinků prokázána nebyla. Přítomnost trombocytopenie může zvýšit riziko krvácení a přítomnost neutropenie nebo leukopenie může zvýšit riziko infekce.
Pohlaví
Do analýzy populační farmakokinetiky založené na zkušenostech z klinického hodnocení bylo zahrnuto 101 žen a 169 mužů, u kterých byla dostupná měření nadiru neutrofilů, a 110 žen a 174 mužů, u kterých byla dostupná měření nadiru krevních destiček. U žen bylo ve srovnání s muži v prvním cyklu terapie zaznamenáno více neutropenií stupně 4 (ANC< 0,5 x 109/l), 12 % vs 5 %, a trombocytopenií (< 20 x 109/l), 9 % vs 3 %. V souboru dat od 400 jedinců s rekurentním gliomem se v prvním cyklu terapie objevila neutropenie stupně 4 u 8 % žen vs 4 % mužů a trombocytopenie stupně 4 u 8 % žen vs 3 % mužů. Ve studii s 288 jedinci s nově diagnostikovaným multiformním glioblastomem se v prvním cyklu terapie vyskytla neutropenie stupně 4 u 3 % žen vs 0 % mužů a trombocytopenie stupně 4 u 1 % žen vs 0 % mužů.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s rekurentním gliomem mozkového kmene nebo rekurentním astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Ačkoliv jsou údaje omezené, tolerance u dětí se očekává být podobná jako u dospělých. Bezpečnost TMZ u dětí ve věku do 3 let nebyla stanovena.
Zkušenosti po uvedení na trh
Po uvedení přípravku na trh byly hlášeny další následující závažné nežádoucí účinky:
|
Tabulka 6. Souhrn příhod hlášených u temozolomidu po jeho uvedení na trh | |
|
Infekce a infestace* | |
|
Méně časté: |
infekce způsobená cytomegaloviry, reaktivace infekce způsobené virem, jako je cytomegalovirus, virus hepatitidy BŤ |
|
Poruchy krve a lymfatického systému | |
|
Velmi vzácné: |
dlouhotrvající pancytopenie, aplastická anémieŤ |
|
Novotvary benigní, maligní a blíže neurčené | |
|
Velmi vzácné: |
myelodysplastický syndrom (MDS), sekundární malignity, včetně myeloidní leukémie |
|
Endokrinní poruchy | |
|
Méně časté: |
diabetes insipidus |
|
Respirační, hrudní a mediastinální poruchy | |
|
Velmi vzácné: |
intersticiální pneumonitida/pneumonitida, plicní fibróza, respirační selhání1 |
|
Poruchy jater a žlučových cest* | |
|
Časté: |
zvýšení jaterních enzymů |
|
Méně časté: |
hyperbilirubinemie, cholestáza, hepatitida, poškození jater, selhání jater1 |
|
Poruchy kůže a podkožní tkáně | |
|
Velmi vzácné: |
toxická epidermální nekrolýza, Stevens-Johnsonův syndrom |
t Včetně smrtelných případů
Frekvence odhadované na základě relevantních klinických studií.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinicky byly u pacientů ověřeny dávky 500, 750, 1 000, a 1 250 mg/m2 (celková dávka za 5-denní cyklus). Toxicita limitující dávku byla toxicita hematologická a byla pozorována u všech dávek, ale lze očekávat, že při vyšších dávkách bude závažnější. U jednoho pacienta došlo k předávkování, když užil dávku 10 000 mg (celková dávka během jednoho 5-denního cyklu), nežádoucí účinky hlášené v tomto případě byly pancytopenie, pyrexie, multiorgánové selhání a smrt. Byly popsány případy, kdy pacienti doporučenou dávku užívali po dobu delší než 5 dnů (až 64 dnů) s hlášenými nežádoucími příhodami včetně suprese kostní dřeně s nebo bez rozvoje infekce, v některých případech závažné, dlouhotrvající a vedoucí ke smrti. V případě předávkování je nezbytné hematologické zhodnocení. Podle potřeby by měla být zajištěna podpůrná léčba.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: protinádorové léky - ostatní alkylační látky, ATC kód: L01A X03 Mechanismus účinku
Temozolomid je triazen, který za fyziologické hodnoty pH prochází rychlou chemickou konverzí na aktivní monometyl-triazenoimidazol-karboxamid (MTIC). Předpokládá se, že cytotoxické účinky MTIC jsou primárně důsledkem alkylace na O6 pozici guaninu, s následnou alkylací rovněž na pozici N7. Následně vznikající cytotoxické léze jsou vysvětlovány nejspíše aberantní reparací methylovaného zbytku.
Klinická účinnost a bezpečnost
Nově diagnostikovaný multiformní glioblastom
Celkem 573 pacientů bylo randomizováno do skupin dostávajících buď TMZ + RT (n=287) nebo RT samotnou (n=286). Pacienti ve větvi TMZ + RT dostávali souběžně TMZ (75 mg/m2) jednou denně od prvního až do posledního dne RT po dobu 42 dnů (maximálně 49 dnů). Pak následovala monoterapie TMZ (150 - 200 mg/m2) ve dnech 1-5 každého 28-denního cyklu, až v 6 cyklech, začínající 4 týdny po ukončení RT. Pacienti v kontrolní větvi dostávali pouze RT. Během RT a souběžné TMZ terapie byla nutná profylaxe pneumonie Pneumocystis jirovecii (PCP).
TMZ byl podáván jako záchranná terapie vnásledné fázi 161 pacientům z 282 (57 %) ve větvi dostávající pouze RT a 62 pacientům z 277 (22 %) ve větvi dostávající TMZ + RT.
Poměr rizika (HR) pro celkové přežívání byl 1,59 (95 % CI pro HR=1,33 - 1,91) s log-rank p < 0,0001 ve prospěch TMZ větve. Odhadnutá pravděpodobnost přežívání 2 roky nebo více (26 % vs 10 %) je vyšší ve větvi RT + TMZ. Přidání souběžně podávaného TMZ k RT následované monoterapií TMZ v léčbě pacientů s nově diagnostikovaným multiformním glioblastomem prokázalo statisticky významné zlepšení v celkovém přežívání (overall survival=OS) ve srovnání s RT samotnou (Graf 1).
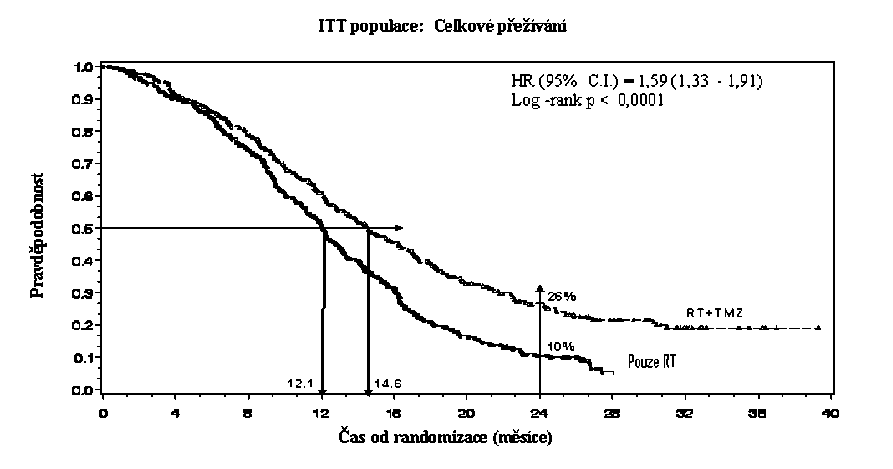
Graf 1 Kaplanovy-Meierovy křivky pro celkové přežívání (ITTpopulace=intent-to- treatpopulation)
Výsledky studie nebyly konzistentní v podskupině pacientů se slabým výkonnostním statusem (WHO PS=2, n=70), kde celkové přežívání a čas do progrese byly podobné v obou větvích. Zdá se však, že u této skupiny pacientů nejsou přítomna žádná nepřijatelná rizika.
Recidivující nebo _progresivní maligní gliom
Data o klinické účinnosti u pacientů s multiformním glioblastomem (Karnofského výkonnostní status [KPS] > 70), u něhož dochází k progresi nebo recidivě po chirurgickém výkonu a RT, vycházela ze dvou klinických studií s perorálním TMZ. V prvním případě šlo o nekomparativní studii u 138 pacientů (29 % bylo předtím léčeno chemoterapií), ve druhém o randomizovanou studii kontrolovanou léčivou látkou s TMZ vs prokarbazinem u celkem 225 pacientů (67 % z nich bylo předtím léčeno chemoterapií na bázi nitrosourey). V obou studiích bylo primárním hodnotícím kritériem přežívání bez známek progrese (PFS - progression-free survival), hodnocených na základě zobrazení magnetickou rezonancí (MRI) nebo na základě zhoršení neurologických funkcí. V nekomparativní studii bylo u 19 % pacientů dosaženo 6měsíční PFS, medián přežívání bez známek progrese byl 2,1 měsíce a medián celkové doby přežívání byl 5,4 měsíce. Objektivní počet terapeutických odpovědí na základě hodnocení MRI dosahoval 8 %.
V randomizované studii kontrolované léčivou látkou bylo šestiměsíční PFS u pacientů léčených TMZ významně vyšší než u těch, kteří dostávali prokarbazin (21 % vs 8 %; chí-kvadrát p=0,008)
s mediánem 2,89, respektive 1,88 měsíce (log rank p=0,0063). Medián přežití byl 7,34 měsíce pro TMZ a 5,66 měsíce pro prokarbazin (log rank p=0,33). Po 6 měsících byl podíl přežívajících pacientů v TMZ skupině významně vyšší (60 %) než ve skupině prokarbazinové (44 %; chí-kvadrát p=0,019). U pacientů, kteří byli předtím léčeni chemoterapií, bylo dosaženo kladného efektu u těch, kteří měli KPS > 80.
Údaje o době do zhoršení neurologického statusu, jakož i o době do zhoršení výkonnostního statusu (pokles na KPS < 70 nebo pokles nejméně o 30 bodů), hovoří ve prospěch TMZ oproti prokarbazinu. Medián doby do progrese v těchto hodnotících kritériích byl u TMZ o 0,7 - 2,1 měsíce delší než u prokarbazinu (log rank p < 0,01 - 0,03).
Recidivující anaplastický astrocytom
V multicentrické prospektivní studii fáze II, v níž byly hodnoceny bezpečnost a účinnost perorálně podávaného TMZ v léčbě pacientů s anaplastickým astrocytomem při prvním relapsu, bylo zjištěno šestiměsíční PSF u 46 % léčených. Medián PSF činil 5,4 měsíce. Medián celkového přežití činil
14,6 měsíce. Procento odpovědí na léčbu, hodnocené centrálně činilo 35 % (13 % kompletních, 43 % částečných odpovědí) pro populaci intent-to-treat (ITT), n=162. U 43 pacientů došlo ke stabilizaci onemocnění. Šestiměsíční přežití bez klinické příhody bylo pro populaci intent-to-treat 44 %, přičemž medián přežití bez klinické příhody činil 4,6 měsíce, což je podobné výsledkům hodnocení přežívání bez známek progrese. Z histologického hlediska byla účinnost obdobná. Objektivní radiologicky prokázaná odpověď na léčbu nebo zachování stavu beze známek progrese velmi dobře korelovaly se zachovanou nebo zlepšenou kvalitou života.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s recidivujícím gliomem mozkového kmene nebo recidivujícím astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Tolerance TMZ byla podobná jako u dospělých.
5.2 Farmakokinetické vlastnosti
TMZ se spontánně hydrolyzuje při fyziologickém pH primárně na aktivní látku, 3-metyl-(triazen-1-yl)imidazol-4-karboxamid (MTIC). MTIC se spontánně hydrolyzuje na 5-amino-imidazol-4-karboxamid (AIC), známý meziprodukt biosyntézy purinů a nukleových kyselin a na metylhydrazin, o kterém se předpokládá, že je aktivní alkylační látkou. Předpokládá se, že cytotoxicita MTIC je
primárně způsobena alkylací DNA zejména na pozicích guaninu O6 a N7' Expozice MTIC a AIC je ~
2,4 %, respektive 23 % v poměru k AUC TMZ. In vivo byl ti/2 MTIC podobný jako pro TMZ,
1,8 hodin.
Absorpce
Po perorálním podání dospělým pacientům se TMZ rychle absorbuje a vrcholné koncentrace dosahuje již za 20 minut po podání (střední doba je mezi 0,5 a 1,5 hodiny). Po perorálním podání 14C-značeného TMZ byla průměrná fekální exkrece 14C v průběhu 7 dnů po podání dávky 0,8 %, což svědčilo o úplné absorpci.
Distribuce v organismu
TMZ vykazuje nízkou vazebnou afinitu k proteinům (10 % až 20 %) proto se neočekává, že by interagoval s látkami s vysokou vazebnou afinitou k proteinům.
PET studie u lidí a preklinické údaje ukazují, že TMZ rychle prostupuje hematoencefalickou bariérou a je přítomný v mozkomíšním moku. Průnik do mozkomíšního moku (CSF) byl potvrzen u jednoho pacienta; expozice CSF na základě AUC TMZ byla přibližně 30 % plazmatické expozice, což odpovídá údajům u zvířat.
Eliminace z organismu
Plazmatický poločas (t1/2) je přibližně 1,8 hodiny. Hlavní způsob eliminace 14C je ledvinami. Po perorálním podání se objevuje přibližně 5 až 10 % dávky v nezměněné podobě v moči v průběhu 24 hodin a zbytek se vylučuje ve formě temozolomidové kyseliny, 5-aminoimidazol-4-karboxamidu (AIC) nebo neidentifikovaných polárních metabolitů.
Plazmatické koncentrace vzrůstají v závislosti na velikosti dávky. Plazmatická clearance, distribuční objem a poločas jsou na velikosti dávky nezávislé.
Zvláštní populace
V analýze populační farmakokinetiky TMZ bylo zjištěno, že plazmatická clearance TMZ nezávisí na věku, renální funkci nebo kouření. V oddělené farmakokinetické studii byly plazmatické farmakokinetické profily u pacientů s mírnou až střední jaterní dysfunkcí stejné jako profily u pacientů s normální funkcí jater.
U dětských pacientů byla AUC větší než u dospělých; nicméně maximální tolerovaná dávka (MTD) činila 1 000 mg/m2 na jeden cyklus jak u dětí, tak u dospělých.
5.3 Předklinické údaje vztahující se k bezpečnosti
U potkanů a psů byly prováděny studie toxicity s jednotlivým cyklem (5 dnů podávání léku, 23 dnů bez léčby) 3 a 6 cyklů léčby. Primární cíle toxických účinků tvořila kostní dřeň, lymforetikulární systém, testes, gastrointestinální ústrojí a při vyšších dávkách, které byly pro 60 % - 100 % testovaných potkanů a psů letální, docházelo k degeneraci retiny. Většina toxických účinků byla reverzibilní, s výjimkou nežádoucích účinků na mužský reprodukční systém a degeneraci retiny. Nicméně vzhledem k tomu, že dávky podmiňující retinální degeneraci se pohybovaly v rozmezí dávek letálních, a v klinických studiích nebyl žádný srovnatelný účinek pozorován, nepřikládá se tomuto nálezu klinická významnost.
TMZ je embryotoxická, teratogenní a genotoxická alkylační látka. TMZ má toxické účinky větší u potkanů a psů než u člověka, a klinická dávka se blíží minimální letální dávce u potkanů a psů. Za citlivé ukazatele toxicity se považují poklesy počtu leukocytů a trombocytů v závislosti na velikosti dávky. Ve studii u potkanů, v níž bylo aplikováno šest cyklů terapie, bylo zjištěno několik druhů novotvarů, včetně karcinomu mléčné žlázy, kožního keratoakanthomu a adenomu bazálních buněk, zatímco ve studiích u psů žádné nádorové ani prenádorové změny pozorovány nebyly. Zdá se, že potkani jsou vůči onkogenním účinkům TMZ mimořádně citliví, neboť první tumory se u nich objevují již po třech měsících od zahájení podávání léku. Jde o dobu krátkou dokonce i pro alkylační látky.
Výsledky aberačních testů s chromosomy Ames/salmonella a lymfocytů z lidské periferní krve prokázaly pozitivní mutagenní reakci.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky: laktóza,
koloidní bezvodý oxid křemičitý, sodná sůl karboxymethylškrobu typu A, kyselina vinná, kyselina stearová.
Obal tobolky: želatina,
oxid titaničitý (E171), natrium-lauryl-sulfát, žlutý oxid železitý (E 172), červený oxid železitý (E 172),
Potisk:
šelak,
propylenglykol, čištěná voda, roztok amoniaku, hydroxid draselný, černý oxid železitý (E 172).
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Balení lahvička
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
Balení sáček
Uchovávejte při teplotě do 30°C.
6.5 Druh obalu a obsah balení
Balení lahvička
Skleněná lahvička třídy I jantarové barvy s polypropylenovými uzávěry jištěnými před dětmi, obsahujícími 5 nebo 20 tvrdých tobolek.
Papírová skládačka (krabička) obsahuje jednu lahvičku.
Balení sáček
Sáčky jsou složené z lineárního nízkohustotního polyethylenu (nejvnitřnější vrstva), aluminia a polyethylen-tereftalátu.
Jeden sáček obsahuje 1 tvrdou tobolku a je dodáván v papírové krabičce.
Papírová skládačka obsahuje 5 nebo 20 tvrdých tobolek, jednotlivě zatavených v sáčcích.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Tobolky by neměly být otevřeny. Pokud dojde k poškození celistvosti tobolky, zabraňte kontaktu jejího práškového obsahu s kůží a sliznicemi. Pokud přijde Temodal do styku s kůží nebo sliznicí, je třeba neprodleně a řádně ho smýt mýdlem a vodou.
Pacientům by mělo být doporučeno uchovávat tobolky mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/98/096/011
EU/1/98/096/012
EU/1/98/096/019
EU/1/98/096/020
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 26. ledna 1999.
Datum posledního prodloužení registrace: 26. ledna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Temodal 250 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje temozolomidum 250 mg.
Pomocná látka se známým účinkem:
Jedna tvrdá tobolka obsahuje 154,3 mg bezvodé laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka (tobolka).
Tvrdé tobolky mají neprůsvitné bílé tělo a víčko a jsou potištěny černým inkoustem. Na víčku je vytištěn nápis „Temodal“. Na těle tobolky je vytištěn nápis „250 mg“, logo společnosti Schering-Plough a dva proužky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Temodal je určen k léčbě:
- dospělých pacientů s nově diagnostikovaným multiformním glioblastomem souběžně s radioterapií (RT) a následně jako monoterapeutická léčba.
- dětí ve věku od tří let, dospívajících a dospělých pacientů s maligním gliomem, jako je multiformní glioblastom nebo anaplastický astrocytom, u nichž se po standardní léčbě projevují recidivy nebo progrese.
4.2 Dávkování a způsob podání
Temodal smí být předepisován výhradně lékařem se zkušenostmi s onkologickou léčbou mozkových
nádorů.
Může se podávat antiemetická léčba (viz bod 4.4).
Dávkování
Dospělí _pacienti s nově diagnostikovaným multiformním glioblastomem
Temodal je podáván v kombinaci s fokální radioterapií (fáze souběžné léčby) s následnými až 6 cykly monoterapie temozolomidem (TMZ) (monoterapeutická fáze).
Fáze souběžné léčby
TMZ se podává perorálně v dávce 75 mg/m2 denně po dobu 42 dnů souběžně s fokální radioterapií (60 Gy podáno ve 30 frakcích). Snižování dávek se nedoporučuje, ale každý týden by se mělo rozhodnout o pozdržení nebo přerušení podávání TMZ podle kritérií hematologické a nehematologické toxicity. Podávání TMZ může pokračovat po celou dobu 42 dnů souběžné fáze (až do 49 dnů), jestliže jsou splněny všechny následující podmínky:
- absolutní počet neutrofilů (ANC) > 1,5 x 109/l
- počet trombocytů > 100 x 109/l
- všeobecná kritéria toxicity (CTC - common toxicity criteria) nehematologické toxicity < stupeň 1 (s výjimkou alopecie, nevolnosti a zvracení).
Během léčby by se měl každý týden vyšetřit celkový krevní obraz. Podávání TMZ by mělo být během fáze souběžné léčby dočasně přerušeno nebo trvale ukončeno podle kritérií hematologické a nehematologické toxicity, jak je uvedeno v Tabulce 1.
|
Tabulka 1. Přerušení nebo ukončení podávání TMZ během souběžné léčby radioterapií a TMZ | ||
|
Toxicita |
TMZ přerušení3 |
TMZ ukončení |
|
Absolutní počet neutrofilů |
> 0,5 a <1,5 x 109/l |
< 0,5 x 109/l |
|
Počet trombocytů |
> 10 a < 100 x 109/l |
<10 x 109/l |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 2 |
CTC stupeň 3 nebo 4 |
a: Léčba se souběžně podávaným TMZ může pokračovat, jestliže jsou splněny všechny následující podmínky: absolutní počet neutrofilů > 1,5 x 109/l; počet trombocytů > 100 x 109/l; CTC nehematologická toxicita < stupeň 1 (s výjimkou alopecie, nausey, zvracení).
