Strensiq 40 Mg/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Strensiq 40 mg/ml injekční roztok Strensiq 100 mg/ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Strensiq 40 mg/ml injekční roztok
Jeden ml roztoku obsahuje asfotasum alfa*40 mg.
Jedna injekční lahvička obsahuje 0,3 ml roztoku a asfotasum alfa 12 mg (40 mg/ml).
Jedna injekční lahvička obsahuje 0,45 ml roztoku a asfotasum alfa 18 mg (40 mg/ml).
Jedna injekční lahvička obsahuje 0,7 ml roztoku a asfotasum alfa 28 mg (40 mg/ml).
Jedna injekční lahvička obsahuje 1,0 ml roztoku a asfotasum alfa 40 mg (40 mg/ml).
Strensiq 100 mg/ml injekční roztok
Jeden ml roztoku obsahuje asfotasum alfa 100 mg
Jedna injekční stříkačka obsahuje 0,8 ml roztoku a asfotasum alfa 80 mg (100 mg/ml).
* vyrobeno rekombinantní DNA technologií za použití savčí buněčné kultury z ovarií čínského křečka. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce).
Čirý, bezbarvý až světle žlutý vodný roztok; pH 7,4.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Strensiq je indikován k dlouhodobé enzymové substituční léčbě u pacientů s hypofosfatázií, u nichž se první příznaky onemocnění objevily do 18 let věku, za účelem léčby kostních projevů tohoto onemocnění (viz bod 5.1).
4.2 Dávkování a způsob podání
Léčbu má zahajovat lékař se zkušenostmi v léčbě pacientů s metabolickými nebo kostními poruchami. Dávkování
Doporučený dávkovací režim asfotázy alfa je 2 mg/kg tělesné hmotnosti podávané subkutánně třikrát za týden nebo dávkovací režim 1 mg/kg tělesné hmotnosti podávané subkutánně šestkrát za týden. Podrobnější informace jsou uvedeny níže v dávkovacím schématu.
|
Tělesná hmotnost (kg) |
Při podávání 3x týdně |
Při |
podávání 6x týdně | |||
|
Podávaná dávka |
Podávaný objem |
Typ injekční lahvičky použitý pro injekci |
Podávaná dávka |
Podávaný objem |
Typ injekční lahvičky použitý pro injekci | |
|
3 |
6 mg |
0,15 ml |
0,3 ml | |||
|
4 |
8 mg |
0,20 ml |
0,3 ml | |||
|
5 |
10 mg |
0,25 ml |
0,3 ml | |||
|
6 |
12 mg |
0,30 ml |
0,3 ml |
6 mg |
0,15 ml |
0,3 ml |
|
7 |
14 mg |
0,35 ml |
0,45 ml |
7 mg |
0,18 ml |
0,3 ml |
|
8 |
16 mg |
0,40 ml |
0,45 ml |
8 mg |
0,20 ml |
0,3 ml |
|
9 |
18 mg |
0,45 ml |
0,45 ml |
9 mg |
0,23 ml |
0,3 ml |
|
10 |
20 mg |
0,50 ml |
0,7 ml |
10 mg |
0,25 ml |
0,3 ml |
|
11 |
22 mg |
0,55 ml |
0,7 ml |
11 mg |
0,28 ml |
0,3 ml |
|
12 |
24 mg |
0,60 ml |
0,7 ml |
12 mg |
0,30 ml |
0,3 ml |
|
13 |
26 mg |
0,65 ml |
0,7 ml |
13 mg |
0,33 ml |
0,45 ml |
|
14 |
28 mg |
0,70 ml |
0,7 ml |
14 mg |
0,35 ml |
0,45 ml |
|
15 |
30 mg |
0,75 ml |
1 ml |
15 mg |
0,38 ml |
0,45 ml |
|
16 |
32 mg |
0,80 ml |
1 ml |
16 mg |
0,40 ml |
0,45 ml |
|
17 |
34 mg |
0,85 ml |
1 ml |
17 mg |
0,43 ml |
0,45 ml |
|
18 |
36 mg |
0,90 ml |
1 ml |
18 mg |
0,45 ml |
0,45 ml |
|
19 |
38 mg |
0,95 ml |
1 ml |
19 mg |
0,48 ml |
0,7 ml |
|
20 |
40 mg |
1,00 ml |
1 ml |
20 mg |
0,50 ml |
0,7 ml |
|
25 |
50 mg |
0,50 ml |
0,80 ml |
25 mg |
0,63 ml |
0,7 ml |
|
30 |
60 mg |
0,60 ml |
0,8 ml |
30 mg |
0,75 ml |
1 ml |
|
35 |
70 mg |
0,70 ml |
0,8 ml |
35 mg |
0,88 ml |
1 ml |
|
40 |
80 mg |
0,80 ml |
0,8 ml |
40 mg |
1,00 ml |
1 ml |
|
50 |
50 mg |
0,50 ml |
0,8 ml | |||
|
60 |
60 mg |
0,60 ml |
0,8 ml | |||
|
70 |
70 mg |
0,70 ml |
0,8 ml | |||
|
80 |
80 mg |
0,80 ml |
0,8 ml | |||
|
90 |
90 mg |
0,90 ml |
0,8 ml (x2) | |||
|
100 |
100 mg |
1,00 ml |
0,8 ml (x2) | |||
Porucha funkce ledvin a jater
Bezpečnost a účinnost přípravku Strensiq u pacientů s poruchou funkce ledvin nebo jater nebyla stanovena a pro tyto pacienty nelze doporučit žádný specifický dávkovací režim.
Dospělí pacienti
Údaje o účinnosti a bezpečnosti u pacientů s hypofosfatázií ve věku > 18 let jsou omezené.
Starší pacienti
Neexistují žádné důkazy vyžadující zvláštní přístup při podávání přípravku Strensiq starším pacientům.
Způsob podání
Přípravek Strensiq je určen pouze k subkutánnímu podání. Není určen pro intravenózní ani intramuskulámí injekci.
Maximální objem léčivého přípravku na injekci nesmí překročit 1 ml. Pokud je nutné podat více než 1 ml, může se podat více injekcí současně.
Přípravek Strensiq se má podávat pomocí sterilních jednorázových injekčních stříkaček a injekčních jehel. Injekční stříkačka má být malého objemu, který je dostačující pro aspiraci předepsané dávky z injekční lahvičky s přijatelnou přesností.
Místa injekcí je třeba střídat a pečlivě sledovat známky potenciálních reakcí (viz bod 4.4).
Pacienti si mohou sami podávat injekce pouze v případě, že byli řádně proškoleni v postupu podávání přípravku.
Informace o zacházení s léčivým přípravkem před podáním jsou uvedeny v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Hypersenzitivní, anafylaktické nebo anafylaktoidní reakce nebyly při léčbě asfotázou alfa v klinických hodnoceních pozorovány. Mohou se vyskytnout těžké hypersenzitivní reakce alergického typu, včetně kopřivky, obtížného dýchání a/nebo kardiovaskulárního kolapsu. V případě výskytu těchto reakcí se doporučuje okamžitě přerušit léčbu a zahájit odpovídající lékařskou péči. Je třeba dodržovat standardní lékařské postupy pro akutní léčbu. V klinických hodnoceních nebyly pozorovány žádné nežádoucí účinky související s hladinou protilátek proti asfotáze alfa.
Kromě toho pacienti, u nichž byla potvrzena přítomnost protilátek proti léku, nevykazovali po podání asfotázy alfa známky hypersenzitivity či tachyfylaxe.
Reakce na injekci
Podání asfotázy alfa může mít za následek lokální reakce v místě injekce (mimo jiné včetně erytému, vyrážky, diskolorace, pruritu, bolesti, papuly, uzlin, atrofie) definované jako jakákoli související nežádoucí reakce objevující se během injekce nebo do konce dne, v němž byla injekce podána (viz bod 4.8). Střídání míst injekce obvykle pomáhá tyto reakce účinně regulovat. Všeobecně byly hodnoceny jako nezávažné, mírné až střední intenzity a spontánně odeznívající.
U všech pacientů s těžkými reakcemi na injekci je třeba přerušit podávání přípravku Strensiq a zahájit odpovídající léčbu.
Kraniosynostóza
V klinických studiích asfotázy alfa byly nežádoucí příhody kraniosynostózy (spojené se zvýšeným intrakraniálním tlakem), včetně zhoršení dříve existující kraniosynostózy, hlášeny u pacientů
s hypofosfatázií ve věku < 5 let. Neexistují dostatečné údaje k prokázání příčinného vztahu mezi expozicí Strensiqu a progresí kraniosynostózy. Kraniosynostóza jako projev hypofosfatázie je zdokumentována v publikované literatuře a vyskytla se u 61,3 % pacientů ve věku od narození do 5 let ve studii přirozené anamnézy u neléčených pacientů s hypofosfatázií s nástupem onemocnění v útlém dětství. Kraniosynostóza může vést ke zvýšení intrakraniálního tlaku. U pacientů s hypofosfatázií ve věku do 5 let se doporučuje pravidelné sledování (včetně fundoskopie ke zjištění známek edému papily) a rychlá intervence v případě zvýšeného intrakraniálního tlaku.
