Respreeza 1000 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Respreeza 1000 mg prášek a rozpouštědlo pro infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička přibližně obsahuje inhibitor alfa-1 proteinasi humani* 1000 mg, což je stanoveno podle kapacity pro neutralizaci elastázy lidských neutrofilů (NE).
Po rekonstituci s použitím 20 ml rozpouštědla roztok obsahuje přibližně 50 mg/ml lidského inhibitoru alfa1-proteáz.
Celkový obsah proteinu je přibližně 1100 mg v jedné injekční lahvičce.
*Vyrobeno z plazmy lidských dárců.
Pomocné látky se známým účinkem:
Přípravek Respreeza obsahuje přibližně 1,9 mg sodíku v jednom ml rekonstituovaného roztoku (81 mmol/l).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro infuzní roztok.
Prášek je bílý až téměř bílý. Rozpouštědlo je čirý bezbarvý roztok.
Přibližná osmolalita rekonstituovaného roztoku je 279 mosmol/kg, pH je 7,0.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Respreeza je indikován k udržovací léčbě, která má zpomalit progresi emfyzému u dospělých se zdokumentovaným těžkým deficitem inhibitoru alfai-proteáz (např. genotypy PiZZ, PiZ(null), Pi(null, null), PiSZ). Pacienti musí mít zavedenu optimální farmakologickou a nefarmakologickou léčbu a vykazovat známky progresivního plicního onemocnění (např. očekávané snížení jednosekundového usilovného výdechového objemu (FEV1), narušenou schopnost chůze nebo zvýšený počet exacerbací) podle hodnocení zdravotnického pracovníka se zkušenostmi s léčbou deficitu inhibitoru alfai-proteáz.
4.2 Dávkování a způsob podání
První infuze by měly být podávány pod dohledem zdravotnického pracovníka se zkušenostmi s léčbou deficitu inhibitoru alfa1-proteáz. Další infuze mohou být podávány pečovatelem nebo si je může podat sám pacient (viz bod 4.4).
Dávkování
Doporučená dávka přípravku Respreeza je 60 mg/kg tělesné hmotnosti, podaných jednou týdně. Pediatrická populace
Bezpečnost a účinnost přípravku Respreeza u pediatrické populace (ve věku do 18 let) nebyla stanovena. Nejsou dostupné žádné údaje.
Starší populace
Bezpečnost a účinnost přípravku Respreeza u starších pacientů (ve věku 65 nebo více let) nebyla dosud stanovena ve specifických klinických hodnoceních.
Pacienti s poruchou funkce ledvin nebo jater
Nebyla provedena žádná zvláštní hodnocení. U těchto pacientů není možné doporučit žádný alternativní režim dávkování.
Způsob podání
Přípravek Respreeza se má podávat pouze intravenózní infuzí po rekonstituci.
Prášek se musí rekonstituovat vodou na injekce (viz návod k rekonstituci v bodě 6.6) a během podávání se přípravek v soupravě pro intravenózní podání musí filtrovat přes vhodný infuzní filtr (doporučená velikost póru je 5 mikrometrů (^m); není součástí balení).
Rekonstituovaný roztok se musí podávat intravenózní infuzí přes samostatnou, pro tento přípravek vyhrazenou infuzní linku rychlostí přibližně 0,08 ml/kg tělesné hmotnosti/min. Rychlost podání infuze má být přizpůsobena podle snášenlivosti pacienta. Infuze doporučené dávky, tj. 60 mg/kg tělesné hmotnosti, bude trvat přibližně 15 minut.
Jedna injekční lahvička přípravku Respreeza je určena pouze k jednorázovému použití.
Podrobné informace o podávání rekonstituovaného roztoku jsou uvedeny v návodu na konci příbalové informace.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 (viz také bod 4.4).
• Pacienti s deficitem IgA, o kterých je známo, že mají protilátky proti IgA, vzhledem k riziku těžké hypersenzitivity a anafylaktických reakcí.
4.4 Zvláštní upozornění a opatření pro použití
Musí se dodržet doporučená rychlost podávání infuze, uvedená v bodě 4.2. Během prvních infuzí musí být po celou dobu podávání infuze pečlivě sledován klinický stav pacienta, včetně životních funkcí. Pokud dojde k jakékoli reakci, která by mohla souviset s podáním přípravku Respreeza, musí se rychlost infuze snížit nebo se musí podávání přípravku zastavit, podle toho, co vyžaduje klinický stav pacienta. Pokud po zastavení infuze příznaky rychle vymizí, lze infuzi obnovit při nižší rychlosti, která je pro pacienta vhodná.
Hypersenzitivita
Mohou se vyskytnout hypersenzitivní reakce, a to i u pacientů, kteří snášeli předchozí léčbu lidským inhibitorem alfai-proteáz.
Respreeza může obsahovat stopové množství IgA. U pacientů se selektivním nebo závažným IgA deficitem se mohou vyvinout IgA protilátky, a proto mají větší riziko vzniku potenciálně závažné hypersenzitivity a anafylaktické reakce.
Při podezření na reakce alergického nebo anafylaktického typu se musí infuze okamžitě přerušit, v závislosti na povaze a závažnosti reakce. Při šoku musí být podána neodkladná léčba.
Podání v domácím prostředí / podání sobě samému
Údaje o podávání tohoto léčivého přípravku v domácím prostředí / podávání sobě samému jsou omezené.
Potenciální rizika spojená s podáním přípravku v domácím prostředí / podáním sobě samému souvisejí s manipulací s přípravkem a s jeho podáním i s léčbou nežádoucích účinků, zejména hypersenzitivity. Pacienti mají být informováni o známkách hypersenzitivních reakcí.
Rozhodnutí, zda je pacient způsobilý pro podávání přípravku v domácím prostředí / podávání sobě samému, rozhodne ošetřující lékař, který zajistí, aby byl pacient správně vyškolen (např. ohledně rekonstituce, použití přenosové pomůcky či filtru, sestavení hadiček infuzní soupravy, postupů při podávání infuzí, vedení deníku léčby, zjištění nežádoucích reakcí a opatření, která je potřeba v případě výskytu takových reakcí provést), a toto rozhodnutí je v pravidelných intervalech přehodnocováno.
Infekční agens
Standardní opatření pro zabránění infekcí následkem podání léčivých přípravků připravovaných z lidské krve či plazmy zahrnují volbu dárců, vyšetření jednotlivých odběrů i směsných jednotek plazmy na specifické infekční markery a zařazení účinných výrobních kroků pro inaktivaci/odstranění virů.