Monoterapeutická fáze
Čtyři týdny po ukončené souběžné fázi TMZ + RT se TMZ podává až v 6 cyklech monoterapeutické léčby. Dávka Cyklu 1 (monoterapie) je 150 mg/m2 jednou denně po dobu 5 dnů, následovaná 23 dny bez léčby. Na začátku Cyklu 2 je dávka zvýšena na 200 mg/m2, jestliže CTC nehematologická toxicita pro Cyklus 1 je stupeň < 2 (s výjimkou alopecie, nausey a zvracení), absolutní počet neutrofilů (ANC) je > 1,5 x 109/l a počet trombocytů je > 100 x 109/l. Jestliže není dávka zvýšena v Cyklu 2, nemělo by být zvýšení prováděno v následných cyklech. Je-li jednou zvýšena, zůstává dávka 200 mg/m2 na den pro prvních 5 dnů každého následujícího cyklu s výjimkou vzniku toxicity. Snížení dávky a ukončení podávání v průběhu monoterapeutické fáze by se mělo provádět podle Tabulek 2 a 3.
V průběhu léčby by měl být ve dni 22 (21 dnů po první dávce TMZ) vyšetřen celkový krevní obraz. Dávka by měla být redukována nebo podávání přípravku ukončeno podle Tabulky 3.
|
Tabulka 2. Úrovně dávky TMZ pro monoterapeutickou léčbu | ||
|
Úroveň dávky |
Dávka TMZ (mg/m2/den) |
Poznámky |
|
-1 |
100 |
Redukce kvůli předchozí toxicitě |
|
0 |
150 |
Dávka během Cyklu 1 |
|
1 |
200 |
Dávka během Cyklů 2-6 při absenci toxicity |
|
Tabulka 3. Snížení dávky nebo ukončení podávání TMZ během monoterapeutické léčby | ||
|
Toxicita |
Redukce TMZ o 1 úroveň dávkya |
Ukončení TMZ |
|
Absolutní počet neutrofilů |
<1,0 x 109/l |
Viz poznámka pod čarou b |
|
Počet trombocytů |
< 50 x 109/l |
Viz poznámka pod čarou b |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 3 |
CTC stupeň 4b |
a: Úrovně dávky TMZ jsou uvedeny v Tabulce 2.
b: TMZ se ukončí jestliže:
• výsledkem úrovně dávky -1 (100 mg/m2) je stále neakceptovatelná toxicita
• jestliže se po redukci dávky znovu objeví stejný stupeň 3 nehematologické toxicity (s výjimkou alopecie, nausey, zvracení).
Dospělí_pacienti a děti ve věku 3 let nebo starší s recidivujícím nebo _progresivním maligním gliomem:
Léčebný cyklus zahrnuje 28 dnů. U pacientů, kteří dosud nebyli léčeni chemoterapií, se TMZ podává perorálně v dávce 200 mg/m2 jedenkrát denně po dobu prvních 5 dnů s následným přerušením léčby po dobu 23 dnů (celkem 28 dnů). U nemocných, kteří již dříve prodělali chemoterapii, činí zahajovací dávka 150 mg/m2 jedenkrát denně, která se pak ve druhém cyklu zvyšuje na 200 mg/m2 jedenkrát denně po dobu 5 dnů, pokud nejsou přítomny známky hematotoxicity (viz bod 4.4).
Zvláštní _ populace
Pediatrická populace
U dětí ve věku 3 let nebo starších by se měl TMZ používat pouze pro léčbu recidivujícího nebo progresivního maligního gliomu. Zkušenosti u těchto dětí jsou velmi omezené (viz body 4.4 a 5.1). Bezpečnost a účinnost TMZ u dětí ve věku do 3 let nebyla stanovena. Nejsou dostupné žádné údaje.
Pacienti s poruchou funkce jater či ledvin
Farmakokinetika TMZ je u pacientů s normální jaterní funkcí a s mírnou nebo středně těžkou jaterní dysfunkcí srovnatelná. Údaje o podání TMZ u pacientů s těžkou jaterní dysfunkcí (Třída C podle Childovy klasifikace) ani s dysfunkcí ledvin nejsou k dispozici. Vzhledem k uvedeným farmakokinetickým vlastnostem TMZ je však pravděpodobné, že ani u nemocných s těžkou jaterní dysfunkcí nebo dysfunkcí ledvin kteréhokoli stupně není snižování jeho dávek nutné. Při podávání TMZ těmto nemocným je však nutná zvýšená opatrnost.
Starší pacienti
Z populační analýzy farmakokinetiky u pacientů ve věku 19-78 let vyplývá, že clearance TMZ není ovlivňována věkem. Nicméně se zdá, že starší pacienti (ve věku > 70 let) mají vyšší riziko vzniku neutropenie a trombocytopenie (viz bod 4.4).
Způsob podání
Temodal tvrdé tobolky by měl podáván nalačno.
Tobolky se musejí polykat vcelku a zapíjet sklenicí vody a nesmějí se otevírat nebo kousat.
Pokud se po podání dávky dostaví zvracení, neměla by být další dávka podána tentýž den.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Hypersenzitivita na dakarbazin (DTIC).
Těžká myelosuprese (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Oportunní infekce a reaktivace infekcí
Během léčby pomocí TMZ byly pozorovány oportunní infekce (jako je pneumonie způsobená Pneumocystis jirovecii) a reaktivace infekcí (jako jsou HBV, CMV) (viz bod 4.8).
Pneumonie způsobená Pneumocystis _ jirovecii
U pacientů, kteří dostávali souběžně TMZ a RT v pilotní studii s prodlouženým harmonogramem 42 dnů, bylo zjištěno zvýšené riziko vzniku pneumonie způsobené Pneumocystis jirovecii (PCP). Proto je nutná profylaxe PCP u všech pacientů, kteří dostávají souběžně TMZ a RT po dobu 42 denního režimu (maximálně 49 dnů) bez ohledu na počet lymfocytů. Jestliže se objeví lymfopenie, je třeba pokračovat v profylaxi, dokud se neobnoví stupeň lymfopenie < 1.
Výskyt PCP může být vyšší, když je TMZ podáván během delšího dávkovacího režimu. Nicméně všichni pacienti, kteří dostávají TMZ, zvláště pacienti dostávající steroidy, by měli být pečlivě sledováni pro vznik PCP, bez ohledu na režim. U pacientů užívajících TMZ, zvláště v kombinaci s dexamethasonem nebo jinými steroidy, byly hlášeny případy fatálního respiračního selhání.
HBV
Byla hlášena hepatitida způsobená reaktivací viru hepatitidy B (HBV), v některých případech vedoucí ke smrti. Před zahájením léčby pacientů s pozitivní sérologií na hepatitidu B (včetně těch s aktivním onemocněním) je nezbytná konzultace s odborníky na onemocnění jater. Během léčby musí být pacienti náležitě monitorováni a vedeni.
Hepatotoxicita
U pacientů léčených TMZ bylo hlášeno poškození jater, včetně smrtelného selhání jater (viz bod 4.8). Před zahájením léčby je nutno provést testy jaterních funkcí. Pokud jsou výsledky abnormální, musí lékař před zahájením léčby temozolomidem vyhodnotit poměr přínosů a rizik, včetně potenciálu ke smrtelnému selhání jater. U pacientů se 42denním léčebným cyklem se musí testy jaterních funkcí opakovat uprostřed tohoto cyklu. Pro všechny pacienty platí, že jaterní funkce je nutno zkontrolovat po každém léčebném cyklu. U pacientů s výraznými abnormalitami jaterních funkcí musí lékař vyhodnotit poměr přínosů a rizik vyplývajících z pokračování v léčbě. Jaterní toxicita se může objevit několik nebo i více týdnů po poslední léčbě temozolomidem.
Malignity
Byly též velmi vzácně hlášeny případy myelodysplastického syndromu a sekundárních malignit, včetně myeloidní leukémie (viz bod 4.8).
Antiemetická léčba:
Nausea a zvracení jsou velmi často spojené s TMZ.
Před nebo po podání TMZ se může podávat antiemetická léčba.
Antiemetická profylaxe se doporučuje před počáteční dávkou souběžné fáze a velmi doporučuje během monoterapeutické fáze.
Pacienti s recidivujícím nebo _progresivním maligním gliomem
Antiemetika může být třeba podat pacientům, u nichž v předchozích léčebných cyklech došlo k těžšímu zvracení (stupeň 3 nebo 4).
Laboratorní parametry
U pacientů léčených TMZ může dojít k myelosupresi, včetně dlouhotrvající pancytopenie, která může vést k aplastické anémii, jež v některých případech skončila fatálně. V některých případech souběžná expozice léčivým přípravkům spojených s výskytem aplastické anémie, zahrnující karbamazepin, fenytoin a sulfamethoxazol/trimethoprim, hodnocení komplikuje. Před zahájením podávání léku by měly být splněny následující laboratorní parametry: ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Kompletní krevní obraz je třeba vyšetřit ve 22. dni (21 dnů po podání 1. dávky) nebo v průběhu 48 hodin po tomto dni, a pak v týdenních intervalech do té doby, dokud není ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Jestliže v průběhu kteréhokoli cyklu poklesne ANC na < 1,0 x 109/l nebo počet trombocytů na < 50 x 109/l, měla by být v následném cyklu dávka o jeden stupeň snížena (viz bod 4.2). Jednotlivé stupně dávky činí 100 mg/m2, 150 mg/m2 a 200 mg/m2. Nejnižší doporučená dávka je 100 mg/m2.
Pediatrická populace
S podáváním TMZ dětem ve věku nižším než 3 roky nejsou dosud žádné klinické zkušenosti. U starších dětí a dospívajících jsou zkušenosti velmi omezené (viz body 4.2 a 5.1).
Starší pacienti (ve věku >70 let)
U starších pacientů je ve srovnání s mladšími osobami pravděpodobně vyšší riziko neutropenie a trombocytopenie. Vzhledem k tomu je při podávání TMZ ve vyšším věku nutná zvýšená opatrnost.
Pacienti mužského pohlaví
Muži léčení TMZ by měli být upozorněni, aby se do 6 měsíců od užití poslední dávky nestali otci a aby se před zahájením léčby informovali o možnosti kryokonzervace spermatu (viz bod 4.6).
Laktóza
Tento léčivý přípravek obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo glukózo-galaktózovou malabsorpcí by proto neměli tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Podávání TMZ spolu s ranitidinem nemělo v oddělené studii fáze I za následek žádné změny rozsahu absorpce temozolomidu nebo expozice jeho aktivnímu metabolitu monomethyl- triazenoimidazol-karboxamidu (MTIC).
Podávání TMZ spolu s jídlem vedlo k poklesu Cmax o 33 % a zmenšení plochy pod křivkou koncentrace (AUC) o 9 %.
Protože nelze vyloučit, že změna Cmax může být klinicky významná, neměl by se Temodal podávat současně s jídlem.
Výsledky analýzy populační farmakokinetiky ve II. fázi klinických studií prokazují, že nebyla clearance TMZ ovlivněna současným podáním dexamethasonu, prochlorperazinu, fenytoinu, karbamazepinu, ondansetronu, antagonistů H2 receptorů nebo fenobarbitalu. Současné podávání kyseliny valproové bylo spojeno s malým, nicméně statisticky významným poklesem clearance TMZ.
Dosud nebyly provedeny žádné studie, které by hodnotily účinek TMZ na metabolismus nebo eliminaci jiných léků. Nicméně vzhledem k tomu, že TMZ neprochází přeměnou v játrech a na plazmatické bílkoviny se váže jen v malé míře, je málo pravděpodobné, že by ovlivňoval farmakokinetiku jiných léčivých přípravků (viz bod 5.2).
V kombinaci s jinými myelosupresivními látkami může TMZ zvýšit pravděpodobnost myelosuprese. Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání léku těhotným ženám nejsou k dispozici. V preklinických studiích u potkanů a králíků, kteří TMZ dostávali v dávce 150 mg/m2, byly prokázány teratogenní a/nebo toxické účinky na plod (viz bod 5.3). Temodal by neměl být podáván těhotným ženám. Pokud je přece jen nutno zvážit použití během těhotenství, je třeba pacientku seznámit s potenciálním rizikem pro plod.
Kojení
Není známo, zda se TMZ vylučuje do mateřského mléka; kojení by se proto mělo po dobu léčby TMZ přerušit.
Ženy ve fertilním věku.
Ženám ve fertilním věku je třeba doporučit používání účinné antikoncepce k zabránění otěhotnění v průběhu užívání TMZ.
Mužská plodnost
TMZ může mít genotoxické účinky. Proto se mužům léčeným temozolomidem nedoporučuje, aby se do 6 měsíců od užití poslední dávky stali otci a naopak se doporučuje, vzhledem k možnosti ireverzibilní neplodnosti po léčbě TMZ, ještě před jejím zahájením konzultovat s lékařem možnost kryokonzervace předem odebraných spermií.
4.7 Účinky na schopnost řídit a obsluhovat stroje
TMZ má malý vliv na schopnost řídit nebo obsluhovat stroje v důsledku únavy a ospalosti (viz bod 4.8.).
4.8 Nežádoucí účinky
Zkušenosti z klinických hodnocení
U pacientů léčených TMZ ať už užívaným v kombinaci s RT nebo ve formě monoterapie následující po RT v případě nově diagnostikovaného multiformního glioblastomu nebo ve formě monoterapie pacientů s recidivujícím nebo progresivním gliomem, byly velmi často hlášené nežádoucí účinky podobné: nausea, zvracení, zácpa, anorexie, bolest hlavy a únava. Křeče byly hlášeny velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených monoterapií, vyrážka byla hlášena velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených TMZ v kombinaci s RT nebo také ve formě monoterapie a často u pacientů s recidivujícím gliomem. Většina hematologických nežádoucích účinků byla hlášena u obou indikací často nebo velmi často (Tabulky 4 a 5); četnost laboratorních nálezů stupně 3 a 4 je uvedena za každou tabulkou.
V tabulkách jsou nežádoucí účinky klasifikovány podle orgánových systémů a četnosti. Zařazení do skupin četností se provádí podle následujícího pravidla: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Nově diagnostikovaný multiformní glioblastom
Tabulka 4 ukazuje nežádoucí účinky, které se objevily při léčbě u pacientů s nově diagnostikovaným multiformním glioblastomem během souběžné a monoterapeutické fáze léčby.
|
Tabulka 4: Nežádoucí účinky, které se objevily během souběžné a monoterapeutické léčby u pacientů s nově diagnostikovaným multiformním glioblastomem | ||
|
Třída orgánového systému |
TMZ + souběžná RT n=288* |
TMZ monoterapie n=224 |
|
Infekce a infestace | ||
|
Časté: |
Infekce, Herpes simplex, infekce rány, faryngitida, orální kandidóza |
Infekce, orální kandidóza |
|
Méně časté: |
Herpes simplex, herpes zoster, symptomy podobné chřipce | |
|
Poruchy krve a lymfatického systému | ||
|
Časté: |
Neutropenie, trombocytopenie, lymfopenie, leukopenie |
Febrilní neutropenie, trombocytopenie, anémie, leukopenie |
|
Méně časté: |
Febrilní neutropenie, anémie |
Lymfopenie, petechie |
|
Endokrinní poruchy | ||
|
Méně časté: |
Cushingoid |
Cushingoid |
|
Poruchy metabolismu a výživy | ||
|
Velmi časté: | ||
|
Časté: |
Hyperglykémie, pokles hmotnosti |
Pokles hmotnosti |
|
Méně časté: |
Hypokalémie, zvýšení alkalické fosfatázy, zvýšení hmotnosti |
Hyperglykémie, zvýšení hmotnosti |
|
Psychiatrické poruchy | ||
|
Časté: |
Úzkost, emoční labilita, nespavost | |
|
Méně časté: |
Neklid, apatie, poruchy chování, deprese, halucinace |
Halucinace, amnézie |
|
Poruchy nervového systému | ||
|
Velmi časté: |
Konvulze, bolest hlavy | |
|
Časté: |
Konvulze, porucha vědomí, somnolence, afázie, porucha rovnováhy, závrať, zmatenost, porucha paměti, porucha koncentrace, neuropatie, parestézie, porucha řeči, třes |
Hemiparéza, afázie, porucha rovnováhy, somnolence, zmatenost, závrať, porucha paměti, porucha koncentrace, dysfázie, neurologická porucha (NOS), neuropatie, periferní neuropatie, parestézie, porucha řeči, třes |
|
Méně časté: |
Status epilepticus, extrapyramidální porucha, hemiparéza, ataxie, poruchy kognice, dysfázie, abnormální chůze, hyperestézie, hypestézie, neurologická porucha (NOS), periferní neuropatie |
Hemiplegie, ataxie, abnormální koordinace, abnormální chůze, hyperestézie, smyslová porucha |
|
Poruchy oka | ||
|
Časté: |
Rozmazané vidění |
Defekt zorného pole, rozmazané vidění, diplopie |
|
Méně časté: |
Hemianopie, zmenšená ostrost vidění, poruchy zraku, defekt zorného pole, bolest očí |
Zmenšená ostrost vidění, bolest očí, suché oči |
|
Poruchy ucha a labyrintu | ||
|
Časté: |
Porucha sluchu |
Porucha sluchu, tinitus |
|
Méně časté: |
Otitis media, tinitus, hyperakuze, bolest ucha |
Hluchota, závrať, bolest ucha |
|
Srdeční poruchy | ||
|
Méně časté: |
Palpitace | |
|
Cévní poruchy | ||
|
Časté: |
Hemoragie, edém, edém nohy |
Hemoragie, hluboká žilní trombóza, edém nohy |
|
Méně časté: |
Cerebrální hemoragie, hypertenze |
Plicní embolie, edém, periferní edém |
|
Respirační, hrudní a mediastinální poruchy | ||
|
Časté: | ||
|
Méně časté: |
Pneumonie, infekce horních cest dýchacích, nosní kongesce |
Pneumonie, sinusitida, infekce horních cest dýchacích, bronchitida |
|
Gastrointestinální poruchy | ||
|
Velmi časté: |
Zácpa, nausea, zvracení |
Zácpa, nausea, zvracení |
|
Časté: |
Stomatitida, průjem, bolest břicha, dyspepsie, dysfagie | |
|
Méně časté: |
Abdominální distenze, fekální inkontinence, gastrointestinální porucha (NOS), gastroenteritida, hemeroidy | |
|
Poruchy kůže a podkožní tkáně | ||
|
Velmi časté: |
Vyrážka, alopecie |
Vyrážka, alopecie |
|
Časté: |
Dermatitida, suchá kůže, erytém, svědění |
Suchá kůže, svědění |
|
Méně časté: |
Exfoliace kůže, fotosenzitivní reakce, abnormální pigmentace |
Erytém, abnormální pigmentace, zvýšené pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Časté: |
Svalová slabost, artralgie |
Svalová slabost, artralgie, muskuloskeletální bolest, myalgie |
|
Méně časté: |
Myopatie, bolest v zádech, muskuloskeletální bolest, myalgie |
Myopatie, bolest v zádech |
|
Poruchy ledvin a močových cest | ||
|
Časté: |
Časté močení, močová inkontinence |
Močová inkontinence |
|
Méně časté: | ||
|
Poruchy reprodukčního systému a prsu | ||
|
Méně časté: |
Impotence |
Vaginální hemoragie, menoragie, amenorea, vaginitida, bolest prsou |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Velmi časté: |
Únava |
Únava |
|
Časté: |
Alergická reakce, horečka, radiační poškození, edém obličeje, bolest, změna chuti |
Alergická reakce, horečka, radiační poškození, bolest, změna chuti |
|
Méně časté: |
Astenie, zrudnutí, návaly horka, zhoršení stavu, rigory, změna barvy jazyka, parosmie, žízeň |
Astenie, edém obličeje, bolest, zhoršení stavu, rigory, zubní potíže |
|
Vyšetření | ||
|
Časté: |
Zvýšené ALT |
Zvýšené ALT |
|
Méně časté: |
Zvýšené jaterní enzymy, zvýšené gama GT, zvýšené AST | |
*Pacient který byl randomizován pouze do větve RT, obdržel Temodal + RT.
Laboratorní výsledky
Byla zjištěna myelosuprese (neutropenie a trombocytopenie), která je známou toxicitou limitující dávku většiny cytotoxických látek včetně TMZ. Když byly zkombinovány abnormality laboratorních testů a nežádoucí účinky v souběžných a monoterapeutických fázích léčby, byly pozorovány u 8 % pacientů abnormality neutrofilů stupeň 3 nebo stupeň 4, včetně neutropenických případů. Stupeň 3 nebo stupeň 4 abnormalit trombocytů, včetně trombocytopenických případů, byl pozorován u 14 % pacientů, kteří dostávali TMZ.
