Sebivo 20 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Sebivo 600 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje telbivudinum 600 mg. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta
Bílá až slabě nažloutlá, oválná potahovaná tableta, na jedné straně s potiskem “LDT”.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Sebivo je indikován k léčbě chronické hepatitidy B u dospělých pacientů s kompenzovaným onemocněním jater a prokázanou replikací viru, s trvale zvýšenými hladinami alaninaminotransferázy (ALT) v séru a histologicky prokázaným aktivním zánětem a/nebo fibrózou.
Zahájení léčby Sebivem má být zvažováno pouze v případě, kdy alternativní antivirová látka s vyšší genetickou bariérou proti vzniku rezistence není k dispozici nebo její použití není vhodné.
Pro detaily studie a specifické charakteristiky pacientů, na kterých je tato indikace založena, viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba musí být zahájena lékařem, který má zkušenosti s léčbou chronické infekční hepatitidy typu B.
Dávkování
Dospělí
Doporučená dávka přípravku Sebivo je 600 mg (jedna tableta) podávaná jednou denně.
U pacientů, kteří mají potíže s polykáním tablet, může být zvažováno podávání přípravku Sebivo perorální roztok.
Sledování během léčby
Odpověď na léčbu ve 24. týdnu se ukázala jako prediktivní pro dlouhodobou odpověď na léčbu (viz tabulka 7 v bodě 5.1). HBV DNA hladiny je třeba monitorovat ve 24. týdnu léčby, aby se zajistila kompletní virová suprese (HBV DNA méně než 300 kopií/ml). U pacientů s detekovatelnou hladinou HBV DNA po 24 týdnech léčby je nutné zvážit úpravu léčby.
HBV DNA je třeba monitorovat každých 6 měsíců, aby se zajistila kontinuální odpověď. Pokud je HBV DNA test pozitivní kdykoliv po první odpovědi, je nutné zvážit úpravu léčby. Optimální léčba musí být provázena testy na rezistenci.
Délka léčby
Optimální délka léčby není známa. O ukončení léčby se rozhoduje následujícím způsobem.
• U HBeAg-pozitivních pacientů bez cirhózy by měla léčba trvat po dobu nejméně 6-12 měsíců po potvrzení sérokonverze HBeAg (vymizení HBeAg a HBV DNA s průkazem anti-HBe) nebo do sérokonverze HBsAg nebo do doby, kdy se prokáže ztráta účinnosti. Sérová hladina ALT a HBV DNA by měla být po ukončení léčby pravidelně sledována z důvodu detekce pozdějšího virologického relapsu.
• U HBeAg-negativních pacientů bez cirhózy by měla léčba pokračovat nejméně do sérokonverze HBsAg, nebo dokud se neprokáže ztráta účinnosti. Při dlouhodobé léčbě, delší než 2 roky, je doporučeno pravidelné přehodnocování léčby, aby bylo zajištěno, že pokračování ve zvolené léčbě je pro pacienta vhodné.
Starší pacienti (více než 65 let)
Údaje pro stanovení specifického dávkovacího režimu u pacientů starších než 65 let nejsou dostupné (viz bod 4.4).
Porucha funkce ledvin
U pacientů s clearance kreatininu >50 ml/min není nutná úprava doporučené dávky telbivudinu. Úprava dávkování je nutná u pacientů s clearance kreatininu <50 ml/min, včetně pacientů v terminálním stadiu onemocnění ledvin (ESRD) na hemodialýze. K podávání snížené denní dávky se doporučuje použít perorální roztok, jak je popsáno níže v tabulce 1. Jestliže není možné použít perorální roztok, mohou se alternativně použít potahované tablety Sebivo a dávkování musí být upraveno tak, že se prodlouží interval mezi dávkami, jak je popsáno v tabulce 1.
Tabulka 1 Úprava režimu dávkování přípravku Sebivo u pacientů se zhoršenou funkcí ledvin
|
Clearance kreatininu (ml/min) |
Telbivudin 20 mg/ml perorální roztok Úprava denní dávky |
Telbivudin 600 mg potahované tablety Alternativní** úprava dávky s prodloužením intervalu mezi dávkami |
|
>50 |
600 mg (30 ml) jednou denně |
600 mg jednou denně |
|
30 - 49 |
400 mg (20 ml) jednou denně |
600 mg jednou za 48 hodin |
|
<30 (nevyžadující dialýzu) |
200 mg (10 ml) jednou denně |
600 mg jednou za 72 hodin |
|
ESRD* |
120 mg (6 ml) jednou denně |
600 mg jednou za 96 hodin |
* Terminální stadium onemocnění ledvin
** V případě, že není možné použít perorální roztok
Navrhované úpravy dávky jsou založeny na extrapolaci a nemusí být optimální. Bezpečnost a účinnost uvedených pokynů pro úpravu dávkování nebyly klinicky hodnoceny. Z tohoto důvodu je u těchto pacientů doporučeno pečlivé klinické sledování.
Pacienti s terminálním stadiem onemocnění ledvin
U pacientů s ESRD by měl být přípravek Sebivo podáván až po hemodialýze (viz bod 5.2).
Porucha funkce jater
U pacientů se zhoršenou funkcí jater není nutná úprava doporučeného dávkování přípravku Sebivo (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Sebiva u pediatrické populace nebyla dosud stanovena.
Způsob podání
Sebivo se užívá perorálně s jídlem nebo bez jídla. Tableta se nežvýká, nedělí ani nedrtí.
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Kombinace telbivudinu s pegylovaným nebo standardním interferonem alfa (viz body 4.4 a 4.5).
4.4 Zvláštní upozornění a opatření pro použití
Těžké akutní exacerbace chronické hepatitidy B jsou relativně časté a jsou charakterizovány přechodným zvýšením sérové ALT. Po zahájení antivirové léčby se mohou u některých pacientů zvýšit hladiny ALT v séru, zatímco hladiny HBV DNA klesnou (viz bod 4.8). U pacientů léčených telbivudinem uplynulo v průměru 4 - 5 týdnů před výskytem exacerbace. Celkově se rychlý vzestup ALT vyskytuje častěji u pacientů s pozitivním nálezem HBeAg, než u pacientů s negativním nálezem HBeAg. U pacientů s kompenzovaným jaterním onemocněním není toto zvýšení sérové ALT obvykle doprovázeno zvýšením hladin sérového bilirubinu nebo jinými známkami jaterní dekompenzace. Riziko jaterní dekompenzace -a následné exacerbace hepatitidy - může být zvýšeno u pacientů s cirhózou. Tito pacienti musí být proto pečlivě sledováni.
Exacerbace hepatitidy byla také hlášena u pacientů, u kterých byla léčba hepatitidy B ukončena. Rychlý vzestup hodnot ALT po léčbě je normálně doprovázen zvýšením hladin HBV DNA v séru a u většiny takovýchto případů bylo prokázáno, že se spontánně upraví. Nicméně byly hlášeny těžké - a někdy fatální - exacerbace po ukončení léčby onemocnění. Z tohoto důvodu mají být jaterní funkce monitorovány v pravidelných intervalech, a to jak klinicky, tak i laboratorně, nejméně po dobu 6 měsíců po ukončení léčby hepatitidy B.
Laktátová acidóza
Při užití nukleosidových/nukleotidových analogů byl hlášen, někdy fatální, výskyt laktátové acidózy (při absenci hypoxie), obvykle doprovázený těžkou hepatomegalií se steatózou. Protože telbivudin je nukleosidový analog, toto riziko nemůže být vyloučeno. Léčba nukleosidovými analogy musí být ukončena, když rychle stoupají hladiny aminotransferáz, dochází k progresivní hepatomegalii nebo výskytu metabolické/laktátové acidózy neznámé etiologie. Benigní zažívací symptomy, jako je nauzea, zvracení a bolesti břicha, mohou indikovat vývoj mléčné acidózy. Těžké případy, někdy s fatálním koncem, byly doprovázeny pankreatitidou, jaterním selháním/steatózou jater, selháním ledvin a vyššími hladinami laktátu v séru. Při předepisování nukleosidových analogů musí být věnována zvýšená opatrnost všem pacientům (především obézním ženám) s hepatomegalií, hepatitidou nebo jinými známými rizikovými faktory onemocnění jater. Tito pacienti musí být pečlivě sledováni.
Vliv na svalstvo
Při léčbě telbivudinem byly hlášeny případy myopatií a myalgií vyskytujících se několik týdnů až měsíců po zahájení terapie (viz bod 4.8). Byly hlášeny případy rabdomyolýzy v období po uvedení telbivudinu na trh (viz bod 4.8).
O myopatii, definované jako přetrvávající nevysvětlitelná svalová bolest a/nebo svalová slabost, bez ohledu na stupeň zvýšení hladin kreatinkinázy (CK), by se mělo uvažovat u kteréhokoliv pacienta s nevysvětlenou difúzní myalgií, svalovou citlivostí na dotek, svalovou slabostí nebo myozitidou (definovanou jako myopatie s histologickým důkazem svalového poškození). Pacienti by měli být upozorněni, aby okamžitě hlásili jakoukoli nevysvětlitelnou trvalou bolest svalů, citlivost na dotek nebo svalovou slabost. Jestliže se objeví jakýkoli z těchto příznaků, mělo by být provedeno podrobné vyšetření svalů za účelem vyhodnocení svalové funkce. Jestliže je diagnostikována myopatie, měla by být léčba telbivudinem ukončena.
Není známo, zda je riziko myopatie během léčby telbivudinem zvýšeno současným podáváním jiných léčivých přípravků, které jsou spojovány se vznikem myopatie (např. statiny, fibráty nebo cyklosporin). Lékaři uvažující o současné léčbě jinými přípravky, které mohou vyvolat myopatii, by měli pečlivě zvážit možné přínosy a rizika léčby, a měli by u pacientů pečlivě sledovat jakékoli příznaky nebo projevy naznačující myopatii.
Periferní neuropatie
Periferní neuropatie byla u pacientů léčených telbivudinem hlášena méně často. V případě podezření na periferní neuropatii by měla být léčba telbivudinem znovu zvážena (viz bod 4.8).
Zvýšené riziko rozvoje periferní neuropatie bylo pozorováno v jedné studii, když byly současně podávány telbivudin a pegylovaný interferon alfa-2a (viz bod 4.5). Takové zvýšené riziko nemůže být vyloučeno v případě podávání jiného interferonu alfa (pegylovaný či standardní). Mimoto není prospěšnost podávání kombinace telbivudinu s interferonem alfa (pegylovaným či standardním) v současné době stanovena. Proto je kombinace telbivudinu s pegylovaným nebo standardním interferonem alfa kontraindikována (viz bod 4.3).
Funkce ledvin
Telbivudin je vylučován primárně ledvinnou exkrecí, proto se u pacientů s clearance kreatininu <50 ml/min, včetně pacientů na hemodialýze, doporučuje úprava intervalu dávkování. Účinnost úpravy intervalu dávkování nebyla klinicky hodnocena. Proto by měla být u pacientů s prodlouženým intervalem dávkování pečlivě sledována virologická odpověď (viz body 4.2 a 5.2).
Pacienti s cirhózou bez dekompenzace
Vzhledem k omezenému dostupnému množství údajů (pouze 3% zařazených pacientů mělo cirhózu) by telbivudin měl být u pacientů s cirhózou používán se zvláštní opatrností. U těchto pacientů je v průběhu léčby a po ukončení léčby nutno důkladně monitorovat klinické, biochemické a virologické parametry spojené s hepatitidou B.
Pacienti s cirhózou s dekompenzací
U pacientů s dekompenzovanou cirhózou nejsou dostačující údaje o bezpečnosti a účinnosti.
Pacienti, kteří již byli nukleosidovvm/nukleotidovvm analogem léčeni
In vitro nebyl telbivudin účinný proti HBV kmenům obsahujícím rtM204V/rtL180M nebo rtM204I mutace (viz bod 5.1). Monoterapie telbivudinem není alternativou pro pacienty s prokázanou virovou hepatitidou B, rezistentní vůči lamivudinu. Pacienti, u kterých nedošlo k virologické odpovědi po více než 24 týdnů trvající léčbě lamivudinem, nebudou mít pravděpodobně z monoterapie telbivudinem prospěch. V současné době nejsou k dispozici klinické údaje, na základě kterých by bylo možno zhodnotit prospěšnost a rizika přechodu na telbivudin u pacientů, u kterých bylo při podávání lamivudinu dosaženo celkové virové suprese.
Nejsou k dispozici údaje o léčbě telbivudinem pacientů s prokázanými jednotlivými mutacemi rtN236T nebo A181V viru hepatitidy B rezistentními k adefoviru. Výsledky založené na buněčných testováních ukázaly, že substituce A181V spojená s rezistencí vůči adefoviru měla 1,5 až přibližně 4krát nižší citlivost na telbivudin.
Pacienti po transplantaci jater
Bezpečnost a účinnost telbivudinu u pacientů po transplantaci jater není známa.
Starší pacienti
Klinické studie s telbivudinem nezahrnovaly dostatečný počet pacientů starších >65 roků, aby bylo možné stanovit, zda odpovídají odlišně od mladých jedinců. Obecně musí být při předepisování přípravku Sebivo starším pacientům věnována zvýšená opatrnost z důvodu zvýšení frekvence výskytu snížené funkce ledvin způsobené souběžným onemocněním nebo souběžným užíváním jiných léčivých přípravků.
Další zvláštní populace
Přípravek Sebivo nebyl studován u pacientů s hepatitidou typu B infikovaných současně jinými viry (např. pacientů infikovaných současně virem lidské imunodeficience [HIV], virovou hepatitidou typu C [HCV] nebo virovou hepatitidou typu D [HDV]).
Obecně
Pacienti by měli být upozorněni, že léčba přípravkem Sebivo neprokázala snížení rizika přenosu HBV pohlavním stykem nebo krví na jiné osoby.
Použití telbivudinu v kombinaci s lamivudinem se nedoporučuje, protože v klinické studii fáze II byla pozorovaná odpověď na léčbu u kombinace telbivudin/lamivudin nižší než u léčby samotným telbivudinem.
Zatím neexistují údaje o bezpečnosti a účinnosti pro další antivirové kombinace s telbivudinem.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vzhledem k tomu, že je telbivudin vylučován především renální exkrecí, může současné podávání přípravku Sebivo s látkami, které ovlivňují funkci ledvin (jako aminoglykosidy, kličková diuretika, sloučeniny platiny, vankomycin, amfotericin B), ovlivnit plazmatické koncentrace telbivudinu a/nebo souběžně podávaných látek. Kombinaci telbivudinu s těmito léčivými přípravky lze užívat pouze s opatrností. Farmakokinetika telbivudinu v ustáleném stavu nebyla změněna při opakovaném podávání v kombinaci s lamivudinem, adefovir-dipivoxilem, tenofovir-disoproxil-fumarátem, cyklosporinem nebo pegylovaným interferonem alfa-2a. Navíc telbivudin nemění farmakokinetiku lamivudinu, adefovir-dipivoxilu, tenofovir-disoproxil-fumarátu nebo cyklosporinu. Definitivní závěr týkající se účinků telbivudinu na farmakokinetiku pegylovaného interferonu nemohl být udělán z důvodu vysoké interindividuální variability koncentrací pegylovaného interferonu alfa-2a. Klinická studie sledující kombinaci telbivudinu 600 mg denně s pegylovaným interferonem alfa-2a 180 mikrogramů podaným subkutánně jednou týdně ukázala, že je tato kombinace spojena se zvýšeným rizikem vzniku periferní neuropatie. Mechanismus těchto účinků není znám (viz bod 4.4). Kombinace telbivudinu s přípravkem obsahujícím jakýkoli interferon alfa je kontraindikována (viz bod 4.3).
Telbivudin není substrát, induktor nebo inhibitor enzymového systému cytochromu P450 (CYP450) (viz bod 5.2). Z tohoto důvodu je možnost lékových interakcí, zprostředkovaných CYP450, které se týkají přípravku Sebivo, nízká.
4.6 Fertilita, těhotenství a kojení
Studie na zvířatech nenaznačují přímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3). Studie na březích potkanech a králících ukázaly, že telbivudin přestupuje přes placentu. Studie na březích králících ukázaly dřívější vrh mláďat a/nebo potrat závisející na toxicitě pro matku.
Omezené klinické údaje (méně než 300 ukončených těhotenství) po expozici telbivudinu během prvního trimestru nenaznačují žádnou malformační toxicitu a údaje od většího počtu pacientek (více než 1000 ukončených těhotenství) po expozici telbivudinu během druhého a třetího trimestru nenaznačují žádnou fetální/neonatální toxicitu.
Přípravek Sebivo by měl být během těhotenství používán pouze v případě, že přínos pro matku převáží potenciální riziko pro plod.
Údaje z literatury ukazují, že expozice telbivudinu ve druhém a třetím trimestru těhotenství snížila riziko přenosu HBV z matky na kojence, pokud byl telbivudin podáván společně s imunoglobulinem a vakcínou proti hepatitidě B.
Kojení
Telbivudin je vylučován do mléka potkanů. Není známo, zda je telbivudin vylučován do lidského mateřského mléka. Ženy, které užívají přípravek Sebivo, by neměly kojit.
Fertilita
Nejsou k dispozici údaje o účincích telbivudinu na mužskou nebo ženskou fertilitu. V reprodukčních toxikologických studiích u dospělých zvířat byla fertilita mírně snížena, když byl samcům i samicím potkanů podán telbivudin. Nežádoucí účinky na fertilitu byly významnější v oddělené studii u nedospělých zvířat, když byl oběma pohlavím podán telbivudin (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie hodnotící účinky na schopnost řídit nebo obsluhovat stroje nebyly provedeny.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Zhodnocení nežádoucích účinků je založeno převážně na dvou studiích, NV-02B-007 (GLOBE) a NV-02B-015, ve kterých 1 699 pacientů s chronickou hepatitidou typu B dostávalo v dvojitě slepém uspořádání telbivudin 600 mg/den (n = 847) nebo lamivudin (n = 852) po dobu 104 týdnů.
Ve 104týdenních klinických studiích byly hlášené nežádoucí účinky obvykle klasifikovány jako mírné nebo středně těžké. Nejčastějšími nežádoucími účinky byly zvýšení kreatinkinázy (6,8%) stupně 3 nebo 4, únava (4,4%), bolest hlavy (3,0%) a nauzea (2,6%).
