Tasigna 200 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Tasigna 150 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje 150 mg nilotinibum (jako monohydrát hydrochloridu). Pomocné látky se známým účinkem
Jedna tvrdá tobolka obsahuje 117,08 mg laktosy (jako monohydrát).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka
Bílý až nažloutlý prášek v červených neprůhledných tvrdých želatinových tobolkách, velikost 1 s černým podélným potiskem „NVR/BCR“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Tasigna je indikován k léčbě dospělých pacientů s nově diagnostikovanou chronickou myeloidní leukemií s přítomností filadelfského chromozomu (Ph chromozom), v chronické fázi.
4.2 Dávkování a způsob podání
Léčba musí být zahájena lékařem, který má zkušenosti s diagnostikou a léčbou pacientů s CML. Dávkování
Doporučená dávka přípravku Tasigna je 300 mg dvakrát denně. Léčba by měla trvat tak dlouho, dokud je přínosem pro pacienta.
Pro podání dávky 400 mg jednou denně (viz úprava dávkování níže) jsou dostupné tvrdé tobolky o síle 200 mg.
Pokud pacient vynechá dávku, neměl by další dávku zdvojovat, ale měl by užít další obvyklou předepsanou dávku.
Úprava nebo modifikace dávkování
Přípravek Tasigna může být dočasně vysazen a/nebo může být snížena dávka z důvodu hematologické toxicity (neutropenie, trombocytopenie), která nesouvisí se základním onemocněním leukémií (viz Tabulka 1).
Tabulka 1 Úprava dávkování při neutropenii a trombocytopenii
|
Nově |
ANC* <1,0 x 109/l a/nebo počet |
1. Léčba přípravkem Tasigna musí být |
|
diagnostikovaná CML v chronické |
destiček <50 x 109/l |
přerušena a musí být monitorován krevní obraz. |
|
fázi při 300 mg dvakrát denně |
2. Léčba musí být obnovena stejnou dávkou, pokud je do 2 týdnů ANC >1,0 x 109/l a/nebo počet destiček >50 x 109/l. 3. Jestliže počet krevních elementů zůstává nízký, je třeba snížit dávku na 400 mg jednou denně. |
*ANC= absolutní počet neutrofilů
Jestliže se vyvine klinicky signifikantní středně závažná nebo závažná nehematologická toxicita, mělo by být podávání přípravku přerušeno a může být znovu zahájeno dávkou 400 mg jednou denně, jakmile toxické příznaky vymizí. Pokud je to klinicky vhodné, je možné uvažovat o zvýšení dávky na 300 mg dvakrát denně.
Zvýšená hladina sérové lipázy: Při zvýšení sérové lipázy stupně 3-4 by měla být dávka snížena na 400 mg jednou denně nebo léčba přerušena. Hladiny sérové lipázy by měly být vyšetřovány jednou měsíčně nebo dle klinické potřeby (viz bod 4.4).
Zvýšení bilirubinu a jaterních transamináz: Při zvýšení bilirubinu a jaterních transamináz na stupeň 3-4 by měla být dávka snížena na 400 mg jednou denně nebo léčba přerušena. Hladiny bilirubinu a jaterních transamináz by měly být vyšetřovány jednou měsíčně nebo dle klinické potřeby.
Starší lidé
V klinické studii bylo přibližně 12 % jedinců ve věku 65 let a starších. Žádné zásadní rozdíly v bezpečnosti a účinnosti nebyly pozorovány u pacientů >65 let ve srovnání s dospělými ve věku mezi 18 a 65 lety.
Porucha funkce ledvin
U pacientů se zhoršenou funkcí ledvin nebyly klinické studie provedeny.
Vzhledem k tomu, že nilotinib ani jeho metabolity nejsou vylučovány ledvinami, nepředpokládá se u pacientů se zhoršenou funkcí ledvin snížení celkové tělesné clearance.
Porucha funkce jater
Zhoršená funkce jater má mírný účinek na farmakokinetiku nilotinibu. Úprava dávky se u pacientů se zhoršenou fukcí jater nepovažuje za nutnou. Nicméně pacienti se zhoršenou funkcí jater by měli být léčeni s opatrností (viz bod 4.4).
Srdeční poruchy
Pacienti s nekompenzovaným nebo závažným srdečním onemocněním (např. nedávný infarkt myokardu, městnavé srdeční selhání, nestabilní angina pectoris nebo klinicky významná bradykardie) byli z klinických studií vyloučeni. Pozornost je třeba věnovat pacientům s významnou srdeční poruchou (viz bod 4.4).
Během léčby přípravkem Tasigna byly hlášeny vzestupy hladin celkového cholesterolu v séru (viz bod 4.4). Lipidový profil by měl být stanoven před zahájením léčby přípravkem Tasigna a vyhodnocován v měsících 3 a 6 po zahájení léčby a dále nejméně jednou za rok během chronické léčby.
Během léčby přípravkem Tasigna byly hlášeny vzestupy hladin glukózy v krvi (viz bod 4.4). Hladiny glukózy v krvi by měly být stanoveny před zahájením léčby přípravkem Tasigna a sledovány během léčby.
Pediatrická populace
Bezpečnost a účinnost přípravku Tasigna u dětí od narození do 18 let nebyla dosud stanovena (viz bod 5.1). Vzhledem k chybějícím údajům o bezpečnosti a účinnosti se proto užívání u pediatrických pacientů nedoporučuje (viz bod 5.1).
Způsob podání
Tasigna by měla být podávána dvakrát denně přibližně po 12 hodinách a nesmí být užívána spolu s jídlem. Tvrdé tobolky musí být spolknuty celé s vodou. 2 hodiny před užitím dávky a alespoň jednu hodinu po užití dávky by neměla být požita žádná potrava.
Pacientům, kteří nejsou schopni tvrdé tobolky spolknout, může být obsah každé tobolky rozmíchán v jedné čajové lžičce jablečné šťávy (jablečného pyré), takto připravený lék se má okamžitě užít. Nesmí být užita více než jedna čajová lžička s každou tobolkou a použita jiná potrava než jablečná šťáva (viz body 4.4 a 5.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Myelosuprese
Léčba přípravkem Tasigna je doprovázena (National Cancer Institute Common Toxicity Criteria stupeň 3-4) trombocytopenií, neutropenií a anemií. Kompletní vyšetření krevního obrazu by mělo být prováděno v prvních 2 měsících léčby každé dva týdny a dále pak jednou měsíčně, nebo podle klinické indikace. Myelosuprese byla zpravidla reverzibilní a obvykle byla zvládnuta dočasným vysazením přípravku Tasigna nebo snížením dávky (viz bod 4.2).
Prodloužení QT intervalu
Ukázalo se, že Tasigna prodlužuje srdeční komorovou repolarizaci; délka naměřeného QT intervalu na EKG byla závislá na koncentraci.
V klinické studii fáze III u pacientů s nově diagnostikovanou CML v chronické fázi, kteří užívali 300 mg nilotinibu dvakrát denně, byla změna zprůměrovaného QTcF intervalu v ustáleném stavu od výchozí hodnoty 6 ms. Žádný pacient neměl QTcF >480 ms. Nebyly pozorovány žádné epizody torsade de pointes.
Ve studii na zdravých dobrovolnících se srovnatelnou expozicí pozorovanou u pacientů byla změna střední zprůměrované hodnoty QTcF od výchozí hodnoty, po odečtení hodnot placeba, 7 ms (CI ± 4 ms). Žádný jedinec neměl hodnoty QTcF >450 ms. Navíc nebyly během studie pozorovány žádné klinicky relevantní arytmie. Zejména nebyly pozorovány žádné epizody torsade de pointes (přechodné ani setrvalé).
Významné prodloužení QT intervalu se může objevit v případě, že je nilotinib nesprávně užíván se silnými inhibitory CYP3A4 a/nebo léčivými přípravky, o kterých je známo, že prodlužují QT interval, a/nebo s potravou (viz bod 4.5). Současná přítomnost hypokalémie nebo hypomagnesémie mohou tento účinek ještě zvyšovat. Prodloužení QT intervalu může vystavit pacienty riziku fatálního konce.
Přípravek Tasigna by měl být užíván opatrně u pacientů s prodlouženým QTc intervalem, nebo u kterých je významné riziko vývoje prodloužení QTc intervalu, jako jsou pacienti:
- s kongenitálním syndromem dlouhého QT intervalu.
- s nekompenzovaným nebo závažným srdečním onemocněním, zahrnujícím nedávný infarkt myokardu, městnavé srdeční selhání, nestabilní anginu pectoris nebo klinicky významnou bradykardii.
- užívající antiarytmika nebo jiné látky, které prodlužují QT interval.
Doporučuje se pečlivé monitorování účinku na QTc interval a provedení výchozího EKG před zahájením léčby a dle klinické potřeby. Hypokalémie nebo hypomagnesémie musí být upraveny před podáním přípravku Tasigna a měly by být pravidelně sledovány během léčby.
Náhlé úmrtí
Méně časté případy (0,1 až 1 %) náhlých úmrtí byly hlášeny u pacientů s imatinib-rezistentní nebo intolerantní CML v chronické fázi nebo akcelerované fázi, u nichž se v minulosti vyskytlo onemocnění srdce nebo měli významné kardiální rizikové faktory. Často se navíc spolu se základním maligním onemocněním vyskytovaly komorbidity léčené doprovodnými léčivými přípravky. Poruchy komorové repolarizace mohly být přispívajícími faktory. V klinické studii fáze III u pacientů s nově diagnostikovanou CML v chronické fázi nebyly hlášené žádné případy náhlých úmrtí.
Zadržování tekutin a edém
Závažné formy zadržování tekutin, jako je pleurální efuze, plicní edém a perikardiální efuze, byly pozorovány méně často (0,1 až 1 %) ve studii fáze III u pacientů s nově diagnostikovanou CML. Podobné případy byly zjištěny v postmarketingových hlášeních. Nečekaný rychlý nárůst tělesné hmotnosti má být pečlivě prověřen. Pokud se během léčby nilotinibem objeví příznaky závažného zadržování tekutin, má být vyhodnocena etiologie a pacienti by měli být léčeni příslušným způsobem (viz bod 4.2 pokyny ke zvládání nehematologických toxicit).
Kardiovaskulární příhody
Kardiovaskulární příhody byly hlášeny v randomizované studii fáze III u pacientů s nově diagnostikovanou CML a zjištěny v postmarketingových hlášeních. V této klinické studii s mediánem doby léčby 60,5 měsíců se vyskytly případy kardiovaskulárních příhod stupně 3-4 zahrnující periferní arteriální okluze (1,4 % při dávce 300 mg nilotinibu dvakrát denně, respektive 1,1 % při dávce 400 mg dvakrát denně), ischemické onemocnění srdce (2,2 % při dávce 300 mg nilotinibu dvakrát denně, respektive 6,1 % při dávce 400 mg dvakrát denně) a ischemické cerebrovaskulární příhody (1,1 % při dávce 300 mg nilotinibu dvakrát denně, respektive 2,2 % při dávce 400 mg dvakrát denně). Pacienti by měli být poučeni o nutnosti okamžitě vyhledat lékaře, pokud se u nich projeví akutní známky nebo příznaky kardiovaskulárních příhod. Měl by být vyhodnocen kardiovaskulární stav pacientů a kardiovaskulární rizikové faktory by měly být během léčby přípravkem Tasigna sledovány a aktivně zvládány podle standardních doporučení. Ke zvládnutí kardiovaskulárních rizikových faktorů by měla být předpsána příslušná léčba (viz bod 4.2 pro pokyny ke zvládání nehematologických toxicit).
Reaktivace hepatitidy B
U pacientů, kteří jsou chronickými nosiči hepatitidy B, dochází k reaktivaci po zahájení léčby inhibitory tyrosinkinázy bcr-abl. Některé případy vyústily v akutní selhání jater nebo ve fulminantní hepatitidu vedoucí k transplantaci jater nebo došlo k úmrtí pacienta.
Před zahájením léčby přípravkem TASIGNA mají být pacienti vyšetřeni na infekci HBV. Před zahájením léčby u pacientů s pozitivní sérologií hepatitidy B (včetně těch s aktivním onemocněním) a u pacientů, u kterých v průběhu léčby vyjde pozitivní test infekce HBV, je třeba se obrátit na odborníky na onemocnění jater a léčbu hepatitidy B. Nosiči HBV, kteří potřebují léčbu přípravkem TASIGNA, mají být po celou dobu léčby a několik měsíců po jejím ukončení pečlivě sledováni s ohledem na možný výskyt známek a příznaků aktivní infekce HBV (viz bod 4.8).
Laboratorní testy a monitorování
Krevní lipidy
Ve studii fáze III u nově diagnostikovaných pacientů s CML vykazovalo 1,1 % pacientů léčených 400 mg nilotinibu dvakrát denně zvýšení hladin celkového cholesterolu stupně 3-4; žádné zvýšení stupně 3-4 však nebylo pozorováno ve skupině léčené 300 mg dvakrát denně (viz bod 4.8).
Doporučuje se stanovit lipidové profily před zahájením léčby přípravkem Tasigna a vyhodnocovat je v měsících 3 a 6 po zahájení léčby a dále nejméně jednou za rok během chronické léčby (viz bod 4.2). Pokud je potřeba podávat inhibitor HMG-CoA reduktázy (léčivo snižující hladinu lipidů), přečtěte si prosím před zahájením léčby bod 4.5, neboť některé inhibitory HMG-CoA reduktázy jsou rovněž metabolizovány systémem CYP3A4.
Glukóza v krvi
Ve studii fáze III u nově diagnostikovaných pacientů s CML vykazovalo 6,9 % pacientů léčených 400 mg nilotinibu dvakrát denně a 7,2 % pacientů léčených 300 mg nilotinibu dvakrát denně zvýšení hladin glukózy stupně 3-4. Před zahájením léčby přípravkem Tasigna se doporučuje stanovit hladinu glukózy a monitorovat ji během léčby, pokud je to klinicky indikováno (viz bod 4.2). Pokud z výsledků testů vyplývá potřeba léčby, měli by se lékaři řídit lokálními standardy a doporučeními léčby.
Interakce s jinými léčivými přípravky
Přípravek Tasigna nemá být podáván s látkami, které jsou silnými inhibitory CYP3A4 (včetně, ale nejen, ketokonazolu, itrakonazolu, vorikonazolu, klarithromycinu, telithromycinu, ritonaviru). V případě, že by léčba těmito látkami byla nezbytná, doporučuje se, pokud je to možné, léčbu přípravkem Tasigna přerušit (viz bod 4.5). Jestliže přechodné přerušení léčby není možné, je třeba nemocného pečlivě sledovat z hlediska prodloužení QT intervalu (viz body 4.2, 4.5 a 5.2).
Při souběžném užívání přípravku Tasigna s léčivými přípravky, které jsou účinnými induktory CYP3A4 (např. fenytoin, rifampicin, karbamazepin, fenobarbital a třezalka), je pravděpodobné snížení expozice nilotinibu až v klinicky významném rozsahu. Z tohoto důvodu by pacientům užívajícím Tasigna měly být souběžně podávány alternativní léčivé látky s nižším potenciálem pro indukci CYP3A4 (viz bod 4.5).
Vliv potravy
Biologická dostupnost nilotinibu je zvýšena příjmem potravy. Přípravek Tasigna nesmí být užíván spolu s jídlem (viz body 4.2 a 4.5) a měl by být užíván 2 hodiny po jídle. Žádná potrava by neměla být přijímána nejméně jednu hodinu po užití dávky léku. Grapefruitová šťáva a jiné potraviny, o kterých je známo, že inhibují CYP3A4, by neměly být požívány. Pacientům, kteří nejsou schopni tvrdé tobolky spolknout, může být obsah každé tvrdé tobolky rozmíchán v jedné čajové lžičce jablečné šťávy (jablečného pyré), takto připravený lék se má okamžitě užít. Nesmí být užita více než jedna čajová lžička s každou tobolkou a použita jiná potrava než jablečná šťáva (viz bod 5.2).
Zhoršená funkce jater
Zhoršená funkce jater má mírný účinek na farmakokinetiku nilotinibu. Výsledkem podání jednorázové dávky 200 mg nilotinibu bylo zvýšení AUC o 35%, 35% a 19% u jedinců s mírným, středně závažným a závažným zhoršením funkce j ater ve srovnání s kontrolní skupinou jedinců s normální j aterní funkcí. Došlo ke zvýšení predikované Cmax nilotinibu v ustáleném stavu o 29%, 18%, respektive 22%.
Do klinických studií nebyli zařazeni pacienti s hodnotami alaninaminotransferázy (ALT) a/nebo aspartátaminotransferázy (AST) >2,5násobku (nebo >5násobku, pokud souvisely s onemocněním) horní hranice normálních hodnot, a/nebo pokud měli celkový bilirubin >1,5násobek horní hranice normálních hodnot. Nilotinib je metabolizován především v játrech. Pacienti se zhoršenou funkcí jater by proto mohli mít zvýšenou expozici nilotinibu a měli by být léčeni s opatrností (viz bod 4.2).
Sérová lipáza
Bylo pozorováno zvýšení lipázy v séru. U pacientů s pankreatitidou v anamnéze se doporučuje opatrnost. Pokud je zvýšení lipázy spojené s břišními příznaky, mělo by být podávání přípravku Tasigna přerušeno a měla by být provedena příslušná diagnostická vyšetření za účelem vyloučení pankreatitidy.
Totální gastrektomie
Biologická dostupnost nilotinibu může být u pacientů s totální gastrektomií omezená (viz bod 5.2). Mělo by být zváženo častější sledování těchto pacientů.
Syndrom nádorového rozpadu
Před započetím léčby přípravkem Tasigna je doporučená úprava klinicky významné dehydratace a léčba vysokých hladin kyseliny močové z důvodu možného výskytu syndromu nádorového rozpadu (TLS) (viz bod 4.8).
Laktóza
Tvrdé tobolky Tasigna obsahují laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Přípravek Tasigna může být podáván v kombinaci s hematopoetickými růstovými faktory, jako je erytropoetin nebo faktor stimulující granulocytární kolonie (G-CSF), pokud je podání klinicky indikováno. Může být podáván s hydroxyureou nebo anagrelidem, pokud je podání klinicky indikováno.
Nilotinib je metabolizován převážně játry a je také substrátem pro efluxní pumpu mnoha léků, P-glykoprotein (P-gp). Proto absorpce a následná eliminace systémově absorbovaného nilotinibu může být ovlivněna látkami, které působí na CYP3A4 a/nebo P-gp.
Látky, které mohou zvyšovat koncentrace nilotinibu v séru
Současné podání nilotinibu a imatinibu (substrát a mírný inhibitor P-gp a CYP3A4) mělo mírný inhibiční účinek na CYP3A4 a/nebo P-gp. Došlo ke zvýšení AUC imatinibu o 18% až 39% a zvýšení AUC nilotinibu o 18% až 40%. Tyto změny pravděpodobně nejsou klinicky významné.
Při současném podání silného inhibitoru CYP3A4, ketokonazolu, zdravým dobrovolníkům byla expozice nilotinibu zvýšena 3krát. Z tohoto důvodu by neměly být současně podávány silné inhibitory CYP3A4, včetně ketokonazolu, itrakonazolu, vorikonazolu, ritonaviru, klarithromycinu a telithromycinu (viz bod 4.4). Zvýšenou expozici nilotinibu je možné také očekávat se středně silnými inhibitory CYP3A4. Mělo by se uvažovat o alternativních léčivých přípravcích, která nemají žádné nebo mají minimální inhibiční účinky na CYP3A4.
Látky, které mohou snižovat koncentrace nilotinibu v séru
Rifampicin, silný induktor CYP3A4, snižuje Cmax nilotinibu o 64% a snižuje AUC nilotinibu o 80%. Rifampicin a nilotinib se nemá užívat současně.
Souběžné podávání jiných léčivých přípravků, které indukují CYP3A4 (např. fenytoin, karbamazepin, fenobarbital a třezalka), může pravděpodobně také v klinicky významném rozsahu snižovat expozici nilotinibu. U pacientů, u kterých jsou induktory CYP3A4 indikovány, by se mělo uvažovat o výběru alternativních přípravků s menším enzymovým indukčním potenciálem.
Rozpustnost nilotinibu je závislá na pH, při vyšším pH je rozpustnost nižší. U zdravých pacientů, kterým bylo podáváno 40 mg esomeprazolu jednou denně po dobu 5 dnů, se výrazně zvýšilo žaludeční pH, ale absorpce nilotinibu se snížila jen mírně (27% snížení Cmax a 34% snížení AUC0-<»). Nilotinib může být v případě potřeby užívána současně s esomeprazolem nebo jinými inhibitory protonové pumpy.
Ve studii u zdravých dobrovolníků nebyly při podání jednorázové dávky 400 mg přípravku Tasigna 10 hodin po a 2 hodiny před podáním famotidinu zjištěny významné změny ve farmakokinetice nilotinibu. V případě nutného souběžného užívání může být H2 blokátor podáván přibližně 10 hodin před a přibližně 2 hodiny po podání přípravku Tasigna.
Ve stejné studii podávání antacid (hydroxid hlinitý/hydroxid hořečnatý/simetikon) 2 hodiny před nebo po jednorázové dávce 400 mg přípravku Tasigna také nezměnilo farmakokinetiku nilotinibu. Pokud je to nutné, mohou být antacida podávána přibližně 2 hodiny před nebo přibližně 2 hodiny po podání přípravku Tasigna.
Látky, jejichž systémové koncentrace mohou být změněny nilotinibem
Nilotinib je in vitro relativně silný inhibitor CYP3A4, CYP2C8, CYP2C9, CYP2D6 a UGT1A1, s nejnižší hodnotou Ki pro CYP2C9 (Ki=0,13 mikromol).
V interakční studii u zdravých dobrovolníků po jednorázovém podání 25 mg warfarinu, citlivého substrátu CYP2C9, a 800 mg nilotinibu nedošlo k žádným změnám farmokokinetických parametrů nebo farmokodynamiky warfarinu, měřených jako protrombinový čas (PT) a mezinárodní normalizovaný poměr (INR). Data o rovnovážném stavu neexistují. Tato studie naznačuje, že při dávce warfarinu do 25 mg je klinicky významná léková interakce mezi nilotinibem a warfarinem méně pravděpodobná. Protože není dostatek dat o rovnovážném stavu, doporučuje se po zahájení léčby nilotinibem (minimálně během prvních 2 týdnů) kontrola farmakodynamických ukazatelů warfarinu (INR nebo PT).
U pacientů s CML zvýšil nilotinib podávaný v dávce 400 mg dvakrát denně po dobu 12 dní systémovou expozici (AUC a Cmax) perorálního midazolamu (substráty CYP3A4) 2,6násobně, respektive 2,0násobně. Nilotinib je středně silný inhibitor CYP3A4. Proto může při souběžném podávání s nilotinibem dojít ke zvýšení systémové expozice dalších léků primárně metabolizovaných CYP3A4 (např. některé inhibitory HMG-CoA reduktázy). Pro léky, které jsou CYP3A4 substráty a které mají úzký terapeutický index (například alfentanil, cyklosporin, dihydroergotamin, ergotamin, fentanyl, sirolimus a takrolimus) může být při souběžném podávání s nilotinibem nezbytné příslušné sledování a úprava dávky.
Antiarytmika a jiné látky, které mohou prodlužovat QT interval
Nilotinib by měl být podáván opatrně pacientům s prodloužením QT intervalu nebo u kterých se může prodloužení QT vyvinout, včetně pacientů, kteří užívají antiarytmika, jako jsou amiodaron, disopyramid, prokainamid, chinidin a sotalol, nebo jiné léčivé látky, které mohou vést k prodloužení QT intervalu, např. chlorochin, halofantrin, klarithromycin, haloperidol, methadon a moxifloxacin (viz bod 4.4).
Interakce s potravou
Absorpce a biodostupnost přípravku Tasigna j sou zvýšeny při současném příjmu potravy s následným zvýšením koncentrace v séru (viz body 4.2, 4.4 a 5.2). Grapefruitová šťáva a jiné potraviny, o kterých je známo, že inhibují CYP3A4, by neměly být požívány.
Ženy ve fertilním věku
Zeny ve fertilním věku musí během léčby a dva měsíce po ukončení léčby přípravkem Tasigna používat vysoce účinnou antikoncepci.
Údaje o podávání nilotinibu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Přípravek Tasigna by neměl být během těhotenství podáván, pokud klinický stav pacientky nevyžaduje léčbu nilotinibem. Pokud je přípravek podáván během těhotenství, musí být pacientka informována o potenciálním riziku pro plod.
Kojení
Není známo, zda se nilotinib vylučuje do lidského mateřského mléka. Dostupné toxikologické údaje u zvířat prokázaly vylučování nilotinibu do mléka (viz bod 5.3). Riziko pro kojené novorozence/děti nelze vyloučit. Přípravek Tasigna se během kojení nemá podávat.
Fertilita
Studie na zvířatech neprokázaly vliv na fertilitu u samců a samic potkanů (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pacienti, kteří pozorují závratě, únavu, zhoršení zraku nebo jiné nežádoucí účinky s potenciálním vlivem na schopnost bezpečně řídit nebo obsluhovat stroje, by neměli tyto činnosti vykonávat, dokud tyto nežádoucí účinky přetrvávají (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Údaje uvedené níže odrážejí expozici přípravkem Tasigna u celkem 279 pacientů v randomizované studii fáze III u pacientů s nově diagnostikovanou Ph+ CML v chronické fázi léčených 300 mg nilotinibu dvakrát denně. Medián trvání expozice byl 60,5 měsíců (rozmezí 0,1-70,8 měsíců).
Nejčastější (>10 %) nehematologické nežádoucí účinky byly vyrážka, svědění, bolest hlavy, nauzea, únava, alopecie, bolest svalů a bolest v nadbřišku. Většina těchto nežádoucích účinků byla mírného až středního stupně závažnosti. Méně často (<10 % a >5 %) byly pozorovány zácpa, suchá kůže, astenie, svalové křeče, průjem, artralgie, bolest břicha, zvracení a periferní otoky, tyto nežádoucí účinky byly mírně až středně závažné, zvládnutelné a obecně nevyžadovaly snížení dávky.
Hematologická toxicita zahrnující myelosupresi vzniklá při léčbě: trombocytopenie (18 %), neutropenie (15 %) a anémie (8 %). Biochemické nežádoucí účinky zahrnují zvýšenou hladinu alaninaminotransferázy (24 %), hyperbilirubinémii (16 %), zvýšenou hladinu
aspartátaminotransferázy (12 %), zvýšenou hladinu lipázy (11 %), zvýšenou hladinu bilirubinu v krvi (10 %), hyperglykémii (4 %), hypercholesterolémii (3 %) a hypertriglyceridémii (<1 %). Pleurální a perikardiální výpotky se bez ohledu na příčinu vyskytly u 2 %, respektive <1 % pacientů, kterým bylo podáváno 300 mg přípravku Tasigna dvakrát denně. Krvácení do zažívacího traktu bylo bez ohledu na příčinu hlášeno u 3 % těchto pacientů.
Změna zprůměrovaného QTcF intervalu v ustáleném stavu od výchozího stavu byla 6 msec. Žádný pacient neměl během studie s léčivým přípravkem absolutní QTcF >500 msec. Prodloužení QTcF přesahující 60 msec bylo pozorováno u <1 % pacientů během studie s léčivým přípravkem. Nebyly zjištěny případy náhlých úmrtí ani epizody torsade de pointes (přechodné ani trvalé). Během léčby nebyl zjištěn pokles průměrné ejekční frakce levé komory (LVEF). Žádný pacient neměl během léčby LVEF <45 % a také žádný pacient neměl pokles LVEF o více než 15 %.