Recidivující nebo progresivní maligní gliom
Nejčastěji se vyskytujícími nežádoucími účinky ve vztahu k léčbě byly v provedených klinických studiích gastrointestinální poruchy, především nausea (43 %) a zvracení (36 %). Tyto reakce dosahovaly obvykle pouze stupně 1 nebo 2 (0 - 5 epizod zvracení během 24 hodin) a buď spontánně odezněly, nebo byly snadno léčitelné standardní antiemetickou léčbou. Incidence těžké nausey a zvracení činila 4 %.
Tabulka 5 obsahuje nežádoucí účinky, které byly hlášeny v průběhu klinických studií pro recidivující nebo progresivní maligní gliom a po uvedení přípravku Temodal na trh.
|
Tabulka 5. Nežádoucí účinky u pacientů s recidivujícím nebo progresivním maligním gliomem | |
|
Infekce a infestace | |
|
Vzácné: |
Oportunní infekce včetně pneumonie způsobené PCP |
|
Poruchy krve a lymfatického systému | |
|
Velmi časté: |
Neutropenie nebo lymfopenie (stupeň 3-4) trombocytopenie (stupeň 3-4) |
|
Méně časté: |
Pancytopenie, anémie (stupeň 3-4), leukopenie |
|
Poruchy metabolismu a výživy | |
|
Velmi časté: | |
|
Časté: |
Pokles hmotnosti |
|
Poruchy nervového systému | |
|
Velmi časté: | |
|
Časté: |
Somnolence, závratě, parestézie |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté: | |
|
Gastrointestinální poruchy | |
|
Velmi časté: |
Zvracení, nausea, zácpa |
|
Časté: | |
|
Poruchy kůže a podkožní tkáně | |
|
Časté: |
Vyrážka, pruritus, alopecie, |
|
Velmi vzácné: |
Multiformní erytém, erytrodermie, urtikarie, exantém |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté: |
Únava |
|
Časté: | |
|
Velmi vzácné: |
Alergické reakce, včetně anafylaxe, angioedém |
Laboratorní výsledky
Trombocytopenie a neutropenie stupně 3 nebo 4 byly zaznamenány u 19 %, resp. 17 % pacientů léčených pro maligní gliom. K hospitalizaci a/nebo přerušení léčby TMZ vedly v 8 %, resp. 4 % případů. Myelosuprese byla předvídatelná (obvykle k ní došlo během několika prvních cyklů, s nadirem mezi 21. dnem a 28. dnem) a úprava k normě byla rychlá, obvykle v průběhu 1-2 týdnů. Kumulace myelosupresivních účinků prokázána nebyla. Přítomnost trombocytopenie může zvýšit riziko krvácení a přítomnost neutropenie nebo leukopenie může zvýšit riziko infekce.
Pohlaví
Do analýzy populační farmakokinetiky založené na zkušenostech z klinického hodnocení bylo zahrnuto 101 žen a 169 mužů, u kterých byla dostupná měření nadiru neutrofilů, a 110 žen a 174 mužů, u kterých byla dostupná měření nadiru krevních destiček. U žen bylo ve srovnání s muži v prvním cyklu terapie zaznamenáno více neutropenií stupně 4 (ANC< 0,5 x 109/l), 12 % vs 5 %, a trombocytopenií < 20 x 109/l), 9 % vs 3 %. V souboru dat od 400 jedinců s rekurentním gliomem se v prvním cyklu terapie objevila neutropenie stupně 4 u 8 % žen vs 4 % mužů a trombocytopenie stupně 4 u 8 % žen vs 3 % mužů. Ve studii s 288 jedinci s nově diagnostikovaným multiformním glioblastomem se v prvním cyklu terapie vyskytla neutropenie stupně 4 u 3 % žen vs 0 % mužů a trombocytopenie stupně 4 u 1 % žen vs 0 % mužů.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s rekurentním gliomem mozkového kmene nebo rekurentním astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Ačkoliv jsou údaje omezené, tolerance u dětí se očekává být podobná jako u dospělých. Bezpečnost TMZ u dětí ve věku do 3 let nebyla stanovena.
Zkušenosti po uvedení na trh
Po uvedení přípravku na trh byly hlášeny další následující závažné nežádoucí účinky:
|
Tabulka 6. Souhrn příhod hlášených u temozolomidu po jeho uvedení na trh | |
|
Infekce a infestace* | |
|
Méně časté: |
infekce způsobená cytomegaloviry, reaktivace infekce způsobené virem, jako je cytomegalovirus, virus hepatitidy BŤ |
|
Poruchy krve a lymfatického systému | |
|
Velmi vzácné: |
dlouhotrvající pancytopenie, aplastická anémieŤ |
|
Novotvary benigní, maligní a blíže neurčené | |
|
Velmi vzácné: |
myelodysplastický syndrom (MDS), sekundární malignity, včetně myeloidní leukémie |
|
Endokrinní poruchy | |
|
Méně časté: |
diabetes insipidus |
|
Respirační, hrudní a mediastinální poruchy | |
|
Velmi vzácné: |
intersticiální pneumonitida/pneumonitida, plicní fibróza, respirační selhání1 |
|
Poruchy jater a žlučových cest* | |
|
Časté: |
zvýšení jaterních enzymů |
|
Méně časté: |
hyperbilirubinemie, cholestáza, hepatitida, poškození jater, selhání jater1 |
|
Poruchy kůže a podkožní tkáně | |
|
Velmi vzácné: |
toxická epidermální nekrolýza, Stevens-Johnsonův syndrom |
t Včetně smrtelných případů
Frekvence odhadované na základě relevantních klinických studií.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinicky byly u pacientů ověřeny dávky 500, 750, 1 000, a 1 250 mg/m2 (celková dávka za 5-denní cyklus). Toxicita limitující dávku byla toxicita hematologická a byla pozorována u všech dávek, ale lze očekávat, že při vyšších dávkách bude závažnější. U jednoho pacienta došlo k předávkování, když užil dávku 10 000 mg (celková dávka během jednoho 5-denního cyklu), nežádoucí účinky hlášené v tomto případě byly pancytopenie, pyrexie, multiorgánové selhání a smrt. Byly popsány případy, kdy pacienti doporučenou dávku užívali po dobu delší než 5 dnů (až 64 dnů) s hlášenými nežádoucími příhodami včetně suprese kostní dřeně s nebo bez rozvoje infekce, v některých případech závažné, dlouhotrvající a vedoucí ke smrti. V případě předávkování je nezbytné hematologické zhodnocení. Podle potřeby by měla být zajištěna podpůrná léčba.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: protinádorové léky - ostatní alkylační látky, ATC kód: L01A X03 Mechanismus účinku
Temozolomid je triazen, který za fyziologické hodnoty pH prochází rychlou chemickou konverzí na aktivní monometyl-triazenoimidazol-karboxamid (MTIC). Předpokládá se, že cytotoxické účinky MTIC jsou primárně důsledkem alkylace na O6 pozici guaninu, s následnou alkylací rovněž na pozici N7. Následně vznikající cytotoxické léze jsou vysvětlovány nejspíše aberantní reparací methylovaného zbytku.
Klinická účinnost a bezpečnost
Nově diagnostikovaný multiformní glioblastom
Celkem 573 pacientů bylo randomizováno do skupin dostávajících buď TMZ + RT (n=287) nebo RT samotnou (n=286). Pacienti ve větvi TMZ + RT dostávali souběžně TMZ (75 mg/m2) jednou denně od prvního až do posledního dne RT po dobu 42 dnů (maximálně 49 dnů). Pak následovala monoterapie TMZ (150 - 200 mg/m2) ve dnech 1-5 každého 28-denního cyklu, až v 6 cyklech, začínající 4 týdny po ukončení RT. Pacienti v kontrolní větvi dostávali pouze RT. Během RT a souběžné TMZ terapie byla nutná profylaxe pneumonie Pneumocystis jirovecii (PCP).
TMZ byl podáván jako záchranná terapie vnásledné fázi 161 pacientům z 282 (57 %) ve větvi dostávající pouze RT a 62 pacientům z 277 (22 %) ve větvi dostávající TMZ + RT.
Poměr rizika (HR) pro celkové přežívání byl 1,59 (95 % CI pro HR=1,33 - 1,91) s log-rank p < 0,0001 ve prospěch TMZ větve. Odhadnutá pravděpodobnost přežívání 2 roky nebo více (26 % vs 10 %) je vyšší ve větvi RT + TMZ. Přidání souběžně podávaného TMZ k RT následované monoterapií TMZ v léčbě pacientů s nově diagnostikovaným multiformním glioblastomem prokázalo statisticky významné zlepšení v celkovém přežívání (overall survival=OS) ve srovnání s RT samotnou (Graf 1).
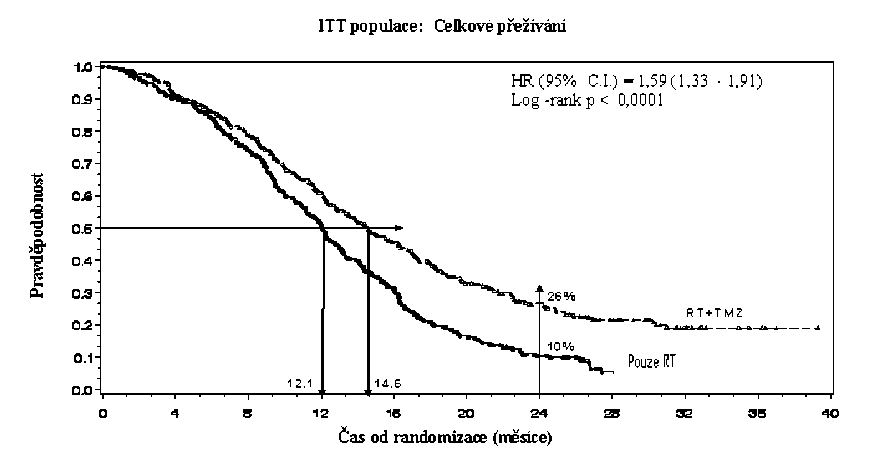
Graf 1 Kaplanovy-Meierovy křivky pro celkové přežívání (ITTpopulace=intent-to- treatpopulation)
Výsledky studie nebyly konzistentní v podskupině pacientů se slabým výkonnostním statusem (WHO PS=2, n=70), kde celkové přežívání a čas do progrese byly podobné v obou větvích. Zdá se však, že u této skupiny pacientů nejsou přítomna žádná nepřijatelná rizika.
Recidivující nebo _progresivní maligní gliom
Data o klinické účinnosti u pacientů s multiformním glioblastomem (Karnofského výkonnostní status [KPS] > 70), u něhož dochází k progresi nebo recidivě po chirurgickém výkonu a RT, vycházela ze dvou klinických studií s perorálním TMZ. V prvním případě šlo o nekomparativní studii u 138 pacientů (29 % bylo předtím léčeno chemoterapií), ve druhém o randomizovanou studii kontrolovanou léčivou látkou s TMZ vs prokarbazinem u celkem 225 pacientů (67 % z nich bylo předtím léčeno chemoterapií na bázi nitrosourey). V obou studiích bylo primárním hodnotícím kritériem přežívání bez známek progrese (PFS - progression-free survival), hodnocených na základě zobrazení magnetickou rezonancí (MRI) nebo na základě zhoršení neurologických funkcí. V nekomparativní studii bylo u 19 % pacientů dosaženo 6měsíční PFS, medián přežívání bez známek progrese byl 2,1 měsíce a medián celkové doby přežívání byl 5,4 měsíce. Objektivní počet terapeutických odpovědí na základě hodnocení MRI dosahoval 8 %.
V randomizované studii kontrolované léčivou látkou bylo šestiměsíční PFS u pacientů léčených TMZ významně vyšší než u těch, kteří dostávali prokarbazin (21 % vs 8 %; chí-kvadrát p=0,008)
s mediánem 2,89, respektive 1,88 měsíce (log rank p=0,0063). Medián přežití byl 7,34 měsíce pro TMZ a 5,66 měsíce pro prokarbazin (log rank p=0,33). Po 6 měsících byl podíl přežívajících pacientů v TMZ skupině významně vyšší (60 %) než ve skupině prokarbazinové (44 %; chí-kvadrát p=0,019). U pacientů, kteří byli předtím léčeni chemoterapií, bylo dosaženo kladného efektu u těch, kteří měli KPS > 80.
Údaje o době do zhoršení neurologického statusu, jakož i o době do zhoršení výkonnostního statusu (pokles na KPS < 70 nebo pokles nejméně o 30 bodů), hovoří ve prospěch TMZ oproti prokarbazinu. Medián doby do progrese v těchto hodnotících kritériích byl u TMZ o 0,7 - 2,1 měsíce delší než u prokarbazinu (log rank p < 0,01 - 0,03).
Recidivující anaplastický astrocytom
V multicentrické prospektivní studii fáze II, v níž byly hodnoceny bezpečnost a účinnost perorálně podávaného TMZ v léčbě pacientů s anaplastickým astrocytomem při prvním relapsu, bylo zjištěno šestiměsíční PSF u 46 % léčených. Medián PSF činil 5,4 měsíce. Medián celkového přežití činil
14,6 měsíce. Procento odpovědí na léčbu, hodnocené centrálně činilo 35 % (13 % kompletních, 43 % částečných odpovědí) pro populaci intent-to-treat (ITT), n=162. U 43 pacientů došlo ke stabilizaci onemocnění. Šestiměsíční přežití bez klinické příhody bylo pro populaci intent-to-treat 44 %, přičemž medián přežití bez klinické příhody činil 4,6 měsíce, což je podobné výsledkům hodnocení přežívání bez známek progrese. Z histologického hlediska byla účinnost obdobná. Objektivní radiologicky prokázaná odpověď na léčbu nebo zachování stavu beze známek progrese velmi dobře korelovaly se zachovanou nebo zlepšenou kvalitou života.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s recidivujícím gliomem mozkového kmene nebo recidivujícím astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Tolerance TMZ byla podobná jako u dospělých.
5.2 Farmakokinetické vlastnosti
TMZ se spontánně hydrolyzuje při fyziologickém pH primárně na aktivní látku, 3-metyl-(triazen-1-yl)imidazol-4-karboxamid (MTIC). MTIC se spontánně hydrolyzuje na 5-amino-imidazol-4-karboxamid (AIC), známý meziprodukt biosyntézy purinů a nukleových kyselin a na metylhydrazin, o kterém se předpokládá, že je aktivní alkylační látkou. Předpokládá se, že cytotoxicita MTIC je
primárně způsobena alkylací DNA zejména na pozicích guaninu O6 a N7' Expozice MTIC a AIC je ~
2,4 %, respektive 23 % v poměru k AUC TMZ. In vivo byl ti/2 MTIC podobný jako pro TMZ,
1,8 hodin.
Absorpce
Po perorálním podání dospělým pacientům se TMZ rychle absorbuje a vrcholné koncentrace dosahuje již za 20 minut po podání (střední doba je mezi 0,5 a 1,5 hodiny). Po perorálním podání 14C-značeného TMZ byla průměrná fekální exkrece 14C v průběhu 7 dnů po podání dávky 0,8 %, což svědčilo o úplné absorpci.
Distribuce v organismu
TMZ vykazuje nízkou vazebnou afinitu k proteinům (10 % až 20 %) proto se neočekává, že by interagoval s látkami s vysokou vazebnou afinitou k proteinům.
PET studie u lidí a preklinické údaje ukazují, že TMZ rychle prostupuje hematoencefalickou bariérou a je přítomný v mozkomíšním moku. Průnik do mozkomíšního moku (CSF) byl potvrzen u jednoho pacienta; expozice CSF na základě AUC TMZ byla přibližně 30 % plazmatické expozice, což odpovídá údajům u zvířat.
Eliminace z organismu
Plazmatický poločas (t1/2) je přibližně 1,8 hodiny. Hlavní způsob eliminace 14C je ledvinami. Po perorálním podání se objevuje přibližně 5 až 10 % dávky v nezměněné podobě v moči v průběhu 24 hodin a zbytek se vylučuje ve formě temozolomidové kyseliny, 5-aminoimidazol-4-karboxamidu (AIC) nebo neidentifikovaných polárních metabolitů.
Plazmatické koncentrace vzrůstají v závislosti na velikosti dávky. Plazmatická clearance, distribuční objem a poločas jsou na velikosti dávky nezávislé.
Zvláštní populace
V analýze populační farmakokinetiky TMZ bylo zjištěno, že plazmatická clearance TMZ nezávisí na věku, renální funkci nebo kouření. V oddělené farmakokinetické studii byly plazmatické farmakokinetické profily u pacientů s mírnou až střední jaterní dysfunkcí stejné jako profily u pacientů s normální funkcí jater.
U dětských pacientů byla AUC větší než u dospělých; nicméně maximální tolerovaná dávka (MTD) činila 1 000 mg/m2 na jeden cyklus jak u dětí, tak u dospělých.
5.3 Předklinické údaje vztahující se k bezpečnosti
U potkanů a psů byly prováděny studie toxicity s jednotlivým cyklem (5 dnů podávání léku, 23 dnů bez léčby) 3 a 6 cyklů léčby. Primární cíle toxických účinků tvořila kostní dřeň, lymforetikulární systém, testes, gastrointestinální ústrojí a při vyšších dávkách, které byly pro 60 % - 100 % testovaných potkanů a psů letální, docházelo k degeneraci retiny. Většina toxických účinků byla reverzibilní, s výjimkou nežádoucích účinků na mužský reprodukční systém a degeneraci retiny. Nicméně vzhledem k tomu, že dávky podmiňující retinální degeneraci se pohybovaly v rozmezí dávek letálních, a v klinických studiích nebyl žádný srovnatelný účinek pozorován, nepřikládá se tomuto nálezu klinická významnost.
TMZ je embryotoxická, teratogenní a genotoxická alkylační látka. TMZ má toxické účinky větší u potkanů a psů než u člověka, a klinická dávka se blíží minimální letální dávce u potkanů a psů. Za citlivé ukazatele toxicity se považují poklesy počtu leukocytů a trombocytů v závislosti na velikosti dávky. Ve studii u potkanů, v níž bylo aplikováno šest cyklů terapie, bylo zjištěno několik druhů novotvarů, včetně karcinomu mléčné žlázy, kožního keratoakanthomu a adenomu bazálních buněk, zatímco ve studiích u psů žádné nádorové ani prenádorové změny pozorovány nebyly. Zdá se, že potkani jsou vůči onkogenním účinkům TMZ mimořádně citliví, neboť první tumory se u nich objevují již po třech měsících od zahájení podávání léku. Jde o dobu krátkou dokonce i pro alkylační látky.
Výsledky aberačních testů s chromosomy Ames/salmonella a lymfocytů z lidské periferní krve prokázaly pozitivní mutagenní reakci.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky: laktóza,
koloidní bezvodý oxid křemičitý, sodná sůl karboxymethylškrobu typu A, kyselina vinná, kyselina stearová.
Obal tobolky: želatina,
oxid titaničitý (E171), natrium-lauryl-sulfát,
Potisk:
šelak,
propylenglykol, čištěná voda, roztok amoniaku, hydroxid draselný, černý oxid železitý (E 172).
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Balení lahvička
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
Balení sáček
Uchovávejte při teplotě do 30°C.
6.5 Druh obalu a obsah balení
Balení lahvička
Skleněná lahvička třídy I jantarové barvy s polypropylenovými uzávěry jištěnými před dětmi, obsahujícími 5 nebo 20 tvrdých tobolek.
Papírová skládačka (krabička) obsahuje jednu lahvičku.
Balení sáček
Sáčky jsou složené z lineárního nízkohustotního polyethylenu (nejvnitřnější vrstva), aluminia a polyethylen-tereftalátu.
Jeden sáček obsahuje 1 tvrdou tobolku a je dodáván v papírové krabičce.
Papírová skládačka obsahuje 5 nebo 20 tvrdých tobolek, jednotlivě zatavených v sáčcích.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Tobolky by neměly být otevřeny. Pokud dojde k poškození celistvosti tobolky, zabraňte kontaktu jejího práškového obsahu s kůží a sliznicemi. Pokud přijde Temodal do styku s kůží nebo sliznicí, je třeba neprodleně a řádně ho smýt mýdlem a vodou.
Pacientům by mělo být doporučeno uchovávat tobolky mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/98/096/007
EU/1/98/096/008
EU/1/98/096/021
EU/1/98/096/022
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 26. ledna 1999.
Datum posledního prodloužení registrace: 26. ledna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Temodal 2,5 mg/ml prášek pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje temozolomidum 100 mg.
Po rekonstituci obsahuje 1 ml infuzního roztoku 2,5 mg temozolomidu.
