Relvar Ellipta 184 Mikrogramů/22 Mikrogramů
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Relvar Ellipta 92 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna inhalace poskytuje dávku (dávka, která vychází z náustku) fluticasoni furoas 92 mikrogramů a vilanterolum 22 mikrogramů (ve formě trifenatas). To odpovídá dávkované dávce fluticasoni furoas 100 mikrogramů a vilanterolum 25 mikrogramů (ve formě trifenatas).
Pomocná látka se známým účinkem:
Jedna dávka obsahuje přibližně 25 mg laktosy (ve formě monohydrátu). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Dávkovaný prášek k inhalaci (prášek k inhalaci).
Bílý prášek ve světle šedém inhalátoru se žlutým krytem náustku a počítadlem dávek.
|
4. |
KLINICKÉ ÚDAJE |
|
4.1 |
Terapeutické indikace |
Přípravek Relvar Ellipta je indikován k pravidelné léčbě astmatu u dospělých a dospívajících od 12 let, kde je vhodné použití kombinovaného léčivého přípravku (agonista beta2-receptorů s dlouhodobým účinkem a inhalační kortikosteroid):
• pacienti, kteří nej sou dostatečně kontrolováni inhalačními kortikosteroidy a kteří jako „léčbu v případě potřeby“ inhalují krátkodobě působící agonisty beta2-receptorů.
CHOPN (chronická obstrukční plicní nemoc)
Přípravek Relvar Ellipta je indikován k symptomatické léčbě dospělých pacientů s CHOPN s FEVi < 70 % náležitých normálních hodnot (po podání bronchodilatancia) s anamnézou exacerbací navzdory pravidelné bronchodilatační léčbě.
4.2 Dávkování a způsob podání
Dávkování
Dospělí a dospívající ve věku 12 let a starší
Jedna inhalace přípravku Relvar Ellipta 92/22 mikrogramů jednou denně.
Pacienti obvykle zaznamenají zlepšení plicních funkcí v průběhu 15 minut po inhalaci přípravku Relvar Ellipta. Pacienti by však měli být informováni, že k udržení kontroly příznaků astmatu je nezbytné pravidelné denní používání, a že léčba by měla pokračovat i v případě, že nemají žádné příznaky.
Pokud se příznaky objeví v období mezi dávkami, je třeba k okamžité úlevě použít inhalačního agonistu-beta2 receptorů s krátkodobým účinkem.
Úvodní dávku přípravku Relvar Ellipta 92/22 mikrogramů je třeba zvážit u dospělých a dospívajících ve věku 12 let a starších, kteří potřebují nízkou až středně vysokou dávku inhalačního kortikosteroidu v kombinaci s dlouhodobě působícím agonistou beta2-receptorů. Pokud není onemocnění při podávání přípravku Relvar Ellipta 92/22 mikrogramů dostatečně kontrolováno, lze dávku zvýšit na 184/22 mikrogramů, což může vést ke zlepšení kontroly příznaků astmatu.
Pacienti by měli být pravidelně kontrolováni lékařem, aby síla flutikason-furoátu/vilanterolu, kterou používají, zůstávala optimální a byla měněna pouze na základě lékařského doporučení. Dávku je třeba titrovat na nejnižší možnou dávku, která bude účinná při udržování kontroly příznaků onemocnění.
Přípravek Relvar Ellipta 184/22 mikrogramů je třeba zvážit u dospělých a dospívajících ve věku 12 let a starších, kteří vyžadují vyšší dávky inhalačního kortikosteroidu v kombinaci s dlouhodobě působícím agonistou beta2-receptorů.
Pacientům s astmatem má být podána síla přípravku Relvar Ellipta obsahující dávku flutikason-furoátu (FF) odpovídající závažnosti jejich onemocnění. Předepisující lékař by si měl být vědom, že u pacientů s astmatem má 100 mikrogramů flutikason-furoátu (FF) podávaných jednou denně podobný účinek jako flutikason-propionát (FP) v dávce 250 mikrogramů podávaný dvakrát denně. FF v dávce 200 mikrogramů podávaný jednou denně pak má podobný účinek jako FP v dávce 500 mikrogramů podávaný dvakrát denně.
Děti mladší 12 let
Bezpečnost a účinnost přípravku Relvar Ellipta u dětí mladších 12 let nebyla v indikaci astmatu stanovena.
K dispozici nejsou žádné údaje.
CHOPN
Dospělí ve věku 18 let a starší
Jedna inhalace přípravku Relvar Ellipta 92/22 mikrogramů jednou denně.
Přípravek Relvar Ellipta 184/22 mikrogramů není indikován k léčbě pacientů s CHOPN. Není zde žádný další prospěch z dávky 184/22 mikrogramů ve srovnání s dávkou 92/22 mikrogramů a existuje potenciální možnost zvýšení rizika pneumonie a systémových nežádoucích účinků spojených s kortikosteroidy (viz body 4.4 a 4.8).
Pacienti zaznamenají zlepšení plicních funkcí obvykle v průběhu 16 - 17 minut po inhalaci přípravku Relvar Ellipta.
Pediatrická populace
U pediatrické populace neexistují žádné relevantní důvody pro užívání přípravku Relvar Ellipta v indikaci CHOPN.
Zvláštní populace
Starší pacienti (> 65 let)
U této populace není nutná úprava dávkování (viz bod 5.2).
Porucha funkce ledvin
U této populace není nutná úprava dávkování (viz bod 5.2).
Porucha funkce jater
Studie u subjektů s lehkou, středně závažnou a závažnou poruchou funkce jater prokázaly zvýšení systémové expozice flutikason-furoátu (Cmax i AUC) (viz bod 5.2).
Opatrnost je třeba při léčbě pacientů s poruchou funkce jater, u kterých může být vyšší riziko systémových nežádoucích účinků souvisejících s podáváním kortikosteroidů.
U pacientů se středně závažnou nebo závažnou poruchou funkce jater je maximální dávka 92/22 mikrogramů (viz bod 4.4).
Způsob podání
Přípravek Relvar Ellipta je určen pouze k inhalačnímu podání.
Přípravek je třeba podávat každý den ve stejnou dobu.
Konečné rozhodnutí o tom, zda se přípravek bude podávat ráno nebo večer, je třeba ponechat na uvážení lékaře.
Pokud dojde k vynechání dávky, následující dávku je třeba použít následující den v obvyklý čas.
Pokud je přípravek uchováván v chladničce, je třeba inhalátor nechat před použitím zahřát na pokojovou teplotu alespoň jednu hodinu před podáním.
Po inhalaci by si pacient měl vypláchnout ústa vodou, aniž by vodu polykal.
Při prvním použití inhalátoru není nutné kontrolovat, zda účinkuje správně, a připravovat ho zvláštním způsobem k použití. Podrobný návod k použití bude následovat.
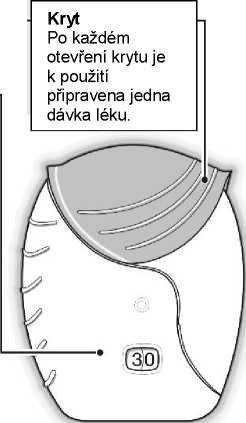
Inhalátor Ellipta je uložený v ochranné vaničce obsahující sáček s vysoušedlem, který snižuje vlhkost. Po otevření je třeba sáček s vysoušedlem vyhodit.
Pacient má být poučen, aby vaničku neotevíral dříve, než bude připraven k inhalaci dávky.
Inhalátor je po vyjmutí z vaničky v „uzavřené“ pozici. Označení “Spotřebujte do“ vyjadřuje datum, které by mělo být zapsáno do štítku inhalátoru. Datum „Spotřebujte do“ je 6 týdnů od data otevření vaničky. Po tomto datu se již nemá inhalátor dále používat. Vanička může být znehodnocena po prvním otevření.
Podrobný návod k použití pro 30dávkový inhalátor Ellipta uvedený níže lze rovněž použít pro 14dávkový inhalátor Ellipta.
Návod k použití
1. Před použitím si přečtěte následující instrukce
Pokud se kryt inhalátoru otevře a zavře bez toho, že by došlo k inhalaci léku, dojde ke ztrátě dávky. Ztracená dávka zůstane bezpečně uzavřená v inhalátoru, ale nebude již dostupná k inhalaci.
Při jedné inhalaci není možné náhodně použít dávku navíc ani dvojitou dávku.
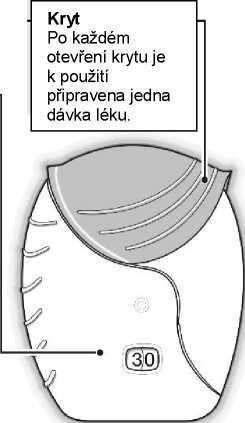
Počítadlo dávek
Počítadlo ukazuje, kolik dávek léku v inhalátoru ještě zbývá.
Před prvním použitím inhalátoru ukazuje počítadlo přesně 30 dávek.
Při každém otevření krytu inhalátoru se 1 dávka odečte.
Pokud v inhalátoru zbývá méně než 10 dávek, polovina počítadla ukazuje červeně.
Po použití poslední dávky ukazuje polovina počítadla červeně a zobrazuje
0. Inhalátor je nyní prázdný.
Pokud poté otevřete kryt inhalátoru, počítadlo změní barvu z původní z poloviny červené na zcela červenou.
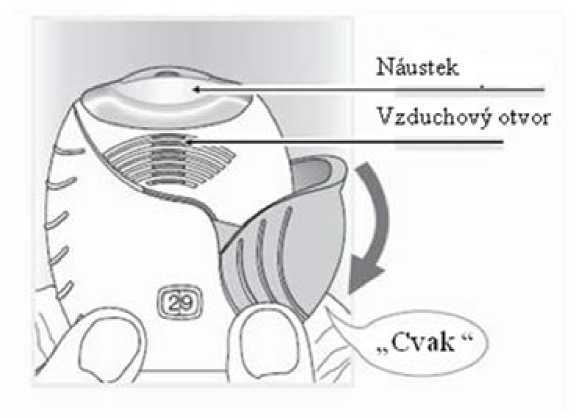
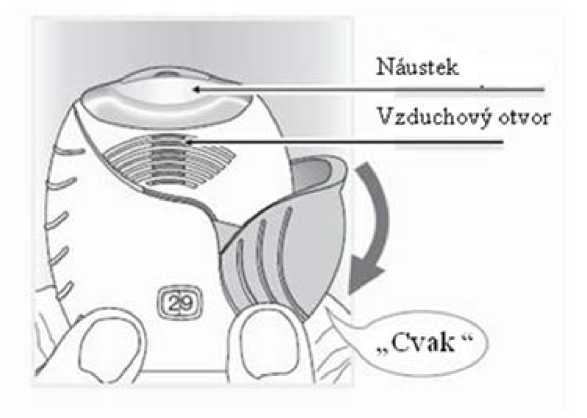
2. Jak připravit dávku
Pokud jste připraven(a) k použití dávky, otevřete kryt inhalátoru. Inhalátorem netřeste.
Stahujte víčko dolů, dokud neuslyšíte „cvaknutí“.
Lék je nyní připraven k inhalaci. Počítadlo dávek pro potvrzení odečetlo 1 dávku.
Pokud počítadlo neodečte dávku v okamžiku, kdy uslyšíte „cvaknutí,“ inhalátor neumožní inhalaci léku. Vezměte jej zpět k lékárníkovi, aby Vám poradil.
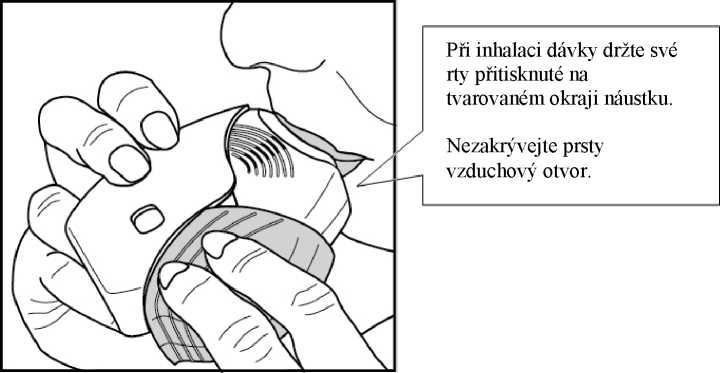
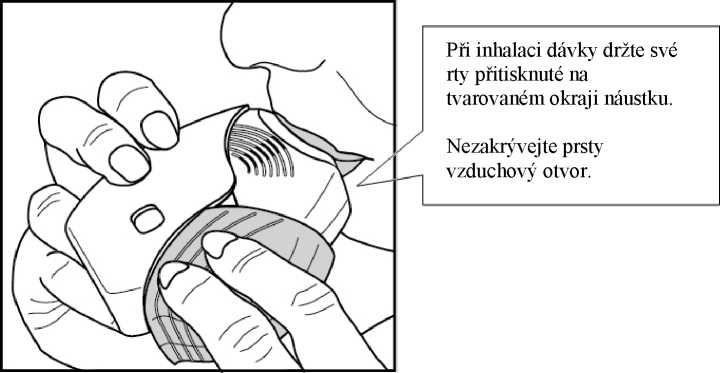
3. Jak se lék inhaluje
Držte inhalátor dále od úst, a co nejvíce vydechněte, jak je Vám pohodlné. Nevydechujte do inhalátoru.
Vložte náustek mezi rty a pevně jej svými rty stiskněte.
Neblokujte vzduchové otvory prsty.
Jednou se dlouze, rovnoměrně a zhluboka nadechněte. Zadržte dech po co nejdelší dobu (alespoň 3 - 4 sekundy).
• Vyjměte inhalátor z úst.
• Pomalu a lehce vydechněte.
Lék by neměl mít žádnou chuť ani by neměl být cítit, a to ani v případě, že se inhalátor použije správně.
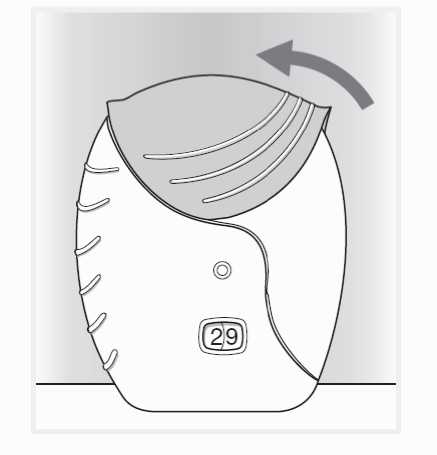
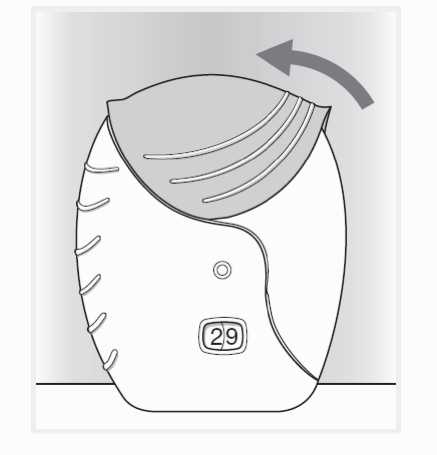
4. Uzavření inhalátoru a vypláchnutí úst
Pokud chcete náustek očistit, otřete jej před uzavřením suchou tkaninou.
Vysuňte kryt zpět nahoru co nejvíce, až je náustek zakrytý.
Po použití inhalátoru si vypláchněte ústa vodou.
To sníží pravděpodobnost, že dojde k rozvoji nežádoucích účinků v podobě bolesti v ústech nebo v krku.
4.3 Kontraindikace
Hypersenzitivita na léčivé látky nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Zhoršení onemocnění
Kombinace flutikason-furoát/vilanterol se nesmí používat k léčbě akutních příznaků astmatu ani akutní exacerbace CHOPN, při kterých je nutné podání krátkodobě působícího bronchodilatancia. Častější používání krátkodobě působících bronchodilatancií k úlevě od příznaků ukazuje na zhoršení kontroly onemocnění a pacient by měl být vyšetřen lékařem.
Pacient by neměl ukončovat terapii kombinací flutikason-furoát/vilanterol u astmatu ani CHOPN bez lékařského dohledu, protože příznaky se mohou po přerušení léčby vrátit.
V průběhu léčby kombinací flutikason-furoát/vilanterol se mohou objevit nežádoucí účinky související s astmatem a exacerbace onemocnění. Pacienti by měli být vyzváni, aby pokračovali v léčbě, ale aby vyhledali lékařskou pomoc, pokud příznaky astmatu zůstanou nekontrolované nebo pokud se po zahájení léčby přípravkem Relvar Ellipta zhorší.
Paradoxní bronchospasmus
Paradoxní bronchospasmus se může projevit okamžitým zhoršením sípotu po podání dávky. Bronchospasmus je třeba léčit okamžitě podáním krátkodobě působícího inhalačního bronchodilatancia. Léčbu přípravkem Relvar Ellipta je třeba okamžitě ukončit, pacienta vyšetřit a v případě potřeby zahájit alternativní léčbu.
Kardiovaskulární účinky
Při podávání sympatomimetických léčivých přípravků, včetně přípravku Relvar Ellipta, se mohou objevit kardiovaskulární nežádoucí účinky, jako srdeční arytmie, např. supraventrikulární tachykardie a extrasystoly. Proto je třeba kombinaci flutikason-furoát/vilanterol používat s opatrností u pacientů se závažným kardiovaskulárním onemocněním nebo poruchami srdečního rytmu, tyreotoxikózou, nekorigovanou hypokalémií nebo u pacientů s predispozicí k nízkým hladinám draslíku v séru.
Pacienti s poruchou funkce jater
U pacientů se středně závažnou až závažnou poruchou funkce jater je třeba používat dávku 92/22 mikrogramů a pacienty pečlivě sledovat s ohledem na možné systémové nežádoucí účinky spojené s podáváním kortikosteroidů (viz bod 5.2).
Systémové účinky kortikosteroidů
Systémové účinky se mohou objevit u jakéhokoli inhalačního kortikosteroidu, zejména při podávání vysokých dávek předepisovaných po dlouhou dobu. Tyto účinky jsou mnohem méně pravděpodobné než u perorálních kortikosteroidů. Možné systémové účinky zahrnují Cushingův syndrom, cushingoidní rysy, adrenální supresi, snížení minerální kostní denzity, růstovou retardaci u dětí a dospívajících, kataraktu a glaukom a vzácněji různé psychologické nebo behaviorální účinky, včetně psychomotorické hyperaktivity, poruch spánku, úzkosti, deprese nebo agresivity (zejména u dětí).
