Tafinlar 50 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Tafinlar 50 mg tvrdé tobolky Tafinlar 75 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Tafinlar 50 mg tvrdé tobolky
Jedna tvrdá tobolka obsahuje dabrafenibum 50 mg (jako dabrafenibi mesilas). Tafinlar 75 mg tvrdé tobolky
Jedna tvrdá tobolka obsahuje dabrafenibum 75 mg (jako dabrafenibi mesilas). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka (tobolka).
Tafinlar 50 mg tvrdé tobolky
Neprůhledná tmavě červená, přibližně 18 mm dlouhá tobolka, s vytištěným „GS TEW“ a „50 mg“ na obalu tobolky.
Tafinlar 75 mg tvrdé tobolky
Neprůhledná tmavě růžová, přibližně 19 mm dlouhá tobolka, s vytištěným „GS LHF“ a „75 mg“ na obalu tobolky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Dabrafenib je indikován v monoterapii nebo v kombinaci s trametinibem k léčbě dospělých pacientů s neresekovatelným nebo metastatickým melanomem s mutací V600 genu BRAF (viz body 4.4 a 5.1).
4.2 Dávkování a způsob podání
Léčba dabrafenibem má být zahájena a vedena kvalifikovaným lékařem se zkušenostmi s používáním protinádorové léčby.
Pacient musí mít před zahájením léčby dabrafenibem potvrzenou mutaci V600 genu BRAF pomocí validovaného testu.
Účinnost a bezpečnost dabrafenibu nebyly stanoveny u pacientů s melanomem s divokým typem genu BRAF, a proto by dabrafenib k léčbě těchto pacientů neměl být používán (viz body 4.4 a 5.1).
Dávkování
Doporučená dávka dabrafenibu, užívaného buď v monoterapii, nebo v kombinaci s trametinibem je 150 mg (dvě 75mg tobolky) dvakrát denně (což odpovídá celkové denní dávce 300 mg). Doporučena dávka trametinibu užívaného v kombinaci s dabrafenibem je 2 mg jednou denně.
Trvání léčby
V léčbě se pokračuje, dokud z ní má pacient prospěch, nebo dokud se neobjeví nepřijatelná toxicita (viz tabulka 2).
Vynechání dávky
Pokud dojde k vynechání dávky dabrafenibu, nemá se tato dávka užívat, pokud do další dávky zbývá méně než 6 hodin.
Pokud dojde k vynechání dávky trametinibu, podávaného v kombinaci s dabrafenibem, má být tato dávka trametinibu užita jenom tehdy, pokud do další dávky zbývá více než 12 hodin.
Úprava dávkování
K úspěšnému zvládnutí požadavků na dávkování je dabrafenib k dispozici ve dvou silách tobolek,
50 mg a 75 mg.
Zvládnutí nežádoucích účinků může vyžadovat přerušení léčby, snížení dávky nebo trvalé ukončení léčby (viz tabulky 1 a 2).
Pokud je nežádoucím účinkem kožní spinocelulární karcinom (cuSCC) nebo nově diagnostikovaný primární melanom (viz bod 4.4), úprava dávky nebo přerušení léčby se nedoporučují.
Pokud u pacienta dojde k vzestupu tělesné teploty na > 38,5 °C, je třeba léčbu přerušit. Pacienti musí být vyšetřeni s ohledem na známky a příznaky infekce (viz bod 4.4).
V případě výskytu uveitidy není nutná úprava dávkování, pokud účinné lokální léčivé přípravky zvládají oční zánět. Pokud není lokální léčba uveitidy účinná, přerušte podávání dabrafenibu až do vymizení očního zánětu a poté opětovně zahajte podávání dabrafenibu sníženou dávkou o jednu dávkovací hladinu. (viz bod 4.4).
Doporučení pro snižování dávek a doporučení pro úpravu dávkování jsou uvedeny v tabulkách 1 a 2. Tabulka 1 Doporučení pro snižování dávek
|
Dávkovači hladina |
Dávka dabrafenibu Užíván v monoterapii nebo v kombinaci s trametinibem |
Dávka trametinibu* Pouze je-li užíván v kombinaci s dabrafenibem |
|
Zahajovací dávka |
150 mg dvakrát denně |
2 mg jednou denně |
|
První snížení |
100 mg dvakrát denně |
1.5 mg jednou denně |
|
Druhé snížení |
75 mg dvakrát denně |
1 mg jednou denně |
|
Třetí snížení |
50 mg dvakrát denně |
1 mg jednou denně |
|
Snížení dávek dabrafenibu, užívaného v monoterapii nebo v kombinaci s trametinibem, pod 50 mg dvakrát denně není doporučeno. Snížení dávek trametinibu, užívaného v kombinaci s dabrafenibem, pod 1 mg jednou denně není doporučeno. | ||
|
* Pokyny k dávkování v případě léčby trametinibem v monoterapii j sou uvedeny v SmPC trametinibu | ||
Tabulka 2
Schéma úpravy dávkování založené na stupni závažnosti nežádoucích účinků
|
Stupeň (CTC-AE)* |
Doporučená úprava dávky dabrafenibu Užívaného v monoterapii nebo v kombinaci s trametinibem |
|
Stupeň 1 nebo 2 (tolerovatelný) |
Pokračujte v léčbě a sledujte pacienta na základě klinického stavu. |
|
Stupeň 2 (netolerovatelný) nebo stupeň 3 |
Přerušte léčbu, dokud toxicita neustoupí na stupeň 0 až 1, při opětovném zahájení léčby snižte dávku o jednu dávkovací hladinu níže. |
|
Stupeň 4 |
Trvale přerušte léčbu, nebo přerušte léčbu, dokud toxicita neustoupí na stupeň 0 až 1, při opětovném zahájení léčby snižte dávku o jednu dávkovací hladinu níže. |
|
* Intenzita klinických nežádoucích účinků je stanovena na základě CTC-AE (Common Terminology Criteria for Adverse Events) verze 4.0. | |
Tam, kde jsou nežádoucí účinky u pacienta úspěšně zvládány, lze zvážit opětovné postupné zvyšování dávky ve stejných krocích, jako probíhalo její snižování. Dávka dabrafenibu by neměla přesáhnout 150 mg denně.
V případě toxicity spojené s užíváním dabrafenibu v kombinaci s trametinibem se musí dávka obou látek snížit, přerušit nebo ukončit současně. Výjimky, kde jsou úpravy dávkování nezbytné jenom pro jednu z těchto dvou látek, jsou popsány níže pro horečku, uveitidu, malignity s RAS pozitivní mutací vyskytující se v jiné lokalitě než kožní, snížení ejekční frakce levé komory (LVEF), okluzi retinální žíly (RVO), ablaci retinálního pigmentového epitelu (RPED) a intersticiální plicní onemocnění (ILD)/pneumonitidu (primárně spojené s trametinibem).
Výjimky ve změně dávkování (kde _je snížena dávka _jenom _jedné ze dvou látek) u vybraných nežádoucích účinků
Pyrexie
Při užívání jak dabrafenibu samotného, tak v kombinaci s trametinibem, je nutné přerušit terapii dabrafenibem, pokud je teplota pacienta > 38.5oC (prostudujte si, prosím, Tabulku 2, která uvádí informace o změně dávkování). Podávání trametinibu má pokračovat ve stejné dávce. Doporučuje se zahájit podávání antipyretik, jako jsou ibuprofen nebo acetaminofenon/paracetamol. V případech, kdy antipyretika nejsou účinná, je nutné zvážit podávání perorálních kortikosteroidů. U pacientů by měly být sledovány známky a příznaky infekce, a pokud je to nezbytné, musí být zahájena vhodná léčba (viz bod 4.4).
Pokud horečka ustoupí, podávání dabrafenibu by mělo být znovu zahájeno spolu s podáváním vhodné antipyretické profylaxe. Podávání dabrafenibu by mělo být zahájeno buď ve stejné dávce, nebo (pokud se horečka opakuje a/nebo byla doprovázena jinými závažnými symptomy včetně dehydratace, hypotenze nebo renálního selhání) v dávce snížené o jednu dávkovací hladinu.
Uveitida
V případě výskytu uveitidy není nutná úprava dávkování, pokud je léčba uveitidy lokálně aplikovanými léky úspěšná. Pokud není lokální léčba uveitidy účinná, přerušte podávání dabrafenibu až do vymizení očního zánětu a poté se má opětovně zahájit podávání dabrafenibu sníženou dávkou o jednu dávkovací hladinu. Změna dávky trametinibu užívaného v kombinaci s dabrafenibem není vyžadována (viz bod 4.4).
Malignity mimo kožní lokalizaci s mutací v RAS
U pacientů s malignitou mimo kožní lokalizaci s mutací v RAS se mají zvážit přínosy a rizika před pokračováním léčby dabrafenibem. Změna dávky trametinibu užívaného v kombinaci s dabrafenibem není vyžadována.
Snížení ejekční frakce levé komory (LVEF)/dysfunkce levé komory
Pokud je dabrafenib užíván v kombinaci s trametinibem a nastane absolutní pokles > 10 % LVEF proti výchozímu stavu a ejekční frakce je pod místní dolní hranicí normy (LLN, Lower limit of normal), prostudujte si, prosím SmPC trametinibu (viz bod 4.2), které uvádí informace o změně dávkování trametinibu. Změna dávky dabrafenibu užívaného v kombinaci s trametinibem není vyžadována.
Okluze retinální žíly (RVO) a odchlípení sítnice (RPED)
Pokud pacienti kdykoli v průběhu léčby dabrafenibem v kombinaci s trametinibem hlásí nově vzniklé poruchy vidění, jako je zhoršení centrálního vidění, rozmazané vidění nebo ztráta vidění, je nutné dále postupovat a upravovat dávku trametinibu tak, jak uvádí SmPC trametinibu (viz bod 4.2). V případě potvrzení okluze retinální žíly a odchlípení sítnice není vyžadována změna dávky dabrafenibu užívaného v kombinaci s trametinibem.
Intersticiální plicní onemocnění (ILD)/pneumonitida
U pacientů léčených kombinací dabrafenibu s trametinibem, u kterých je podezření na intersticiální plicní onemocnění nebo pneumonitidu, včetně pacientů s novými nebo progredujícími plicními symptomy a nálezy zahrnujícími kašel, dušnost, hypoxii, pleurální výpotek nebo infiltráty, je nutné postupovat dle SmPC trametinibu (viz bod 4.2), které uvádí informace o změně dávkování trametinibu. V případě intersticiálního plicního onemocnění nebo pneumonitidy se nevyžaduje změna dávky dabrafenibu užívaného v kombinaci s trametinibem.
Pacienti _jiné než bělošské rasy
Bezpečnost a účinnost dabrafenibu u pacientů jiné než bělošské rasy nebyly stanoveny. Nejsou dostupné žádné údaje.
Starší _ pacienti
U pacientů nad 65 let není nutná úprava počáteční dávky.
Porucha _ funkce ledvin
U pacientů s mírnou nebo středně závažnou poruchou funkce ledvin není nutná úprava dávkování.
U pacientů se závažnou poruchou funkce ledvin nejsou k dispozici žádné klinické údaje a potenciální nutnost úpravy dávkování nelze stanovit (viz bod 5.2). Dabrafenib je třeba u pacientů se závažnou poruchou funkce ledvin používat s opatrností, a to jak při užívání v monoterapii, tak v kombinaci s trametinibem.
Porucha funkce jater
U pacientů s mírnou poruchou funkce jater není nutná úprava dávkování. U jedinců se středně závažnou až závažnou poruchou funkce jater nejsou k dispozici klinické údaje a potenciální nutnost úpravy dávkování nelze stanovit (viz bod 5.2). Jaterní metabolismus a biliární sekrece jsou primárními cestami eliminace dabrafenibu a jeho metabolitů a u pacientů se středně závažnou až závažnou poruchou funkce jater tak může docházet ke zvýšení expozice. U pacientů se středně závažnou až závažnou poruchou funkce jater je třeba dabrafenib používat s opatrností, a to jak při užívání v monoterapii, tak v kombinaci s trametinibem.
Pediatrická populace
Bezpečnost a účinnost dabrafenibu nebyly zatím u dětí a dospívajících (< 18 let) stanoveny. Klinické údaje nejsou k dispozici. Studie u mláďat zvířat prokázaly nežádoucí účinky dabrafenibu, které nebyly u dospělých zvířat pozorovány (viz bod 5.3).
Způsob podání
Tobolky dabrafenibu se polykají celé a zapíjejí vodou. Tobolky se nesmí žvýkat ani otevírat a z důvodu chemické nestability dabrafenibu se nesmí míchat s potravou ani nápoji.
Dabrafenib se doporučuje užívat ve stejnou dobu každý den, s intervalem přibližně 12 hodin mezi jednotlivými dávkami. Pokud se užívá dabrafenib a trametinib v kombinaci, trametinib se má podávat jednou denně ve stejnou dobu každý den buď s ranní dávkou, nebo s večerní dávkou dabrafenibu.
Dabrafenib se užívá nejméně 1 hodinu před jídlem nebo nejméně 2 hodiny po jídle.
Pokud pacient po užití dabrafenibu zvrací, nemá užít dávku přípravku znovu, ale má užít až další plánovanou dávku.
Informace o způsobu podání trametinibu v kombinaci s dabrafenibem jsou uvedeny v SmPC trametinibu.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Při podávání dabrafenibu v kombinaci s trametinibem je nutné před zahájením léčby prostudovat SmPC trametinibu. V SmPC trametinibu jsou uvedeny další informace týkající se upozornění a opatření spojených s léčbou dabrafenibem.
Testování mutace V600 v genu BRAF
Bezpečnost a účinnost dabrafenibu nebyly stanoveny u pacientů s melanomem s divokým typem genu BRAF, proto se dabrafenib u těchto pacientů nemá používat (viz body 4.2 a 5.1).
Dabrafenib a trametinib u pacientů s progresí během léčby inhibitorem BRAF
Údaje u pacientů, kteří užívali dabrafenib v kombinaci s trametinibem po předchozí progresi onemocnění na léčbě inhibitorem BRAF, jsou omezené, ukazují však na sníženou účinnost kombinace dabrafenibu s trametinibem u těchto pacientů (viz bod 5.1). Z toho důvodu by před léčbou touto kombinací měly být zváženy u pacientů po předchozí léčbě BRAF inhibitory jiné léčebné možnosti. Pořadí různých typů léčby u pacientů, u nichž došlo k progresi onemocnění na předcházející léčbě inhibitorem BRAF, nebyla dosud stanovena.
Trametinib v kombinaci s dabrafenibem u pacientů s metastázami v mozku
Bezpečnost a účinnost kombinace dabrafenibu a trametinibu nebyla hodnocena u pacientů s melanomem s mutací V600 v genu BRAF, u kterých došlo k rozšíření nádorového onemocnění do mozku.
Nové malignity
Při podávání dabrafenibu jak v monoterapii, tak v kombinaci s trametinibem, se mohou vyskytovat nové malignity, a to jak kožní, tak mimo kožní lokalizaci.
Kožní spinocelulární karcinom (cuSCC)
U pacientů léčených dabrafenibem samotným nebo v kombinaci s trametinibem byly zaznamenány případy cuSCC (včetně keratoakantomu) (viz bod 4.8). V studii MEK115306 fáze III se spinocelulární karcinom objevil u 3 % (6/209) pacientů užívajících trametinib v kombinaci s dabrafenibem a u 10 % pacientů (22/211) užívajících dabrafenib samostatně. V studii MEK116513 fáze III, se spinocelulární karcinom objevil u 1 % (5/350) pacientů užívajících trametinib v kombinaci s dabrafenibem a u 18 % pacientů (63/349) užívajících vemurafenib samostatně. Medián doby do výskytu prvního spinocelulárního karcinomu v studii MEK115306 byl 223 dní (rozmezí od 56 do 510 dní) v rameni kombinované terapie a 60 dní (rozmezí od 9 do 653 dní) v rameni s dabrafenibem v monoterapii.
Před zahájením léčby dabrafenibem a poté každý měsíc v průběhu léčby a po dobu až 6 měsíců od ukončení léčby se doporučuje provádět kožní vyšetření s ohledem na možný cuSCC. Ve sledování se má pokračovat po dobu 6 měsíců po ukončení léčby dabrafenibem nebo do zahájení další protinádorové léčby.
Kožní SCC je třeba léčit kožní excizí, v léčbě dabrafenibem, případně v léčbě kombinací dabrafenibu s trametinibem, se pokračuje bez úpravy dávkování. Pacienty je třeba poučit, aby okamžitě informovali svého lékaře, pokud se u nich objeví nové kožní léze.
