Relistor 12 Mg/0,6 Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Relistor 12 mg/0,6 ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička 0,6 ml obsahuje 12 mg methylnaltrexonii bromidům. Jeden mililitr roztoku obsahuje 20 mg methylnaltrexonii bromidům.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Čirý roztok, bezbarvý až světle žlutý, bez viditelných částic.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Relistor je určen k léčbě obstipace vyvolané opioidy, jestliže odpověď na léčbu laxativy není dostatečná u dospělých pacientů ve věku 18 let a starších.
4.2 Dávkování a způsob podání
Dávkování
Obstipace vyvolaná opioidy u dospělých pacientů s chronickou bolestí (kroměpacientů s pokročilým onemocněním, kteří dostávají paliativní péči)
Doporučená dávka methylnaltrexonium-bromidu je 12 mg (0,6 ml roztoku) subkutánně, podle potřeby, která je podávána jako minimálně 4 dávky týdně až maximálně jedenkrát denně (7 dávek za týden).
U těchto pacientů se má při zahájení léčby Relistorem ukončit léčba obvykle používanými laxativy (viz bod 5.1).
Obstipace vyvolaná opioidy u dospělých pacientů s pokročilým onemocněním (pacienti, kteří dostávají paliativní péči)
Doporučená dávka methylnaltrexonium-bromidu je 8 mg (0,4 ml roztoku ) (pro pacienty o hmotnosti 38-61 kg) nebo 12 mg (0,6 ml roztoku) (pro pacienty o hmotnosti 62-114 kg).
Obvyklé dávkovací schéma je jedna samostatná dávka každý druhý den. Dávky mohou být podány také v delších intervalech, podle klinické potřeby.
Pouze pokud se nedostavila odpověď (defekace/zrychlení střevní peristaltiky) po dávce podané předcházející den, mohou pacienti dostat dvě následné, po sobě jdoucí dávky, mezi nimiž je interval 24 hodin.
Pacienti, jejichž hmotnost spadá mimo uvedené rozmezí hmotností, by měli dostat dávku 0,15 mg/kg. Objem podané injekce musí být těmto pacientům vypočítán následovně:
Dávka (ml) = hmotnost pacienta (kg) x 0,0075
U pacientů s paliativní péčí se Relistor přidá k obvyklé léčbě laxativy (viz bod 5.1).
Speciální populace
Starší populace
Není třeba úprava dávek v závislosti na věku (viz bod 5.2).
Pacienti s renálním poškozením
U pacientů s těžkým renálním poškozením (clearance kreatininu nižší než 30 ml/min.) má být snížena dávka methylnaltrexonium-bromidu z 12 mg na 8 mg (0,4 ml roztoku) u pacientů o hmotnosti 62 až 114 kg.). U pacientů s těžkým renálním poškozením, jejichž hmotnost je mimo rozmezí od 62 do 114 kg (viz bod 5.2), musí být jejich dávka v mg/kg snížena o 50 %. Tito pacienti by měli používat přípravek Relistor v injekční lahvičce a nikoli v předplněné injekční stříkačce. Nejsou dostupné údaje o pacientech s renálním poškozením v terminálním stádiu na dialýze a podání methylnaltrexonium-bromidu těmto pacientům se nedoporučuje (viz bod 4.4).
Pacienti s jaterním poškozením
U pacientů s mírným až středně těžkým jaterním poškozením není třeba úprava dávek (viz bod 5.2).
Nejsou dostupné údaje o pacientech s těžkým jaterním poškozením (Child-Pugh C) a podání methylnaltrexonium-bromidu těmto pacientům se nedoporučuje (viz bod 4.4).
Pediatrická populace
Účinnost a bezpečnost methylnaltrexonium-bromidu u dětí ve věku do 18 let nebyla stanovena. Nejsou k dispozici žádné údaje.
Způsob podání
Přípravek Relistor se podává jako subkutánní injekce.
Doporučuje se pravidelně měnit místa podání injekce. Nedoporučuje se podávat injekci do oblastí, kde je jemná kůže, podlitiny, zarudnutí nebo tvrdá kůže. Vyhněte se místům s jizvami nebo s narušenou strukturou kůže.
Tři doporučené oblasti těla pro injekci přípravku Relistor jsou horní část dolních končetin, břicho a horní část paží.
Přípravek Relistor může být podáván bez ohledu na příjem potravy.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Podávání methylnaltrexonium-bromidu pacientům se známou nebo suspektní mechanickou gastrointestinální obstrukcí nebo po akutní břišní chirurgické operaci je kontraindikováno.
4.4 Zvláštní upozornění a opatření pro použití
Závažnost a zhoršení příznaků
Pacienti mají být poučeni, aby neprodleně hlásili závažné, přetrvávající a/nebo zhoršující se potíže.
Dojde-li v průběhu léčení k vážnému nebo přetrvávajícímu průjmu, pacientovi má být doporučeno, aby nepokračoval v terapii methylnaltrexonium-bromidem a poradil se se svým lékařem.
Obstipace nesouvisející s používáním opioidů
Účinnost methylnaltrexonium-bromidu byla studována u pacientů s obstipací vyvolanou opioidy. Přípravek Relistor by neměl být proto používán u pacientů k léčbě obstipace, která s použitím opioidů nesouvisí.
Rychlý nástup defekace
Údaje z klinických studií svědčí o tom, že léčení methylnaltrexonium-bromidem může vést k rychlému nástupu (v průměru během 30 až 60 minut) defekace.