Kombinaci flutikason-furoát/vilanterol je třeba podávat s opatrností u pacientů s plicní tuberkulózou nebo u pacientů s chronickou nebo neléčenou infekcí.
Hyperglykémie
U diabetiků byly zaznamenány případy zvýšení hladiny glukózy v krvi, což je třeba zvážit při předepisování přípravku pacientům s diabetes mellitus v anamnéze.
Pneumonie u pacientů s CHOPN
U pacientů s CHOPN, kterým byly podávány inhalační glukokortikoidy, byl pozorován vyšší výskyt pneumonie, včetně pneumonie vyžadující hospitalizaci. Existují určité důkazy o tom, že zvýšené riziko pneumonie souvisí se zvyšováním dávky steroidu, avšak tuto závislost se nepodařilo definitivně prokázat ve všech studiích.
Neexistují jednoznačné klinické důkazy o rozdílech mezi léčivými přípravky ze skupiny inhalačních glukokortikoidů ohledně výše rizika pneumonie.
Lékaři mají sledovat možný vývoj pneumonie u pacientů s CHOPN, neboť klinické známky těchto infekcí se mohou překrývat se symptomy, které doprovázejí exacerbaci CHOPN.
Rizikovými faktory pro pneumonii u pacientů s CHOPN jsou současné kouření, vyšší věk, nízký index tělesné hmotnosti (BMI) a těžká CHOPN.
Přípravek Relvar Ellipta 184/22 mikrogramů není indikován k léčbě pacientů s CHOPN. Nebyl zaznamenán dodatečný prospěch z podávání dávky 184/22 mikrogramů ve srovnání s dávkou 92/22 mikrogramů a naopak bylo zaznamenáno potenciální zvýšení rizika výskytu systémových nežádoucích účinků souvisejících s podáváním kortikosteroidů (viz bod 4.8).
Pneumonie u pacientů s astmatem
Incidence pneumonie u pacientů s astmatem byla častá při vyšší dávce. Incidence pneumonie u pacientů s astmatem užívajících kombinaci flutikason-furoát/vilanterol 184/22 mikrogramů byla numericky vyšší ve srovnání s incidencí zaznamenanou u pacientů léčených kombinací flutikason-furoát/vilanterol v dávce 92/22 mikrogramů nebo placebem (viz bod 4.8). Žádné rizikové faktory nebyly zaznamenány.
Pomocné látky
Pacienti se vzácnými dědičnými poruchami galaktózové intolerance, vrozené deficience laktázy nebo malabsorpce glukózy-galaktózy by tento přípravek neměli používat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Klinicky významné lékové interakce zprostředkované kombinací flutikason-furoát/vilanterol při klinických dávkách jsou považovány za nepravděpodobné z důvodu nízkých plazmatických koncentrací dosahovaných po inhalaci.
Interakce s beta-blokátory
Blokátory beta2-adrenergních receptorů mohou účinky agonistů beta2-adrenergních receptorů oslabit nebo antagonizovat. Současného podávání neselektivních i selektivních blokátorů beta2-adrenergních receptorů je třeba se vyvarovat, pokud pro jejich použití není závažný důvod.
Interakce s inhibitory CYP3A4
Obě látky, flutikason-furoát i vilanterol, jsou rychle odstraňovány při prvním průchodu játry výraznou metabolizací, která je zprostředkována jaterním enzymem CYP3A4.
Při společném podávání se silnými inhibitory CYP3A4 (např. ketokonazol, ritonavir) je třeba opatrnosti, protože je zde riziko zvýšení systémové expozice flutikason-furoátu i vilanterolu;
současného používání je třeba se vyvarovat. U zdravých subjektů byla provedena studie lékových interakcí s CYP3A4 po opakované dávce s kombinací flutikason-furoát/vilanterol (184/22 mikrogramů) a silným inhibitorem CYP3A4 ketokonazolem (400 mg). Společné podání zvýšilo průměrné AUC(0-24) flutikason-furoátu o 36 % a Cmax flutikason-furoátu o 33 %. Zvýšení expozice flutikason-furoátu bylo spojeno s 27 % snížením váženého průměru sérových hladin kortizolu za 24 hodin. Společné podávání zvýšilo průměrné AUC(0-t) vilanterolu o 65 % a Cmax vilanterolu o 22 %. Zvýšení expozice vilanterolu nebylo spojeno se zvýšením systémových účinků agonistů beta2-receptorů na srdeční frekvenci, hladinu draslíku v krvi ani QTcF interval.
Interakce s inhibitory glykoproteinu P
Flutikason-furoát i vilanterol jsou substráty glykoproteinu P (P-gp). Klinická farmakologická studie u zdravých subjektů, kterým byl společně podáván vilanterol a verapamil (silný inhibitor P-gp a středně silný inhibitor CYP3A4), neprokázala výrazný účinek na farmakokinetiku vilanterolu. Klinické farmakologické studie se specifickým inhibitorem P-gp a flutikason-furoátem nebyly provedeny.
Sympatomimetické léčivé přípravky
Společné podávání dalších sympatomimetik (samotných nebo jako součásti kombinované léčby) může zvyšovat riziko nežádoucích účinků kombinace flutikason-furoát/vilanterol. Přípravek Relvar Ellipta se nesmí používat společně s jinými dlouhodobě působícími agonisty beta2-adrenergních receptorů ani léčivými přípravky obsahujícími dlouhodobě působící agonisty beta2-adrenergních receptorů.
Pediatrická populace.
Studie lékových interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Studie se zvířaty prokázaly reprodukční toxicitu při expozicích, které nejsou klinicky relevantní (viz bod 5.3). K dispozici nejsou žádné nebo pouze omezené údaje týkající se používání flutikason-furoátu a vilanterol-trifenatátu u těhotných žen.
Podávání flutikason-furoátu/vilanterolu těhotným ženám je třeba zvážit, pouze pokud prospěch z léčby pro matku převáží možná rizika pro plod.
Kojení
Údaje týkající se vylučování flutikason-furoátu nebo vilanterol-trifenatátu a/nebo jejich metabolitů do lidského mateřského mléka jsou nedostatečné. Jiné kortikosteroidy a agonisté beta2-receptorů jsou však v mateřském mléce člověka detekovány (viz bod 5.3). Riziko pro kojené novorozence/kojence nelze vyloučit.
Při rozhodování, zda přerušit kojení nebo přerušit léčbu kombinací flutikason-furoát/vilanterol, je třeba vzít v úvahu prospěch z kojení pro dítě a prospěch z léčby pro ženu.
Fertilita
K dispozici nejsou žádné údaje týkající se fertility u člověka. Studie na zvířatech neprokázaly žádné účinky flutikason-furoátu/vilanterol-trifenatátu na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Flutikason-furoát ani vilanterol nemají žádný nebo mají pouze zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Ke stanovení četností nežádoucích účinků souvisejících s podáváním kombinace flutikason-furoát/vilanterol byly použity údaje z rozsáhlých klinických studií s astmatem a CHOPN. V klinickém vývojovém programu astmatu bylo v jednotném hodnocení nežádoucích účinků zahrnuto celkem 7 034 pacientů. V klinickém vývojovém programu CHOPN bylo v jednotném hodnocení nežádoucích účinků zahrnuto celkem 6 237 pacientů.
Nejčastěji hlášené nežádoucí účinky související s podáváním flutikason-furoátu a vilanterolu byly bolest hlavy a nazofaryngitida. S výjimkou pneumonie a zlomenin byl bezpečnostní profil přípravku podobný u pacientů s astmatem i CHOPN. V průběhu klinických studií byly pneumonie a zlomeniny častěji pozorovány u pacientů s CHOPN.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů a frekvence. Ke klasifikaci četností byla použita následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Třída orgánových systémů |
Nežádoucí účinky |
Četnost |
|
Infekce a infestace |
Pneumonie* Infekce horních cest dýchacích Bronchitida Chřipka Kandidóza úst a hrdla |
Časté |
|
Poruchy imunitního systému |
Reakce přecitlivělosti, včetně anafylaxe, angioedému, vyrážky a kopřivky. |
Vzácné |
|
Poruchy nervového systému |
Velmi časté | |
|
Vzácné | ||
|
Psychiatrické poruchy |
Úzkost |
Vzácné |
|
Srdeční poruchy |
Extrasystoly |
Méně časté |
|
Palpitace |
Vzácné | |
|
Vzácné | ||
|
Respirační, hrudní |
Nazofaryngitida |
Velmi časté |
|
a mediastinální poruchy |
Orofaryngeální bolest Sinusitida Faryngitida Dysfonie |
Časté |
|
Gastrointestinální poruchy |
Časté | |
|
Poruchy svalové a kosterní |
Artralgie |
Časté |
|
soustavy a pojivové tkáně |
Bolest zad Zlomeniny** Svalové křeče | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
*, ** viz níže „Popis vybraných nežádoucích účinků“
Popis vybraných nežádoucích účinků
V integrované analýze dvou zdvojených jeden rok trvajících studií, do kterých bylo zahrnuto celkem 3 255 pacientů s CHOPN s exacerbací, byl počet výskytu pneumonie na 1 000 pacientoroků 97,9 ve skupině s FF/VI 184/22; 85,7 ve skupině s FF/VI 92/22 a 42,3 ve skupině VI 22. U těžké formy pneumonie odpovídající počet případů na 1 000 pacientoroků byl 33,6; 35,5 a 7,6, zatímco závažná forma pneumonie odpovídající případům na 1 000 pacientoroků byla 35,1 ve skupině FF/VI 184/22, 42,9 ve skupině FF/VI 92/22 a 12,1 ve skupině VI 22. Závěrem, případů pneumonie s fatálním průběhem s přihlédnutím k expozici bylo 8,8 pro FF/VI 184/22 ve srovnání s 1,5 pro FF/VI 92/22 a 0 pro VI 22.
Ve společné analýze 11 studií s astmatem (7 034 pacientů) byla incidence pneumonie na 1 000 pacientoroků 18,4 ve skupině FF/VI 184/22 proti 9,6 ve skupině s FF/VI 92/22 a 8,0 ve skupině s placebem.
Zlomeniny
Ve dvou opakovaných 12měsíčních studiích, do kterých bylo zahrnuto celkem 3 255 pacientů s CHOPN, byla celková incidence kostních zlomenin nízká ve všech léčebných skupinách, s vyšší incidencí ve všech skupinách s přípravkem Relvar Ellipta (2 %) ve srovnání se skupinou s vilanterolem v dávce 22 mikrogramů (< 1 %). Ačkoli ve skupinách s přípravkem Relvar Ellipta bylo zaznamenáno více zlomenin ve srovnání se skupinou se samotným vilanterolem v dávce 22 mikrogramů, zlomeniny typicky související s používáním kortikosteroidů (např. kompresivní zlomeniny obratlů/zlomeniny obratlů v thorakolumbální oblasti, zlomeniny proximálního femuru a acetabula) se vyskytovaly ve skupině s přípravkem Relvar Ellipta i vilanterolem s četností < 1 %.
Ve společné analýze 11 studií s astmatem (7 034 pacientů) byla incidence zlomenin < 1 % a zlomeniny obvykle souvisely s traumatem.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Známky a příznaky
Předávkování kombinací flutikason-furoát/vilanterol může vést k subjektivním a objektivním příznakům způsobeným účinky jednotlivých složek, zahrnující nežádoucí účinky pozorované při předávkování jinými agonisty beta2-receptorů a konzistentní se známými nežádoucími účinky třídy inhalačních kortikosteroidů (viz bod 4.4).
Léčba
Pro předávkování kombinací flutikason-furoát/vilanterol není k dispozici žádná specifická léčba. Pokud dojde k předávkování, je třeba podle potřeby zahájit podpůrnou léčbu s odpovídajícím sledováním pacienta.
Kardioselektivní beta-blokáda se má zvážit pouze u těžkého předávkování vilanterolem, pokud je klinicky závažné a pacient nereaguje na podpůrná opatření. Kardioselektivní beta-blokátory je třeba používat s opatrností u pacientů s anamnézou bronchospasmu.
Další léčba by měla odpovídat klinickým příznakům nebo doporučením národního toxikologického centra, jsou-li k dispozici.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii onemocnění spojených s obstrukcí dýchacích cest, Sympatomimetika a jiná léčiva onemocnění spojených s obstrukcí dýchacích cest, ATC kód: R03AK10
Mechanismus účinku
Flutikason-furoát a vilanterol představují dvě třídy léků (syntetický kortikosteroid a selektivní agonista beta2-receptorů s dlouhodobým účinkem).
Farmakodynamické účinky
Flutikason-furoát
Flutikason-furoát je syntetický trifluorovaný kortikosteroid se silnou protizánětlivou aktivitou. Přesný mechanismus, jakým flutikason-furoát ovlivňuje příznaky astmatu a CHOPN, není znám. Bylo prokázáno, že kortikosteroidy mají široké spektrum účinku na mnoho různých druhů buněk (např. eozinofily, makrofágy, lymfocyty) a mediátorů (např. cytokiny a chemokiny zahrnuté do procesů zánětu).
Vilanterol-trifenatát
Vilanterol-trifenatát je selektivní agonista beta2-adrenergních receptorů s dlouhodobým účinkem (LABA).
Farmakologické účinky agonistů beta2-adrenergních receptorů, mezi něž patří i vilanterol-trifenatát, jsou alespoň zčásti způsobené stimulací intracelulární adenylátcyklázy, enzymu, který katalyzuje přeměnu adenosin-trifosfátu (ATP) na cyklický 3‘,5‘-adenosinmonofosfát (cAMP). Zvýšení hladin cAMP vede k relaxaci bronchiální hladké svaloviny a inhibici uvolňování mediátorů z buněk okamžité přecitlivělosti, zejména z mastocytů.
Mezi kortikosteroidy a LABA dochází k molekulárním interakcím, zatímco steroidy aktivují gen pro beta2-receptor, zvyšují počet receptorů a jejich senzitivitu, LABA připravuje receptor pro glukokortikoidy k aktivaci závislé na steroidech a zrychluje přesun steroidů do buněčného jádra. Tyto synergické interakce se odrážejí ve zvýšení protizánětlivé aktivity, která byla prokázána in vitro i in vivo u širokého spektra zánětlivých buněk, které se podílejí na patofyziologii astmatu i CHOPN.
Studie hodnotící biopsii dýchacích cest po podání flutikason-furoátu a vilanterolu rovněž prokázaly synergický účinek kortikosteroidů a LABA, který se objevuje při klinických dávkách léků u pacientů s CHOPN.
Klinická účinnost a bezpečnost
Tři randomizované, dvojitě zaslepené studie fáze III (HZA106827, HZA106829 a HZA106837) různé délky trvání hodnotily bezpečnost a účinnost kombinace flutikason-furoát/vilanterol u dospělých a dospívajících pacientů s perzistentním astmatem. Všechny subjekty používaly IKS (inhalační kortikosteroid) s nebo bez LABA po dobu alespoň 12 týdnů před první kontrolou. Ve studii HZA106837 prodělali všichni pacienti v průběhu jednoho roku před první kontrolou alespoň jednu exacerbaci, která vyžadovala léčbu perorálními kortikosteroidy. Studie HZA106827 trvala 12 týdnů a hodnotila účinnost kombinace flutikason-furoát/vilanterol v dávce 92/22 mikrogramů (n = 201) a FF (flutikason-furoát) v dávce 92 mikrogramů (n = 205) ve srovnání s placebem (n = 203); léčba byla u všech skupin podávána jednou denně. Studie HZA106829 trvala 24 týdnů a hodnotila účinnost kombinace flutikason-furoát/vilanterol v dávce 184/22 mikrogramů (n = 197) a FF v dávce 184 mikrogramů (n = 194), oba podávané jednou denně, ve srovnání s flutikason-propionátem (FP) v dávce 500 mikrogramů dvakrát denně (n = 195).
Ve studiích HZA106827/HZA106829 byla koprimárním cílovým parametrem účinnosti změna od výchozích hodnot při klinických kontrolách na konci léčby hodnocená u všech subjektů pomocí tzv. „trough“ čili prebronchodilatační hodnoty FEV1 (před aplikací bronchodilatancia a před podáním dávky) a vážený průměr série měření FEV1 v průběhu 0 - 24 hodin po podání dávky vypočtený u podskupiny subjektů na konci období léčby. Sekundárním cílovým parametrem v průběhu léčby bylo procento změny 24hodinových období bez nutnosti podání záchranné léčby oproti počátku léčby. Výsledky pro primární a klíčové sekundární cílové parametry těchto studií jsou shrnuty v tabulce 1.
Tabulka 1 - Výsledky primárních a klíčových sekundárních cílových parametrů ve studiích HZA106827 a HZA106829
|
Číslo studie |
HZA106829 |
HZA] |
106827 | |
|
Léčebná dávka FF/VI*(mikrogramy) |
FF/VI 184/22 jednou denně vs. FF 184 jednou denně |
FF/VI 184/22 jednou denně vs. FP 500 dvakrát denně |
FF/VI 92/22 jednou denně vs. FF 92 jednou denně |
FF/VI/92/22 jednou denně vs. placebo jednou denně |
|
Změna od výchozích hor Forward) |
not v trough FEV1 při LOCF (Last Observation Carried | |||
|
Rozdíl v léčbě p-hodnota (95% CI) |
193 ml p < 0,001 (108, 277) |
210 ml p < 0,001 (127, 294) |
36 ml p = 0,405 (-48, 120) |
172 ml p < 0,001 (87, 258) |
|
Vážený průměr série měření FEV1 v průběhu 0 - 24 hodin po podání dávky | ||||
|
Rozdíl v léčbě p-hodnota (95% CI) |
136 ml p = 0,048 (1, 270) |
206 ml p = 0,003 (73, 339) |
116 ml p = 0,06 (-5, 236) |
302 ml p < 0,001 (178, 426) |
|
Změna od výchozích hor záchranné léčby |
not v procentu 24hodinových období bez nutnosti podání | |||
|
Rozdíl v léčbě p-hodnota (95% CI) |
11,7 % p < 0,001 (4,9; 18,4) |
6,3 % p = 0,067 (-0,4; 13,1) |
10,6 % p < 0,001 (4,3; 16,8) |
19,3 % p < 0,001 (13,0; 25,6) |
|
Změna od výchozích hor |
not v procentu 24hodinového období bez příznaků | |||
|
Rozdíl v léčbě p-hodnota (95% CI) |
8,4 % p = 0,010 (2,0; 14,8) |
4,9 % p = 0,137 (-1,6; 11,3 |
12,1 % p < 0,001 (6,2; 18,1) |
18,0 % p < 0,001 (12,0; 23,9) |
|
Změna hodnoty ranní vrcholové výdechové rychlosti (PEF , peak expiratory flow) | ||||
|
Rozdíl v léčbě p-hodnota (95% CI) |
33,5 l/min p < 0,001 (22,3; 41,7) |
32,9 l/min p < 0,001 (24,8; 41,1) |
14,6 l/min p < 0,001 (7,9; 21,3) |
33,3 l/min p < 0,001 (26,5; 40,0) |
|
Změna hodnoty večerní vrcholové výdec |
íové rychlosti (PEF) | |||
|
Rozdíl v léčbě p-hodnota (95% CI) |
30,7 l/min p < 0,001 (22,5; 38,9) |
26,2 l/min p < 0,001 (18,0; 34,3) |
12,3 l/min p < 0,001 (5,8; 18,8) |
28,2 l/min p < 0,001 (21,7; 34,8) |
*FF/VI = flutikason-furoát/vilanterol
Studie HZA106837 měla variabilní délku trvání (od minimálně 24 týdnů do maximálně 76 týdnů, přičemž většina pacientů byla léčena po dobu alespoň 52 týdnů léčby). Ve studii HZA106837 byli pacienti randomizováni k léčbě buď kombinací flutikason-furoát/vilanterol 92/22 mikrogramů (n = 1 009), nebo FF v dávce 92 mikrogramů (n = 1 010), oboje podáváno jednou denně. Ve studii HZA106837 byl primární cílový parametr doba do první závažné exacerbace astmatu. Závažná exacerbace astmatu byla definována jako zhoršení astmatu, které vyžadovalo podávání systémových kortikosteroidů po dobu alespoň 3 dnů, nebo hospitalizaci pacienta, nebo návštěvu pohotovosti kvůli astmatu, které vyžadovalo systémové podání kortikosteroidů. Upravená průměrná změna od výchozích hodnot u trough FEV1 byla hodnocena jako sekundární cílový parametr.
