Saxenda 6 Mg/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Saxenda 6 mg/ml injekční roztok v předplněném peru
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml roztoku obsahuje liraglutidum 6 mg *. Jedno předplněné pero obsahuje liraglutidum 18 mg ve 3 ml.
* Analog lidského glukagonu podobného peptidu-1 (GLP-1) vyrobený rekombinantní DNA technologií v Saccharomyces cerevisiae.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce).
Čirý bezbarvý či téměř bezbarvý izotonický roztok; pH = 8,15.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Saxenda je indikován jako doplňková léčba k dietě se sníženým obsahem kalorií a zvýšené fyzické aktivitě za účelem úpravy hmotnosti u dospělých pacientů s počáteční hodnotou indexu tělesné hmotnosti (Body Mass Index, BMI)
• > 30 kg/m2 (obézních) nebo
• > 27 kg/m2 až <30 kg/m2 (s nadváhou) za přítomnosti alespoň jedné komorbidity související
s hmotností, např. s dysglykemií (prediabetes nebo diabetes mellitus 2. typu), hypertenzí, dyslipidemií nebo obstrukční spánkovou apnoí.
Pokud u pacientů při dávce 3,0 mg/den nedojde po 12 týdnech k poklesu počáteční tělesné hmotnosti alespoň o 5 %, léčba přípravkem Saxenda má být ukončena.
4.2 Dávkování a způsob podání
Dávkování
Počáteční dávka je 0,6 mg denně. Dávka má být zvýšena na 3,0 mg denně v přírůstcích po 0,6 mg v nejméně jednotýdenních intervalech, aby se zlepšila gastrointestinální snášenlivost (viz tabulka 1). Pokud není navýšení na vyšší dávku během dvou po sobě jdoucích týdnů dobře snášeno, zvažte ukončení léčby. Denní dávky vyšší než 3,0 mg se nedoporučují.
Tabulka 1 Schéma navyšování dávky
|
Dávka |
Dávka |
Týdny |
|
Postup při navyšování dávky během 4 týdnů |
0,6 mg |
1 |
|
1,2 mg |
1 |
|
1,8 mg |
1 | |
|
2,4 mg |
1 | |
|
Udržovací dávka |
3,0 mg | |
Léčebný efekt byl doložen pouze pro 1 rok. Každoročně je třeba vyhodnotit, zda je nutné v léčbě dále pokračovat.
Pacienti s onemocněním diabetes mellitus 2. typu
Přípravek Saxenda se nemá používat v kombinaci s jinými agonisty GLP-1 receptoru.
Při zahájení podávání přípravku Saxenda zvažte snížení dávky souběžně podávaného inzulinu nebo sekretagog inzulinu (např. sulfonylurey), aby se snížilo riziko hypoglykémie.
Zvláštní skupiny pacientů
Starší pacienti (> 65 let)
Z důvodu věku není nutná žádná úprava dávkování. Zkušenosti s léčbou pacientů ve věku > 75 let jsou omezené a použití u těchto pacientů se nedoporučuje (viz body 4.4 a 5.2).
Pacienti s poruchou funkce ledvin
U pacientů s lehkou či středně těžkou poruchou funkce ledvin (clearance kreatininu >30 ml/min) není nutná žádná úprava dávkování. Přípravek Saxenda není doporučen pro použití u pacientů s těžkou poruchou funkce ledvin (clearance kreatininu <30 ml/min) včetně pacientů v konečném stadiu selhání ledvin (viz body 4.4, 4.8 a 5.2).
Pacienti s poruchou funkce jater
U pacientů s lehkou nebo středně těžkou poruchou funkce jater není doporučena žádná úprava dávkování. Přípravek Saxenda se nedoporučuje používat u pacientů s těžkou poruchou funkce jater a u pacientů s lehkou či středně těžkou poruchou funkce jater se musí používat s opatrností (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Saxenda u dětí a dospívajících do 18 let nebyly stanoveny (viz bod 5.1). Nejsou dostupné žádné údaje. Tento léčivý přípravek se nedoporučuje používat u pediatrických pacientů.
Způsob podání
Přípravek Saxenda je určen pouze pro subkutánní podání. Nesmí být podán intravenózně nebo intramuskulárně.
Přípravek Saxenda se podává jedenkrát denně kdykoli v průběhu dne, nezávisle na jídle. Aplikuje se injekčně do břicha, stehna nebo horní části paže. Místo aplikace i doba aplikace injekce mohou být změněny bez úpravy dávkování. Je však vhodnější aplikovat přípravek Saxenda přibližně ve stejnou denní dobu, která je jako nejvhodnější denní doba stanovena.
Přípravek Saxenda se nesmí mísit s jinými injekčními přípravky (např. inzuliny).
Pokud dojde k vynechání dávky do 12 hodin od obvyklé doby podání, má si ji pacient aplikovat co nejdříve. Pokud do další dávky zbývá méně než 12 hodin, pacient si nemá vynechanou dávku aplikovat a musí pokračovat v režimu podávání jednou denně další obvyklou dávkou. Nesmí docházet k nahrazování vynechané dávky či k navyšování dávky za účelem úpravy vynechané dávky. Další pokyny o podávání viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na liraglutid nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
U pacientů s diabetes mellitus se nesmí liraglutid používat jako náhrada za inzulin.
S podáváním pacientům s městnavým srdečním selháním třídy I-II podle New York Heart Association (NYHA) jsou pouze omezené zkušenosti. Liraglutid proto musí být používán s opatrností. Nejsou žádné zkušenosti s podáváním pacientům s městnavým srdečním selháním třídy III-IV podle NYHA. Liraglutid proto není u těchto pacientů doporučován.
Bezpečnost a účinnost liraglutidu v souvislosti s úpravou hmotnosti nebyly stanoveny u pacientů:
- ve věku 75 let nebo výše,
- léčených dalšími přípravky k úpravě hmotnosti,
- se sekundární obezitou při endokrinologických chorobách či při poruchách příjmu potravy nebo u pacientů léčených přípravky, které mohou způsobovat nárůst tělesné hmotnosti,
- s těžkou poruchou funkce ledvin,
- s těžkou poruchou funkce jater.
Použití u těchto pacientů se nedoporučuje (viz bod 4.2).
Vzhledem k tomu, že použití liraglutidu v souvislosti s úpravou hmotnosti nebylo studováno u pacientů s mírnou nebo se středně těžkou poruchou funkce jater, musí být u těchto pacientů liraglutid používán s opatrností (viz body 4.2 a 5.2).
S podáváním pacientům se zánětlivým onemocněním střev a diabetickou gastroparézou j sou pouze omezené zkušenosti. Používání liraglutidu není u těchto pacientů doporučeno, neboť je spojeno s přechodnými gastrointestinálními nežádoucími účinky, včetně nauzey, zvracení a průjmu.
Pankreatitida
Použití agonistů GLP-1 receptoru bylo spojeno s rizikem vzniku akutní pankreatitidy. U liraglutidu bylo hlášeno několik případů akutní pankreatitidy. Pacienti musí být informováni o charakteristických příznacích akutní pankreatitidy. Je-li podezření na pankreatitidu, musí být liraglutid vysazen. Pokud je akutní pankreatitida potvrzena, nesmí být léčba liraglutidem znovu zahájena. U pacientů s prodělanou pankreatitidou je nutno dbát zvláštní opatrnosti.
Cholelitiáza a cholecystitida
V klinických hodnoceních, prováděných v souvislosti s úpravou hmotnosti, byla u pacientů léčených liraglutidem pozorována vyšší četnost výskytu cholelitiázy a cholecystitidy než u pacientů užívajících placebo. Skutečnost, že značný hmotnostní úbytek může zvýšit riziko cholelitiázy (a tím
i cholecystitidy), je vysvětlena vyšší četností výskytu těchto onemocnění u liraglutidu pouze částečně. Cholelitiáza a cholecystitida mohou vést k hospitalizaci a cholecystektomii. Pacienti musí být informováni o charakteristických příznacích cholelitiázy a cholecystitidy.
Onemocnění štítné žlázy
V klinických hodnoceních u diabetes mellitus 2. typu byly hlášeny nežádoucí účinky na štítnou žlázu, včetně zvýšené hladiny kalcitoninu v krvi, strumy a neoplazie štítné žlázy, a to zvláště u pacientů s již dříve existujícím onemocněním štítné žlázy. V klinických hodnoceních týkajících se úpravy hmotnosti byly rovněž pozorovány případy zvýšení kalcitoninu v krvi. Liraglutid proto musí být u pacientů
s onemocněním štítné žlázy používán s opatrností.
Srdeční frekvence
V klinických hodnoceních bylo u liraglutidu pozorováno zvýšení srdeční frekvence (viz bod 5.1). Klinický význam zvýšení srdeční frekvence při léčbě liraglutidem je nejasný, především u pacientů se srdečním a cerebrovaskulárním onemocněním, a to z důvodu omezené expozice u těchto pacientů
v klinických hodnoceních. Srdeční frekvenci je nutné pravidelně sledovat v souladu s běžnou klinickou praxí. Pacienti musí být informováni o příznacích zvýšené srdeční frekvence (palpitacích nebo pocitu prudkého bušení srdce v klidu). U pacientů, kteří mají klinicky významné trvalé zvýšení klidové srdeční frekvence, je nutné léčbu přípravkem liraglutid ukončit.
Dehydratace
U pacientů léčených agonisty GLP-1 receptoru byly hlášeny známky a příznaky dehydratace, včetně poruchy funkce ledvin a akutního selhání ledvin. Pacienti používající liraglutid musí být upozorněni na potenciální riziko dehydratace v případě gastrointestinálních nežádoucích účinků a musí být seznámeni s bezpečnostními opatřeními, která mají provést, aby zabránili úbytku tekutin.
Hypoglykemie u pacientů s diabetes mellitus 2. typu
U pacientů s diabetes mellitus 2. typu, kterým je podáván liraglutid v kombinaci se sulfonylureou, může být zvýšené riziko hypoglykemie. Riziko hypoglykemie lze zmenšit snížením dávky sulfonylurey. U pacientů léčených inzulinem nebylo přidání přípravku Saxenda hodnoceno.