Seznam nežádoucích účinků seřazený v tabulce
V tabulce 2 j sou uvedeny nežádoucí účinky tříděné dle tříd orgánových systémů podle MedDRA a frekvence výskytu dle následujících kriterií: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000); není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 2 Nežádoucí účinky
|
Poruchy metabolismu a výživy | |
|
Vzácné* |
Laktátová acidóza jako sekundární příhoda často spojená se závažnými stavy (např. multiorgánové selhání nebo sepse) |
|
Poruchy nervového systému | |
|
Časté |
Závratě, bolesti hlavy |
|
Méně časté |
Periferní neuropatie, porucha chuti, hypestezie, parestezie, ischias |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté | |
|
Gastrointestinální poruchy | |
|
Časté | |
|
Poruchy kůže a podkožní tkáně | |
|
Časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Méně časté |
Myopatie/myozitida, artralgie, myalgie, bolesti končetin, bolesti zad, spasmy svalů, bolesti krku, bolesti v boku (mezi hrudníkem a iliem) |
|
Vzácné* |
Rabdomyolýza |
|
Celkové poruchy a reakce v místě aplikace | |
|
Časté |
Únava |
|
Méně časté |
|
Vyšetření | |
|
Časté |
Zvýšení kreatinfosfokinázy v krvi, zvýšení alaninaminotransferázy v krvi, zvýšení krevní amylázy |
|
Méně časté |
Zvýšení aspartátaminotransferázy |
|
* Tyto nežádoucí účinky byly popsány po uvec |
ení přípravku na trh, ale nebyly pozorovány |
v kontrolovaných klinických studiích. Četnost výskytu byla odhadnuta ze statistického výpočtu na základě celkového počtu pacientů léčených telbivudinem v klinických studiích (n = 8 914).
Popis vybraných nežádoucích reakcí
Zvýšení kreatinkinázy
Ve sloučené analýze NV-02B-007 (GLOBE) a NV-02B-015 se do 104. týdne léčby se vyskytl stupeň 3 nebo 4 zvýšení CK (>7x ULN) u 12,6% pacientů léčených telbivudinem (n = 847) a u 4,0% pacientů léčených lamivudinem (n = 846). Zvýšení CK bylo ve většině případů asymptomatické a hodnoty CK obvykle klesly do příští návštěvy při pokračování v léčbě.
Rychlé zvýšení ALT
Incidence rychlého vzestupu alaninaminotransferázy (ALT) při léčbě v obou léčených větvích podle definice AASLD (American Association for the Study of Liver Disease) (elevace ALT >2x výchozí hodnota a >10x horní hranice normy (ULN)) je dále popsána v níže uvedené tabulce 3.
Tabulka 3 Přehled vzestupu ALT při léčbě - sloučené studie NV-02B-007 (GLOBE) a NV-02B-015
|
Vzestup ALT: |
Lamivudin |
Telbivudin |
|
elevace ALT >2x výchozí a >10x ULN |
n/N (%) |
n/N (%) |
|
Celkově |
67/852 (7,9) |
41/847 (4,8) |
|
Od výchozí hodnoty do 24. týdne |
25/852 (2,9) |
25/847 (3,0) |
|
Od 24. týdne do konce studie |
44/837 (5,3) |
17/834 (2,0) |
Během léčby se doporučuje pravidelné sledování jaterních funkcí (viz bod 4.4).
Exacerbace hepatitidy B po ukončení léčby
U pacientů, kteří ukončili léčbu hepatitidy B, byly hlášeny případy těžké akutní exacerbace hepatitidy B (viz bod 4.4), včetně pacientů léčených telbivudinem.
Incidence rychlého vzestupu alaninaminotransferázy (ALT) po léčbě v obou léčených větvích je dále popsána v níže uvedenéých tabulce 4.
Tabulka 4 Přehled vzestupu ALT po léčbě - sloučené studie NV-02B-007 (GLOBE) a NV-02B-015
|
Lamivudin |
Telbivudin | |
|
Vzestup ALT |
n/N (%) |
n/N (%) |
|
elevace ALT >2x výchozí hodnota a >10x ULN |
10/180 (5,6) |
9/154 (5,8) |
Výsledky ve 208. týdnu
Po 104 týdnech léčby telbivudinem bylo 78% pacientů (530/680) ze studie NV-02B-007 (GLOBE) a 82% pacientů (137/167) ze studie NV-02B-015 převedeno do extenzní studie CLDT600A2303 (viz bod 5.1), aby pokračovalo v léčbě až do 208. týdne. Soubor pacientů pro sledování dlouhodobé bezpečnosti se skládal z 655 pacientů, včetně 518 z NV-02B-007 (GLOBE) a 137 z NV-02B-015. Celkový bezpečnostní profil ze sloučené analýzy až do 104. a 208. týdne byl podobný. Zvýšení CK stupně 3 nebo 4 se nově vyskytlo u 15,9% pacientů léčených telbivudinem 208 týdnů. Většina případů zvýšení CK stupně 3 nebo 4 byla asymptomatická a přechodná.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nejsou informace o záměrném předávkování telbivudinem, ale u jednoho jedince došlo neúmyslně k předávkování, které bylo asymptomatické. Testované dávky až do dávky 1 800 mg/den, třikrát vyšší než doporučená denní dávka, byly dobře tolerovány. Maximální tolerovaná dávka telbivudinu nebyla stanovena. V případě předávkování musí být podávání přípravku Sebivo přerušeno a podle potřeby zahájena odpovídající podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antivirotika k systémovému užití, nukleosidové a nukletidové inhibitory reverzní transkriptázy, ATC kód: J05AF11
Mechanismus účinku
Telbivudin je syntetický analog nukleosidu thymidinu s účinností proti HBV DNA polymeráze. Je účinně fosforylován buněčnými kinázami na účinnou trifosfátovou formu, která má nitrobuněčný poločas 14 hodin. Telbivudin-5'-trifosfát inhibuje HBV DNA polymerázu (reverzní transkriptázu) kompeticí s přirozeným substrátem, thymidin 5'-trifosfátem. Inkorporace telbivudin-5'-trifosfátu do virové DNA vyvolá ukončení řetězce DNA, což vede k inhibici replikace HBV.
Farmakodynamické účinky
Telbivudin je inhibitor syntézy jak prvního vlákna HBV (EC50 = 0,4 - 1,3 ^M), tak i druhého vlákna HBV (EC50 = 0,12 - 0,24 ^M), a vykazuje výraznou preferenci pro inhibici produkce druhého vlákna. Oproti tomu telbivudin-5-trifosfát neinhiboval v koncentracích do 100 buněčnou DNA polymerázu a, p nebo y. Při sledování mitochondriální struktury, funkce a obsahu DNA, nevykazoval telbivudin prokazatelný toxický účinek v koncentracích až do 10 a nezvyšoval tvorbu kyseliny mléčné in vitro.
Antivirová účinnost telbivudinu in vitro byla hodnocena u HBV-exprimujících buněčných linií lidského hepatomu 2.2.15. Koncentrace telbivudinu, která účinně inhibovala 50% virové syntézy (EC50), byla přibližně 0,2 ^M. Antivirová účinnost telbivudinu je specifická pro virus hepatitidy B a příbuzné hepadnaviry. Telbivudin není účiný proti HIV in vitro. V klinických studiích nebyla absence účinnosti telbivudinu proti HIV hodnocena.
Klinické zkušenosti
Bezpečnost a účinnost dlouhodobé léčby (104 týdnů) přípravkem Sebivo byly hodnoceny ve dvou, léčivou látkou kontrolovaných klinických studiích, do kterých bylo zařazeno 1 699 pacientů s chronickou hepatitidou B (NV-02B-007 (GLOBE) a NV-02B-015).
Studie NV-02B-007 (GLOBE)
NV-02B-007 GLOBE studie je randomizovaná, dvojitě-slepá mezinárodní studie fáze III, kde byl telbivudin srovnáván s lamivudinem po dobu 104 týdnů u 1 367 pacientů, kteří dosud nedostávali žádné nukleosidy a měli chronickou hepatitidu B a byli jak HBeAg-pozitivní, tak i HBeAg-negativní. Většina zahrnuté populace byli asiaté. Nejčastějšími HBV genotypy byly B (26%) a C (51%). Telbivudinem bylo léčeno malé množství pacientů bělošské rasy (celkem 98). Primární analýza dat byla provedena po dosažení 52. týdne u všech pacientů.
HBeAg-pozitivnípacienti: Střední věk pacientů byl 32 let, 74% byli muži, 82% byli asiaté, 12% byli běloši a 6% bylo dříve léčeno interferonem alfa.
HBeAg-negativnípacienti: Střední věk pacientů byl 43 let, 79% byli muži, 62% asiaté, 23% běloši a 11% bylo dříve léčeno interferonem alfa.
Klinické výsledky v 52.týdnu
Klinické a virologické cílové parametry účinnosti byly hodnoceny odděleně u HBeAg-pozitivních a HBeAg-negativních pacientů. Primární cílový parametr terapeutické odpovědi byl složen ze serologických cílových parametrů vyžadujících supresi HBV DNA na <5 log10 kopií/ml ve spojení buď se ztrátou sérového HBeAg, nebo normalizací ALT. Sekundární cílové parametry zahrnovaly histologickou odpověď, normalizaci ALT a různé změny antivirové účinnosti.
Bez ohledu na charakteristiku výchozích hodnot se u většiny pacientů užívajících přípravek Sebivo projevila histologická, virologická, biochemická a serologická odpověď na léčbu. Výchozí hladiny ALT >2x ULN a výchozí HBV DNA <9 log10 kopií/ml byly spojeny s vyšší rychlostí sérokonverze HBeAg u HBeAg-pozitivních pacientů. Pacienti, kteří dosáhli hladin HBV DNA <3 log10 kopií/ml do 24. týdne, odpovídali na léčbu optimálně; naopak u pacientů s hladinami HBV DNA ve 24. týdnu >4 log10 kopií/ml bylo v 52. týdnu dosaženo méně příznivých výsledků.
U HBeAg-pozitivních pacientů byl telbivudin účinnější než lamivudin v terapeutické odpovědi (75,3% vs 67,0% reagujících pacientů; p = 0,0047). U HBeAg-negativních pacientů, telbivudin nebyl lepší než lamivudin (75,2% a 77,2% reagujících pacientů; p = 0,6187). U bělochů byla léčebná odpověď u obou antivirových látek použitých ve studii NV-02B-007 (GLOBE) nižší; počet bělošských pacientů byl však velmi omezený (n = 98).
Ve 24. týdnu dosáhlo 203 HBeAg-pozitivních pacientů a 177 HBeAg-negativních pacientů nedetekovatelných hladin HBV DNA. Z HBeAg-pozitivních pacientů 95% dosáhlo nedetekovatelných hladin HBV DNA, 39% dosáhlo HBeAg sérokonverze, 90% dosáhlo normalizace ALT v 52.týdnu a 0,5% bylo ve 48. týdnu rezistentních. Podobně z HBeAg-negativních pacientů 96% dosáhlo nedetekovatelné hladiny HBV DNA, 79% dosáhlo v 52.týdnu normalizace ALT a 0% bylo rezistentních ve 48. týdnu.
Vybrané virologické, biochemické a sérologické výsledky vyšetření jsou uvedeny v tabulce 5 a histologické odpovědi v tabulce 6.
|
Parametr odpovědi |
HBeAg-pozitivní (n = 921) |
HBeAg-negativní (n = 446) | ||
|
Telbivudin 600 mg (n = 458) |
Lamivudin 100 mg (n = 463) |
Telbivudin 600 mg (n = 222) |
Lamivudin 100 mg (n = 224) | |
|
Střední hodnota snížení HBV DNA od výchozí hodnoty (log10 kopií/ml) ± SE M1,2,3 |
-6,45 (0,11) * |
-5,54 (0,11) |
-5,23 (0,13) * |
-4,40 (0,13) |
|
% pacientů HBV DNA nedetekovatelných metodou PCR |
60%* |
40% |
88%* |
71% |
|
normalizace ALT 4 |
77% |
75% |
74% |
79% |
|
sérokonverze HBeAg 4 |
23% |
22% |
- |
- |
|
vymizení HBeAg 5 |
26% |
23% |
- |
- |
1 SEM: Standard error of mean (Standardní odchylka od střední hodnoty).
2 Roche COBAS Amplicor® PCR test (dolní limit kvantifikace <300 kopií/ml).
3 HBeAg-pozitivní n = 443 ve skupině telbivudinu a 444 ve skupině s lamivudinem; HBeAg-negativní n = 219 ve skupině s telbivudinem a 219 ve skupině s lamivudinem. Rozdíly ve skupinách jsou způsobeny ukončením léčby pacientů v průběhu studie a chybějícím hodnocením HBV DNA v 52.týdnu
4 HBeAg-pozitivní n = 440 ve skupině telbivudinu a 446 ve skupině s lamivudinem; HBeAg-negativní n = 203 ve skupině telbivudinu a 207 ve skupině s lamivudinem. ALT normalizace byla stanovena pouze u pacientů s ALT > ULN ve výchozích hodnotách.
5 n = 432 ve skupině telbivudinu a 442 ve skupině s lamivudinem. HBeAg sérokonverze a vymizení bylo sledováno pouze u pacientů s detekovatelným HBeAg při výchozím vyšetření.
*p < 0,0001
|
HBeAg-pozitivní (n = 921) |
HBeAg-negativní (n = 446) | |||
|
Telbivudin 600 mg (n = 384)1 |
Lamivudin 100 mg (n = 386)1 |
Telbivudin 600 mg (n = 199)1 |
Lamivudin 100 mg (n = 207)1 | |
|
Histologická odpověď2 | ||||
|
Zlepšení |
71%* |
61% |
71% |
70% |
|
Žádné zlepšení |
17% |
24% |
21% |
24% |
|
Ishakovo skóre fibrózy (Ishak Fibrosis Score)3 | ||||
|
Zlepšení |
42% |
47% |
49% |
45% |
|
Beze změn |
39% |
32% |
34% |
43% |
|
Zhoršení |
8% |
7% |
9% |
5% |
|
Chybějící biopsie v 52.týdnu |
12% |
15% |
9% |
7% |
|
1 Pacienti s > než jednou dávkou studijní medi hodnotou Knodellova indexu histologické akti 2 Histologická odpověď definovaná jako >2bo< skóre od výchozí hodnoty bez zhoršení Knode 3 Zlepšení Ishakova skóre fibrózy, bylo stanov výchozí hodnoty v 52. týdnu. *p = 0,0024 |
kace s hodnotitelnou výchozí biopsií jater a výchozí vity (HAI) skóre >3. ový pokles Knodellova zánětlivě nekrotického lova skóre fibrózy. eno jako snížení o >1 bod v Ishakově skóre od | |||
Klinické výsledky ve 104.týdnu
Klinické výsledky ve 104. týdnu byly u pacientů léčených telbivudinem konzistentní s výsledky v 52.týdnu a ukázaly přetrvávání účinnosti telbivudinu při pokračování v léčbě.
Mezi HBeAg-pozitivními pacienty byl mezi telbivudinem a lamivudinem ve 104. týdnu demonstrován zvětšující se rozdíl v terapeutické odpovědi (63% vs 48%; p < 0,0001) a klíčových sekundárních cílových parametrech (průměrná log10 snížení HBV DNA: -5,74 vs -4,42; p < 0,0001, nedetekovatelnost HBV DNA: 56% vs 39%; p < 0,0001 a normalizace ALT 70% vs 62%). Pro telbivudin byl také pozorován trend směrem k vyššímu výskytu ztrát HBeAg (35% vv s 29%) a vyšší frekvenci sérokonverze (30% vs 25%). Navíc, v podskupině pacientů s výchozí hodnotou ALT >2x ULN (320), výrazně vyšší podíl pacientů léčených telbivudinem oproti pacientům léčených lamivudinem dosáhl ve 104. týdnu sérokonverze HBeAg (36% oproti 28%).
U HBeAg-negativních pacientů byly rozdíly v terapeutické odpovědi (78% vs 66%) a klíčových sekundárních cílových parametrech (průměrný logJ0 snížení hladin HBV DNA: -5,00 vs -4,17, a nedetekovatelnost HBV DNA: 82% vs. 57%; p < 0,0001) vyšší pro telbivudin až do 104. týdne. Normalizace ALT (78% vs 70%) pokračovala a byla ve 104. týdnu vyšší.
Předpověditelnost v 24. týdnu
Ve 24. týdnu 203 telbivudinem léčených pacientů HBeAg-pozitivních (44%) a 177 pacientů HBeAg-negativních (80%) dosáhlo nedetekovatelných hladin HBV DNA.
Výsledky HBV DNA ve 24. týdnu byly pro obě skupiny HBeAg-pozitivních i HBeAg-negativních pacientů prediktorem dlouhodobého příznivého výsledku. Telbuvidinem léčení pacienti, kteří dosáhli nedetekovatelných hladin HBV DNA metodou PCR ve 24. týdnu, měli nejvyšší poměr nedetekovatelnosti HBV DNA a sérokonverze HBeAg (u HBeAg-pozitivních pacientů) a nejnižší celkový výskyt virologického vzplanutí (breakthrough) ve 104. týdnu.
Závěrečné výsledky ve 104. týdnu, založené na hladině HBV DNA ve 24. týdnu jsou, jak pro HBeAg-pozitivní nebo HBeAg-negativní pacienty, uvedeny v tabulce 7.