Ve studii HZA106837 bylo u pacientů léčených kombinací flutikason-furoát/vilanterol v dávce 92/22 mikrogramů riziko prodělání závažné exacerbace astmatu sníženo o 20 % ve srovnání se samotným FF v dávce 92 mikrogramů (poměr rizik 0,795, p = 0,036 95% CI 0,642; 0,985). Výskyt závažné exacerbace astmatu na jednoho pacienta za rok byl 0,19 ve skupině léčené FF dávkou 92 mikrogramů (přibližně 1 pacient každých 5 let) a 0,14 ve skupině léčené kombinací flutikason-furoát/vilanterol v dávce 92/22 mikrogramů (přibližně 1 pacient každých 7 let). Poměr výskytu exacerbací u kombinace flutikason-furoát/vilanterol 92/22 mikrogramů oproti FF 92 mikrogramů byl 0,755 (95% CI 0,603; 0,945). To představuje 25 % snížení výskytu závažných exacerbací astmatu u subjektů léčených kombinací flutikason-furoát/vilanterol 92/22 mikrogramů ve srovnání s FF 92 mikrogramů (p = 0,014). Dvacetičtyřhodinový bronchodilatační účinek po podání kombinace flutikason-furoát/vilanterol byl udržován v průběhu jednoho roku léčby bez průkazu ztráty účinnosti (žádná tachyfylaxe). Kombinace flutikason-furoát/vilanterol 92/22 mikrogramů vykazovala setrvalé zlepšení z 83 ml na 95 ml u trough FEV1 ve 12., 36. a 52. týdnu a na konci léčby ve srovnání s FF v dávce 92 mikrogramů (p < 0,001 95% CI 52, 126 ml na konci léčby). Čtyřicet čtyři procent pacientů ve skupině léčené kombinací flutikason-furoát/vilanterol 92/22 mikrogramů bylo na konci léčby dobře kontrolováno (ACQ7 < 0,75) ve srovnání s 36 % subjektů ve skupině léčené FF v dávce 92 mikrogramů (p < 0,001 95% CI 1,23; 1,82).
Srovnávací studie s kombinací salmeterol/flutikason-propionát
Ve 24týdenní studii (HZA113091) u dospělých a dospívajících pacientů s perzistentním astmatem bylo zlepšení plicních funkcí oproti výchozímu stavu prokázáno při podávání kombinace flutikason-furoát/vilanterol 92/22 mikrogramů jednou denně večer i při podávání kombinace salmeterol/FP v dávce 50/250 mikrogramů dvakrát denně. Upravené průměrné zlepšení v průběhu léčby oproti výchozím hodnotám hodnocené pomocí váženého průměru FEVj v průběhu 0 - 24 hodin bylo 341 ml (flutikason-furoát/vilanterol) a 377 ml (salmeterol/FP) a prokázalo celkové zlepšení plicních funkcí v průběhu 24 hodin u obou druhů léčby. Upravený průměrný rozdíl v průběhu léčby 37 ml mezi jednotlivými skupinami nebyl statisticky významný (p = 0,162). Subjekty ve skupině léčené kombinací flutikason-furoát/vilanterol dosáhly průměrné změny (vypočítané pomocí metody nejmenších čtverců) od výchozích hodnot 281 ml u trough FEVi a subjekty léčené kombinací salmeterol/FP změny 300 ml; rozdíl v upraveném průměru 19 ml (95% CI: -0,073; 0,034) nebyl statisticky významný (p = 0,485).
Srovnávací studie se salmeterol/FP nebo s jinými kombinacemi IKS/LABA nebyly provedeny ke srovnání účinků na exacerbace astma bronchiale.
Flutikason-furoát v monoterapii
Dvacetičtyřtýdenní randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie (FFA112059) hodnotila bezpečnost a účinnost FF v dávce 92 mikrogramů podávaného jednou denně (n = 114) a FP v dávce 250 mikrogramů podávaného dvakrát denně (n = 114) ve srovnání s placebem (n = 115) u dospělých a dospívajících pacientů s persistentním astmatem. Všechny subjekty musely být léčeny stabilní dávkou IKS po dobu alespoň 4 týdnů před první návštěvou (screening) a naopak nebylo povoleno užívání LABA v průběhu 4 týdnů před 1. návštěvou. Primárním cílovým parametrem účinnosti byla změna od výchozích hodnot v trough FEV1 (před podáním bronchodilatancia a před podáním dávky) při klinické návštěvě na konci léčby. Změna od výchozích hodnot v procentech 24hodinových období bez nutnosti podání záchranné léčby v průběhu 24 týdnů léčby byla sekundárním parametrem. Ve 24. týdnu zvýšily FF i FP trough FEVi o 146 ml (95% CI 36, 257 ml, p = 0,009), resp. 145 ml (95% CI 33, 257 ml, p = 0,011) ve srovnání s placebem. FF i FP zvyšovaly procento 24hodinových období bez nutnosti podání záchranné léčby o 14,8 % (95% CI 6,9; 22,7, p < 0,001), resp. 17,9 % (95% CI 10,0; 25,7; p < 0,001) ve srovnání s placebem.
Studie vystavení alergenu
Bronchoprotektivní účinky kombinace flutikason-furoát/vilanterol 92/22 mikrogramů na časnou i pozdní astmatickou odpověď na inhalační alergen byly hodnoceny v placebem kontrolované, 4krát zkřížené studii s opakovanou dávkou (HZA113126) u pacientů s lehkým astmatem. Pacienti byli randomizováni k léčbě kombinací flutikason-furoát/vilanterol 92/22 mikrogramů, FF v dávce 92 mikrogramů, vilanterolem v dávce 22 mikrogramů nebo placebem jednou denně po dobu 21 dnů, po které následovalo vystavení alergenu 1 hodinu po podání poslední dávky. Alergeny byly roztoči z domácího prachu, kočičí srst nebo pyl z břízy; výběr byl založen na individuálních screeningových testech. Série měření FEVi byla porovnávána s hodnotami před vystavením alergenu, které byly získány po inhalaci solného roztoku (výchozí hodnoty). Celkově největší účinek na časnou astmatickou odpověď byl pozorován ve skupině léčené kombinací flutikason-furoát/vilanterol v dávce 92/22 mikrogramů ve srovnání se samotným FF v dávce 92 mikrogramů nebo samotným vilanterolem v dávce 22 mikrogramů. U skupin léčených kombinací flutikason-furoát/vilanterol (92/22 mikrogramů) a FF v dávce 92 mikrogramů byla prakticky zrušena pozdní astmatická odpověď ve srovnání s podáváním samotného vilanterolu. Kombinace flutikason-furoát/vilanterol
92/22 mikrogramů poskytla významně vyšší ochranu proti bronchiální hyperreaktivitě indukované
alergenem ve srovnání s monoterapií FF a vilanterolem hodnocené 22. den od vystavení metacholinu.
Chronická obstrukční _plicní nemoc
Klinický vývojový program CHOPN zahrnoval jednu 12týdenní (HZC113107), dvě 6měsíční (HZC112206 a HZC112207) a dvě jednoroční (HZC102970, HZC102871) randomizované kontrolované studie prováděné u pacientů s klinicky diagnostikovanou CHOPN. Tyto studie zahrnovaly hodnocení parametrů plicních funkcí, dušnosti a středně závažných až závažných exacerbací.
Šestiměsíční studie
Studie HZC112206 a HZC112207 byly 24týdenní randomizované, dvojitě zaslepené, placebem kontrolované studie paralelních skupin porovnávající účinek kombinace flutikason-furoát/vilanterol se samotným vilanterolem a FF a placebem. Studie HZC112206 hodnotila účinnost kombinace flutikason-furoát/vilanterol v dávce 46 mikrogramů/22 mikrogramů (n = 206) flutikason-furoát/vilanterol v dávce 92/22 mikrogramů (n = 206) oproti FF 92 mikrogramů (n = 206), vilanterolu 22 mikrogramů (n = 205) a placebu (n = 207); ve všech případech byla léčba podávána jednou denně. Studie HZC112207 hodnotila účinnost flutikason-furoátu/vilanterolu v dávce 92/22 mikrogramů (n = 204) a flutikason-furoátu/vilanterolu v dávce 184/22 mikrogramů (n = 205) oproti FF 92 mikrogramů (n = 204) a FF 184 mikrogramů (n = 203), vilanterolu 22 mikrogramů (n = 203) a placebu (n = 205); ve všech případech byla léčba podávaná jednou denně.
U všech pacientů bylo vyžadováno, aby měli v době screeningu v anamnéze kouření (alespoň 10 krabiček za rok), poměr FEVýFVC po podání salbutamolu nižší nebo rovný 0,70, FEVi po podání salbutamolu nižší nebo rovnou 70 % náležité hodnoty a aby měli na modifikované škále dušnosti mMRC (modified Medical Research Council) hodnoty > 2 (škála 0 - 4). Při screeningu bylo průměrné FEVi před podáním bronchodilatancia 42,6 % náležitých hodnot ve studii HZC112206 resp. 43,6 % náležitých hodnot ve studii HZC112207 a průměrná reverzibilita byla 15,9 % ve studii HZC112206 resp. 12,0 % ve studii HZC112207. Koprimární cílové parametry v obou studiích byly vážené průměry FEVi 0 - 4 hodiny po podání dávky 168. den a změny od výchozích hodnot u trough FEVi před podáním dávky 169. den.
Ve společné analýze obou studií bylo prokázáno, že kombinace flutikason-furoát/vilanterol v dávce 92/22 mikrogramů vedla ke klinicky významnému zlepšení plicních funkcí. Kombinace flutikason-furoát/vilanterol 92/22 mikrogramů i vilanterol zvýšily upravený průměrný trough FEV1 o 129 ml (95% CI: 91, 167 ml, p < 0,001); resp. 83 ml (95% CI: 46, 121 ml, p < 0,001) ve srovnání s placebem, hodnoceno 169. den léčby. Flutikason-furoát/vilanterol 92/22 mikrogramů zvýšil trough FEV1 o 46 ml ve srovnání s vilanterolem (95% CI: 8, 83ml, p = 0,017). Flutikason-furoát/vilanterol 92/22 mikrogramů i vilanterol zvýšily upravený průměrný vážený průměr FEV1 v průběhu 0 - 4 hodin o 193 ml (95% CI: 156, 230 ml, p < 0,001) resp. 145 ml (95% CI: 108, 181 ml, p < 0,001) ve srovnání s placebem, hodnoceno 168. den léčby. Flutikason-furoát/vilanterol 92/22 mikrogramů zvyšoval upravený průměrný vážený průměr FEV1 v průběhu 0 - 4 hodin o 148 ml ve srovnání se samotným FF (95% CI: 112, 84 ml, p< 0,001).
Dvanáctiměsíční studie
Studie HZC102970 a HZC102871 byly 52týdenní randomizované, dvojitě zaslepené studie paralelních skupin porovnávající účinky flutikason-furoátu/vilanterolu v dávce 184/22 mikrogramů, flutikason-furoátu/vilanterolu v dávce 92/22 mikrogramů a flutikason-furoátu/vilanterolu v dávce 46/22 mikrogramů s vilanterolem v dávce 22 mikrogramů, vše podáváno jednou denně. Hodnocen byl roční výskyt středně těžké/těžké exacerbace u subjektů s CHOPN s anamnézou kouření (alespoň 10 krabiček za rok), poměrem FEV1/FVC po podání salbutamolu nižším nebo rovným 0,70 a FEV1 po podání salbutamolu nižším nebo rovným 70 % náležitých hodnot a anamnesticky zaznamenaným výskytem > 1 exacerbace CHOPN, která vyžadovala podání antibiotik a/nebo perorálních kortikosteroidů nebo hospitalizaci v posledních 12 měsících před 1. návštěvou. Primárním cílovým parametrem byl roční výskyt středně těžké a těžké exacerbace. Středně těžká/těžká exacerbace byla definována jako zhoršení příznaků, které vyžadovalo léčbu perorálními kortikosteroidy a/nebo antibiotiky nebo hospitalizaci pacienta na lůžkovém oddělení. Obě studie měly 4týdenní zaváděcí období (run-in period), během kterého dostávaly všechny subjekty kombinaci salmeterol/FP v dávce 50/250 dvakrát denně ke standardizaci farmakoterapie CHOPN a stabilizaci onemocnění před randomizací k zaslepené studijní léčbě, která trvala 52 týdnů. Před zaváděcím obdobím přerušily subjekty svoji předchozí medikaci CHOPN, s výjimkou krátkodobě působících bronchodilatancií. Současné používání inhalačních bronchodilatancií s dlouhodobým účinkem (agonisté beta2 receptorů a anitcholinergika), kombinovaných přípravků obsahujících ipratropium/salbutamol, perorálních agonistů beta2 receptorů a teofylinových přípravků nebylo v průběhu období léčby dovoleno. K akutní léčbě exacerbací CHOPN bylo podle specifických doporučení pro použití povoleno podání perorálních kortikosteroidů a antibiotik. V průběhu studie používaly subjekty v případě potřeby salbutamol.
Výsledky obou studií prokázaly, že léčba kombinací flutikason-furoát/vilanterol v dávce
92/22 mikrogramů jednou denně vede ve srovnání s vilanterolem k nižšímu ročnímu výskytu středně
těžkých/těžkých exacerbací CHOPN (tabulka 2).
Tabulka 2: Analýza výskytu exacerbací po 12 měsících léčby
|
HZC102970 |
HZC102871 |
HZC102970 a HZC102871 společně | ||||
|
Cílový parametr |
Vilanterol (n = 409) |
Flutikason-furoát/ vilanterol 92/22 (n = 403) |
Vilanterol (n = 409) |
Flutikason-furoát/ vilanterol 92/22 (n = 403) |
Vilanterol (n = 818) |
Flutikason-furoát/ vilanterol 92/22 (n = 806) |
|
Středně těžké až těžké exacerbace | ||||||
|
Upravený průměrný roční výskyt |
1,14 |
0,90 |
1,05 |
0,70 |
1,11 |
0,81 |
|
Poměr vs. VI 95% CI p-hodnota % snížení (95% CI) |
0,79 (0,64 ; 0,97) 0,024 21 (3, 36) |
0,66 (0,54; 0,81) < 0,001 34 (19,46) |
0,73 (0,63; 0,84) < 0,001 27 (16, 37) | |||
|
Absolutní rozdíl v počtu za rok vs VI (95% CI) |
0,24 (0,03; 0,41) |
0,36 (0,20; 0,48) |
0,30 (0,18; 0,41) | |||
|
Doba do první exacerbace: Poměr rizik (95% CI) % snížení rizika |
0,80 (0,66; 0,99) 20 |
0,72 (0,59; 0,89) 28 |
0,76 (0,66; 0,88) 24 | |||
|
p-hodnota |
0,036 |
0,002 |
p< 0,001 | |||
Ve společné analýze studií HZC102970 a HZC102871 v 52. týdnu bylo pozorováno zlepšení upraveného průměrného trough FEV1, pokud byla kombinace flutikason-furoát/vilanterol v dávce 92/22 mikrogramů porovnávána se samotným vilanterolem v dávce 22 mikrogramů (42 ml 95% CI: 19; 64 ml, p < 0,001). Dvacetičtyřhodinový bronchodilatační účinek kombinace flutikason-furoát/vilanterol byl udržován od první dávky po dobu celého období léčby trvající jeden rok bez průkazu ztráty účinnosti (žádná tachyfylaxe).
Ve dvou kombinovaných studiích mělo celkem 2009 (62 %) pacientů při screeningu anamnézu/rizikové faktory kardiovaskulárního onemocnění. Incidence anamnézy/rizikových faktorů kardiovaskulárních onemocnění byla podobná napříč léčebnými skupinami, přičemž nejčastěji byla zaznamenána hypertenze (46 %), dále pak hypercholesterolémie (29 %) a diabetes mellitus (12 %).
U této podskupiny byly pozorovány podobné účinky na snížení výskytu středně závažné a závažné exacerbace ve srovnání s celkovou populací. U pacientů s anamnézou/rizikovými faktory kardiovaskulárních onemocnění vedlo podávání kombinace flutikason-furoát/vilanterol 92/22 mikrogramů k významnému snížení ročního výskytu středně závažné/závažné exacerbace CHOPN ve srovnání s vilanterolem [upravený průměrný roční výskyt 0,83 resp. 1,18, 30 % snížení (95% CI: 16, 42%, p< 0,001)]. Zlepšení bylo u této podskupiny pozorováno rovněž v 52. týdnu při hodnocení upravené průměrné trough FEVi, pokud byla porovnávána kombinace flutikason-furoát/vilanterol v dávce 92/22 mikrogramů s vilanterolem v dávce 22 mikrogramů [44 ml 95% CI:
15, 73 ml, (p = 0,003)].