Ektopická kalcifikace
V klinických studiích asfotázy alfa byla u pacientů s hypofosfatázií hlášena oční kalcifikace (spojivky a rohovky) a nefrokalcinóza. Neexistují dostatečné údaje k prokázání příčinného vztahu mezi expozicí Strensiqu a ektopickou kalcifikací. Oční kalcifikace (spojivky a rohovky) a nefrokalcinóza jako projevy hypofosfatázie jsou zdokumentovány v publikované literatuře. Nefrokalcinóza se vyskytla
u 51,6 % pacientů ve věku od narození do 5 let ve studii přirozené anamnézy u neléčených pacientů
s hypofosfatázií s nástupem onemocnění v útlém dětství. U pacientů s hypofosfatázií se doporučují pravidelné oftalmologické kontroly a ultrazvuková vyšetření ledvin.
Parathormon a kalcium v séru
Koncentrace parathormonu v séru u pacientů s hypofosfatázií se může po podání asfotázy alfa zvýšit, nejvýrazněji během prvních 12 týdnů léčby. U pacientů léčených asfotázou alfa se doporučuje monitorovat sérové hladiny parathormonu a kalcia. Může být nutná suplementace kalcia a perorální suplementace vitaminu D. Viz bod 5.1.
Nepřiměřený nárůst hmotnosti
U pacientů může dojít k nepřiměřenému zvýšení hmotnosti. Doporučuje se dietologický dohled. Pomocné látky
Tento přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné injekční lahvičce, tj. přípravek je v podstatě „bez sodíku“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí s asfotázou alfa. Vzhledem ke struktuře a farmakokinetice asfotázy alfa je nepravděpodobné, že by ovlivňovala metabolismus související s cytochromem P-450.
Asfotáza alfa obsahuje katalytickou doménu tkáňově nespecifické alkalické fosfatázy. Podání asfotázy alfa ovlivní rutinní měření alkalické fosfatázy v séru v nemocničních laboratořích, v důsledku čehož bude naměřená aktivita alkalické fosfatázy v séru odpovídat několika tisícům jednotek na litr. Výsledky měření aktivity asfotázy alfa nelze interpretovat jako stejnou hodnotu, která byla naměřena pro alkalickou fosfatázu v séru, kvůli rozdílům v parametrech enzymů.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání asfotázy alfa těhotným ženám nejsou k dispozici.
Po opakovaném subkutánním podání březím myším v terapeutickém dávkovém rozmezí (> 0,5 mg/kg) byly hladiny asfotázy alfa u plodů kvantifikovatelné ve všech hodnocených dávkách, což napovídá o přestupu asfotázy alfa placentou. Studie reprodukční toxicity na zvířatech jsou nedostatečné (viz bod 5.3). Podávání asfotázy alfa se v těhotenství a u žen v reprodukčním věku, které nepoužívají antikoncepci, nedoporučuje.
Kojení
Informace o vylučování asfotázy alfa do lidského mateřského mléka jsou nedostatečné. Riziko pro kojené novorozence/děti nelze vyloučit.
Kojení má být během léčby asfotázou alfa přerušeno.
Fertilita
Byly provedeny předklinické studie fertility, které nepřinesly žádný průkaz účinku na fertilitu a embryofetální vývoj.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Strensiq nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Nejčastěji pozorovanými nežádoucími účinky byly reakce v místě injekce a nežádoucí účinky související s injekcí. Většina těchto účinků byla nezávažná, mírné až střední intenzity. Závažné účinky související s injekcí byly hlášeny u 2 pacientů bez přerušení léčby asfotázou alfa: u 1 pacienta s hypofosfatázií s nástupem onemocnění v útlém dětství byla zaznamenána horečka a zimnice a u 1 pacienta s hypofosfatázií s nástupem onemocnění v době dospívání byla zaznamenána orální hypestezie, bolest v končetině, zimnice a bolest hlavy.
Souhrn nežádoucích účinků v tabulce
V tabulce 1 jsou uvedeny nežádoucí účinky pozorované v klinických hodnoceních u 71 pacientů (ve věku od 1 dne do 66 let). Nežádoucí účinky asfotázy alfa jsou uvedeny podle tříd orgánových systémů a preferovaných termínů a jejich četnost je klasifikována podle pravidel databáze MedDRA: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 1: Nežádoucí účinky hlášené v klinických hodnoceních u pacientů s hypofosfatázií (ve věku od 1 dne do 66 let)_
|
Třída orgánového systému |
Kategorie četnosti |
Nežádoucí účinek |
|
Infekce a infestace |
časté |
Cellulitis (flegmóna) v místě injekce |
|
Poruchy krve a lymfatického systému |
časté |
Zvýšená náchylnost k tvoření modřin |
|
Poruchy nervového systému |
velmi časté | |
|
Cévní poruchy |
časté |
Nával horka |
|
Gastrointestinální poruchy |
časté |
Orální hypestezie Pocit na zvracení |
|
Poruchy kůže a podkožní tkáně |
velmi časté |
Erytém |
|
časté |
Lipohypertrofie Syndrom ochablé kůže Kožní diskolorace včetně hypopigmentace Kožní porucha (napnutá kůže) | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
velmi časté |
Bolest v končetině |
|
časté |
Myalgie | |
|
Celkové poruchy a reakce v místě aplikace |
velmi časté |
Reakce v místě injekce1 Pyrexie |
|
časté | ||
|
Poranění, otravy a procedurální komplikace |
velmi časté |
Kontuze |
|
časté |
Jizva |
1- Preferované termíny považované za reakce v místě injekce jsou uvedeny v bodě níže.
Popis vybraných nežádoucích účinků
Reakce v místě injekce
Reakce v místě injekce (včetně erytému v místě injekce, diskolorace, bolesti, pruritu, makuly, zduření, zhmoždění, hypertrofie, indurace, reakce, atrofie, uzliny, vyrážky, papuly, hematomu, zánětu, kopřivky, tepla, krvácení, cellulitis (flegmóny) a rezistence) patří mezi nejčastější nežádoucí účinky pozorované přibližně u 73 % pacientů v klinických studiích. Četnost reakcí v místě injekce byla vyšší u pacientů s hypofosfatázií s nástupem onemocnění v době dospívání a u pacientů, kteří dostávali injekce 6krát týdně (v porovnání s podáváním 3krát týdně). Většina reakcí v místě injekce byla mírná a spontánně odezněla a žádný případ nebyl hlášen jako závažná nežádoucí příhoda. U dvou pacientů byly zaznamenány reakce v místě injekce, které vedly ke snížení jejich dávky asfotázy alfa.
U jednoho ze 71 pacientů léčených v klinických hodnoceních došlo k těžké reakci v místě injekce ve formě diskolorace v místě injekce, což vedlo k ukončení léčby.
Imunogenicita
Existuje potenciál k imunogenicitě. Mezi 69 pacienty s hypofosfatázií, kteří byli zařazeni do klinických hodnocení a u nichž byly získány údaje v průběhu studie, mělo 56 (81,2 %) pacientů pozitivní test na protilátky proti léku v některém časovém bodě po zahájení léčby přípravkem Strensiq. Z těchto 56 pacientů jich 25 (44,6 %) také vykazovalo přítomnost neutralizujících protilátek. Odpověď protilátek (s přítomností nebo bez přítomnosti neutralizujících protilátek) měla časově proměnný charakter. Nebyl prokázán vliv rozvoje protilátek na klinickou účinnost nebo bezpečnost (viz bod 5.2).
V klinických hodnoceních nebyl u nežádoucích příhod pozorován žádný trend související s hladinou protilátek. Kromě toho pacienti, u nichž byla potvrzena přítomnost protilátek, nevykazovali po subkutánním podání asfotázy alfa známky hypersenzitivity či tachyfylaxe.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
S předávkováním asfotázou alfa nejsou žádné zkušenosti. Pro zvládnutí nežádoucích účinků viz body
4.4 a 4.8.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: <dosud nepřidělena>, ATC kód: <dosud nepřidělen>
Asfotáza alfa je lidský rekombinantní tkáňově nespecifický fúzní protein alkalická fosfatáza-Fc-dekaaspartát, který je exprimovaný v geneticky upravené buněčné linii ovarií čínského křečka. Asfotáza alfa je rozpustný glykoprotein složený ze dvou identických polypeptidových řetězců, každý o délce 726 aminokyselin, který tvoří (i) katalytická doména lidské tkáňově nespecifické alkalické fosfatázy, (ii) Fc doména lidského imunoglobulinu-G1 a (iii) peptidová doména dekaaspartátu.
Hypofosfatázie
Hypofosfatázie je vzácná, těžká a potenciálně fatální genetická porucha způsobená mutací (mutacemi) genu kódujícího tkáňově nespecifickou alkalickou fosfatázu s následkem ztráty jeho funkce. Hypofosfatázie je spojena s mnohočetnými kostními projevy včetně rachitidy/osteomalacie, změněného metabolismu kalcia a fosfátů, poruchy růstu a pohyblivosti, s respiračními potížemi, které mohou vyžadovat ventilaci, a záchvaty reagujícími na vitamin B6.
Mechanismus účinku
Asfotáza alfa, lidský rekombinantní tkáňově nespecifický fúzní protein alkalická fosfatáza-Fc-dekaaspartát, vykazuje enzymovou aktivitu a podporuje mineralizaci skeletu u pacientů s hypofosfatázií.