Tabulka 7 Klíčové cílové parametry účinnosti ve 104.týdnu při sérových hladinách HBV DNA ve 24.týdnu u pacientů léčených telbivudinem ve studii NV-02B-007 (GLOBE)
|
HBV DNA ve 24.týdnu |
Výsledky klíčových cílových parametrů účinnosti ve 104. týdnu založené na výsledcích ve 24. týdnu | ||||
|
Terapeutická odpověď n/N (%) |
nedetekovateln á hladina HBV DNA n/N (%) |
sérokonverze HBeAg n/N (%) |
normalizace ALT n/N (%) |
Virologický breakthrough* n/N (%) | |
|
HBeAg-pozitivní | |||||
|
<300 kopií/ml |
172/203 (85) |
166/203 (82) |
84/183 (46) |
160/194 (82) |
22/203 (11) |
|
300 kopií/ml až <3 log10 kopií/ml |
36/57 (63) |
35/57 (61) |
21/54 (39) |
40/54 (74) |
18/57 (32) |
|
>3 log10 kopií/ml |
82/190 (43) |
54/190 (28) |
23/188(12) |
106/184 (58) |
90/190 (47) |
|
HBeAg-negativní | |||||
|
<300 kopií/ml |
146/177 (82) |
156/177 (88) |
N/A |
131/159 (82) |
11/177 (6) |
|
300 kopií/ml až <3 log10 kopií/ml |
13/18 (72) |
14/18 (78) |
N/A |
13/17 (76) |
4/18 (22) |
|
>3 log10 kopií/ml |
13/26 (50) |
12/26 (46) |
N/A |
14/26 (54) |
12/26 (46) |
N/A = neuplatňuje se
* virologický breakthrough: “1 log nad dolní hodnotu nadir” definice stanovené ve 104. týdnu
Studie NV-02B-015
Výsledky účinnosti a bezpečnosti studie NV-02B-007 (GLOBE) byly potvrzeny ve studii NV-02B-015. Tato studie je studie fáze III, randomizovaná, dvojitě slepá, srovnávající telbivudin 600 mg podávaný jednou denně s lamivudinem 100 mg, podávaným jednou denně po dobu 104 týdnů, 332 čínským pacientům s chronickou hepatitidou B, HBeAg-pozitivním a HBeAg-negativním, kteří nebyli předtím nukleosidy léčeni.
Studie CLDT600A2303 - klinické výsledky po 208 týdnech
Studie CLDT600A2303 byla otevřená 104týdenní extenzní studie u pacientů s kompenzovanou chronickou hepatitidou B, kteří byli předtím léčeni telbivudinem po dobu 2 let, včetně pacientů ze studií NV-02B-007 (GLOBE) a NV-02B-015, poskytující údaje o účinnosti a bezpečnosti telbivudinu po 156 a 208 týdnech kontinuální léčby. Pacienti s nedetekovatelnou hladinou HBV DNA ve 24. týdnu léčby měli lepší výsledky ve 156. a 208. týdnu (Tabulka 8).
Tabulka 8 Analýza účinnosti ze sloučených údajů studií NV-02B-007 (GLOBE), NV-02B-015 a CLDT600A2303
|
Týden 52 |
Týden 104 |
Týden 156 |
Týden 208 | |
|
HBeAg-pozitivní pacienti (n = 293*) | ||||
|
Průběžně nedetekovatelné hladiny HBV DNA (<300 kopií/ml) |
70,3% (206/293) |
77,3% (218/282) |
75,0% (198/264) |
76,2% (163/214) |
|
Průběžně nedetekovatelné hladiny HBV DNA (<300 kopií/ml) s nedetekovatelnou hladinou HBV DNA v týdnu 24 |
99,4% (161/162) |
94,9% (150/158) |
86,7% (130/150) |
87,9% (109/124) |
|
Kumulativní poměr HBeAg sérokonverze (%) |
27,6% (81/293) |
41,6% (122/293) |
48,5% (142/293) |
53,2% (156/293) |
|
Kumulativní poměr HBeAg sérokonverze u pacientů s nedetekovatelnou hladinou HBV DNA v týdnu 24 (%) |
40,1% (65/162) |
52,5% (85/162) |
59,3% (96/162) |
65,4% (106/162) |
|
Průběžná ALT normalizace |
81,4% (228/280) |
87,5% (237/271) |
82,9% (209/252) |
86,4% (178/106) |
|
HBeAg-negativnípacienti (n = 209*) | ||||
|
Průběžně nedetekovatelné hladiny HBV DNA (<300 kopií/ml) |
95,2% (199/209) |
96,5% (195/202) |
84,7% (160/189) |
86,0% (141/164) |
|
Průběžně nedetekovatelné hladiny HBV DNA (<300 kopií/ml) s nedetekovatelnou hladinou HBV DNA v týdnu 24 |
97,8% (175/179) |
96,5% (166/172) |
86,7% (143/165) |
87,5% (126/144) |
|
Průběžná ALT normalizace |
80,3% (151/188) |
89,0% (161/181) |
83,5% (142/170) |
89,6% (129/144) |
* Soubor pacientů bez virové rezistence na začátku studie CLDT600A2303 se skládal z 502 pacientů (293 HBeAg-pozitivních a 209 HBeAg-negativních).
Studie CLDT600ACN04E1 - vliv léčby na histologii _jater
Ve studii CLDT600ACN04E1 byly u 57 pacientů s dostupnou jatemí biopsií na počátku léčby a po průměrně 260,8 týdnech léčby hodnoceny změny v histologii jater (38 HBeAg-pozitivních a 19 HBeAg-negativních pacientů).
• Průměrná výchozí hodnota Knodellova zánětlivě nekrotického skóre 7,6 (SD 2,9) se zlepšila
(p < 0,0001) na 1,4 (SD 0,9) s průměrnou změnou -6,3 (SD 2,8). Knodellovo zánětlivě nekrotické skóre <3 (žádné nebo minimální zánětlivě nekrotické změny) bylo pozorováno u 98,2% (56/57) pacientů.
• Průměrná výchozí hodnota Ishakova skóre 2,2 (SD 1,1) se zlepšila (p < 0,0001) na 0,9 (SD 1,0)
s průměrnou změnou -1,3 (SD 1,3). Ishakovo skóre fibrózy <1 (žádná nebo minimální fibróza) bylo pozorováno u 84,2% (48/57) pacientů.
Změny Knodellova zánětlivě nekrotického skóre a Ishakova skóre byly podobné u HBeAg-pozitivních i HBeAg-negativních pacientů.
CLDT600A2303 - trvání HBeAg odpovědi po ukončení léčby
Studie CLDT600A2303 zahrnovala HBeAg-pozitivní pacienty ze studií NV-02B-007 (GLOBE) nebo NV-02B-015, kteří byli sledováni po ukončení léčby. Tito pacienti dokončili >52týdenní léčbu telbivudinem a vykazovali pokles HBeAg po dobu >24 týdnů s HBV DNA <5 logi0 kopiemi/ml při poslední kontrole. Střední délka léčby byla 104 týdnů. Po střední délce sledování 120 týdnů po ukončení léčby prokázala většina HBeAg-pozitivních pacientů léčených telbivudinem trvalý pokles HBeAg (83,3%; 25/30) a trvalou HBeAg sérokonverzi (79,2%; 19/24). Pacienti s trvalou HBeAg sérokonverzí měli střední hodnotu HBV DNA 3,3 log10 kopií/ml; a 73,7% mělo HBV DNA <4 log10 kopií/ml.
Klinická, rezistence
Test genotypové rezistence byl proveden ve studii NV-02B-007 (GLOBE; n = 680) u pacientů s virologickým reboundem (potvrzeno zvýšení o >1 log10 kopií/ml HBV DNA od nejnižší hodnoty/nadir).
Ve 48. týdnu mezi pacienty léčenými telbivudinem mělo 5% (23/458) HBeAg-pozitivních pacientů a 2% (5/222) HBeAg-negativních pacientů virologický rebound s detekovatelnými rezistentními mutacemi HBV.
Studie NV-02B-007 (GLOBE) a CLDT600A2303 - kumulativní poměry genotypové rezistence Prvotní analýza kumulativní genotypové rezistence ve 104. a 208. týdnu byla založena na souboru ITT a zahrnovala všechny pacienty, kteří pokračovali v léčbě do 4. roku bez ohledu na HBV DNA hladiny.
Z 680 pacientů léčených telbivudinem, kteří byli od počátku zařazeni do pivotní studie NV-02B-007 (GLOBE), bylo 517 pacientů (76%) zahrnuto do studie CLDT600A2303 s pokračující léčbou telbivudinem po dobu až 208 týdnů. Z těchto 517 pacientů mělo 159 pacientů (HBeAg-pozitivní=135, HBeAg-negativní=24) detekovatelnou hladinu HBV DNA.
Kumulativní genotypové četnosti ve 104. týdnu byly 25,1% (115/458) pro HBeAg-pozitivní pacienty a 10,8% (24/222) pro HBeAg-negativní pacienty.
V celkovém ITT souboru byly poměry kumulativní rezistence ve 4. roce 40,8% (131/321) u HBeAg-pozitivních pacientů a 18,9% (37/196) HBeAg-negativních pacientů.
Kumulativní poměry genotypové rezistence byly také stanoveny pomocí matematického modelu, který zvažoval pouze pacienty s nedetekovatelnou hladinou HBV DNA počátkem příslušného roku. V této analýze byly poměry kumulativní rezistence ve 4. roce 22,3% u HBeAg-pozitivních pacientů a 16,0% u HBeAg-negativních pacientů.
Jestliže zhodnotíme pacienty, u kterých došlo do 104. týdne ve studii NV-02B-007 (GLOBE) k virologickému breakthrough, byla četnost rezistence nižší u pacientů s HBV DNA <300 kopií/ml ve 24. týdnu, než u pacientů s HBV DNA >300 kopií/ml ve 24. týdnu. U HBeAg-pozitivních pacientů s HBV DNA <300 kopií/ml ve 24. týdnu byla rezistence 1% (3/203) ve 48. týdnu a 9% (18/203) ve 104. týdnu, zatímco u pacientů s HBV DNA >300 kopií/ml byla rezistence 8% (20/247) ve 48. týdnu a 39% (97/247) ve 104. týdnu. U HBeAg-negativních pacientů s HBV DNA <300 kopií/ml ve 24. týdnu byla rezistence 0% (0/177) ve 48. týdnu a 5% (9/177) ve 104. týdnu, zatímco u pacientů s HBV DNA >300 kopií/ml byla rezistence 11% (5/44) ve 48. týdnu a 34% (15/44) ve 104. týdnu.
Charakter genotvpovvch mutací a zkřížená rezistence
Genotypové analýzy 203 hodnotitelných párových vzorků s HBV DNA >1 000 kopií/ml ve 104. týdnu (NV-02B-007 (GLOBE)) ukázaly, že primární mutace, která souvisela s rezistencí k telbivudinu byla rtM204I. Často byla doprovázena mutacemi rtL180M a rtL80I/V a občas rtV27A, rtL82M, rtV173L, rtT184I a rtA200V. Základní faktory (přítomné při vstupu do studie) sdružené s vývojem genotypové lékové rezistence zahrnovaly: léčbu lamivudinem, vyšší výchozí HBV DNA, nižší výchozí hodnoty sérové ALT a zvýšenou tělesnou hmotnost/BMI. Parametry léčebné odpovědi ve 24. týdnu, které předpovídaly vývoj rezistence viru na léky do 104. týdne, byly HBV DNA >300 kopií/ml a zvýšení ALT v séru.
Genotypová analýza 50 HBV izolátů od pacientů léčených telbivudinem ve 208. týdnu (CLDT600A2303) odhalila podobný profil rezistence, jaký byl hlášen ve 104. týdnu. Konverze na pozici 80, 180 a polymorfních pozicích 91, 229 byly vždy zjištěny v sekvencích, které obsahovaly mutaci M204I, která vyvolává genotypovou rezistenci. Jde velmi pravděpodobně o mutace kompenzační. U pacientů léčených telbivudinem, u kterých došlo ke vzniku virologického breakthrough do 208. týdne, byla hlášena jedna izolovaná rtM204V mutace a dvě rtM204I/V/M mutace. Nebyla hlášená žádná nová mutace.
Zkřížená rezistence byla pozorována mezi nukleosidovými analogy HBV (viz bod 4.4). HBV kmeny rezistentní vůči lamivudinu, které obsahovaly buď mutaci rtM204I nebo dvojitou mutaci rtL180M/rtM204V, měly v buněčných testech >1 000násobně sníženou citlivost k telbivudinu. HBV kódující substituce rtN236T nebo rtA181V, které souvisejí s rezistencí vůči adefoviru, měly v buněčné kultuře přibližně 0,3, resp. 4násobné změny citlivosti k telbivudinu (viz bod 4.4).
5.2 Farmakokinetické vlastnosti
Farmakokinetika telbivudinu po jednorázovém a opakovaném podávání byla sledována u zdravých jedinců i u pacientů s chronickou hepatitidou B. Farmakokinetika telbivudinu v doporučené dávce 600 mg nebyla hodnocena u pacientů s chronickou hepatitidou B. Farmakokinetika telbivudinu je však u obou těchto populací podobná.
Absorpce
Po perorálním podání jedné dávky 600 mg telbivudinu zdravým jedincům (n = 42), byla vrcholová koncentrace (Cmax) telbivudinu v plazmě 3,2 ± 1,1 ^g/ml (střední hodnota ± SD) a bylo jí dosaženo za 3 hodiny (medián) po podání dávky. Pro telbivudin byla plocha pod křivkou plazmatické koncentrace v čase (AUC0-<X)) 28,0 ± 8,5 ^g*h/ml (střední hodnota ± SD). Interindividuální variabilita (CV%) pro měření systémové expozice (Cmax, AUC) byla typicky přibližně 30%.
Vliv potravy na absorpci po perorálním podání
Po jednorázovém podání 600 mg telbivudinu s potravou nebyla jeho absorpce a expozice ovlivněna. Distribuce
In vitro je vazba telbivudinu na lidské plazmatické proteiny nízká (3,3%).
Biotransformace
Po aplikaci 14C-telbivudinu lidem nebyly detekovány žádné metabolity. Telbivudin není ani substrát, inhibitor ani induktor enzymatického systému cytochromu P450 (CYP450).
Eliminace
Po dosažení vrcholu plazmatické koncentrace telbivudinu dochází k jeho poklesu biexponenciálně s terminálním eliminačním poločasem (t1/2) 41,8 ± 11,8 hodin. Telbivudin je primárně vylučován v nezměněné formě močí. Renální clearance telbivudinu dosahuje hodnot normální glomerulární filtrace, což ukazuje, že filtrace je hlavním mechanizmem exkrece. Přibližně 42% z podané dávky se objeví v moči v průběhu 7 dnů po perorálním podání jedné dávky 600 mg telbivudinu. Protože je renální exkrece hlavní cestou vylučování, vyžadují pacienti se střední až těžkou renální dysfunkcí a dialyzovaní pacienti úpravu dávkovacího intervalu (viz bod 4.2).
Linearita/nelinearita
Farmakokinetika telbivudinu je závislá na dávce v rozmezí dávek od 25 do 1 800 mg. Rovnovážného stavu bylo dosaženo po 5 až 7 dnech při podávání jednou denně, s přibližně 1,5násobnou akumulací v systémové expozici, což naznačuje, že efektivní akumulační poločas je přibližně 15 hodin. Po podání telbivudinu v dávce 600 mg jednou denně byla dolní hodnota koncentrace v rovnovážném stavu přibližně 0,2 - 0,3 ^g/ml.
Zvláštní populace
Pohlaví
Ve farmakokinetice telbivudinu není signifikantní rozdíl v závislosti na pohlaví.
Lidská rasa
Ve farmakokinetice telbivudinu není signifikantní rozdíl v závislosti na rase.
Děti a starší pacienti (více než 65 let)
U dětí a u starších pacientů nebyly provedeny studie farmakokinetiky.
Porucha funkce ledvin
Farmakokinetika telbivudinu po jednorázovém podání dávek (200, 400 a 600 mg) byla hodnocena u pacientů (bez chronické hepatitidy B) s různým stupněm poškození renálních funkcí (hodnoceno clearance kreatininu). Na základě výsledků, uvedených v tabulce 9, je doporučeno upravit dávkové intervaly telbivudinu u pacientů s clearance kreatininu <50 ml/min (viz body 4.2 a 4.4).
Tabulka 9 Farmakokinetické parametry (střední hodnota ± SD) telbivudinu u osob s různým stupněm poškození renálních funkcí
|
Renální funkce (clearance kreatininu v ml/min) | |||||
|
Normální (>80) (n = 8) 600 mg |
Mírné (50-80) (n = 8) 600 mg |
Střední (30-49) (n = 8) 400 mg |
Těžké (<30) (n = 6) 200 mg |
ESRD/ hemodialýza (n = 6) 200 mg | |
|
Cmax (Mg/ml) |
3,4 ± 0,9 |
3,2 ± 0,9 |
2,8 ± 1,3 |
1,6 ± 0,8 |
2,1 ± 0,9 |
|
AUC0-X, (pg^h/ml) |
28,5 ± 9,6 |
32,5 ± 10,1 |
36,0 ± 13,2 |
32,5 ± 13,2 |
67,4 ± 36,9 |
|
CLrenal (ml/min) |
126,7 ± 48,3 |
83,3 ± 20,0 |
43,3 ± 20,0 |
11,7 ± 6,7 |
- |
Pacienti s poškozenou funkcí ledvin na hemodialýze
Hemodialýza (do 4 hodin) snižuje systémovou expozici telbivudinu přibližně o 23%. Po úpravě dávkových intervalů podle clearance kreatininu není další úprava dávky během rutinní dialýzy nutná (viz 4.2). Telbivudin má být podáván po hemodialýze.
Zhoršená funkce jater
Farmakokinetika telbivudinu byla studována u pacientů (bez chronické hepatitidy B) s různým stupněm zhoršené funkce jater a u některých pacientů s dekompenzovanou chorobou jater. Nebyly pozorovány významné změny farmakokinetiky telbivudinu u osob se zhoršenou funkcí jater ve srovnání s jedinci bez zhoršené funkce jater. Výsledky těchto studií ukazují, že není nezbytné upravovat dávku u pacientů se zhoršenou funkcí jater (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání a genotoxicity neodhalily žádné zvláštní riziko pro člověka. Telbivudin neukázal žádný karcinogenní potenciál pro člověka. Standardními testy reprodukční toxicity nebyl zjištěn žádný důkaz přímého toxického účinku telbivudinu. U králíků byly dávky telbivudinu, po kterých dosahovaly expozice 37násobku expozice u člověka po podání terapeutické dávky (600 mg), spojeny se zvýšením incidence potratu a předčasného porodu. Tento účinek byl považován za důsledek toxicity pro matku.
Fertilita byla hodnocena v konvenčních studiích u dospělých potkanů, a jako součást toxikologické studie na nedospělých jedincích.
