Stivarga 40 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Stivarga 40 mg potahované tablety.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje regorafenibum 40 mg.
Pomocné látky se známým účinkem:
Jedna denní dávka 160 mg obsahuje 2,427 mmol (nebo 55,8 mg) sodíku (viz bod 4.4). Jedna denní dávka 160 mg obsahuje 1,68 mg sójového lecithinu (viz bod 4.4).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta.
Světle růžové potahované tablety oválného tvaru o délce 16 mm a šířce 7 mm, na jedné straně s označením "BAYER", na druhé straně „40“
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Stivarga je indikován k léčbě dospělých pacientů s:
- metastazujícím kolorektálním karcinomem (CRC), kteří byli dříve léčeni dostupnými typy léčby nebo kteří nejsou vhodnými pro dostupné typy léčby. Tyto typy léčby zahrnují chemoterapii na bázi fluoropyrimidinů, anti-VEGF léčbu a anti-EGFR léčbu (viz bod 5.1).
- neresekovatelnými nebo metastazujícími gastrointestinálními stromálními nádory (GIST), u kterých došlo k progresi na předchozí léčbě imatinibem a sunitinibem nebo kteří tuto léčbu netolerovali.
4.2 Dávkování a způsob podání
Přípravek Stivarga byl měl být předepisován lékaři, kteří mají zkušenosti s podáváním protinádorové
léčby.
Dávkování
Doporučená dávka regorafenibu je 160 mg (4 tablety obsahující 40 mg) užívaná jednou denně po dobu
3 týdnů s následujícím 1týdenním obdobím bez léčby. Toto 4týdenní období je považováno za léčebný
cyklus.
Pokud je dávka přípravku vynechána, měla by se užít ve stejný den, co nejdříve si pacient vzpomene. Pacient by neměl užít dvě dávky ve stejný den, aby nahradil vynechanou dávku. V případě zvracení po podání regorafenibu by pacient neměl užívat další tablety.
Léčba by měla pokračovat tak dlouho, dokud je pozorován přínos nebo dokud se neobjeví nepřijatelná toxicita (viz bod 4.4).
Pacienti s výkonnostním stavem (PS) 2 nebo vyšším byli vyřazeni z klinických studií. U pacientů s PS > 2 jsou k dispozici omezené údaje.
Úpravy dávkování
Přerušení léčby a/nebo snížení dávky může být nutné podle individuální bezpečnosti a tolerance. Úpravy dávky se mají provádět v krocích po 40 mg (jedna tableta). Nejnižší doporučená denní dávka je 80 mg. Maximální denní dávka je 160 mg.
Pro doporučené úpravy dávky a opatření v případě kožních reakcí ruka-noha (hand-foot skin reaction, HFSR) / syndrom palmoplantární erytrodysestézie viz Tabulka 1.
Tabulka 1: Doporučené úpravy dávky a opatření pro HFSR
|
Stupeň kožní toxicity |
Výskyt |
Doporučená úprava dávky a opatření |
|
Stupeň 1 |
Jakýkoli |
Udržte hladinu dávky a okamžitě proveďte podpůrná opatření pro symptomatickou úlevu. |
|
Stupeň 2 |
1. výskyt |
Snižte dávku o 40 mg (jedna tableta) a okamžitě proveďte podpůrná opatření. Pokud nedojde ke zlepšení i přes snížení dávky, přerušte léčbu na dobu minimálně 7 dnů, dokud toxicita nedosáhne stupně 0-1. Opětovné zvýšení dávky je přípustné na základě rozhodnutí lékaře. |
|
Bez zlepšení během 7 dnů nebo 2. Výskyt |
Přerušte léčbu, dokud toxicita nedosáhne stupně 0-1. Při znovuzahájení léčby snižte dávku o 40 mg (jedna tableta). Opětovné zvýšení dávky je přípustné na základě rozhodnutí lékaře. | |
|
3. výskyt |
Přerušte léčbu do toxicity na stupeň 0-1. Při znovuzahájení léčby snižte dávku o 40 mg (jedna tableta). Opětovné zvýšení dávky je přípustné na základě rozhodnutí lékaře. | |
|
4. výskyt |
Trvale ukončete léčbu přípravkem Stivarga. | |
|
Stupeň 3 |
1. výskyt |
Zahajte ihned podpůrná opatření. Přerušte léčbu na dobu minimálně 7 dnů, dokud toxicita nedosáhne stupně 0-1. Při znovuzahájení léčby snižte dávku o 40 mg (jedna tableta). Opětovné zvýšení dávky je přípustné na základě rozhodnutí lékaře. |
|
Stupeň kožní toxicity |
Výskyt |
Doporučená úprava dávky a opatření |
|
2. výskyt |
Zahajte ihned podpůrná opatření. Přerušte léčbu na dobu minimálně 7 dnů, dokud toxicita nedosáhne stupně 0-1. Při znovuzahájení léčby snižte dávku o 40 mg (jedna tableta). | |
|
3. výskyt |
Trvale ukončete léčbu přípravkem Stivarga. |
Pro doporučená opatření a změny dávek v případě zhoršení funkce jatemích testů, u kterých se předpokládá souvislost s přípravkem Stivarga, viz Tabulka 2 (viz také bod 4.4).
Tabulka 2: Doporučená opatření a úpravy dávky v případě abnormalit funkcí jaterních testů souvisejících s lékem
|
Pozorovaná zvýšení ALT a/nebo AST |
Výskyt |
Doporučená opatření a úprava dávky |
|
< 5násobek horní hranice normálních hodnot (ULN) (maximálně stupeň 2) |
Jakýkoli výskyt |
Pokračujte s léčbou přípravkem Stivarga. Sledujte jaterní funkce týdně až do doby, kdy se hladiny transamináz vrátí na < 3násobek ULN (stupeň 1) nebo na výchozí stav. |
|
> 5násobek < 20násobek ULN (Stupeň 3) |
1. výskyt |
Přerušte léčbu přípravkem Stivarga. Sledujte transaminázy týdně až do doby, kdy se vrátí na < 3násobek ULN nebo na výchozí stav. Znovuzahájení: Pokud potenciální přínos převáží riziko hepatotoxicity, zahajte znovu léčbu přípravkem Stivarga, snižte dávku o 40 mg (jedna tableta) a sledujte týdně jaterní funkce po dobu minimálně 4 týdnů. |
|
Opakovaný výskyt |
Trvale ukončete léčbu přípravkem Stivarga. | |
|
> 20násobek ULN (Stupeň 4) |
Jakýkoli výskyt |
Trvale ukončete léčbu přípravkem Stivarga. |
|
> 3násobek ULN (stupeň 2 nebo vyšší) se současnou hladinou bilirubinu > 2násobek ULN |
Jakýkoli výskyt |
Trvale ukončete léčbu přípravkem Stivarga. Sledujte týdně jaterní funkce až do vyřešení nebo navrácení k výchozímu stavu. Výjimka: pacienti s Gilbertovým syndromem, u kterých se objeví zvýšené hladiny transamináz, by měli být léčeni podle výše uvedených doporučení pro příslušné pozorované zvýšení ALT a/nebo AST. |
Porucha funkce jater
Regorafenib je eliminován hlavně v játrech.
V klinických studiích nebyly pozorovány žádné významné rozdíly v expozici, bezpečnosti nebo účinnosti mezi pacienty s mírnou poruchou funkce jater (Child-Pugh A) a pacienty s normální funkcí jater. U pacientů s mírnou poruchou funkce jater není nutná žádná úprava dávkování. Protože jsou k dispozici pouze omezené údaje pro pacienty se středně závažnou poruchou funkce jater (Child-Pugh B), není možné poskytnout doporučení pro dávkování. U těchto pacientů je doporučeno pečlivé sledování celkové bezpečnosti (viz body 4.4 a 5.2).
Přípravek Stivarga není doporučován u pacientů se závažnou poruchou funkce jater (Child-Pugh C), protože u této populace nebyl přípravek Stivarga hodnocen.
Porucha funkce ledvin
V klinických studiích nebyly pozorovány žádné významné rozdíly v expozici, bezpečnosti nebo účinnosti mezi pacienty s mírnou poruchou funkce ledvin (odhad rychlosti glomerulární filtrace [eGFR] 60-89 ml/min/1,73m2) a pacienty s normální funkcí ledvin. Omezené farmakokinetické údaje neukazují žádný rozdíl v expozici u pacientů se středně závažnou poruchou funkce ledvin (eGFR 30-59 ml/min/1,73 m2). U pacientů s mírnou nebo středně závažnou poruchou funkce ledvin není nutná žádná úprava dávkování (viz také bod 5.2). U pacientů se závažnou poruchou funkce ledvin (eGFR < 30 ml/min/1,73 m2) nejsou k dispozici žádné klinické údaje.
Starší populace
V klinických studiích nebyly pozorovány žádné významné rozdíly v expozici, bezpečnosti nebo účinnosti mezi staršími (65 let a více) a mladšími pacienty (viz také bod 5.2).
Pohlaví
V klinických studiích nebyly pozorovány žádné významné rozdíly v expozici, bezpečnosti nebo účinnosti mezi muži a ženami. V závislosti na pohlaví není nutná žádná úprava dávkování (viz také bod 5.2).
Etnické rozdíly
V klinických studiích nebyly pozorovány žádné významné rozdíly v expozici nebo účinnosti mezi pacienty různých etnických skupin. Vyšší incidence kožních reakcí ruka-noha (hand-foot skin reaction, HFSR) / syndrom palmoplantární erytrodysestézie, závažných abnormalit funkčních jaterních testů a hepatální dysfunkce byla pozorována u asijských (zvláště japonských) pacientů léčených přípravkem Stivarga v porovnání s bělošskou populací. Asijští pacienti léčení přípravkem Stivarga
v klinických studiích byli primárně z východní Asie (přibližně 90 %).
K dispozici jsou omezené údaje u regorafenibu u černošské populace pacientů.
V závislosti na etnickém původu není nutná žádná úprava dávkování (viz bod 5.2).
Pediatrická populace
Pro použití přípravku Stivarga u pediatrické populace v indikaci metastazujícího kolorektálního karcinomu nejsou relevantní důvody.
Bezpečnost a účinnost regorafenibu u pacientů ve věku do 18 let v indikaci gastrointestinálních stromálních nádorů (GIST) nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Stivarga je určen pro perorální podání.
Přípravek Stivarga by měl být užíván ve stejnou denní dobu. Tablety by se měly polykat celé a zapíjet vodou po lehkém jídle, které neobsahuje více než 30 % tuku. Příkladem lehkého (nízkotučného) jídla je 1 porce cereálií (asi 30 g), 1 sklenice odstředěného mléka, 1 plátek toastového chleba s džemem,
1 sklenice jablečného džusu a 1 šálek kávy nebo čaje (520 kalorií, 2 g tuku).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Účinky na játra
U pacientů léčených přípravkem Stivarga byly často pozorovány abnormality funkčních jaterních testů (alaninaminotransferáza [ALT], aspartátaminotransferáza [AST] a bilirubin). Závažné abnormality vyšetření jaterních funkcí (stupeň 3 až 4) a jaterní dysfunkce s klinickými manifestacemi (včetně fatálních následků) byly hlášeny u malé části pacientů (viz bod 4.8).
V klinických studiích byla pozorována vyšší incidence závažných abnormalit funkčních jatemích testů a hepatální dysfunkce u asijských (zvláště japonských) pacientů léčených přípravkem Stivarga v porovnání s bělošskou populací (viz bod 4.2).
Doporučuje se provést vyšetření jaterních funkcí (ALT, AST a bilirubin) před zahájením léčby přípravkem Stivarga a sledovat je pečlivě (minimálně každé dva týdny) během prvních 2 měsíců léčby. Poté by mělo pokračovat pravidelné sledování minimálně každý měsíc a podle klinické potřeby.
Regorafenib je inhibitor uridindifosfát-glukuronyltransferázy (UGT) 1A1 (viz bod 4.5). U pacientů s Gilbertovým syndromem se může vyskytnout mírná nepřímá (nekonjugovaná) hyperbilirubinémie.