Klinická účinnost a bezpečnost
Studie ENB-006-09/ENB-008-10
Do otevřené, nerandomizované studie ENB-006-09/ENB-008-10 bylo zařazeno 13 pacientů. Pět (5) pacientů se dostavilo k lékaři s hypofosfatázií dříve, než dosáhli věku 6 měsíců, a 8 pacientů se dostavilo po dosažení věku 6 měsíců. Věk při zařazení do studie byl 6 až 12 let. Dvanáct (12) pacientů ve studii nadále pokračuje. Ve studii byla použita historická kontrolní skupina ze stejného centra jako pacienti, kteří dostávali asfotázu alfa a byli léčeni podle obdobného protokolu klinické léčby.
Účinky asfotázy alfa na rentgenový snímek
Vyškolení radiologové hodnotili rentgenové snímky zápěstí a kolen pacientů pořízené před zahájením studie a po jejím zahájení z hlediska následujících známek: zjevné rozšíření epifyzární štěrbiny, pohárkovité rozšíření metafýzy, nepravidelné zóny provizorní kalcifikace, projasnění v metafýze, skleróza metadiafýzy, osteopenie, „popcorn“ kalcifikace v metadiafýze, demineralizace distální metafýzy, transverzální pruh projasnění pod epifyzární štěrbinou a podélně probíhající zóny projasnění (,jazyky“). Změny na rentgenových snímcích oproti počátečnímu stavu byly poté zhodnoceny podle hodnoticí stupnice pro globální radiografické hodnocení změny (RGI-C - Radiographic Global Impression of Change) následovně: -3 = těžké zhoršení, -2 = střední zhoršení, -1 = minimální zhoršení, 0 = beze změny, +1 = minimální zhojení, +2 = výrazné zhojení, +3 = téměř úplné nebo úplné zhojení. Pacienti, kteří dostávali asfotázu alfa, dosáhli za prvních 6 měsíců expozice skóre +2 a +3 a tento stav byl při pokračování léčby zachován. V historické kontrolní skupině nebyla změna v průběhu času pozorována.
Kostní biopsie
Před provedením kostní biopsie byl za účelem značení kosti podáván tetracyklin ve dvou 3denních cyklech (oddělených 14denním odstupem). Transiliakální biopsie ze hřebene kyčelní kosti byly provedeny standardním postupem. K histologické analýze odebraných vzorků byl použit software Osteomeasure (Osteometrics, USA). Nomenklatura, symboly a jednotky splňovaly doporučení Americké společnosti pro výzkum kostí a minerálů (American Society for Bone andMineral Research). U 10 pacientů v souboru subjektů léčených dle protokolu (byli vyloučeni ti pacienti, kterým byl perorálně podáván vitamin D v období od počátku studie do 24. týdne), kteří podstoupili transiliakální biopsii ze hřebene kyčelní kosti před podáním asfotázy alfa i po jejím podání:
- střední (směrodatná odchylka [SD]) tloušťka osteoidu byla 12,8 (3,5) ^m na počátku studie
a 9,5 (5,1) ^m ve 24. týdnu;
- střední (SD) objem osteoidu / objem kosti byl 11,8 (5,9) % na počátku studie a 8,6 (7,2) % ve
24. týdnu;
- střední (SD) doba mineralizace byla 93 (70) dní na počátku studie a 119 (225) dní ve 24. týdnu.
Růst
Výška, hmotnost a obvod hlavy byly zaznamenány do růstových grafů (série percentilových křivek znázorňujících distribuci) získaných ze středisek pro kontrolu a prevenci nemocí (CDC - Centers for Disease Control andPrevention) v USA. Tyto referenční údaje byly převzaty od reprezentativního vzorku zdravých dětí a nejsou specifické pro děti, které vyžadují speciální zdravotní péči: byly použity bez růstových grafů dětí s hypofosfatázií.
U pacientů, kteří dostávali asfotázu alfa: 9 ze 13 pacientů vykázalo perzistentní zjevný dorovnávací výškový přírůstek, jak ukazuje přechod v průběhu času do vyššího percentilu na růstových grafech CDC. Tři (3) ze 13 pacientů nevykázali zjevný dorovnávací výškový přírůstek a u 1 pacienta nebyly údaje pro posouzení dostačující. Vývoj podle stadií Tannerovy stupnice se zdál být odpovídající.
U historické kontrolní skupiny během doby sledování: 1 ze 16 pacientů vykázal zjevný dorovnávací výškový přírůstek, 12 ze 16 pacientů nevykázalo zjevný dorovnávací výškový přírůstek a u 3 ze 16 pacientů nebylo možné údaje vyhodnotit.
U některých pacientů byla v průběhu studie nutná perorální suplementace vitaminu D (viz body 4.4 a 4.8).
Studie ENB-002-08/ENB-003-08
Do otevřené, nerandomizované, nekontrolované studie ENB-002-08/ENB-003-08 bylo zařazeno 11 pacientů a 9 pacientů ve studii nadále pokračuje. U všech pacientů došlo k nástupu hypofosfatázie ve věku do 6 měsíců. Věk při zařazení do studie byl 0,5 až 35 měsíců.
Sedm (7) z 11 pacientů v úplném analyzovaném souboru dosáhlo ve 24. týdnu skóre RGI-C +2 v porovnání s rentgenovými snímky na počátku studie.
Pět (5) z 11 subjektů vykázalo zjevný dorovnávací výškový přírůstek. Byla zjevná fluktuace ve výškovém přírůstku, což může odrážet těžší onemocnění a vyšší míru morbidity u těchto mladších pacientů.
Studie ENB-009-10
Do otevřené, randomizované studie ENB-009-10 bylo zařazeno 19 pacientů a 18 pacientů ve studii nadále pokračuje. U 4 pacientů došlo k nástupu hypofosfatázie ve věku do 6 měsíců, u 12 pacientů ve věku od 6 měsíců do 18 let a u 2 pacientů ve věku nad 18 let. U 1 pacienta nebyl věk při nástupu onemocnění znám. Věk při zařazení do studie byl 13 až 66 let.
Dospívající (a dospělí) pacienti v této studii nevykázali zjevný výškový přírůstek.
Pacienti podstoupili transiliakální biopsii ze hřebene kyčelní kosti buď v rámci kontrolní skupiny, nebo před expozicí asfotázy alfa i po její expozici:
- kontrolní skupina, standardní léčba (5 hodnotitelných pacientů): střední (SD) doba mineralizace
byla 226 (248) dní na počátku studie a 304 (211) dní ve 24. týdnu;
- skupina užívající 0,3 mg/kg/den asfotázy alfa (4 hodnotitelní pacienti): střední (SD) doba
mineralizace byla 1236 (1468) dní na počátku studie a 328 (200) dní ve 48. týdnu;
- skupina užívající 0,5 mg/kg/den asfotázy alfa (5 hodnotitelných pacientů): střední (SD) doba
mineralizace byla 257 (146) dní na počátku studie a 130 (142) dní ve 48. týdnu.
Po 48. týdnu bylo dávkování u všech pacientů upraveno na doporučenou dávku 1,0 mg/kg/den.
Ventilační podpora
Ve dvou otevřených, nerandomizovaných, nekontrolovaných studiích ENB-002-08/ENB-003-08 (11 pacientů) a ENB-010-10 (26 pacientů) s pacienty ve věku 0,1 až 310 týdnů na počátku studie byla u 21 z 37 pacientů nutná ventilační podpora:
• U 14 pacientů byla na počátku studie nutná invazivní ventilační podpora (intubace nebo tracheostomie; u jednoho pacienta bylo zaznamenáno na počátku studie před přemístěním krátké období s neinvazivní ventilací).
- Sedm (7) pacientů bylo odpojeno od ventilace (doba ventilace od 24 do 168 týdnů), všichni dosáhli skóre RGI-C (Radiographic Global Impression of Change) >2.
- U 3 pacientů se pokračovalo s ventilační podporou, skóre RGI-C <2.
- Tři (3) pacienti na ventilační podpoře zemřeli.
- Jeden (1) pacient odvolal souhlas.
• U 7 pacientů byla po začátku studie zahájena neinvazivní ventilace (BiPAP nebo CPAP; u 2 pacientů byla nutná krátkodobá podpora pomocí invazivní ventilace).
- Pět (5) pacientů bylo odpojeno od ventilace (doba ventilace od 4 týdnů do 48 týdnů).
- Dva (2) pacienti zemřeli.
Přirozená anamnéza u pediatrických pacientů s neléčenou hypofosfatázií naznačuje vysokou mortalitu v případech, pokud je zapotřebí ventilace.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Strensiq u jedné nebo více podskupin pediatrické populace v indikaci hypofosfatázie (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován za „výjimečných okolností“.
Znamená to, že vzhledem ke vzácné povaze onemocnění, pro které je indikován, nebylo možné získat úplné informace o přínosech a rizicích tohoto léčivého přípravku.
