Removab 10 Mikrogramů
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Removab 10 mikrogramů koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka obsahuje 10 mikrogramů catumaxomabum* v 0,1 ml roztoku. Odpovídá 0,1 mg/ml.
* hybridní potkaní-myší IgG2 monoklonální protilátka produkovaná potkaní-myší buněčnou linií hybrid-hybridom
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok.
Čirý a bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Removab je indikován k intraperitoneální léčbě maligního ascitu u dospělých s EpCAM pozitivními karcinomy, kde není k dispozici standardní terapie nebo již není dále použitelná.
4.2 Dávkování a způsob podání
Removab musí být podáván pod dohledem lékaře, který má zkušenosti s používáním antineoplastických léčivých přípravků.
Dávkování
Před intraperitoneální infuzí se doporučuje premedikace analgetiky/antipyretiky/nesteroidními antiflogistiky (viz bod 4.4).
Harmonogram dávkování Removabu zahrnuje následující čtyři intraperitoneální infuze:
1. dávka 10 mikrogramů v den 0
2. dávka 20 mikrogramů v den 3
3. dávka 50 mikrogramů v den 7
4. dávka 150 mikrogramů v den 10
Removab musí být podáván intraperitoneálními infuzemi o konstantním průtoku po dobu nejméně tří hodin. V klinických studiích bylo zkoumáno podání infuzí po dobu 3 a 6 hodin. Pro první čtyři dávky infuze se musí uvažovat s dobou v délce 6 hodin v závislosti na zdravotním stavu pacienta.
Mezi dny s infuzí musí uplynout nejméně dva kalendářní dny bez infuze. Interval ve dnech mezi infuzemi lze prodloužit v případě významných nežádoucích účinků. Celková doba léčby by neměla překročit 20 dnů.
Sledování
Doporučuje se odpovídající sledování pacienta po ukončení infuze Removabu. V pivotních studiích byli pacienti sledování 24 hodin po každé infuzi.
Zvláštní populace
Poškození funkce jater
Nebyli zkoumáni pacienti se závažnějším než středně závažným poškozením funkce jater, a/nebo kdy je nejméně 70% jater zasaženo metastázami, a/nebo trombózou/obstrukcí portální žíly. Léčba těchto pacientů Removabem by se měla zvažovat pouze po důkladném vyhodnocení poměru přínosu a rizika (viz bod 4.4).
Poškození funkce ledvin
Pacienti se závažnějším než mírným poškozením funkce ledvin nebyli zkoumáni. Léčba těchto pacientů katumaxomabem by se měla zvažovat pouze po důkladném vyhodnocení poměru přínosu a rizika (viz bod 4.4).
Pediatrická populace
Neexistuje žádné relevantní použití přípravku Removab u pediatrické populace v povolené indikaci. Způsob podání
Removab se musí podávat pouze jako intraperitoneální infuze.
Removab nesmí být podáván jako intraperitoneální bolus, ani žádným jiným způsobem podání. Informace k použití pro perfuzní systém viz bod 4.4.
Opatření, která je nutné učinit před podáním léčivého přípravku
Před podáním se koncentrát Removabu pro infuzní roztok naředí v injekčním roztoku chloridu sodného 9 mg/ml (0,9%) podle příslušné potřebné dávky. Naředěný roztok je poté podáván intraperitoneální infuzí o konstantním průtoku s vhodným systémem infuzní pumpy.
Návod k naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
Hypersenzitivita na murinní (potkaní a/nebo myší) proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Removab nesmí být podáván jako bolus ani žádnou jinou cestou podání než intraperitoneálně. Příznaky související s uvolňováním cytokinů
Protože vázání katumaxomabu na imunitní buňky a buňky tumoru iniciuje prozánětlivé a cytotoxické cytokiny, byly během podání Removabu a po něm velmi často hlášeny klinické příznaky spojené s uvolňováním cytokinů, jako je například horečka, nauzea, zvracení a zimnice,(viz bod 4.8). Často byla pozorována dyspnoe a hypo/hypertenze. V klinických studiích u pacientů s maligním ascitem bylo před infuzí Removabu intravenózně podáváno pravidelně 1000 mg paracetamolu pro kontrolu bolesti a pyrexie, a proto se jeho podávání doporučuje. Navzdory této premedikaci se u pacientů objevovaly shora uvedené nežádoucí účinky, jejichž intenzita dosahovala až stupně 3 podle Obecných terminologických kritérií pro nežádoucí účinky (CTCAE) U.S. National Cancer Institute, verze 3.0. Doporučuje se další nebo dodatečná standardní premedikace analgetiky/antipyretiky/nesteroidními antiflogistiky.
Syndrom systémové zánětlivé odpovědi (SIRS), který může rovněž často vzniknout díky mechanismu působení katumaxomabu, se obecně rozvíjí během 24 hodin po infuzi Removabu, kdy se projevují příznaky horečky, tachykardie, tachypnoe a leukocytózy (viz bod 4.8). K omezení rizika je vhodná standardní terapie nebo premedikace, napři analgetika/antipyretika/nesteroidní antiflogistika.
Bolesti v břišní krajině
Jako nežádoucí účinek byla často hlášena bolest v krajině břišní. Má se za to, že tento přechodný účinek je částečně důsledkem intraperitoneálního podání.
Tělesná zdatnost a BMI
Před zahájením léčby Removabem je nutná solidní tělesná zdatnost vyjádřená jako index tělesné hmotnosti (BMI) > 17 (bude stanovena po drenáži tekutiny ascitu) a ^varnofského index > 60.
Akutní infekce
Podávání Removabu se nedoporučuje za přítomnosti faktorů narušujících imunitní systém, zejména akutních infekcí.
Drenáž ascitu
Pro zajištění stabilních funkcí krevního oběhu a ledvin je při léčbě Removabem nezbytným předpokladem vhodné lékařské zvládnutí drenáže ascitu. To musí zahrnovat drenáž ascitu do zastavení spontánního odtoku nebo známek úlevy a, bude-li to vhodné, podpůrnou náhradní léčbu krystaloidy a/nebo koloidy.
Pacienti s hemodynamickou insuficiencí, edémem nebo hypoproteinemií
Před každou infuzí Removabu musí být změřen objem krve, krevní proteiny, krevní tlak, puls a funkce ledvin. Před každou infuzí Removabu se musí vyřešit stavy, jako jsou hypovolemie, hypoproteinemie, hypotenze, oběhová dekompenzace a akutní zhoršení funkce ledvin.
Poškození funkce jater či trombóza/obstrukce portální žíly
Pacienti s poškozením funkce jater vyššího stupně závažnosti, než je střední, a/nebo kdy je nejméně 70 % jater zasaženo metastázami, a/nebo trombózou/obstrukcí portální žíly nebyli zkoumání. Léčba těchto pacientů Removabem by se měla zvážit po důkladném vyhodnocení poměru mezi přínosem a riziky.
Poškození funkce ledvin
Pacienti se závažnějším než mírným poškozením funkce ledvin nebyli zkoumáni. Léčba těchto pacientů katumaxomabem by se měla zvažovat pouze po důkladném vyhodnocení poměru přínosu a rizika.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání katumaxomabu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie reprodukční toxicity na zvířatech jsou nedostatečné (viz bod 5.3). Podávání Removabu se v těhotenství a u žen ve fertilním věku, které nepoužívají antikoncepci, nedoporučuje.
Kojení
Není známo, zda se katumaxomab/metabolity vylučují do lidského mateřského mléka. Riziko pro kojené novorozence/děti nelze vyloučit. Na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit podávání přípravku Removab.
Fertilita
K dispozici nejsou žádné údaje zkoumající vliv catumaxomabu na plodnost.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Removab má malý až mírný vliv na schopnost řídit nebo obsluhovat stroje.
Pacienty, u nichž se objeví symptomy spojené s infuzí, je nutno upozornit na to, aby neřídili a neobsluhovali stroje, dokud příznaky neodezní.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nežádoucí účinky uvedené níže jsou odvozeny od integrované bezpečnostní analýzy zahrnující 12 klinických studií. Catumaxomab byl podán intraperitoneálně 728 pacientům, 293 pacientům byl podán v 6 hodinových infuzích a 435 pacientům v 3 hodinových infuzích.
Celkový bezpečnostní profil Removabu je charakterizován symptomy souvisejícími s uvolňováním cytokinů a gastrointestinálními reakcemi.
Symptomy související s uvolňováním cytokinů: SIRS, potenciálně život ohrožující kombinace tachykardie, horečky a/nebo dušnosti, se objevuje během prvních 24 hodin po infuzi catumaxomabu a odezní po vhodné symptomatické léčbě. Další symptomy související s uvolňováním cytokinů, jako jsou horečka, zimnice, nauzea a zvracení, jsou velmi často hlášené účinky se stupněm intenzity 1 a 2 podle CTCAE (US National Cancer Institute, verze 4.0). Tyto symptomy odrážejí mechanizmus účinku katumaxomabu a jsou obvykle zcela reverzibilní. .
Gastrointestinální reakce, jako abdominální bolest, nauzea zvracení a průjem jsou velmi časté, nejčastěji se vyskytují se stupněm intenzity CTCAE 1 nebo 2, byly však zaznamenány i stupně vyšší. Tyto reakce se řeší vhodnou symptomatickou léčbou.
Bezpečnostní profil katumaxomabu při porovnání infuze v délce 3 hodin s infuzí v délce 6 hodin s ohledem na povahu, četnost a závažnost, v podstatě srovnatelný. V případě 3hodinové infuze byla pozorována zvýšená četnost některých nežádoucích účinků, jako jsou zimnice a hypotenze (stupně 1/2), průjem (všechny stupně) a únava (stupně 1/2).
Tabulkový přehled nežádoucích účinků
V tabulce 1 jsou uvedeny nežádoucí účinky rozdělené podle tříd orgánových systémů. Četnost je definována následovně: velmi časté (> 1/10); časté (> 1/100 až <1/10); méně časté (>1/1.000 až<1/100).
Tabulka 1 Nežádoucí účinky hlášené u pacientů léčených katumaxomabem
|
Infekce a infestace | |
|
Časté |
Infekce. |
|
Méně časté |
Erythema induratum*, infekce spojené s přístroji*. |
|
Poruchy krve a lymfatického systému | |
|
Časté |
Anémie*, lymfopenie, leukocytóza, neutrofilie. |
|
Méně časté |
Trombocytopenie*, koagulopatie*. |
|
Poruchy imunitního systému | |
|
Časté S |
>yndromy spojené s uvolňováním cytokinů*, hypersenzitivita* |
|
Poruchy metabolismu a výživy | |
|
Časté |
Snížená chuť k jídlu*/ anorexie, dehydratace*, hypokalémie, hypoalbuminémie, hyponatrémie*, hypokalcémie*, hypoproteinémie. |
|
Psychiatrické poruchy | |
|
Časté |
Úzkost, nespavost. |
|
Poruchy nervového |
systému |
|
Časté |
Bolesti hlavy, závratě. |
|
Méně časté |
Konvulze *. |
|
Poruchy ucha a labyrintu | |
|
Časté | |
|
Srdeční poruchy | |
|
Časté |
Tachykardie*, včetně sinusové tachykardie. |
|
Cévní poruchy | |
|
Časté |
Hypotenze*, hypertenze*, zčervenání. |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté |
Dyspnoe*, pleurální výpotek*, kašel. |
|
Méně časté |
Plicní embolie*, hypoxie*. |
|
Gastrointestinální poruchy | |
|
Velmi časté |
Bolest v kraiině břišní*, nauzea*, zvracení*, průjem*. |
|
Časté |
Zácpa*, dyspepsie, abdominální distenze, subileus*, flatulence, gastrická porucha, ileus*, gastroezofageální reflux, sucho v ústech. |
|
Méně časté |
Gastrointestinální krvácení*, intestinální obstrukce*. |
|
Poruchy jater a žlučových cest | |
|
Časté |
Cholangitida*, hyperbilirubinemie. |
|
Poruchy kůže a pot |
kožní tkáně |
|
Časté |
Vyrážka*, erytém*, hyperhidróza, pruritus. |
|
Méně časté |
Kožní reakce*, alergická dermatitida*. |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Časté |
Bolesti zad, myalgie, artralgie. |
|
Poruchy ledvin a močových cest | |
|
Časté |
Proteinurie. |
|
Méně časté |
Akutní selhání ledvin*. |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté |
Pyrexie*, únava*, zimnice*. |
|
Časté |
Bolesti, astenie*, syndrom systémové zánětlivé odpovědi*, edém včetně |
|
periferního edému*, celkové zhoršení zdravotního stavu*, bolest na hrudi, chřipce podobné onemocnění, malátnost*, erytém v místě vstupu katétru. | |
|
Méně časté |
Extravazace*, reakce v místě aplikace* . |
* byly rovněž hlášeny jako závažný nežádoucí účinek léku. podtržené: viz bod ,Popis vybraných nežádoucích účinků’
Popis vybraných nežádoucích účinků
Platí následující definice kritérií CTCAE US National Cancer Institute (verze 4.0):
CTCAE stupeň 1 = mírný, CTCAE stupeň 2 = středně závažný, CTCAE stupeň 3 = závažný, CTCAE stupeň 4 = život ohrožující.