V klinických studiích byly hlášeny u pacientů léčených dabrafenibem případy nově diagnostikovaného primárního melanomu. Tyto případy byly zaznamenány v průběhu prvních 5 měsíců léčby dabrafenibem v monoterapii.Případy nově diagnostikovaného melanomu se léčí excizí a nevyžadují úpravu léčby. Monitorování kožních lézí má probíhat stejně, jak je popsáno u kožního spinocelulárního karcinomu.
Sekundární/rekurentní malignity mimo kožní lokalizaci
Experimenty in vitro prokázaly paradoxní aktivaci signalizace MAP-kinázy (mitogen-activated protein kinase) u buněk s divokým typem genu BRAF s mutacemi RAS, pokud j sou vystaveny účinkům inhibitorů BRAF. To může u pacientů s mutacemi RAS vést ke zvýšení rizika výskytu malignit v jiných než kožních lokalitách při expozici dabrafenibu (viz bod 4.8). Malignity související s RAS byly hlášeny v klinických studiích jak při léčbě jinými inhibitory BRAF (chronická myelomonocytová leukémie a SCC hlavy a krku mimo kožní lokalizaci), tak při léčbě dabrafenibem v monoterapii (adenokarcinom pankreatu, adenokarcinom žlučovodu) i při léčbě dabrafenibem v kombinaci s inhibitorem MEK, trametinibem (kolorektální karcinom, karcinom pankreatu).
Před zahájením léčby má být u pacientů provedeno vyšetření hlavy a krku minimálně s vizuální kontrolou ústní sliznice a vyšetřením lymfatických uzlin pohmatem, dále pak CT vyšetření hrudní a abdominální oblasti. Během léčby mají být pacienti odpovídajícím způsobem monitorováni, což může zahrnovat vyšetření hlavy a krku každé 3 měsíce a CT vyšetření hrudní/abdominální oblasti každých 6 měsíců. Doporučuje se provést anální vyšetření a vyšetření pánevní oblasti (u žen) před zahájením a na konci léčby nebo kdykoli je to považováno za klinicky indikované. Vyšetření krevního obrazu má být provedeno dle klinické indikace.
Před zahájením podávání dabrafenibu pečlivě zvažte přínosy a rizika u pacientů s předchozím nebo stávajícím nádorovým onemocněním spojeným s mutacemi v RAS. Při užívání trametinibu v kombinaci s dabrafenibem se změna dávky trametinibu nevyžaduje.
Po ukončení léčby dabrafenibem má pokračovat monitorování sekundárních/rekurentních malignit v jiné než kožní lokalizaci po dobu až 6 měsíců nebo do zahájení další protinádorové léčby. Abnormální nálezy mají být léčeny dle klinické praxe.
Hemoragie
U pacientů užívajících kombinaci dabrafenibu s trametinibem se vykytly hemoragické příhody, včetně těžkých hemoragických příhod a fatálních hemoragií (viz 4.8). Pro další informace se, prosím, podívejte do SmPC trametinibu (viz bod 4.4.)
Poruchy vidění
V klinických studiích byly hlášeny u pacientů léčených dabrafenibem jak v monoterapii, tak v kombinaci trametinibem oční reakce, včetně uveitidy, iridocyklitidy a iriditidy.
Pacienti by měli být v průběhu léčby rutinně sledováni s ohledem na subjektivní a objektivní oční příznaky (jako jsou změny zraku, fotofobie a bolest očí).
V případě výskytu uveitidy není nutná úprava dávkování, pokud účinné lokální léčivé přípravky zvládají oční zánět. Pokud není lokální léčba uveitidy účinná, přerušte podávání dabrafenibu až do vymizení očního zánětu a poté opětovně zahajte podávání dabrafenibu sníženou dávkou o jednu dávkovací hladinu. Změna dávky trametinibu se, je-li užíván v kombinaci s dabrafenibem, po stanovení diagnózy uveitidy nevyžaduje.
V průběhu užívání dabrafenibu v kombinaci s trametinibem může dojít k odchlípení sítnice a okluzi retinální žíly. Další informace jsou uvedeny v SmPC trametinibu (viz bod 4.4). Změna dávky dabrafenibu se, je-li užíván v kombinaci s trametinibem, po stanovení diagnózy odchlípení sítnice a okluze retinální žíly, nevyžaduje.
Pyrexie
Horečka byla hlášena v klinických studiích s dabrafenibem v monoterapii a v kombinaci dabrafenibu s trametinibem (viz bod 4.8). U 1 % pacientů v klinických studiích byly zaznamenány závažné neinfekční febrilie, které byly definovány jako horečky doprovázené těžkým rigorem, dehydratací, hypotenzí a/nebo akutním renálním selháním prerenálního původu u jedinců s původně normálními renálními funkcemi (viz bod 4.8). Nástup těchto závažných neinfekčních febrilií byl typicky zaznamenán v průběhu prvního měsíce léčby dabrafenibem v monoterapii. Pacienti s těžkými neinfekčními febriliemi dobře reagovali na přerušení léčby a/nebo snížení dávky a podpůrnou léčbu.
Výskyt a závažnost horečky jsou vyšší v případě kombinační terapie. Ve studii MEK115306 byly v rameni s kombinační terapií hlášeny případy horečky u 57 % (119/209) pacientů, 7 % z nich bylo stupně 3, ve srovnání s ramenem dabrafenibu v monoterapii, kde byla horečka hlášena u 33 %
(69/211) pacientů, z toho stupně 3 u 2 % případů.
U zhruba poloviny pacientů užívajících dabrafenib v kombinaci s trametinibem, u kterých se rozvinula horečka, došlo k jejímu prvnímu výskytu v prvním měsící užívání léků, a zhruba třetina pacientů měla horečku třikrát či vícekrát.
Pokud má pacient tělesnou teplotu > 38,5 °C, je třeba léčbu dabrafenibem přerušit (informace o změně dávkování naleznete v tabulce 2). Pacienty je třeba vyšetřit s ohledem na známky a příznaky infekce. Pokud horečka ustoupí, lze léčbu dabrafenibem zahájit znovu, a to společně s podáním vhodné profylaxe ve formě nesteroidních protizánětlivých léčivých přípravků nebo paracetamolu. Pokud je horečka spojena s dalšími závažnými známkami nebo příznaky, lze léčbu po poklesu horečky a na základě klinického obrazu znovu zahájit, ale s použitím nižší dávky dabrafenibu (viz bod 4.2). Je-li v kombinaci s dabrafenibem užíván trametinib, změna dávky trametinibu se nevyžaduje.
Snížení ejekční frakce levé komory (LVEF)/dysfunkce levé komory
Snížení ejekční frakce levé komory bylo hlášeno u pacientů léčených dabrafenibem v kombinaci s trametinibem. Další informace jsou uvedeny v SmPC trametinibu (viz bod 4.4). Je-li dabrafenib užíván v kombinaci s trametinibem, změna dávky dabrafenibu se nevyžaduje.
Renální selhání
Renální selhání bylo hlášeno u < 1 % pacientů léčených dabrafenibem samostatně a u <1 % pacientů léčených dabrafenibem v kombinaci s trametinibem. Hlášené případy byly obecně spojeny s pyrexií a dehydratací a dobře reagovaly na přerušení léčby a obecná podpůrná opatření. Byla hlášena granulomatózní nefritida (viz bod 4.8). V průběhu léčby má být u pacientů rutinně monitorován sérový kreatinin. Při zvýšení hladiny kreatininu může být, pokud je to klinicky odpovídající, zapotřebí léčbu dabrafenibem přerušit. Použití dabrafenibu nebylo studováno u pacientů s renální insuficiencí (definovanou jako kreatinin > 1,5x ULN), proto je v těchto případech zapotřebí opatrnost (viz bod 5.2).
Účinky na játra
V klinických studiích s dabrafenibem užívaným v kombinaci s trametinibem byly hlášeny nežádoucí účinky na játra (viz bod 4.8). Doporučuje se, aby u pacientů léčených dabrafenibem v kombinaci s trametinibem bylo prováděno monitorování jaterních funkcí každé čtyři týdny po dobu 6 měsíců po zahájení terapie trametinibem. V monitorování jaterních funkcí je možno dále pokračovat podle klinické potřeby. Další informace naleznete v SmPC trametinibu.
Hypertenze
V souvislosti s dabrafenibem užívaným v kombinaci s trametinibem bylo hlášeno zvýšení krevního tlaku, a to u pacientů s preexistující hypertenzí i bez ní (viz bod 4.8). Další informace jsou uvedeny v SmPC trametinibu.
Intersticiální plicní onemocnění (ILD)/Pneumonitida
V klinických studiích s dabrafenibem užívaným v kombinaci s trametinibem byly hlášeny případy pneumonitidy nebo intersticiálního plicního onemocnění. Další informace jsou uvedeny vbodě 4.4 SmPC trametinibu.
Vyrážka byla pozorována u přibližně 25 % pacientů v klinických studiích s dabrafenibem užívaným v kombinaci s trametinibem. Další informace jsou uvedeny v bodě 4.4 SmPC trametinibu.
Rhabdomyolýza
Rhabdomyolýza byla hlášena u pacientů užívajících dabrafenib v kombinaci s trametinibem (viz bod 4.8). Další informace jsou uvedeny v bodě 4.4 SmPC trametinibu.
Pankreatitida
Pankreatitida byla hlášena u méně než 1 % jedinců léčených dabrafenibem v monoterapii a v kombinaci s trametinibem. Jeden z těchto případů se vyskytl první den léčby dabrafenibem a znovu se vrátil i po opakovaném zahájení léčby nižší dávkou. Nevysvětlitelné bolesti břicha je nutné neprodleně vyšetřit, včetně stanovení hladiny sérových amyláz a lipáz. Při opětovném zahájení léčby po atace pankreatitidy je třeba pacienty pečlivě monitorovat.
Hluboká žilní trombóza (DVT)/Plicní embolie (PE)
Plicní embolie nebo hluboká žilní trombóza se mohou vyskytnout v průběhu užívání dabrafenibu v kombinaci s trametinibem. Pokud se u pacientů objeví symptomy plicní embolie nebo hluboké žilní trombózy, jako jsou dušnost, bolest na hrudi nebo otok rukou či nohou, musí ihned vyhledat lékařskou pomoc. V případě život ohrožující plicní embolie léčbu trametinibem a dabrafenibem trvale ukončete.
Účinky jiných látek na dabrafenib
Dabrafenib je substrátem CYP2C8 a CYP3A4. Je-li to možné, je třeba se vyvarovat podávání silných induktorů těchto enzymů, protože tyto látky mohou snižovat účinnost dabrafenibu (viz bod 4.5).
Látky, které zvyšují pH žaludku, mohou snižovat biologickou dostupnost dabrafenibu a jejich podávání je třeba se vyvarovat, je-li to možné (viz bod 4.5).
Účinky dabrafenibu na jiné látky
Dabrafenib je induktor metabolizujících enzymů, což může vést ke ztrátě účinnosti mnoha běžně užívaných léčivých přípravků (viz příklady v bodě 4.5). Při zahájení léčby dabrafenibem je proto zásadní hodnocení DUR (drug utilisation review). Současného užívání dabrafenibu s léčivými přípravky, které jsou citlivými substráty určitých metabolických enzymů nebo transportérů (viz bod 4.5), je obecně třeba se vyvarovat, pokud není možné monitorování účinnosti a úprava dávky.
Současné podávání dabrafenibu s warfarinem vede ke snížení expozice warfarinu. Při současném podávání dabrafenibu s warfarinem a při ukončení léčby dabrafenibem je třeba postupovat s opatrností a doporučuje se monitorovat INR (International Normalized Ratio) (viz bod 4.5).
Současné podávání dabrafenibu a digoxinu může vést ke snížení expozice digoxinu. Při současném podávání dabrafenibu s digoxinem (substrátem přenašeče) a při ukončení léčby dabrafenibem je třeba postupovat s opatrností a doporučuje se dodatečné monitorování digoxinu (viz bod 4.5).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Účinky jiných léčivých přípravků na dabrafenib
Dabrafenib je substrátem enzymů CYP2C8 a CYP3A4, zatímco aktivní metabolity hydroxy-dabrafenib a desmethyl-dabrafenib jsou substráty CYP3A4. Léčivé přípravky, které jsou silnými inhibitory nebo induktory CYP2C8 nebo CYP2A4, proto budou s velkou pravděpodobností zvyšovat resp. snižovat koncentrace dabrafenibu. Při podávání s dabrafenibem je třeba zvažovat alternativní látky, je-li to možné. Pokud se společně s dabrafenibem podávají silné inhibitory (např. ketokonazol, gemfibrozil, nefazodon, klarithromycin, ritonavir, sachinavir, telithromycin, itrakonazol, vorikonazol, posakonazol, atazanavir), je třeba postupovat s opatrností. Je nutné se vyvarovat současného podávání dabrafenibu se silnými induktory CYP2C8 a CYP3A4 [např. rifampicin, fenytoin, karbamazepin, fenobarbital nebo třezalka tečkovaná (Hypericum perforatum)].
Podávání ketokonazolu (inhibitor CYP3A4) v dávce 400 mg jednou denně s dabrafenibem v dávce 75 mg dvakrát denně vedlo k 71 % zvýšení AUC dabrafenibu a 33 % zvýšení Cmax dabrafenibu v porovnání s hodnotami při podávání samotného dabrafenibu v dávce 75 mg dvakrát denně. Současné podávání vedlo k 82% zvýšení AUC hydroxy-dabrafenibu a 68 % zvýšení AUC desmethyl-dabrafenibu. U karboxy-dabrafenibu bylo zaznamenáno 16 % snížení AUC.
Podávání gemfibrozilu (inhibitor CYP2C8) v dávce 600 mg dvakrát denně s dabrafenibem v dávce 75 mg dvakrát denně vedlo k 47 % zvýšení AUC dabrafenibu, ale neovlivnilo Cmax dabrafenibu v porovnání s hodnotami při podávání samotného dabrafenibu v dávce 75 mg dvakrát denně. Gemfibrozil neměl klinicky významný účinek na systémovou expozici metabolitům dabrafenibu (< 13 %).
Rozpustnost dabrafenibu je závislá na pH, při vyšším pH se rozpustnost snižuje. Léčivé přípravky, jako jsou inhibitory protonové pumpy, které inhibují sekreci kyselé žaludeční kyseliny, mohou zvyšovat žaludeční pH a snižovat rozpustnost dabrafenibu a tím snížit jeho biologickou dostupnost. Nebyly provedeny žádné klinické studie, které by hodnotily vliv pH na farmakokinetiku dabrafenibu. Vzhledem k teoretickému riziku, že látky, které zvyšují pH, mohou snižovat biologickou dostupnost dabrafenibu po perorálním podání a jeho expozici, je třeba se v průběhu léčby dabrafenibem vyvarovat současného podávání léčivých přípravků, které zvyšují žaludeční pH.
Účinky dabrafenibu na jiné léčivé přípravky
Dabrafenib je induktor enzymů a zvyšuje syntézu enzymů, které metabolizují léky, včetně CYP3A4, CYP2C a CYP2B6 a může zvyšovat syntézu transportérů. Toto vede ke snížení plazmatických hladin léčivých přípravků metabolizovaných těmito enzymy a může ovlivnit některé transportované léčivé přípravky. Snížení plazmatických koncentrací může vést ke ztrátě nebo snížení klinického účinku těchto léčivých přípravků. Existuje rovněž riziko zvýšení tvorby aktivních metabolitů těchto léčivých přípravků. Enzymy, které mohou být indukovány, zahrnují CYP3A v játrech a ve střevě, CYP2B6, CYP2C8, CYP2C9, CYP2C19 a UGT (glukuronid-konjugující enzymy). Transportní protein Pgp může být rovněž indukován stejně jako další transportéry, např. MRP-2, BCRP a OATP1B1/1B3.
In vitro způsoboval dabrafenib na dávce závislé zvýšení CYP3B6 a CYP3A4. Ve studii klinických lékových interakcí klesaly Cmax a AUC perorálně podaného midazolamu (substrát CYP3A4) o 61 %, resp. 74 %, pokud byl midazolam podávaný společně s opakovanými dávkami dabrafenibu při použití lékové formy s nižší biologickou dostupností, než má léková forma dabrafenibu.
Podávání dabrafenibu v dávce 150 mg dvakrát denně spolu s warfarinem vedlo ke snížení AUC S-warfarinu o 37 % a snížení AUC R-warfarinu o 33 % v porovnání s hodnotami při podávání samotného warfarinu. Cmax S-warfarinu se zvýšila o 18 % a Cmax R-warfarinu se zvýšila o 19 %.