Srovnávací studie s kombinací salmeterol/flutikason-propionát
Ve 12týdenní studii (HZC113107) u pacientů s CHOPN vykazovala léčba kombinací flutikason-furoát/vilanterol 92/22 mikrogramů podávaná jednou denně ráno a kombinací salmeterol/FP v dávce 50/500 mikrogramů podávaná dvakrát denně zlepšení plicních funkcí oproti výchozím hodnotám. Upravené průměrné zlepšení v průběhu léčby od výchozích hodnot, pokud jde o vážený průměr FEV1 v průběhu 0 - 24 hodin, bylo 130 ml (flutikason-furoát/vilanterol) a 108 ml (salmeterol/FP), což ukazuje na celkové zlepšení plicních funkcí v průběhu 24 hodin u obou typů léčby. Upravený průměrný rozdíl 22 ml (95% CI: -18, 63 ml) pozorovaný mezi skupinami nebyl statisticky významný (p = 0,282). Upravená průměrná změna trough FEV1 85. den byla v porovnání s výchozími hodnotami 111 ml ve skupině s kombinací flutikason-furoát/vilanterol a 88 ml ve skupině s kombinací salmeterol /FP; rozdíl 23 ml (95% CI: -20, 66) mezi léčebnými skupinami nebyl klinicky ani statisticky významný (p = 0,294).
Nebyly provedeny žádné komparativní studie srovnávající kombinaci flutikason-furoát/vilanterol oproti kombinaci salmeterol/FP nebo oproti jiným používaným bronchodilatanciím, pokud jde o účinek na exacerbace CHOPN.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Relvar Ellipta u všech podskupin pediatrické populace s CHOPN (informace o použití u dětí viz bod 4.2).
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Relvar Ellipta u jedné nebo více podskupin pediatrické populace s astmatem (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Absolutní biologická dostupnost flutikason-furoátu a vilanterolu, pokud jsou podány inhalačně jako kombinace flutikason-furoát/vilanterol, je průměrně 15,2 % resp. 27,3 %. Perorální biologická dostupnost flutikason-furoátu i vilanterolu byla nízká, průměrně 1,26 % resp. < 2 %. Vzhledem k této nízké perorální biologické dostupnosti je systémová expozice flutikason-furoátu a vilanterolu po inhalačním podání primárně důsledkem absorpce části inhalované dávky, která se dostala do plic.
Distribuce
Po intravenózním podání jsou flutikason-furoát i vilanterol široce distribuovány s průměrným distribučním objemem v ustáleném stavu 661 l, resp. 165 l. Flutikason-furoát i vilanterol mají nízkou vazbu na červené krvinky. In vitro je vazba flutikason-furoátu i vilanterolu na plazmatické bílkoviny u člověka vysoká, v průměru > 99,6 % resp. 93,9 %. U subjektů s poruchou funkce ledvin nebo jater nebylo zaznamenáno snížení rozsahu vazby na plazmatické bílkoviny.
Flutikason-furoát i vilanterol jsou substráty glykoproteinu P (P-gp); je však nepravděpodobné, že by současné podávání kombinace flutikason-furoát/vilanterol a inhibitorů P-gp ovlivnilo expozici flutikason-furoátu nebo vilanterolu, protože obě látky jsou dobře absorbovatelné molekuly.
Biotransformace
Na základě údajů získaných in vitro jsou hlavní cesty metabolismu flutikason-furoátu i vilanterolu u člověka zprostředkovány převážně CYP3A4.
Flutikason-furoát je primárně metabolizován hydrolýzou S-fluoromethyl-karbothioátové skupiny na metabolity s výrazně redukovanou kortikosteroidní aktivitou. Vilanterol je primárně metabolizován O-dealkylací na široké spektrum metabolitů s výrazně sníženou agonistickou aktivitou na p1- a p2-adrenergní receptory.
Eliminace
Po perorálním podání je flutikason-furoát u člověka eliminován převážně metabolizací, přičemž metabolity jsou vylučovány téměř výlučně stolicí, s vyloučením < 1 % radioaktivní dávky močí.
Ve studii hodnotící perorální podání radioaktivně značené látky byl vilanterol po perorálním podání u člověka eliminován převážně metabolizací, s následnou exkrecí metabolitů močí, cca 70 % podané radioaktivní dávky, a stolicí, cca 30 % podané radioaktivní dávky. Zdánlivý plazmatický eliminační poločas vilanterolu po jedné inhalaci kombinace flutikason-furoát/vilanterol byl v průměru 2,5 hodiny. Účinný poločas kumulace vilanterolu, stanovený po inhalačním podání opakované dávky 25 mikrogramů vilanterolu, je 16,0 hodin u subjektů s astmatem a 21,3 hodiny u subjektů s CHOPN.
Pediatrická populace
U dospívajících (12 let a starších) se nedoporučuje úprava dávky.
Farmakokinetika flutikason-furoátu/vilanterolu u pacientů mladších 12 let nebyla hodnocena. Bezpečnost a účinnost flutikason-furoátu/vilanterolu u dětí mladších 12 let nebyla zatím stanovena.
Zvláštní populace
Starší pacienti (> 65 let)
Vliv věku na farmakokinetiku flutikason-furoátu a vilanterolu byl zjišťován ve studiích fáze III u CHOPN i astmatu. U subjektů s astmatem nebylo prokázáno, že by věk (12 - 84 let) ovlivňoval farmakokinetiku flutikason-furoátu a vilanterolu.
U subjektů s CHOPN nebylo prokázáno, že by věk ovlivňoval farmakokinetiku flutikason-furoátu, zatímco u vilanterolu bylo zaznamenáno zvýšení (37 %) AUC(0-24) ve věkovém rozmezí 41 až 84 let.
U starších subjektů (ve věku 84 let) s nízkou tělesnou hmotností (35 kg) se předpokládá o 35 % vyšší AUC(0-24) vilanterolu, než je odhad pro průměrnou populaci (subjekty s CHOPN ve věku 60 let a s tělesnou hmotností 70 kg), zatímco Cmax zůstávala nezměněná. Není pravděpodobné, že by tento rozdíl měl klinický význam.
U subjektů s astmatem i subjektů s CHOPN nejsou žádná doporučení pro úpravu dávky.
Porucha funkce ledvin
Klinická farmakologická studie s kombinací flutikason-furoát/vilanterol prokázala, že závažná porucha funkce ledvin (clearance kreatininu < 30 ml/min) nevedla k významně vyšší expozici flutikason-furoátu ani vilanterolu ani k výrazněj ším systémovým účinkům kortikosteroidů nebo agonistů beta2-receptorů ve srovnání se zdravými subjekty.
U pacientů s poruchou funkce ledvin není nutná úprava dávkování.
Vliv hemodialýzy nebyl hodnocen.
Porucha funkce jater
Po opakovaném podávání kombinace flutikason-furoát/vilanterol po dobu 7 dnů bylo zaznamenáno zvýšení systémové expozice flutikason-furoátu (až trojnásobné na základě AUC(0-24)) u subjektů s poruchou funkce jater (Child-Pugh A, B nebo C) ve srovnání se zdravými subjekty. Zvýšení systémové expozice flutikason-furoátu u subjektů se středně závažnou poruchou funkce jater (Child-Pugh B, flutikason-furoát/vilanterol 184/22 mikrogramů) bylo spojeno s průměrně 34% snížením sérových hladin kortizolu ve srovnání se zdravými subjekty. Systémová expozice flutikason-furoátu normalizovaná podle dávky byla podobná u subjektů se středně závažnou a závažnou poruchou funkce jater (Child-Pugh B nebo C).
Po opakovaném podávání kombinace flutikason-furoát/vilanterol po dobu 7 dnů nebylo zaznamenáno významné zvýšení systémové expozice vilanterolu (Cmax a AUC) u subjektů s mírnou, středně závažnou ani závažnou poruchou funkce jater (Child-Pugh A, B nebo C).
U subjektů s mírnou nebo středně závažnou poruchou funkce jater (vilanterol, 22 mikrogramů) ani se závažnou poruchou funkce jater (vilanterol, 12,5 mikrogramů) nebyl zaznamenán žádný klinicky významný vliv kombinace flutikason-furoát/vilanterol na beta-adrenergní systémové účinky (srdeční frekvence nebo hladina draslíku v séru) ve srovnání se zdravými subjekty.
Další zvláštní populace
Odhadnutá AUC(0-24) flutikason-furoátu byla u subjektů s astmatem z východní Asie, Japonska a jihovýchodní Asie (12 - 13 % subjektů) průměrně o 33 % až 53 % vyšší ve srovnání s dalšími etnickými skupinami. Nebylo však zaznamenáno, že by vyšší systémová expozice u této populace byla spojená s vyšším vlivem na 24hodinovou exkreci kortizolu močí. Předpokládá se, že Cmax vilanterolu bude průměrně o 220 až 287 % vyšší a AUC(0-24) bude srovnatelná u subjektů asijského původu ve srovnání se subjekty jiných etnik. Nebylo však zaznamenáno, že by vyšší Cmax vilanterolu vedla ke klinicky významným účinkům na srdeční frekvenci. Odhadnutá AUC(0-24) flutikason-furoátu byla u subjektů s CHOPN z východní Asie, Japonska a jihovýchodní Asie (13 - 14 % subjektů) průměrně o 23 % až 30 % vyšší ve srovnání s bělochy. Nebylo však zaznamenáno, že by vyšší systémová expozice u této populace byla spojená s vyšším vlivem na 24hodinovou exkreci kortizolu močí. Nebyl zaznamenán vliv rasy na odhady farmakokinetických parametrů vilanterolu u subjektů s CHOPN.
Pohlaví, tělesná hmotnost a BMI
Údaje získané z populační farmakokinetické analýzy fáze III u 1 213 subjektů s astmatem (712 žen) a 1 225 subjektů s CHOPN (392 žen) neprokázaly, že by pohlaví, tělesná hmotnost nebo BMI (body mass index) měly vliv na farmakokinetiku flutikason-furoátu.
Údaje získané z populační farmakokinetické analýzy u 856 subjektů s astmatem (500 žen) a 1 091 subjektů s CHOPN (340 žen) neprokázaly, že by pohlaví, tělesná hmotnost nebo BMI měly vliv na farmakokinetiku vilanterolu.
Na základě pohlaví, tělesné hmotnosti ani BMI není nutná úprava dávkování.
5.3 Předklinické údaje vztahující se k bezpečnosti
Farmakologické a toxikologické účinky pozorované při podávání flutikason-furoátu nebo vilanterolu v neklinických studiích byly účinky typické pro glukokortikoidy nebo agonisty beta2-receptorů. Podávání flutikason-furoátu v kombinaci s vilanterolem nevedlo k žádné významné nové toxicitě.
Genotoxicita a kancerogenita
Flutikason-furoát nebyl ve standardní baterii studií genotoxický a nebyl ani kancerogenní ve studiích celoživotní inhalace u potkanů ani myší při expozicích podobných, jakých se dosahuje u člověka při podávání maximální doporučené dávky (na základě AUC).
Vilanterol-trifenatát
Ve studiích genetické toxicity nebyly vilanterol (ve formě alfa-fenylcinamátu) ani kyselina trifenyloctová genotoxické, což naznačuje, že vilanterol (ve formě trifenatátu) nepředstavuje genotoxické riziko pro člověka.
V souladu s nálezy u dalších agonistů beta2-receptorů způsoboval vilanterol-trifenatát ve studiích celoživotní inhalace proliferační změny na reprodukčních orgánech u samic potkanů a myší a na hypofýze u potkanů. U potkanů ani myší nebylo prokázáno zvýšení incidence tumorů při expozicích 2-resp. 30násobně vyšších, než jsou dosahovány při podávání maximální doporučené dávky u člověka (na základě AUC).
Reprodukční toxicita
Flutikason-furoát
Účinky pozorované po inhalačním podání flutikason-furoátu v kombinaci s vilanterolem u potkanů byly podobné účinkům pozorovaným u samotného flutikason-furoátu.
Flutikason-furoát nebyl u potkanů ani králíků teratogenní, ale opožďoval vývoj u potkanů a způsoboval potraty u králíků při dávkách toxických pro matku. Při expozicích přibližně 3násobně vyšších, než jaké jsou dosahovány po podání maximální doporučené dávky u člověka (na základě AUC), nebyly zaznamenány žádné účinky na vývoj potkanů.
Vilanterol-trifenatát
Vilanterol-trifenatát nebyl u potkanů teratogenní. Ve studiích inhalace u králíků způsoboval vilanterol-trifenatát podobné účinky, jaké byly pozorovány u ostatních agonistů beta2 receptorů (rozštěp patra, otevření očních víček, srůst jednotlivých částí hrudní kosti a flexury/malformace končetin). Při podkožním podávání nebyly při expozicích 84násobně vyšších, než jaké jsou dosahovány při podávání maximální doporučené dávky u člověka (na základě AUC), pozorovány žádné účinky. Flutikason-furoát ani vilanterol-trifenatát neměly nežádoucí účinky na fertilitu ani prenatální nebo postnatální vývoj u potkanů.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Monohydrát laktosy Magnesium-stearát
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
Doba použitelnosti po otevření vaničky: 6 týdnů.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C. Pokud je přípravek uchováván v chladničce, nechte inhalátor alespoň jednu hodinu před použitím ohřát na pokojovou teplotu.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Po prvním otevření vaničky, využijte v průběhu 6 týdnů.
Na štítek inhalátoru napište datum, do kdy má být inhalátor spotřebován. Datum má být zapsáno ihned, jakmile byl inhalátor vyndán z vaničky.
6.5 Druh obalu a obsah balení
Inhalátor se skládá ze světle šedého těla, žlutého krytu náustku a počítadla dávek. Přípravek je uložen v ochranné vaničce z laminované fólie, která obsahuje vysoušedlo. Tato vanička je zatavena odlupovacím fóliovým víčkem.
Inhalátor obsahuje dva hliníkové stripy z laminované fólie obsahující 14 nebo 30 dávek.
Inhalátor je zařízení složené z několika komponent, které jsou vyrobeny z polypropylenu, polyetylenu s vysokou hustotou, polyoxymetylenu, polybutylen-teraftalátu, akrylonitril-butadien-styrenu, polykarbonátu a nerezové oceli.
Balení obsahuje inhalátor se 14 nebo 30 dávkami. Multipack obsahuje 3 inhalátory po 30 dávkách.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Instrukce pro použití viz bod 4.2.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Glaxo Group Limited
980 Great West Road, Brentford, Middlesex TW8 9GS, Velká Británie.
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/886/001
EU/1/13/886/002
EU/1/13/886/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. listopadu 2013.
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
23
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Relvar Ellipta 184 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna inhalace poskytuje dávku (dávka, která vychází z náustku) fluticasoni furoas 184 mikrogramů a vilanterolum 22 mikrogramů (ve formě trifenatas). To odpovídá dávkované dávce fluticasoni furoas 200 mikrogramů a vilanterolum 25 mikrogramů (ve formě trifenatas).
Pomocná látka se známým účinkem:
Jedna dávka obsahuje přibližně 25 mg laktosy (ve formě monohydrátu). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Dávkovaný prášek k inhalaci (prášek k inhalaci).
Bílý prášek ve světle šedém inhalátoru se žlutým krytem náustku a počítadlem dávek.
|
4. |
KLINICKÉ ÚDAJE |
|
4.1 |
Terapeutické indikace |
Přípravek Relvar Ellipta je indikován k pravidelné léčbě astmatu u dospělých a dospívajících od 12 let, kde je vhodné použití kombinovaného léčivého přípravku (agonista beta2 receptorů s dlouhodobým účinkem a inhalační kortikosteroid):
• pacienti, kteří nejsou dostatečně kontrolováni inhalačními kortikosteroidy, a kteří jako „léčbu v případě potřeby“ inhalují krátkodobě působící agonisty beta2-receptorů.
4.2 Dávkování a způsob podání
Dávkování
Dospělí a dospívající ve věku 12 let a starší
Jedna inhalace přípravku Relvar Ellipta 184/22 mikrogramů jednou denně.
Pacienti obvykle zaznamenají zlepšení plicních funkcí v průběhu 15 minut po inhalaci přípravku Relvar Ellipta. Pacienti by však měli být informováni, že k udržení kontroly příznaků astmatu je nezbytné pravidelné denní používání a že léčba by měla pokračovat i v případě, že nemají žádné příznaky.
Pokud se příznaky objeví v období mezi dávkami, je třeba k okamžité úlevě použít inhalačního agonistu beta2-receptorů s krátkodobým účinkem.
Úvodní dávku přípravku Relvar Ellipta 92/22 mikrogramů je třeba zvážit u dospělých a dospívajících ve věku 12 let a starších, kteří potřebují nízkou až středně vysokou dávku inhalačního kortikosteroidu v kombinaci s dlouhodobě působícím agonistou beta2-receptorů. Pokud není onemocnění při podávání přípravku Relvar Ellipta 92/22 mikrogramů dostatečně kontrolováno, lze dávku zvýšit na 184/22 mikrogramů, což může vést ke zlepšení kontroly příznaků astmatu.
Pacienti by měli být pravidelně kontrolováni lékařem, aby síla flutikason-furoátu/vilanterolu, kterou používají, zůstávala optimální a byla měněna pouze na základě lékařského doporučení. Dávku je třeba titrovat na nejnižší možnou dávku, která bude účinná při udržování kontroly příznaků onemocnění.
Přípravek Relvar Ellipta 184/22 mikrogramů je třeba zvážit u dospělých a dospívajících ve věku 12 let a starších, kteří vyžadují vyšší dávky inhalačního kortikosteroidu v kombinaci s dlouhodobě působícím agonistou beta2-receptorů.
Maximální doporučená dávka přípravku Relvar Ellipta 184/22 mikrogramů je jedna inhalace jednou denně.
Pacientům s astmatem má být podána síla přípravku Relvar Ellipta obsahující dávku flutikason-furoátu (FF) odpovídající závažnosti jejich onemocnění. Předepisující lékař by si měl být vědom, že u pacientů s astmatem má 100 mikrogramů flutikason-furoátu (FF) podávaných jednou denně podobný účinek jako flutikason-propionát (FP) v dávce 250 mikrogramů podávaný dvakrát denně. FF v dávce 200 mikrogramů podávaný jednou denně pak má podobný účinek jako FP v dávce 500 mikrogramů podávaný dvakrát denně.
Děti mladší 12 let
Bezpečnost a účinnost přípravku Relvar Ellipta u dětí mladších 12 let nebyla v indikaci astmatu stanovena.
K dispozici nejsou žádné údaje.
Zvláštní populace
Starší pacienti (> 65 let)
U této populace není nutná úprava dávkování (viz bod 5.2).