Příznaky související s uvolňováním cytokinů s vyšším stupněm intenzity
U 5,1 % pacientů dosáhla pyrexie stupně intenzity 3 (CTCAE), stejný stupeň byl zaznamenán i pro příznaky spojené s uvolňováním cytokinů (1,0 %), zimnici (0,8 %), nauzea (3,4 %), zvracení (4,4 %), dušnost (1,6 %) a hypo/hypertenzi (2,1 % / 0,8 %). U j ednoho pacienta (0,1 %) s dušností a u 3 pacientů (0,4 %) s hypotenzí byl naměřen stupeň intenzity CTCAE v hodnotě 4.
Příznaky bolesti a pyrexie lze zmírnit nebo odstranit premedikací (viz bod 4.2 a 4.4).
Syndrom systémové zánětlivé odpovědi (SIRS)
U 3,8 % pacientů byly pozorovány příznaky SIRS během 24 hodin po infuzi catumaxomabu
U 3 pacientů (0,4 %) šlo o stupeň intenzity 4 (CTCAE). Tyto reakce byly odstraněny symptomatickou
léčbou.
Bolesti v krajině břišní
U 43,7 % pacientů byla jako nežádoucí účinek hlášena bolest v krajině břišní, která dosáhla stupně 3 u 8,2 % pacientů, ale byla odstraněna symptomatickou léčbou.
Jaterní enzymy
Po podávání Removabu bylo často pozorováno přechodné zvýšení hladiny jaterních enzymů. Změny laboratorních parametrů obecně nebyly klinicky významné a po skončení léčby se většinou vrátily na výchozí hodnoty.
Pouze v případě klinicky významného nebo přetrvávajícího zvýšení by měla být zvážena další diagnostika nebo léčba.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyl hlášen žádný případ předávkování. Pacienti dostávající vyšší než doporučenou dávku katumaxomabu uváděli závažnější (vyšší než stupeň 3) nežádoucí účinky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antineoplastická léčiva, monoklonální protilátky, ATC kód: L01XC09 Mechanismus účinku
Katumaxomab je trojfunkční potkaní-myší hybridní monoklonální protilátka, která je specificky zaměřena proti adhezní molekule epitelových buněk (EpCAM) a antigenu CD3.
Antigen EpCAM je nadměrně exprimován u většiny karcinomů (tabulka 2). CD3 je exprimován u zralých T buněk jako součást receptoru T buněk. Třetí funkční vazebné místo v regionu Fc katumaxomabu umožňuje interakci s akcesorními imunitními buňkami prostřednictvím receptorů Fcy. Díky vazebným vlastnostem katumaxomabu se buňky tumoru, T buňky a akcesorní imunitní buňky dostávají do těsné blízkosti. Tím se vyvolá koordinovaná imunitní reakce proti buňkám tumoru, což zahrnuje různé mechanismy působení, například aktivaci T buněk, buněčnou cytotoxicitu závislou na protilátkách (ADCC), cytotoxicitu závislou na komplementu (CDC) a fagocytózu. To má za následek destrukci buněk tumoru.
Tabulka 2 Exprese antigenu EpCAM u nejrelevantnějších typů karcinomů způsobujících ascites
|
Údaje z literatury |
Údaje z retrospektivní studie IP-CAT-AC-03 | ||
|
Karcinom |
Podíl nádorů s expresí EpCAM v procentech |
Podíl výpotků pozitivních na EpCAM v procentech |
Podíl výpotků pozitivních na EpCAM v procentech |
|
Vaječníku |
90-92 |
79-100 |
98 |
|
Žaludku |
96 |
75-100 |
100 |
|
Tlustého střeva |
100 |
87-100 |
100 |
|
Slinivky |
98 |
83-100 |
80 |
|
Prsu |
00 1 * in |
71-100 |
86 |
|
Endometriální |
94 |
100 |
100 |
*= lobulární karcinom prsu
Farmakodynamické účinky
Antitumorová aktivita katumaxomabu byla prokázána in vitro a in vivo. Efektivní zabíjení buněk tumoru zprostředkované katumaxomabem in vitro bylo pozorováno u cílových buněk s nízkou a vysokou expresí antigenu EpCAM nezávisle na typu tumoru. Antitumorová aktivita katumaxomabu in vivo byla potvrzena v imunologicky narušeném myším modelu karcinomu ovaria, kde byl rozvoj tumoru opožděn intraperitoneální léčbou katumaxomabem a lidskými mononukleárními buňkami z periferní krve.
Klinická, účinnost.
Účinnost katumaxomabu byla prokázána ve dvou klinických studiích fáze III. Do těchto klinických studií nebyli zahrnuti jiní pacienti než běloši.
IP-REM-AC-01
Jednalo se o základní randomizované, otevřené klinické hodnocení fáze II/III se dvěma rameny, do něhož bylo zařazeno 258 pacientů se symptomatickým maligním ascitem způsobeným EpCAM pozitivními karcinomy, z nichž 170 bylo randomizováno pro léčbu katumaxomabem. Tato studie porovnávala paracentézu plus katumaxomab versus paracentézu samotnou (kontrola).
Katumaxomab byl podáván pacientům, pro něž nebyla dostupná standardní terapie nebo již dále nebyla proveditelná, a kteří měli Karnofského stav tělesné zdatnosti nejméně 60. Katumaxomab byl podáván jako čtyři intraperitoneální infuze se zvyšujícími se dávkami: 10 mikrogramů v den 0, 20 mikrogramů v den 3, 50 mikrogramů v den 7 a 150 mikrogramů v den 10 (viz bod 4.2). V pivotní studii IP-REM-AC-01 bylo hospitalizováno 98,1 % pacientů po medián 11 dnů.
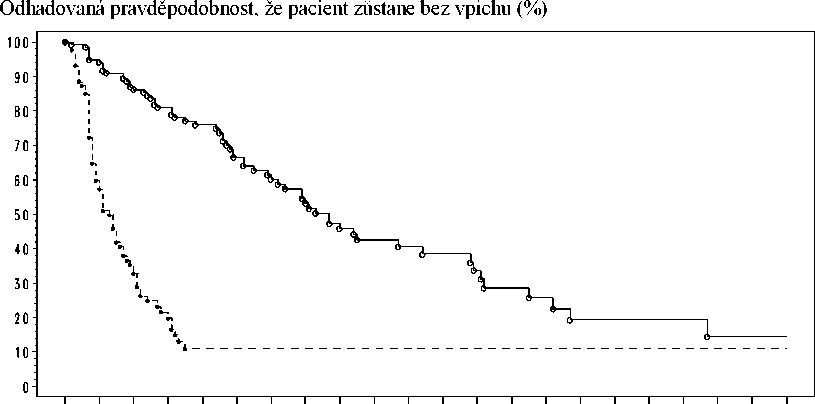
V této studii bylo primárním cílovým parametrem účinnosti doba přežití bez punkce, což je složený cílový parametr definovaný jako doba do první potřeby terapeutické punkce ascitu nebo úmrtí, podle toho, co nastane jako první. Výsledky doby přežití bez punkce a doby do první potřeby léčebné punkce ascitu jako střední doby, a relativní riziko jsou uvedeny v tabulce 3. Kaplanův-Meierův odhad doby do první potřeby terapeutické punkce ascitu je zachycen na obrázku 1.
Tabulka 3 Výsledky účinnosti ve studii IP-REM-AC-01 (doba přežití bez punkce a doba do první potřeby terapeutické punkce ascitu)
|
Proměnná |
Paracentéza + katumaxomab (N=170) |
Paracentéza (kontrola) (N=88) |
|
Doba přežití bez punkce | ||
|
Medián doby přežití bez punkce (dny) |
44 |
11 |
|
95% IS pro medián (dny) |
[31; 49] |
[9; 16] |
|
Hodnota p (test v logaritmickém vyjádření) |
< 0,0001 | |
|
Relativní riziko (HR) |
0,310 | |
|
95% IS pro HR |
[0,228; 0,423] | |
|
Doba do první potřeby terapeutické punkce ascitu | ||
|
Medián doby do první potřeby terapeutické punkce ascitu (dny) |
77 |
13 |
|
95% IS pro medián (dny) |
[62; 104] |
[9; 17] |
|
Hodnota p (test v logaritmickém vyjádření) |
< 0,0001 | |
|
Relativní riziko (HR) |
0,169 | |
|
95% IS pro HR |
[0,114; 0,251] | |
Doba (dny) do události
Katumaxomab (N=170) Kontrola (N=88)
Obrázek 1 Kaplanův-Meierův odhad doby do první potřeby terapeutické punkce ascitu ze studie IP-REM-AC-01
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210
Léčba:
-• — -• — -•
N: počet pacientů v léčené skupině.
Účinnost léčby pacientů s maligním ascitem způsobeným EpCAM pozitivními karcinomy paracentézou a katumaxomabem byla statisticky významně lepší ve srovnání s léčbou paracentézou samotnou, z hlediska doby přežití bez punkce a doby do první potřeby terapeutické punkce ascitu.
Po dokončení studie byli pacienti dále sledování až do konce jejich života, za účelem vyhodnocení celkového přežití (Tabulka 4).
Tabulka 4 Celkové přežití ve studi IP-REM-AC-01 ve fázi po ukončení studie
|
Paracentéza + katumaxomab (N=170) |
Paracentéza (kontrola) (N=88) | |
|
Míra rizika (HR) |
0,798 | |
|
95% CI pro HR |
[0,606; 1.0511 | |
|
Míra přežití 6 měsíců |
27.5% |
17.1% |
|
Míra přežití 1 roku |
11.4% |
2.6% |
|
Medián celkového přežití (dny) |
72 |
71 |
|
95% CI pro medián (dny) |
[61; 98] |
[54; 89] |
|
Hodnota p (test v logaritmickém vyjádření) |
0.1064 | |
Celkem 45 z 88 (51 %) pacientů z kontrolní skupiny přešlo na aktivní léčbu katumaxomabem. IP-CAT-AC-03
Tohoto potvrzujícího randomizovaného otevřeného klinického hodnocení fáze IIIb se dvěma rameny se účastnilo 219 pacientů s epitelovým karcinomem se symptomatickým maligním ascitem vyžadujícím terapeutickou punkci ascitu. Byla porovnávána léčba katumaxomabem s premedikací 25 mg prednisolonem a léčba katumaxomabem samotným. V obou skupinách byl katumaxomab podáván formou čtyř 3hodinových intraperitoneálních infuzí při konstantní rychlosti v den 0, 3, 7 a 10 v dávkách 10, 20, 50, resp. 150 mikrogramů. Soubor pacientů byl srovnatelný se souborem v základním klinickém hodnocení.
Aby bylo možné posoudit účinek premedikace prednisolonem na bezpečnost a účinnost, bylo jako hlavní cílový parametr bezpečnosti hodnoceno tzv. složené skóre bezpečnosti a jako vedlejší cílový parametr účinnosti doba přežití bez punkce.
Složeným skóre bezpečnosti byla v obou skupinách léčby hodnocena četnost a závažnost hlavních známých nežádoucích účinků, tj. pyrexie, nevolnosti, zvracení a bolesti břicha. Podávání prednisolonu jako premedikace nevedlo ke snížení výskytu těchto nežádoucích reakcí.
Primárním cílovým parametrem účinnosti byla doba přežití bez punkce, což je složený cílový parametr definovaný jako doba do první potřeby terapeutické punkce ascitu nebo úmrtí, podle toho, co nastane jako první (stejný parametr jako v základním klinickém hodnocení).
