Rixubis 2000 Iu
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
RIXUBIS 250 IU - prášek a rozpouštědlo pro injekční roztok RIXUBIS 500 IU - prášek a rozpouštědlo pro injekční roztok RIXUBIS 1000 IU - prášek a rozpouštědlo pro injekční roztok RIXUBIS 2000 IU - prášek a rozpouštědlo pro injekční roztok RIXUBIS 3000 IU - prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
RIXUBIS 250 IU - prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nonakogum gama, nominálně 250 IU, rekombinantní humánní koagulační faktor IX (rDNA), odpovídající koncentraci 50 IU/ml po rekonstituci s 5 ml rozpouštědla.
RIXUBIS 500 IU - prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nonakogum gama, nominálně 500 IU, rekombinantní humánní koagulační faktor IX (rDNA), odpovídající koncentraci 100 IU/ml po rekonstituci s 5 ml rozpouštědla.
RIXUBIS 1000 IU - prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nonakogum gama, nominálně 1000 IU, rekombinantní humánní koagulační faktor IX (rDNA), odpovídající koncentraci 200 IU/ml po rekonstituci s 5 ml rozpouštědla.
RIXUBIS 2000 IU - prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nonakogum gama, nominálně 2000 IU, rekombinantní humánní koagulační faktor IX (rDNA), odpovídající koncentraci 400 IU/ml po rekonstituci s 5 ml rozpouštědla.
RIXUBIS 3000 IU - prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nonakogum gama, nominálně 3000 IU, rekombinantní humánní koagulační faktor IX (rDNA), odpovídající koncentraci 600 IU/ml po rekonstituci s 5 ml rozpouštědla.
Účinnost (IU) je stanovena pomocí jednostupňového testu srážlivosti podle evropského lékopisu. Specifická aktivita přípravku RIXUBIS je přibližně 200-390 IU/mg proteinu.
Nonakog gama (rekombinantní koagulační faktor IX) je jednořetězcový purifikovaný glykoprotein, který má 415 aminokyselin. Je produkován technologií rekombinantní DNA v buňkách vaječníku čínského křečíka (CHO).
Pomocná látka/Pomocné látky se známým účinkem:
Jedna injekční lahvička obsahuje 19 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek je bílý až šedobílý. Rozpouštědlo je čiré a bezbarvé.
KLINICKÉ ÚDAJE
4.
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií B (vrozený deficit faktoru IX).
Přípravek RIXUBIS je indikován u pacientů všech věkových skupin.
4.2 Dávkování a způsob podání
Léčba má probíhat pod dohledem lékaře se zkušenostmi s léčbou hemofilie.
Monitorování léčby
V průběhu léčby se doporučuje patřičným způsobem stanovit hladinu faktoru IX a podle této informace upravit podávanou dávku a četnost opakovaných infuzí. Pacienti se mohou individuálně lišit ve své odpovědi na faktor IX, s různými délkami poločasu a recovery. Dávky stanovené na základě tělesné hmotnosti může být zapotřebí upravit u pacientů s podváhou či nadváhou. Zejména v případě rozsáhlejších chirurgických zákroků je nezbytné pečlivé monitorování průběhu substituční terapie, a to pomocí koagulační analýzy (aktivita faktoru IX v plazmě).
Aby bylo dosaženo požadované hladiny aktivity faktoru IX v plazmě, doporučuje se důsledně sledovat aktivitu faktoru IX pomocí vhodného testu a dle potřeby provádět i odpovídající úpravy dávky a četnosti opakovaných infuzí. Při použití jednostupňového testu srážlivosti na základě tromboplastinového času (aPTT) in vitro ke stanovení aktivity faktoru IX v krevních vzorcích pacienta mohou být výsledky aktivity faktoru IX v plazmě významně ovlivněny typem činidla aPTT použitého pro test a referenčním standardem použitým při testu. Toto má význam zejména při změně laboratoře a/nebo činidel používaných při testu.
Dávkování
Dávka a délka trvání substituční terapie závisí na závažnosti deficitu faktoru IX, na místě a rozsahu krvácení a na klinickém stavu pacienta, jeho věku a farmakokinetických parametrech faktoru IX, např. na přírůstku recovery a poločasu.
Počet podaných jednotek faktoru IX je vyjádřen v mezinárodních jednotkách (IU), které odpovídají aktuálnímu mezinárodnímu standardu WHO pro přípravky s faktorem IX. Aktivita faktoru IX v plazmě je vyjádřena buď v procentech (vzhledem k normální lidské plazmě), nebo v mezinárodních jednotkách (vzhledem k mezinárodnímu standardu pro faktor IX v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru IX odpovídá množství faktoru IX v jednom ml normální lidské plazmy.
Léčba podle potřeby
Výpočet požadované dávky faktoru IX je založen na empirickém zjištění, že 1 mezinárodní jednotka (IU) faktoru IX na kg tělesné hmotnosti zvýší aktivitu faktoru IX v plazmě o 0,9 IU/dl (rozmezí od 0,5 do 1,4 IU/dl) nebo o 0,9 % normální aktivity u pacientů ve věku 12 let a starších (další informace viz bod 5.2).
Požadovaná dávka se stanoví pomocí následujícího vzorce:
Pacienti ve věku 12 let a starší
Potřebné = tělesná hmotnost (kg) x požadovaný nárůst x recipročně
jednotky faktoru IX pozorovaná recovery
(%) nebo (IU/dl) (dl/kg)
Pro postupnou recovery o 0,9 IU/dl na IU/kg se dávka vypočítá následovně:
Potřebné = tělesná hmotnost (kg) x požadovaný nárůst x 1,1 dl/kg
jednotky faktoru IX
(%) nebo (IU/dl)
Množství přípravku, které je třeba podat, a četnost podání by měly vždy záviset na klinickém účinku v jednotlivém případě.
V případě následujících krvácivých příhod by aktivita faktoru IX v odpovídajícím časovém úseku neměla klesnout pod danou hladinu plazmatické aktivity (v % normální hodnoty nebo IU/dl). Následující tabulku lze použít jako pomůcku dávkování u krvácivých příhod a operací.
|
Stupeň krvácení / Typ chirurgické procedury |
Požadovaná hladina faktoru IX v (%) nebo (IU/dl) |
Četnost dávek (hodiny) / Trvání léčby (dny) |
|
Krvácení Časná hemartróza, krvácení do svalů nebo z ústní dutiny |
20 - 40 |
Opakujte každých 24 hodin. Nejméně 1 den, dokud trvá krvácivá příhoda, dokud nedojde k ústupu bolesti nebo není dosaženo zahojení. |
|
Intenzivnější hemartróza, krvácení do svalů nebo hematom |
30 - 60 |
Opakujte infuzi každých 24 hodin po dobu 3-4 dnů nebo déle, dokud nedojde k ústupu bolesti a postižení. |
|
Život ohrožující krvácení |
60 - 100 |
Opakujte infuzi každých 8 až 24 hodin, dokud nedojde k ústupu ohrožení. |
|
Operace Malé operace včetně extrakcí zubu |
30 - 60 |
Každých 24 hodin, nejméně 1 den, dokud nedojde ke zhojení. |
|
Velké operace |
80 - 100 (před a po operaci) |
Opakujte infuzi každých 8 až 24 hodin, dokud nedojde k odpovídajícímu zhojení rány, poté pokračujte v léčbě nejméně 7 dalších dnů, aby se udržela aktivita faktoru IX mezi 30 % až 60 % (IU/dl). |
Důsledné monitorování substituční léčby je důležité především v případě velkých operací nebo život ohrožujících krvácení.
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů s těžkou hemofilií B jsou obvyklé dávky u pacientů ve věku 12 let a starších 40 až 60 IU faktoru IX na kilogram tělesné hmotnosti v intervalech 3 až 4 dnů. V některých případech mohou být v závislosti na farmakokinetice jednotlivých pacientů, jejich věku, fenotypu krvácení a fyzické aktivitě nutné kratší intervaly dávkování nebo vyšší dávky.
Kontinuální infuze
Přípravek RIXUBIS nepodávejte v kontinuální infuzi.
Pediatrická _ populace Léčba podle potřeby:
Výpočet požadované dávky faktoru IX je založen na empirickém zjištění, že 1 mezinárodní jednotka (IU) faktoru IX na kg tělesné hmotnosti zvýší aktivitu faktoru IX v plazmě o 0,7 IU/dl (rozmezí od 0,31 do 1,0 IU/dl) nebo o 0,7 % normální aktivity u pacientů mladších 12 let (další informace viz bod 5.2).