Pomocné látky
Přípravek Saxenda obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. v podstatě je „bez sodíku“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
In vitro je u liraglutidu prokázán velmi nízký potenciál pro farmakokinetické interakce s jinými léčivými látkami s vlivem na cytochrom P450 (CYP) a vazbu na plasmatické proteiny.
Malé zpoždění ve vyprazdňování žaludku při používání liraglutidu může ovlivnit absorpci současně podávaných perorálních léčivých přípravků. Studie interakcí neprokázaly žádné klinicky významné zpoždění absorpce, a proto není vyžadována úprava dávky.
Byly provedeny studie interakcí s 1,8 mg liraglutidu. Účinek na rychlost vyprazdňování žaludku u liraglutidu v dávce 1,8 mg a v dávce 3 mg (AUC0-300 min paracetamolu) byl stejný. Několik pacientů léčených liraglutidem hlásilo nejméně jednu epizodu těžkého průjmu. Průjem může ovlivnit absorpci současně podávaných perorálních léčivých přípravků.
Warfarin a další deriváty kumarinu
Nebyly provedeny žádné studie interakcí. Klinicky významné interakce s léčivými látkami se špatnou rozpustností nebo s úzkým terapeutickým indexem, jako je warfarin, nemohou být vyloučeny. Po zahájení léčby liraglutidem se u pacientů užívajících warfarin nebo další deriváty kumarinu doporučuje častější monitorování INR (International Normalised Ratio).
Paracetamol (acetaminofen)
Liraglutid neměnil celkovou expozici paracetamolu po podání 1 000 mg v jedné dávce. Hodnota Cmax paracetamolu byla snížena o 31 % a střední hodnota Cax byla zpožděna na 15 min. Při současném podávání paracetamolu není nutná žádná úprava dávkování.
Atorvastatin
Liraglutid neměnil celkovou expozici atorvastatinu po podání 40 mg atorvastatinu v jedné dávce. Proto při podávání s liraglutidem není nutná žádná úprava dávky atorvastatinu. Při podávání s liraglutidem se hodnota Cmax atorvastatinu snížila o 38 % a střední hodnota tmax se zpozdila z 1 hod na 3 hod.
Griseofulvin
Liraglutid neměnil celkovou expozici griseofulvinu po podání 500 mg griseofulvinu v jedné dávce. Hodnota Cmax griseofulvinu byla zvýšena o 37 %, zatímco ke změně střední hodnoty tmax nedošlo. Úprava dávky griseofulvinu a jiných látek s nízkou rozpustností a vysokou permeabilitou není nutná.
Digoxin
Podání 1 mg digoxinu v jedné dávce spolu s liraglutidem vedlo ke snížení AUC digoxinu o 16 %, hodnota Cmax byla snížena o 31 %. Medián tmax digoxinu byl zpožděn z 1 hod na 1,5 hod. Na základě těchto výsledků není nutná úprava dávky digoxinu.
Lisinopril
Podání 20 mg lisinoprilu v jedné dávce spolu s liraglutidem vedlo ke snížení AUC lisinoprilu o 15 %, hodnota Cmax byla snížena o 27 %. Medián tmax lisinoprilu byl při podávání liraglutidu zpožděn z 6 hod na 8 hod. Na základě těchto výsledků není nutná úprava dávky lisinoprilu.
Perorální antikoncepční přípravky
Po podání jednorázové dávky perorálního antikoncepčního přípravku snižoval liraglutid hodnotu Cmax ethinylestradiolu o 12 % a levonorgestrelu o 13 %. Hodnota tmax byla u obou látek při podání liraglutidu zpožděna o 1,5 hod. Nedošlo k žádnému klinicky významnému účinku na celkovou expozici ethinylestradiolu ani levonorgestrelu. Předpokládá se proto, že antikoncepční účinek není při současném podávání liraglutidu ovlivněn.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání liraglutidu těhotným ženám jsou omezené. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Potenciální riziko pro člověka není známé.
Liraglutid se nemá v těhotenství užívat. Pokud si pacientka přeje otěhotnět nebo otěhotní, má být léčba liraglutidem přerušena.
Kojení
Není známo, zda se liraglutid vylučuje do lidského mateřského mléka. Studie na zvířatech ukázaly, že přenos liraglutidu a strukturně blízkých metabolitů do mléka je nízký. Neklinické studie ukázaly snížení neonatálního růstu u kojených potkaních mláďat spojené s léčbou (viz bod 5.3). Vzhledem k chybějícím zkušenostem se přípravek Saxenda během kojení nemá podávat.
Fertilita
S výjimkou lehkého snížení počtu živých nidovaných vajíček neprokázaly studie na zvířatech škodlivé účinky v souvislosti s fertilitou (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Saxenda nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu:
Program klinického vývoje pro přípravek Saxenda tvoří 6 ukončených klinických hodnocení, kterých se účastnilo 5 813 obézních pacientů nebo pacientů s nadváhou a nejméně jednou komorbiditou související s tělesnou hmotností. Celkově byly nejčastěji hlášenými nežádoucími účinky během léčby přípravkem Saxenda gastrointestinální reakce (viz bod „Popis vybraných nežádoucích účinků“).
Seznam nežádoucích účinků v tabulce
V tabulce 2 jsou uvedeny nežádoucí účinky hlášené během fáze 3 dlouhodobých kontrolovaných klinických hodnocení. Nežádoucí účinky jsou uvedeny podle třídy orgánových systémů a frekvence výskytu. Kategorie frekvence výskytu jsou definovány takto: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000). V každé skupině frekvence výskytu jsou nežádoucí účinky seřazeny v pořadí podle klesající závažnosti.
Tabulka 2 Nežádoucí účinky hlášené ve fázi 3 kontrolovaných klinických hodnocení
|
Třídy orgánových systémů podle databáze MedDRA |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Poruchy imunitního systému |
Anafylaktická reakce |
|
Poruchy metabolismu a výživy |
Hypoglykemie* |
Dehydratace | ||
|
Psychiatrické poruchy |
Nespavost** | |||
|
Poruchy nervového systému |
Závratě Dysgeuzie | |||
|
Srdeční poruchy | ||||
|
Gastrointestinální poruchy |
Zácpa |
Sucho v ústech Gastritida Gastroezofageální refluxní onemocnění v epigastriu Nadýmání Říhání Břišní distenze |
Pankreatitida** * | |
|
Poruchy jater a žlučových cest |
Cholelitiáza*** |
Cholecystitida*** | ||
|
Poruchy kůže a podkožní tkáně |
Kopřivka | |||
|
Poruchy ledvin a močových cest |
Akutní selhání ledvin Porucha funkce ledvin | |||
|
Celkové poruchy a reakce v místě aplikace |
Reakce v místě vpichu Astenie Únava | |||
|
Vyšetření |
Zvýšené hladiny lipázy Zvýšené hladiny amylázy |
*Hypoglykemie (na základě příznaků hlášených samotnými pacienty a nepotvrzených měřením hladiny glukózy v krvi) hlášená u pacientů bez onemocnění diabetes mellitus 2. typu léčených přípravkem Saxenda v kombinaci s dietou a cvičením. Další informace naleznete v části „Popis vybraných nežádoucích účinků“.
**Nespavost byla většinou pozorována během prvních 3 měsíců léčby.
*** Viz bod 4.4.
Popis vybraných nežádoucích účinků:
Hypoglykemie u pacientů bez diabetes mellitus 2. typu
V klinických hodnoceních u pacientů s nadváhou nebo obézních pacientů bez diabetes mellitus 2. typu léčených přípravkem Saxenda v kombinaci s dietou a cvičením nebyly hlášeny žádné závažné hypoglykemické příhody (vyžadující pomoc další osoby). Příznaky hypoglykemických příhod byly hlášeny u 1,6 % pacientů léčených přípravkem Saxenda a 1,1 % pacientů užívajících placebo. Tyto příhody však nebyly potvrzeny měřením hladiny glukózy v krvi. Většina příhod byla mírná.
Hypoglykemie u pacientů s onemocněním diabetes mellitus 2. typu
V klinickém hodnocení u pacientů s nadváhou nebo obézních pacientů s diabetes mellitus 2. typu léčených přípravkem Saxenda v kombinaci s dietou a cvičením byla hlášena těžká hypoglykemie (vyžadující pomoc další osoby) u 0,7 % pacientů léčených přípravkem Saxenda a pouze u pacientů léčených souběžně sulfonylureou. Rovněž byla u těchto pacientů zdokumentována symptomatická hypoglykemie (u 43,6 % pacientů léčených přípravkem Saxenda a u 27,3 % pacientů užívajících placebo). U pacientů, kteří nebyli souběžně léčeni sulfonylureou, hlásilo 15,7 % pacientů léčených přípravkem Saxenda a 7,6 % pacientů užívajících placebo zdokumentované symptomatické hypoglykemické příhody (definované jako hladina glukózy v plazmě < 3,9 mmol/l doprovázená příznaky).
Gastrointestinální nežádoucí účinky
Většina epizod gastrointestinálních nežádoucích účinků byla mírná až středně závažná, přechodná a většina z nich nevedla k přerušení léčby. Reakce se obvykle objevily během prvních týdnů léčby a zeslábly během několika dnů nebo týdnů pokračující léčby.
U pacientů ve věku > 65 let se může při léčbě přípravkem Saxenda projevit více gastrointestinálních účinků.
U pacientů s lehkou nebo středně těžkou poruchou funkce ledvin (clearance kreatininu >30 ml/min) se může při léčbě přípravkem Saxenda projevit více gastrointestinálních účinků.
Akutní selhání ledvin
U pacientů léčených agonisty GLP-1 receptoru byla hlášena akutní renální selhání. Většina hlášených příhod se objevila u pacientů, kteří trpěli nauzeou, zvracením nebo průjmem vedoucím k depleci objemu (viz bod 4.4).
Alergické reakce
Po uvedení liraglutidu na trh bylo hlášeno několik případů anafylaktických reakcí s příznaky jako např. hypotenzí, palpitacemi, dušností a edémem. Anafylaktické reakce mohou být potenciálně život ohrožující. Je-li podezření na anafylaktickou reakci, musí být liraglutid vysazen a léčba již nesmí být znovu zahájena (viz bod 4.3).
Reakce v místě vpichu
U pacientů léčených přípravkem Saxenda byly hlášeny reakce v místě vpichu. Tyto reakce byly obvykle mírné a přechodné a většina z nich v průběhu léčby vymizela.