Ukončení léčby z důvodu výskytu nežádoucích účinků bylo pozorováno u 10 % pacientů.
Nejčastěji hlášené nežádoucí účinky u přípravku Tasigna v klinických studiích Nehematologické nežádoucí účinky (kromě laboratorních abnormalit), které byly hlášeny u nejméně 5 % pacientů léčených 300 mg nilotinibu dvakrát denně v randomizované studii fáze III jsou uvedeny v Tabulce 2. Jsou řazeny podle frekvence výskytu, s nějčastějším na prvním místě, uvedené v záhlaví při použití přesnosti na jedno desetinné místo a následující konvence: velmi časté (>1/10) nebo časté (>1/100 až <1/10). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 2 Nehematologické nežádoucí účinky (>5 % všech pacientů)*
|
Třídy orgánových systémů |
Četnost |
Nežádoucí účinek |
Všechny stupně % |
Stupeň 3-4 % |
|
Poruchy nervového systému |
Velmi časté |
Bolesti hlavy |
16 |
2 |
|
Gastrointestinální poruchy |
Velmi časté |
14 |
<1 | |
|
Velmi časté |
Bolest v nadbřišku |
10 |
1 | |
|
Časté |
Zácpa |
10 |
0 | |
|
Časté |
9 |
<1 | ||
|
Časté |
6 |
0 | ||
|
Časté |
6 |
0 | ||
|
Časté |
5 |
0 | ||
|
Poruchy kůže a podkožní tkáně |
Velmi časté |
33 |
<1 | |
|
Velmi časté |
18 |
<1 | ||
|
Velmi časté |
Alopecie |
10 |
0 | |
|
Časté |
Suchá kůže |
10 |
0 | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Velmi časté |
Myalgie |
10 |
<1 |
|
Časté |
Spasmy svalů |
9 |
0 | |
|
Časté |
Artralgie |
8 |
<1 | |
|
Časté |
Bolest končetin |
5 |
<1 | |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Únava |
12 |
0 |
|
Časté |
Astenie |
9 |
<1 | |
|
Časté |
Periferní otoky |
5 |
<1 |
*Pro prezentaci v této tabulce jsou procenta zaokrouhlená na celá čísla. Nicméně k identifikaci nežádoucích účinků s četností nejméně 5 % a ke klasifikace nežádoucích účinků podle kategorií četností jsou použita procenta s přesností na jedno desetinné místo.
Následující nežádoucí účinky byly hlášeny ve studii fáze III s přípravkem Tasigna s frekvencí nižší než 5 % U laboratorních změn jsou uvedené také velmi časté případy (>1/10), nezahrnuté do Tabulky 2. Tyto nežádoucí účinky jsou zahrnuty podle klinické významnosti a řazeny v každé skupině podle klesající závažnosti a následující konvence: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), není známo (z dostupných údajů nelze určit).
Infekce a infestace:
Časté: folikulitida, infekce horních cest dýchacích (zahrnující faryngitidu, nazofaryngitidu, rhinitidu). Není známo: infekce herpes viry, ústní kandidóza, subkutánní absces, absces konečníku, tinea pedis, reaktivace hepatitidy B.
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy):
Časté: kožní papilom.
Není známo: orální papilom, paraproteinémie.
Poruchy krve a lymfatického sytému:
Časté: leukopenie, eozinofilie, lymfopenie.
Méně časté: pancytopenie.
Není známo: febrilní neutropenie.
Poruchy imunitního systému:
Není známo: hypersenzitivita.
Endokrinní poruchy:
Není známo: sekundární hyperparatyreóza.
Poruchy metabolismu a výživy:
Velmi časté: hypofosfatémie (včetně snížení hladiny fosforu v krvi).
Časté: diabetes mellitus, hypercholesterolémie, hyperlipidémie, hypertriacylglycerolémie, hyperglykémie, snížení chuti k jídlu, hypokalcémie, hypokalémie.
Méně časté: hyperkalémie, dyslipidémie, dna.
Není známo: hyperurikémie, hypoglykémie, poruchy chuti k jídlu.
Psychiatrické poruchy:
Časté: nespavost, deprese, stavy úzkosti.
Není známo: amnezie, stav úzkosti.
Poruchy nervového systému:
Časté: závratě, hypoestezie, periferní neuropatie.
Méně časté: ischemická cerebrovaskulární příhoda. mozkový infarkt, migréna, parestezie.
Není známo: cerebrovaskulární příhoda, stenóza bazilární arterie, synkopa, třes, letargie, dysestezie, syndrom neklidných nohou, hyperestezie.
Poruchy oka:
Časté: svědění očí, konjunktivitida, suchost očí (včetně xeroftalmie).
Méně časté: otok očních víček, fotopsie, spojivkové krvácení, hyperémie (bělma, spojivek, očí). Není známo: periorbitální otok, blefaritida, bolest očí, chorioretinopatie, alergický zánět spojivek, poškození povrchu oka, rozostřené vidění.
Poruchy ucha a lybyrintu:
Časté: vertigo.
Srdeční poruchy*:
Časté: angina pectoris, arytmie (zahrnující atrioventrikulární blok, tachykardii, fibrilaci síní, ventrikulární extrasystoly, bradykardie), prodloužení QT intervalu na EKG, palpitace, infarkt myokardu.
Méně časté: srdeční selhání, cyanóza.
Není známo: snížení ejekční frakce, výpotek v osrdečníku, perikarditida, diastolická dysfunkce, blokáda levého Tawarova raménka.
*hlášeno ve studii fáze III v rameni s léčbou 300 mg dvakrát denně a/nebo 400 mg dvakrát denně Cévní poruchy:
Časté: hypertenze, návaly (zčervenání).
Méně časté: intermitentní klaudikace, periferní arteriální okluzivní choroba, arterioskleróza.
Není známo: hematomy, periferní arteriální stenóza.
Respirační, hrudní a mediastinální poruchy:
Méně časté: pleurální výpotek.
Není známo: námahová dušnost, zánět pohrudnice, epistaxe, orofaryngeální bolest.
Gastrointestinálni poruchy:
Časté: nadýmání, abdominální diskomfort, poruchy chuti, plynatost.
Méně časté: pankreatitida, gastritida, citlivost zubů.
Není známo: ezofageální vřed, žaludeční vřed, ezofageální bolest, stomatitida, sucho v ústech, enterokolitida, hemoroidy, hiátová hernie, krvácení z konečníku, zánět dásní.
Poruchy jater a žlučových cest:
Velmi časté: hyperbilirubinémie (včetně zvýšení hladiny bilirubinu v krvi).
Časté: abnormální jaterní funkce.
Méně časté: žloutenka.
Není známo: toxická hepatitida.
Poruchy kůže a podkožní tkáně:
Časté: erytém, hyperhidróza, pohmožděniny, akné, dermatitida (včetně alergické, exfoliativní a akneiformní), noční pocení, ekzém.
Méně časté: poléková vyrážka, bolest kůže.
Není známo: multiformní erytém, kopřivka, puchýře, kožní cysty, hyperplázie mazových žláz, otok obličeje, atrofie kůže, hypertrofie kůže, loupání kůže, hyperpigmentace kůže, vyblednutí kůže, hyperkeratóza, psoriáza.
Poruchy svalové a kosterní soustavy a pojivové tkáně:
Časté: bolest kostí, bolest zad, svalová slabost.
Méně časté: bolest svalů a kostí, bolest ve slabinách.
Poruchy ledvin a močových cest:
Není známo: dysurie, polakisurie, chromaturie.
Poruchy reprodukčního systému a prsu:
Méně časté: erektilní dysfunkce.
Není známo: gynekomastie, tvrdnutí prsu, menoragie, otok bradavek.
Celkové poruchy a reakce v místě aplikace:
Časté: horečka, bolest na prsou (zahrnující bolesti na prsou jiného než srdečního původu), hrudní dyskomfort.
Méně časté: bolest, zimnice, pocit změny tělesné teploty (zahrnující pocit horka, pocit chladu), malátnost.
Není známo: otoky obličeje, lokalizovaný edém.
Vyšetření:
Velmi časté: zvýšená hladina alaninaminotransferázy, zvýšená hladina aspartátaminotransferázy, zvýšená hladina lipázy, zvýšená hladina lipoproteinů (včetně lipoproteinů s nízkou denzitou a s vysokou denzitou), zvýšená hladina celkového cholesterolu, zvýšená hladina triglyceridů.
Časté: snížení hladiny hemoglobinu, zvýšená amyláza v krvi, zvýšená alkalická fosfatáza v krvi, zvýšená hladina gamaglutamyltransferázy, zvýšená tělesná hmotnost, zvýšená hladina inzulinu v krvi, snížená hladina globulinů.
Není známo: zvýšený paratyreoidální hormon v krvi, snížená hladina inzulinu v krvi, snížená hladina C-peptidu, snížená tělesná hmotnost.
Klinicky relevantní nebo závažné abnormality rutinních hematologických nebo biochemických laboratorních hodnot jsou uvedeny v Tabulce 3.
Tabulka 3 Laboratorní abnormality stupně 3-4*
|
n=279 (%) | |
|
Hematologické parametry | |
|
Myelosuprese | |
|
- Neutropenie |
12 |
|
- Trombocytopenie |
10 |
|
- Anémie |
4 |
|
Biochemické parametry | |
|
- Zvýšený kreatinin |
0 |
|
- Zvýšená lipáza |
9 |
|
- Zvýšená SGOT (AST) |
1 |
|
- Zvýšená SGPT (ALT) |
4 |
|
- Hypofosfatémie |
7 |
|
- Zvýšený bilirubin (celkový) |
4 |
|
- Zvýšená glukóza |
7 |
|
- Zvýšený cholesterol (celkový) |
0 |
|
- Zvýšené triglyceridy |
0 |
*Pro prezentaci v této tabulce jsou použitá procenta s přesností na jedno desetinné místo a zaokrouhlená na celé číslo.
Popis vybraných nežádoucích účinků
Reaktivace hepatitidy B
V souvislosti s tyrosinkinázou bcr-abl byla zaznamenána reaktivace hepatitidy B. Některé případy vyústily v akutní selhání jater nebo ve fulminantní hepatitidu vedoucí k transplantaci jater nebo došlo k úmrtí pacienta (viz bod 4.4).
Zkušenosti po uvedení na trh
Následující nežádoucí účinky byly získány z postmarketingové zkušenosti s přípravkem Tasigna prostřednictvím spontánních hlášení, literárních případů, rozšířených programů přístupu (Extended Access Programs) a klinických studií odlišných od globálních registračních studií. Protože byly tyto účinky hlášeny dobrovolně u populací o neznámé velikosti, není vždy možné spolehlivě stanovit jejich frekvenci nebo stanovit příčinný vztah k expozici nilotinibem.
Frekvence vzácná: U pacientů léčených přípravkem Tasigna byly hlášené případy syndromu nádorového rozpadu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Byly hlášeny ojedinělé případy záměrného předávkování nilotinibem, kdy byl požit nespecifikovaný počet tvrdých tobolek Tasigny v kombinaci s alkoholem a jinými léčivými přípravky. Objevila se neutropenie, zvracení a ospalost. Nebyly hlášené změny EKG nebo hepatotoxicita. Bylo hlášeno úplné uzdravení pacienta.
V případě předávkování by měl být pacient pozorován a měla by mu být poskytnuta vhodná podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antineoplastika, inhibitory proteinkinázy, ATC kód: L01XE08
Nilotinib je účinný inhibitor aktivity ABL tyrozinkinázy BCR-ABL onkoproteinu v buněčných liniích i v primárně leukemických buňkách s filadelfským chromozomem. Přípravek se s vysokou afinitou váže na vazebná místa ATP, a tím účinně inhibuje divoký typ BCR-ABL. Přípravek je účinný proti 32 ze 33 imatinib-rezistentních mutantních forem BCR-ABL. V důsledku této biochemické aktivity nilotinib selektivně inhibuje proliferaci a vyvolává apoptózu buněčných linií a u primárně leukemických buněk s filadelfským chromozomem pacientů s CML. U myších modelů CML nilotinib po perorálním podání samostatně redukuje objem nádoru a prodlužuje přežití.
Nilotinib má malý nebo žádný účinek na většinu dalších hodnocených proteinkináz, včetně Src, s výjimkou PDGF, KIT a Ephrin receptorových kináz, které jsou inhibovány v rozmezí koncentrací dosažených po perorálním podání terapeutických dávek doporučených k léčbě CML (viz Tabulka 4).
Tabulka 4 Kinázový profil nilotinibu (fosforylace IC50 nM)
|
BCR-ABL |
PDGFR |
KIT |
|
20 |
69 |
210 |
Klinické studie u nově diagnostikovaných CML v chronické fázi
Byla provedena otevřená, multicentrická, randomizovaná studie fáze III za účelem stanovení účinnosti nilotinibu oproti imatinibu u 846 dospělých pacientů s cytogeneticky potvrzenou nově diagnostikovanou CML s přítomností filadelfského chromosomu v chronické fázi. Pacienti byli diagnostikováni maximálně 6 měsíců před zařazením do studie a nebyli dříve léčeni s výjimkou hydroxyurey a/nebo anagrelidu. Pacienti byli randomizováni 1:1:1 k podávání nilotinibu 300 mg dvakrát denně (n=282), nilotinibu 400 mg dvakrát denně (n=281) nebo imatinibu 400 mg jednou denně (n=283). Randomizace byla stratifikována Sokalovým skóre v době diagnózy.
Základní charakteristiky tří terapeutických ramen byly dobře vyvážené. Průměrný věk byl 47 let v obou ramenech s nilotinibem a 46 let v rameni s imatinibem. 12,8 % pacientů bylo starších 65 let v rameni s nilotinibem 300 mg dvakrát denně, 10,0 % pacientů bylo starších 65 let v rameni s nilotinibem 400 mg dvakrát denně a 12,4 % pacientů bylo starších 65 let v rameni s imatinibem 400 mg jednou denně. Počet mužů byl mírně vyšší než počet žen (56,0 % v rameni s nilotinibem 300 mg dvakrát denně, 62,3 % v rameni s nilotinibem 400 mg dvakrát denně a 55,8 % v rameni s imatinibem 400 mg jednou denně). Více než 60 % pacientů byli běloši a 25 % všech pacientů byli Asiaté.
Primární analýza dat byla provedena při dokončení 12měsíční léčby všech 846 pacientů (či dříve při předčasném ukončení léčby pacienta). Následné analýzy hodnotí stav pacientů po dosažení 24, 36, 48, 60 a 72 měsíců léčby (nebo ukončení léčby dříve). Medián doby léčby byl přibližně 70 měsíců vterapeutických skupinách s nilotinibem a 64 měsíců ve skupinách s imatinibem. Medián dávky byl 593 mg/den pro nilotinib 300 mg dvakrát denně, 772 mg/den pro nilotinib 400 mg dvakrát denně a 400 mg/den pro imatinib 400 mg jednou denně. Studie stále probíhá.
Primárním cílovým parametrem účinnosti bylo dosažení velké molekulární odpovědi (MMR) ve 12 měsících. MMR byla definovaná jako <0,1 % BCR-ABL/ABL % dle mezinárodní stupnice (IS) měřená RQ-PCR, což odpovídá >3 log snížení BCR-ABL transkriptu od standardizované výchozí úrovně. Výskyt MMR ve 12 měsících byl statisticky významně vyšší pro nilotinib 300 mg dvakrát denně ve srovnání s imatinibem 400 mg jednou denně (44,3 % oproti 22,3 %, p<0,0001). Výskyt MMR ve 12 měsících byl také statisticky významně vyšší pro nilotinib 400 mg dvakrát denně ve srovnání s imatinibem 400 mg jednou denně (42,7 % oproti 22,3 %, p<0,0001).
Procento výskytu MMR ve 3, 6, 9 a 12 měsících byly 8,9 %, 33,0 %, 43,3 % a 44,3 % pro nilotinib 300 mg dvakrát denně, 5,0 %, 29,5 %, 38,1 % a 42,7 % pro nilotinib 400 mg dvakrát denně a 0,7 %, 12,0 %, 18,0 % a 22,3 % pro imatinib 400 mg jednou denně.
Výskyty MMR ve 12, 24, 36, 48 60 a 72 měsících jsou uvedené v Tabulce 5.
Tabulka 5 Výskyt MMR
|
Tasigna 300 mg dvakrát denně n=282 (%) |
Tasigna 400 mg dvakrát denně n=281 (%) |
Imatinib 400 mg jednou denně n=283 (%) | |
|
MMR ve 12 měsících | |||
|
Odpověď (95% CI) |
44,3' (38,4; 50,3) |
42J1 (36,8; 48,7) |
22,3 (17,6; 27,6) |
|
MMR ve 24 měsících | |||
|
Odpověď (95% CI) |
61,7' (55,8; 67,4) |
59,1 (53,1; 64,9) |
37,5 (31,8; 43,4) |
|
MMR ve 36 měsících2 | |||
|
Odpověď (95% CI) |
58,52 (52,5; 64,3) |
57,3: (51,3; 63,2) |
38,5 (32,8; 44,5) |
|
MMR ve 48 měsících3 | |||
|
Odpověď (95% CI) |
59V1 (54,0; 65,7) |
55,2 (49,1; 61,1) |
43,8 (38,0; 49,8) |
|
MMR v 60 měsících4 | |||
|
Odpověď (95% CI) |
62,8 (56,8; 68,4) |
61,2 (55,2; 66,9) |
49,1 (43,2; 55,1) |
|
MMR v 72 měsících5 | |||
|
Odpověď (95% CI) |
52,5 (46,5; 58,4) |
57,7 (51,6; 63,5) |
41,7 (35,9; 47,7) |
|
1 Cochran-Mantel-Haenszelův (C |
MH) test p-hodnoty pro výskyt odpovědi (oproti imatinibu 400 mg) | ||
<0,0001
2 Jako respondéři byli uvedeni pouze pacienti, kteří dosáhli MMR ve specifickém časovém bodě. Ve 36 měsících nebylo pro MMR hodnotitelných celkem 199 (35,2%) pacientů (87 ve skupině
s nilotinibem 300 mg dvakrát denně a 112 ve skupině s imatinibem) z důvodu
chybějících/nehodnotitelných stanovení PCR (n=17), atypických transkriptů ve výchozím stavu (n=7) nebo ukončení před 36 měsíci léčby (n=175).
3 Jako respondéři j sou pro určitý časový bod zahrnuti pouze pacienti, kteří dosáhli MMR v tomto časovém bodě. Celkem u 305 (36,1 %) pacientů nebyla ve 48 měsících MMR vyhodnotitelná (98 ve skupině s nilotinibem 300 mg podávaným dvakrát denně, 88 ve skupině s nilotinibem 400 mg dvakrát denně a 119 ve skupině s imatinibem) z důvodu chybějících/nevyhodnotitelných PCR vyšetření (n=18), atypických transkriptů ve výchozím stavu (n=8), nebo pro ukončení před časovým bodem
48 měsíců (n=279).
4 Jako respondéři jsou pro určitý časový bod zahrnuti pouze pacienti, kteří dosáhli MMR v tomto časovém bodě. Celkem u 332 (38,1 %) pacientů nebyla v 60 měsících MMR vyhodnotitelná (99 ve skupině s nilotinibem 300 mg podávaným dvakrát denně, 93 ve skupině s nilotinibem 400 mg dvakrát denně a 130 ve skupině s imatinibem) z důvodu chybějících/nevyhodnotitelných PCR vyšetření (n=9), atypických transkriptů ve výchozím stavu (n=8), nebo pro ukončení před časovým bodem 60 měsíců (n=305).
Jako respondéři jsou pro určitý časový bod zahrnuti pouze pacienti, kteří dosáhli MMR v tomto časovém bodě. Celkem u 395 (46,7 %) pacientů nebyla v 72 měsících MMR vyhodnotitelná (130 ve skupině s nilotinibem 300 mg podávaným dvakrát denně, 110 ve skupině s nilotinibem 400 mg dvakrát denně a 155 ve skupině s imatinibem) z důvodu chybějících/nevyhodnotitelných PCR vyšetření (n=25), atypických transkriptů ve výchozím stavu (n=8), nebo pro ukončení před časovým bodem 72 měsíců (n=362).
Kumulativní výskyt MMR v různých časových bodech (mezi pacienty s odpovědí na léčbu j sou zahrnuti pacienti, kteří dosáhli MMR v časových bodech nebo před uplynutím těchto časových bodů) (viz Obrázek 1).
Obrázek 1 Kumulativní výskyt MMR
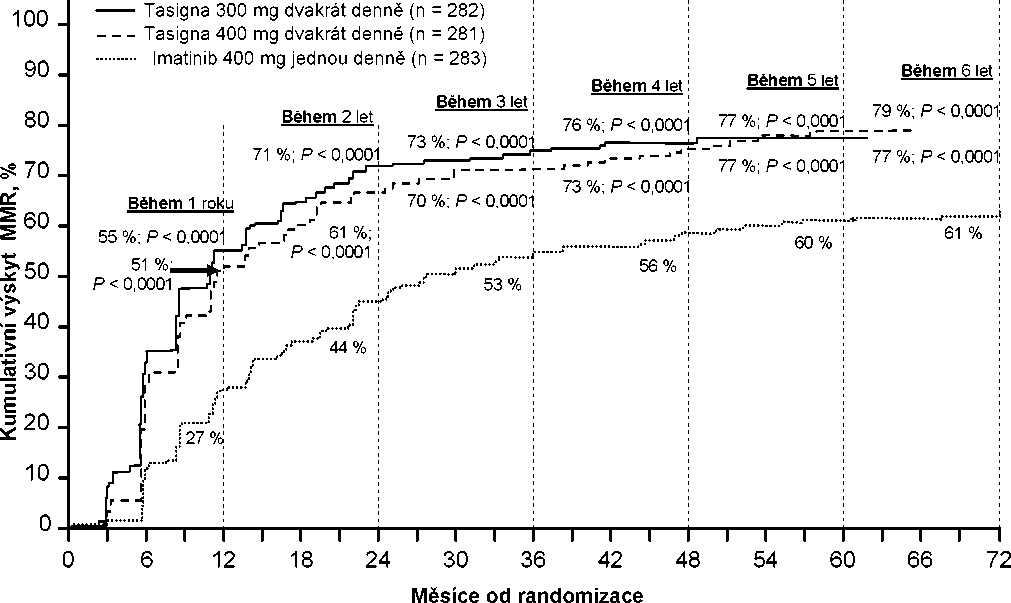
Pro všechny rizikové skupiny dle Sokalova skóre zůstal ve všech časových bodech výskyt MMR konzistentně vyšší v obou skupinách s nilotinibem oproti skupině s imatinibem.
V retrospektivních analýzách dosáhlo 91 % (234/258) pacientů na nilotinibu 300 mg dvakrát denně hodnot BCR-ABL <10 % ve 3 měsících léčby oproti 67 % (176/264) pacientů na imatinibu 400 mg jednou denně. Pacienti s hodnotami BCR-ABL <10 % ve 3 měsících léčby vykazovali vyšší poměr celkového přežití v 72 měsících v porovnání s pacienty kteří nedosáhli této hodnoty molekulární odpovědi (94,5 % oproti 77,1 % [p=0,0005]).
Na základě Kaplan-Meierovy analýzy doby do první MMR byla pravděpodobnost dosažení MMR v různých časových úsecích vyšší pro nilotinib v dávkách 300 mg a 400 mg dvakrát denně v porovnání s imatinibem 400 mg jednou denně (HR=2,17 a stratifikovaná hodnota log-rank p<0,0001 mezi nilotinibem 300 mg dvakrát denně a imatinibem 400 mg jednou denně, HR=1,88 a stratifikovaná hodnota log-rank p<0,0001 mezi nilotinibem 400 mg dvakrát denně a imatinibem 400 mg jednou denně).
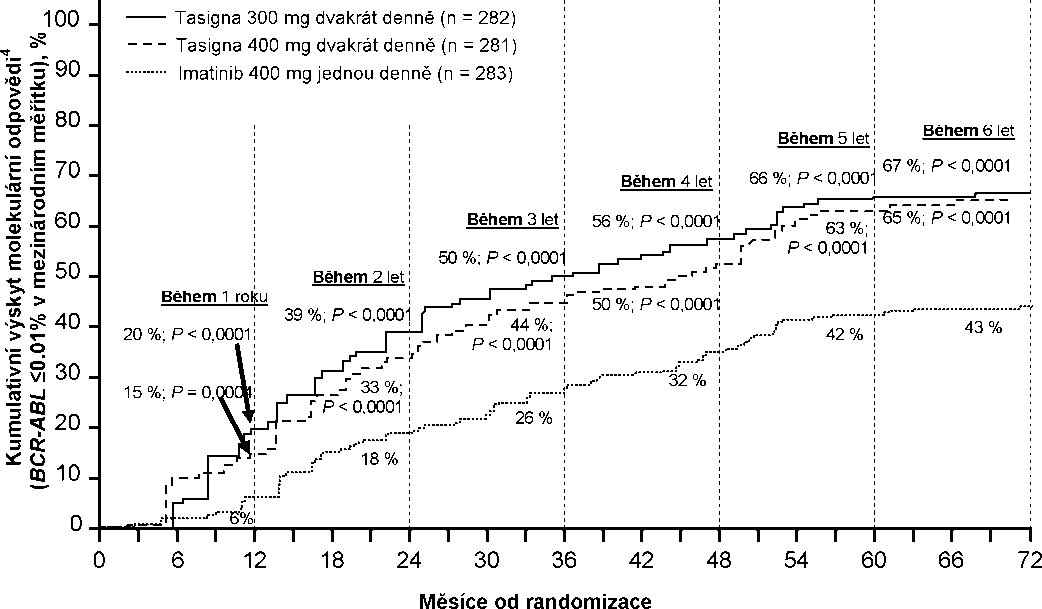
Podíly pacientů, kteří dosáhli molekulárních odpovědí <0,01 % a <0,0032 % podle IS v různých časových bodech, jsou uvedené v Tabulce 6 a poměry pacientů, kteří dosáhli molekulárních odpovědí <0,01 % a <0,0032 % podle IS během různých časových období, jsou uvedeny na Obrázcích 2 a 3. Molekulární odpověď <0,01 % a <0,0032 % podle IS odpovídá poklesu hladiny BCR-ABL transkriptů o >4 log, respektive pokles o >4,5 log od standardizovaného výchozího stavu.
