Somavert 25 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
SOMAVERT 10 mg prášek a rozpouštědlo pro injekční roztok SOMAVERT 15 mg prášek a rozpouštědlo pro injekční roztok SOMAVERT 20 mg prášek a rozpouštědlo pro injekční roztok SOMAVERT 25 mg prášek a rozpouštědlo pro injekční roztok SOMAVERT 30 mg prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
SOMAVERT 10 mg prášek a rozpouštědlo pro injekční roztok Jedna injekční lahvička obsahuje pegvisomantum 10 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 10 mg.*
Pomocná látka se známým účinkem
Léčivý přípravek o síle 10 mg obsahuje 0,4 mg sodíku v každé injekční lahvičce.
SOMAVERT 15 mg prášek a rozpouštědlo pro injekční roztok Jedna injekční lahvička obsahuje pegvisomantum 15 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 15 mg. *
Pomocná látka se známým účinkem
Léčivý přípravek o síle 15 mg obsahuje 0,4 mg sodíku v každé injekční lahvičce.
SOMAVERT 20 mg prášek a rozpouštědlo pro injekční roztok Jedna injekční lahvička obsahuje pegvisomantum 20 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 20 mg. *
Pomocná látka se známým účinkem
Léčivý přípravek o síle 20 mg obsahuje 0,4 mg sodíku v každé injekční lahvičce.
SOMAVERT 25 mg prášek a rozpouštědlo pro injekční roztok Jedna injekční lahvička obsahuje pegvisomantum 25 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 25 mg. *
Pomocná látka se známým účinkem:
Léčivý přípravek o síle 25 mg obsahuje 0,5 mg sodíku v každé injekční lahvičce.
SOMAVERT 30 mg prášek a rozpouštědlo pro injekční roztok Jedna injekční lahvička obsahuje pegvisomantum 30 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 30 mg. *
Pomocná látka se známým účinkem:
Léčivý přípravek o síle 30 mg obsahuje 0,6 mg sodíku v každé injekční lahvičce. * Je vyráběn v buňkách Escherichia coli technologií rekombinantní DNA.
Úplný seznam pomocných látek viz bod 6.1
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok (prášek na injekci). Prášek má bílou nebo mírně našedlou barvu.
KLINICKÉ ÚDAJE
4.
4.1. Terapeutické indikace
Léčba dospělých pacientů s akromegalií, kteří měli nedostatečnou odpověď na chirurgickou a/nebo radiační terapii, a u kterých odpovídající konzervativní léčba analogy somatostatinu nevedla k normalizaci koncentrace IGF-I nebo nebyla snášena.
4.2. Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře se zkušenostmi s léčbou akromegalie.
Dávkování
Nárazová dávka 80 mg pegvisomantu se má podat subkutánně pod dohledem lékaře. Poté se přípravek SOMAVERT 10 mg rekonstituovaný v 1 ml rozpouštědla podává 1x denně ve formě subkutánní injekce.
Úprava dávky se provádí na základě koncentrací IGF-I v séru. Koncentrace IGF-I v séru má být stanovována každých 4-6 týdnů a příslušná úprava dávek se má provádět postupně po 5 mg denně, aby byla koncentrace IGF-I v séru udržena v normálním rozmezí upraveném podle věku a aby byla udržena optimální terapeutická odpověď.
Maximální dávka nesmí překročit 30 mg/den.
Pro různé dávkovací režimy jsou k dispozici následující síly přípravku: SOMAVERT 10 mg, SOMAVERT 15 mg, SOMAVERT 20 mg, SOMAVERT 25 mg a SOMAVERT 30 mg.
Pediatrická populace
Bezpečnost a účinnost přípravku SOMAVERT u dětí ve věku 0 až 7 let nebyla stanovena. Nejsou dostupné žádné údaje.
Použití u starších pacientů Není potřebná žádná úprava dávek.
Porucha funkce jater nebo ledvin
Bezpečnost a účinnost přípravku SOMAVERT u pacientů se sníženou funkcí ledvin nebo jater nebyla stanovena.
Způsob podání
Pegvisomant se má podávat subkutánní injekcí.
Místo vpichu injekce je vhodné denně měnit, aby se zabránilo vzniku lipohypertrofie.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3. Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Tumory secemující růstový hormon
Někdy může dojít k expanzi tumorů hypofýzy secernujících růstový hormon, což může způsobovat závažné komplikace (např. poruchy zorného pole). Léčba pegvisomantem nesnižuje velikost tumoru. Všichni pacienti s těmito tumory musí být pečlivě sledováni s cílem zabránit případné progresi velikosti tumoru během léčby.
Sledování koncentrace IGF-I v séru
Pegvisomant je silným antagonistou účinku růstového hormonu. Po podávání tohoto léčivého přípravku může dojít ke stavu s deficitem růstového hormonu, bez ohledu na přítomnost zvýšených koncentrací růstového hormonu v séru. Koncentrace IGF-I v séru musí být sledovány a udržovány v normálním rozmezí upraveném podle věku úpravou dávky pegvisomantu.
Zvýšení koncentrace ALT nebo AST
V prvních šesti měsících léčby pegvisomantem musí být ve čtyřtýdenních až šestitýdenních intervalech monitorovány koncentrace alanin aminotransferázy (ALT) a aspartát transaminázy (AST) v séru a dále kdykoliv, pokud se u pacientů vyvinou příznaky naznačující hepatitidu. U pacientů se zvýšením koncentrací ALT a AST nebo u pacientů s anamnézou léčby analogem somatostatinu je nutné vyloučit obstrukční onemocnění žlučových cest. V případě přetrvávání známek jaterního onemocnění má být léčba pegvisomantem přerušena.
Hypoglykémie
Ve studii prováděné s pegvisomantem u pacientů s diabetem léčených inzulínem nebo perorálními antidiabetiky bylo u této populace nemocných zjištěno riziko hypoglykémie. U pacientů s akromegalií a diabetes mellitus může být proto nezbytné snížit dávky inzulínu nebo perorálních antidiabetik (viz bod 4.5).
Zvýšená fertilita
Terapeutický přínos snížení koncentrace IGF-I, který vede ke zlepšení klinického stavu pacienta, by mohl potenciálně zvýšit fertilitu u pacientek. Pacientky musí být upozorněny, aby v případě potřeby používaly přiměřenou antikoncepční metodu. Pegvisomant není doporučován v těhotenství (viz bod 4.6).
4.5. Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí. Je zapotřebí zvážit, zda pokračovat v léčbě analogy somatostatinu. Použití tohoto přípravku v kombinaci s jinými přípravky na léčbu akromegalie nebylo studováno.
U pacientů užívajících inzulín nebo perorální antidiabetika může být potřebné snížení dávky těchto léků vzhledem k účinku pegvisomantu na senzitivitu inzulínu (viz bod 4.4).
Pegvisomant je strukturálně významně podobný růstovému hormonu, což způsobuje zkříženou reakci s komerčně dostupnými analýzami na růstový hormon. Protože sérové koncentrace terapeuticky účinných dávek tohoto přípravku jsou obvykle 100 - 1000x vyšší než skutečné koncentrace růstového hormonu v séru, které lze vidět u akromegaliků, stanovení koncentrace růstového hormonu v séru komerčně dostupnými analýzami je zkresleno. Léčbu pegvisomantem nelze proto sledovat nebo upravovat na základě koncentrací růstového hormonu v séru vyšetřeného pomocí těchto analýz.
4.6. Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Viz bod 4.4. Těhotenství
Nejsou k dispozici žádné klinické údaje o působení pegvisomantu v těhotenství.
Z hlediska účinku pegvisomantu v těhotenství, na vývoj embrya či plodu, porod nebo postnatální vývoj jsou studie na zvířatech nedostatečné (viz bod 5.3). Potenciální riziko pro člověka není známo.