V klinických hodnoceních byla u pacientů léčených přípravkem Saxenda hlášena tachykardie u 0,6 % pacientů a u 0,1 % pacientů užívajících placebo. Většina příhod byla mírná nebo středně závažná. Tyto příhody byly izolované a většina v průběhu léčby přípravkem Saxenda odezněla.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Z klinických hodnocení a použití liraglutidu po uvedení na trh bylo hlášeno předávkování až 72 mg přípravku (24násobek doporučené dávky k úpravě hmotnosti). Hlášené příhody zahrnovaly těžkou nauzeu a těžké zvracení, což jsou rovněž očekávané příznaky předávkování liraglutidem. Žádné z hlášení nezahrnovalo těžkou hypoglykemii. Všichni pacienti se zotavili bez komplikací.
V případě předávkování má být zahájena vhodná podpůrná léčba podle klinických známek a příznaků, které se u pacienta vyskytnou. Pacienta je nutno pozorovat s ohledem na klinické příznaky dehydratace a musí být monitorována hladina glukózy v krvi.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: léčiva k terapii diabetů, jiná antidiabetika, kromě inzulínů. ATC kód: A10BX07
Mechanismus účinku
Liraglutid je acylovaný analog lidského glukagonu podobného peptidu 1 (GLP-1) se sekvencí aminokyselin s 97% homologií k endogennímu lidskému GLP-1. Liraglutid se váže na receptor GLP-1 (GLP- 1R) a aktivuj e jej.
GLP-1 je fyziologický regulátor chuti k jídlu a příjmu potravy, ale přesný mechanismus účinku není zcela jasný. Ve studiích na zvířatech vedla periferní aplikace liraglutidu k vychytávání ve specifických oblastech mozku zapojených do regulace chuti k jídlu, kdy liraglutid prostřednictvím specifické aktivace GLP-1R zvyšoval klíčové signály sytosti a snižoval klíčové signály hladu, čímž docházelo ke snižování tělesné hmotnosti.
Farmakodynamické účinky
Liraglutid snižuje tělesnou hmotnost u lidí především prostřednictvím ztráty tukové hmoty s relativní redukcí viscerálního tuku, která je větší než ztráta podkožního tuku. Liraglutid reguluje chuť k jídlu zvýšením pocitu plnosti a sytosti, zatímco snižuje pocit hladu a potenciální konzumaci jídla, čímž dochází ke snížení příjmu potravy. Liraglutid nezvyšuje v porovnání s placebem výdej energie.
Liraglutid stimuluje sekreci inzulinu a snižuje sekreci glukagonu v závislosti na koncentraci glukózy, což vede ke snížení hladiny glukózy na lačno i postprandiálně. Účinek na snížení hladiny glukózy je více patrný u pacientů s prediabetem a diabetem v porovnání s pacienty s normoglykemií. Klinická hodnocení naznačují, že liraglutid zlepšuje a udržuje funkce beta buněk podle modelu HOMA-B a poměru proinzulinu k inzulinu.
Klinická účinnost a bezpečnost
Účinnost a bezpečnost liraglutidu pro úpravu hmotnosti ve spojení se sníženým příjmem kalorií a zvýšenou fyzickou aktivitou byly studovány ve 4 randomizovaných, dvojitě zaslepených, placebem kontrolovaných klinických hodnoceních fáze 3, které se účastnilo celkem 5 358 pacientů.
• Klinické hodnocení 1 (SCALE Obesity & prediabetes - 1839): 56týdenní klinické hodnocení posuzující hmotnostní úbytek u 3 731 randomizovaných (2 590 dokončivších) obézních pacientů a pacientů s nadváhou s jednou z následujících chorob: prediabetem, hypertenzí nebo dyslipidemií. 61 % mělo na začátku prediabetes.
• Klinické hodnocení 2 (SCALE Diabetes - 1922): 56týdenní klinické hodnocení posuzující hmotnostní úbytek u 846 randomizovaných (628 dokončivších) obézních pacientů a pacientů s nadváhou s nedostatečně kontrolovaným diabetes mellitus 2. typu (HbA1c v rozmezí 7 -
10 %). Základní léčba na začátku klinického hodnocení byla buď pouze dieta a cvičení samotné či v kombinaci s metforminem, sulfonylureou, glitazonem a to buď s jediným z těchto přípravků, nebo v jakékoliv kombinaci zde uvedeného.
• Klinické hodnocení 3 (SCALE Sleep apnoea - 3970): 32týdenní klinické hodnocení posuzující závažnost spánkové apnoe a hmotnostní úbytek u 359 randomizovaných
(276 dokončivších) obézních pacientů se středně těžkou nebo těžkou obstrukční spánkovou apnoí.
• Klinické hodnocení 4 (SCALE Maintenance - 1923): 56týdenní klinické hodnocení posuzující zachování tělesné hmotnosti a hmotnostní úbytek u 422 randomizovaných
(305 dokončivších) obézních pacientů a pacientů s nadváhou a hypertenzí nebo dyslipidemií s předcházejícím úbytkem hmotnosti > 5 % vyvolaným nízkokalorickou dietou.
Tělesná hmotnost
Ve všech studovaných skupinách bylo u obézních pacientů/pacientů s nadváhou dosaženo většího hmotnostního úbytku s liraglutidem v porovnání s placebem. Napříč hodnocenými populacemi dosáhlo větší procento pacientů > 5% a > 10% hmotnostního úbytku s liraglutidem než s placebem (tabulky 3 - 5). V klinickém hodnocení 4 si hmotnostní úbytek dosažený před zahájením léčby liraglutidem udrželo více pacientů (81,4 %) než u placeba (48,9 %). Konkrétní údaje o hmotnostním úbytku, respondérech, časovém průběhu a kumulativní distribuci změny hmotnosti (%) pro klinická hodnocení 1 - 4 jsou uvedeny v tabulkách 3 - 6 a na obrázcích 1, 2 a 3.
Hmotnostní úbytek po 12 týdnech léčby 3,0 mg liraglutidu
Časní respondéři byli definováni jako pacienti, kteří léčebnou dávkou liraglutidu (4 týdny nárůstu dávky a 12 týdnů na léčebné dávce) dosáhli > 5% hmotnostního úbytku po 12 týdnech. V klinickém hodnocení 1 dosáhlo 67,5 % účastníků hmotnostního úbytku > 5 % po 12 týdnech. V klinickém hodnocení 2 dosáhlo 50,4 % účastníků hmotnostního úbytku > 5 % po 12 týdnech. S pokračující léčbou liraglutidem se u 86,2 % těchto časných respondérů předpovídá dosažení hmotnostního úbytku > 5 % a u 51 % se předpovídá dosažení hmotnostního úbytku > 10 % po 1 roce léčby. Predikovaný průměrný hmotnostní úbytek u časných respondérů, kteří dokončili 1 rok léčby, je 11,2 % jejich výchozí tělesné hmotnosti (9,7 % u mužů a 11,6 % u žen). U pacientů, kteří dosáhli hmotnostního úbytku < 5 % po 12 týdnech na léčebné dávce liraglutidu bylo procento pacientů nedosahujících hmotnostního úbytku > 10 % po 1 roce 93,4 %.
Kontrola hladin glukózy v krvi
Léčba liraglutidem významně zlepšila glykemické parametry napříč podskupinami populací s normoglykemií, prediabetem a diabetes mellitus 2. typu. V klinickém hodnocení 1 se u pacientů léčených liraglutidem vyvinul diabetes mellitus 2. typu u menšího počtu (0,2 %) v porovnání s pacienty užívajícími placebo (1,1 %). U pacientů s prediabetem na začátku léčby se tento stav zvrátil u většího počtu (69,2 %) v porovnání s pacienty užívajícími placebo (32,7 %).
Kardiometabolické rizikové _faktory
Léčba liraglutidem významně zlepšila systolický krevní tlak a obvod pasu v porovnání s placebem (tabulka 3 a 4).
Apnoe-hypopnoe index (AHI)
Léčba liraglutidem významně snížila závažnost obstrukční spánkové apnoe vyhodnocené na základě změny z výchozí hodnoty AHI v porovnání s placebem (tabulka 5).
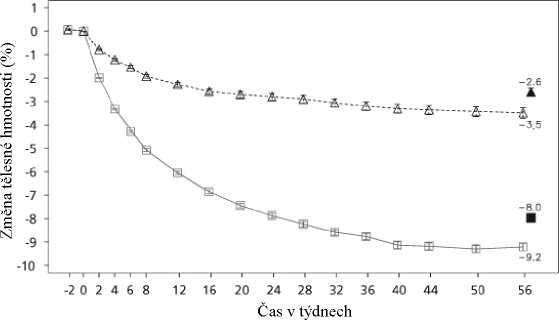
Tabulka 3 Klinické hodnocení 1: Změny oproti výchozí hodnotě u tělesné hmotnosti, hladiny glukózy v krvi a kardiometabolických parametrů v 56. týdnu_
|
Saxenda (N = 2 437) |
Placebo (N = 1 225) |
Saxenda vs. placebo | ||
|
Tělesná hmotnost | ||||
|
Výchozí hodnota, kg (SD) |
106,3 (21,2) |
106,3 (21,7) |
- | |
|
Průměrná změna v 56. týdnu, % (95% CI) |
-8,0 |
-2,6 |
-5,4** (-5,8; -5,0) | |
|
Průměrná změna v 56. týdnu, kg (95 % CI) |
-8,4 |
-2,8 |
-5,6** (-6,0; -5,1) | |
|
Procento pacientů s úbytkem > 5 % tělesné hmotnosti v 56. týdnu, % (95 % CI) |
63,5 |
26,6 |
4,8** (4,1; 5,6) | |
|
Procento pacientů s úbytkem > 10 % tělesné hmotnosti v 56. týdnu, % (95 % CI) |
32,8 |
10,1 |
4,3** (3,5; 5,3) | |
|
Hladina glukózy v krvi a kardiometabolické faktory |
Výchozí Změna hodnota |
Výchozí Změna hodnota | ||
|
HbA1C, % |
5,6 |
-0,3 |
5,6 -0,1 |
-0,23** (-0,25; -0,21) |
|
FPG, mmol/l |
5,3 |
-0,4 |
5,3 -0,01 |
-0,38** (-0,42; -0,35) |
|
Systolický krevní tlak, mmHg |
123,0 |
-4,3 |
123,3 -1,5 |
-2,8** (-3,6; -2,1) |
|
Diastolický krevní tlak, mmHg |
78,7 |
-2,7 |
78,9 -1,8 |
-0,9* (-1,4; -0,4) |
|
Obvod pasu, cm |
115,0 |
-8,2 |
114,5 -4,0 |
-4,2** (-4,7; -3,7) |
Analýza celého souboru. U tělesné hmotnosti, HbA1c, FPG, krevního tlaku a obvodu pasu jsou výchozími hodnotami průměry, změny od výchozí hodnoty v 56. týdnu jsou odhadované průměry (nejmenší čtverce) a léčebné kontrasty v 56. týdnu jsou odhadované léčebné rozdíly. U části pacientů s úbytkem tělesné hmotnosti > 5 /> 10 % jsou uváděny odhadované poměry pravděpodobnosti. Chybějící hodnoty po začátku byly přiřazeny
pomocí přenesených posledních pozorování. *p < 0,05. **p < 0,0001. CI = konfidenční intervaly. FPG = hladina glukózy v plazmě na lačno. SD = směrodatná odchylka.