Tabulka 6 Podíl pacientů, kteří dosáhli molekulární odpovědi <0,01% (pokles o 4 log) a <0,0032% (pokles o 4,5 log)
|
Ta 300 mg d n= |
signa vakrát denně 282 To) |
Ta 400 mg d n= |
signa vakrát denně =281 To) |
Im 400 mg j( n= |
atinib sdnou denně =283 í%) | |
|
<0,01 % |
<0,0032 % |
<0,01 % |
< 0,0032 % |
<0,01 % |
<0,0032 % | |
|
Ve 12 měsících |
11,7 |
4,3 |
8,5 |
4,6 |
3,9 |
0,4 |
|
Ve 24 měsících |
24,5 |
12,4 |
22,1 |
7,8 |
10,2 |
2,8 |
|
Ve 36 měsících |
29,4 |
13,8 |
23,8 |
12,1 |
14,1 |
8,1 |
|
Ve 48 měsících |
33,0 |
16,3 |
29,9 |
17,1 |
19,8 |
10,2 |
|
V 60 měsících |
47,9 |
32,3 |
43,4 |
29,5 |
31,1 |
19,8 |
|
V 72 měsících |
44,3 |
31,2 |
45,2 |
28,8 |
27,2 |
18,0 |
Obrázek 2 Kumulativní výskyt molekulární odpovědi <0,01% (pokles o 4-log)
100 _
40 _
30 -
3 CQ
E ^
0
10 -
- Tasigna 300 mg dvakrát denně (n = 282)
---Tasigna 400 mg dvakrát denně (n = 281)
.......... Imatinib 400 mg jednou denně (n = 283)
T^í
0 6
7
12
18
T-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1
24 30 36 42 48 54 60 66 72
Měsíce od randomizace
Na základě Kaplan-Meierových analýz trvání první MMR, byl podíl pacientů, kteří udržovali odpověď po dobu 72 měsíců mezi pacienty, kteří dosáhli MMR, 92,5 % (95% CI: 88,6-96,4 %) ve skupině s nilotinibem 300 mg dvakrát denně, 92,2 % (95% CI: 88,5-95,9 %) ve skupině s nilotinibem 400 mg dvakrát denně a 88,0 % (95% CI: 83,0-93,1 %) ve skupině s imatinibem 400 mg jednou denně.
Kompletní cytogenetická odpověď (CCyR) byla definována jako 0 % Ph+ metafáze v kostní dřeni na základě minimálně 20 vyhodnocených metafází. Nejlepší dosažený výskyt CCyR ve 12 měsících (zahrnuje na léčbu odpovídající pacienty, kteří dosáhli CCyR během 12 měsíců) byl statisticky vyšší pro nilotinib 300 mg a 400 mg dvakrát denně oproti imatinibu 400 mg jednou denně, viz Tabulka 7.
Podíl dosažení CCyR během 24 měsíců (zahrnuje pacienty, kteří dosáhli CCyR ve 24 měsících nebo dříve) byl statisticky významně vyšší pro obě skupiny s nilotinibem 300 mg dvakrát denně a 400 mg dvakrát denně v porovnání se skupinou s imatinibem 400 mg jednou denně.
Tabulka 7 Nejlepší celkový výskyt cytogenetické odpovědi (CCyR)
|
Tasigna (nilotinib) 300 mg dvakrát denně n=282 (%) |
Tasigna (nilotinib) 400 mg dvakrát denně n=281 (%) |
Glivec (imatinib) 400 mg jednou denně n=283 (%) | |
|
Během 12 měsíců | |||
|
Odpověď (95 % CI) |
80,1 (75,0; 84,6) |
77,9 (72,6; 82,6) |
65,0 (59,2; 70,6) |
|
Žádná odpověď |
19,9 |
22,1 |
35,0 |
|
CMH test p-hodnoty pro výskyt odpovědi (vs. imatinib 400 mg jednou denně) |
<0,0001 |
0,0005 | |
|
Během 24 měsíců | |||
|
Odpověď (95% CI) |
86,9 (82,4; 90,6) |
84,7 (79,9; 88,7) |
77,0 (71,7; 81,8) |
|
Žádná odpověď |
13,1 |
15,3 |
23,0 |
|
CMH test p-hodnoty pro výskyt odpovědi (vs. imatinib 400 mg jednou denně) |
0,0018 |
0,0160 |
Na základě Kaplan-Meierových analýz byl podíl pacientů, kteří udržovali odpověď po dobu 72 měsíců mezi pacienty, kteří dosáhli CCyR, 99,1 % (95% CI: 97,9-100 %) ve skupině s nilotinibem 300 mg dvakrát denně, 98,7 % (95% CI: 97,1-100 %) ve skupině s nilotinibem 400 mg dvakrát denně a 97,0 % (95% CI: 94,7-99,4 %) ve skupině s imatinibem 400 mg jednou denně.
Progrese do akcelerované fáze (AP) nebo blastické krize (BC) během léčby je definovaná jako doba od data randomizace do první dokumentované progrese onemocnění do akcelerované fáze nebo blastické krize nebo úmrtí v důsledku CML. Progrese do akcelerované fáze nebo blastické krize v průběhu léčby byla zjištěna celkem u 17 pacientů: u 2 pacientů na nilotinibu 300 mg dvakrát denně, u 3 pacientů na nilotinibu 400 mg dvakrát denně a u 12 pacientů na imatinibu 400 mg jednou denně. Odhadované podíly pacientů bez progrese do akcelerované fáze nebo blastické krize v 72 měsících byly 99,3 %, 98,7 % a 95,2 % (HR=0,1599 a stratifikovaný log-rank p=0,0059 mezi nilotinibem 300 mg dvakrát denně a imatinibem jednou denně, HR=0,2457 a stratifikovaný log-rank p=0,0185 mezi nilotinibem 400 mg dvakrát denně a imatinibem jednou denně). Do 2-letých analýz nebyly při léčbě hlášené nové případy progrese do AP/BC.
Při zahrnutí klonální evoluce jako kriteria progrese progredovalo do akcelerované fáze nebo blastické krize během léčby v době hodnocení celkem 25 pacientů (3 ve skupině s nilotinibem 300 mg dvakrát denně, 5 ve skupině s nilotinibem 400 mg dvakrát denně a 17 ve skupině s imatinibem 400 mg jednou denně). Odhadované podíly pacientů bez progrese do akcelerované fáze nebo blastické krize zahrnující klonální evoluci v 72 měsících byly 98,7 %, 97,9 % a 93,2 % (HR=0,1626 a stratifikovaný log-rank p=0,0009 mezi nilotinibem 300 mg dvakrát denně a imatinibem jednou denně, HR = 0,2848 a stratifikovaný log-rank p=0,0085 mezi nilotinibem 400 mg dvakrát denně a imatinibem jednou denně).
Během léčby nebo během doby sledování po ukončení léčby zemřelo celkem 55 pacientů (21 ve skupině s nilotinibem 300 mg dvakrát denně, 11 ve skupině s nilotinibem 400 mg dvakrát denně a 23 ve skupině s imatinibem 400 mg jednou denně). Dvacet šest (26) z těchto 55 úmrtí souviselo s CML (6 ve skupině s nilotinibem 300 mg dvakrát denně, 4 ve skupině s nilotinibem 400 mg dvakrát denně a 16 ve skupině s imatinibem 400 mg jednou denně). Odhadovaná procenta přeživších pacientů v 72 měsících byly 91,6 %, 95,8 % a 91,4 % (HR=0,8934 a stratifikovaný log-rank p=0,7085 mezi nilotinibem 300 mg dvakrát denně a imatinibem, HR=0,4632 a stratifikovaný log-rank p=0,0314 mezi nilotinibem 400 mg dvakrát denně a imatinibem). Vezmou-li se v úvahu pouze úmrtí související s CML, odhadovaná procenta celkového přežití v 72 měsících byla 97,7 %, 98,5 % a 93,9 % (HR=0,3694 a stratifikovaný log-rank p=0,0302 mezi nilotinibem 300 mg dvakrát denně a imatinibem, HR=0,2433 a stratifikovaný log-rank p=0,0061 mezi nilotinibem 400 mg dvakrát denně a imatinibem).
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Tasigna u pediatrických pacientů od narození do 18 let při léčbě chronické myeloidní leukémie (CML) s přítomností filadelfského chromozomu (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Vrcholové koncentrace nilotinibu je dosaženo za 3 hodiny po perorálním podání. Absorpce nilotinibu po perorálním podání byla přibližně 30 %. Absolutní biologická dostupnost nilotinibu nebyla stanovena. V porovnání s perorálním roztokem (pH 1,2 až 1,3) je relativní biologická dostupnost tobolky s nilotinibem přibližně 50 %. Pokud byl zdravým dobrovolníkům podán přípravek Tasigna s jídlem, zvýšila se Cmax nilotinibu o 112 % a plocha pod křivkou koncentrace v séru (AUC) o 82 %, ve srovnání s podáním přípravku Tasigna nalačno. Po podání přípravku Tasigna 30 minut nebo 2 hodiny po jídle stoupla biologická dostupnost nilotinibu o 29 %, respektive o 15 % (viz body 4.2, 4.4 a 4.5).
Absorpce nilotinibu (relativní biologická dostupnost) může být snížena přibližně o 48 % u pacientů s totální gastrektomií a přibližně o 22 % u pacientů s částečnou gastrektomií.
Distribuce
Poměr krev/plazma nilotinibu je 0,71. Na základě in vitro experimentů je vazba na plazmatické proteiny přibližně 98 %.
Biotransformace
Hlavní metabolické cesty, zjištěné u zdravých dobrovolníků, jsou oxidace a hydroxylace. Nilotinib je hlavní cirkulující složkou v séru. Žádný z metabolitů nepřispívá významným způsobem k farmakologické aktivitě nilotinibu. Nilotinib je primárně metabolizován CYP3A4 s možným menším přispěním CYP2C8.
Eliminace
Po jednorázové dávce radioaktivně značeného nilotinibu zdravým dobrovolníkům bylo více než 90 % dávky vyloučeno během 7 dnů, a to převážně stolicí (94 % dávky). Nezměněný nilotinib odpovídal 69 % dávky.
Zdánlivý eliminační poločas stanovený z farmakokinetiky po opakovaném denním podávání byl přibližně 17 hodin. Variabilita farmakokinetiky mezi pacienty byla střední až vysoká.
Linearita/nelinearita
Expozice nilotinibu v rovnovážném stavu byla závislá na dávce, při dávkách vyšších než 400 mg podávaných jednou denně byla systémová expozice méně úměrná dávce. Denní systémová expozice nilotinibu se 400 mg podávanými dvakrát denně byla v rovnovážném stavu o 35 % vyšší než s dávkou 800 mg podanou jednou denně. Systémová expozice (AUC) nilotinibu v rovnovážném stavu při dávce 400 mg dvakrát denně byla přibližně o 13,4 % vyšší oproti dávce 300 mg dvakrát denně. Průměrné minimální a maximální koncentrace nilotinibu po dobu 12 měsíců byly přibližně o 15,7 % a 14,8 % vyšší při dávkování 400 mg dvakrát denně oproti dávkování 300 mg dvakrát denně. Při zvýšení dávky ze 400 mg dvakrát denně na 600 mg dvakrát denně nebylo zvýšení expozice nilotinibu odpovídající.
Podmínek rovnovážného stavu bylo v zásadě dosaženo za 8 dnů. Zvýšení sérové expozice nilotinibu mezi první dávkou a rovnovážným stavem bylo přibližně 2násobné při dávkování jednou denně a 3,8násobné při dávkování dvakrát denně.
Biodostupnost/bioekvivalenční studie
Bylo prokázáno, že jednorázové podání 400 mg nilotinibu ve 2 tvrdých tobolkách o síle 200 mg, kde obsah každé tvrdé tobolky byl rozmíchaný v jedné lžičce jablečné šťávy, bylo bioekvivalentní jednorázovému podání 2 neporušených tvrdých tobolek o síle 200 mg.
5.3 Předklinické údaje vztahující se k bezpečnosti
Nilotinib byl hodnocen farmakologickými studiemi na bezpečnost, toxicitu po opakovaném podání, genotoxicitu, reprodukční toxicitu, fototoxicitu a kancerogenitu u potkanů a myší.
Nilotinib neměl účinky na CNS nebo respirační funkce. Výsledky in vitro studie srdeční bezpečnosti provedené s nilotinibem na izolovaných králičích srdcích předklinicky naznačovaly možnost prodloužení QT intervalu: byla zřejmá blokáda hERG proudů a prodloužení trvání akčního potenciálu. Žádné účinky nebyly pozorovány při vyšetření EKG u psů nebo opic léčených až 39 týdnů nebo ve speciální telemetrické studii u psů.
Studie toxicity po opakovaném podávání psům až po dobu 4 týdnů a makakům jávským po podávání až 9 týdnů ukázaly, že játra jsou primárním orgánem toxicity nilotinibu. Změny zahrnují zvýšení alaninaminotransferázy a aktivity alkalické fosfatázy a histopatologické nálezy (především hyperplazie/hypertrofie sinusoidálních buněk a Kupfferových buněk, hyperplazie žlučovodů a periportální fibróza). Po čtyřech týdnech rekonvalescence byly obvykle změny klinické biochemie plně reverzibilní a histologické změny částečně reverzibilní. Expozice při nejnižších dávkových hladinách, při kterých byly pozorovány účinky na játra, byly nižší než expozice u lidí při dávce 800 mg/den. U myší nebo potkanů, léčených až 26 týdnů, byly pozorovány malé změny na játrech. U potkanů, psů a opic bylo pozorováno převážně reverzibilní zvýšení hladin cholesterolu.
Studie genotoxicity na bakteriálních systémech in vitro a na savčích modelech in vitro a in vivo s metabolickou aktivací a bez ní nepřinesly žádný důkaz mutagenního potenciálu nilotinibu.
Ve 2leté studii kancerogenity u potkanů byla hlavním orgánem, kde vznikaly jiné než neoplastické léze, děloha (dilatace, vaskulární ektázie, hyperplázie endoteliálních buněk, zánět a/nebo epiteliální hyperplázie). Při podání nilotinibu v dávkách 5, 15 a 40 mg/kg/den nebyla kancerogenita prokázána. Expozice (vyjádřená AUC) při nejvyšší dávce představovaly přibližně 2 až 3násobek denní expozice nilotinibu v ustáleném stavu u lidí (na základě AUC) při dávce 800 mg/den.
Ve studii kancerogenity u myší (Tg.rasH2) trvající 26 týdnů, při které byl nilotinib podáván v dávkách 30, 100 a 300 mg/kg/den, byly pozorovány kožní papilomy/karcinomy při dávce 300 mg/kg, což představuje přibližně 30 až 40násobek expozice u člověka (na základě AUC) při maximální schválené dávce 800 mg/den (při dávkování 400 mg 2x denně). Hladina, při které nebyl pozorován žádný účinek (No-Observed-Effect-Levels) na kožní neoplastické léze byla 100 mg/kg/den, což představuje přibližně 10 až 20násobek expozice u člověka při maximální schválené dávce 800 mg/den (při dávkování 400 mg 2x denně). Hlavními cílovými orgány non-neoplastických lézí byly kůže (epidermální hyperplasie), zuby (degenerace/atrofie skloviny horních řezáků a zánět dásní/odontogenního epitelu řezáků) a brzlík (zvýšený výskyt a/nebo závažnost úbytku lymfocytů).
Nilotinib neindukoval teratogenitu, ale v dávkách toxických pro matku byl embryo a fetotoxický. Zvýšení postimplantační ztráty bylo pozorováno jak ve studii fertility, která zahrnovala léčbu samců i samic, tak ve studii embryotoxicity, která zahrnovala léčbu samic. Úmrtnost embryí a účinky na plod (především snížení hmotnosti plodu, předčasný srůst obličejových kostí (fúze maxily a jařmové kosti), viscerální a skeletální změny) u potkanů a zvýšení resorpce plodů a změny na skeletu u králíků byly přítomny ve studiích embryotoxicity. V pre- a postnatální výzkumné studii u potkanů expozice nilotinibu u matek způsobila snížení tělesné hmotnosti u mláďat spojené se změnami fyzických vývojových parametrů stejně jako snížení ukazatelů páření a fertility u potomků. Expozice nilotinibu u samic, při hladině bez nežádoucích účinků (No-Observed-Adverse-Effect-Levels), byla obvykle menší nebo stejná jako u lidí při dávce 800 mg/den.
Ve studii zaměřené na sledování vývoje v juvenilním období byl podáván nilotinib perorální sondou juvenilním potkanům od prvního týdne po narození do časné dospělosti (den 70 po narození) v dávkách 2, 6 a 20 mg/kg/den. Vedle standardních parametů studie bylo provedeno hodnocení vývojových mezníků, vlivu na CNS, páření a fertilitu. Na základě snížení tělesné hmotnosti obou pohlaví a zpoždění prepuciální separace u samců (které může být spojeno se snížením tělesné hmotnosti) byla stanovena výše dávky bez pozorovatelného efektu u juvenilních potkanů na 6 mg/kg/den. Juvenilní zvířata nevykazovala zvýšenou citlivost na nilotinib při porovnání s dospělými. Navíc byl profil toxicity u juvenilních potkanů srovnatelný s profilem toxicity zjištěným u dospělých potkanů.
Až do nejvyšší testované dávky, přibližně 5násobku doporučené dávky pro člověka, nebyly u samců a samic potkanů zaznamenány účinky na počet/motilitu spermií ani na fertilitu.
Bylo zjištěno, že nilotinib, který absorbuje světlo v pásmu UV-B a UV-A, je distribuován do kůže a má fototoxický potenciál in vitro, ale tento účinek nebyl pozorován in vivo. Proto se riziko, že nilotinib bude příčinou fotosenzitivity u pacientů, považuje ze velmi nízké.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tvrdé tobolky Monohydrát laktosy Krospovidon Poloxamer 188
Oxid křemičitý, koloidní bezvodý Magnesium-stearát
Obal tvrdé tobolky Želatina
Oxid titaničitý (E 171)
Červený oxid železitý (E 172)
Žlutý oxid železitý (E 172)
Tiskařský inkoust:
Šelak
Černý oxid železitý (E 172)
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30°C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení PVC/PVDC/Al blistry.
Přípravek Tasigna je dostupný v následujících velikostech balení:
• Jednotková balení obsahující 28 tvrdých tobolek (7 denních blistrů, jeden obsahuje 4 tvrdé tobolky) nebo 40 tvrdých tobolek (5 blistrů, jeden obsahuje 8 tvrdých tobolek).
• Vícečetná balení obsahující 112 (4 balení po 28) tvrdých tobolek, 120 (3 balení po 40) tvrdých tobolek nebo 392 (14 balení po 28) tvrdých tobolek.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky na likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/07/422/005-006
EU/1/07/422/009-010
EU/1/07/422/013
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 19. listopad 2007
Datum posledního prodloužení registrace: 19. listopad 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Tasigna 200 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje 200 mg nilotinibum (jako monohydrát hydrochloridu). Pomocné látky se známým účinkem
Jedna tvrdá tobolka obsahuje 156,11 mg laktosy (jako monohydrát).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka
Bílý až nažloutlý prášek ve světle žlutých neprůhledných tvrdých želatinových tobolkách, velikost 0 s červeným podélným potiskem „NVR/TKI“
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Tasigna je indikován k léčbě dospělých pacientů s:
- nově diagnostikovanou chronickou myeloidní leukemií s přítomností filadelfského chromozomu (Ph chromozom) v chronické fázi.
- chronickou a akcelerovanou fází CML s přítomností filadelfského chromozomu, kteří j sou rezistentní nebo nesnášeli předcházející léčbu zahrnující imatinib. Data o účinnosti u pacientů s CML v blastické krizi nejsou k dispozici.
4.2 Dávkování a způsob podání
Léčba musí být zahájena lékařem, který má zkušenosti s diagnostikou a léčbou pacientů s CML.
Dávkování
Doporučená dávka přípravku Tasigna je:
- 300 mg dvakrát denně u nově diagnostikovaných pacientů s CML v chronické fázi.
- 400 mg dvakrát denně u pacientů s chronickou nebo akcelerovanou fází CML, kteří j sou rezistentní nebo nesnášeli předcházející léčbu.
Léčba by měla trvat tak dlouho, dokud je přínosem pro pacienta.
Pro podání dávky 300 mg dvakrát denně jsou dostupné tvrdé tobolky o síle 150 mg.
Pokud pacient vynechá dávku, neměl by další dávku zdvojovat, ale měl by užít další obvyklou
předepsanou dávku.
Úprava nebo modifikace dávkování
Přípravek Tasigna může být dočasně vysazen a/nebo může být snížena dávka z důvodu hematologické toxicity (neutropenie, trombocytopenie), která nesouvisí se základním onemocněním leukemií (viz Tabulka 1).
Tabulka 1 Úprava dávkování při neutropenii a trombocytopenii
|
Nově diagnostikovaná CML v chronické fázi při 300 mg dvakrát denně a imatinib-rezistentní nebo intolerantní CML v chronické fázi při 400 mg dvakrát denně |
ANC* <1,0 x 109/l a/nebo počet destiček <50 x 109/l |
1. Léčba přípravkem Tasigna musí být přerušena a musí být monitorován krevní obraz. 2. Léčba musí být obnovena stejnou dávkou, pokud je do 2 týdnů ANC >1,0 x 109/l a/nebo počet destiček >50 x 109/l. 3. Jestliže počet krevních elementů zůstává nízký, může být třeba snížit dávku na 400 mg jednou denně. |
|
Imatinib-rezistentní nebo intolerantní CML v akcelerované fázi při 400 mg dvakrát denně |
ANC* <0,5 x 109/l a/nebo počet destiček <10 x 109/l |
1. Léčba přípravkem Tasigna musí být přerušena a musí být monitorován krevní obraz. 2. Léčba musí být obnovena stejnou dávkou, pokud je do 2 týdnů ANC >1,0 x 109/l a/nebo počet destiček >20 x 109/l. 3. Jestliže počet krevních elementů zůstává nízký, může být třeba snížit dávku na 400 mg jednou denně. |
*ANC= absolutní počet neutrofilů
Jestliže se vyvine klinicky signifikantní středně závažná nebo závažná nehematologická toxicita, mělo by být podávání přípravku přerušeno a může být znovu zahájeno dávkou 400 mg jednou denně, jakmile toxické příznaky vymizí. Pokud je to klinicky vhodné, mělo by se zvážit opětovné navýšení dávky dávky na počátečních 300 mg dvakrát denně u nově diagnostikovaných pacientů s CML v chronické fázi nebo na 400 mg dvakrát denně u pacientů s imatinib-rezistentní nebo intolerantní CML v chronické fázi a akcelerované fázi.
Zvýšená hladina sérové lipázy: Při zvýšení sérové lipázy stupně 3-4 by měla být dávka snížena na 400 mg jednou denně nebo léčba přerušena. Hladiny sérové lipázy by měly být vyšetřovány jednou měsíčně nebo dle klinické potřeby (viz bod 4.4).
Zvýšení bilirubinu a jaterních transamináz: Při zvýšení bilirubinu a jaterních transamináz na stupeň 3-4 by měla být dávka snížena na 400 mg jednou denně nebo léčba přerušena. Hladiny bilirubinu a jaterních transamináz by měly být vyšetřovány jednou měsíčně nebo dle klinické potřeby.
Starší lidé
V klinických studiích bylo přibližně 12 % jedinců ve studii fáze III u pacientů s nově diagnostikovanou CML v chronické fázi a přibližně 30% jedinců ve studii fáze II u pacientů s imatinib-rezistentní nebo intolerantní CML v chronické fázi a akcelerované fázi ve věku 65 let a starších. Žádné zásadní rozdíly v bezpečnosti a účinnosti nebyly pozorovány u pacientů >65 let ve srovnání s dospělými ve věku mezi 18 a 65 lety.
Porucha funkce ledvin
U pacientů se zhoršenou funkcí ledvin nebyly klinické studie provedeny.
Vzhledem k tomu, že nilotinib ani jeho metabolity nejsou vylučovány ledvinami, nepředpokládá se u pacientů se zhoršenou funkcí ledvin snížení celkové tělesné clearance.
Porucha funkce jater
Zhoršená funkce jater má mírný účinek na farmakokinetiku nilotinibu. Úprava dávky se u pacientů se zhoršenou fukcí jater nepovažuje za nutnou. Nicméně pacienti se zhoršenou funkcí jater by měli být léčeni s opatrností (viz bod 4.4).
Srdeční poruchy
Pacienti s nekompenzovaným nebo závažným srdečním onemocněním (např. nedávný infarkt myokardu, městnavé srdeční selhání, nestabilní angina pectoris nebo klinicky významná bradykardie) byli z klinických studií vyloučeni. Pozornost je třeba věnovat pacientům s významnou srdeční poruchou (viz bod 4.4).
Během léčby přípravkem Tasigna byly hlášeny vzestupy hladin celkového cholesterolu v séru (viz bod 4.4). Lipidový profil by měl být stanoven před zahájením léčby přípravkem Tasigna a vyhodnocován v měsících 3 a 6 po zahájení léčby a dále nejméně jednou za rok během chronické léčby.
Během léčby přípravkem Tasigna byly hlášeny vzestupy hladin glukózy v krvi (viz bod 4.4). Hladiny glukózy v krvi by měly být stanoveny před zahájením léčby přípravkem Tasigna a sledovány během léčby.
Pediatrická populace
Bezpečnost a účinnost přípravku Tasigna u dětí od narození do 18 let nebyla dosud stanovena (viz bod 5.1). Vzhledem k chybějícím údajům o bezpečnosti a účinnosti se proto užívání u pediatrických pacientů nedoporučuje (viz bod 5.1).
Způsob podání
Tasigna by měla být podávána dvakrát denně přibližně po 12 hodinách a nesmí být užívána spolu s jídlem. Tvrdé tobolky musí být spolknuty celé s vodou. Dvě hodiny před užitím dávky a alespoň jednu hodinu po užití dávky by neměla být požita žádná potrava.
Pacientům, kteří nejsou schopni tvrdé tobolky spolknout, může být obsah každé tobolky rozmíchán v jedné čajové lžičce jablečné šťávy (jablečného pyré), takto připravený lék se má okamžitě užít. Nesmí být užita více než jedna čajová lžička s každou tobolkou a použita jiná potrava než jablečná šťáva (viz body 4.4 a 5.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Myelosuprese
Léčba přípravkem Tasigna je doprovázena (National Cancer Institute Common Toxicity Criteria stupeň 3-4) trombocytopenií, neutropenií a anemií. Výskyt je častější u pacientů s imatinib-rezistentní nebo intolerantní CML, zejména u pacientů s akcelerovanou fází CML. Kompletní vyšetření krevního obrazu by mělo být prováděno v prvních 2 měsících léčby každé dva týdny a dále pak jednou měsíčně, nebo podle klinické indikace. Myelosuprese byla zpravidla reverzibilní a obvykle byla zvládnuta dočasným vysazením přípravku Tasigna nebo snížením dávky (viz bod 4.2).
Prodloužení QT intervalu
Ukázalo se, že Tasigna prodlužuje srdeční komorovou repolarizaci; délka naměřeného QT intervalu na EKG byla závislá na koncentraci.
V klinické studii fáze III u pacientů s nově diagnostikovanou CML v chronické fázi, kteří užívali 300 mg nilotinibu dvakrát denně, byla změna zprůměrovaného QTcF intervalu v ustáleném stavu od výchozí hodnoty 6 ms. Žádný pacient neměl QTcF >480 ms. V klinických studiích nebyly pozorovány epizody torsade de pointes.
V klinické studii fáze II u pacientů s CML v chronické a akcelerované fázi, kteří byli rezistentní a netolerující léčbu imatinibem a užívali 400 mg nilotinibu dvakrát denně, byla změna zprůměrovaného QTcF intervalu v ustáleném stavu od výchozí hodnoty 5 a 8 ms. U <1% těchto pacientů byl pozorován QTcF interval >500 ms. V klinických studiích nebyly pozorovány žádné epizody torsade de pointes.
Ve studii na zdravých dobrovolnících se srovnatelnou expozicí pozorovanou u pacientů byla změna střední zprůměrované hodnoty QTcF od výchozí hodnoty, po odečtení hodnot placeba, 7 ms (CI ± 4 ms). Žádný jedinec neměl hodnoty QTcF >450 ms. Navíc nebyly během studie pozorovány žádné klinicky relevantní arytmie. Zejména nebyly pozorovány žádné epizody torsade de pointes (přechodné ani setrvalé).