Požadované dávkování se stanoví pomocí následujícího vzorce:
Pacienti mladší 12 let:
Potřebné = tělesná hmotnost (kg) x požadovaný nárůst x recipročně
jednotky faktoru IX pozorovaná recovery
(%) nebo (IU/dl) (dl/kg)
Pro postupnou recovery o 0,7 IU/dl na IU/kg se dávka vypočítá následovně:
Potřebné = tělesná hmotnost (kg) x požadovaný nárůst x 1,4 dl/kg
jednotky faktoru IX
(%) nebo (IU/dl)
Jako pomůcku dávkování u krvácivých příhod a operací lze použít stejnou tabulku jako u dospělých (viz výše).
Profylaxe:
Doporučené rozmezí dávkování u pediatrických pacientů mladších 12 let je 40 až 80 IU/kg v intervalech 3 až 4 dny. V některých případech mohou být v závislosti na farmakokinetice jednotlivých pacientů, jejich věku, fenotypu krvácení a fyzické aktivitě nutné kratší intervaly dávkování nebo vyšší dávky.
Způsob podání
Intravenózní podání.
V případě aplikace samotným pacientem nebo prostřednictvím poskytovatele péče je třeba jejich odpovídající vyškolení.
Přípravek RIXUBIS musí být podáván rychlostí zajišťující komfort pacienta, maximálně až 10 ml/min.
Po rekonstituci je roztok čirý, bezbarvý, neobsahuje částice a má pH 6,8 až 7,2. Osmolalita je větší než 240 mosmol/kg.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
S tímto přípravkem se smí používat pouze plastové injekční stříkačky typu luer-lock.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Známá alergická reakce na křeččí protein.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita:
U přípravku RIXUBIS byly hlášeny reakce přecitlivělosti alergického typu. Přípravek obsahuje stopy křečcích proteinů. Pokud se objeví příznaky přecitlivělosti, pacienti nebo jejich poskytovatelé péče musí okamžitě přerušit podávání léčivého přípravku a kontaktovat svého lékaře. Pacienty je nutno informovat o časných známkách reakcí přecitlivělosti, mezi něž patří kopřivka, generalizovaná kopřivka, tíseň na hrudi, sípot, hypotenze a anafylaxe.
Riziko je nejvyšší během časných fází zahájení expozice koncentrátům faktoru IX u dříve neléčených pacientů, především u pacientů s vysokým rizikem genových mutací. V literatuře bývá uváděna souvislost mezi výskytem inhibitoru faktoru IX a alergickými reakcemi, především u pacientů s vysokým rizikem genových mutací. Proto by pacienti, kteří mají alergické reakce, měli být vyšetřeni na přítomnost inhibitoru.
V případě šoku je zapotřebí dodržovat standardní lékařský postup pro léčbu šoku.
Inhibitory:
Po opakované léčbě přípravky humánního koagulačního faktoru IX (rDNA) musí být pacienti monitorováni z hlediska vývoje neutralizačních protilátek (inhibitorů), které musí být kvantifikovány v jednotkách Bethesda (BU) pomocí odpovídajícího biologického testování.
V literatuře bývá uváděna korelace mezi výskytem inhibitoru faktoru IX a alergickými reakcemi.
Proto by pacienti, kteří mají alergické reakce, měli být vyšetřeni na přítomnost inhibitoru. Je třeba si uvědomit, že pacienti s inhibitory faktoru IX mohou mít zvýšené riziko anafylaxe při následném vystavení faktoru IX.
Kvůli riziku alergických reakcí na koncentráty faktoru IX je vhodné dle uvážení lékaře provádět počáteční podání faktoru IX pod lékařským dohledem, aby mohla být při alergických reakcích poskytnuta odpovídající lékařská péče.
Nefrotický syndrom:
U pacientů s hemofilií B s inhibitory faktoru IX byl hlášen nefrotický syndrom v návaznosti na snahu o navození imunitní tolerance.
Tromboembolismus:
Pokud je tento přípravek podáván pacientům s onemocněním jater, pacientům po operaci, novorozencům nebo pacientům s rizikem trombotického jevu nebo DIC, je kvůli možnému riziku trombotických komplikací nutné zahájit klinické sledování časných známek trombotické a konzumpční koagulopatie pomocí odpovídajících biologických testů. V každé z těchto situací je třeba zvážit přínos léčby přípravkem RIXUBIS vůči riziku těchto komplikací.
Kardiovaskulární příhody
U pacientů s existujícími kardiovaskulárními rizikovými faktory může substituční terapie faktorem IX zvýšit kardiovaskulární riziko.
Komplikace spojené s katétrem
Pokud je potřeba zavedení centrálního žilního katétru (CŽK), je nutné zvážit riziko s ním spojených komplikací, včetně lokálních infekcí, bakteriemie a trombózy v místě katétru.
Informace v souvislosti s pomocnou látkou
Po rekonstituci tento léčivý přípravek obsahuje 0,83 mmol (19 mg) sodíku v jedné injekční lahvičce. Tato informace může být důležitá u pacientů na dietě s kontrolovaným příjmem sodíku.
Důrazně se doporučuje zaznamenat při každém podání přípravku RIXUBIS pacientovi název a číslo šarže přípravku takovým způsobem, aby bylo možné zpětně přiřadit k pacientovi použitou šarži léčivého přípravku.
Pediatrická populace
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí.
Osoby pokročilejšího věku
Klinické studie s přípravkem RIXUBIS nezahrnují subjekty ve věku 65 let a starší. Není známo, zda odpovídají jinak než mladší subjekty. Tak jako u všech pacientů je třeba volbu dávky u starších pacientů individualizovat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly hlášeny žádné interakce přípravků s humánním koagulačním faktorem IX (rDNA) a jiných léčivých přípravků.
4.6 Fertilita, těhotenství a kojení
U faktoru IX nebyly provedeny studie na zvířatech. Vzhledem ke vzácnému výskytu hemofilie B u žen nejsou k dispozici žádné zkušenosti týkající se použití faktoru IX během těhotenství a kojení. Proto je faktor IX možné užívat během těhotenství a kojení pouze v případě, že to je jasně indikováno. Nejsou k dispozici žádné údaje o účincích faktoru IX na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek RIXUBIS nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Vzácně byla pozorována přecitlivělost nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, třesavku, zrudnutí, generalizovanou kopřivku, bolest hlavy, kopřivku, hypotenzi, letargii, nevolnost, neklid, tachykardii, tíseň na hrudi, brnění, zvracení, sípot), které mohou v některých případech přejít do těžké anafylaxe (zahrnující šok). V některých případech tyto reakce progredovaly do těžké anafylaxe a vyskytovaly se v těsné časové souvislosti s vývojem inhibitorů faktoru IX (také viz 4.4).
U pacientů s hemofilií B s inhibitory faktoru IX a s alergickými reakcemi v anamnéze byl hlášen nefrotický syndrom v návaznosti na snahu o navození imunitní tolerance.
Velmi vzácně byl pozorován vývoj protilátek proti křeččímu proteinu se související reakcí přecitlivělosti.
U pacientů s hemofilií B se mohou vyvinout neutralizační protilátky (inhibitory) proti faktoru IX. Pokud se takové inhibitory objeví, tento stav se projeví jako nedostatečná klinická odpověď.
V takových případech se doporučuje kontaktovat specializované centrum pro hemofilii.
Existuje potenciální riziko tromboembolických příhod po podání léčivých přípravků s faktorem IX s vyšším rizikem u preparátů s nízkou čistotou. Použití přípravků s faktorem IX s nízkou čistotou bývá spojeno např. s infarktem myokardu, diseminovanou intravaskulární koagulací, žilní trombózou a plicní embolií. Použití faktoru IX s vysokou čistotou bývá s takovými nežádoucími účinky spojeno vzácně.
Seznam nežádoucích účinků v tabulce
Klinické studie s přípravkem RIXUBIS zahrnovaly 99 subjektů s alespoň jednou hlášenou expozicí přípravku RIXUBIS z celkem 5 nežádoucích účinků. Níže uvedená tabulka je v souladu s klasifikací třídy orgánových systémů MedDRA (úroveň preferovaných termínů a SOC).
Četnosti byly stanoveny podle následující konvence: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10 000 až <1/1000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Nežádoucí účinky léku, z klinických studií a spontánních hlášení | ||
|
Třída orgánových systémů MedDRA |
Nežádoucí účinky |
Četnost dle pacientů |
|
Poruchy imunitního systému |
Hypersenzitivita a) |
Není známo |
|
Poruchy nervového systému |
Dysgeuzie |
Časté |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolest v končetině |
Časté |
a) NÚ popsán níže.