U pacientů s pozorovaným zhoršením jaterních testů, u nichž je předpokládána souvislost s léčbou přípravkem Stivarga (tj. kde není jasná jiná příčina, jako je posthepatální cholestáza nebo progrese onemocnění), by mělo být přistoupeno k úpravě dávky a sledování podle instrukcí v Tabulce 2 (viz bod 4.2).
Regorafenib je eliminován hlavně hepatální cestou.
Pečlivé sledování celkové bezpečnosti je doporučováno u pacientů s mírnou nebo středně závažnou poruchou funkce jater (viz také body 4.2 a 5.2). Přípravek Stivarga není doporučen u pacientů se závažnou poruchou funkce jater (Child-Pugh C), protože nebyl u této populace hodnocený a expozice může být u těchto pacientů zvýšena.
Krvácení
Přípravek Stivarga byl spojen se zvýšeným výskytem krvácivých příhod, z nichž některé byly fatální (viz bod 4.8). U pacientů se stavy predisponujícími ke krvácení a u těch, kteří jsou léčeni antikoagulačními léky (např. warfarin a fenprokumon) nebo jinými souběžně užívanými léčivými přípravky, které zvyšují riziko krvácení, by měl být sledován krevní obraz a koagulační parametry. V případě závažného krvácení vyžadujícího urgentní lékařský zákrok by mělo být zváženo trvalé vysazení přípravku Stivarga.
Srdeční ischémie a infarkt
Přípravek Stivarga byl spojen se zvýšeným výskytem ischémie myokardu a infarktu (viz bod 4.8). Pacienti s nestabilní anginou pectoris nebo nově se vyskytující anginou pectoris (během 3 měsíců od zahájení léčby přípravkem Stivarga), nedávno prodělaným infarktem myokardu (během 6 měsíců od zahájení léčby přípravkem Stivarga) a pacienti se srdečním selháním stupně 2 nebo vyšším podle klasifikace New York Hart Asssociation (NYHA) byli z klinických studií vyřazeni.
Pacienti s anamnézou ischemické choroby srdeční by měli být sledováni s ohledem na klinické známky a příznaky ischémie myokardu. U pacientů, u kterých se vyvine ischémie myokardu a/nebo infarkt, je doporučeno přerušení léčby přípravkem Stivarga až do vyřešení. Rozhodnutí znovu zahájit léčbu přípravkem Stivarga by mělo být založeno na pečlivém zvážení možných přínosů a rizik u jednotlivého pacienta. Přípravek Stivarga by měl být trvale vysazen, pokud nedojde k žádnému vyřešení.
Syndrom reverzibilní zadní encefalopatie (PRES)
PRES byl hlášen v souvislosti s léčbou přípravkem Stivarga (viz bod 4.8). Známky a příznaky PRES zahrnují křeče, bolest hlavy, změnu duševního stavu, poruchu zraku nebo kortikální slepotu, s přidruženou hypertenzí nebo bez ní. Diagnóza PRES vyžaduje potvrzení pomocí vyšetření mozku zobrazovací metodou. U pacientů, u kterých se vyvine PRES, je doporučeno ukončit léčbu přípravkem Stivarga a provádět kontrolu hypertenze a podpůrnou léčbu dalších příznaků.
Gastrointestinální perforace a pištěl
U pacientů léčených přípravkem Stivarga (viz bod 4.8) byly hlášeny gastrointestinální perforace (včetně fatálního výsledku) a píštěle. Tyto příhody jsou také známy jako časté komplikace související s onemocněním u pacientů s nitrobřišními malignitami. Přerušení léčby přípravkem Stivarga je doporučeno u pacientů, u kterých se vyvine gastrointestinální perforace nebo píštěl.
Arteriální hypertenze
Přípravek Stivarga byl spojen se zvýšeným výskytem arteriální hypertenze (viz bod 4.8). Krevní tlak by měl být pod kontrolou před zahájením léčby přípravkem Stivarga. Je doporučeno sledovat krevní tlak a léčit hypertenzi v souladu se standardní lékařskou praxí. V případech závažné nebo i přes odpovídající léčbu přetrvávající hypertenze by měla být léčba dočasně vysazena a/nebo by měla být snížena dávka podle rozhodnutí lékaře (viz bod 4.2). V případě hypertenzní krize by měl být přípravek Stivarga vysazen.
Komplikace hojení ran
Protože léčivé přípravky s antiangiogenními vlastnostmi mohou potlačovat nebo ovlivňovat hojení ran, je doporučeno dočasné přerušení léčby přípravkem Stivarga z preventivních důvodů u pacientů podstupujících větší chirurgický výkon. Rozhodnutí o znovuzahájení léčby přípravkem Stivarga po větším chirurgickém výkonu mělo být založeno na posouzení odpovídajícího hojení rány.
Dermatologická toxicita
Kožní reakce ruka-noha (HFSR) neboli syndrom palmo-plantární erytrodysestézie a vyrážka představují nejčastěji pozorované dermatologické nežádoucí účinky přípravku Stivarga (viz bod 4.8). V klinických studiích byla pozorována vyšší incidence HFSR u asijských (zvláště japonských) pacientů léčených přípravkem Stivarga v porovnání s bělošskou populací (viz bod 4.2). Opatření pro prevenci HFSR zahrnují kontrolu ztvrdlé kůže a použití vložek do bot a rukavic pro zabránění otlaků chodidel a dlaní. Léčba HFSR může zahrnovat použití keratolytických krémů (např. na bázi urey, kyseliny salicylové nebo alfahydroxykyselin aplikovaných střídmě pouze na postižené plochy) a zvlhčujících krémů (aplikovaných hojně) pro symptomatickou úlevu. Mělo by být zváženo snížení dávky a/nebo dočasné přerušení léčby přípravkem Stivarga nebo v závažných a neustupujících případech trvalé vysazení léčby přípravkem Stivarga (viz bod 4.2).
Abnormality biochemických a metabolických laboratorních vyšetření
Přípravek Stivarga byl spojen se zvýšeným výskytem iontových abnormalit (včetně hypofosfatémie, hypokalcémie, hyponatrémie a hypokalémie) a metabolických abnormalit (včetně zvýšení hladiny TSH, lipázy a amylázy). Abnormality jsou obecně mírné až středně závažné, nesouvisí s klinickými projevy a obvykle nevyžadují přerušení podávání nebo snížení dávkování. Doporučuje se sledovat biochemické a metabolické parametry během léčby přípravkem Stivarga a zahájit vhodnou substituční léčbu podle standardní klinické praxe, je-li potřeba. Přerušení podávání nebo snížení dávkování nebo trvalé vysazení léčby přípravkem Stivarga by mělo být zváženo v případě trvalých nebo recidivujících významných abnormalit (viz bod 4.2).
Důležité informace o některých složkách přípravku
Jedna denní dávka 160 mg obsahuje 2,427 mmol (nebo 55,8 mg) sodíku. Toto je nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku. Jedna denní dávka 160 mg obsahuje 1,68 mg sójového lecithinu.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Inhibitory CYP3A4 a UGT1A9 / induktory CYP3A4
In vitro údaje ukazují, že regorafenib je metabolizovaný cytochromem CYP3A4 a uridindifosfát-glukuronyltransferázou UGT1A9.
Podání ketokonazolu (400 mg po dobu 18 dnů), což je silný inhibitor CYP3A4, s jednou dávkou regorafenibu (160 mg v den 5) vedlo ke zvýšení průměrné expozice (AUC) regorafenibu o přibližně 33 % a snížení průměrné expozice aktivním metabolitům, M-2 (N-oxid) a M-5 (N-oxid a N-desmethyl), asi o 90 %. Doporučuje se vyhnout se současnému podávání silných inhibitorů aktivity CYP3A4 (např. klaritromycin, grapefruitová šťáva, itrakonazol, ketokonazol, posakonazol, telithromycin a vorikonazol), protože jejich vliv na expozici regorafenibu a jeho metabolitům v ustáleném stavu nebyl hodnocen.
Současné podávání silného inhibitoru UGT1A9 (např. kyseliny mefenamové, diflunisalu a kyseliny niflumové) během léčby regorafenibem by nemělo být prováděno, protože jejich vliv na expozici regorafenibu a jeho metabolitům v ustáleném stavu nebyl hodnocen.
Podání rifampicinu (600 mg po dobu 9 dnů), silného induktoru CYP3A4, s jednou dávkou regorafenibu (160 mg v den 7) vedlo ke snížení AUC regorafenibu asi o 50 %, 3 až 4násobnému zvýšení průměrné expozice aktivnímu metabolitu M-5 a k žádné změně v expozici aktivnímu metabolitu M-2. Další silné induktory CYP3A4 (např. fenytoin, karbamazepin a fenobarbital a třezalka tečkovaná) mohou také zvyšovat metabolismus regorafenibu. Silné induktory CYP3A4 by neměly být používány nebo by měla být zvážena volba náhradního, současně podávaného léčivého přípravku s žádným nebo s minimálním potenciálem indukce CYP3A4.
Substráty pro UGT1A1 a UGT1A9
In vitro údaje ukazují, že regorafenib a rovněž jeho aktivní metabolit M-2 inhibují glukuronidaci zprostředkovanou UGT1A1 a UGT1A9, zatímco M-5 inhibuje pouze UGT1A1, při koncentracích, které jsou dosaženy in vivo v ustáleném stavu. Podání regorafenibu s 5denní pauzou před podáním irinotekanu vedlo přibližně k 44 % zvýšení AUC u SN-38, což je substrát UGT1A1 a aktivní metabolit irinotekanu. Bylo také pozorováno zvýšení AUC irinotekanu asi o 28 %. To ukazuje, že současné podávání regorafenibu může zvýšit systémovou expozici substrátů pro UGT1A1 a UGT1A9.
Substráty pro protein rezistence karcinomu prsu (BCRP) a P-glykoprotein
Podávání regorafenibu (160 mg 14 dní) před podáním jednotlivé dávky rosuvastatinu (5 mg), BCRP substrátu, mělo za následek 3,8krát zvýšení průměrné expozice (AUC) rosuvastatinu a 4,6krát zvýšení
Cmax.
To znamená, že současné podávání regorafenibu může zvýšit plazmatickou koncentraci souběžných BCRP substrátů (např. metotrexátu, fluvastatinu, atorvastatinu). Proto se doporučuje u pacientů pozorně sledovat známky a příznaky zvýšené expozice BCRP substrátům.
Klinické údaje ukazují, že regorafenib nemá vliv na farmakokinetiku digoxinu, proto může být současně podáván se substráty P-glykoproteinu, jako je digoxin, bez klinicky významných lékových interakcí.
Inhibitory P-glykoproteinu a BCRP / induktory P-glykoproteinu a BCRP In vitro studie ukazují, že aktivní metabolity M-2 a M-5 jsou substráty P-glykoproteinu a BCRP. Inhibitory a induktory BCRP a P-glykoproteinu mohou ovlivňovat expozici M-2 a M-5. Klinický význam těchto zjištění není znám.
Selektivní substráty pro isoformy CYP
In vitro údaje ukazují, že regorafenib je kompetitivní inhibitor cytochromů CYP2C8 (Ki hodnota 0,6 ^mol/l), CYP2C9 (Ki hodnota 4,7 ^mol/l), CYP2B6 (Ki hodnota 5,2 ^mol/l) při koncentracích, které jsou dosaženy in vivo v ustáleném stavu (maximální plasmatická koncentrace 8,1 ^mol/l). In vitro inhibiční potenciál vůči CYP3A4 (Ki hodnota 11,1 ^mol/l) a CYP2C19 (Ki hodnota 16,4 ^mol/l) byl méně výrazný.
Byla provedena studie hodnotící účinek 14denního podávání dávky 160 mg regorafenibu na farmakokinetiku zkoumaných substrátů CYP2C8 (rosiglitazon), (CYP2C9) (S-warfarin), CYP2C19 (omeprazol) a CYP3A4 (midazolam).