Významné prodloužení QT intervalu se může objevit v případě, že je nilotinib nesprávně užíván se silnými inhibitory CYP3A4 a/nebo léčivými přípravky, o kterých je známo, že prodlužují QT interval, a/nebo s potravou (viz bod 4.5). Současná přítomnost hypokalémie nebo hypomagnesémie mohou tento účinek ještě zvyšovat. Prodloužení QT intervalu může vystavit pacienty riziku fatálního konce.
Přípravek Tasigna by měl být užíván opatrně u pacientů s prodlouženým QTc intervalem, nebo u kterých je významné riziko vývoje prodloužení QTc intervalu, jako jsou pacienti:
- s kongenitálním syndromem dlouhého QT intervalu.
- s nekompenzovaným nebo závažným srdečním onemocněním, zahrnujícím nedávný infarkt myokardu, městnavé srdeční selhání, nestabilní anginu pectoris nebo klinicky významnou bradykardii.
- užívající antiarytmika nebo jiné látky, které prodlužují QT interval.
Doporučuje se pečlivé monitorování účinku na QTc interval a provedení výchozího EKG před zahájením léčby a dle klinické potřeby. Hypokalémie nebo hypomagnesémie musí být upraveny před podáním přípravku Tasigna a měly by být pravidelně sledovány během léčby.
Náhlé úmrtí
Méně časté případy (0,1 až 1%) náhlých úmrtí byly hlášeny u pacientů s imatinib-rezistentní nebo intolerantní CML v chronické fázi nebo akcelerované fázi, u nichž se v minulosti vyskytlo onemocnění srdce nebo měli významné kardiální rizikové faktory. Často se navíc spolu se základním maligním onemocněním vyskytovaly komorbidity léčené jinými současně podávanými léčivými přípravky. Poruchy komorové repolarizace mohly být přispívajícími faktory. V klinické studii fáze III u pacientů s nově diagnostikovanou CML v chronické fázi nebyly hlášené žádné případy náhlých úmrtí.
Zadržování tekutin a edém
Závažné formy zadržování tekutin, jako je pleurální efuze, plicní edém a perikardiální efuze, byly pozorovány méně často (0,1 až 1 %) ve studii fáze III u pacientů s nově diagnostikovanou CML. Podobné případy byly zjištěny v postmarketingových hlášeních. Nečekaný rychlý nárůst tělesné hmotnosti má být pečlivě prověřen. Pokud se během léčby nilotinibem objeví příznaky závažného zadržování tekutin, má být vyhodnocena etiologie a pacienti by měli být léčeni příslušným způsobem (viz bod 4.2 pokyny ke zvládání nehematologických toxicit).
Kardiovaskulární příhody
Kardiovaskulární příhody byly hlášeny v randomizované studii fáze III u pacientů s nově diagnostikovanou CML a zjištěny v postmarketingových hlášeních. V této klinické studii s mediánem doby léčby 60,5 měsíců se vyskytly případy kardiovaskulárních příhod stupně 3-4 zahrnující periferní arteriální okluze (1,4 % při dávce 300 mg nilotinibu dvakrát denně, respektive 1,1 % při dávce 400 mg dvakrát denně), ischemické onemocnění srdce (2,2 % při dávce 300 mg nilotinibu dvakrát denně, respektive 6,1 % při dávce 400 mg dvakrát denně) a ischemické cerebrovaskulární příhody (1,1 % při dávce 300 mg nilotinibu dvakrát denně, respektive 2,2 % při dávce 400 mg dvakrát denně). Pacienti by měli být poučeni o nutnosti okamžitě vyhledat lékaře, pokud se u nich projeví akutní známky nebo příznaky kardiovaskulárních příhod. Měl by být vyhodnocen kardiovaskulární stav pacientů a kardiovaskulární rizikové faktory by měly být během léčby přípravkem Tasigna sledovány a aktivně zvládány podle standardních doporučení. Ke zvládnutí kardiovaskulárních rizikových faktorů by měla být předpsána příslušná léčba (viz bod 4.2 pro pokyny ke zvládání nehematologických toxicit).
Reaktivace hepatitidy B
U pacientů, kteří jsou chronickými nosiči hepatitidy B, dochází k reaktivaci po zahájení léčby inhibitory tyrosinkinázy bcr-abl. Některé případy vyústily v akutní selhání jater nebo ve fulminantní hepatitidu vedoucí k transplantaci jater nebo došlo k úmrtí pacienta.
Před zahájením léčby přípravkem TASIGNA mají být pacienti vyšetřeni na infekci HBV. Před zahájením léčby u pacientů s pozitivní sérologií hepatitidy B (včetně těch s aktivním onemocněním) a u pacientů, u kterých v průběhu léčby vyjde pozitivní test infekce HBV, je třeba se obrátit na odborníky na onemocnění jater a léčbu hepatitidy B. Nosiči HBV, kteří potřebují léčbu přípravkem TASIGNA, mají být po celou dobu léčby a několik měsíců po jejím ukončení pečlivě sledováni s ohledem na možný výskyt známek a příznaků aktivní infekce HBV (viz bod 4.8).
Laboratorní testy a monitorování
Krevní lipidy
Ve studii fáze III u nově diagnostikovaných pacientů s CML vykazovalo 1,1 % pacientů léčených 400 mg nilotinibu dvakrát denně zvýšení hladin celkového cholesterolu stupně 3-4; žádné zvýšení stupně 3-4 však nebylo pozorováno ve skupině léčené 300 mg dvakrát denně (viz bod 4.8). Doporučuje se stanovit lipidové profily před zahájením léčby přípravkem Tasigna a vyhodnocovat je v měsících 3 a 6 po zahájení léčby a dále nejméně jednou za rok během chronické léčby (viz bod 4.2). Pokud je potřeba podávat inhibitor HMG-CoA reduktázy (léčivo snižující hladinu lipidů), přečtěte si prosím před zahájením léčby bod 4.5, neboť některé inhibitory HMG-CoA reduktázy jsou rovněž metabolizovány systémem CYP3A4.
Glukóza v krvi
Ve studii fáze III u nově diagnostikovaných pacientů s CML vykazovalo 6,9 % pacientů léčených 400 mg nilotinibu dvakrát denně a 7,2 % pacientů léčených 300 mg nilotinibu dvakrát denně zvýšení hladin glukózy stupně 3-4. Před zahájením léčby přípravkem Tasigna se doporučuje stanovit hladinu glukózy a monitorovat ji během léčby, pokud je to klinicky indikováno (viz bod 4.2). Pokud z výsledků testů vyplývá potřeba léčby, měli by se lékaři řídit lokálními standardy a doporučeními léčby.
Interakce s jinými léčivými přípravky
Přípravek Tasigna nemá být podáván s látkami, které jsou silnými inhibitory CYP3A4 (včetně, ale nejen, ketokonazolu, itrakonazolu, vorikonazolu, klarithromycinu, telithromycinu, ritonaviru). V případě, že by léčba těmito látkami byla nezbytná, doporučuje se, pokud je to možné, léčbu přípravkem Tasigna přerušit (viz bod 4.5). Jestliže přechodné přerušení léčby není možné, je třeba nemocného pečlivě sledovat z hlediska prodloužení QT intervalu (viz body 4.2, 4.5 a 5.2).
Při souběžném užívání přípravku Tasigna s léčivými přípravky, které jsou účinnými induktory CYP3A4 (např. fenytoin, rifampicin, karbamazepin, fenobarbital a třezalka), je pravděpodobné snížení expozice nilotinibu až v klinicky významném rozsahu. Z tohoto důvodu by pacientům užívajícím Tasigna měly být souběžně podávány alternativní léčivé látky s nižším potenciálem pro indukci CYP3A4 (viz bod 4.5).
Vliv potravy
Biologická dostupnost nilotinibu je zvýšena příjmem potravy. Přípravek Tasigna nesmí být užíván spolu s jídlem (viz body 4.2 a 4.5) a měl by být užíván 2 hodiny po jídle. Žádná potrava by neměla být přijímána nejméně jednu hodinu po užití dávky léku. Grapefruitová šťáva a jiné potraviny, o kterých je známo, že inhibují CYP3A4, by neměly být požívány. Pacientům, kteří nejsou schopni tvrdé tobolky spolknout, může být obsah každé tvrdé tobolky rozmíchán v jedné čajové lžičce jablečné šťávy (jablečného pyré), takto připravený lék se má okamžitě užít. Nesmí být užita více než jedna čajová lžička s každou tobolkou a použita jiná potrava než jablečná šťáva (viz bod 5.2).
Zhoršená funkce jater
Zhoršená funkce jater má mírný účinek na farmakokinetiku nilotinibu. Výsledkem podání jednorázové dávky 200 mg nilotinibu bylo zvýšení AUC o 35%, 35% a 19% u jedinců s mírným, středně závažným a závažným zhoršením funkce j ater ve srovnání s kontrolní skupinou jedinců s normální j aterní funkcí. Došlo ke zvýšení predikované Cmax nilotinibu v ustáleném stavu o 29%, 18%, respektive 22%.
Do klinických studií nebyli zařazeni pacienti s hodnotami alaninaminotransferázy (ALT) a/nebo aspartátaminotransferázy (AST) >2,5násobku (nebo >5násobku, pokud souvisely s onemocněním) horní hranice normálních hodnot, a/nebo pokud měli celkový bilirubin >1,5násobek horní hranice normálních hodnot. Nilotinib je metabolizován především v játrech. Pacienti se zhoršenou funkcí jater by proto mohli mít zvýšenou expozici nilotinibu a měli by být léčeni s opatrností (viz bod 4.2).
Sérová lipáza
Bylo pozorováno zvýšení lipázy v séru. U pacientů s pankreatitidou v anamnéze se doporučuje opatrnost. Pokud je zvýšení lipázy spojené s břišními příznaky, mělo by být podávání přípravku Tasigna přerušeno a měla by být provedena příslušná diagnostická vyšetření za účelem vyloučení pankreatitidy.
Totální gastrektomie
Biologická dostupnost nilotinibu může být u pacientů s totální gastrektomií omezená (viz bod 5.2). Mělo by být zváženo častější sledování těchto pacientů.
Syndrom nádorového rozpadu
Před započetím léčby přípravkem Tasigna je doporučená úprava klinicky významné dehydratace a léčba vysokých hladin kyseliny močové z důvodu možného výskytu syndromu nádorového rozpadu (TLS) (viz bod 4.8).
Laktóza
Tvrdé tobolky Tasigna obsahují laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Přípravek Tasigna může být podáván v kombinaci s hematopoetickými růstovými faktory, jako je erytropoetin nebo faktor stimulující granulocytární kolonie (G-CSF), pokud je podání klinicky indikováno. Může být podáván s hydroxyureou nebo anagrelidem, pokud je podání klinicky indikováno.
Nilotinib je metabolizován převážně játry a je také substrátem pro efluxní pumpu mnoha léků, P-glykoprotein (P-gp). Proto absorpce a následná eliminace systémově absorbovaného nilotinibu může být ovlivněna látkami, které působí na CYP3A4 a/nebo P-gp.
Látky, které mohou zvyšovat koncentrace nilotinibu v séru
Současné podání nilotinibu a imatinibu (substrát a mírný inhibitor P-gp a CYP3A4) mělo mírný inhibiční účinek na CYP3A4 a/nebo P-gp. Došlo ke zvýšení AUC imatinibu o 18% až 39% a zvýšení AUC nilotinibu o 18% až 40%. Tyto změny pravděpodobně nejsou klinicky významné.
Při současném podání silného inhibitoru CYP3A4, ketokonazolu, zdravým dobrovolníkům byla expozice nilotinibu zvýšena 3krát. Z tohoto důvodu by neměly být současně podávány silné inhibitory CYP3A4, včetně ketokonazolu, itrakonazolu, vorikonazolu, ritonaviru, klarithromycinu a telithromycinu (viz bod 4.4). Zvýšenou expozici nilotinibu je možné také očekávat se středně silnými inhibitory CYP3A4. Mělo by se uvažovat o alternativních léčivých přípravcích, která nemají žádné nebo mají minimální inhibiční účinky na CYP3A4.
Látky, které mohou snižovat koncentrace nilotinibu v séru
Rifampicin, silný induktor CYP3A4, snižuje Cmax nilotinibu o 64% a snižuje AUC nilotinibu o 80%. Rifampicin a nilotinib se nemá užívat současně.
Souběžné podávání jiných léčivých přípravků, které indukují CYP3A4 (např. fenytoin, karbamazepin, fenobarbital a třezalka), může pravděpodobně také v klinicky významném rozsahu snižovat expozici nilotinibu. U pacientů, u kterých jsou induktory CYP3A4 indikovány, by se mělo uvažovat o výběru alternativních přípravků s menším enzymovým indukčním potenciálem.
Rozpustnost nilotinibu je závislá na pH, při vyšším pH je rozpustnost nižší. U zdravých pacientů, kterým bylo podáváno 40 mg esomeprazolu jednou denně po dobu 5 dnů, se výrazně zvýšilo žaludeční pH, ale absorpce nilotinibu se snížila jen mírně (27% snížení Cmax a 34% snížení AUC0-<»). Nilotinib může být v případě potřeby užívána současně s esomeprazolem nebo jinými inhibitory protonové pumpy.
Ve studii u zdravých dobrovolníků nebyly při podání jednorázové dávky 400 mg přípravku Tasigna 10 hodin po a 2 hodiny před podáním famotidinu zjištěny významné změny ve farmakokinetice nilotinibu. V případě nutného souběžného užívání může být H2 blokátor podáván přibližně 10 hodin před a přibližně 2 hodiny po podání přípravku Tasigna.
Ve stejné studii podávání antacid (hydroxid hlinitý/hydroxid hořečnatý/simetikon) 2 hodiny před nebo po jednorázové dávce 400 mg přípravku Tasigna také nezměnilo farmakokinetiku nilotinibu. Pokud je to nutné, mohou být antacida podávána přibližně 2 hodiny před nebo přibližně 2 hodiny po podání přípravku Tasigna.
Látky, jejichž systémové koncentrace mohou být změněny nilotinibem
Nilotinib je in vitro relativně silný inhibitor CYP3A4, CYP2C8, CYP2C9, CYP2D6 a UGT1A1, s nejnižší hodnotou Ki pro CYP2C9 (Ki=0,13 mikromol).
V interakční studii u zdravých dobrovolníků po jednorázovém podání 25 mg warfarinu, citlivého substrátu CYP2C9, a 800 mg nilotinibu nedošlo k žádným změnám farmokokinetických parametrů nebo farmokodynamiky warfarinu, měřených jako protrombinový čas (PT) a mezinárodní normalizovaný poměr (INR). Data o rovnovážném stavu neexistují. Tato studie naznačuje, že při dávce warfarinu do 25 mg je klinicky významná léková interakce mezi nilotinibem a warfarinem méně pravděpodobná. Protože není dostatek dat o rovnovážném stavu, doporučuje se po zahájení léčby nilotinibem (minimálně během prvních 2 týdnů) kontrola farmakodynamických ukazatelů warfarinu (INR nebo PT).
U pacientů s CML zvýšil nilotinib podávaný v dávce 400 mg dvakrát denně po dobu 12 dní systémovou expozici (AUC a Cmax) perorálního midazolamu (substráty CYP3A4) 2,6násobně, respektive 2,0násobně. Nilotinib je středně silný inhibitor CYP3A4. Proto může při souběžném podávání s nilotinibem dojít ke zvýšení systémové expozice dalších léků primárně metabolizovaných CYP3A4 (např. některé inhibitory HMG-CoA reduktázy). Pro léky, které jsou CYP3A4 substráty a které mají úzký terapeutický index (například alfentanil, cyklosporin, dihydroergotamin, ergotamin, fentanyl, sirolimus a takrolimus) může být při souběžném podávání s nilotinibem nezbytné příslušné sledování a úprava dávky.
Antiarytmika a jiné látky, které mohou prodlužovat QT interval
Nilotinib by měl být podáván opatrně pacientům s prodloužením QT intervalu nebo u kterých se může prodloužení QT vyvinout, včetně pacientů, kteří užívají antiarytmika, jako jsou amiodaron, disopyramid, prokainamid, chinidin a sotalol, nebo jiné léčivé látky, které mohou vést k prodloužení QT intervalu, např. chlorochin, halofantrin, klarithromycin, haloperidol, methadon a moxifloxacin (viz bod 4.4).
Interakce s potravou
Absorpce a biodostupnost přípravku Tasigna jsou zvýšeny při současném příjmu potravy s následným zvýšením koncentrace v séru (viz body 4.2, 4.4 a 5.2). Grapefruitová šťáva a jiné potraviny, o kterých je známo, že inhibují CYP3A4, by neměly být požívány.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Zeny ve fertilním věku musí během léčby a dva měsíce po ukončení léčby přípravkem Tasigna používat vysoce účinnou antikoncepci.
Údaje o podávání nilotinibu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Přípravek Tasigna by neměl být během těhotenství podáván, pokud klinický stav pacientky nevyžaduje léčbu nilotinibem. Pokud je přípravek podáván během těhotenství, musí být pacientka informována o potenciálním riziku pro plod.
Kojení
Není známo, zda se nilotinib vylučuje do lidského mateřského mléka. Dostupné toxikologické údaje u zvířat prokázaly vylučování nilotinibu do mléka (viz bod 5.3). Riziko pro kojené novorozence/děti nelze vyloučit. Přípravek Tasigna se během kojení nemá podávat.
Fertilita
Studie na zvířatech neprokázaly vliv na fertilitu u samců a samic potkanů (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pacienti, kteří pozorují závratě, únavu, zhoršení zraku nebo jiné nežádoucí účinky s potenciálním vlivem na schopnost bezpečně řídit nebo obsluhovat stroje, by neměli tyto činnosti vykonávat, dokud tyto nežádoucí účinky přetrvávají (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Údaje uvedené níže odrážejí expozici přípravkem Tasigna u celkem 717 pacientů v randomizované studii fáze III u nově diagnostikovaných pacientů s Ph+ CML v chronické fázi léčených doporučenou dávkou 300 mg dvakrát denně (n=279) a v otevřené multicentrické studii fáze II u pacientů s imatinib-rezistentní nebo intolerantní CML v chronické fázi (n=321) a akcelerované fázi (n=137) léčených doporučenou dávkou 400 mg dvakrát denně.
Pacienti s nově diagnostikovanou CML v chronické fázi Medián trvání expozice byl 60,5 měsíců (rozmezí 0,1-70,8 měsíců).
Nejčastější (>10 %) nehematologické nežádoucí účinky byly vyrážka, svědění, bolest hlavy, nauzea, únava, alopecie, bolest svalů a bolest v nadbřišku. Většina těchto nežádoucích účinků byla mírného až středního stupně závažnosti. Méně často (<10 % a >5 %) byly pozorovány zácpa, suchá kůže, astenie, svalové křeče, průjem, artralgie, bolest břicha, zvracení a periferní otoky, tyto nežádoucí účinky byly mírně až středně závažné, zvládnutelné a obecně nevyžadovaly snížení dávky.
Hematologická toxicita zahrnující myelosupresi vzniklá při léčbě: trombocytopenie (18 %), neutropenie (15 %) a anémie (8 %). Biochemické nežádoucí účinky zahrnují zvýšenou hladinu alaninaminotransferázy (24 %), hyperbilirubinémii (16 %), zvýšenou hladinu
aspartátaminotransferázy (12 %), zvýšenou hladinu lipázy (11 %), zvýšenou hladinu bilirubinu v krvi (10 %), hyperglykémii (4 %), hypercholesterolémii (3 %) a hypertriglyceridémii (<1 %). Pleurální a perikardiální výpotky, bez ohledu na příčinu, se vyskytly u 2 % pacientů, respektive <1 % pacientů, kterým bylo podáváno 300 mg přípravku Tasigna dvakrát denně. Krvácení do zažívacího traktu, bez ohledu na příčinu, bylo hlášeno u 3 % těchto pacientů.
Změna zprůměrovaného QTcF intervalu v ustáleném stavu od výchozího stavu byla 6 msec. Žádný pacient neměl během studie s léčivým přípravkem absolutní QTcF >500 msec. Prodloužení QTcF přesahující 60 msec bylo pozorováno u <1 % pacientů během studie s léčivým přípravkem. Nebyly zjištěny případy náhlých úmrtí ani epizody torsade de pointes (přechodné ani trvalé). Během léčby nebyl zjištěn pokles průměrné ejekční frakce levé komory (LVEF). Žádný pacient neměl během léčby LVEF <45 % a také žádný pacient neměl pokles LVEF o více než 15 %.
Ukončení léčby z důvodu výskytu nežádoucích účinků bylo pozorováno u 10 % pacientů.
Pacienti s imatinib-rezistentní nebo intolerantní CML v chronické fázi a akcelerované fázi Údaje uvedené níže odrážejí expozici přípravku Tasigna u 458 pacientů v otevřené multicentrické studii fáze II u pacientů s imatinib-rezistentní nebo intolerantní CML v chronické fazi (n=321) a akcelerované fázi (n=137) léčených doporučenou dávkou 400 mg dvakrát denně.
Nejčastější (>10%) nehematologické nežádoucí účinky, které souvisely s podávaným přípravkem, byly vyrážka, svědění, nauzea, únava, bolest hlavy, zvracení, bolest svalů, zácpa a průjem. Většina těchto nežádoucích účinků byla mírná až středně závažná. Méně často (<10 % a >5 %) bylo pozorováno vypadávání vlasů, svalové křeče, snížená chuťk jídlu, bolesti kloubů, bolest břicha, bolesti kostí, periferní otoky, celková tělesná slabost (astenie), bolest v nadbřišku, suchá kůže, erytém a bolest končetin, tyto nežádoucí účinky byly mírné až středně závažné (stupeň 1 nebo 2). Ukončení léčby pro nežádoucí účinky bylo pozorováno u 16 % pacientů v chronické fázi a u 10 % pacientů v akcelerované fázi.
Při léčbě vzniklé hematologické toxicity zahrnovaly myelosupresi: trombocytopenii (31 %), neutropenii (17 %) a anemii (14 %). Pleurální a perikardiální výpotek, obdobně i komplikace s retencí tekutin se vyskytly u <1 % pacientů léčených přípravkem Tasigna. Srdeční selhání bylo pozorováno u <1 % pacientů. Krvácení do zažívacího traktu bylo hlášeno u 1 % pacientů a krvácení do CNS u <1 % pacientů.
QTcF interval delší než 500 ms byl pozorován u <1% pacientů. Žádné epizody torsade de pointes (transientní nebo setrvalé) nebyly pozorovány.
Nejčastěji hlášené nežádoucí účinky u přípravku Tasigna v klinických studiích
Nehematologické nežádoucí účinky (kromě laboratorních abnormalit), které byly hlášeny v klinických studiích u nejméně 5% pacientů užívajících Tasignu, jsou uvedeny v Tabulce 2. Jsou řazeny podle frekvence výskytu, s nejčastějším na prvním místě, uvedené v záhlaví při použití přesnosti na jedno desetinné místo a následující konvence: velmi časté (>1/10) nebo časté (>1/100 až <1/10) V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Nově diagnostikovaná |
Imatinib-rezistentní nebo intolerantní | |||||||
|
CML-CP |
CML-CP a CML-AP | |||||||
|
300 mg dvakrát denně |
400 mg dvakrát denně | |||||||
|
n |
=279 |
n=458 | ||||||
|
60-měsíční analýza |
24-měsíční analýza | |||||||
|
CML- |
CML- | |||||||
|
Třídy |
Četnost |
Všech |
Stupeň |
Četnost |
Všech |
Stupeň |
CP |
AP |
|
orgánových |
ny |
3-4 |
ny |
3-4 |
n=321 |
n=137 | ||
|
systémů/ nežádoucí |
stupně |
stupně |
Stupeň 3-4 |
Stupeň 3-4 | ||||
|
účinky |
% |
% |
% |
% |
% |
% | ||
|
Poruchy metabolismu a výživy | ||||||||
|
Snížená chuť k jídlu** |
Časté |
4 |
0 |
Časté |
8 |
<1 |
<1 |
0 |
|
Poruchy nervového systému | ||||||||
|
Bolesti hlavy |
Velmi časté |
16 |
2 |
Velmi časté |
15 |
1 |
2 |
<1 |
|
Gastrointestiná |
ní poruchy | |||||||
|
Velmi časté |
14 |
<1 |
Velmi časté |
20 |
<1 |
<1 |
<1 | |
|
Zácpa |
Časté |
10 |
0 |
Velmi časté |
12 |
<1 |
<1 |
0 |
|
Časté |
9 |
<1 |
Velmi časté |
11 |
2 |
2 |
<1 | |
|
Časté |
6 |
0 |
Velmi časté |
10 |
<1 |
<1 |
0 | |
|
Bolesti v nadbřišku |
Velmi časté |
10 |
1 |
Časté |
5 |
<1 |
<1 |
0 |
|
Bolesti břícha |
Časté |
6 |
0 |
Časté |
6 |
<1 |
<1 |
<1 |
|
Časté |
5 |
0 |
Časté |
3 |
0 |
0 |
0 | |
|
Poruchy kůže a |
podkožní tkáně | |||||||
|
Velmi časté |
33 |
<1 |
Velmi časté |
28 |
1 |
2 |
0 | |
|
Velmi časté |
18 |
<1 |
Velmi časté |
24 |
<1 |
<1 |
0 | |
|
Alopecie |
Velmi časté |
10 |
0 |
Časté |
9 |
0 |
0 |
0 |
|
Suchá kůže |
Časté |
10 |
0 |
Časté |
5 |
0 |
0 |
0 |
|
Erytém |
Časté |
3 |
0 |
Časté |
5 |
<1 |
<1 |
0 |
|
Poruchy svalové a kosterní soustavy a |
pojivové tkáně | |||||||
|
Myalgie |
Velmi časté |
10 |
<1 |
Velmi časté |
10 |
<1 |
<1 |
<1 |
|
Spasmy svalů |
Časté |
9 |
0 |
Časté |
8 |
<1 |
<1 |
0 |
|
Artralgie |
Časté |
8 |
<1 |
Časté |
7 |
<1 |
1 |
0 |
|
Bolesti kostí |
Časté |
4 |
0 |
Časté |
6 |
<1 |
<1 |
0 |
|
Bolesti končetin |
Časté |
5 |
<1 |
Časté |
5 |
<1 |
<1 |
<1 |
|
Celkové poruchy a reakce v místě aplikace | ||||||||
|
Únava |
Velmi časté |
12 |
0 |
Velmi časté |
17 |
1 |
1 |
<1 |
|
Astenie |
Časté |
9 |
<1 |
Časté |
6 |
<1 |
0 |
<1 |
|
Periferní otoky |
Časté |
5 |
0 |
Časté |
6 |
0 |
0 |
0 |
* Pro prezentaci v této tabulce j sou procenta zaokrouhlená na celá čísla. Nicméně k identifikaci nežádoucích účinků s četností nejméně 5 % a ke klasifikace nežádoucích účinků podle kategorií četností jsou použita procenta s přesností na jedno desetinné místo.
** Rovněž zahrnuje přednostní název anorexie
Následující nežádoucí účinky byly hlášeny v klinických studiích u pacientů s přípravkem Tasigna s frekvencí nižší než 5% U laboratorních změn jsou uvedené také velmi časté případy (>1/10), nezahrnuté do Tabulky 2. Tyto nežádoucí účinky jsou zahrnuty podle klinické významnosti a řazeny v každé skupině podle klesající závažnosti a následující konvence: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), není známo (z dostupných údajů nelze určit).