Trvání léčby
Obstipace vyvolaná opioidy u dospělých pacientů s pokročilým onemocněním Podávání methylnaltrexonium-bromidu nebylo u dospělých pacientů s pokročilým onemocněním v klinických studiích studováno déle než po dobu 4 měsíců, a proto by měl být podáván pouze po časově omezené období (viz bod 5.1).
Pacienti s poškozením funkce jater nebo ledvin
Podávání methylnaltrexonium-bromidu pacientům s těžkou poruchou funkce jater nebo pacientům s poruchou funkce ledvin v terminálním stádiu vyžadujícím dialýzu se nedoporučuje (viz bod 4.2).
Gastrointestinální (GI) onemocnění a GI perforace
Methylnaltrexonium-bromid se má používat s opatrností u pacientů s potvrzenými lézemi GI traktu nebo s podezřením na ně.
Použití methylnaltrexonium-bromidu u pacientů s kolostomií, peritoneálním katetrem, aktivní divertikulární chorobou nebo s fekálním zaklíněním nebylo studováno. Proto má být přípravek Relistor podáván těmto pacientům s opatrností.
Po uvedení na trh byly u pacientů, kterým byl methylnaltrexonium-bromid podáván, hlášeny případy gastrointestinální perforace. Přestože onemocnění, pro která byli pacienti léčeni, mohla být spojena s lokalizovaným nebo difúzním snížením strukturální integrity stěny gastrointestinálního traktu (např. maligní onemocnění, peptický vřed, pseudoobstrukce), methylnaltrexonium-bromid mohl k těmto příhodám přispět.
Obsah sodíku
Tento lék obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce (tzn. je v podstatě sodíku prostý).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Methylnaltrexonium-bromid neovlivňuje farmakokinetiku léčivých přípravků metabolizovaných isoenzymy cytochromu P450 (CYP). Methylnaltrexonium-bromid je minimálně metabolizován isoenzymy CYP. Metabolické studie in vitro ukazují, že methylnaltrexonium-bromid neinhibuje aktivitu CYP1A2, CYP2E1, CYP2B6, CYP2A6, CYP2C9, CYP2C19 ani CYP3A4, zatímco je slabým inhibitorem metabolizmu modelového substrátu CYP2D6. V klinické studii interakcí u zdravých dospělých mužských subjektů neovlivnila dávka methylnaltrexonium-bromidu 0,3 mg/kg s.c. signifikantně metabolizmus substrátu CYP2D6 dextrometorfanu.
Interakční potenciál transportérů organických kationtů (OCT) vztahující se k transportérům OCT methylnaltrexonium-bromidu a OCT inhibitoru byl studován u 18 zdravých subjektů porovnáním farmakokinetických profilů po jednotlivé dávce methylnaltrexonium-bromidu před podáním a po podání vícenásobných dávek 400 mg cimetidinu. Renální clearance methylnaltrexonium-bromidu byla po podání vícenásobných dávek cimetidinu snížena (z 31 l/hod. na 18 l/hod.). To však vedlo jen k malému snížení celkové clearance (ze 107 l/hod. na 95 l/hod.). Návazně nebyla pozorována žádná významná změna AUC methylnaltrexonium-bromidu ani Cmax před podáním a po podání vícenásobných dávek cimetidinu.
4.6 Fertilita, těhotenství a kojení
Nejsou dostatečné údaje o podávání methylnaltrexonium-bromidu těhotným ženám. Studie na zvířatech prokázaly reprodukční toxicitu při podávání vysokých dávek (viz bod 5.3.). Potenciální riziko pro člověka není známo. Methylnaltrexonium-bromid nemá být podáván v průběhu těhotenství, pokud to není zcela nezbytné.
Kojení
Není známo, zda je methylnalteroxonium-bromid vylučován do mateřského mléka. Studie na zvířatech prokázaly vylučování methylnaltrexonium-bromidu do mateřského mléka. Je proto třeba rozhodnout, zda pokračovat/přerušit kojení, nebo pokračovat/přerušit podávání methylnaltrexonium-bromidu, a přitom vzít v úvahu přínos kojení pro dítě a přínos terapie methylnaltrexonium-bromidem pro matku.
Fertilita
Subkutánní injekce přípravku Relistor v dávce 150 mg/kg/den snížily fertilitu u potkanů. Dávky do 25 mg/kg/den (18ti násobek expozice [AUC] u lidí při subkutánní dávce 0,3 mg/kg) neovlivnily fertilitu nebo celkovou schopnost reprodukce.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Methylnaltrexonium-bromid ma malý vliv na schopnost řídit nebo obsluhovat stroj.
Může se vyskytnout závrať, a ta může ovlivnit schopnost řídit a obsluhovat stroje (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nejčastějšími nežádoucími účinky u všech pacientů léčených methylnaltrexonium-bromidem v průběhu všech fází placebem kontrolovaných studií byly bolest břicha, nauzea, průjem a flatulence. Tyto účinky byly obecně mírné až středně silné.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky jsou klasifikovány jako: velmi časté (>1/10); časté (>1/100 až <1/10) ; méně časté (>1/1000 až <1/100); vzácné (>1/10 000 až <1/1000); velmi vzácné (<1/10 000) a frekvence neznámá (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti:
Poruchy nervového systému
Časté: Závrať
Časté: Mírné příznaky jako při náhlém přerušení léčby opioidy (kterýkoliv z uvedených příznaků: zimnice, třes, rhinorea, piloerekce, návaly horka, palpitace, hyperhidróza)
Gastrointestinální poruchy
Neznámé: Gastrointestinální perforace (viz bod 4.4)
Časté: Zvracení
Velmi časté: Bolest břicha, nauzea, flatulence, průjem
Poruchy kůže a _podkožní tkáně
Časté: Reakce v místě podání (např. píchání, pálení, bolest, zčervenání, edém)
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Zdraví dobrovolníci ve studii zaznamenali ortostatickou hypotenzi po dávce 0,64 mg/kg podané jako intravenózní bolus.