Farmakokinetické údaje ukazují, že regorafenib může být podáván současně se substráty CYP2C8, CYP2C9, CYP3A4 a CYP2C19 bez klinicky významné lékové interakce (viz také bod 4.4).
Antibiotika
Profil koncentrace-čas ukazuje, že regorafenib a jeho metabolity mohou procházet enterohepatální cirkulací (viz bod 5.2). Současné podávání s neomycinem, špatně vstřebatelnou antimikrobiální látkou, používanou k eradikaci gastrointestinální mikroflóry (která může ovlivnit enterohepatální cirkulaci regorafenibu) nemělo vliv na expozici regorafenibu,ale došlo přibližně k 80 % poklesu expozice aktivních metabolitů M-2 a M-5, které ukázaly in vitro a in vivo farmakologickou aktivitu srovnatelnou s regorafenibem. Klinický význam této možné interakce s neomycinem není znám, ale může vést ke snížení účinnosti regorafenibu. Farmakologické interakce jiných antibiotik nebyly studovány.
Látky sekvestrující žlučové kyseliny
Regorafenib, metabolity M-2 a M-5 pravděpodobně podléhají enterohepatální cirkulaci (viz bod 5.2). Látky sekvestrující žlučové kyseliny, jako cholestyramin a cholestagel, mohou reagovat s regorafenibem tvorbou nerozpustných komplexů, které mohou mít vliv na absorpci (nebo reabsorpci), což má za následek potenciálně sníženou expozici. Klinický význam těchto potenciálních interakcí není znám, ale následkem by mohla být snížená účinnost regorafenibu.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku / Antikoncepce u mužů a žen
Ženy ve fertilním věku musí být informovány, že regorafenib může způsobit poškození plodu.
Ženy ve fertilním věku a muži by měli používat účinnou antikoncepci během léčby a až 8 týdnů po ukončení terapie.
Údaje o podávání regorafenibu těhotným ženám nejsou k dispozici.
Na základě mechanismu účinku regorafenibu se předpokládá, že působí poškození plodu, pokud je podáván během těhotenství. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Přípravek Stivarga lze v těhotenství použít pouze tehdy, pokud to je nezbytně nutné a po pečlivém zvážení přínosů pro matku a rizika pro plod.
Kojení
Není známo, zda se regorafenib nebo jeho metabolity vylučují do lidského mateřského mléka. Regorafenib nebo jeho metabolity se u potkanů vylučují do mateřského mléka. Riziko pro kojené dítě nelze vyloučit. Regorafenib by mohl narušit růst a vývoj kojence (viz bod 5.3).
Kojení musí být během léčby přípravkem Stivarga přerušeno.
Fertilita
Nejsou k dispozici žádné údaje o účinku přípravku Stivarga na fertilitu u člověka. Výsledky ze studií na zvířatech ukazují, že regorafenib může poškodit mužskou a ženskou fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly provedeny žádné studie hodnotící účinky přípravku Stivarga na schopnost řídit nebo obsluhovat stroje. Pokud se u pacientů objeví během léčby přípravkem Stivarga příznaky, které ovlivňují jejich schopnost se soustředit a reagovat, doporučuje se, aby neřídili nebo neobsluhovali stroje, dokud příznaky neodezní.
Souhrn bezpečnostního profilu
Celkový bezpečnostní profil přípravku Stivarga vychází z údajů od více než 1 200 pacientů v klinických studiích zahrnujících data z placebem kontrolovaných studií fáze III u 500 pacientů s metastazujícím kolorektálním karcinomem (CRC) a 132 pacientů s gastrointestinálními stromálními nádory (GIST).
Bezpečnostní profil regorafenibu v těchto studiích byl v souladu s bezpečnostními výsledky studie fáze III B provedené u 2872 pacientů s metastatickým kolorektálním karcinomem, jejichž nemoc progredovala po léčbě standardními terapiemi.
Nejzávažnějšími nežádoucími účinky u pacientů léčených přípravkem Stivarga jsou závažné poškození jater, krvácení a gastrointestinální perforace.
Nej častěji pozorovanými nežádoucími účinky (> 30 %) u pacientů léčených přípravkem Stivarga jsou astenie/únava, kožní reakce ruka-noha, průjem, snížení chuti k jídlu a příjmu potravy, hypertenze, dysfonie a infekce.
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky hlášené v klinických studiích u pacientů léčených přípravkem Stivarga jsou uvedeny v tabulce 3. Jsou rozděleny podle třídy orgánových systémů a pro popis určitého účinku, jeho synonym a přidružených stavů se používá nejvhodnější termín MedDRA.
Nežádoucí účinky jsou seskupeny podle svých frekvencí. Skupiny četnosti jsou definované podle následující konvence: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100) a vzácné (> 1/10 000 až < 1/1 000).
V každé skupině četnosti jsou nežádoucí účinky uvedeny sestupně podle klesající závažnosti.
Tabulka 3: Nežádoucí účinky (NÚ) hlášené v klinických studiích u pacientů léčených přípravkem Stivarga
|
Třídy orgánových systémů (MedDRA) |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Infekce a infestace |
Infekce | |||
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a p°lypy) |
Keratoakanthom / spinocelulární karcinom kůže | |||
|
Poruchy krve a lymfatického systému |
Trombocytopenie |
Leukopenie | ||
|
Poruchy imunitního systému |
Hypersenzitivní reakce | |||
|
Endokrinní poruchy |
Hypotyreóza | |||
|
Poruchy metabolismu a výživy |
Snížená chuť k jídlu a snížený příjem potravy |
Hypokalémie Hypofosfatémie Hypokalcémie Hyponatrémie |
|
Třídy orgánových systémů (MedDRA) |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Hypomagnesémie Hyperurikémie | ||||
|
Poruchy nervového systému |
Syndrom reverzibilní zadní encefalopatie (PRES) | |||
|
Srdeční poruchy |
Infarkt myokardu Ischémie myokardu | |||
|
Cévní poruchy |
Krvácení* Hypertenze |
Hypertenzní krize | ||
|
Respirační, hrudní a mediastinální poruchy |
Dysfonie | |||
|
Gastrointe stinální poruchy |
Stomatitida Nausea |
Poruchy chuti Sucho v ústech Gastroesofageální reflux Gastroenteritida |
Gastrointestinální perforace* Gastrointestinální píštěl | |
|
Poruchy jater a žlučových cest |
Hyperbilirubinémie |
Zvýšení hladiny transamináz |
Závažné poškození jater*# | |
|
Poruchy kůže a podkožní tkáně |
Kožní reakce ruka-noha** Vyrážka Alopecie |
Suchá kůže Exfoliativní vyrážka |
Poruchy nehtů Erythema multiforme |
Stevens-Johnsonův syndrome Toxická epidermální nekrolýza |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Muskuloskeletální ztuhlost | |||
|
Poruchy ledvin a močových cest |
Proteinurie | |||
|
Celkové poruchy a reakce v místě aplikace |
Astenie/únava Bolest Horečka Zánět sliznic | |||
|
Vyšetření |
Úbytek tělesné hmotnosti |
Zvýšení hladiny amylázy Zvýšení hladiny lipázy Abnormální INR (International normalised ratio) |
* byly hlášeny fatální případy
** syndrom palmoplantární erytrodysestézie v terminologii MedDRA
# podle kritérií léky indukovaného poškození jater (DILI) mezinárodní odborné pracovní skupiny
DILI
Popis vybraných nežádoucích účinků
Ve většině případů závažného poškození jater se jaterní dysfunkce objevila během prvních 2 měsíců léčby a byla charakterizována hepatocelulárním typem poškození se zvýšením hladiny transamináz > 20násobek horní hranice normálních hodnot s následným zvýšením hladiny bilirubinu. V klinických studiích byla pozorována vyšší incidence závažných jaterních poškození s fatálními následky u
japonských pacientů (~1,5%) léčených přípravkem Stivarga ve srovnání s jinými (ne japonskými) pacienty (<0,1%).
Ve dvou placebem kontrolovaných studiích fáze III byl celkový výskyt hemorhagie 19,3 % u pacientů léčených přípravkem Stivarga. Většina případů výskytu krvácivých příhod u pacientů léčených přípravkem Stivarga byla mírná až středně závažná (stupně 1 a 2: 16,9 %), nejvýznamněji epistaxe (7,6 %). Fatální příhody u pacientů léčených přípravkem Stivarga byly méně časté (0,6 %) a zahrnovaly respirační, gastrointestinální a urogenitální trakt.
Ve dvou placebem kontrolovaných studiích fáze III byly infekce častěji pozorovány u pacientů léčených přípravkem Stivarga ve srovnání s pacienty na placebu (všechny stupně: 31,0 % vs. 14,4 %). Většina infekcí u pacientů léčených přípravkem Stivarga byla mírná až středně závažná (stupně 1 a 2: 22,9 %) a zahrnovaly infekce močového traktu (6,8 %), nasofaryngitidu (4,2 %) a rovněž mukokutánní a systémové mykotické infekce (2,4 %). Mezi léčebnými skupinami nebyl pozorován žádný rozdíl ve fatálních výsledcích souvisejících s infekcí (0,6 %, rameno přípravku Stivarga vs. 0,6 %, placebové rameno).
V placebem kontrolované studii fáze III u pacientů s metastazujícím kolorektálním karcinomem byl celkový výskyt kožní reakce ruka-noha 45,2 % u pacientů léčených přípravkem Stivarga ve srovnání se 7,1 % u pacientů užívajících placebo. V placebem kontrolované studii fáze III u GIST nádorů byl celkový výskyt infekce ruka-noha 66,7 % u pacientů léčených přípravkem Stivarga ve srovnání
s 15,2 % pacientů užívajících placebo. V obou studiích se většina případů kožní reakce ruka-noha objevila během prvního cyklu léčby a byla mírná až středně závažná (stupně 1 a 2: 28,6 % u CRC a
44,7 % u GIST). Výskyt kožní reakce ruka-noha stupně 3 byl 16,6 % (CRC) a 22,0 % (GIST). V obou klinických studiích byl celkový výskyt kožní reakce ruka-noha (78,4 % u CRC a 88,2 % u GIST) vyšší u pacientů Asiatů léčených přípravkem Stivarga ve srovnání s pacienty jiných etnických příslušností. Výskyt kožní reakce ruka-noha stupně 3 byl u Asiatů 28,4% (CRC) a 23,5% (GIST) (viz body 4.2 a 4.4).
V placebem kontrolované studii fáze III u pacientů s metastazujícím kolorektálním karcinomem byl celkový výskyt hypertenze 30,4 % u pacientů léčených přípravkem Stivarga ve srovnání se 7,9 % u pacientů užívajících placebo. V placebem kontrolované studii fáze III u GIST nádorů byl celkový výskyt hypertenze 59,1 % u pacientů léčených přípravkem Stivarga ve srovnání s 27,3 % u pacientů užívajících placebo. V obou studiích se většina případů hypertenze u pacientů léčených přípravkem Stivarga objevila během prvního cyklu léčby a byla mírná až středně závažná (stupně 1 a 2: 22,8 % u CRC a 31,1 % u GIST). Výskyt hypertenze stupně 3 byl 7,6 % (CRC) a 27,3 % (GIST). Ve studii u GIST nádorů byl zaznamenán jeden případ hypertenze stupně 4.
V placebem kontrolované klinické studii fáze III u pacientů s metastazujícím kolorektálním karcinomem byl celkový výskyt proteinurie, která se objevila během léčby, 7,4 % u pacientů léčených přípravkem Stivarga ve srovnání s 2,4 % pacientů užívajících placebo. Z těchto příhod bylo 40,5 %
v ramenu přípravku Stivarga a 66,7 % v ramenu placeba hlášeno jako neustupující/nevyřešené.
V placebem kontrolované studii fáze III u GIST nádorů byl celkový výskyt proteinurie 6,8 % u pacientů léčených přípravkem Stivarga ve srovnání s 1,5 % u pacientů užívajících placebo.