Evropská agentura pro léčivé přípravky každoročně vyhodnotí jakékoli nově dostupné informace a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Farmakokinetika asfotázy alfa byla hodnocena v 1měsíční multicentrické otevřené studii, s eskalací dávky u dospělých s hypofosfatázií. Kohorta 1 (n = 3) ve studii dostávala první týden asfotázu alfa v dávce 3 mg/kg intravenózně a poté od 2. do 4. týdne 3 dávky 1 mg/kg subkutánně v týdenních intervalech. Kohorta 2 (n = 3) ve studii dostávala první týden asfotázu alfa v dávce 3 mg/kg intravenózně a poté od 2. do 4. týdne 3 dávky 2 mg/kg subkutánně v týdenních intervalech. Medián doby (Tmax) po intravenózní infuzi v dávce 3 mg/kg po dobu 1,08 hodin se pohyboval v rozmezí 1,25 až 1,50 hodin a střední (SD) hodnota Cmax se pohybovala v rozmezí od 42 694 (8 443) do 46 890 (6 635) j./l v hodnocených kohortách. Absolutní biologická dostupnost po prvním a třetím subkutánním podání se pohybovala v rozmezí 45,8 až 98,4 % s mediánem doby Tmax v rozmezí
24,2 až 48,1 hodin. Po subkutánním podání 1 mg/kg týdně v kohortě 1 byla střední (SD) hodnota AUC v průběhu dávkovacího intervalu (AUCt) 66 034 (19 241) a 40 444 (N = 1) j.*h/l po první, resp. po třetí dávce. Po subkutánním podání 2 mg/kg týdně v kohortě 2 byla střední (SD) hodnota AUCt 138 595 (6 958) a 136 109 (41 875) po první, resp. po třetí dávce.
Farmakokinetické údaje ze všech klinických hodnocení asfotázy alfa byly analyzovány metodami populační farmakokinetiky. Farmakokinetické proměnné charakterizované na základě populační farmakokinetické analýzy reprezentují celkovou populaci pacientů s hypofosfatázií ve věkovém rozmezí 1 den až 66 let, subkutánní dávky až 28 mg/kg/týden a rozmezí kohort dle nástupu onemocnění. Dvacet pět procent (15 ze 60) z celkové populace pacientů bylo na počátku studie dospělých (> 18 let). Absolutní biologická dostupnost a míra absorpce po subkutánním podání podle odhadu činí 0,602 (95% interval spolehlivosti [CI]: 0,567; 0,638) nebo 60,2 %, resp. 0,572 (95% CI: 0,338; 0,967) nebo 57,2 % za den. Odhadované centrální a periferní distribuční objemy u pacienta s tělesnou hmotností 70 kg (a 95% CI) byly 5,66 (2,76; 11,6) l, resp. 44,8 (33,2; 60,5) l. Odhadovaná centrální a periferní clearance u pacienta s tělesnou hmotností 70 kg (a 95% CI) byla 15,8 (13,2;
18,9) l/den, resp. 51,9 (44,0; 61,2) l/den. Mezi vnější faktory ovlivňující farmakokinetickou expozici asfotázy alfa patřily specifická aktivita lékové formy a celkový obsah N-acetylneuraminové kyseliny. Průměrný ± SD eliminační poločas po subkutánním podání byl 2,28 ± 0,58 dní.
Linearita/nelinearita
Na základě výsledků populační farmakokinetické analýzy lze potvrdit, že asfotáza alfa vykazuje lineární farmakokinetiku při subkutánních dávkách až do 28 mg/kg/týden. Na základě modelace bylo zjištěno, že tělesná hmotnost ovlivňuje clearance a parametry distribučního objemu asfotázy alfa. Předpokládá se, že farmakokinetická expozice se bude s tělesnou hmotností zvyšovat. Vliv imunogenicity na farmakokinetiku asfotázy alfa se v průběhu času liší v důsledku časově proměnného charakteru imunogenicity a obecně se odhaduje, že snižuje farmakokinetickou expozici o méně než 20 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
V neklinickém testování bezpečnosti na potkanech nebyly při žádných dávkách či cestách podání zjištěny specifické nežádoucí účinky na orgánové systémy.
U potkanů byly po intravenózním podání dávek 1 až 180 mg/kg pozorovány akutní reakce na injekci závislé na dávce a čase, které měly přechodný charakter a spontánně odezněly.
U opic byly pozorovány ektopické kalcifikace a reakce v místě injekce po subkutánním podávání asfotázy alfa v denních dávkách až 10 mg/kg po dobu 26 týdnů. Tyto účinky byly omezeny na místa injekcí a byly částečně nebo zcela reverzibilní.
Nebyl pozorován žádný důkaz ektopické kalcifikace v žádných jiných vyšetřovaných tkáních.
Předklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání nebo reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka. U březích samic králíků, kterým byly intravenózně podávány dávky asfotázy alfa až do 50 mg/kg/den, však byly protilátky proti léku detekovány až u 75 % zvířat, což mohlo ovlivnit zjištění reprodukční toxicity.
Nebyly provedeny žádné studie na zvířatech, které by hodnotily genotoxicitu a kancerogenní potenciál asfotázy alfa.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný
Heptahydrát hydrogenfosforečnanu sodného Monohydrát dihydrogenfosforečnanu sodného Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
Chemická a fyzikální stabilita přípravku připraveného k použití byla prokázána až na 1 hodinu při teplotě 23 °C až 27 °C.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Podmínky uchovávání před podáním léčivého přípravku viz bod 6.3.
6.5 Druh obalu a obsah balení
2ml injekční lahvička (sklo třídy I) se zátkou (butylová pryž) a uzávěrem (hliník) s odtrhovacím víčkem (polypropylen).
Strensiq 40 mg/ml injekční roztok
Plnicí objemy injekčních lahviček jsou: 0,3 ml, 0,45 ml, 0,7 ml a 1,0 ml
Strensiq 100 mg/ml injekční roztok
Plnicí objemy injekčních lahviček jsou: 0,8 ml.
Velikost balení: krabička s 1 nebo 12 injekčními lahvičkami Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Každá injekční lahvička je určena pouze k jednorázovému použití a smí se propíchnout jen jednou. Veškerý nepoužitý roztok v injekční lahvičce musí být zlikvidován.
Přípravek Strensiq se má podávat pomocí sterilních jednorázových injekčních stříkaček a injekčních jehel. Injekční stříkačka má být malého objemu, který je dostačující pro aspiraci předepsané dávky z injekční lahvičky s přijatelnou přesností. Je nutné dodržovat aseptické postupy.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Alexion Europe SAS 1-15, Avenue Edouard Belin 92500 Rueil-Malmaison Francie
8. REGISTRAČNÍ ČÍSLO(A)
Strensiq 40 mg/ml injekční roztok
EU/1/15/1015/001
EU/1/15/1015/002
EU/1/15/1015/005
EU/1/15/1015/006
EU/1/15/1015/007
EU/1/15/1015/008
EU/1/15/1015/009
EU/1/15/1015/010
Strensiq 100 mg/ml injekční roztok
EU/1/15/1015/003
EU/1/15/1015/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
28/08/2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO REGISTRACI PŘÍPRAVKU ZA VÝJIMEČNÝCH OKOLNOSTÍ
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Lonza Biologics
101 International Drive
Pease International Tradeport
03801 Portsmouth
USA
Název a adresa výrobce odpovědného za propouštění šarží
Alexion Pharma International Trading
College Business and Technology Park, Blanchardstown
Dublin 15
Irsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Před uvedením přípravku Strensiq na trh v každém členském státě musí držitel rozhodnutí o registraci s příslušným národním orgánem odsouhlasit obsah a formát vzdělávacího programu, včetně komunikačních médií, způsobů distribuce a jakýchkoliv dalších aspektů programu.
Vzdělávací program je určen k poskytování pokynů pacientům a jejich ošetřovatelům týkajících se
správné techniky podání přípravku, s důrazem na rizika chyb v medikaci a reakcí v místě podání injekce.