Infekce a infestace:
Časté: folikulitida infekce horních cest dýchacích (zahrnují faryngitidu, nazofaryngitidu, rhinitidu). Méně časté: pneumonie, infekce močových cest, gastroenteritida, bronchitida, herpesvirové infekce, kandidózy (zahrnují orální kandidózy).
Není známo: sepse, subkutánní absces, absces konečníku, furunkl, tinea pedis, reaktivace hepatitidy B.
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy):
Časté: kožní papilom.
Není známo: orální papilom, paraproteinémie.
Poruchy krve a lymfatického sytému:
Časté: leukopenie, eozinofilie, febrilní neutropenie, pancytopenie, lymfopenie.
Méně časté: trombocytemie, leukocytóza.
Poruchy imunitního systému:
Není známo: hypersenzitivita.
Endokrinní poruchy:
Méně časté: hypertyreóza, hypotyreóza.
Není známo: sekundární hyperparatyreóza, thyreoiditis.
Poruchy metabolismu a výživy:
Velmi časté: hypofosfatémie (včetně snížení krevního fosforu)
Časté: dysbalance elektrolytů (zahrnuje hypomagnesémii, hyperkalémii, hypokalémii, hyponatrémii, hypokalcémii, hyperkalcémii, hyperfosfatémii), diabetes mellitus, hyperglykémie, hypercholesterolémie, hyperlipidémie, hypertriacylglycerolémie.
Méně časté:dehydratace, zvýšená chuť k jídlu, dna, dyslipidémie.
Není známo: hyperurikémie, hypoglykémie.
Psychiatrické poruchy:
Časté: deprese, nespavost, stavy úzkosti.
Není známo: dezorientace, stavy zmatenosti, amnezie, dysforie.
Poruchy nervového systému:
Časté: závratě, periferní neuropatie, hypestezie, parestezie.
Méně časté: nitrolební krvácení, ischemická cerebrovaskulární příhoda, tranzitorní ischemická ataka, mozkový infarkt, migréna, ztráta vědomí (včetně krátkodobé ztráty vědomí), třes, poruchy pozornosti, hyperestezie.
Není známo: cerebrovaskulární příhoda, edém mozku, zánět optického nervu, otupělost, dysestezie, syndrom neklidných nohou.
Poruchy oka:
Časté: oční krvácení, periorbitální otok, svědění očí, konjunktivitida, suchost očí (včetně xeroftalmie). Méně časté: poruchy vidění, neostré vidění, spojivkové krvácení, snížená ostrost vidění, otok očních víček. fotopsie, hyperémie (bělma, spojivek, očí), oční podráždění.
Není známo: edém papily, chorioretinopatie, diplopie, fotofobie, otok očí, blefaritida, bolest očí, alergie spojivek, poškození povrchu oka.
Poruchy ucha a labyrintu:
Časté: vertigo.
Není známo: zhoršení sluchu, bolest uší, ušní šelest.
Srdeční poruchy:
Časté: angina pectoris, arytmie (včetně atrioventrikulárního bloku, srdečního flutteru, extrasystol, tachykardie, fibrilace síní, bradykardie), palpitace, prodloužení QT intervalu na EKG.
Méně časté: srdeční selhání, infarkt myokardu, onemocnění věnčitých tepen, srdeční šelest, perikardiální výpotek, cyanóza.
Není známo: komorová dysfunkce, perikarditida, snížení ejekční frakce
Cévní poruchy:
Časté: hypertenze, návaly, periferní arteriální stenóza.
Méně časté: hypertenzní krize, periferní arteriální okluzivní choroba, intermitentní klaudikace, stenóza končetinové arterie, hematomy, arterioskleróza.
Není známo: hemoragický šok, hypotenze, trombóza.
Respirační, hrudní a mediastinální poruchy:
Časté: dušnost, námahová dušnost, epistaxe, kašel, dysfonie.
Méně časté: plicní edém, pleurální výpotek, intersticiální plicní onemocnění, pleurální bolest, zánět pohrudnice, faryngolaryngeální bolest, podráždění hrdla.
Není známo: plicní hypertenze, sípání, orofaryngeální bolest.
Gastrointestinální poruchy:
Časté: pankreatitida, abdominální diskomfort, nadýmání, dyspepsie, poruchy chuti, plynatost.
Méně časté: gastrointestinální krvácení, meléna, ulcerace v ústech, gastroezofageální reflux, stomatitida, ezofageální bolest, sucho v ústech, gastritida, citlivost zubů.
Není známo: perforace gastrointestinálního vředu, retroperitoneální hemoragie, hematemeza, žaludeční vřed, ulcerózní ezofagitida, subileus, enterokolitida, hemoroidy, hiátová hernie, krvácení z konečníku, zánět dásní.
Poruchy jater a žlučových cest:
Velmi časté: hyperbilirubinémie (včetně zvýšení krevního bilirubinu).
Časté: abnormální jaterní funkce.
Méně časté: hepatotoxicita, toxická hepatitida, žloutenka.
Není známo: cholestáza, hepatomegalie.
Poruchy kůže a podkožní tkáně:
Časté: noční pocení, ekzém, kopřivka, hyperhidróza, pohmožděniny, akné, dermatitida (včetně alergické, exfoliativní a akneiformní).
Méně časté: exfoliativní vyrážka, poléková vyrážka, bolest kůže, ekchymóza, otok obličeje.
Není známo: multiformní erytém, erythema nodosum, kožní ulcerace, syndrom palmárně-plantární erytrodysestezie, petechie, fotosenzitivita, puchýře, kožní cysty, hyperplázie mazových žláz, atrofie kůže, vyblednutí kůže, loupání kůže, hyperpigmentace kůže, hypertrofie kůže, hyperkeratóza, psoriáza.
Poruchy svalové a kosterní soustavy a pojivové tkáně:
Časté: svalové bolesti hrudníku, bolest svalů a kostí, bolest zad, bolest v boku, bolest šíje, svalová slabost.
Méně časté: svalová ztuhlost, otok kloubů.
Není známo: artritida.
Poruchy ledvin a močových cest:
Časté: polakisurie
Méně časté: dysurie, urgentní močení, nykturie.
Není známo: renální selhání, hematurie, močová inkontinence, chromaturie.
Poruchy reprodukčního systému a prsu:
Méně časté: bolesti prsou, gynekomastie, erektilní dysfunkce.
Není známo: tvrdnutí prsu, menoragie, otok bradavek.
Celkové poruchy a reakce v místě aplikace:
Časté: bolest na prsou (včetně bolesti na prsou jiného než srdečního původu), bolest, horečka, bolest na prsou, malátnost.
Méně časté: otoky obličeje, gravitační otoky, chřipce podobné onemocnění, zimnice, pocit změny tělesné teploty (zahrnující pocit horka, pocit chladu).
Není známo: lokalizovaný edém.
Vyšetření:
Velmi časté: zvýšená alanin aminotransferáza, zvýšená aspartát aminotransferáza, zvýšená lipáza, zvýšená hladina lipoproteinů (včetně lipoproteinů s nízkou denzitou a s vysokou denzitou), zvýšená hladina celkového cholesterolu, zvýšená hladina triglyceridů.
Časté: snížení hladiny hemoglobinu, zvýšená amyláza v krvi, zvýšená alkalická fosfatáza v krvi, zvýšená gamaglutamyltransferáza, zvýšená kreatinfosfokináza v krvi, snížená tělesná hmotnost, zvýšená tělesná hmotnost, zvýšená hladina inzulinu v krvi, snížená hladina globulinů.
Méně časté: zvýšená hladina laktátdehydrogenázy v krvi, snížená hladina glukózy v krvi, zvýšená hladina močoviny v krvi.
Není známo: zvýšená hladina troponinu, zvýšená hladina nekonjugovaného bilirubinu v krvi, snížená hladina inzulinu v krvi, snížená hladina inzulin C-peptidu, zvýšený paratyroidální hormon v krvi.
Klinicky relevantní nebo závažné abnormality rutinních hematologických nebo biochemických laboratorních hodnot jsou uvedeny v Tabulce 3.
Tabulka 3 Laboratorní abnormality stupně 3-4*
|
Nově diagnostikovaná CML-CP 300 mg dvakrát denně |
Imatinib-rezistentní nebo intolerantní CML-CP a CML-AP 400 mg dvakrát denně | ||
|
n=279 (%) |
CML-CP n=321 (%) |
CML-AP n=137 (%) | |
|
Hematologické parametry | |||
|
Myelosuprese | |||
|
- Neutropenie |
12 |
31 |
42 |
|
- Trombocytopenie |
10 |
30 |
42 |
|
- Anémie |
4 |
11 |
27 |
|
Biochemické parametry | |||
|
- Zvýšený kreatinin |
0 |
1 |
<1 |
|
- Zvýšená lipáza |
9 |
18 |
18 |
|
- Zvýšená SGOT (AST) |
1 |
3 |
2 |
|
- Zvýšená SGPT (ALT) |
4 |
4 |
4 |
|
- Hypofosfatémie |
7 |
17 |
15 |
|
- Zvýšený bilirubin (celkový) |
4 |
7 |
9 |
|
- Zvýšená glukóza |
7 |
12 |
6 |
|
- Zvýšený cholesterol (celkový) |
0 |
** |
** |
|
- Zvýšené triglyceridy |
0 |
** |
** |
*Pro prezentaci v této tabulce jsou použitá procenta s přesností na jedno desetinné místo a zaokrouhlená na celé číslo.
**Parametry, které nebyly sledované
Popis vybraných nežádoucích účinků
Náhlé úmrtí
Méně časté případy (0,1 až 1 %) náhlých úmrtí byly hlášeny v klinických studiích s Tasignou a/nebo v soucitných programech u pacientů s imatinib-rezistentní nebo intolerantní CML v chronické nebo akcelerované fázi, u nichž se v minulosti vyskytlo onemocnění srdce nebo u pacientů s významnými kardiálními rizikovými faktory (viz bod 4.4).
Reaktivace hepatitidy B
V souvislosti s tyrosinkinázou bcr-abl byla zaznamenána reaktivace hepatitidy B. Některé případy vyústily v akutní selhání jater nebo ve fulminantní hepatitidu vedoucí k transplantaci jater nebo došlo k úmrtí pacienta (viz bod 4.4).
Zkušenosti po uvedení na trh
Následující nežádoucí účinky byly získány z postmarketingové zkušenosti s přípravkem Tasigna prostřednictvím spontánních hlášení, literárních případů, rozšířených programů přístupu (Extended Access Programs) a klinických studií odlišných od globálních registračních studií. Protože byly tyto účinky hlášeny dobrovolně u populací o neznámé velikosti, není vždy možné spolehlivě stanovit jejich frekvenci nebo stanovit příčinný vztah k expozici nilotinibem.
Frekvence vzácná: U pacientů léčených přípravkem Tasigna byly hlášené případy syndromu nádorového rozpadu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Byly hlášeny ojedinělé případy záměrného předávkování nilotinibem, kdy byl požit nespecifikovaný počet tvrdých tobolek Tasigny v kombinaci s alkoholem a jinými léčivými přípravky. Objevila se neutropenie, zvracení a ospalost. Nebyly hlášené změny EKG nebo hepatotoxicita. Bylo hlášeno úplné uzdravení pacienta.
V případě předávkování by měl být pacient pozorován a měla by mu být poskytnuta vhodná podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antineoplastika, inhibitory proteinkinázy, ATC kód: L01XE08
Nilotinib je účinný inhibitor aktivity ABL tyrozinkinázy BCR-ABL onkoproteinu v buněčných liniích i v primárně leukemických buňkách s filadelfským chromozomem. Přípravek se s vysokou afinitou váže na vazebná místa ATP, a tím účinně inhibuje divoký typ BCR-ABL. Přípravek je účinný proti 32 ze 33 imatinib-rezistentních mutantních forem BCR-ABL. V důsledku této biochemické aktivity nilotinib selektivně inhibuje proliferaci a vyvolává apoptózu buněčných linií a u primárně leukemických buněk s filadelfským chromozomem pacientů s CML. U myších modelů CML nilotinib po perorálním podání samostatně redukuje objem nádoru a prodlužuje přežití.
Nilotinib má malý nebo žádný účinek na většinu dalších hodnocených proteinkináz, včetně Src, s výjimkou PDGF, KIT a Ephrin receptorových kináz, které jsou inhibovány v rozmezí koncentrací dosažených po perorálním podání terapeutických dávek doporučených k léčbě CML (viz Tabulka 4).
Tabulka 4 Kinázový profil nilotinibu (fosforylace IC50 nM)
|
BCR-ABL |
PDGFR |
KIT |
|
20 |
69 |
210 |
Klinické studie u nově diagnostikovaných CML v chronické fázi
Byla provedena otevřená, multicentrická, randomizovaná studie fáze III za účelem stanovení účinnosti nilotinibu oproti imatinibu u 846 dospělých pacientů s cytogeneticky potvrzenou nově diagnostikovanou CML s přítomností filadelfského chromosomu v chronické fázi. Pacienti byli diagnostikováni maximálně 6 měsíců před zařazením do studie a nebyli dříve léčeni s výjimkou hydroxyurey a/nebo anagrelidu. Pacienti byli randomizováni 1:1:1 k podávání nilotinibu 300 mg dvakrát denně (n=282), nilotinibu 400 mg dvakrát denně (n=281) nebo imatinibu 400 mg jednou denně (n=283). Randomizace byla stratifikována Sokalovým skóre v době diagnózy.
Základní charakteristiky tří terapeutických ramen byly dobře vyvážené. Průměrný věk byl 47 let v obou ramenech s nilotinibem a 46 let v rameni s imatinibem. 12,8 % pacientů bylo starších 65 let v rameni s nilotinibem 300 mg dvakrát denně, 10,0 % pacientů bylo starších 65 let v rameni s nilotinibem 400 mg dvakrát denně a 12,4 % pacientů bylo starších 65 let v rameni s imatinibem 400 mg jednou denně. Počet mužů byl mírně vyšší než počet žen (56,0 % v rameni s nilotinibem 300 mg dvakrát denně, 62,3 % v rameni s nilotinibem 400 mg dvakrát denně a 55,8 % v rameni s imatinibem 400 mg jednou denně). Více než 60 % pacientů byli běloši a 25 % všech pacientů byli Asiaté.
Primární analýza dat byla provedena při dokončení 12měsíční léčby všech 846 pacientů (či dříve při předčasném ukončení léčby pacienta). Následné analýzy hodnotí stav pacientů po dosažení 24, 36, 48, 60 a 72 měsíců léčby (nebo ukončení léčby dříve). Medián doby léčby byl přibližně 70 měsíců v terapeutických skupinách s nilotinibem a 64 měsíců ve skupinách s imatinibem. Medián dávky byl 593 mg/den pro nilotinib 300 mg dvakrát denně, 772 mg/den pro nilotinib 400 mg dvakrát denně a 400 mg/den pro imatinib 400 mg jednou denně. Studie stále probíhá.
Primárním cílovým parametrem účinnosti bylo dosažení velké molekulární odpovědi (MMR) ve 12 měsících. MMR byla definovaná jako <0,1 % BCR-ABL/ABL % dle mezinárodní stupnice (IS) měřená RQ-PCR, což odpovídá >3 log snížení BCR-ABL transkriptu od standardizované výchozí úrovně. Výskyt MMR ve 12 měsících byl statisticky významně vyšší pro nilotinib 300 mg dvakrát denně ve srovnání s imatinibem 400 mg jednou denně (44,3 % oproti 22,3 %, p<0,0001). Výskyt MMR ve 12 měsících byl také statisticky významně vyšší pro nilotinib 400 mg dvakrát denně ve srovnání s imatinibem 400 mg jednou denně (42,7 % oproti 22,3 %, p<0,0001).
Procento výskytu MMR ve 3, 6, 9 a 12 měsících byly 8,9 %, 33,0 %, 43,3 % a 44,3 % pro nilotinib 300 mg dvakrát denně, 5,0 %, 29,5 %, 38,1 % a 42,7 % pro nilotinib 400 mg dvakrát denně a 0,7 %, 12,0 %, 18,0 % a 22,3 % pro imatinib 400 mg jednou denně.
Výskyty MMR ve 12, 24, 36, 48, 60 a 72 měsících jsou uvedené v Tabulce 5.
Tabulka 5 Výskyt MMR
|
Tasigna 300 mg dvakrát denně n=282 (%) |
Tasigna 400 mg dvakrát denně n=281 (%) |
Imatinib 400 mg jednou denně n=283 (%) | |
|
MMR ve 12 měsících | |||
|
Odpověď (95% CI) |
44,3' (38,4; 50,3) |
42J1 (36,8; 48,7) |
22,3 (17,6; 27,6) |
|
MMR ve 24 měsících | |||
|
Odpověď (95% CI) |
61,7' (55,8; 67,4) |
59,1 (53,1; 64,9) |
37,5 (31,8; 43,4) |
|
MMR ve 36 měsících2 | |||
|
Odpověď (95% CI) |
58,5a (52,5; 64,3) |
57,31 (51,3; 63,2) |
38,5 (32,8; 44,5) |
|
MMR ve 48 měsících3 | |||
|
Odpověď (95% CI) |
59V1 (54,0; 65,7) |
55,2 (49,1; 61,1) |
43,8 (38,0; 49,8) |
|
MMR v 60 měsících4 | |||
|
Odpověď (95% CI) |
62,8 (56,8; 68,4) |
61,2 (55,2; 66,9) |
49,1 (43,2; 55,1) |
|
MMR v 72 měsících5 | |||
|
Odpověď (95% CI) |
52,5 (46,5; 58,4) |
57,7 (51,6; 63,5) |
41,7 (35,9; 47,7) |
|
1 Cochran-Mantel-Haenszelův (C |
MH) test p-hodnoty pro výskyt odpovědi (oproti imatinibu 400 mg) | ||
<0,0001
2 Jako respondéři byli uvedeni pouze pacienti, kteří dosáhli MMR ve specifickém časovém bodě. Ve 36 měsících nebylo pro MMR hodnotitelných celkem 199 (35,2%) pacientů (87 ve skupině
s nilotinibem 300 mg dvakrát denně a 112 ve skupině s imatinibem) z důvodu
chybějících/nehodnotitelných stanovení PCR (n=17), atypických transkriptů ve výchozím stavu (n=7) nebo ukončení před 36 měsíci léčby (n=175).
3 Jako respondéři jsou pro určitý časový bod zahrnuti pouze pacienti, kteří dosáhli MMR v tomto časovém bodě. Celkem u 305 (36,1 %) pacientů nebyla ve 48 měsících MMR vyhodnotitelná (98 ve skupině s nilotinibem 300 mg podávaným dvakrát denně, 88 ve skupině s nilotinibem 400 mg dvakrát denně a 119 ve skupině s imatinibem) z důvodu chybějících/nevyhodnotitelných PCR vyšetření (n=18), atypických transkriptů ve výchozím stavu (n=8), nebo pro ukončení před časovým bodem
48 měsíců (n=279).
4 Jako respondéři jsou pro určitý časový bod zahrnuti pouze pacienti, kteří dosáhli MMR v tomto časovém bodě. Celkem u 332 (38,1 %) pacientů nebyla v 60 měsících MMR vyhodnotitelná (99 ve skupině s nilotinibem 300 mg podávaným dvakrát denně, 93 ve skupině s nilotinibem 400 mg dvakrát denně a 130 ve skupině s imatinibem) z důvodu chybějících/nevyhodnotitelných PCR vyšetření (n=9), atypických transkriptů ve výchozím stavu (n=8), nebo pro ukončení před časovým bodem 60 měsíců (n=305).
5Jako respondéři jsou pro určitý časový bod zahrnuti pouze pacienti, kteří dosáhli MMR v tomto časovém bodě. Celkem u 395 (46,7 %) pacientů nebyla v 72 měsících MMR vyhodnotitelná (130 ve skupině s nilotinibem 300 mg podávaným dvakrát denně, 110 ve skupině s nilotinibem 400 mg dvakrát denně a 155 ve skupině s imatinibem) z důvodu chybějících/nevyhodnotitelných PCR vyšetření (n=25), atypických transkriptů ve výchozím stavu (n=8), nebo pro ukončení před časovým bodem 72 měsíců (n=362).
Kumulativní výskyt MMR v různých časových bodech (mezi pacienty s odpovědí na léčbu jsou zahrnuti pacienti, kteří dosáhli MMR v časových bodech nebo před uplynutím těchto časových bodů) (viz Obrázek 1).
Obrázek 1 Kumulativní výskyt MMR
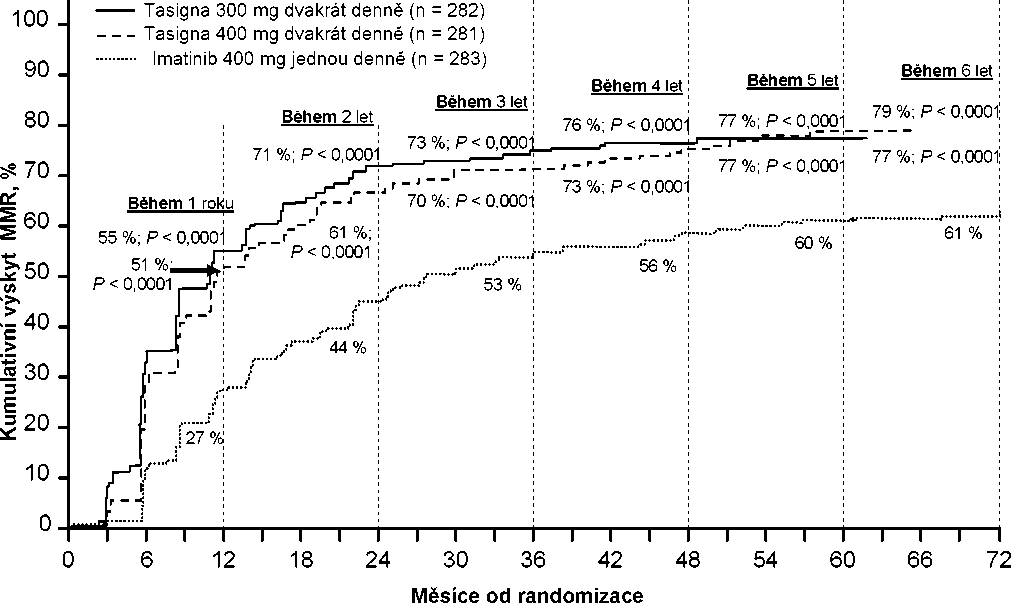
Pro všechny rizikové skupiny dle Sokalova skóre zůstal ve všech časových bodech výskyt MMR konzistentně vyšší v obou skupinách s nilotinibem oproti skupině s imatinibem.
V retrospektivních analýzách dosáhlo 91 % (234/258) pacientů na nilotinibu 300 mg dvakrát denně hodnot BCR-ABL <10 % ve 3 měsících léčby oproti 67 % (176/264) pacientů na imatinibu 400 mg jednou denně. Pacienti s hodnotami BCR-ABL <10 % ve 3 měsících léčby vykazovali vyšší poměr celkového přežití v 72 měsících v porovnání s pacienty kteří nedosáhli této hodnoty molekulární odpovědi (94,5 % oproti 77,1 % [p=0,0005]).
Na základě Kaplan-Meierovy analýzy doby do první MMR byla pravděpodobnost dosažení MMR v různých časových úsecích vyšší pro nilotinib v dávkách 300 mg a 400 mg dvakrát denně v porovnání s imatinibem 400 mg jednou denně (HR=2,17 a stratifikovaná hodnota log-rank p<0,0001 mezi nilotinibem 300 mg dvakrát denně a imatinibem 400 mg jednou denně, HR=1,88 a stratifikovaná hodnota log-rank p<0,0001 mezi nilotinibem 400 mg dvakrát denně a imatinibem 400 mg jednou denně).
Podíly pacientů, kteří dosáhli molekulárních odpovědí <0,01 % a <0,0032 % podle IS v různých časových bodech, jsou uvedené v Tabulce 6 a poměry pacientů, kteří dosáhli molekulárních odpovědí <0,01 % a <0,0032 % podle IS během různých časových období, jsou uvedeny na Obrázcích 2 a 3. Molekulární odpověď <0,01 % a <0,0032 % podle IS odpovídá poklesu hladiny BCR-ABL transkriptů o >4 log, respektive pokles o >4,5 log od standardizovaného výchozího stavu.
Tabulka 6 Podíl pacientů, kteří dosáhli molekulární odpovědi <0,01% (pokles o 4 log) a <0,0032% (pokles o 4,5 log)
|
Ta 300 mg d n= |
signa vakrát denně 282 To) |
Ta 400 mg d n= |
signa vakrát denně =281 To) |
Im 400 mg j( n= |
atinib sdnou denně =283 í%) | |
|
<0,01 % |
<0,0032 % |
<0,01 % |
< 0,0032 % |
<0,01 % |
<0,0032 % | |
|
Ve 12 měsících |
11,7 |
4,3 |
8,5 |
4,6 |
3,9 |
0,4 |
|
Ve 24 měsících |
24,5 |
12,4 |
22,1 |
7,8 |
10,2 |
2,8 |
|
Ve 36 měsících |
29,4 |
13,8 |
23,8 |
12,1 |
14,1 |
8,1 |
|
Ve 48 měsících |
33,0 |
16,3 |
29,9 |
17,1 |
19,8 |
10,2 |
|
V 60 měsících |
47,9 |
32,3 |
43,4 |
29,5 |
31,1 |
19,8 |
|
V 72 měsících |
44,3 |
31,2 |
45,2 |
28,8 |
27,2 |
18,0 |
Obrázek 2 Kumulativní výskyt molekulární odpovědi <0,01% (pokles o 4-log)
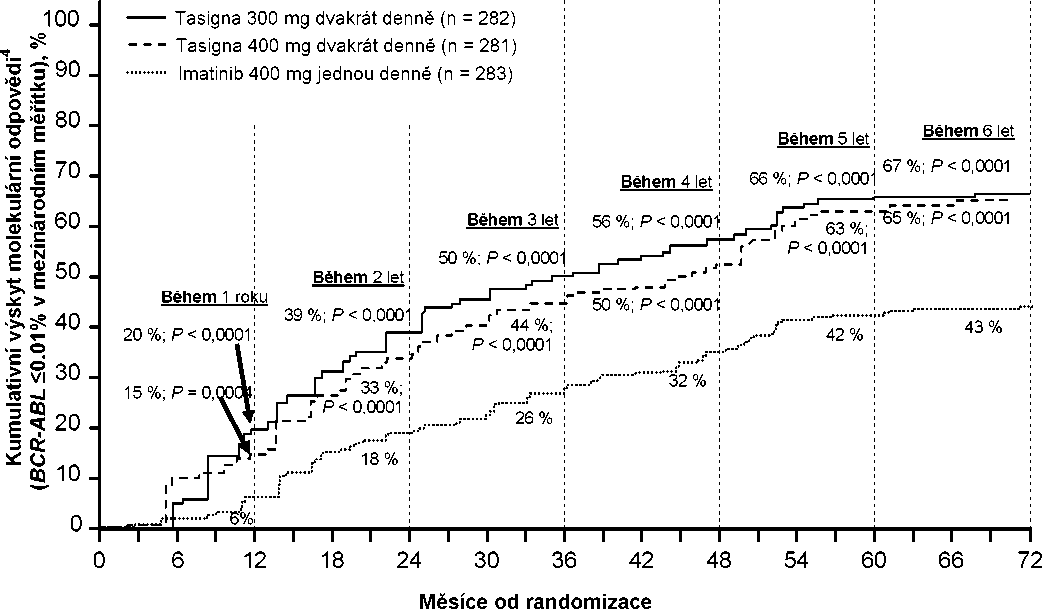
100 _
- Tasigna 300 mg dvakrát denně (n = 282)
---Tasigna 400 mg dvakrát denně (n = 281)
.......... Imatinib 400 mg jednou denně (n = 283)
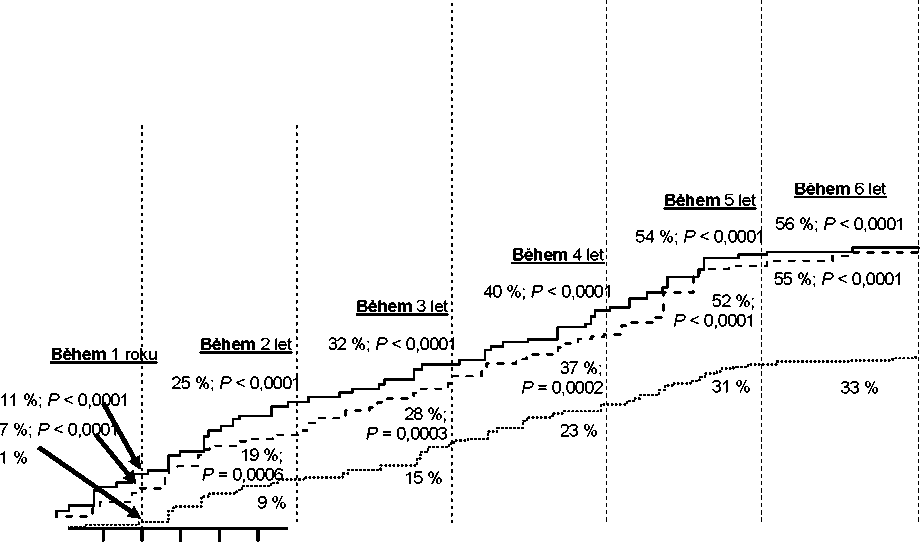
T 1
0 6
7
12
18
T-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1
24 30 36 42 48 54 60 66 72
Měsíce od randomizace
Na základě Kaplan-Meierových analýz trvání první MMR, byl podíl pacientů, kteří udržovali odpověď po dobu 72 měsíců mezi pacienty, kteří dosáhli MMR, 92,5 % (95% CI: 88,6-96,4 %) ve skupině s nilotinibem 300 mg dvakrát denně, 92,2 % (95% CI: 88,5-95,9 %) ve skupině s nilotinibem 400 mg dvakrát denně a 88,0 % (95% CI: 83,0-93,1 %) ve skupině s imatinibem 400 mg jednou denně.