V případě předávkování mají být známky a příznaky ortostatické hypotenze monitorovány a hlášeny lékaři. Léčení by mělo být zahájeno podle potřeby.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Laxativa, Antagonisté periferních opioidních receptorů, ATC kód: A06AH01
Mechanismus účinku
Methylnaltrexonium-bromid je selektivní antagonista vazby opioidů na mu-receptor. In vitro studie prokázaly, že methylnaltrexonium-bromid je antagonista opioidního mu-receptoru (inhibiční konstanta [Ki] = 28 nM) s 8 x nižším účinkem na kappa opioidní receptory (Ki = 230 nM) a mnohokrát nižší afinitou k opioidním delta receptorům.
Jako kvarterní amin má methylnaltrexonium-bromid omezenou schopnost přecházet přes hematoencefalickou bariéru. To umožňuje methylnaltrexonium-bromidu fungovat jako periferně účinkující mu-opioidní antagonista ve tkáních, jako je gastrointestinální trakt, aniž by ovlivnil analgetický účinek vyvolaný opioidy v centrálním nervovém systému.
Klinická účinnost a bezpečnost
Obstipace vyvolaná opioidy u dospělých pacientů s chronickou nenádorovou bolestí Účinnost a bezpečnost methylnaltrexonium-bromidu při léčbě obstipace vyvolané opioidy u pacientů s chronickou nenádorovou bolestí byla prokázána v randomizované, dvojitě zaslepené, placebem kontrolované studii (Studie 3356). V této studii byl medián věku pacientů 49 let (rozsah 23-83); 60 % bylo žen. Primární diagnózou většiny pacientů byla bolest zad.
Studie 3356 porovnávala 4-týdenní podávání 12 mg methylnaltrexonium-bromidu jedenkrát denně a 12 mg methylnaltrexonium-bromidu každý druhý den s placebem. Po 4-týdenním, dvojitě zaslepeném období následovala otevřená 8 týdenní fáze, ve které se methylnaltrexonium-bromid měl používat podle potřeby, ale ne víc než 1 dávka denně. Celkově bylo v dvojitě zaslepené fázi léčeno 460 pacientů (12 mg methylnaltrexonium-bromidu jedenkrát denně, n=150, 12 mg methylnaltrexonium-bromidu každý druhý den, n=148, placebo, n=162). Pacienti trpěli v minulosti chronickou nenádorovou bolestí a užívali stabilní dávku opioidu alespoň 50 mg perorálního ekvivalentu morfinu denně. Pacienti měli obstipaci vyvolanou opioidy (během období screeningu měli <3 stolice týdně bez podání záchranné léčby). Pacienti museli splnit požadavek, aby vysadili všechna dříve užívaná laxativa.
Prvním koprimárním ukazatelem byl podíl pacientů s defekací bez záchranných laxativ (RFBM) během 4 hodin po podání první dávky a druhým byl percentuelní podíl aktivních injekcí, po kterých došlo k RFBM během 4 hodin v průběhu dvojitě zaslepené fáze. RFBM byla definována jako stolice, ke které došlo bez použití laxativ během předchozích 24 hodin.
Podíl pacientů, u kterých došlo k RFBM během 4 hodin po podání první dávky, byl 34,2 % v kombinované skupině s methylnaltrexonium-bromidem versus 9,9 % ve skupině s placebem (p<0,001). Průměrný percentuelní podíl podání methylnaltrexonium-bromidu, jehož důsledkem byla RFBM během 4 hodin, byl 28,9 % ve skupině s podáváním jedenkrát denně a 30,2 % ve skupině s podáváním každý druhý den v porovnání s 9,4 % a 9,3 % při podávání odpovídajícího režimu s placebem (p < 0,001).
Klíčový sekundární ukazatel upravené průměrné změny týdenních RFBM v porovnání s hodnotou při vstupu byl 3,1 v léčebné skupině s 12 mg methylnaltrexonium-bromidu jedenkrát denně, 2,1 v léčebné skupině s 12 mg methylnaltrexonium-bromidu každý druhý den a 1,5 v léčebné skupině s placebem během 4 týdenního dvojitě zaslepeného období. Rozdíl 1,6 RFBM za týden mezi 12 mg methylnaltrexonium-bromidu jedenkrát denně a placebem je statisticky významný (p < 0,001) a má význam i v klinické praxi.
Další sekundární ukazatel hodnotil podíl pacientů s >3 RFBM za týden během 4-týdenní dvojitě zaslepené fáze. Ten byl dosažen u 59 % pacientů ve skupině, která dostávala 12 mg methylnatrexonu denně (p<0,001 versus placebo), u 61 % pacientů, kteří ho dostávali každý druhý den (p<0,001 versus placebo), a u 38 % pacientů, kteří dostávali placebo. Doplňková analýza hodnotila percentuelní podíl pacientů, kteří dosáhli >3 úplných RFBM za týden a zvýšení o >1 úplných RFBM za týden alespoň ve 3 týdnech ze 4 týdnů léčby. Toto bylo dosaženo u 28,7 % pacientů ve skupině s podáváním 12 mg methylnatrexonium-bromidu denně (p<0,001 versus placebo), u 14,9 % pacientů, kteří ho dostávali každý druhý den (p=0,012 versus placebo), a u 6,2 % pacientů léčených placebem.