Držitel rozhodnutí o registraci zajistí, aby byl v každém členském státě, ve kterém se bude přípravek Strensiq prodávat, všem pacientům/rodičům nebo ošetřovatelům, u nichž se předpokládá, že budou přípravek Strensiq používat, poskytnut následující vzdělávací balíček:
• Informace o přípravku pro pacienta
• Návod k samopodání injekce pro pacienta
• Návod k podání injekce pro rodiče dětských pacientů nebo jejich ošetřovatele Návod pro pacienta/rodiče nebo ošetřovatele musí obsahovat následující klíčové body:
• Upozornění a opatření týkající se možných rizik chyb v medikaci a reakcí v místě podání injekce spojených s použitím přípravku Strensiq
• Pokyny ke správnému dávkování
• Pokyny k výběru místa vpichu injekce, způsobu injekce a k jejímu záznamu
• Podrobný popis způsobu podání injekce přípravku Strensiq za použití aseptických technik
• Informace o řízení chladicího teplotního režimu přípravku Strensiq během skladování a přepravy
• Informace o hlášení nežádoucích účinků
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Držitel rozhodnutí o registraci uskuteční multicentrickou, randomizovanou, otevřenou studii fáze 2a přípravku Strensiq u pacientů s hypofosfatázií (HPP) za účelem: (i) vyhodnocení farmakokinetiky (PK) přípravku Strensiq u dospělých po podání dávky doporučené u dětí; (ii) poskytnutí údajů o odezvě dávky na plazmatický anorganický pyrofosfát (PPi) a pyridoxal-5-fosfát (PLP) a prozkoumání důkazů o klinickém prospěchu. V rámci zajištění spolehlivosti údajů předloží držitel rozhodnutí o registraci protokol studie zahrnující odpovídající techniky odběru krve, skladování a testy biomarkerů PPi a PLP, které mají být před začátkem studie odsouhlaseny CHMP. |
31. března 2017 |
|
Držitel rozhodnutí o registraci zajistí prodloužení studií ENB-008-10 a ENB-009-10 za účelem zajištění údajů o účinnosti (jako jsou mimo jiné, skóre RGI-C, změna výšky a hmotnosti, stanovení biomarkerů) u pacientů ve věku 13 až 18 let. |
31. března 2017 |
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO REGISTRACI PŘÍPRAVKU ZA VÝJIMEČNÝCH OKOLNOSTÍ
Tato registrace byla schválena za „výjimečných okolností“, a proto podle článku 14(8) nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
Držitel rozhodnutí registraci zřídí observační, longitudinální, prospektivní, dlouhodobý registr pacientů s HPP za účelem sběru informací týkajících se epidemiologie onemocnění, včetně klinických výstupů a kvality života, a vyhodnocení údajů o bezpečnosti a účinnosti u pacientů léčených přípravkem Strensiq. |
Jednou ročně v rámci pravidelného ročního hodnocení |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Strensiq 40 mg/ml injekčního roztoku asfotasum alfa
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml roztoku obsahuje asfotasum alfa 40 mg.
Jedna injekční lahvička obsahuje asfotasum alfa 12 mg (12 mg/0,3 ml). Jedna injekční lahvička obsahuje asfotasum alfa 18 mg (18 mg/0,45 ml). Jedna injekční lahvička obsahuje asfotasum alfa 28 mg (28 mg/0,7 ml). Jedna injekční lahvička obsahuje asfotasum alfa 40 mg (40 mg/1 ml).
3. SEZNAM POMOCNÝCH LÁTEK
Chlorid sodný, heptahydrát hydrogenfosforečnanu sodného, monohydrát dihydrogenfosforečnanu sodného, voda na injekci.
Podrobnější informace naleznete v příbalové informaci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok 1 injekční lahvička s 0,3[0,45; 0,7; 1] ml
12 injekčních lahviček s 0,3[0,45; 0,7; 1] ml
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý roztok musí být zlikvidován.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Alexion Europe SAS 1-15, Avenue Edouard Belin 92500 Rueil-Malmaison Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1015/001
EU/1/15/1015/002
EU/1/15/1015/005
EU/1/15/1015/006
EU/1/15/1015/007
EU/1/15/1015/008
EU/1/15/1015/009
EU/1/15/1015/010
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
STRENSIQ 40 mg/ml 12 mg/0,3 ml 18 mg/0,45 ml 28 mg/0,7 ml 40 mg/1 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Strensiq 40 mg/ml injekce Strensiq 40 mg/ml injekce Strensiq 40 mg/ml injekce Strensiq 40 mg/ml injekce asfotasum alfa s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
12 mg/0,3 ml 18 mg/0,45 ml 28 mg/0,7 ml 40 mg/1 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Strensiq 100 mg/ml injekčního roztoku asfotasum alfa
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml roztoku obsahuje asfotasum alfa 100 mg.
Jedna injekční lahvička obsahuje asfotasum alfa 80 mg (80 mg/0,8 ml).
3. SEZNAM POMOCNÝCH LÁTEK
Chlorid sodný, heptahydrát hydrogenfosforečnanu sodného, monohydrát dihydrogenfosforečnanu sodného, voda na injekci.
Podrobnější informace naleznete v příbalové informaci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok 1 injekční lahvička s 0,8 ml 12 injekčních lahviček s 0,8 ml
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý roztok musí být zlikvidován.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Alexion Europe SAS 1-15, Avenue Edouard Belin 92500 Rueil-Malmaison Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1015/003
EU/1/15/1015/004
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
STRENSIQ 100 mg/ml 80 mg/0,8 ml
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK INJEKČNÍ LAHVIČKY 100 mg/ml_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Strensiq 100 mg/ml injekce
asfotasum alfa
s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
80 mg/0,8 ml
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Strensiq 40 mg/ml injekční roztok (12 mg/0,3 ml 18 mg/0,45 ml 28 mg/0,7 ml 40 mg/1 ml)
Asfotasum alfa
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Strensiq a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Strensiq používat
3. Jak se přípravek Strensiq používá
4. Možné nežádoucí účinky
5. Jak přípravek Strensiq uchovávat
6. Obsah balení a další informace
1. Co je přípravek Strensiq a k čemu se používá Co je přípravek Strensiq
Přípravek Strensiq je lék, který se používá k léčbě dědičného onemocnění hypofosfatázie. Obsahuje léčivou látku asfotáza alfa.
Co je hypofosfatázie
Pacienti s hypofosfatázií mají nízké hladiny enzymu nazývaného alkalická fosfatáza, který je důležitý pro různé tělesné funkce, včetně řádného zpevnění kostí a zubů. Pacienti mívají potíže s růstem a pevností kostí, což může vést ke zlomeninám a bolestem kostí a obtížné chůzi, a také k potížím s dýcháním a riziku záchvatů (křečí).
K čemu se přípravek Strensiq používá
Léčivá látka v přípravku Strensiq může při hypofosfatázii nahradit chybějící enzym (alkalickou fosfatázu). Používá se jako dlouhodobá enzymová substituční léčba ke zvládání příznaků.
Jaké přínosy přípravku Strensiq byly prokázány v klinických studiích
Bylo prokázáno, že přípravek Strensiq má pro pacienty přínosy z hlediska mineralizace kostry a růstu kostí.
2. Čemu musíte věnovat pozornost, než začnete přípravek Strensiq používat Nepoužívejte přípravek Strensiq:
- jestliže jste alergický(á) na asfotázu alfa nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
• Přípravek Strensiq může u některých osob způsobovat alergické reakce. Mezi příznaky takových reakcí patří zvracení, dýchací potíže, rychlá srdeční frekvence, náhlý pokles krevního tlaku, kopřivka nebo vyrážka. Pokud se u Vás vyskytne kterýkoli z těchto příznaků, sdělte to okamžitě svému lékaři. Možná budete muset dostávat další léky k prevenci alergické reakce (antihistaminika nebo kortikosteroidy).
• Ve studiích byly hlášeny některé nežádoucí účinky postihující oči, které pravděpodobně souvisely s hypofosfatázií, jak u pacientů používajících přípravek Strensiq, tak u těch, kteří tento přípravek nepoužívali. V případě problémů s viděním se poraďte se svým lékařem.
• V klinických hodnoceních s přípravkem Strensiq i bez něj, která zahrnovala děti
s hypofosfatázií, byl hlášen časný srůst lebečních kostí u dětí ve věku do 5 let. Poraďte se se svým lékařem, jestliže si všimnete jakékoli změny tvaru hlavy svého dítěte.
• Jestliže se léčíte přípravkem Strensiq, mohou se u Vás při injekčním podání přípravku nebo během několika hodin po podání objevit reakce v místě injekce (bolest, uzlík, vyrážka, změna barvy). Pokud se u Vás vyskytne jakákoli těžká reakce v místě injekce, sdělte to okamžitě svému lékaři.
• Ve studiích bylo hlášeno zvýšení koncentrace parathormonu a nízké hladiny vápníku. Z tohoto důvodu Vás může lékař požádat, abyste v případě potřeby užíval(a) doplňky vápníku a perorální vitamin D.
• Během léčby přípravkem Strensiq se může objevit nárůst hmotnosti. Dle potřeby Vám lékař poskytne dietologické poradenství.
Další léčivé přípravky a přípravek Strensiq
Informujte svého lékaře nebo lékárníka o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
Těhotenství a kojení
Přípravek Strensiq se smí v průběhu těhotenství nebo kojení použít pouze tehdy, když je to z lékařského hlediska nezbytné.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Řízení dopravních prostředků a obsluha strojů
Nepředpokládá se, že by tento přípravek měl jakýkoli vliv na schopnost řídit nebo obsluhovat stroje. Důležité informace o některých složkách přípravku Strensiq
Tento přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné injekční lahvičce; to znamená, že přípravek je v podstatě „bez sodíku“.
3. Jak se přípravek Strensiq používá
Vždy používejte tento přípravek přesně v souladu s příbalovou informací nebo podle pokynů svého lékaře, lékárníka nebo zdravotní sestry. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Použití přípravku Strensiq Vám vysvětlí lékař, který má zkušenosti v léčbě pacientů s metabolickým nebo kostním onemocněním. Po proškolení lékařem nebo odbornou sestrou si můžete podávat injekce přípravku Strensiq sám (sama) v domácím prostředí.
Dávkování
• Dávka, kterou dostáváte, vychází z Vaší tělesné hmotnosti.
• Správnou dávku určí Váš lékař. Celkem budete injekcí pod kůži (subkutánně) každý týden dostávat 6 mg asfotázy alfa na kg tělesné hmotnosti (pro detailní informace o podávaném objemu a typu lahvičky, která má být použita na základě Vaší hmotnosti, viz dávkovací tabulka níže).