Přesto však při podávání léčivých přípravků z lidské krve nebo plazmy nelze zcela vyloučit možnost přenosu infekčních agens. Týká se to i neznámých virů, nově objevených virů a jiných patogenů. Přijatá opatření se považují za účinná pro obalené viry, jako je virus lidské imunodeficience (HIV), virus hepatitidy B (HBV) a virus hepatitidy C (HCV), a pro neobalené viry, jako je virus hepatitidy A (HAV) a parvovirus B19.
U pacientů, kteří dostávají pravidelně/opakovaně inhibitory proteáz vyráběné z lidské plazmy, by se mělo zvážit vhodné očkování (proti hepatitidě A a B).
Důrazně se doporučuje, aby byl při každém podání přípravku Respreeza pacientovi zaznamenán název a číslo šarže přípravku, aby byla zachována znalost souvislosti mezi pacientem a šarží přípravku.
Kouření
Kouření tabákových výrobků je významným rizikovým faktorem pro vznik a progresi emfyzému. Proto se důrazně doporučuje, aby pacient přestal kouřit a vyhýbal se prostředí, kde se vyskytuje tabákový dým.
Obsah sodíku
Přípravek Respreeza obsahuje přibližně 1,9 mg (<1 mmol) sodíku v jednom ml rekonstituovaného roztoku. To je třeba vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
S přípravkem Respreeza nebyly provedeny žádné reprodukční studie na zvířatech a bezpečnost jeho použití během těhotenství u člověka nebyla zjišťována v kontrolovaných klinických hodnoceních. Protože inhibitor alfa1-proteáz je endogenní lidský protein, považuje se za nepravděpodobné, že by přípravek Respreeza poškozoval plod, pokud je podáván v doporučených dávkách. Těhotným ženám se však přípravek Respreeza musí podávat s opatrností.
Kojení
Není známo, zda se přípravek Respreeza / metabolity vylučují do lidského mateřského mléka. Vylučování lidského inhibitoru alfai-proteáz do mateřského mléka zvířat nebylo studováno. Na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby lidským inhibitorem alfa 1-proteáz pro matku je nutno rozhodnout, zda ukončit/přerušit kojení nebo ukončit/přerušit léčbu přípravkem Respreeza.
Fertilita
S přípravkem Respreeza nebyly provedeny žádné studie fertility na zvířatech a jeho vliv na fertilitu člověka nebyl posuzován v kontrolovaných klinických studiích. Protože lidský inhibitor alfai-proteáz je endogenní lidský protein, neočekává se, že by měl nežádoucí účinky na fertilitu, pokud je podáván v doporučených dávkách.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Po podání přípravku Respreeza se mohou vyskytovat závratě (viz bod 4.8). Proto přípravek Respreeza může mít malý vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Během léčby byla pozorována hypersenzitivita nebo alergické reakce. V nejzávažnějších případech se mohou alergické reakce stupňovat až k těžkým anafylaktickým reakcím i v případech, kdy pacient nevykazoval hypersenzitivitu při předchozích podáních (viz bod 4.4).
Tabulkový přehled nežádoucích účinků
Nežádoucí účinky (AR), zjištěné v šesti klinických studiích u 221 pacientů a ze zkušeností po uvedení na trh, jsou uvedeny v tabulce níže podle klasifikace orgánových systémů MedDRA (System organ classification, SOC a úroveň preferovaných termínů, PT). Frekvence výskytu připadající na jednoho pacienta (na základě šestiměsíční expozice v klinických studiích) byla vyhodnocena podle následující konvence: časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100) a velmi vzácné (<1/10000). Frekvence výskytu nežádoucích účinků zjištěných pouze v období po uvedení na trh je hodnocena jako „není známo (z dostupných údajů nelze určit)“.
V každé skupině frekvencí jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Frekvence výskytu nežádoucích účinků (AR) přípravku Respreeza v klinických studiích a po uvedení na trh
|
Třída orgánového systému (SOC) |
Frekvence výskytu AR | |||
|
Časté (> 1/100 až < 1/10) |
Méně časté (> 1/1000 až < 1/100) |
Velmi vzácné (< 1/10000) |
Není známo | |
|
Poruchy krve a lymfatického systému |
Bolesti lymfatických uzlin | |||
|
Poruchy imunitního systému |
Hypersenzitivní reakce (zahrnující tachykardii, hypotenzi, zmatenost, synkopu, sníženou spotřebu kyslíku a edém hltanu) |
Anafylaktické reakce | ||
|
Poruchy nervového systému |
Závrať, bolest hlavy |
Parestezie |
Hypestezie | |
|
Poruchy oka |
Otok oka | |||
|
Cévní poruchy |
Návaly | |||
|
Třída orgánového systému (SOC) |
Frekvence výskytu AR | |||
|
Respirační, hrudní a mediastinální poruchy | ||||
|
Gastrointestinální poruchy |
Otok rtů | |||
|
Poruchy kůže a podkožní tkáně |
Kopřivka, vyrážka (včetně exfoliativní a generalizované) |
Hyperhidrosis, |
Otok obličeje | |
|
Celkové poruchy a reakce v místě aplikace |
Slabost, reakce v místě infuze (včetně hematomu v místě podání infuze), |
Bolest na hrudi, zimnice, pyrexie | ||
Pediatrická populace
Bezpečnost a účinnost u pediatrických pacientů nebyla stanovena. Nejsou k dispozici žádné údaje. Starší pacienti
Bezpečnost a účinnost přípravku Respreeza u starších pacientů (65 let a starších), nebyly stanoveny v klinických studiích.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Následky předávkování nejsou známé.
Při předávkování je nutné pacienta pečlivě sledovat, zda se u něj nevyskytují nežádoucí účinky, a podle potřeby musí být k dispozici možnost provést podpůrná opatření.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Hemostyptika, hemostatika, inhibitory proteáz, ATC kód: B02AB02
Lidský inhibitor alfa1-proteáz je normální složkou lidské krve. Lidský inhibitor alfa1-proteáz má molekulovou hmotnost 51 kDa a patří do skupiny inhibitorů serinových proteáz.
Mechanismus účinku
Lidský inhibitor alfa1-proteáz je považován za hlavní antiproteázu v dolních dýchacích cestách, kde inhibuje elastázu neutrofilů (NE). Normální zdraví jedinci vytvářejí dostatek inhibitoru alfa1-proteáz pro kontrolu NE vytvářené aktivovanými neutrofily, a proto jsou schopni bránit nežádoucí proteolýze plicní tkáně, které je způsobena NE. Podmínky, které zvyšují hromadění a aktivaci neutrofilů v plicích, jako jsou respirační infekce a kouření, hladiny NE naopak zvyšují. Jedinci s deficitem endogenního inhibitoru alfa1-proteáz však nejsou schopni udržet si správnou obranu proti proteázám a dochází u nich k rychlejší proteolýze alveolárních stěn, která začíná ještě před rozvojem klinicky zjevné chronické obstrukční plicní onemocnění ve třetí nebo čtvrté dekádě.