Tabulka 5 Výsledky účinnosti ve studii IP-REM-AC-03 (doba přežití bez punkce a doba do první potřeby terapeutické punkce ascitu) ___
|
Proměnná |
Katumaxomab + prednisolon (N = 111) |
Katumaxomab (N = 108) |
Souhrnný soubor (N = 219) |
|
Doba přežití bez punkce | |||
|
Medián doby přežití bez punkce (dny) |
30 |
37 |
35 |
|
95% IS pro medián (dny) |
[23; 67j |
[24; 61] |
[26; 59] |
|
Hodnota p (test v logaritmickém vyjádření) |
0,402 | ||
|
Relativní riziko (HR, katumaxomab v porovnání s katumaxomabem a prednisolonem) |
1,130 | ||
|
95% IS pro HR |
[0,845; 1,511] | ||
|
Doba do první potřeby terapeutické punkce ascitu | |||
|
Medián doby do první potřeby terapeutické punkce ascitu (dny) |
78 |
102 |
97 |
|
95% IS pro medián (dny) |
[30; 223] |
[69; 159] |
[67; 155] |
|
Hodnota p (test v logaritmickém vyjádření) |
0,599 | ||
|
Relativní riziko (HR, katumaxomab v porovnání s katumaxomabem a prednisolonem) |
0,901 | ||
|
95% IS pro HR |
[0,608; 1,335] | ||
Jako vedlejší cílový parametr účinnosti byla hodnocena celková doba přežití (viz tabulka 6).
Tabulka 6 Celkové přežití po skončení klinického hodnocení IP-CAT-AC-03
|
Katumaxomab + prednisolon (N = 111) |
Katumaxomab (N = 108) |
Souhrnný soubor (N = 219) | |
|
Medián celkového přežití (dny) |
124 |
86 |
103 |
|
95% IS pro medián (dny) |
[97,0; 169,0] |
(72,0; 126,0) |
[82; 133] |
|
Hodnota p (test v logaritmickém vyjádření) |
0,186 | ||
|
Relativní riziko (HR, katumaxomab v porovnání s katumaxomabem a prednisolonem) |
1,221 | ||
|
95% IS pro HR |
[0,907;1,645] | ||
Imunogenicita
Indukce humánních antimurinních (potkaních a/nebo myších) protilátek (HAMA/HARA) je vlastním účinkem murinních monoklonálních protilátek. Současné údaje o katumaxomabu odvozené z pivotní studie ukazují, že pouze 5,6 % pacientů (7/124 pacientů) bylo HAMA pozitivních před 4. infuzí. Antimurinní protilátky (HAMAs) byly přítomny u 94 % pacientů jeden měsíc po poslední infuzi katumaxomabu. Nebyly pozorovány žádné reakce hypersenzitivity.
Pacienti, u kterých se do 8 dnů po léčbě katumaxomabem vytvořily protilátky na HAMA, prokázali lepší klinické výsledky z hlediska doby přežití bez punkce, doby do první potřeby terapeutické punkce a celkového přežití, v porovnání s pacienty HAMA negativními.
Ve studii proveditelnosti léčby, která posuzovala druhý cyklus intraperitoneální infuze 10, 20, 50 a 150 mikrogramů katumaxomabu u 8 pacientů s maligními ascity způsobenými karcinomem (studie IP-CAT-AC-04), byly ve všech získaných ascitech a ve všech vzorcích plazmy odebraných při zařazování do studie zjištěny protilátky proti hodnocenému přípravku. Protilátky proti hodnocenému přípravku vykazovali pacienti i během fáze léčby a následné kontrolní fáze. Přestože si tito pacienti již vytvořili protilátky proti hodnocenému přípravku, dostali všichni z nich všechny čtyři infuze katumaxomabu. Medián doby přežití bez punkce činil 47,5 dnů, medián doby do první potřeby terapeutické punkce 60,0 dnů a medián celkového přežití 406,5 dnů. U všech pacientů se projevily příznaky související se způsobem účinkování katumaxomabu. Bezpečnostní profil byl co do povahy srovnatelný s prvním cyklem intraperitoneálních infuzí. Nebyla zaznamenána žádná přecitlivělost.
5.2 Farmakokinetické vlastnosti
U 13 pacientů se symptomatickým maligním ascitem způsobeným EpCAM pozitivními karcinomy byla zkoumána farmakokinetika katumaxomabu během a po čtyřech intraperitoneálních infuzích 10, 20, 50 a 150 mikrogramů katumaxomabu.
Variabilita mezi subjekty byla vysoká. Geometrický průměr plazmatické Cmax byl přibližně 0,5 ng/ml (od 0 do 2,3) a geometrický průměr plazmatické AUC byl přibližně 1,7 dne1ng/ml (od < LLOQ (dolní mez kvantifikace) do 13,5). Geometrický průměr zdánlivého terminálního plazmatického eliminačního poločasu (f/2) byl přibližně 2,5 dne (od 0,7 do 17).
Katumaxomab byl detekovatelný v tekutině ascitu a v plazmě.
U většiny pacientů rostly koncentrace s počtem infuzí a podaných dávek. Hladiny v plazmě měly sklon klesat po dosažení maxima po každé dávce.
Speciální populace
Žádné studie nebyly provedeny.
5.3 Předklinické údaje vztahující se k bezpečnosti
Podání katumaxomabu u zvířecích modelů nepřineslo žádné známky abnormální nebo s léčivem související akutní toxicity nebo známky lokální nesnášenlivosti v místě vpichu injekce/infuze. Ovšem tyto nálezy mají omezenou hodnotu kvůli vysoké druhové specifitě katumaxomabu.
Studie toxicity po opakovaném podávání, genotoxicity, kancerogenity, reprodukční a vývojové toxicity nebyly prováděny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Citronan sodný
Monohydrát kyseliny citronové Polysorbát 80 Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
2 roky
Po naředění
Připravený roztok pro infuzi je fyzikálně a chemicky stabilní po 48 hodin při teplotě od 2°C do 8°C a 24 hodin při teplotě do 25 °C. Z mikrobiologického hlediska má být přípravek použit okamžitě.
Není-li použit okamžitě, doba a podmínky uchovávání před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při 2°C až 8°C, pokud ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po j eho naředění j sou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
0,1 ml koncentrátu pro infuzní roztok v předplněné injekční stříkačce (sklo typu I, silikonizované) se zátkou na pístu (brombutylový kaučuk) a systémem uzávěru luer (silikonizovaný polypropylén a polykarbonát) s víčkem na hrotu (styren butadienový kaučuk) a s jehlou; velikost balení: 1 předplněná injekční stříkačka, jedna jehla.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Likvidace
Žádné zvláštní požadavky.
Požadovaný materiál a vybavení
K naředění a podání Removabu se musí používat následující komponenty, protože Removab je kompatibilní pouze s:
• 50ml polypropylenové injekční stříkačky
• polyethylenové perfuzní hadičky o vnitřním průměru 1 mm a délce 150 cm
• polykarbonátové infuzní ventily/spojky typu Y
• polyuretanové katétry se silikonovým povlakem nebo bez něho
Dále se požaduje následující:
• izotonický injekční roztok chloridu sodného 9 mg/ml (0,9%)
• přesná perfuzní pumpa
Pokyny k naředění před podáním
Je nutné, aby Removab připravoval zdravotnický pracovník za použití vhodné aseptické techniky. Vnější povrch předplněné injekční stříkačky není sterilní. 1
propláchli, aby tak nedošlo ke kontaminaci a aby bylo zajištěno vypuzení správného objemu.
• Uzavřete 50ml injekční stříkačku víčkem a roztok jemně promíchejte. Z 50ml injekční stříkačky odstraňte všechny vzduchové bubliny.
• Odlupovací štítek, který se nachází na vnitřní straně krabičky s přípravkem Removab a obsahuje text „Naředěný přípravek Removab. Pouze pro intraperitoneální podání.“ se musí přilepit na 50 ml injekční stříkačku obsahující naředěný roztok přípravku Removab pro intraperitoneální infuzi. Jedná se o preventivní opatření, aby bylo jisté, že se přípravek Removab použije jako infuze pouze intraperitoneální cestou podání.
• 50ml injekční stříkačku vložte do infuzní pumpy.
Tabulka 7 Příprava roztoku Removab pro intraperitoneální infuzi
|
Počet infuzí/ Dávka |
Počet předplněných injekčních stříkaček |
Celkový objem koncentrátu Removabu pro infuzní roztok |
Injekční roztok chloridu sodného 9 mg/ml (0,9 %) |
Konečný objem k podání | |
|
Předplněná injekční stříkačka 10 mikrogramů |
Předplněná injekční stříkačka 50 mikrogramů | ||||
|
1.infuze 10 mikrogramů |
1 |
0,1 ml |
10 ml |
10,1 ml | |
|
2. infuze 20 mikrogramů |
2 |
0,2 ml |
20 ml |
20,2 ml | |
|
3.infuze 50 mikrogramů |
1 |
0,5 ml |
49,5 ml |
50 ml | |
|
4. infuze 150 mikrogramů |
3 |
1,5 ml |
48,5 ml |
50 ml | |
Obrázek 2 Schéma přenosu Removabu z předplněné injekční stříkačky do 50ml injekční stříkačky
Předplněná injekční stříkačka
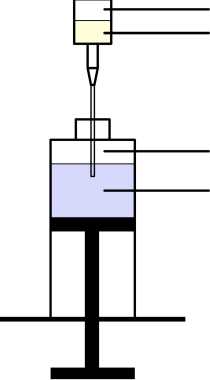
Vzduchový polštář Roztok Removabu
Vzduchový polštář Chlorid sodný 9 mg/ml (0,9 %)
50ml injekční stříkačka
Způsob podání
Katétr k intraperitoneálnímu podání zavádí za kontroly ultrazvukem lékař, který má zkušenosti s postupy intraperitoneálního podávání. Katétr se používá k drenáži ascitu a k infuzi roztoku Removabu a injekčního roztoku chloridu sodného 9 mg/ml (0,9 %). Doporučuje se, aby katétr zůstal
v dutině břišní po celou dobu léčby. Lze jej vyjmout den po poslední infuzi.
Před každým podáním Removabu se musí provést drenáž ascitu až do zastavení spontánního odtoku nebo do známek zlepšení (viz bod 4.4). Následně, před každým podáním Removabu, se podá infuzí 500 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9 %), aby se podpořila distribuce protilátky v dutině břišní.
Removab se musí podávat intraperitoneálně po dobu infuze nejméně 3 hodiny pomocí systému infuzní pumpy o konstantním průtoku, který je popsán dále:
• Nainstalujte perfuzní injekční stříkačku obsahující roztok Removabu pro infuzi do pumpy s přesným dávkováním.
• Předplňte připojenou soustavu perfuzních hadičky pumpy s přesným dávkováním roztokem Removabu pro infuzi. Musíte používat perfuzní hadičky o vnitřním průměru 1 mm a délce 150 cm.
• Připojte perfuzní hadičku ke spojce typu Y.
• Souběžně s každým podáním Removabu podávejte infuzí 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9 %) přes infuzní ventil/spojku typu Y v perfuzním vývodu katétru.
• Nastavte rychlost dávkování pumpy podle objemu, který se má podávat, a plánovanou dobu infuze.
• Když je 50ml stříkačka obsahující zředěný infuzní roztok Removabu prázdná, nahradí se 50ml stříkačkou s 20 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9 %), až do konce plánované doby infuze, aby se odstranil mrtvý objem v perfuzním vývodu (přibližně 2 ml) při nezměněných podmínkách. Zbývající roztok chloridu sodného 9 mg/ml (0,9 %) se může zlikvidovat.
• Do další infuze udržujte katétr v uzavřeném stavu.
• Den po poslední infuzi proveďte drenáž ascitu až do zastavení spontánního odtoku. Poté lze katétr vyjmout.
Obrázek 3 Schematické znázornění infuzního systému
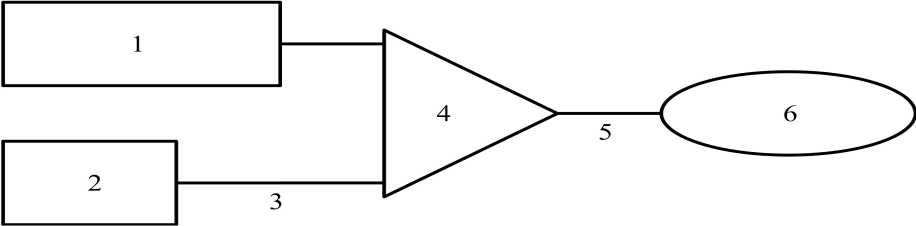
1 250 ml chlorid sodný 9 mg/ml (0,9 %)
2 Roztok Removabu pro i.p. infuzi
3 Perfuzní hadička (1 mm vnitřní průměr, délka 150 cm)
4 Infuzní ventil
5 Perfuzní vývod
6 Katétr
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Neovii Biotech GmbH Am Haag 6-7 82166 Graefelfing Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/09/512/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 20. dubna 2009
Datum posledního prodloužení registrace: 18. prosinec 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Removab 50 mikrogramů koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka obsahuje 50 mikrogramů catumaxomabum* v 0,5 ml roztoku. Odpovídá 0,1 mg/ml.