Kompletní cytogenetická odpověď (CCyR) byla definována jako 0 % Ph+ metafáze v kostní dřeni na základě minimálně 20 vyhodnocených metafází. Nejlepší dosažený výskyt CCyR ve 12 měsících (zahrnuje na léčbu odpovídající pacienty, kteří dosáhli CCyR během 12 měsíců) byl statisticky vyšší pro nilotinib 300 mg a 400 mg dvakrát denně oproti imatinibu 400 mg jednou denně, viz Tabulka 7.
Podíl dosažení CCyR během 24 měsíců (zahrnuje pacienty, kteří dosáhli CCyR ve 24 měsících nebo dříve) byl statisticky významně vyšší pro obě skupiny s nilotinibem 300 mg dvakrát denně a 400 mg dvakrát denně v porovnání se skupinou s imatinibem 400 mg jednou denně.
Tabulka 7 Nejlepší celkový výskyt cytogenetické odpovědi (CCyR)
|
Tasigna (nilotinib) 300 mg dvakrát denně n=282 (%) |
Tasigna (nilotinib) 400 mg dvakrát denně n=281 (%) |
Glivec (imatinib) 400 mg jednou denně n=283 (%) | |
|
Během 12 měsíců | |||
|
Odpověď (95 % CI) |
80,1 (75,0; 84,6) |
77,9 (72,6; 82,6) |
65,0 (59,2; 70,6) |
|
Žádná odpověď |
19,9 |
22,1 |
35,0 |
|
CMH test p-hodnoty pro výskyt odpovědi (vs. imatinib 400 mg jednou denně) |
<0,0001 |
0,0005 | |
|
Během 24 měsíců | |||
|
Odpověď (95% CI) |
86,9 (82,4; 90,6) |
84,7 (79,9; 88,7) |
77,0 (71,7; 81,8) |
|
Žádná odpověď |
13,1 |
15,3 |
23,0 |
|
CMH test p-hodnoty pro výskyt odpovědi (vs. imatinib 400 mg jednou denně) |
0,0018 |
0,0160 |
Na základě Kaplan-Meierových analýz byl podíl pacientů, kteří udržovali odpověď po dobu 72 měsíců mezi pacienty, kteří dosáhli CCyR, 99,1 % (95% CI: 97,9-100 %) ve skupině s nilotinibem 300 mg dvakrát denně, 98,7 % (95% CI: 97,1-100 %) ve skupině s nilotinibem 400 mg dvakrát denně a 97,0 % (95% CI: 94,7-99,4 %) ve skupině s imatinibem 400 mg jednou denně.
Progrese do akcelerované fáze (AP) nebo blastické krize (BC) během léčby je definovaná jako doba od data randomizace do první dokumentované progrese onemocnění do akcelerované fáze nebo blastické krize nebo úmrtí v důsledku CML. Progrese do akcelerované fáze nebo blastické krize v průběhu léčby byla zjištěna celkem u 17 pacientů: u 2 pacientů na nilotinibu 300 mg dvakrát denně, u 3 pacientů na nilotinibu 400 mg dvakrát denně a u 12 pacientů na imatinibu 400 mg jednou denně. Odhadované podíly pacientů bez progrese do akcelerované fáze nebo blastické krize v 72 měsících byly 99,3 %, 98,7 % a 95,2 % (HR=0,1599 a stratifikovaný log-rank p=0,0059 mezi nilotinibem 300 mg dvakrát denně a imatinibem jednou denně, HR=0,2457 a stratifikovaný log-rank p=0,0185 mezi nilotinibem 400 mg dvakrát denně a imatinibem jednou denně). Do 2-letých analýz nebyly při léčbě hlášené nové případy progrese do AP/BC.
Při zahrnutí klonální evoluce jako kriteria progrese progredovalo do akcelerované fáze nebo blastické krize během léčby v době hodnocení celkem 25 pacientů (3 ve skupině s nilotinibem 300 mg dvakrát denně, 5 ve skupině s nilotinibem 400 mg dvakrát denně a 17 ve skupině s imatinibem 400 mg jednou denně). Odhadované podíly pacientů bez progrese do akcelerované fáze nebo blastické krize zahrnující klonální evoluci v 72 měsících byly 98,7 %, 97,9 % a 93,2 % (HR=0,1626 a stratifikovaný log-rank p=0,0009 mezi nilotinibem 300 mg dvakrát denně a imatinibem jednou denně, HR = 0,2848 a stratifikovaný log-rank p=0,0085 mezi nilotinibem 400 mg dvakrát denně a imatinibem jednou denně).
Během léčby nebo během doby sledování po ukončení léčby zemřelo celkem 55 pacientů (21 ve skupině s nilotinibem 300 mg dvakrát denně, 11 ve skupině s nilotinibem 400 mg dvakrát denně a 23 ve skupině s imatinibem 400 mg jednou denně). Dvacet šest (26) z těchto 55 úmrtí souviselo s CML (6 ve skupině s nilotinibem 300 mg dvakrát denně, 4 ve skupině s nilotinibem 400 mg dvakrát denně a 16 ve skupině s imatinibem 400 mg jednou denně). Odhadovaná procenta přeživších pacientů v 72 měsících byly 91,6 %, 95,8 % a 91,4 % (HR=0,8934 a stratifikovaný log-rank p=0,7085 mezi nilotinibem 300 mg dvakrát denně a imatinibem, HR=0,4632 a stratifikovaný log-rank p=0,0314 mezi nilotinibem 400 mg dvakrát denně a imatinibem). Vezmou-li se v úvahu pouze úmrtí související s CML, odhadovaná procenta celkového přežití v 72 měsících byla 97,7 %, 98,5 % a 93,9 % (HR=0,3694 a stratifikovaný log-rank p=0,0302 mezi nilotinibem 300 mg dvakrát denně a imatinibem, HR=0,2433 a stratifikovaný log-rank p=0,0061 mezi nilotinibem 400 mg dvakrát denně a imatinibem).
Klinické studie u imatinib-rezistentní nebo intolerantní CML v chronické fázi a akcelerované fázi Otevřená, nekontrolovaná, multicentrická studie fáze II byla provedena s cílem stanovit účinnost přípravku Tasigna u pacientů s CML rezistentních nebo netolerujících imatinib, s odděleně léčenými větvemi pro chronické a akcelerované fáze onemocnění. Účinnost byla stanovena na 321 pacientech v chronické fázi (CP) a 137 pacientech v akcelerované fázi (AP) zařazených do studie. Medián trvání léčby byl 561 dnů u pacientů v CP a 264 dnů u pacientů v AP (viz Tabulka 8). Přípravek Tasigna byl podáván kontinuálně (dvakrát denně 2 hodiny po jídle a žádné jídlo nebylo podáváno nejméně jednu hodinu po aplikaci přípravku), dokud nebyla prokázána inadekvátní odpověď nebo progrese onemocnění. Byla povolena dávka 400 mg dvakrát denně a navýšení dávky na 600 mg dvakrát denně bylo povoleno.
Tabulka 8 Trvání expozice přípravku Tasigna
|
Chronická fáze |
Akcelerovaná fáze | |
|
n=321 |
n=137 | |
|
Medián trvání léčby ve dnech |
561 |
264 |
|
(25.-75. percentil) |
(196-852) |
(115-595) |
Rezistence na imatinib zahrnovala nedosažení kompletní hematologické odpovědi (do 3 měsíců), cytogenetické odpovědi (do 6 měsíců) nebo velké cytogenetické odpovědi (do 12 měsíců) nebo progresi onemocnění po předchozí cytogenetické nebo hematologické odpovědi. Intolerance imatinibu zahrnovala pacienty, kteří přerušili léčbu imatinibem z důvodu toxicity a nedosáhli velké cytogenetické odpovědi v době vstupu do studie.
Celkem bylo 73 % pacientů rezistentních na imatinib, zatímco 27 % pacientů imatinib nesnášelo. Většina pacientů měla dlouhodobou anamnézu CML, včetně extenzivní předchozí léčby jinými antineoplastickými látkami, zahrnujícími imatinib, hydroxyureu a interferon, a rovněž byli zařazeni nemocní, u kterých selhala orgánová transplantace (Tabulka 9). Medián nejvyšší předchozí dávky imatinibu byl 600 mg/den. Nejvyšší předchozí dávka imatinibu byla >600 mg/den u 74 % všech pacientů a 40 % pacientů dostávalo dávky imatinibu >800 mg/den.
Tabulka 9 Charakteristiky průběhu CML u zařazených pacientů
|
Chronická fáze |
Akcelerovaná fáze | |
|
(n=321) |
(n=137)* | |
|
Medián doby od diagnózy v měsících |
58 |
71 |
|
(rozmezí) |
(5-275) |
(2-298) |
|
Imatinib | ||
|
Rezistentní |
226(70 %) |
109 (80 %) |
|
Netolerující bez MCyR |
95 (30 %) |
27 (20 %) |
|
Medián doby léčby imatinibem ve dnech |
975 |
857 |
|
(25.-75. percentil) |
(519-1 488) |
(424-1 497) |
|
Předchozí hydroxyurea |
83 % |
91 % |
|
Předchozí interferon |
58 % |
50 % |
|
Předchozí transplantace kostní dřeně |
7 % |
8 % |
* Chybějící informace o stavu rezistence/intolerance u jednoho pacienta.
Primárním cílovým parametrem účinnosti u pacientů v chronické fázi (CP) byla velká cytogenetická odpověď (MCyR) definovaná jako eliminace (CCyR, kompletní cytogenetická odpověď) nebo signifikantní snížení na <35% Ph+ metafází (částečná cytogenetická odpověď) Ph+ hematopoetických buněk. Kompletní hematologická odpověď (CHR) byla u pacientů v CP hodnocena jako sekundární cíl. Primárním cílovým parametrem účinnosti u pacientů v akcelerované fázi (AP) byla úplná, potvrzená hematologická odpověď (HR), definovaná buď jako kompletní hematologická odpověď, žádný průkaz leukémie, nebo návrat do chronické fáze onemocnění.
Chronická fáze
Výskyt MCyR byl u 321 pacientů v CP 51 %. Většina respondentů dosáhla své MCyR rychle, během 3 měsíců (medián 2,8 měsíce) od zahájení léčby přípravkem Tasigna a tito pacienti byli stabilizováni. Medián doby do dosažení CCyR lehce přesáhl 3 měsíce (medián 3,4 měsíce). Ve 24. měsíci stále vykazovalo MCyR 77 % (95 %CI: 70 % - 84 %) pacientů, kteří této odpovědi dosáhli. Medián trvání MCyR nebyl dosažen. Ve 24. měsíci stále vykazovalo CCyR 85 % (95 %CI: 78 % - 93 %) pacientů, kteří této odpovědi dříve dosáhli. Medián trvání CCyR nebyl dosažen. Pacienti s výchozí CHR dosáhli MCyR rychleji (1,9 vs 2,8 měsíce). Z pacientů v CP bez výchozí CHR dosáhlo CHR 70 %, medián doby dosažení CHR byl 1 měsíc a medián trvání CHR byl 32,8 měsíce. Odhadované procento 24měsíčního celkového přežití pacientů s CML-CP bylo 87 %.
Akcelerovaná fáze
Celkový potvrzený výskyt HR u 137 pacientů v AP byl 50 %. Většina respondentů dosáhla HR při léčbě přípravkem Tasigna časně (medián 1,0 měsíc) a tato byla trvalá (medián trvání potvrzené HR byl 24,2 měsíce). Ve 24. měsíci stále vykazovalo HR 53 % (95 %CI: 39 % - 67 %) pacientů, kteří této odpovědi dříve dosáhli.Výskyt MCyR byl 30 % s mediánem doby do odpovědi 2,8 měsíce. Ve 24. měsíci stále vykazovalo MCyR 63 % (95 %CI: 45 % - 80 %) pacientů, kteří této odpovědi dříve dosáhli. Medián trvání MCyR byl 32,7 měsíce. Odhadované procento 24-měsíčního celkového přežití pacientů s CML-AP bylo70 %.
Výskyty odpovědí v obou větvích léčby jsou uvedeny v Tabulce 10.
Tabulka 10 Odpovědi u CML
|
(Výskyt nejlepší odpovědi) |
Chronická fáze |
Akcelerovaná fáze | ||||
|
Netolerují cí (n=95)_ |
Rezistentn í (n=226) |
Celkem (n=321) |
Netolerují cí (n=27)_ |
Rezistentn í (n=109) |
Celkem* (n=137) | |
|
Hematologická odpověď (%) | ||||||
|
Souhrnně (95%CI) Kompletní NEL Návrat do CP |
87 (74-94) |
65 (56-72) |
701 (63-76) |
48 (29-68) 37 7 4 |
51(42-61) 28 10 13 |
50 (42-59) 30 9 11 |
|
Cytogenetická odpověď (%) | ||||||
|
Velká (95%CI) Kompletní Částečná |
57 (46-67) 41 16 |
49 (42-56) 35 14 |
51 (46-57) 37 15 |
33 (17-54) 22 11 |
29 (21-39) 19 10 |
30 (22-38) 20 10 |
|
NEL = žádný průkaz leukemie/odpověď kostní c |
řeně | |||||
I 114 CP pacientů mělo výchozí CHR a nebyli proto hodnotitelní z hlediska kompletní hematologické odpovědi
* Chybějící informace o stavu rezistence/intolerance u jednoho pacienta.
Data o účinnosti u pacientů s CML v blastické krizi nejsou dosud k dispozici. Do studie fáze II byly také zahrnuty samostatné léčebné větve za účelem hodnotit Tasigna ve skupině pacientů v CP a AP, kteří byli extenzivně předtím léčeni četnými druhy léčby zahrnujícími vedle imatinibu jiný inhibitor tyrosinkinázy. Z těchto pacientů bylo 30 ze 36 (83%) rezistentních, ne intolerantních. U 22 pacientů v CP hodnocených pro účinnost vedla léčba přípravkem Tasigna u 32% k MCyR a u 50% ke CHR. U
II pacientů v AP, hodnocených z hlediska účinnosti, vedla léčba k HR celkově u 36% nemocných.
Po selhání léčby imatinibem bylo zaznamenáno 24 různých BCR-ABL mutací u 42 % pacientů v chronické fázi a 54 % pacientů v akcelerované fázi CML, u kterých byl hodnocen výskyt mutací. Přípravek Tasigna prokázal účinnost u pacientů majících různé BCR-ABL mutace související s rezistencí na imatinib, s výjimkou T315I.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Tasigna u pediatrických pacientů od narození do 18 let při léčbě chronické myeloidní leukémie (CML) s přítomností filadelfského chromozomu (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Vrcholové koncentrace nilotinibu je dosaženo za 3 hodiny po perorálním podání. Absorpce nilotinibu po perorálním podání byla přibližně 30 %. Absolutní biologická dostupnost nilotinibu nebyla stanovena. V porovnání s perorálním roztokem (pH 1,2 až 1,3) je relativní biologická dostupnost tobolky s nilotinibem přibližně 50 %. Pokud byl zdravým dobrovolníkům podán přípravek Tasigna s jídlem, zvýšila se Cmax nilotinibu o 112 % a plocha pod křivkou koncentrace v séru (AUC) o 82 %, ve srovnání s podáním přípravku Tasigna nalačno. Po podání přípravku Tasigna 30 minut nebo 2 hodiny po jídle stoupla biologická dostupnost nilotinibu o 29 %, respektive o 15 % (viz body 4.2, 4.4 a 4.5).
Absorpce nilotinibu (relativní biologická dostupnost) může být snížena přibližně o 48 % u pacientů s totální gastrektomií a přibližně o 22 % u pacientů s částečnou gastrektomií.
Distribuce
Poměr krev/plazma nilotinibu je 0,71. Na základě in vitro experimentů je vazba na plazmatické proteiny přibližně 98 %.
Biotransformace
Hlavní metabolické cesty, zjištěné u zdravých dobrovolníků, jsou oxidace a hydroxylace. Nilotinib je hlavní cirkulující složkou v séru. Žádný z metabolitů nepřispívá významným způsobem k farmakologické aktivitě nilotinibu. Nilotinib je primárně metabolizován CYP3A4 s možným menším přispěním CYP2C8.
Eliminace
Po jednorázové dávce radioaktivně značeného nilotinibu zdravým dobrovolníkům bylo více než 90 % dávky vyloučeno během 7 dnů, a to převážně stolicí (94 % dávky). Nezměněný nilotinib odpovídal 69 % dávky.
Zdánlivý eliminační poločas stanovený z farmakokinetiky po opakovaném denním podávání byl přibližně 17 hodin. Variabilita farmakokinetiky mezi pacienty byla střední až vysoká
Linearita/nelinearita
Expozice nilotinibu v rovnovážném stavu byla závislá na dávce, při dávkách vyšších než 400 mg podávaných jednou denně byla systémová expozice méně úměrná dávce. Denní systémová expozice nilotinibu se 400 mg podávanými dvakrát denně byla v rovnovážném stavu o 35 % vyšší než s dávkou 800 mg podanou jednou denně. Systémová expozice (AUC) nilotinibu v rovnovážném stavu při dávce 400 mg dvakrát denně byla přibližně o 13,4 % vyšší oproti dávce 300 mg dvakrát denně. Průměrné minimální a maximální koncentrace nilotinibu po dobu 12 měsíců byly přibližně o 15,7 % a 14,8 % vyšší při dávkování 400 mg dvakrát denně oproti dávkování 300 mg dvakrát denně. Při zvýšení dávky ze 400 mg dvakrát denně na 600 mg dvakrát denně nebylo zvýšení expozice nilotinibu odpovídající.
Podmínek rovnovážného stavu bylo v zásadě dosaženo za 8 dnů. Zvýšení sérové expozice nilotinibu mezi první dávkou a rovnovážným stavem bylo přibližně 2násobné při dávkování jednou denně a 3,8násobné při dávkování dvakrát denně.
Biodostupnost/bioekvivalenční studie
Bylo prokázáno, že jednorázové podání 400 mg nilotinibu ve 2 tvrdých tobolkách o síle 200 mg, kde obsah každé tvrdé tobolky byl rozmíchaný v jedné lžičce jablečné šťávy, bylo bioekvivalentní jednorázovému podání 2 neporušených tvrdých tobolek o síle 200 mg.
5.3 Předklinické údaje vztahující se k bezpečnosti
Nilotinib byl hodnocen farmakologickými studiemi na bezpečnost, toxicitu po opakovaném podání, genotoxicitu, reprodukční toxicitu, fototoxicitu a kancerogenitu u potkanů a myší.
Nilotinib neměl účinky na CNS nebo respirační funkce. Výsledky in vitro studie srdeční bezpečnosti provedené s nilotinibem na izolovaných králičích srdcích předklinicky naznačovaly možnost prodloužení QT intervalu: byla zřejmá blokáda hERG proudů a prodloužení trvání akčního potenciálu. Žádné účinky nebyly pozorovány při vyšetření EKG u psů nebo opic léčených až 39 týdnů nebo ve speciální telemetrické studii u psů.
Studie toxicity po opakovaném podávání psům až po dobu 4 týdnů a makakům jávským po podávání až 9 týdnů ukázaly, že játra jsou primárním orgánem toxicity nilotinibu. Změny zahrnují zvýšení alaninaminotransferázy a aktivity alkalické fosfatázy a histopatologické nálezy (především hyperplazie/hypertrofie sinusoidálních buněk a Kupfferových buněk, hyperplazie žlučovodů a periportální fibróza). Po čtyřech týdnech rekonvalescence byly obvykle změny klinické biochemie plně reverzibilní a histologické změny částečně reverzibilní. Expozice při nejnižších dávkových hladinách, při kterých byly pozorovány účinky na játra, byly nižší než expozice u lidí při dávce 800 mg/den. U myší nebo potkanů, léčených až 26 týdnů, byly pozorovány malé změny na játrech. U potkanů, psů a opic bylo pozorováno převážně reverzibilní zvýšení hladin cholesterolu.
Studie genotoxicity na bakteriálních systémech in vitro a na savčích modelech in vitro a in vivo s metabolickou aktivací a bez ní nepřinesly žádný důkaz mutagenního potenciálu nilotinibu.
Ve 2leté studii kancerogenity u potkanů byla hlavním orgánem, kde vznikaly jiné než neoplastické léze, děloha (dilatace, vaskulární ektázie, hyperplázie endoteliálních buněk, zánět a/nebo epiteliální hyperplázie). Při podání nilotinibu v dávkách 5, 15 a 40 mg/kg/den nebyla kancerogenita prokázána. Expozice (vyjádřená AUC) při nejvyšší dávce představovaly přibližně 2 až 3násobek denní expozice nilotinibu v ustáleném stavu u lidí (na základě AUC) při dávce 800 mg/den.
Ve studii kancerogenity u myší (Tg.rasH2) trvající 26 týdnů, při které byl nilotinib podáván v dávkách 30, 100 a 300 mg/kg/den, byly pozorovány kožní papilomy/karcinomy při dávce 300 mg/kg, což představuje přibližně 30 až 40 násobek expozice u člověka (na základě AUC) při maximální schválené dávce 800 mg/den (při dávkování 400 mg 2x denně). Hladina, při které nebyl pozorován žádný účinek (No-Observed-Effect-Levels) na kožní neoplastické léze byla 100 mg/kg/den, což představuje přibližně 10 až 20 násobek expozice u člověka při maximální schválené dávce 800 mg/den (při dávkování 400 mg 2x denně). Hlavními cílovými orgány non-neoplastických lézí byly: kůže (epidermální hyperplasie), zuby (degenerace/atrofie skloviny horních řezáků a zánět dásní/odontogenního epitelu řezáků) a brzlík (zvýšený výskyt a/nebo závažnost úbytku lymfocytů).
Nilotinib neindukoval teratogenitu, ale v dávkách toxických pro matku byl embryo a fetotoxický. Zvýšení postimplantační ztráty bylo pozorováno jak ve studii fertility, která zahrnovala léčbu samců i samic, tak ve studii embryotoxicity, která zahrnovala léčbu samic. Úmrtnost embryí a účinky na plod (především snížení hmotnosti plodu, předčasný srůst obličejových kostí (fúze maxily a jařmové kosti), viscerální a skeletální změny) u potkanů a zvýšení resorpce plodů a změny na skeletu u králíků byly přítomny ve studiích embryotoxicity. V pre- a postnatální výzkumné studii u potkanů expozice nilotinibu u matek způsobila snížení tělesné hmotnosti u mláďat spojené se změnami fyzických vývojových parametrů stejně jako snížení ukazatelů páření a fertility u potomků. Expozice nilotinibu u samic, při hladině bez nežádoucích účinků (No-Observed-Adverse-Effect-Levels), byla obvykle menší nebo stejná jako u lidí při dávce 800 mg/den.
Ve studii zaměřené na sledování vývoje v juvenilním období byl podáván nilotinib perorální sondou juvenilním potkanům od prvního týdne po narození do časné dospělosti (den 70 po narození) v dávkách 2, 6 a 20 mg/kg/den. Vedle standardních parametů studie bylo provedeno hodnocení vývojových mezníků, vlivu na CNS, páření a fertilitu. Na základě snížení tělesné hmotnosti obou pohlaví a zpoždění prepuciální separace u samců (které může být spojeno se snížením tělesné hmotnosti) byla stanovena výše dávky bez pozorovatelného efektu u juvenilních potkanů na 6 mg/kg/den. Juvenilní zvířata nevykazovala zvýšenou citlivost na nilotinib při porovnání s dospělými. Navíc byl profil toxicity u juvenilních potkanů srovnatelný s profilem toxicity zjištěným u dospělých potkanů.
Až do nejvyšší testované dávky, přibližně 5násobku doporučené dávky pro člověka, nebyly u samců a samic potkanů zaznamenány účinky na počet/motilitu spermií ani na fertilitu.
Bylo zjištěno, že nilotinib, který absorbuje světlo v pásmu UV-B a UV-A, je distribuován do kůže a má fototoxický potenciál in vitro, ale tento účinek nebyl pozorován in vivo. Proto se riziko, že nilotinib bude příčinou fotosenzitivity u pacientů, považuje ze velmi nízké.
FARMACEUTICKÉ ÚDAJE
6.
6.1 Seznam pomocných látek
Obsah tvrdé tobolky Monohydrát laktosy Krospovidon Poloxamer 188
oxid křemičitý, koloidní bezvodý Magnesium-stearát
Obal tvrdé tobolky Želatina
Oxid titaničitý (E 171)
Žlutý oxid železitý (E 172)
Tiskařský inkoust Šelak (E 904)
Červený oxid železitý (E 172)
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30°C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení PVC/PVDC/Al a PA/Al/PVC/Al blistry.