Očekávají se interakce s mnoha léčivými přípravky, které jsou eliminovány prostřednictvím metabolismu nebo aktivního transportu. Pokud jsou jejich terapeutické účinky pro pacienty velmi důležité a pokud úprava dávky není snadno proveditelná na základně monitorování účinnosti nebo plazmatických koncentrací, je třeba se podávání těchto léčivých přípravků vyvarovat nebo je používat s opatrností. Očekává se vyšší riziko jaterního poškození po podání paracetamolu u pacientů, kteří jsou současně léčeni induktory jaterních enzymů.
Očekává se velký počet ovlivněných léčivých přípravků, i když rozsah interakcí se bude různit. Skupiny léčivých přípravků, které mohou být ovlivněné, zahrnují, ale nejsou omezeny pouze na:
• Analgetika (např. fentanyl, methadon);
• Antibiotika (např. klarithromycin, doxycyklin);
• Protinádorová léčiva (např. kabazitaxel);
• Antikoagulancia (např. acenokumarol, warfarin, viz bod 4.4);
• Antiepileptika (např. karbamazepin, fenytoin, primidon, kyselina valproová);
• Antipsychotika (např. haloperidol);
• Blokátory kalciových kanálů (např. diltiazem, felodipin, nikardipin, nifedipin, verapamil);
• Srdeční glykosidy (např. digoxin, viz bod 4.4);
• Kortikosteroidy (např. dexamethazon, methylprednisolon);
• HIV antivirotika (např. amprenavir, atazanavir, darunavir, delavirdin, efavirenz, fosamprenavir, indinavir, lopinavir, nelfinavir, sachinavir, tipranavir);
• Hormonální antikoncepce (viz bod 4.6);
• Hypnotika (např. diazepam, midazolam, zolpidem);
• Imunosupresiva (např. cyklosporin, takrolimus, sirolimus);
• Statiny metabolizované prostřednictvím CYP3A4 (např. atorvastatin, simvastatin).
K nástupu indukce dojde pravděpodobně po 3 dnech opakovaného podávání dabrafenibu. Po přerušení léčby dabrafenibem je ústup indukce postupný, koncentrace citlivých substrátů CYP3A4, CYP2B6, CYP2C8, CYP2C9 a CYP2C19, UDP-glukuronyltransferázy (UGT) a transportérů může být zvýšena a tyto látky je třeba monitorovat pro možnou toxicitu a může být nutná úprava dávkování těchto látek.
In vitro je dabrafenib inhibitorem CYP3A4. Proto může být během prvních několika dnů léčby pozorována přechodná inhibice CYP3A4.
Účinky dabrafenibu na transportní systém látek
Dabrafenib je in vitro inhibitor lidských transportérů organických aniontů (OATP) 1B1 (OATP1B1) a OATP1B3 a klinický význam nelze vyloučit. Proto je při současném podávání dabrafenibu se substráty OATP1B1 nebo OATP1B3, jako jsou statiny, nutná opatrnost.
Ačkoli byly dabrafenib a jeho metabolity, hydroxy-dabrafenib, karboxy-dabrafenib a desmethyl-dabrafenib, inhibitory lidských transportérů organických aniontů (OAT) 1 a OAT3 in vitro, riziko lékových interakcí je na základě klinických expozic minimální. U dabrafenibu a desmethyl-dabrafenibu byla rovněž pozorována středně významná inhibice lidských proteinů nádorové rezistence BCRP (breast cancer resistence protein), avšak na základě klinické expozice je riziko lékových interakcí minimální.
Léčba v kombinaci s trametinibem
Opakované podávání dávky trametinibu 2 mg jednou denně společně s dabrafenibem 150 mg dvakrát denně nevedlo ke klinicky významným změnám Cmax a AUC trametinibu nebo dabrafenibu se zvýšením o 16 % Cmax a 23 % AUC dabrafenibu. Nepatrné snížení biodostupnosti trametinibu, což odpovídá snížení 12 % AUC, se odhadovalo při podávání v kombinaci s dabrafenibem, CYP3A4 induktorem, za použití PK analýzy.
Informace o interakcích při užívání dabrafenibu v kombinaci s trametinibem jsou uvedeny v bodech 4.4 a 4.5 v SmPC dabrafenibu a trametinibu.
Vliv jídla na účinky dabrafenibu
Pacienti by měli užívat dabrafenib v monoterapii nebo v kombinaci s trametinibem alespoň jednu hodinu před jídlem nebo dvě hodiny po jídle, vzhledem k vlivu jídla na absorpci dabrafenibu (viz bod 5.2).
Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/antikoncepce u žen
Ženy ve fertilním věku musí používat účinnou antikoncepci v průběhu léčby a po dobu 4 týdnů po ukončení léčby dabrafenibem a 4 měsíce po poslední dávce trametinibu, je-li podáván v kombinaci s dabrafenibem.Dabrafenib může snižovat účinnost hormonální antikoncepce, a proto je třeba používat alternativní metodu antikoncepce, jako je např. bariérová metoda (viz bod 4.5).
Údaje o používání dabrafenibu u těhotných žen nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu a toxicity s ohledem na embryofetální vývoj, včetně teratogenních účinků (viz bod 5.3). Dabrafenib se nemá podávat těhotným ženám, pokud prospěch pro matku nepřeváží možná rizika pro plod. Pokud žena otěhotní v průběhu léčby dabrafenibem, musí být informována o možném riziku pro plod. Další informace o užívání trametinibu v kombinaci s dabrafenibem jsou uvedeny v SmPC trametinibu (viz bod 4.6).
Kojení
Není známo, zda se dabrafenib vylučuje do mateřského mléka. Protože se do mateřského mléka vylučuje mnoho léčivých přípravků, nelze riziko pro kojené dítě vyloučit. Rozhodnutí, zda kojení přerušit, nebo zda přerušit léčbu dabrafenibem, je třeba učinit s přihlédnutím k prospěchu kojení pro dítě a prospěchu z léčby pro ženu.
Fertilita
Údaje týkající se člověka pro užívání dabrafenibu buď v monoterapii, nebo v kombinaci s trametinibem nejsou k dispozici. Dabrafenib může narušit fertilitu u mužů i žen, protože nežádoucí účinky na samičí a samčí reprodukční orgány byly pozorovány u zvířat (viz bod 5.3). Pacienti (muži) užívající dabrafenib buď v monoterapii, nebo v kombinaci s trametinibem musí být informováni o možném riziku poškození spermatogeneze, které může být ireverzibilní.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Dabrafenib má malý vliv na schopnost řídit a obsluhovat stroje. Při zvažování, zda je pacient schopný provádět činnosti, které vyžadují úsudek a motorické nebo kognitivní schopnosti, je třeba vzít v úvahu klinický stav pacienta a profil nežádoucích účinků dabrafenibu. Pacienti by měli být poučeni o možné únavě a potížích se zrakem, které mohou tyto činnosti ovlivnit.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnostní profil monoterapie dabrafenibem je založen na údajích z pěti klinických studií, které zahrnovaly 578 pacientů s melanomem. Nejčastější nežádoucí účinky (s četností 15 % a více) hlášené v souvislosti s léčbou dabrafenibem byly hyperkeratóza, bolest hlavy, pyrexie, artralgie, únava, nauzea, papilom, alopecie, vyrážka a zvracení.
Bezpečnost dabrafenibu v kombinaci s trametinibem se hodnotila ve dvou studiích fáze III MEK115306 a MEK116513, kde byly provedeny analýzy bezpečnosti dabrafenibu v kombinaci s trametinibem u 209, respektive 350 pacientů, kteří měli neresektovatelný nebo metastatický melanom pozitivní na mutaci V600 genu BRAF a užívali dabrafenib (150 mg dvakrát denně) a trametinib (2 mg jednou denně) v kombinované terapii (viz informace týkající se kombinované terapie v bodě 5.1). Nejčastější nežádoucí účinky (> 20 %) dabrafenibu a trametinibu v kombinaci zahrnují horečku, únavu, bolest hlavy, zimnici, průjem, vyrážku, bolest kloubů, hypertenzi, zvracení a kašel.
Souhrn nežádoucích účinků v tabulce
Nežádoucí účinky, které byly hlášeny, jsou shrnuty níže podle tříd orgánů MedDRA a podle jejich četnosti. Ke klasifikaci byla použita následující konvence:
Velmi časté Časté
Méně časté Vzácné Není známo
> 1/10
> 1/100 až < 1/10
> 1/1 000 až < 1/100
> 1/10 000 až < 1/1 000
(z dostupných údajů nelze určit)
Monoterapie dabrafenibem
Tabulka 3 Nežádoucí účinky hlášené ve studiích s melanomem
|
Třída orgánových systémů |
Četnost (všechny stupně) |
Nežádoucí účinky |
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy) |
Velmi časté |
Papilom |
|
Časté |
Kožní spinocelulární karcinom | |
|
Seborhoická keratóza | ||
|
Akrochordon (kožní přívěsky) | ||
|
Bazocelulární karcinom | ||
|
Méně časté |
Nově diagnostikovaný primární melanom | |
|
Poruchy imunitního systému |
Méně časté |
Hypersenzitivita |
|
Poruchy metabolismu a výživy |
Velmi časté |
Snížení chuti k jídlu |
|
Časté |
Hypofosfatémie | |
|
Hyperglykémie | ||
|
Poruchy nervového systému |
Velmi časté | |
|
Poruchy oka |
Méně časté |
Uveitida |
|
Respirační, hrudní a mediastinální poruchy |
Velmi časté | |
|
Gastrointestinální poruchy |
Velmi časté | |
|
Časté |
Zácpa | |
|
Méně časté |
Pankreatitida |
|
Poruchy kůže a podkožní tkáně |
Velmi časté |
Hyperkeratóza |
|
Alopecie | ||
|
Syndrom palmo-plantární erythrodyzestezie | ||
|
Časté |
Suchá kůže | |
|
Pruritus | ||
|
Aktinická keratóza | ||
|
Erytém | ||
|
Méně časté |
Panikulitida | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Velmi časté |
Artralgie |
|
Myalgie | ||
|
Bolest končetin | ||
|
Poruchy ledvin a močových cest |
Méně časté |
Selhání ledvin, akutní renální selhání |
|
Nefritida | ||
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Pyrexie |
|
Únava | ||
|
Astenie | ||
|
Časté |
Příznaky podobné chřipce | |
|
Vyšetření |
Časté |
Pokles LVEF |
|
Méně časté |
Prodloužení QT intervalu |
Terapie kombinací dabrafenib a trametinib
Tabulka 4 Nežádoucí účinky, které se vyskytly ve dvou randomizovaných studiích fáze III
(kombinace trametinibu s dabrafenibem) - MEK115306 (n = 209) a MEK116513 a (n = 350)
|
Třída orgánových systémů |
Četnost (všechny stupně) |
Nežádoucí účinky |
|
Infekce a infestace |
Velmi časté |
Infekce močových cest |
|
Nazofaryngitida | ||
|
Časté |
Celulitida | |
|
Folikulitida | ||
|
Paronychium | ||
|
Pustulární vyrážka | ||
|
Novotvary benigní, maligní a nespecifikované (včetně cyst a polypů) |
Časté |
Spinocelulární karcinomb |
|
Papilomc | ||
|
Seboroická keratóza | ||
|
Akrochordon (kožní přívěsky) | ||
|
Méně časté |
Nový primární melanom | |
|
Poruchy krve a lymfatického systému |
Velmi časté |
Neutropenie |
|
Časté |
Anemia | |
|
T rombocytopenie | ||
|
Leukopenie | ||
|
Poruchy imunitního systému |
Méně časté |
Hypersenzitivita na lék |
|
Poruchy metabolismu a výživy |
Velmi časté |
Snížena chuť k jídlu |
|
Časté |
Dehydratace | |
|
Hyponatremie | ||
|
Hypofosfatemie | ||
|
Hyperglykémie |
|
Poruchy nervového systému |
Velmi časté | |
|
Závratě | ||
|
Poruchy oka |
Časté |
Rozmazané vidění |
|
Poruchy vidění | ||
|
Méně časté |
Chorioretinopatie | |
|
Uveitida | ||
|
Odchlípení sítnice | ||
|
Periorbitální edém | ||
|
Srdeční poruchy |
Časté |
Snížení ejekční frakce |
|
Bradykardie | ||
|
Cévní poruchy |
Velmi časté |
Hypertenze |
|
Hemoragied | ||
|
Časté | ||
|
Méně časté |
Lymfedéma | |
|
Respirační, hrudní a mediastinální poruchy |
Velmi časté | |
|
Časté | ||
|
Méně časté |
Pneumonitida | |
|
Gastrointestinální poruchy |
Velmi časté |
Abdominální bolest |
|
Zácpa | ||
|
Časté |
Sucho v ústech | |
|
Stomatitida | ||
|
Méně časté |
Pankreatitida | |
|
Hepatobiliární poruchy |
Velmi časté |
Zvýšení ALT |
|
Zvýšení AST | ||
|
Časté |
Zvýšení alkalické fosfatázy v krvi | |
|
Zvýšení gamaglutamyl transferázy v krvi | ||
|
Poruchy kůže a podkožní tkáně |
Velmi časté |
Suchá kůže |
|
Pruritus | ||
|
Akneiformní dermatitida | ||
|
Časté |
Erytém | |
|
Aktinická keratóza | ||
|
Noční pocení | ||
|
Hyperkeratóza | ||
|
Alopecie | ||
|
Syndrom palmo-plantární erythrodysestezie | ||
|
Hyperhidróza | ||
|
Panikulitida | ||
|
Kožní fisury | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Velmi časté |
Artralgie |
|
Myalgie | ||
|
Bolest končetin | ||
|
Časté |
Svalové spazmya | |
|
Zvýšení kreatinkinázy v krvi |
|
Poruchy ledvin a močových cest |
Méně časté |
Renální selhání3 |
|
Nefritida | ||
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Únava |
|
Astenie | ||
|
Periferní otok | ||
|
Časté |
Zánět sliznice | |
|
Nemoc podobná chřipke | ||
|
Otok obličeje | ||
|
Vyšetření |
Časté |
Snížená tepová frekvence |
|
a Bezpečnostní profil ze studie MEK116513 je obecně podobný profilu ze studie MEK115306 s následujícími výjimkami:1) následující nežádoucí účinky mají vyšší stupeň četnosti v porovnání se studii MEK115306: svalový spasmus (velmi časté), renální selhání a lymfedém (časté), akutní renální selhání (méně časté); 2) následující nežádoucí účinky se vyskytly v MEK116513 studii, nikoliv ve studii MEK115306: srdeční selhání, dysfunkce levé srdeční komory, intersticiální plicní onemocnění, rhabdomyolýza (méně časté). bcuSCC : SCC kůže, SCC in situ (Bowenova nemoc) a keratoakantom c Papilom, kožní papilom d Krvácení z různých míst, včetně intrakraniálního krvácení a fatálního krvácení | ||
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Popis vybraných nežádoucích účinků
Kožní spinocelulární karcinom
Kožní spinocelulární karcinom (který zahrnuje léze klasifikované jako podtyp keratoakantomu nebo smíšeného keratoakantomu) byl zaznamenán u 9 % pacientů léčených monoterapií dabrafenibem v integrované bezpečnostní populaci a u 3 % pacientů léčených dabrafenibem v kombinaci s trametinibem v studii MEK115306. Přibližně 70 % příhod se objevilo v průběhu prvních 12 týdnů léčby monoterapií dabrafenibem, s mediánem doby do vzniku obtíží 8 týdnů. U pacientů, kteří užívali kombinaci dabrafenibu s trametinibem, se příhody objevily později s mediánem doby do vzniku obtíží 22 týdnů. 96 % pacientů, užívajících dabrafenib v monoterapii v integrované bezpečnostní populaci a všichni pacienti s kombinační terapií ve studiích fáze III, u kterých došlo k vývoji kožního spinocelulárního karcinomu, pokračovalo v léčbě bez úpravy dávky.
Nově diagnostikovaný _primární melanom
Nově diagnostikovaný primární melanom byl hlášený v klinických studiích s dabrafenibem jak v monoterapii, tak v kombinaci s trametinibem. Případy byly léčeny excizí a nevyžadovaly úpravu dávkování (viz bod 4.4).