Farmakodynamické účinky
Podání přípravku Respreeza zvyšuje a udržuje koncentrace inhibitoru alfai-proteáz v séru a v tekutině kryjící epitel respiračního systému (epithelial lining fluid, ELF), což zpomaluje progresi emfyzému.
Klinická bezpečnost a účinnost
Studie RAPID
Bezpečnost a účinnost přípravku Respreeza byla hodnocena v randomizované, dvojitě zaslepené, placebem kontrolované, multicentrické studii (RAPID) následované 2letou prodlouženou otevřenou studií (prodloužená studie RAPID). Celkem 180 pacientů s deficitem inhibitoru alfa1-proteáz charakterizovaným hladinou inhibitoru alfa1-proteáz v séru < 11 pmol/l (tj. < 50 mg/dl při nefelometrickém stanovení) a klinicky prokázaným emfyzémem bylo randomizováno pro používání intravenózní týdenní dávky 60 mg/kg tělesné hmotnosti přípravku Respreeza (93 pacientů) nebo placeba (87 pacientů) po dobu nejvýše 24 měsíců. Pacienti byli ve věku od 31 do 67 let (průměrný věk byl 54 let) při průměrné výchozí hladině inhibitoru alfa1-proteáz přibližně 6,15 pM a průměrné denzitě plicní tkáně podle CT s objemovou korekcí 47 g/l pro přípravek Respreeza a 50 g/l pro pacienty používající placebo.
Sto čtyřicet pacientů (76 pacientů léčených přípravkem Respreeza a 64 pacientů s placebem ve studii RAPID) pokračovalo do rozšířené studie RAPID a byli léčeni jednou týdně intravenózní dávkou 60 mg/kg tělesné hmotnosti přípravku Respreeza po dobu až 24 měsíců.
Studie hodnotily vliv přípravku Respreeza na progresi emfyzému, která byla posuzována podle poklesu hustoty plicní tkáně, měřeného počítačovou tomografií (CT).
U pacientů léčených přípravkem Respreeza byl konzistentně pozorován pomalejší pokles hustoty plicní tkáně než u pacientů používajících placebo (viz Obrázek 1). Roční rychlost poklesu hustoty plicní tkáně, jejímž měřítkem byla celková kapacita plic (total lung capacity, TLC) podle vyšetření CT, za dobu 2 let, byla při používání přípravku Respreeza nižší (-1,45 g/l) než u placeba (-2,19 g/l), přičemž rozdíl v poklesu činil 34 % (p = 0,017, 1-stranný).
Rozšířená studie RAPID prokázala, že byla zachována snížená hodnota poklesu denzity plicní tkáně u pacientů kontinuálně léčených přípravkem Respreeza po dobu 4 let. (viz obrázek 1).
Obrázek 1: Změny hustoty plicní tkáně (TLC) oproti výchozímu stavu ve studii RAPID a rozšířené studii RAPID
■0.18
- Sít DO —Hjcsnriíeza
■0.5Ů
Sk
pi na
časným slaném
95
■>;
Sk
pna
odloženým slartem
Dí
Měsíc
lecby
Jednotlivé dávky 120 mg/kg tělesné hmotnosti byly podány 137 pacientům léčeným přípravkem Respreeza.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Respreeza u všech podskupin pediatrické populace v léčbě chronické obstrukční plicní onemocnění (CHOPN) způsobené deficitem inhibitoru alfai-proteáz (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Byly provedeny čtyři klinické studie s přípravkem Respreeza u 89 pacientů (59 mužů a 30 žen), aby zhodnotily vliv přípravku Respreeza na hladiny inhibitoru alfai-proteáz v séru. Věk pacientů byl v rozsahu od 29 do 68 let (průměrná hodnota věku byla 49 let). Při screeningu byly hladiny inhibitoru alfa1-proteáz v séru mezi 3,2 a 10,1 pmol/l (střední hodnota 5,6 pmol/l).
Byla provedena dvojitě zaslepená, randomizovaná, aktivní látkou kontrolovaná farmakokinetická studie se zkříženým uspořádáním zahrnující 13 mužů a 5 žen s deficitem inhibitoru alfa1-proteáz a s věkovým rozsahem od 36 do 66 let. 9 pacientů dostalo jednu dávku 60 mg/kg tělesné hmotnosti přípravku Respreeza a poté srovnávací přípravek a 9 pacientů dostalo srovnávací přípravek a poté jednu dávku 60 mg/kg tělesné hmotnosti přípravku Respreeza. Mezi dávkami byl vyplavovací („wash-out“) interval 35 dní. Do 21. dne po infuzi bylo v různých okamžicích odebráno celkem 13 vzorků séra. Tabulka 1 uvádí střední hodnoty výsledků pro farmakokinetické parametry přípravku Respreeza.
Tabulka č. 1: Farmakokinetické parametry pro inhibitor alfai-proteáz po podání jedné dávky 60 mg/kg tělesné hmotnosti přípravku Respreeza
|
Farmakokinetický parametr |
Střední hodnota (směrodatná odchylka)* |
|
Plocha pod křivkou (AUC0.^) |
144 (±27) pmol/l x den |
|
Maximální koncentrace (Cmax) |
44,1 (±10,8) pmol/l l |
|
Terminální poločas (t1/2e) |
5,1 (±2,4) dny |
|
Celková clearance |
603 (±129) ml/den |
|
Distribuční objem v ustáleném stavu |
3,8 (±1,3) l |
* n=18 pacientů.
Farmakokinetická analýza populací byla provedena na údajích od 90 pacientů léčených přípravkem Respreeza z klinického hodnocení RAPID. Odhad střední hodnoty poločasu pro tuto populaci byl 6,8 dne. Střední koncentrace po dávce 60 mg/kg tělesné hmotnosti/týden v ustáleném stavu, předpovězená na základě tohoto modelu, byla 21,8 pmol/l. Farmakokinetická analýza populací nenaznačila žádné významné vlivy věku, pohlaví, tělesné hmotnosti nebo výchozích koncentrací antigenu inhibitoru alfa1-proteáz v séru na clearanci přípravku Respreeza.
FarmakokinetickV/farmakodynamickv vztah
Ve dvojitě zaslepené kontrolované klinické studii hodnotící bezpečnost a biochemickou účinnost přípravku Respreeza bylo 44 pacientů randomizováno pro používání intravenózní dávky 60 mg/kg tělesné hmotnosti přípravku Respreeza, podávané jednou týdně, po dobu 24 týdnů. Střední hodnota minimální (trough) koncentrace hladin inhibitoru alfa1-proteáz v séru v ustáleném stavu (týden 7-11) byla udržována nad 11 pmol/l. Střední hodnota (směrodatná odchylka) minimální (trough) koncentrace hladin inhibitoru alfa1-proteáz v séru v ustáleném stavu u pacientů léčených přípravkem Respreeza byla 17,7 pmol/l (2,5).