Porucha funkce ledvin
U této populace není nutná úprava dávkování (viz bod 5.2).
Porucha funkce jater
Studie u subjektů s lehkou, středně závažnou a závažnou poruchou funkce jater prokázaly zvýšení systémové expozice flutikason-furoátu (Cmax i AUC) (viz bod 5.2).
Opatrnost je třeba při léčbě pacientů s poruchou funkce jater, u kterých může být vyšší riziko systémových nežádoucích účinků souvisejících s podáváním kortikosteroidů.
U pacientů se středně závažnou nebo závažnou poruchou funkce jater je maximální dávka 92/22 mikrogramů (viz bod 4.4).
Způsob podání
Přípravek Relvar Ellipta je určen pouze k inhalačnímu podání.
Přípravek je třeba podávat každý den ve stejnou dobu.
Konečné rozhodnutí o tom, zda se přípravek bude podávat ráno nebo večer, je třeba ponechat na uvážení lékaře.
Pokud dojde k vynechání dávky, následující dávku je třeba použít následující den v obvyklý čas.
Pokud je přípravek uchováván v chladničce, je třeba inhalátor nechat před použitím zahřát na pokojovou teplotu alespoň jednu hodinu před podáním.
Po inhalaci by si pacient měl vypláchnout ústa vodou, aniž by vodu polykal.
Při prvním použití inhalátoru není nutné kontrolovat, zda účinkuje správně, a připravovat ho zvláštním způsobem k použití. Podrobný návod k použití bude následovat.
Inhalátor Ellipta je uložený v ochranné vaničce obsahující sáček s vysoušedlem, který snižuje vlhkost. Po otevření je třeba sáček s vysoušedlem vyhodit.
Pacient má být poučen, aby vaničku neotevíral dříve, než bude připraven k inhalaci dávky.
Inhalátor je po vyjmutí z vaničky v „uzavřené“ pozici. Označení “Spotřebujte do“ vyjadřuje datum, které by mělo být zapsáno do štítku inhalátoru. Datum „Spotřebujte do“ je 6 týdnů od data otevření vaničky. Po tomto datu se již nemá inhalátor dále používat. Vanička může být znehodnocena po prvním otevření.
Podrobný návod k použití pro 30dávkový inhalátor Ellipta uvedený níže lze rovněž použít pro 14dávkový inhalátor Ellipta.
Návod k použití
1. Před použitím si přečtěte následující instrukce
Pokud se kryt inhalátoru otevře a zavře bez toho, že by došlo k inhalaci léku, dojde ke ztrátě dávky. Ztracená dávka zůstane bezpečně uzavřená v inhalátoru, ale nebude již dostupná k inhalaci.
Při jedné inhalaci není možné náhodně použít dávku navíc ani dvojitou dávku.
Počítadlo dávek
Počítadlo ukazuje, kolik dávek léku v inhalátoru ještě zbývá.
Před prvním použitím inhalátoru ukazuje počítadlo přesně 30 dávek.
Při každém otevření krytu inhalátoru se 1 dávka odečte.
Pokud v inhalátoru zbývá méně než 10 dávek, polovina počítadla ukazuje červeně.
Po použití poslední dávky ukazuje polovina počítadla červeně a zobrazuje
0. Inhalátor je nyní prázdný.
Pokud poté otevřete kryt inhalátoru, počítadlo změní barvu z původní z poloviny červené na zcela červenou.
2. Jak připravit dávku
Pokud jste připraven(a) k použití dávky, otevřete kryt inhalátoru. Inhalátorem netřeste.
Stahujte víčko dolů, dokud neuslyšíte „cvaknutí“.
Lék je nyní připraven k inhalaci. Počítadlo dávek pro potvrzení odečetlo 1 dávku.
Pokud počítadlo neodečte dávku v okamžiku, kdy uslyšíte „cvaknutí,“ inhalátor neumožní inhalaci léku. Vezměte jej zpět k lékárníkovi, aby Vám poradil.
3. Jak se lék ínhaluje
Držte inhalátor dále od úst, a co nejvíce vydechněte, jak je Vám pohodlné.
Nevydechujte do inhalátoru.
Vložte náustek mezi rty a pevně jej svými rty stiskněte.
Neblokujte vzduchové otvory prsty.
Jednou se dlouze, rovnoměrně a zhluboka nadechněte. Zadržte dech po co nejdelší dobu (alespoň 3 -4 sekundy).
• Vyjměte inhalátor z úst.
• Pomalu a lehce vydechněte.
Lék by neměl mít žádnou chuť ani by neměl být cítit, a to ani v případě, že se inhalátor použije správně.
4. Uzavření inhalátoru a vypláchnutí úst
Pokud chcete náustek očistit, otřete jej před uzavřením suchou tkaninou.
Vysuňte kryt zpět nahoru co nejvíce, až je náustek zakrytý.
Po použití inhalátoru si vypláchněte ústa vodou.
To sníží pravděpodobnost, že dojde k rozvoji nežádoucích účinků v podobě bolesti v ústech nebo v krku.
4.3 Kontraindikace
Hypersenzitivita na léčivé látky nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. 4.4 Zvláštní upozornění a opatření pro použití
Zhoršení onemocnění
Kombinace flutikason-furoát/vilanterol se nesmí používat k léčbě akutních příznaků astmatu, při kterých je nutné podání krátkodobě působícího bronchodilatancia. Častější používání krátkodobě působících bronchodilatancií k úlevě od příznaků ukazuje na zhoršení kontroly onemocnění a pacient by měl být vyšetřen lékařem.
Pacient by neměl ukončovat terapii kombinací flutikason-furoát/vilanterol u astmatu bez lékařského dohledu, protože příznaky se mohou po přerušení léčby vrátit.
V průběhu léčby kombinací flutikason-furoát/vilanterol se mohou objevit nežádoucí účinky související s astmatem a exacerbace onemocnění. Pacienti by měli být vyzváni, aby pokračovali v léčbě, ale aby vyhledali lékařskou pomoc, pokud příznaky astmatu zůstanou nekontrolované nebo pokud se po zahájení léčby přípravkem Relvar Ellipta zhorší.
Paradoxní bronchospasmus
Paradoxní bronchospasmus se může projevit okamžitým zhoršením sípotu po podání dávky. Bronchospasmus je třeba léčit okamžitě podáním krátkodobě působícího inhalačního bronchodilatancia. Léčbu přípravkem Relvar Ellipta je třeba okamžitě ukončit, pacienta vyšetřit a v případě potřeby zahájit alternativní léčbu.
Kardiovaskulární účinky
Při podávání sympatomimetických léčivých přípravků, včetně přípravku Relvar Ellipta, se mohou objevit kardiovaskulární nežádoucí účinky, jako srdeční arytmie, např. supraventrikulární tachykardie a extrasystoly. Proto je třeba kombinaci flutikason-furoát/vilanterol používat s opatrností u pacientů se závažným kardiovaskulárním onemocněním nebo poruchami srdečního rytmu, tyreotoxikózou, nekorigovanou hypokalémií nebo u pacientů s predispozicí k nízkým hladinám draslíku v séru.
Pacienti s poruchou funkce jater
U pacientů se středně závažnou až závažnou poruchou funkce jater je třeba používat dávku 92/22 mikrogramů a pacienty pečlivě sledovat s ohledem na možné systémové nežádoucí účinky spojené s podáváním kortikosteroidů (viz bod 5.2).
Systémové účinky kortikosteroidů
Systémové účinky se mohou objevit u jakéhokoli inhalačního kortikosteroidu, zejména při podávání vysokých dávek předepisovaných po dlouhou dobu. Tyto účinky jsou mnohem méně pravděpodobné než u perorálních kortikosteroidů. Možné systémové účinky zahrnují Cushingův syndrom, cushingoidní rysy, adrenální supresi, snížení minerální kostní denzity, růstovou retardaci u dětí a dospívajících, kataraktu a glaukom a vzácněji různé psychologické nebo behaviorální účinky, včetně psychomotorické hyperaktivity, poruch spánku, úzkost, deprese nebo agresivity (zejména u dětí).
Kombinaci flutikason-furoát/vilanterol je třeba podávat s opatrností u pacientů s plicní tuberkulózou nebo u pacientů s chronickou nebo neléčenou infekcí.
Hyperglykémie
U diabetiků byly zaznamenány případy zvýšení hladiny glukózy v krvi, což je třeba zvážit při předepisování přípravku pacientům s diabetes mellitus v anamnéze.
Pneumonie u pacientů s CHOPN
U pacientů s CHOPN léčených kombinací flutikason-furoát/vilanterol byl zaznamenán zvýšený výskyt pneumonie. Zaznamenána byla rovněž zvýšená incidence pneumonie, která vyžadovala hospitalizaci. V některých případech byly tyto případy pneumonie fatální (viz bod 4.8). Lékaři by měli u pacientů s CHOPN pečlivě sledovat možný rozvoj pneumonie, protože klinické rysy takových infekcí se překrývají s příznaky exacerbace CHOPN. Rizikové faktory pneumonie u pacientů s CHOPN léčených kombinací flutikason-furoát/vilanterol zahrnují současné kouření, anamnézu předchozí pneumonie, BMI < 25 kg/m2 a FEV1 (forced expiratory volume) < 50 % náležité hodnoty. Tyto rizikové faktory je třeba vzít v úvahu při předepisování kombinace flutikason-furoát/vilanterol a při výskytu pneumonie je třeba léčbu znovu přehodnotit.
Přípravek Relvar Ellipta 184/22 mikrogramů není indikován k léčbě pacientů s CHOPN. Nebyl zaznamenán dodatečný prospěch z podávání dávky 184/22 mikrogramů ve srovnání s dávkou 92/22 mikrogramů a naopak bylo zaznamenáno potenciální zvýšení rizika výskytu systémových nežádoucích účinků souvisejících s podáváním kortikosteroidů (viz bod 4.8).
Incidence pneumonie u pacientů s astmatem byla častá při vyšší dávce. Incidence pneumonie u pacientů s astmatem užívajících kombinaci flutikason-furoát/vilanterol 184/22 mikrogramů byla numericky vyšší ve srovnání s incidencí zaznamenanou u pacientů léčených kombinací flutikason-furoát/vilanterol v dávce 92/22 mikrogramů nebo placebem (viz bod 4.8). Žádné rizikové faktory nebyly zaznamenány.
Pomocné látky
Pacienti se vzácnými dědičnými poruchami galaktózové intolerance, vrozené deficience laktázy nebo malabsorpce glukózy-galaktózy by tento přípravek neměli používat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Klinicky významné lékové interakce zprostředkované kombinací flutikason-furoát/vilanterol při klinických dávkách jsou považovány za nepravděpodobné z důvodu nízkých plazmatických koncentrací dosahovaných po inhalaci.
Interakce s beta-blokátory
Blokátory beta2-adrenergních receptorů mohou účinky agonistů beta2-adrenergních receptorů oslabit nebo antagonizovat. Současného podávání neselektivních i selektivních blokátorů beta2-adrenergních receptorů je třeba se vyvarovat, pokud pro jejich použití není závažný důvod.
Interakce s inhibitory CYP3A4
Obě látky, flutikason-furoát i vilanterol, jsou rychle odstraňovány při prvním průchodu játry výraznou metabolizací, která je zprostředkována jaterním enzymem CYP3A4.
Při společném podávání se silnými inhibitory CYP3A4 (např. ketokonazol, ritonavir) je třeba opatrnosti, protože je zde riziko zvýšení systémové expozice flutikason-furoátu i vilanterolu; současného používání je třeba se vyvarovat. U zdravých subjektů byla provedena studie lékových interakcí s CYP3A4 po opakované dávce s kombinací flutikason-furoát/vilanterol (184/22 mikrogramů) a silným inhibitorem CYP3A4 ketokonazolem (400 mg). Společné podání zvýšilo průměrné AUC(0-24) flutikason-furoátu o 36 % a Cmax flutikason-furoátu o 33 %. Zvýšení expozice flutikason-furoátu bylo spojeno s 27% snížením váženého průměru sérových hladin kortizolu za 24 hodin. Společné podávání zvýšilo průměrné AUC(0-t) vilanterolu o 65 % a Cmax vilanterolu o 22 %. Zvýšení expozice vilanterolu nebylo spojeno se zvýšením systémových účinků agonistů beta2-receptorů na srdeční frekvenci, hladinu draslíku v krvi ani QTcF interval.
Interakce s inhibitory glykoproteinu P
Flutikason-furoát i vilanterol jsou substráty glykoproteinu P (P-gp). Klinická farmakologická studie u zdravých subjektů, kterým byl společně podáván vilanterol a verapamil (silný inhibitor P-gp a středně silný inhibitor CYP3A4), neprokázala výrazný účinek na farmakokinetiku vilanterolu. Klinické farmakologické studie se specifickým inhibitorem P-gp a flutikason-furoátem nebyly provedeny.
Sympatomimetické léčivé přípravky
Společné podávání dalších sympatomimetik (samotných nebo jako součásti kombinované léčby) může zvyšovat riziko nežádoucích účinků kombinace flutikason-furoát/vilanterol. Přípravek Relvar Ellipta se nesmí používat společně s jinými dlouhodobě působícími agonisty beta2-adrenergních receptorů ani léčivými přípravky obsahujícími dlouhodobě působící agonisty beta2-adrenergních receptorů.
Pediatrická populace.
Studie lékových interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Studie se zvířaty prokázaly reprodukční toxicitu při expozicích, které nejsou klinicky relevantní (viz bod 5.3). K dispozici nejsou žádné, nebo pouze omezené údaje týkající se používání flutikason-furoátu a vilanterol-trifenatátu u těhotných žen.
Podávání flutikason-furoátu/vilanterolu těhotným ženám je třeba zvážit, pouze pokud prospěch z léčby pro matku převáží možná rizika pro plod.
Kojení
Údaje týkající se vylučování flutikason-furoátu nebo vilanterol-trifenatátu a/nebo jejich metabolitů do lidského mateřského mléka jsou nedostatečné. Jiné kortikosteroidy a agonisté beta2-receptorů jsou však v mateřském mléce člověka detekovány (viz bod 5.3). Riziko pro kojené novorozence/kojence nelze vyloučit.
Při rozhodování, zda přerušit kojení nebo přerušit léčbu kombinací flutikason-furoát/vilanterol, je třeba vzít v úvahu prospěch z kojení pro dítě a prospěch z léčby pro ženu.
Fertilita
K dispozici nejsou žádné údaje týkající se fertility u člověka. Studie na zvířatech neprokázaly žádné účinky flutikason-furoátu/vilanterol-trifenatátu na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Flutikason-furoát ani vilanterol nemají žádný nebo mají pouze zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Ke stanovení četností nežádoucích účinků souvisejících s podáváním kombinace flutikason-furoát/vilanterol byly použity údaje z rozsáhlých klinických studií s astmatem a CHOPN. V klinickém vývojovém programu astmatu bylo v jednotném hodnocení nežádoucích účinků zahrnuto celkem 7 034 pacientů. V klinickém vývojovém programu CHOPN bylo v jednotném hodnocení nežádoucích účinků zahrnuto celkem 6 237 pacientů.
Nejčastěji hlášené nežádoucí účinky související s podáváním flutikason-furoátu a vilanterolu byly bolest hlavy a nazofaryngitida. S výjimkou pneumonie a zlomenin byl bezpečnostní profil přípravku podobný u pacientů s astmatem i CHOPN. V průběhu klinických studií byly pneumonie a zlomeniny častěji pozorovány u pacientů s CHOPN.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů a frekvence. Ke klasifikaci četností byla použita následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Třída orgánových systémů |
Nežádoucí účinky |
Četnost |
|
Infekce a infestace |
Pneumonie* Infekce horních cest dýchacích Bronchitida Chřipka Kandidóza úst a hrdla |
Časté |
|
Poruchy imunitního systému |
Reakce přecitlivělosti, včetně anafylaxe, angioedému, vyrážky a kopřivky. |
Vzácné |
|
Poruchy nervového systému |
Velmi časté | |
|
Vzácné | ||
|
Psychiatrické poruchy |
Úzkost |
Vzácné |
|
Srdeční poruchy |
Extrasystoly |
Méně časté |
|
Palpitace |
Vzácné | |
|
Vzácné | ||
|
Respirační, hrudní |
Nazofaryngitida |
Velmi časté |
|
a mediastinální poruchy |
Orofaryngeální bolest Sinusitida Faryngitida Dysfonie |
Časté |
|
Gastrointestinální poruchy |
Časté | |
|
Poruchy svalové a kosterní |
Artralgie |
Časté |
|
soustavy a pojivové tkáně |
Bolest zad Zlomeniny** Svalové křeče | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
*, ** viz níže „Popis vybraných nežádoucích účinků“
Popis vybraných nežádoucích účinků
V integrované analýze dvou zdvojených jeden rok trvajících studií, do kterých bylo zahrnuto celkem 3 255 pacientů s CHOPN s exacerbací, byl počet výskytu pneumonie na 1 000 pacientoroků 97,9 ve skupině s FF/VI 184/22; 85,7 ve skupině s FF/VI 92/22 a 42,3 ve skupině VI 22. U těžké formy pneumonie odpovídající počet případů na 1 000 pacientoroků byl 33,6; 35,5 a 7,6, zatímco závažná forma pneumonie odpovídající případům na 1 000 pacientoroků byla 35,1 ve skupině FF/VI 184/22,
42,9 ve skupině FF/VI 92/22 a 12,1 ve skupině VI 22. Závěrem, případů pneumonie s fatálním průběhem s přihlédnutím k expozici bylo 8,8 pro FF/VI 184/22 ve srovnání s 1,5 pro FF/VI 92/22 a 0 pro VI 22.
Ve společné analýze 11 studií s astmatem (7 034 pacientů) byla incidence pneumonie na 1 000 pacientoroků 18,4 ve skupině FF/VI 184/22 proti 9,6 ve skupině s FF/VI 92/22 a 8,0 ve skupině s placebem.
Zlomeniny
Ve dvou opakovaných 12měsíčních studiích, do kterých bylo zahrnuto celkem 3 255 pacientů s CHOPN, byla celková incidence kostních zlomenin nízká ve všech léčebných skupinách, s vyšší incidencí ve všech skupinách s přípravkem Relvar Ellipta (2 %) ve srovnání se skupinou s vilanterolem v dávce 22 mikrogramů (< 1 %). Ačkoli ve skupinách s přípravkem Relvar Ellipta bylo zaznamenáno více zlomenin ve srovnání se skupinou se samotným vilanterolem v dávce 22 mikrogramů, zlomeniny typicky související s používáním kortikosteroidů (např. kompresivní zlomeniny obratů/zlomeniny obratlů v thorakolumbální oblasti, zlomeniny proximálního femuru a acetabula) se vyskytovaly ve skupině s přípravkem Relvar Ellipta i vilanterolem s četností < 1 %.