Malignity mimo kožní lokalizaci
Aktivace signalizace MAP-kinázy u buněk s divokým typem genu BRAF, které jsou vystavené účinku inhibitorů BRAF, může vést ke zvýšení rizika vzniku malignit mimo kožní lokalizaci, včetně malignit obsahujících mutaci RAS (viz bod 4.4). V klinických studiích byly hlášeny případy malignit mimo kožní lokalizaci u 1 % (6/586) pacientů léčených dabrafenibem v monoterapii a u 1 % (3/209) pacientů v studii MEK115306 a méně než 1 % (3/350) pacientů v studii MEK116513 s dabrafenibem v kombinaci s trametinibem. Při léčbě dabrafenibem jak v monoterapii, tak v kombinaci s trametinibem byly pozorovány případy malignit odvozených od RAS. Pacienti by měli být monitorováni klinicky odpovídajícím způsobem.
U pacientů užívajících dabrafenib v kombinaci s trametinibem, se vyskytly hemoragické příhody, včetně těžkých hemoragických příhod a fatálních hemoragií. Další informace jsou uvedeny v SmPC trametinibu.
Snížení ejekční_frakce levé komory (LVEF)/dysfunkce levé komory
Ve dvou klinických studiích fáze III v integrované bezpečnostní populaci byl hlášen pokles LVEF u 1 % pacientů léčených dabrafenibem v monoterapii a u 6 až 8 % pacientů léčených dabrafenibem v kombinaci s trametinibem, většina případů byla asymptomatická a reverzibilní. Pacienti s LVEF nižším, než je institucionální dolní limit normálních hodnot, nebyli do klinických studií s dabrafenibem zařazeni. U pacientů se stavy, které mohou ovlivnit funkci levé komory, má být dabrafenib v kombinaci s trametinibem používán s opatrností.
Pyrexie
Horečka byla hlášena v klinických studiích s dabrafenibem jak v monoterapii, tak v kombinaci s trametinibem (viz bod 4.8), výskyt a závažnost horečky jsou vyšší v případě kombinační terapie (viz SmPC dabrafenibu bod 4.4). U zhruba poloviny pacientů užívajících dabrafenib v kombinaci s trametinibem, u kterých se rozvinula horečka, došlo k jejímu prvnímu výskytu v prvním měsící užívání léků, a zhruba třetina pacientů měla horečku třikrát či vícekrát. V integrované bezpečností populaci pacientů užívajících monoterapii dabrafenibem byly u 1 % pacientů v klinických studiíchzaznamenány závažné neinfekční febrilie, které byly definovány jako horečky doprovázené těžkou ztuhlostí, dehydratací, hypotenzí a/nebo akutním renálním selháním prerenálního původu u jedinců s původně normálními renálními funkcemi. Nástup těchto závažných neinfekčních febrilií byl typicky zaznamenán v průběhu prvního měsíce léčby. Pacienti s těžkými neinfekčními febriliemi dobře reagovali na přerušení léčby a/nebo snížení dávky a podpůrnou léčbu (viz body 4.2 a 4.4).
Účinky na játra
V klinických studiích s dabrafenibem v kombinaci s trametinibem (viz bod 4.8) byly hlášeny jaterní nežádoucí účinky. Další informace jsou uvedeny v SmPC trametinibu.
Hypertenze
V souvislosti s dabrafenibem užívaným v kombinaci s trametinibem bylo hlášeno zvýšení krevního tlaku, a to u pacientů s preexistující hypertenzí i bez ní. Krevní tlak má být měřen před zahájením léčby a monitorován v jejím průběhu, hypertenze má být léčena odpovídající standardní terapií.
Artralgie
Artralgie byla v klinických studiích s dabrafenibem jak v monoterapii, tak v kombinované terapii s trametinibem, hlášená velmi často (přibližně 25 %), ačkoli tyto příhody byly převážně 1. a 2. stupně závažnosti, 3. stupeň se vyskytoval méně často (méně než 1 %) a stupeň 4 nebyl zaznamenán vůbec.
Hypofosfatémie
Hypofosfatémie byla v integrované bezpečnostní populaci pacientů léčených monoterapií dabrafenibem v klinických studiích fáze III hlášena u 7 % pacientů, při léčbě kombinací s trametinibem u 3 až 4 % pacientů. Je třeba poznamenat, že přibližně polovina těchto případů hlášených u pacientů léčených dabrafenibem v monoterapii (4 %) a < 1 % případů hlášených u pacientů léčených dabrafenibem v kombinaci s trametinibem byly stupně závažnosti 3.
Pankreatitida byla hlášena u pacientů léčených monoterapií dabrafenibem a kombinací dabrafenibu s trametinibem. Nevysvětlitelná bolest břicha musí být neprodleně vyšetřena, včetně stanovení hladiny sérové amylázy a lipázy. Při opětovném zahajování léčby dabrafenibem po prodělané epizodě pankreatitidy by pacienti měli být pečlivě monitorováni (viz bod 4.4).
Renální selhání
Renální selhání způsobené s pyrexií související prerenální azotémií nebo granulomatózní nefritidou bylo méně časté, dabrafenib však nebyl hodnocen u pacientů s renální insuficiencí (definovanou jako kreatinin > 1,5x ULN). V těchto případech je při léčbě třeba postupovat s opatrností (viz bod 4.4).
Zvláštní populace
Starší _ pacienti
Ze všech pacientů zařazených do klinických studií s dabrafenibem (n = 578) bylo 22 % ve věku 65 let a starších, 6 % bylo ve věku 75 let a starších. Ve srovnání s mladšími jedinci (< 65 let) mělo více jedinců > 65 let nežádoucí účinky, které vedly ke snížení dávky studijního léku (22 % vs. 12 %) nebo přerušení léčby (39 % vs. 27 %). U starších pacientů byly navíc ve srovnání s mladšími pacienty častěji zaznamenávány závažnější nežádoucí účinky (41 % vs. 22 %). Žádné celkové rozdíly v účinnosti nebyly mezi mladšími a staršími jedinci pozorovány.
V klinických studiích fáze III MEK115306 (n = 209) a MEK116513 (n = 350) s dabrafenibem v kombinaci s trametinibem u pacientů s neresektovatelným nebo metastatickým melanomem 56 pacientů (27 %) (studie MEK115306), respektive 77 pacientů (22 %) (studie MEK116513) bylo > 65 let, 11 pacientů (5 %), respektive 21 pacientů (6 %), bylo > 75 let. Podíl pacientů, u kterých se objevily nežádoucí účinky, byl podobný u skupiny pacientů ve věku < 65 let a u skupiny pacientů ve věku > 65 let v obou studiích. U pacientů > 65 let vedly nežádoucí účinky a závažné nežádoucí účinky s větší četností k trvalému vysazení léčivého přípravku, snížení dávky a přerušení léčby, než u pacientů < 65 let.
4.9 Předávkování
Pro předávkování dabrafenibem není k dispozici žádná specifická léčba. V případě předávkování je třeba zahájit podpůrnou léčbu a odpovídající monitoraci pacienta.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, inhibitory proteinkinázy, ATC kód: L01XE23 Mechanismus účinku
Dabrafenib je inhibitor RAF kináz. Onkogenní mutace genu BRAF vedou ke konstitutivní aktivaci dráhy RAS/RAF/MEK/ERK. Mutace genu BRAF byly zaznamenány s vysokou frekvencí výskytu u specifických nádorových onemocnění, včetně asi u 50 % melanomů. Nejčastější mutací genu BRAF je V600E, která tvoří přibližně 90 % mutací genu BRAF pozorovaných u melanomu.
Preklinické údaje získané při biochemických studiích ukázaly, že dabrafenib inhibuje BRAF kinázu, přičemž aktivuje mutace kodonu 600 (tabulka 5).
Tabulka 5 Kinázová inhibiční aktivita dabrafenibu ve srovnání s RAF kinázami
|
Kináza |
Inhibiční koncentrace 50 (nmol/l) |
|
BRAF V600E |
0,65 |
|
BRAF V600K |
0,50 |
|
BRAF V600D |
1,8 |
|
BRAF WT |
3,2 |
|
CRAF WT |
5,0 |
Dabrafenib vykazoval supresi následných farmakodynamických biomarkerů (fosforylovaných ERK) a inhiboval buněčný růst buněčných linií melanomu s mutací V600 genu BRAF, a to in vitro i na zvířecích modelech.
U jedinců s melanomem s pozitivní mutací V600 genu BRAF vedlo podávání dabrafenibu k inhibici nádorového fosforylovaného ERK ve srovnání s výchozími hodnotami.
Kombinace s trametinibem
Trametinib je reverzibilní, vysoce selektivní, alosterický inhibitor mitogenem aktivované, mimobuněčným signálem regulované kinázy 1 (MEK1) a aktivace MEK2 a kinázové aktivity.
Proteiny MEK jsou součástí kaskády kináz regulovaných mimobuněčným signálem (ERK).
Trametinib a dabrafenib tedy inhibují dvě různé kinázy této dráhy, MEK a RAF, a kombinace těchto inhibitorů tudíž poskytuje současnou inhibici těchto dvou kináz. Kombinace trametinibu s dabrafenibem je synergická v buněčných liniích in vitro pozitivních na mutaci V600 v genech BRAF a oddaluje vznik rezistence in vivo v buňkách melanomu xenograftů pozitivních na mutaci V600 v genech BRAF.
Stanovení mutace genu BRAF
Před zahájením léčby dabrafenibem nebo kombinací dabrafenibu s trametinibem musí být u pacientů potvrzena pozitivní mutace V600 genu BRAF pomocí validovaného testu. V klinických studiích fáze II a III vyžadoval screening vhodnosti k léčbě centrální provedení testu mutace V600 genu BRAF za použití testu mutace BRAF provedeného na co nejčerstvějších dostupných vzorcích tumoru. Primární tumor nebo tumor z metastatického ložiska byl testován testem pouze pro výzkumné použití IUO (investigational use only assay). IUO je polymerázová řetězová reakce (PCR) specifická pro určitou alelu prováděná na DNA extrahované z formalinem fixované nádorové tkáně zalité v parafinu (FFPE, formalin-fixed paraffin-embedded). Test byl specificky navržený k odlišení mezi mutacemi V600E a V600K. Pouze jedinci s pozitivní mutací V600E nebo V600K genu BRAF byli vhodní k účasti ve studii.
Všechny vzorky od pacientů byly následně znovu testovány za použití validovaného textu bioMerieux (bMx) THxID BRAF, který má certifikaci CE. Test bMx THxID BRAF je PCR specifická pro určitou alelu provedená na DNA extrahované z nádorové tkáně fixované formalinem zalité v parafinu (FFPE). Tato zkouška byla navržena ke stanovení mutací V600E a V600K genu BRAF s vysokou senzitivitou (méně než 5 % sekvence V600E a V600K na pozadí sekvence z divokých typů s použitím DNA extrahované z FFPE tkáňových bloků). Neklinické a klinické studie s retrospektivní dvousměrnou sekvenční analýzou dle Sangera prokázaly, že tyto testy rovněž detekovaly nejméně častou mutaci V600D a V600E/K601E genu BRAF s nižší senzitivitou. U vzorků z neklinických i klinických studií (n = 876), ve kterých byla prokázána pozitivní mutace pomocí testu THxID BRAF a které byly následně sekvenovány s použitím referenční metody, byla specificita této zkoušky 94 %.
Klinická účinnost a bezpečnost
Dabrafenib v kombinaci s trametinibem Pacienti, kteří nebyli dříve léčeni
Bezpečnost a účinnost doporučené dávky trametinibu (2 mg jednou denně) v kombinaci s dabrafenibem (150 mg dvakrát denně) k léčbě dospělých pacientů s neresktovatelným nebo metastatickým melanomem s mutací V600 v genu BRAF byla hodnocena ve dvou studiích fáze III a v jedné podpůrné studii fáze I/II.
MEK115306 (COMBI-d):
MEK115306 byla randomizovaná, dvojitě zaslepená studie fáze III, porovnávající kombinaci dabrafenibu a trametinibu s dabrafenibem a placebem v první linii terapie u pacientů s neresektovatelným (stupeň III) nebo metastatickým (stupeň IV) kožním melanomem s pozitivní mutací V600E/K genu BRAF. Primárním cílovým parametrem bylo přežití bez progrese (progression-free survival, PFS), s klíčovým sekundárním cílovým parametrem celkového přežití (overall survival, OS). Pacienti byli stratifikováni podle hladiny laktát dehydrogenázy (LDH) (> horní limit normálu (ULN) versus < ULN) a mutace v genu BRAF (V600E versus V600K).
Všech 423 pacientů bylo randomizováno 1:1, a to na léčbu kombinovanou terapií (N = 211), nebo dabrafenibem (N = 212). Většina pacientů byla bělošské rasy (> 99 %) a mužského pohlaví (53 %), medián věku činil 56 let (28 % byli pacienti > 65 let). Většina případů (67 %) měla stupeň nemoci IVM1c. Většina pacientů měla LDH <ULN (65 %), ECOG výkonnostní status 0 (72 %) a viscerální postižení (73 %) ve výchozím stavu. Většina pacientů měla mutaci V600E v genu BRAF (85 %). Do studie nebyli zahrnuti pacienti s metastázami v mozku.
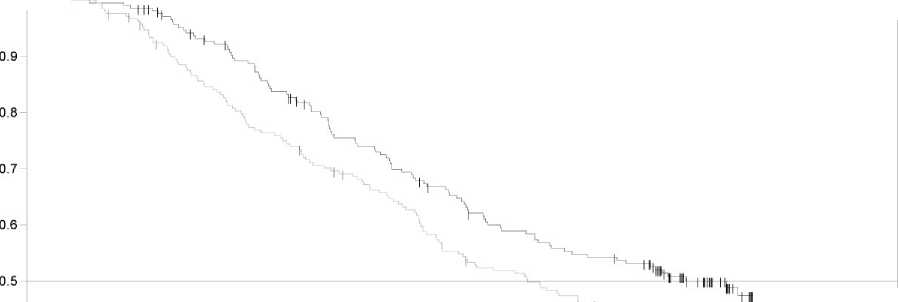
Finální analýza celkového přežití (12. leden 2015) prokázala statisticky významné zlepšení celkového přežití v kombinované terapii ve srovnání s monoterapií dabrafenibem (Obrázek 1). Výskyt celkového přežití v 1 roce po zahájení léčby (74 %) a 2 roky po zahájení léčby (51 %) v rameni kombinované terapie byl vyšší v porovnání s monoterapií dabrafenibem (68 %, respektive 42 %).
Obrázek 1 Kaplan-Meierovy křivky celkového přežití pro studii MEK115306 (ITT populace)
Page 1 of 1
1.0
Dabrafenib + Trametinib Dabrafenib + Placebo
0.4
1 tamti
w tiii um f h i
BliliŤI—1-1-
0.3
Dabrafenib + Trametinib
Dabrafenib + Placebo
0.1
|
Dabrafenib + trametinib (N = 211) |
Dabrafenib + placebo (N = 212) | |||
|
Celkové přežití 12 leden 2015 | ||||
|
Počet případů ( %) |
99 (47 %) |
123 (58 %) | ||
|
Medián celkového přežití (měsíce) |
25,1 |
18,7 | ||
|
Upravený poměr rizik (95 % CI) |
0,71 (0,55 ; 0,92) | |||
|
Stratifikovaná Log-Rank P-hodnota |
0,011 | |||
Number at risk 211 208
212 206
200
191
187
175
174
159
159
147
144
138
135
127
124
111
112
104
106
95
103
88
88
53
21
12 14 16 18 20
Time from Randomization (Months)
Vysvětlivky:
Proportion Alive - Poměr přežívajících pacientů Number at risk - Počet v riziku
Time from Randomization (Months) - Doba od randomizace pacientů (měsíce)
Tabulka 6 Výsledky účinnosti v studii MEK115306 (COMBI-d)
|
Cílový parametr |
Dabrafenib + Trametinib (N = 211) |
Dabrafenib + Placebo (N = 212) |
Dabrafenib + Trametinib (N = 211) |
Dabrafenib + Placebo (N = 212) |
|
Ukončení sběru dat k |
26. srpnu 2013 |
12. lednu 2015 | ||
|
PFSa | ||||
|
Progrese onemocnění nebo úmrtí, n (%) |
102 (48) |
109 (51) |
139 (66) |
162 (76) |
|
Medián PFS (měsíce) (95 % CI) |
9,3 (7,7; 11,1) |
8,8 (5,9; 10,9) |
11,0 (8,0; 13,9) |
8,8 (5,9; 9,3) |
|
Poměr rizik (95 % CI) |
0,75 (0,57; 0,99) |
0,67 (0,53; 0,84) | ||
|
P hodnota |
0,035 |
< 0,001 | ||
|
ORRb (95 % CI) |
67 (59,9; 73,0) |
51 (44,5; 58,4) |
69 (61,8; 74,8) |
53 (46,3; 60,2) |
|
ORR rozdíl (95 % CI) |
15e (5,9; 24,5) |
15e (6,0; 24,5) | ||
|
P hodnota |
0,0015 |
0,0014 | ||
|
DoRc (měsíce) Medián (95 % CI) |
9,2d (7,4; NR) |
10,2d (7,5; NR) |
12,9 (9,4; 19,5) |
10,6 (9,1; 13,8) |
|
a - Přežití bez progrese (hodnoceno investigátorem) b - Četnost celkové odpovědi = Celková odpověď + parciální odpověď c - Trvání odpovědi d - V čase hlášení většina (>59 %) investigátorem hodnocených odpovědí ještě stále probíhala e - ORR rozdíl počítán na základě nezaokrouhleného ORR výsledku NR = Nedosáhnuto | ||||
MEK116513 (COMBI-v):
Studie MEK116513 (COMBI-v) byla 2-ramenná, randomizovaná, otevřená, studie fáze III porovnávající kombinaci dabrafenibu a trametinibu s vemurafenibem v monoterapii u melanomu s pozitivní mutací V600 v genu BRAF. Primárním cílovým parametrem studie bylo celkové přežití s klíčovým sekundárním cílovým parametrem PFS. Pacienti byli stratifikováni podle hladiny laktát dehydrogenázy (LDH) (> horní limit normálu (ULN) versus < ULN) a mutace v genu BRAF (V600E versus V600K).