•=• Saxenda Placebo ■ ± Přenesené poslední pozorování (LOCF) Pozorované hodnoty u pacientů, kteří dokončili každou plánovanou návštěvu
Obrázek 1 Změna oproti výchozí hodnotě u tělesné hmotnosti (%) v čase v klinickém hodnocení 1
Změna tělesné hmotnosti (%) ..... Saxenda ---Placebo
Přenesené poslední pozorování
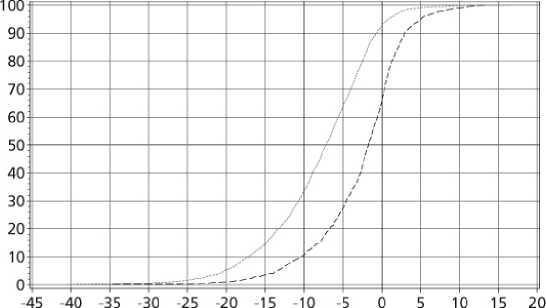
Obrázek 2 Kumulativní distribuce změny hmotnosti (%) po 56 týdnech léčby v klinickém hodnocení 1
Tabulka 4 Klinické hodnocení 2: Změny oproti výchozí hodnotě u tělesné hmotnosti, hladiny glukózy v krvi a kardiometabolických parametrů v 56. týdnu_
|
Saxenda (N = 412) |
Placebo (N = 211) |
Saxenda vs. placebo | |
|
Tělesná hmotnost | |||
|
Výchozí hodnota, kg (SD) |
105,6 (21,9) |
106,7 (21,2) |
- |
|
Průměrná změna v 56. týdnu, % (95% CI) |
-5,9 |
-2,0 |
-4,0** (-4,8; -3,1) |
|
Průměrná změna v 56. týdnu, kg (95 % CI) |
-6,2 |
-2,2 |
-4,1** (-5,0; -3,1) |
|
Procento pacientů s úbytkem > 5 % tělesné hmotnosti v 56. týdnu, % (95 % CI) |
49,8 |
13,5 |
6,4** (4,1; 10,0) |
|
Procento pacientů s úbytkem > 10 % tělesné hmotnosti v 56. týdnu, % (95 % CI) |
22,9 |
4,2 |
6,8** (3,4; 13,8) |
|
Hladina glukózy v krvi a kardiometabolické faktory |
Výchozí Změna hodnota |
Výchozí Změna hodnota | |
|
HbAjc, % |
7,9 -1,3 |
7,9 -0,4 |
-0,9** (-1,1; -0,8) |
|
FPG, mmol/l |
8,8 |
-1,9 |
8,6 |
-0,1 |
-1,8** (-2,1; -1,4) |
|
Systolický krevní tlak, mmHg |
128,9 |
-3,0 |
129,2 |
-0,4 |
-2,6* (-4,6; -0,6) |
|
Diastolický krevní tlak, mmHg |
79,0 |
-1,0 |
79,3 |
-0,6 |
-0,4 (-1,7; 1,0) |
|
Obvod pasu, cm |
118,1 |
-6,0 |
117,3 |
-2,8 |
-3,2** (-4,2; -2,2) |
Analýza celého souboru. U tělesné hmotnosti, HbAJc, FPG, krevního tlaku a obvodu pasu jsou výchozími hodnotami průměry, změny od výchozí hodnoty v 56. týdnu jsou odhadované průměry (nejmenší čtverce) a léčebné kontrasty v 56. týdnu jsou odhadované léčebné rozdíly. U části pacientů s úbytkem tělesné hmotnosti > 5 /> 10 % jsou uváděny odhadované poměry pravděpodobnosti. Chybějící hodnoty po začátku byly přiřazeny pomocí přenesených posledních pozorování. *p < 0,05. **p < 0,0001. CI = konfidenční intervaly. FPG = hladina glukózy v plazmě na lačno. SD = směrodatná odchylka.
Tabulka 5 Klinické hodnocení 3: Změny oproti výchozí hodnotě u tělesné hmotnosti a indexu apnoe-hypopnoe ve 32. týdnu_
|
Saxenda (N = 180) |
Placebo (N = 179) |
Saxenda vs. placebo | |
|
Tělesná hmotnost | |||
|
Výchozí hodnota, kg (SD) |
116,5 (23,0) |
118,7 (25,4) |
- |
|
Průměrná změna ve 32. týdnu, % (95% CI) |
-5,7 |
-1,6 |
-4,2** (-5,2; -3,1) |
|
Průměrná změna ve 32. týdnu, kg (95 % CI) |
-6,8 |
-1,8 |
-4,9** (-6,2; -3,7) |
|
Procento pacientů s úbytkem > 5 % tělesné hmotnosti ve 32. týdnu, % (95 % CI) |
46,4 |
18,1 |
3,9** (2,4; 6,4) |
|
Procento pacientů s úbytkem > 10 % tělesné hmotnosti ve 32. týdnu, % (95 % CI) |
22,4 |
1,5 |
19,0** (5,7; 63,1) |
|
Výchozí Změna hodnota |
Výchozí Změna hodnota | ||
|
Index apnoe-hypopnoe, příhody/hodina |
49,0 -12,2 |
49,3 -6,1 |
-6,1* (-11,0; -1,2) |
Analýza celého souboru. Výchozími hodnotami jsou průměry, změny od výchozí hodnoty ve 32. týdnu jsou odhadované průměry (nejmenší čtverce) a léčebné kontrasty ve 32. týdnu jsou odhadované léčebné rozdíly (95% CI). U části pacientů s úbytkem tělesné hmotnosti > 5 /> 10 % jsou uváděny odhadované poměry pravděpodobnosti. Chybějící hodnoty po začátku byly přiřazeny pomocí přenesených posledních pozorování.
*p < 0,05. **p < 0,0001. CI = konfidenční intervaly. SD = směrodatná odchylka.
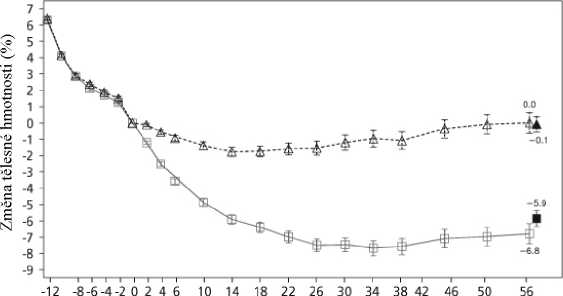
Tabulka 6 Klinické hodnocení 4: Změny oproti výchozí hodnotě u tělesné hmotnosti v 56. týdnu
Saxenda (N = 207) Placebo (N = 206) Saxenda vs. placebo
|
Výchozí hodnota, kg (SD) |
100,7 (20,8) |
98,9 (21,2) |
- |
|
Průměrná změna v 56. týdnu, % (95% CI) |
-6,3 |
-0,2 |
-6,1** (-7,5; -4,6) |
|
Průměrná změna v 56. týdnu, kg (95 % CI) |
-6,0 |
-0,2 |
-5,9** (-7,3; -4,4) |
|
Procento pacientů s úbytkem > 5 % tělesné hmotnosti v 56. týdnu, % (95 % CI) |
50,7 |
21,3 |
3,8** (2,4; 6,0) |
|
Procento pacientů s úbytkem > 10 % tělesné hmotnosti v 56. týdnu, % (95 % CI) |
27,4 |
6,8 |
5,1** (2,7; 9,7) |
Analýza celého souboru. Výchozími hodnotami jsou průměry, změny od výchozí hodnoty v 56. týdnu jsou odhadované průměry (nejmenší čtverce) a léčebné kontrasty v 56. týdnu jsou odhadované léčebné rozdíly.
U části pacientů s úbytkem tělesné hmotnosti > 5 /> 10 % jsou uváděny odhadované poměry pravděpodobnosti. Chybějící hodnoty po začátku byly přiřazeny pomocí přenesených posledních pozorování. **p < 0,0001.
CI = konfidenční intervaly. SD = směrodatná odchylka.
Čas v týdnech
Saxenda t Placebo ■ á Přenesené poslední pozorování (LOCF)
Pozorované hodnoty u pacientů, kteří dokončili každou plánovanou návštěvu
Obrázek 3 Změna od randomizace (0. týden) u tělesné hmotnosti (%) v čase v klinickém hodnocení 4
Před 0. týdnem byli pacienti léčeni pouze nízkokalorickou dietou a cvičením. V 0. týdnu byli pacienti randomizováni do skupin užívajících buď přípravek Saxenda, nebo placebo.
Imunogenita
V souvislosti s potenciálně imunogenními vlastnostmi proteinových a peptidových léčivých přípravků se mohou u pacientů léčených liraglutidem tvořit protilátky proti liraglutidu. V klinických hodnoceních se u 2,5 % pacientů léčených liraglutidem vytvořily proti této látce protilátky. Tvorba protilátek nebyla spojena se sníženou účinností liraglutidu.