U dospělých potkanů byla fertilita snížena v případech, kdy byl samci i samici potkana podáván telbivudin v dávkách 500 nebo 1000 mg/kg/den (nižší index fertility ve srovnání se souběžnými kontrolami). Neprojevily se žádné známky abnormality v morfologii nebo funkci spermatu a varlata a vaječníky byly bez histologických pozoruhodností.
Nebylo prokázáno zhoršení fertility v dalších studiích, kdy byly buď samcům nebo samicím potkanů podávány dávky až 2000 mg/kg/den a kteří byli spářeni s potkany bez léčby (systémová expozice přibližně 6-14násobně vyšší, než jakých bylo dosaženo u člověka).
V toxikologických studiích byli nedospělí jedinci léčeni od 14. do 70. dne po porodu a byli pářeni s potkany, kterým byla podávána stejná léčba (bez páření mezi sourozenci). Fertilita byla snížena u párů s dávkami >1000 mg/kg/den, jak ukazují snížené indexy fertility a páření a snížený počet početí. Ovariální a děložní parametry potkaních samic, které se úspešně spářily, však nebyly podáváním telbivudinu ovlivněny.
NOAEL (no observed adverse effect level) účinků na parametry fertility nebo páření se rovná dávce 250 mg/kg/den, jejíž podávání vede k 2,5 až 2,8krát vyšší expozici, než jaké je dosaženo při podávání terapeutické dávky u člověka s normální funkcí ledvin.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety
Mikrokrystalická celulosa Povidon
Sodná sůl karboxymethylškrobu Koloidní bezvodý oxid křemičitý Magnesium-stearát
Potahová vrstva tablety Oxid titaničitý (E171)
Makrogol
Mastek
Hypromelosa
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení PVC/Al blistry
Velikosti balení: 28 nebo 98 potahovaných tablet Na trhu nemusí být k dispozici všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/07/388/001
EU/1/07/388/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 24. dubna 2007
Datum posledního prodloužení registrace: 26. dubna 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Sebivo 20 mg/ml perorální roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje telbivudinum 20 mg.
Pomocné látky se známým účinkem: Jedna 600 mg dávka (30 ml) perorálního roztoku obsahuje asi 47 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Perorální roztok
Čirý, bezbarvý až slabě nažloutlý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Sebivo je indikován k léčbě chronické hepatitidy B u dospělých pacientů s kompenzovaným onemocněním jater a prokázanou replikací viru, s trvale zvýšenými hladinami alaninaminotransferázy (ALT) v séru a histologicky prokázaným aktivním zánětem a/nebo fibrózou.
Zahájení léčby Sebivem má být zvažováno pouze v případě, kdy alternativní antivirová látka s vyšší genetickou bariérou proti vzniku rezistence není k dispozici nebo její použití není vhodné.
Pro detaily studie a specifické charakteristiky pacientů, na kterých je tato indikace založena, viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba musí být zahájena lékařem, který má zkušenosti s léčbou chronické infekční hepatitidy typu B.
Dávkování
Dospělí
Doporučená dávka přípravku Sebivo je 30 ml, což je dávka ekvivalentní 600 mg, užívaná jednou denně.
Sledování během léčby
Odpověď na léčbu ve 24. týdnu se ukázala jako prediktivní pro dlouhodobou odpověď na léčbu (viz tabulka 7 v bodě 5.1). HBV DNA hladiny je třeba monitorovat ve 24. týdnu léčby, aby se zajistila kompletní virová suprese (HBV DNA méně než 300 kopií/ml). U pacientů s detekovatelnou hladinou HBV DNA po 24 týdnech léčby je nutné zvážit úpravu léčby.
HBV DNA je třeba monitorovat každých 6 měsíců, aby se zajistila kontinuální odpověď. Pokud je HBV DNA test pozitivní kdykoliv po první odpovědi, je nutné zvážit úpravu léčby. Optimální léčba musí být provázena testy na rezistenci.
Délka léčby
Optimální délka léčby není známa. O ukončení léčby se rozhoduje následujícím způsobem:
• U HBeAg-pozitivních pacientů bez cirhózy by měla léčba trvat po dobu nejméně 6-12 měsíců po potvrzení sérokonverze HBeAg (vymizení HBeAg a HBV DNA s průkazem anti-HBe) nebo do sérokonverze HBsAg nebo do doby, kdy se prokáže ztráta účinnosti. Sérová hladina ALT a HBV DNA by měla být po ukončení léčby pravidelně sledována z důvodu detekce pozdějšího virologického relapsu.
• U HBeAg-negativních pacientů bez cirhózy by měla léčba pokračovat nejméně do sérokonverze HBsAg, nebo dokud se neprokáže ztráta účinnosti. Při dlouhodobé léčbě, delší než 2 roky, je doporučeno pravidelné přehodnocování léčby, aby bylo zajištěno, že pokračování ve zvolené léčbě je pro pacienta vhodné.
Starší pacienti (více než 65 let)
Údaje pro stanovení specifického dávkovacího režimu u pacientů starších než 65 let nejsou dostupné (viz bod 4.4).
Porucha funkce ledvin
U pacientů s clearance kreatininu >50 ml/min není nutná úprava doporučené dávky telbivudinu. Úprava dávkování je nutná u pacientů s clearance kreatininu <50 ml/min, včetně pacientů v terminálním stadiu onemocnění ledvin (ESRD) na hemodialýze. K podávání snížené denní dávky se doporučuje použít perorální roztok, jak je popsáno níže v tabulce 1. Jestliže není možné použít perorální roztok, mohou se alternativně použít potahované tablety Sebivo a dávkování musí být upraveno tak, že se prodlouží interval mezi dávkami, jak je popsáno v tabulce 1.
Tabulka 1 Úprava režimu dávkování přípravku Sebivo u pacientů se zhoršenou funkcí ledvin
|
Clearance kreatininu (ml/min) |
Telbivudin 20 mg/ml perorální roztok Úprava denní dávky |
Telbivudin 600 mg potahované tablety Alternativní** úprava dávky s prodloužením intervalu mezi dávkami |
|
>50 |
600 mg(30 ml) jednou denně |
600 mg jednou denně |
|
30 - 49 |
400 mg (20 ml) jednou denně |
600 mg jednou za 48 hodin |
|
<30 (nevyžadující dialýzu) |
200 mg(10 ml) jednou denně |
600 mg jednou za 72 hodin |
|
ESRD* |
120 mg (6 ml) jednou denně |
600 mg jednou za 96 hodin |
* Terminální stadium onemocnění ledvin
** V případě, že není možné použít perorální roztok
Navrhované úpravy dávky jsou založeny na extrapolaci a nemusí být optimální. Bezpečnost a účinnost uvedených pokynů pro úpravu dávkování nebyly klinicky hodnoceny. Z tohoto důvodu je u těchto pacientů doporučeno pečlivé klinické sledování.
Pacienti s terminálním stadiem onemocnění ledvin
U pacientů s ESRD by měl být přípravek Sebivo podáván až po hemodialýze (viz bod 5.2).
Porucha funkce jater
U pacientů se zhoršenou funkcí jater není nutná úprava doporučeného dávkování přípravku Sebivo (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Sebiva u pediatrické populace nebyla dosud stanovena.
Způsob podání
Sebivo se užívá perorálně s jídlem nebo bez jídla.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Kombinace telbivudinu s pegylovaným nebo standardním interferonem alfa (viz body 4.4 a 4.5).
4.4 Zvláštní upozornění a opatření pro použití
Těžké akutní exacerbace chronické hepatitidy B jsou relativně časté a jsou charakterizovány přechodným zvýšením sérové ALT. Po zahájení antivirové léčby se mohou u některých pacientů zvýšit hladiny ALT v séru, zatímco hladiny HBV DNA klesnou (viz bod 4.8). U pacientů léčených telbivudinem uplynulo v průměru 4 - 5 týdnů před výskytem exacerbace. Celkově se rychlý vzestup ALT vyskytuje častěji u pacientů s pozitivním nálezem HBeAg, než u pacientů s negativním nálezem HBeAg. U pacientů s kompenzovaným jaterním onemocněním není toto zvýšení sérové ALT obvykle doprovázeno zvýšením hladin sérového bilirubinu nebo jinými známkami jaterní dekompenzace. Riziko jaterní dekompenzace -a následné exacerbace hepatitidy - může být zvýšeno u pacientů s cirhózou. Tito pacienti musí být proto pečlivě sledováni.
Exacerbace hepatitidy byla také hlášena u pacientů, u kterých byla léčba hepatitidy B ukončena. Rychlý vzestup hodnot ALT po léčbě je normálně doprovázen zvýšením hladin HBV DNA v séru a u většiny takovýchto případů bylo prokázáno, že se spontánně upraví. Nicméně byly hlášeny těžké - a někdy fatální - exacerbace po ukončení léčby onemocnění. Z tohoto důvodu mají být jaterní funkce monitorovány v pravidelných intervalech, a to jak klinicky, tak i laboratorně, nejméně po dobu 6 měsíců po ukončení léčby hepatitidy B.
Laktátová acidóza
Při užití nukleosidových/nukleotidových analogů byl hlášen, někdy fatální, výskyt laktátové acidózy (při absenci hypoxie), obvykle doprovázený těžkou hepatomegalií se steatózou. Protože telbivudin je nukleosidový analog, toto riziko nemůže být vyloučeno. Léčba nukleosidovými analogy musí být ukončena, když rychle stoupají hladiny aminotransferáz, dochází k progresivní hepatomegalii nebo výskytu metabolické/laktátové acidózy neznámé etiologie. Benigní zažívací symptomy, jako je nauzea, zvracení a bolesti břicha, mohou indikovat vývoj mléčné acidózy. Těžké případy, někdy s fatálním koncem, byly doprovázeny pankreatitidou, jaterním selháním/steatózou jater, selháním ledvin a vyššími hladinami laktátu v séru. Při předepisování nukleosidových analogů musí být věnována zvýšená opatrnost všem pacientům (především obézním ženám) s hepatomegalií, hepatitidou nebo jinými známými rizikovými faktory onemocnění jater. Tito pacienti musí být pečlivě sledováni.
Vliv na svalstvo
Při léčbě telbivudinem byly hlášeny případy myopatií a myalgií vyskytujících se několik týdnů až měsíců po zahájení terapie (viz bod 4.8). Byly hlášeny případy rabdomyolýzy v období po uvedení telbivudinu na trh (viz bod 4.8).
O myopatii, definované jako přetrvávající nevysvětlitelná svalová bolest a/nebo svalová slabost, bez ohledu na stupeň zvýšení hladin kreatinkinázy (CK), by se mělo uvažovat u kteréhokoliv pacienta s nevysvětlenou difúzní myalgií, svalovou citlivostí na dotek, svalovou slabostí nebo myozitidou (definovanou jako myopatie s histologickým důkazem svalového poškození). Pacienti by měli být upozorněni, aby okamžitě hlásili jakoukoli nevysvětlitelnou trvalou bolest svalů, citlivost na dotek nebo svalovou slabost. Jestliže se objeví jakýkoli z těchto příznaků, mělo by být provedeno podrobné vyšetření svalů za účelem vyhodnocení svalové funkce. Jestliže je diagnostikována myopatie, měla by být léčba telbivudinem ukončena.
Není známo, zda je riziko myopatie během léčby telbivudinem zvýšeno současným podáváním jiných léčivých přípravků, které jsou spojovány se vznikem myopatie (např. statiny, fibráty nebo cyklosporin). Lékaři uvažující o současné léčbě jinými přípravky, které mohou vyvolat myopatii, by měli pečlivě zvážit možné přínosy a rizika léčby, a měli by u pacientů pečlivě sledovat jakékoli příznaky nebo projevy naznačující myopatii.
Periferní neuropatie
Periferní neuropatie byla u pacientů léčených telbivudinem hlášena méně často. V případě podezření na periferní neuropatii by měla být léčba telbivudinem znovu zvážena (viz bod 4.8).
Zvýšené riziko rozvoje periferní neuropatie bylo pozorováno v jedné studii, když byly současně podávány telbivudin a pegylovaný interferon alfa-2a (viz bod 4.5). Takové zvýšené riziko nemůže být vyloučeno v případě podávání jiného interferonu alfa (pegylovaný či standardní). Mimoto není prospěšnost podávání kombinace telbivudinu s interferonem alfa (pegylovaným či standardním) v současné době stanovena. Proto je kombinace telbivudinu s pegylovaným nebo standardním interferonem alfa kontraindikována (viz bod 4.3).
Funkce ledvin
Telbivudin je vylučován primárně ledvinnou exkrecí, proto se u pacientů s clearance kreatininu <50 ml/min, včetně pacientů na hemodialýze, doporučuje úprava intervalu dávkování. Účinnost úpravy intervalu dávkování nebyla klinicky hodnocena. Proto by měla být u pacientů s prodlouženým intervalem dávkování pečlivě sledována virologická odpověď (viz body 4.2 a 5.2).
Pacienti s cirhózou bez dekompenzace
Vzhledem k omezenému dostupnému množství údajů (pouze 3% zařazených pacientů mělo cirhózu) by telbivudin měl být u pacientů s cirhózou používán se zvláštní opatrností. U těchto pacientů je v průběhu léčby a po ukončení léčby nutno důkladně monitorovat klinické, biochemické a virologické parametry spojené s hepatitidou B.
Pacienti s cirhózou s dekompenzací
U pacientů s dekompenzovanou cirhózou nejsou dostačující údaje o bezpečnosti a účinnosti.
Pacienti, kteří již byli nukleosidovvm/nukleotidovvm analogem léčeni
In vitro nebyl telbivudin účinný proti HBV kmenům obsahujícím rtM204V/rtL180M nebo rtM204I mutace (viz bod 5.1). Monoterapie telbivudinem není alternativou pro pacienty s prokázanou virovou hepatitidou B, rezistentní vůči lamivudinu. Pacienti, u kterých nedošlo k virologické odpovědi po více než 24 týdnů trvající léčbě lamivudinem, nebudou mít pravděpodobně z monoterapie telbivudinem prospěch. V současné době nejsou k dispozici klinické údaje, na základě kterých by bylo možno zhodnotit prospěšnost a rizika přechodu na telbivudin u pacientů, u kterých bylo při podávání lamivudinu dosaženo celkové virové suprese.
Nejsou k dispozici údaje o léčbě telbivudinem pacientů s prokázanými jednotlivými mutacemi rtN236T nebo A181V viru hepatitidy B rezistentními k adefoviru. Výsledky založené na buněčných testováních ukázaly, že substituce A181V spojená s rezistencí vůči adefoviru měla 1,5 až přibližně 4krát nižší citlivost na telbivudin.
Pacienti po transplantaci jater
Bezpečnost a účinnost telbivudinu u pacientů po transplantaci jater není známa.
Starší pacienti
Klinické studie s telbivudinem nezahrnovaly dostatečný počet pacientů starších >65 roků, aby bylo možné stanovit, zda odpovídají odlišně od mladých jedinců. Obecně musí být při předepisování přípravku Sebivo starším pacientům věnována zvýšená opatrnost z důvodu zvýšení frekvence výskytu snížené funkce ledvin způsobené souběžným onemocněním nebo souběžným užíváním jiných léčivých přípravků.
Další zvláštní populace
Přípravek Sebivo nebyl studován u pacientů s hepatitidou typu B infikovaných současně jinými viry (např. pacientů infikovaných současně virem lidské imunodeficience [HIV], virovou hepatitidou typu C [HCV] nebo virovou hepatitidou typu D [HDV]).
Obecně
Pacienti by měli být upozorněni, že léčba přípravkem Sebivo neprokázala snížení rizika přenosu HBV pohlavním stykem nebo krví na jiné osoby.
Použití telbivudinu v kombinaci s lamivudinem se nedoporučuje, protože v klinické studii fáze II byla pozorovaná odpověď na léčbu u kombinace telbivudin/lamivudin nižší než u léčby samotným telbivudinem.
Zatím neexistují údaje o bezpečnosti a účinnosti pro další antivirové kombinace s telbivudinem.
Sebivo perorální roztok obsahuje v dávce 600 mg (30 ml) asi 47 mg sodíku, který musí být brán do úvahy u pacientů na kontrolované sodíkové dietě.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vzhledem k tomu, že je telbivudin vylučován především renální exkrecí, může současné podávání přípravku Sebivo s látkami, které ovlivňují funkci ledvin (jako aminoglykosidy, kličková diuretika, sloučeniny platiny, vankomycin, amfotericin B), ovlivnit plazmatické koncentrace telbivudinu a/nebo souběžně podávaných látek. Kombinaci telbivudinu s těmito léčivými přípravky lze užívat pouze s opatrností. Farmakokinetika telbivudinu v ustáleném stavu nebyla změněna při opakovaném podávání v kombinaci s lamivudinem, adefovir-dipivoxilem, tenofovir-disoproxil-fumarátem, cyklosporinem nebo pegylovaným interferonem alfa-2a. Navíc telbivudin nemění farmakokinetiku lamivudinu, adefovir-dipivoxilu, tenofovir-disoproxil-fumarátu nebo cyklosporinu. Definitivní závěr týkající se účinků telbivudinu na farmakokinetiku pegylovaného interferonu nemohl být udělán z důvodu vysoké interindividuální variability koncentrací pegylovaného interferonu alfa-2a. Klinická studie sledující kombinaci telbivudinu 600 mg denně s pegylovaným interferonem alfa-2a 180 mikrogramů podaným subkutánně jednou týdně ukázala, že je tato kombinace spojena se zvýšeným rizikem vzniku periferní neuropatie. Mechanismus těchto účinků není znám (viz bod 4.4). Kombinace telbivudinu s přípravkem obsahujícím jakýkoli interferon alfa je kontraindikována (viz bod 4.3).
Telbivudin není substrát, induktor nebo inhibitor enzymového systému cytochromu P450 (CYP450) (viz bod 5.2). Z tohoto důvodu je možnost lékových interakcí, zprostředkovaných CYP450, které se týkají přípravku Sebivo, nízká.
4.6 Fertilita, těhotenství a kojení
Studie na zvířatech nenaznačují přímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3). Studie na březích potkanech a králících ukázaly, že telbivudin přestupuje přes placentu. Studie na březích králících ukázaly dřívější vrh mláďat a/nebo potrat závisející na toxicitě pro matku.