Pomocná látka se známým účinkem:
Jedna injekční lahvička obsahuje 2,4 mmol sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro infuzní roztok. Bílý prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Temodal je určen k léčbě:
- dospělých pacientů s nově diagnostikovaným multiformním glioblastomem souběžně s radioterapií (RT) a následně jako monoterapeutická léčba.
- dětí ve věku od tří let, dospívajících a dospělých pacientů s maligním gliomem, jako je multiformní glioblastom nebo anaplastický astrocytom, u nichž se po standardní léčbě projevují recidivy nebo progrese.
4.2 Dávkování a způsob podání
Temodal smí být předepisován výhradně lékařem se zkušenostmi s onkologickou léčbou mozkových
nádorů.
Může se podávat antiemetická léčba (viz bod 4.4).
Dávkování
Dospělí _pacienti s nově diagnostikovaným multiformním glioblastomem
Temodal je podáván v kombinaci s fokální radioterapií (fáze souběžné léčby) s následnými až 6 cykly monoterapie temozolomidem (TMZ) (monoterapeutická fáze).
Fáze souběžné léčby
TMZ se podává v dávce 75 mg/m2 denně po dobu 42 dnů souběžně s fokální radioterapií (60 Gy podáno ve 30 frakcích). Snižování dávek se nedoporučuje, ale každý týden by se mělo rozhodnout o pozdržení nebo přerušení podávání TMZ podle kritérií hematologické a nehematologické toxicity. Podávání TMZ může pokračovat po celou dobu 42 dnů souběžné fáze (až do 49 dnů), jestliže jsou splněny všechny následující podmínky:
• absolutní počet neutrofilů (ANC) > 1,5 x 109/l
• počet trombocytů > 100 x 109/l
• všeobecná kritéria toxicity (CTC - common toxicity criteria) nehematologické toxicity < stupeň 1 (s výjimkou alopecie, nevolnosti a zvracení).
Během léčby by se měl každý týden vyšetřit celkový krevní obraz. Podávání TMZ by mělo být během fáze souběžné léčby dočasně přerušeno nebo trvale ukončeno podle kritérií hematologické a nehematologické toxicity, jak je uvedeno v Tabulce 1.
|
Tabulka 1. Přerušení nebo ukončení podávání TMZ během souběžné léčby radioterapií a TMZ | ||
|
Toxicita |
TMZ přerušení3 |
TMZ ukončení |
|
Absolutní počet neutrofilů |
> 0,5 a <1,5 x 109/l |
< 0,5 x 109/l |
|
Počet trombocytů |
> 10 a < 100 x 109/l |
<10 x 109/l |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 2 |
CTC stupeň 3 nebo 4 |
a: Léčba se souběžně podávaným TMZ může pokračovat, jestliže jsou splněny všechny následující podmínky: absolutní počet neutrofilů > 1,5 x 109/l; počet trombocytů > 100 x 109/l; CTC nehematologická toxicita < stupeň 1 (s výjimkou alopecie, nausey, zvracení).
Monoterapeutická fáze
Čtyři týdny po ukončené souběžné fázi TMZ + RT se TMZ podává až v 6 cyklech monoterapeutické léčby. Dávka Cyklu 1 (monoterapie) je 150 mg/m2 jednou denně po dobu 5 dnů, následovaná 23 dny bez léčby. Na začátku Cyklu 2 je dávka zvýšena na 200 mg/m2, jestliže CTC nehematologická toxicita pro Cyklus 1 je stupeň < 2 (s výjimkou alopecie, nausey a zvracení), absolutní počet neutrofilů (ANC) je > 1,5 x 109/l a počet trombocytů je > 100 x 109/l. Jestliže není dávka zvýšena v Cyklu 2, nemělo by být zvýšení prováděno v následných cyklech. Je-li jednou zvýšena, zůstává dávka 200 mg/m2 na den pro prvních 5 dnů každého následujícího cyklu s výjimkou vzniku toxicity. Snížení dávky a ukončení podávání v průběhu monoterapeutické fáze by se mělo provádět podle Tabulek 2 a 3.
V průběhu léčby by měl být ve dni 22 (21 dnů po první dávce TMZ) vyšetřen celkový krevní obraz. Dávka by měla být redukována nebo podávání přípravku ukončeno podle Tabulky 3.
|
Tabulka 2. Dávky TMZ pro monoterapeutickou léčbu | ||
|
Dávka |
Dávka TMZ (mg/m2/den) |
Poznámky |
|
-1 |
100 |
Redukce kvůli předchozí toxicitě |
|
0 |
150 |
Dávka během Cyklu 1 |
|
1 |
200 |
Dávka během Cyklů 2-6 při absenci toxicity |
|
Tabulka 3. Snížení dávky nebo ukončení podávání TMZ během monoterapeutické léčby | ||
|
Toxicita |
Redukce TMZ o 1 úroveň dávkya |
Ukončení TMZ |
|
Absolutní počet neutrofilů |
<1,0 x 109/l |
Viz poznámka pod čarou b |
|
Počet trombocytů |
< 50 x 109/l |
Viz poznámka pod čarou b |
|
CTC nehematologická toxicita (s výjimkou alopecie, nausey, zvracení) |
CTC stupeň 3 |
CTC stupeň 4b |
a: Dávky TMZ jsou uvedeny v Tabulce 2. b: TMZ se ukončí jestliže:
• výsledkem úrovně dávky -1 (100 mg/m2) je stále neakceptovatelná toxicita
• jestliže se po redukci dávky znovu objeví stejný stupeň 3 nehematologické toxicity (s výjimkou alopecie, nausey, zvracení).
Léčebný cyklus zahrnuje 28 dnů. U pacientů, kteří dosud nebyli léčeni chemoterapií, se TMZ podává v dávce 200 mg/m2 jedenkrát denně po dobu prvních 5 dnů s následným přerušením léčby po dobu 23 dnů (celkem 28 dnů). U nemocných, kteří již dříve prodělali chemoterapii, činí zahajovací dávka 150 mg/m2 jedenkrát denně, která se pak ve druhém cyklu zvyšuje na 200 mg/m2 jedenkrát denně po dobu 5 dnů, pokud nejsou přítomny známky hematotoxicity (viz bod 4.4).
Zvláštní _ populace
Pediatrická populace
U dětí ve věku 3 let nebo starších by se měl TMZ používat pouze pro léčbu recidivujícího nebo progresivního maligního gliomu. Zkušenosti u těchto dětí jsou velmi omezené (viz body 4.4 a 5.1). Bezpečnost a účinnost TMZ u dětí ve věku do 3 let nebyla stanovena. Nejsou dostupné žádné údaje.
Pacienti s poruchou funkce jater či ledvin
Farmakokinetika TMZ je u pacientů s normální jaterní funkcí a s mírnou nebo středně těžkou jaterní dysfunkcí srovnatelná. Údaje o podání TMZ u pacientů s těžkou jaterní dysfunkcí (Třída C podle Childovy klasifikace) ani s dysfunkcí ledvin nejsou k dispozici. Vzhledem k uvedeným farmakokinetickým vlastnostem TMZ je však pravděpodobné, že ani u nemocných s těžkou jaterní dysfunkcí nebo dysfunkcí ledvin kteréhokoli stupně není snižování jeho dávek nutné. Při podávání TMZ těmto nemocným je však nutná zvýšená opatrnost.
Starší pacienti
Z populační analýzy farmakokinetiky u pacientů ve věku 19-78 let vyplývá, že clearance TMZ není ovlivňována věkem. Nicméně se zdá, že starší pacienti (ve věku > 70 let) mají vyšší riziko vzniku neutropenie a trombocytopenie (viz bod 4.4).
Způsob podání
Temodal 2,5 mg/ml prášek pro infuzní roztok by se měl podávat pouze intravenózní infuzí. Nesmí se podávat jinými způsoby aplikace, například intratekálně, nitrosvalově nebo do podkoží. Temodal
2,5 mg/ml prášek pro infuzní roztok lze podávat stejnou i.v. linkou s injekcí 0,9 % roztoku chloridu sodného. Není kompatibilní s roztoky glukózy.
Náležitá dávka přípravku Temodal by se měla podávat intravenózní infuzí pomocí pumpy během 90 minut.
Stejně jako u jiných podobných chemoterapeutik se doporučuje postupovat opatrně, aby se zabránilo extravazaci. U pacientů, kterým byl podáván Temodal 2,5 mg/ml prášek pro infuzní roztok, byly hlášeny lokální nežádoucí reakce v místě vpichu, většinou mírného stupně a krátkého trvání. Preklinické studie neprokázaly trvalé poškození tkáně (viz body 4.8 a 5.3).
Temodal je také k dispozici v lékové formě tvrdé tobolky (perorální podání). Temodal 2,5 mg/ml prášek pro infuzní roztok podaný intravenózní infuzí během 90 minut je bioekvivalentní lékové formě tvrdé tobolky (viz bod 5.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Hypersenzitivita na dakarbazin (DTIC).
Těžká myelosuprese (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Oportunní infekce a reaktivace infekcí
Během léčby pomocí TMZ byly pozorovány oportunní infekce (jako je pneumonie způsobená Pneumocystis jirovecii) a reaktivace infekcí (jako jsou HBV, CMV) (viz bod 4.8).
Pneumonie způsobená Pneumocystis _ jirovecii
U pacientů, kteří dostávali souběžně TMZ a RT v pilotní studii s prodlouženým harmonogramem 42 dnů, bylo zjištěno zvýšené riziko vzniku pneumonie způsobené Pneumocystis jirovecii (PCP). Proto je nutná profylaxe PCP u všech pacientů, kteří dostávají souběžně TMZ a RT po dobu 42 denního režimu (maximálně 49 dnů) bez ohledu na počet lymfocytů. Jestliže se objeví lymfopenie, je třeba pokračovat v profylaxi, dokud se neobnoví stupeň lymfopenie < 1.
Výskyt PCP může být vyšší, když je TMZ podáván během delšího dávkovacího režimu. Nicméně všichni pacienti, kteří dostávají TMZ, zvláště pacienti dostávající steroidy, by měli být pečlivě sledováni pro vznik PCP, bez ohledu na režim. U pacientů užívajících TMZ, zvláště v kombinaci s dexamethasonem nebo jinými steroidy, byly hlášeny případy fatálního respiračního selhání.
HBV
Byla hlášena hepatitida způsobená reaktivací viru hepatitidy B (HBV), v některých případech vedoucí ke smrti. Před zahájením léčby pacientů s pozitivní sérologií na hepatitidu B (včetně těch s aktivním onemocněním) je nezbytná konzultace s odborníky na onemocnění jater. Během léčby musí být pacienti náležitě monitorováni a vedeni.
Hepatotoxicita
U pacientů léčených TMZ bylo hlášeno poškození jater, včetně smrtelného selhání jater (viz bod 4.8). Před zahájením léčby je nutno provést testy jaterních funkcí. Pokud jsou výsledky abnormální, musí lékař před zahájením léčby temozolomidem vyhodnotit poměr přínosů a rizik, včetně potenciálu ke smrtelnému selhání jater. U pacientů se 42denním léčebným cyklem se musí testy jaterních funkcí opakovat uprostřed tohoto cyklu. Pro všechny pacienty platí, že jaterní funkce je nutno zkontrolovat po každém léčebném cyklu. U pacientů s výraznými abnormalitami jaterních funkcí musí lékař vyhodnotit poměr přínosů a rizik vyplývajících z pokračování v léčbě. Jaterní toxicita se může objevit několik nebo i více týdnů po poslední léčbě temozolomidem.
Malignity
Byly též velmi vzácně hlášeny případy myelodysplastického syndromu a sekundárních malignit, včetně myeloidní leukémie (viz bod 4.8).
Antiemetická léčba
Nausea a zvracení jsou velmi často spojené s TMZ.
Před nebo po podání TMZ se může podávat antiemetická léčba.
Dospělí _pacienti s nově diagnostikovaným multiformním glioblastomem
Antiemetická profylaxe se doporučuje před počáteční dávkou souběžné fáze a velmi doporučuje během monoterapeutické fáze.
Antiemetika může být třeba podat pacientům, u nichž v předchozích léčebných cyklech došlo k těžšímu zvracení (stupeň 3 nebo 4).
Laboratorní parametry
U pacientů léčených TMZ může dojít k myelosupresi, včetně dlouhotrvající pancytopenie, která může vést k aplastické anémii, jež v některých případech skončila fatálně. V některých případech souběžná expozice léčivým přípravkům spojených s výskytem aplastické anémie, zahrnující karbamazepin, fenytoin a sulfamethoxazol/trimethoprim, hodnocení komplikuje. Před zahájením podávání léku by měly být splněny následující laboratorní parametry: ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Kompletní krevní obraz je třeba vyšetřit ve 22. dni (21 dnů po podání první dávky) nebo v průběhu 48 hodin po tomto dni, a pak v týdenních intervalech do té doby, dokud není ANC > 1,5 x 109/l a počet trombocytů > 100 x 109/l. Jestliže v průběhu kteréhokoli cyklu poklesne ANC na < 1,0 x 109/l nebo počet trombocytů na < 50 x 109/l, měla by být v následném cyklu dávka o jeden stupeň snížena (viz bod 4.2). Jednotlivé stupně dávky činí 100 mg/m2, 150 mg/m2 a 200 mg/m2. Nejnižší doporučená dávka je 100 mg/m2.
Pediatrická populace
S podáváním TMZ dětem ve věku nižším než 3 roky nejsou dosud žádné klinické zkušenosti. U starších dětí a dospívajících jsou zkušenosti velmi omezené (viz body 4.2 a 5.1).
Starší pacienti (ve věku >70 let)
U starších pacientů je ve srovnání s mladšími osobami pravděpodobně vyšší riziko neutropenie a trombocytopenie. Vzhledem k tomu je při podávání TMZ ve vyšším věku nutná zvýšená opatrnost.
Pacienti mužského pohlaví
Muži léčení TMZ by měli být upozorněni, aby se do 6 měsíců od užití poslední dávky nestali otci a aby se před zahájením léčby informovali o možnosti kryokonzervace spermatu (viz bod 4.6).
Sodík
Tento léčivý přípravek obsahuje 2,4 mmol sodíku v jedné injekční lahvičce. To by měli brát v potaz pacienti dodržující dietu s omezením sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Podávání TMZ spolu s ranitidinem nemělo v oddělené studii fáze I za následek žádné změny rozsahu absorpce temozolomidu nebo expozice jeho aktivnímu metabolitu monomethyl- triazenoimidazol-karboxamidu (MTIC).
Výsledky analýzy populační farmakokinetiky ve II. fázi klinických studií prokazují, že nebyla clearance TMZ ovlivněna současným podáním dexamethasonu, prochlorperazinu, fenytoinu, karbamazepinu, ondansetronu, antagonistů H2 receptorů nebo fenobarbitalu. Současné podávání kyseliny valproové bylo spojeno s malým, nicméně statisticky významným poklesem clearance TMZ.
Dosud nebyly provedeny žádné studie, které by hodnotily účinek TMZ na metabolismus nebo eliminaci jiných léků. Nicméně vzhledem k tomu, že TMZ neprochází přeměnou v játrech a na plazmatické bílkoviny se váže jen v malé míře, je málo pravděpodobné, že by ovlivňoval farmakokinetiku jiných léčivých přípravků (viz bod 5.2).
V kombinaci s jinými myelosupresivními látkami může TMZ zvýšit pravděpodobnost myelosuprese.
Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání léku těhotným ženám nejsou k dispozici. V preklinických studiích u potkanů a králíků, kteří TMZ dostávali v dávce 150 mg/m2, byly prokázány teratogenní a/nebo toxické účinky na plod (viz bod 5.3). Temodal by neměl být podáván těhotným ženám. Pokud je přece jen nutno zvážit použití během těhotenství, je třeba pacientku seznámit s potenciálním rizikem pro plod.
Kojení
Není známo, zda se TMZ vylučuje do mateřského mléka; kojení by se proto mělo po dobu léčby TMZ přerušit.
Ženy ve fertilním věku.
Ženám ve fertilním věku je třeba doporučit používání účinné antikoncepce k zabránění otěhotnění v průběhu užívání TMZ.
Mužská plodnost
TMZ může mít genotoxické účinky. Proto se mužům léčeným temozolomidem nedoporučuje, aby se do 6 měsíců od užití poslední dávky stali otci a naopak se doporučuje, vzhledem k možnosti ireverzibilní neplodnosti po léčbě TMZ, ještě před zahájením léčby konzultovat s lékařem možnost kryokonzervace předem odebraných spermií.
4.7 Účinky na schopnost řídit a obsluhovat stroje
TMZ má malý vliv na schopnost řídit nebo obsluhovat stroje v důsledku únavy a ospalosti (viz bod 4.8.).
4.8 Nežádoucí účinky
Zkušenosti z klinických hodnocení tvrdých tobolek
U pacientů léčených TMZ, ať už užívaným v kombinaci s RT nebo ve formě monoterapie následující po RT v případě nově diagnostikovaného multiformního glioblastomu, nebo ve formě monoterapie pacientů s recidivujícím nebo progresivním gliomem, byly velmi často hlášené nežádoucí účinky podobné: nausea, zvracení, zácpa, anorexie, bolest hlavy a únava. Křeče byly hlášeny velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených monoterapií, vyrážka byla hlášena velmi často u nově diagnostikovaných pacientů s multiformním glioblastomem léčených TMZ v kombinaci s RT nebo také ve formě monoterapie a často u pacientů s recidivujícím gliomem.
Většina hematologických nežádoucích účinků byla hlášena u obou indikací často nebo velmi často (Tabulky 4 a 5); četnost laboratorních nálezů stupně 3-4 je uvedena za každou tabulkou.
V tabulkách jsou nežádoucí účinky klasifikovány podle orgánových systémů a četnosti. Zařazení do skupin četností se provádí podle následujícího pravidla: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 4 ukazuje nežádoucí účinky, které se objevily při léčbě u pacientů s nově diagnostikovaným multiformním glioblastomem během souběžné a monoterapeutické fáze léčby.
|
Tabulka 4. Nežádoucí účinky, které se objevily během souběžné a monoterapeutické léčby u pacientů s nově diagnostikovaným multiformním glioblastomem | ||
|
Třída orgánového systému |
TMZ + souběžná RT n=288* |
TMZ monoterapie n=224 |
|
Infekce a infestace | ||
|
Časté: |
Infekce, Herpes simplex, infekce rány, faryngitida, orální kandidóza |
Infekce, orální kandidóza |
|
Méně časté: |
Herpes simplex, herpes zoster, symptomy podobné chřipce | |
|
Poruchy krve a lymfatického systému | ||
|
Časté: |
Neutropenie, trombocytopenie, lymfopenie, leukopenie |
Febrilní neutropenie, trombocytopenie, anémie, leukopenie |
|
Méně časté: |
Febrilní neutropenie, anémie |
Lymfopenie, petechie |
|
Endokrinní poruchy | ||
|
Méně časté: |
Cushingoid |
Cushingoid |
|
Poruchy metabolismu a výživy | ||
|
Velmi časté: | ||
|
Časté: |
Hyperglykémie, pokles hmotnosti |
Pokles hmotnosti |
|
Méně časté: |
Hypokalémie, zvýšení alkalické fosfatázy, zvýšení hmotnosti |
Hyperglykémie, zvýšení hmotnosti |
|
Psychiatrické poruchy | ||
|
Časté: |
Úzkost, emoční labilita, nespavost | |
|
Méně časté: |
Neklid, apatie, poruchy chování, deprese, halucinace |
Halucinace, amnézie |
|
Poruchy nervového systému | ||
|
Velmi časté: |
Konvulze, bolest hlavy | |
|
Časté: |
Konvulze, porucha vědomí, somnolence, afázie, porucha rovnováhy, závrať, zmatenost, porucha paměti, porucha koncentrace, neuropatie, parestézie, porucha řeči, třes |
Hemiparéza, afázie, porucha rovnováhy, somnolence, zmatenost, závrať, porucha paměti, porucha koncentrace, dysfázie, neurologická porucha (NOS), neuropatie, periferní neuropatie, parestézie, porucha řeči, třes |
|
Méně časté: |
Status epilepticus, extrapyramidální porucha, hemiparéza, ataxie, poruchy kognice, dysfázie, abnormální chůze, hyperestézie, hypestézie, neurologická porucha (NOS), periferní neuropatie |
Hemiplegie, ataxie, abnormální koordinace, abnormální chůze, hyperestézie, smyslová porucha |
|
Poruchy oka | ||
|
Časté: |
Rozmazané vidění |
Defekt zorného pole, rozmazané vidění, diplopie |
|
Méně časté: |
Hemianopie, zmenšená ostrost vidění, poruchy zraku, defekt zorného pole, bolest očí |
Zmenšená ostrost vidění, bolest očí, suché oči |
|
Poruchy ucha a labyrintu | ||
|
Časté: |
Porucha sluchu |
Porucha sluchu, tinitus |
|
Méně časté: |
Otitis media, tinitus, hyperakuze, bolest ucha |
Hluchota, závrať, bolest ucha |
|
Srdeční poruchy | ||
|
Méně časté: |
Palpitace | |
|
Cévní poruchy | ||
|
Časté: |
Hemoragie, edém, edém nohy |
Hemoragie, hluboká žilní trombóza, edém nohy |
|
Méně časté: |
Cerebrální hemoragie, hypertenze |
Plicní embolie, edém, periferní edém |
|
Respirační, hrudní a mediastinální poruchy | ||
|
Časté: | ||
|
Méně časté: |
Pneumonie, infekce horních cest dýchacích, nosní kongesce |
Pneumonie, sinusitida, infekce horních cest dýchacích, bronchitida |
|
Gastrointestinální poruchy | ||
|
Velmi časté: |
Zácpa, nausea, zvracení |
Zácpa, nausea, zvracení |
|
Časté: |
Stomatitida, průjem, bolest břicha, dyspepsie, dysfagie | |
|
Méně časté: |
Abdominální distenze, fekální inkontinence, gastrointestinální porucha (NOS), gastroenteritida, hemeroidy | |
|
Poruchy kůže a podkožní tkáně | ||
|
Velmi časté: |
Vyrážka, alopecie |
Vyrážka, alopecie |
|
Časté: |
Dermatitida, suchá kůže, erytém, svědění |
Suchá kůže, svědění |
|
Méně časté: |
Exfoliace kůže, fotosenzitivní reakce, abnormální pigmentace |
Erytém, abnormální pigmentace, zvýšené pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Časté: |
Svalová slabost, artralgie |
Svalová slabost, artralgie, muskuloskeletální bolest, myalgie |
|
Méně časté: |
Myopatie, bolest v zádech, muskuloskeletální bolest, myalgie |
Myopatie, bolest v zádech |
|
Poruchy ledvin a močových cest | ||
|
Časté: |
Časté močení, močová inkontinence |
Močová inkontinence |
|
Méně časté: | ||
|
Poruchy reprodukčního systému a prsu | ||
|
Méně časté: |
Impotence |
Vaginální hemoragie, menoragie, amenorea, vaginitida, bolest prsou |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Velmi časté: |
Únava |
Únava |
|
Časté: |
Alergická reakce, horečka, radiační poškození, edém obličeje, bolest, změna chuti |
Alergická reakce, horečka, radiační poškození, bolest, změna chuti |
|
Méně časté: |
Astenie, zrudnutí, návaly horka, zhoršení stavu, rigory, změna barvy jazyka, parosmie, žízeň |
Astenie, edém obličeje, bolest, zhoršení stavu, rigory, zubní potíže |
|
Vyšetření | ||
|
Časté: |
Zvýšené ALT |
Zvýšené ALT |
|
Méně časté: |
Zvýšené jaterní enzymy, zvýšené gama GT, zvýšené AST | |
*Pacient který byl randomizován pouze do větve RT, obdržel Temodal + RT.