Popis vybraných nežádoucích účinků
Hypersenzitivita
Alergický typ reakce se projevil dyspnoí, pruritem, generalizovanou kopřivkou a vyrážkou. Pediatrická populace
Četnost, typ a závažnost nežádoucích účinků u dětí se očekává stejná jako u dospělých. Nicméně nejsou k dispozici žádné údaje u dříve neléčených pacientů, protože do klinických studií byli zahrnuti pouze dříve léčení pacienti. U této rizikové populace proto nebylo provedeno žádné vyšetření imunogenicity stran vývoje inhibitorů.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Účinky vyšších než doporučených dávek přípravku RIXUBIS nebyly popsány.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: hemostyptika, koagulační faktor IX. ATC kód: B02BD04.
Přípravek RIXUBIS obsahuje rekombinantní koagulační faktor IX (nonakog gama). Faktor IX je jednořetězcový glykoprotein s molekulovou hmotností přibližně 68 000 Daltonů. Je to koagulační faktor závislý na vitaminu K a je syntetizován játry. Faktor IX je aktivován faktorem XIa ve vnitřní koagulační kaskádě a komplexem faktoru VII / tkáňového faktoru ve vnější koagulační kaskádě. Aktivovaný faktor IX v kombinaci s aktivovaným faktorem VIII aktivuje faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin poté konvertuje fibrinogen na fibrin a vytvoří se sraženina.
Hemofilie B je dědičná porucha krevní koagulace vázaná na pohlaví způsobující snížení hladin faktoru IX a vedoucí k silnému krvácení do kloubů, svalů a vnitřních orgánů, buď spontánně, nebo v důsledku úrazu či poranění během operace. Pomocí substituční léčby se zvyšují plazmatické hladiny faktoru IX, čímž je umožněna dočasná korekce deficitu faktoru a korekce krvácivých stavů.
Klinická účinnost a bezpečnost:
Profylaxe a kontrola krvácení u dříve léčených pacientů ve věku 12 let a starších:
Účinnost přípravku RIXUBIS byla hodnocena v otevřené, nekontrolované části kombinované studie fáze 1/3, v níž celkem 73 dříve léčených pacientů mužského pohlaví ve věku mezi 12 a 59 lety dostávalo přípravek RIXUBIS jako profylaxi a/nebo jako léčbu krvácivých příhod léčených v případě potřeby. Všechny subjekty měly těžkou (hladina faktoru IX <1 %) nebo středně těžkou (hladina faktoru IX <2 %) hemofilii B. Padesát devět dříve léčených pacientů dostávalo přípravek RIXUBIS jako profylaxi. Padesát šest z těchto pacientů, kteří dostávali přípravek RIXUBIS po dobu minimálně 3 měsíce, bylo zahrnuto do hodnocení účinnosti v profylaxi. Dalších 14 dříve léčených pacientů dostávalo přípravek RIXUBIS pouze při léčbě krvácivých příhod. Subjekty v kohortě léčby dle potřeby musely mít alespoň 12 zdokumentovaných krvácivých příhod vyžadujících léčbu v průběhu 12 měsíců před zahrnutím do studie. Průměrné trvání léčby v kohortě léčby dle potřeby bylo 3,5 ±1,00 měsíce (medián 3,4, rozpětí od 1,2 do 5,1 měsíce), průměrná celková roční četnost krvácení byla 33,9 ±17,37 s mediánem 27,0 a rozpětím od 12,9 do 73,1.
Medián roční četnosti krvácení u profylaxe přípravkem RIXUBIS byl u všech krvácení 2,0, u spontánních krvácení 0,0 a u krvácení do kloubů 0,0. 24 subjektů (42,9 %) neprodělalo žádné krvácení.
Celkem bylo přípravkem RIXUBIS ošetřeno 249 krvácivých příhod, z nichž 197 bylo krvácení do kloubů a 52 krvácení mimo kloub (měkká tkáň, sval, tělní dutina, intrakraniální a jiné).
Z celkem 249 krvácivých epizod bylo 163 středních, 71 malých a 15 velkých. Léčba byla individualizována podle závažnosti, příčiny a místa krvácení. Z 249 krvácivých příhod byla většina (211; 84,7 %) léčena 1-2 infuzemi. Účinnost hemostázy při řešení krvácení byla hodnocena jako výborná nebo dobrá u 96 % všech léčených krvácivých příhod.
Profylaxe a kontrola krvácení u dříve léčených pacientů mladších 12 let:
Účinnost přípravku RIXUBIS byla hodnocena v kombinované studii fáze 2/3, v níž celkem 23 dříve léčených pacientů mužského pohlaví ve věku mezi 1,8 a 11,8 let (medián věku 7,10 let), s 11 pacienty <6 let, dostávalo přípravek RIXUBIS jako profylaxi a kontrolu krvácivých příhod. Všechny subjekty měly těžkou (hladina faktoru IX <1 %) nebo středně těžkou (hladina faktoru IX <2 %) hemofilii B. Všech 23 subjektů dostávalo profylaktickou léčbu přípravkem RIXUBIS po dobu minimálně 3 měsíce a byly zahrnuty do hodnocení účinnosti profylaxe.
Medián roční četnosti krvácení byl 2,0, u spontánních krvácení 0,0 a u krvácení do kloubů 0,0.
Devět subjektů (39,1 %) nemělo žádné krvácení.
Přípravkem RIXUBIS bylo léčeno celkem 26 krvácivých příhod, z nichž 23 krvácení bylo z důvodu poranění, 2 spontánní a 1 z neznámé příčiny. 19 krvácení bylo mimokloubních (měkká tkáň, sval, tělní dutina, intrakraniální a jiné) a 7 bylo krvácení do kloubu, z nichž 1 bylo krvácení do cílového kloubu. Z 26 krvácivých příhod bylo 15 malých, 9 středních a 2 velké. Léčba byla individualizována podle závažnosti, příčiny a místa krvácení. Většina (23; 88,5 %) byla léčena 1-2 infuzemi. Účinnost hemostázy při řešení krvácení byla hodnocena jako výborná nebo dobrá u 96,2 % všech léčených krvácivých příhod.
Peroperační péče:
Bezpečnost a účinnost při peroperačním použití byla hodnocena v prospektivní otevřené, nekontrolované, multicentrické studii fáze 3 u dříve neléčených pacientů mužského pohlaví s těžkou až středně těžkou hemofilií B používajících přípravek RIXUBIS. Analýza účinnosti podle protokolu zahrnovala 37 operací provedených u 27 pacientů ve věku mezi 17 a 57 lety, kteří podstoupili velkou nebo malou chirurgickou, zubní nebo jinou invazivní chirurgickou proceduru. Dvacet procedur bylo velkých, z toho 13 ortopedických a 3 zubní operace. Celkem 17 procedur, včetně 10 zubních extrakcí, bylo považováno za malé. U pacientů, kteří podstoupili velkou operaci, muselo být provedeno farmakokinetické (FK) vyhodnocení. Všichni pacienti dostávali dávky na základě jejich poslední individuální postupné recovery. Doporučená úvodní dávka přípravku RIXUBIS měla zajistit, aby byly během operace udržovány úrovně aktivity faktoru IX 80-100 % u velkých operací a 30-60 % u malých operací. Přípravek RIXUBIS byl podáván bolusovou infuzí.
Hemostáze byla v průběhu trvání studie udržována.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem RIXUBIS u dříve neléčených pacientů při léčbě a profylaxi krvácení u hemofilie B (informace o použití u dětí viz bod 4.2)
5.2 Farmakokinetické vlastnosti
Dříve neléčení pacienti ve věku 12 let a starší:
Randomizovaná, zaslepená, kontrolovaná, zkřížená farmakokinetická studie s přípravkem RIXUBIS a komparátorem byla provedena u nekrvácejících subjektů mužského pohlaví (>15 let) jako součást kombinované pivotní studie fáze 1/3. Subjekty dostávaly některý z přípravků jako jednorázovou intravenózní infuzi. Průměrná (± SD) dávka přípravku RIXUBIS v sadě analýz podle protokolu (n=25) byla 74,69 ±2,37 a její medián 74,25 IU/kg, s rozsahem od 71,27 do 79,38 IU/kg. Farmakokinetické parametry byly vypočítány z měření aktivity faktoru IX u vzorků krve získaných až 72 hodin po každé infuzi.
Farmakokinetické vyhodnocení bylo u přípravku RIXUBIS opakováno v otevřené, nekontrolované studii s přípravkem RIXUBIS u subjektů mužského pohlaví, které se účastnily úvodní FK zkřížené studie a dostávaly profylaxi přípravkem RIXUBIS po dobu 26 ±1 týdnů (průměr ± SD) a nahromadily alespoň 30 expozičních dnů (ED) přípravku RIXUBIS. Rozsah dávky přípravku RIXUBIS v opakované farmakokinetické studii byl 64,48 až 79,18 IU/kg (n=23).