Kardiovaskulární zhodnocení
Závažné kardiovaskulární nežádoucí účinky (major adverse cardiovascular events, MACE) byly posouzeny externí nezávislou skupinou odborníků a definovány jako nefatální infarkt myokardu, nefatální cévní mozková příhoda a úmrtí z kardiovaskulární příčiny. Ve všech dlouhodobých klinických hodnoceních s přípravkem Saxenda došlo k 6 MACE u pacientů léčených liraglutidem a 10 MACE u pacientů užívajících placebo. Poměr rizik a 95% CI je 0,33 [0,12; 0,90] u liraglutidu oproti placebu. V klinických hodnoceních fáze 3 bylo u liraglutidu pozorováno průměrné zvýšení srdeční frekvence o 2,5 tepů za minutu (v rozmezí od 1,6 do 3,6 tepů za minutu napříč klinickými hodnoceními) oproti výchozí hodnotě. Srdeční frekvence dosáhla vrcholu po cca 6 týdnech. Dlouhodobý klinický dopad tohoto průměrného zvýšení srdeční frekvence nebyl stanoven. Změna srdeční frekvence byla po vysazení liraglutidu reverzibilní (viz bod 4.4).
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Saxenda u jedné nebo více podskupin pediatrické populace v léčbě obezity a léčbě Prader-Williho syndromu (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Absorpce liraglutidu po subkutánním podání byla pomalá a dosahuje maximální koncentrace za cca 11 hodin po podání. Průměrná koncentrace liraglutidu v ustáleném stavu (AUCt/24) dosáhla u obézních pacientů (BMI 30 - 40 kg/m2) po podání 3 mg liraglutidu přibližně 31 nmol/l. Expozice liraglutidu se zvyšovala proporčně s dávkou. Absolutní biologická dostupnost liraglutidu po subkutánním podání je přibližně 55 %.
Distribuce
Zdánlivý průměrný distribuční objem po subkutánním podání je 20 - 25 l (u osob s tělesnou hmotností přibližně 100 kg). Liraglutid se značně váže na plasmatické proteiny (> 98 %).
Biotransformace
Během 24 hodin po podání jednorázové dávky liraglutidu s [3H] zdravým subjektům byl hlavní složkou v plasmě intaktní liraglutid. Byly zjištěny dva méně významné metabolity v plasmě (< 9 % a < 5 % celkové expozice plasmatické radioaktivity).
Eliminace
Liraglutid je endogenně metabolizován podobným způsobem jako velké bílkoviny, aniž by byl zjištěn určitý orgán jako hlavní cesta eliminace. Po podání dávky liraglutidu značeného [3H] nebyl intaktní liraglutid zjištěn v moči ani ve stolici. Močí nebo stolicí byla jako metabolity liraglutidu vyloučena pouze menší část podané radioaktivity (6 % a 5 %). Radioaktivita v moči a stolici byla vylučována hlavně během prvních 6 - 8 dnů a odpovídala třem méně významným metabolitům liraglutidu.
Průměrná clearance po subkutánním podání liraglutidu je přibližně 0,9 - 1,4 l/h s poločasem eliminace přibližně 13 hodin.
Zvláštní skupiny pacientů
Starší pacienti
Podle výsledků dat populační farmakokinetické analýzy u pacientů s nadváhou a obézních pacientů (18 až 82 let) nemá věk žádný klinicky významný vliv na farmakokinetiku liraglutidu. Z důvodu věku není nutná žádná úprava dávkování.
Pohlaví
Na základě výsledků populačních farmakokinetických analýz měly ženy o 24 % nižší clearance liraglutidu upravenou podle tělesné hmotnosti v porovnání s muži. Na základě dat o odpovědi na expozici není nutná žádná úprava dávky podle pohlaví.
Etnický _ původ
Podle výsledků populační farmakokinetické analýzy, která zahrnovala pacienty s nadváhou a obézní pacienty z bělošských, černošských, asijských a hispánských/nehispánských skupin, nemá etnický původ žádný klinicky významný vliv na farmakokinetiku liraglutidu.
Tělesná hmotnost
Expozice liraglutidu se snižuje se vzrůstající výchozí tělesnou hmotností. 3mg denní dávka liraglutidu poskytovala odpovídající systémové expozice pro tělesnou hmotnost v rozmezí 60 - 234 kg, což bylo hodnoceno z hlediska odpovědi na expozici v klinických hodnoceních. Expozice liraglutidu nebyla studována u pacientů s tělesnou hmotností > 234 kg.
Porucha _ funkce _ jater
Farmakokinetika liraglutidu byla hodnocena u pacientů s různým stupněm poruchy funkce jater v hodnocení s jednorázovou dávkou (0,75 mg). U pacientů s lehkou až středně těžkou poruchou funkce jater byla expozice liraglutidu snížena ve srovnání se zdravými pacienty o 13 - 23 %. Expozice byla významně nižší (44 %) u pacientů s těžkou poruchou funkce jater (skóre Child Pugh > 9).
Porucha _ funkce ledvin
U pacientů s poruchou funkce ledvin byla expozice liraglutidu ve srovnání s jedinci s normální funkcí ledvin v klinickém hodnocení s jednorázovou dávkou (0,75 mg) snížena. U pacientů s lehkou (clearance kreatininu CrCl 50 - 80 ml/min), středně těžkou (CrCl 30 - 50 ml/min) a těžkou (CrCl < 30 ml/min) poruchou funkce ledvin a u konečného stadia renálního onemocnění vyžadujícího dialýzu byla expozice liraglutidem snížena o 33 %, 14 %, 27 % a 26 %.
Pediatrická populace
Přípravek Saxenda nebyl studován u pediatrických pacientů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Ve dvouletých studiích kancerogenity na potkanech a myších byly pozorovány nonletální tumory C-buněk štítné žlázy. U potkanů nebyl pozorován NOAEL (No observed adverse effect level). Tyto tumory nebyly pozorovány u opic léčených po dobu 20 měsíců. Tyto nálezy u hlodavců jsou způsobeny negenotoxickým a receptory zprostředkovaným mechanismem specifickým pro GLP-1, na který jsou hlodavci zvláště citliví. Význam pro člověka je pravděpodobně nízký, ale nemůže být zcela vyloučen. Žádné jiné tumory spojené s léčbou nebyly zjištěny.
Studie na zvířatech neprokázaly přímé škodlivé účinky týkající se fertility, ale lehce zvýšenou míru časných úmrtí embryí po nejvyšší dávce. Podávání liraglutidu ve střední fázi březosti vedlo ke snížení tělesné hmotnosti matky a zpomalení růstu plodu s neprůkazným vlivem na žebra u potkanů a kosterní změny u králíků. Růst novorozenců byl u potkanů vystavených působení liraglutidu snížen a tento efekt přetrvával u skupiny s vysokou dávkou i po odstavení. Není známo, zda je redukovaný růst mláďat způsoben sníženým příjmem mléka v důsledku přímého vlivu GLP-1, nebo sníženou tvorbou mateřského mléka při sníženém kalorickém příjmu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát hydrogenfosforečnanu sodného
Propylenglykol
Fenol
Kyselina chlorovodíková (k úpravě pH)
Hydroxid sodný (k úpravě pH)
Voda na injekci
6.2 Inkompatibility
Látky přidané k přípravku Saxenda mohou způsobit degradaci liraglutidu. Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
30 měsíců
Po prvním použití: 1 měsíc
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem.
Neuchovávejte v blízkosti mrazicího oddílu.
Po prvním použití: Uchovávejte při teplotě do 30 C nebo v chladničce (2°C - 8°C).
Ponechávejte uzávěr na peru, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
Zásobní vložka (sklo třídy 1) s pístem (bromobutyl) a zátkou (bromobutyl/polyisopren) obsažená v jednorázovém předplněném vícedávkovém peru zhotoveném z polypropylenu, polyacetalu, polykarbonátu a akrylonitrilbutadienstyrenu.
Jedno pero obsahuje 3 ml roztoku a lze jej použít k aplikaci dávky 0,6 mg, 1,2 mg, 1,8 mg, 2,4 mg a 3,0 mg.
Velikost balení po 1, 3 nebo 5 předplněných perech.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Roztok nesmí být použit v případě, že není čirý, bezbarvý či téměř bezbarvý.
Přípravek Saxenda nesmí být použit v případě, že byl zmražen.
Pero je určené k použití s jednorázovými jehlami NovoFine nebo NovoTwist o délce do 8 mm a síle do 32 G.
Jehly nejsou součástí balení.
Pacient musí být poučen, že má injekční jehlu po každé aplikaci zlikvidovat a pero má uchovávat bez nasazené jehly. Tím se zabrání kontaminaci, infekci a vytékání přípravku. Také se tím zajistí přesnost dávkování.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
8. REGISTRAČNÍ ČÍSLA
EU/1/15/992/001-003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 23. března 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců biologické léčivé látky
Novo Nordisk A/S
Hallas Allé
4400 Kalundborg
Dánsko
Novo Nordisk A/S Novo Allé 2880 Bagsv^rd Dánsko
Název a adresa výrobce odpovědného za propouštění šarží
Novo Nordisk A/S
Novo Allé
2880 Bagsv^rd
Dánsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Saxenda 6 mg/ml injekční roztok v předplněném peru Liraglutidum
2. OBSAH LÉČIVÉ LÁTKY
Jeden ml obsahuje liraglutidum 6 mg. Jedno předplněné pero obsahuje liraglutidum 18 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: dihydrát hydrogenfosforečnanu sodného, propylenglykol, fenol, kyselina chlorovodíková/hydroxid sodný (k úpravě pH), voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 pero 3 pera 5 per
Jedno pero obsahuje 3 ml roztoku a lze jej použít k aplikaci dávky 0,6 mg, 1,2 mg, 1,8 mg, 2,4 mg a 3,0 mg.
5. ZPŮSOB A CESTA PODÁNÍ
Pero je určeno k použití s jednorázovými jehlami NovoFine nebo NovoTwist. Jehly nejsou součástí balení.
Před použitím si přečtěte příbalovou informaci.
Subkutánní podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Neuchovávejte pero s nasazenou jehlou. Určeno pouze pro jednu osobu.