Napříč všemi klinickými studiemi byly příhody srdečních poruch (všechny stupně) hlášeny častěji (20,5 % vs. 10,4 %) u pacientů léčených přípravkem Stivarga ve věku 75 let nebo starších (N=78) ve srovnání s pacienty léčenými přípravkem Stivarga mladšími než 75 let (N=995).
Abnormality jaterních testů
Laboratorní abnormality objevující se během léčby pozorované v placebem kontrolovaných studiích fáze III jsou uvedeny v Tabulce 4, 4a a Tabulce 5 (viz také bod 4.4).
|
Laboratorní parametr, (v % hodnocených vzorků) |
Stivarga plus BSC§ (N=500) |
Placebo plus BSC§ (N=253) | ||||
|
Všechny stupně* |
Stupeň 3* |
Stupeň 4* |
Všechny stupně* |
Stupeň 3* |
Stupeň 4* | |
|
Poruchy krve a lymfatického systému Snížení hladiny hemoglobinu |
78,5 |
4,7 |
0,6 |
66,3 |
2,8 |
0 |
|
Snížení počtu trombocytů |
40,5 |
2,4 |
0,4 |
16,8 |
0,4 |
0 |
|
Snížení počtu neutrofilů |
2,8 |
0,6 |
0 |
0 |
0 |
0 |
|
Snížení počtu lymfocytů |
54,1 |
9,3 |
0 |
34,4 |
3,2 |
0 |
|
Poruchy metabolismu a výživy Snížení hladiny vápníku |
59,3 |
1,0 |
0,2 |
18,3 |
1,2 |
0 |
|
Snížení hladiny draslíku |
25,7 |
4,3 |
0 |
8,3 |
0,4 |
0 |
|
Snížení hladiny fosfátu |
57,4 |
30,5 |
0,6 |
11,1 |
3,6 |
0 |
|
Poruchy jater a žlučových cest Zvýšení hladiny bilirubinu |
44,6 |
9,6 |
2,6 |
17,1 |
5,2 |
3,2 |
|
Zvýšení hladiny AST |
65,0 |
5,3 |
0,6 |
45,6 |
4,4 |
0,8 |
|
Zvýšení hladiny ALT |
45,2 |
4,9 |
0,6 |
29,8 |
2,8 |
0,4 |
|
Poruchy ledvin a močových cest Proteinurie |
59,7 |
0,4 |
0 |
34,1 |
0,4 |
0 |
|
Vyšetření Zvýšený INR** |
23,7 |
4,2 |
_# |
16,6 |
1,6 |
_# |
|
Zvýšení hladiny lipázy |
46,0 |
9,4 |
2,0 |
18,7 |
2,8 |
1,6 |
|
Zvýšení hladiny amylázy |
25,5 |
2,2 |
0,4 |
16,7 |
2,0 |
0,4 |
§ Nej lepší podpůrná péče
* CTC kritéria pro nežádoucí příhody (Common Terminology Criteria for Adverse Events, CTCAE), verze 3.0
** Mezinárodní normalizovaný poměr
# V CTCAE, verze 3.0 není stupeň 4 uveden
V porovnání s globální studií fáze III u CRC (CORRECT), do které byli zařazeni převáženě (přibližně 80 %) bělošští pacienti, byla pozorována vyšší incidence zvýšení jaterních enzymů u pacientů léčených přípravkem Stivarga v asijské studii fáze III u CRC (CONCUR), do které byli zařazeni převážně (> 90 %) východoasijští pacienti.
|
Laboratorní parametr, (v % hodnocených vzorků) |
Stivarga plus BSC§ (N=136) |
Placebo plus BSC§ (N=68) | ||||
|
Všechny stupně* |
Stupeň 3* |
Stupeň 4* |
Všechny stupně* |
Stupeň 3* |
Stupeň 4* | |
|
Zvýšení hladiny bilirubinu |
66,7 |
7,4 |
4,4 |
32,8 |
4,5 |
0,0 |
|
Zvýšení hladiny AST |
69,6 |
10,4 |
0,7 |
47,8 |
3,0 |
0,0 |
|
Zvýšení hladiny ALT |
54,1 |
8,9 |
0,0 |
29,9 |
1,5 |
0,0 |
§ Nejlepší podpůrná péče
* CTC kritéria pro nežádoucí příhody (Common Terminology Criteria for Adverse Events, CTCAE), verze 4.0
Tabulka 5: Laboratorní abnormality vyskytující se během léčby hlášené v placebem kontrolované studii fáze III (dvojitě zaslepená fáze) u pacientů s GIST (GRID)
|
Laboratorní parametr, (v % vyhodnocených vzorků) |
Stivarga plus BSC§ (N=132) |
Placebo plus BSC§ (N=66) | ||||
|
Všechny stupně* |
Stupeň 3* |
Stupeň 4* |
Všechny stupně* |
Stupeň 3* |
Stupeň 4* | |
|
Poruchy krve a lymfatického systému Snížení hladiny hemoglobinu |
75,0 |
3,0 |
0 |
72,7 |
1,5 |
0 |
|
Snížení počtu trombocytů |
12,9 |
0,8 |
0 |
1,5 |
0 |
1,5 |
|
Snížení počtu neutrofilů |
15,9 |
2,3 |
0 |
12,1 |
3,0 |
0 |
|
Snížení počtu lymfocytů |
29,5 |
7,6 |
0 |
24,2 |
3,0 |
0 |
|
Poruchy metabolismu a výživy Snížení hladiny vápníku |
16,7 |
1,5 |
0 |
4,5 |
0 |
0 |
|
Snížení hladiny draslíku |
20,5 |
3,0 |
0 |
3,0 |
0 |
0 |
|
Snížení hladiny fosfátu |
54,5 |
19,7 |
1,5 |
3,1 |
1,5 |
0 |
|
Poruchy jater a žlučových cest Zvýšení hladiny bilirubinu |
33,3 |
3,0 |
0,8 |
12,1 |
1,5 |
0 |
|
Zvýšení hladiny AST |
58,3 |
3,0 |
0,8 |
47,0 |
3,0 |
0 |
|
Zvýšení hladiny ALT |
39,4 |
3,8 |
0,8 |
39,4 |
1,5 |
0 |
|
Poruchy ledvin a močových cest Proteinurie |
38,5 |
1,5 |
39,0 |
1,7 | ||
|
Vyšetření Zvýšený INR** |
9,3 |
1,6 |
12,5 |
4,7 | ||
|
Zvýšení hladiny lipázy |
14,4 |
0 |
0,8 |
4,6 |
0 |
0 |
§ Nejlepší podpůrná péče
* CTC kritéria pro nežádoucí příhody (Common Terminology Criteria for Adverse Events, CTCAE), Verze 4.0
** Mezinárodní normalizovaný poměr - V CTCAE, verze 4.0 není stupeň 4 uveden
Ve dvou placebem kontrolovaných studiích fáze III testy tyreotropního hormonu hypofýzy (TSH) celkově prokázaly hladinu TSH po výchozím stavu vyšší než horní hranice normálních hodnot u 26,1 % pacientů léčených přípravkem Stivarga a u 15,1 % pacientů užívajících placebo. U 6,9 % pacientů léčených přípravkem Stivarga a u 0,7 % pacientů užívajících placebo byly hlášeny hladiny TSH po výchozím stavu >4krát vyšší než horní hranice normálních hodnot. Koncentrace volného trijodtyroninu (FT3) po výchozím stavu pod dolní hranicí normálních hodnot (< LLN) byla hlášena u 25,6 % pacientů léčených přípravkem Stivarga a u 20,9 % pacientů užívajících placebo. Koncentrace volného tyroxinu (FT4) po výchozím stavu nižší než LLN byla hlášena u 8,0 % pacientů léčených přípravkem Stivarga a u 6,6 % pacientů užívajících placebo. Celkově se u přibližně 7 % pacientů léčených přípravkem Stivarga objevila hypothyreóza vyžadující hormonální substituční léčbu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nejvyšší dávka přípravku Stivarga, která byla klinicky hodnocená, byla 220 mg denně. Nejčastěji pozorované nežádoucí účinky při této dávce byly dermatologické příhody, dysfonie, průjem, zánět sliznice, sucho v ústech, snížená chuť k jídlu, hypertenze a únava.
Při předávkování přípravkem Stivarga neexistuje žádné specifické antidotum. V případě suspektního předávkování by měl být přípravek Stivarga okamžitě vysazen, měla by být zahájena lékařem prováděná nejlepší podpůrná péče a pacient by měl být sledován až do klinické stabilizace.
5. FARMAKOLOGICKÉ VLASTNOSTI 5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antineoplastické léky, inhibitor proteinkinázy;
ATC kód: L01XE21
Mechanismus účinku a farmakodynamické účinky
Regorafenib je perorálně podávaná látka deaktivující nádor, která silně blokuje vícečetné proteinkinázy, včetně kináz účastnících se nádorové angiogeneze (VEGFR1, -2, -3, TIE2), onkogeneze (KIT, RET, RAF-1, BRAF, BRAFV600E) a mikroprostředí nádoru (PDGFR, FGFR). Regorafenib inhibuje zvláště mutovaný KIT, hlavní onkogenní složku u gastrointestinálních stromálních nádorů, a tím blokuje proliferaci nádorových buněk. V preklinických studiích prokázal regorafenib silnou protinádorovou aktivitu v širokém spektru nádorových modelů, včetně modelů kolorektálního karcinomu a gastrointestinálního stromálního nádoru, která je zprostředkována jeho antiangiogenními a antiproliferativními účinky. Kromě toho regorafenib prokázal antimetastatické účinky in vivo. Hlavní metabolity u člověka (M-2 a M-5) vykazovaly v modelech in vitro a in vivo při srovnání s regorafenibem podobnou účinnost.
Metastazující kolorektální karcinom (CRC)
Klinická účinnost a bezpečnost přípravku Stivarga byla hodnocena v mezinárodní, multicentrické, randomizované, dvojitě zaslepené, placebem kontrolované studii fáze III (CORRECT) u pacientů s metastazujícím kolorektálním karcinomem, u kterých došlo k progresi po selhání standardní léčby.
Primární cílový ukazatel účinnosti bylo celkové přežití (OS). Sekundární cílové ukazatele byly přežití bez progrese (PFS), výskyt objektivní nádorové odpovědi a kontrola onemocnění.
Celkem bylo randomizováno 760 pacientů v poměru 2:1 do léčby pomocí 160 mg regorafenibu (4 tablety přípravku Stivarga, každá o obsahu 40 mg regorafenibu) perorálně jednou denně (N=505) plus nejlepší podpůrná péče (BSC) nebo do odpovídajícího placeba (N=255) plus BSC po dobu 3 týdnů léčby s následným týdnem bez léčby. Průměrná denní dávka regorafenibu byla 147 mg.
Pacienti pokračovali v léčbě až do progrese onemocnění nebo nepřijatelné toxicity. Předem plánovaná průběžná analýza účinnosti byla provedena při výskytu 432 úmrtí. Studie byla odslepena poté, co tato plánovaná průběžná analýza celkového přežití překročila předem specifikovanou hranici účinnosti.
U 760 randomizovaných pacientů byl medián věku 61 let. 61 % byli muži, 78 % byli běloši a všichni pacienti měli výchozí výkonnostní stav ECOG (PS) 0 nebo 1. PS >2 byl hlášen během léčby přípravkem Stivarga u 11,4 % pacientů. Medián doby léčby a denní dávky a rovněž výskytu modifikace a snížení dávky byly podobné jako u pacientů s hlášeným PS >2, kteří užívali placebo (8,3 %). Většina pacientů s PS >2 ukončila léčbu pro progresi onemocnění. Primární lokalizace onemocnění bylo tlusté střevo (65 %), rektum (29 %) nebo obě (6 %). Mutace KRAS byla hlášena u 57 % pacientů při vstupu do studie.