Nebyl získaný důkaz o rozdílech v účinnosti a bezpečnosti způsobených pohlavím. Vliv etnického původu nebylo možné analyzovat, protože populace byla především bělošská (90 %). Medián denní dávky opioidů se nijak významně neodlišoval od vstupních hodnot ani u pacientů užívajících methylnaltrexonium-bromid ani u pacientů dostávajících placebo.
Nezjistili se žádné klinicky významné změny v hodnocení bolesti v porovnání se vstupními hodnotami ani u pacientů užívajících methylnaltrexonium-bromid, ani u pacientů dostávajících placebo.
V klinických hodnoceních se nehodnotilo používání methylnaltrexonium-bromidu při léčbě obstipace vyvolané opioidy delší než 48 týdnů.
Obstipace vyvolaná opioidy u dospělých pacientů s pokročilým onemocněním
Účinnost a bezpečnost methylnaltrexonium-bromidu při léčbě obstipace vyvolané opioidy u pacientů, kteří dostávají paliativní péči, byla prokázána ve dvou randomizovaných dvojitě zaslepených, placebem kontrolovaných studiích. V těchto studiích byl střední věk 68 let (rozmezí 21-100); 51 % byly ženy. Pacienti v obou studiích měli pokročilé terminální onemocnění s omezenou předpokládanou délkou přežití, ve většině případů s primární diagnózou nevyléčitelné rakoviny; ostatní primární diagnózy zahrnovaly konečné stádium CHOPN/emfyzém, onemocnění srdce/srdeční selhání, Alzheimerovu chorobu/demenci, HIV/AIDS a jiná onemocnění v pokročilém stádiu. Před screeningem měli pacienti opioidy vyvolanou obstipaci definovanou buď jako <3 stolice v předchozím týdnu, nebo žádná stolice po >2 dny.
Studie 301 porovnávala jednorázovou dvojitě zaslepenou subkutánní dávku methylnaltrexonium-bromidu 0,15 mg/kg, nebo 0,3 mg/kg versus placebo. Po dvojitě zaslepené dávce následovala otevřená 4-týdenní perioda, během které mohl být podáván methylnaltrexonium-bromid podle potřeby, avšak ne častěji než 1 x za 24 hodin. Během obou period studie udržovali pacienti svůj obvyklý laxativní režim. V periodě dvojitě zaslepené studie bylo celkově zařazeno a léčeno 154 pacientů (47 dostalo methylnaltrexonium-bromid 0,15 mg/kg, 55 dostalo methylnaltrexonium-bromid 0,3 mg/kg, 52 dostalo placebo). Primárním cílovým parametrem byl podíl pacientů s defekací bez záchranné laxativní léčby do 4 hodin po podání dvojitě zaslepené dávky studovaného léčivého přípravku. Ve skupině léčené methylnaltrexonium-bromidem byl signifikantně vyšší podíl pacientů s defekací do 4 hodin po dvojitě zaslepené dávce (62 % u 0,15 mg/kg a 58 % u 0,3 mg/kg), než ve skupině léčené placebem (14 %); p<0,0001 pro každou dávku versus placebo.
Studie 302 porovnávala dvojitě zaslepené subkutánní dávky methylnaltrexonium-bromidu podané každý druhý den po 2 týdny proti placebu. V průběhu prvního týdne (dny 1., 3., 5., 7.) dostali pacienti buď 0,15 mg/kg methylnaltrexonium-bromidu, nebo placebo. Ve druhém týdnu mohla být dávka zvýšena na 0,30 mg/kg, pokud měl pacient do 8. dne bez použití záchranné dávky projímadla 2 nebo méně stolic. Předepsaná dávka mohla být pacientovi kdykoli snížena v závislosti na toleranci. Byly analyzovány údaje od 133 pacientů (62 užívalo methylnaltrexonium-bromidu, 71 užívalo placebo). Studie měla dva primární cíle: Podíl pacientů s defekací bez záchranné laxativní medikace do 4 hodin po první dávce léčivého přípravku a podíl pacientů s defekací bez záchranné laxativní medikace do 4 hodin po nejméně 2 z prvních 4 dávek léčivého přípravku. U pacientů léčených methylnaltrexonium-bromidem byl vyšší podíl defekací do 4 hodin po první dávce (48 %) než u pacientů léčených placebem (16 %); p<0,0001. Pacienti léčeni methylnaltrexonium-bromidem měli také signifikantně vyšší počet defekací do 4 hodin po podání alespoň 2 ze 4 dávek (52 %) než pacienti léčení placebem (9 %); p<0,0001. Konsistence stolice u pacientů, kteří měli zpočátku měkkou stolici, se významně nezlepšila.
V obou studiích nebyl prokázán rozdíl v účinnosti ani v bezpečnosti v závislosti na věku nebo pohlaví. Účinek nebylo možno analyzovat podle rasy, protože populace ve studii byla převážně bělošská
(88 %).
Trvání odpovědi bylo demonstrováno ve studii 302, v níž byl počet laxačních odpovědí konzistentní od dávky 1 až po dávku 7 v průběhu celé dvoutýdenní dvojitě zaslepené periody.
Účinnost a bezpečnost methylnaltrexonium-bromidu byly demonstrovány také v otevřené studii 301, při níž bylo léčivo podáváno od 2. dne po 4 týdny, a ve 2 otevřených rozšířených studiích (301EXT a 302EXT), v nichž byl methylnaltrexonium-bromid podáván podle potřeby po dobu až 4 měsíců (po tuto dobu pouze 8 pacientů). Celkově 136 pacientů ve studii 301, 21 pacientů ve studii 301EXT a 82 pacientů ve studii 302EXT dostalo nejméně jednu léčebnou dávku v otevřené studii. Přípravek Relistor byl podáván každých 3,2 dnů (medián dávkovacího intervalu s rozmezím 1-39 dnů).