Přípravek SOMAVERT lze v těhotenství použít pouze tehdy, když je to nezbytné (viz bod 4.4).
Kojení
Vylučování pegvisomantu do lidského mateřského mléka nebylo u zvířat zkoumáno. Klinické údaje jsou příliš omezené (jeden hlášený případ), aby bylo možné činit závěry o vylučování pegvisomantu do lidského mateřského mléka. Proto nesmí být pegvisomant u kojících žen používán. V kojení je možné pokračovat po vysazení tohoto přípravku: při tomto rozhodnutí je třeba vzít v úvahu přínos léčby pegvisomantem pro matku a přínos kojení pro dítě.
Fertilita
Nejsou dostupné žádné údaje o účincích pegvisomantu na fertilitu.
4.7. Účinky na schopnost řídit a obsluhovat stroje
Nebyly prováděny žádné studie sledující účinky pegvisomantu na schopnost řídit motorová vozidla nebo obsluhovat stroje.
4.8. Nežádoucí účinky
Shrnutí bezpečnostního profilu
Níže uvedený seznam obsahuje nežádoucí účinky pozorované v klinických studiích s přípravkem SOMAVERT.
V klinických studiích u pacientů léčených pegvisomantem (n=550) byla většina nežádoucích účinků z hlediska intenzity mírné až středně těžké povahy, měla pouze omezené trvání a nevyžadovala přerušení léčby.
Nejčastěji hlášenými nežádoucími účinky během klinických studií, které se vyskytly u > 10% pacientů s akromegalií léčených pegvisomantem, byly: bolesti hlavy (25%), artralgie (16%) a průjem (13%).
Seznam nežádoucích účinků
Seznam níže obsahuje nežádoucí účinky pozorované v klinických studiích nebo spontánně hlášené, zařazené podle tříd orgánových systémů a četnosti.
Nežádoucí účinky jsou uvedeny podle následujících kategorií:
Velmi časté: >1/10
Časté: >1/100 až <1/10
Méně časté: >1/1000 až <1/100
Není známo (z dostupných údajů nelze určit)
|
Třídy orgánových systémů |
Velmi časté: >1/10 |
Časté: >1/100 až <1/10 |
Méně časté: >1/1000 až <1/100 |
Není známo (z dostupných údajů nelze určit) |
|
Poruchy krve a lymfatického systému |
trombocytopenie, leukopenie, leukocytóza, hemoragická diatéza | |||
|
Poruchy imunitního systému |
reakce z přecitlivělostib |
anafylaktická reakceb, anafylaktoidní reakceb | ||
|
Poruchy metabolismu a výživy |
hypercholesterolemie, hyperglykemie, hypoglykemie, zvýšení tělesné hmotnosti |
hypertriglyceridemie | ||
|
Psychiatrické poruchy |
abnormální sny |
panická ataka, krátkodobá ztráta paměti, apatie, zmatenost, porucha spánku, zvýšené libido |
hněv | |
|
Poruchy nervového systému |
bolest hlavy |
somnolence, tremor, závratě, hypestezie |
narkolepsie, migréna, poruchy chuti | |
|
Poruchy oka |
bolest oka |
astenopie | ||
|
Poruchy ucha a labyrintu |
Meniérova choroba | |||
|
Srdeční poruchy |
periferní edém | |||
|
Cévní poruchy |
hypertenze | |||
|
Respirační, hrudní a mediastinální poruchy |
laryngospazmusb | |||
|
Gastrointestinální poruchy |
zvracení, zácpa, nauzea, břišní distenze, dyspepsie, flatulence |
hemeroidy, hypersekrece slin, sucho v ústech, onemocnění zubů | ||
|
Poruchy jater a žlučových cest |
abnormální funkční jaterní testy (např. zvýšení transamináz) (viz bod 4.4) | |||
|
Poruchy kůže a podkožní tkáně |
hyperhidróza, kontuze, svěděníb, vyrážkab |
otok obličeje, suchá kůže, zvýšená náchylnost k tvorbě modřin, noční pocení, erytémb, kopřivkab |
angioedémb | |
|
Poruchy svalové a |
artralgie |
myalgie, artritida |
|
Třídy orgánových systémů |
Velmi časté: >1/10 |
Časté: >1/100 až <1/10 |
Méně časté: >1/1000 až <1/100 |
Není známo (z dostupných údajů nelze určit) |
|
kosterní soustavy a pojivové tkáně | ||||
|
Poruchy ledvin a močových cest |
hematurie |
proteinurie, polyurie, zhoršení funkce ledvin | ||
|
Celkové poruchy a reakce v místě aplikace |
reakce v místě vpichu injekce (včetně hypersenzitivity v místě vpichu), hematom nebo krvácení v místě vpichu injekce, hypertrofie v místě vpichu injekce (např. lipohypertrofie)3, onemocnění podobné chřipce, únava, asténie, horečka |
abnormální pocity, zhoršené hojení, hlad |
a viz Popis vybraných nežádoucích účinků níže b NÚ související s reakcí z přecitlivělosti
Popis vybraných nežádoucích účinků
Většina reakcí v místě vpichu injekce, které měly charakter lokalizovaných erytémů a bolestivosti, spontánně při lokální symptomatické léčbě ustoupila, přičemž se v léčbě pegvisomantem pokračovalo. Byl pozorován výskyt hypertrofie v místě vpichu injekce, včetně lipohypertrofie.
U 16,9% pacientů léčených pegvisomantem byl pozorován vývoj izolovaných protilátek s nízkým titrem proti růstovému hormonu. Klinický význam těchto protilátek není znám.
Po uvedení přípravku na trh byly hlášeny systémové reakce z přecitlivělosti zahrnující anafylaktické/anafylaktoidní reakce, laryngospazmus, angioedém, generalizované kožní reakce (vyrážka, erytém, pruritus, kopřivka). Někteří pacienti vyžadovali hospitalizaci. Po opětovném podání se příznaky neobjevily u všech pacientů.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9. Předávkování
S předávkováním pegvisomantem jsou pouze omezené zkušenosti. V jednom popsaném případě akutního předávkování, kdy byla po dobu 7 dní podávána dávka 80 mg denně, se u pacienta objevila mírná únava a sucho v ústech. Za týden po vysazení léku byly pozorovány tyto nežádoucí účinky: insomnie, zvýšená únava, periferní edém, tremor a zvýšení tělesné hmotnosti. Za dva týdny po vysazení léčby byly pozorovány leukocytóza a středně silné krvácení z místa vpichu injekce a z místa odběru krve, které byly považovány za účinky s možnou souvislostí s léčbou pegvisomantem.
V případě předávkování musí být léčba tímto přípravkem vysazena a obnovena až po návratu koncentrace IGF-I do rozmezí normálních hodnot nebo vyšších hodnot.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1. Farmakodynamické vlastnosti
Farmakoterapeutická skupina: jiné hormony předního laloku hypofýzy a analogy, ATC kód: H01AX01
Mechanismus účinku
Pegvisomant je geneticky modifikovaný analog lidského růstového hormonu, působící jako antagonista receptoru růstového hormonu. Pegvisomant se váže na receptory růstového hormonu na povrchu buněk, kde blokuje vazbu růstového hormonu, a tak interferuje s intracelulární signální transdukcí růstového hormonu. Pegvisomant je vysoce selektivní pro GH receptor a nevykazuje zkříženou reakci s receptory jiných cytokinů včetně prolaktinu.
Farmakodynamické účinky
Inhibice účinku růstového hormonu pegvisomantem vede k poklesu sérových koncentrací růstového faktoru-I podobného inzulínu (insulin-like growth factor-I, IGF-I), jakož i ostatních sérových bílkovin citlivých na růstový hormon, jako je volný IGF-I, podjednotka IGF-I labilní na kyseliny (ALS) a vazebný protein-3 růstového faktoru podobnému inzulínu (insulin-like growth factor binding protein-3, IGFBP-3).