Přípravek Tasigna je dostupný v následujících velikostech balení:
• Jednotková balení obsahující 28 tvrdých tobolek v pouzdře.
• Jednotková balení obsahující 28 tvrdých tobolek (7 denních blistrů, jeden obsahuje 4 tvrdé
tobolky) nebo 40 tvrdých tobolek (5 blistrů, jeden obsahuje 8 tvrdých tobolek).
• Vícenásobná balení obsahující 112 (4 pouzdra po 28) tvrdých tobolek.
• Vícenásobná balení obsahující 112 (4 balení po 28) tvrdých tobolek, 120 (3 balení po 40)
tvrdých tobolek nebo 392 (14 balení po 28) tvrdých tobolek.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky na likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/07/422/001 -004 EU/1/07/422/007-008 EU/1/07/422/011-012 EU/1/07/422/014
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 19. listopad 2007
Datum posledního prodloužení registrace: 19. listopad 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://ema.europa.eu
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Novartis Pharma GmbH RoonstraBe 25 D-90429 Norimberk Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci (MAH) zajistí před uvedením přípravku do oběhu, že všichni lékaři, u kterých se očekává, že budou tento přípravek předepisovat, a všichni lékárníci, kteří mohou tento přípravek vydávat, obdrží soubor zdravotnických odborných informací, který obsahuje následující:
• Vzdělávací brožuru
• Souhrn údajů o přípravku, příbalovou informaci a označení na obalu
Klíčové informace obsažené ve vzdělávací brožuře
• Stručné informace o přípravku Tasigna, jeho schválených indikacích a dávkování
• Informace o kardiálních rizicích spojených s užíváním přípravku Tasigna
o že Tasigna může způsobit prodloužení QT intervalu a že Tasigna může být užívána s opatrností u pacientů u nichž došlo k prodloužení QT intervalu, nebo u pacientů s významným rizikem prodloužení QT intevalu. Souběžné užívání Tasigny s antiarytmiky nebo jinými léčivými přípravky, které mohou prodloužit QT interval by mělo být prováděno s opatrností.
o s rizikem arytmií, zvláště torsades de pointes, by neměl být přípravek Tasigna předepisován.
o O potřebě vyhnout se současnému předepisování jiných léků, které mohou prodlužovat QT interval
o O pozornosti při předepisování pacientům s anamnézou koronárních srdečních onemocnění nebo rizikovými faktory pro ně
o že Tasigna může způsobit retenci tekutin, srdeční selhání a plicní otok
• Že Tasigna je metabolizována CYP3A4 a že silné inhibitory a induktory tohoto enzymu mohou významně ovlivnit expozici přípravku Tasigna.
o Tyto inhibitory mohou zvýšit potenciál vzniku nežádoucích účinků léku, zvláště prodloužení QT intervalu.
o Varovat pacienty před užíváním volně prodejných léků, zvláště třezalky tečkované.
• O potřebě informovat pacienty o účincích potravy na Tasigna
o nejíst 2 hodiny před a hodinu po užití přípravku Tasigna.
o potřeba vyvarovat se potravin jako grapefruitový džus, který inhibuje CYP3A4 enzymy.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA JEDNOTKOVÉHO BALENÍ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Tasigna 150 mg tvrdé tobolky Nilotinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje 150 mg nilotinibu (jako monohydrát hydrochloridu).
3. SEZNAM POMOCNÝCH LÁTEK
Přípravek obsahuje laktózu, další informace - viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdé tobolky
28 tvrdých tobolek 40 tvrdých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Neuchovávejte při teplotě nad 30°C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/07/422/005 28 tvrdých tobolek
EU/1/07/422/009 40 tvrdých tobolek
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Tasigna 150 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTRY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Tasigna 150 mg tvrdé tobolky Nilotinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
KRABIČKA VÍCEČETNÉHO BALENÍ (VČETNĚ BLUE BOXU)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Tasigna 150 mg tvrdé tobolky Nilotinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje 150 mg nilotinibu (jako monohydrát hydrochloridu).
3. SEZNAM POMOCNÝCH LÁTEK
Přípravek obsahuje laktózu, další informace - viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdé tobolky
Vícečetné balení: 112 (4 balení po 28) tvrdých tobolek. Vícečetné balení: 120 (3 balení po 40) tvrdých tobolek. Vícečetné balení: 392 (14 balení po 28) tvrdých tobolek.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Neuchovávejte při teplotě nad 30°C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/07/422/006
EU/1/07/422/010
EU/1/07/422/013
112 tvrdých tobolek 120 tvrdých tobolek 392 tvrdých tobolek
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Tasigna 150 mg
VNITŘNÍ KRABIČKA VÍCEČETNÉHO BALENÍ (BEZ BLUE BOXU)
1. NÁZEV LÉČIVÉHO PŘÍPRVKU
Tasigna 150 mg tvrdé tobolky Nilotinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje 150 mg nilotinibu (jako monohydrát hydrochloridu).
3. SEZNAM POMOCNÝCH LÁTEK
Přípravek obsahuje laktózu, další informace - viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdé tobolky
28 tvrdých tobolek. Součást vícečetného balení. Neprodávat samostatně 40 tvrdých tobolek. Součást vícečetného balení. Neprodávat samostatně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Neuchovávejte při teplotě nad 30°C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/07/422/006
EU/1/07/422/010
EU/1/07/422/013
112 tvrdých tobolek 120 tvrdých tobolek 392 tvrdých tobolek
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský přepis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Tasigna 150 mg
|
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA, JEDNOTKOVÉ BALENÍ (POUZDRO) | |
|
KRABIČKA, JEDNOTKOVÉ BALENÍ (KRABIČKA) | |
|
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU | |
Tasigna 200 mg tvrdé tobolky Nilotinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje 200 mg nilotinibu (jako monohydrát hydrochloridu).
3. SEZNAM POMOCNÝCH LÁTEK
Přípravek obsahuje laktózu, další informace - viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdé tobolky
28 tvrdých tobolek 40 tvrdých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Neuchovávejte při teplotě nad 30°C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/07/422/001
EU/1/07/422/002
EU/1/07/422/007
EU/1/07/422/011
PVC/PVDC/Al [v pouzdře] 28 tvrdých tobolek PA/Al/PVC/Al [v pouzdře] 28 tvrdých tobolek PVC/PVDC/Al [v krabičce] 28 tvrdých tobolek PVC/PVDC/Al [v krabičce] 40 tvrdých tobolek
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Tasigna 200 mg

Tasigna 200 mg tvrdé tobolky Nilotinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
KRABIČKA VÍCEČETNÉHO BALENÍ (POUZDRO) (VČETNĚ BLUE BOXU) KRABIČKA VÍCEČETNÉHO BALENÍ (KRABIČKA) (VČETNĚ BLUE BOXU)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Tasigna 200 mg tvrdé tobolky Nilotinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje 200 mg nilotinibu (jako monohydrát hydrochloridu).
3. SEZNAM POMOCNÝCH LÁTEK
Přípravek obsahuje laktózu, další informace - viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdé tobolky
Vícečetné balení: 112 (4 pouzdra po 28) tvrdých tobolek. Vícečetné balení: 112 (4 balení po 28) tvrdých tobolek. Vícečetné balení: 120 (3 balení po 40) tvrdých tobolek. Vícečetné balení: 392 (14 balení po 28) tvrdých tobolek.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
Neuchovávejte při teplotě nad 30°C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/07/422/003
EU/1/07/422/004
EU/1/07/422/008
EU/1/07/422/012
EU/1/07/422/014
PVC/PVDC/Al [v pouzdře] 112 tvrdých tobolek PA/Al/PVC/Al [v pouzdře] 112 tvrdých tobolek PVC/PVDC/Al [v krabičce] 112 tvrdých tobolek PVC/PVDC/Al [v krabičce] 120 tvrdých tobolek PVC/PVDC/Al [v krabičce] 392 tvrdých tobolek
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Tasigna 200 mg
VNITŘNÍ POUZDRO VÍCEČETNÉHO BALENÍ (BEZ BLUE BOXU) VNITŘNÍ KRABIČKA VÍCEČETNÉHO BALENÍ (BEZ BLUE BOXU)
1. NÁZEV LÉČIVÉHO PŘÍPRVKU
Tasigna 200 mg tvrdé tobolky Nilotinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje 200 mg nilotinibu (jako monohydrát hydrochloridu).
3. SEZNAM POMOCNÝCH LÁTEK
Přípravek obsahuje laktózu, další informace - viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdé tobolky
28 tvrdých tobolek. Součást vícečetného balení 28 tvrdých tobolek. Součást vícečetného balení 40 tvrdých tobolek. Součást vícečetného balení 28 tvrdých tobolek. Součást vícečetného balení
obsahujícího 4 pouzdra. Neprodávat samostatně. obsahujícího 4 krabičky. Neprodávat samostatně. obsahujícího 3 krabičky. Neprodávat samostatně. obsahujícího 14 krabiček. Neprodávat samostatně.
5. ZPŮSOB A CESTA/CESTY-PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
Neuchovávejte při teplotě nad 30°C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/07/422/003
EU/1/07/422/004
EU/1/07/422/008
EU/1/07/422/012
EU/1/07/422/014
PVC/PVDC/Al [v pouzdře] 112 tvrdých tobolek PA/Al/PVC/Al [v pouzdře] 112 tvrdých tobolek PVC/PVDC/Al [v krabičce] 112 tvrdých tobolek PVC/PVDC/Al [v krabičce] 120 tvrdých tobolek PVC/PVDC/Al [v krabičce] 392 tvrdých tobolek
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský přepis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Tasigna 200 mg
B. PŘÍBALOVÁ INFORMACE
Tasigna 150 mg tvrdé tobolky
Nilotinibum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat,
protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Tasigna a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Tasigna užívat
3. Jak se přípravek Tasigna užívá
4. Možné nežádoucí účinky
5. Jak přípravek Tasigna uchovávat
6. Obsah balení a další informace
1. Co je přípravek Tasigna a k čemu se používá Co je Tasigna
Tasigna je lék, který obsahuje léčivou látku nazvanou nilotinib.
K čemu se Tasigna používá
Tasigna se používá k léčbě leukémie nazývané chronická myeloidní leukémie s přítomností filadelfského chromozomu (Ph-pozitivní CML). CML je nádorové onemocnění krve, kdy tělo tvoří příliš mnoho abnormálních bílých krvinek.
Tasigna se používá u pacientů s nově diagnostikovanou CML.
Jak Tasigna účinkuje
U pacientů s CML spouští změna v DNA (genetickém materiálu) signál, který tělu říká, aby tvořilo abnormální bílé krvinky. Přípravek Tasigna tento signál blokuje, a tak zastavuje produkci těchto buněk.
Sledování Vaší léčby přípravkem Tasigna
Během léčby budete pravidelně vyšetřován/a včetně vyšetření krve. Tyto testy budou sledovat:
- počet krevních elementů ve Vašem těle (bílé krvinky, červené krvinky, krevní destičky), aby se zjistilo, jak je přípravek Tasigna snášen.
- funkci slinivky břišní a jater ve Vašem těle, aby se zjistilo, jak je přípravek Tasigna snášen.
- hladinu iontů ve Vašem těle (draslík, hořčík). Ty jsou velmi důležité pro funkci Vašeho srdce.
- hladinu cukru a tuků ve Vaší krvi.
Rovněž Vám bude kontrolován srdeční rytmus pomocí přístroje, který měří elektrickou aktivitu srdce (zvaný EKG).
Jestliže máte jakékoli dotazy týkající se toho, jak Tasigna působí, nebo proč Vám byl předepsán, zeptejte se svého lékaře.
Dodržujte pečlivě všechna doporučení lékaře. Mohou se lišit od obecných informací uvedených v této
příbalové informaci.
Neužívejte Tasignu
- jestliže j ste alergický/á na nilotinib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Jestliže si myslíte, že byste mohl/a být alergický/á, řekněte to svému lékaři před užíváním přípravku
Tasigna.
Upozornění a opatření
Před užitím přípravku Tasigna se poraďte se svým lékařem nebo lékárníkem:
- pokud u Vás dříve došlo ke kardiovaskulárním příhodám, jako je srdeční infarkt, bolest na hrudi (angina pectoris), potíže s krevním zásobením Vašeho mozku (mozková mrtvice) nebo potíže s přívodem krve do vašich dolních končetin (kulhání či námahová bolest), nebo pokud máte rizikové faktory pro kardiovaskulární onemocnění, jako je vysoký krevní tlak (hypertenze), cukrovka nebo potíže s hladinou tuků ve Vaší krvi (poruchy metabolizmu tuků).
- jestliže trpíte srdeční poruchou, jako je abnormální vedení elektrického signálu nazývaného „prodloužení QT intervalu“.
- jestliže jste léčen/a léky, které ovlivňují tepovou frekvenci srdce (antiarytmika) nebo játra (viz Další léčivé přípravky a Tasigna).
- jestliže trpíte nedostatkem draslíku nebo hořčíku.
- jestliže trpíte onemocněním jater nebo slinivky břišní.
- jestliže trpíte příznaky snadného vzniku modřin, pocitem únavy nebo dýchavičností nebo jste prodělal/a opakované infekce.
- jestliže jste prodělal/a chirurgický zákrok zahrnující odstranění celého žaludku (celková gastrektomie).
- pokud jste někdy měl (a) infekční hepatitidu B (žloutenka typu B) nebo toto onemocnění máte v současné době. Přípravek TASIGNA může hepatitidu B znovu aktivovat, což může v některých případech vést k úmrtí. Před zahájením léčby lékař pacienty pečlivě vyšetří s ohledem na možný výskyt známek této infekce.
Pokud se Vás cokoli z výše uvedeného týká, řekněte to svému lékaři.
Během léčby Tasignou
- pokud omdléváte (ztráta vědomí) nebo máte nepravidelný srdeční tep během léčby Tasignou, řekněte to okamžitě svému lékaři, protože může jít o příznak vážné srdeční choroby. Prodloužení QT intervalu nebo nepravidelný srdeční tep mohou vést k náhlému úmrtí. Méně časté případy náhlého úmrtí byly hlášeny u pacientů, kterým byla podávána Tasigna.
- pokud u Vás došlo k náhlému bušení srdce, závažné svalové slabosti nebo ochrnutí, záchvatům nebo náhlým změnám v myšlení nebo míře bdělosti, řekněte to okamžitě svému lékaři, protože může jít o příznak rychlého ničení nádorových buněk zvaný syndrom nádorového rozpadu. U pacientů léčených přípravkem Tasigna byly zjištěny vzácné případy syndromu nádorového rozpadu.
- pokud se u Vás objevila bolest nebo nepříjemný pocit na hrudi, strnulost nebo slabost, potíže
s chůzí nebo řečí, bolest, bledost nebo pocit chladu v končetinách, okamžitě informujte svého lékaře, protože může jít o příznak kardiovaskulární příhody. Závažné kardiovaskulární příhody zahrnující potíže s přívodem krve do dolních končetin (periferní arteriální okluzivní onemocnění), ischemické srdeční onemocnění a potíže s přívodem krve do mozku (ischemické onemocnění mozku) byly hlášeny u pacientů užívajících přípravek Tasigna. Váš lékař by měl vyhodnotit hladinu tuků (lipidů) a cukru ve Vaší krvi před zahájením léčby a během léčby přípravkem Tasigna.
- pokud u Vás dojde k otokům rukou nebo nohou, celkovým otokům nebo k rychlému nárůstu hmotnosti, sdělte to svému lékaři, protože může jít o příznaky závažného zadržování tekutin. Příznaky závažného zadržování tekutin byly hlášeny u pacientů léčených přípravkem Tasigna méně často.
Další léčivé přípravky a Tasigna
Tasigna může interferovat s některými jinými léky.
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době
užíval(a) nebo které možná budete užívat. Toto se týká především:
- antiarytmik - užívaných k léčbě nepravidelného tlukotu srdce;
- chlorochin, halofantrin, klarithromycin, haloperidol, methadon, moxifloxacin - léčivé přípravky, které mohou mít nechtěný účinek na srdeční činnost;
- ketokonazol, itrakonazol, vorikonazol, klarithromycin, telithromycin - užívané k léčbě infekcí;
- ritonavir - lék ze skupiny „antiproteáz“, užívané k léčbě HIV;
- karbamazepin, fenobarbital, fenytoin - užívané k léčbě epilepsie;
- rifampicin - užívaný k léčbě tuberkulózy;
- třezalka tečkovaná - rostlinný lék, užívaný k léčbě depresí a jiných stavů (také známý jako Hypericum perforatum);
- midazolam - užívaný ke zmírnění úzkosti před chirurgickým zákrokem;
- alfentanil a fentanyl - užívané k léčbě bolesti a jako sedativa před a během chirurgického zákroku nebo léčebných procedur;
- cyklosporin, sirolimus a takrolimus - léky, které potlačují “sebeobrannou” schopnost těla a boj proti infekcím a jsou často užívané k prevenci odmítnutí (rejekce) transplantovaných orgánů, jako jsou játra, srdce a ledviny;
- dihydroergotamin a ergotamin - užívané k léčbě demence;
- lovastatin, simvastatin - užívané k léčbě vysoké hladiny tuků v krvi;
- warfarin - užívaný k léčbě poruchy krevní srážlivosti (např. krevní sraženiny nebo trombózy);
- astemizol, terfenadin, cisaprid, pimozid, chinidin, bepridil nebo námelové alkaloidy (ergotamin, dihydroergotamin).
Tyto léky byste neměl/a užívat během Vaší léčby přípravkem Tasigna. Pokud užíváte kterýkoli z
těchto léků, může Vám lékař předepsat jiný alternativní lék.
Před započetím léčby přípravkem Tasigna informujte svého lékaře nebo lékárníka, pokud užíváte antacida, což jsou léky proti pálení žáhy. Tyto léky musí být užívány odděleně od přípravku Tasigna:
- blokátory H2, které snižují tvorbu kyseliny v žaludku. Blokátory H2 by měly být užívány přibližně 10 hodin před a přibližně 2 hodiny poté, kdy jste užil(a) přípravek Tasigna;
- antacida, např. antacida, která obsahují hydroxid hlinitý, hydroxid hořečnatý a simetikon, které neutralizují vysokou kyselost v žaludku. Tato antacida by měla být užívána přibližně 2 hodiny před nebo přibližně 2 hodiny poté, kdy jste užil(a) přípravek Tasigna.
Jestliže již užíváte Tasigna a bude Vám předepisován k užívání nový (další) lék, měl/a byste lékaře informovat o tom, že jste ještě tento lék společně s přípravkem Tasigna v minulosti neužíval/a.
Přípravek Tasigna s jídlem a pitím
Přípravek Tasigna neužívejte s jídlem. Jídlo může zvýšit absorpci přípravku Tasigna, a tak zvýšit množství přípravku Tasigna v krvi až na škodlivou hladinu. Nepijte grapefruitovou šťávu ani nejezte grapefruit. To může zvýšit množství přípravku Tasigna v krvi až ke škodlivé hladině.
Starší lidé (věk 65 let a více)
Tasigna mohou užívat lidé ve věku 65 let a starší ve stejných dávkách jako ostatní dospělí jedinci. Těhotenství, kojení a plodnost
- Tasigna se nedoporučuje podávat během těhotenství, pokud to není nezbytně nutné. Pokud jste těhotná, nebo si myslíte, že můžete být těhotná, řekněte to svému lékaři, který s Vámi prodiskutuje, zda můžete Tasigna užívat během těhotenství.
- Ženám, které mohou otěhotnět, se doporučuje užívat během léčby a dva týdny po ukončení léčby velmi účinné prostředky proti možnosti početí.
- Během léčby přípravkem Tasigna se kojení nedoporučuje. Pokud kojíte, řekněte to svému lékaři.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Pokud se u Vás objeví po užití přípravku Tasigna nežádoucí účinky (např. závratě nebo poruchy zraku), které by u Vás mohly ovlivnit schopnost bezpečně řídit nebo používat nástroje nebo obsluhovat stroje, měl/a byste se těchto činností vyvarovat, dokud tyto účinky nevymizí.
Tasigna obsahuje laktózu
Tento přípravek obsahuje laktózu (také známou jako mléčný cukr). Pokud Vám Váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
3. Jak se přípravek Tasigna užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Jaké množství přípravku Tasigna se užívá
- Doporučená dávka je 600 mg denně. Této dávky je dosaženo užitím dvou 150 mg tvrdých tobolek dvakrát denně.
Lékař Vám může předepsat nižší dávku v závislosti na Vaši odpověď na léčbu.
Kdy užívat přípravek Tasigna
Tvrdé tobolky užívejte:
- dvakrát denně (přibližně každých 12 hodin);
- nej méně 2 hodiny po j akémkoli jídle;
- potom počkejte 1 hodinu, než budete znovu jíst.
Pokud máte dotazy, kdy Tasigna užívat, zeptejte se svého lékaře nebo lékárníka. Užívání přípravku Tasigna každý den ve stejnou dobu Vám pomůže si zapamatovat, kdy máte tvrdé tobolky užít.
Jak se Tasigna užívá
- Tvrdé tobolky polykejte celé a zapíjejte je vodou.
- Tvrdé tobolky neužívejte dohromady s žádnou potravou.
- Tvrdé tobolky otevřete pouze v případě, kdy je nemůžete spolknout. V tomto případě můžete rozmíchat obsah každé tvrdé tobolky v jedné čajové lžičce jablečné šťávy a okamžitě užít. Neužívejte více než jednu čajovou lžičku s každou tvrdou tobolkou a nepoužívejte jinou potravu než jablečnou šťávu.
Jak dlouho se Tasigna užívá
Pokračujte v užívání přípravku Tasigna tak dlouho, jak Vám řekl lékař. Toto je dlouhodobá léčba. Váš lékař bude pravidelně sledovat Váš zdravotní stav, aby zjistil, zda má léčba požadovaný účinek.
Jestliže máte dotazy, jak dlouho bude užívání přípravku Tasigna trvat, zeptejte se svého lékaře.
Jestliže jste užil(a) více přípravku Tasigna, než jste měl(a)
Jestliže jste užil(a) více přípravku Tasigna, než jste měl(a), nebo pokud někdo jiný náhodně užil Vaše tvrdé tobolky, okamžitě vyhledejte lékaře nebo nemocniční zařízení, abyste se mohl(a) o této věci poradit. Ukažte jim balení tvrdých tobolek a tuto příbalovou informaci. Může být nutné lékařské ošetření.
Jestliže jste zapomněl(a) užít přípravek Tasigna
Pokud jste vynechal(a) dávku, vezměte si následující dávku v obvyklou dobu. Nezdvoj násobujte následující dávku, abyste nahradil(a) vynechanou tvrdou tobolku.
Jestliže jste přestal(a) užívat přípravek Tasigna
Nepřestávejte užívat Tasignu, dokud Vám to Váš lékař neřekne. Při ukončení léčby přípravkem Tasigna bez doporučení lékaře riskujete zhoršení svého onemocnění, což může mít život ohrožující následky. Pokud zvažujete ukončení léčby Tasignou, poraďte se se svým lékařem, zdravotní sestrou a/nebo lékárníkem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého. Většina nežádoucích účinků jsou mírné až středně závažné a obvykle vymizí za
několik dnů až týdnů léčby.
Některé nežádoucí účinky by mohly být závažné.
Nežádoucí účinky jsou časté (mohou postihnout až 1 z 10 lidí), méně časté (mohou postihnout až 1 ze
100 lidí) nebo byly hlášeny jen u velmi malého počtu pacientů.
- rychlé přibývání tělesné hmotnosti, otoky rukou, kotníků, nohou nebo obličeje (příznaky zadržování vody)
- bolest na hrudi, vysoký krevní tlak, nepravidelný tlukot srdce, modré zabarvení rtů, jazyka nebo kůže (příznaky srdeční poruchy)
- potíže při dýchání, kašel, sípání s horečkou nebo bez horečky, otoky nohou (příznaky onemocnění plic)
- horečka, snadný vznik modřin, časté infekce (příznaky poruchy krve)
- neostré vidění, ztráta zraku, krvácení v oku (příznaky oční poruchy)
- otoky a bolest v j edné části těla (příznaky ucpání žíly sraženinou)
- bolest břicha, nevolnost, zácpa, zduření břicha (příznaky poruchy zažívacího systému)
- silná bolest v nadbřišku (příznak zánětu slinivky břišní)
- zežloutnutí kůže a očí, nevolnost, ztráta chuti k jídlu, tmavě zbarvená moč (příznaky poruchy jater)
- vyrážka, bolestivé červené opuchliny, bolesti kloubů a svalů (příznaky poruchy kůže)
- nadměrná žízeň, velké množství moči, zvýšená chuť k jídlu s úbytkem tělesné hmotnosti, únava (příznaky vysoké hladiny krevního cukru)
- nevolnost, zkrácený dech, nepravidelný srdeční tep, zakalená moč, únava a/nebo kloubní potíže spojené s neobvyklými výsledky krevních testů (např. vysoká hladina draslíku, kyseliny močové a fosforu a nízké hladiny vápníku)
- bolest, nepohodlí, ochablost nebo křeče svalů nohou, které mohou být způsobeny sníženým průtokem krve, vředy na nohou nebo rukách, které se hojí pomalu nebo nehojí vůbec a výrazná změna barvy (zmodrání nebo zblednutí) nebo teploty (chlad) nohou a rukou, protože tyto příznaky mohou být příznaky překážky v tepnách v postižených končetinách (noha nebo ruka) a prstech (na nohou a rukou)
- recidiva (reaktivace) hepatitidy B, pokud j ste v minulosti měl(a) toto onemocnění (infekce jater).
Jestliže se u Vás cokoli z tohoto objeví, okamžitě to řekněte svému lékaři.
Některé nežádoucí účinky jsou velmi časté (mohou postihnout více než 1 z 10 lidí)
- bolest hlavy
- únava
- bolest svalů
- svědění, vyrážka, kopřivka
- nevolnost
- padání vlasů
- vysoká hladina bilirubinu v krvi (funkce jater)
- vysoká hladina lipázy v krvi (funkce slinivky břišní)
Pokud Vás cokoli z tohoto postihne v závažné míře, řekněte to svému lékaři.
Některé nežádoucí účinky jsou časté (mohou postihnout až 1 z 10 lidí)
- průjem, zvracení, břišní potíže, žaludeční nevolnost po jídle, plynatost, otok nebo nadýmání břicha
- bolest kostí, bolest kloubů, křeče svalů, bolest končetin, bolest zad, bolest nebo diskomfort v bocích
- podráždění očí, otoky, výtok, svědění nebo zarudnutí, suchost očí (příznaky onemocnění očí)
- zčervenání kůže, suchá kůže, akne, bradavice, snížená citlivost kůže
- ztráta chuti k jídlu, porušené vnímání chuti, zvýšení tělesné hmotnosti
-- nespavost, úzkost
- noční poty, nadměrné pocení, návaly horka
- závratě, pocit točení
- palpitace (pocit rychlého bušení srdce)
Pokud Vás cokoli z tohoto postihne v závažné míře, řekněte to svému lékaři.
Některé nežádoucí účinky jsou méně časté (mohou postihnout až 1 ze 100 lidí)
- bolest kůže
- otok očních víček
- krvácení z nosu
- chřipce podobné příznaky
- brnění nebo znecitlivění
- poruchy zraku
- pocit změn tělesné teploty (zahrnující pocit horka, pocit chladu)
- ztluštělá ložiska červené/stříbřité kůže (příznaky lupénky)
- citlivost zubů
Pokud Vás cokoliv z tohoto postihne v závažné míře, řekněte to svému lékaři.