U podskupiny pacientů zařazených do této studie (10 pacientů léčených přípravkem Respreeza) byl proveden bronchoalveolární výplach. Měření hladin inhibitoru alfa1-proteáz v tekutině kryjící epitel respiračního systému (epithelial lining fluid, ELF) po léčbě prokázalo jejich konzistentní zvýšení. Hladiny při stanovení antigenu inhibitoru alfa1-proteáz a inhibitoru alfa1-proteáz v ELF: Hladiny komplexů s elastázou neutrofilů (NE) se oproti výchozí hodnotě zvýšily. Volná elastáza byla u všech vzorků neměřitelně nízká.
Po dokončení studie RAPID byla provedena analýza dosažených mediánů hladin inhibitoru alfa1-proteáz a poklesu hustoty plicní tkáně. Tato analýza prokázala inverzní lineární vztah mezi minimálními (trough) hladinami inhibitoru alfa1-proteáz v séru a poklesem hustoty plicní tkáně za jeden rok, měřeným podle hustoty plicní tkáně na CT skenech s objemovou korekcí, u pacientů užívajících intravenózní dávku přípravku Respreeza 60 mg/kg tělesné hmotnosti.
5.3 Předklinické údaje vztahující se k bezpečnosti
Bezpečnost přípravku Respreeza byla hodnocena v několika předklinických studiích. Neklinické údaje získané na základě farmakologických studií bezpečnosti a toxicity po krátkodobém podávání neodhalily žádné zvláštní riziko pro člověka. Studie toxicity po opakovaném podávání po dobu delší než 5 dní, studie reprodukční toxicity a studie karcinogenicity nebyly provedeny. Tyto studie nejsou považovány za smysluplné vzhledem k tvorbě protilátek proti heterologním lidským proteinům u zvířat. Protože lidský inhibitor alfai-proteáz je protein a fyziologická složka lidské krve, neočekává se, že by měl karcinogenní, genotoxické nebo teratogenní účinky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Chlorid sodný
Monohydrát dihydrogenfosforečnanu sodného
Manitol
Rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
3 roky.
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Chemická a fyzikální stabilita během používání při pokojové teplotě (do 25 °C) byla prokázána po dobu 3 hodin. Rekonstituovaný roztok chraňte před mrazem.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 25 °C. Chraňte před mrazem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení a zvláštní vybavení pro podání
1000 mg prášku ve skleněné injekční lahvičce (typu I), uzavřené zátkou z (brombutylové) pryže a hliníkovým uzávěrem s plastovým odtrhávacím („flip off") víčkem.
20 ml vody na injekce ve skleněné injekční lahvičce (typu I), uzavřené zátkou z (chlorbutylové) pryže a hliníkovým uzávěrem s plastovým odtrhávacím („flip off") víčkem.
Jedno balení obsahuje:
Jednu injekční lahvičku s práškem
Jednu injekční lahvičku s rozpouštědlem
Jedno převodní zařízení s filtrem přívodu vzduchu
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Při rekonstituci, podávání přípravku a zacházení s ním se musí dbát opatrnosti a používat aseptické postupy, aby byla zachována sterilita přípravku.
Rekonstituce za použití převodního zařízení a injekční lahvičky s rozpouštědlem:
K rekonstituci prášku se musí použít 20 ml rozpouštědla (vody na injekci). Úplné rekonstituce by mělo být dosaženo za 5 minut.
Postupujte podle níže uvedených pokynů:
Poznámky k použití převodního zařízení:
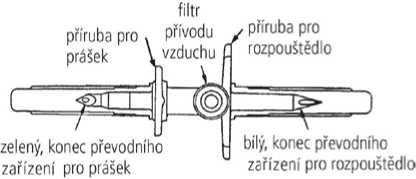
• Převodní zařízení přibalené do krabičky s přípravkem Respreeza má bílý konec (pro rozpouštědlo) se dvěma otvory a zelený konec (pro prášek) s jediným otvorem.
• Při nesprávném použití převodního zařízení se zruší podtlak, takže nemůže dojít k převedení rozpouštědla a tím se rekonstituce přípravku Respreeza prodlouží nebo se jí zabrání.
• Převodní zařízení je sterilní. Po odstranění chráničů (krok 3 a 4) se nedotýkejte obnažených zakončení bodců.
1. Vytemperujte injekční lahvičku s práškem (zelené víčko) a injekční lahvičku s rozpouštědlem (modré víčko) na pokojovou teplotu (do 25°C).
To můžete provést tak, že ponecháte injekční lahvičky při pokojové teplotě po dobu přibližně jedné hodiny, nebo tak, že je několik minut zahřejete v rukách._
2. Sejměte plastová odtrhávací („flip off“) víčka z obou použitých injekčních lahviček.
Vydezinfikujte každou z pryžových zátek antiseptickým roztokem a nechte je uschnout.
3. Sejměte chránič z bílého konce převodního zařízení. Postavte injekční lahvičku s rozpouštědlem na rovný povrch a zasuňte bílý konec převodního zařízení do středu pryžové zátky stojící injekční lahvičky s rozpouštědlem (modré víčko).
filtr
přívodu
vzduchu
příruba pro rozpouštědlo
zelený, konec převodního zařízení pro prášek
bílý, konec převodního zařízení pro rozpouštědlo
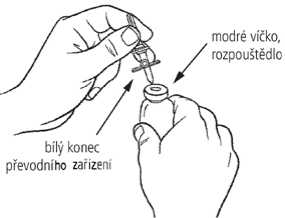
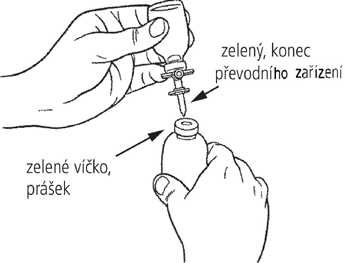
4. Postavte injekční lahvičku s práškem (zelené víčko) na rovný povrch. Sejměte chránič ze zeleného konce převodního zařízení. Převraťte injekční lahvičku s rozpouštědlem se zasunutým převodním zařízením a jemně zasuňte zelený konec převodního zařízení do středu pryžové zátky stojící injekční lahvičky s práškem (zelené víčko). Příruba převodního zařízení by měla spočívat na povrchu zátky tak, aby rozpouštědlo odteklo do injekční lahvičky s práškem.