Klinická účinnost a bezpečnost
Pacienti s akromegalií (n=112) byli léčeni ve 12-týdenní randomizované, dvojitě zaslepené, multicentrické studii porovnávající placebo a pegvisomant. Při všech návštěvách po zařazení do studie bylo ve skupinách léčených pegvisomantem pozorováno statisticky významné, na dávce závislé snížení IGF-I (p<0,0001), volného IGF-I (p<0,05), IGFBP-3 (p<0,05) a ALS (p<0,05). Sérový IGF-I se znormalizoval na konci studie (týden 12) u 9,7% pacientů léčených placebem, u 38,5% pacientů léčených pegvisomantem 20 mg denně, u 75% pacientů léčených dávkou 15 mg denně a u 82% nemocných léčených dávkou 20 mg denně.
Statisticky významné rozdíly oproti placebu (p<0,05) byly pozorovány při hodnocení zlepšení skóre celkových příznaků u všech dávkovacích skupin v porovnání s placebem.
V dlouhodobé, otevřené studii s titrací dávek byla sledována kohorta 38 pacientů s akromegalií, kterým byl podáván denně pegvisomant po dobu nejméně 12 měsíců (průměr = 55 týdnů). Průměrná koncentrace IGF-I v této kohortě poklesla při podávání pegvisomantu z 917 ng/ml na 299 ng/ml, přičemž 92% pacientů dosáhlo normální (upravenou k věku) koncentraci IGF-I.
5.2. Farmakokinetické vlastnosti
Absorpce
Absorpce pegvisomantu po jeho subkutánním podání je pomalá a protrahovaná. Vrcholových koncentrací pegvisomantu není obecně dosaženo do 33-77 hodin po jeho podání. Průměrný rozsah absorpce po subkutánní dávce byl 57% v porovnání s intravenózní dávkou.
Distribuce
Zdánlivý distribuční objem pegvisomantu je relativně malý (7-12 l).
Biotransformace
Metabolismus pegvisomantu nebyl studován. Eliminace
Průměrná tělesná systémová clearance pegvisomantu po vícenásobných dávkách se odhaduje na 28 ml/hod. pro subkutánní dávky v rozmezí 10-20 mg denně. Renální clearance pegvisomantu je zanedbatelná a činí méně než 1% celkové tělesné clearance. Pegvisomant je jen pomalu eliminován ze séra, průměrný odhad poločasu se obecně pohybuje mezi 74-172 hodinami po jednorázové dávce i vícenásobných dávkách.
Linearita/nelinearita
Po jednorázovém subkutánním podání pegvisomantu nebyla pozorována žádná linearita při stoupajících dávkách 10, 15 nebo 20 mg. V populačních farmakokinetických studiích byla pozorována přibližně lineární farmakokinetika v ustáleném stavu. Údaje od 145 pacientů ze dvou dlouhodobých studií ukazují průměrné koncentrace pegvisomantu v séru (± SD) 8800 ± 6300 ng/ml při podávání dávky 10 mg denně, 13200 ± 8000 ng/ml při dávce 15 mg denně a 15600 ± 10300 ng/ml při dávce 20 mg denně.
Farmakokinetika pegvisomantu je obdobná u normálních zdravých dobrovolníků a u pacientů s akromegalií, i když pacienti s vyšší tělesnou hmotností mají sklon k vyšší celkové tělesné clearanci pegvisomantu než pacienti s menší váhou a silnější pacienti mohou tedy potřebovat vyšší dávky pegvisomantu.
5.3. Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě studií toxicity po opakovaném podávání u potkanů a opic neodhalily žádné zvláštní riziko pro člověka. Vzhledem k výrazné farmakologické odpovědi u opic však nebylo studováno vyšší systémové vystavení léku než to, které bylo dosaženo u pacientů v terapeutických dávkách. Až na jeden segmentový test II u králíka, nebyly prováděny žádné další studie sledující reprodukční toxicitu. Relevantní odpověď u člověka není v současné době známa.
Ve studii sledující kancerogenitu u potkanů byly při expozici srovnatelné s trojnásobnou expozicí u člověka (odvozené od středních plazmatických koncentrací ve 2 dlouhodobých studiích) při denní dávce 30 mg pozorovány u samců potkanů v místě podání injekce maligní fibrózní histiocytomy spojené s fibrózou a histiocytárním zánětem.
6. FARMACEUTICKÉ ÚDAJE
6.1. Seznam pomocných látek
Prášek:
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný
Monohydrát dihydrogenfosforečnanu sodného
Rozpouštědlo:
Voda na injekci
6.2. Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3. Doba použitelnosti
3 roky
Přípravek musí být podán bezprostředně po rekonstituci.
6.4. Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem. Uchovávejte injekční lahvičku (lahvičky) a předplněnou injekční stříkačku (stříkačky) v krabičce, aby byl přípravek chráněn před světlem.
Po rekonstituci:
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5. Druh obalu a obsah balení
10 mg nebo 15 mg nebo 20 mg nebo 25 mg nebo 30 mg pegvisomantu v prášku v injekční lahvičce (ze skla typu I) se zátkou (chlorobutylová pryž) a 1 ml rozpouštědla (voda na injekci) v předplněné stříkačce (z borosilikátového skla typu I) se zarážkou pístu (bromobutylová pryž) a víčkem (bromobutylová pryž). Barva ochranného plastového víčka odpovídá příslušné síle přípravku.
SOMAVERT 10 mg a 15 mg
Balení obsahující 30 injekčních lahviček, předplněných injekčních stříkaček a bezpečnostních jehel. SOMAVERT 20 mg, 25 mg a 30 mg
Balení obsahující 1 injekční lahvičku, předplněnou injekční stříkačku a bezpečnostní jehlu a 30 injekčních lahviček, předplněných injekčních stříkaček a bezpečnostních jehel.
Na trhu nemusí být všechny velikosti balení.
6.6. Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
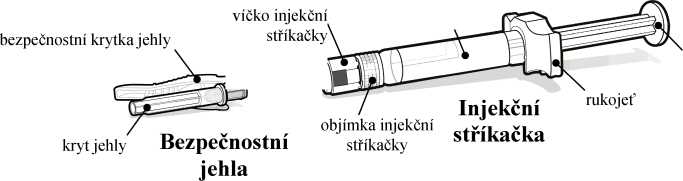
Injekční stříkačka a bezpečnostní jehla, které se používají k podání injekce, jsou součástí balení léčivého přípravku.
Před nasazením dodané bezpečnostní jehly je nutné nejprve z předplněné injekční stříkačky odstranit víčko. To se odstraní odlomením. Injekční stříkačku držte svisle, aby nedošlo k úniku tekutiny; konec injekční stříkačky se nesmí ničeho dotknout.
Prášek se má rekonstituovat v 1 ml rozpouštědla. Při přidávání rozpouštědla z injekční stříkačky se mají injekční lahvička a injekční stříkačka držet zešikma tak, jak je znázorněno na obrázku níže.
Do injekční lahvičky s práškem přidejte rozpouštědlo. Rozpouštědlo se má do injekční lahvičky vytlačit pomalu, aby se nemohla vytvořit pěna. Pak by byl přípravek nepoužitelný. Prášek pozvolna rozpouštějte pomalými krouživými pohyby. Netřepejte zprudka, protože to může způsobit denaturaci léčivé látky.
Po rekonstituci je nutné rekonstituovaný roztok před podáním pohledem zkontrolovat, zda neobsahuje nežádoucí (nebo jakékoli cizorodé) částice nebo zda se nezměnil jeho vzhled. V případě, že něco takového zpozorujete, léčivý přípravek zlikvidujte.