Všech 704 pacientů bylo randomizováno 1:1, a to buď na léčbu kombinovanou terapií, nebo na léčbu vemurafenibem. Většina pacientů byla bělošské rasy (> 96 %) a mužského pohlaví (55 %), medián věku činil 55 let (24 % byli > 65 let). Většina pacientů (61 % celkově) měla stupeň nemoci IVMlc. U většiny pacientů bylo LDH <ULN (67 %), ECOG výkonnostní status 0 (70 %) a viscerální postižení (78 %) ve výchozím stavu. Většina pacientů měla mutaci V600E v genu BRAF (89 %). Do studie nebyli zahrnuti pacienti s metastázami v mozku.
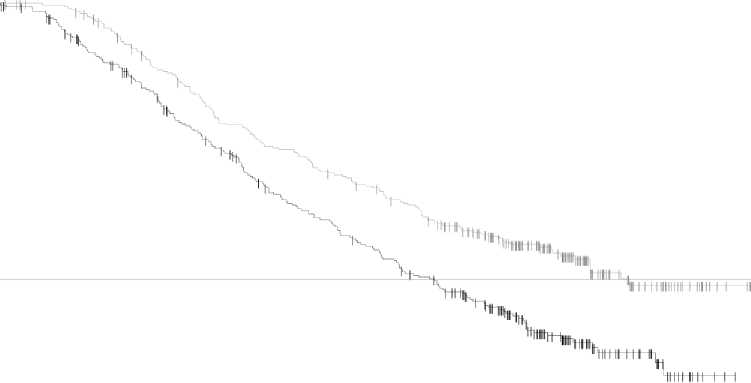
Aktualizovaná analýza celkového přežití (13. Březen 2015) vykazovala statisticky významné zlepšení celkového přežití pro kombinovanou terapii ve srovnání s vemurafenibem v monoterapii (obrázek 2). Výskyt celkového přežití ve 12 měsících byl 72 % u kombinované terapie a 65 % u vemurafenibu.
Obrázek 2 Kaplan-Meierovy křivky aktualizovaných analýz celkového přežití studie MEK116513
Page 1 of1
0.8
0.7
0.3
0.2
Vemurafenib
Dabrafenib + Trametinib
Vemurafenib Dabrafenib + Trametinib
0.0
|
Dabrafenib + trametinib (N = 352) |
Vemurafenib (N = 352) | |||||
|
Celkové přežití 13 Březen 2015 | ||||||
|
Počet případů (%) |
155 (44 %) |
195 (55 %) | ||||
|
Medián celkového přežití (měsíce) |
25,6 |
18,0 | ||||
|
Upravený poměr rizik (95 % CI) |
0,66 (0,53; 0,81) | |||||
|
Stratifikovaná Log-Rank P- |
< 0,001 | |||||
|
hodnota | ||||||
Number at risk 352 341
352 342
315
336
286
311
252
286
231
260
201
245
187
230
166
217
152
198
129
173
88
128
28
38
7
16
12 14 16 18 20
Time from Randomization (Months)
Vysvětlivky:
Proportion Alive - Poměr přežívajících pacientů Number at risk - Počet v riziku
Time from Randomization (Months) - Doba od randomizace pacientů (měsíce)
Tabulka 7 Výsledky účinnosti v studii MEK116513 (COMBI-v)
|
Cílový parametr |
Dabrafenib + Trametinib (N = 352) |
Vemurafenib (N = 352) |
|
PFS | ||
|
Progrese onemocnění nebo |
166 (47) |
217 (62) |
|
úmrtí, | ||
|
n (%) | ||
|
Medián PFS (měsíce) |
11,4 |
7,3 |
|
(95 % CI) |
(9,9; 14,9) |
(5,8; 7,8) |
|
Poměr rizik |
0,56 | |
|
(95 % CI) |
(0,46; 0,69) | |
|
P value |
< 0,001 | |
|
ORRb |
226 (64) |
180 (51) |
|
(95 % CI) |
(59,1; 69,4) |
(46,1; 56,8) |
|
ORR rozdíl |
13 | |
|
(95 % CI) |
(5,7; 20,2) | |
|
P hodnota |
0,0005 | |
|
DoR (měsíce) | ||
|
Medián |
13,8 |
7,5 |
|
(95 % CI) |
(11,0; NR) |
(7,3; 9,3) |
Předchozí léčba BRAF inhibitorem
Údaje o pacientech užívajících kombinaci dabrafenibu s trametinibem, u nichž došlo k progresi onemocnění po předcházející léčbě inhibitorem BRAF, jsou omezené.
Část B studie BRF113220 zahrnovala kohortu 26 pacientů, kteří zprogredovali na léčbě BRAF inhibitorem. Kombinace trametinibu v dávce 2 mg jednou denně s dabrafenibem v dávce 150 mg dvakrát denně vykazovala omezenou klinickou aktivitu u pacientů, kteří zprogredovali na léčbě BRAF inhibitorem. Četnost odpovědi hodnocená zkoušejícími byla 15 % (95 % CI: 4,4; 34,9) a medián PFS byl 3,6 měsíce (95 % CI: 1,9; 5,2). Podobné výsledky byly hlášeny u 45 pacientů, kteří přestoupili z monoterapie dabrafenibem na léčbu kombinací trametinibu 2 mg jednou denně s dabrafenibem 150 mg dvakrát denně v části C této studie. Výskyt odpovědi u těchto pacientů byl 13 % (95 % CI:
5,0; 27,0), medián PFS byl 3,6 měsíce (95 % CI: 2, 4).
Monoterapie dabrafenibem
Účinnost dabrafenibu v léčbě dospělých pacientů s neresekovatelným nebo metastatickým melanomem s pozitivitou mutace V600 genu BRAF byla hodnocena ve 3 studiích [BRF113683 (BREAK-3), BRF113929 (BREAK-MB) a BRF113710 (BREAK-2)], které zahrnovaly pacienty s pozitivitou mutací V600E a/nebo V600K genu BRAF.
Do těchto studií bylo zahrnuto celkem 402 jedinců s mutací V600E genu BRAF a 49 jedinců s mutací V600K genu BRAF. Pacienti s melanomem způsobeným jinou mutací genu BRAF než V600E byli vyloučeni z potvrzovací studie a u pacientů s mutací V600K ve studii s jedním ramenem se účinnost zdála nižší než u pacientů s nádory s mutací V600E.
U pacientů s melanomem nesoucím jiné mutace V600 genu BRAF než V600E a V600K nejsou k dispozici žádné údaje. Účinnost dabrafenibu u jedinců dříve léčených inhibitory proteinkinázy nebyla hodnocena.
Dříve neléčení pacienti [výsledky ze studie fáze III (BREAK-3)]
Účinnost a bezpečnost dabrafenibu byly hodnoceny v randomizované, otevřené studii fáze III (BREAK-3), která porovnávala léčbu dabrafenibem oproti dakarbazinu (DTIC) u dříve neléčených pacientů s pokročilým (neresekovatelným, stupeň III) nebo metastatickým (stupeň IV) melanomem s pozitivní mutací V600E genu BRAF. Pacienti s melanomem způsobeným jinou mutací genu BRAF než V600E byli ze studie vyloučeni.
Primárním cílem této studie bylo zhodnocení účinnosti dabrafenibu ve srovnání s DTIC s ohledem na přežití bez progrese (PFS), které bylo hodnoceno zkoušejícím. Pacientům v rameni s léčbou DTIC bylo umožněno přejít do ramene s léčbou dabrafenibem po prokázání progrese původního onemocnění pomocí nezávislého radiografického vyšetření. Základní charakteristiky byly mezi jednotlivými skupinami dobře vyvážené. Šedesát procent pacientů byli muži a 99,6 % byli běloši, medián věku byl 52 let, přičemž 21 % pacientů bylo > 65 let, 98,4 % pacientů mělo ECOG status 0 nebo 1 a 97 % pacientů mělo metastatické onemocnění.
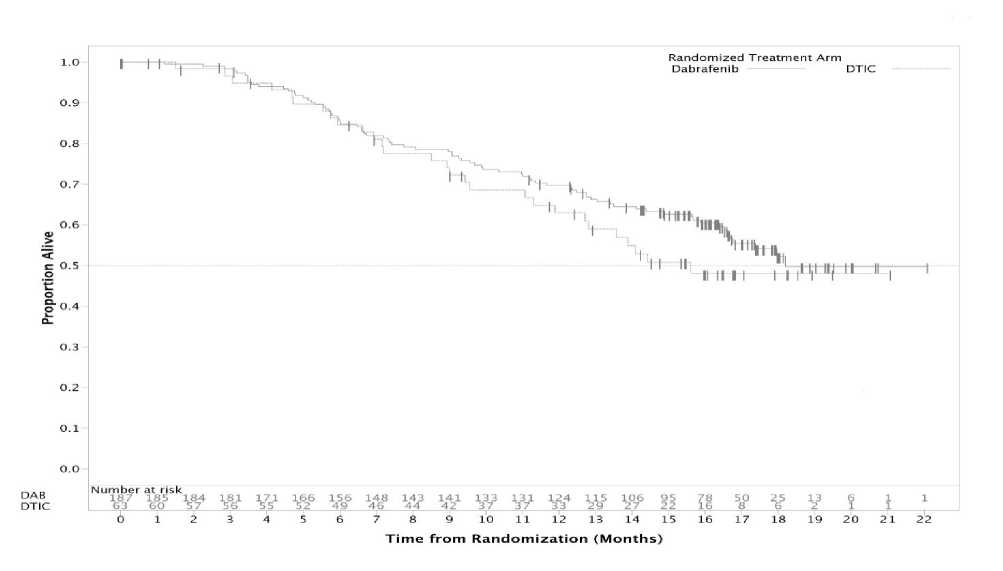
V prespecifické analýze s ukončením sběru údajů ke dni 19. prosince 2011 bylo dosaženo významného zlepšení primárního cílového parametru PFS (HR = 0,30, 95 % IS 0,18; 0,51, p < 0,0001). Výsledky účinnosti z primární analýzy a post-hoc analýzy s 6měsíčním následným sledováním jsou shrnuty v tabulce 8. Údaje o celkovém přežití z další post-hoc analýzy s ukončením sběru údajů ke dni 18. prosince 2012 jsou zobrazeny na obrázku 3.
Tabulka 8 Účinnost u dříve neléčených pacientů (studie BREAK-3, 25. červen 2012)
|
Údaje k 19. prosinci 2011 |
Údaje k 25. červnu 2012 | |||
|
Dabrafenib n = 187 |
DTIC n = 63 |
Dabrafenib n = 187 |
DTIC n = 63 | |
|
Přežití bez progrese onemocnění | ||||
|
Medián, měsíce (95 % IS) HR (95 % IS) |
5,1 (4,9; 6,9) 0,30 (0, p < 0 |
2,7 (1,5; 3,2) 8; 0,51) ,0001 |
6,9 (5,2; 9,0) 0,37 (0,2 p < 0 |
2,7 (1,5; 3,2) !4; 0,58) 0001 |
|
Celkový výskyt odpovědi3 | ||||
|
% (95% IS) |
53 (45,5; 60,3) |
19 (10,2; 30,9) |
59 (51,4; 66,0) |
24 (14; 36,2) |
|
Trvání odpovědi | ||||
|
Medián, měsíce (95 % IS) |
n = 99 5,6 (4,8; NR) |
n = 12 NR (5,0; NR) |
n = 110 8,0 (6,6; 11,5) |
n = 15 7,6 (5,0; 9,7) |
|
Zkratky: IS: interval spolehlivosti; DTIC: dakarbazin; HR: poměr rizik; NR: nedosaženo a Definována jako potvrzená kompletní + parciální odpověď. | ||||
Při ukončení sběru údajů ke dni 25. června 2012 přešlo 35 jedinců (55,6 %) z 63 původně randomizovaných do ramene s léčbou DTIC do ramene s léčbou dabrafenibem a u 63 % jedinců původně randomizovaných do ramene s léčbou dabrafenibem a u 79 % jedinců randomizovaných do ramene s léčbou DTIC došlo k progresi onemocnění nebo pacienti zemřeli. Medián PFS po změně léčby byl 4,4 měsíce.
Tabulka 9 Data týkající se přežití pacientů z primární a post-hoc analýzy
|
Ukončení sběru dat ke dni |
Léčba |
Počet úmrtí (%) |
Poměr rizik (95 % IS) |
|
19. prosince 2011 |
DTIC |
9 (14 %) |
0,61 (0,25; 1,48) (a) |
|
dabrafenib |
21 (11 %) | ||
|
25. června 2012 |
DTIC |
21 (33 %) |
0,75 (0,44; 1,29) (a) |
|
dabrafenib |
55 (29 %) | ||
|
18. prosince 2012 |
DTIC |
28 (44 %) |
0,76 (0,48; 1,21) (a) |
|
dabrafenib |
78 (42 %) |
(a) Pacienti nebyli v době změny léčby kontrolováni
Údaje o celkovém přežití z další post-hoc analýzy založené na údajích získaných před ukončením sběru dat ke dni 18. prosince 2012 vykazují výskyt 12měsíčního celkového přežití 63 % u DTIC a 70 % u dabrafenibu.
Obrázek 3: Kaplan-Meierova křivka celkového přežití (BREAK-3) (18. prosinec 2012)
Pacienti s mozkovými metastázami [výsledky ze studie fáze II (BREAK-MB)]
Studie BREAK-MB byla multicentrická, otevřená, dvoukohortová studie fáze II navržená k hodnocení intrakraniální odpovědi na dabrafenib u jedinců s histologicky potvrzenými mozkovými metastázami melanomu s pozitivitou mutace genu BRAF (V600E nebo V600K) (stadium IV). Jedinci byli zařazeni do kohorty A (jedinci bez předchozí lokální léčby mozkových metastáz) nebo kohorty B (jedinci, kteří byli již dříve lokálně léčeni pro mozkové metastázy).
Primárním cílovým parametrem studie byl celkový výskyt intrakraniální odpovědi (OIRR, overall intracranial response rate) u pacientů s mutací V600E, na základě hodnocení zkoušejícího. Potvrzený OIRR a další výsledky účinnosti založené na posouzení zkoušejícím jsou shrnuty v tabulce 10.