8. POUŽITELNOST
Použitelné do:
1 měsíc po prvním použití pero zlikvidujte.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Po prvním použití pera uchovávejte při teplotě do 30°C nebo v chladničce. Ponechávejte uzávěr na peru, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
12. REGISTRAČNÍ ČÍSLO
EU/1/15/992/001 1 x 3 ml EU/1/15/992/002 3 x 3 ml EU/1/15/992/003 5 x 3 ml
13. ČÍSLO ŠARŽE
č.S.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Saxenda
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK NA PŘEDPLNĚNÉ PERO
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
Saxenda 6 mg/ml injekce Liraglutidum Subkutánní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE
c.s.
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
3 ml
6. JINÉ
Novo Nordisk A/S
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Saxenda 6 mg/ml injekční roztok v předplněném peru
liraglutidum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Saxenda a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Saxenda používat
3. Jak se přípravek Saxenda používá
4. Možné nežádoucí účinky
5. Jak přípravek Saxenda uchovávat
6. Obsah balení a další informace
1. Co je přípravek Saxenda a k čemu se používá Co je přípravek Saxenda
Přípravek Saxenda je léčivý přípravek sloužící k dosažení úbytku hmotnosti, který obsahuje léčivou látku liraglutid. Je podobný přirozeně se vyskytujícímu hormonu zvanému GLP-1, který se po jídle uvolňuje ze střev. Přípravek Saxenda působí na receptory v mozku, které ovlivňují chuť k jídlu, a způsobuje, že se cítíte nasycenější a méně hladový(á). Tak Vám může pomoci jíst méně a zredukovat tělesnou hmotnost.
K čemu se přípravek Saxenda používá
Přípravek Saxenda se používá k dosažení hmotnostního úbytku jako doplňková léčba k dietě a cvičení u dospělých ve věku 18 let a starších, kteří mají:
• BMI 30 nebo vyšší (obézní) nebo
• BMI 27 nebo nižší než 30 (s nadváhou) a trpí zdravotními problémy souvisejícími s tělesnou hmotností (např. diabetes, vysoký krevní tlak, abnormální hladiny tuků v krvi nebo potíže
s dýcháním během spánku zvané „obstrukční spánková apnoe“).
BMI (index tělesné hmotnosti, Body Mass Index) je měřítko tělesné hmotnosti vztažené k výšce pacienta.
V léčbě přípravkem Saxenda smíte pokračovat pouze, pokud u vás došlo k minimálně 5% úbytku Vaší počáteční tělesné hmotnosti, a to po 12 týdnech užívání dávky 3 mg denně (viz bod 3). Poraďte se s lékařem před tím, než budete s léčbou dále pokračovat.
Dieta a cvičení
Lékař Vám nejprve stanoví dietní a cvičební program. Během užívání přípravku Saxenda dodržujte pokyny, které jste v rámci tohoto programu obdržel(a).
2. Čemu musíte věnovat pozornost, než začnete přípravek Saxenda používat Nepoužívejte přípravek Saxenda:
- jestliže j ste alergický(á) na liraglutid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Saxenda se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
S použitím tohoto přípravku u pacientů se srdečním selháním jsou jen malé či žádné zkušenosti. Trpíte-li závažným srdečním selháním, není používání tohoto přípravku doporučeno.
S použitím tohoto přípravku u pacientů ve věku 75 let a starších jsou jen malé zkušenosti. Pokud je Vám 75 let a více, použití se nedoporučuje.
S použitím tohoto přípravku u pacientů s ledvinovými problémy jsou jen malé zkušenosti. Trpíte-li onemocněním ledvin, či pokud podstupujete dialýzu, poraďte se s lékařem
S použitím tohoto přípravku u pacientů s jaterními problémy jsou jen malé zkušenosti. Pokud máte potíže s játry, poraďte se s lékařem.
Použití tohoto přípravku není doporučeno, pokud máte závažné žaludeční nebo střevní potíže, které mají za následek zpožděné vyprazdňování žaludku (tzv. gastroparéza), či pokud máte zánětlivé onemocnění střev.
Lidé s diabetem
Pokud máte diabetes, nepoužívejte přípravek Saxenda jako náhradu za inzulin.
Zánět slinivky břišní
Informujte svého lékaře, pokud trpíte onemocněním slinivky břišní nebo jste je někdy prodělal(a). Zánět žlučníku a žlučové kameny
Pokud značně ztratíte na hmotnosti, existuje riziko tvorby žlučových kamenů, a tím i zánět žlučníku. Pokud pociťujete silnou bolest v horní části břicha, obvykle silnější na pravé straně pod žebry, přestaňte přípravek Saxenda používat a kontaktujte okamžitě lékaře. Bolest se může rozšiřovat až do zad nebo do pravého ramene. Viz bod 4.
Onemocnění štítné žlázy
Pokud trpíte onemocněním štítné žlázy včetně uzlovitého zbytnění štítné žlázy a zvětšení štítné žlázy, obraťte se na lékaře.
Srdeční frekvence
Pokud máte během léčby přípravkem Saxenda palpitace, (pociťujete bušení srdce) nebo pokud pociťujete v klidu prudké bušení srdce, sdělte to svému lékaři.
Ztráta tekutin a dehydratace
Při zahájení léčby přípravkem Saxenda může dojít ke ztrátě tělesných tekutin nebo dehydrataci (odvodnění). K tomu může dojít vlivem nevolnosti (pocitu na zvracení) či následkem zvracení a průjmu. Je důležité, abyste zabránil(a) dehydrataci pitím dostatečného množství tekutin. Máte-li jakékoli otázky nebo obavy, promluvte si se svým lékařem, lékárníkem nebo zdravotní sestrou. Viz bod 4.
Děti a dospívající
Přípravek Saxenda se nemá používat u dětí a dospívajících mladších 18 let. Je tomu tak proto, že účinky tohoto léku nebyly u těchto věkových skupin studovány.
Další léčivé přípravky a přípravek Saxenda
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Zvláště byste měl(a) svému lékaři, lékárníkovi nebo zdravotní sestře oznámit, pokud:
• užíváte antidiabetika (přípravky k léčbě cukrovky) zvaná „sulfonylurea“ (např. glimepirid nebo glibenklamid) - při užívání těchto léčiv spolu s přípravkem Saxenda Vám může poklesnout hladina cukru v krvi (hypoglykemie). Lékař Vám může upravit dávku antidiabetik tak, aby zabránil poklesu hladiny cukru v krvi. Varovné příznaky nízké hladiny cukru v krvi najdete
v bodě 4.
• užíváte warfarin nebo jiná perorální léčiva (užívaná ústy), která snižují srážlivost krve (antikoagulancia). Lékař může požadovat častější provádění krevních testů k určení schopnosti vytvářet krevní sraženinu.
Těhotenství a kojení
Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, přípravek Saxenda nepoužívejte. Není totiž známo, zda přípravek Saxenda může mít vliv dítě.
Pokud používáte přípravek Saxenda, nekojte. Není totiž známo, zda přípravek Saxenda přechází do mateřského mléka.
Řízení dopravních prostředků a obsluha strojů
Přípravek Saxenda pravděpodobně neovlivňuje Vaši schopnost řídit dopravní prostředky a obsluhovat stroje. Pokud potřebujete jakékoli další informace, poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Důležitá informace o některých složkách přípravku Saxenda
Tento přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. v podstatě je „bez sodíku“.
3. Jak se přípravek Saxenda používá
Vždy používejte přípravek Saxenda přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Váš lékař Vám nejprve stanoví dietní a cvičební program. Během užívání přípravku Saxenda dodržujte pokyny, které jste v rámci tohoto programu obdržel(a).
Jaké množství přípravku Saxenda si aplikovat
Léčba započne nízkou dávkou, která se bude postupně v průběhu prvních pěti týdnů léčby zvyšovat.
• Při prvním zahájení používání přípravku Saxenda je počáteční dávka je 0,6 mg jedenkrát denně po dobu minimálně jednoho týdne.
• Dávku je třeba každý týden zvyšovat o 0,6 mg, dokud nedosáhnete doporučené dávky 3,0 mg jedenkrát denně.
Váš lékař Vám sdělí, kolik přípravku Saxenda máte každý týden použít. Obvykle se doporučuje postupovat podle níže uvedené tabulky.
|
Týden |
Aplikovaná dávka |
|
1. týden |
0,6 mg jedenkrát denně |
|
2. týden |
1,2 mg jedenkrát denně |
|
3. týden |
1,8 mg jedenkrát denně |
|
4. týden |
2,4 mg jedenkrát denně |
3,0 mg jedenkrát denně
Jakmile dosáhnete doporučené dávky 3,0 mg v 5. týdnu léčby, pokračujte v podávání této dávky až do skončení léčebného období. Dále svou dávku nezvyšujte.
Váš lékař bude pravidelně Vaši léčbu hodnotit.
Jak a kdy se přípravek Saxenda používá
• Před prvním použitím pera Vám lékař nebo zdravotní sestra ukážou, jak se pero používá.
• Přípravek Saxenda můžete použít kdykoli během dne, s jídlem a pitím nebo bez nich.
• Používejte přípravek Saxenda každý den přibližně ve stejnou dobu - vyberte si denní dobu, která je pro Vás nejlepší.
Kam si aplikovat injekci
Přípravek Saxenda se podává jako injekce pod kůži (subkutánní injekce).
• Nejlepší místa k aplikaci jsou přední část pasu (břicho), přední strana stehen nebo horní část paže.
• Neaplikujte si injekci do žíly ani do svalu.
Podrobné pokyny k použití jsou uvedeny na druhé straně této příbalové informace.
Lidé s diabetem
Sdělte svému lékaři, že máte diabetes. Váš lékař může upravit dávku antidiabetik tak, aby se zabránilo poklesu hladiny cukru v krvi.
• Nemíchejte přípravek Saxenda s jinými injekčními přípravky (např. inzuliny).
• Nepoužívejte přípravek Saxenda v kombinaci s jinými přípravky , které obsahují agonisty GLP-1 receptoru (např. exenatid, lixisenatid).
Jestliže jste použil(a) více přípravku Saxenda, než jste měl(a)
Pokud použijete více přípravku Saxenda, než jste měl(a), sdělte to okamžitě svému lékaři nebo ihned navštivte nemocnici. Balení léku si vezměte s sebou. Můžete vyžadovat léčbu. Může dojít k následujícím účinkům:
• nevolnost (pocit na zvracení)
• zvracení.