• Tuto celkovou dávku lze podávat buď jako injekci 1 mg/kg asfotázy alfa 6krát týdně, nebo jako 2 mg/kg asfotázy alfa 3krát týdně podle doporučení Vašeho lékaře.
• Maximální objem na injekci nesmí překročit 1 ml. Pokud je nutné podat více než 1 ml, musíte podat více injekcí ihned za sebou.
Při podávání 3krát týdně Při podávání 6krát týdně
|
Tělesná hmotnost (kg) |
Podávaný objem |
Barva injekční lahvičky, která se má použít |
|
3 |
0,15 ml |
tmavě modrá |
|
4 |
0,20 ml |
tmavě modrá |
|
5 |
0,25 ml |
tmavě modrá |
|
6 |
0,30 ml |
tmavě modrá |
|
7 |
0,35 ml |
oranžová |
|
8 |
0,40 ml |
oranžová |
|
9 |
0,45 ml |
oranžová |
|
10 |
0,50 ml |
světle modrá |
|
11 |
0,55 ml |
světle modrá |
|
12 |
0,60 ml |
světle modrá |
|
13 |
0,65 ml |
světle modrá |
|
14 |
0,70 ml |
světle modrá |
|
15 |
0,75 ml |
růžová |
|
16 |
0,80 ml |
růžová |
|
17 |
0,85 ml |
růžová |
|
18 |
0,90 ml |
růžová |
|
19 |
0,95 ml |
růžová |
|
20 |
1 ml |
růžová |
|
25 |
0,50 ml |
zelená |
|
30 |
0,60 ml |
zelená |
|
35 |
0,70 ml |
zelená |
|
40 |
0,80 ml |
zelená |
|
Tělesná hmotnost (kg) |
Podávaný objem |
Barva injekční lahvičky, která se má použít |
|
6 |
0,15 ml |
tmavě modrá |
|
7 |
0,18 ml |
tmavě modrá |
|
8 |
0,20 ml |
tmavě modrá |
|
9 |
0,23 ml |
tmavě modrá |
|
10 |
0,25 ml |
tmavě modrá |
|
11 |
0,28 ml |
tmavě modrá |
|
12 |
0,30 ml |
tmavě modrá |
|
13 |
0,33 ml |
oranžová |
|
14 |
0,35 ml |
oranžová |
|
15 |
0,38 ml |
oranžová |
|
16 |
0,40 ml |
oranžová |
|
17 |
0,43 ml |
oranžová |
|
18 |
0,45 ml |
oranžová |
|
19 |
0,48 ml |
světle modrá |
|
20 |
0,50 ml |
světle modrá |
|
25 |
0,63 ml |
světle modrá |
|
30 |
0,75 ml |
růžová |
|
35 |
0,88 ml |
růžová |
|
40 |
1 ml |
růžová |
|
50 |
0,50 ml |
zelená |
|
60 |
0,60 ml |
zelená |
|
70 |
0,70 ml |
zelená |
|
80 |
0,80 ml |
zelená |
|
90 |
0,90 ml |
zelená (x2) |
|
100 |
1 ml |
zelená (x2) |
Použití u dětí a dospívajících
Doporučená dávka přípravku Strensiq u dětí a dospívajících je stejně jako u dospělých 6 mg asfotázy
alfa na kg týdně. Váš lékař bude muset dávky pravidelně upravovat podle změn tělesné hmotnosti.
Doporučení týkající se injekcí
• Můžete zaznamenat reakci v místě injekce. Před použitím přípravku si pozorně přečtěte bod 4, abyste věděl(a), jaké nežádoucí účinky se mohou vyskytnout.
• Při pravidelném podávání je pro aplikace injekce nutné měnit různé oblasti těla; tak lze omezit případnou bolest a podráždění.
• Pro aplikaci injekce jsou nejvhodnější oblasti s větším objemem tuku pod kůží (stehno, paže). Poraďte se se svým lékařem nebo zdravotní sestrou, která místa jsou ve Vašem případě nejvhodnější.
Před injekčním podáním přípravku Strensiq si pozorně přečtěte následující pokyny
• Každá injekční lahvička je určena k jednorázovému použití a smí se propíchnout jen jednou. Použít se smí pouze čirý a bezbarvý až světle žlutý vodný roztok bez viditelných známek zhoršené kvality. Veškerý nepoužitý léčivý přípravek nebo odpad musí být okamžitě zlikvidován.
• Jestliže si aplikujete injekci tohoto přípravku sám (sama), Váš lékař, lékárník nebo zdravotní sestra Vám ukáže, jak lék připravit a aplikovat injekci. Injekce tohoto léčivého přípravku si smíte podávat sám (sama) pouze v případě, že jste byl(a) poučen(a) a postup aplikace jste pochopil(a).
Jak podat injekci přípravku Strensiq
Důkladně si umyjte ruce mýdlem a vodou.
Z injekční lahvičky přípravku Strensiq sejměte ochranné víčko.
Přípravek Strensiq je třeba použít nejpozději do 1 hodiny po jeho vyjmutí z chladničky.
Z injekční stříkačky, kterou použijete k aplikaci přípravku, sejměte ochranný plastový obal.
Vždy použijte novou injekční stříkačku zabalenou v ochranném plastovém obalu.
Dbejte, abyste se jehlou neporanil(a).
Do injekční stříkačky natáhněte správnou dávku přípravku Strensiq. Pro natažení celého potřebného
objemu možná budete muset použít několik injekčních lahviček, abyste připravil(a) správnou dávku.
Pohledem zkontrolujte, zda je v injekční stříkačce správný objem.
Objem na injekci nesmí překročit 1 ml. Pokud je nutné podat více než 1 ml, musíte aplikovat více
injekcí do různých míst.
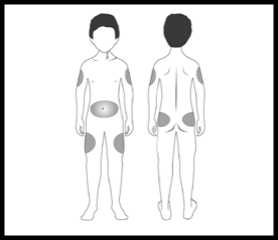
Zvolte místo injekce (stehna, břicho, paže, hýždě). Nejvhodnější oblasti pro podání injekce jsou na obrázku označeny šedou barvou. Lékař Vám poradí, do jakých míst je možné injekci aplikovat.
POZNÁMKA: neaplikujte přípravek do míst, kde nahmatáte bulky, tvrdé uzlíky nebo cítíte bolest; o všech zjištěných abnormalitách informujte svého lékaře.
Kůži na vybraném místě injekce jemně uchopte mezi palec a ukazováček.
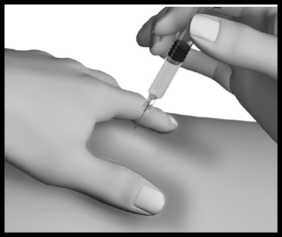
Injekční stříkačku držte stejně jako tužku nebo šipku a vpíchněte jehlu do kožní řasy tak, aby s povrchem kůže svírala úhel 45° až 90°.
U pacientů s malým objemem tuku pod kůží nebo tenkou kůží může být vhodnější úhel 45°.
Stále držte kožní řasu a zároveň stlačte píst injekční stříkačky; přitom pomalu počítejte do 10.
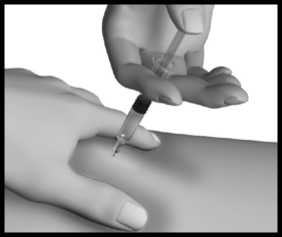
Jehlu vytáhněte, pusťte kožní řasu a na místo injekce jemně přiložte kousek vaty nebo gázy a několik sekund přidržte.
To urychlí zacelení propíchnuté tkáně a zabrání případnému úniku přípravku. Místo injekce po podání přípravku neškrábejte. Injekční stříkačky, injekční lahvičky a jehly ukládejte do nádoby na ostré předměty. Váš lékař, lékárník nebo zdravotní sestra Vám poradí, jak si můžete nádobu na ostré předměty obstarat.
Jestliže jste použil(a) více přípravku Strensiq, než jste měl(a)
Máte-li podezření, že Vám byla nedopatřením podána vyšší dávka přípravku Strensiq, než jaká Vám byla předepsána, poraďte se se svým lékařem.
Jestliže jste zapomněl(a) použít přípravek Strensiq
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku, a požádejte o radu svého lékaře.
Více informací naleznete na webových stránkách: asfotazaalfa-pacient.cz.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud si nejste jistý(á), jak se projevují níže uvedené nežádoucí účinky, požádejte svého lékaře o vysvětlení.
Velmi časté: mohou postihnout více než 1 pacienta z 10
Reakce v místě injekce během injekčního podávání přípravku nebo v průběhu několika hodin po injekci (které mohou vést k zarudnutí, změně barvy, svědění, bolesti a/nebo zduření)
Horečka (pyrexie), podrážděnost Zarudnutí kůže (erytém)
Bolest rukou a nohou Podlitina (zhmožděnina)
Časté: mohou postihnout až 1 pacienta z 10
Tukové bulky na povrchu kůže (lipohypertrofie), volná kůže (cutis laxa), napnutá kůže, změna barvy kůže (kožní diskolorace) včetně světlejších skvrn na kůži (kožní hypopigmentace)
Pocit na zvracení (nauzea), snížená citlivost úst (orální hypestezie)
Bolestivé svaly (myalgie)
Jizva
Zvýšená náchylnost k tvoření modřin Nával horka
Kožní infekce v místě injekce (cellulitis (flegmóna) v místě injekce)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Strensiq uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku injekční lahvičky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Přípravek se musí použít ihned po otevření injekční lahvičky (nejpozději do 1 hodiny).