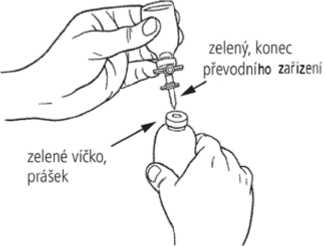
5. Nechte rozpouštědlo odtéct do injekční lahvičky s práškem. To proběhne automaticky, protože v
injekční lahvičce s práškem je podtlak. Pokud v lahvičce s práškem není podtlak, rozpouštědlo nenateče do injekční lahvičky s práškem. V takovém případě přípravek nepoužívejte._
6. Opatrným nakláněním injekční lahvičky s práškem během převodu rozpouštědla zajistěte, aby
se všechen prášek zvlhčil._
7. Až bude všechno rozpouštědlo převedeno, vytáhněte převodní zařízení z injekční lahvičky s
práškem a injekční lahvičku od rozpouštědla a převodní zařízení zlikvidujte._
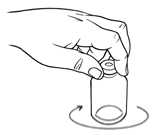
8. Jemným kroužením lahvičkou s práškem rozpusťte všechen prášek.
Netřepejte - zabraňte vzniku pěny.
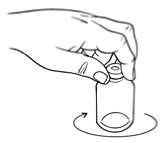
9. Prohlédněte rekonstituovaný roztok. Roztok musí být čirý, bezbarvý až nažloutlý a nesmí
obsahovat viditelné částice. Nepoužívejte roztoky, které nejsou bezbarvé, jsou zakalené nebo obsahují částice._
10. Pokud pro dosažení požadované dávky bude potřebná více než 1 injekční lahvička prášku, opakujte
pokyny 1 až 9 výše s dalším balením obsahujícím převodní zařízení. Převodní zařízení nepoužívejte opakovaně._
11. Za použití aseptického postupu převeďte rekonstituované roztoky z injekčních lahviček do obalu,
ze kterého bude podáván (např. prázdný infuzní vak nebo skleněná lahev; není součástí balení), přes komerčně dostupnou převodní soupravu intravenózních hadiček (není součástí balení)._
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH Emil-von-Behring-Strasse 76 D-35041 Marburg Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1006/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 20.08.2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY /BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY /BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
CSL Behring LLC
Route 50 North 1201 N. Kinzie
Bradley, IL 60915
Spojené státy americké
Název a adresa výrobce odpovědného za propouštění šarží
CSL Behring GmbH Emil-von-Behring-Strasse 76 35041 Marburg Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• Při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Poregistrační studie účinnosti (PAES): Byla dohodnuta realizace randomizovaného, dlouhodobé hodnocení PAES k ověření účinku dávek, tj. vlivu vyšších krevních hladin API na rychlost řídnutí plicní tkáně a vhodnosti vyšší dávky, 120 mg/kg.Držitel rozhodnutí o registraci by měl realizovat randomizované, dlouhodobé klinické hodnocení účinnosti podle dohodnutého protokolu a předložit jeho výsledky. |
Předložení konečného zprávy z klinického hodnocení do 31. 3. 2025 |
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Respreeza 1000 mg prášek a rozpouštědlo pro infuzní roztok Inhibitor alfa-1 proteinasi humani
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Inhibitor alfa-1 proteinasi humani 1000 mg
Po rekonstituci za použití 20 ml rozpouštědla roztok obsahuje přibližně 50 mg/ml lidského inhibitoru alfa1-proteáz.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, manitol.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro infuzní roztok
1 injekční lahvička s práškem 1 injekční lahvička s rozpouštědlem
1 převodní zařízení s filtrem přívodu vzduchu pro rekonstituci
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 25 °C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH, 35041 Marburg, Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1006/001
13. ČÍSLO ŠARŽE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Respreeza
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Respreeza 1000 mg prášek pro infuzní roztok Inhibitor alfa-1 proteinasi humani
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Inhibitor alfa-1 proteinasi humani 1000 mg
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, manitol.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro infuzní roztok 1000 mg
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 25 °C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
CSL Behring
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1006/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU INJEKČNÍ LAHVIČKA S ROZPOUŠTĚDLEM 1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Rozpouštědlo pro přípravek Respreeza
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Voda na injekci
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
20 ml
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
CSL Behring
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1006/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: Informace pro pacienta
Respreeza 1000 mg
Prášek a rozpouštědlo pro infuzní roztok
Inhibitor alfa-1 proteinasi humani
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotnického pracovníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotnickému pracovníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Respreeza a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Respreeza používat
3. Jak se přípravek Respreeza používá
4. Možné nežádoucí účinky
5. Jak přípravek Respreeza uchovávat
6. Obsah balení a další informace
1. Co je přípravek Respreeza a k čemu se používá Co je přípravek Respreeza
Tento přípravek obsahuje léčivou látku lidský inhibitor alfa1-proteáz, který je normální složkou krve a nalézá se v plicích. Na místě výskytu je jeho hlavní funkcí chránit plicní tkáň omezením účinku enzymu nazývaného elastáza neutrofilů. Elastáza neutrofilů může způsobit poškození, pokud její účinek není kontrolovaný (například jestliže máte deficit inhibitoru alfa1-proteáz).
K čemu se přípravek Respreeza používá
Tento léčivý přípravek se používá u dospělých pacientů se známým těžkým deficitem inhibitoru alfa1-proteáz (vrozené onemocnění, nazývané také deficit alfa1 antitrypsinu), u kterých vzniklo onemocnění plic nazývané rozedma (emfyzém).
Rozedma vznikne tehdy, když nedostatek inhibitoru alfa1-proteáz způsobí stav, v němž není správně kontrolována elastáza neutrofilů, takže poškozuje drobné vzduchové váčky v plicích (plicní sklípky), jimiž přechází do organismu kyslík. Vzhledem k tomuto poškození plíce nepracují správně.
Pravidelné používání tohoto léčivého přípravku zvyšuje hladinu inhibitoru alfa1-proteáz v krvi a v plicích a tedy zpomaluje postup rozedmy plic.
2. Čemu musíte věnovat pozornost, než začnete přípravek Respreeza používat NEPOUŽÍVEJTE přípravek Respreeza
• jestliže jste alergický(á) na lidský inhibitor alfai-proteáz nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
• jestliže bylo zjištěno, že máte nedostatek jistých krevních proteinů nazývaných imunoglobulin typu A (IgA) a vytvořil(a) jste si proti nim protilátky.
Upozornění a opatření
Před použitím přípravku Respreeza se poraďte se svým lékařem nebo zdravotnickým pracovníkem.
Informace o alergických reakcích: kdy může být nutné zpomalení nebo zastavení infuze?