Injekční stříkačku ponechejte zasunutou do injekční lahvičky a před natažením roztoku přípravku Somavert ji obraťte tak, aby byl vidět otvor v zátce, jak je znázorněno na obrázku níže:
Stáhněte jehlu dolů, aby byl její hrot co nejníže v tekutině. Pomalým tažením pístu odeberte z injekční lahvičky přípravek do injekční stříkačky. Jsou-li v injekční stříkačce vidět vzduchové bubliny, poklepejte na tělo stříkačky, aby vypluly na hladinu, a poté je opatrně vytlačte do injekční lahvičky.
Než injekční stříkačku a jehlu vyhodíte, přiklopte jehlu bezpečnostní krytkou a ujistěte se, že se zacvakla na místo. Injekční stříkačku a jehlu nikdy nepoužívejte opakovaně.
Pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/02/240/001 10 mg 30 injekčních lahviček EU/1/02/240/002 15 mg 30 injekčních lahviček EU/1/02/240/003 20 mg 30 injekčních lahviček EU/1/02/240/004 20 mg 1 injekční lahvička EU/1/02/240/009 25 mg 1 injekční lahvička EU/1/02/240/010 25 mg 30 injekčních lahviček EU/1/02/240/011 30 mg 1 injekční lahvička EU/1/02/240/012 30 mg 30 injekčních lahviček
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. listopadu 2002
Datum posledního prodloužení registrace: 20. září 2007
10. DATUM REVIZE TEXTU
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY /BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY /BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa vvrobce/vvrobců biologické léčivé látky/biologických léčivých látek
Pfizer Health AB Mariefredsvagen 37 645 41 Strángnás Švédsko
Pfizer Ireland Pharmaceuticals
Grange Castle Business Park
Clondalkin
Dublin 22
Irsko
Název a adresa výrobce odpovědného/vvrobců odpovědných za propouštění šarží
Pfizer Manufacturing Belgium NV Rijksweg 12 2870 Puurs Belgie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
SOMAVERT 10 mg prášek a rozpouštědlo pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 10 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 10 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný
Monohydrát dihydrogenfosforečnanu sodného
Voda na injekci
Prášek a rozpouštědlo pro injekční roztok
30 injekčních lahviček s práškem
30 předplněných injekčních stříkaček s rozpouštědlem
30 injekčních jehel
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/001
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 10 mg
SOMAVERT 10 mg prášek pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 10 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 10 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný bezvodý Monohydrát dihydrogenfosforečnanu sodného
Prášek pro injekční roztok 10 injekčních lahviček s práškem
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Použitelné do:
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/001
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 10 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU INJEKČNÍ LAHVIČKA S PRÁŠKEM
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
SOMAVERT 10 mg prášek na injekci pegvisomantum s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
10 mg
6. JINÉ
SOMAVERT 15 mg prášek a rozpouštědlo pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 15 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 15 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný
Monohydrát dihydrogenfosforečnanu sodného
Voda na injekci
Prášek a rozpouštědlo pro injekční roztok
30 injekčních lahviček s práškem
30 předplněných injekčních stříkaček s rozpouštědlem
30 injekčních jehel
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/002
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 15 mg
SOMAVERT 15 mg prášek pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 15 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 15 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný bezvodý Monohydrát dihydrogenfosforečnanu sodného
Prášek pro injekční roztok 10 injekčních lahviček s práškem
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Použitelné do:
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/002
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 15 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU INJEKČNÍ LAHVIČKA S PRÁŠKEM
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
SOMAVERT 15 mg prášek na injekci pegvisomantum. s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
15 mg
6. JINÉ
SOMAVERT 20 mg prášek a rozpouštědlo pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 20 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 20 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný
Monohydrát dihydrogenfosforečnanu sodného
Voda na injekci
Prášek a rozpouštědlo pro injekční roztok
30 injekčních lahviček s práškem
30 předplněných injekčních stříkaček s rozpouštědlem
30 injekčních jehel
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/003
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 20 mg
SOMAVERT 20 mg prášek pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 20 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 20 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný bezvodý Monohydrát dihydrogenfosforečnanu sodného
Prášek pro injekční roztok 10 injekčních lahviček s práškem
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Použitelné do:
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/003
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 20 mg
SOMAVERT 20 mg prášek a rozpouštědlo pro injekční roztok pegvisomant
Jedna injekční lahvička obsahuje pegvisomantum 20 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 20 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný
Monohydrát dihydrogenfosforečnanu sodného
Voda na injekci
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s práškem 1 předplněná injekční stříkačka s rozpouštědlem 1 injekční jehla
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/004
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 20 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU INJEKČNÍ LAHVIČKA S PRÁŠKEM
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
SOMAVERT 20 mg prášek na injekci pegvisomantum s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
20 mg
6. JINÉ
SOMAVERT 25 mg prášek a rozpouštědlo pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 25 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 25 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný
Monohydrát dihydrogenfosforečnanu sodného
Voda na injekci
Prášek a rozpouštědlo pro injekční roztok
30 injekčních lahviček s práškem
30 předplněných injekčních stříkaček s rozpouštědlem
30 injekčních jehel
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/010
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 25 mg
SOMAVERT 25 mg prášek pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 25 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 25 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný bezvodý Monohydrát dihydrogenfosforečnanu sodného
Prášek pro injekční roztok 10 injekčních lahviček s práškem
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Použitelné do:
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/010
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 25 mg
SOMAVERT 25 mg prášek a rozpouštědlo pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 25 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 25 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný
Monohydrát dihydrogenfosforečnanu sodného
Voda na injekci
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s práškem 1 předplněná injekční stříkačka s rozpouštědlem 1 injekční jehla
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/009
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 25 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU INJEKČNÍ LAHVIČKA S PRÁŠKEM
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
SOMAVERT 25 mg prášek na injekci pegvisomantum s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
25 mg
6. JINÉ
SOMAVERT 30 mg prášek a rozpouštědlo pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 30 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 30 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný
Monohydrát dihydrogenfosforečnanu sodného
Voda na injekci
Prášek a rozpouštědlo pro injekční roztok
30 injekčních lahviček s práškem
30 předplněných injekčních stříkaček s rozpouštědlem
30 injekčních jehel
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/012
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 30 mg
SOMAVERT 30 mg prášek pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 30 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 30 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný bezvodý Monohydrát dihydrogenfosforečnanu sodného
Prášek pro injekční roztok 10 injekčních lahviček s práškem
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Použitelné do:
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/012
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 30 mg
SOMAVERT 30 mg prášek a rozpouštědlo pro injekční roztok pegvisomantum
Jedna injekční lahvička obsahuje pegvisomantum 30 mg.
Po rekonstituci 1 ml roztoku obsahuje pegvisomantum 30 mg.
Glycin
Mannitol (E421)
Hydrogenfosforečnan sodný
Monohydrát dihydrogenfosforečnanu sodného
Voda na injekci
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s práškem 1 předplněná injekční stříkačka s rozpouštědlem 1 injekční jehla
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
Uchovávejte mimo dohled a dosah dětí.
Použijte ihned po rekonstituci. Pouze k jednorázovému použití.