Farmakokinetické parametry u hodnotitelných subjektů (analýza dle protokolu) jsou uvedeny v následující tabulce.
|
Parametr |
RIXUBIS Úvodní zkřížená studie (N=25) |
RIXUBIS Opakované hodnocení (N=23) |
|
AUCc.72h (IU.h/dl)a Průměr ± SD Medián (rozsah) |
1067,81±238,42 1108,35 (696,07-1571,16) |
1156,15±259,44 1170,26 (753,85-1626,81) |
|
Postupná recovery při Cmax (IU/dl:IU/kg)b Průměr ± SD Medián (rozsah) |
0,87±0,22 0,88 (0,53-1,35) |
0,95±0,25 0,93 (0,52-1,38) |
|
Poločas (h) Průměr ± SD Medián (rozsah) |
26,70±9,55 24,58 (15,83-52,34) |
25,36±6,86 24,59 (16,24-42,20) |
|
Cmax (IU/dl) Průměr ± SD Medián (rozsah) |
66,22±15,80 68,10 (41,70-100,30) |
72,75±19,73 72,40 (38,50-106,30) |
|
Průměrný rezidenční čas (h) Průměr ± SD Medián (rozsah) |
30,82±7,26 28,93 (22,25-47,78) |
29,88±4,16 29,04 (21,32-37,52) |
|
Vssc (dl/kg) Průměr ± SD Medián (rozsah) |
2,02±0,77 1,72 (1,10-3,94) |
1,79±0,45 1,74 (1,12-2,72) |
|
Clearance (dl/(kg.h)) Průměr ± SD Medián (rozsah) |
0,0644±0,0133 0,0622 (0,0426-0,0912) |
0,0602±0,0146 0,0576 (0,0413-0,0945) |
a
b
Plocha pod křivkou plazmatické koncentrace závislé na čase v době 0-72 hodin po infuzi. Vypočítá se jako (Cmax - výchozí faktor IX) děleno dávkou v IU/kg, kde Cmax je maximální měření faktoru IX po infuzi.
Distribuční objem v rovnovážném stavu
c
Postupná recovery 30 minut po infuzi byla stanovena u všech subjektů v kombinované studii fáze 1/3 v 1. den expozice, při návštěvách v 5., 13. a 26. týdnu a v čase kompletace nebo ukončení studie, pokud toto nekolidovalo s návštěvou ve 26. týdnu. Data ukázala, že postupná recovery je konzistentní v čase (viz následující tabulka).
|
1. den expozice (N=73) |
5. týden (N=71) |
13. týden (N=68) |
26. týden (N=55) |
Při kompletaci/ ukončení studieb (N=23) | |
|
Postupná recovery 30 min po infuzi (IU/dl: IU/kg)a Průměr ± SD Medián (rozsah) |
0,79±0,20 0,78 (0,26-1,35) |
0,83±0,21 0,79 (0,46-1,48) |
0,85±0,25 0,83 (0,14-1,47) |
0,89±0,12 0,88 (0,52-1,29) |
0,87±0,20 0,89 (0,52-1,32) |
a
b
Vypočítá se jako (C30min - výchozí faktor IX) děleno dávkou v IU/kg, kde C30min je měření faktoru IX 30 minut po infuzi.
Pokud toto nekolidovalo s návštěvou ve 26. týdnu.
Pediatrická populace (dříve léčení pacienti mladší 12 let)
Všech 23 subjektů mužského pohlaví podstoupilo úvodní farmakokinetické hodnocení přípravku RIXUBIS ve stavu bez krvácení jako součást kombinované pediatrické studie fáze 2/3. Subjekty byly randomizovány do jedné ze dvou skupin četnosti odběru krve, aby se snížila zátěž častými odběry krve u jednotlivých subjektů. Průměrná (± SD) dávka přípravku RIXUBIS v kompletní sadě analýz (n=23) byla 75,50 ±3,016 a její medián 75,25 IU/kg, s rozsahem od 70,0 do 83,6 IU/kg. Farmakokinetické parametry byly vypočítány z měření aktivity faktoru IX u vzorků krve získaných až 72 hodin po infuzi.
Farmakokinetické parametry u všech subjektů (úplná sada analýz) jsou uvedeny v následující tabulce.
|
Parametr |
<6 let (N=11) |
6 - <12 let (N=12) |
Všichni (N=23) |
|
AUCinf (IU.h/dl)a Průměr ± SD Medián (rozsah) |
723,7 ± 119,00 717,2 (488-947) |
886,0 ± 133,66 863,7 (730-1138) |
808,4 ± 149,14 802,9 (488-1138) |
|
Poločas (h) Průměr ± SD Medián (rozsah) |
27,67 ± 2,66 27,28 (24,0-32,2) |
23,15 ± 1,58 22,65 (21,8-27,4) |
25,31 ± 3,13 24,48 (21,8-32,2) |
|
Průměrný rezidenční čas (h) Průměr ± SD Medián (rozsah) |
30,62 ±3,27 30,08 (26,2-36,2) |
25,31 ± 1,83 24,74 (23,7-30,3) |
27,85 ± 3,73 26,77 (23,7-36,2) |
|
Vssb (dl/kg) Průměr ± SD Medián (rozsah) |
3,22 ± 0,52 3,16 (2,65-4,42) |
2,21 ± 0,32 2,185 (1,70-2,70) |
2,7 ± 0,67 2,69 (1,70-4,42) |
|
Clearance (dl/(kg.h)) Průměr ± SD Medián (rozsah) |
0,1058 ± 0,01650 0,1050 (0,081-0,144) |
0,0874 ± 0,01213 0,0863 (0,069-0,108) |
0,0962 ± 0,01689 0,0935 (0,069-0,144) |
a
b
Plocha pod křivkou plazmatické koncentrace závislé na čase od okamžiku 0 do konce. Distribuční objem v rovnovážném stavu
Postupná recovery 30 minut po infuzi byla stanovena u všech subjektů v kombinované studii fáze 2/3 na začátku farmakokinetického vyhodnocení (v 1. den expozice), při návštěvách v 5., 13. a 26. týdnu a v čase kompletace nebo ukončení studie, pokud toto nekolidovalo s návštěvou ve 26. týdnu. Data ukázala, že postupná recovery je konzistentní v čase napříč všemi pediatrickými věkovými skupinami. Viz tabulky níže.
Postupná recovery u přípravku RIXUBIS za 30 minut po infuzi, obě pediatrické věkové skupiny:
|
Postupná recovery 30 min po infuzi |
FK (ED 1) Všichni (N=22) |
5. týden Všichni (N=23) |
13. týden Všichni (N=21) |
26. týden Všichni (N=21) |
|
(IU/dl: IU/kg)a Průměr ± SD Medián (rozsah) |
0,67 ±0,16 0,69 (0,31 - 1,00) |
0,68 ± 0,12 0,66 (0,48 - 0,92) |
0,71 ± 0,13 0,66 (0,51-1,00) |
0,72 ± 0,15 0,734 (0,51-1,01) |
|
a Vypočítá se jako (C30min - výchozí faktor IX) děleno dáv |
kou v IU/kg, kde C30min je měření | |||
faktoru IX 30 minut po infuzi.
Postupná recovery u přípravku RIXUBIS za 30 minut po infuzi, pediatričtí pacienti <6 let:
|
Postupná recovery 30 min po infuzi |
FK (ED 1) Všichni (N=10) |
5. týden Všichni (N=11) |
13. týden Všichni (N=10) |
26. týden Všichni (N=10) |
|
(IU/dl: IU/kg)a Průměr ± SD Medián (rozsah) |
0,59 ± 0,13 0,59 (0,31-0,75) |
0,63 ± 0,10 0,6 (0,49-0,80) |
0,68 ± 0,12 0,66 (0,51-0,84) |
0,65 ± 0,13 0,61 (0,51-0,84) |
|
a Vypočítá se jako (C30min - výchozí faktor IX) děleno dáv faktoru IX 30 minut po infuzi. Postupná recovery u přípravku RIXUBIS za 30 minut po infuz |
kou v IU/kg, kde C30min je měření i, pediatričtí pacienti 6 až <12 let: | |||
|
Postupná recovery 30 min po infuzi |
FK (ED 1) Všichni (N=12) |
5. týden Všichni (N=12) |
13. týden Všichni (N=11) |
26. týden Všichni (N=11) |
|
(IU/dl: IU/kg)a Průměr ± SD Medián (rozsah) |
0,73 ± 0,16 0,71 (0,51-1,00) |
0,73 ± 0,13 0,70 (0,48-0,92) |
0,73 ± 0,14 0,70 (0,54 - 1,00) |
0,8 ± 0,14 0,78 (0,56-1,01) |
|
a Vypočítá se jako (C30min - výchozí faktor IX) děleno dáv |
kou v IU/kg, kde C30min je měření | |||
faktoru IX 30 minut po infuzi.