Můžete mít alergii na lidský inhibitor alfai-proteáz i tehdy, když jste již dříve lidské inhibitory alfar proteáz užíval(a) a snášel(a) jste je dobře. V některých případech může docházet k těžkým alergickým reakcím. Váš lékař Vás informuje o známkách alergických reakcí (například zimnice, návaly, rychlejší srdeční činnost, pokles krevního tlaku, pocity na omdlení, vyrážka, kopřivka, svědění, potíže s dýcháním nebo s polykáním a otok rukou, obličeje nebo úst) (viz také bod 4).
Ihned sdělte svému lékaři nebo zdravotnickému pracovníkovi, jestliže si během infuze tohoto přípravku všimnete takových reakcí. Podle povahy a závažnosti takové reakce se Váš lékař může rozhodnout, zda infuzi zpomalí nebo zcela zastaví a zahájí příslušnou léčbu.
V případě podávání v domácím prostředí / podávání sobě samému infuzi ihned zastavte a kontaktujte svého lékaře nebo zdravotnického pracovníka.
Informace o bezpečnosti v souvislosti s infekcemi
Přípravek Respreeza se vyrábí z lidské krevní plazmy (tekuté složky krve, z níž byly odstraněny buněčné elementy).
Protože krví se mohou přenášet infekce, jsou-li léky připravovány z lidské krve či plazmy, přijímají se určitá opatření k zabránění jejich výskytu v krvi a přenosu na pacienty. Tato opatření jsou následující:
• pečlivý výběr dárců krve či plazmy zajišťující vyloučení osob s rizikem přenosu infekce,
• testování vzorků darované krve a plazmy tak, aby se pokud možno zabránilo použití materiálů se známkami virů/infekcí,
• zařazení takových kroků při zpracování krve či plazmy, které jsou schopny inaktivovat či odstranit viry.
Přijatá opatření se považují za účinná pro viry, jako je virus lidské imunodeficience (HIV), virus hepatitidy A, virus hepatitidy B, virus hepatitidy C a parvovirus B19.
Přes tato opatření však nelze při podání léků připravených z lidské krve či plazmy zcela vyloučit možnost přenosu infekce.
Pokud pravidelně/opakovaně dostáváte inhibitory proteáz vyrobené z lidské plazmy, může Váš lékař doporučit, abyste zvážil(a) očkování proti hepatitidě typu A a B.
Důrazně se doporučuje poznamenat si při každém podání dávky přípravku Respreeza název přípravku a číslo šarže pro uchování záznamu o použitých šaržích.
Kouření
Protože tabákový dým je významným rizikovým faktorem rozvoje a postupu rozedmy plic, důrazně se doporučuje, abyste přestal(a) kouřit a vyhýbal(a) se pobytu v prostředí, kde se kouří (pasivní kouření).
Děti a dospívající
Tento léčivý přípravek není určen k podávání dětem nebo dospívajícím ve věku do 18 let.
Další léčivé přípravky a přípravek Respreeza
Informujte svého lékaře nebo zdravotnického pracovníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství, kojení a plodnost
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná nebo plánujete otěhotnět, poraďte se se svým lékařem nebo zdravotnickým pracovníkem dříve, než začnete tento přípravek používat.
Protože inhibitor alfai-proteáz je inhibitor normální složkou lidské krve, nepředpokládá se, že by doporučená dávka tohoto přípravku mohla mít škodlivé účinky na vyvíjející se plod. Protože však informace o bezpečnosti použití přípravku Respreeza během těhotenství nejsou k dispozici, jestliže jste těhotná, tento přípravek Vám smí být podáván pouze s opatrností.
Není známo, zda přípravek Respreeza přechází do lidského mateřského mléka. Jestliže kojíte, Váš lékař s Vámi projedná rizika a přínosy používání tohoto přípravku.
Údaje o vlivu na plodnost nejsou k dispozici, ale vzhledem k tomu, že inhibitor alfai-proteáz je normální složkou lidské krve, nepředpokládají se žádné nežádoucí účinky na plodnost, pokud používáte přípravek Respreeza v doporučené dávce.
Řízení dopravních prostředků a obsluha strojů
Po podání tohoto přípravku se mohou vyskytovat závratě. Jestliže máte závratě, nesmíte řídit nebo obsluhovat stroje, dokud závratě nezmizí (viz bod 4).
Přípravek Respreeza obsahuje sodík
Tento přípravek obsahuje 1,9 mg sodíku v jednom ml rekonstituovaného roztoku. Váš lékař nebo zdravotnický pracovník k tomu bude přihlížet, pokud jste na dietě s kontrolovaným příjmem sodíku.
3. Jak se přípravek Respreeza používá
Po rekonstituci se Respreeza podává infuzí do žíly. Na první infuze přípravku bude dohlížet zdravotnický pracovník se zkušenostmi s léčbou deficitu inhibitoru alfa1-proteáz.
Podávání v domácím prostředí / podávání sobě samému
Po prvních infuzích si můžete podávat přípravek Respreeza sám(sama) nebo Vám jej může podávat Váš pečovatel, ale pouze po příslušném zaškolení. Pokud Váš lékař rozhodne, že jste způsobilou osobou pro takové podávání v domácím prostředí / podávání sobě samému, dá Vám pokyny ohledně:
• přípravy a podávání tohoto přípravku (viz pokyny s obrázky na konci této příbalové informace v bodě „Informace pro zdravotnické pracovníky a pacienty způsobilé pro podávání v domácím prostředí / podávání sobě samému“),
• zachování sterility přípravku (aseptických postupů při podávání infuze),
• vedení deníku léčby,
• rozpoznání vedlejších účinků, včetně známek alergických reakcí, a opatření v případě takových účinků (viz také bod 2 a bod 4).
Váš lékař nebo Váš zdravotnický pracovník budou pravidelně kontrolovat způsob, jakým si sám (sama) podáváte infuze / jakým Vám podává infuze Váš pečovatel, aby zajistil, že způsob podání bude stále správný.
Dávka
Množství přípravku Respreeza, které Vám bude podáno, bude založeno na Vaší tělesné hmotnosti. Doporučená dávka je 60 mg na kilogram tělesné hmotnosti a měla by se podávat jednou týdně. Infuzní roztok se normálně podává přibližně po dobu 15 minut (přibližně 0,08 ml roztoku na jeden kg tělesné hmotnosti za minutu). Váš lékař pro Vás stanoví vhodnou rychlost infuze s přihlédnutím k Vaší hmotnosti a snášenlivosti infuze.
Jestliže jste užil(a) více přípravku Respreeza, než jste měl(a)
Následky předávkování nejsou známé.
Pokud se domníváte, že jste užil(a) více přípravku Respreeza, než jste měl(a), sdělte to svému lékaři nebo zdravotnickému pracovníkovi. Ten provede vhodná opatření.