Ve společné analýze 11 studií s astmatem (7 034 pacientů) byla incidence zlomenin < 1 % a zlomeniny obvykle souvisely s traumatem.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Známky a příznaky
Předávkování kombinací flutikason-furoát/vilanterol může vést k subjektivním a objektivním příznakům způsobeným účinky jednotlivých složek, zahrnující nežádoucí účinky pozorované při předávkování jinými agonisty beta2-receptorů a konzistentní se známými nežádoucími účinky třídy inhalačních kortikosteroidů (viz bod 4.4).
Léčba
Pro předávkování kombinací flutikason-furoát/vilanterol není k dispozici žádná specifická léčba. Pokud dojde k předávkování, je třeba, podle potřeby zahájit podpůrnou léčbu s odpovídajícím sledováním pacienta.
Kardioselektivní beta-blokáda se má zvážit pouze u těžkého předávkování vilanterolem, pokud je klinicky závažné a pacient nereaguje na podpůrná opatření. Kardioselektivní beta-blokátory je třeba používat s opatrností u pacientů s anamnézou bronchospasmu.
Další léčba by měla odpovídat klinickým příznakům nebo doporučením národního toxikologického centra, jsou-li k dispozici.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii onemocnění spojených s léčbou obstrukcí dýchacích cest, Sympatomimetika a jiná léčiva onemocnění spojených s obstrukcí dýchacích cest, ATC kód: R03AK10
Mechanismus účinku
Flutikason-furoát a vilanterol představují dvě třídy léků (syntetický kortikosteroid a selektivní agonista beta2-receptorů s dlouhodobým účinkem).
Farmakodynamické účinky
Flutikason-furoát
Flutikason-furoát je syntetický trifluorovaný kortikosteroid se silnou protizánětlivou aktivitou. Přesný mechanismus, jakým flutikason-furoát ovlivňuje příznaky astmatu a CHOPN, není znám. Bylo prokázáno, že kortikosteroidy mají široké spektrum účinku na mnoho různých druhů buněk (např. eozinofily, makrofágy, lymfocyty) a mediátorů (např. cytokiny a chemokiny zahrnuté do procesů zánětu).
Vilanterol-trifenatát
Vilanterol-trifenatát je selektivní agonista beta2-adrenergních receptorů s dlouhodobým účinkem (LABA).
Farmakologické účinky agonistů beta2-adrenergních receptorů, mezi něž patří i vilanterol-trifenatát, jsou alespoň zčásti způsobené stimulací intracelulární adenylátcyklázy, enzymu, který katalyzuje přeměnu adenosin-trifosfátu (ATP) na cyklický 3‘,5‘-adenosinmonofosfát (cAMP). Zvýšení hladin cAMP vede k relaxaci bronchiální hladké svaloviny a inhibici uvolňování mediátorů z buněk okamžité přecitlivělosti, zejména z mastocytů.
Mezi kortikosteroidy a LABA dochází k molekulárním interakcím, zatímco steroidy aktivují gen pro beta2-receptor, zvyšují počet receptorů a jejich senzitivitu, LABA připravuje receptor pro glukokortikoidy k aktivaci závislé na steroidech a zrychluje přesun steroidů do buněčného jádra. Tyto synergické interakce se odrážejí ve zvýšení protizánětlivé aktivity, která byla prokázána in vitro i in vivo u širokého spektra zánětlivých buněk, které se podílejí na patofyziologii astmatu i CHOPN.
Studie hodnotící biopsii dýchacích cest po podání flutikason-furoátu a vilanterolu rovněž prokázaly synergický účinek kortikosteroidů a LABA, který se objevuje při klinických dávkách léků u pacientů s CHOPN.
Klinická účinnost a bezpečnost
Tři randomizované, dvojitě zaslepené studie fáze III (HZA106827, HZA106829 a HZA106837) různé délky trvání hodnotily bezpečnost a účinnost kombinace flutikason-furoát/vilanterol u dospělých a dospívajících pacientů s perzistentním astmatem. Všechny subjekty používaly IKS (inhalační kortikosteroid) s nebo bez LABA po dobu alespoň 12 týdnů před první kontrolou. Ve studii HZA106837 prodělali všichni pacienti v průběhu jednoho roku před první kontrolou alespoň jednu exacerbaci, která vyžadovala léčbu perorálními kortikosteroidy. Studie HZA106827 trvala 12 týdnů a hodnotila účinnost kombinace flutikason-furoát/vilanterov dávce 92/22 mikrogramů (n = 01) a FF (flutikason-furoát) v dávce 92 mikrogramů (n = 205) ve srovnání s placebem (n = 203), léčba byla u všech skupin podávána jednou denně. Studie HZA106829 trvala 24 týdnů a hodnotila účinnost kombinace flutikason-furoát/vilanterov dávce 184/22 mikrogramů (n = 197) a FF v dávce 184 mikrogramů (n = 194), oba podávané jednou denně, ve srovnání s flutikason-propionátem (FP) v dávce 500 mikrogramů dvakrát denně (n = 195).
Ve studiích HZA106827/HZA106829 byla koprimárním cílovým parametrem účinnosti změna od výchozích hodnot při klinických kontrolách na konci léčby hodnocená u všech subjektů pomocí tzv. „trough“ čili prebronchodilatační hodnoty FEV1 (před aplikací bronchodilatancia a před podáním dávky) a vážený průměr série měření FEV 1 v průběhu 0 - 24 hodin po podání dávky vypočtený u podskupiny subjektů na konci období léčby. Sekundárním cílovým parametrem v průběhu léčby bylo procento změny 24hodinových období bez nutnosti podání záchranné léčby oproti počátku léčby. Výsledky pro primární a klíčové sekundární cílové parametry těchto studií jsou shrnuty v tabulce 1.
Tabulka 1 - Výsledky primárních a klíčových sekundárních cílových parametrů ve studiích HZA106827 a HZA106829
|
Číslo studie |
HZA106829 |
HZA] |
106827 | |
|
Léčebná dávka FF/VI* (mikrogramy) |
FF/VI 184/22 jednou denně vs. FF 184 jednou denně |
FF/VI 184/22 jednou denně vs. FP 500 dvakrát denně |
FF/VI 92/22 jednou denně vs. FF 92 jednou denně |
FF/VI 92/22 jednou denně vs. placebo jednou denně |
|
Změna od výchozích hor Forward) |
not v trough FEV1 při LOCF (Last Observation Carried | |||
|
Rozdíl v léčbě p-hodnota (95% CI) |
193 ml p < 0,001 (108, 277) |
210 ml p < 0,001 (127, 294) |
36 ml p = 0,405 (-48, 120) |
172 ml p < 0,001 (87, 258) |
|
Vážený průměr série měření FEV1 v průběhu 0 - 24 hodin po podání dávky | ||||
|
Rozdíl v léčbě p-hodnota (95% CI) |
136 ml p = 0,048 (1, 270) |
206 ml p = 0,003 (73, 339) |
116 ml p = 0,06 (-5, 236) |
302 ml p < 0,001 (178, 426) |
|
Změna od výchozích hor záchranné léčby |
not v procentu 24hodinových období bez nutnosti podání | |||
|
Rozdíl v léčbě p-hodnota (95% CI) |
11,7 % p < 0,001 (4,9; 18,4) |
6,3 % p = 0,067 (-0,4; 13,1) |
10,6 % p < 0,001 (4,3; 16,8) |
19,3 % p < 0,001 (13,0; 25,6) |
|
Změna od výchozích hod |
not v procentu 24hodinového období bez příznaků | |||
|
Rozdíl v léčbě p-hodnota (95% CI) |
8,4 % p = 0,010 (2,0; 14,8) |
4,9 % p = 0,137 (-1,6; 11,3 |
12,1 % p < 0,001 (6,2; 18,1) |
18,0 % p < 0,001 (12,0; 23,9) |
|
Změna hodnoty ranní vrcholové výdechové rychlosti (PEF , peak expiratory flow) | ||||
|
Rozdíl v léčbě p-hodnota (95% CI) |
33,5 l/min p < 0,001 (22,3; 41,7) |
32,9 l/min p < 0,001 (24,8; 41,1) |
14,6 l/min p < 0,001 (7,9; 21,3) |
33,3 l/min p < 0,001 (26,5; 40,0) |
|
Změna hodnoty večerní vrcholové výdec |
íové rychlosti (PEF) | |||
|
Rozdíl v léčbě p-hodnota (95% CI) |
30,7 l/min p < 0,001 (22,5; 38,9) |
26,2 l/min p < 0,001 (18,0; 34,3) |
12,3 l/min p < 0,001 (5,8; 18,8) |
28,2 l/min p < 0,001 (21,7; 34,8) |
*FF/VI = flutikason-furoát/vilanterol
Studie HZA106837 měla variabilní délku trvání (od minimálně 24 týdnů do maximálně 76 týdnů, přičemž většina pacientů byla léčena po dobu alespoň 52 týdnů léčby). Ve studii HZA106837 byli pacienti randomizováni k léčbě buď kombinací flutikason-furoát/vilanterol 92/22 mikrogramů (n = 1 009) nebo FF v dávce 92 mikrogramů (n = 1 010), oboje podáváno jednou denně. Ve studii HZA106837 byl primární cílový parametr doba do první závažné exacerbace astmatu. Závažná exacerbace astmatu byla definována jako zhoršení astmatu, které vyžadovalo podávání systémových kortikosteroidů po dobu alespoň 3 dnů, nebo hospitalizaci pacienta nebo návštěvu pohotovosti kvůli astmatu, které vyžadovalo systémové podání kortikosteroidů. Upravená průměrná změna od výchozích hodnot u trough FEV1 byla hodnocena jako sekundární cílový parametr.
Ve studii HZA106837 bylo u pacientů léčených kombinací flutikason-furoát/vilanterol v dávce 92/22 mikrogramů riziko prodělání závažné exacerbace astmatu sníženo o 20 % ve srovnání se samotným FF v dávce 92 mikrogramů (poměr rizik 0,795, p = 0,036; 95% CI: 0,642; 0,985). Výskyt závažné exacerbace astmatu na jednoho pacienta za rok byl 0,19 ve skupině léčené FF dávkou 92 mikrogramů (přibližně 1 pacient každých 5 let) a 0,14 ve skupině léčené kombinací flutikason-furoát/vilanterol v dávce 92/22 mikrogramů (přibližně 1 pacient každých 7 let). Poměr výskytu exacerbací u kombinace flutikason-furoát/vilanterol 92/22 mikrogramů oproti FF 92 mikrogramů byl 0,755 (95% CI: 0,603; 0,945). To představuje 25 % snížení výskytu závažných exacerbací astmatu u subjektů léčených kombinací flutikason-furoát/vilanterol 92/22 mikrogramů ve srovnání s FF 92 mikrogramů (p = 0,014). Dvaceti čtyřhodinový bronchodilatační účinek po podání kombinace flutikason-furoát/vilanterol byl udržován v průběhu jednoho roku léčby bez průkazu ztráty účinnosti (žádná tachyfylaxe). Kombinace flutikason-fUroát/vilanterol 92/22 mikrogramů vykazovala setrvalé zlepšení z 83 ml na 95 ml u trough FEV1 ve 12., 36. a 52. týdnu a na konci léčby ve srovnání s FF v dávce 92 mikrogramů (p < 0,001 95% CI: 52, 126 ml na konci léčby). Čtyřicet čtyři procent pacientů ve skupině léčené kombinací flutikason-furoát/vilanterol 92/22 mikrogramů bylo na konci léčby dobře kontrolováno (ACQ7 < 0,75) ve srovnání s 36 % subjektů ve skupině léčené FF v dávce 92 mikrogramů (p < 0,001 95% CI: ,23; 1,82).
Srovnávací studie s kombinací salmeterol/flutikason-propionát
Ve 24týdenní studii (HZA113091) u dospělých a dospívajících pacientů s perzistentním astmatem bylo zlepšení plicních funkcí oproti výchozímu stavu prokázáno při podávání kombinace flutikason-furoát/vilanterol 92/22 mikrogramů jednou denně večer i při podávání kombinace salmeterol/FP v dávce 50/250 mikrogramů dvakrát denně. Upravené průměrné zlepšení v průběhu léčby oproti výchozím hodnotám hodnocené pomocí váženého průměru FEVj v průběhu 0 - 24 hodin bylo 341 ml (flutikason-furoát/vilanterol) a 377 ml (salmeterol/FP) a prokázalo celkové zlepšení plicních funkcí v průběhu 24 hodin u obou druhů léčby. Upravený průměrný rozdíl v průběhu léčby 37 ml mezi jednotlivými skupinami nebyl statisticky významný (p = 0,162). Subjekty ve skupině léčené kombinací flutikason-furoát/vilanterol dosáhly průměrné změny (vypočítané pomocí metody nejmenších čtverců) od výchozích hodnot 281 ml u trough FEVi a subjekty léčené kombinací salmeterol/FP změny 300 ml; (rozdíl v upraveném průměru 19 ml (95% CI: -0,073; 0,034) nebyl statisticky významný (p = 0,485).
Srovnávací studie se salmeterol/FP nebo s jinými kombinacemi IKS/LABA nebyly provedeny ke srovnání účinků na exacerbace astma bronchiale.
Flutikason-furoát v monoterapii
Dvaceti čtyřtýdenní randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie (studie FFA112059) hodnotila bezpečnost a účinnost FF v dávce 92 mikrogramů podávaného jednou denně (n = 114) a FP v dávce 250 mikrogramů podávaného dvakrát denně (n = 114) ve srovnání s placebem (n = 115) u dospělých a dospívajících pacientů s persistentním astmatem. Všechny subjekty musely být léčeny stabilní dávkou IKS po dobu alespoň 4 týdnů před první návštěvou (screening) a naopak nebylo povoleno užívání LABA v průběhu 4 týdnů před 1. návštěvou. Primárním cílovým parametrem účinnosti byla změna od výchozích hodnot v trough FEV1 (před podáním bronchodilatancia a před podáním dávky) při klinické návštěvě na konci léčby. Změna od výchozích hodnot v procentech 24hodinových období bez nutnosti podání záchranné léčby v průběhu 24 týdnů léčby byla sekundárním parametrem. Ve 24. týdnu zvýšily FF i FP trough FEVi o 146 ml (95% CI: 36, 257 ml, p = 0,009), resp. 145 ml (95% CI: 33, 257 ml, p = 0,011) ve srovnání s placebem. FF i FP zvyšovaly procento 24hodinových období bez nutnosti podání záchranné léčby o 14,8 % (95% CI: 6,9; 22,7, p < 0,001), resp. 17,9 % (95% CI: 10,0; 25,7; p < 0,001) ve srovnání s placebem.
Studie vystavení alergenu
Bronchoprotektivní účinky kombinace flutikason-furoát/vilanterol 92/22 mikrogramů na časnou i pozdní astmatickou odpověď na inhalační alergen byly hodnoceny v placebem kontrolované, 4krát zkřížené studii s opakovanou dávkou (HZA113126) u pacientů s lehkým astmatem. Pacienti byli randomizováni k léčbě kombinací flutikason-furoát/vilanterol 92/22 mikrogramů, FF v dávce 92 mikrogramů, vilanterolem v dávce 22 mikrogramů nebo placebem jednou denně po dobu 21 dnů, po které následovalo vystavení alergenu 1 hodinu po podání poslední dávky. Alergeny byly roztoči z domácího prachu, kočičí srst nebo pyl z břízy; výběr byl založen na individuálních screeningových testech. Série měření FEVi byla porovnávána s hodnotami před vystavením alergenu, které byly získány po inhalaci solného roztoku (výchozí hodnoty). Celkově největší účinek na časnou astmatickou odpověď byl pozorován ve skupině léčené kombinací flutikason-furoát/vilanterol v dávce 92/22 mikrogramů ve srovnání se samotným FF v dávce 92 mikrogramů nebo samotným vilanterolem v dávce 22 mikrogramů. U skupin léčených kombinací flutikason-furoát/vilanterol (92/22 mikrogramů) a FF v dávce 92 mikrogramů byla prakticky zrušena pozdní astmatická odpověď ve srovnání s podáváním samotného vilanterolu. Kombinace flutikason-furoát/vilanterol
92/22 mikrogramů poskytla významně vyšší ochranu proti bronchiální hyperreaktivitě indukované
alergenem ve srovnání s monoterapií FF a vilanterolem hodnocené 22. den od vystavení metacholinu.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Relvar Ellipta u jedné nebo více podskupin pediatrické populace s astmatem (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Absolutní biologická dostupnost flutikason-furoátu a vilanterolu, pokud jsou podány inhalačně jako kombinace flutikason-furoát/vilanterol, je průměrně 15,2 % resp. 27,3 %. Perorální biologická dostupnost flutikason-furoátu i vilanterolu byla nízká, průměrně 1,26 % resp. < 2 %. Vzhledem k této nízké perorální biologické dostupnosti je systémová expozice flutikason-furoátu a vilanterolu po inhalačním podání primárně důsledkem absorpce části inhalované dávky, která se dostala do plic.
Distribuce
Po intravenózním podání jsou flutikason-furoát i vilanterol široce distribuovány s průměrným distribučním objemem v ustáleném stavu 661 l, resp. 165 l. Flutikason-furoát i vilanterol mají nízkou vazbu na červené krvinky. In vitro je vazba flutikason-furoátu i vilanterolu na plazmatické bílkoviny u člověka vysoká, v průměru > 99,6 % resp. 93,9 %. U subjektů s poruchou funkce ledvin nebo jater nebylo zaznamenáno snížení rozsahu vazby na plazmatické bílkoviny.
Flutikason-furoát i vilanterol jsou substráty glykoproteinu P (P-gp); je však nepravděpodobné, že by současné podávání kombinace flutikason-furoát/vilanterol a inhibitorů P-gp ovlivnilo expozici flutikason-furoátu nebo vilanterolu, protože obě látky jsou dobře absorbovatelné molekuly.
Biotransformace
Na základě údajů získaných in vitro jsou hlavní cesty metabolismu flutikason-furoátu i vilanterolu u člověka zprostředkovány převážně CYP3A4.
Flutikason-furoát je primárně metabolizován hydrolýzou S-fluoromethyl-karbothioátové skupiny na metabolity s výrazně redukovanou kortikosteroidní aktivitou. Vilanterol je primárně metabolizován O-dealkylací na široké spektrum metabolitů s výrazně sníženou agonistickou aktivitou na Pr a P2-adrenergní receptory.