Tabulka 10 Údaje týkající se účinnosti u pacientů s mozkovými metastázami (studie BREAK-MB)
|
Populace všech léčených jedinců | ||||
|
BRAF V600E (primární) |
BRAF V600K | |||
|
Kohorta A n = 74 |
Kohorta B n = 65 |
Kohorta A n = 15 |
Kohorta B n = 18 | |
|
Celkový výskyt intrakraniální odpovědi, % (95 % IS)a | ||||
|
39 % (28,0; 51,2) p < 0,001b |
31 % (19,9; 43,4) p < 0,001b |
7 % (0,2; 31,9) |
22 % (6,4; 47,6) | |
|
Trvání intrakraniální odpovědi, medián, měsíce (95 % IS) | ||||
|
n = 29 4,6 (2,8; NR) |
n = 20 6,5 (4,6; 6,5) |
n = 1 2,9 (NR; NR) |
n = 4 3,8 (NR; NR) | |
|
Celková odpověď, % (95 % IS)a | ||||
|
38 % (26,8; 49,9) |
31 % (19,9; 43,4) |
0 (0; 21,8) |
28 % (9,7; 53,5) | |
|
Trvání odpovědi, medián, měsíce (95 % IS' | ||||
|
n = 28 5,1 (3,7; NR) |
n = 20 4,6 (4,6; 6,5) |
NA |
n = 5 3,1 (2,8; NR) | |
|
Přežití bez progrese, medián, měsíce (95 % IS) | ||||
|
3,7 (3,6; 5,0) |
3,8 (3,6; 5,5) |
1,9 (0,7; 3,7) |
3,6 (1,8; 5,2) | |
|
Celkové přežití, medián, měsíce (95 % IS) | ||||
|
Medián, měsíce |
7,6 (5,9; NR) |
7,2 (5,9; NR) |
3,7 (1,6; 5,2) |
5,0 (3,5; NR) |
|
Zkratky: IS: interval spolehlivosti; NR: nedosaženo; NA: není relevantní a - Potvrzená odpověď. b - Tato studie byla navržena k podpoření nebo zamítnutí nulové hypotézy OIRR < 10 % (na základě historických výsledků) ve prospěch alternativní hypotézy OIRR > 30 % u jedinců s mutací V600E genu BRAF. | ||||
Pacienti dříve neléčení nebo pacienti, u kterých selhala alespoň jedna předchozí systémová léčba [výsledky ze studie fáze II (BREAK-2)]
Studie BRF113710 (BREAK-2) byla multicentrická, jednoramenná studie, do které bylo zařazeno 92 jedinců s metastatickým melanomem (stupeň IV) s potvrzenou pozitivitou mutace V600E nebo V600K genu BRAF.
Zkoušejícím potvrzený výskyt odpovědi u pacientů s metastazujícím melanomem s pozitivitou mutace V600E genu BRAF (n = 76) byl 59 % (95 % IS: 48,2; 70,3) a medián trvání odpovědi byl 5,2 měsíce (95 % IS: 3,9; nepočitatelný) na základě mediánu sledování 6,5 měsíce. U pacientů s metastatickým melanomem s pozitivitou mutace V600K genu BRAF (n = 16) byl výskyt odpovědi 13 % (95 % IS: 0,0; 28,7) s mediánem trvání odpovědi 5,3 měsíce (95 % IS: 3,7; 6,8). Ačkoli byly údaje limitované malým počtem pacientů, medián OS se zdál konzistentní s údaji od pacientů s nádory s pozitivitou mutace V600E genu BRAF.
Prodloužení QT intervalu
Nejzávažnější případy prodloužení QTc intervalu o > 60 milisekund (ms) byly pozorovány u 3 % jedinců léčených dabrafenibem (jeden jedinec > 500 ms ve sloučené bezpečnostní populaci). Ve studii fáze III MEK115306 nedošlo u žádného z pacientů léčených trametinibem v kombinaci s dabrafenibem k prodloužení QTcB intervalu nad 500 ms; QTcB interval byl prodloužen o více než 60 ms oproti výchozímu stavu u 1 % (3/209) pacientů. Ve studii fáze III MEK116513 měli čtyři pacienti (1 %) léčení trametinibem v kombinaci s dabrafenibem prodloužení QTcB intervalu stupně 3 (> 500 ms). Dva z těchto pacientů, kteří měli prodloužení QTcB intervalu stupně 3 (> 500 ms), měli zároveň prodloužení QTcB o > 60 ms oproti výchozímu stavu.
Potenciální účinek dabrafenibu na prodloužení QT intervalu byl hodnocen v QT studii zaměřené na podávání vícenásobné dávky. Supraterapeutická dávka 300 mg dabrafenibu dvakrát denně byla podávána 32 pacientům s nádory s přítomnou mutací V600 genu BRAF. Žádný klinicky relevantní účinek dabrafenibu nebo jeho metabolitů na QTc interval nebyl pozorován.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s dabrafenibem u jedné nebo více podskupin pediatrické populace s melanomem (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Dabrafenib je po perorálním podání absorbován s mediánem doby do dosažení maximálních plazmatických koncentrací 2 hodiny po podání dávky. Průměrná absolutní biologická dostupnost dabrafenibu po perorálním podání je 95 % (90 % IS: 81; 110 %). Expozice dabrafenibu (Cmax a AUC) se zvyšovala úměrně podané dávce v rozmezí od 12 do 300 mg po podání jednotlivé dávky, ale při opakovaném dávkování dvakrát denně bylo zvyšování nižší než proporcionální. Snížení expozice bylo pozorováno při opakovaném dávkování, pravděpodobně v důsledku indukce vlastního metabolismu. Poměr průměrné akumulace AUC den 18/den 1 byl 0,73. Po podání 150 mg 2x denně byl geometrický průměr Cmax 1 478 ng/ml, AUC(0-X) 4 341 ng*h/ml a koncentrace před podáním dávky (Cx) 26 ng/ml.
Podání dabrafenibu s jídlem snižovalo jeho biologickou dostupnost (snížení Cmax o 51 % a AUC o 31 %) a opožďovalo vstřebávání dabrafenibu z tobolek ve srovnání s podáním nalačno.
Distribuce
Dabrafenib je z 99,7 % navázán na lidské plazmatické proteiny. Distribuční objem v rovnovážném stavu po intravenózním podání je 46 l.
Dabrafenib je in vitro substrátem lidského P-glykoproteinu (Pgp) a myšího BCRP. Tyto transportéry však mají minimální dopad na biologickou dostupnost dabrafenibu po perorálním podání a jeho eliminaci a riziko klinicky relevantních lékových interakcí s Pgp nebo BCRP je nízké. Dabrafenib není in vitro substrátem transportérů OATP1B1, OATP1B3 nebo OATP2B1.
Ani dabrafenib ani jeho 3 hlavní metabolity neinhibují Pgp in vitro.
Biotransformace
Metabolismus dabrafenibu je primárně zprostředkován CYP2C8 a CYP3A4 za tvorby hydroxy-dabrafenibu, který je dále oxidován prostřednictvím CYP3A4 za tvorby karboxy-dabrafenibu. Karboxy-dabrafenib může být dekarboxylován neenzymatickým procesem na desmethyl-dabrafenib. Karboxy-dabrafenib je vylučován žlučí a močí. Desmethyl-dabrafenib může být rovněž vytvořen ve střevě a může se reabsorbovat. Desmethyl-dabrafenib je metabolizován CYP3A4 na oxidativní metabolity. Terminální poločas hydroxy-dabrafenibu odpovídá původní látce s poločasem 10 hodin, zatímco karboxy- a desmethyl-metabolity mají delší poločas (21 - 22 hodin). Průměrný poměr AUC metabolitů k původní látce po opakovaném podání je 0,9 pro hydroxy-dabrafenib, 11 pro karboxy-dabrafenib a 0,7 pro desmethyl-dabrafenib. Na základě expozice, relativní síly a farmakokinetických vlastností je pravděpodobné, že se jak hydroxy-dabrafenib tak desmethyl-dabrafenib budou podílet na klinickém účinku dabrafenibu, zatímco aktivita karboxy-dabrafenibu je pravděpodobně nevýznamná.
Terminální poločas po intravenózním podání jednotlivé mikrodávky je 2,6 hodiny. Terminální poločas dabrafenibu po podání jednotlivé dávky je 8 hodin v důsledku eliminace omezené vstřebáváním po perorálním podání (flip-flop farmakokinetika). Intravenózní plasmatická clearance je 12 l/hodinu.
Po perorální dávce je hlavní cestou eliminace dabrafenibu metabolizace zprostředkovaná CYP3A4 a CYP2C8. Látky odvozené od dabrafenibu jsou vylučovány primárně stolicí, 71 % perorální dávky se vyloučí stolicí a 23 % močí pouze ve formě metabolitů.
Zvláštní populace pacientů
Porucha funkce jater
Populační farmakokinetická analýza naznačuje, že mírné zvýšení hladin bilirubinu a/nebo AST [na základě NCI (National Cancer Institute) klasifikace] signifikantně neovlivňuje perorální clearance dabrafenibu. Mírná porucha funkce jater, jak je definována na základě hladin bilirubinu a AST, navíc neměla významný vliv na plazmatické koncentrace metabolitů dabrafenibu. U pacientů se středně závažnou až závažnou poruchou funkce jater nejsou dostupné žádné údaje. Protože jaterní metabolismus a biliární sekrece jsou primárními cestami eliminace dabrafenibu a jeho metabolitů, mělo by podávání dabrafenibu pacientům se středně závažnou až závažnou poruchou funkce jater probíhat s opatrností (viz bod 4.2).
Porucha funkce ledvin
Populační farmakokinetická analýza naznačuje, že mírná porucha funkce ledvin neovlivňuje perorální clearance dabrafenibu. Ačkoli údaje u středně závažné poruchy funkce ledvin jsou omezené, tato data nenaznačují klinicky významný vliv. U jedinců se závažnou poruchou funkce ledvin nejsou k dispozici žádné údaje (viz bod 4.2).
Starší pacienti
Na základě populační farmakokinetické analýzy neměl věk významný vliv na farmakokinetiku dabrafenibu. Vyšší věk než 75 let byl významným prediktorem plazmatické koncentrace karboxy- a desmethyl-dabrafenibu, přičemž u jedinců > 75 let byla expozice o 40 % vyšší ve srovnání s jedinci < 75 let.
Tělesná hmotnost a pohlaví
Na základě populační farmakokinetické analýzy má pohlaví i tělesná hmotnost vliv na perorální clearance dabrafenibu; tělesná hmotnost má rovněž vliv na distribuční objem po perorálním podání a distribuční clearance. Tyto farmakokinetické rozdíly však nebyly považovány za významné.
Rasa
Ke stanovení vlivu rasy na farmakokinetiku dabrafenibu není k dispozici dostatek údajů.
Pediatrická populace
Nebyly provedeny žádné studie, které by hodnotily farmakokinetiku dabrafenibu u pediatrických pacientů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Studie kancerogenity nebyly s dabrafenibem provedeny. Dabrafenib nebyl mutagenní ani klastrogenní v testech na bakteriích ani kulturách savčích buněk in vitro a ani v testu tvorby mikrojader u hlodavců
in vivo.
V kombinovaných studiích fertility samic a časného embryonálního a embryofetálního vývoje u potkanů byl snížen počet žlutých tělísek v ovariích u březích samic při dávkách 300 mg/kg/den (přibližně trojnásobek klinické expozice u člověka na základě AUC), ale nebyl pozorován vliv na estrální cyklus, páření a parametry fertility. Vývojová toxicita, včetně embryoletality a defektů ventrikulárního septa, byla pozorována při dávkách 300 mg/kg/den a opožděný vývoj skeletu a snížení tělesné hmotnosti plodu při dávkách > 20 mg/kg/den (což odpovídá > 0,5násobku klinické expozice u člověka na základě AUC).
Studie hodnotící fertilitu samců nebyla s dabrafenibem provedena. Ve studiích toxicity po opakované dávce byla u potkanů a psů pozorována testikulární degenerace/deplece (> 0,2násobek klinické expozice u člověka na základě AUC). Testikulární změny u potkanů a psů přetrvávaly i po 4 týdnech rekonvalescence (viz bod 4.6).
U psů byly (při dávkách > 2násobek klinické expozice na základě AUC) pozorovány kardiovaskulární účinky, včetně degenerace/nekrózy koronárních arterií a/nebo krvácení, hypertrofie srdeční atrioventrikulární chlopně/krvácení do srdeční atrioventrikulární chlopně a fibrovaskulární proliferace síní. U myší byl pozorován fokální arteriální/perivaskulární zánět v různých tkáních a u potkanů byla pozorována zvýšená incidence hepatální arteriální degenerace a spontánní kardiomyocytární degenerace se zánětem (spontánní kardiomyopatie) (při dávkách > 0,5násobku klinické expozice u potkanů a > 0,6násobku klinické expozice u myší). U myší byly pozorovány účinky na játra, včetně hepatocelulární nekrózy a zánětu (při dávkách > 0,6násobku klinické expozice). U několika psů byl při dávkách > 20 mg/kg/den (odpovídá > 9násobku klinické expozici na základně AUC) pozorován bronchoalveolární zánět plic, který byl spojen s oslabeným a/nebo ztíženým dýcháním.
U psů a potkanů, kterým byl podáván dabrafenib, byly pozorovány reverzibilní hematologické účinky. Ve studiích hodnotících až 13 týdnů léčby bylo pozorováno snížení počtu retikulocytů a/nebo červených krvinek (při dávkách > 10- resp. 1,4násobek klinické expozice).
Ve studii juvenilní toxicity u potkanů byly pozorovány účinky na růst (zkrácení délky dlouhých kostí), renální toxicita (tubulární depozita, zvýšení incidence kortikálních cyst a tubulární bazofilie a reverzibilní zvýšení koncentrace urey a/nebo kreatininu) a testikulární toxicita (degenerace a dilatace tubulů) (při dávkách > 0,2násobku klinické expozice u dospělých pacientů na základě AUC).
Dabrafenib je fototoxický v testu 3T3 NRU (Neutral Red Uptake) na myších fibroblastech in vitro a in vivo v dávce > 100 mg/kg (> 44násobek klinické expozice na základě Cmax) v studii perorální fototoxicity u bezsrstých myší.
Léčba v kombinaci s trametinibem
Ve studii na psech, ve které byla podávaná kombinace trametinibu a dabrafenibu po dobu 4 týdnů, byly pozorovány příznaky gastrointestinální toxicity a snížení množství lymfatických buněk v thymu při užití nižších dávek, než u psů, kterým byl podáván trametinib samostatně. Jinak byla pozorována obdobná toxicita jako u srovnatelných studií s monoterapií.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky Mikrokrystalická celulosa Magnesium-stearát Koloidní bezvodý oxid křemičitý
Obal tobolky
Červený oxid železitý (E172)
Oxid titaničitý (E171)
Hypromelosa (E464)
Inkoust
Černý oxid železitý (E172)
Šelak
Propylenglykol
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Neprůhledná bílá lahev z polyethylenu o vysoké hustotě (HDPE) s polypropylenovým šroubovacím uzávěrem a silikagelovým desikantem.
Jedna lahev obsahuje 28 nebo 120 tvrdých tobolek.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
Tafinlar 50 mg tvrdé tobolky
EU/1/13/865/001
EU/1/13/865/002
Tafinlar 75 mg tvrdé tobolky
EU/1/13/865/003
EU/1/13/865/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
26. srpna 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCI ODPOVĚDNI ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI ODPOVĚDNI ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců odpovědných za propouštění šarží
GLAXO WELLCOME, S.A.
Avda. Extremadura, 3, Pol. Ind. Allendeduero 09400, Aranda de Duero (Burgos)
Španělsko
Novartis Pharmaceuticals UK Limited
Frimley Business Park
Frimley
Camberley, Surrey GU16 7SR Velká Británie
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Neuplatňuje se.
• Povinnost uskutečnit poregistrační opatření
Neuplatňuje se.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Tafínlar 50 mg tvrdé tobolky dabrafenibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje dabrafenibi mesilas v množství odpovídajícím 50 mg dabrafenibu.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdá tobolka
28 tobolek 120 tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Lahev obsahuje vysoušedlo, neodstraňujte jej ani jej nejezte.
8. POUŽITELNOST
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/865/001 28 tobolek
EU/1/13/865/002 120 tobolek
13. ČÍSLO ŠARŽE
Lot:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
tafinlar 50 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Tafinlar 50 mg tobolky dabrafenibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje dabrafenibi mesilas v množství odpovídajícím 50 mg dabrafenibu.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdá tobolka
28 tobolek 120 tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/865/001 28 tobolek
EU/1/13/865/002 120 tobolek
13. ČÍSLO ŠARŽE
Lot:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Tafínlar 75 mg tvrdé tobolky dabrafenibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje dabrafenibi mesilas v množství odpovídajícím 75 mg dabrafenibu.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdá tobolka
28 tobolek 120 tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Lahev obsahuje vysoušedlo, neodstraňujte jej ani jej nejezte.