Omezené klinické údaje (méně než 300 ukončených těhotenství) po expozici telbivudinu během prvního trimestru nenaznačují žádnou malformační toxicitu a údaje od většího počtu pacientek (více než 1000 ukončených těhotenství) po expozici telbivudinu během druhého a třetího trimestru nenaznačují žádnou fetální/neonatální toxicitu.
Přípravek Sebivo by měl být během těhotenství používán pouze v případě, že přínos pro matku převáží potenciální riziko pro plod.
Údaje z literatury ukazují, že expozice telbivudinu ve druhém a třetím trimestru těhotenství snížila riziko přenosu HBV z matky na kojence, pokud byl telbivudin podáván společně s imunoglobulinem a vakcínou proti hepatitidě B.
Kojení
Telbivudin je vylučován do mléka potkanů. Není známo, zda je telbivudin vylučován do lidského mateřského mléka. Ženy, které užívají přípravek Sebivo, by neměly kojit.
Fertilita
Nejsou k dispozici údaje o účincích telbivudinu na mužskou nebo ženskou fertilitu. V reprodukčních toxikologických studiích u dospělých zvířat byla fertilita mírně snížena, když byl samcům i samicím potkanů podán telbivudin. Nežádoucí účinky na fertilitu byly významnější v oddělené studii u nedospělých zvířat, když byl oběma pohlavím podán telbivudin (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie hodnotící účinky na schopnost řídit nebo obsluhovat stroje nebyly provedeny.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Zhodnocení nežádoucích účinků je založeno převážně na dvou studiích, NV-02B-007 (GLOBE) a NV-02B-015, ve kterých 1 699 pacientů s chronickou hepatitidou typu B dostávalo v dvojitě slepém uspořádání telbivudin 600 mg/den (n = 847) nebo lamivudin (n = 852) po dobu 104 týdnů.
Ve 104týdenních klinických studiích byly hlášené nežádoucí účinky obvykle klasifikovány jako mírné nebo středně těžké. Nejčastějšími nežádoucími účinky byly zvýšení kreatinkinázy (6,8%) stupně 3 nebo 4, únava (4,4%), bolest hlavy (3,0%) a nauzea (2,6%).
Seznam nežádoucích účinků seřazený v tabulce
V tabulce 2 j sou uvedeny nežádoucí účinky tříděné dle tříd orgánových systémů podle MedDRA a frekvence výskytu dle následujících kriterií: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000); není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 2 Nežádoucí účinky
|
Poruchy metabolismu a výživy | |
|
Vzácné* |
Laktátová acidóza jako sekundární příhoda často spojená se závažnými stavy (např. multiorgánové selhání nebo sepse) |
|
Poruchy nervového systému | |
|
Časté |
Závratě, bolesti hlavy |
|
Méně časté |
Periferní neuropatie, porucha chuti, hypestezie, parestezie, ischias |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté | |
|
Gastrointestinální poruchy | |
|
Časté | |
|
Poruchy kůže a podkožní tkáně | |
|
Časté |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Méně časté |
Myopatie/myozitida, artralgie, myalgie, bolesti končetin, bolesti zad, spasmy svalů, bolesti krku, bolesti v boku (mezi hrudníkem a iliem) |
|
Vzácné* |
Rabdomyolýza |
|
Celkové poruchy a reakce v místě aplikace | |
|
Časté |
Únava |
|
Méně časté | |
|
Vyšetření | |
|
Časté |
Zvýšení kreatinfosfokinázy v krvi, zvýšení alaninaminotransferázy v krvi, zvýšení krevní amylázy |
|
Méně časté |
Zvýšení aspartátaminotransferázy |
|
* Tyto nežádoucí účinky byly popsány po uvec |
ení přípravku na trh, ale nebyly pozorovány |
v kontrolovaných klinických studiích. Četnost výskytu byla odhadnuta ze statistického výpočtu na základě celkového počtu pacientů léčených telbivudinem v klinických studiích (n = 8 914).
Popis vybraných nežádoucích reakcí
Zvýšení kreatinkinázy
Ve sloučené analýze NV-02B-007 (GLOBE) a NV-02B-015 se do 104. týdne léčby se vyskytl stupeň 3 nebo 4 zvýšení CK (>7x ULN) u 12,6% pacientů léčených telbivudinem (n = 847) a u 4,0% pacientů léčených lamivudinem (n = 846). Zvýšení CK bylo ve většině případů asymptomatické a hodnoty CK obvykle klesly do příští návštěvy při pokračování v léčbě.
Rychlé zvýšení ALT
Incidence rychlého vzestupu alaninaminotransferázy (ALT) při léčbě v obou léčených větvích podle definice AASLD (American Association for the Study of Liver Disease) (elevace ALT >2x výchozí hodnota a >10x horní hranice normy (ULN)) je dále popsána v níže uvedené tabulce 3.
Tabulka 3 Přehled vzestupu ALT při léčbě - sloučené studie NV-02B-007 (GLOBE) a NV-02B-015
|
Vzestup ALT: |
Lamivudin |
Telbivudin |
|
elevace ALT >2x výchozí a >10x ULN |
n/N (%) |
n/N (%) |
|
Celkově |
67/852 (7,9) |
41/847 (4,8) |
|
Od výchozí hodnoty do 24. týdne |
25/852 (2,9) |
25/847 (3,0) |
|
Od 24. týdne do konce studie |
44/837 (5,3) |
17/834 (2,0) |
Během léčby se doporučuje pravidelné sledování jaterních funkcí (viz bod 4.4).
Exacerbace hepatitidy B po ukončení léčby
U pacientů, kteří ukončili léčbu hepatitidy B, byly hlášeny případy těžké akutní exacerbace hepatitidy B (viz bod 4.4), včetně pacientů léčených telbivudinem.
Incidence rychlého vzestupu alaninaminotransferázy (ALT) po léčbě v obou léčených větvích je dále popsána v níže uvedenéých tabulce 4.
Tabulka 4 Přehled vzestupu ALT po léčbě - sloučené studie NV-02B-007 (GLOBE) a NV-02B-015
|
Lamivudin |
Telbivudin | |
|
Vzestup ALT |
n/N (%) |
n/N (%) |
|
elevace ALT >2x výchozí hodnota a >10x ULN |
10/180 (5,6) |
9/154 (5,8) |
Výsledky ve 208. týdnu
Po 104 týdnech léčby telbivudinem bylo 78% pacientů (530/680) ze studie NV-02B-007 (GLOBE) a 82% pacientů (137/167) ze studie NV-02B-015 převedeno do extenzní studie CLDT600A2303 (viz bod 5.1), aby pokračovalo v léčbě až do 208. týdne. Soubor pacientů pro sledování dlouhodobé bezpečnosti se skládal z 655 pacientů, včetně 518 z NV-02B-007 (GLOBE) a 137 z NV-02B-015. Celkový bezpečnostní profil ze sloučené analýzy až do 104. a 208. týdne byl podobný. Zvýšení CK stupně 3 nebo 4 se nově vyskytlo u 15,9% pacientů léčených telbivudinem 208 týdnů. Většina případů zvýšení CK stupně 3 nebo 4 byla asymptomatická a přechodná.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nejsou informace o záměrném předávkování telbivudinem, ale u jednoho jedince došlo neúmyslně k předávkování, které bylo asymptomatické. Testované dávky až do dávky 1 800 mg/den, třikrát vyšší než doporučená denní dávka, byly dobře tolerovány. Maximální tolerovaná dávka telbivudinu nebyla stanovena. V případě předávkování musí být podávání přípravku Sebivo přerušeno a podle potřeby zahájena odpovídající podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antivirotika k systémovému užití, nukleosidové a nukletidové inhibitory reverzní transkriptázy, ATC kód: J05AF11
Mechanismus účinku
Telbivudin je syntetický analog nukleosidu thymidinu s účinností proti HBV DNA polymeráze. Je účinně fosforylován buněčnými kinázami na účinnou trifosfátovou formu, která má nitrobuněčný poločas 14 hodin. Telbivudin-5'-trifosfát inhibuje HBV DNA polymerázu (reverzní transkriptázu) kompeticí s přirozeným substrátem, thymidin 5'-trifosfátem. Inkorporace telbivudin-5'-trifosfátu do virové DNA vyvolá ukončení řetězce DNA, což vede k inhibici replikace HBV.
Farmakodynamické účinky
Telbivudin je inhibitor syntézy jak prvního vlákna HBV (EC50 = 0,4 - 1,3 pM), tak i druhého vlákna HBV (EC50 = 0,12 - 0,24 pM), a vykazuje výraznou preferenci pro inhibici produkce druhého vlákna. Oproti tomu telbivudin-5'-trifosfát neinhiboval v koncentracích do 100 pM buněčnou DNA polymerázu a, p nebo y. Při sledování mitochondriální struktury, funkce a obsahu DNA, nevykazoval telbivudin prokazatelný toxický účinek v koncentracích až do 10 pM a nezvyšoval tvorbu kyseliny mléčné in vitro.
Antivirová účinnost telbivudinu in vitro byla hodnocena u HBV-exprimujících buněčných linií lidského hepatomu 2.2.15. Koncentrace telbivudinu, která účinně inhibovala 50% virové syntézy (EC50), byla přibližně 0,2 pM. Antivirová účinnost telbivudinu je specifická pro virus hepatitidy B a příbuzné hepadnaviry. Telbivudin není účiný proti HIV in vitro. V klinických studiích nebyla absence účinnosti telbivudinu proti HIV hodnocena.
Klinické zkušenosti
Bezpečnost a účinnost dlouhodobé léčby (104 týdnů) přípravkem Sebivo byly hodnoceny ve dvou, léčivou látkou kontrolovaných klinických studiích, do kterých bylo zařazeno 1 699 pacientů s chronickou hepatitidou B (NV-02B-007 (GLOBE) a NV-02B-015).
Studie NV-02B-007 (GLOBE)
NV-02B-007 GLOBE studie je randomizovaná, dvojitě-slepá mezinárodní studie fáze III, kde byl telbivudin srovnáván s lamivudinem po dobu 104 týdnů u 1 367 pacientů, kteří dosud nedostávali žádné nukleosidy a měli chronickou hepatitidu B a byli jak HBeAg-pozitivní, tak i HBeAg-negativní. Většina zahrnuté populace byli asiaté. Nejčastějšími HBV genotypy byly B (26%) a C (51%). Telbivudinem bylo léčeno malé množství pacientů bělošské rasy (celkem 98). Primární analýza dat byla provedena po dosažení 52. týdne u všech pacientů.
HBeAg-pozitivní pacienti: Střední věk pacientů byl 32 let, 74% byli muži, 82% byli asiaté, 12% byli běloši a 6% bylo dříve léčeno interferonem alfa.
HBeAg-negativní pacienti: Střední věk pacientů byl 43 let, 79% byli muži, 62% asiaté, 23% běloši a 11% bylo dříve léčeno interferonem alfa.
Klinické výsledky v 52.týdnu
Klinické a virologické cílové parametry účinnosti byly hodnoceny odděleně u HBeAg-pozitivních a HBeAg-negativních pacientů. Primární cílový parametr terapeutické odpovědi byl složen ze serologických cílových parametrů vyžadujících supresi HBV DNA na <5 log10 kopií/ml ve spojení buď se ztrátou sérového HBeAg, nebo normalizací ALT. Sekundární cílové parametry zahrnovaly histologickou odpověď, normalizaci ALT a různé změny antivirové účinnosti.
Bez ohledu na charakteristiku výchozích hodnot se u většiny pacientů užívajících přípravek Sebivo projevila histologická, virologická, biochemická a serologická odpověď na léčbu. Výchozí hladiny ALT >2x ULN a výchozí HBV DNA <9 log10 kopií/ml byly spojeny s vyšší rychlostí sérokonverze HBeAg u HBeAg-pozitivních pacientů. Pacienti, kteří dosáhli hladin HBV DNA <3 log10 kopií/ml do 24. týdne, odpovídali na léčbu optimálně; naopak u pacientů s hladinami HBV DNA ve 24. týdnu >4 logi0 kopií/ml bylo v 52. týdnu dosaženo méně příznivých výsledků.
U HBeAg-pozitivních pacientů byl telbivudin účinnější než lamivudin v terapeutické odpovědi (75,3% vs 67,0% reagujících pacientů; p = 0,0047). U HBeAg-negativních pacientů, telbivudin nebyl lepší než lamivudin (75,2% a 77,2% reagujících pacientů; p = 0,6187). U bělochů byla léčebná odpověď u obou antivirových látek použitých ve studii NV-02B-007 (GLOBE) nižší; počet bělošských pacientů byl však velmi omezený (n = 98).
Ve 24. týdnu dosáhlo 203 HBeAg-pozitivních pacientů a 177 HBeAg-negativních pacientů nedetekovatelných hladin HBV DNA. Z HBeAg-pozitivních pacientů 95% dosáhlo nedetekovatelných hladin HBV DNA, 39% dosáhlo HBeAg sérokonverze, 90% dosáhlo normalizace ALT v 52.týdnu a 0,5% bylo ve 48. týdnu rezistentních. Podobně z HBeAg-negativních pacientů 96% dosáhlo nedetekovatelné hladiny HBV DNA, 79% dosáhlo v 52.týdnu normalizace ALT a 0% bylo rezistentních ve 48. týdnu.
Vybrané virologické, biochemické a sérologické výsledky vyšetření jsou uvedeny v tabulce 5 a histologické odpovědi v tabulce 6.
|
Parametr odpovědi |
HBeAg-pozitivní (n = 921) |
HBeAg-negativní (n = 446) | ||
|
Telbivudin 600 mg (n = 458) |
Lamivudin 100 mg (n = 463) |
Telbivudin 600 mg (n = 222) |
Lamivudin 100 mg (n = 224) | |
|
Střední hodnota snížení HBV DNA od výchozí hodnoty (log10 kopií/ml) ± SE M1,2,3 |
-6,45 (0,11) * |
-5,54 (0,11) |
-5,23 (0,13) * |
-4,40 (0,13) |
|
% pacientů HBV DNA nedetekovatelných metodou PCR |
60%* |
40% |
88%* |
71% |
|
normalizace ALT 4 |
77% |
75% |
74% |
79% |
|
sérokonverze HBeAg 4 |
23% |
22% |
- |
- |
|
vymizení HBeAg 5 |
26% |
23% |
- |
- |
1 SEM: Standard error of mean (Standardní odchylka od střední hodnoty).
2 Roche COBAS Amplicor® PCR test (dolní limit kvantifikace <300 kopií/ml).
3 HBeAg-pozitivní n = 443 ve skupině telbivudinu a 444 ve skupině s lamivudinem; HBeAg-negativní n = 219 ve skupině s telbivudinem a 219 ve skupině s lamivudinem. Rozdíly ve skupinách jsou způsobeny ukončením léčby pacientů v průběhu studie a chybějícím hodnocením HBV DNA v 52.týdnu
4 HBeAg-pozitivní n = 440 ve skupině telbivudinu a 446 ve skupině s lamivudinem; HBeAg-negativní n = 203 ve skupině telbivudinu a 207 ve skupině s lamivudinem. ALT normalizace byla stanovena pouze u pacientů s ALT > ULN ve výchozích hodnotách.
5 n = 432 ve skupině telbivudinu a 442 ve skupině s lamivudinem. HBeAg sérokonverze a vymizení bylo sledováno pouze u pacientů s detekovatelným HBeAg při výchozím vyšetření.
*p < 0,0001
|
HBeAg-pozitivní (n = 921) |
HBeAg-negativní (n = 446) | |||
|
Telbivudin 600 mg (n = 384)1 |
Lamivudin 100 mg (n = 386)1 |
Telbivudin 600 mg (n = 199)1 |
Lamivudin 100 mg (n = 207)1 | |
|
Histologická odpověď2 | ||||
|
Zlepšení |
71%* |
61% |
71% |
70% |
|
Žádné zlepšení |
17% |
24% |
21% |
24% |
|
Ishakovo skóre fibrózy (Ishak Fibrosis Score)3 | ||||
|
Zlepšení |
42% |
47% |
49% |
45% |
|
Beze změn |
39% |
32% |
34% |
43% |
|
Zhoršení |
8% |
7% |
9% |
5% |
|
Chybějící biopsie v 52.týdnu |
12% |
15% |
9% |
7% |
|
1 Pacienti s > než jednou dávkou studijní medi hodnotou Knodellova indexu histologické akti 2 Histologická odpověď definovaná jako >2bo< skóre od výchozí hodnoty bez zhoršení Knode 3 Zlepšení Ishakova skóre fibrózy, bylo stanov výchozí hodnoty v 52. týdnu. *p = 0,0024 |
kace s hodnotitelnou výchozí biopsií jater a výchozí vity (HAI) skóre >3. ový pokles Knodellova zánětlivě nekrotického lova skóre fibrózy. eno jako snížení o >1 bod v Ishakově skóre od | |||
Klinické výsledky ve 104.týdnu
Klinické výsledky ve 104. týdnu byly u pacientů léčených telbivudinem konzistentní s výsledky v 52.týdnu a ukázaly přetrvávání účinnosti telbivudinu při pokračování v léčbě.
Mezi HBeAg-pozitivními pacienty byl mezi telbivudinem a lamivudinem ve 104. týdnu demonstrován zvětšující se rozdíl v terapeutické odpovědi (63% vs 48%; p < 0,0001) a klíčových sekundárních cílových parametrech (průměrná log10 snížení HBV DNA: -5,74 vs -4,42; p < 0,0001, nedetekovatelnost HBV DNA: 56% vs 39%; p < 0,0001 a normalizace ALT 70% vs 62%). Pro telbivudin byl také pozorován trend směrem k vyššímu výskytu ztrát HBeAg (35% vv s 29%) a vyšší frekvenci sérokonverze (30% vs 25%). Navíc, v podskupině pacientů s výchozí hodnotou ALT >2x ULN (320), výrazně vyšší podíl pacientů léčených telbivudinem oproti pacientům léčených lamivudinem dosáhl ve 104. týdnu sérokonverze HBeAg (36% oproti 28%).