Eliminace
Po perorálním podání je flutikason-furoát u člověka eliminován převážně metabolizací, přičemž metabolity jsou vylučovány téměř výlučně stolicí, s vyloučením < 1 % radioaktivní dávky močí.
Ve studii hodnotící perorální podání radioaktivně značené látky byl vilanterol po perorálním podání u člověka eliminován převážně metabolizací, s následnou exkrecí metabolitů močí, cca 70 % podané radioaktivní dávky, a stolicí, cca 30 % podané radioaktivní dávky. Zdánlivý plazmatický eliminační poločas vilanterolu po jedné inhalaci kombinace flutikason-furoát/vilanterol byl v průměru 2,5 hodiny. Účinný poločas kumulace vilanterolu, stanovený po inhalačním podání opakované dávky 25 mikrogramů vilanterolu, je 16,0 hodin u subjektů s astmatem a 21,3 hodiny u subjektů s CHOPN.
Pediatrická populace
U dospívajících (12 let a starších) se nedoporučuje úprava dávky.
Farmakokinetika flutikason-furoátu/vilanterolu u pacientů mladších 12 let nebyla hodnocena. Bezpečnost a účinnost flutikason-furoátu/vilanterolu u dětí mladších 12 let nebyla zatím stanovena.
Zvláštní populace
Starší pacienti (> 65 let)
Vliv věku na farmakokinetiku flutikason-furoátu a vilanterolu byl zjišťován ve studiích fáze III u CHOPN i astmatu. U subjektů s astmatem nebylo prokázáno, že by věk (12 - 84 let) ovlivňoval farmakokinetiku flutikason-furoátu a vilanterolu.
U subjektů s astmatem i subjektů s CHOPN nejsou žádná doporučení pro úpravu dávky.
Porucha funkce ledvin
Klinická farmakologická studie s kombinací flutikason-furoát/vilanterol prokázala, že závažná porucha funkce ledvin (clearance kreatininu < 30 ml/min) nevedla k významně vyšší expozici flutikason-furoátu ani vilanterolu ani k výraznějším systémovým účinkům kortikosteroidů nebo agonistů beta2-receptorů ve srovnání se zdravými subjekty.
U pacientů s poruchou funkce ledvin není nutná úprava dávkování.
Vliv hemodialýzy nebyl hodnocen.
Porucha funkce jater
Po opakovaném podávání kombinace flutikason-furoát/vilanterol po dobu 7 dnů bylo zaznamenáno zvýšení systémové expozice flutikason-furoátu (až trojnásobné na základě AUC(0-24)) u subjektů s poruchou funkce jater (Child-Pugh A, B nebo C) ve srovnání se zdravými subjekty. Zvýšení systémové expozice flutikason-furoátu u subjektů se středně závažnou poruchou funkce jater (Child-Pugh B, flutikason-furoát/vilanterol 184/22 mikrogramů) bylo spojeno s průměrně 34 % snížením sérových hladin kortizolu ve srovnání se zdravými subjekty. Systémová expozice flutikason-furoátu normalizovaná podle dávky byla podobná u subjektů se středně závažnou a závažnou poruchou funkce jater (Child-Pugh B nebo C).
Po opakovaném podávání kombinace flutikason-furoát/vilanterol po dobu 7 dnů nebylo zaznamenáno významné zvýšení systémové expozice vilanterolu (Cmax a AUC) u subjektů s mírnou, středně závažnou ani závažnou poruchou funkce jater (Child-Pugh A, B nebo C).
U subjektů s mírnou nebo středně závažnou poruchou funkce jater (vilanterol, 22 mikrogramů) ani se závažnou poruchou funkce jater (vilanterol, 12,5 mikrogramů) nebyl zaznamenán žádný klinicky významný vliv kombinace flutikason-furoát/vilanterol na beta-adrenergní systémové účinky (srdeční frekvence nebo hladina draslíku v séru) ve srovnání se zdravými subjekty.
Další zvláštní populace
Odhadnutá AUC(0_24) flutikason-furoátu byla u subjektů s astmatem z východní Asie, Japonska a jihovýchodní Asie (12 - 13 % subjektů) průměrně o 33 % až 53 % vyšší ve srovnání s dalšími etnickými skupinami. Nebylo však zaznamenáno, že by vyšší systémová expozice u této populace byla spojená s vyšším vlivem na 24hodinovou exkreci kortizolu močí. Předpokládá se, že Cmax vilanterolu bude průměrně o 220 až 287 % vyšší a AUC(0-24) bude srovnatelná u subjektů asijského původu ve srovnání se subjekty jiných etnik. Nebylo však zaznamenáno, že by vyšší Cmax vilanterolu vedla ke klinicky významným účinkům na srdeční frekvenci.
Pohlaví, tělesná hmotnost a BMI
Údaje získané z populační farmakokinetické analýzy fáze III u 1 213 subjektů s astmatem (712 žen) neprokázaly, že by pohlaví, tělesná hmotnost nebo BMI (body mass index) měly vliv na farmakokinetiku flutikason-furoátu.
Údaje získané z populační farmakokinetické analýzy u 856 subjektů s astmatem (500 žen) neprokázaly, že by pohlaví, tělesná hmotnost nebo BMI měly vliv na farmakokinetiku vilanterolu.
Na základě pohlaví, tělesné hmotnosti ani BMI není nutná úprava dávkování.
5.3 Předklinické údaje vztahující se k bezpečnosti
Farmakologické a toxikologické účinky pozorované při podávání flutikason-furoátu nebo vilanterolu v neklinických studiích byly účinky typické pro glukokortikoidy nebo agonisty beta2-receptorů. Podávání flutikason-furoátu v kombinaci s vilanterolem nevedlo k žádné významné nové toxicitě.
Genotoxicita a kancerogenita
Flutikason-furoát
Flutikason-furoát nebyl ve standardní baterii studií genotoxický a nebyl ani kancerogenní ve studiích celoživotní inhalace u potkanů ani myší při expozicích podobných, jakých se dosahuje u člověka při podávání maximální doporučené dávky (na základě AUC).
Vilanterol-trifenatát
Ve studiích genetické toxicity nebyly vilanterol (ve formě alfa-fenylcinamátu) ani kyselina trifenyloctová genotoxické, což naznačuje, že vilanterol (ve formě trifenatátu) nepředstavuje genotoxické riziko pro člověka.
V souladu s nálezy u dalších agonistů beta2-receptorů, způsoboval vilanterol-trifenatát ve studiích celoživotní inhalace proliferační změny na reprodukčních orgánech u samic potkanů a myší a na hypofýze u potkanů. U potkanů ani myší nebylo prokázáno zvýšení incidence tumorů při expozicích 2- resp. 30násobně vyšších, než jsou dosahovány při podávání maximální doporučené dávky u člověka (na základě AUC).
Reprodukční toxicita
Flutikason-furoát
Účinky pozorované po inhalačním podání flutikason-furoátu v kombinaci s vilanterolem u potkanů byly podobné účinkům pozorovaným u samotného flutikason-furoátu.
Flutikason-furoát nebyl u potkanů ani králíků teratogenní, ale opožďoval vývoj u potkanů a způsoboval potraty u králíků při dávkách toxických pro matku. Při expozicích přibližně 3násobně vyšších, než jaké jsou dosahovány po podání maximální doporučené dávky u člověka (na základě AUC), nebyly zaznamenány žádné účinky na vývoj potkanů.
Vilanterol-trifenatát
Vilanterol-trifenatát nebyl u potkanů teratogenní. Ve studiích inhalace u králíků způsoboval vilanterol-trifenatát podobné účinky, jaké byly pozorovány u ostatních agonistů beta2-receptorů (rozštěp patra, otevření očních víček, srůst jednotlivých částí hrudní kosti a flexury/malformace končetin). Při podkožním podávání nebyly při expozicích 84násobně vyšších, než jaké jsou dosahovány při podávání maximální doporučené dávky u člověka (na základě AUC), pozorovány žádné účinky. Flutikason-furoát ani vilanterol-trifenatát neměly nežádoucí účinky na fertilitu ani prenatální nebo postnatální vývoj u potkanů.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Monohydrát laktosy Magnesium-stearát
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
Doba použitelnosti po otevření vaničky: 6 týdnů.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C. Pokud je přípravek uchováván v chladničce, nechte inhalátor alespoň jednu hodinu před použitím ohřát na pokojovou teplotu.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Po prvním otevření vaničky, využijte v průběhu 6 týdnů.
Na štítek inhalátoru napište datum, do kdy má být inhalátor spotřebován. Datum má být zapsáno ihned, jakmile byl inhalátor vyndán z vaničky.
6.5 Druh obalu a obsah balení
Inhalátor se skládá ze světle šedého těla, žlutého krytu náustku a počítadla dávek. Přípravek je uložen v ochranné vaničce z laminované fólie, která obsahuje vysoušedlo. Tato vanička je zatavena odlupovacím fóliovým víčkem.
Inhalátor obsahuje dva hliníkové stripy z laminované fólie obsahující 14 nebo 30 dávek.
Inhalátor je zařízení složené z několika komponent, které jsou vyrobeny z polypropylenu, polyetylenu s vysokou hustotou, polyoxymetylenu, polybutylen-tereftalátu, akrylonitril-butadien-styrenu, polykarbonátu a nerezové oceli.
Balení obsahuje inhalátor se 14 nebo 30 dávkami. Multipack obsahuje 3 inhalátory po 30 dávkách.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Instrukce pro použití viz bod 4.2.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Glaxo Group Limited
980 Great West Road, Brentford, Middlesex TW8 9GS, Velká Británie.
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/886/004
EU/1/13/886/005
EU/1/13/886/006
10. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. listopadu 2013.
11. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
Název a adresa výrobce odpovědného za propouštění šarží
Glaxo Operations UK Ltd. (trading as Glaxo Wellcome Operations)
Priory Street
Ware, Hertfordshire SG12 0DJ Velká Británie
Glaxo Operations UK Ltd. (trading as Glaxo Wellcome Operations),
Harmire Road,
Barnard Castle,
County Durham DL12 8DT,
Velká Británie.
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Podat závěrečnou zprávu o klinické studii k intervenční post-marketingové bezpečnostní studii s dalším sledováním rizika výskytu pneumonie při podávání přípravku Relvar Ellipta ve srovnání s jiným fixními kombinacemi IKS/LABA v léčbě CHOPN, dle Výborem schváleného protokolu. |
31. prosince 2016 |
|
Podat závěrečnou zprávu o klinické studii k intervenční post-marketingové bezpečnostní studii s dalším sledováním rizika výskytu pneumonie při podávání přípravku Relvar Ellipta ve srovnání s jiným fixními kombinacemi IKS/LABA v léčbě astmatu, dle Výborem schváleného protokolu. |
30. června 2018 |
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
92/22 mikrogramů_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Relvar Ellipta 92 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci fluticasoni furoas/vilanterolum
Jedna podaná dávka obsahuje fluticasoni furoas 92 mikrogramů a vilanterolum 22 mikrogramů (ve formě trifenatas).
Obsahuje rovněž laktosu a magnesium-stearát.
Dávkovaný prášek k inhalaci
14 dávek 30 dávek 3x 30 dávek
1 inhalátor po 14 dávkách 1 inhalátor po 30 dávkách Multipack: 90 dávek (3 balení po 30 dávkách)
Inhalační podání JEDNOU DENNĚ
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP
Doba použitelnosti po prvním otevření: 6 týdnů.
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10 ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, Velká Británie
EU/1/13/886/001
EU/1/13/886/002
EU/1/13/886/003
Lot
relvar ellipta 92:22
184/22 mikrogramů_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Relvar Ellipta 184 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci fluticasoni furoas/vilanterolum
Jedna podaná dávka obsahuje fluticasoni furoas 184 mikrogramů a vilanterolum 22 mikrogramů (ve formě trifenatas).
Obsahuje rovněž laktosu a magnesium-stearát.
Dávkovaný prášek k inhalaci
14 dávek 30 dávek 3x 30 dávek
1 inhalátor po 14 dávkách 1 inhalátor po 30 dávkách Multipack: 90 dávek (3 balení po 30 dávkách)
Inhalační podání JEDNOU DENNĚ
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP
Doba použitelnosti po prvním otevření: 6 týdnů.
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10 ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, Velká Británie
EU/1/13/886/004
EU/1/13/886/005
EU/1/13/886/006
Lot
relvar ellipta 184:22
Relvar Ellipta 92 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci fluticasoni furoas/vilanterolum
Jedna podaná dávka obsahuje fluticasoni furoas 92 mikrogramů a vilanterolum 22 mikrogramů (ve formě trifenatas).
Obsahuje rovněž laktosu a magnesium-stearát.
30 dávek
1 inhalátor po 30 dávkách
Součást multipacku, samostatně neprodejné.
Inhalační podání JEDNOU DENNĚ
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP
Doba použitelnosti po prvním otevření: 6 týdnů.
9 ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH
PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/886/003
13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis. 15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
relvar ellipta 92:22
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VNITŘNÍ KRABIČKA (POUZE MULTIPACK BEZ BLUE-BOXU) 184/22 mikrogramů_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Relvar Ellipta 184 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci fluticasoni furoas/vilanterolum
Jedna podaná dávka obsahuje fluticasoni furoas 184 mikrogramů a vilanterolum 22 mikrogramů (ve formě trifenatas).
Obsahuje rovněž laktosu a magnesium-stearát.
30 dávek
1 inhalátor po 30 dávkách
Součást multipacku, samostatně neprodejné.
Inhalační podání JEDNOU DENNĚ
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP
Doba použitelnosti po prvním otevření: 6 týdnů.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH
PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/886/006
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
relvar ellipta 184:22
92/22 mikrogramů_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Relvar Ellipta 92 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci fluticasoni furoas/vilanterolum
GSK Logo
EXP
Lot
Neotevírejte, dokud nejste připraven(a) k inhalaci. Doba použitelnosti po prvním otevření: 6 týdnů.
14 dávek 30 dávek
184/22 mikrogramů_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Relvar Ellipta 184 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci fluticasoni furoas/vilanterolum
2 NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
GSK Logo
3 POUŽITELNOST
EXP
4. ČÍSLO SARZE
Lot
5. JINÉ
Neotevírejte, dokud nejste připraven(a) k inhalaci. Doba použitelnosti po prvním otevření: 6 týdnů.
14 dávek 30 dávek
92/22 mikrogramů_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Relvar Ellipta 92 mikrogramů/22 mikrogramů prášek k inhalaci fluticasoni furoas/vilanterolum Inhalační podání
2. POUŽITELNOST
EXP
Doba použitelnosti po prvním otevření: 6 týdnů. Spotřebujte do:
3. ČÍSLO ŠARŽE
Lot
4. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
14 dávek 30 dávek
184/22 mikrogramů_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Relvar Ellipta 184 mikrogramů/22 mikrogramů prášek k inhalaci fluticasoni furoas/vilanterolum Inhalační podání
2. POUŽITELNOST
EXP
Doba použitelnosti po prvním otevření: 6 týdnů. Spotřebujte do:
3. ČÍSLO SARZE
Lot
4. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
14 dávek 30 dávek
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Relvar Ellipta 92 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci Relvar Ellipta 184 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci
Fluticasoni furoas/vilanterolum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Relvar Ellipta a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Relvar Ellipta používat
3. Jak se přípravek Relvar Ellipta používá
4. Možné nežádoucí účinky
5. Jak přípravek Relvar Ellipta uchovávat
6. Obsah balení a další informace Podrobný návod k použití
1. Co je přípravek Relvar Ellipta a k čemu se používá
Přípravek Relvar Ellipta obsahuje dvě léčivé látky: flutikason-furoát a vilanterol. Přípravek Relvar Ellipta je dostupný ve dvou rozdílných silách: flutikason-furoát 92 mikrogramů/vilanterol 22 mikrogramů a flutikason-furoát 184 mikrogramů/vilanterol 22 mikrogramů.
Síla 92/22 mikrogramů se používá k pravidelné léčbě chronické obstrukční plicní nemoci (CHOPN) u dospělých a k léčbě astmatu u dospělých a dospívajících starších 12 let.
Síla 184/22 mikrogramů se používá k léčbě astmatu u dospělých a dospívajících ve věku 12 let a starších.
Přípravek Relvar Ellipta se má používat denně a ne pouze tehdy, když máte potíže s dýcháním nebo jiné příznaky CHOPN a astmatu. Přípravek se nesmí používat k úlevě od náhlého záchvatu dušnosti nebo sípotu. Pokud se u Vás objeví tento druh záchvatu, musíte použít inhalátor s rychle účinkujícím přípravkem (jako je např. salbutamol).
Flutikason-furoát patří do skupiny léků zvaných kortikosteroidy, často zkráceně nazývaných steroidy. Kortikosteroidy snižují zánět. Snižují otok a podráždění malých dýchacích cest v plicích a tím postupně zmírňují obtíže s dýcháním. Kortikosteroidy rovněž pomáhají v předcházení astmatických záchvatů a zhoršení CHOPN.
Vilanterol patří do skupiny léků nazývaných bronchodilatancia s dlouhodobým účinkem. Uvolňuje svaly malých dýchacích cest v plicích. Tím pomáhá otevření dýchacích cest a usnadňuje průchod vzduchu do plic i z plic. Pokud se používá pravidelně, pomáhá, aby malé dýchací cesty zůstaly otevřené.
Pokud užíváte tyto dvě léčivé látky společně pravidelně, pomohou Vám při kontrole obtíží s dýcháním více, než když je užívána pouze jedna nebo druhá látka samostatně.
Astma je závažné dlouhodobé plicní onemocnění, které způsobuje stažení svalů, které obklopují malé dýchací cesty (bronchokonstrikce), jejich otok a podráždění (zánět). Příznaky přicházejí a odcházejí a zahrnují dušnost, sípot, pocit tísně na hrudi a kašel.
Chronická obstrukční plicní nemoc (CHOPN) je závažné dlouhodobé plicní onemocnění, při kterém dochází k zánětu dýchacích cest a jejich ztluštění. Příznaky zahrnují dušnost, kašel, nepříjemný pocit na hrudi a vykašlávání hlenu. Přípravek Relvar Ellipta snižuje počet vzplanutí příznaků CHOPN.
2. Čemu musíte věnovat pozornost, než začnete přípravek Relvar Ellipta používat Nepoužívejte přípravek Relvar Ellipta
- jestliže jste alergický(á) na flutikason-furoát, vilanterol nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- pokud si myslíte, že výše popsané se Vás týká, nepoužívejte přípravek Relvar Ellipta, dokud se neporadíte se svým lékařem.