* hybridní potkaní-myší IgG2 monoklonální protilátka produkovaná potkaní-myší buněčnou linií hybrid-hybridom
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok.
Čirý a bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Removab je indikován k intraperitoneální léčbě maligního ascitu u dospělých s EpCAM pozitivními karcinomy, kde není k dispozici standardní terapie nebo již není dále použitelná.
4.2 Dávkování a způsob podání
Removab musí být podáván pod dohledem lékaře, který má zkušenosti s používáním antineoplastických léčivých přípravků.
Dávkování
Před intraperitoneální infuzí se doporučuje premedikace analgetiky/antipyretiky/nesteroidními antiflogistiky (viz bod 4.4).
Harmonogram dávkování Removabu zahrnuje následující čtyři intraperitoneální infuze:
1. dávka 10 mikrogramů v den 0
2. dávka 20 mikrogramů v den 3
3. dávka 50 mikrogramů v den 7
4. dávka 150 mikrogramů v den 10
Removab musí být podáván intraperitoneálními infuzemi o konstantním průtoku po dobu nejméně tří hodin. V klinických studiích bylo zkoumáno podání infuzí po dobu 3 a 6 hodin. Pro první čtyři dávky infuze se musí uvažovat s dobou v délce 6 hodin v závislosti na zdravotním stavu pacienta.
Mezi dny s infuzí musí uplynout nejméně dva kalendářní dny bez infuze. Interval ve dnech mezi infuzemi lze prodloužit v případě významných nežádoucích účinků. Celková doba léčby by neměla překročit 20 dnů.
Sledování
Doporučuje se odpovídající sledování pacienta po ukončení infuze Removabu. V pivotních studiích byli pacienti sledování 24 hodin po každé infuzi.
Zvláštní populace
Poškození funkce jater
Nebyli zkoumáni pacienti se závažnějším než středně závažným poškozením funkce jater, a/nebo kdy je nejméně 70% jater zasaženo metastázami, a/nebo trombózou/obstrukcí portální žíly. Léčba těchto pacientů Removabem by se měla zvažovat pouze po důkladném vyhodnocení poměru přínosu a rizika (viz bod 4.4).
Poškození funkce ledvin
Pacienti se závažnějším než mírným poškozením funkce ledvin nebyli zkoumáni. Léčba těchto pacientů katumaxomabem by se měla zvažovat pouze po důkladném vyhodnocení poměru přínosu a rizika (viz bod 4.4).
Pediatrická populace
Neexistuje žádné relevantní použití přípravku Removab u pediatrické populace v povolené indikaci. Způsob podání
Removab se musí podávat pouze jako intraperitoneální infuze.
Removab nesmí být podáván jako intraperitoneální bolus, ani žádným jiným způsobem podání. Informace k použití pro perfuzní systém viz bod 4.4.
Opatření, která je nutné učinit před podáním léčivého přípravku
Před podáním se koncentrát Removabu pro infuzní roztok naředí v injekčním roztoku chloridu sodného 9 mg/ml (0,9%) podle příslušné potřebné dávky. Naředěný roztok je poté podáván intraperitoneální infuzí o konstantním průtoku s vhodným systémem infuzní pumpy.
Návod k naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
Hypersenzitivita na murinní (potkaní a/nebo myší) proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Removab nesmí být podáván jako bolus ani žádnou jinou cestou podání než intraperitoneálně. Příznaky související s uvolňováním cytokinů
Protože vázání katumaxomabu na imunitní buňky a buňky tumoru iniciuje prozánětlivé a cytotoxické cytokiny, byly během podání Removabu a po něm velmi často hlášeny klinické příznaky spojené s uvolňováním cytokinů, jako je například horečka, nauzea, zvracení a zimnice,(viz bod 4.8). Často byla pozorována dyspnoe a hypo/hypertenze. V klinických studiích u pacientů s maligním ascitem bylo před infuzí Removabu intravenózně podáváno pravidelně 1000 mg paracetamolu pro kontrolu bolesti a pyrexie, a proto se jeho podávání doporučuje. Navzdory této premedikaci se u pacientů objevovaly shora uvedené nežádoucí účinky, jejichž intenzita dosahovala až stupně 3 podle Obecných terminologických kritérií pro nežádoucí účinky (CTCAE) U.S. National Cancer Institute, verze 3.0. Doporučuje se další nebo dodatečná standardní premedikace analgetiky/antipyretiky/nesteroidními antiflogistiky.
Syndrom systémové zánětlivé odpovědi (SIRS), který může rovněž často vzniknout díky mechanismu působení katumaxomabu, se obecně rozvíjí během 24 hodin po infuzi Removabu, kdy se projevují příznaky horečky, tachykardie, tachypnoe a leukocytózy (viz bod 4.8). K omezení rizika je vhodná standardní terapie nebo premedikace, napři analgetika/antipyretika/nesteroidní antiflogistika.
Bolesti v břišní krajině
Jako nežádoucí účinek byla často hlášena bolest v krajině břišní. Má se za to, že tento přechodný účinek je částečně důsledkem intraperitoneálního podání.
Tělesná zdatnost a BMI
Před zahájením léčby Removabem je nutná solidní tělesná zdatnost vyjádřená jako index tělesné hmotnosti (BMI) > 17 (bude stanovena po drenáži tekutiny ascitu) a ^varnofského index > 60.
Akutní infekce
Podávání Removabu se nedoporučuje za přítomnosti faktorů narušujících imunitní systém, zejména akutních infekcí.
Drenáž ascitu
Pro zajištění stabilních funkcí krevního oběhu a ledvin je při léčbě Removabem nezbytným předpokladem vhodné lékařské zvládnutí drenáže ascitu. To musí zahrnovat drenáž ascitu do zastavení spontánního odtoku nebo známek úlevy a, bude-li to vhodné, podpůrnou náhradní léčbu krystaloidy a/nebo koloidy.
Pacienti s hemodynamickou insuficiencí, edémem nebo hypoproteinemií
Před každou infuzí Removabu musí být změřen objem krve, krevní proteiny, krevní tlak, puls a funkce ledvin. Před každou infuzí Removabu se musí vyřešit stavy, jako jsou hypovolemie, hypoproteinemie, hypotenze, oběhová dekompenzace a akutní zhoršení funkce ledvin.
Poškození funkce jater či trombóza/obstrukce portální žíly
Pacienti s poškozením funkce jater vyššího stupně závažnosti, než je střední, a/nebo kdy je nejméně 70 % jater zasaženo metastázami, a/nebo trombózou/obstrukcí portální žíly nebyli zkoumání. Léčba těchto pacientů Removabem by se měla zvážit po důkladném vyhodnocení poměru mezi přínosem a riziky.
Poškození funkce ledvin
Pacienti se závažnějším než mírným poškozením funkce ledvin nebyli zkoumáni. Léčba těchto pacientů katumaxomabem by se měla zvažovat pouze po důkladném vyhodnocení poměru přínosu a rizika.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání katumaxomabu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie reprodukční toxicity na zvířatech jsou nedostatečné (viz bod 5.3). Podávání Removabu se v těhotenství a u žen ve fertilním věku, které nepoužívají antikoncepci, nedoporučuje.
Kojení
Není známo, zda se katumaxomab/metabolity vylučují do lidského mateřského mléka. Riziko pro kojené novorozence/děti nelze vyloučit. Na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit podávání přípravku Removab.
Fertilita
K dispozici nejsou žádné údaje zkoumající vliv catumaxomabu na plodnost.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Removab má malý až mírný vliv na schopnost řídit nebo obsluhovat stroje.
Pacienty, u nichž se objeví symptomy spojené s infuzí, je nutno upozornit na to, aby neřídili a neobsluhovali stroje, dokud příznaky neodezní.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nežádoucí účinky uvedené níže jsou odvozeny od integrované bezpečnostní analýzy zahrnující 12 klinických studií. Catumaxomab byl podán intraperitoneálně 728 pacientům, 293 pacientům byl podán v 6 hodinových infuzích a 435 pacientům v 3 hodinových infuzích.
Celkový bezpečnostní profil Removabu je charakterizován symptomy souvisejícími s uvolňováním cytokinů a gastrointestinálními reakcemi.
Symptomy související s uvolňováním cytokinů: SIRS, potenciálně život ohrožující kombinace tachykardie, horečky a/nebo dušnosti, se objevuje během prvních 24 hodin po infuzi catumaxomabu a odezní po vhodné symptomatické léčbě. Další symptomy související s uvolňováním cytokinů, jako jsou horečka, zimnice, nauzea a zvracení, jsou velmi často hlášené účinky se stupněm intenzity 1 a 2 podle CTCAE (US National Cancer Institute, verze 4.0). Tyto symptomy odrážejí mechanizmus účinku katumaxomabu a jsou obvykle zcela reverzibilní. .
Gastrointestinální reakce, jako abdominální bolest, nauzea zvracení a průjem jsou velmi časté, nejčastěji se vyskytují se stupněm intenzity CTCAE 1 nebo 2, byly však zaznamenány i stupně vyšší. Tyto reakce se řeší vhodnou symptomatickou léčbou.
Bezpečnostní profil katumaxomabu při porovnání infuze v délce 3 hodin s infuzí v délce 6 hodin s ohledem na povahu, četnost a závažnost, v podstatě srovnatelný. V případě 3hodinové infuze byla pozorována zvýšená četnost některých nežádoucích účinků, jako jsou zimnice a hypotenze (stupně 1/2), průjem (všechny stupně) a únava (stupně 1/2).
Tabulkový přehled nežádoucích účinků
V tabulce 1 jsou uvedeny nežádoucí účinky rozdělené podle tříd orgánových systémů. Četnost je definována následovně: velmi časté (> 1/10); časté (> 1/100 až <1/10); méně časté (>1/1.000 až<1/100).
Tabulka 1 Nežádoucí účinky hlášené u pacientů léčených katumaxomabem
|
Infekce a infestace | |
|
Časté |
Infekce. |
|
Méně časté |
Erythema induratum*, infekce spojené s přístroji*. |
|
Poruchy krve a lymfatického systému | |
|
Časté |
Anémie*, lymfopenie, leukocytóza, neutrofilie. |
|
Méně časté |
Trombocytopenie*, koagulopatie*. |
|
Poruchy imunitního systému | |
|
Časté S |
>yndromy spojené s uvolňováním cytokinů*, hypersenzitivita* |
|
Poruchy metabolismu a výživy | |
|
Časté |
Snížená chuť k jídlu*/ anorexie, dehydratace*, hypokalémie, hypoalbuminémie, hyponatrémie*, hypokalcémie*, hypoproteinémie. |
|
Psychiatrické poruchy | |
|
Časté |
Úzkost, nespavost. |
|
Poruchy nervového |
systému |
|
Časté |
Bolesti hlavy, závratě. |
|
Méně časté |
Konvulze *. |
|
Poruchy ucha a labyrintu | |
|
Časté | |
|
Srdeční poruchy | |
|
Časté |
Tachykardie*, včetně sinusové tachykardie. |
|
Cévní poruchy | |
|
Časté |
Hypotenze*, hypertenze*, zčervenání. |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté |
Dyspnoe*, pleurální výpotek*, kašel. |
|
Méně časté |
Plicní embolie*, hypoxie*. |
|
Gastrointestinální poruchy | |
|
Velmi časté |
Bolest v kraiině břišní*, nauzea*, zvracení*, průjem*. |
|
Časté |
Zácpa*, dyspepsie, abdominální distenze, subileus*, flatulence, gastrická porucha, ileus*, gastroezofageální reflux, sucho v ústech. |
|
Méně časté |
Gastrointestinální krvácení*, intestinální obstrukce*. |
|
Poruchy jater a žlučových cest | |
|
Časté |
Cholangitida*, hyperbilirubinemie. |
|
Poruchy kůže a pot |
kožní tkáně |
|
Časté |
Vyrážka*, erytém*, hyperhidróza, pruritus. |
|
Méně časté |
Kožní reakce*, alergická dermatitida*. |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Časté |
Bolesti zad, myalgie, artralgie. |
|
Poruchy ledvin a močových cest | |
|
Časté |
Proteinurie. |
|
Méně časté |
Akutní selhání ledvin*. |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté |
Pyrexie*, únava*, zimnice*. |
|
Časté |
Bolesti, astenie*, syndrom systémové zánětlivé odpovědi*, edém včetně |
|
periferního edému*, celkové zhoršení zdravotního stavu*, bolest na hrudi, chřipce podobné onemocnění, malátnost*, erytém v místě vstupu katétru. | |
|
Méně časté |
Extravazace*, reakce v místě aplikace* . |
* byly rovněž hlášeny jako závažný nežádoucí účinek léku. podtržené: viz bod ,Popis vybraných nežádoucích účinků’
Popis vybraných nežádoucích účinků
Platí následující definice kritérií CTCAE US National Cancer Institute (verze 4.0):
CTCAE stupeň 1 = mírný, CTCAE stupeň 2 = středně závažný, CTCAE stupeň 3 = závažný, CTCAE stupeň 4 = život ohrožující.