Jestliže jste zapomněl(a) použít přípravek Saxenda
• Pokud zapomenete jednu dávku a vzpomenete si v průběhu 12 hodin od obvyklé doby podání, aplikujte si ji hned, jak si na to vzpomenete.
• Pokud však od doby, kdy jste měl(a) přípravek Saxenda použít, uplynulo více než 12 hodin, vynechejte zmeškanou dávku a aplikujte si injekčně další dávku následující den v obvyklou dobu.
• Nezdvojnásobujte dávku ani dávku nezvyšujte následující den, abyste vynechanou dávku nahradil(a).
Jestliže jste přestal(a) používat přípravek Saxenda
Nepřestávejte přípravek Saxenda používat, aniž byste si o tom promluvil(a) se svým lékařem.
Máte-li jakékoli další otázky, týkající se používání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
r-g r V r V a I r r V • I
Závažné nežádoucí účinky
U pacientů používajících přípravek Saxenda bylo vzácně hlášeno několik těžkých alergických reakcí (anafylaxe). Pokud máte příznaky, jakými jsou např. potíže s dýcháním, otok obličeje a hrdla a rychlý srdeční tep, musíte ihned vyhledat lékaře.
U pacientů používajících přípravek Saxenda byly méně často hlášeny případy zánětu slinivky břišní (pankreatitidy). Pankreatitida je závažný, potencionálně život ohrožující stav.
Pocítíte-li následující závažné příznaky, ukončete užívání přípravku Saxenda a okamžitě kontaktujte lékaře:
• Silná a přetrvávající bolest břicha (v oblasti žaludku), která může zasahovat až do zad, rovněž pocit na zvracení a zvracení. Mohou to být příznaky zánětu slinivky břišní (pankreatitidy).
Další nežádoucí účinky
Velmi časté: mohou se projevit u více než 1 pacienta z 10
• nevolnost (pocit na zvracení), zvracení, průjem, zácpa - tyto obvykle po několika dnech nebo týdnech odezní.
Časté: mohou se projevit až u 1 z 10 pacientů
• žaludeční a střevní potíže, jako jsou: porucha trávení (dyspepsie), zánět žaludeční sliznice (gastritida), nepříjemný pocit v oblasti žaludku, bolest v horní části břicha, pálení žáhy, pocit nadmutí, větry (flatulence), říhání, sucho v ústech
• pocit slabosti nebo únavy
• změna vnímání chuti
• závrať
• problémy se spaním (nespavost). Ta se obvykle objevuje první 3 měsíce léčby.
• žlučové kameny
• reakce v místě vpichu (jako podlitiny, bolest, podráždění, svědění a vyrážka)
• nízká hladina cukru v krvi (hypoglykemie). Varovné příznaky nízké hladiny cukru v krvi se mohou objevit náhle a mohou zahrnovat: studený pot, chladnou bledou kůži, bolest hlavy, rychlý srdeční tep, pocit na zvracení, pocit velkého hladu, změny vidění, ospalost, pocit slabosti, nervozitu, pocit úzkosti, zmatenost, potíže s koncentrací a třes (tremor). Lékař Vám řekne, jak nízkou hladinu cukru v krvi léčit a co dělat, když zaznamenáte tyto varovné příznaky
• zvýšení hladin pankreatických enzymů (např. lipázy a amylázy).
Méně časté: mohou se projevit až u 1 ze 100 pacientů
• ztráta tekutin (dehydratace). Ta je pravděpodobnější na začátku léčby a může k ní docházet kvůli zvracení, nevolnosti (pocitu na zvracení) a průjmu
• zánět žlučníku
• alergické reakce včetně kožní vyrážky
• pocit celkové nepohody
• rychlejší tep.
Vzácné: mohou se projevit až u 1 pacienta z 1 000
• snížená funkce ledvin
• akutní selhání ledvin. Příznaky mohou zahrnovat snížení objemu moči, kovovou chuť v ústech a snadnou tvorbu modřin.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Saxenda uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte přípravek Saxenda po uplynutí doby použitelnosti uvedené na štítku pera a na krabičce za „Použitelné do“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Před, prvním použitím:
Uchovávejte v chladničce (2°C až 8°C). Chraňte před mrazem. Neuchovávejte v blízkosti mrazicího oddílu.
Jakmile začnete pero používat:
Pero můžete uchovávat při teplotě do 30°C nebo v chladničce (2°C - 8°C) po dobu 1 měsíce. Chraňte před mrazem. Neuchovávejte v blízkosti mrazicího oddílu.
Pokud pero nepoužíváte, ponechejte uzávěr na peru, aby byl přípravek chráněn před světlem.
Nepoužívejte tento přípravek, pokud není roztok čirý a bezbarvý či téměř bezbarvý.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Saxenda obsahuje
- Léčivou látkou je liraglutidum. Jeden ml injekčního roztoku obsahuje liraglutidum 6 mg. Jedno předplněné pero obsahuje liraglutidum 18 mg.
- Dalšími složkami jsou dihydrát hydrogenfosforečnanu sodného, propylenglykol, fenol, kyselina chlorovodíková a hydroxid sodný (k úpravě pH) a voda na injekci.
Jak přípravek Saxenda vypadá a co obsahuje toto balení
Přípravek Saxenda je dodáván jako čirý bezbarvý či téměř bezbarvý injekční roztok v předplněném peru. Jedno pero obsahuje 3 ml roztoku a lze jej použít k aplikaci dávky 0,6 mg, 1,2 mg, 1,8 mg,
2,4 mg a 3,0 mg.
Přípravek Saxenda je dostupný ve velikostech balení 1, 3 nebo 5 per. Na trhu nemusí být všechny velikosti balení.
Jehly nejsou součástí balení.
Držitel rozhodnutí o registraci a výrobce
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
|
Pokyny k použití přípravku Saxenda 6 mg/ml injekční roztok v předplněném peru | |
|
Před použitím předplněného pera Saxenda si pečlivě přečtěte tyto pokyny. Nepoužívejte pero bez odpovídajícího proškolení lékařem nebo zdravotní | |
|
sestrou. | |
|
Nejprve pero zkontrolujte a ujistěte se, že obsahuje přípravek Saxenda 6 mg/ml. Poté si prostudujte následující obrázky a seznamte se se všemi částmi pera a jehly. Pokud jste slepý(á) či slabozraký(á) a nejste schopen (schopna) přečíst údaje na počítadle dávky svého pera, nepoužívejte toto pero bez pomoci. Požádejte o pomoc druhou osobu, která má dobrý zrak a je proškolena v používání předplněného pera Saxenda. Vaše pero je předplněné dávkovací pero. Obsahuje 18 mg liraglutidu a aplikuje dávky 0,6 mg, 1,2 mg, 1,8 mg, 2,4 mg a 3,0 mg. Pero je určeno k použití s jednorázovými jehlami NovoFine nebo NovoTwist o délce do 8 mm a síle do 32 G. Jehly nejsou součástí balení. | |
|
A Důležité upozornění Věnujte těmto poznámkám zvláštní pozornost, neboť jsou důležité pro bezpečné | |
|
používání pera. |
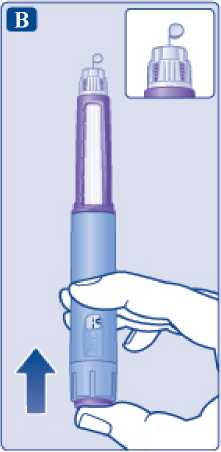
Předplněné pero Saxenda a jehla (příklad)
Uzávěr
pera
au ».»,
Vnější kryt jehly ■HP
í. Vnitřní kryt jehly
Stupnice
pera
Okénko
pera

Papírový
kryt
Štítek
pera
Počítadlo
dávky
Ukazatel
dávky
Volič dávky
r
Symbol
kontroly
průtoku
Dávkovací tlačítko
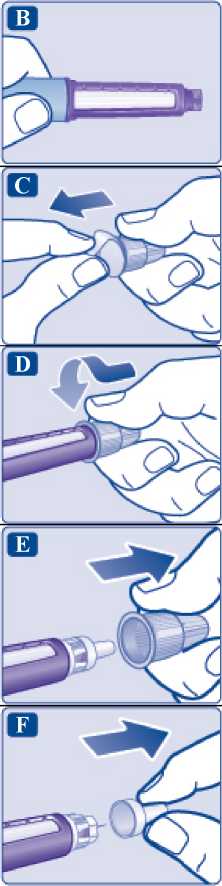
1 Připravte si pero a novou jehlu
• Zkontrolujte název a barevný štítek pera a ujistěte se, že pero obsahuje přípravek Saxenda. To je obzvlášť důležité, pokud užíváte více než jeden typ léku aplikovaného injekčně. Použití špatného léku může poškodit Vaše zdraví.
• Sejměte uzávěr pera.
Zkontrolujte, zda je roztok v peru čirý a bezbarvý. Podívejte se přes okénko pera. Pokud je roztok zakalený, pero nepoužívejte.
Vezměte si novou jehlu a odtrhněte papírový kryt.
Sejměte vnější kryt jehly a ponechejte si jej pro pozdější potřebu.
Budete jej potřebovat po podání injekce, abyste mohl(a) jehlu bezpečně sejmout z pera.
Sejměte vnitřní kryt jehly a vyhoďte jej. Pokud byste se pokusil(a) jej opět nasadit, mohl(a) byste se o jehlu nechtěně píchnout.
Na hrotu jehly se může objevit kapka roztoku. To je normální, ale i přesto musíte zkontrolovat průtok, pokud používáte nové pero poprvé. Nenasazujte na pero novou jehlu, dokud nejste připraven(a) si injekci aplikovat.
A Pro každou aplikaci vždy použijte novou jehlu.
Tím se může předejít ucpání jehel, kontaminaci, infekci a nepřesnému dávkování.
A Nikdy nepoužívejte ohnutou či poškozenou jehlu._
2 Zkontrolujte průtok
• Před první aplikací injekce novým perem vždy zkontrolujte průtok.
Pokud již pero používáte, přejděte ke kroku 3, „Nastavení dávky“.