Většina pacientů (52 %) dostala 3 nebo méně předchozích linií léčby metastazujícího onemocnění. Léčba zahrnovala chemoterapii na bázi fluoropyrimidinu, anti-VEGF léčbu a v případě divokého typu (wild type) onkogenu KRAS anti-EGFR léčbu.
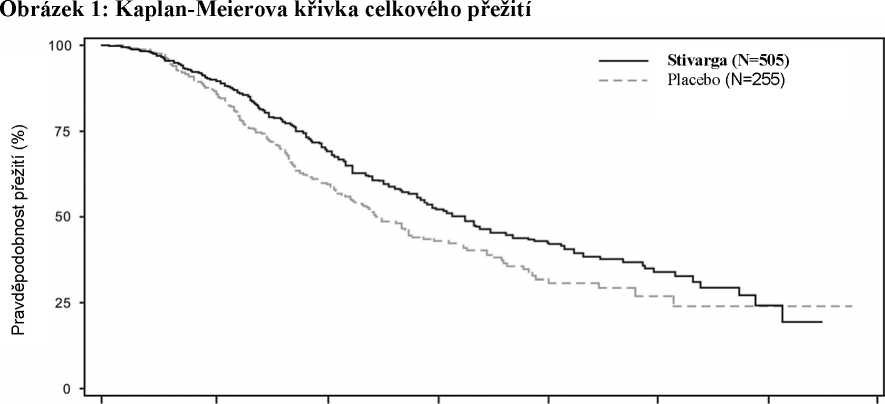
Přidání přípravku Stivarga k BSC vedlo k významně delšímu přežití ve srovnání s placebem plus BSC s poměrem rizik 0,774 (p=0,005178 stratifikovaný log rank test) a mediánem OS 6,4 měsíců vs. 5,0 měsíců [95 % CI 0,636, 0,942] (viz Tabulka 6 a Obrázek 1). PFS bylo významně delší u pacientů, kteří byli léčeni přípravkem Stivarga plus nejlepší podpůrnou péčí (BSC) (poměr rizik: 0,494, p < 0,000001, viz Tabulka 6). Výskyt odpovědi (kompletní nebo částečná odpověď) byl 1 % pro pacienty léčené přípravkem Stivarga a 0,4 % pro pacienty užívající placebo (p=0,188432, 1stranný). Výskyt kontroly onemocnění (kompletní nebo částečná odpověď nebo stabilizace onemocnění) byl významně vyšší u pacientů léčených přípravkem Stivarga (41,0 % vs 14,9 %, p < 0,000001, 1stranný).
|
Parametr účinnosti |
Poměr rizik* (95% CI) |
P-hodnota (jednostranný) |
Medián (95% CI) | |
|
Stivarga plus BSC§ (N=505) |
Placebo plus BSC§ (N=255) | |||
|
Celkové přežití |
0,774 (0,636, 0,942) |
0,005178 |
6,4 měsíců (5,9, 7,3) |
5,0 měsíců (4,4, 5,8) |
|
Doba přežití bez progrese** |
0,494 (0,419, 0,582) |
< 0,000001 |
1,9 měsíců (1,9, 2,1) |
1,7 měsíců (1,7, 1,7) |
§ Nejlepší podpůrná péče
* Poměr rizik < 1 ve prospěch přípravku Stivarga
** na základě hodnocení nádorové odpovědi zkoušejícím
Analýza celkového přežití a přežití bez progrese u podskupin podle věku (< 65; >65), pohlaví, výkonnostního stavu (ECOG PS), místa primárního výskytu onemocnění, doby od první diagnózy metastazujícího onemocnění, předchozí protinádorové léčby, předchozích linií léčby metastazujícího onemocnění a podle mutace KRAS prokázala účinek léčby regorafenibem oproti léčbě placebem.
Výsledky analýzy podskupin podle původního výskytu mutace KRAS ukázaly léčebný účinek pro celkové přežití ve prospěch léčby regorafenibem oproti placebu pro pacienty s nádory s genem KRAS divokého typu, zatímco pro pacienty s nádory s genem KRAS mutovaného typu byl hlášen početně nižší účinek; pozitivní účinek léčby regorafenibem na přežití bez progrese byl pozorován bez ohledu na stav mutace KRAS. Poměr rizik (95% CI) pro celkové přežití byl 0,653 (0,476 až 0,895) pro pacienty s nádory s genem KRAS divokého typu a 0,867 (0,670 až 1,123) pro pacienty s nádory s genem KRAS mutovaného typu bez známek heterogenity účinku léčby (nevýznamný test interakce). Poměr rizik (95% CI) pro přežití bez progrese byl 0,475 (0,362 až 0,623) pro pacienty s nádory s genem KRAS divokého typu a 0,525 (0,425 až 0,649) pro pacienty s nádory s genem KRAS mutovaného typu.
0 2 4 6 8 10 12 14
Měsíce od randomizace
Pacienti s rizikem
Stl.aiga 452 352 182 93 33 7
Flaceba 221 1 50 75 32 9 3
Druhá mezinárodní, multicentrická, randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie fáze III (CONCUR) hodnotila účinnost a bezpečnost přípravku Stivarga u 204 dříve léčených asijských pacientů (> 90 % východoasijská populace) s metastazujícím kolorektálním karcinomem, u kterých došlo k progresi po selhání chemoterapie na bázi fluoropyrimidinu. Pouze 59,5 % pacientů zařazených do studie CONCUR bylo také dříve léčeno pomocí VEGF nebo EGFR cílené léčby. Primárním cílovým ukazatelem účinnosti bylo celkové přežití (OS). Přidání přípravku Stivarga k BSC vedlo k významně delšímu přežití v porovnání s placebem plus BSC s poměrem rizik 0,550 (p = 0,000159 stratifikovaný log-rank test) a středním OS 8,8 měsíce oproti 6,3 měsíce [95% IS 0,395, 0,765]. PFS byl také významně delší u pacientů léčených přípravkem Stivarga plus BSC (poměr rizik: 0,311, p<0,000001), medián PFS 3,2 měsíce u přípravku Stivarga oproti 1,7 měsíce u placeba. Bezpečnostní profil přípravku Stivarga plus BSC ve studii CONCUR odpovídal bezpečnostnímu profilu pozorovanému ve studii CORRECT.
Gastrointestinální stromální nádory (GIST)
Klinická účinnost a bezpečnost přípravku Stivarga byla hodnocena v mezinárodní, multicentrické, randomizované, dvojitě zaslepené, placebem kontrolované studii fáze III (GRID) u pacientů s gastrointestinálními stromálními nádory (GIST), kteří byli dříve léčeni 2 inhibitory tyrosinkinázy (imatinib a sunitinib).
Analýza primárního cílového parametru účinnosti přežití bez progrese (PFS) byla provedena po 144 příhodách PFS (centrální zaslepené hodnocení). Byly také hodnoceny sekundární cílové parametry účinnosti zahrnující dobu do progrese (TTP) a celkové přežití (OS).
Celkem bylo randomizováno 199 pacientů s GIST v poměru 2:1 do léčby buď 160 mg regorafenibu plus nejlepší podpůrnou péčí (BSC; N=133) perorálně jednou denně nebo do odpovídajícího placebo plus BSC (N=66) po dobu 3 týdnů léčby s následnou dobou 1 týdne bez léčby. Průměrná denní podaná dávka regorafenibu byla 140 mg.
Pacienti byli léčeni do progrese onemocnění nebo nepřijatelné toxicity. Pacientům na placebu, u kterých došlo k progresi onemocnění, byl nabídnut odslepený regorafenib (překřížená možnost). Pacientům na regorafenibu, u kterých došlo k progresi onemocnění a u kterých podle názoru zkoušejícího poskytoval regorafenib klinický přínos, byla nabídnuta možnost pokračovat v léčbě odslepeným regorafenibem.
Průměrný věk u 199 randomizovaných pacientů byl 58 let, 64 % byli muži, 68 % byli běloši a všichni pacienti měli výchozí výkonnostní stav ECOG (ECOG Performance Status -PS) 0 nebo 1. Celková střední doba od poslední progrese nebo relapsu do randomizace byla 6 týdnů.
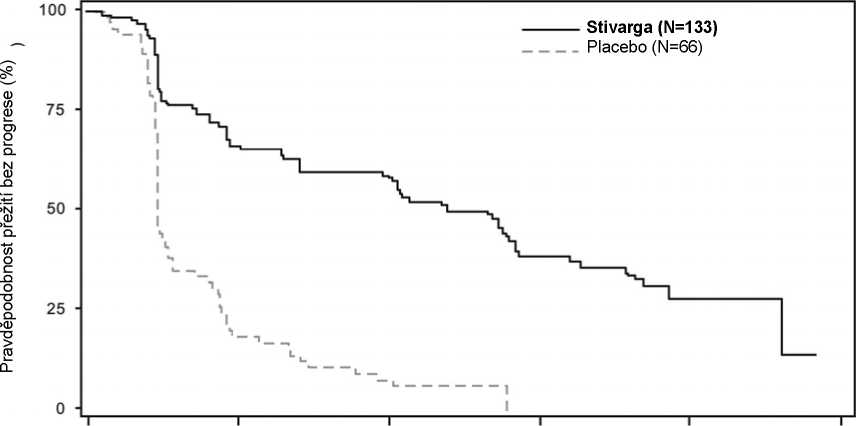
Regorafenib plus BSC vedly k významně delšímu PFS ve srovnání s placebem plus BSC s poměrem rizik 0,268 [95% CI 0,185; 0,388] a středním PFS 4,8 měsíce vs. 0,9 měsíce (p<0,000001). Relativní riziko progrese onemocnění nebo úmrtí byla snížena přibližně o 73,2 % u pacientů léčených regorafenibem ve srovnání s pacienty na placebu (viz Tabulka 7, Obrázek 2). Prodloužení PFS bylo trvale nezávislé na věku, pohlaví, geografické oblasti, předchozích liniích léčby a výkonnostním stavu ECOG.
TTP se významně prodloužila u pacientů léčených regorafenibem plus BSC ve srovnání s pacienty užívajícími placebo plus BSC s poměrem rizik 0,248 [95% IS 0,170; 0,364] a střední TTP 5,4 měsíců versus 0,9 měsíců (p<0,000001) (viz Tabulka 7).