Příznaky související s uvolňováním cytokinů s vyšším stupněm intenzity
U 5,1 % pacientů dosáhla pyrexie stupně intenzity 3 (CTCAE), stejný stupeň byl zaznamenán i pro příznaky spojené s uvolňováním cytokinů (1,0 %), zimnici (0,8 %), nauzea (3,4 %), zvracení (4,4 %), dušnost (1,6 %) a hypo/hypertenzi (2,1 % / 0,8 %). U jednoho pacienta (0,1 %) s dušností a u 3 pacientů (0,4 %) s hypotenzí byl naměřen stupeň intenzity CTCAE v hodnotě 4.
Příznaky bolesti a pyrexie lze zmírnit nebo odstranit premedikací (viz bod 4.2 a 4.4).
Syndrom systémové zánětlivé odpovědi (SIRS)
U 3,8 % pacientů byly pozorovány příznaky SIRS během 24 hodin po infuzi catumaxomabu
U 3 pacientů (0,4 %) šlo o stupeň intenzity 4 (CTCAE). Tyto reakce byly odstraněny symptomatickou
léčbou.
Bolesti v krajině břišní
U 43,7 % pacientů byla jako nežádoucí účinek hlášena bolest v krajině břišní, která dosáhla stupně 3 u 8,2 % pacientů, ale byla odstraněna symptomatickou léčbou.
Jaterní enzymy
Po podávání Removabu bylo často pozorováno přechodné zvýšení hladiny jaterních enzymů. Změny laboratorních parametrů obecně nebyly klinicky významné a po skončení léčby se většinou vrátily na výchozí hodnoty.
Pouze v případě klinicky významného nebo přetrvávajícího zvýšení by měla být zvážena další diagnostika nebo léčba.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyl hlášen žádný případ předávkování. Pacienti dostávající vyšší než doporučenou dávku katumaxomabu uváděli závažnější (vyšší než stupeň 3) nežádoucí účinky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antineoplastická léčiva, monoklonální protilátky, ATC kód: L01XC09 Mechanismus účinku
Katumaxomab je trojfunkční potkaní-myší hybridní monoklonální protilátka, která je specificky zaměřena proti adhezní molekule epitelových buněk (EpCAM) a antigenu CD3.
Antigen EpCAM je nadměrně exprimován u většiny karcinomů (tabulka 2). CD3 je exprimován u zralých T buněk jako součást receptoru T buněk. Třetí funkční vazebné místo v regionu Fc katumaxomabu umožňuje interakci s akcesorními imunitními buňkami prostřednictvím receptorů Fcy. Díky vazebným vlastnostem katumaxomabu se buňky tumoru, T buňky a akcesorní imunitní buňky dostávají do těsné blízkosti. Tím se vyvolá koordinovaná imunitní reakce proti buňkám tumoru, což zahrnuje různé mechanismy působení, například aktivaci T buněk, buněčnou cytotoxicitu závislou na protilátkách (ADCC), cytotoxicitu závislou na komplementu (CDC) a fagocytózu. To má za následek destrukci buněk tumoru.
Tabulka 2 Exprese antigenu EpCAM u nejrelevantnějších typů karcinomů způsobujících ascites
|
Údaje z literatury |
Údaje z retrospektivní studie IP-CAT-AC-03 | ||
|
Karcinom |
Podíl nádorů s expresí EpCAM v procentech |
Podíl výpotků pozitivních na EpCAM v procentech |
Podíl výpotků pozitivních na EpCAM v procentech |
|
Vaječníku |
90-92 |
79-100 |
98 |
|
Žaludku |
96 |
75-100 |
100 |
|
Tlustého střeva |
100 |
87-100 |
100 |
|
Slinivky |
98 |
83-100 |
80 |
|
Prsu |
00 1 * in |
71-100 |
86 |
|
Endometriální |
94 |
100 |
100 |
*= lobulární karcinom prsu
Farmakodynamické účinky
Antitumorová aktivita katumaxomabu byla prokázána in vitro a in vivo. Efektivní zabíjení buněk tumoru zprostředkované katumaxomabem in vitro bylo pozorováno u cílových buněk s nízkou a vysokou expresí antigenu EpCAM nezávisle na typu tumoru. Antitumorová aktivita katumaxomabu in vivo byla potvrzena v imunologicky narušeném myším modelu karcinomu ovaria, kde byl rozvoj tumoru opožděn intraperitoneální léčbou katumaxomabem a lidskými mononukleárními buňkami z periferní krve.
Klinická, účinnost.
Účinnost katumaxomabu byla prokázána ve dvou klinických studiích fáze III. Do těchto klinických studií nebyli zahrnuti jiní pacienti než běloši.
IP-REM-AC-01
Jednalo se o základní randomizované, otevřené klinické hodnocení fáze II/III se dvěma rameny, do něhož bylo zařazeno 258 pacientů se symptomatickým maligním ascitem způsobeným EpCAM pozitivními karcinomy, z nichž 170 bylo randomizováno pro léčbu katumaxomabem. Tato studie porovnávala paracentézu plus katumaxomab versus paracentézu samotnou (kontrola).
Katumaxomab byl podáván pacientům, pro něž nebyla dostupná standardní terapie nebo již dále nebyla proveditelná, a kteří měli Karnofského stav tělesné zdatnosti nejméně 60. Katumaxomab byl podáván jako čtyři intraperitoneální infuze se zvyšujícími se dávkami: 10 mikrogramů v den 0, 20 mikrogramů v den 3, 50 mikrogramů v den 7 a 150 mikrogramů v den 10 (viz bod 4.2). V pivotní studii IP-REM-AC-01 bylo hospitalizováno 98,1 % pacientů po medián 11 dnů.
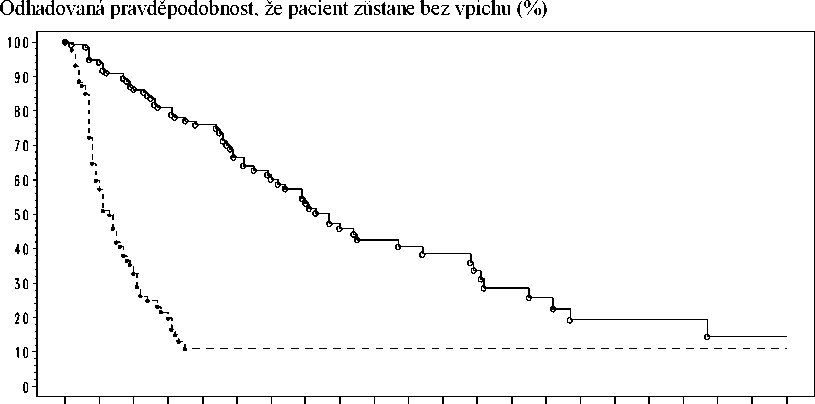
V této studii bylo primárním cílovým parametrem účinnosti doba přežití bez punkce, což je složený cílový parametr definovaný jako doba do první potřeby terapeutické punkce ascitu nebo úmrtí, podle toho, co nastane jako první. Výsledky doby přežití bez punkce a doby do první potřeby léčebné punkce ascitu jako střední doby, a relativní riziko jsou uvedeny v tabulce 3. Kaplanův-Meierův odhad doby do první potřeby terapeutické punkce ascitu je zachycen na obrázku 1.
Tabulka 3 Výsledky účinnosti ve studii IP-REM-AC-01 (doba přežití bez punkce a doba do první potřeby terapeutické punkce ascitu)
|
Proměnná |
Paracentéza + katumaxomab (N=170) |
Paracentéza (kontrola) (N=88) |
|
Doba přežití bez punkce | ||
|
Medián doby přežití bez punkce (dny) |
44 |
11 |
|
95% IS pro medián (dny) |
[31; 49] |
[9; 16] |
|
Hodnota p (test v logaritmickém vyjádření) |
< 0,0001 | |
|
Relativní riziko (HR) |
0,310 | |
|
95% IS pro HR |
[0,228; 0,423] | |
|
Doba do první potřeby terapeutické punkce ascitu | ||
|
Medián doby do první potřeby terapeutické punkce ascitu (dny) |
77 |
13 |
|
95% IS pro medián (dny) |
[62; 104] |
[9; 17] |
|
Hodnota p (test v logaritmickém vyjádření) |
< 0,0001 | |
|
Relativní riziko (HR) |
0,169 | |
|
95% IS pro HR |
[0,114; 0,251] | |
Doba (dny) do události
Katumaxomab (N=170) Kontrola (N=88)
Obrázek 1 Kaplanův-Meierův odhad doby do první potřeby terapeutické punkce ascitu ze studie IP-REM-AC-01
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210
Léčba:
-• — -• — -•
N: počet pacientů v léčené skupině.
Účinnost léčby pacientů s maligním ascitem způsobeným EpCAM pozitivními karcinomy paracentézou a katumaxomabem byla statisticky významně lepší ve srovnání s léčbou paracentézou samotnou, z hlediska doby přežití bez punkce a doby do první potřeby terapeutické punkce ascitu.
Po dokončení studie byli pacienti dále sledování až do konce jejich života, za účelem vyhodnocení celkového přežití (Tabulka 4).
Tabulka 4 Celkové přežití ve studi IP-REM-AC-01 ve fázi po ukončení studie
|
Paracentéza + katumaxomab (N=170) |
Paracentéza (kontrola) (N=88) | |
|
Míra rizika (HR) |
0,798 | |
|
95% CI pro HR |
[0,606; 1.0511 | |
|
Míra přežití 6 měsíců |
27.5% |
17.1% |
|
Míra přežití 1 roku |
11.4% |
2.6% |
|
Medián celkového přežití (dny) |
72 |
71 |
|
95% CI pro medián (dny) |
[61; 98] |
[54; 89] |
|
Hodnota p (test v logaritmickém vyjádření) |
0.1064 | |
Celkem 45 z 88 (51 %) pacientů z kontrolní skupiny přešlo na aktivní léčbu katumaxomabem. IP-CAT-AC-03
Tohoto potvrzujícího randomizovaného otevřeného klinického hodnocení fáze IIIb se dvěma rameny se účastnilo 219 pacientů s epitelovým karcinomem se symptomatickým maligním ascitem vyžadujícím terapeutickou punkci ascitu. Byla porovnávána léčba katumaxomabem s premedikací 25 mg prednisolonem a léčba katumaxomabem samotným. V obou skupinách byl katumaxomab podáván formou čtyř 3hodinových intraperitoneálních infuzí při konstantní rychlosti v den 0, 3, 7 a 10 v dávkách 10, 20, 50, resp. 150 mikrogramů. Soubor pacientů byl srovnatelný se souborem v základním klinickém hodnocení.
Aby bylo možné posoudit účinek premedikace prednisolonem na bezpečnost a účinnost, bylo jako hlavní cílový parametr bezpečnosti hodnoceno tzv. složené skóre bezpečnosti a jako vedlejší cílový parametr účinnosti doba přežití bez punkce.
Složeným skóre bezpečnosti byla v obou skupinách léčby hodnocena četnost a závažnost hlavních známých nežádoucích účinků, tj. pyrexie, nevolnosti, zvracení a bolesti břicha. Podávání prednisolonu jako premedikace nevedlo ke snížení výskytu těchto nežádoucích reakcí.
Primárním cílovým parametrem účinnosti byla doba přežití bez punkce, což je složený cílový parametr definovaný jako doba do první potřeby terapeutické punkce ascitu nebo úmrtí, podle toho, co nastane jako první (stejný parametr jako v základním klinickém hodnocení).