5.3 Předklinické údaje vztahující se k bezpečnosti
Přípravek RIXUBIS nebyl trombogenní v dávce 750 IU/kg v modelu stáze u králíka (Wessler-Test). Přípravek RIXUBIS nezpůsoboval žádné nežádoucí klinické, respirační ani kardiovaskulární účinky až do dávky 450 IU/kg u opic druhu cynomolgus.
Nebyla provedena žádná šetření týkající se kancerogenity, poškození plodnosti a fetálního vývoje. Přípravek RIXUBIS byl dobře tolerován ve studiích toxicity po jednorázovém podání i opakovaném podávání prováděných u myší, potkanů a opic druhu cynomolgus až do dávek 7500 IU/kg (jednorázová dávka) a 750 IU/kg (opakované podávání).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek Sacharóza Mannitol Chlorid sodný Chlorid vápenatý Histidin Polysorbát 80
Rozpouštědlo
Sterilizovaná voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky
S tímto přípravkem se smí používat pouze plastové injekční stříkačky typu luer-lock. V důsledku adsorpce humánního koagulačního faktoru IX na vnitřní povrchy některých infuzních zařízení se může projevit nesprávné dávkování.
6.3 Doba použitelnosti
3 roky.
Chemická a fyzikální stabilita po otevření před použitím byla prokázána na dobu 3 hodin při teplotě nepřekračující 30 oC. Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele, pokud rekonstituce neproběhla za kontrolovaných a validovaných aseptických podmínek. Chraňte před chladem.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Chraňte před mrazem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení a zvláštní vybavení pro použití
Jedno balení obsahuje jednu injekční lahvičku s práškem (sklo typu I) se zátkou (butylová pryž) a odklápěcím víčkem, injekční lahvičku obsahující 5 ml rozpouštědla (sklo typu I) se zátkou (chlorobutylová pryž) a odklápěcím víčkem a bezjehlové rekonstituční zařízení (BAXJECT II). Obsahuje 1 balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek RIXUBIS je nutné po rekonstituci prášku s dodaným rozpouštědlem podat intravenózně.
- K rekonstituci používejte pouze rozpouštědlo a rekonstituční zařízení (BAXJECT II) dodané v balení.
- K podání je nutné použít stříkačku typu luer lock.
- Nepoužívejte, je-li zařízení BAXJECT II, jeho sterilní ochranný systém nebo jeho balení poškozeno, případně pokud vykazuje známky opotřebení.
Rekonstituce
Použijte aseptickou techniku
1. Pokud je léčivý přípravek uložen v chladničce, vyjměte injekční lahvičky s práškem
a rozpouštědlem RIXUBIS z chladničky a nechejte je ohřát na pokojovou teplotu (mezi 15 °C a 30 °C).
2. Důkladně si umyjte ruce za použití mýdla a teplé vody.
3. Sundejte z injekčních lahviček s práškem a rozpouštědlem víčka.
4. Očistěte zátky tampony namočenými v alkoholu. Injekční lahvičky umístěte na rovný a čistý povrch.
5. Otevřete obal zařízení BAXJECT II odloupnutím papírového víčka, aniž byste se dotkli vnitřní části (obr. a). Zařízení z obalu nevyjímejte.
6. Obal převraťte a zaveďte přes zátku rozpouštědla čistý plastový hrot. Uchopte obal na jeho kraji a ze zařízení BAXJECT II jej stáhněte (obr. b). Ze zařízení BAXJECT II nesundávejte modrý kryt.
7. Když je zařízení BAXJECT II připojeno k injekční lahvičce s rozpouštědlem, otočte systém tak, aby injekční lahvička s rozpouštědlem byla na zařízení shora. Zaveďte bílý plastový hrot přes zátku RIXUBIS. Podtlak vtáhne rozpouštědlo do injekční lahvičky RIXUBIS (obr. c).
8. Jemně kružte lahvičkou, dokud se veškerý materiál nerozpustí. Přípravek se rozpustí rychle (během 2 minut). Ujistěte se, že se přípravek RIXUBIS úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Rekonstituované léčivé přípravky před podáváním prohlédněte, zda neobsahují žádné částečky a nejsou barevně změněné. Roztok má být čirý nebo světle žlutý. Nepoužívejte roztoky, které jsou zakalené, nebo roztoky, v nichž se nacházejí usazeniny.
Obr. a

Obr. b

Obr. c

Po rekonstituci přípravek chraňte před chladem.
Použijte ihned.
Podávání
Použijte aseptickou techniku
1. Ze zařízení BAXJECT II sundejte modrý kryt. Do injekční stříkačky nenatahujte vzduch. Připojte injekční stříkačku k zařízení BAXJECT II (obr. d).
2. Systém převraťte (injekční lahvička s rekonstituovaným roztokem musí být nahoře). Natáhněte rekonstituovaný roztok do injekční stříkačky pomalým zatažením za píst (obr. e).
3. Injekční stříkačku odpojte.
4. K injekční stříkačce připojte motýlkovou jehlu. Podejte intravenózní injekcí. Roztok se musí podávat pomalu, rychlostí podle určené úrovně komfortu pacienta, nesmí překročit rychlost 10 ml za minutu.
Obr. d
Obr. e
Kdykoli je to možné, zaznamenejte název léčivého přípravku a číslo šarže při každém použití přípravku RIXUBIS (např. do svého deníčku), aby bylo možné zpětně zjistit použitý léčivý přípravek a jeho šarži.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxalta Innovations GmbH Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/970/001
EU/1/14/970/002
EU/1/14/970/003
EU/1/14/970/004
EU/1/14/970/005
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 19. prosince 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ
ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Baxter AG Uferstrasse 15 A-2304 Orth an der Donau Rakousko
Název a adresa výrobců odpovědných za propouštění šarží
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Belgie
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Belgie
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
RIXUBIS 250 IU - prášek a rozpouštědlo pro injekční roztok Nonakogum gama (rekombinantní humánní koagulační faktor IX)
1 injekční lahvička: nonakogum gama 250 IU, cca 50 IU/ml po rekonstituci s 5 ml rozpouštědla.
Pomocná látka se známým účinkem: sodík (v chloridu sodném)
Pomocné látky: sacharóza, chlorid vápenatý, histidin, mannitol, polysorbát 80.
Rozpouštědlo: sterilizovaná voda na injekci
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s práškem, 1 injekční lahvička s rozpouštědlem, 1 zařízení BAXJECT II
Před použitím si přečtěte příbalovou informaci.
K intravenóznímu podání, pouze k jednorázovému použití.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Uchovávejte při teplotě do 30 °C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxalta Innovations GmbH A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/970/001
13. ČÍSLO ŠARŽE
Č. š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
RIXUBIS 250
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
RIXUBIS 250 IU - prášek na injekci Nonakogum gama Intravenózní podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. K jednorázovému injekčnímu podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
250 IU
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 ml
6. JINÉ
RIXUBIS 500 IU - prášek a rozpouštědlo pro injekční roztok Nonakogum gama (rekombinantní humánní koagulační faktor IX)
1 injekční lahvička: nonakogum gama 500 IU, cca 100 IU/ml po rekonstituci s 5 ml rozpouštědla.
Pomocná látka se známým účinkem: sodík (v chloridu sodném)
Pomocné látky: sacharóza, chlorid vápenatý, histidin, mannitol, polysorbát 80.
Rozpouštědlo: sterilizovaná voda na injekci
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s práškem, 1 injekční lahvička s rozpouštědlem, 1 zařízení BAXJECT II
Před použitím si přečtěte příbalovou informaci.
K intravenóznímu podání, pouze k jednorázovému použití.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Uchovávejte při teplotě do 30 °C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxalta Innovations GmbH A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/970/002
13. ČÍSLO ŠARŽE
Č. š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
RIXUBIS 500
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
RIXUBIS 500 IU - prášek na injekci Nonakogum gama Intravenózní podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. K jednorázovému injekčnímu podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
500 IU
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 ml
6. JINÉ
RIXUBIS 1000 IU - prášek a rozpouštědlo pro injekční roztok Nonakogum gama (rekombinantní humánní koagulační faktor IX)
1 injekční lahvička: nonakogum gama 1000 IU, cca 200 IU/ml po rekonstituci s 5 ml rozpouštědla.
Pomocná látka se známým účinkem: sodík (v chloridu sodném)
Pomocné látky: sacharóza, chlorid vápenatý, histidin, mannitol, polysorbát 80.
Rozpouštědlo: sterilizovaná voda na injekci
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s práškem, 1 injekční lahvička s rozpouštědlem, 1 zařízení BAXJECT II
Před použitím si přečtěte příbalovou informaci.