U HBeAg-negativních pacientů byly rozdíly v terapeutické odpovědi (78% vs 66%) a klíčových sekundárních cílových parametrech (průměrný log10 snížení hladin HBV DNA: -5,00 vs -4,17, a nedetekovatelnost HBV DNA: 82% vs. 57%; p < 0,0001) vyšší pro telbivudin až do 104. týdne. Normalizace ALT (78% vs 70%) pokračovala a byla ve 104. týdnu vyšší.
Předpověditelnost v 24.týdnu
Ve 24. týdnu 203 telbivudinem léčených pacientů HBeAg-pozitivních (44%) a 177 pacientů HBeAg-negativních (80%) dosáhlo nedetekovatelných hladin HBV DNA.
Výsledky HBV DNA ve 24. týdnu byly pro obě skupiny HBeAg-pozitivních i HBeAg-negativních pacientů prediktorem dlouhodobého příznivého výsledku. Telbuvidinem léčení pacienti, kteří dosáhli nedetekovatelných hladin HBV DNA metodou PCR ve 24. týdnu, měli nejvyšší poměr nedetekovatelnosti HBV DNA a sérokonverze HBeAg (u HBeAg-pozitivních pacientů) a nejnižší celkový výskyt virologického vzplanutí (breakthrough) ve 104. týdnu.
Závěrečné výsledky ve 104. týdnu, založené na hladině HBV DNA ve 24. týdnu jsou, jak pro HBeAg-pozitivní nebo HBeAg-negativní pacienty, uvedeny v tabulce 7.
Tabulka 7 Klíčové cílové parametry účinnosti ve 104.týdnu při sérových hladinách HBV DNA ve 24.týdnu u pacientů léčených telbivudinem ve studii NV-02B-007 (GLOBE)
|
HBV DNA ve 24.týdnu |
Výsledky klíčových cílových parametrů účinnosti ve 104. týdnu založené na výsledcích ve 24. týdnu | ||||
|
Terapeutická odpověď n/N (%) |
nedetekovateln á hladina HBV DNA n/N (%) |
sérokonverze HBeAg n/N (%) |
normalizace ALT n/N (%) |
Virologický breakthrough* n/N (%) | |
|
HBeAg-pozitivní | |||||
|
<300 kopií/ml |
172/203 (85) |
166/203 (82) |
84/183 (46) |
160/194 (82) |
22/203 (11) |
|
300 kopií/ml až <3 log10 kopií/ml |
36/57 (63) |
35/57 (61) |
21/54 (39) |
40/54 (74) |
18/57 (32) |
|
>3 log10 kopií/ml |
82/190 (43) |
54/190 (28) |
23/188(12) |
106/184 (58) |
90/190 (47) |
|
HBeAg-negativní | |||||
|
<300 kopií/ml |
146/177 (82) |
156/177 (88) |
N/A |
131/159 (82) |
11/177 (6) |
|
300 kopií/ml až <3 log10 kopií/ml |
13/18 (72) |
14/18 (78) |
N/A |
13/17 (76) |
4/18 (22) |
|
>3 log10 kopií/ml |
13/26 (50) |
12/26 (46) |
N/A |
14/26 (54) |
12/26 (46) |
N/A = neuplatňuje se
* virologický breakthrough: “1 log nad dolní hodnotu nadir” definice stanovené ve 104. týdnu
Studie NV-02B-015
Výsledky účinnosti a bezpečnosti studie NV-02B-007 (GLOBE) byly potvrzeny ve studii NV-02B-015. Tato studie je studie fáze III, randomizovaná, dvojitě slepá, srovnávající telbivudin 600 mg podávaný jednou denně s lamivudinem 100 mg, podávaným jednou denně po dobu 104 týdnů, 332 čínským pacientům s chronickou hepatitidou B, HBeAg-pozitivním a HBeAg-negativním, kteří nebyli předtím nukleosidy léčeni.
Studie CLDT600A2303 - klinické výsledky po 208 týdnech
Studie CLDT600A2303 byla otevřená 104týdenní extenzní studie u pacientů s kompenzovanou chronickou hepatitidou B, kteří byli předtím léčeni telbivudinem po dobu 2 let, včetně pacientů ze studií NV-02B-007 (GLOBE) a NV-02B-015, poskytující údaje o účinnosti a bezpečnosti telbivudinu po 156 a 208 týdnech kontinuální léčby. Pacienti s nedetekovatelnou hladinou HBV DNA ve 24. týdnu léčby měli lepší výsledky ve 156. a 208. týdnu (Tabulka 8).
Tabulka 8 Analýza účinnosti ze sloučených údajů studií NV-02B-007 (GLOBE), NV-02B-015 a CLDT600A2303
|
Týden 52 |
Týden 104 |
Týden 156 |
Týden 208 | |
|
HBeAg-pozitivní pacienti (n = 293*) | ||||
|
Průběžně nedetekovatelné hladiny HBV DNA (<300 kopií/ml) |
70,3% (206/293) |
77,3% (218/282) |
75,0% (198/264) |
76,2% (163/214) |
|
Průběžně nedetekovatelné hladiny HBV DNA (<300 kopií/ml) s nedetekovatelnou hladinou HBV DNA v týdnu 24 |
99,4% (161/162) |
94,9% (150/158) |
86,7% (130/150) |
87,9% (109/124) |
|
Kumulativní poměr HBeAg sérokonverze (%) |
27,6% (81/293) |
41,6% (122/293) |
48,5% (142/293) |
53,2% (156/293) |
|
Kumulativní poměr HBeAg sérokonverze u pacientů s nedetekovatelnou hladinou HBV DNA v týdnu 24 (%) |
40,1% (65/162) |
52,5% (85/162) |
59,3% (96/162) |
65,4% (106/162) |
|
Průběžná ALT normalizace |
81,4% (228/280) |
87,5% (237/271) |
82,9% (209/252) |
86,4% (178/106) |
|
HBeAg-negativnípacienti (n = 209*) | ||||
|
Průběžně nedetekovatelné hladiny HBV DNA (<300 kopií/ml) |
95,2% (199/209) |
96,5% (195/202) |
84,7% (160/189) |
86,0% (141/164) |
|
Průběžně nedetekovatelné hladiny HBV DNA (<300 kopií/ml) s nedetekovatelnou hladinou HBV DNA v týdnu 24 |
97,8% (175/179) |
96,5% (166/172) |
86,7% (143/165) |
87,5% (126/144) |
|
Průběžná ALT normalizace |
80,3% (151/188) |
89,0% (161/181) |
83,5% (142/170) |
89,6% (129/144) |
* Soubor pacientů bez virové rezistence na začátku studie CLDT600A2303 se skládal z 502 pacientů (293 HBeAg-pozitivních a 209 HBeAg-negativních).
Studie CLDT600ACN04E1 - vliv léčby na histologii _jater
Ve studii CLDT600ACN04E1 byly u 57 pacientů s dostupnou jatemí biopsií na počátku léčby a po průměrně 260,8 týdnech léčby hodnoceny změny v histologii jater (38 HBeAg-pozitivních a 19 HBeAg-negativních pacientů).
• Průměrná výchozí hodnota Knodellova zánětlivě nekrotického skóre 7,6 (SD 2,9) se zlepšila
(p < 0,0001) na 1,4 (SD 0,9) s průměrnou změnou -6,3 (SD 2,8). Knodellovo zánětlivě nekrotické skóre <3 (žádné nebo minimální zánětlivě nekrotické změny) bylo pozorováno u 98,2% (56/57) pacientů.
• Průměrná výchozí hodnota Ishakova skóre 2,2 (SD 1,1) se zlepšila (p < 0,0001) na 0,9 (SD 1,0)
s průměrnou změnou -1,3 (SD 1,3). Ishakovo skóre fibrózy <1 (žádná nebo minimální fibróza) bylo pozorováno u 84,2% (48/57) pacientů.
Změny Knodellova zánětlivě nekrotického skóre a Ishakova skóre byly podobné u HBeAg-pozitivních i HBeAg-negativních pacientů.
CLDT600A2303 - trvání HBeAg odpovědi po ukončení léčby
Studie CLDT600A2303 zahrnovala HBeAg-pozitivní pacienty ze studií NV-02B-007 (GLOBE) nebo NV-02B-015, kteří byli sledováni po ukončení léčby. Tito pacienti dokončili >52týdenní léčbu telbivudinem a vykazovali pokles HBeAg po dobu >24 týdnů s HBV DNA <5 logi0 kopiemi/ml při poslední kontrole. Střední délka léčby byla 104 týdnů. Po střední délce sledování 120 týdnů po ukončení léčby prokázala většina HBeAg-pozitivních pacientů léčených telbivudinem trvalý pokles HBeAg (83,3%; 25/30) a trvalou HBeAg sérokonverzi (79,2%; 19/24). Pacienti s trvalou HBeAg sérokonverzí měli střední hodnotu HBV DNA 3,3 logJ0 kopií/ml; a 73,7% mělo HBV DNA <4 logJ0 kopií/ml.
Klinická, rezistence
Test genotypové rezistence byl proveden ve studii NV-02B-007 (GLOBE; n = 680) u pacientů s virologickým reboundem (potvrzeno zvýšení o >1 log10 kopií/ml HBV DNA od nejnižší hodnoty/nadir).
Ve 48. týdnu mezi pacienty léčenými telbivudinem mělo 5% (23/458) HBeAg-pozitivních pacientů a 2% (5/222) HBeAg-negativních pacientů virologický rebound s detekovatelnými rezistentními mutacemi HBV.
Studie NV-02B-007 (GLOBE) a CLDT600A2303 - kumulativní poměry genotypové rezistence Prvotní analýza kumulativní genotypové rezistence ve 104. a 208. týdnu byla založena na souboru ITT a zahrnovala všechny pacienty, kteří pokračovali v léčbě do 4. roku bez ohledu na HBV DNA hladiny.
Z 680 pacientů léčených telbivudinem, kteří byli od počátku zařazeni do pivotní studie NV-02B-007 (GLOBE), bylo 517 pacientů (76%) zahrnuto do studie CLDT600A2303 s pokračující léčbou telbivudinem po dobu až 208 týdnů. Z těchto 517 pacientů mělo 159 pacientů (HBeAg-pozitivní=135, HBeAg-negativní=24) detekovatelnou hladinu HBV DNA.
Kumulativní genotypové četnosti ve 104. týdnu byly 25,1% (115/458) pro HBeAg-pozitivní pacienty a 10,8% (24/222) pro HBeAg-negativní pacienty.
V celkovém ITT souboru byly poměry kumulativní rezistence ve 4. roce 40,8% (131/321) u HBeAg-pozitivních pacientů a 18,9% (37/196) HBeAg-negativních pacientů.
Kumulativní poměry genotypové rezistence byly také stanoveny pomocí matematického modelu, který zvažoval pouze pacienty s nedetekovatelnou hladinou HBV DNA počátkem příslušného roku. V této analýze byly poměry kumulativní rezistence ve 4. roce 22,3% u HBeAg-pozitivních pacientů a 16,0% u HBeAg-negativních pacientů.
Jestliže zhodnotíme pacienty, u kterých došlo do 104. týdne ve studii NV-02B-007 (GLOBE) k virologickému breakthrough, byla četnost rezistence nižší u pacientů s HBV DNA <300 kopií/ml ve 24. týdnu, než u pacientů s HBV DNA >300 kopií/ml ve 24. týdnu. U HBeAg-pozitivních pacientů s HBV DNA <300 kopií/ml ve 24. týdnu byla rezistence 1% (3/203) ve 48. týdnu a 9% (18/203) ve 104. týdnu, zatímco u pacientů s HBV DNA >300 kopií/ml byla rezistence 8% (20/247) ve 48. týdnu a 39% (97/247) ve 104. týdnu. U HBeAg-negativních pacientů s HBV DNA <300 kopií/ml ve 24. týdnu byla rezistence 0% (0/177) ve 48. týdnu a 5% (9/177) ve 104. týdnu, zatímco u pacientů s HBV DNA >300 kopií/ml byla rezistence 11% (5/44) ve 48. týdnu a 34% (15/44) ve 104. týdnu.
Charakter genotvpovvch mutací a zkřížená rezistence
Genotypové analýzy 203 hodnotitelných párových vzorků s HBV DNA >1 000 kopií/ml ve 104. týdnu (NV-02B-007 (GLOBE)) ukázaly, že primární mutace, která souvisela s rezistencí k telbivudinu byla rtM204I. Často byla doprovázena mutacemi rtL180M a rtL80I/V a občas rtV27A, rtL82M, rtV173L, rtT184I a rtA200V. Základní faktory (přítomné při vstupu do studie) sdružené s vývojem genotypové lékové rezistence zahrnovaly: léčbu lamivudinem, vyšší výchozí HBV DNA, nižší výchozí hodnoty sérové ALT a zvýšenou tělesnou hmotnost/BMI. Parametry léčebné odpovědi ve 24. týdnu, které předpovídaly vývoj rezistence viru na léky do 104. týdne, byly HBV DNA >300 kopií/ml a zvýšení ALT v séru.
Genotypová analýza 50 HBV izolátů od pacientů léčených telbivudinem ve 208. týdnu (CLDT600A2303) odhalila podobný profil rezistence, jaký byl hlášen ve 104. týdnu. Konverze na pozici 80, 180 a polymorfních pozicích 91, 229 byly vždy zjištěny v sekvencích, které obsahovaly mutaci M204I, která vyvolává genotypovou rezistenci. Jde velmi pravděpodobně o mutace kompenzační. U pacientů léčených telbivudinem, u kterých došlo ke vzniku virologického breakthrough do 208. týdne, byla hlášena jedna izolovaná rtM204V mutace a dvě rtM204I/V/M mutace. Nebyla hlášená žádná nová mutace.
Zkřížená rezistence byla pozorována mezi nukleosidovými analogy HBV (viz bod 4.4). HBV kmeny rezistentní vůči lamivudinu, které obsahovaly buď mutaci rtM204I nebo dvojitou mutaci rtL180M/rtM204V, měly v buněčných testech >1 000násobně sníženou citlivost k telbivudinu. HBV kódující substituce rtN236T nebo rtA181V, které souvisejí s rezistencí vůči adefoviru, měly v buněčné kultuře přibližně 0,3, resp. 4násobné změny citlivosti k telbivudinu (viz bod 4.4).
5.2 Farmakokinetické vlastnosti
Farmakokinetika telbivudinu po jednorázovém a opakovaném podávání byla sledována u zdravých jedinců i u pacientů s chronickou hepatitidou B. Farmakokinetika telbivudinu v doporučené dávce 600 mg nebyla hodnocena u pacientů s chronickou hepatitidou B. Farmakokinetika telbivudinu je však u obou těchto populací podobná.
Absorpce
Po perorálním podání jedné dávky 600 mg telbivudinu zdravým jedincům (n = 42), byla vrcholová koncentrace (Cmax) telbivudinu v plazmě 3,2 ± 1,1 ^g/ml (střední hodnota ± SD) a bylo jí dosaženo za 3 hodiny (medián) po podání dávky. Pro telbivudin byla plocha pod křivkou plazmatické koncentrace v čase (AUC0-<X)) 28,0 ± 8,5 ^g*h/ml (střední hodnota ± SD). Interindividuální variabilita (CV%) pro měření systémové expozice (Cmax, AUC) byla typicky přibližně 30%. Potahované tablety obsahující 600 mg telbivudinu jsou bioekvivalentní 30 ml perorálního roztoku telbivudinu (20 mg/ml).
Vliv potravy na absorpci po perorálním podání
Po jednorázovém podání 600 mg telbivudinu s potravou nebyla jeho absorpce a expozice ovlivněna. Distribuce
In vitro je vazba telbivudinu na lidské plazmatické proteiny nízká (3,3%).
Biotransformace
Po aplikaci 14C-telbivudinu lidem nebyly detekovány žádné metabolity. Telbivudin není ani substrát, inhibitor ani induktor enzymatického systému cytochromu P450 (CYP450).
Eliminace
Po dosažení vrcholu plazmatické koncentrace telbivudinu dochází k jeho poklesu biexponenciálně s terminálním eliminačním poločasem (t1/2) 41,8 ± 11,8 hodin. Telbivudin je primárně vylučován v nezměněné formě močí. Renální clearance telbivudinu dosahuje hodnot normální glomerulární filtrace, což ukazuje, že filtrace je hlavním mechanizmem exkrece. Přibližně 42% z podané dávky se objeví v moči v průběhu 7 dnů po perorálním podání jedné dávky 600 mg telbivudinu. Protože je renální exkrece hlavní cestou vylučování, vyžadují pacienti se střední až těžkou renální dysfunkcí a dialyzovaní pacienti úpravu dávkovacího intervalu (viz bod 4.2).
Linearita/nelinearita
Farmakokinetika telbivudinu je závislá na dávce v rozmezí dávek od 25 do 1 800 mg. Rovnovážného stavu bylo dosaženo po 5 až 7 dnech při podávání jednou denně, s přibližně 1,5násobnou akumulací v systémové expozici, což naznačuje, že efektivní akumulační poločas je přibližně 15 hodin. Po podání telbivudinu v dávce 600 mg jednou denně byla dolní hodnota koncentrace v rovnovážném stavu přibližně 0,2 - 0,3 ^g/ml.
Zvláštní populace
Pohlaví
Ve farmakokinetice telbivudinu není signifikantní rozdíl v závislosti na pohlaví.
Lidská rasa
Ve farmakokinetice telbivudinu není signifikantní rozdíl v závislosti na rase.
Děti a starší pacienti (více než 65 let)
U dětí a u starších pacientů nebyly provedeny studie farmakokinetiky.
Porucha funkce ledvin
Farmakokinetika telbivudinu po jednorázovém podání dávek (200, 400 a 600 mg) byla hodnocena u pacientů (bez chronické hepatitidy B) s různým stupněm poškození renálních funkcí (hodnoceno clearance kreatininu). Na základě výsledků, uvedených v tabulce 9, je doporučeno upravit dávkové intervaly telbivudinu u pacientů s clearance kreatininu <50 ml/min (viz body 4.2 a 4.4).