Počet laxačních odpovědí byl udržován po dobu rozšířené studie u těch pacientů, kteří pokračovali v léčení.
V těchto studiích nebyl zjištěn žádný signifikantní vztah mezi úvodní dávkou opiátu a laxační odpovědí u pacientů léčených methylnaltrexonium-bromidem. Navíc střední dávka opiátu ve skupině pacientů léčených methylnaltrexonium-bromidem a pacientů léčených placebem se podstatně nelišila od úvodních dávek. Nedošlo ke klinicky relevantní změně skóre bolesti od úvodních hodnot ani ve skupině léčené methylnaltrexonium-bromidem, ani ve skupině léčené placebem.
Účinek na srdeční repolarizaci
Ve dvojitě zaslepené randomizované EKG studii s paralelní skupinou jednotlivá dávka methylnaltrexonium-bromidu (0,15; 0,30 a 0,50 mg/kg) u 207 zdravých dobrovolníků neprodloužila interval QT/QTc ani neovlivnila sekundární parametry EKG a tvar křivky byl srovnatelný s placebem a s pozitivní kontrolou (perorálně podaný moxifloxacin 400 mg).
5.2 Farmakokinetické vlastnosti
Absorpce
Methylnaltrexonium-bromid se po subkutánním podání rychle absorbuje a dosahuje maximální koncentrace (Cmax) přibližně po 0,5 hodině. Cmax a plocha pod křivkou koncentrace (AUC) se zvyšují úměrně se zvyšující se dávkou od 0,15 mg/kg do 0,5 mg/kg. Absolutní biologická dostupnost subkutánní dávky 0,30 mg/kg ve srovnání s intravenozní dávkou 0,30 mg/kg je 82 %.
Distribuce
Methylnaltrexonium-bromid podléhá středně silné distribuci do tkání. Distribuční objem v ustáleném stavu (Vss) je přibližně 1,1 l/kg. Methylnaltrexonium-bromid se minimálně váže na lidské plazmatické proteiny (11,0 % až 15,3 %), jak bylo zjištěno rovnovážnou dialýzou.
Biotransformace
Podle množství metabolitů získaných z exkretů je methylnaltrexonium-bromid u lidí metabolizován v malé míře. Zdá se, že primární metabolickou cestou je přeměna na isomery metyl-6-naltrexolu a na methylnatrexon-sulfát. Každý z izomerů metyl-6-naltrexolu má poněkud nižší antagonistický účinek než mateřská sloučenina a nižší expozici v plazmě ve výši přibližně 8 % z původního léčiva. Methylnaltrexon-sulfát je neúčinný metabolit a v plazmě je přítomný v hladině přibližně 25 % z původního léčiva. N-demetylace methylnaltrexonu na naltrexon není významná a činí asi 0,06 % podané dávky.
Eliminace
Methylnaltrexonium-bromid je vylučován převážně ve formě nezměněné léčivé látky. Přibližně polovina dávky je vyloučena močí a o něco méně stolicí. Konečný poločas vylučování1/2) je přibližně 8 hodin.
Zvláštní populace
Pacienti s jaterním poškozením
Celkový účinek methylnaltrexonium-bromidu u mírného a středně těžkého jaterního poškození byl studován u 8 pacientů s klasifikací Child-Pugh A a B a porovnáván se zdravými subjekty. Výsledky ukázaly, že jaterní poškození nemá podstatný vliv na AUC ani na Cmax methylnaltrexonium-bromidu. Vliv těžkého jaterního poškození na farmakokinetiku methylnaltrexonium-bromidu nebyl studován.
Pacienti s renálním poškozením
Ve studii s dobrovolníky s různým stupněm renálního poškození byla podávána jednotlivá dávka methylnaltrexonium-bromidu 0,30 mg/kg; renální poškození mělo značný účinek na renální vylučování methylnaltrexonium-bromidu. Renální clearance methylnaltrexonium-bromidu se snížila se zvýšenou závažností renálního poškození. Těžké renální poškození snížilo renální clearance methylnaltrexonium-bromidu 8 až 9-krát; to však vedlo pouze ke dvojnásobnému zvýšení celkové expozice (AUC) methylnaltrexonium-bromidu. Cmax se významně nezměnila. Nebyla provedena žádná studie u pacientů s renálním poškozením v terminálním stádiu vyžadujícím dialýzu.
Pediatrická populace
Nebyly provedeny žádné studie u pediatrické populace (viz bod 4.2).
Starší populace
Ve studii porovnávající farmakokinetické profily jednotlivé dávky a opakovaných dávek intravenozně podaného methylnaltrexonium-bromidu v dávce 24 mg zdravým mladým (věk 18-45 let, n=10) a starším (věk 65 let a více, n=10) subjektům byl shledán minimální vliv věku na účinky methylnatrexonium-bromidu. Střední Cmax a AUC v ustáleném stavu u starších subjektů byly 545 ng/ ml a 412 ng^hod/ml tj. přibližně o 8,1 % resp. o 20 % vyšší než u zdravých subjektů. Proto se nedoporučuje upravovat dávkování v závislosti na věku.
Pohlaví
Nebyly pozorovány podstatné rozdíly v závislosti na pohlaví.