Jestliže jste zapomněl(a) užít přípravek Respreeza
Ihned pokračujte následující dávkou a pokračujte v pravidelných intervalech tak, jak Vám poradil Váš lékař nebo zdravotnický pracovník.
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat přípravek Respreeza
Nepřestávejte používat tento přípravek, pokud se nejprve neporadíte se svým lékařem nebo zdravotnickým pracovníkem. Je-li léčba přípravkem Respreeza zastavena, může se Váš stav zhoršit.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Tyto vedlejší účinky se mohou vyskytnout i tehdy, když jste již dříve lidské inhibitory alfa1-proteáz užíval(a) a snášel(a) jste je dobře.
Některé vedlejší účinky mohou být závažné:
Méně často byly pozorovány alergické reakce (mohou se vyskytnout nejvýše u 1 ze 100 osob).
V některých velmi vzácných případech (mohou se vyskytnout nejvýše u 1 z 10 000 osob) se mohou rozvinout závažné alergické reakce i v případech, kdy se u Vás nevyskytly žádné známky alergie při předchozích infuzích.
Ihned sdělte svému lékaři nebo zdravotnickému pracovníkovi, jestliže si všimnete jakékoli známky alergické reakce (například zimnice, návaly, zrychlený srdeční tep, pokles krevního tlaku, pocity na omdlení, vyrážka, kopřivka, svědění, potíže s dýcháním nebo polykáním a otok rukou, obličeje nebo úst) během podávání přípravku Respreeza. Podle povahy a závažnosti takové reakce se lékař nebo zdravotnický pracovník může rozhodnout, zda podávání zpomalí nebo zcela zastaví a provede příslušnou léčbu reakce.
V případě podávání v domácím prostředí / podávání sobě samému infuzi ihned zastavte a kontaktujte svého lékaře nebo zdravotnického pracovníka.
Další vedlejší účinky mohou být:
Časté (mohou se vyskytovat až u 1 ze 10 osob)
Závratě, bolesti hlavy, dušnost, pocit na zvracení
Méně časté (mohou se vyskytovat až u 1 ze 100 osob)
Změněné vnímání dotyku, pocity jako pálení, mravenčení nebo necitlivost v rukách, pažích, nohách nebo chodidlech (parestezie), návaly, vyrážka (kopřivka), šupinatá vyrážka a vyrážka po celém těle, fyzická slabost (astenie), reakce v místě infuze (například pálení, bodání, bolest, otok nebo zarudnutí v místě infuze (hematom).
Velmi vzácné (mohou se vyskytovat až u 1 z 10 000 osob)
Snížené vnímání dotyku, pocity jako pálení, mravenčení nebo necitlivost v rukách, pažích, nohách nebo chodidlech (hypestezie), nadměrné pocení (hyperhidróza), svědění, bolesti na hrudi, zimnice, horečka (pyrexie).
Frekvence není známá (frekvenci z dostupných údajů nelze určit)
Bolest lymfatických uzlin (oválné shluky tkáně, které se vyskytují po celém těle a které lze nahmatat například v podpaží, tříslech nebo na krku), otoky obličeje, otoky očí a rtů.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotnickému pracovníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Respreeza uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na vnější krabičce a štítcích na injekční lahvičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce. Neuchovávejte při teplotě nad 25 °C. Chraňte před mrazem.
Po rekonstituci se roztok má ihned spotřebovat. Pokud to není možné, lze roztoky uchovávat po dobu až 3 hodin při pokojové teplotě (do 25 °C). Rekonstituovaný roztok chraňte před mrazem.
6. Obsah balení a další informace Co přípravek Respreeza obsahuje
Léčivou látkou je inhibitor alfa-1 proteinasi humani. Jedna injekční lahvička přibližně obsahuje inhibitor alfa__-1 proteinasi humani 1000 mg.
Dalšími složkami jsou chlorid sodný, monohydrát dihydrogenfosforečnanu sodného a manitol (viz poslední odstavec bodu 2).
Rozpouštědlo: Voda na injekci.
Jak přípravek Respreeza vypadá a co obsahuje toto balení
Tento přípravek je bílý až téměř bílý prášek.
Po rekonstituci vodou na injekce má být roztok čirý, bezbarvý až nažloutlý a bez viditelných částic.
Jedno balení obsahuje:
• 1 injekční lahvičku s práškem
• 1 injekční lahvičku s 20 ml vody na injekci
• 1 převodní zařízení pro rekonstituci
Držitel rozhodnutí o registraci a výrobce
CSL Behring GmbH
Emil-von-Behring-Strasse 76 D-35041 Marburg Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
CSL Behring NV CSL Behring AB
Tél/Tel: +32 15 28 89 20 Tel: +46 8 544 966 70
|
Btarapna HoBHMeg OOfl Ten: +359 2 850 86 17 |
Luxembourg/Luxemburg CSL Behring NV Tél/Tel: +32 15 28 89 20 |
|
Česká republika CSL Behring s.r.o. Tel: +420 702 137 233 |
Magyarország CSL Behring Kft. Tel.: +36 1 213 4290 |
|
Danmark CSL Behring AB Tel: +46 8 544 966 70 |
Malta AM Mangion Ltd. Tel: +356 2397 6333 |
|
Deutschland CSL Behring GmbH Tel: +49 69 30584437 |
Nederland CSL Behring BV Tel: +31 85 111 96 00 |
|
Eesti CSL Behring AB Tel: +46 8 544 966 70 |
Norge CSL Behring AB Tlf: +46 8 544 966 70 |
|
EXláSa CSL Behring MEnE T^: +30 210 7255 660 |
Osterreich CSL Behring GmbH Tel: +43 1 80101 2463 |
|
Espaňa CSL Behring S.A. Tel: +34 933 67 1870 |
Polska CSL Behring Sp. z.o.o. Tel.: +48 22 213 22 65 |
|
France CSL Behring SA Tél: +33 1 53 58 54 00 |
Portugal CSL Behring Lda Tel: +351 21 782 62 30 |
|
Hrvatska PharmaSwiss d.o.o. Tel: +385 1 631 1833 |
Románia Nicopharma Distribution Group Tel.: +40 21 327 2614 |
|
Ireland CSL Behring UK Ltd. Tel: +44 1444 447405 |
Slovenija MediSanus d.o.o. Tel: +386 1 25 71 496 |
|
Ísland CSL Behring AB Sími: +46 8 544 966 70 |
Slovenská republika CSL Behring s.r.o. Tel: +421 911 653 862 |
|
Italia CSL Behring S.p.A. Tel: +39 02 34964 200 |
Suomi/Finland CSL Behring AB Puh/Tel: +46 8 544 966 70 |
|
Kúnpoq AKH! nANAriQTOY & YIO! ATA T^: +357 22677038 |
Sverige CSL Behring AB Tel: +46 8 544 966 70 |
|
Latvija CSL Behring AB Tel: +46 8 544 966 70 |
United Kingdom CSL Behring UK Ltd. Tel: +44 1444 447405 |
Tato příbalová informace byla naposledy revidována MM/RRRR.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pro zdravotnické pracovníky a pacienty způsobilé pro podávání přípravku v domácím prostředí / podávání sobě samému
Rekonstituce a podávání přípravku Respreeza
Při rekonstituci, podávání přípravku a zacházení s ním se musí dbát opatrnosti a používat aseptické postupy, aby byla zachována sterilita přípravku.