Tabulka 5 Výsledky účinnosti ve studii IP-REM-AC-03 (doba přežití bez punkce a doba do první potřeby terapeutické punkce ascitu) ___
|
Proměnná |
Katumaxomab + prednisolon (N = 111) |
Katumaxomab (N = 108) |
Souhrnný soubor (N = 219) |
|
Doba přežití bez punkce | |||
|
Medián doby přežití bez punkce (dny) |
30 |
37 |
35 |
|
95% IS pro medián (dny) |
[23; 67j |
[24; 61] |
[26; 59] |
|
Hodnota p (test v logaritmickém vyjádření) |
0,402 | ||
|
Relativní riziko (HR, katumaxomab v porovnání s katumaxomabem a prednisolonem) |
1,130 | ||
|
95% IS pro HR |
[0,845; 1,511] | ||
|
Doba do první potřeby terapeutické punkce ascitu | |||
|
Medián doby do první potřeby terapeutické punkce ascitu (dny) |
78 |
102 |
97 |
|
95% IS pro medián (dny) |
[30; 223] |
[69; 159] |
[67; 155] |
|
Hodnota p (test v logaritmickém vyjádření) |
0,599 | ||
|
Relativní riziko (HR, katumaxomab v porovnání s katumaxomabem a prednisolonem) |
0,901 | ||
|
95% IS pro HR |
[0,608; 1,335] | ||
Jako vedlejší cílový parametr účinnosti byla hodnocena celková doba přežití (viz tabulka 6).
Tabulka 6 Celkové přežití po skončení klinického hodnocení IP-CAT-AC-03
|
Katumaxomab + prednisolon (N = 111) |
Katumaxomab (N = 108) |
Souhrnný soubor (N = 219) | |
|
Medián celkového přežití (dny) |
124 |
86 |
103 |
|
95% IS pro medián (dny) |
[97,0; 169,0] |
(72,0; 126,0) |
[82; 133] |
|
Hodnota p (test v logaritmickém vyjádření) |
0,186 | ||
|
Relativní riziko (HR, katumaxomab v porovnání s katumaxomabem a prednisolonem) |
1,221 | ||
|
95% IS pro HR |
[0,907;1,645] | ||
Imunogenicita
Indukce humánních antimurinních (potkaních a/nebo myších) protilátek (HAMA/HARA) je vlastním účinkem murinních monoklonálních protilátek. Současné údaje o katumaxomabu odvozené z pivotní studie ukazují, že pouze 5,6 % pacientů (7/124 pacientů) bylo HAMA pozitivních před 4. infuzí. Antimurinní protilátky (HAMAs) byly přítomny u 94 % pacientů jeden měsíc po poslední infuzi katumaxomabu. Nebyly pozorovány žádné reakce hypersenzitivity.
Pacienti, u kterých se do 8 dnů po léčbě katumaxomabem vytvořily protilátky na HAMA, prokázali lepší klinické výsledky z hlediska doby přežití bez punkce, doby do první potřeby terapeutické punkce a celkového přežití, v porovnání s pacienty HAMA negativními.
Ve studii proveditelnosti léčby, která posuzovala druhý cyklus intraperitoneální infuze 10, 20, 50 a 150 mikrogramů katumaxomabu u 8 pacientů s maligními ascity způsobenými karcinomem (studie IP-CAT-AC-04), byly ve všech získaných ascitech a ve všech vzorcích plazmy odebraných při zařazování do studie zjištěny protilátky proti hodnocenému přípravku. Protilátky proti hodnocenému přípravku vykazovali pacienti i během fáze léčby a následné kontrolní fáze. Přestože si tito pacienti již vytvořili protilátky proti hodnocenému přípravku, dostali všichni z nich všechny čtyři infuze katumaxomabu. Medián doby přežití bez punkce činil 47,5 dnů, medián doby do první potřeby terapeutické punkce 60,0 dnů a medián celkového přežití 406,5 dnů. U všech pacientů se projevily příznaky související se způsobem účinkování katumaxomabu. Bezpečnostní profil byl co do povahy srovnatelný s prvním cyklem intraperitoneálních infuzí. Nebyla zaznamenána žádná přecitlivělost.
5.2 Farmakokinetické vlastnosti
U 13 pacientů se symptomatickým maligním ascitem způsobeným EpCAM pozitivními karcinomy byla zkoumána farmakokinetika katumaxomabu během a po čtyřech intraperitoneálních infuzích 10, 20, 50 a 150 mikrogramů katumaxomabu.
Variabilita mezi subjekty byla vysoká. Geometrický průměr plazmatické Cmax byl přibližně 0,5 ng/ml (od 0 do 2,3) a geometrický průměr plazmatické AUC byl přibližně 1,7 dne2ng/ml (od < LLOQ (dolní mez kvantifikace) do 13,5). Geometrický průměr zdánlivého terminálního plazmatického eliminačního poločasu (tJ/2) byl přibližně 2,5 dne (od 0,7 do 17).
Katumaxomab byl detekovatelný v tekutině ascitu a v plazmě.
U většiny pacientů rostly koncentrace s počtem infuzí a podaných dávek. Hladiny v plazmě měly sklon klesat po dosažení maxima po každé dávce.
Speciální populace
Žádné studie nebyly provedeny.
5.3 Předklinické údaje vztahující se k bezpečnosti
Podání katumaxomabu u zvířecích modelů nepřineslo žádné známky abnormální nebo s léčivem související akutní toxicity nebo známky lokální nesnášenlivosti v místě vpichu injekce/infuze. Ovšem tyto nálezy mají omezenou hodnotu kvůli vysoké druhové specifitě katumaxomabu.
Studie toxicity po opakovaném podávání, genotoxicity, kancerogenity, reprodukční a vývojové toxicity nebyly prováděny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Citronan sodný
Monohydrát kyseliny citronové Polysorbát 80 Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
2 roky
Po naředění
Připravený roztok pro infuzi je fyzikálně a chemicky stabilní po 48 hodin při teplotě od 2°C do 8°C a 24 hodin při teplotě do 25 °C. Z mikrobiologického hlediska má být přípravek použit okamžitě.
Není-li použit okamžitě, doba a podmínky uchovávání před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při 2°C až 8°C, pokud ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
0,5 ml koncentrátu pro infuzní roztok v předplněné injekční stříkačce (sklo typu I, silikonizované) se zátkou na pístu (brombutylový kaučuk) a systémem uzávěru luer (silikonizovaný polypropylén a polykarbonát) s víčkem na hrotu (styren butadienový kaučuk) a s jehlou; velikost balení: 1 předplněná injekční stříkačka, jedna jehla.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Likvidace
Žádné zvláštní požadavky.
Požadovaný materiál a vybavení
K naředění a podání Removabu se musí používat následující komponenty, protože Removab je kompatibilní pouze s:
• 50ml polypropylenové injekční stříkačky
• polyethylenové perfuzní hadičky o vnitřním průměru 1 mm a délce 150 cm
• polykarbonátové infuzní ventily/spojky typu Y
• polyuretanové katétry se silikonovým povlakem nebo bez něho
Dále se požaduje následující:
• izotonický injekční roztok chloridu sodného 9 mg/ml (0,9%)
• přesná perfuzní pumpa
Pokyny k naředění před podáním
Je nutné, aby Removab připravoval zdravotnický pracovník za použití vhodné aseptické techniky. Vnější povrch předplněné injekční stříkačky není sterilní. 2
propláchli, aby tak nedošlo ke kontaminaci a aby bylo zajištěno vypuzení správného objemu.
• Uzavřete 50ml injekční stříkačku víčkem a roztok jemně promíchejte. Z 50ml injekční stříkačky odstraňte všechny vzduchové bubliny.
• Odlupovací štítek, který se nachází na vnitřní straně krabičky s přípravkem Removab a obsahuje text „Naředěný přípravek Removab. Pouze pro intraperitoneální podání.“ se musí přilepit na 50 ml injekční stříkačku obsahující naředěný roztok přípravku Removab pro intraperitoneální infuzi. Jedná se o preventivní opatření, aby bylo jisté, že se přípravek Removab použije jako infuze pouze intraperitoneální cestou podání.
• 50ml injekční stříkačku vložte do infuzní pumpy.
Tabulka 7 Příprava roztoku Removab pro intraperitoneální infuzi
|
Počet infuzí/ Dávka |
Počet předplněných injekčních stříkaček |
Celkový objem koncentrátu Removabu pro infuzní roztok |
Injekční roztok chloridu sodného 9 mg/ml (0,9 %) |
Konečný objem k podání | |
|
Předplněná injekční stříkačka 10 mikrogramů |
Předplněná injekční stříkačka 50 mikrogramů | ||||
|
1.infuze 10 mikrogramů |
1 |
0,1 ml |
10 ml |
10,1 ml | |
|
2. infuze 20 mikrogramů |
2 |
0,2 ml |
20 ml |
20,2 ml | |
|
3.infuze 50 mikrogramů |
1 |
0,5 ml |
49,5 ml |
50 ml | |
|
4. infuze 150 mikrogramů |
3 |
1,5 ml |
48,5 ml |
50 ml | |
Obrázek 2 Schéma přenosu Removabu z předplněné injekční stříkačky do 50ml injekční stříkačky
Předplněná injekční stříkačka
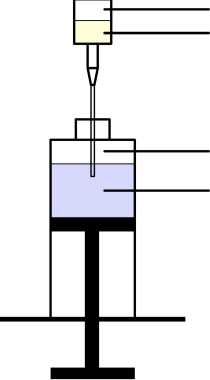
Vzduchový polštář Roztok Removabu
Vzduchový polštář Chlorid sodný 9 mg/ml (0,9 %)
50ml injekční stříkačka
Způsob podání
Katétr k intraperitoneálnímu podání zavádí za kontroly ultrazvukem lékař, který má zkušenosti s postupy intraperitoneálního podávání. Katétr se používá k drenáži ascitu a k infuzi roztoku Removabu a injekčního roztoku chloridu sodného 9 mg/ml (0,9 %). Doporučuje se, aby katétr zůstal
v dutině břišní po celou dobu léčby. Lze jej vyjmout den po poslední infuzi.
Před každým podáním Removabu se musí provést drenáž ascitu až do zastavení spontánního odtoku nebo do známek zlepšení (viz bod 4.4). Následně, před každým podáním Removabu, se podá infuzí 500 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9 %), aby se podpořila distribuce protilátky v dutině břišní.
Removab se musí podávat intraperitoneálně po dobu infuze nejméně 3 hodiny pomocí systému infuzní pumpy o konstantním průtoku, který je popsán dále:
• Nainstalujte perfuzní injekční stříkačku obsahující roztok Removabu pro infuzi do pumpy s přesným dávkováním.
• Předplňte připojenou soustavu perfuzních hadičky pumpy s přesným dávkováním roztokem Removabu pro infuzi. Musíte používat perfuzní hadičky o vnitřním průměru 1 mm a délce 150 cm.
• Připojte perfuzní hadičku ke spojce typu Y.
• Souběžně s každým podáním Removabu podávejte infuzí 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9 %) přes infuzní ventil/spojku typu Y v perfuzním vývodu katétru.
• Nastavte rychlost dávkování pumpy podle objemu, který se má podávat, a plánovanou dobu infuze.
• Když je 50ml stříkačka obsahující zředěný infuzní roztok Removabu prázdná, nahradí se 50ml stříkačkou s 20 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9 %), až do konce plánované doby infuze, aby se odstranil mrtvý objem v perfuzním vývodu (přibližně 2 ml) při nezměněných podmínkách. Zbývající roztok chloridu sodného 9 mg/ml (0,9 %) se může zlikvidovat.
• Do další infuze udržujte katétr v uzavřeném stavu.
• Den po poslední infuzi proveďte drenáž ascitu až do zastavení spontánního odtoku. Poté lze katétr vyjmout.
Obrázek 3 Schematické znázornění infuzního systému
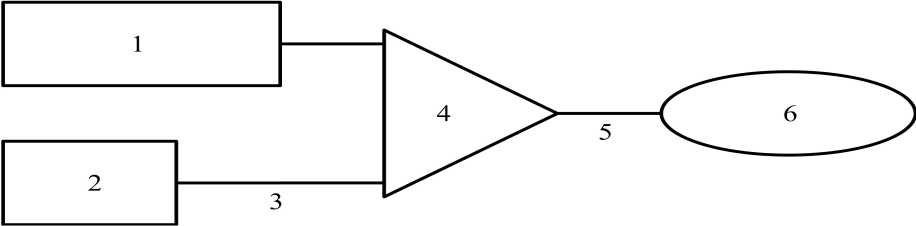
1 250 ml chlorid sodný 9 mg/ml (0,9 %)
2 Roztok Removabu pro i.p. infuzi
3 Perfuzní hadička (1 mm vnitřní průměr, délka 150 cm)
4 Infuzní ventil
5 Perfuzní vývod
6 Katétr
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Neovii Biotech GmbH Am Haag 6-7 82166 Graefelfing Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/09/512/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 20. dubna 2009
Datum posledního prodloužení registrace: 18. prosinec 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Trion Pharma GmbH Frankfurter Ring 193a DE-80807 Mnichov Německo
Název a adresa výrobce odpovědnéhoza propouštění šarží
Neovii Biotech GmbH Am Haag 6-7 82166 Graefelfing Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Removab 10 mikrogramů koncentrát pro infuzní roztok catumaxomabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka obsahuje 10 mikrogramů katumaxomabu v 0,1 ml roztoku, odpovídá 0,1 mg/ml.