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/240/011
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
SOMAVERT 30 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
SOMAVERT 30 mg prášek na injekci pegvisomantum s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
30 mg
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Rozpouštědlo pro SOMAVERT Subkutánní podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
Voda na injekci 8 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
SOMAVERT 10 mg prášek a rozpouštědlo pro injekční roztok SOMAVERT 15 mg prášek a rozpouštědlo pro injekční roztok SOMAVERT 20 mg prášek a rozpouštědlo pro injekční roztok SOMAVERT 25 mg prášek a rozpouštědlo pro injekční roztok SOMAVERT 30 mg prášek a rozpouštědlo pro injekční roztok
Pegvisomantum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek SOMAVERT a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek SOMAVERT používat
3. Jak se přípravek SOMAVERT používá
4. Možné nežádoucí účinky
5. Jak přípravek SOMAVERT uchovávat
6. Obsah balení a další informace
1. Co je přípravek SOMAVERT a k čemu se používá
Přípravek SOMAVERT se používá k léčbě akromegalie, což je hormonální porucha, která je důsledkem zvýšené sekrece růstového hormonu (GH) a IGF-I (inzulínu podobný růstový faktor I) a je charakterizována nadměrným růstem kostí, měkkých tkání, otoky, onemocněním srdce a příbuznými chorobami.
Léčivou látkou přípravku SOMAVERT je pegvisomant, což je antagonista receptoru růstového hormonu. Tyto látky snižují účinek GH a hladinu inzulínu podobných růstových faktorů I v krvi.
2. Čemu musíte věnovat pozornost, než začnete přípravek SOMAVERT používat
Nepoužívejte přípravek SOMAVERT:
• jestliže jste alergický(á) na pegvisomant nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku SOMAVERT se poraďte se svým lékařem, lékárníkem nebo zdravotní
sestrou.
- pokud budete při používání přípravku SOMAVERT pozorovat poruchy zraku nebo bolesti hlavy, obraťte se ihned na svého lékaře
- Váš lékař nebo sestra budou monitorovat hladiny IGF-I (inzulínu podobný růstový faktor I) obíhajících v krvi a podle potřeby přizpůsobovat dávku přípravku SOMAVERT
- Váš lékař bude sledovat Váš adenom (nezhoubný nádor)
- Váš lékař nebo sestra budou monitorovat hladiny jatemích enzymů v krvi každých 4-6 týdnů během prvních 6 měsíců léčby přípravkem SOMAVERT. Podání přípravku SOMAVERT musí být při prvních známkách onemocnění jater ukončeno
- pokud máte cukrovku, Váš lékař Vám může upravit dávku inzulínu nebo jiných léků, které užíváte
- pacientky musí užívat vhodnou kontracepci, protože se může zvýšit plodnost. Viz též níže - bod Těhotenství.
Další léčivé přípravky a přípravek SOMAVERT
Oznamte svému lékaři, pokud jste v minulosti užíval(a) nějaké jiné léky k léčbě akromegalie nebo cukrovky.
Informujte svého lékaře nebo lékárníka o všech lécích, které používáte, nebo které jste v nedávné době používal(a).
Součástí Vaší léčby může být i užívání dalších léků. Je důležité, abyste užíval(a) všechny léky, včetně přípravku SOMAVERT, pokud Vám nedoporučí Váš lékař, lékárník nebo zdravotní sestra jinak.
Těhotenství, kojení a plodnost
Účinky přípravku SOMAVERT u těhotných žen nejsou známy, proto se užívání tohoto přípravku u těhotných žen nedoporučuje. Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat
Není známo, zda se pegvisomant vylučuje do lidského mateřského mléka. Během léčby přípravkem SOMAVERT nesmíte kojit, pokud s Vámi Váš lékař tuto otázku neprojedná.
Řízení dopravních prostředků a obsluha strojů
Nebyly provedeny žádné studie týkající se účinků na schopnost řídit motorová vozidla nebo obsluhovat stroje.
Přípravek SOMAVERT obsahuje sodík
Tento přípravek obsahuje méně než 1 mg sodíku v jedné dávce, tj. v podstatě „bez sodíku”.
3. Jak se přípravek SOMAVERT používá
Vždy aplikujte injekci tohoto přípravku přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Zahajovací dávku 80 mg pegvisomantu Vám podá podkožně Váš lékař. Poté je obvyklá denní dávka pegvisomantu 10 mg, která se podává podkožní injekcí.
Každých 4 až 6 týdnů provede Váš lékař příslušnou úpravu dávky, která se provádí postupným zvyšováním dávky pegvisomantu o 5 mg denně na základě koncentrací tzv. IGF-I tak, aby se udržela optimální léčebná odpověď.
Způsob a cesta podání léku
Přípravek SOMAVERT se podává podkožně. Injekci si můžete aplikovat sám(sama), případně Vám ji může aplikovat jiná osoba, např. Váš lékař nebo zdravotní sestra. Je zapotřebí, abyste dodržoval(a) podrobné pokyny ohledně postupu při podávání injekce, které jsou uvedeny na konci této příbalové informace. V podávání injekcí tohoto přípravku pokračujte po celou dobu, kterou Vám určil Váš lékař.
Před použitím musí být tento přípravek rozpuštěn. Injekce se nesmí míchat ve stejné stříkačce nebo lahvičce jako jiné léky.
V místě vpichu injekce může dojít ke zmnožení podkožní tukové tkáně (nahromadění podkožního
tuku). Abyste tomu zabránil(a), použijte při každém podání odlišné místo vpichu, jak je uvedeno v Kroku 2 „Návod na přípravu a podání injekce přípravku SOMAVERT“ na konci této příbalové informace. Tím umožníte kůži a podkoží, aby se po podání injekce regenerovaly dříve, než bude do stejného místa podána další injekce.
Pokud máte dojem, že účinek tohoto přípravku je příliš silný nebo slabý, oznamte to svému lékaři, lékárníkovi nebo zdravotní sestře.
Jestliže jste použila(a) více přípravku SOMAVERT, než jste měl(a)
Pokud jste si náhodně aplikoval(a) větší množství přípravku SOMAVERT než Vám naordinoval Váš lékař, není pravděpodobné, že by se jednalo o závažnou příhodu, měl(a) byste však ihned kontaktovat svého lékaře, lékárníka nebo zdravotní sestru.
Jestliže jste zapomněl(a) použít přípravek SOMAVERT
Pokud jste si zapomněl(a) aplikovat injekci přípravku SOMAVERT, měl(a) byste si aplikovat další dávku co nejdříve, jakmile si vzpomenete a dále pokračovat v injekcích přípravku SOMAVERT podle doporučení Vašeho lékaře. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
U některých pacientů léčených přípravkem SOMAVERT byly hlášeny mírné až vážné alergické (anafylaktické) reakce. Příznaky závažné alergické reakce mohou zahrnovat 1 nebo více z následujících: otok obličeje, jazyka, rtů nebo hrdla; sípot nebo obtížné dýchání (stažení hrtanu); celková kožní vyrážka, kopřivka nebo svědění; závrať. Při výskytu některého z těchto příznaků kontaktujte ihned svého lékaře.
Velmi časté: mohou postihnout více než 1 pacienta z 10:
- Bolesti hlavy
- Průjem
- Bolest kloubů
Časté: mohou postihnout až 1 pacienta z 10:
- Pocit krátkého dechu
- Zvýšení hladin látek, podle kterých se měří funkce jater. Lze je nalézt ve výsledcích krevních testů
- Krev v moči
- Zvýšení krevního tlaku
- Zácpa, pocit na zvracení nebo zvracení, pocit nadmutí, špatné trávení, plynatost
- Závratě, ospalost, nekontrolovaný třes, snížená citlivost
- Tvorba modřin nebo krvácení v místě vpichu injekce, bolestivost nebo otok v místě vpichu injekce, nahromadění tuku v podkoží v místě vpichu injekce, otok končetin, slabost, horečka
- Pocení, svědění, vyrážka, sklon k tvorbě modřin
- Bolest svalů, zánět kloubu
- Zvýšení hladiny cholesterolu v krvi, přírůstek tělesné hmotnosti, zvýšení hladiny krevní glukózy, snížení hladiny glukózy v krvi
- Příznaky podobné chřipce, únava
- Nepřirozené sny
Bolest očí
Méně časté: mohou postihnout až 1 pacienta ze 100:
- Alergické reakce po podání (horečka, vyrážka, pruritus a ve velmi závažných případech obtíže s dýcháním, rychlý otok kůže vyžadující neodkladné lékařské ošetření). Mohou se objevit okamžitě nebo několik dnů po podání.