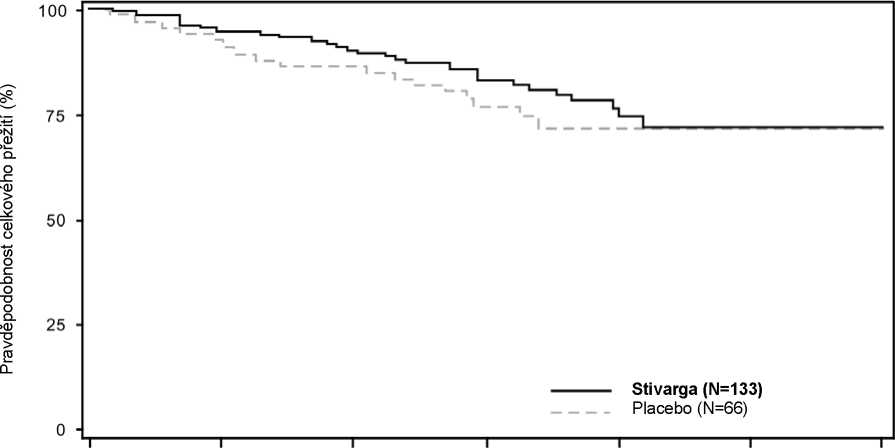
HR pro OS byl 0,772 (95% CI, 0,423; 1,408; p = 0,199; středního OS nebylo dosaženo v žádném ramenu); 85 % z pacientů, kteří byli v úvodu randomizováni do placebového ramena, dostávalo po progresi léčbu regorafenibem (viz Tabulka 7, Obrázek 3).
|
Parametr účinnosti |
Poměr rizik* (95% CI) |
P-hodnota (jednostranný) |
Medián (95% CI) | |
|
Stivarga plus BSC§ (N=133) |
Placebo plus BSC§ (N=66) | |||
|
Přežití bez progrese |
0,268 (0,185; 0,388) |
<0,000001 |
4,8 měsíců (4,0; 5,7) |
0,9 měsíců (0,9; 1,1) |
|
Doba do progrese |
0,248 (0,170; 0,364) |
<0,000001 |
5,4 měsíců (4,1; 5,7) |
0,9 měsíců (0,9; 1,1) |
|
Celkové přežití |
0,772 (0,423; 1,408) |
0,199 |
NR** |
NR** |
§ Nejlepší podpůrná péče
* Poměr rizik < 1 ve prospěch přípravku Stivarga ** NR: nebylo dosaženo
|
0 |
2 |
4 6 Měsíce od randomizace |
8 |
10 |
|
Rizikoví pacienti | ||||
|
Stivarga |
82 |
72 27 |
9 | |
|
Placebo |
12 |
5 0 |
0 | |
Obrázek 3: Kaplan-Meierovy křivky celkového přežití
|
0 |
2 |
4 6 Měsíce od randomizace |
8 |
10 |
12 |
|
Rizikoví pacienti | |||||
|
Stivarga |
126 |
119 94 |
39 |
10 |
1 |
|
Placebo |
61 |
57 41 |
16 |
3 |
1 |
Kromě toho dostalo 56 pacientů na placebu plus BSC odslepený přípravek Stivarga po překřížení po progresi onemocnění a celkem 41 pacientů na přípravku Stivarga plus BSC pokračovalo v léčbě přípravkem Stivarga po progresi onemocnění. Střední sekundární PFS (měřeno podle hodnocení zkoušejícím) bylo 5,0 a 4,5 měsíců, v uvedeném pořadí.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Stivarga u všech podskupin pediatrické populace v léčbě adenokarcinomu tlustého střeva a rekta (informace o použití u dětí viz bod 4.2).
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Stivarga u jedné nebo více podskupin pediatrické populace v léčbě solidních maligních nádorů (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Regorafenib dosahuje průměrných maximálních plasmatických hladin 2,5 mg/l přibližně 3 až 4 hodiny po jednorázové perorální dávce 160 mg podávané jako 4 tablety o obsahu 40 mg v jedné tabletě. Po jednorázových dávkách 60 mg nebo 100 mg byla průměrná relativní biologická dostupnost tablet ve srovnání s perorálním roztokem 69%, respektive 83 %.
Koncentrace regorafenibu a jeho hlavních farmakologicky aktivních metabolitů (M-2 a M-5) byly nejvyšší, když byl podáván po nízkotučné (lehké) snídani ve srovnání buď se snídaní o vysokém obsahu tuku, nebo se stavem nalačno. Expozice regorafenibu se zvýšila o 48 %, když byl podáván se snídaní o vysokém obsahu tuku, a o 36 %, když byl podáván po nízkotučné snídani, ve srovnání se
Obrázek 2: Kaplan-Meierovy křivky přežití bez progrese
stavem nalačno. Expozice metabolitům M-2 (N-oxid) a M-5 (N-oxid a N-desmethyl) je vyšší, když je regorafenib podáván s nízkotučnou snídaní, ve srovnání se stavem nalačno, a nižší, když je podáván se snídaní o vysokém obsahu tuku, ve srovnání se stavem nalačno.
Distribuce v organismu
Profily koncentrace v plasmě-čas pro regorafenib a rovněž pro hlavní cirkulující metabolity prokázaly mnohočetná maxima v průběhu 24hodinového dávkovacího intervalu, což je připisováno enterohepatální cirkulaci. Vazba regorafenibu na proteiny lidské plasmy je in vitro vysoká (99,5%). In vitro vazba na proteiny u M-2 a M-5 je vyšší (99,8 %, respektive 99,95 %) než u regorafenibu. Metabolity M-2 a M-5 jsou slabými substráty P-gp. Metabolit M-5 je slabý substrát BCRP.
Biotransformace
Regorafenib je metabolizován primárně v játrech oxidativním metabolismem zprostředkovaným CYP3A4 a rovněž glukuronidací zprostředkovanou UGT1A9. V plasmě byly identifikovány dva hlavní a šest vedlejších metabolitů regorafenibu. Hlavní cirkulující metabolity regorafenibu v plasmě u člověka jsou M-2 (N-oxid) a M-5 (N-oxid a N-desmethyl), které jsou farmakologicky aktivní a mají podobné koncentrace jako regorafenib v ustáleném stavu. M-2 je dále metabolizován oxidativní cestou zprostředkovanou CYP3A4 a rovněž glukuronidací zprostředkovanou UGT1A9.
Metabolity mohou být redukovány nebo hydrolyzovány mikrobiální flórou v trávicím traktu, což umožňuje reabsorpci nekonjugované léčivé látky a metabolitů (enterohepatální cirkulace).
Eliminace z organismu
Po perorálním podání v různých studiích se průměrný biologický poločas regorafenibu a jeho metabolitu M-2 v plasmě pohybuje od 20 do 30 hodin. Průměrný biologický poločas metabolitu M-5 je asi 60 hodin (rozmezí od 40 do 100 hodin).
Přibližně 90 % radioaktivní dávky se objevilo během 12 dnů po podání a asi 71 % dávky se vyloučilo ve stolici (47 % jako mateřská látka, 24 % jako metabolity) a asi 19 % dávky se vyloučilo močí ve formě glukuronidů. Vylučování glukuronidů močí se snížilo pod 10 % v ustálených stavech. Původní látka nalezená ve stolici by mohla pocházet z degradace glukuronidů nebo redukce metabolitu M-2 (N-oxid) ve střevě a rovněž neabsorbovaného regorafenibu.
M-5 může být redukován na M-4 mikrobiální flórou v trávicím traktu, což umožní reabsorpci M-4 (enterohepatální cirkulace). M-5 je nakonec vyloučen prostřednictvím M-4 jako M-6 (kyselina karboxylová) ve stolici.
Linearita/nelinearita
Systémová expozice regorafenibu v ustáleném stavu se zvyšuje proporcionálně až do dávky 60 mg a méně proporcionálně při dávkách nad 60 mg. Akumulace regorafenibu v ustáleném stavu vede asi k 2násobnému zvýšení plasmatických koncentrací, což odpovídá biologickému poločasu a frekvenci dávkování. V ustáleném stavu dosahuje regorafenib průměrných maximálních hladin v plasmě asi 3,9 mg/l (8,1 pmol/l) po perorálním podání 160 mg regorafenibu a poměr maximálních a minimálních plasmatických koncentrací v ustáleném stavu je menší než 2.
Oba metabolity, M-2 a M-5, vykazují nelineární akumulaci, která může být způsobena enterohepatální recyklací nebo saturací UGT1A9 cesty. Zatímco plasmatické koncentrace M-2 a M-5 po jednorázové dávce regorafenibu jsou daleko nižší než koncentrace mateřské látky, jsou plasmatické koncentrace M-2 a M-5 v ustáleném stavu srovnatelné s koncentracemi regorafenibu.
Porucha funkce jater
Expozice regorafenibu a jeho metabolitům M-2 a M-5 je srovnatelná u pacientů s mírnou poruchou funkce jater (Child-Pugh A) a u pacientů s normální funkcí jater.
Omezené údaje u pacientů se středně závažnou poruchou funkce jater (Child-Pugh B) ukazují po jednorázové dávce 100 mg regorafenibu podobnou expozici ve srovnání s pacienty s normální funkcí jater. Nejsou k dispozici žádné údaje u pacientů se poruchou funkce jater skóre C podle
Child-Pughovy klasifikace (závažná). Regorafenib je hlavně eliminován v játrech a expozice může být zvýšena u této populace pacientů.
Porucha funkce ledvin
Dostupné klinické údaje a farmakokinetické modelování za fyziologických podmínek ukazují podobnou expozici regorafenibu a jeho metabolitů M-2 a M-5 v ustáleném stavu u pacientů s mírnou a středně závažnou poruchou funkce ledvin ve srovnání s pacienty s normální funkcí ledvin. Farmakokinetika regorafenibu nebyla hodnocena u pacientů se závažnou poruchou funkce ledvin nebo v konečném stadiu renálního onemocnění. Farmakokinetické modelování za fyziologických podmínek však nepředpovídá žádnou významnou změnu expozice u těchto pacientů.
Starší pacienti
Věk neovlivňoval farmakokinetiku regorafenibu v hodnoceném věkovém rozmezí (29-85 let).
Pohlaví
Farmakokinetika regorafenibu není ovlivněna pohlavím.
Etnické rozdíly
Expozice regorafenibu pozorovaná u různých asijských populací (čínská, japonská, korejská) je ve stejném rozmezí jako u bělochů.
Srdeční elektrofyziologie/prodloužení QT
Nebyly pozorovány žádné účinky na prodloužení QTc intervalu po podání 160 mg regorafenibu v ustáleném stavu ve zvláštní QT studii u mužů a žen s nádorovým onemocněním.
5.3 Předklinické údaje vztahující se k bezpečnosti
Systémová toxicita
Po opakovaném podávání myším, potkanům a psům byly pozorovány nežádoucí účinky na mnoho orgánů, přednostně na ledviny, játra, trávicí trakt, štítnou žlázu, lymfatický/hematopoetický systém, endokrinní systém, reprodukční systém a kůži. Mírně zvýšená incidence ztluštění atrioventrikulárních srdečních chlopní byla pozorována ve 26týdenní studii opakované toxicity u potkanů. To může být způsobeno akcelerací fyziologického procesu souvisejícího s věkem. Tyto účinky se objevily při systémových expozicích v rozsahu expozice u člověka nebo pod očekávanou expozicí u člověka (podle srovnání AUC).
Změny zubů a kostí a nežádoucí účinky v reprodukčním systému byly výraznější u mladých a dospívajících zvířat a rovněž u mladých potkanů a ukazují na možné riziko pro děti a dospívající.
Reprodukční a vývojová toxicita
Specifické studie hodnotící fertilitu nebyly provedeny. Musí však být zvážen potenciál regorafenibu nežádoucím způsobem ovlivnit mužskou a ženskou reprodukci na základě morfologických změn varlat, vaječníků a dělohy pozorovaných po opakovaném podávání u potkanů a psů při expozicích pod očekávanou expozicí u člověka (na základě AUC). Pozorované změny byly pouze částečně reverzibilní.
Účinek regorafenibu na nitroděložní vývoj byl prokázán u králíků při expozicích pod očekávanou expozicí u člověka (na základě srovnání AUC). Hlavní nálezy zahrnovaly malformace močového systému, srdce a velkých cév a kostry.
Genotoxicita a kancerogenita
Nebyly zaznamenány žádné známky genotoxického potenciálu regorafenibu hodnoceného ve standardních zkouškách in vitro a in vivo u myší.
Studie kancerogenního potenciálu regorafenibu nebyly provedeny.
Posouzení rizika pro životní prostředí
Studie posuzující riziko pro životní prostředí ukázaly, že regorafenib má potenciál být stálý, biokumulativní a toxický k životnímu prostředí a může představovat riziko pro povrchové vody a pro sediment (viz bod 6.6).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety
Mikrokrystalická celulóza Sodná sůl kroskarmelózy Magnesium-stearát Povidon 25
Koloidní bezvodý oxid křemičitý Potah
Červený oxid železitý (E172)
Žlutý oxid železitý (E172)
Sójový lecithin Makrogol 3350
Částečně hydrolyzovaný polyvinylalkohol Mastek
Oxid titaničitý (E171)
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
Po prvním otevření lahvičky byla prokázána stabilita léčivého přípravku po dobu 7 týdnů. Poté má být léčivý přípravek zlikvidován.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Lahvičku uchovávejte dobře uzavřenou a vysoušedlo ponechte v lahvičce.
6.5 Druh obalu a obsah balení
Bílá, neprůhledná HDPE lahvička uzavřená PP/PP (polypropylen) šroubovacím víčkem s těsnicí vložkou a vysoušedlem s molekulárním sítem.