Upozornění a opatření u přípravku Relvar Ellipta
Před použitím přípravku Relvar Ellipta se poraďte se svým lékařem:
- Jestliže máte onemocnění jater, protože pak můžete být náchylnější k nežádoucím účinkům. Jestliže máte středně závažné nebo závažné onemocnění jater, Váš lékař omezí Vaši dávku na nižší sílu přípravku Relvar Ellipta (92/22 mikrogramů jednou denně).
- Jestliže máte problémy se srdcem nebo vysoký krevní tlak.
- Jestliže máte plicní tuberkulózu (TBC) nebo jinou dlouhodobou nebo neléčenou infekci.
- Jestliže máte v anamnéze diabetes (cukrovku).
- Jestliže máte potíže se štítnou žlázou.
- Jestliže máte nízkou hladinu draslíku v krvi.
^ Pokud si myslíte, že se Vás cokoli z výše uvedeného týká, poraďte se se svým lékařem dříve, než začnete používat tento lék.
Okamžité dýchací potíže
Pokud se po použití přípravku Relvar Ellipta dýchání nebo sípot zhorší, přestaňte přípravek používat a neprodleně vyhledejte lékařskou pomoc.
Infekce plic
Pokud používáte tento lék k léčbě CHOPN, může u Vás být zvýšené riziko rozvoje plicní infekce, známé jako pneumonie. Informace týkající se příznaků, kterým je třeba věnovat pozornost v průběhu používání tohoto léku, viz bod 4 „Možné nežádoucí účinky“. Pokud se u Vás jakýkoli z těchto příznaků objeví, sdělte to co nejdříve svému lékaři.
Děti a dospívající
Tento přípravek se nedoporučuje dětem mladším 12 let k léčbě astmatu ani dětem a dospívajícím jakéhokoli věku k léčbě CHOPN.
Další léčivé přípravky a přípravek Relvar Ellipta
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Některé léky mohou ovlivnit účinek tohoto léku, nebo mohou zvýšit pravděpodobnost, že se u Vás objeví nežádoucí účinky.
Mezi tyto léky patří:
- beta-blokátory, jako je metoprolol, používaný k léčbě vysokého krevního tlaku nebo onemocnění srdce.
- ketokonazol, používaný k léčbě plísňových infekcí.
- ritonavir, používaný k léčbě HIV infekcí.
- agonisté beta2-adrenergních receptorů s dlouhodobým účinkem, jako je salmeterol.
^ Pokud některý z těchto léků užíváte, sdělte to svému lékaři nebo lékárníkovi.
Podání přípravku Relvar Ellipta těhotným ženám připadá v úvahu pouze, pokud očekávaný přinos převáži nad možnými riziky.
Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Kojení
Není známo, zda tento lék může procházet do mateřského mléka. Riziko pro kojené dítě proto nelze vyloučit.
Pokud kojíte, poraďte se před použitím přípravku Relvar Ellipta se svým lékařem.
Řízení dopravních prostředků a obsluha strojů
Není pravděpodobné, že by tento lék ovlivnil schopnost řídit nebo obsluhovat stroje.
Přípravek Relvar Ellipta obsahuje laktózu
Pokud Vám někdy lékař řekl, že trpíte nesnášenlivostí některých cukrů nebo mléčných bílkovin, sdělte to svému lékaři dříve, než začnete používat tento lék.
3. Jak se přípravek Relvar Ellipta používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Jaké množství přípravku se používá
Doporučená dávka přípravku k léčbě astmatu je jedna inhalace (92 mikrogramů flutikason-furoátu a 22 mikrogramů vilanterolu) jednou denně, každý den ve stejný čas.
Pokud máte těžkou formu astmatu, lékař může rozhodnout, že budete k inhalaci používat vyšší sílu inhalátoru (184 mikrogramů flutikason-furoátu a 22 mikrogramů vilanterolu). Tato dávka se rovněž používá jednou denně, každý den ve stejnou dobu.
CHOPN
Doporučená dávka k léčbě CHOPN je jedna inhalace (92 mikrogramů flutikason-furoátu a 22 mikrogramů vilanterolu) jednou denně, každý den ve stejnou dobu.
Vyšší síla přípravku Relvar Ellipta není k léčbě CHOPN vhodná.
Používejte přípravek Relvar Ellipta denně ve stejnou dobu, jelikož je účinný přes 24 hodin
Je velmi důležité, abyste tento lék používal(a) každý den tak, jak Vám doporučil Váš lékař. To pomáhá zajistit odstranění příznaků onemocnění v průběhu dne i noci.
Přípravek Relvar Ellipta se nesmí používat k úlevě od náhlého záchvatu dusnosti nebo sípotu.
Pokud se u Vás objeví tento druh záchvatu, musíte použít inhalátor s rychle účinkujícím přípravkem (jako je např. salbutamol).
Pokud cítíte, že jste častěji dušný(á) nebo se u Vás častěji objevuje sípot, než obvykle, nebo pokud musíte používat inhalátor s rychle účinkujícím přípravkem častěji než obvykle, poraďte se se svým lékařem.
Jak se přípravek Relvar Ellipta používá
Úplné informace naleznete v části Podrobný návod k použití za bodem 6 této příbalové informace. Přípravek Relvar Ellipta nemusíte zvlášť připravovat, dokonce ani před prvním použitím.
Pokud se příznaky nezlepšují
Pokud se Vaše příznaky (dušnost, sípot, kašel) nezlepšují, nebo pokud se zhoršují, nebo pokud musíte používat inhalátor s rychle účinkujícím přípravkem častěji, kontaktujte co nejdříve svého lékaře.
Jestliže jste použil(a) více přípravku Relvar Ellipta, než jste měl(a)
Pokud omylem použijete více přípravku Relvar Ellipta, než Vám doporučil lékař, sdělte to svému lékaři nebo lékárníkovi. Můžete zaznamenat rychlejší tlukot srdce, než obvykle, třes nebo bolest hlavy.
Pokud jste používal(a) více přípravku po delší dobu, je velmi důležité, abyste požádal(a) svého lékaře nebo lékárníka o radu. To je proto, že vyšší dávky přípravku Relvar Ellipta mohou snížit množství steroidních hormonů, které Vaše tělo tvoří.
Jestliže jste zapomněl(a) použít přípravek Relvar Ellipta Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Vezměte si pouze následující dávku v obvyklý čas.
Pokud se objeví dušnost nebo sípot, nebo pokud se objeví příznaky astmatického záchvatu, použijte inhalátor s rychle účinkujícím přípravkem (např. salbutamol) a poté vyhledejte lékaře.
Jestliže jste přestal(a) používat přípravek Relvar Ellipta
Používejte tento přípravek tak dlouho, jak Vám doporučil Váš lékař. Tento přípravek bude účinný pouze tak dlouho, jak dlouho jej budete používat. Nepřestávejte používat tento přípravek dříve, než Vám to doporučí lékař, a to ani v případě, že se budete cítit lépe.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Alergické reakce
Alergické reakce na přípravek Relvar Ellipta jsou vzácné (mohou postihnout méně než 1 osobu z 1 000).
Pokud se u Vás při používání přípravku Relvar Ellipta objeví následující příznaky, přestaňte tento lék používat a ihned vyhledejte svého lékaře.
• kožní vyrážka (kopřivka) nebo zarudnutí;
• otok, někdy obličeje nebo úst (angioedém);
• zhoršující se sípání, kašel nebo potíže s dýcháním;
• náhlý pocit slabosti nebo závratě (což může vést ke kolapsu nebo ztrátě vědomí).
Okamžité dýchací potíže
Pokud se ihned po použití tohoto přípravku dušnost nebo sípot zhorší, přestaňte tento lék používat a ihned vyhledejte lékařskou pomoc.
Pneumonie (infekční onemocnění plic) u pacientů s CHOPN (častý nežádoucí účinek)
Pokud se u Vás při používání přípravku Relvar Ellipta objeví následující příznaky, sdělte to svému lékaři - mohou to být příznaky plicní infekce:
• horečka nebo zimnice;
• zvýšená tvorba hlenu, změna barvy hlenu;
• zhoršení kašle nebo zhoršení dýchacích obtíží.
Mezi další nežádoucí účinky patří:
Velmi časté nežádoucí účinky
Mohou postihnout více než 1 osobu z 10:
• bolest hlavy;
• nachlazení.
Časté nežádoucí účinky
Mohou postihnout až 1 osobu z 10:
• bolestivé vystouplé skvrny v ústech nebo hrdle způsobené plísňovou infekcí (kandidóza). Výplach úst vodou ihned po použití přípravku Relvar Ellipta může pomoci snížit výskyt tohoto nežádoucího účinku.
• zánět plic (bronchitida);
• infekce vedlejších dutin nosních nebo hrdla;
• chřipka;
• bolest a podráždění zadní části úst a hrdla;
• zánět vedlejších dutin nosních;
• svědění v nose, rýma nebo ucpaný nos;
• kašel;
• poruchy hlasu;
• oslabení kostí, vedoucí ke zlomeninám;
• bolest břicha;
• bolest zad;
• vysoká těle sná teplota (horečka);
• bolest kloubů;
• svalové křeče.
Méně časté nežádoucí účinky
Mohou postihnout až 1 osobu ze 100:
- nepravidelný tlukot srdce.
Vzácné nežádoucí účinky
Mohou postihnout až 1 osobu z 1 000:
• alergické reakce;
• zrychlený tlukot srdce (tachykardie);
• nepravidelný tlukot srdce (palpitace);
• třes;
• úzkost.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Relvar Ellipta uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 25 °C.
Uchovávejte inhalátor v původním obalu, aby byl přípravek chráněn před vlhkostí. Neotevírejte fóliové víčko ochranné vaničky, dokud nejste připraven(a) k inhalaci. Jakmile je vanička otevřena, inhalátor může být použit po dobu 6 týdnů od data otevření. Napište na štítek inhalátoru datum, do kdy má být inhalátor spotřebován. Datum zapište ihned, jakmile vyjmete inhalátor z vaničky.
Pokud přípravek uchováváte v chladničce, nechejte jen alespoň hodinu před použitím ohřát na pokojovou teplotu.
Nevyhazujte žádné léčivé přípravky do domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Relvar Ellipta obsahuje
• Léčivými látkami jsou fluticasoni furoas a vilanterolum. Jedna inhalace poskytne dávku (dávka, která vyjde z náustku) fluticasoni furoas 92 nebo 184 mikrogramů a vilanterolum 22 mikrogramů (ve formě trifenatas).
• Dalšími složkami jsou monohydrát laktosy a magnesium-stearát.
Jak přípravek Relvar Ellipta vypadá a co obsahuje toto balení
Přípravek Relvar Ellipta je bílý prášek. Samotný přístroj je světle šedý inhalátor se žlutým krytem náustku a počítadlem dávek. Je zabalený ve vaničce z laminované fólie s odlupovacím fóliovým víčkem. Vanička obsahuje vysoušedlo, které snižuje vlhkost uvnitř balení. Po otevření víčka vaničky vysoušedlo vyhoďte - nejezte ho ani neinhalujte. Inhalátor není nutné po otevření uchovávat ve vaničce z laminované fólie.
Inhalátor obsahuje dva hliníkové stripy z laminované fólie obsahující 14 nebo 30 dávek. Multipack obsahuje 3 inhalátory po 30 dávkách.
Podrobný návod k použití
Co je inhalátor Ellipta?
Při prvním použití inhalátoru Ellipta není potřeba kontrolovat, zda funguje správně, nemusíte ho nijak zvlášť k použití připravovat. Pouze postupujte podle následujících podrobných instrukcí.
Inhalátor je zabalen v ochranné vaničce obsahující sáček s vysoušedlem, které snižuje vlhkost. Tento sáček vyhoďte - nejezte ho ani neinhalujte.
Když vyndáte inhalátor z vaničky, je v uzavřené pozici. Neotevírejte jej, dokud nejste připraven(a) k inhalaci léku. Jakmile je vanička otevřena, napište na štítek inhalátoru datum „Spotřebujte do“, do kdy má být inhalátor spotřebován. Datum „Spotřebujte do“ je 6 týdnů od data otevření vaničky. Po tomto datu se již nemá inhalátor dále používat. Vaničku lze po prvním otevření vyhodit.
1. Před použitím si přečtěte následující informace
Pokud kryt inhalátoru otevřete a zavřete bez toho, že byste inhaloval(a) lék, dojde ke ztrátě dávky. Ztracená dávka zůstane bezpečně uzavřená v inhalátoru, ale nebude již dostupná k inhalaci. Při jedné inhalaci není možné náhodně použít dávku navíc ani dvojitou dávku.
Počítadlo dávek
Počítadlo ukazuje, kolik dávek léku v inhalátoru ještě zbývá.
Před prvním použitím inhalátoru ukazuje počítadlo přesně 30 dávek.
Při každém otevření krytu inhalátoru se 1 dávka odečte.
Pokud v inhalátoru zbývá méně než 10 dávek, polovina počítadla ukazuje červeně.
Po použití poslední dávky ukazuje polovina počítadla červeně a zobrazuje
0. Inhalátor je nyní prázdný.
Pokud poté otevřete kryt inhalátoru, počítadlo změní barvu z původní z poloviny červené na zcela červenou.

2. Příprava dávky
Dokud nejste připraven(a) k použití své dávky, inhalátor neotevírejte. Inhalátorem netřeste. • Stahujte víčko dolů, dokud neuslyšíte „cvaknutí“.

Lék je nyní připraven k inhalaci.
Počítadlo dávky pro potvrzení odečetlo 1 dávku.
• Pokud počítadlo neodečte dávku v okamžiku, kdy uslyšíte „cvaknutí,“ inhalátor neumožní inhalaci léku. Vezměte jej zpět k lékárníkovi, aby Vám poradil.
3. Inhalace léku
• Držte inhalátor dále od úst a co nejvíce vydechněte (jak je Vám pohodlné). Nevydechujte do inhalátoru.
• Vložte náustek mezi rty a pevně jej svými rty stiskněte.
Neblokujte vzduchový otvor prsty.

• Jednou se dlouze, rovnoměrně a zhluboka nadechněte. Zadržte dech po co nejdelší dobu (alespoň 3 - 4 sekundy).
• Vyjměte inhalátor z úst.
• Pomalu a lehce vydechněte.
Lék by neměl mít žádnou chuť ani byste jej neměl(a) cítit, a to ani v případě, že jste inhalátor použil(a) správně.
4. Uzavření inhalátoru a vypláchnutí úst
Pokud chcete náustek očistit, otřete jej před uzavřením suchou tkaninou.
• Vysuňte kryt zpět nahoru co nejvíce, až je náustek zakrytý.

• Po použití inhalátoru si vypláchněte ústa vodou.
To sníží pravděpodobnost, že dojde k rozvoji nežádoucích účinků v podobě bolesti v ústech nebo v krku.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci:
Glaxo Group Limited,
980 Great West Road,
Brentford,
Middlesex TW8 9GS.
Velká Británie.
Výrobce:
Glaxo Operations UK Limited (trading as Glaxo Wellcome Operations),
Priory Street,
Ware,
Hertfordshire, SG12 0DJ Velká Británie
Glaxo Operations UK Limited (trading as Glaxo Wellcome Operations),
Harmire Road,
Barnard Castle,
County Durham, DL12 8DT Velká Británie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
GlaxoSmithKline Pharmaceuticals s.a./n.v. GlaxoSmithKline Lietuva UAB
Tél/Tel: + 32 (0) 10 85 52 00
Tel: + 370 5 264 90 00
Btnrapnn
rnaKCoCMHTKnaňH EOOfl
Ten.: + 359 2 953 10 34
Luxembourg/Luxemburg
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Belgique/Belgien
Tél/Tel: + 32 (0) 10 85 52 00
Česká republika
GlaxoSmithKline s.r.o. Tel: + 420 222 001 111
Magyarország
GlaxoSmithKline Kft. Tel.: + 36 1 225 5300
Danmark
GlaxoSmithKline Pharma A/S Tlf: + 45 36 35 91 00 dk-info@gsk.com
Malta
GlaxoSmithKline Malta Tel: + 356 21 238131
Deutschland
GlaxoSmithKline GmbH & Co. KG Tel.: + 49 (0)89 36044 8701 produkt.info@gsk.com
Nederland
GlaxoSmithKline BV Tel: + 31 (0)30 6938100 nlinfo@gsk.com
Eesti
GlaxoSmithKline Eesti OU Tel: + 372 6676 900
Norge
GlaxoSmithKline AS Tlf: + 47 22 70 20 00
ELXáSa
GlaxoSmithKline A.E.B.E. Tr(k: + 30 210 68 82 100
Osterreich
GlaxoSmithKline Pharma GmbH Tel: + 43 (0)1 97075 0
Espaňa
GlaxoSmithKline, S.A. Tel: + 34 902 202 700 es-ci@gsk.com
Polska
GSK Services Sp. z o.o. Tel.: + 48 (0)22 576 9000
France
Laboratoire GlaxoSmithKline Tél.: + 33 (0)1 39 17 84 44 diam@gsk.com
Hrvatska
GlaxoSmithKline d.o.o. Tel: + 385 1 6051 999
Portugal
GlaxoSmithKline - Produtos Farmaceuticos, Lda.
Tel: + 351 21 412 95 00
Románia
GlaxoSmithKline (GSK) S.R.L. Tel: + 4021 3028 208
Ireland
GlaxoSmithKline (Ireland) Limited Tel: + 353 (0)1 4955000
Ísland
Vistor hf.
Sími: + 354 535 7000
Italia
GlaxoSmithKline S.p.A. Tel: + 39 (0)45 9218 111
Kónpoq
GlaxoSmithKline (Cyprus) Ltd T@: + 357 22 39 70 00 gskcyprus@gsk.com
Latvija
GlaxoSmithKline Latvia SIA Tel: + 371 67312687 lv-epasts@gsk.com
Slovenija
GlaxoSmithKline d.o.o.
Tel: + 386 (0)1 280 25 00 medical.x.si@gsk.com
Slovenská republika
GlaxoSmithKline Slovakia s. r. o. Tel: + 421 (0)2 48 26 11 11 recepcia.sk@gsk.com
Suomi/Finland
GlaxoSmithKline Oy Puh/Tel: + 358 (0)10 30 30 30 Finland.tuoteinfo@gsk.com
Sverige
GlaxoSmithKline AB Tel: + 46 (0)8 638 93 00 info.produkt@gsk.com
United Kingdom GlaxoSmithKline UK Tel: + 44 (0)800 221441 customercontactuk@gsk.com
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
69