- Bílkovina v moči, zvýšené množství moči, onemocnění ledvin
- Nedostatek zájmu, pocit zmatenosti, zvýšení sexuální touhy, panika, ztráta paměti, problémy se spánkem
- Snížený počet krevních destiček, zvýšený nebo snížený počet bílých krvinek v krvi, sklon ke krvácení
- Abnormální pocity, špatné hojení
- Únava očí, potíže vnitřního ucha
- Otok obličeje, suchá kůže, noční pocení, zčervenání kůže (erytém), vystouplé, svědivé hrbolky na kůži (kopřivka)
- Zvýšená hladina tuků v krvi, zvýšená chuť k jídlu
- Sucho v ústech, zvýšené slinění, problémy se zuby, hemoroidy
- Abnormální vnímání chutí, migréna
Není známo: z dostupných údajů nelze určit četnost výskytu
- Zlost
- Závažná dušnost (laryngospasmus)
- Rychlý otok kůže, podkožní tkáně a sliznic (mukózy) orgánů (angioedém)
U přibližně 17% pacientů se během léčby vytvořily protilátky na růstový hormon. Tyto protilátky zřejmě nebrání působení tohoto přípravku.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek SOMAVERT uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na injekční lahvičce a krabičce za „Použitelné do:“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem. Uchovávejte injekční lahvičku (lahvičky) a předplněnou injekční stříkačku (stříkačky) v krabičce, aby byl přípravek chráněn před světlem.
Po naředění musí být přípravek SOMAVERT použit okamžitě.
Nepoužívejte tento přípravek, pokud si všimnete, že je roztok zakalený nebo obsahuje částice.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek SOMAVERT obsahuje
- Léčivou látkou je pegvisomantum.
- SOMAVERT 10 mg: Jedna injekční lahvička s práškem obsahuje pegvisomantum 10 mg. Po rozpuštění v 1 ml rozpouštědla 1 ml roztoku obsahuje pegvisomantum 10 mg.
- SOMAVERT 15 mg: Jedna injekční lahvička s práškem obsahuje pegvisomantum 15 mg. Po rozpuštění v 1 ml rozpouštědla 1 ml roztoku obsahuje pegvisomantum 15 mg.
- SOMAVERT 20 mg: Jedna injekční lahvička s práškem obsahuje pegvisomantum 20 mg. Po rozpuštění v 1 ml rozpouštědla 1 ml roztoku obsahuje pegvisomantum 20 mg.
- SOMAVERT 25 mg: Jedna injekční lahvička s práškem obsahuje pegvisomantum 25 mg. Po rozpuštění v 1 ml rozpouštědla 1 ml roztoku obsahuje pegvisomantum 25 mg.
- SOMAVERT 30 mg: Jedna injekční lahvička s práškem obsahuje pegvisomantum 30 mg. Po rozpuštění v 1 ml rozpouštědla 1 ml roztoku obsahuje pegvisomantum 30 mg.
- Dalšími složkami jsou glycin, mannitol (E421), hydrogenfosforečnan sodný a monohydrát dihydrogenfosforečnanu sodného.
- Rozpouštědlem je voda na inj ekci.
Jak přípravek SOMAVERT vypadá a co obsahuje toto balení
Přípravek SOMAVERT je ve formě prášku a rozpouštědla na injekci (10 mg, 15 mg, 20 mg, 25 mg nebo 30 mg pegvisomantu v injekční lahvičce a 1 ml rozpouštědla v předplněné injekční stříkačce) Velikost balení je 1 a/nebo 30. Na trhu nemusí být všechny velikosti balení. Prášek je bílý a rozpouštědlo je čiré a bezbarvé.
Držitel rozhodnutí o registraci a výrobce:
Držitel rozhodnutí o registraci
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
Výrobce
Pfizer Manufacturing Belgium NV Rijksweg 12 2870 Puurs Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje
Tel. +3705 2514000
Eunrapnu Luxembourg/Luxemburg
n$aň3ep HrnKceMÓypr CAPH, KnoH Etnrapnn Pfizer S.A.
Ten.: +359 2 970 4333 Tél/Tel: +32 (0)2 554 62 11
|
Česká republika Pfizer PFE spol. s r.o. Tel: +420 283 004 111 |
Magyarország Pfizer Kft. Tel.: + 36 1 488 37 00 |
|
Danmark Pfizer ApS Tlf: +45 44 20 11 00 |
Malta V.J. Salomone Pharma Ltd. Tel: + 356 21 22 01 74 |
|
Deutschland Pfizer Pharma GmbH Tel: +49 (0)30 550055 51000 |
Nederland Pfizer bv Tel: +31 (0)10 406 43 01 |
|
Eesti Pfizer Luxembourg SARL Eesti filiaal Tel: +372 666 7500 |
Norge Pfizer Norge AS Tlf: +47 67 52 61 00 |
|
EXXáSa PFIZER EAAAI A.E. Tn^: +30 210 6785800 |
Osterreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0 |
|
Espaňa Pfizer S.L. Tel: +34 91 490 99 00 |
Polska Pfizer Polska Sp. z o.o. Tel.: +48 22 335 61 00 |
|
France Pfizer Tél: +33 (0)1 58 07 34 40 |
Portugal Pfizer Biofarmaceutica, Sociedade Unipessoal Lda Tel: +351 21 423 5500 |
|
Hrvatska Pfizer Croatia d.o.o. Tel: + 385 1 3908 777 |
Románia Pfizer Románia S.R.L. Tel: +40 (0)21 207 28 00 |
|
Ireland Pfizer Healthcare Ireland Tel: 1800 633 363 (toll free) +44 (0)1304 616161 |
Slovenija Pfizer Luxembourg SARL, Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana Tel: + 386 (0)1 52 11 400 |
|
Ísland Icepharma hf. Sími: + 354 540 8000 |
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel: +421 2 3355 5500 |
|
Italia Pfizer S.r.l. Tel: +39 06 33 18 21 |
Suomi/Finland Pfizer Oy Puh/Tel: +358 (0)9 43 00 40 |
|
Kúnpoq PFIZER EAAAE A.E. (Cyprus Branch) Tn^: +357 22 817690 |
Sverige Pfizer Innovations AB Tel: +46 (0)8 550 520 00 |
|
Latvija Pfizer Luxembourg SARL filiale Latvija Tel: +371 670 35 775 |
United Kingdom Pfizer Limited Tel: +44 (0)1304 616161 |
Tato příbalová informace byla naposledy revidována {MM.RRRR}
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
NÁVOD K POUŽITÍ
Prášek přípravku Somavert v injekční lahvičce s rozpouštědlem v předplněné injekční stříkačce
Pegvisomant k injekčnímu podání Pouze pro subkutánní injekci Jednodávková injekční lahvička
Přípravek Somavert je balený v injekční lahvičce ve formě bílého hrubého prášku. Přípravek Somavert můžete použít až poté, co jej smísíte s tekutinou (rozpouštědlem).
Tekutina je balená v předplněných injekčních stříkačkách s označením „1 ml sterilního rozpouštědla k jednorázovému použití“.
Přípravek Somavert se nesmí mísit s žádnou jinou tekutinou.
Je důležité, abyste nezkoušel(a) podat injekci sobě ani nikomu jinému, dokud Vás to nenaučí Váš lékař nebo zdravotní sestra.
Celé balení uchovávejte v chladničce při teplotě 2 °C až 8 °C. Chraňte před přímým slunečním světlem. Uchovávejte mimo dosah dětí.