• Otáčejte voličem dávky, dokud počítadlo dávky neukazuje symbol kontroly průtoku
|
j :irnr:’ | |
|
Nastaven | |
|
3 1 |
symbol |
|
kontroly | |
|
průtoku | |
• Držte pero s jehlou směrem vzhůru.
Stiskněte a podržte dávkovači tlačítko, dokud se počítadlo dávky nevrátí na 0. 0 musí být zarovnána s ukazatelem dávky.
Na hrotu jehly se musí objevit kapka roztoku.
Na hrotu jehly může zůstat malá kapka, kterou si však neaplikujete.
Pokud se kapka neobjeví, opakujte krok 2 „Zkontrolujte průtok“, a to až 6krát. Pokud se stále žádná kapka neobjevuje, vyměňte jehlu a zopakujte krok 2 „Zkontrolujte průtok“ ještě jednou.
Pokud se kapka ani poté neobjeví, pero zlikvidujte a použijte nové.
A Vždy před prvním použitím nového pera zkontrolujte, zda se na hrotu jehly objeví kapka. Tak se ujistíte, že průtoku roztoku nic nebrání.
Pokud se neobjeví žádná kapka, nedoje k aplikaci žádného léku, i kdyby se počítadlo dávky pohybovalo. Může to znamenat, že došlo k ucpání nebo poškození jehly.
Pokud před první injekcí pomocí každého nového pera nezkontrolujete průtok, může se stát, že nedojde k aplikaci předepsané dávky, a tedy ani
_k zamýšlenému účinku přípravku Saxenda._
3 Nastavení dávky
• Otáčejte voličem dávky, dokud počítadlo dávky nezobrazí Vaši dávku (0,6 mg, 1,2 mg, 1,8 mg, 2,4 mg nebo 3,0 mg).
Pokud zvolíte špatnou dávku, můžete ji opravit otočením voliče dávky směrem dopředu nebo dozadu.
Na peru lze nastavit maximálně 3,0 mg.
Volič dávky mění dávku. Pouze počítadlo dávky a ukazatel dávky ukazují, kolik mg na dávku jste zvolil(a).
Můžete zvolit až 3,0 mg na dávku. Pokud pero obsahuje méně než 3,0 mg, počítadlo dávky se zastaví, než se zobrazí 3,0.
Volič dávky cvaká jiným způsobem při otáčení dopředu, zpět nebo přes počet zbývajících mg. Cvakání pera nepočítejte.
A Před aplikací tohoto léku vždy použijte počítadlo dávky a ukazatel dávky k ověření, kolik mg jste zvolil(a).
Cvakání pera nepočítejte.
Stupnici pera nepoužívejte. Ta pouze přibližně ukazuje, kolik roztoku v peru zbývá.
Voličem dávky lze nastavit pouze dávku 0,6 mg, 1,2 mg, 1,8 mg, 2,4 mg nebo 3,0 mg. Aby bylo zajištěno, že si aplikujete správnou dávku, musí být
_nastavená dávka zarovnána přesně proti ukazateli dávky._
Kolik roztoku zbývá?
Stupnice pera přibližně ukazuje, kolik roztoku v peru zbývá.
Příklad nastaveno 0,6 mg
Kolik
roztoku cca
zbývá

|
• Přesné množství zbývajícího roztoku lze zjistit pomocí počítadla dávky: Otáčejte voličem dávky, dokud se počítadlo dávky nezastaví. Pokud se zobrazí hodnota 3,0, zbývají v peru alespoň 3,0 mg. Pokud se počítadlo dávky zastaví před 3,0 mg, nezbývá dost roztoku na úplnou dávku 3,0 mg. Pokud potřebujete více léku, než v peru zbývá Rozdělit dávku mezi staré a nové pero můžete, pouze pokud jste proškolen(a) či pokud Vám to lékař nebo zdravotní sestra doporučí. K propočtu dávky použijte kalkulačku tak, jak jste byl(a) poučen(a) lékařem nebo zdravotní sestrou. A Při výpočtu buďte velmi opatrní. Pokud si nejste jistý(á), jak svou dávku za použití dvou per rozdělit, pak si zvolte a injekčně aplikujte potřebnou dávku pomocí pera nového. |
■B * Příklad 1.4 |
i Počítadlo dávky se zastavilo: zbývá 2,4 mg | ||
|
4 Aplikace dávky • Zaveďte jehlu pod kůži, jak Vám ukázal lékař nebo zdravotní sestra. • Ujistěte se, že vidíte na počítadlo dávky. Nezakrývejte je prsty. Mohlo by dojít k přerušení aplikace. |
O i |
‘S 1 Ljr _j | ||
|
• Stiskněte a podržte dávkovací tlačítko, dokud počítadlo dávky nezobrazí 0. 0 musí být zarovnána s ukazatelem dávky. Můžete poté uslyšet nebo cítit cvaknutí. |
O * |
L i 1 v íj^ |
l | |
|
• Po návratu počítadla dávky na 0 přidržte jehlu v kůži a pomalu počítejte do 6. • Pokud jehlu vytáhnete dřív, můžete vidět proud roztoku vytékající z hrotu jehly. Pokud se tak stane, nebude aplikována celá dávka. |
C Pomal IJ v * ' C i~c f i C, 1 H 1——■* |
u počítejte: 1-2-3-4-5-6) | ||
|
• Vytáhněte jehlu z kůže. Pokud se v místě vpichu objeví krev, jemně je stiskněte. Oblast netřete. Po podání injekce se na hrotu jehly může objevit kapka roztoku. To je běžné a nemá to žádný vliv na podanou dávku. A Vždy sledujte počítadlo dávky, abyste věděl(a), kolik mg aplikujete. Podržte dávkovací tlačítko, dokud počítadlo dávky neukazuje 0. Jak lze zjistit, že je jehla ucpaná nebo poškozená? • Pokud se při nepřerušovaném stisknutí dávkovacího tlačítka na počítadle dávky neobjeví 0, je možné, že jste použil(a) ucpanou nebo poškozenou jehlu. • V takovém případě jste si neaplikoval(a) žádný lék, ačkoli se počítadlo dávky posunulo z původní dávky, kterou jste nastavil(a). Jak nakládat s ucpanou jehlou? Vyměňte jehlu podle popisu v kroku 5 „Po aplikaci“ a opakujte všechny |
0 t |
r7 | ||
|
kroky počínaje krokem 1 „Připravte si pero a novou jehlu“. Ujistěte se, že jste zvolil(a) celou potřebnou dávku. Nikdy se při aplikaci nedotýkejte počítadla dávky. Mohlo by dojít k přerušení aplikace. | ||
|
5 Po aplikaci • Na hladkém povrchu zaveďte hrot jehly do vnějšího krytu jehly. Nedotýkejte se při tom jehly ani jejího vnějšího krytu. |
O ^ | |
|
• Po zakrytí jehly opatrně vnější kryt jehly zcela dotlačte. • Odšroubujte jehlu a opatrně ji zlikvidujte. |
i>-Jr" ■ 1'9 . i „ | |
|
• Uzávěr pera nasaďte na pero po každém použití, aby byl roztok chráněn před světlem. Po každé aplikaci vždy jehlu zlikvidujte. Zajistí se tím správná aplikace a zabrání se ucpání jehel. Pokud je jehla ucpaná, nelze aplikovat žádný lék. Prázdné pero bez nasazené jehly zlikvidujte dle pokynů svého lékaře, zdravotní sestry, lékárníka nebo místních úřadů. A Nikdy se nepokoušejte nasadit vnitřní kryt jehly zpět na jehlu. Mohl(a) byste se o jehlu píchnout. A Po každé aplikaci vždy jehlu z pera sejměte. Tím se může předejít ucpání jehel, kontaminaci, infekci, úniku roztoku a nepřesnému dávkování. |
“ v v | |
|
Další důležité informace • Pero a jehly vždy uchovávejte mimo dohled a dosah ostatních, zejména pak dětí. • Pero ani jehly nikdy s nikým nesdílejte. • Pečující osoby musí být při manipulaci s použitými jehlami velice opatrné, aby nedošlo k poranění jehlou a přenesení infekce. | ||
|
Péče o pero • Pero nenechávejte v autě ani na jiném místě, kde může být příliš vysoká nebo příliš nízká teplota. • Neaplikujte si přípravek Saxenda, který zmrzl. Pokud se tak stane, nemusíte dosáhnout zamýšleného účinku tohoto léku. • Nevystavujte pero prachu, nečistotám ani tekutinám. • Pero neumývejte, nenamáčejte ani nepromazávejte. V případě potřeby je očistěte hadříkem navlhčeným ve slabém čisticím prostředku. • Nenechte pero spadnout na tvrdý povrch ani s ním o takový povrch neklepejte. Pokud pero upustíte nebo máte podezření, že se poškodilo, našroubujte na něj novou jehlu a před aplikací zkontrolujte průtok roztoku. • Nepokoušejte se pero znovu naplnit. Po vypotřebování je nutné je zlikvidovat. • Nepokoušejte se pero opravovat, ani je rozebírat. | ||
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY V REGISTRACI
Vědecké závěry
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizovaných zpráv o bezpečnosti (PSUR) pro liraglutid dospěl výbor CHMP k těmto vědeckým závěrům:
Na základě signálu zvýšené hladiny lipázy a případů zvýšení průměrných hodnot lipázy získaných z klinických studií a z poregistračních zdrojů výbor PRAC zastává stanovisko, že informace o přípravku mají být aktualizovány. Do bodu 4.8 souhrnu údajů o přípravku mají být přidány nežádoucí účinky „zvýšené hladiny lipázy“ a „zvýšené hladiny amylázy“ s častou četností výskytu. Příbalová informace má být aktualizována odpovídajícím způsobem.
Proto s ohledem na údaje uvedené v hodnocené zprávě PSUR výbor PRAC dospěl k závěru, že změny informací o přípravku u léčivých přípravků obsahujících liraglutid jsou opodstatněné.
Výbor CHMP souhlasí s vědeckými závěry výboru PRAC.
Zdůvodnění změny v registraci
Na základě vědeckých závěrů týkajících se liraglutidu výbor CHMP zastává stanovisko, že poměr přínosů a rizik léčivých přípravků obsahujících liraglutid zůstává nezměněný, a to pod podmínkou, že v informacích o přípravku budou provedeny navrhované změny.
Výbor CHMP doporučuje změnu v registraci.
38