Laboratorní výsledky
Byla zjištěna myelosuprese (neutropenie a trombocytopenie), která je známou toxicitou limitující dávku většiny cytotoxických látek včetně TMZ. Když byly zkombinovány abnormality laboratorních testů a nežádoucí účinky v souběžných a monoterapeutických fázích léčby, byly pozorovány u 8 % pacientů abnormality neutrofilů stupeň 3 nebo stupeň 4, včetně neutropenických případů. Stupeň 3 nebo stupeň 4 abnormalit trombocytů, včetně trombocytopenických případů, byl pozorován u 14 % pacientů, kteří dostávali TMZ.
Recidivující nebo _progresivní maligní gliom
Nejčastěji se vyskytujícími nežádoucími účinky ve vztahu k léčbě byly v provedených klinických studiích gastrointestinální poruchy, především nausea (43 %) a zvracení (36 %). Tyto reakce dosahovaly obvykle pouze stupně 1 nebo 2 (0 - 5 epizod zvracení během 24 hodin) a buď spontánně odezněly, nebo byly snadno léčitelné standardní antiemetickou léčbou. Incidence těžké nausey a zvracení činila 4 %.
Tabulka 5 obsahuje nežádoucí účinky, které byly hlášeny v průběhu klinických studií pro recidivující nebo progresivní maligní gliom a po uvedení přípravku Temodal na trh.
|
Tabulka 5. Nežádoucí účinky u pacientů s recidivujícím nebo progresivním maligním gliomem | |
|
Infekce a infestace | |
|
Vzácné: |
Oportunní infekce včetně pneumonie způsobené PCP |
|
Poruchy krve a lymfatického systému | |
|
Velmi časté: |
Neutropenie nebo lymfopenie (stupeň 3-4) trombocytopenie (stupeň 3-4) |
|
Méně časté: |
Pancytopenie, anémie (stupeň 3-4), leukopenie |
|
Poruchy metabolismu a výživy | |
|
Velmi časté: | |
|
Časté: |
Pokles hmotnosti |
|
Poruchy nervového systému | |
|
Velmi časté: | |
|
Časté: |
Somnolence, závratě, parestézie |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté: | |
|
Gastrointestinální poruchy | |
|
Velmi časté: |
Zvracení, nausea, zácpa |
|
Časté: | |
|
Poruchy kůže a podkožní tkáně | |
|
Časté: |
Vyrážka, pruritus, alopecie, |
|
Velmi vzácné: |
Multiformní erytém, erytrodermie, urtikarie, exantém |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté: |
Únava |
|
Časté: | |
|
Velmi vzácné: |
Alergické reakce, včetně anafylaxe, angioedém |
Laboratorní výsledky
Trombocytopenie a neutropenie stupně 3 nebo 4 byly zaznamenány u 19 %, resp. 17 % pacientů léčených pro maligní gliom. K hospitalizaci a/nebo přerušení podávání TMZ vedly v 8 %, resp. 4 % případů. Myelosuprese byla předvídatelná (obvykle k ní došlo během několika prvních cyklů, s nadirem mezi 21. dnem a 28. dnem) a úprava k normě byla rychlá, obvykle v průběhu 1-2 týdnů. Kumulace myelosupresivních účinků prokázána nebyla. Přítomnost trombocytopenie může zvýšit riziko krvácení a přítomnost neutropenie nebo leukopenie může zvýšit riziko infekce.
Pohlaví
Do analýzy populační farmakokinetiky založené na zkušenostech z klinického hodnocení bylo zahrnuto 101 žen a 169 mužů, u kterých byla dostupná měření nadiru neutrofilů, a 110 žen a 174 mužů, u kterých byla dostupná měření nadiru krevních destiček. U žen bylo ve srovnání s muži v prvním cyklu terapie zaznamenáno více neutropenií stupně 4 (ANC < 0,5 x 109/l), 12 % vs5 %, a trombocytopenií (< 20 x 109/l), 9 % vs 3 %. V souboru dat od 400 jedinců s rekurentním gliomem se v prvním cyklu terapie objevila neutropenie stupně 4 u 8 % žen vs 4 % mužů a trombocytopenie stupně 4 u 8 % žen vs 3 % mužů. Ve studii s 288 jedinci s nově diagnostikovaným multiformním glioblastomem se v prvním cyklu terapie vyskytla neutropenie stupně 4 u 3 % žen vs 0 % mužů a trombocytopenie stupně 4 u 1 % žen vs 0 % mužů.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s rekurentním gliomem mozkového kmene nebo rekurentním astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Ačkoliv jsou údaje omezené, tolerance u dětí se očekává být podobná jako u dospělých. Bezpečnost TMZ u dětí ve věku do 3 let nebyla stanovena.
Zkušenosti z klinických hodnocení lékové formy i.v.
Přípravkem Temodal 2,5 mg/ml prášek pro infuzní roztok se do těla dostává ekvivalentní dávka TMZ jako u odpovídajících tobolek Temodal, se stejnou expozicí jak TMZ, tak účinnému metabolitu MTIC (viz bod 5.2). Nežádoucí účinky, které byly hlášeny ze dvou klinických hodnocení s intravenózní formou (n=35), ale nebyly hlášeny z klinických hodnocení s užíváním tvrdých tobolek, byly reakce v místě aplikace infuze: bolest, podráždění, svědění, horkost, otok a erytém a rovněž hematom.
Zkušenosti po uvedení na trh
Po uvedení přípravku na trh byly hlášeny další následující závažné nežádoucí účinky:
|
Tabulka 6. Souhrn příhod hlášených u temozolomidu po jeho uvedení na trh | |
|
Infekce a infestace* | |
|
Méně časté: |
infekce způsobená cytomegaloviry, reaktivace infekce způsobené virem, jako je cytomegalovirus, virus hepatitidy BŤ |
|
Poruchy krve a lymfatického systému | |
|
Velmi vzácné: |
dlouhotrvající pancytopenie, aplastická anémieŤ |
|
Novotvary benigní, maligní a blíže neurčené | |
|
Velmi vzácné: |
myelodysplastický syndrom (MDS), sekundární malignity, včetně myeloidní leukémie |
|
Endokrinní poruchy | |
|
Méně časté: |
diabetes insipidus |
|
Respirační, hrudní a mediastinální poruchy | |
|
Velmi vzácné: |
intersticiální pneumonitida/pneumonitida, plicní fibróza, respirační selhání1 |
|
Poruchy jater a žlučových cest* | |
|
Časté: |
zvýšení jaterních enzymů |
|
Méně časté: |
hyperbilirubinemie, cholestáza, hepatitida, poškození jater, selhání jater1 |
|
Poruchy kůže a podkožní tkáně | |
|
Velmi vzácné: |
toxická epidermální nekrolýza, Stevens-Johnsonův syndrom |
t Včetně smrtelných případů
Frekvence odhadované na základě relevantních klinických studií.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinicky byly u pacientů ověřeny dávky 500, 750, 1 000, a 1 250 mg/m2 (celková dávka za 5-denní cyklus). Toxicita limitující dávku byla toxicita hematologická a byla pozorována u všech dávek, ale lze očekávat, že při vyšších dávkách bude závažnější. U jednoho pacienta došlo k předávkování, když užil dávku 10 000 mg (celková dávka během jednoho 5-denního cyklu), nežádoucí účinky hlášené v tomto případě byly pancytopenie, pyrexie, multiorgánové selhání a smrt. Byly popsány případy, kdy pacienti doporučenou dávku užívali po dobu delší než 5 dnů (až 64 dnů) s hlášenými nežádoucími příhodami včetně suprese kostní dřeně s nebo bez rozvoje infekce, v některých případech závažné, dlouhotrvající a vedoucí ke smrti. V případě předávkování je nezbytné hematologické zhodnocení. Podl e potřeby by měla být zajištěna podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: protinádorové léky - ostatní alkylační látky, ATC kód: L01A X03 Mechanismus účinku
Temozolomid je triazen, který za fyziologické hodnoty pH prochází rychlou chemickou konverzí na aktivní monometyl-triazenoimidazol-karboxamid (MTIC). Předpokládá se, že cytotoxické účinky MTIC jsou primárně důsledkem alkylace na O6 pozici guaninu, s následnou alkylací rovněž na pozici N7. Následně vznikající cytotoxické léze jsou vysvětlovány nejspíše aberantní reparací methylovaného zbytku.
Klinická účinnost a bezpečnost
Nově diagnostikovaný multiformní glioblastom
Celkem 573 pacientů bylo randomizováno do skupin dostávajících buď TMZ + RT (n=287) nebo RT samotnou (n=286). Pacienti ve větvi TMZ + RT dostávali souběžně TMZ (75 mg/m2) jednou denně od prvního až do posledního dne RT po dobu 42 dnů (maximálně 49 dnů). Pak následovala monoterapie TMZ (150 - 200 mg/m2) ve dnech 1-5 každého 28-denního cyklu, až v 6 cyklech, začínající 4 týdny po ukončení RT. Pacienti v kontrolní větvi dostávali pouze RT. Během RT a souběžné TMZ terapie byla nutná profylaxe pneumonie Pneumocystis jirovecii (PCP).
TMZ byl podáván jako záchranná terapie vnásledné fázi 161 pacientům z 282 (57 %) ve větvi dostávající pouze RT a 62 pacientům z 277 (22 %) ve větvi dostávající TMZ + RT.
Poměr rizika (HR) pro celkové přežívání byl 1,59 (95 % CI pro HR=1,33 - 1,91) s log-rank p < 0,0001 ve prospěch TMZ větve. Odhadnutá pravděpodobnost přežívání 2 roky nebo více (26 % vs 10 %) je vyšší ve větvi RT + TMZ. Přidání souběžně podávaného TMZ k RT následované monoterapií TMZ v léčbě pacientů s nově diagnostikovaným multiformním glioblastomem prokázalo statisticky významné zlepšení v celkovém přežívání (overall survival=OS) ve srovnání s RT samotnou (Graf 1).

Čas od randomizace (měsíce)
Graf 1 Kaplanovy-Meierovy křivky pro celkové přežívání (ITTpopulace=intent-to- treatpopulation)
Výsledky studie nebyly konzistentní v podskupině pacientů se slabým výkonnostním statusem (WHO PS=2, n=70), kde celkové přežívání a čas do progrese byly podobné v obou větvích. Zdá se však, že u této skupiny pacientů nejsou přítomna žádná nepřijatelná rizika.
Recidivující nebo _progresivní maligní gliom
Data o klinické účinnosti u pacientů s multiformním glioblastomem (Karnofského výkonnostní status [KPS] > 70), u něhož dochází k progresi nebo recidivě po chirurgickém výkonu a RT, vycházela ze dvou klinických studií s perorálním TMZ. V prvním případě šlo o nekomparativní studii u 138 pacientů (29 % bylo předtím léčeno chemoterapií), ve druhém o randomizovanou studii kontrolovanou léčivou látkou s TMZ vs prokarbazinem u celkem 225 pacientů (67 % z nich bylo předtím léčeno chemoterapií na bázi nitrosourey). V obou studiích bylo primárním hodnotícím kritériem přežívání bez známek progrese (PFS - progression-free survival), hodnocených na základě zobrazení magnetickou rezonancí (MRI) nebo na základě zhoršení neurologických funkcí. V nekomparativní studii bylo u 19 % pacientů dosaženo 6měsíční PFS, medián přežívání bez známek progrese byl 2,1 měsíce a medián celkové doby přežívání byl 5,4 měsíce. Objektivní počet terapeutických odpovědí na základě hodnocení MRI dosahoval 8 %.
V randomizované studii kontrolované léčivou látkou bylo šestiměsíční PFS u pacientů léčených TMZ významně vyšší než u těch, kteří dostávali prokarbazin (21 % vs 8 %; chí-kvadrát p=0,008) s mediánem PFS 2,89, respektive 1,88 měsíce (log rank p=0,0063). Medián přežití byl 7,34 měsíce pro TMZ a 5,66 měsíce pro prokarbazin (log rank p=0,33). Po 6 měsících byl podíl přežívajících pacientů v TMZ skupině významně vyšší (60 %) než ve skupině prokarbazinové (44 %; chí-kvadrát p=0,019). U pacientů, kteří byli předtím léčeni chemoterapií, bylo dosaženo kladného efektu u těch, kteří měli KPS > 80.
Údaje o době do zhoršení neurologického statusu, jakož i o době do zhoršení výkonnostního statusu (pokles na KPS < 70 nebo pokles nejméně o 30 bodů), hovoří ve prospěch TMZ oproti prokarbazinu. Medián doby do progrese v těchto hodnotících kritériích byl u TMZ o 0,7 - 2,1 měsíce delší než u prokarbazinu (log rank p < 0,01 - 0,03).
Recidivující anaplastický astrocytom
V multicentrické prospektivní studii fáze II, v níž byly hodnoceny bezpečnost a účinnost perorálně podávaného TMZ v léčbě pacientů s anaplastickým astrocytomem při prvním relapsu, bylo zjištěno šestiměsíční PFS u 46 % léčených. Medián PFS činil 5,4 měsíce. Medián celkového přežití činil
14,6 měsíce. Procento odpovědí na léčbu, hodnocené centrálně činilo 35 % (13 % kompletních, 43 % částečných odpovědí) pro populaci intent-to-treat (ITT), n = 162. U 43 pacientů došlo ke stabilizaci onemocnění. Šestiměsíční přežití bez klinické příhody bylo pro populaci intent-to-treat 44 %, přičemž medián přežití bez klinické příhody činil 4,6 měsíce, což je podobné výsledkům hodnocení přežívání bez známek progrese. Z histologického hlediska byla účinnost obdobná. Objektivní radiologicky prokázaná odpověď na léčbu nebo zachování stavu beze známek progrese velmi dobře korelovaly se zachovanou nebo zlepšenou kvalitou života.
Pediatrická populace
Perorální TMZ byl hodnocen u dětských pacientů (ve věku 3-18 let) s recidivujícím gliomem mozkového kmene nebo recidivujícím astrocytomem vysokého stupně v režimu denního podávání po dobu 5 dnů každých 28 dnů. Tolerance TMZ byla podobná jako u dospělých.
5.2 Farmakokinetické vlastnosti
TMZ se spontánně hydrolyzuje při fyziologickém pH primárně na aktivní látku, 3-metyl-(triazen-1-yl)imidazol-4-karboxamid (MTIC). MTIC se spontánně hydrolyzuje na 5-amino-imidazol-4-karboxamid (AIC), známý meziprodukt biosyntézy purinů a nukleových kyselin a na metylhydrazin, o kterém se předpokládá, že je aktivní alkylační látkou. Předpokládá se, že cytotoxicita MTIC je primárně způsobena alkylací DNA zejména na pozicích guaninu O6 a N7' Expozice MTIC a AIC je ~
2.4 %, respektive 23 % v poměru k AUC TMZ. In vivo byl t1/2 MTIC podobný jako pro TMZ,
1,8 hodin.
V otevřeném, dvojitě zkříženém bioekvivalentním klinickém hodnocení farmakokinetiky perorálního a intravenózního TMZ u pacientů s primárními malignitami CNS bylo zjištěno, že je Temodal
2.5 mg/ml prášek pro infuzní roztok v dávce 150 mg/m2 aplikované během 90 minut biologicky ekvivalentní tvrdým tobolkám Temodal v Cmax i AUC TMZ a MTIC. Průměrné hodnoty Cmax u TMZ a MTIC byly po 90-minutové intravenózní infuzi 7,4 pg/ml, respektive 320 ng/ml. Průměrné hodnoty AUC (0 ^ <x>) u TMZ a MTIC byly 25 pg^h/ml, respektive 1 004 ng^h/ml.
Absorpce
Po perorálním podání dospělým pacientům se TMZ rychle absorbuje a vrcholné koncentrace dosahuje již za 20 minut po podání (střední doba je mezi 0,5 a 1,5 hodiny). Po perorálním podání 14C-značeného TMZ byla průměrná fekální exkrece 14C v průběhu 7 dnů po podání dávky 0,8 %, což svědčilo o úplné absorpci.
Distribuce v organismu
TMZ vykazuje nízkou vazebnou afinitu k proteinům (10 % až 20 %) proto se neočekává, že by interagoval s látkami s vysokou vazebnou afinitou k proteinům.
PET studie u lidí a preklinické údaje ukazují, že TMZ rychle překračuje hematoencefalickou bariérou a je přítomný v mozkomíšním moku. Průnik do mozkomíšního moku (CSF) byl potvrzen u jednoho pacienta; expozice CSF na základě AUC TMZ byla přibližně 30 % plazmatické expozice, což odpovídá údajům u zvířat.
Eliminace z organismu
Plazmatický poločas (ti/2) je přibližně 1,8 hodin. Hlavní způsob eliminace 14C je ledvinami. Po perorálním podání se objevuje přibližně 5 až 10 % dávky v nezměněné podobě v moči v průběhu 24 hodin a zbytek se vylučuje ve formě temozolomidové kyseliny, 5-aminoimidazol-4-karboxamidu (AIC) nebo neidentifikovaných polárních metabolitů.
Plazmatické koncentrace vzrůstají v závislosti na velikosti dávky. Plazmatická clearance, distribuční objem a poločas jsou na velikosti dávky nezávislé.
Zvláštní populace
V analýze populační farmakokinetiky TMZ bylo zjištěno, že plazmatická clearance TMZ nezávisí na věku, renální funkci nebo kouření. V oddělené farmakokinetické studii byly plazmatické farmakokinetické profily u pacientů s mírnou až střední jaterní dysfunkcí stejné jako profily u pacientů s normální funkcí jater.
U dětských pacientů byla AUC větší než u dospělých; nicméně maximální tolerovaná dávka (MTD) činila 1 000 mg/m2 na jeden cyklus jak u dětí, tak u dospělých.