1. Co potřebujete
Jedno balení přípravku Somavert obsahuje:
• injekční lahvičku s práškem přípravku Somavert,
• předplněnou injekční stříkačku s rozpouštědlem,
• bezpečnostní jehlu.
Dále budete potřebovat:
• vatový tampon,
• tampon s alkoholem,
• nádobu vhodnou na ostré předměty.
Injekční
lahvička
víčko injekční lahvičky

datum v použitelnosti
zátka injekční lahvičky (po odstranění víčka)
otvor v zátce
tělo
píst
Než začnete:
• Prášek přípravku Somavert smíchejte s rozpouštědlem až tehdy, když budete připraven(a) si injekční dávku podat.
• Vyndejte jedno balení přípravku Somavert z chladničky a na bezpečném místě nechte ohřát na pokojovou teplotu.
• Mýdlem a vodou si umyjte ruce a dobře je osušte.
• Sloupněte fólii z obalu injekční stříkačky a bezpečnostní jehly, abyste si je mohl(a) snadno vzít, až si budete připravovat injekci.
• Injekční stříkačka a injekční lahvička se nesmí používat, jestliže:
o jsou poškozené nebo vadné, o vypršela doba jejich použitelnosti,
o injekční stříkačka zmrzla, a to i v případě, že následně roztála (pouze injekční stříkačka).
3. Vyberte oblast pro aplikaci injekce
Vyberte oblast pro aplikaci injekce
Paže nebo dolní část zad:
Zadní část paží (pouze lékař nebo zdravotní sestra)
Břicho:
Dodržujte vzdálenost alespoň 5 cm od pupíku.
Stehna
• V jednotlivých oblastech podávejte injekce vždy do jiného místa.
• Vyhýbejte se oblastem, kde jsou kosti, nebo oblastem s podlitinami, zarudnutím, boláky či ztvrdlinami nebo oblastem s jizvami či kožními útvary.
• Oblast injekce očistěte tamponem s alkoholem, jak Vás poučil lékař.
• Oblast injekce nechejte oschnout.
(S^TSejmětevíčko^^^^^^
• Sejměte víčko z injekční lahvičky.
• Víčko vyhoďte; už jej nebudete potřebovat.
Upozornění: Zátky injekční lahvičky se nesmí nic dotknout.
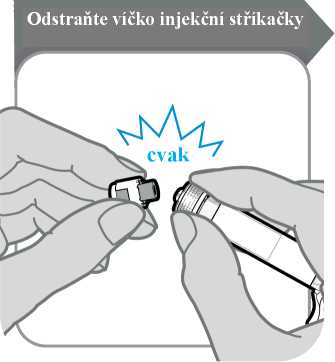
5. Odstraňte víčko injekční stříkačky
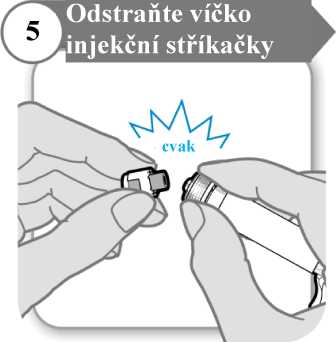
• Odlomte víčko injekční stříkačky. Odlomení může vyžadovat větší sílu, než možná čekáte.
• Víčko injekční stříkačky vyhoďte; už jej nebudete potřebovat.
• Injekční stříkačku držte svisle, aby nedošlo k úniku tekutiny.
Upozornění: Po odlomení víčka se nesmíte koncem injekční stříkačky ničeho dotknout.
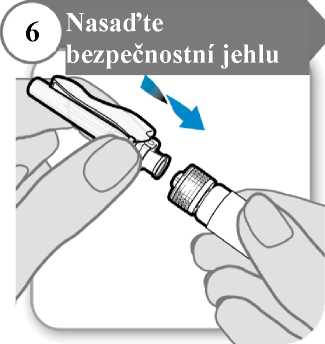
Šroubovitým pohybem nasaďte bezpečnostní jehlu co nejhlouběji na injekční stříkačku.
7. Odstraňte kryt jehly
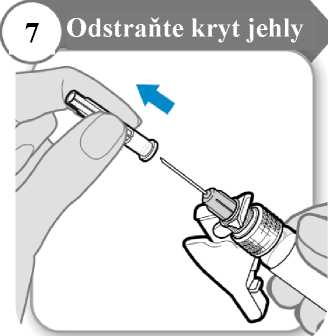
• Odklopte bezpečnostní krytku jehly, pod níž se nachází kryt jehly.
• Opatrně stáhněte kryt jehly.
• Kryt jehly vyhoďte; už jej nebudete potřebovat.
Upozornění: Jehlou se nesmíte ničeho dotknout.
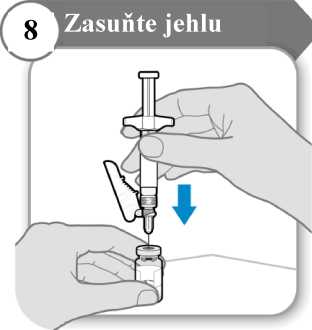
• Jehlu protlačte středem zátky injekční lahvičky, jak je znázorněno na obrázku.
• Přidržujte injekční stříkačku, aby se jehla neohnula, když je zasunutá v zátce injekční lahvičky.
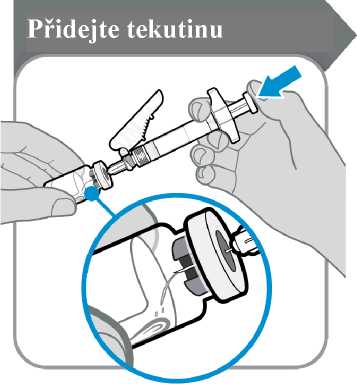
9. Přidejte tekutinu
Přidejte tekutinu
• Injekční lahvičku s injekční stříkačkou nakloňte, jak je znázorněno na obrázku.
• Píst stříkačky pomalu tlačte dolů, dokud nevytlačíte veškerou tekutinu do injekční lahvičky.
• Upozornění: Tekutinu nevstřikujte přímo na prášek, protože by došlo k tvorbě pěny. Zpěněný přípravek nelze použít.
• Jehlu zatím nevytahujte.
Kružte injekční lahvičkou
• Injekční stříkačku i injekční lahvičku přidržujte jednou rukou, jak je znázorněno na obrázku.
• Tekutinu jemně míchejte krouživým pohybem injekční lahvičkou po rovném povrchu.
• V kroužení pokračujte do té doby, než se v tekutině zcela rozpustí všechen prášek. Poznámka: To může trvat až 5 minut.
11. Zkontrolujte přípravek
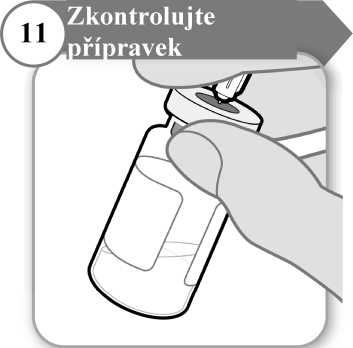
• Jehlu ponechejte zasunutou do injekční lahvičky a pečlivě si prohlédněte přípravek. Musí být čirý a bez pevných částic.
• Přípravek nepoužívejte, jestliže:
o je přípravek zkalený nebo zamlžený;
o je přípravek jakkoli zabarvený;
o jsou v injekční lahvičce jakékoli pevné částice nebo vrstva pěny.