K intravenóznímu podání, pouze k jednorázovému použití.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Uchovávejte při teplotě do 30 °C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxalta Innovations GmbH A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/970/003
13. ČÍSLO ŠARŽE
Č. š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
RIXUBIS 1000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
RIXUBIS 1000 IU - prášek na injekci Nonakogum gama Intravenózní podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. K jednorázovému injekčnímu podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1000IU
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 ml
6. JINÉ
RIXUBIS 2000 IU - prášek a rozpouštědlo pro injekční roztok Nonakogum gama (rekombinantní humánní koagulační faktor IX)
1 injekční lahvička: nonakogum gama 2000 IU, cca 400 IU/ml po rekonstituci s 5 ml rozpouštědla.
Pomocná látka se známým účinkem: sodík (v chloridu sodném)
Pomocné látky: sacharóza, chlorid vápenatý, histidin, mannitol, polysorbát 80.
Rozpouštědlo: sterilizovaná voda na injekci
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s práškem, 1 injekční lahvička s rozpouštědlem, 1 zařízení BAXJECT II
Před použitím si přečtěte příbalovou informaci.
K intravenóznímu podání, pouze k jednorázovému použití.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Uchovávejte při teplotě do 30 °C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxalta Innovations GmbH A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/970/004
13. ČÍSLO ŠARŽE
Č. š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
RIXUBIS 2000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
RIXUBIS 2000 IU - prášek na injekci Nonakogum gama Intravenózní podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. K jednorázovému injekčnímu podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2000 IU
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 ml
6. JINÉ
RIXUBIS 3000 IU - prášek a rozpouštědlo pro injekční roztok Nonakogum gama (rekombinantní humánní koagulační faktor IX)
1 injekční lahvička: nonakogum gama 3000 IU, cca 600 IU/ml po rekonstituci s 5 ml rozpouštědla.
Pomocná látka se známým účinkem: sodík (v chloridu sodném)
Pomocné látky: sacharóza, chlorid vápenatý, histidin, mannitol, polysorbát 80.
Rozpouštědlo: sterilizovaná voda na injekci
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s práškem, 1 injekční lahvička s rozpouštědlem, 1 zařízení BAXJECT II
Před použitím si přečtěte příbalovou informaci.
K intravenóznímu podání, pouze k jednorázovému použití.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Uchovávejte při teplotě do 30 °C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxalta Innovations GmbH A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/970/005
13. ČÍSLO ŠARŽE
Č. š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
RIXUBIS 3000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
RIXUBIS 3000 IU - prášek na injekci Nonakogum gama Intravenózní podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. K jednorázovému injekčnímu podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
3000 IU
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
RIXUBIS 250 IU - prášek a rozpouštědlo pro injekční roztok RIXUBIS 500 IU - prášek a rozpouštědlo pro injekční roztok RIXUBIS 1000 IU - prášek a rozpouštědlo pro injekční roztok RIXUBIS 2000 IU - prášek a rozpouštědlo pro injekční roztok RIXUBIS 3000 IU - prášek a rozpouštědlo pro injekční roztok
Nonakogum gama (rekombinantní humánní koagulační faktor IX)
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek RIXUBIS a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek RIXUBIS používat
3. Jak se přípravek RIXUBIS používá
4. Možné nežádoucí účinky
5. Jak přípravek RIXUBIS uchovávat
6. Obsah balení a další informace
1. Co je přípravek RIXUBIS a k čemu se používá
Přípravek RIXUBIS obsahuje léčivou látku nonakog gama. Je to koagulační faktor IX. Faktor IX je normální složkou lidské krve nezbytnou k účinnému srážení krve. Přípravek RIXUBIS se používá u pacientů s hemofilií B (Christmasova choroba, dědičná porucha krvácení způsobená nedostatkem faktoru IX). Přípravek funguje jako náhrada chybějícího faktoru IX a umožňuje krvi pacienta se srážet.
Přípravek RIXUBIS se používá k léčbě a prevenci krvácení u pacientů s hemofilií B všech věkových skupin.
2. Čemu musíte věnovat pozornost, než začnete přípravek RIXUBIS používat
Nepoužívejte přípravek RIXUBIS
- jestliže jste alergický(á) na nonakog gama nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže jste alergický(á) na křeččí proteiny.
Upozornění a opatření
U přípravku RIXUBIS jsou možné reakce přecitlivělosti alergického typu. Pokud zaznamenáte časné známky přecitlivělosti / alergické reakce, např. kopřivku, vyrážku, tíseň na hrudi, sípot, nízký krevní tlak nebo anafylaxi (těžká alergická reakce, která může způsobit potíže s polykáním a/nebo dýcháním,
zarudlý nebo oteklý obličej a/nebo ruce), zastavte infuzi a okamžitě kontaktujte svého lékaře nebo vyhledejte lékařskou pohotovost. Lékař může tyto reakce začít ihned léčit. Lékař Vám rovněž provede krevní test, aby zkontroloval, zda se u Vás nevyvinulyaktivitu neutralizující protilátky (inhibitory) proti léku, protože inhibitory se mohou rozvíjet společně s alergiemi. Pacienti s inhibitory faktoru IX mohou mít zvýšené riziko anafylaxe při budoucí léčbě faktorem IX.
Pokud se krvácení podle očekávání nezastaví, nebo pokud zaznamenáte významné zvýšení používání přípravku RIXUBIS k zástavě krvácení, ihned to sdělte svému lékaři. Lékař Vám provede krevní test, aby zkontroloval, zda se u Vás nevyvinuly aktivitu neutralizující protilátky (inhibitory) proti přípravku RIXUBIS. Riziko vývoje inhibitorů je nejvyšší u pacientů, kteří nebyli ještě léčeni náhradou faktoru IX, nebo během časných fází léčby, tj. u malých dětí.
Tvorba faktoru IX v těle je řízena genem pro faktor IX. Pacienti s určitými mutacemi genu pro faktor IX, např. s velkou delecí, mohou být náchylnější k tvorbě inhibitorů faktoru IX a k alergické reakci během časného období používání jakéhokoli koncentrátu faktoru IX. Pokud tedy víte, že takovou mutaci máte, bude Vás lékař z hlediska známek alergické reakce sledovat důkladněji.
Pokud trpíte jaterním nebo srdečním onemocněním nebo jste nedávno prodělal(a) velkou operaci, sdělte to svému lékaři, protože existuje zvýšené riziko komplikací krevního srážení (koagulace).
Po vysokých dávkách faktoru IX u pacientů s hemofilií B a s inhibitory faktoru IX a anamnézou alergických reakcí byly hlášeny poruchy ledvin (nefrotický syndrom).
Kdykoli je to možné, zaznamenejte název léčivého přípravku a číslo šarže při každém použití přípravku RIXUBIS (např. do svého deníčku), aby bylo možné zpětně zjistit použitý přípravek a jeho šarži.
Další léčivé přípravky a RIXUBIS
Informujte svého lékaře o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat. Nejsou známy žádné interakce přípravku RIXUBIS s jinými léčivými přípravky.
Těhotenství, kojení a plodnost
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat. Hemofilie B se u žen vyskytuje velmi vzácně.
Řízení dopravních prostředků a obsluha strojů
Přípravek RIXUBIS nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
Přípravek RIXUBIS obsahuje sodík
Tento léčivý přípravek obsahuje 19 mg sodíku na injekční lahvičku. Tato informace může být důležitá u pacientů na dietě s kontrolovaným příjmem sodíku.
3. Jak se přípravek RIXUBIS používá
Léčba přípravkem RIXUBIS bude zahájena lékařem se zkušenostmi v péči o pacienty s hemofilií B.
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Váš lékař rozhodne o dávce přípravku RIXUBIS, kterou dostanete. Tato dávka a trvání budou záviset na závažnosti deficitu faktoru IX, na místě a rozsahu krvácení a na klinickém stavu, věku a na tom, jak rychle tělo faktor IX spotřebovává, což bude nutné pravidelně kontrolovat.
Přípravek RIXUBIS podává lékař nebo zdravotní sestra intravenózní (i.v.) injekcí po rekonstituci (rozpuštění) prášku dodaným rozpouštědlem. Přípravek RIXUBIS si můžete rovněž injekčně aplikovat sám (sama) nebo Vám jej může podat někdo jiný, ale pouze po odpovídajícím vyškolení.
Rekonstituce a podávání
- K rekonstituci používejte pouze rozpouštědlo a rekonstituční zařízení (BAXJECT II) dodané v balení.
- K podání je nutné použít stříkačku typu luer lock.
- Nepoužívejte, je-li zařízení BAXJECT II, jeho sterilní ochranný systém nebo jeho balení poškozeno, případně pokud vykazuje známky opotřebení.