Postupujte podle níže uvedených pokynů:
Rekonstituce
K rekonstituci prášku se musí použít 20 ml rozpouštědla (vody na injekci). Úplné rekonstituce by mělo být dosaženo za 5 minut.
Poznámky k použití převodního zařízení:
• Převodní zařízení přibalené do krabičky s přípravkem Respreeza má bílý konec (pro rozpouštědlo) se dvěma otvory a zelený konec (pro prášek) s jediným otvorem.
• Při nesprávném použití převodního zařízení se zruší podtlak, takže nemůže dojít k převedení rozpouštědla a tím se rekonstituce přípravku Respreeza prodlouží nebo se jí zabrání.
• Převodní zařízení je sterilní. Po odstranění chráničů (krok 3 a 4) se nedotýkejte obnažených zakončení bodců.
1. Vytemperujte injekční lahvičku s práškem (zelené víčko) a injekční lahvičku s rozpouštědlem (modré víčko) na pokojovou teplotu (do 25°C).
To můžete provést tak, že ponecháte injekční lahvičky při pokojové teplotě po dobu přibližně jedné hodiny, nebo tak, že je několik minut zahřejete v rukách._
2. Sejměte plastová odtrhávací („flip off“) víčka z obou použitých injekčních lahviček.
Vydezinfikujte každou z pryžových zátek antiseptickým^ roztokem a nechte je uschnout._
3. Sejměte chránič z bílého konce převodního zařízení. Postavte injekční lahvičku s rozpouštědlem na rovný povrch a zasuňte bílý konec převodního zařízení do středu pryžové zátky stojící injekční lahvičky s rozpouštědlem (modré víčko).
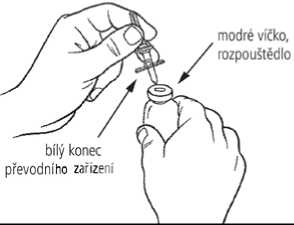
4. Postavte injekční lahvičku s práškem (zelené víčko) na rovný povrch. Sejměte chránič ze zeleného konce převodního zařízení. Převraťte injekční lahvičku s rozpouštědlem se zasunutým převodním zařízením a jemně zasuňte zelený konec převodního zařízení do středu pryžové zátky stojící injekční lahvičky s práškem (zelené víčko). Příruba převodního zařízení by měla spočívat na povrchu zátky tak, aby rozpouštědlo odteklo do injekční lahvičky s práškem.
5. Nechte rozpouštědlo odtéct do injekční lahvičky s práškem. To proběhne automaticky, protože v
injekční lahvičce s práškem je podtlak. Pokud v lahvičce s práškem není podtlak, rozpouštědlo nenateče do injekční lahvičky s práškem. V takovém případě přípravek nepoužívejte._
6. Opatrným nakláněním injekční lahvičky s práškem během převodu rozpouštědla zajistěte, aby se
všechen prášek zvlhčil._
7. Až bude všechno rozpouštědlo převedeno, vytáhněte převodní zařízení z injekční lahvičky s
práškem a injekční lahvičku od rozpouštědla a převodní zařízení zlikvidujte._
8. Jemným kroužením lahvičkou s práškem rozpusťte všechen prášek.
Netřepejte - zabraňte vzniku pěny.
9. Prohlédněte rekonstituovaný roztok. Roztok musí být čirý, bezbarvý až nažloutlý a nesmí obsahovat viditelné částice. Nepoužívejte roztoky, které nejsou bezbarvé, jsou zakalené nebo obsahují částice.
10. Pokud pro dosažení požadované dávky bude potřebná více než 1 injekční lahvička prášku, opakujte pokyny 1 až 9 výše s dalším balením obsahujícím převodní zařízení. Převodní zařízení nepoužívejte opakovaně.
11. Za použití aseptického postupu převeďte rekonstituované roztoky z injekčních lahviček do obalu, ze kterého bude přípravek podáván (např. prázdný infuzní vak nebo skleněná lahev; není součástí balení), přes komerčně dostupnou převodní soupravu intravenózních hadiček (není součástí balení).
Podávání
Rekonstituovaný roztok musí být během podávání filtrován přes vhodný infuzní filtr (doporučená velikost póru je 5 mikrometrů; není součástí balení) a podáván soupravou pro nitrožilní podávání (není součástí balení).
1. Připojte soupravu pro podávání k obalu, ze kterého je přípravek podáván.
Ujistěte se, že regulační svorka na soupravě pro podávání je zavřená.
Zavěste obal, ze kterého je přípravek podáván, do vyvýšené polohy (pokud se jedná o infuzní vak, zavěste jej na infuzní stojan).
Naplňte kapací komoru tak, že ji stisknete a necháte do poloviny naplnit přípravkem Respreeza. Pomalu otevřete regulační svorku na soupravě pro podávání a nechte přípravek Respreeza téci, až dosáhne konce hadičky. V hadičce nesmí být bubliny vzduchu.
Zavřete regulační svorku._
2. Připojte na konec soupravy pro podávání filtr s velikostí póru 5 mikrometrů.
Znovu otevřete regulační svorku a nechte přípravek Respreeza téci, až bude filtr navlhčen._
3. Připojte druhý konec filtru k injekční soupravě (např. k motýlkové infuzní jehle / infuzní jehle s
křidélky nebo k infuzní kanyle)._
4. Podejte (si) injekci/infuzi rekonstituovaného roztoku do žíly podle pokynů Vašeho lékaře. Roztok
musí být podáván přibližně rychlostí 0,08 ml na kilogram tělesné hmotnosti za minutu, podle Vaší reakce a tak, aby bylo zajištěno Vaše pohodlí. Infuze doporučené dávky, tj._
60 mg na kilogram tělesné hmotnosti, bude trvat přibližně 15 minut._
5. Pokud se infuze zpomalí nebo zastaví, možná budete muset vyměnit filtr, který může být ucpaný. V takovém případě opakujte krok 2-4._
Jedna injekční lahvička přípravku Respreeza je určena pouze pro jednorázové použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s pokyny Vašeho lékaře nebo zdravotnického pracovníka.
32