Následující další nežádoucí účinky byly hlášeny u velmi malého počtu pacientů léčených přípravkem Tasigna:
- ztráta paměti, poruchy nálady nebo depresivní nálada, ztráta energie, slabost
- ústní moučnivka, bakteriální infekce kůže
- puchýře, kožní cysty, mastná kůže, ztenčování kůže, tmavé skvrny na kůži, vyblednutí kůže
- zvýšená citlivost kůže
- krvácení, citlivé nebo zvětšené dásně
- výtok z nosu nebo ucpaný nos, kýchání
- sucho v ústech, bolest v krku, vřídky v ústech
- třes
- bolest očí nebo zarudnutí očí, bolest, svědění očních víček
- bolestivé a oteklé klouby (dna), svalová slabost
- bezvědomí
- obtížné a bolestivé močení, zvýšený pocit nutkání na močení
- časté močení, neobvyklé zbarvení moči
- hemoroidy
- pocit ztvrdnutí prsů, poruchy menstruace, otoky bradavek
- porucha chuti, snížení tělesné hmotnosti
silné bolesti hlavy často doprovázené pocitem nevolnosti, zvracením a citlivostí na světlo
- pálení žáhy
- zvětšení prsů u mužů
- příznaky syndromu neklidných nohou (neodolatelné nutkání pohybovat částí těla, obvykle nohou spojené s pocity nepohodlí)
Pokud Vás cokoliv z tohoto postihne v závažné míře, řekněte to svému lékaři.
Během léčby přípravkem Tasigna mohou být zjištěny abnormální výsledky krevních testů jako je nízký počet krevních elementů (bílé krvinky, červené krvinky, krevní destičky), vysoká hladina lipázy nebo amylázy v krvi (funkce slinivky břišní), vysoká hladina bilirubinu v krvi (funkce jater) nebo vysoká hladina kreatininu v krvi (funkce ledvin), nízká nebo vysoká hladina inzulinu v krvi (hormonu, který reguluje hladinu cukru v krvi), nízká nebo vysoká hladina cukru nebo vysoká hladina tuků v krvi.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Tasignu uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a blistru. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
- Neuchovávejte při teplotě nad 30°C.
- Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
- Nepoužívejte tento přípravek, pokud si všimnete, že je obal poškozený nebo s viditelnými známkami porušení.
- Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Tasigna obsahuje
- Léčivou látkou je nilotinib. Jedna tvrdá tobolka obsahuje 150 mg nilotinibu (jako monohydrát hydrochloridu).
- Dalšími složkami jsou monohydrát laktosy, krospovidon, poloxamer 188, koloidní bezvodý oxid křemičitý a magnesium-stearát. Pouzdro tvrdé tobolky je složeno ze želatiny, oxidu titaničitého (E 171), červeného a žlutého oxidu železitého (E 172), šelaku a černého oxidu železitého (E 172) pro razítkování potisku.
Jak Tasigna vypadá a co obsahuje toto balení
Přípravek Tasigna je dodáván jako tvrdé tobolky. Tvrdé tobolky jsou červené. Na každé tvrdé tobolce je černý potisk („NVR/BCR“).
Tasigna je dostupná v baleních obsahujících 28 nebo 40 tvrdých tobolek a ve vícečetných baleních 112 tvrdých tobolek (obsahujících 4 krabičky, každá obsahuje 28 tvrdých tobolek), 120 tvrdých tobolek (obsahujících 3 krabičky, každá obsahuje 40 tvrdých tobolek) nebo 392 tvrdých tobolek (obsahujících 14 krabiček, každá obsahuje 28 tvrdých tobolek).
Ve Vaší zemi na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH RoonstraBe 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Bt^rapnn Novartis Pharma Services Inc. Ten.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Česká republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 555 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EkXába Novartis (Hellas) A.E.B.E. Tq^: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Island Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kónpoq Novartis Pharma Services Inc. TpA,: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
Příbalová informace: informace pro uživatele
Tasigna 200 mg tvrdé tobolky
Nilotinibum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat,
protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Tasigna a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Tasigna užívat
3. Jak se přípravek Tasigna užívá
4. Možné nežádoucí účinky
5. Jak přípravek Tasigna uchovávat
6. Obsah balení a další informace
1. Co je přípravek Tasigna a k čemu se používá Co je Tasigna
Tasigna je lék, který obsahuje léčivou látku nazvanou nilotinib.
K čemu se Tasigna používá
Tasigna se používá k léčbě leukemie nazývané chronická myeloidní leukemie s přítomností filadelfského chromozomu (Ph-pozitivní CML). CML je nádorové onemocnění krve, kdy tělo tvoří příliš mnoho abnormálních bílých krvinek.
Tasigna se používá u pacientů s nově diagnostikovanou CML nebo u pacientů s CML, u kterých již není předchozí léčba zahrnující imatinib účinná. Používá se také u pacientů, u kterých se při předchozí léčbě objevily závažné nežádoucí účinky a nemohou v této léčbě pokračovat.
Jak Tasigna účinkuje
U pacientů s CML spouští změna v DNA (genetickém materiálu) signál, který tělu říká, aby tvořilo abnormální bílé krvinky. Přípravek Tasigna tento signál blokuje, a tak zastavuje produkci těchto buněk.
Sledování Vaší léčby přípravkem Tasigna
Během léčby budete pravidelně vyšetřován/a včetně vyšetření krve. Tyto testy budou sledovat:
- počet krevních elementů ve Vašem těle (bílé krvinky, červené krvinky, krevní destičky), aby se zjistilo, jak je přípravek Tasigna snášen.
- funkci slinivky břišní a jater ve Vašem těle, aby se zjistilo, jak je přípravek Tasigna snášen.
- hladinu iontů ve Vašem těle (draslík, hořčík). Ty jsou velmi důležité pro funkci Vašeho srdce.
- hladinu cukru a tuků ve Vaší krvi.
Rovněž Vám bude kontrolován srdeční rytmus pomocí přístroje, který měří elektrickou aktivitu srdce (zvaný EKG).
Jestliže máte jakékoli dotazy týkající se toho, jak Tasigna působí, nebo proč Vám byl předepsán, zeptejte se svého lékaře.
2. Čemu musíte věnovat pozornost, než začnete přípravek Tasigna užívat
Dodržujte pečlivě všechna doporučení lékaře. Mohou se lišit od obecných informací uvedených v této
příbalové informaci.
Neužívejte Tasignu
- jestliže jste alergický/á na nilotinib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Jestliže si myslíte, že byste mohl/a být alergický/á, řekněte to svému lékaři před užíváním přípravku
Tasigna.
Upozornění a opatření
Před užitím přípravku Tasigna se poraďte se svým lékařem nebo lékárníkem:
- pokud u Vás dříve došlo ke kardiovaskulárním příhodám, jako je srdeční infarkt, bolest na hrudi (angina pectoris), potíže s krevním zásobením Vašeho mozku (mozková mrtvice) nebo potíže s přívodem krve do vašich dolních končetin (kulhání či námahová bolest), nebo pokud máte rizikové faktory pro kardiovaskulární onemocnění, jako je vysoký krevní tlak (hypertenze), cukrovka nebo potíže s hladinou tuků ve Vaší krvi (poruchy metabolizmu tuků).
- jestliže trpíte srdeční poruchou, jako je abnormální vedení elektrického signálu nazývaného „prodloužení QT intervalu“.
- jestliže jste léčen/a léky, které ovlivňují tepovou frekvenci srdce (antiarytmika) nebo játra (viz Další léčivé přípravky a Tasigna).
- jestliže trpíte nedostatkem draslíku nebo hořčíku.
- jestliže trpíte onemocněním jater nebo slinivky břišní.
- jestliže trpíte příznaky snadného vzniku modřin, pocitem únavy nebo dýchavičností nebo jste prodělal/a opakované infekce.
- jestliže jste prodělal/a chirurgický zákrok zahrnující odstranění celého žaludku (celková gastrektomie).
- pokud jste někdy měl (a) infekční hepatitidu B (žloutenka typu B) nebo toto onemocnění máte v současné době. Přípravek TASIGNA může hepatitidu B znovu aktivovat, což může v některých případech vést k úmrtí. Před zahájením léčby lékař pacienty pečlivě vyšetří s ohledem na možný výskyt známek této infekce.
Pokud se Vás cokoli z výše uvedeného týká, řekněte to svému lékaři.
Během léčby Tasignou
- pokud omdléváte (ztráta vědomí) nebo máte nepravidelný srdeční tep během léčby Tasignou, řekněte to okamžitě svému lékaři, protože může jít o příznak vážné srdeční choroby. Prodloužení QT intervalu nebo nepravidelný srdeční tep mohou vést k náhlému úmrtí. Méně časté případy náhlého úmrtí byly hlášeny u pacientů, kterým byla podávána Tasigna.
- pokud u Vás došlo k náhlému bušení srdce, závažné svalové slabosti nebo ochrnutí, záchvatům nebo náhlým změnám v myšlení nebo míře bdělosti, řekněte to okamžitě svému lékaři, protože může jít o příznak rychlého ničení nádorových buněk zvaný syndrom nádorového rozpadu. U pacientů léčených přípravkem Tasigna byly zjištěny vzácné případy syndromu nádorového rozpadu.
- pokud se u Vás objevila bolest nebo nepříjemný pocit na hrudi, strnulost nebo slabost, potíže
s chůzí nebo řečí, bolest, bledost nebo pocit chladu v končetinách, okamžitě informujte svého lékaře, protože může jít o příznak kardiovaskulární příhody. Závažné kardiovaskulární příhody zahrnující potíže s přívodem krve do dolních končetin (periferní arteriální okluzivní onemocnění), ischemické srdeční onemocnění a potíže s přívodem krve do mozku (ischemické onemocnění mozku) byly hlášeny u pacientů užívajících přípravek Tasigna. Váš lékař by měl vyhodnotit hladinu tuků (lipidů) a cukru ve Vaší krvi před zahájením léčby a během léčby přípravkem Tasigna.
- pokud u Vás dojde k otokům rukou nebo nohou, celkovým otokům nebo k rychlému nárůstu hmotnosti, sdělte to svému lékaři, protože může jít o příznaky závažného zadržování tekutin. Příznaky závažného zadržování tekutin byly hlášeny u pacientů léčených přípravkem Tasigna méně často.
Další léčivé přípravky a Tasigna
Tasigna může interferovat s některými jinými léky.
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době
užíval(a) nebo které možná budete užívat. Toto se týká především:
- antiarytmik - užívaných k léčbě nepravidelného tlukotu srdce;
- chlorochin, halofantrin, klarithromycin, haloperidol, methadon, moxifloxacin - léčivé přípravky, které mohou mít nechtěný účinek na srdeční činnost;
- ketokonazol, itrakonazol, vorikonazol, klarithromycin, telithromycin - užívané k léčbě infekcí;
- ritonavir - lék ze skupiny „antiproteáz“, užívané k léčbě HIV;
- karbamazepin, fenobarbital, fenytoin - užívané k léčbě epilepsie;
- rifampicin - užívaný k léčbě tuberkulózy;
- třezalka tečkovaná - rostlinný lék, užívaný k léčbě depresí a jiných stavů (také známý jako Hypericum perforatum);
- midazolam - užívaný ke zmírnění úzkosti před chirurgickým zákrokem;
- alfentanil a fentanyl - užívané k léčbě bolesti a jako sedativa před a během chirurgického zákroku nebo léčebných procedur;
- cyklosporin, sirolimus a takrolimus - léky, které potlačují “sebeobrannou” schopnost těla a boj proti infekcím a jsou často užívané k prevenci odmítnutí (rejekce) transplantovaných orgánů, jako jsou játra, srdce a ledviny;
- dihydroergotamin a ergotamin - užívané k léčbě demence;
- lovastatin, simvastatin - užívané k léčbě vysoké hladiny tuků v krvi;
- warfarin - užívaný k léčbě poruchy krevní srážlivosti (např. krevní sraženiny nebo trombózy);
- astemizol, terfenadin, cisaprid, pimozid, chinidin, bepridil nebo námelové alkaloidy (ergotamin, dihydroergotamin).
Tyto léky byste neměl/a užívat během Vaší léčby přípravkem Tasigna. Pokud užíváte kterýkoli z
těchto léků, může Vám lékař předepsat jiný alternativní lék.
Před započetím léčby přípravkem Tasigna informujte svého lékaře nebo lékárníka, pokud užíváte antacida, což jsou léky proti pálení žáhy. Tyto léky musí být užívány odděleně od přípravku Tasigna:
- blokátory H2, které snižují tvorbu kyseliny v žaludku. Blokátory H2 by měly být užívány přibližně 10 hodin před a přibližně 2 hodiny poté, kdy jste užil(a) přípravek Tasigna;
- antacida, např. antacida, která obsahují hydroxid hlinitý, hydroxid hořečnatý a simetikon, které neutralizují vysokou kyselost v žaludku. Tato antacida by měla být užívána přibližně 2 hodiny před nebo přibližně 2 hodiny poté, kdy jste užil(a) přípravek Tasigna.
Jestliže již užíváte Tasigna a bude Vám předepisován k užívání nový (další) lék, měl/a byste lékaře informovat o tom, že jste ještě tento lék společně s přípravkem Tasigna v minulosti neužíval/a.
Přípravek Tasigna s jídlem a pitím
Přípravek Tasigna neužívejte s jídlem. Jídlo může zvýšit absorpci přípravku Tasigna, a tak zvýšit množství přípravku Tasigna v krvi až na škodlivou hladinu. Nepijte grapefruitovou šťávu ani nejezte grapefruit. To může zvýšit množství přípravku Tasigna v krvi až ke škodlivé hladině.
Starší lidé (věk 65 let a více)
Tasigna mohou užívat lidé ve věku 65 let a starší ve stejných dávkách jako ostatní dospělí jedinci. Těhotenství, kojení a plodnost
- Tasigna se nedoporučuje podávat během těhotenství, pokud to není nezbytně nutné. Pokud jste těhotná, nebo si myslíte, že můžete být těhotná, řekněte to svému lékaři, který s Vámi prodiskutuje, zda můžete Tasigna užívat během těhotenství.
- Ženám, které mohou otěhotnět, se doporučuje užívat během léčby a dva týdny po ukončení léčby velmi účinné prostředky proti možnosti početí.
- Během léčby přípravkem Tasigna se kojení nedoporučuje. Pokud kojíte, řekněte to svému lékaři.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Pokud se u Vás objeví po užití přípravku Tasigna nežádoucí účinky (např. závratě nebo poruchy zraku), které by u Vás mohly ovlivnit schopnost bezpečně řídit nebo používat nástroje nebo obsluhovat stroje, měl/a byste se těchto činností vyvarovat, dokud tyto účinky nevymizí.
Tasigna obsahuje laktózu
Tento přípravek obsahuje laktózu (také známou jako mléčný cukr). Pokud Vám Váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
3. Jak se přípravek Tasigna užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Jaké množství přípravku Tasigna se užívá
- Doporučená dávka je 800 mg denně. Této dávky je dosaženo užitím dvou 200 mg tvrdých tobolek dvakrát denně.
Lékař Vám může předepsat nižší dávku v závislosti na Vaši odpověď na léčbu.
Kdy užívat přípravek Tasigna
Tvrdé tobolky užívejte:
- dvakrát denně (přibližně každých 12 hodin);
- nej méně 2 hodiny po j akémkoli jídle;
- potom počkejte 1 hodinu, než budete znovu jíst.
Pokud máte dotazy, kdy Tasigna užívat, zeptejte se svého lékaře nebo lékárníka. Užívání přípravku Tasigna každý den ve stejnou dobu Vám pomůže si zapamatovat, kdy máte tvrdé tobolky užít.
Jak se Tasigna užívá
- Tvrdé tobolky polykejte celé a zapíjejte je vodou.
- Tvrdé tobolky neužívejte dohromady s žádnou potravou.
- Tvrdé tobolky otevřete pouze v případě, kdy je nemůžete spolknout. V tomto případě můžete rozmíchat obsah každé tvrdé tobolky v jedné čajové lžičce jablečné šťávy (jablečného pyré) a okamžitě užít. Neužívejte více než jednu čajovou lžičku s každou tvrdou tobolkou a nepoužívejte jinou potravu než jablečnou šťávu.
Jak dlouho se Tasigna užívá
Pokračujte v užívání přípravku Tasigna tak dlouho, jak Vám řekl lékař. Toto je dlouhodobá léčba. Váš lékař bude pravidelně sledovat Váš zdravotní stav, aby zjistil, zda má léčba požadovaný účinek.
Jestliže máte dotazy, jak dlouho bude užívání přípravku Tasigna trvat, zeptejte se svého lékaře.
Jestliže jste užil(a) více přípravku Tasigna, než jste měl(a)
Jestliže jste užil(a) více přípravku Tasigna, než jste měl(a), nebo pokud někdo jiný náhodně užil Vaše tvrdé tobolky, okamžitě vyhledejte lékaře nebo nemocniční zařízení, abyste se mohl(a) o této věci poradit. Ukažte jim balení tvrdých tobolek a tuto příbalovou informaci. Může být nutné lékařské ošetření.
Jestliže jste zapomněl(a) užít přípravek Tasigna
Pokud jste vynechal(a) dávku, vezměte si následující dávku v obvyklou dobu. Nezdvoj násobujte následující dávku, abyste nahradil(a) vynechanou tvrdou tobolku.
Jestliže jste přestal(a) užívat přípravek Tasigna
Nepřestávejte užívat Tasigna, dokud Vám to Váš lékař neřekne. Při ukončení léčby přípravkem Tasigna bez doporučení lékaře riskujete zhoršení svého onemocnění, což může mít život ohrožující následky. Pokud zvažujete ukončení léčby Tasignou, poraďte se se svým lékařem, zdravotní sestrou a/nebo lékárníkem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého. Většina nežádoucích účinků jsou mírné až středně závažné a obvykle vymizí za
několik dnů až týdnů léčby.
Některé nežádoucí účinky by mohly být závažné.
Nežádoucí účinky jsou časté (mohou postihnout až 1 z 10 lidí),, méně časté (mohou postihnout až 1 ze
100 lidí) nebo byly hlášeny jen u velmi malého počtu pacientů.
- rychlé přibývání tělesné hmotnosti, otoky rukou, kotníků, nohou nebo obličeje, (příznaky zadržování vody)
- bolest na hrudi, vysoký krevní tlak, nepravidelný tlukot srdce, modré zabarvení rtů, jazyka nebo kůže (příznaky srdeční poruchy)
- potíže při dýchání, kašel, sípání s horečkou nebo bez horečky, otoky nohou (příznaky onemocnění plic)
- horečka, snadný vznik modřin, časté infekce (příznaky poruchy krve)
- slabost nebo ochrnutí končetin nebo obličeje, obtíže při mluvení, silná bolest hlavy, vidění, cítění nebo slyšení věcí, které nejsou (příznaky poruchy nervového systému)
- žízeň, suchá kůže, podrážděnost, tmavá moč, pokles množství moče (příznaky poruchy ledvin)
- neostré vidění, ztráta zraku, krvácení v oku (příznaky oční poruchy)
- otoky a bolest v jedné části těla (příznaky ucpání žíly sraženinou)
- bolest břicha, nevolnost, zvracení krve, černá stolice, zácpa, zduření břicha (příznaky poruchy zažívacího systému)
- silná bolest v nadbřišku (příznak zánětu slinivky břišní)
- zežloutnutí kůže a očí, nevolnost, ztráta chuti k jídlu, tmavě zbarvená moč (příznaky poruchy jater)
- vyrážka, bolestivé červené opuchliny, bolesti kloubů a svalů (příznaky poruchy kůže)
- nadměrná žízeň, velké množství moči, zvýšená chuť k jídlu s úbytkem tělesné hmotnosti, únava (příznaky vysoké hladiny krevního cukru)
- rychlý tlukot srdce, vystouplé oči, snížení tělesné hmotnosti, otoky přední strany krku (příznaky zvýšené činnosti štítné žlázy)
- nevolnost, zkrácený dech, nepravidelný srdeční tep, zakalená moč, únava a/nebo kloubní potíže spojené s neobvyklými výsledky krevních testů (např. vysoká hladina draslíku, kyseliny močové a fosforu a nízké hladiny vápníku)
- bolest, nepohodlí, ochablost nebo křeče svalů nohou, které mohou být způsobeny sníženým průtokem krve, vředy na nohou nebo rukách, které se hojí pomalu nebo nehojí vůbec a výrazná změna barvy (zmodrání nebo zblednutí) nebo teploty (chlad) nohou a rukou, protože tyto příznaky mohou být příznaky překážky v tepnách v postižených končetinách (noha nebo ruka) a prstech (na nohou a rukou)
- recidiva (reaktivace) hepatitidy B, pokud jste v minulosti měl(a) toto onemocnění (infekce jater).
Jestliže se u Vás cokoli z tohoto objeví, okamžitě to řekněte svému lékaři.
Některé nežádoucí účinky jsou velmi časté (mohou postihnout více než 1 z 10 lidí)
- průjem
- bolest hlavy
- únava
- bolest svalů
- svědění, vyrážka, kopřivka
- nevolnost
- zvracení
- padání vlasů
- vysoká hladina bilirubinu v krvi (funkce jater)
- vysoká hladina lipázy v krvi (funkce slinivky břišní)
Pokud Vás cokoli z tohoto postihne v závažné míře, řekněte to svému lékaři.
Některé nežádoucí účinky jsou časté (mohou postihnout až 1 z 10 lidí)
- břišní potíže, žaludeční nevolnost po jídle, plynatost, otok nebo nadýmání břicha
- bolest kostí, bolest kloubů, křeče svalů
- bolest včetně bolesti zad, bolesti za krkem a bolesti končetin, bolest nebo diskomfort v bocích
- podráždění očí, otoky, výtok, svědění nebo zarudnutí, suchost očí (příznaky onemocnění očí)
- zčervenání kůže, suchá kůže, akne, bradavice, snížená citlivost kůže
- ztráta chuti k jídlu, porušené vnímání chuti, snížení nebo zvýšení tělesné hmotnosti
- noční poty, nadměrné pocení, návaly horka
- závratě, celkový pocit nevolnosti, pocit točení
- brnění nebo znecitlivění
- poruchy hlasu
- krvácení z nosu
- časté močení
- palpitace (pocit rychlého bušení srdce)
Pokud Vás cokoli z tohoto postihne v závažné míře, řekněte to svému lékaři.
Některé nežádoucí účinky jsou méně časté (mohou postihnout až 1 ze 100 lidí)
- zvýšení kožní citlivosti, bolest kůže
- otok očních víček
- sucho v ústech, bolesti v krku, vřídky v ústech chuti
- pálení žáhy
- bolest prsou
- zvýšení chuti k jídlu
- poruchy pozornosti
- obtíže a bolest při močení, zvýšení pocitu nutkání na močení
- neschopnost dosáhnout nebo udržet erekci
- zvětšení prsů u mužů
- chřipce podobné příznaky, svalová slabost
- třes
- pokles ostrosti zraku
- silné bolesti hlavy často doprovázené pocitem na zvracení, zvracením a citlivostí na světlo
- poruchy zraku
- ústní nebo poševní moučnivka (soor)
- svalová a kloubní ztuhlost
- bezvědomí
- zvýšení tělesné hmotnosti
- pocit změn tělesné teploty (zahrnující pocit horka, pocit chladu)
- ztluštělá ložiska červené/stříbřité kůže (příznaky lupénky)
- citlivost zubů
Pokud Vás cokoliv z tohoto postihne v závažné míře, řekněte to svému lékaři.
Následující další nežádoucí účinky byly hlášeny u velmi malého počtu pacientů léčených přípravkem Tasigna:
- zmatenost a dezorientace, ztráta paměti, změny nálad, slabost
- bakteriální infekce kůže
- puchýře, kožní cysty, mastná kůže, ztenčování kůže, tmavé skvrny na kůži, odbarvování kůže
- krvácení, měkké nebo zvětšené dásně
- výtok z nosu nebo ucpaný nos, kýchání
- zarudnutí a/nebo otok a možné loupání kůže na dlaních a chodidlech (takzvaný syndrom ruka-noha)
- zvýšení citlivosti očí nebo kůže na světlo
- bolest očí nebo zarudnutí očí, bolest, svědění očních víček
- zhoršení sluchu, bolest ucha, šelest (zvonění) v uších
- bolestivé a oteklé klouby (dna)
- krev v moči, neobvyklé zbarvení moči, samovolný únik moči
- hemoroidy
- pocit ztvrdnutí prsů, poruchy menstruace, otoky bradavek
- příznaky syndromu neklidných nohou (neodolatelné nutkání pohybovat částí těla, obvykle nohou spojené s pocity nepohodlí)
Pokud Vás cokoliv z tohoto postihne v závažné míře, řekněte to svému lékaři.
Během léčby přípravkem Tasigna mohou být zjištěny abnormální výsledky krevních testů jako je nízký počet krevních elementů (bílé krvinky, červené krvinky, krevní destičky), vysoká hladina lipázy nebo amylázy v krvi (funkce slinivky břišní), vysoká hladina bilirubinu v krvi (funkce jater) nebo vysoká hladina kreatininu v krvi (funkce ledvin), nízká nebo vysoká hladina inzulinu v krvi (hormonu, který reguluje hladinu cukru v krvi), nízká nebo vysoká hladina cukru nebo vysoká hladina tuků v krvi.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Tasignu uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a blistru. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
- Neuchovávejte při teplotě nad 30°C.
- Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
- Nepoužívejte tento přípravek, pokud si všimnete, že je obal poškozený nebo s viditelnými známkami porušení.
- Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
Co Tasigna obsahuje
- Léčivou látkou je nilotinib. Jedna tvrdá tobolka obsahuje 200 mg nilotinibu (jako monohydrát hydrochloridu).
- Dalšími složkami jsou monohydrát laktosy, krospovidon, poloxamer 188, koloidní bezvodý oxid křemičitý a magnesium-stearát. Pouzdro tvrdé tobolky je složeno ze želatiny, oxidu titaničitého (E 171), žlutého oxidu železitého (E 172) a šelaku (E 904) a červeného oxidu železitého (E 172) pro razítkování potisku.
Jak Tasigna vypadá a co obsahuje toto balení
Přípravek Tasigna je dodáván jako tvrdé tobolky. Tvrdé tobolky jsou světle žluté. Na každé tvrdé
tobolce je červený potisk („NVR/TKI“).
Přípravek Tasigna je dostupný v pouzdře obsahujícím 28 tvrdých tobolek a v krabičce obsahující 28
nebo 40 tvrdých tobolek.
Přípravek Tasigna je také dostupný ve vícečetných baleních:
- 112 (4 pouzdra po 28) tvrdých tobolek.
- 112 (4 balení po 28) tvrdých tobolek.
- 120 (3 balení po 40) tvrdých tobolek.
- 392 (14 balení po 28) tvrdých tobolek.
Ve Vaší zemi na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH RoonstraBe 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Etarapnu
Novartis Pharma Services Inc. Tea.: +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Lietuva
Novartis Pharma Services Inc. Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
|
Eesti Novartis Pharma Services Inc Tel: +372 66 30 810 |
Norge . Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáSa Novartis (Hellas) A.E.B.E. Tn^: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A. Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kónpoq Novartis Pharma Services Inc Tn^: +357 22 690 690 |
Sverige . Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc Tel: +371 67 887 070 |
United Kingdom . Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Nederland
Novartis Pharma B.V. Tel: +31 26 37 82 555
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
91