8. POUŽITELNOST
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/865/003 28 tobolek
EU/1/13/865/004 120 tobolek
13. ČÍSLO ŠARŽE
Lot:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
tafinlar 75 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Tafinlar 75 mg tobolky dabrafenibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje dabrafenibi mesilas v množství odpovídajícím 75 mg dabrafenibu.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdá tobolka
28 tobolek 120 tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/865/003 28 tobolek
EU/1/13/865/004 120 tobolek
13. ČÍSLO ŠARŽE
Lot:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Tafinlar 50 mg tvrdé tobolky Tafinlar 75 mg tvrdé tobolky
dabrafenibum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Tafinlar a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Tafinlar užívat
3. Jak se přípravek Tafinlar užívá
4. Možné nežádoucí účinky
5. Jak přípravek Tafinlar uchovávat
6. Obsah balení a další informace
1. Co je přípravek Tafinlar a k čemu se používá
Přípravek Tafinlar je lék, který obsahuje léčivou látku dabrafenib. Používá se buď samostatně, nebo v kombinaci s jiným lékem obsahujícím trametinib, u dospělých k léčbě určitého typu nádorového onemocnění kůže nazývaného melanom,
• který má určitou změnu (mutaci) v genu nazývaném BRAF, a
• který se rozšířil do dalších částí těla a nelze jej odstranit chirurgicky.
Tato mutace v genu mohla způsobit, že melanom vznikl. Přípravek Tafinlar se zaměřuje na bílkoviny vytvořené tímto pozměněným genem BRAF a zpomaluje nebo zastavuje vývoj nádorového onemocnění.
2. Čemu musíte věnovat pozornost, než začnete přípravek Tafinlar užívat
Přípravek Tafinlar lze používat pouze k léčbě melanomu, který má změnu (mutaci) v genu BRAF, proto Vám lékař před zahájením léčby musí odebrat tkáňové vzorky, aby zjistil, zda je pro Vás přípravek Tafinlar vhodný.
Pokud Váš lékař rozhodne, že budete užívat přípravek Tafinlar v kombinaci s trametinibem, přečtěte si příbalovou informaci dabrafenibu stejně pečlivě jako tuto příbalovou informaci.
Neužívejte přípravek Tafinlar:
• jestliže jste alergický(á) na dabrafenib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6). Pokud si myslíte, že se Vás to týká, poraďte se se svým lékařem.
Upozornění a opatření
Před užitím přípravku Tafinlar se poraďte se svým lékařem. Váš lékař potřebuje vědět, jestli:
• máte problémy s játry;
• máte nebo jste někdy měl(a) problémy s ledvinami;
Lékař Vám může v průběhu léčby přípravkem Tafinlar odebírat krevní vzorky, aby sledoval činnost jater a ledvin.
• máte nebo jste měl(a) jiný typ nádoru než melanom, protože u Vás může být při užívání přípravku Tafinlar větší riziko vzniku nádorů vyskytujících se jinde než na kůži.
Než začnete užívat přípravek Tafinlar v kombinaci s trametinibem, Váš lékař potřebuje vědět, jestli:
• máte problémy se srdcem jako srdeční selhání nebo problémy se srdeční činností.
• máte problémy s očima, včetně ucpání sítnicové žíly nebo otoku v oku, který může být způsoben onemocněním sítnice, kdy dochází k hromadění tekutiny pod sítnicí, zvaným chorioretinopatie.
• máte plicní nebo dýchací problémy, včetně problémů s dýcháním často doprovázené suchým kašlem, dušností a únavou.
Pokud si myslíte, že se Vás něco z tohoto týká, poraďte se se svým lékařem.
Stavy, kterým musíte věnovat pozornost
U některých osob, které užívají přípravek Tafinlar, se mohou rozvinout další stavy (onemocnění), které mohou být závažné. Musíte vědět o důležitých známkách a příznacích, kterým máte v průběhu užívání tohoto léku věnovat pozornost. Některé z těchto příznaků (krvácení, horečka, změny na kůži a problémy s očima) jsou krátce uvedeny v tomto bodě, ale podrobnější informace naleznete v bodě 4 „Možné nežádoucí účinky“.
Krvácení
Užívání přípravku Tafinlar v kombinaci s trametinibem může způsobit vážné krvácení, včetně krvácení do mozku, do zažívacího ústrojí (např. žaludku, střev, konečníku), nebo do plic a dalších orgánů, které může vést k úmrtí. Příznaky mohou zahrnovat:
• bolest hlavy, závratě, nebo pocit slabosti
• krev ve stolici nebo černou stolici
• krev v moči
• bolest žaludku
• vykašlávání / zvracení krve
Pokud se u Vás objeví některý z těchto příznaků, co nejdříve informujte svého lékaře.
Horečka (vysoká teplota)
Tento lék může způsobovat horečku (viz rovněž bod 4). V některých případech se u lidí s horečkou může objevit nízký krevní tlak, závratě nebo jiné příznaky. Pokud se u Vás v průběhu užívání tohoto léku objeví horečka, sdělte to neprodleně svému lékaři, lékárníkovi nebo zdravotní sestře.
Porucha srdeční činnosti
Přípravek Tafinlar může způsobovat problémy se srdcem nebo může zhoršovat již přítomné srdeční problémy u lidí užívajících Tafinlar v kombinaci s trametinibem.
Pokud máte poruchu srdeční činnosti, sdělte to svému lékaři. Váš lékař bude před zahájením léčby a v průběhu léčby přípravkem Tafinlar v kombinaci s trametinibem provádět vyšetření, aby zkontroloval, zda Vaše srdce pracuje správně. Neprodleně sdělte svému lékaři, pokud máte pocity bušení srdce či rychlé nebo nepravidelné činnosti srdce, nebo pokud se u Vás vyskytnou závratě, únava, pocit na omdlení, dušnost, nebo otoky nohou. Pokud to bude nutné, Váš lékař může rozhodnout o přerušení léčby nebo o jejím trvalém ukončení.
Změny na kůži v průběhu léčby a po léčbě
Lékař Vám kůži zkontroluje před zahájením léčby tímto lékem a poté ji bude pravidelně kontrolovat i v jejím průběhu.
Pokud zaznamenáte v průběhu užívání tohoto léku nebo po ukončení léčby jakékoli změny na kůži, sdělte to neprodleně svému lékaři (viz rovněž bod 4).
Problémy s očima
Při užívání tohoto léku je nutné podstoupit lékařské oční vyšetření.
Pokud v průběhu léčby zaznamenáte zarudnutí nebo podráždění očí, rozmazané vidění, bolest očí nebo další změny zraku, sdělte to ihned svému lékaři (viz rovněž bod 4).
Užívání přípravku Tafinlar v kombinaci s trametinibem může způsobovat problémy s očima, včetně slepoty. Užívání trametinibu se nedoporučuje, pokud u Vás kdykoli v minulosti došlo k ucpání žíly, která odvádí krev z oka (okluze retinální žíly). Neprodleně sdělte svému lékaři, pokud se u Vás v průběhu léčby vyskytnou následující příznaky: rozmazané vidění, ztráta vidění nebo jiné změny zraku, barevné body v zorném poli nebo vidění rozostřeného obrysu kolem předmětů. Pokud to bude nutné, Váš lékař může rozhodnout o přerušení léčby nebo o jejím trvalém ukončení.
^ Přečtěte si informace týkající se horečky, změn na kůži a problémů s očima v bodě 4 této příbalové informace. Pokud se u Vás kterékoli z těchto známek a příznaků objeví, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře.
Problémy s játry
Přípravek Tafinlar v kombinaci s trametinibem, může způsobit problémy s játry, které se mohou
vyvinout do tak závažných stavů, jako je hepatitida a jaterní selhání, jež mohou být fatální. Lékař Vás
bude pravidelně kontrolovat. Příznaky toho, že Vaše játra nepracují správně, mohou zahrnovat:
ztrátu chuti k jídlu
pocit nevolnosti (pocit na zvracení)
nevolnost (zvracení)
bolest žaludku (břicha)
zežloutnutí kůže nebo očního bělma (žloutenka) tmavou moč svědění kůže
Pokud se objeví některý z těchto příznaků, co nejdříve informujte svého lékaře.
Bolest svalů
Přípravek Tafinlar v kombinaci s trametinibem může vést k rozpadu svalů (rabdomyolýza).
Informujte svého lékaře co nejdříve, pokud máte některý z těchto příznaků:
• bolest svalů
• tmavou moč v důsledku poškození ledvin
Pokud je to nutné, může lékař rozhodnout o přerušení léčby nebo ji úplně ukončit.
Děti a dospívající
Přípravek Tafinlar není doporučen pro děti a dospívající. Účinky přípravku Tafinlar na osoby mladší 18 let nejsou známy.
Další léčivé přípravky a přípravek Tafinlar
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. To se týká i léků dostupných bez lékařského předpisu.
Některé léky mohou ovlivnit účinek přípravku Tafinlar, nebo mohou zvýšit pravděpodobnost, že se u Vás objeví nežádoucí účinky. Přípravek Tafinlar rovněž může ovlivnit způsob, jakým účinkují některé jiné léky. Ty zahrnují:
• léky ke kontrole početí (antikoncepce) obsahující hormony, jako jsou tablety, injekce nebo náplasti;
• warfarin nebo acenokumarol, léky používané na ředění krve;
• digoxin, užívaný k léčbě srdečních obtíží;
• léky k léčbě plísňových infekcí, jako j sou ketokonazol, itrakonazol, vorikonazol a posakonazol;
• některé blokátory kalciových kanálů, používané k léčbě vysokého krevního tlaku, jako jsou diltiazem, felodipin, nikardipin, nifedipin nebo verapamil;
• léky k léčbě nádorových onemocnění, jako je kabazitaxel;
• některé léky ke snížení hladiny tuků (lipidů) v krvi, jako je gemfibrozil;
• některé léky používané k léčbě určitých psychiatrických stavů, jako je haloperidol;
• některá antibiotika, j ako j e klarithromycin, doxycyklin a telithromycin;
• některé léky k léčbě tuberkulózy (TBC), jako je rifampicin;
• některé léky používané ke snížení hladiny cholesterolu, jako je atorvastatin a simvastatin;
• některá imunosupresiva, jako je cyklosporin, takrolimus a sirolimus;
• léky, které snižují kyselost žaludku, jako je omeprazol;
• některé protizánětlivé léky, jako je dexamethazon a methylprednisolon;
• některé léky používané k léčbě infekce virem HIV, jako je ritonavir, amprenavir, indinavir, darunavir, delavirdin, efavirenz, fosamprenavir, lopinavir, nelfinavir, tipranavir, sachinavir a atazanavir;
• některé léky používané k úlevě od bolesti, jako je fentanyl a methadon;
• léky k léčbě záchvatů křečí (epilepsie), jako je fenytoin, fenobarbital, primidon, kyselina valproová nebo karbamazepin;
• léky k léčbě deprese, jako je nefazodon, a rostlinný přípravek obsahující třezalku tečkovanou (Hypericum perforatum).
Pokud některé z těchto léků užíváte (nebo pokud si nejste jistý(á)), sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Váš lékař může rozhodnout o úpravě dávky.
Mějte u sebe vždy seznam léků, které užíváte, abyste ho mohl(a) ukázat lékaři, lékárníkovi nebo zdravotní sestře.
Těhotenství, kojení a plodnost
Přípravek Tafinlar se nedoporučuje v průběhu těhotenství.
• Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou dříve, než začnete tento přípravek užívat. Přípravek Tafinlar se nedoporučuje v průběhu těhotenství, protože může poškodit nenarozené dítě.
• Pokud jste žena, která může otěhotnět, musíte v průběhu léčby přípravkem Tafinlar a po dobu 4 týdnů po ukončení léčby a po dobu 4 následujících měsíců po poslední dávce trametinibu podávaného v kombinaci s přípravkem Tafinlar používat vhodný způsob antikoncepce.
• Hormonální antikoncepce (jako jsou tablety, injekce nebo náplasti) nemusí účinkovat správně, pokud zároveň užíváte přípravek Tafinlar nebo kombinační terapii (přípravek Tafinlar spolu s trametinibem). Může být nutné, abyste používala další vhodnou metodu kontroly početí, jako je barierová metoda (např. prezervativ), abyste v průběhu léčby tímto přípravkem neotěhotněla. Poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
• Pokud v průběhu léčby tímto léčivým přípravkem otěhotníte, sdělte to neprodleně svému lékaři.
Přípravek Tafinlar se nedoporučuje v průběhu kojení.
Není známo, zda složky tohoto léku mohou procházet do mateřského mléka.
Pokud kojíte nebo kojení plánujete, musíte to sdělit svému lékaři. Vy a Váš lékař pak společně rozhodnete, zda budete užívat tento lék, nebo kojit.
Plodnost - muži i ženy
Studie na zvířatech prokázaly, že léčivá látka dabrafenib může trvale snížit plodnost u mužů. Muži, kteří užívají přípravek Tafinlar, navíc mohou mít snížený počet spermií a počet spermií se po ukončení léčby tímto přípravkem nemusí vrátit na původní normální hodnoty.
Před zahájením léčby přípravkem Tafinlar si promluvte se svým lékařem o způsobech zachování možnosti mít v budoucnu děti.
Užívání přípravku Tafinlar s trametinibem: trametinib může narušit plodnost u mužů i u žen.
Pokud máte další otázky týkající se účinků tohoto léku na počet spermií, poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Řízení dopravních prostředků a obsluha strojů
Přípravek Tafinlar může mít nežádoucí účinky, které mohou ovlivnit schopnost řídit nebo obsluhovat stroje.
Vyvarujte se řízení dopravních prostředků a obsluhy strojů, pokud máte problémy se zrakem, nebo pokud se cítíte unavený(á) nebo slabý(á), nebo pokud máte pocit, že máte nedostatek energie.
Popis těchto účinků naleznete v bodech 2 a 4.
Promluvte si se svým lékařem, lékárníkem nebo zdravotní sestrou, pokud si čímkoli nejste jistý(á). Schopnost řídit nebo obsluhovat stroje mohou ovlivnit Vaše onemocnění, příznaky i léčba.
3. Jak se přípravek Tafinlar užívá
Vždy užívejte přípravek Tafinlar přesně podle pokynů svého lékaře, lékárníka nebo zdravotní sestry. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Kolik přípravku se užívá
Obvyklá dávka přípravku Tafinlar užívaného buď samostatně nebo v kombinaci s trametinibem jsou dvě 75mg tobolky dvakrát denně (což odpovídá denní dávce 300 mg). Doporučená dávka trametinibu, který se užívá v kombinaci s přípravkem Tafinlar, je 2 mg jedenkrát denně.
Váš lékař může rozhodnout, zda máte užívat nižší dávku, pokud se u Vás objeví nežádoucí účinky.
Přípravek Tafinlar je rovněž dostupný jako 50 mg tobolky, pokud je doporučeno snížení dávky.
Neužívejte více přípravku Tafinlar, než Vám doporučil Váš lékař, protože to může zvýšit riziko nežádoucích účinků.
Jak se přípravek užívá
Tobolky spolkněte celé a zapijte vodou, jednu po druhé.
Tobolky nežvýkejte ani nedrťte, jinak by se snížil jejich účinek.
Užívejte přípravek Tafinlar dvakrát denně, nalačno. To znamená:
• po užití přípravku Tafinlar musíte počkat alespoň 1 hodinu, než se můžete najíst, nebo
• naopak po jídle musíte počkat alespoň 2 hodiny, než můžete užít přípravek Tafinlar.
Přípravek Tafinlar užívejte ráno a večer, přibližně s 12hodinovým odstupem. Ranní a večerní dávku přípravku Tafinlar užívejte vždy ve stejnou denní dobu. Tím se zvýší pravděpodobnost, že si vzpomenete tobolku užít.
Neužívejte ranní a večerní dávku přípravku Tafinlar současně.
Jestliže jste užil(a) více přípravku Tafinlar, než jste měl(a)
Jestliže jste užil(a) příliš mnoho tobolek přípravku Tafinlar, poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou. Je-li to možné, ukažte jim balení přípravku Tafinlar a tuto příbalovou informaci.
Jestliže jste zapomněl(a) užít přípravek Tafinlar
Pokud od vynechané dávky uplynulo méně než 6 hodin, dávku si vezměte, jakmile si vzpomenete.
Pokud od vynechané dávky uplynulo více než 6 hodin, dávku vynechejte a vezměte si až následující dávku v obvyklý čas. Poté pokračujte v užívání tobolek pravidelně dále jako obvykle.
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Tafinlar
Užívejte přípravek Tafinlar tak dlouho, jak Vám doporučil Váš lékař. Nepřestávejte s léčbou, dokud Vám lékař, lékárník nebo zdravotní sestra neřeknou, že ji máte ukončit.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Jak byste měl(a) užívat přípravek Tafinlar v kombinaci s trametinibem
• Užívejte přípravek Tafinlar v kombinaci s trametinibem přesně dle pokynů Vašeho lékaře, lékárníka nebo zdravotní sestry. Neměňte dávku nebo nepřestávejte užívat přípravek Tafinlar nebo trametinib, dokud Vám to Váš lékař, zdravotní sestra nebo lékárník neřekne.