Hmotnost
Integrovaná analýza farmakokinetických údajů od zdravých subjektů prokázala, že expozice upravené dávky methylnaltrexonium-bromidu mg/kg se zvyšuje se zvyšující se hmotností. Průměrná expozice methylnaltrexonium-bromidu v dávce 0,15 mg/kg při hmotnosti v rozmezí od 38 do 114 kg byla 179 (rozmezí = 139-240) ng^hod/ml. Této expozice při dávce 0,15 mg/kg je možno dosáhnout úpravou dávky podle hmotnosti, použitím dávky 8 mg při tělesné hmotnosti od 38 do méně než 62 kg a dávky 12 mg při tělesné hmotnosti od 62 do 114 kg; průměrná dosažená expozice činí 187 (rozmezí =148-220) ng^hod/ml. Analýza údajů založená na rozdělení pacientů ve studii 301 a 302 podle hmotnosti dále ukázala, že dávka 8 mg pro tělesnou hmotnost od 38 do méně než 62 kg odpovídá průměrné dávce 0,16 (rozmezí = 0,21-0,13) mg/kg a dávka 12 mg pro tělesnou hmotnost od 62 do 114 kg odpovídá průměrné dávce 0,16 (rozmezí = 0,19-0,11) mg/kg.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a kancerogenního potenciálu neodhalily žádné zvláštní riziko pro člověka. V některých neklinických studiích na psech byly pozorovány účinky na srdce (prodloužení akčního potenciálu Purkyňových vláken nebo prodloužení QTc intervalu). Mechanizmus tohoto účinku není znám; zdá se však, že lidský srdeční kaliový iontový kanál (hERG) se na tom nepodílí.
Subkutánní injekce přípravku Relistor v dávce 150 mg/kg/den vedly ke snížení fertility u potkanů. Dávky až do 25 mg/kg/den (18-krát vyšší než expozice [AUC] u člověka po subkutánní dávce 0,3 mg/kg) neovlivňovaly fertilitu ani celkovou reprodukční schopnost.
Nebyla prokázána teratogenita u potkanů ani králíků. Subkutánní injekce přípravku Relistor v dávce 150/100 mg/kg/den u potkanů vedly ke snížení porodní hmotnosti mláďat; dávky až do 25 mg/kg/den (18-krát vyšší než expozice [AUC] u člověka po subkutánní dávce 0,3 mg/kg) neměly žádný vliv na průběh porodu, porod, přežívání ani na růst mláďat.
Methylnaltrexonium-bromid se vylučuje do mateřského mléka u potkanů.
Studie byly provedeny u mláďat potkanů a psů. Po intravenózní injekci methylnaltrexonium-bromidu bylo zjištěno, že mláďata potkanů jsou citlivější na toxicitu související s methylnaltrexonium-bromidem než dospělí potkani. U mláďat potkanů, kterým byl podáván intravenózně methylnaltrexonium-bromid po dobu 13 týdnů, se nežádoucí klinické příznaky (incidence křečí a namáhavý dech) objevily při dávkách (> 3 mg/kg/den) a expozicích (5,4 krát vyšší než expozice {AUC} u dospělého člověka při subkutánní dávce 0,15 mg/kg) nižších než byly ty, které způsobily podobnou toxicitu u dospělých potkanů (20 mg/kg/den). U mláďat potkanů při dávce 1 mg/kg/den nebo u dospělých potkanů při dávce 5 mg/kg/den se nevyskytly žádné nežádoucí účinky (1,6násobek resp. 7,8násobek expozice {AUC} u dospělého člověka při subkutánní dávce 0,15 mg/kg).
Po podávání intravenózních injekcí methylnaltrexonium-bromidu po dobu 13 týdnů byla u mladých i dospělých psů pozorována podobná toxicita související s methylnaltrexonium- bromidem. U dospělých a mladých psů, kteří dostávali methylnaltrexonium-bromid v dávce 20 mg/kg/den, byly pozorovány klinické příznaky CNS toxicity a prodloužení intervalu QTc. U mláďat ani u dospělých psů se při dávce 5 mg/kg/den (44násobek expozice {AUC} u dospělého člověka při subkutánní dávce 0,15 mg/kg) nevyskytly žádné nežádoucí účinky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
chlorid sodný natrium-kalcium-edetát glycin-hydrochlorid voda na injekci
kyselina chlorovodíková (k úpravě pH) hydroxid sodný (k úpravě pH)
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mí sen s žádnými dalšími léčivými přípravky.
6.3 Doba použitelnosti
4 roky
Po natažení do injekční stříkačky:
Injekční roztok má být spotřebován do 24 hodin z důvodu citlivosti na světlo.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní teplotní podmínky uchovávání.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku v injekční stříkačce, viz bod 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička pro jedno použití z čirého křemenného skla, typ I, šedá pryžová zátka a hliníkový kryt s odtrhávacím víčkem.
Jedna injekční lahvička obsahuje 0,6 ml injekčního roztoku.
Velikosti balení
1 injekční lahvička
2 injekční lahvičky, 2 sterilní injekční stříkačky o objemu 1 ml se zasunovatelnou injekční jehlou a 4 alkoholové tampony; nebo
7 injekčních lahviček, 7 sterilních injekčních stříkaček o objemu 1 ml se zasunovatelnou injekční jehlou a 14 alkoholových tamponů;
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o.
Jankovcova 1569/2c 170 00, Praha 7 Česká republika
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/463/001
EU/1/08/463/002
EU/1/08/463/003
DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
9.
Datum první registrace: 2. července 2008
Datum posledního prodloužení registrace: 27. května 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Relistor 8 mg injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka 0,4 ml obsahuje 8 mg methylnaltrexonii bromidům. Jeden mililitr roztoku obsahuje 20 mg methylnaltrexonii bromidum.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce).