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Strensiq obsahuje
Léčivou látkou je asfotasum alfa. Jeden mililitr roztoku obsahuje asfotasum alfa 40 mg.
Jedna injekční lahvička s 0,3 ml roztoku (40 mg/ml) obsahuje asfotasum alfa 12 mg.
Jedna injekční lahvička s 0,45 ml roztoku (40 mg/ml) obsahuje asfotasum alfa 18 mg.
Jedna injekční lahvička s 0,7 ml roztoku (40 mg/ml) obsahuje asfotasum alfa 28 mg.
Jedna injekční lahvička s 1 ml roztoku (40 mg/ml) obsahuje asfotasum alfa 40 mg.
Dalšími složkami jsou chlorid sodný, heptahydrát hydrogenfosforečnanu sodného, monohydrát dihydrogenfosforečnanu sodného a voda na injekci.
Jak přípravek Strensiq vypadá a co obsahuje toto balení
Přípravek Strensiq je dostupný ve formě čirého bezbarvého až světle žlutého vodného injekčního roztoku v injekčních lahvičkách s obsahem 0,3 ml, 0,45 ml, 0,7 ml a 1 ml roztoku.
Velikosti balení obsahující 1 nebo 12 injekčních lahviček.
Ve Vaší zemi nemusí být na trhu všechny velikosti balení.
Držitel rozhodnutí o registraci
Alexion Europe SAS 1-15, Avenue Edouard Belin 92500 Rueil-Malmaison Francie
Telefon: +33 1 47 32 36 03
Výrobce
Alexion Pharma International Trading
College Business and Technology Park, Blanchardstown
Dublin 15
Irsko
Tato příbalová informace byla naposledy revidována
Tento léčivý přípravek byl registrován za „výjimečných okolností“.
Znamená to, že vzhledem ke vzácné povaze tohoto onemocnění nebylo možné získat o tomto léčivém přípravku úplné informace.
Evropská agentura pro léčivé přípravky každoročně vyhodnotí jakékoli nové informace týkající se tohoto léčivého přípravku a tato příbalová informace bude podle potřeby aktualizována.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
Příbalová informace: informace pro uživatele
Strensiq 100 mg/ml injekční roztok (80 mg/0,8 ml)
Asfotasum alfa
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Strensiq a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Strensiq používat
3. Jak se přípravek Strensiq používá
4. Možné nežádoucí účinky
5. Jak přípravek Strensiq uchovávat
6. Obsah balení a další informace
1. Co je přípravek Strensiq a k čemu se používá Co je přípravek Strensiq
Přípravek Strensiq je lék, který se používá k léčbě dědičného onemocnění hypofosfatázie. Obsahuje léčivou látku asfotáza alfa.
Co je hypofosfatázie
Pacienti s hypofosfatázií mají nízké hladiny enzymu nazývaného alkalická fosfatáza, který je důležitý pro různé tělesné funkce, včetně řádného zpevnění kostí a zubů. Pacienti mívají potíže s růstem a pevností kostí, což může vést ke zlomeninám a bolestem kostí a obtížné chůzi, a také k potížím s dýcháním a riziku záchvatů (křečí).
K čemu se přípravek Strensiq používá
Léčivá látka v přípravku Strensiq může při hypofosfatázii nahradit chybějící enzym (alkalickou fosfatázu). Používá se jako dlouhodobá enzymová substituční léčba ke zvládání příznaků.
Jaké přínosy přípravku Strensiq byly prokázány v klinických studiích
Bylo prokázáno, že přípravek Strensiq má pro pacienty přínosy z hlediska mineralizace kostry a růstu kostí.
2. Čemu musíte věnovat pozornost, než začnete přípravek Strensiq používat Nepoužívejte přípravek Strensiq:
- jestliže jste alergický(á) na asfotázu alfa nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
• Přípravek Strensiq může u některých osob způsobovat alergické reakce. Mezi příznaky takových reakcí patří zvracení, dýchací potíže, rychlá srdeční frekvence, náhlý pokles krevního tlaku, kopřivka nebo vyrážka. Pokud se u Vás vyskytne kterýkoli z těchto příznaků, sdělte to okamžitě svému lékaři. Možná budete muset dostávat další léky k prevenci alergické reakce (antihistaminika nebo kortikosteroidy).
• Ve studiích byly hlášeny některé nežádoucí účinky postihující oči, které pravděpodobně souvisely s hypofosfatázií, jak u pacientů používajících přípravek Strensiq, tak u těch, kteří tento přípravek nepoužívali. V případě problémů s viděním se poraďte se svým lékařem.
• V klinických hodnoceních s přípravkem Strensiq i bez něj, která zahrnovala děti
s hypofosfatázií, byl hlášen časný srůst lebečních kostí u dětí ve věku do 5 let. Poraďte se se svým lékařem, jestliže si všimnete jakékoli změny tvaru hlavy svého dítěte.
• Jestliže se léčíte přípravkem Strensiq, mohou se u Vás při injekčním podání přípravku nebo během několika hodin po podání objevit reakce v místě injekce (bolest, uzlík, vyrážka, změna barvy). Pokud se u Vás vyskytne jakákoli těžká reakce v místě injekce, sdělte to okamžitě svému lékaři.
• Ve studiích bylo hlášeno zvýšení koncentrace parathormonu a nízké hladiny vápníku. Z tohoto důvodu Vás může lékař požádat, abyste v případě potřeby užíval(a) doplňky vápníku a perorální vitamin D.
• Během léčby přípravkem Strensiq se může objevit nárůst hmotnosti. Dle potřeby Vám lékař poskytne dietologické poradenství.
Další léčivé přípravky a přípravek Strensiq
Informujte svého lékaře nebo lékárníka o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
Těhotenství a kojení
Přípravek Strensiq se smí v průběhu těhotenství nebo kojení použít pouze tehdy, když je to z lékařského hlediska nezbytné.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Řízení dopravních prostředků a obsluha strojů
Nepředpokládá se, že by tento přípravek měl jakýkoli vliv na schopnost řídit nebo obsluhovat stroje. Důležité informace o některých složkách přípravku Strensiq
Tento přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné injekční lahvičce; to znamená, že přípravek je v podstatě „bez sodíku“.
3. Jak se přípravek Strensiq používá
Vždy používejte tento přípravek přesně v souladu s příbalovou informací nebo podle pokynů svého lékaře, lékárníka nebo zdravotní sestry. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Použití přípravku Strensiq Vám vysvětlí lékař, který má zkušenosti v léčbě pacientů s metabolickým nebo kostním onemocněním. Po proškolení lékařem nebo odbornou sestrou si můžete podávat injekce přípravku Strensiq sám (sama) v domácím prostředí.
Dávkování
• Dávka, kterou dostáváte, vychází z Vaší tělesné hmotnosti.
• Správnou dávku určí Váš lékař. Celkem budete injekcí pod kůži (subkutánně) každý týden dostávat 6 mg asfotázy alfa na kg tělesné hmotnosti (pro detailní informace o podávaném objemu a typu lahvičky, která má být použita na základě Vaší hmotnosti, viz dávkovací tabulka níže).
• Tuto celkovou dávku lze podávat buď jako injekci 1 mg/kg asfotázy alfa 6krát týdně, nebo jako 2 mg/kg asfotázy alfa 3krát týdně podle doporučení Vašeho lékaře.
• Maximální objem na injekci nesmí překročit 1 ml. Pokud je nutné podat více než 1 ml, musíte podat více injekcí ihned za sebou.