Tabulka 9 Farmakokinetické parametry (střední hodnota ± SD) telbivudinu u osob s různým stupněm poškození renálních funkcí
|
Renální funkce (clearance kreatininu v ml/min) | |||||
|
Normální (>80) (n = 8) 600 mg |
Mírné (50-80) (n = 8) 600 mg |
Střední (30-49) (n = 8) 400 mg |
Těžké (<30) (n = 6) 200 mg |
ESRD/ hemodialýza (n = 6) 200 mg | |
|
Cmax (Mg/ml) |
3,4 ± 0,9 |
3,2 ± 0,9 |
2,8 ± 1,3 |
1,6 ± 0,8 |
2,1 ± 0,9 |
|
AUC0-X, (pg^h/ml) |
28,5 ± 9,6 |
32,5 ± 10,1 |
36,0 ± 13,2 |
32,5 ± 13,2 |
67,4 ± 36,9 |
|
CLrenal (ml/min) |
126,7 ± 48,3 |
83,3 ± 20,0 |
43,3 ± 20,0 |
11,7 ± 6,7 |
- |
Pacienti s poškozenou funkcí ledvin na hemodialýze
Hemodialýza (do 4 hodin) snižuje systémovou expozici telbivudinu přibližně o 23%. Po úpravě dávkových intervalů podle clearance kreatininu není další úprava dávky během rutinní dialýzy nutná (viz 4.2). Telbivudin má být podáván po hemodialýze.
Zhoršená funkce jater
Farmakokinetika telbivudinu byla studována u pacientů (bez chronické hepatitidy B) s různým stupněm zhoršené funkce jater a u některých pacientů s dekompenzovanou chorobou jater. Nebyly pozorovány významné změny farmakokinetiky telbivudinu u osob se zhoršenou funkcí jater ve srovnání s jedinci bez zhoršené funkce jater. Výsledky těchto studií ukazují, že není nezbytné upravovat dávku u pacientů se zhoršenou funkcí jater (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání a genotoxicity neodhalily žádné zvláštní riziko pro člověka. Telbivudin neukázal žádný karcinogenní potenciál pro člověka. Standardními testy reprodukční toxicity nebyl zjištěn žádný důkaz přímého toxického účinku telbivudinu. U králíků byly dávky telbivudinu, po kterých dosahovaly expozice 37násobku expozice u člověka po podání terapeutické dávky (600 mg), spojeny se zvýšením incidence potratu a předčasného porodu. Tento účinek byl považován za důsledek toxicity pro matku.
Fertilita byla hodnocena v konvenčních studiích u dospělých potkanů, a jako součást toxikologické studie na nedospělých jedincích.
U dospělých potkanů byla fertilita snížena v případech, kdy byl samci i samici potkana podáván telbivudin v dávkách 500 nebo 1000 mg/kg/den (nižší index fertility ve srovnání se souběžnými kontrolami). Neprojevily se žádné známky abnormality v morfologii nebo funkci spermatu a varlata a vaječníky byly bez histologických pozoruhodností.
Nebylo prokázáno zhoršení fertility v dalších studiích, kdy byly buď samcům nebo samicím potkanů podávány dávky až 2000 mg/kg/den a kteří byli spářeni s potkany bez léčby (systémová expozice přibližně 6-14násobně vyšší, než jakých bylo dosaženo u člověka).
V toxikologických studiích byli nedospělí jedinci léčeni od 14. do 70. dne po porodu a byli pářeni s potkany, kterým byla podávána stejná léčba (bez páření mezi sourozenci). Fertilita byla snížena u párů s dávkami >1000 mg/kg/den, jak ukazují snížené indexy fertility a páření a snížený počet početí. Ovariální a děložní parametry potkaních samic, které se úspešně spářily, však nebyly podáváním telbivudinu ovlivněny.
NOAEL (no observed adverse effect level) účinků na parametry fertility nebo páření se rovná dávce 250 mg/kg/den, jejíž podávání vede k 2,5 až 2,8krát vyšší expozici, než jaké je dosaženo při podávání terapeutické dávky u člověka s normální funkcí ledvin.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Kyselina benzoová (E210)
Dihydrát sodné soli sacharinu Mučenkové aroma Hydroxid sodný Kyselina citronová Čištěná voda
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
Po otevření lahve spotřebujte do 2 měsíců.
6.4 Zvláštní opatření pro uchovávání Neuchovávejte při teplotě nad 30oC. Chraňte před mrazem.
6.5 Druh obalu a obsah balení
300 ml hnědá skleněná lahev s dětským bezpečnostním uzávěrem, včetně polyethylenového těsnícího kroužku a jistícího kroužku polypropylenové odměrky s vyznačeným dávkováním od 5 do 30 ml se stupnicí po 5 ml, a polypropylenová stříkačka pro perorální podání s vyznačeným dávkováním od 1 do 10 ml se stupnicí po 0,5 ml.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/07/388/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 24. dubna 2007
Datum posledního prodloužení registrace: 26. dubna 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Novartis Pharma GmbH Roonstrasse 25 90429 Norimberk Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční činnosti v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Sebivo 600 mg potahované tablety telbivudinum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje telbivudinum 600 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 potahovaných tablet 98 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Perorální podání. Tabletu nežvýkejte, nedělte ani nedrťte.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/07/388/001 28 potahovaných tablet
EU/1/07/388/002 98 potahovaných tablet
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Sebivo 600 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTRY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Sebivo 600 mg potahované tablety telbivudinum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Pondělí
Úterý
Středa
Čtvrtek
Pátek
Sobota
Neděle
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Sebivo 20 mg/ml perorální roztok telbivudinum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje telbivudinum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje také sodík. Bližší informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
1 lahev obsahující 300 ml perorálního roztoku [pouze krabička] 1 odměrka + 1 stříkačka pro perorální podání [pouze krabička]
300 ml [pouze štítek lahvičky]
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
Po otevření lahve spotřebujte do 2 měsíců
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 30oC. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/07/388/003
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Sebivo 20 mg/ml [pouze krabička]
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Sebivo 600 mg potahované tablety
Telbivudinum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit,
a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Sebivo a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Sebivo užívat
3. Jak se Sebivo užívá
4. Možné nežádoucí účinky
5. Jak Sebivo uchovávat
6. Obsah balení a další informace
1. Co je Sebivo a k čemu se používá
Sebivo obsahuje léčivou látku telbivudin. Sebivo patří do skupiny léčivých přípravků nazývaných antivirotika, která se používají k léčbě infekcí vyvolaných viry.
Sebivo se užívá k léčbě dospělých pacientů s chronickým zánětem jater typu B (hepatitida B).
Chronický zánět jater typu B je způsoben infekcí virem hepatitidy B, který se množí v játrech a je příčinou poškození jater. Léčba přípravkem Sebivo snižuje množství virů hepatitidy B v těle tím, že brání jeho rozmnožování, což má za následek menší poškození jater a zlepšení jaterních funkcí.
2. Čemu musíte věnovat pozornost, než začnete Sebivo užívat Neužívejte Sebivo
- jestliže jste alergický/á na telbivudin nebo kteroukoli další složku tohoto přípravku (uvedenou v bodě 6),
- jestliže jste léčen(a) pegylovaným nebo standardním interferonem alfa (viz „Vzájemné působení s dalšími léčivými přípravky“).
Jestliže toto pro Vás platí, Sebivo neužívejte. Poraďte se se svým lékařem.
Upozornění a opatření
Před užitím přípravku Sebivo se poraďte se svým lékařem:
- jestliže máte nebo jste měl(a) jakékoli problémy s ledvinami. Před léčbou a během léčby Vás lékař může poslat na laboratorní testy, kterými se zjistí, zda Vaše ledviny pracují správně. Podle výsledků těchto testů může lékař změnit dávkování přípravku Sebivo.
- jestliže máte cirhózu jater (vážný stav, který způsobuje “ztvrdnutí” jater). V takovém případě Vás bude lékař velmi pečlivě sledovat.
- jestliže j ste po transplantaci jater.
- jestliže užíváte jakékoliv léky, které mohou způsobit svalové obtíže (pokud si nejste jistý/á, poraďte se se svým lékařem nebo lékárníkem).
- jestliže máte infekci virem HIV, hepatitidou C nebo D, nebo jste léčen(a) jakýmikoli antivirovými léky.
Jestliže toto pro Vás platí, řekněte to svému lékaři dříve, než začnete přípravek Sebivo užívat.
Během léčby přípravkem Sebivo:
- Sebivo může způsobit přetrvávající nevysvětlitelnou svalovou slabost nebo svalovou bolest (myopatii). Svalové příznaky se mohou zhoršovat a stát se závažnými. Někdy může dojít k rozpadu svalu (rabdomyolýza), což může způsobit poškození ledvin.
- Sebivo může vzácně vyvolat pocit necitlivosti, brnění, bolest a/nebo pocit pálení v pažích nebo dolních končetinách (periferní neuropatie).
Jestliže se u Vás objeví některý z těchto příznaků během léčby přípravkem Sebivo, kontaktujte okamžitě svého lékaře.
Další nežádoucí účinky tohoto typu léku
Sebivo patří do skupiny léčivých přípravků (nukleosidový analog), které mohou způsobit nadměrné zvýšení množství kyseliny mléčné v krvi (laktátová acidóza) a zvětšení jater (hepatomegalie) provázené ztučněním jater (steatózou). Laktátová acidóza je vzácný, ale závažný nežádoucí účinek, který může být někdy i smrtelný. Laktátová acidóza se vyskytuje častěji u žen, zvláště pokud mají výraznou nadváhu. Pokud budete užívat Sebivo, lékař Vás bude pravidelně kontrolovat. Pokud se u Vás v průběhu léčby přípravkem Sebivo objeví svalová bolest, těžká a přetrvávající bolest žaludku spojená s pocitem na zvracení a zvracením, těžkým a přetrvávajícím ztíženým dýcháním, únava a nadýmání a/nebo nevolnost, kontaktujte okamžitě lékaře.
U některých lidí se mohou objevit velmi závažné jaterní příznaky, když přestanou užívat léky jako Sebivo. Po ukončení léčby Sebivem bude Váš lékař sledovat Váš zdravotní stav a pravidelně provádět krevní testy na kontrolu Vašich jater. Informujte okamžitě svého lékaře o nových nebo neobvyklých příznacích, kterých si všimnete po ukončení léčby (viz „Jestliže jste přestal(a) užívat Sebivo“ v bodě 3 této informace).
Dávejte pozor, abyste nenakazil(a) jiné lidi
Sebivo nesnižuje riziko nakažení jiných lidí virem hepatitidy B (HBV) sexuálním stykem nebo kontaminovanou krví nebo jinými tělními tekutinami. Pokud máte sexuální styk s partnerem, který není imunní vůči hepatitidě B, vždy použijte kondom a zabraňte jakékoli jiné výměně tělních tekutin. Nikdy nesdílejte s nikým Vámi použité injekční jehly. Nesdílejte s nikým Vaše osobní předměty, na kterých může být Vaše krev nebo jiné tělní tekutiny, např. zubní kartáčky, žiletky na holení. K prevenci infekce HBV je dostupné očkování.
Děti a dospívající
Sebivo se nedoporučuje podávat dětem a dospívajícím.
Další léčivé přípravky a Sebivo
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Váš lékař nebo lékárník potřebuje vědět o ostatních lécích, které užíváte, protože některé léky by mohly ovlivňovat funkci ledvin a protože je přípravek Sebivo vylučován z těla ledvinami do moči.
Neužívejte Sebivo, jestliže je Vám podáván / podáváte si pegylovaný nebo standardní interferon alfa (viz „Neužívejte Sebivo“), protože kombinace těchto přípravků může u Vás zvýšit riziko vzniku periferní neuropatie (pocity znecitlivění, brnění a/nebo pálení v rukou a/nebo nohou). Pokud jste léčen(a) interferonem, sdělte to svému lékaři nebo lékárníkovi.
Těhotenství a kojení
- Během těhotenství neužívejte Sebivo, aniž by Vám to lékař doporučil. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat. Váš lékař s Vámi prodiskutuje možná rizika užívání přípravku Sebivo během těhotenství.
- Jestliže máte hepatitidu B a otěhotníte, zeptejte se svého lékaře, jak nejlépe můžete chránit své dítě. Sebivo může snížit riziko přenosu viru Vaší hepatitidy B na Vaše nenarozené dítě, pokud je užíván v kombinaci s imunoglobulinem a vakcínou proti hepatitidě B.
- Během léčby přípravkem Sebivo nekojte. Pokud kojíte, řekněte to svému lékaři.
3. Jak se Sebivo užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý/á, poraďte se se svým lékařem nebo lékárníkem.
Kolik se přípravku Sebivo užívá
Doporučená dávka přípravku je jedna 600miligramová tableta jednou denně. Tabletu užívejte každý den přibližně ve stejnou dobu.
Tabletu můžete užívat s jídlem nebo bez jídla. Polykejte ji celou a zapíjejte ji trochou vody. Tabletu nekousejte, nedělte ani ji nedrťte.
Jestliže trpíte onemocněním ledvin, budete možná muset užívat Sebivo méně často (v delších časových intervalech). Pokud máte nebo jste někdy měl(a) problémy s ledvinami, řekněte to svému lékaři.
Jak dlouho se Sebivo užívá
Pokračujte v každodenním užívání Sebiva tak dlouho, jak Vám řekl lékař. Neměňte svou dávku nebo nepřestávejte Sebivo užívat, aniž byste se o tom poradil(a) s lékařem. Tento přípravek je určen k dlouhodobé léčbě, která může trvat měsíce nebo roky. Váš lékař bude pravidelně sledovat Váš stav, aby zkontroloval, zda má léčba požadovaný účinek.
Jestliže jste užil(a) více přípravku Sebivo, než jste měl(a)
Jestliže jste užil(a) příliš mnoho přípravku Sebivo nebo někdo jiný náhodně užil Vaše tablety, vyhledejte okamžitě lékaře nebo jděte do nemocnice. Vezměte s sebou krabičku s léky a ukažte ji lékaři.
Jestliže jste zapomněl(a) užít Sebivo
- Jestliže jste zapomněl(a) užít Sebivo, vezměte si ho co nejdříve, jakmile si na to vzpomenete. Další dávku užijte v obvyklém čase.
- Pokud do užití další dávky zbývá méně než 4 hodiny, vynechanou dávku přeskočte a následující dávku užijte v obvyklém čase.
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou tabletu. To u Vás může zvýšit možnost výskytu nežádoucích účinků. Pokud si nejste jistý/á, co máte dělat, poraďte se se svým lékařem nebo lékárníkem.
Jestliže jste přestal(a) užívat Sebivo
Ukončení léčby Sebivem může mít za následek zhoršení Vaší hepatitidy B, tj. progrese onemocnění a abnormální výsledky testů (zvýšení virového zatížení, vzestup ALT). Sebivo nepřestávejte užívat, aniž by Vám to řekl lékař. Při užívání Sebiva dbejte na to, aby Vám Sebivo nedošlo.
Po ukončení léčby Sebivem bude lékař kontrolovat Váš stav a bude pravidelně provádět krevní jaterní testy, jelikož Vaše infekce hepatitidou B se může po ukončení léčby zhoršovat nebo se stát velmi závažnou. Informujte okamžitě svého lékaře o všech nových či neobvyklých příznacích, kterých si po ukončení léčby všimnete.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Některé nežádoucí účinky by mohly být závažné:
- přetrvávající svalová slabost nebo svalová bolest
- pocit necitlivosti, brnění, bolest a/nebo pocit pálení v pažích nebo dolních končetinách Jestliže se u Vás objeví některý z těchto příznaků, kontaktujte okamžitě svého lékaře.
Sebivo může také vyvolat další nežádoucí účinky:
Časté (mohou postihnout až 1 z 10 pacientů)
- Závratě, bolest hlavy
- Kašel
- Průjem, pocit nevolnosti (nauzea), bolest žaludku (břicha)
- Kožní vyrážka
- Únava (vyčerpanost)
- Při výsledcích vyšetření krve se zjistí zvýšení hladin některých jaterních enzymů (např. ALT, AST), amylázy, lipázy nebo kreatinkinázy
Méně časté (mohou postihnout až 1 ze 100 pacientů)
- Bolest kloubů
- Přetrvávající svalová slabost nebo bolest svalů (myopatie/myozitida), svalová křeč
- Bolest zad, krku, bolest v boku
- Snížená citlivost, brnění, bolest a/nebo palčivý pocit v pažích a/nebo dolních končetinách nebo v okolí úst
- Ischias (bolest v dolní části zad nebo v oblasti kyčle, která se může šířit do dolních končetin)
- Porucha chuti
- Stav, kdy se necítíte dobře (malátnost)
Vzácné (mohou postihnout až 1 z 1 000 pacientů)
- Nadbytek kyseliny mléčné v krvi (laktátová acidóza)
- Poškození (rozpadu) svalů (rabdomyolýza)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za Použitelné do:. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nepoužívejte tento přípravek, pokud si všimnete, že je obal poškozen nebo přípravek jeví známky manipulace (otevření).
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Sebivo obsahuje
- Léčivou látkou je telbivudinum. Jedna tableta obsahuje telbivudinum 600 mg.
- Dalšími složkami jsou: mikrokrystalická celulosa, povidon, sodná sůl karboxymethylškrobu, koloidní bezvodý oxid křemičitý, magnesium-stearát, hypromelosa, oxid titaničitý (E171), mastek, makrogol.
Jak Sebivo vypadá a co obsahuje toto balení
Sebivo jsou bílé až slabě nažloutlé, oválné potahované tablety, na jedné straně s potiskem “LDT”.
Sebivo potahované tablety jsou dodávány v balení 28 nebo 98 tablet. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
EtnrapHH
Novartis Pharma Services Inc. Ten.: +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Lietuva
Novartis Pharma Services Inc. Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma GmbH Tel: +49 911 273 0
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Novartis Pharma Services Inc. Tel: +356 2122 2872
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáSa Novartis (Hellas) A.E.B.E. Tn^: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kónpoq Novartis Pharma Services Inc. Tn^: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Sebivo 20 mg /ml perorální roztok
Telbivudinum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit,
a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Sebivo a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Sebivo užívat
3. Jak se Sebivo užívá
4. Možné nežádoucí účinky
5. Jak Sebivo uchovávat
6. Obsah balení a další informace
1. Co je Sebivo a k čemu se používá
Sebivo obsahuje léčivou látku telbivudin. Sebivo patří do skupiny léčivých přípravků nazývaných antivirotika, která se používají k léčbě infekcí vyvolaných viry.