Rekonstituce
Použijte aseptickou techniku
1. Pokud je léčivý přípravek uložen v chladničce, vyjměte injekční lahvičky s práškem
a rozpouštědlem RIXUBIS z chladničky a nechejte je ohřát na pokojovou teplotu (mezi 15 °C a 30 °C).
2. Důkladně si umyjte ruce za použití mýdla a teplé vody.
3. Sundejte z injekčních lahviček s práškem a rozpouštědlem víčka.
4. Očistěte zátky tampony namočenými v alkoholu. Injekční lahvičky umístěte na rovný a čistý povrch.
5. Otevřete obal zařízení BAXJECT II odloupnutím papírového víčka, aniž byste se dotkli vnitřní části (obr. a). Zařízení z obalu nevyjímejte.
6. Obal převraťte a zaveďte přes zátku rozpouštědla čistý plastový hrot. Uchopte obal na jeho kraji
a ze zařízení BAXJECT II jej stáhněte (obr. b). Ze zařízení BAXJECT II nesundávejte modrý kryt.
7. Když je zařízení BAXJECT II připojeno k injekční lahvičce s rozpouštědlem, otočte systém tak, aby injekční lahvička s rozpouštědlem byla na zařízení shora. Zaveďte bílý plastový hrot přes zátku RIXUBIS. Podtlak vtáhne rozpouštědlo do injekční lahvičky RIXUBIS (obr. c).
8. Jemně kružte lahvičkou, dokud se veškerý materiál nerozpustí. Přípravek se rozpustí rychle (během 2 minut). Ujistěte se, že se přípravek RIXUBIS úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Rekonstituované léčivé přípravky před podáváním prohlédněte, zda neobsahují žádné částečky a nejsou barevně změněné. Roztok má být čirý nebo světle žlutý. Nepoužívejte roztoky, které jsou zakalené, nebo roztoky, v nichž se nacházejí usazeniny.
Obr. a
Obr. b
Obr. c

Po rekonstituci přípravek chraňte před chladem. Použijte ihned.
Podávání
Použijte aseptickou techniku
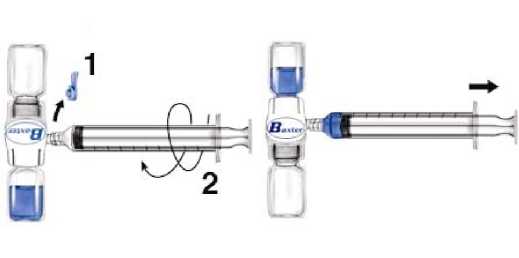
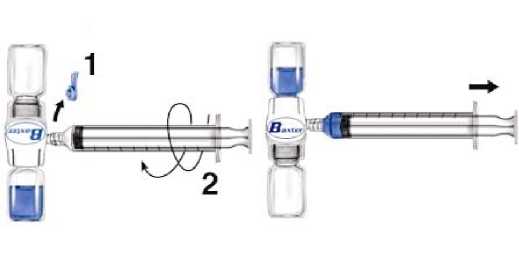
1. Ze zařízení BAXJECT II sundejte modrý kryt. Do injekční stříkačky nenatahujte vzduch. Připojte injekční stříkačku k zařízení BAXJECT II (obr. d).
2. Systém převraťte (injekční lahvička s rekonstituovaným roztokem musí být nahoře). Natáhněte rekonstituovaný roztok do injekční stříkačky pomalým zatažením za píst (obr. e).
3. Inj ekční stříkačku odpojte.
4. K injekční stříkačce připojte motýlkovou jehlu. Podejte intravenózní injekcí. Roztok se musí podávat pomalu, rychlostí podle určené úrovně komfortu pacienta, nesmí překročit rychlost 10 ml za minutu.
Obr. d
Obr. e
Kdykoli je to možné, zaznamenejte název léčivého přípravku a číslo šarže při každém použití přípravku RIXUBIS (např. do svého deníčku), aby bylo možné zpětně zjistit použitý přípravek a jeho šarži.
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. Jestliže jste použil(a) více přípravku RIXUBIS, než jste měl(a)
Vždy používejte přípravek RIXUBIS přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem. Pokud jste si injekčně aplikoval(a) více přípravku RIXUBIS, než je doporučeno, sdělte to co nejdříve svému lékaři.
Jestliže jste zapomněl(a) použít přípravek RIXUBIS
Nezdvojnásobujte následující injekci, abyste nahradil(a) vynechanou dávku. Přejděte k další plánované injekci a pokračujte dle pokynů svého lékaře.
Jestliže jste přestal(a) používat přípravek RIXUBIS
Nepřestávejte používat přípravek RIXUBIS, aniž byste to předem probrali se svým lékařem.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
U přípravku RIXUBIS jsou možné reakce přecitlivělosti alergického typu. Takové reakce mohou zahrnovat pálivé pocity a bodání v místě infuze, třesavku, zrudnutí, letargii, neklid, brnění, kopřivku, svědění a vyrážku, nízký krevní tlak, rychlou srdeční frekvenci, tíseň na hrudi, sípot, otok krku, anafylaxi (těžkou alergickou reakci), bolest hlavy, nevolnost a zvracení. Pokud se u Vás takovéto známky objeví, ihned kontaktujte svého lékaře. Lékař může tyto reakce začít ihned léčit (viz bod 2 „Upozornění a opatření“).
Po podání přípravku RIXUBIS byly pozorovány následující nežádoucí účinky:
Časté nežádoucí účinky (mohou postihnout až 1 pacienta z 10)
- změněné vnímání chutí
- bolest v končetinách
Nežádoucí účinky s neznámou četností (četnost nelze z dostupných údajů určit)
- alergické reakce (hypersenzitivita)
Potíže s přehnaným srážením krve (tromboembolické příhody) nebyly u tohoto léčivého přípravku pozorovány, ale mohou se objevit u kteréhokoli z přípravků s faktorem IX. Mohou zahrnovat srdeční infarkt, krevní sraženiny v žilách nebo v plicích.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek RIXUBIS uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku injekční lahvičky za značkou Použitelné do. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 30 °C.
Chraňte před mrazem.
Rekonstituovaný roztok použijte ihned.
Přípravek RIXUBIS nepoužívejte, pokud roztok není čirý a bezbarvý.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek RIXUBIS obsahuje
- Léčivou látkou je nonakogum gama (rekombinantní humánní koagulační faktor IX). Jedna injekční lahvička prášku obsahuje nominálně 250, 500, 1000, 2000 nebo 3000 IU, odpovídající koncentraci 50, 100, 200, 400 nebo 600 IU/ml po rekonstituci s 5 ml rozpouštědla.
- Dalšími složkami v prášku jsou sacharóza, mannitol, chlorid sodný, chlorid vápenatý, histidin a polysorbát 80.
Injekční lahvička s rozpouštědlem: 5 ml sterilizované vody na injekci.
Jak přípravek RIXUBIS vypadá a co obsahuje toto balení
Přípravek RIXUBIS je dodáván jako prášek a rozpouštědlo pro injekční roztok.