Jedna lahvička obsahuje 28 potahovaných tablet.
Velikosti balení:
Balení po 28 potahovaných tabletách.
Balení obsahující 84 (3 lahvičky po 28) potahovaných tablet.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Tento léčivý přípravek může představovat riziko pro životní prostředí (viz bod 5.3). Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/858/001
EU/1/13/858/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 26. srpna 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Bayer Pharma AG Kaiser-Wilhelm-Allee 51368 Leverkusen Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky na předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a následných aktualizací zveřejněných na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Předložit předem definovanou výzkumnou analýzu genetických (NRAS, KRAS, BRAF a PIK3CA) a negenetických (ANG-2, IL-6, IL-8, P1GF, VEGFR-1, TIE1, VEGF-A, VEGF-C, VEGF-D, VEGF-A-121, BMP-7, VWF, M-CSF, SDF-1) biomarkerů ze studie 15983 (randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie fáze III s adjuvantním regorafenibem versus placebem pro pacienty s kolorektálním karcinomem ve stadiu IV po léčbě jaterních metastáz). Analýza genetických a negenetických biomarkerů musí být nedílnou součástí pro všechny zařazené pacienty. |
31/12/2020 |
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU VNĚJŠÍ KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Stivarga 40 mg potahované tablety regorafenibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje regorafenibum 40 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sodík a sójový lecithin, pro další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 potahovaných tablet
84 (3 x 28) potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí. Uchovávejte v dobře uzavřené lahvičce. Vysoušedlo ponechte v lahvičce.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/858/001
EU/1/13/858/002
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
stivarga 40 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU ŠTÍTEK
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Stivarga 40 mg potahované tablety regorafenibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje regorafenibum 40 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sodík a sójový lecithin.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6 ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí. Lahvičku uchovávejte dobře uzavřenou. Vysoušedlo ponechte v lahvičce.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
12 REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/858/001
EU/1/13/858/002 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
B. PŘÍBALOVÁ INFORMACE
Stivarga 40 mg potahované tablety
regorafenibum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat,
protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli možných nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Stivarga a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Stivarga užívat
3. Jak se přípravek Stivarga užívá
4. Možné nežádoucí účinky
5. Jak přípravek Stivarga uchovávat
6. Obsah balení a další informace
1. Co je přípravek Stivarga a k čemu se používá
Stivarga obsahuje léčivou látku regorafenib. Je to lék používaný k léčbě rakoviny pomocí zpomalení
růstu a šíření nádorových buněk a přerušení zásobení krví, které udržuje růst nádorových buněk.
Přípravek Stivarga se používá pro léčbu:
- karcinomu tlustého střeva nebo konečníku (kolorektálního karcinomu), který se rozšířil do dalších částí těla, u dospělých pacientů, kteří byli léčeni jinými typy léčby nebo kteří nemohou být léčeni jinými léky (chemoterapie na bázi fluoropyrimidinů, anti-VEGF léčba a anti-EGFR léčba).
- gastrointestinálních stromálních nádorů (GIST), což je typ rakoviny žaludku a střeva, který se rozšířil do dalších částí těla nebo není léčitelný chirurgicky, u dospělých pacientů, kteří byli dříve léčeni jinými protinádorovými léky (imatinib a sunitinib).
Zeptejte se, prosím, svého lékaře, pokud budete mít jakékoli otázky ohledně toho, jak přípravek
Stivarga účinkuje nebo proč Vám byl předepsán.
2. Čemu musíte věnovat pozornost, než začnete přípravek Stivarga užívat Neužívejte přípravek Stivarga
- jestliže jste alergický(á) na regorafenib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před užitím přípravku Stivarga se poraďte se svým lékařem nebo lékárníkem.
Zvláštní opatrnosti při užívání přípravku Stivarga je zapotřebí
- pokud máte jakékoli jaterní problémy, včetně Gilbertova syndromu se známkami jako je: zežloutnutí kůže a bělma očí, tmavá moč a zmatenost a/nebo dezorientace. Léčba přípravkem Stivarga může vést k vyššímu riziku jaterních problémů. Před léčbou a během léčby přípravkem Stivarga provede Váš lékař krevní testy pro sledování funkce Vašich jater. Pokud je funkce Vašich jater závažně porušena, neměl/a byste být léčen/a přípravkem Stivarga, protože nejsou k dispozici žádné údaje o použití přípravku Stivarga u pacientů se závažnou poruchou funkce jater.
- pokud jste měl/a nebo máte jakékoli krvácivé problémy a pokud užíváte warfarin, fenprokumon nebo jiný lék, který ředí krev pro zabránění tvorby krevních sraženin. Léčba přípravkem Stivarga může vést k vyššímu riziku krvácení. Před zahájením užívání přípravku Stivarga se Váš lékař může rozhodnout provést krevní testy. Přípravek Stivarga může způsobit závažné krvácení v trávicím traktu, jako například v žaludku, krku, konečníku nebo ve střevech nebo v plicích, ledvinách, ústech, pochvě a/nebo mozku. Pokud se u Vás objeví následující příznaky, vyhledejte ihned lékařskou pomoc: krev ve stolici nebo tmavá stolice, krev v moči, bolest břicha, vykašlávání/zvracení krve.
- pokud se u Vás objeví bolest na hrudi nebo máte srdeční potíže. Před zahájením užívání přípravku Stivarga a během léčby lékař zkontroluje, jak dobře pracuje Vaše srdce. Pokud se u Vás objeví následující příznaky, vyhledejte ihned lékařskou pomoc, protože to mohou být známky srdečního záchvatu nebo snížení průtoku krve srdcem: nepříjemný pocit nebo bolest na hrudi, které se mohou šířit mimo Váš hrudník do ramen, paží, zad, krku, zubů, čelisti nebo žaludku a mohou přijít a odeznít; dušnost, náhlé pocení s chladnou, vlhkou kůží, pocit závratí nebo mdloby.
- pokud se u Vás objeví závažná a trvalá bolest hlavy, poruchy zraku, záchvaty nebo změna duševního stavu (jako je zmatenost, ztráta paměti nebo orientace), kontaktujte, prosím, ihned svého lékaře.
- pokud máte závažné problémy s žaludkem a střevy (proděravění stěny trávicího traktu nebo píštěl), měl by se Váš lékař rozhodnout ukončit léčbu přípravkem Stivarga. Pokud se u Vás objeví následující příznaky, vyhledejte ihned lékařskou pomoc: silná bolest břicha nebo bolest břicha, které neodeznívá, zvracení krve, červená nebo černá stolice.
- pokud máte vysoký krevní tlak - přípravek Stivarga může zvyšovat krevní tlak. Váš lékař bude sledovat Váš krevní tlak před léčbou a během léčby a může Vám dát lék na léčbu zvýšeného krevního tlaku.
- pokud jste v nedávné době prodělal(a) nebo se chystáte podstoupit chirurgickou léčbu.
Přípravek Stivarga může ovlivnit způsob hojení ran a může být nutné zastavit léčbu přípravkem Stivarga, dokud se nezhojí Vaše rána.
- pokud se u Vás objeví kožní potíže. Přípravek Stivarga může způsobit zarudnutí, bolest, otok nebo tvorbu puchýřů na dlaních nebo ploskách nohou. Pokud si všimnete jakýchkoli změn, kontaktujte svého lékaře. Pro léčbu příznaků může lékař doporučit použití krémů a/nebo vložek do bot a rukavic. Pokud se u Vás tento nežádoucí účinek objeví, může Váš lékař změnit dávku nebo ukončit léčbu až do zlepšení Vašeho stavu.
Předtím, než užijete přípravek Stivarga, řekněte svému lékaři, zda se Vás jakýkoli z těchto stavů
týká. Může být nutná léčba těchto stavů a také mohou být provedena dodatečná vyšetření (viz také bod 4 „Možné nežádoucí účinky“).
Děti a dospívající
Pro použití přípravku Stivarga u dětí a dospívajících v indikaci karcinomu tlustého střeva nebo konečníku, který se rozšířil do dalších částí těla, nejsou relevantní důvody.
Bezpečnost a účinnost přípravku Stivarga u dětí a dospívajících v indikaci gastrointestinálních stromálních nádorů (GIST) nebyla stanovena. Nejsou dostupné žádné údaje.
Další léčivé přípravky a přípravek Stivarga
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte nebo jste v nedávné době užíval(a) nebo které možná budete užívat. To se týká i léků, které jsou dostupné bez lékařského předpisu nebo dostupné mimo lékárnu, jako jsou vitaminy, potravinové doplňky nebo rostlinné přípravky. Některé léky mohou ovlivnit způsob účinku přípravku Stivarga nebo přípravek Stivarga může ovlivnit účinky jiných léků a může vést k závažným nežádoucím účinkům. Zvláště informujte svého lékaře, pokud užíváte některé z následujícího seznamu léků či jakékoli jiné léky:
- některé léky pro léčbu infekcí způsobených houbami (např. ketokonazol, itrakonazol, posakonazol a vorikonazol),
- některé léky pro léčbu bolesti (např. kyselina mefenamová, diflunisal a kyselina niflumová),
- některé léky pro léčbu bakteriálních infekcí (např. rifampicin, klaritromycin, telitromycin),
- léky obvykle používané k léčbě epilepsie (záchvatů) (např. fenytoin, karbamazepin nebo fenobarbital),
- methotrexát, což je lék obvykle používaný k léčbě rakoviny,
- rosuvastatin, fluvastatin, atorvastatin, což j sou léky obvykle používané k léčbě vysoké hladiny cholesterolu,
- warfarin nebo fenprokumon, což jsou léky obvykle používané pro ředění krve,
- třezalka tečkovaná (lék dostupný bez lékařského předpisu), rostlinný lék pro léčbu deprese.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete užívat jakýkoli lék.
Užívání přípravku Stivarga s jídlem a pitím
Vyhněte se pití grapefruitové šťávy během užívání přípravku Stivarga. To může ovlivnit způsob účinku přípravku Stivarga.
Těhotenství, kojení a plodnost
Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, informujte svého lékaře, protože přípravek Stivarga by měl být používán během těhotenství pouze, pokud je to nezbytně nutné. Váš lékař s Vámi prodiskutuje možná rizika užívání přípravku Stivarga během těhotenství.
Zabraňte otěhotnění během léčby přípravkem Stivarga, protože tento lék může poškodit Vaše nenarozené dítě. Jak ženy ve fertilním věku tak muži by měli během léčby a minimálně osm týdnů po dokončení léčby používat účinnou antikoncepci.
Během léčby přípravkem Stivarga nesmíte kojit své dítě, protože tento lék může ovlivnit růst a vývoj Vašeho dítěte. Informujte svého lékaře, pokud kojíte nebo plánujete kojit.
Přípravek Stivarga může snižovat fertilitu (plodnost) u mužů a žen. Poraďte se s lékařem dříve, než začnete přípravek Stivarga užívat.
Řízení dopravních prostředků a obsluha strojů
Není známo, zda přípravek Stivarga mění schopnost řídit nebo obsluhovat stroje. Neřiďte vozidlo ani neobsluhujte žádné přístroje nebo stroje, pokud se u Vás objeví příznaky související s léčbou, které ovlivní Vaši schopnost soustředit se a reagovat.
Důležité informace o některých složkách přípravku Stivarga
Tento lék obsahuje 2,427 mmol (nebo 55,8 mg) sodíku na denní dávku (4 tablety). Toto množství je nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
Tento lék obsahuje 1,68 mg sójového lecithinu na denní dávku (4 tablety).
3. Jak se přípravek Stivarga užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená denní dávka u dospělých je 4 tablety přípravku Stivarga 40 mg (160 mg regorafenibu). Váš lékař může změnit Vaši dávku. Užívejte dávku přípravku Stivarga, kterou Vám lékař předepíše. Váš lékař Vás obvykle požádá, abyste užíval(a) přípravek Stivarga 3 týdny a pak ukončil(a) léčbu na 1 týden. To je 1 cyklus léčby.