5.3 Předklinické údaje vztahující se k bezpečnosti
U potkanů a psů byly prováděny studie toxicity s jednotlivým cyklem (5 dnů podávání léku, 23 dnů bez léčby) 3 a 6 cykly léčby. Primární cíle toxických účinků tvořila kostní dřeň, lymforetikulární systém, testes, gastrointestinální ústrojí a při vyšších dávkách, které byly pro 60 % - 100 % testovaných potkanů a psů letální, docházelo k degeneraci retiny. Většina toxických účinků byla reverzibilní, s výjimkou nežádoucích účinků na mužský reprodukční systém a degeneraci retiny. Nicméně vzhledem k tomu, že dávky podmiňující retinální degeneraci se pohybovaly v rozmezí dávek letálních, a v klinických studiích nebyl žádný srovnatelný účinek pozorován, nepřikládá se tomuto nálezu klinická významnost.
TMZ je embryotoxická, teratogenní a genotoxická alkylační látka. TMZ má toxické účinky větší u potkanů a psů než u člověka, a klinická dávka se blíží minimální letální dávce u potkanů a psů. Za citlivé ukazatele toxicity se považují poklesy počtu leukocytů a trombocytů v závislosti na velikosti dávky. Ve studii u potkanů, v níž bylo aplikováno šest cyklů terapie, bylo zjištěno několik druhů novotvarů, včetně karcinomu mléčné žlázy, kožního keratoakanthomu a adenomu bazálních buněk, zatímco ve studiích u psů žádné nádorové ani prenádorové změny pozorovány nebyly. Zdá se, že potkani jsou vůči onkogenním účinkům TMZ mimořádně citliví, neboť první tumory se u nich objevují již po třech měsících od zahájení podávání léku. Jde o dobu krátkou dokonce i pro alkylační látky.
Výsledky aberačních testů s chromosomy Ames/salmonella a lymfocytů z lidské periferní krve prokázaly pozitivní mutagenní reakci.
Intravenózní léková forma vedla k místní iritaci v místě aplikace injekce jak u králíků, tak potkanů. Iritace byla přechodná a nebyla spojena s trvalým poškozením tkáně.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
mannitol (E421) threonin polysorbát 80
natrium-citrát (pro úpravu pH)
koncentrovaná kyselina chlorovodíková (pro úpravu pH)
6.2 Inkompatibility
Vzhledem k tomu, že studie kompatibility nejsou k dispozici, nesmí být tento léčivý přípravek kombinován s jinými léčivými přípravky.
6.3 Doba použitelnosti
Neotevřená injekční lahvička: 4 roky Rekonstituovaný roztok:
Po rekonstituci byla prokázána chemická a fyzikální stabilita pro použití po dobu 14 hodin při teplotě 25°C, včetně doby infuze.
Z mikrobiologického hlediska by se měl přípravek použít okamžitě. Jestliže není použit okamžitě, je doba a podmínky skladování před použitím zodpovědností uživatele a za normálních okolností by neměla přesahovat 24 hodin při teplotě 2 až 8°C, jestliže příprava neproběhla za kontrolovaných a ověřených aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C).
Podmínky uchovávání rekonstituovanélo léčivého přípravku viz bod 6.3.
6.5 Druh obalu a obsah balení
Čirá injekční lahvička, sklo třídy I, uzavřená bromobutylovou gumovou zátkou a hliníkovým uzávěrem s odstranitelným krytem broskvové barvy. Jedna injekční lahvička obsahuje 100 mg TMZ.
Temodal 2,5 mg/ml se dodává v balení po jedné injekční lahvičce.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Při zacházení s přípravkem Temodal 2,5 mg/ml prášek po přípravu infuzního roztoku je třeba postupovat opatrně. Je nutné použít rukavice a aseptickou techniku. Pokud přijde Temodal 2,5 mg/ml do styku s kůží nebo sliznicí, je třeba neprodleně a řádně ho smýt mýdlem a vodou.
Obsah jedné injekční lahvičky musí být smísen se 41 ml sterilizované vody na injekci. Výsledný roztok obsahuje 2,5 mg/ml TMZ. Injekčními lahvičkami by se mělo jemně kroužit a netřepat. Roztok by se měl prohlédnout a neměla by se použít žádná injekční lahvička, která obsahuje viditelné částice. Podle celkové předepsané dávky se z injekční lahvičky vytáhne připravený roztok v objemu až 40 ml, který se přenese do prázdného 250ti mililitrového infuzního vaku (z PVC nebo polyolefínu). Hadičky pumpy je třeba připojit k vaku, propláchnout je a zakrýt. Temodal 2,5 mg/ml musí být podáván pouze intravenózní infuzí trvající 90 minut.
Temodal 2,5 mg/ml prášek pro infuzní roztok lze podávat stejnou i.v. linkou s injekcí 0,9 % roztoku chloridu sodného. Není kompatibilní s roztoky glukózy.
Protože nejsou k dispozici žádné další údaje, nesmí se tento léčivý přípravek kombinovat s jinými léčivými přípravky nebo podávat současně stejnou intravenózní cestou.
Tento léčivý přípravek je určen pouze pro jednorázové použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/98/096/023
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 26. ledna 1999.
Datum posledního prodloužení registrace: 26. ledna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
SP Labo N.V.
Industriepark 30
2220 Heist op den Berg
Belgie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném včl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
Temodal 5 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 5 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci.
5 tvrdých tobolek 20 tvrdých tobolek
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je nekousejte, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/001 (5 tvrdých tobolek) EU/1/98/096/002 (20 tvrdých tobolek)
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 5 mg
Temodal 20 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 20 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci.
5 tvrdých tobolek 20 tvrdých tobolek
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je nekousejte, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/003 (5 tvrdých tobolek) EU/1/98/096/004 (20 tvrdých tobolek)
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 20 mg
Temodal 100 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 100 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci.
5 tvrdých tobolek 20 tvrdých tobolek
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je nekousejte, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/005 (5 tvrdých tobolek) EU/1/98/096/006 (20 tvrdých tobolek)
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 100 mg
Temodal 140 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 140 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci.
5 tvrdých tobolek 20 tvrdých tobolek
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je nekousejte, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/009 (5 tvrdých tobolek) EU/1/98/096/010 (20 tvrdých tobolek)
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 140 mg
Temodal 180 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 180 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci.
5 tvrdých tobolek 20 tvrdých tobolek
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/011 (5 tvrdých tobolek) EU/1/98/096/012 (20 tvrdých tobolek)
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 180 mg
Temodal 250 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 250 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci.
5 tvrdých tobolek 20 tvrdých tobolek
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je nekousejte, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/007 (5 tvrdých tobolek) EU/1/98/096/008 (20 tvrdých tobolek)
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 250 mg
ETIKETA PRO LAHVIČKU OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 5 mg_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 5 mg tvrdé tobolky temozolomidum Perorální podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 tvrdých tobolek 20 tvrdých tobolek
6. JINÉ
ETIKETA PRO LAHVIČKU OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 20 mg_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 20 mg tvrdé tobolky temozolomidum Perorální podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 tvrdých tobolek 20 tvrdých tobolek
6. JINÉ
ETIKETA PRO LAHVIČKU OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 100 mg_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 100 mg tvrdé tobolky
temozolomidum
Perorální podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 tvrdých tobolek 20 tvrdých tobolek
6. JINÉ
ETIKETA PRO LAHVIČKU OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 140 mg_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 140 mg tvrdé tobolky
temozolomidum
Perorální podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 tvrdých tobolek 20 tvrdých tobolek
6. JINÉ
ETIKETA PRO LAHVIČKU OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 180 mg_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 180 mg tvrdé tobolky
temozolomidum
Perorální podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 tvrdých tobolek 20 tvrdých tobolek
6. JINÉ
ETIKETA PRO LAHVIČKU OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 250 mg_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 250 mg tvrdé tobolky
temozolomidum
Perorální podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 tvrdých tobolek 20 tvrdých tobolek
6. JINÉ
KRABIČKA OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 5 mg JEDNOTLIVĚ ZATAVENÝCH V SÁČCÍCH_
Temodal 5 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 5 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci.
5 tvrdých tobolek v sáčcích 20 tvrdých tobolek v sáčcích
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je nekousejte, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/024 (5 tvrdých tobolek) EU/1/98/096/025 (20 tvrdých tobolek)
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 5 mg
KRABIČKA OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 20 mg JEDNOTLIVĚ ZATAVENÝCH V SÁČCÍCH_
Temodal 20 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 20 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci.
5 tvrdých tobolek v sáčcích 20 tvrdých tobolek v sáčcích
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je nekousejte, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/013 (5 tvrdých tobolek) EU/1/98/096/014 (20 tvrdých tobolek)
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 20 mg
KRABIČKA OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 100 mg JEDNOTLIVĚ ZATAVENÝCH V SÁČCÍCH_
Temodal 100 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 100 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci.
5 tvrdých tobolek v sáčcích 20 tvrdých tobolek v sáčcích
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je nekousejte, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/015 (5 tvrdých tobolek) EU/1/98/096/016 (20 tvrdých tobolek)
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 100 mg
KRABIČKA OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 140 mg JEDNOTLIVĚ ZATAVENÝCH V SÁČCÍCH_
Temodal 140 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 140 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci.
5 tvrdých tobolek v sáčcích 20 tvrdých tobolek v sáčcích
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je nekousejte, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/017 (5 tvrdých tobolek) EU/1/98/096/018 (20 tvrdých tobolek)
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 140 mg
KRABIČKA OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 180 mg JEDNOTLIVĚ ZATAVENÝCH V SÁČCÍCH_
Temodal 180 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 180 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci.
5 tvrdých tobolek v sáčcích 20 tvrdých tobolek v sáčcích
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je nekousejte, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/019 (5 tvrdých tobolek) EU/1/98/096/020 (20 tvrdých tobolek)
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 180 mg
KRABIČKA OBSAHUJÍCÍ 5 NEBO 20 TVRDÝCH TOBOLEK PŘÍPRAVKU TEMODAL 250 mg JEDNOTLIVĚ ZATAVENÝCH V SÁČCÍCH_
Temodal 250 mg tvrdé tobolky temozolomidum
Jedna tvrdá tobolka obsahuje temozolomidum 250 mg.
Obsahuje laktózu. Přečtěte si více v příbalové informaci
5 tvrdých tobolek v sáčcích 20 tvrdých tobolek v sáčcích
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Cytotoxický
Tobolky neotvírejte, nedrťte je nebo je nekousejte, polykejte je celé. Pokud je tobolka poškozená, zabraňte kontaktu s kůží, očima či nosem.
Uchovávejte při teplotě do 30°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/021 (5 tvrdých tobolek) EU/1/98/096/022 (20 tvrdých tobolek)
Výdej léčivého přípravku vázán na lékařský předpis.
Temodal 250 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 5 mg tobolky temozolomidum Perorální podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 tobolka
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 20 mg tobolky temozolomidum Perorální podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 tobolka
6. JINÉ
SÁČEK OBSAHUJÍCÍ 1 TVRDOU TOBOLKU PŘÍPRAVKU TEMODAL 100 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 100 mg tobolky temozolomidum Perorální podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 tobolka
6. JINÉ
SÁČEK OBSAHUJÍCÍ 1 TVRDOU TOBOLKU PŘÍPRAVKU TEMODAL 140 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 140 mg tobolky temozolomidum Perorální podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 tobolka
6. JINÉ
SÁČEK OBSAHUJÍCÍ 1 TVRDOU TOBOLKU PŘÍPRAVKU TEMODAL 180 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 180 mg tobolky temozolomidum Perorální podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 tobolka
6. JINÉ
SÁČEK OBSAHUJÍCÍ 1 TVRDOU TOBOLKU PŘÍPRAVKU TEMODAL 250 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Temodal 250 mg tobolky temozolomidum Perorální podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 tobolka
6. JINÉ
Temodal 2,5 mg/ml prášek pro infuzní roztok temozolomidum
Jedna injekční lahvička obsahuje temozolomidum 100 mg.
Po rekonstituci obsahuje 1 ml infuzního roztoku temozolomidum 2,5 mg.
Pomocné látky: mannitol (E421), threonin, polysorbát 80, natrium-citrát a koncentrovaná kyselina chlorovodíková pro úpravu pH.
Více informací ohledně sodíku je uvedeno v příbalové informaci.
Prášek pro infuzní roztok 1 injekční lahvička 100 mg
Pouze pro intravenózní podání.
Pouze pro jednorázové použití.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP
Uchovávejte v chladničce.
Po rekonstituci roztok použijte do 14 hodin při 25°C, včetně doby infuze.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
EU/1/98/096/023
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato
Temodal 2,5 mg/ml prášek pro infuzní roztok temozolomidum
Jedna injekční lahvička obsahuje temozolomidum 100 mg.
Po rekonstituci obsahuje 1 ml infuzního roztoku temozolomidum 2,5 mg.
Mannitol (E421), threonin, polysorbát 80, natrium-citrát a koncentrovaná kyselina chlorovodíková. Více informací ohledně sodíku je uvedeno v příbalové informaci.
Prášek pro infuzní roztok 100 mg
Intravenózní podání, jednorázové použití.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP
Uchovávejte v chladničce.
Po rekonstituci: 14 hodin při 25°C, včetně doby infuze.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
Likvidace v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/98/096/023
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
Příbalová informace: informace pro uživatele
Temodal 5 mg tvrdé tobolky Temodal 20 mg tvrdé tobolky Temodal 100 mg tvrdé tobolky Temodal 140 mg tvrdé tobolky Temodal 180 mg tvrdé tobolky Temodal 250 mg tvrdé tobolky temozolomidum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Temodal a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Temodal užívat
3. Jak se Temodal užívá
4. Možné nežádoucí účinky
5. Jak Temodal uchovávat
6. Obsah balení a další informace
1. Co je Temodal a k čemu se používá
Přípravek Temodal obsahuje léčivou látku zvanou temozolomid. Toto léčivo je protinádorová látka.
Temodal se používá k léčbě specifických forem mozkových nádorů:
- u dospělých s nově diagnostikovaným multiformním glioblastomem. Temodal se používá nejprve v kombinaci s léčbou ozařováním (souběžná fáze léčby) a následně samostatně (monoterapeutická fáze léčby).
- u dětí ve věku 3 let a starších a u dospělých pacientů s maligním gliomem, jako je multiformní glioblastom nebo anaplastický astrocytom. Temodal se u těchto nádorů používá, jestliže se tyto nádory objeví znovu nebo se po standardní léčbě zhorší.
2. Čemu musíte věnovat pozornost, než začnete Temodal užívat
Neužívejte Temodal
- jestliže jste alergický(á) na temozolomid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže jste prodělali alergickou reakci na dakarbazin (protinádorové léčivo, někdy nazývané DTIC). Známky alergické reakce zahrnují svědění, dušnost nebo sípání, otok obličeje, rtů, jazyka nebo hrdla.
- jestliže máte významně snížený počet určitých typů krevních buněk (myelosuprese), jako je počet bílých krvinek a počet krevních destiček. Tyto krevní buňky jsou důležité v boji proti infekci a pro správnou krevní srážlivost. Váš lékař Vám před zahájením léčby zkontroluje krevní obraz, aby se ujistil, že máte dostatečný počet těchto buněk.
Upozornění a opatření
Před užitím přípravku Temodal se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou,
- jelikož máte být pečlivě sledován(a), zda se u Vás nevyvíjí závažná forma hrudní infekce (zápal plic vyvolanýPneumocystis jirovecii - PCP). Jestliže jste nově diagnostikovaný(á) pacient(pacientka) (s multiformním glioblastomem), můžete Temodal užívat po dobu 42 dnů v kombinaci s radioterapií. V tomto případě Vám lékař rovněž předepíše lék, který Vám pomůže předejít tomuto typu zápalu plic (PCP).
- pokud jste někdy měl(a) nebo možná nyní máte infekci způsobenou virem hepatitidy B. To proto, že přípravek Temodal by mohl způsobit, že se hepatitida B znovu aktivuje, což může být v některých případech smrtelné. Pacienti budou před začátkem léčby svým doktorem pečlivě zkontrolováni na příznaky této infekce.
- jestliže před zahájením léčby máte nízký počet červených krvinek (anémie), bílých krvinek nebo krevních destiček, nebo máte problémy s krevní srážlivostí, nebo pokud se tyto poruchy objeví v průběhu léčby. Váš lékař se může rozhodnout snížit dávku, přerušit, ukončit nebo Vaši léčbu změnit. Můžete také potřebovat další způsoby léčby. V některých případech může být nutné léčbu přípravkem Temodal ukončit. V průběhu léčby budou prováděna častá vyšetření krve, aby se sledovaly nežádoucí účinky přípravku Temodal na Vaše krevní buňky.
- jelikož můžete mít malé riziko jiných změn krevních buněk, včetně leukémie.
- jestliže se u Vás objeví nausea (nevolnost od žaludku) a/nebo zvracení, což jsou velmi časté nežádoucí účinky přípravku Temodal (viz bod 4), Váš lékař Vám může předepsat léčivo (antiemetikum), které napomáhá předcházet zvracení.
Jestliže zvracíte často před zahájením léčby nebo v průběhu léčby, požádejte svého lékaře o doporučení nejlepší doby pro užití přípravku Temodal, dokud zvracení není pod kontrolou. Pokud se dostaví zvracení po požití dávky léku, neužívejte již tentýž den další dávku.
- jestliže se u Vás objeví horečka nebo příznaky infekce, kontaktujte ihned svého lékaře.
- jestliže je Vám více než 70 let, můžete být náchylnější k infekcím, tvorbě modřin nebo krvácení.
- jestliže máte problémy s játry nebo ledvinami, může být nutné Vaši dávku přípravku Temodal upravit.
Děti a dospívající
Nepodávejte tento přípravek dětem mladším 3 let věku, jelikož nebyl u této skupiny hodnocen.
O pacientech ve věku 3 let a starších, kteří užívali přípravek Temodal, jsou k dispozici omezené údaje.
Další léčivé přípravky a Temodal
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství, kojení a fertilita
Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat. Je to proto, že přípravkem Temodal nesmíte být během těhotenství léčena, pokud to přímo nedoporučí Váš lékař.
Pacienti užívající Temodal, muži i ženy, musí užívat účinné prostředky proti početí (viz též „Mužská plodnost“ níže).
Kojení je nutné po dobu léčby přípravkem Temodal přerušit.
Mužská plodnost
Temodal může způsobit trvalou neplodnost. Mužští pacienti mají používat účinnou antikoncepci a nestát se otci po dobu až 6 měsíců od ukončení léčby. Doporučuje se, aby se před léčbou informovali o možnosti konzervace spermatu.
Řízení dopravních prostředků a obsluha strojů
V průběhu léčby přípravkem Temodal se můžete cítit unavení či ospalí. V takovém případě neřiďte dopravní prostředek ani neobsluhujte žádné nástroje nebo stroje nebo jízdní kolo, dokud nezjistíte, jak na Vás tento přípravek působí (viz bod 4).
Temodal obsahuje laktózu
Temodal obsahuje laktózu (druh cukru). Pokud Vám Váš lékař v minulosti sdělil, že trpíte nesnášenlivostí některých cukrů, kontaktujte svého lékaře před použitím tohoto přípravku.
3. Jak se Temodal užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Dávkování a doba trvání léčby
Vaši dávku přípravku Temodal stanoví Váš lékař. Je to na základě Vaší velikosti (výšky a hmotnosti) a v závislosti na tom, zda se u Vás nádor objevil znovu a zda jste již v minulosti podstoupil(a) chemoterapeutickou léčbu. Můžete dostat i další léky (antiemetika) k užívání před a/nebo po užití přípravku Temodal, které zabraňují nebo potlačují nevolnost a zvracení.
Pacienti s nově diagnostikovaným multiformním glioblastomem:
Jestliže jste nově diagnostikovaný(á) pacient(pacientka), bude léčba probíhat ve dvou fázích:
- nejprve léčba společně s ozařováním (fáze souběžné léčby)
- následovaná léčbou pouze přípravkem Temodal (monoterapeutická fáze léčby).
Během souběžné fáze léčby Váš lékař zahájí léčbu přípravkem Temodal v dávce 75 mg/m2 (obvyklá dávka). Tuto dávku budete užívat každý den po dobu 42 dnů (až 49 dnů) v kombinaci s léčbou ozařováním. Užití dávky přípravku Temodal může být oddáleno nebo užívání přerušeno v závislosti na Vašem krevním obraze a na tom, jak tento přípravek v souběžné fázi léčby snášíte.
Jakmile bude léčba ozařováním ukončena, přerušíte léčbu na 4 týdny. To Vašemu tělu umožní, aby se zotavilo.
Potom zahájíte monoterapeutickou fázi léčby.
V průběhu monoterapeutické fáze léčby se dávka a způsob užívání přípravku Temodal budou lišit.