Obsahem balení jsou:
• jedna injekční lahvička prášku RIXUBIS 250, 500, 1000, 2000 nebo 3000 IU ve skleněné injekční lahvičce s pryžovou zátkou
• jedna injekční lahvička 5 ml sterilizované vody na injekci ve skleněné injekční lahvičce s pryžovou zátkou
• jedno zařízení BAXJECT II (bezjehlové rekonstituční zařízení)
Držitel rozhodnutí o registraci
Baxalta Innovations GmbH Industriestrasse 67 A-1221 Vídeň
Výrobce
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Belgie
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
Lietuva UAB "Baxter Lithuania'' Tel: +370 5 269 16 90/+370 5 252 71 00 |
|
Etarapnu EaKCTep Etarapna EOOfl Tea.: +359 2 9808482 |
Luxembourg/Luxemburg Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
|
Česká republika Baxter Czech spol.s.r.o. Tel.: +420 225774111 |
Magyarország Baxter Hungary Kft. Tel.: +361 202 19 80 |
|
Danmark Baxalta Denmark A/S Tlf: +45 32 70 12 00 |
Malta Baxalta UK Limited Tel.: +44 1 635 798 777 |
|
Deutschland Baxalta Deutschland GmbH Tel: +49 89 262077-011 |
Nederland Baxalta Netherlands B.V. Tel: +31 30 799 27 77 |
|
Eesti OU Baxter Estonia Tel.: +372 6 515 120 |
Norge Baxalta AS Tlf: +47 22 585 000 |
|
EXlába Baxter (Hellas) E.n.E. TqL: +30 210 28 80 000 |
Osterreich Baxalta Osterreich GmbH Tel.: +43 1 20100-0 |
|
Espaňa Baxalta Spain S.L. Tel: +34 91 790 42 22 |
Polska Baxter Polska Sp. z o.o. Tel.: +48 22 4883 777 |
|
France Baxalta France S.A.S. Tél: +33 1 70 96 06 00 |
Portugal Baxalta Portugal, Unipessoal, Lda. Tel: +351 21 122 03 00 |
|
Hrvatska Baxter d.o.o. Tel: +386 1 420 16 80 |
Románia FARMACEUTICA REMEDIA SA Tel.: +40 21 321 16 40 |
|
Ireland Baxalta UK Limited Tel: +44 1 635 798 777 |
Slovenija Baxter d.o.o. Tel.: +386 1 420 16 80 |
|
Ísland Lyfjaver ehf. Sími: +354 533 6100 |
Slovenská republika Baxter Slovakia s.r.o. Tel: +421 2 3210 1150 |
|
Italia Baxalta Italy S.r.l. Tel: +39 06 45224 600 |
Suomi/Finland Baxalta Finland Oy Puh/Tel: +358 201478200 |
|
Kónpoq Baxter (Hellas) E.n.E. TqA.: +30 210 28 80 000 |
Sverige Baxalta Sweden AB Tel: +46 8 50 53 26 00 |
|
Latvija SIA BAXTER Latvia Tel.: +371 67 784 784 |
United Kingdom Baxalta UK Limited Tel: +44 1 635 798 777 |
Tato příbalová informace byla naposledy revidována MM/RRRR.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky: Monitorování léčby
V průběhu léčby se doporučuje patřičným způsobem stanovit hladinu faktoru IX a podle této informace upravit podávanou dávku a četnost opakovaných infuzí. Pacienti se mohou individuálně lišit ve své odpovědi na faktor IX, s různými délkami poločasu a recovery. Dávky stanovené na základě tělesné hmotnosti může být zapotřebí upravit u pacientů s podváhou či nadváhou. Zejména v případě rozsáhlejších chirurgických zákroků je nezbytné pečlivé monitorování průběhu substituční terapie, a to pomocí koagulační analýzy (aktivita faktoru IX v plazmě).
Aby bylo dosaženo požadované hladiny aktivity faktoru IX v plazmě, je doporučováno důsledně sledovat aktivitu faktoru IX pomocí vhodného testu a dle potřeby provádět i odpovídající úpravy dávky a četnosti opakovaných injekcí. Při použití jednostupňového testu srážlivosti na základě tromboplastinového času (aPTT) in vitro ke stanovení aktivity faktoru IX v krevních vzorcích pacienta mohou být výsledky aktivity faktoru IX v plazmě významně ovlivněny typem činidla aPTT použitého pro test a referenčním standardem použitým při testu. Toto má význam zejména při změně laboratoře a/nebo činidel používaných při testu.
Dávkování
Dávka a délka trvání substituční terapie závisí na závažnosti deficitu faktoru IX, na místě a rozsahu krvácení a na klinickém stavu pacienta, jeho věku a farmakokinetických parametrech faktoru IX, např. na přírůstku recovery a poločasu.
Počet podaných jednotek faktoru IX je vyjádřen v mezinárodních jednotkách (IU), které odpovídají aktuálnímu mezinárodnímu standardu WHO pro léčivé přípravky s faktorem IX. Aktivita faktoru IX v plazmě je vyjádřena buď v procentech (vzhledem k normální lidské plazmě), nebo v mezinárodních jednotkách (vzhledem k mezinárodnímu standardu pro faktor IX v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru IX odpovídá množství faktoru IX v jednom ml normální lidské plazmy.
Léčba podle potřeby
Výpočet požadované dávky faktoru IX je založen na empirickém zjištění, že 1 mezinárodní jednotka (IU) faktoru IX na kg tělesné hmotnosti zvýší aktivitu faktoru IX v plazmě o 0,9 IU/dl (rozmezí od 0,5 do 1,4 IU/dl) nebo o 0,9 % normální aktivity u pacientů ve věku 12 let a starších (další informace viz bod 5.2).
Požadovaná dávka se stanoví pomocí následujícího vzorce:
Pacienti ve věku 12 let a starší
Potřebné = tělesná hmotnost (kg) x požadovaný nárůst x recipročně
jednotky faktoru IX pozorovaná recovery
(%) nebo (IU/dl) (dl/kg)
Pro postupnou recovery o 0,9 IU/dl na IU/kg se dávka vypočítá následovně:
Potřebné = tělesná hmotnost (kg) x požadovaný nárůst x 1,1 dl/kg
jednotky faktoru IX
(%) nebo (IU/dl)
Množství přípravku, které je třeba podat, a četnost podání by měly vždy záviset na klinickém účinku v jednotlivém případě.
V případě následujících krvácivých příhod by aktivita faktoru IX v odpovídajícím časovém úseku neměla klesnout pod danou hladinu plazmatické aktivity (v % normální hodnoty nebo IU/dl). Následující tabulku lze použít jako pomůcku dávkování u krvácivých příhod a operací.
|
Stupeň krvácení / Typ chirurgické procedury |
Požadovaná hladina faktoru IX v (%) nebo (IU/dl) |
Četnost dávek (hodiny) / Trvání léčby (dny) |
|
Krvácení Časná hemartróza, krvácení do svalů nebo z ústní dutiny |
20 - 40 |
Opakujte každých 24 hodin. Nejméně 1 den, dokud trvá krvácivá příhoda, dokud nedojde k ústupu bolesti nebo není dosaženo zahojení. |
|
Intenzivnější hemartróza, krvácení do svalů nebo hematom |
30 - 60 |
Opakujte infuzi každých 24 hodin po dobu 3-4 dnů nebo déle, dokud nedojde k ústupu bolesti a postižení. |
|
Život ohrožující krvácení |
60 - 100 |
Opakujte infuzi každých 8 až 24 hodin, dokud nedojde k ústupu ohrožení. |
|
Operace Malé operace včetně extrakcí zubu |
30 - 60 |
Každých 24 hodin, nejméně 1 den, dokud nedojde ke zhojení. |
|
Stupeň krvácení / Typ chirurgické procedury |
Požadovaná hladina faktoru IX v (%) nebo (IU/dl) |
Četnost dávek (hodiny) / Trvání léčby (dny) |
|
Velké operace |
80 - 100 (před a po operaci) |
Opakujte infuzi každých 8 až 24 hodin, dokud nedojde k odpovídajícímu zhojení rány, poté pokračujte v léčbě nejméně 7 dalších dnů, aby se udržela aktivita faktoru IX mezi 30 % až 60 % (IU/dl). |
Důsledné monitorování substituční léčby je důležité především v případě velkých operací nebo život ohrožujících krvácení.
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů s těžkou hemofilií B jsou obvyklé dávky u pacientů ve věku 12 let a starších 40 až 60 IU faktoru IX na kilogram tělesné hmotnosti v intervalech 3 až 4 dnů. V některých případech mohou být v závislosti na farmakokinetice jednotlivých pacientů, jejich věku, fenotypu krvácení a fyzické aktivitě nutné kratší intervaly dávkování nebo vyšší dávky.
Kontinuální infuze
Přípravek RIXUBIS nepodávejte v kontinuální infuzi.
Pediatrická populace Léčba podle potřeby:
1 mezinárodní jednotka 0,7 IU/dl (rozmezí let (další informace viz
Výpočet požadované dávky faktoru IX je založen na empirickém zjištění, že (IU) faktoru IX na kg tělesné hmotnosti zvýší aktivitu faktoru IX v plazmě o od 0,31 do 1,0 IU/dl) nebo o 0,7 % normální aktivity u pacientů mladších 12 bod 5.2).
Požadované dávkování se stanoví pomocí následujícího vzorce:
Pacienti mladší 12 let:
Potřebné = tělesná hmotnost (kg) x požadovaný nárůst x recipročně
jednotky faktoru IX pozorovaná recovery
(%) nebo (IU/dl) (dl/kg)
Pro postupnou recovery o 0,7 IU/dl na IU/kg se dávka vypočítá následovně:
Potřebné = tělesná hmotnost (kg) x požadovaný nárůst x 1,4 dl/kg
jednotky faktoru IX
(%) nebo (IU/dl)
Jako pomůcku dávkování u krvácivých příhod a operací lze použít stejnou tabulku jako u dospělých (viz výše).
Profylaxe:
Doporučené rozmezí dávkování u pediatrických pacientů mladších 12 let je 40 až 80 IU/kg v intervalech 3 až 4 dny. V některých případech mohou být v závislosti na farmakokinetice jednotlivých pacientů, jejich věku, fenotypu krvácení a fyzické aktivitě nutné kratší intervaly dávkování nebo vyšší dávky.
49