• Užívejte přípravek Tafinlar dvakrát denně a trametinib užívejte jedenkrát denně. Užívat oba léky ve stejnou dobu může být vhodné k osvojení si návyku na každý den. Dávky přípravku Tafinlar by měly být užívány v odstupu 12 hodin. Trametinib užívaný v kombinaci s přípravkem Tafinlar by měl být podáván společně s ranní dávkou přípravku Tafinlar nebo
s večerní dávkou.
• Užívejte Tafinlar a trametinib nalačno nejméně hodinu před jídlem, nebo dvě hodiny po jídle. Zapíjejte plnou sklenicí vody.
• Jestliže zapomenete užít dávku přípravku Tafinlar nebo trametinibu, vezměte si ji, jakmile si vzpomenete: Tuto zapomenutou dávku si neberte v případě, že:
o Je to méně než 6 hodin do následující dávky přípravku Tafinlar, který se užívá dvakrát denně.
o Jestliže je to méně než 12 hodin do následující dávky trametinibu, který se užívá jednou denně.
• Jestliže jste užil(a) více přípravku Tafinlar nebo trametinibu, okamžitě kontaktujte Vašeho lékaře, zdravotní sestru nebo lékárníka. Vezměte si Tafinlar tobolky a trametinib tablety
s sebou, pokud to bude možné. Jestliže to bude možné, ukažte lékaři, zdravotní sestře či lékárníkovi, které navštívíte, balení přípravku Tafinlar a trametinib s jednotlivými příbalovými informacemi.
• Jestliže se u Vás objeví nežádoucí účinky, Váš lékař by měl rozhodnout, zda snížit dávku přípravku Tafinlar nebo trametinibu. Užívejte dávku přípravku Tafinlar a trametinibu přesně dle pokynů Vašeho lékaře, zdravotní sestry nebo lékárníka.
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
T-W r V r Vil i i V* 1
Závažné nežádoucí účinky Problémy s krvácením
Přípravek Tafinlar podáván v kombinaci s trametinibem může způsobit závažné problémy s krvácením, zejména v mozku. Neprodleně kontaktujte svého lékaře nebo zdravotní sestru a zajistěte si lékařskou pomoc, pokud máte jakékoli neobvyklé příznaky krvácení:
• bolesti hlavy, závratě nebo slabost,
• vykašlávání krve nebo krevních sraženin,
• zvracení krve nebo hmoty, která vypadá jako kávová sedlina,
• červenou nebo černou stolici, která má dehtovitý vzhled.
Horečka (vysoká tělesná teplota)
Užívání přípravku Tafinlar může způsobit horečku u více než 1 osoby z 10. Pokud se u Vás při užívání tohoto léku objeví horečka (tělesná teplota 38,5 °C nebo vyšší), sdělte to neprodleně svému lékaři, lékárníkovi nebo zdravotní sestře. Provedou Vám testy, aby zjistili, zda horečka není způsobena jinou příčinou, a případný problém budou léčit.
V některých případech se může u osob s horečkou vyvinout nízký krevní tlak a závrať. Pokud je horečka závažná, Váš lékař Vám může doporučit, abyste přestal(a) přípravek Tafinlar užívat v době, kdy bude léčit horečku jiným lékem. Jakmile je horečka pod kontrolou, lékař Vám může doporučit, abyste znovu zahájil(a) léčbu přípravkem Tafinlar.
Srdeční choroby
Přípravek Tafinlar podáván v kombinaci s trametinibem může ovlivnit schopnost srdce pumpovat krev. To může s větší pravděpodobností nastat u lidí, kteří již mají nějaké srdeční problémy. Během léčby přípravkem Tafinlar v kombinaci s trametinibem budete podstupovat vyšetření, aby byly odhaleny jakékoli případné srdeční problémy. Známky a příznaky srdečních problémů zahrnují:
• pocit bušení srdce, rychlé nebo nepravidelné činnosti srdce,
• závratě,
• únavu,
• pocit na omdlení,
• dušnost,
• otoky nohou.
Neprodleně informujte svého lékaře, pokud se u Vás vyskytnou tyto příznaky, ať už poprvé nebo se zhoršují.
Změny na kůži
Pokud při užívání tohoto léku zaznamenáte jakékoli změny na kůži, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře co nejdříve.
Až u 1 z 10 osob, které užívají přípravek Tafinlar, se může objevit jiný typ kožního nádoru nazývaný kožní spinocelulární karcinom (cuSCC). U jiných osob se může objevit typ kožního nádoru nazývaný bazocelulární karcinom (BCC). Tyto změny obvykle zůstanou omezené na jedno místo na kůži, lze je odstranit chirurgicky a léčba přípravkem Tafinlar může pokračovat bez přerušení.
Některé osoby užívající přípravek Tafinlar mohou rovněž zaznamenat nově vzniklý melanom. Takovéto melanomy lze obvykle odstranit chirurgicky a léčba přípravkem Tafinlar může pokračovat bez přerušení.
Váš lékař Vám před zahájením léčby přípravkem Tafinlar zkontroluje kůži a poté ji bude opakovaně kontrolovat každý měsíc v průběhu léčby a po dobu 6 měsíců po ukončení léčby. To je proto, že bude hledat případné nové kožní nádory.
Váš lékař Vám bude rovněž kontrolovat hlavu, krk, ústa, lymfatické uzliny a bude Vám pravidelně prováděno CT vyšetření (vyšetření pomocí počítačové tomografie) hrudníku a břicha. Mohou Vám rovněž být prováděny krevní testy. Tato vyšetření slouží k zjištění, zda se ve Vašem těle nevytváří případný jiný nádor, včetně spinocelulárního karcinomu. Rovněž je doporučeno provést na začátku a na konci Vaší léčby vyšetření pánve (u žen) a vyšetření konečníku.
V průběhu léčby přípravkem Tafinlar si pravidelně kontrolujte kůži
Pokud zaznamenáte cokoli z níže zmíněného:
• nově vzniklé bradavice;
• kožní vřed nebo zarudlá bulka, která krvácí a nehojí se;
• změny velikosti nebo barvy kožních znamének.
Pokud se u Vás nově objeví některý z těchto příznaků, nebo pokud se tyto příznaky zhorší, sdělte to co nejdříve svému lékaři, lékárníkovi nebo zdravotní sestře.
V průběhu užívání přípravku Tafinlar v kombinaci s trametinibem se můžou objevit kožní reakce (vyrážka). Informujte svého lékaře, pokud se u Vás vyskytne vyrážka v průběhu užívání přípravku Tafinlar v kombinaci s trametinibem.
Oční problémy
Až u 1 ze 100 osob užívajících přípravek Tafinlar buď samostatně, nebo v kombinaci s trametinibem se může objevit oční problém zvaný uveitida (zánět živnatky), který může poškodit zrak, pokud se neléčí. Uveitida se může rozvíjet rychle a příznaky zahrnují:
• zarudnutí a podráždění očí;
• rozmazané vidění;
• bolest oka;
• zvýšení citlivosti na světlo;
• plovoucí skvrny před očima.
^ Pokud se u Vás tyto příznaky objeví, kontaktujte neprodleně svého lékaře, lékárníka nebo zdravotní sestru.
Přípravek Tafinlar užíván v kombinaci s trametinibem může způsobit problémy s očima. Užívání trametinibu se nedoporučuje, pokud u Vás kdykoli v minulosti došlo k ucpání žíly, která odvádí krev z oka (okluze retinální žíly). Před zahájením léčby přípravkem Tafinlar v kombinaci s trametinibem a v jejím průběhu Vám může lékař doporučit oční vyšetření. Váš lékař Vám může nařídit, abyste přestal(a) užívat trametinib nebo Vás poslat ke specializovanému lékaři, pokud se u Vás vyskytnou známky a příznaky týkající se vidění, které zahrnují:
• ztrátu vidění,
• zarudnutí a podráždění očí,
• barevné body v zorném poli,
• vidění rozostřeného obrysu kolem předmětů,
• rozmazané vidění.
^ Pokud se u Vás tyto příznaky objeví, kontaktujte neprodleně svého lékaře, lékárníka nebo zdravotní sestru.
Je velmi důležité, abyste svému lékaři, lékárníkovi nebo zdravotní sestře ihned řekl(a), pokud se u Vás tyto příznaky objeví, zejména pokud pociťujete bolest a zarudnutí očí, které rychle neustupuje. Mohou Vám domluvit vyšetření u očního lékaře, který provede kompletní vyšetření.
Kromě závažných nežádoucích účinků zmíněných výše se mohou u osob užívajících přípravek Tafinlar rovněž objevit další nežádoucí účinky.
Ty zahrnují následující:
Velmi časté nežádoucí účinky mohou postihnout více než 1 osobu z 10:
• ztluštění zevní vrstvy kůže;
• kožní reakce jako je vyrážka, bradavičnaté výrůstky, zarudnutí nebo otok dlaní, prstů a chodidel (viz „Změny na kůži“ výše v bodě 4);
• bolest hlavy;
• nevolnost (pocit na zvracení), zvracení, průjem;
• snížení chuti k jídlu;
• zimnice;
• pocit slabosti;
• nedostatek energie;
• horečka (viz „Horečka (vysoká tělesná teplota)“ výše v bodě 4);
• bolesti kloubů, svalů nebo bolesti rukou nebo nohou;
• kašel;
• neobvyklá ztráta vlasů nebo jejich řídnutí.
Časté nežádoucí účinky mohou postihnout až 1 osobu z 10
• zácpa;
• příznaky podobné chřipce;
• nízká hladina fosforu v krvi, pozorovaná v krevních testech;
• zvýšení hladiny cukru (glukózy) v krvi, pozorované v krevních testech;
• změny v čerpání krve srdcem;
• kožní změny, zahrnující hrubá šupinatá místa na kůži, hnědě nebo žlutě zbarvené ztluštění kůže, kožní přívěsky, suchou kůži, lesklé pupínky, otevřené boláky, svědění nebo zarudnutí kůže (viz „Změny na kůži“ výše v bodě 4).
Méně časté nežádoucí účinky mohou postihnout až 1 osobu ze 100
• zánět oka (uveitida, viz „Oční problémy“ výše v bodě 4);
• zánět slinivky břišní (způsobující silnou bolest břicha);
• zánět podkožní tukové tkáně, příznaky zahrnují citlivé kožní uzlíky;
• alergická reakce;
• nově vzniklý melanom;
• problémy s ledvinami, selhání ledvin, pozorované v krevních testech;
• poruchy srdečního rytmu.
Nežádoucí účinky při užívání přípravku Tafinlar dohromady s trametinibem
Budete-li užívat Tafinlar dohromady s trametinibem, může u Vás dojít k některému z nežádoucích účinků uvedených v seznamu výše, i když frekvence se může být změněna (zvýšena nebo snížena).
Může u Vás dojít také k dalším nežádoucím účinkům v důsledku současného užívání trametinibu
s přípravkem Tafinlar, které jsou zmíněny v níže uvedeném seznamu.
Jestliže se u Vás objeví některý z těchto symptomů, sdělte to co nejdříve svému lékaři, lékárníkovi nebo zdravotní sestře, a to jak při prvních, tak i opakovaných projevech.
Pro získání detailní informace o nežádoucích účincích trametinibu si, prosím, přečtěte příbalovou informaci tohoto přípravku.
Nežádoucí účinky, které se u vás mohou projevit při užívání přípravku Tafinlar v kombinaci s trametinibem, jsou následující:
Velmi časté nežádoucí účinky (mohou postihnout více než 1 osobu z 10):
• závratě
• zimnice
• vysoká teplota
• vyrážka, suchá kůže, svědění, kožní problémy typu akné,
• snížená chuť k j ídlu
• bolest hlavy
• vysoký krevní tlak
• kašel
• bolesti žaludku
• pocit nevolnosti (nausea), nevolnost (zvracení)
• průjem
• zácpa
• bolest kloubů, bolest svalů, nebo bolest v rukou nebo nohou
• nedostatek energie, pocit slabosti
• otoky rukou nebo nohou
• nosní a krční zánět
• krvácení (hemoragie)
• infekce močového ústrojí
Velmi časté nežádoucí účinky, které se mohou projevit v krevních testech
• nízká hladina bílých krvinek
• abnormální výsledky krevních testů vztahující se k játrům
Časté nežádoucí účinky (mohou postihnout až 1 osobu z 10)
• nízký krevní tlak
• nadměrné pocení
• kožní účinky včetně hrubých šupinatých skvrn na kůži, kožní vyrážka s puchýřky naplněnými hnisem, hnědé nebo nažloutlé zesílení kůže, hrbolky na kůži, praskání kůže, výrůstky na kůži podobné bradavicím nebo zarudnutí a otok dlaní, prstů a chodidel, kožní karcinom skvamózních buněk (typ rakoviny kůže), zánět tukové vrstvy pod kůží, papilom (typ nádoru kůže, který není obvykle škodlivý), infekce kůže (celulitida), zánět vlasových váčků v kůži
• poruchy nehtů, jako jsou změny nehtového lůžka, bolest nehtů, infekce a otok okolní kůže
• neobvyklá ztráta vlasů nebo řídnutí
• dehydratace (nízká hladina vody nebo tekutiny)
• rozmazané vidění, problémy se zrakem
• dušnost
• bolest v ústech nebo vředy v ústech, zánět sliznice
• sucho v ústech
• chřipce podobné onemocnění
• svalové křeče
• otok obličeje
• noční pocení
• méně výkonná činnost srdce
• nižší tepová frekvence než je normální rozmezí a/nebo pokles tepové frekvence
Časté nežádoucí účinky, které se mohou projevit v krevních testech
• snížení počtu krevních destiček (buňky, které pomáhají srážení krve)
• snížení počtu červených krvinek (anémie) a určitého typu bílých krvinek (leukopenie)
• nízká hladina sodíku v krvi
• zvýšení některých látek (enzymů), produkovaných játry
• zvýšení kreatinfosfokinázy, enzymu, který se vyskytuje převážně v srdci, mozku a kosterním svalstvu
• zvýšení hladiny cukru v krvi
• nízká hladina fosfátů v krvi
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 osob)
• alergické reakce
• oční změny včetně otoku v oku způsobeného změnami na sítnici (chorioretinopatie), zánět oka (uveitida), oddělení světlocitlivé membrány v zadní části oka (sítnice) z jeho podpůrné vrstvy (odchlípení sítnice) a otoky kolem očí
• otok obličeje, lokalizovaný otok tkáně
• zánět slinivky břišní
• selhání ledvin, zánět ledvin
• zánět plic (pneumonie)
• nový primární melanom
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Tafinlar uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na lahvi a krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Tafinlar obsahuje
- Léčivou látkou je dabrafenibum. Jedna tvrdá tobolka obsahuje dabrafenibi mesilas v množství odpovídajícím 50 mg nebo 75 mg dabrafenibu.
- Dalšími složkami jsou mikrokrystalická celulosa, magnesium-stearát, koloidní bezvodý oxid křemičitý, červený oxid železitý (E172), oxid titaničitý (E171) a hypromelosa. Dále jsou tobolky potištěny černým inkoustem, který obsahuje černý oxid železitý (E172), šelak a propylenglykol.
Jak přípravek Tafinlar vypadá a co obsahuje toto balení
Tafinlar 50 mg tvrdé tobolky jsou neprůhledné tmavě červené s potiskem „GS TEW“ a „50 mg“. Tafinlar 75 mg tvrdé tobolky jsou neprůhledné tmavě růžové s potiskem „GS LHF“ a „75 mg“
Lahve jsou neprůhledné bílé z plastu s plastovým šroubovacím uzávěrem.
Lahev rovněž obsahuje jako vysoušedlo silikagel v malém obalu tvaru válce. Toto vysoušedlo musíte ponechat uvnitř lahve a nesmíte ho sníst.
Přípravky Tafinlar 50 mg a Tafinlar 75 mg tvrdé tobolky jsou dostupné v baleních obsahujících 28 nebo 120 tobolek. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Glaxo Wellcome, S.A., Avda. Extremadura, 3, 09400 Aranda De Duero, Burgos, Španělsko Novartis Pharmaceuticals UK Limited, Frimley Business Park, Frimley, Camberley, Surrey GU16 7SR, Velká Británie
Novartis Pharma GmbH, Roonstrasse 25, D-90429 Norimberk, Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11 Tel: +370 5 269 16 50
Btarapnu
Novartis Pharma Services Inc. Tem +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
EkXúba
Novartis (Hellas) A.E.B.E. Tpk: +30 210 281 17 12
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 555
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Osterreich
Novartis Pharma GmbH Tel: +43 1 86 6570
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Island Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kúnpoq Novartis Pharma Services Inc. TpA.: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
60