3. SEZNAM POMOCNÝCH LÁTEK
Citronan sodný, monohydrát kyseliny citronové, polysorbát 80, voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro infuzní roztok.
1 předplněná injekční stříkačka. 1 sterilní jehla
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze intraperitoneální podání po naředění.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Neovii Biotech GmbH Am Haag 6-7 82166 Graefelfing Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/09/512/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Removab 10 mikrogramů koncentrát pro infuzní roztok catumaxomabum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Neovii Biotech GmbH
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1 předplněná injekční stříkačka.
Pouze intraperitoneální podání po naředění. Před použitím si přečtěte příbalovou informaci.
Uchovávejte v chladničce. Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Removab 10 mikrogramů koncentrát pro infuzní roztok catumaxomabum
Pouze intraperitoneální podání po naředění.
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,1 ml
6. JINÉ
Neovii Biotech GmbH
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Removab 50 mikrogramů koncentrát pro infuzní roztok catumaxomabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka obsahuje 50 mikrogramů katumaxomabu v 0,5 ml roztoku, odpovídá 0,1 mg/ml.
3. SEZNAM POMOCNÝCH LÁTEK
Citronan sodný, monohydrát kyseliny citronové, polysorbát 80, voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro infuzní roztok.
1 předplněná injekční stříkačka. 1 sterilní jehla
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze intraperitoneální podání po naředění.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Neovii Biotech GmbH Am Haag 6-7 82166 Graefelfing Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/09/512/002
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Removab 50 mikrogramů koncentrát pro infuzní roztok catumaxomabum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Neovii Biotech GmbH
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1 předplněná injekční stříkačka.
Pouze intraperitoneální podání po naředění. Před použitím si přečtěte příbalovou informaci.
Uchovávejte v chladničce. Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Removab 50 mikrogramů koncentrát pro infuzní roztok catumaxomabum
Pouze intraperitoneální podání po naředění.
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,5 ml
6. JINÉ
Neovii Biotech GmbH
TEXT VAROVÁNÍ, KTERÝ MÁ BÝT UVEDEN NA ODLUPOVACÍM ŠTÍTKU K PŘILEPENÍ NA 50ml INJEKČNÍ STŘÍKAČKU OBSAHUJÍCÍ NAŘEDĚNÝ INFUZNÍ ROZTOK PŘÍPRAVKU REMOVAB
(část vnějšího obalu)_
Naředěný přípravek Removab. Pouze pro intraperitoneální podání.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Removab 10 mikrogramů koncentrát pro infuzní roztok
Catumaxomabum
Přečtěte si, pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat,
protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Removab a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Removab používat
3. Jak se Removab používá
4. Možné nežádoucí účinky
5. Jak Removab uchovávat
6. Obsah balení a další informace
1. Co je Removab a k čemu se používá
Removab obsahuje léčivou látku katumaxomab,monoklonální protilátku. Rozpoznává protein na povrchu buněk nádoru a povolává imunitní buňky k jejich zničení.
Removab se používá k léčbě maligního ascitu, kdy není k dispozici žádná standardní terapie nebo již není dále použitelná. Maligní ascites je nahromadění tekutiny v břišním prostoru (peritoneální dutině) v důsledku určitých typů nádorových onemocnění.
2. Čemu musíte věnovat pozornost, než začnete Removab používat Nepoužívejte Removab
- jestliže jste alergický/á na katumaxomab nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6)
- j estliže j ste alergický/á na murinní bílkoviny (z myší či potkanů)
Upozornění a opatření
Před použitím Removabu se poraďte se svým lékařem nebo zdravotní sestrou.
Je důležité, abyste informovali svého lékaře, jestliže máte jakékoliv z následujících onemocnění.
- Neodstraněná tekutina v břišní dutině
- Studené ruce a nohy, točí se Vám hlava, máte potíže s močením, zvýšený srdeční tep a slabost (příznaky nízkého krevního objemu)
- Zvýšená tělesná hmotnost, trpíte slabostí, dechovou nedostatečností a zadržováním tekutin (příznaky nízkých hladin bílkovin v krvi).
- Závratě a mdloby (příznaky nízkého krevního tlaku)
- Problémy se srdcem a krevním oběhem.
- Poruchy funkce ledvin nebo jater
- Infekce
Předtím, než začnete Removab používat, ošetřující lékař zkontroluje:
- index tělesné hmotnosti (BMI), který závisí na hmotnosti a výšce
- Karnofského index, ukazatel celkového stavu výkonnosti
Abyste mohli toto léčivo používat, je zapotřebí mít BMI nad 17 (po drenáži tekutiny z ascitu) a Karnofského index nad 60.
Nežádoucí účinky související s infuzí a bolesti břicha jsou velmi časté (viz bod 4. Budete dostávat další léky na snižení horečky, bolesti nebo infekce způsobené přípravkem Removab (viz bod 3).
Děti a dospívající
Removab by neměl být podáván dětem a dospívajícím mladším 18 let.
Další léčivé přípravky a Removab
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval/a nebo které možná budete užívat.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo těhotenství plánujete, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat. Removab byste neměla používat, pokud jste těhotná, pokud to není zcela nezbytné.
Řízení dopravních prostředků a obsluha strojů
Pokud se však setkáte s nežádoucími účinky, jako jsou závratě nebo zimnice, během nebo po podání, neměli byste řídit ani obsluhovat stroje, dokud nezmizí.
3. Jak se Removab používá
Removab vám bude podáván za dohledu kvalifikovaného lékaře, který má zkušenosti s léčbou zhoubných nádorů. Po infuzi Removabu budete sledováni podle rozhodnutí lékaře.
Před zahájením léčby Removabem a během ní Vám mohou být podána jiná léčiva, aby snížila horečku nebo zánět způsobený Removabem.
Removab se podává jako 4 intraperitoneální infuze se zvyšující se dávkou (10, 20, 50 a 150 mikrogramů), oddělené intervalem nejméně 2 kalendářních dnů bez infuze (například dostanete infuzi v den 0, 3, 7, 10). Infuze se musí aplikovat konstantní rychlostí s dobou trvání alespoň 3 hodiny. Celková doba léčby nemá přesáhnout 20 dní.
Do břišní dutiny bude umístěn katétr (intraperitoneálně) po celou dobu léčby až do jednoho dne po poslední infuzi.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nejčastějšími závažnými nežádoucími účinky Removabu jsou nežádoucí účinky související s infuzí a nežádoucí účinky postihující trávicí soustavu (žaludek a střeva).
Nežádoucí účinky související s infuzí
Nežádoucí účinky související s infuzí se během infuze Removabu nebo po ní projeví pravděpodobně u víc než 1 pacienta z 10 (velmi časté). K nejčastějším nežádoucím účinkům souvisejícím s infuzí, které většinou bývají mírné až středně závažné, patří horečka, zimnice, nevolnost a zvracení.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to co nejdříve svému lékaři. Lékař poté případně zváží snížení rychlosti infuze Removabu nebo Vám k potlačení těchto účinků podá nějakou další léčbu.
Až u 4 pacientů ze 100 se může projevit komplex příznaků zahrnující velmi rychlý srdeční tep, horečku a dušnost. Tyto příznaky se projevují hlavně do 24 hodin po infuzi Removabu a mohou být i životu nebezpečné, dají se ale dobře léčit další léčbou.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, vyhledejte okamžitě svého lékaře,
protože některé z těchto nežádoucích účinků mohou vyžadovat okamžitou péči a léčbu.
Nežádoucí účinky související s trávicí soustavou
Zažívací potíže jako bolest břicha, nevolnost, zvracení nebo průjem mohou postihovat víc než 1 člověka z 10 (velmi časté), jsou ale většinou mírné až středně závažné a dají se dobře léčit další léčbou.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to co nejdříve svému lékaři.
Lékař poté případně zváží snížení rychlosti infuze Removabu nebo Vám k potlačení těchto účinků podá nějakou další léčbu.
Další závažné nežádoucí účinky
Velmi časté závažné nežádoucí účinky (mohou se projevit u víc než 1 člověka z 10):
- v únava
Časté závažné nežádoucí účinky (mohou se projevit až u 1 člověka z 10):
- ztráta chuti k jídlu
- dehydratace
- pokles počtu červených krvinek (chudokrevnost)
- pokles hladiny vápníku a sodíku v krvi
- velmi rychlý srdeční tep
- vysoký nebo nízký krevní tlak
- bolest v krajině břišní doprovázená obtížemi nebo blokádou při vypuzování stolice, zácpa
- dušnost
- zadržování vody v plicích, které způsobuje bolest na hrudi a dechovou nedostatečnost
- zánět žlučovodů
- zarudnutí kůže, vyrážka
- velmi rychlý srdeční tep, horečka, obtížné dýchání, pocit na omdlení nebo omámenost
- komplex reakcí v důsledku uvolnění mediátorů zánětu
- zhoršení celkového zdravotního stavu, celkový pocit nevolnosti a slabosti
- zadržování tekutin
- přecitlivělost
Méně časté závažné nežádoucí účinky (mohou se projevit až u 1 člověka ze 100):
- bulky pod kůží na zadní straně nohou, které se mohou změnit v boláky a zanechat jizvy
- zánět a bolest nebo pálení a bodání okolo katétru
- snížení počtu krevních destiček, problémy se srážlivostí krve
- krvácení do žaludku nebo střeva, které se projevuje zvracením krve nebo červenou či černou stolicí
- kožní reakce, závažná alergická kožní reakce (zánět kůže)
- křeče
- plicní obtíže včetně krevní sraženiny v plicích
- nízká hladina kyslíku v krvi
- závažné ledvinové obtíže
- extravazace (nechtěný únik podávaného léčivého přípravku z intraperitoneálního katétru do
okolní tkáně)
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to co nejdříve svému lékaři.
Některé z těchto nežádoucích účinků mohou vyžadovat léčbu.
Ostatní nežádoucí účinky
Časté nežádoucí účinky (mohou se projevit u víc než 1 člověka z 10):
- bolest
- úbytek nebo zvýšení počtu bílých krvinek
- snížené krevní hladiny draslíku
- snížené hladiny bílkovin
- zvýšené krevní hladiny bilirubinu
- závratě
- špatné trávení, problémy se žaludkem, pálení žáhy, nadýmání, plynatost,sucho v ústech
- příznaky podobné chřipce
- závratě nebo bolest hlavy
- bolest na hrudníku
- zvýšené pocení
- infekce
- zvýšené hladiny bílkovin v moči
- bolest v zádech, bolesti svalů a kloubů
- pocit úzkosti a obtíže se spánkem
- svědivá vyrážka nebo kopřivka, zarudnutí kůže okolo katétru
- zčervenání
- kašel
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Removab uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2°C-8°C). Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Připravený infuzní roztok je zapotřebí ihned použit.
6. Obsah balení a další informace Co Removab obsahuje
- Léčivou látkou je katumaxomab (10 mikrogramů v 0,1 ml, což odpovídá 0,1 mg/ml).
- Dalšími složkami jsou citronan sodný, monohydrát kyseliny citronové, polysorbát 80 a voda na injekci.
Jak Removab vypadá a co obsahuje toto balení
Removab je dodáván jako čirý a bezbarvý koncentrát pro infuzní roztok v předplněné injekční stříkačce s jehlou. Velikost balení: 1 předplněná injekční stříkačka a jedna jehla.
Držitel rozhodnutí o registraci a výrobce
Neovii Biotech GmbH Am Haag 6-7 82166 Graefelfing Německo
S žádostí o jakékoliv informace o tomto léčivu se prosím obracejte na držitele rozhodnutí o registraci. Tato příbalová informace byla naposledy revidována v MM/RRRR.
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Informace o ředění a podávání Removabu naleznete v bodě 6.6 v souhrnu údajů o přípravku (SmPC), a dále v každém balení Removabu 10 mikrogramů a Removabu 50 mikrogramů.