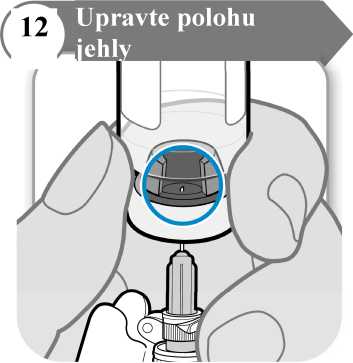
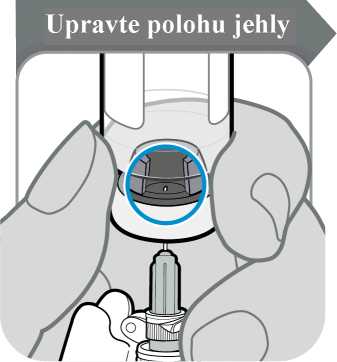
• Injekční lahvičku obraťte tak, abyste viděl(a) otvor v zátce, jak je znázorněno na obrázku.
• Stáhněte jehlu dolů, aby byl její hrot co nejníže v tekutině. Tak se vám podaří odebrat co nejvíce tekutiny.
• Zkontrolujte, jestli se píst stříkačky nepohnul. Pokud ano, zatlačte celý píst zpět do injekční stříkačky. Tím z injekční stříkačky odstraníte veškerý vzduch předtím, než do ní odeberete dávku.
13. Odeberte dávku
Odeberte dávku
• Pomalu vytahujte píst ze stříkačky a odeberte co nejvíce přípravku z injekční lahvičky. Poznámka: Jsou-li v injekční stříkačce vidět vzduchové bubliny, poklepejte na tělo stříkačky, aby vypluly nahoru, a poté je opatrně vytlačte do injekční lahvičky.
• Vytáhněte jehlu z injekční lahvičky.
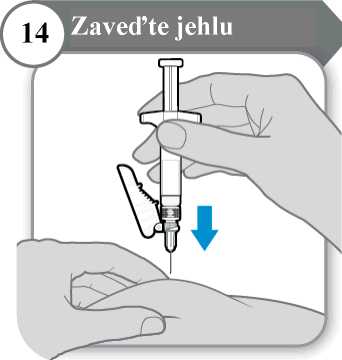
• Jemně uchopte kůži v místě vpichu injekce.
• Zaveďte celou jehlu do kožní řasy.
15. Vpíchněte přípravek
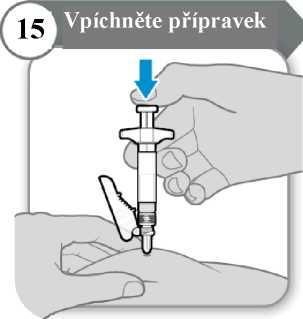
• Píst pomalu tlačte dolů, dokud nebude tělo stříkačky prázdné. Poznámka: Pod kůží musí být zavedena celá délka jehly.
• Uvolněte kožní řasu a jehlu zpříma vytáhněte.
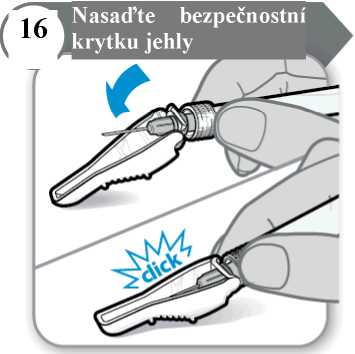
• Jehlu přiklopte bezpečnostní krytkou.
• Jemným tlakem na pevný povrch zacvakněte bezpečnostní krytku na místo. Poznámka: Při zajištění bezpečnostní krytky uslyšíte cvaknutí.
17. Zlikvidujte
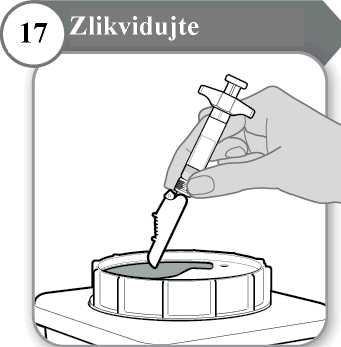
• Injekční stříkačku a jehlu NIKDY nepoužívejte opakovaně. Jehlu a injekční stříkačku
zlikvidujte podle pokynů svého lékaře, zdravotní sestry nebo lékárníka a v souladu s místními hygienickými a bezpečnostními předpisy.
• V případě potřeby použijte čistý vatový tampon a jemně jej přitlačte na oblast injekce.
• Ošetřovanou oblast netřete.
OTÁZKY A ODPOVĚDI
Co mám dělat, když se nedopatřením něčím dotknu zátky injekční lahvičky?
• Očistěte zátku injekční lahvičky čistým tamponem s alkoholem a nechte dobře oschnout. Jestliže se Vám nedaří zátku očistit, injekční lahvičku nepoužívejte.
Co mám dělat s injekční stříkačkou, když mi spadla?
• Nepoužívejte ji, a to ani v případě, že vypadá nepoškozeně. Zlikvidujte ji stejným způsobem jako použitou injekční stříkačku. Budete potřebovat novou injekční stříkačku.
Kolikrát mohu bezpečně zasunout jehlu do zátky injekční lahvičky?
• Pouze jednou. Vytahováním a opakovaným zasunováním jehly se značně zvýší riziko
jejího poškození a jehla se ztupí. Podání přípravku pak může být nepříjemné a zvyšuje se riziko poškození kůže a infekce. Rovněž existuje riziko, že přijdete o část přípravku.
Mohu s injekční lahvičkou zatřepat, pokud se prášek nerozpouští?
• Ne, injekční lahvičkou nikdy netřepejte. Třepáním můžete přípravek znehodnotit a vytvořit pěnu. Úplné rozpuštění prášku může trvat několik minut, pokračujte tedy v kroužení injekční lahvičkou, dokud nezískáte naprosto čirou tekutinu.
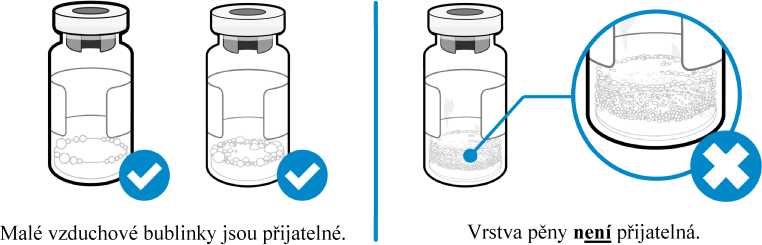
Jak poznám, zda je v injekční lahvičce nějaká pěna?
• Pěna vypadá jako spousta malých bublinek, které vytvářejí plovoucí vrstvu na hladině tekutiny. Jestliže je přípravek Somavert zpěněný, injekci nepodávejte.
Jak mohu zabránit zpěnění přípravku?
• Píst tlačte velmi pomalu tak, aby tekutina v injekční lahvičce pozvolna stékala dolů. Nestříkejte tekutinu přímo na prášek, protože tím by se vytvořila pěna. Tento postup také zkracuje dobu kroužení a umožňuje odebrat více přípravku.
V injekční stříkačce vidím nějaký vzduch. Je to v pořádku?
• Malé vzduchové bublinky v tekutině jsou normální a injekci přípravku lze bezpečně použít. Je však možné, že nedopatřením natáhnete do injekční stříkačky vzduch, který je třeba před podáním injekce odstranit. Vzduchové bubliny, které vyplavou na hladinu kapaliny, je třeba vytlačit zpět do injekční lahvičky.
Proč se mi nedaří odebrat z injekční lahvičky veškerý přípravek?
• Tvar injekční lahvičky způsobuje, že velmi malý objem přípravku v ní zůstane. To je normální. Aby v ní zůstal jen minimální objem přípravku, musí být při odebírání dávky hrot jehly zasunutý do injekční lahvičky co nejníže.
Co mám dělat v případě jakýchkoli pochybností ohledně mého přípravku?
• Na všechny otázky by měl odpovědět lékař, zdravotní sestra nebo lékárník, který(á) je seznámen(a) s přípravkem SOMAVERT.
67