Při podávání 3krát týdně Při podávání 6krát týdně
|
Tělesná hmotnost (kg) |
Podávaný objem |
Barva injekční lahvičky, která se má použít |
|
3 |
0,15 ml |
tmavě modrá |
|
4 |
0,20 ml |
tmavě modrá |
|
5 |
0,25 ml |
tmavě modrá |
|
6 |
0,30 ml |
tmavě modrá |
|
7 |
0,35 ml |
oranžová |
|
8 |
0,40 ml |
oranžová |
|
9 |
0,45 ml |
oranžová |
|
10 |
0,50 ml |
světle modrá |
|
11 |
0,55 ml |
světle modrá |
|
12 |
0,60 ml |
světle modrá |
|
13 |
0,65 ml |
světle modrá |
|
14 |
0,70 ml |
světle modrá |
|
15 |
0,75 ml |
růžová |
|
16 |
0,80 ml |
růžová |
|
17 |
0,85 ml |
růžová |
|
18 |
0,90 ml |
růžová |
|
19 |
0,95 ml |
růžová |
|
20 |
1 ml |
růžová |
|
25 |
0,50 ml |
zelená |
|
30 |
0,60 ml |
zelená |
|
35 |
0,70 ml |
zelená |
|
40 |
0,80 ml |
zelená |
|
Tělesná hmotnost (kg) |
Podávaný objem |
Barva injekční lahvičky, která se má použít |
|
6 |
0,15 ml |
tmavě modrá |
|
7 |
0,18 ml |
tmavě modrá |
|
8 |
0,20 ml |
tmavě modrá |
|
9 |
0,23 ml |
tmavě modrá |
|
10 |
0,25 ml |
tmavě modrá |
|
11 |
0,28 ml |
tmavě modrá |
|
12 |
0,30 ml |
tmavě modrá |
|
13 |
0,33 ml |
oranžová |
|
14 |
0,35 ml |
oranžová |
|
15 |
0,38 ml |
oranžová |
|
16 |
0,40 ml |
oranžová |
|
17 |
0,43 ml |
oranžová |
|
18 |
0,45 ml |
oranžová |
|
19 |
0,48 ml |
světle modrá |
|
20 |
0,50 ml |
světle modrá |
|
25 |
0,63 ml |
světle modrá |
|
30 |
0,75 ml |
růžová |
|
35 |
0,88 ml |
růžová |
|
40 |
1 ml |
růžová |
|
50 |
0,50 ml |
zelená |
|
60 |
0,60 ml |
zelená |
|
70 |
0,70 ml |
zelená |
|
80 |
0,80 ml |
zelená |
|
90 |
0,90 ml |
zelená (x2) |
|
100 |
1 ml |
zelená (x2) |
Použití u dětí a dospívajících
Doporučená dávka přípravku Strensiq u dětí a dospívajících je stejně jako u dospělých 6 mg asfotázy
alfa na kg týdně. Váš lékař bude muset dávky pravidelně upravovat podle změn tělesné hmotnosti.
Doporučení týkající se injekcí
• Můžete zaznamenat reakci v místě injekce. Před použitím přípravku si pozorně přečtěte bod 4, abyste věděl(a), jaké nežádoucí účinky se mohou vyskytnout.
• Při pravidelném podávání je pro aplikace injekce nutné měnit různé oblasti těla; tak lze omezit případnou bolest a podráždění.
• Pro aplikaci injekce jsou nejvhodnější oblasti s větším objemem tuku pod kůží (stehno, paže). Poraďte se se svým lékařem nebo zdravotní sestrou, která místa jsou ve Vašem případě nejvhodnější.
Před injekčním podáním přípravku Strensiq si pozorně přečtěte následující pokyny
• Každá injekční lahvička je určena k jednorázovému použití a smí se propíchnout jen jednou. Použít se smí pouze čirý a bezbarvý až světle žlutý vodný roztok bez viditelných známek zhoršené kvality. Veškerý nepoužitý léčivý přípravek nebo odpad musí být okamžitě zlikvidován.
• Jestliže si aplikujete injekci tohoto přípravku sám (sama), Váš lékař, lékárník nebo zdravotní sestra Vám ukáže, jak lék připravit a aplikovat injekci. Injekce tohoto léčivého přípravku si smíte podávat sám (sama) pouze v případě, že jste byl(a) poučen(a) a postup aplikace jste pochopil(a).
Jak podat injekci přípravku Strensiq
Důkladně si umyjte ruce mýdlem a vodou.
Z injekční lahvičky přípravku Strensiq sejměte ochranné víčko.
Přípravek Strensiq je třeba použít nejpozději do 1 hodiny po jeho vyjmutí z chladničky.
Z injekční stříkačky, kterou použijete k aplikaci přípravku, sejměte ochranný plastový obal.
Vždy použijte novou injekční stříkačku zabalenou v ochranném plastovém obalu.
Dbejte, abyste se jehlou neporanil(a).
Do injekční stříkačky natáhněte správnou dávku přípravku Strensiq. Pro natažení celého potřebného
objemu možná budete muset použít několik injekčních lahviček, abyste připravil(a) správnou dávku.
Pohledem zkontrolujte, zda je v injekční stříkačce správný objem.
Objem na injekci nesmí překročit 1 ml. Pokud je nutné podat více než 1 ml, musíte aplikovat více
injekcí do různých míst.
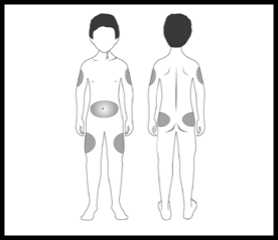
Zvolte místo injekce (stehna, břicho, paže, hýždě). Nejvhodnější oblasti pro podání injekce jsou na obrázku označeny šedou barvou. Lékař Vám poradí, do jakých míst je možné injekci aplikovat.
POZNÁMKA: neaplikujte přípravek do míst, kde nahmatáte bulky, tvrdé uzlíky nebo cítíte bolest; o všech zjištěných abnormalitách informujte svého lékaře.
Kůži na vybraném místě injekce jemně uchopte mezi palec a ukazováček.
Injekční stříkačku držte stejně jako tužku nebo šipku a vpíchněte jehlu do kožní řasy tak, aby s povrchem kůže svírala úhel 45° až 90°.
U pacientů s malým objemem tuku pod kůží nebo tenkou kůží může být vhodnější úhel 45°.
Stále držte kožní řasu a zároveň stlačte píst injekční stříkačky; přitom pomalu počítejte do 10.
Jehlu vytáhněte, pusťte kožní řasu a na místo injekce jemně přiložte kousek vaty nebo gázy a několik sekund přidržte.
To urychlí zacelení propíchnuté tkáně a zabrání případnému úniku přípravku. Místo injekce po podání přípravku neškrábejte. Injekční stříkačky, injekční lahvičky a jehly ukládejte do nádoby na ostré předměty. Váš lékař, lékárník nebo zdravotní sestra Vám poradí, jak si můžete nádobu na ostré předměty obstarat.
Jestliže jste použil(a) více přípravku Strensiq, než jste měl(a)
Máte-li podezření, že Vám byla nedopatřením podána vyšší dávka přípravku Strensiq, než jaká Vám byla předepsána, poraďte se se svým lékařem.
Jestliže jste zapomněl(a) použít přípravek Strensiq
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku, a požádejte o radu svého lékaře.
Více informací naleznete na webových stránkách: asfotazaalfa-pacient.cz.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud si nejste jistý(á), jak se projevují níže uvedené nežádoucí účinky, požádejte svého lékaře o vysvětlení.
Velmi časté: mohou postihnout více než 1 pacienta z 10
Reakce v místě injekce během injekčního podávání přípravku nebo v průběhu několika hodin po injekci (které mohou vést k zarudnutí, změně barvy, svědění, bolesti a/nebo zduření)
Horečka (pyrexie), podrážděnost Zarudnutí kůže (erytém)
Bolest rukou a nohou Podlitina (zhmožděnina)
Časté: mohou postihnout až 1 pacienta z 10
Tukové bulky na povrchu kůže (lipohypertrofie), volná kůže (cutis laxa), napnutá kůže, změna barvy kůže (kožní diskolorace) včetně světlejších skvrn na kůži (kožní hypopigmentace)
Pocit na zvracení (nauzea), snížená citlivost úst (orální hypestezie)
Bolestivé svaly (myalgie)
Jizva
Zvýšená náchylnost k tvoření modřin Nával horka
Kožní infekce v místě injekce (cellulitis (flegmóna) v místě injekce)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Strensiq uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku injekční lahvičky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Přípravek se musí použít ihned po otevření injekční lahvičky (nejpozději do 1 hodiny).
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Strensiq obsahuje
Léčivou látkou je asfotasum alfa. Jeden mililitr roztoku obsahuje asfotasum alfa 100 mg.
Jedna injekční lahvička s 0,8 ml roztoku (100 mg/ml) obsahuje asfotasum alfa 80 mg.
Dalšími složkami jsou chlorid sodný, heptahydrát hydrogenfosforečnanu sodného, monohydrát dihydrogenfosforečnanu sodného a voda na injekci.
Jak přípravek Strensiq vypadá a co obsahuje toto balení
Přípravek Strensiq je dostupný ve formě čirého bezbarvého až světle žlutého vodného injekčního roztoku v injekčních lahvičkách s obsahem 0,8 ml roztoku.
Velikosti balení obsahující 1 nebo 12 injekčních lahviček.
Ve Vaší zemi nemusí být na trhu všechny velikosti balení.
Držitel rozhodnutí o registraci
Alexion Europe SAS 1-15, Avenue Edouard Belin 92500 Rueil-Malmaison Francie
Telefon: +33 1 47 32 36 03
Výrobce
Alexion Pharma International Trading
College Business and Technology Park, Blanchardstown
Dublin 15
Irsko
Tato příbalová informace byla naposledy revidována
Tento léčivý přípravek byl registrován za „výjimečných okolností“.
Znamená to, že vzhledem ke vzácné povaze tohoto onemocnění nebylo možné získat o tomto léčivém přípravku úplné informace.
Evropská agentura pro léčivé přípravky každoročně vyhodnotí jakékoli nové informace týkající se tohoto léčivého přípravku a tato příbalová informace bude podle potřeby aktualizována.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
Příloha IV
• Registrace přípravku za výjimečných okolností
Výbor CHMP posoudil žádost a je toho názoru, že poměr přínosů a rizik je příznivý, a proto doporučuje, aby přípravku byla udělena registrace za výjimečných okolností, jak je podrobněji popsáno v Evropské veřejné zprávě o hodnocení.
40