Váš lékař stanoví Vaši přesnou dávku. Léčebných období (cyklů) může být až 6. Každé trvá 28 dnů. Vaši novou dávku přípravku Temodal samotného budete užívat jedenkrát denně po dobu prvních 5 dnů („dávkovací dny“) každého cyklu. První dávka bude 150 mg/m2. Poté dalších 23 dnů nebudete přípravek Temodal užívat. To je dohromady 28 dnů léčebného cyklu.
Po 28. dni začne další cyklus. Budete opět užívat přípravek Temodal jedenkrát denně po dobu 5 dnů následovaných 23 dny bez přípravku Temodal. Dávka přípravku Temodal se může upravit, její podání oddálit nebo přerušit v závislosti na Vašem krevním obraze a na tom, jak léčivý přípravek v průběhu každého léčebného cyklu snášíte.
Pacienti s nádory, které se objevily znovu nebo se zhoršily (maligní gliom, _jako například multiformní glioblastom nebo anaplastický astrocytom), užívající pouze Temodal:
Léčebný cyklus s přípravkem Temodal trvá 28 dnů.
Budete užívat pouze přípravek Temodal jedenkrát denně po dobu prvních 5 dnů. Tato denní dávka závisí na tom, zda jste předtím užíval(a) chemoterapii.
Jestliže jste předtím nebyl(a) léčen(a) chemoterapií, Vaše první dávka přípravku Temodal bude 200 mg/m2 jedenkrát denně prvních 5 dnů. Jestliže jste byl(a) předtím léčen(a) chemoterapií, Vaše první dávka přípravku Temodal bude 150 mg/m2 jedenkrát denně po dobu prvních 5 dnů. Poté nebudete dalších 23 dnů přípravek Temodal užívat. To je dohromady 28 dní léčebného cyklu.
Po 2 8. dni začne další cyklus. Budete opět užívat přípravek Temodal jedenkrát denně po dobu 5 dnů, s následujícími 23 dny bez přípravku Temodal.
Před každým novým léčebným cyklem bude vyšetřena krev, aby se zjistilo, zda nemá být dávka přípravku Temodal upravena. Podle výsledků krevních zkoušek může Váš lékař pro následující cyklus Vaši dávku upravit.
Jak se přípravek Temodal užívá
Užívejte předepsanou dávku přípravku Temodal jedenkrát denně, nejlépe každý den ve stejnou dobu.
Tobolky užívejte nalačno; například nejméně jednu hodinu před plánovanou snídaní. Tobolku(tobolky) polykejte celou(celé) a zapijte sklenicí vody. Tobolky neotvírejte, nedrťte nebo nekousejte. Pokud je tobolka poškozená, zabraňte kontaktu prášku s kůží, očima nebo nosem. Pokud se vám omylem nějaký dostane do očí nebo nosu, opláchněte oblast vodou.
Podle Vaší předepsané dávky můžete užívat více než jednu tobolku naráz, případně tobolky s různými silami (obsah léčivé látky, v mg). Barva víčka tobolky je odlišná pro každou sílu (viz tabulka níže).
|
Síla |
Barva víčka tobolky |
|
Temodal 5 mg tvrdé tobolky |
zelená |
|
Temodal 20 mg tvrdé tobolky |
žlutá |
|
Temodal 100 mg tvrdé tobolky |
růžová |
|
Temodal 140 mg tvrdé tobolky |
modrá |
|
Temodal 180 mg tvrdé tobolky |
oranžová |
|
Temodal 250 mg tvrdé tobolky |
bílá |
Měl(a) byste se ubezpečit, že plně rozumíte a pamatujete si následující:
• kolik tobolek potřebujete každý dávkovací den užívat. Požádejte svého lékaře či lékárníka, aby Vám rozepsal počet tobolek (včetně barvy).
• které dny jsou Vaše dávkovací dny.
Ujistěte se se svým lékařem pokaždé, když začínáte nový cyklus, jak je upraveno dávkování, protože může být jiné než v minulém cyklu.
Vždy užívejte Temodal přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), porad’te se se svým lékařem nebo lékárníkem. Chyba při užívání tohoto přípravku může mít závažné důsledky pro Vaše zdraví.
Jestliže jste užil(a) více přípravku Temodal, než jste měl(a)
Pokud omylem užijete vyšší než doporučený počet tobolek přípravku Temodal, okamžitě kontaktujte svého lékaře, lékárníka nebo zdravotní sestru.
Jestliže jste zapomněl(a) užít Temodal
Užijte zapomenutou dávku co nejdříve tentýž den. Pokud jste zmeškal(a) celý den, poraďte se se svým lékařem. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku, pokud Vám to lékař neurčí.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Možné nežádoucí účinky
4.
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Neprodleně kontaktujte svého lékaře, jestliže se u Vás objeví kterýkoli z následujících příznaků:
- závažná alergická (hypersenzitivní) reakce (kopřivka, sípaní nebo jiné dýchací potíže),
- nekontrolované krvácení,
- záchvaty (křeče),
- horečka,
- silná neustupující bolest hlavy.
Podávání přípravku Temodal může způsobit snížení počtu některých typů krvinek. To může vést ke zvýšenému vzniku podlitin nebo krvácení, anémii (chudokrevnosti, tj. sníženému počtu červených krvinek), horečce a snížené odolnosti vůči infekcím. Toto snížení počtu krvinek je obvykle krátkodobé. V některých případech ale může být dlouhotrvající a může vést k velmi závažné formě anémie (aplastická anémie). Váš lékař zajistí pravidelné vyšetřování Vaší krve, aby se zjistily jakékoli změny, a rozhodne o tom, zda je nutná specifická léčba. V některých případech může být nutné snížit dávky přípravku Temodal nebo ukončit jeho podávání.
Nežádoucí účinky zjištěné v klinických hodnoceních:
Temodal v kombinované léčbě s ozařováním u nově diagnostikovaného glioblastomu Pacienti užívající přípravek Temodal v kombinaci s ozařováním mohou zaznamenat odlišné nežádoucí účinky než pacienti léčení pouze přípravkem Temodal. Mohou se vyskytnout následující nežádoucí účinky, které mohou vyžadovat lékařský zásah.
Velmi časté (mohou postihnout více než 1 z 10 osob): ztráta chuti k jídlu, bolest hlavy, zácpa (problém s vyprazdňováním), nausea (nevolnost), zvracení, vyrážka, vypadávání vlasů, utahanost.
Časté (mohou postihnout až 1 z 10 osob): infekce v ústech, infekce rány, snížení počtu krvinek (neutropenie, trombocytopenie, lymfopenie, leukopenie), zvýšení cukru v krvi, pokles hmotnosti, změna duševního stavu nebo bdělosti, úzkost/deprese, ospalost, potíže s mluvením, porucha rovnováhy, závrať, zmatenost, zapomnětlivost, potíže s koncentrací, neschopnost usnout nebo spát, pocit mravenčení, podlitiny, třes, abnormální nebo rozmazané vidění, dvojité vidění, porucha sluchu, dušnost, kašel, krevní sraženina v nohách, zadržování tekutiny, otok dolních končetin, průjem, bolest žaludku nebo břicha, pálení žáhy, žaludeční potíže, potíže s polykáním, sucho v ústech, podráždění nebo zčervenání kůže, suchá kůže, svědění, svalová slabost, bolestivé klouby, pobolívání a bolesti svalů, časté močení, potíže s udržením moče, alergická reakce, horečka, poškození ozářením, otok obličeje, bolest, změny chuti, abnormality testů funkce jater.
Méně časté (mohou postihnout až 1 ze 100 osob): příznaky podobné chřipce, červené skvrny pod kůží, nízká hladina draslíku v krvi, zvýšení hmotnosti, změny nálady, halucinace a porucha paměti, částečné ochrnutí, poruchy koordinace, poruchy vnímání, částečná ztráta zraku, suché oči nebo bolest očí, hluchota, infekce středního ucha, zvonění v uších, bolest ucha, palpitace (bušení srdce), krevní sraženina v plicích, vysoký krevní tlak, zápal plic, zánět nosních dutin, zánět průdušek, nachlazení nebo chřipka, otok žaludku, potíže s kontrolou vyprazdňování střev, hemeroidy, odlupování kůže, zvýšená citlivost kůže na sluneční záření, změna barvy kůže, zvýšené pocení, poškození svalů, bolest v zádech, potíže s močením, poševní krvácení, sexuální impotence, vynechání menstruace nebo silné menstruační krvácení, podráždění pochvy, bolest prsou, návaly horka, třesení, změny barvy jazyka, změna čichového smyslu, žízeň, zubní potíže.
Monoterapie _přípravkem Temodal u opakovaného výskytu nebo zhoršení nádoru Mohou se vyskytnout následující nežádoucí účinky, které mohou vyžadovat lékařský zásah.
Velmi časté (mohou postihnout více než 1 z 10 osob): snížený počet krevních buněk (bílých krvinek-neutropenie nebo lymfopenie, krevních destiček- trombocytopenie), ztráta chuti k jídlu, bolest hlavy, zvracení, nausea (nevolnost od žaludku), zácpa (problém s vyprazdňováním), únava.
Časté (mohou postihnout až 1 z 10 osob): pokles hmotnosti, ospalost, závrať, pocit mravenčení, dušnost, průjem, bolest břicha, žaludeční potíže, vyrážka, svědění, vypadávání vlasů, horečka, slabost, třesení, celkový pocit nepohody, bolest, změny chuti.
Méně časté (mohou postihnout až 1 ze 100 osob): snížený počet krevních buněk (všech buněk-pancytopenie, červených krvinek-anémie, bílých krvinek-leukopenie).
Vzácné (mohou postihnout až 1 z 1 000 osob): kašel, infekce včetně zápalu plic.
Velmi vzácné (mohou postihnout až 1 z 10 000 osob): zčervenání kůže, kopřivka, kožní výsev, alergické reakce.
Jiné nežádoucí účinky:
Často byly hlášeny případy zvýšených hladin jaterních enzymů. Méně často byly hlášeny případy zvýšené hladiny bilirubinu, problémů s odtokem žluče (cholestáza), zánětu jater a poškození jater, včetně smrtelného selhání jater.
Ve velmi vzácných případech byla pozorována závažná vyrážka s otokem kůže, včetně dlaní a chodidel, nebo bolestivé zarudnutí kůže a/nebo puchýřky na těle nebo v ústech. Jestliže se tyto příznaky vyskytnou, oznamte to neprodleně svému lékaři.
U přípravku Temodal byly pozorovány velmi vzácné případy plicních nežádoucích účinků. Pacienti obvykle udávali dušnost a kašel. Pokud si všimnete některého z těchto příznaků, sdělte to svému lékaři.
Velmi vzácně existuje u pacientů užívajících Temodal a jemu podobná léčiva malé riziko vzniku druhotných nádorů, včetně leukémie.
Méně často byly hlášeny nové nebo reaktivované (opakující se) infekce způsobené cytomegaloviry a reaktivované infekce způsobené virem heapatitidy B.
Méně často byly hlášeny případy diabetes insipidus. Příznaky diabetes insipidus zahrnují nadměrné močení a pocit žízně.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Temodal uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí, nejlépe v uzamčené skříni. Náhodné požití může být pro děti smrtelné.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Balení v lahvičce
Uchovávejte při teplotě do 30°C.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před vlhkostí.
Lahvičku uchovávejte dobře uzavřenou.
Balení v sáčcích
Uchovávejte při teplotě do 30°C.
Pokud zjistíte změny ve vzhledu tobolek, oznamte to lékárníkovi.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak máte naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co Temodal obsahuje
Léčivou látkou je temozolomidum.
Temodal 5 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 5 mg.
Temodal 20 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 20 mg.
Temodal 100 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 100 mg.
Temodal 140 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 140 mg.
Temodal 180 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 180 mg.
Temodal 250 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 250 mg.
Pomocnými látkami jsou: obsah tobolky:
laktóza, koloidní bezvodý oxid křemičitý, sodná sůl karboxymethylškrobu typu A, kyselina vinná, kyselina stearová (viz bod 2 "Temodal obsahuje laktózu"). obal tobolky:
Temodal 5 mg tvrdé tobolky: želatina, oxid titaničitý (E 171), natrium-lauryl-sulfát, žlutý oxid železitý (E 172), indigokarmín (E 132),
Temodal 20 mg tvrdé tobolky: želatina, oxid titaničitý (E 171), natrium-lauryl-sulfát, žlutý oxid železitý (E 172),
Temodal 100 mg tvrdé tobolky: želatina, oxid titaničitý (E171), natrium-lauryl-sulfát, červený oxid železitý (E 172),
Temodal 140 mg tvrdé tobolky: želatina, oxid titaničitý (E 171), natrium-lauryl-sulfát, indigokarmín (E 132),
Temodal 180 mg tvrdé tobolky: želatina, oxid titaničitý (E 171), natrium-lauryl-sulfát, žlutý oxid železitý (E 172), červený oxid železitý (E 172),
Temodal 250 mg tvrdé tobolky: želatina, oxid titaničitý (E 171), natrium-lauryl-sulfát. inkoustový potisk:
šelak, propylenglykol, čištěná voda, roztok amoniaku, hydroxid draselný a černý oxid železitý (E 172).
Jak Temodal vypadá a co obsahuje toto balení
Temodal 5 mg tvrdé tobolky mají neprůsvitné bílé tělo, neprůsvitné zelené víčko a jsou potištěny černým inkoustem.
Temodal 20 mg tvrdé tobolky mají neprůsvitné bílé tělo, neprůsvitné žluté víčko a jsou potištěny černým inkoustem.
Temodal 100 mg tvrdé tobolky mají neprůsvitné bílé tělo, neprůsvitné růžové víčko a jsou potištěny černým inkoustem.
Temodal 140 mg tvrdé tobolky mají neprůsvitné bílé tělo, modré víčko a jsou potištěny černým inkoustem.
Temodal 180 mg tvrdé tobolky mají neprůsvitné bílé tělo, neprůsvitné oranžové víčko a jsou potištěny černým inkoustem.
Temodal 250 mg tvrdé tobolky mají neprůsvitné bílé tělo a víčko a jsou potištěny černým inkoustem.
Balení v lahvičce
Tvrdé tobolky (tobolky) pro perorální podání jsou dodávány ve skleněných lahvičkách jantarové barvy, obsahujících 5 nebo 20 tobolek.
Krabička obsahuje jednu lahvičku.
Balení v sáčcích
Tvrdé tobolky pro perorální podání jsou jednotlivě uzavřené v sáčcích a vydáváné v krabičkách obsahujících 5 nebo 20 tvrdých tobolek.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci: Merck Sharp & Dohme Limited, Hertford Road, Hoddesdon, Hertfordshire EN11 9BU, Velká Británie
Výrobce: SP Labo N.V., Industriepark 30, B-2220 Heist-op-den-Berg, Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgique/Belgie/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Bt^rapnn
MepK fflapn h floyM Etnrapna EOOfl Ten.: +359 2 819 3737 info-msdbg@merck.com
Česká republika
Merck Sharp & Dohme s.r.o.
Tel.: +420 233 010 111 dpoc_czechslovak@merck.com
Danmark
MSD Danmark ApS Tlf: +45 4482 4000 dkmail@merck.com
Deutschland
MSD SHARP & DOHME GMBH
Tel: 0800 673 673 673 (+49 (0) 89 4561 2612)
Eesti
Merck Sharp & Dohme OU Tel.: +372 6144 200 msdeesti@merck.com
Lietuva
UAB Merck Sharp & Dohme Tel.:+370 5 278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Magyarország
MSD Pharma Hungary Kft.
Tel.: +36 1 888 53 00 hungary_msd@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Nederland
Merck Sharp & Dohme BV
Tel: 0800 9999000 (+31 23 5153153)
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
|
EXXáSa MSD A.OB.E.E. TpA,: +30 210 98 97 300 dpoc_greece@merck.com |
Osterreich Merck Sharp & Dohme Ges.m.b.H. Tel: +43 (0) 1 26 044 msd-medizin@merck.com |
|
Espaňa Merck Sharp & Dohme de Espana, S.A. Tel: +34 91 321 06 00 msd info@merck.com |
Polska MSD Polska Sp. z o.o. Tel:+48 22 549 51 00 msdpolska@merck.com |
|
France MSD France Tél: +33 (0) 1 80 46 40 40 |
Portugal Merck Sharp & Dohme, Lda. Tel: +351 21 446 57 00 clic@merck.com |
|
Hrvatska Merck Sharp & Dohme d.o.o. Tel: + 385 1 6611 333 croatia info@merck.com |
Románia Merck Sharp & Dohme Romania S.R.L. Tel: + 4021 529 29 00 msdromania@merck.com |
Ireland Slovenija
Merck Sharp & Dohme Ireland (Human Health) Merck Sharp & Dohme, inovativna zdravila
|
Limited Tel: +353 (0)1 2998700 medinfo ireland@merck.com |
d.o.o. Tel: +386 1 5204 201 msd_slovenia@merck.com |
|
Ísland Vistor hf. Simi: +354 535 7000 |
Slovenská republika Merck Sharp & Dohme, s. r. o. Tel.: +421 2 58282010 dpoc_czechslovak@merck.com |
|
Italia MSD Italia S.r.l. Tel: +39 06 361911 medicalinformation.it@merck.com |
Suomi/Finland MSD Finland Oy Puh/Tel: +358 (0)9 804650 info@msd.fi |
|
Kúnpoq Merck Sharp & Dohme Cyprus Limited TpA,: 800 00 673 (+357 22866700) cyprus info@merck.com |
Sverige Merck Sharp & Dohme (Sweden) AB Tel: +46 (0) 77 5700488 medicinskinfo@merck.com |
|
Latvija SIA Merck Sharp & Dohme Latvija Tel: +371 67364224 msd lv@merck.com |
United Kingdom Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Příbalová informace: informace pro uživatele
Temodal 2,5 mg/ml prášek pro infuzní roztok
temozolomidum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Temodal a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Temodal používat
3. Jak se Temodal používá
4. Možné nežádoucí účinky
5. Jak Temodal uchovávat
6. Obsah balení a další informace
1. Co je Temodal a k čemu se používá
Přípravek Temodal obsahuje léčivou látku zvanou temozolomid. Toto léčivo je protinádorová látka.
Temodal se používá k léčbě specifických forem mozkových nádorů:
- u dospělých s nově diagnostikovaným multiformním glioblastomem. Temodal se používá nejprve v kombinaci s léčbou ozařováním (souběžná fáze léčby) a následně samostatně (monoterapeutická fáze léčby).
- u dětí ve věku 3 let a starších a u dospělých pacientů s maligním gliomem, jako je multiformní glioblastom nebo anaplastický astrocytom. Temodal se u těchto nádorů používá, jestliže se tyto nádory objeví znovu nebo se po standardní léčbě zhorší.
2. Čemu musíte věnovat pozornost, než začnete Temodal používat
Nepoužívejte Temodal
- jestliže jste alergický(á) na temozolomid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže jste prodělali alergickou reakci na dakarbazin (protinádorové léčivo, někdy nazývané DTIC). Známky alergické reakce zahrnují svědění, dušnost nebo sípání, otok obličeje, rtů, jazyka nebo hrdla.
- jestliže máte významně snížený počet určitých typů krevních buněk (myelosuprese), jako je počet bílých krvinek a počet krevních destiček. Tyto krevní buňky jsou důležité v boji proti infekci a pro správnou krevní srážlivost. Váš lékař Vám před zahájením léčby zkontroluje krevní obraz, aby se ujistil, že máte dostatečný počet těchto buněk.
Upozornění a opatření
Před použitím přípravku Temodal se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou,
- jelikož máte být pečlivě sledován(a), zda se u Vás nevyvíjí závažná forma hrudní infekce (zápal plic vyvolanýPneumocystis jirovecii - PCP. Jestliže jste nově diagnostikovaný(á) pacient(pacientka) (s multiformním glioblastomem), můžete Temodal dostávat po dobu 42 dnů v kombinaci s radioterapií. V tomto případě Vám lékař rovněž předepíše lék, který Vám pomůže předejít tomuto typu zápalu plic (PCP).
- pokud jste někdy měl(a) nebo možná nyní máte infekci způsobenou virem hepatitidy B. To proto, že přípravek Temodal by mohl způsobit, že se hepatitida B znovu aktivuje, což může být v některých případech smrtelné. Pacienti budou před začátkem léčby svým doktorem pečlivě zkontrolováni na příznaky této infekce.
- jestliže před zahájením léčby máte nízký počet červených krvinek (anémie), bílých krvinek nebo krevních destiček nebo máte problémy s krevní srážlivostí, nebo pokud se tyto poruchy objeví v průběhu léčby. Váš lékař se může rozhodnout snížit dávku, přerušit, ukončit nebo Vaši léčbu změnit. Můžete také potřebovat další způsoby léčby. V některých případech může být nutné léčbu přípravkem Temodal ukončit. V průběhu léčby budou prováděna častá vyšetření krve, aby se sledovaly nežádoucí účinky přípravku Temodal na Vaše krevní buňky.