Užívejte přípravek Stivarga ve stejnou denní dobu po lehkém (nízkotučném) jídle. Polykejte tabletu celou a zapíjejte ji vodou po lehkém jídle, které neobsahuje více než 30 % tuku. Příkladem lehkého (nízkotučného) jídla je 1 porce cereálií (asi 30 g), 1 sklenice odstředěného mléka, 1 plátek toastového chleba s džemem, 1 sklenice jablečného džusu a 1 šálek kávy nebo čaje (520 kalorií, 2 g tuku). Přípravek Stivarga neužívejte spolu s grapefruitovou šťávou (viz také část „Užívání přípravku Stivarga s jídlem a pitím“).
V případě zvracení po podání regorafenibu byste neměl(a) užívat další tablety a měl(a) byste informovat svého lékaře.
Může být nutné, že lékař sníží Vaši dávku nebo se rozhodne přerušit nebo ukončit trvale léčbu, pokud to je nutné. Obvykle budete pokračovat v léčbě přípravkem Stivarga, dokud budete mít přínos z léčby a nebudete mít nepřijatelné nežádoucí účinky.
Pokud máte mírnou poruchu funkce jater, není nutná úprava dávkování. Pokud máte mírnou nebo středně závažnou poruchu funkce jater během léčby přípravkem Stivarga, měl by Vás Váš lékař pečlivě sledovat. Pokud máte závažnou poruchu funkce jater, neměl(a) byste být léčen(a) přípravkem Stivarga, protože u pacientů se závažnou poruchou funkce jater nejsou k dispozici žádné údaje.
Není nutná úprava dávkování, pokud máte mírnou nebo středně závažnou poruchu funkce ledvin. U pacientů se závažnou poruchou funkce ledvin nejsou k dispozici žádné údaje o použití přípravku Stivarga.
Jestliže jste užil(a) více přípravku Stivarga, než jste měl(a)
Informujte ihned svého lékaře, pokud jste užil(a) více přípravku, než je předepsáno. Může být nutné lékařské ošetření a Váš lékař může ukončit léčbu přípravkem Stivarga.
Užití příliš velkého množství přípravku Stivarga může vyvolat některé nežádoucí účinky pravděpodobněji nebo s vyšší závažností, zvláště:
- kožní reakce (vyrážka, puchýře, zarudnutí, bolest, otok, svědění nebo olupování kůže),
- změny hlasu nebo chrapot (dysfonie),
- časté nebo řídké stolice (průjem),
- vředy v ústech (zánět sliznice),
- sucho v ústech,
- snížení chuti k jídlu,
- vysoký krevní tlak (hypertenze)
- nadměrná unavenost (vyčerpání).
Jestliže jste zapomněl(a) užít přípravek Stivarga
Pokud zapomenete užít dávku, užijte ji co nejdříve, jak si vzpomenete, ve stejný den. Neužívejte dvě dávky přípravku Stivarga ve stejný den, abyste nahradil(a) vynechanou dávkou z předchozího dne. Informujte svého lékaře o každé vynechané dávce.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Tento lék může také ovlivnit výsledky některých krevních testů.
Nejzávažnějšími nežádoucími účinky, u kterých byl pozorován fatální výsledek, jsou:
- závažné problémy s játry, krvácení a gastrointestinální perforace (proděravění stěny trávicího traktu).
Informujte ihned svého lékaře, pokud se u Vás objeví některé z následujících příznaků:
Problémy s játry:
Léčba přípravkem Stivarga může vést k vyššímu riziku závažných jaterních problémů. Vyhledejte ihned lékařskou pomoc, pokud si všimnete následujících příznaků:
- žlutavé zbarvení kůže a bělma očí,
- tmavá moč,
- zmatenost a/nebo dezorientace.
Mohou to být známky závažného poškození jater.
Krvácení:
Přípravek Stivarga může způsobit závažné krvácení v trávicím traktu, jako je žaludek, jícen, konečník nebo střevo, nebo v plicích, ledvinách, ústech, pochvě a/nebo v mozku. Vyhledejte ihned lékařskou pomoc, pokud si všimnete následujících příznaků:
- krev ve stolici nebo tmavá stolice,
- krev v moči,
- bolest břicha,
- vykašlávání/zvracení krve.
To mohou být známky krvácení.
Závažné žaludeční a střevní potíže (gastrointestinální perforace nebo pištěl):
Vyhledejte ihned lékařskou pomoc, pokud si všimnete následujících příznaků:
- silná bolest břicha nebo žaludku, která neodeznívá,
- zvracení krve,
- červená nebo černá stolice.
To mohou být známky závažných žaludečních nebo střevních potíží.
Další nežádoucí účinky přípravku Stivarga uvedené podle četnosti:
Velmi časté nežádoucí účinky (mohou postihovat více než 1 z 10 uživatelů):
- infekce
- snížení počtu krevních destiček s příznaky charakterizovanými snadnou tvorbou podlitin nebo krvácení (trombocytopenie)
- snížení počtu červených krvinek (anémie)
- snížení chuti k j ídlu a příj mu potravy
- bolest hlavy
- zvýšený krevní tlak (hypertenze)
- změny hlasu nebo chrapot (dysfonie)
- časté nebo řídké stolice (průjem)
- bolestivá nebo suchá ústa, bolestivý jazyk, vředy v dutině ústní (stomatitida a/nebo zánět sliznice)
- nevolnost (nauzea)
- zvracení
- vysoké hladiny bilirubinu, což je látka vytvářená v játrech (hyperbilirubinémie)
- zarudnutí, bolest, puchýře a otok dlaní nebo plosek nohou (kožní reakce ruka-noha)
- vyrážka
- vypadávání vlasů (alopecie)
- slabost, nedostatek síly a energie, nadměrná únava a neobvyklá spavost (asthenie/únava)
- bolest
- horečka
- snížení tělesné hmotnosti.
Časté nežádoucí účinky (mohou postihovat až 1 z 10 uživatelů):
- snížení počtu bílých krvinek (leukopenie)
- snížená aktivita štítné žlázy (hypotyreóza)
- nízké hladiny draslíku, fosfátu, vápníku, sodíku nebo hořčíku v krvi (hypokalémie, hypofosfatémie, hypokalcémie, hyponatrémie a hypomagnezémie)
- vysoká hladina kyseliny močové v krvi (hyperurikémie)
- třes (tremor)
- poruchy chuti
- sucho v ústech
- pálení žáhy (gastroesofageální reflux)
- infekce nebo podráždění žaludku a střev (gastroenteritida)
- změny v enzymech vytvářených v j átrech, které mohou ukazovat na poruchu v j átrech (zvýšení transamináz)
- suchá kůže
- vyrážka s olupováním kůže (exfoliativní vyrážka)
- ztuhlost svalů nebo kloubů
- bílkovina v moči (proteinurie)
- vysoké hladiny určitých enzymů účastnících se trávení (zvýšení amylázy a lipázy)
- abnormální stav srážení krve (abnormální mezinárodní normalizovaný poměr INR).
Méně časté nežádoucí účinky (mohou postihovat až 1 ze 100 uživatelů):
- známky/příznaky alergické reakce mohou zahrnovat rozsáhlou těžkou vyrážku, pocit nemoci, horečku, dušnost, žloutenku, změny v látkách, které vytvářejí játra (hypersenzitivní reakce)
- srdeční záchvat, bolest na hrudi (infarkt myokardu a ischémie)
- závažné zvýšení krevního tlaku způsobující bolest hlavy, zmatenost, rozmazané vidění, nevolnost, zvracení a záchvaty (hypertenzní krize)
- poruchy nehtů (změny nehtu j ako j sou rýhy a/nebo třepení)
- mnohočetné erupce na kůži (erythema multiforme).
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1 000 uživatelů):
- určitý typ rakoviny kůže (keratoakanthom/spinocelulární karcinom kůže)
- bolest hlavy, zmatenost, záchvaty a ztráta zraku spojené s vysokým krevním tlakem nebo bez vysokého krevního tlaku (syndrom reverzibilní zadní leukoencefalopatie/PRES)
- závažné reakce na kůži a/nebo sliznicích, které mohou zahrnovat bolestivé puchýře a horečku, včetně rozsáhlého odlupování kůže (Stevens-Johnsonův syndrom a toxická epidermální nekrolýza).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Stivarga uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku lahvičky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Lahvičku uchovávejte dobře uzavřenou a vysoušedlo ponechte v lahvičce. Vysoušedlo je materiál absorbující vlhkost vyplňující malý obal, aby chránil tablety před vlhkostí.
Po otevření lahvičky má být lék zlikvidován po 7 týdnech.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Stivarga obsahuje
- Léčivou látkou je regorafenibum. Jedna potahovaná tableta obsahuje regorafenibum 40 mg.
- Dalšími složkami jsou: mikrokrystalická celulóza, sodná sůl kroskarmelózy, magnesium-stearát, povidon 25 a koloidní bezvodý oxid křemičitý, červený oxid železitý (E172), žlutý oxid železitý (E172), sójový lecithin, makrogol 3350, částečně hydrolyzovaný polyvinylalkohol, mastek a oxid titaničitý (E171).
Jak přípravek Stivarga vypadá a co obsahuje toto balení
Tablety přípravku Stivarga 40 mg jsou světle růžové a oválné s označením "BAYER" na jedné straně
a "40" na druhé straně.
Každá lahvička obsahuje 28 potahovaných tablet.
Přípravek Stivarga 40 mg tablety je k dispozici v balení obsahujícím jednu lahvičku s 28 tabletami a v balení obsahujícím 3 lahvičky, každá po 28 tabletách.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Bayer Pharma AG 13342 Berlin Německo
Výrobce
Bayer Pharma AG 51368 Leverkusen Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie / Belgique / Belgien
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Btarapna
Eaňep Etnrapua EOOfl Ten. +359 02 81 401 01 Česká republika Bayer s.r.o.
Tel: +420 266 101 111
Danmark
Bayer A/S
Tlf: +45-45 23 50 00
Deutschland
Bayer Vital GmbH
Tel: +49-(0)214-30 513 48
Eesti
Bayer OU
Tel: +372 655 85 65
EXlába
Bayer EAAág ABEE Tqk+30 210 618 75 00 Espaňa
Bayer Hispania S.L.
Tel: +34-93-495 65 00 France
Bayer HealthCare Tél: +33-(0)800 87 54 54 Hrvatska Bayer d.o.o.
Tel: + 385-(0)1-6599 900
Ireland
Bayer Limited
Tel: +353 1 299 93 13
Island
Icepharma hf.
Sími: +354 540 80 00
Italia
Bayer S.p.A.
Tel: +39-02-397 81 Kúrcpog
NOVAGEM Limited T^: +357 22 48 38 58 Latvija SIA Bayer
Tel: +371 67 84 55 63
Lietuva
UAB Bayer
Tel. +370 5 23 36 868
Luxembourg / Luxemburg
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Magyarország
Bayer Hungária Kft.
Tel.:+36-14 87-41 00
Malta
Alfred Gera and Sons Ltd. Tel: +356-21 44 62 05 Nederland Bayer B.V.
Tel: +31-(0)297-28 06 66
Norge
Bayer AS
Tlf. +47 23 13 05 00 Osterreich
Bayer Austria Ges. m. b. H. Tel: +43-(0)1-711 46-0 Polska
Bayer Sp. z o.o.
Tel.: +48-22-572 35 00 Portugal
Bayer Portugal Lda.
Tel: +351-21-416 42 00
Románia
SC Bayer SRL
Tel: +40 21 528 59 00
Slovenija
Bayer d. o. o.
Tel.: +386-(0)1-58 14 400 Slovenská republika Bayer, spol. s r.o.
Tel: +421 2 59 21 31 11 Suomi/Finland Bayer Oy
Puh/Tel: +358-20 785 21
Sverige
Bayer AB
Tel: +46-(0)8-580 223 00 United Kingdom Bayer plc
Tel: +44 (0)1 635-56 30 00
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
43