Sebivo se užívá k léčbě dospělých pacientů s chronickým zánětem jater typu B (hepatitida B).
Chronický zánět jater typu B je způsoben infekcí virem hepatitidy B, který se množí v játrech a je příčinou poškození jater. Léčba přípravkem Sebivo snižuje množství virů hepatitidy B v těle tím, že brání jeho rozmnožování, což má za následek menší poškození jater a zlepšení jaterních funkcí.
2. Čemu musíte věnovat pozornost, než začnete Sebivo užívat Neužívejte Sebivo
- jestliže j ste alergický/á na telbivudin nebo kteroukoli další složku tohoto přípravku (uvedenou v bodě 6),
- jestliže jste léčen(a) pegylovaným nebo standardním interferonem alfa (viz „Vzájemné působení s dalšími léčivými přípravky“).
Jestliže toto pro Vás platí, Sebivo neužívejte. Poraďte se se svým lékařem.
Upozornění a opatření
Před užitím přípravku Sebivo se poraďte se svým lékařem:
- jestliže máte nebo jste měl(a) jakékoli problémy s ledvinami. Před léčbou a během léčby Vás lékař může poslat na laboratorní testy, kterými se zjistí, zda Vaše ledviny pracují správně. Podle výsledků těchto testů může lékař změnit dávkování přípravku Sebivo.
- jestliže máte cirhózu jater (vážný stav, který způsobuje “ztvrdnutí” jater). V takovém případě Vás bude lékař velmi pečlivě sledovat.
- jestliže jste po transplantaci jater.
- jestliže užíváte jakékoliv léky, které mohou způsobit svalové obtíže (pokud si nejste jistý/á, poraďte se se svým lékařem nebo lékárníkem).
- jestliže máte infekci virem HIV, hepatitidou C nebo D, nebo jste léčen(a) jakýmikoli antivirovými léky.
Jestliže toto pro Vás platí, řekněte to svému lékaři dříve, než začnete přípravek Sebivo užívat.
Během léčby přípravkem Sebivo:
- Sebivo může způsobit přetrvávající nevysvětlitelnou svalovou slabost nebo svalovou bolest (myopatii). Svalové příznaky se mohou zhoršovat a stát se závažnými. Někdy může dojít k rozpadu svalu (rabdomyolýza), což může způsobit poškození ledvin.
- Sebivo může vzácně vyvolat pocit necitlivosti, brnění, bolest a/nebo pocit pálení v pažích nebo dolních končetinách (periferní neuropatie).
Jestliže se u Vás objeví některý z těchto příznaků během léčby přípravkem Sebivo, kontaktujte okamžitě svého lékaře.
Další nežádoucí účinky tohoto typu léku
Sebivo patří do skupiny léčivých přípravků (nukleosidový analog), které mohou způsobit nadměrné zvýšení množství kyseliny mléčné v krvi (laktátová acidóza) a zvětšení jater (hepatomegalie) provázené ztučněním jater (steatózou). Laktátová acidóza je vzácný, ale závažný nežádoucí účinek, který může být někdy i smrtelný. Laktátová acidóza se vyskytuje častěji u žen, zvláště pokud mají výraznou nadváhu. Pokud budete užívat Sebivo, lékař Vás bude pravidelně kontrolovat. Pokud se u Vás v průběhu léčby přípravkem Sebivo objeví svalová bolest, těžká a přetrvávající bolest žaludku spojená s pocitem na zvracení a zvracením, těžkým a přetrvávajícím ztíženým dýcháním, únava a nadýmání a/nebo nevolnost, kontaktujte okamžitě lékaře.
U některých lidí se mohou objevit velmi závažné jaterní příznaky, když přestanou užívat léky jako Sebivo. Po ukončení léčby Sebivem bude Váš lékař sledovat Váš zdravotní stav a pravidelně provádět krevní testy na kontrolu Vašich jater. Informujte okamžitě svého lékaře o nových nebo neobvyklých příznacích, kterých si všimnete po ukončení léčby (viz „Jestliže jste přestal(a) užívat Sebivo“ v bodě 3 této informace).
Dávejte pozor, abyste nenakazil(a) jiné lidi
Sebivo nesnižuje riziko nakažení jiných lidí virem hepatitidy B (HBV) sexuálním stykem nebo kontaminovanou krví nebo jinými tělními tekutinami. Pokud máte sexuální styk s partnerem, který není imunní vůči hepatitidě B, vždy použijte kondom a zabraňte jakékoli jiné výměně tělních tekutin. Nikdy nesdílejte s nikým Vámi použité injekční jehly. Nesdílejte s nikým Vaše osobní předměty, na kterých může být Vaše krev nebo jiné tělní tekutiny, např. zubní kartáčky, žiletky na holení. K prevenci infekce HBV je dostupné očkování.
Děti a dospívající
Sebivo se nedoporučuje podávat dětem a dospívajícím.
Další léčivé přípravky a Sebivo
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Váš lékař nebo lékárník potřebuje vědět o ostatních lécích, které užíváte, protože některé léky by mohly ovlivňovat funkci ledvin a protože je přípravek Sebivo vylučován z těla ledvinami do moči.
Neužívejte Sebivo, jestliže je Vám podáván / podáváte si pegylovaný nebo standardní interferon alfa (viz „Neužívejte Sebivo“), protože kombinace těchto přípravků může u Vás zvýšit riziko vzniku periferní neuropatie (pocity znecitlivění, brnění a/nebo pálení v rukou a/nebo nohou). Pokud jste léčen(a) interferonem, sdělte to svému lékaři nebo lékárníkovi.
Sebivo s jídlem a pitím
Sebivo můžete užívat s jídlem i bez jídla.
Těhotenství a kojení
- Během těhotenství neužívejte Sebivo, aniž by Vám to lékař doporučil. Pokud jste těhotná, domníváte se, že můžete být těhotná,nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat. Váš lékař s Vámi prodiskutuje možná rizika užívání přípravku Sebivo během těhotenství.
- Jestliže máte hepatitidu B a otěhotníte, zeptejte se svého lékaře, jak nejlépe můžete chránit své dítě. Sebivo může snížit riziko přenosu viru Vaší hepatitidy B na Vaše nenarozené dítě, pokud je užíván v kombinaci s imunoglobulinem a vakcínou proti hepatitidě B.
- Během léčby přípravkem Sebivo nekojte. Pokud kojíte, řekněte to svému lékaři.
Sebivo obsahuje sodík
Sebivo perorální roztok obsahuje v dávce 600 mg (30 ml) asi 47 mg sodíku. Jestliže držíte kontrolovanou sodíkovou dietu, požádejte svého lékaře o radu.
3. Jak se Sebivo užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý/á, poraďte se se svým lékařem nebo lékárníkem.
Kolik se přípravku Sebivo užívá
Doporučená dávka Sebiva je 30 ml perorálního roztoku (600 mg telbivudinu) jednou denně. Užívejte Sebivo každý den přibližně ve stejnou dobu. Přípravek může být užíván s jídlem i bez jídla.
Úplné instrukce, jak používat Sebivo, viz bod „Návod k použití“ na konci této informace.
Sejměte odměrku a otevřete lahvičku. Pomalu a pečlivě nalijte roztok z lahvičky do odměrky po značku předepsaného množství. Ihned všechen odměřený roztok z odměrky vypijte.
V případě, že nemůžete přesně odměřit předepsané množství pomocí odměrky, použijte stříkačku pro perorální podání. Podrobné instrukce jak použít stříkačku pro perorální podání, viz bod „Návod k použití“.
Jestliže trpíte onemocněním ledvin, bude možná Vaše dávka snížena. Pokud máte nebo jste někdy měl(a) problémy s ledvinami, řekněte to svému lékaři.
Jak dlouho se Sebivo užívá
Pokračujte v každodenním užívání Sebiva tak dlouho, jak Vám řekl lékař. Neměňte svou dávku nebo nepřestávejte Sebivo užívat, aniž byste se o tom poradil(a) s lékařem. Tento přípravek je určen k dlouhodobé léčbě, která může trvat měsíce nebo roky. Váš lékař bude pravidelně sledovat Váš stav, aby zkontroloval, zda má léčba požadovaný účinek.
Jestliže jste užil(a) více přípravku Sebivo, než jste měl(a)
Jestliže jste užil(a) příliš mnoho přípravku Sebivo nebo někdo jiný náhodně užil Váš perorální roztok, vyhledejte okamžitě lékaře nebo jděte do nemocnice. Vezměte s sebou balení léku a ukažte ho lékaři.
Jestliže jste zapomněl(a) užít Sebivo
- Jestliže jste zapomněl(a) užít Sebivo, vezměte si ho co nejdříve, jakmile si na to vzpomenete. Další dávku užijte v obvyklém čase.
- Pokud do užití další dávky zbývá méně než 4 hodiny, vynechanou dávku přeskočte a následující dávku užijte v obvyklém čase.
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. To u Vás může zvýšit možnost výskytu nežádoucích účinků. Pokud si nejste jistý/á, co máte dělat, poraďte se se svým lékařem nebo lékárníkem.
Jestliže jste přestal(a) užívat Sebivo
Ukončení léčby Sebivem může mít za následek zhoršení Vaší hepatitidy B, tj. progrese onemocnění a abnormální výsledky testů (zvýšení virového zatížení, vzestup ALT). Sebivo nepřestávejte užívat, aniž by Vám to řekl lékař. Při užívání Sebiva dbejte na to, aby Vám Sebivo nedošlo.
Po ukončení léčby Sebivem bude lékař kontrolovat Váš stav a bude pravidelně provádět krevní jaterní testy, jelikož Vaše infekce hepatitidou B se může po ukončení léčby zhoršovat nebo se stát velmi závažnou. Informujte okamžitě svého lékaře o všech nových či neobvyklých příznacích, kterých si po ukončení léčby všimnete.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Některé nežádoucí účinky by mohly být závažné:
- přetrvávající svalová slabost nebo svalová bolest
- pocit necitlivosti, brnění, bolest a/nebo pocit pálení v pažích nebo dolních končetinách Jestliže se u Vás objeví některý z těchto příznaků, kontaktujte okamžitě svého lékaře.
Sebivo může také vyvolat další nežádoucí účinky:
Časté (mohou postihnout až 1 z 10 pacientů)
- Závratě, bolest hlavy
- Kašel
- Průjem, pocit nevolnosti (nauzea), bolest žaludku (břicha)
- Kožní vyrážka
- Únava (vyčerpanost)
- Při výsledcích vyšetření krve se zjistí zvýšení hladin některých jaterních enzymů (např. ALT, AST), amylázy, lipázy nebo kreatinkinázy
Méně časté (mohou postihnout až 1 ze 100 pacientů)
- Bolest kloubů
- Přetrvávající svalová slabost nebo bolest svalů (myopatie/myozitida), svalová křeč
- Bolest zad, krku, bolest v boku
- Snížená citlivost, brnění, bolest a/nebo palčivý pocit v pažích a/nebo dolních končetinách nebo v okolí úst
- Ischias (bolest v dolní části zad nebo v oblasti kyčle, která se může šířit do dolních končetin)
- Porucha chuti
- Stav, kdy se necítíte dobře (malátnost)
Vzácné (mohou postihnout až 1 z 1 000 pacientů)
- Nadbytek kyseliny mléčné v krvi (laktátová acidóza)
- Poškození (rozpadu) svalů (rabdomyolýza)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Sebivo uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce a nálepce na lahvičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Neuchovávejte při teplotě nad 30oC. Chraňte před mrazem.
Po otevření lahvičky spotřebujte do 2 měsíců.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Sebivo obsahuje
- Léčivou látkou je telbivudinum. 30 ml perorálního roztoku obsahuje telbivudinum 600 mg.
- Dalšími složkami jsou: kyselina benzoová, dihydrát sodné soli sacharinu, mučenkové aroma, hydroxid sodný, kyselina citronová, čištěná voda.
Jak Sebivo vypadá a co obsahuje toto balení
Sebivo 20 mg/ml perorální roztok je dodáván jako 300 ml čirého, bezbarvého až světle žlutého roztoku v hnědé skleněné lahvičce s dětským bezpečnostním uzávěrem, včetně polyethylenového těsnícího kroužku a jistícího kroužku. Balení obsahuje polypropylenovou odměrku s vyznačeným dávkováním od 5 do 30 ml se stupnicí po 5 ml a polypropylenová stříkačka pro perorální podání s vyznačeným dávkováním od 1 do 10 ml se stupnicí 0,5 ml.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Btarapna Novartis Pharma Services Inc. Tea.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma GmbH Tel: +49 911 273 0 |
|
Česká republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáSa Novartis (Hellas) A.E.B.E. Tqk: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Přečtěte si prosím pečlivě tento návod k použití, abyste věděli, jak používat roztok správně.
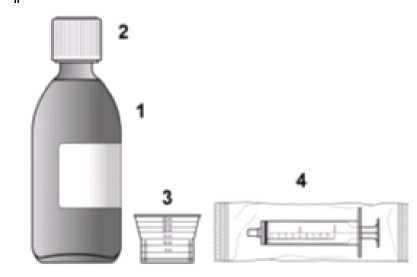
1. Lahvička s perorálním roztokem.
2. Dětský bezpečnostní uzávěr s jistícím kroužkem. Vždy po použití lahvičku uzavřete uzávěrem.
3. Odměrka k dávkování perorálního roztoku. Po použití odměrku vyčistěte a vždy ji vraťte zpět na uzávěr lahvičky.
4. Stříkačka pro perorální podání pro odměření dávek, které nemohou být přesně odměřeny použitím odměrky.
Příprava dávky léku při použití odměrky
1. Sejměte odměrku.
2. Zatlačte na uzávěr (2a) a současně otočte doleva dětským bezpečnostním uzávěrem (2b).
3. Před nalitím roztoku do odměrky, prosím, zkontrolujte
správný dílek stupnice, abyste se vyhnuli případnému plýtvání nebo rozlití roztoku.
Odměrku držte ve výšce očí, pečlivě a pomalu lijte předepsané množství roztoku z lahvičky do odměrky, až roztok dosáhne vrcholku příslušného dílku.
Poznámka: Jestliže množství nalité do odměrky přesáhne požadovanou dávku, zlikvidujte přebytečné množství vylitím do dřezu. Nelijte je zpět do lahvičky.
4. Roztok vypijte nebo ho podejte pacientovi ihned.
5. Lahvičku pevně uzavřete zašroubováním uzávěru.
Ihned vypláchněte odměrku vodou.
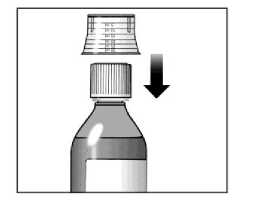
6.
7.
Odstraňte vodu z odměrky utřením čistým papírovým kapesníkem a vraťte odměrku zpět na uzávěr.
Příprava 6 ml dávky léku při použití stříkačky pro perorální podání
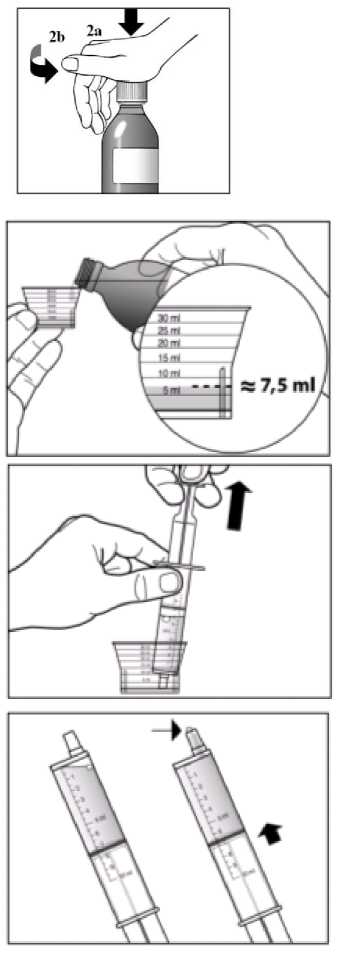
1. Sejměte odměrku.
2. Zatlačte na uzávěr (2a) a současně otočte doleva dětským bezpečnostním uzávěrem (2b).
3. Před nalitím roztoku do odměrky, prosím,
zkontrolujte polohu dílků 5 ml a 10 ml, abyste se vyhnuli případnému plýtvání nebo rozlití roztoku. Odměrku držte ve výšce očí, pečlivě a pomalu lijte předepsané množství roztoku z lahvičky do odměrky, až hladina roztoku dosáhne přesně mezi dílek 5 ml a dílek 10 ml.
4. Natáhněte veškerý roztok z odměrky do stříkačky pro perorální podání.
5. Obraťte stříkačku do svislé polohy a mírně j í nakloňte tak, aby se vzduchové bubliny dostaly k hornímu konci stříkačky.
6. Tlačte opatrně a pomalu na píst tak, abyste odstranili vzduch ze stříkačky, přičemž se objeví první kapka roztoku.
7. Držte stříkačku nad odměrkou.
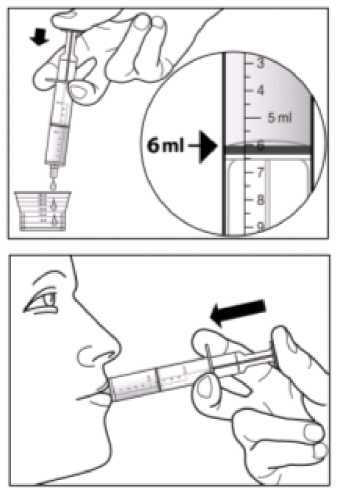
8. Posunujte pomalu a opatrně píst tak, dokud množství roztoku nedosáhne dílku 6 ml.
9. Roztok ihned polkněte přímo ze stříkačky.
10. Roztok zbylý v odměrce vylijte do dopadu. Nevracejte ho zpátky do lahve, protože by mohlo dojít ke kontaminaci.
11. Láhev pevně uzavřete.
12. Vypláchněte odměrku a stříkačku v čisté vodě.
13. Osušte odměrku čistým kusem látky a vraťte ji zpět na hrdlo lahve.
14. Stříkačku ponechte, aby se vysušila vzduchem a uchovávejte ji spolu s lahví.
63