Příbalová informace: informace pro pacienta
Removab 50 mikrogramů koncentrát pro infuzní roztok
Catumaxomabum
Přečtěte si, pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat,
protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Removab a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Removab používat
3. Jak se Removab používá
4. Možné nežádoucí účinky
5. Jak Removab uchovávat
6. Obsah balení a další informace
1. Co je Removab a k čemu se používá
Removab obsahuje léčivou látku katumaxomab,monoklonální protilátku. Rozpoznává protein na povrchu buněk nádoru a povolává imunitní buňky k jejich zničení.
Removab se používá k léčbě maligního ascitu, kdy není k dispozici žádná standardní terapie nebo již není dále použitelná. Maligní ascites je nahromadění tekutiny v břišním prostoru (peritoneální dutině) v důsledku určitých typů nádorových onemocnění.
2. Čemu musíte věnovat pozornost, než začnete Removab používat Nepoužívejte Removab
- j estliže j ste alergický/á na katumaxomab nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6)
- jestliže jste alergický/á na murinní bílkoviny (z myší či potkanů)
Upozornění a opatření
Před použitím Removabu se poraďte se svým lékařem nebo zdravotní sestrou.
Je důležité, abyste informovali svého lékaře, jestliže máte jakékoliv z následujících onemocnění.
- Neodstraněná tekutina v břišní dutině
- Studené ruce a nohy, točí se Vám hlava, máte potíže s močením, zvýšený srdeční tep a slabost (příznaky nízkého krevního objemu)
- Zvýšená tělesná hmotnost, trpíte slabostí, dechovou nedostatečností a zadržováním tekutin (příznaky nízkých hladin bílkovin v krvi).
- Závratě a mdloby (příznaky nízkého krevního tlaku)
- Problémy se srdcem a krevním oběhem.
- Poruchy funkce ledvin nebo jater
- Infekce
Předtím, než začnete Removab používat, ošetřující lékař zkontroluje:
- index tělesné hmotnosti (BMI), který závisí na hmotnosti a výšce
- Karnofského index, ukazatel celkového stavu výkonnosti
Abyste mohli toto léčivo používat, je zapotřebí mít BMI nad 17 (po drenáži tekutiny z ascitu) a Karnofského index nad 60.
Nežádoucí účinky související s infuzí a bolesti břicha jsou velmi časté (viz bod 4. Budete dostávat další léky na snižení horečky, bolesti nebo infekce způsobené přípravkem Removab (viz bod 3).
Děti a dospívající
Removab by neměl být podáván dětem a dospívajícím mladším 18 let.
Další léčivé přípravky a Removab
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval/a nebo které možná budete užívat.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo těhotenství plánujete, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat. Removab byste neměla používat, pokud jste těhotná, pokud to není zcela nezbytné.
Řízení dopravních prostředků a obsluha strojů
Pokud se však setkáte s nežádoucími účinky, jako jsou závratě nebo zimnice, během nebo po podání, neměli byste řídit ani obsluhovat stroje, dokud nezmizí.
3. Jak se Removab používá
Removab vám bude podáván za dohledu kvalifikovaného lékaře, který má zkušenosti s léčbou zhoubných nádorů. Po infuzi Removabu budete sledováni podle rozhodnutí lékaře.
Před zahájením léčby Removabem a během ní Vám mohou být podána jiná léčiva, aby snížila horečku nebo zánět způsobený Removabem.
Removab se podává jako 4 intraperitoneální infuze se zvyšující se dávkou (10, 20, 50 a 150 mikrogramů), oddělené intervalem nejméně 2 kalendářních dnů bez infuze (například dostanete infuzi v den 0, 3, 7, 10). Infuze se musí aplikovat konstantní rychlostí s dobou trvání alespoň 3 hodiny. Celková doba léčby nemá přesáhnout 20 dní.
Do břišní dutiny bude umístěn katétr (intraperitoneálně) po celou dobu léčby až do jednoho dne po poslední infuzi.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nejčastějšími závažnými nežádoucími účinky Removabu jsou nežádoucí účinky související s infuzí a nežádoucí účinky postihující trávicí soustavu (žaludek a střeva).
Nežádoucí účinky související s infuzí
Nežádoucí účinky související s infuzí se během infuze Removabu nebo po ní projeví pravděpodobně u víc než 1 pacienta z 10 (velmi časté). K nejčastějším nežádoucím účinkům souvisejícím s infuzí, které většinou bývají mírné až středně závažné, patří horečka, zimnice, nevolnost a zvracení.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to co nejdříve svému lékaři. Lékař poté případně zváží snížení rychlosti infuze Removabu nebo Vám k potlačení těchto účinků podá nějakou další léčbu.
Až u 4 pacientů ze 100 se může projevit komplex příznaků zahrnující velmi rychlý srdeční tep, horečku a dušnost. Tyto příznaky se projevují hlavně do 24 hodin po infuzi Removabu a mohou být i životu nebezpečné, dají se ale dobře léčit další léčbou.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, vyhledejte okamžitě svého lékaře,
protože některé z těchto nežádoucích účinků mohou vyžadovat okamžitou péči a léčbu.
Nežádoucí účinky související s trávicí soustavou
Zažívací potíže jako bolest břicha, nevolnost, zvracení nebo průjem mohou postihovat víc než 1 člověka z 10 (velmi časté), jsou ale většinou mírné až středně závažné a dají se dobře léčit další léčbou.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to co nejdříve svému lékaři.
Lékař poté případně zváží snížení rychlosti infuze Removabu nebo Vám k potlačení těchto účinků podá nějakou další léčbu.
Další závažné nežádoucí účinky
Velmi časté závažné nežádoucí účinky (mohou se projevit u víc než 1 člověka z 10):
- v únava
Časté závažné nežádoucí účinky (mohou se projevit až u 1 člověka z 10):
- ztráta chuti k jídlu
- dehydratace
- pokles počtu červených krvinek (chudokrevnost)
- pokles hladiny vápníku a sodíku v krvi
- velmi rychlý srdeční tep
- vysoký nebo nízký krevní tlak
- bolest v krajině břišní doprovázená obtížemi nebo blokádou při vypuzování stolice, zácpa
- dušnost
- zadržování vody v plicích, které způsobuje bolest na hrudi a dechovou nedostatečnost
- zánět žlučovodů
- zarudnutí kůže, vyrážka
- velmi rychlý srdeční tep, horečka, obtížné dýchání, pocit na omdlení nebo omámenost
- komplex reakcí v důsledku uvolnění mediátorů zánětu
- zhoršení celkového zdravotního stavu, celkový pocit nevolnosti a slabosti
- zadržování tekutin
- přecitlivělost
Méně časté závažné nežádoucí účinky (mohou se projevit až u 1 člověka ze 100):
- bulky pod kůží na zadní straně nohou, které se mohou změnit v boláky a zanechat jizvy
- zánět a bolest nebo pálení a bodání okolo katétru
- snížení počtu krevních destiček, problémy se srážlivostí krve
- krvácení do žaludku nebo střeva, které se projevuje zvracením krve nebo červenou či černou stolicí
- kožní reakce, závažná alergická kožní reakce (zánět kůže)
- křeče
- plicní obtíže včetně krevní sraženiny v plicích
- nízká hladina kyslíku v krvi
- závažné ledvinové obtíže
- extravazace (nechtěný únik podávaného léčivého přípravku z intraperitoneálního katétru do
okolní tkáně)
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to co nejdříve svému lékaři.
Některé z těchto nežádoucích účinků mohou vyžadovat léčbu.
Ostatní nežádoucí účinky
Časté nežádoucí účinky (mohou se projevit u víc než 1 člověka z 10):
- bolest
- úbytek nebo zvýšení počtu bílých krvinek
- snížené krevní hladiny draslíku
- snížené hladiny bílkovin
- zvýšené krevní hladiny bilirubinu
- závratě
- špatné trávení, problémy se žaludkem, pálení žáhy, nadýmání, plynatost,sucho v ústech
- příznaky podobné chřipce
- závratě nebo bolest hlavy
- bolest na hrudníku
- zvýšené pocení
- infekce
- zvýšené hladiny bílkovin v moči
- bolest v zádech, bolesti svalů a kloubů
- pocit úzkosti a obtíže se spánkem
- svědivá vyrážka nebo kopřivka, zarudnutí kůže okolo katétru
- zčervenání
- kašel
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Removab uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2°C-8°C). Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Připravený infuzní roztok je zapotřebí ihned použit.
6. Obsah balení a další informace Co Removab obsahuje
- Léčivou látkou je katumaxomab (50 mikrogramů v 0,5 ml, což odpovídá 0,1 mg/ml).
- Dalšími složkami j sou citronan sodný, monohydrát kyseliny citronové, polysorbát 80 a voda na injekci.
Jak Removab vypadá a co obsahuje toto balení
Removab je dodáván jako čirý a bezbarvý koncentrát pro infuzní roztok v předplněné injekční stříkačce s jehlou. Velikost balení: 1 předplněná injekční stříkačka a jedna jehla.
Držitel rozhodnutí o registraci a výrobce
Neovii Biotech GmbH Am Haag 6-7 82166 Graefelfing Německo
S žádostí o jakékoliv informace o tomto léčivu se prosím obracejte na držitele rozhodnutí o registraci. Tato příbalová informace byla naposledy revidována v MM/RRRR.
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Informace o ředění a podávání Removabu naleznete v bodě 6.6 v souhrnu údajů o přípravku (SmPC), a dále v každém balení Removabu 10 mikrogramů a Removabu 50 mikrogramů.
PŘÍLOHA IV
ZDŮVODNĚNÍ NUTNOSTI POŽÁDAT O JEDNO DALŠÍ PRODLOUŽENÍ REGISTRACE
• Zdůvodnění nutnosti požádat o jedno další prodloužení registrace
Na základě údajů, které byly dodány po udělení původního rozhodnutí o registraci, výbor CHMP usoudil, že poměr přínosů a rizik přípravku Removab sice zůstává příznivý, avšak zároveň se domnívá, že bezpečnostní profil přípravku je třeba pečlivě sledovat z těchto důvodů:
• Nejisté poznatky o výskytu vzácných nežádoucích účinků, protože databáze údajů o bezpečnosti je ještě stále velmi omezená vzhledem k nízkému počtu pacientů léčených přípravkem Removab.
Proto na základě bezpečnostního profilu přípravku Removab, kdy je požadováno předkládání zpráv PSUR každý rok, dospěl výbor CHMP k závěru, že držitel rozhodnutí o registraci musí za pět let požádat o jedno další prodloužení registrace.
55
Na základě dávky natáhněte do 50ml injekční stříkačky (tabulka 7) příslušné množství injekčního roztoku chloridu sodného 9 mg/ml (0,9 %).
• Do 50ml injekční stříkačky nasajte nejméně další 3 ml vzduchového polštáře.
• Sejměte víčko hrotu z předplněné injekční stříkačky s Removabem, hrot je otočen nahoru.
• Přiloženou jehlu připojte k předplněné injekční stříkačce s Removabem. Pro každou injekční stříkačku se používá nová jehla.
• Protáhněte jehlu předplněné injekční stříkačky otvorem 50 ml injekční stříkačky tak, aby byla jehla ponořena v injekčním roztoku chloridu sodného 9 mg/ml (0,9 %) (Obrázek 2).
• Injikujte celý obsah injekční stříkačky (koncentrát Removabu plus vzduchový polštář)
z předplněné injekční stříkačky přímo do injekčního roztoku chloridu sodného 9 mg/ml (0,9 %).
• NESMÍTE stahovat píst injekční stříkačky dozadu, abyste předplněnou injekční stříkačku
Na základě dávky natáhněte do 50ml injekční stříkačky (tabulka 7) příslušné množství injekčního roztoku chloridu sodného 9 mg/ml (0,9 %).
• Do 50ml injekční stříkačky nasajte nejméně další 3 ml vzduchového polštáře.
• Sejměte víčko hrotu z předplněné injekční stříkačky s Removabem, hrot je otočen nahoru.
• Přiloženou jehlu připojte k předplněné injekční stříkačce s Removabem. Pro každou injekční stříkačku se používá nová jehla.
• Protáhněte jehlu předplněné injekční stříkačky otvorem 50 ml injekční stříkačky tak, aby byla jehla ponořena v injekčním roztoku chloridu sodného 9 mg/ml (0,9 %) (Obrázek 2).
• Injikujte celý obsah injekční stříkačky (koncentrát Removabu plus vzduchový polštář)
z předplněné injekční stříkačky přímo do injekčního roztoku chloridu sodného 9 mg/ml (0,9 %).
• NESMÍTE stahovat píst injekční stříkačky dozadu, abyste předplněnou injekční stříkačku