- jelikož můžete mít malé riziko jiných změn krevních buněk, včetně leukémie.
- jestliže se u Vás objeví nausea (nevolnost od žaludku) a/nebo zvracení, což jsou velmi časté nežádoucí účinky přípravku Temodal (viz bod 4), Váš lékař Vám může předepsat léčivo (antiemetikum), které napomáhá předcházet zvracení.
- jestliže se u Vás objeví horečka nebo příznaky infekce, kontaktujte ihned svého lékaře.
- jestliže je Vám více než 70 let, můžete být náchylnější k infekcím, tvorbě modřin nebo krvácení.
- jestliže máte problémy s játry nebo ledvinami, může být nutné Vaši dávku přípravku Temodal upravit.
Děti a dospívající
Nepodávejte tento přípravek dětem mladším 3 let věku, jelikož nebyl u této skupiny hodnocen.
O pacientech ve věku 3 let a starších, kteří používali přípravek Temodal, jsou k dispozici omezené údaje.
Další léčivé přípravky a Temodal
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství, kojení a fertilita
Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat. Je to proto, že přípravkem Temodal nesmíte být během těhotenství léčena, pokud to přímo nedoporučí Váš lékař.
Pacienti používající Temodal, muži i ženy, musí užívat účinné prostředky proti početí (viz též „Mužská plodnost“ níže).
Kojení je nutné po dobu léčby přípravkem Temodal přerušit.
Mužská plodnost
Temodal může způsobit trvalou neplodnost. Mužští pacienti mají používat účinnou antikoncepci a nestát se otci po dobu až 6 měsíců od ukončení léčby. Doporučuje se, aby se informovali o možnosti konzervace spermatu před léčbou.
Řízení dopravních prostředků a obsluha strojů
V průběhu léčby přípravkem Temodal se můžete cítit unavení či ospalí. V takovém případě neřiďte dopravní prostředek ani neobsluhujte žádné nástroje nebo stroje nebo jízdní kolo, dokud nezjistíte, jak na Vás tento přípravek působí (viz bod 4).
Temodal obsahuje sodík
Tento přípravek obsahuje 2,4 mmol sodíku v jedné injekční lahvičce. To by měli brát v úvahu pacienti dodržující dietu s omezením sodíku.
Jak se Temodal používá
3.
Vždy používejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Vaši dávku přípravku Temodal stanoví Váš lékař. Je to na základě Vaší velikosti (výšky a hmotnosti) a v závislosti na tom, zda se u Vás nádor objevil znovu a zda jste již v minulosti podstoupil(a) chemoterapeutickou léčbu. Můžete dostat i další léky (antiemetika) k užívání před a/nebo po použití přípravku Temodal, které zabraňují nebo potlačují nevolnost a zvracení.
Pacienti s nově diagnostikovaným multiformním glioblastomem:
Jestliže jste nově diagnostikovaný(á) pacient(pacientka), bude léčba probíhat ve dvou fázích:
- nejprve léčba společně s ozařováním (fáze souběžné léčby)
- následovaná léčbou pouze přípravkem Temodal (monoterapeutická fáze léčby).
Během souběžné fáze léčby Váš lékař zahájí léčbu přípravkem Temodal v dávce 75 mg/m2 (obvyklá dávka). Tuto dávku budete dostávat každý den po dobu 42 dnů (až 49 dnů) v kombinaci s léčbou ozařováním. Užití dávky přípravku Temodal může být oddáleno nebo používání přerušeno v závislosti na Vašem krevním obraze a na tom, jak tento přípravek v souběžné fázi léčby snášíte.
Jakmile bude léčba ozařováním ukončena, přerušíte léčbu na 4 týdny. To Vašemu tělu umožní, aby se zotavilo.
Poté zahájíte monoterapeutickou fázi léčby.
V průběhu monoterapeutické fáze léčby se dávka a způsob používání přípravku Temodal budou lišit. Váš lékař stanoví Vaši přesnou dávku. Léčebných období (cyklů) může být až 6. Každé trvá 28 dnů. Vaši novou dávku přípravku Temodal samotného budete dostávat jedenkrát denně po dobu prvních 5 dnů („dávkovací dny“) každého cyklu. První dávka bude 150 mg/m2. Poté dalších 23 dnů nebudete přípravek Temodal používat. To je dohromady 28 dnů léčebného cyklu.
Po 28. dni začne další cyklus. Budete opět dostávat přípravek Temodal jedenkrát denně po dobu 5 dnů následovaných 23 dny bez přípravku Temodal. Dávka přípravku Temodal se může upravit, její podání oddálit nebo přerušit v závislosti na Vašem krevním obraze a na tom, jak léčivý přípravek v průběhu každého léčebného cyklu snášíte.
Pacienti s nádory, které se objevily znovu nebo se zhoršily (maligní gliom, _jako například multiformní glioblastom nebo anaplastický astrocytom), používající pouze Temodal:
Léčebný cyklus s přípravkem Temodal trvá 28 dnů.
Budete dostávat pouze přípravek Temodal jedenkrát denně po dobu prvních 5 dnů. Tato denní dávka závisí na tom, zda jste předtím užíval(a) chemoterapii.
Jestliže jste předtím nebyl(a) léčen(a) chemoterapií, Vaše první dávka přípravku Temodal bude 200 mg/m2 jedenkrát denně po dobu prvních 5 dnů. Jestliže jste byl(a) předtím léčen(a) chemoterapií, Vaše první dávka přípravku Temodal bude 150 mg/m2 jedenkrát denně po dobu prvních 5 dnů.
Poté nebudete dalších 23 dnů přípravek Temodal používat. To je dohromady 28 dní léčebného cyklu.
Po 28. dni začne další cyklus. Budete opět dostávat přípravek Temodal jedenkrát denně po dobu pěti dnů s následujícími 23 dny bez přípravku Temodal.
Před každým novým léčebným cyklem bude vyšetřena krev, aby se zjistilo, zda nemá být dávka přípravku Temodal upravena. Podle výsledku krevních zkoušek může Váš lékař pro následující cyklus Vaši dávku upravit.
Jak se přípravek Temodal podává
Temodal Vám bude podán Vaším lékařem ve formě žilní infuze (intravenózní infuze), která trvá přibližně 90 minut. Jiné místo infuze vyjma žíly není přijatelné.
Jestliže jste užil(a) více přípravku Temodal, než jste měl(a)
Protože Vám tento lék podávají odborní zdravotničtí pracovníci, není pravděpodobné, že by Vám podávali více přípravku Temodal, než by měli. Pokud však k tomu dojde, lékař nebo zdravotní sestra Vás budou léčit, jak bude třeba.
Máte-li jakékoli další otázky, týkající se používání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Neprodleně kontaktujte svého lékaře, jestliže se u Vás objeví kterýkoli z následujících příznaků:
- závažná alergická (hypersenzitivní) reakce (kopřivka, sípaní nebo jiné dýchací potíže),
- nekontrolované krvácení,
- záchvaty (křeče),
- horečka,
- silná neustupující bolest hlavy.
Podávání přípravku Temodal může způsobit snížení počtu některých typů krvinek. To může vést ke zvýšenému vzniku podlitin nebo krvácení, anémii (chudokrevnosti, tj. sníženému počtu červených krvinek), horečce a snížené odolnosti vůči infekcím. Toto snížení počtu krvinek je obvykle krátkodobé. V některých případech ale může být dlouhotrvající a může vést k velmi závažné formě anémie (aplastická anémie). Váš lékař zajistí pravidelné vyšetřování Vaší krve, aby se zjistily jakékoli změny, a rozhodne o tom, zda je nutná specifická léčba. V některých případech může být nutné snížit dávky přípravku Temodal nebo ukončit jeho podávání.
Nežádoucí účinky zjištěné v klinických hodnoceních:
Temodal prášek pro infuzní roztok
Kromě níže uvedených nežádoucích účinků se při používání přípravku Temodal prášek pro infuzní roztok mohou objevit také následující účinky: bolest, podráždění, svědění, horkost, otok nebo zarudnutí v místě vpichu; také modřiny (hematomy).
Temodal v kombinované léčbě s ozařováním u nově diagnostikovaného glioblastomu
Pacienti používající přípravek Temodal v kombinaci s ozařováním mohou zaznamenat odlišné nežádoucí účinky než pacienti léčení pouze přípravkem Temodal. Mohou se vyskytnout následující nežádoucí účinky, které mohou vyžadovat lékařský zásah.
Velmi časté (mohou postihnout více než 1 z 10 osob): ztráta chuti k jídlu, bolest hlavy, zácpa (problém s vyprazdňováním), nausea (nevolnost od žaludku), zvracení, vyrážka, vypadávání vlasů, utahanost.
Časté (mohou postihnout až 1 z 10 osob): infekce v ústech, infekce rány, snížení počtu krvinek (neutropenie, trombocytopenie, lymfopenie, leukopenie), zvýšení cukru v krvi, pokles hmotnosti, změna duševního stavu nebo bdělosti, úzkost/deprese, ospalost, potíže s mluvením, porucha rovnováhy, závrať, zmatenost, zapomnětlivost, potíže s koncentrací, neschopnost usnout nebo spát, pocit mravenčení, podlitiny, třes, abnormální nebo rozmazané vidění, dvojité vidění, porucha sluchu, dušnost, kašel, krevní sraženina v nohách, zadržování tekutiny, otok dolních končetin, průjem, bolest žaludku nebo břicha, pálení žáhy, žaludeční potíže, potíže s polykáním, sucho v ústech, podráždění nebo zčervenání kůže, suchá kůže, svědění, svalová slabost, bolestivé klouby, pobolívání a bolesti svalů, časté močení, potíže s udržením moče, alergická reakce, horečka, poškození ozářením, otok obličeje, bolest, změny chuti, abnormality testů funkce jater.
Méně časté (mohou postihnout až 1 ze 100 osob): příznaky podobné chřipce, červené skvrny pod kůží, nízká hladina draslíku v krvi, zvýšení hmotnosti, změny nálady, halucinace a porucha paměti, částečné ochrnutí, poruchy koordinace, poruchy vnímání, částečná ztráta zraku, suché oči nebo bolest očí, hluchota, infekce středního ucha, zvonění v uších, bolest ucha, palpitace (bušení srdce), krevní sraženina v plicích, vysoký krevní tlak, zápal plic, zánět nosních dutin, zánět průdušek, nachlazení nebo chřipka, otok žaludku, potíže s kontrolou vyprazdňování střev, hemeroidy, odlupování kůže, zvýšená citlivost kůže na sluneční záření, změna barvy kůže, zvýšené pocení, poškození svalů, bolest v zádech, potíže s močením, poševní krvácení, sexuální impotence, vynechání menstruace nebo silné menstruační krvácení, podráždění pochvy, bolest prsou, návaly horka, třesení, změny barvy jazyka, změna čichového smyslu, žízeň, zubní potíže.
Monoterapie _přípravkem Temodal u opakovaného výskytu nebo zhoršení nádoru
Mohou se vyskytnout následující nežádoucí účinky, které mohou vyžadovat lékařský zásah.
Velmi časté (mohou postihnout více než 1 z 10 osob): snížený počet krevních buněk (bílých krvinek-neutropenie nebo lymfopenie, krevních destiček- trombocytopenie), ztráta chuti k jídlu, bolest hlavy, zvracení, nausea (nevolnost od žaludku), zácpa (problém s vyprazdňováním), únava.
Časté (mohou postihnout až 1 z 10 osob): pokles hmotnosti, ospalost, závrať, pocit mravenčení, dušnost, průjem, bolest břicha, žaludeční potíže, vyrážka, svědění, vypadávání vlasů, horečka, slabost, třesení, celkový pocit nepohody, bolest, změny chuti.
Méně časté (mohou postihnout až 1 ze 100 osob): snížený počet krevních buněk (všech buněk-pancytopenie, červených krvinek-anémie, bílých krvinek-leukopenie).
Vzácné (mohou postihnout až 1 z 1 000 osob): kašel, infekce včetně zápalu plic.
Velmi vzácné (mohou postihnout až 1 z 10 000 osob): zčervenání kůže, kopřivka, kožní výsev, alergické reakce.
Jiné nežádoucí účinky:
Často byly hlášeny případy zvýšených hladin jaterních enzymů. Méně často byly hlášeny případy zvýšené hladiny bilirubinu, problémů s odtokem žluče (cholestáza), zánětu jater a poškození jater, včetně smrtelného selhání jater.
Ve velmi vzácných případech byla pozorována závažná vyrážka s otokem kůže, včetně dlaní a chodidel, nebo bolestivé zarudnutí kůže a/nebo puchýřky na těle nebo v ústech. Jestliže se tyto příznaky vyskytnou, oznamte to neprodleně svému lékaři.
U přípravku Temodal byly pozorovány velmi vzácné případy plicních nežádoucích účinků. Pacienti obvykle udávali dušnost a kašel. Pokud si všimnete některého z těchto příznaků, sdělte to svému lékaři
Velmi vzácně existuje u pacientů užívajících Temodal a jemu podobná léčiva malé riziko vzniku druhotných nádorů, včetně leukémie.
Méně často byly hlášeny nové nebo reaktivované (opakující se) infekce způsobené cytomegaloviry a reaktivované infekce způsobené virem heapatitidy B.
Méně často byly hlášeny případy diabetes insipidus. Příznaky diabetes insipidus zahrnují nadměrné močení a pocit žízně.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Temodal uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 - 8 °C).
Jakmile je přípravek připraven k infuzi (rekonstituován/rozpuštěn), lze roztok uchovávat při pokojové teplotě (25 °C) po dobu až 14 hodin, včetně doby infuze.
Připravený roztok by se neměl používat, jestliže má změněnou barvu nebo obsahuje částice.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak máte naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Temodal obsahuje
Léčivou látkou je temozolomidum. Jedna injekční lahvička obsahuje temozolomidum 100 mg. Po rekonstituci/rozpuštění obsahuje 1 ml infuzního roztoku 2,5 mg temozolomidu.
Pomocnými látkami jsou mannitol (E421), threonin, polysorbát 80, natrium-citrát (pro úpravu pH) a koncentrovaná kyselina chlorovodíková (pro úpravu pH) (viz bod 2).
Jak Temodal vypadá a co obsahuje toto balení
Prášek pro infuzní roztok je bílé barvy. Temodal je dostupný ve skleněné injekční lahvičce s butylovou gumovou zátkou a hliníkovým uzávěrem s odstranitelným krytem.
Jedno balení obsahuje 1 injekční lahvičku se 100 mg temozolomidu.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci: Merck Sharp & Dohme Limited, Hertford Road, Hoddesdon, Hertfordshire EN11 9BU, Velká Británie
Výrobce: SP Labo N.V., Industriepark 30, B-2220 Heist-op-den-Berg, Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgique/Belgie/Belgien MSD Belgium BVBA/SPRL Tél/Tel: 0800 38 693 (+32(0)27766211) |
Lietuva UAB Merck Sharp & Dohme Tel.:+370 5 278 02 47 msd_lietuva@merck.com |
|
BtnrapHH MepK fflapn h floyM Etarapna EOOfl Ten.: +359 2 819 3737 info-msdbg@merck.com |
Luxembourg/Luxemburg MSD Belgium BVBA/SPRL Tél/Tel: 0800 38 693 (+32(0)27766211) |
|
Česká republika Merck Sharp & Dohme s.r.o. Tel.: +420 233 010 111 dpoc_czechslovak@merck.com |
Magyarország MSD Pharma Hungary Kft. Tel.: +36 1 888 53 00 hungary_msd@merck.com |
|
Danmark MSD Danmark ApS Tlf: +45 4482 4000 dkmail@merck.com |
Malta Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta info@merck.com |
|
Deutschland MSD SHARP & DOHME GMBH Tel: 0800 673 673 673 (+49 (0) 89 4561 2612) |
Nederland Merck Sharp & Dohme BV Tel: 0800 9999000 (+31 23 5153153) |
|
Eesti Merck Sharp & Dohme OU Tel.: +372 6144 200 msdeesti@merck.com |
Norge MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no |
|
EXXáSa MSD A.OB.E.E. TpA,: +30 210 98 97 300 dpoc_greece@merck.com |
Osterreich Merck Sharp & Dohme Ges.m.b.H. Tel: +43 (0) 1 26 044 msd-medizin@merck.com |
|
Espaňa Merck Sharp & Dohme de Espana, S.A. Tel: +34 91 321 06 00 msd info@merck.com |
Polska MSD Polska Sp. z o.o. Tel:+48 22 549 51 00 msdpolska@merck.com |
|
France MSD France Tél: +33 (0) 1 80 46 40 40 |
Portugal Merck Sharp & Dohme, Lda. Tel: +351 21 446 57 00 clic@merck.com |
|
Hrvatska Merck Sharp & Dohme d.o.o. Tel: + 385 1 6611 333 croatia_info@merck.com |
Románia Merck Sharp & Dohme Romania S.R.L. Tel: + 4021 529 29 00 msdromania@merck.com |
Ireland Slovenija
Merck Sharp & Dohme Ireland (Human Health) Merck Sharp & Dohme, inovativna zdravila
|
Limited Tel: +353 (0)1 2998700 medinfo_ireland@merck.com |
d.o.o. Tel: +386 1 5204 201 msd_slovenia@merck.com |
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúrcpog
Merck Sharp & Dohme Cyprus Limited Tpk: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364224 msd_lv@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel.: +421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy Puh/Tel: +358 (0)9 804650 info@msd.fi
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 (0) 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Při zacházení s přípravkem Temodal 2,5 mg/ml prášek po přípravu infuzního roztoku je třeba postupovat opatrně. Je nutné použít rukavice a aseptickou techniku. Pokud přijde Temodal 2,5 mg/ml do styku s kůží nebo sliznicí, je třeba neprodleně a řádně ho smýt mýdlem a vodou.
Obsah jedné injekční lahvičky musí být smísen se 41 ml sterilizované vody na injekci: Výsledný roztok bude obsahovat 2,5 mg/ml TMZ. Injekčními lahvičkami by se mělo jemně kroužit a netřepat. Roztok by se měl prohlédnout a neměla by se použít žádná injekční lahvička, která obsahuje viditelné částice. Rekonstituovaný přípravek se musí použít do 14 hodin, včetně doby infuze.
Podle celkové předepsané dávky můžete z injekční lahvičky vytáhnout rekonstituovaný roztok v objemu až 40 ml, který přenesete do prázdného 250 mililitrového infuzního vaku (z PVC nebo polyolefínu). Hadičky pumpy je třeba připojit k vaku, propláchnout je a zakrýt. Temodal 2,5 mg/ml musí být podáván pouze intravenózní infuzí trvající 90 minut.
Temodal 2,5 mg/ml prášek pro infuzní roztok lze podávat stejnou i.v. linkou s injekcí 0,9 % roztoku chloridu sodného. Není kompatibilní s roztoky glukózy.
Protože nejsou k dispozici žádné další studie, nesmí se tento léčivý přípravek kombinovat s jinými léčivými přípravky nebo podávat současně stejnou intravenózní cestou.
Tento léčivý přípravek je určen pouze pro jednorázové použití. Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY PODMÍNEK ROZHODNUTÍ O REGISTRACI
Vědecké závěry
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizovaných zpráv o bezpečnosti (PSUR) temozolomidu dospěl výbor CHMP k těmto vědeckým závěrům:
Kumulativní revize údajů o infekci způsobené cytomegalovirem, hepatitidě B, herpes simplex, herpes zoster a tuberkulóze odhalila případy reaktivace cytomegalovirové infekce a hepatitidy B. Vyhodnocení dalších zpráv o bezpečnosti o cytomegalovirové infekci odhalilo, že vedle reaktivované cytomegalovirové infekce rovněž nelze vyloučit infekci primární. Dále byly dodatečně z databáze zjištěny tři fatální případy reaktivace hepatitidy B. Proto je nutno do aktualizované informace o přípravcích obsahujících temozolomid zahrnout informace o reaktivaci hepatitidy B, včetně smrtelných následků.
Navíc po zpracování signálu týkajícího se diabetes insipidus vedly čtyři z pěti literárních zpráv k možné asociaci s léčbou temozolomidem, proto musí být odpovídajícím způsobem aktualizována informace o přípravku.
Proto má s ohledem na dostupné údaje o temozolomidu výbor PRAC za to, že změny informací o přípravku jsou nutné.
Výbor CHMP souhlasí s vědeckými závěry předloženými výborem PRAC.
Zdůvodnění doporučující změnu podmínek rozhodnutí o registraci
Na základě vědeckých závěrů týkajících se temozolomidu zastává výbor CHMP stanovisko, že poměr přínosů a rizik léčivého přípravku obsahujícího (léčivých přípravků obsahujících) temozolomid je příznivý pod podmínkou, že v údajích o přípravku budou provedeny navržené změny.
Výbor CHMP doporučuje změnit podmínky rozhodnutí o registraci.
185