Čirý roztok, bezbarvý až světle žlutý, bez viditelných částic.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Relistor je určen k léčbě obstipace vyvolané opioidy, jestliže odpověď na léčbu laxativy není dostatečná u dospělých pacientů ve věku 18 let a starších.
4.2 Dávkování a způsob podání
Dávkování
Obstipace vyvolaná opioidy u dospělých pacientů s chronickou bolestí (kroměpacientů s pokročilým onemocněním, kteří dostávají paliativní péči)
Doporučená dávka methylnaltrexonium-bromidu je 12 mg (0,6 ml roztoku) subkutánně, podle potřeby, která je podávána jako minimálně 4 dávky týdně až maximálně jedenkrát denně (7 dávek za týden).
U těchto pacientů se má při zahájení léčby Relistorem ukončit léčba obvykle používanými laxativy (viz bod 5.1).
8mg předplněná injekční stříkačka Relistor se má použít jen na léčbu těch pacientů, jejichž aktuální zdravotní stav si vyžaduje snížení dávky na 8 mg (0,4 ml roztoku), viz Speciální populace.
Obstipace vyvolaná opioidy u dospělých pacientů s pokročilým onemocněním (pacienti, kteří dostávají paliativní péči)
Doporučená dávka methylnaltrexonium-bromidu je 8 mg (0,4 ml roztoku) (pro pacienty o hmotnosti 38-61 kg) nebo 12 mg (0,6 ml roztoku) (pro pacienty o hmotnosti 62-114 kg).
Obvyklé dávkovací schéma je jedna samostatná dávka každý druhý den. Dávky mohou být podány také v delších intervalech, podle klinické potřeby.
Pouze pokud se nedostavila odpověď (defekace/zrychlení střevní peristaltiky) po dávce podané předcházející den, mohou pacienti dostat dvě následné, po sobě jdoucí dávky, mezi nimiž je interval 24 hodin.
Pacienti o hmotnosti nižší než 38 kg nebo vyšší než 114 kg by měli použít přípravek Relistor injekční lahvičky, protože doporučená dávka mg/kg nemůže být přesně podána předplněnou injekční stříkačkou.
U pacientů s paliativní péčí se Relistor přidá k obvyklé léčbě laxativy (viz bod 5.1).
Speciální populace
Starší populace
Není třeba úprava dávek v závislosti na věku (viz bod 5.2).
Pacienti s renálním poškozením
U pacientů s těžkým renálním poškozením (clearance kreatininu nižší než 30 ml/min.) má být snížena dávka methylnaltrexonium-bromidu z 12 mg na 8 mg (0,4 ml roztoku) u pacientů o hmotnosti 62 až 114 kg. U pacientů s těžkým renálním poškozením, jejichž hmotnost je mimo rozmezí od 62 do 114 kg (viz bod 5.2), musí být jejich dávka v mg/kg snížena o 50 %. Tito pacienti by měli používat přípravek Relistor v injekční lahvičce a nikoli v předplněné injekční stříkačce. Nejsou dostupné údaje o pacientech s renálním poškozením v terminálním stádiu na dialýze a podání methylnaltrexonium-bromidu těmto pacientům se nedoporučuje (viz bod 4.4).
Pacienti s jaterním poškozením
U pacientů s mírným až středně těžkým jaterním poškozením není třeba úprava dávek (viz bod 5.2).
Nejsou dostupné údaje o pacientech s těžkým jaterním poškozením (Child-Pugh C) a podání methylnaltrexonium-bromidu těmto pacientům se nedoporučuje (viz bod 4.4).
Pediatrická populace
Účinnost a bezpečnost methylnaltrexonium-bromidu u dětí ve věku do 18 let nebyla stanovena. Nejsou k dispozici žádné údaje.
Způsob podání
Přípravek Relistor se podává jako subkutánní injekce.
Doporučuje se pravidelně měnit místa podání injekce. Nedoporučuje se podávat injekci do oblastí, kde je jemná kůže, podlitiny, zarudnutí nebo tvrdá kůže. Vyhněte se místům s jizvami nebo s narušenou strukturou kůže.
Tři doporučené oblasti těla pro injekci přípravku Relistor jsou horní část dolních končetin, břicho a horní část paží.
Přípravek Relistor může být podáván bez ohledu na příjem potravy.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Podávání methylnaltrexonium-bromidu pacientům se známou nebo suspektní mechanickou gastrointestinální obstrukcí nebo po akutní břišní chirurgické operaci je kontraindikováno.
4.4 Zvláštní upozornění a opatření pro použití
Závažnost a zhoršení příznaků
Pacienti mají být poučeni, aby neprodleně hlásili závažné, přetrvávající a/nebo zhoršující se potíže.
Dojde-li v průběhu léčení k vážnému nebo přetrvávajícímu průjmu, pacientovi má být doporučeno, aby nepokračoval v terapii methylnaltrexonium-bromidem a poradil se se svým lékařem.
Obstipace nesouvisející s používáním opioidů
Účinnost methylnaltrexonium-bromidu byla studována u pacientů s obstipací vyvolanou opioidy. Přípravek Relistor by neměl být proto používán u pacientů k léčbě obstipace, která s použitím opioidů nesouvisí.
Rychlý nástup defekace
Údaje z klinických studií svědčí o tom, že léčení methylnaltrexonium-bromidem může vést k rychlému nástupu (v průměru během 30 až 60 minut) defekace.
Trvání léčby
Obstipace vyvolaná opioidy u dospělých pacientů s pokročilým onemocněním Podávání methylnaltrexonium-bromidu nebylo u dospělých pacientů s pokročilým onemocněním v klinických studiích studováno déle než po dobu 4 měsíců, a proto by měl být podáván pouze po časově omezené období (viz bod 5.1).
Pacienti s poškozením funkce jater nebo ledvin
Podávání methylnaltrexonium-bromidu pacientům s těžkou poruchou funkce jater nebo pacientům s poruchou funkce ledvin v terminálním stádiu vyžadujícím dialýzu se nedoporučuje (viz bod 4.2).
Gastrointestinální (GI) onemocnění a GI perforace
Methylnaltrexonium-bromid se má používat s opatrností u pacientů s potvrzenými lézemi GI traktu nebo s podezřením na ně.
Použití methylnaltrexonium-bromidu u pacientů s kolostomií, peritoneálním katetrem, aktivní divertikulární chorobou nebo s fekálním zaklíněním nebylo studováno. Proto má být přípravek Relistor podáván těmto pacientům s opatrností.
Po uvedení na trh byly u pacientů, kterým byl methylnaltrexonium-bromid podáván, hlášeny případy gastrointestinální perforace. Přestože onemocnění, pro která byli pacienti léčeni, mohla být spojena s lokalizovaným nebo difúzním snížením strukturální integrity stěny gastrointestinálního traktu (např. maligní onemocnění, peptický vřed, pseudoobstrukce), methylnaltrexonium-bromid mohl k těmto příhodám přispět.
Obsah sodíku
Tento lék obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce (tzn. je v podstatě sodíku prostý).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Methylnaltrexonium-bromid neovlivňuje farmakokinetiku léčivých přípravků metabolizovaných isoenzymy cytochromu P450 (CYP). Methylnaltrexonium-bromid je minimálně metabolizován isoenzymy CYP. Metabolické studie in vitro ukazují, že methylnaltrexonium-bromid neinhibuje aktivitu CYP1A2, CYP2E1, CYP2B6, CYP2A6, CYP2C9, CYP2C19 ani CYP3A4, zatímco je slabým inhibitorem metabolizmu modelového substrátu CYP2D6. V klinické studii interakcí u zdravých dospělých mužských subjektů neovlivnila dávka methylnaltrexonium-bromidu 0,3 mg/kg s.c. signifikantně metabolizmus substrátu CYP2D6 dextrometorfanu.
Interakční potenciál transportérů organických kationtů (OCT) vztahující se k transportérům OCT methylnaltrexonium-bromidu a OCT inhibitoru byl studován u 18 zdravých subjektů porovnáním farmakokinetických profilů po jednotlivé dávce methylnaltrexonium-bromidu před podáním a po podání vícenásobných dávek 400 mg cimetidinu. Renální clearance methylnaltrexonium-bromidu byla po podání vícenásobných dávek cimetidinu snížena (z 31 L/hod. na 18 L/hod.). To však vedlo jen k malému snížení celkové clearance (ze 107 L/hod. na 95 L/hod.). Návazně nebyla pozorována žádná významná změna AUC methylnaltrexonium-bromidu ani Cmax před podáním a po podání vícenásobných dávek cimetidinu.
4.6 Fertilita, těhotenství a kojení
Nejsou dostatečné údaje o podávání methylnaltrexonium-bromidu těhotným ženám. Studie na zvířatech prokázaly reprodukční toxicitu při podávání vysokých dávek (viz bod 5.3.). Potenciální riziko pro člověka není známo. Methylnaltrexonium-bromid nemá být podáván v průběhu těhotenství, pokud to není zcela nezbytné.
Kojení
Není známo, zda je methylnalteroxonium-bromid vylučován do mateřského mléka. Studie na zvířatech prokázaly vylučování methylnaltrexonium-bromidu do mateřského mléka. Je proto třeba rozhodnout, zda pokračovat/přerušit kojení, nebo pokračovat/přerušit podávání methylnaltrexonium-bromidu, a přitom vzít v úvahu přínos kojení pro dítě a přínos terapie methylnaltrexonium-bromidem pro matku.
Fertilita
Subkutánní injekce přípravku Relistor v dávce 150 mg/kg/den snížily fertilitu u potkanů. Dávky do 25 mg/kg/den (18ti násobek expozice [AUC] u lidí při subkutánní dávce 0,3 mg/kg) neovlivnily fertilitu nebo celkovou schopnost reprodukce.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Methylnaltrexonium-bromid ma malý vliv na schopnost řídit nebo obsluhovat.
Může se vyskytnout závrať, a ta může ovlivnit schopnost řídit a obsluhovat stroje (viz bod 4.8).
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejčastějšími nežádoucími účinky vztaženými k léku u všech pacientů léčených methylnaltrexonium-bromidem v průběhu všech fází placebem kontrolovaných studií byly bolest břicha, nauzea, průjem a flatulence. Tyto účinky byly obecně mírné až středně silné.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky jsou klasifikovány jako: velmi časté (>1/10); časté (>1/100 až <1/10) ; méně časté (>1/1000 až <1/100); vzácné (>1/10 000 až <1/1000); velmi vzácné (<1/10 000) a frekvence neznámá (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti:
Poruchy nervového systému
Časté: Závrať
Časté: Mírné příznaky jako při náhlém přerušení léčby opioidy (kterýkoliv z uvedených příznaků: zimnice, třes, rhinorea, piloerekce, návaly horka, palpitace, hyperhidróza)
Gastrointestinální _ poruchy
Neznámé: Gastrointestinální perforace (viz bod 4.4)
Časté: Zvracení
Velmi časté: Bolest břicha, nauzea, flatulence, průjem
Poruchy kůže a podkožní tkáně
Časté: Reakce v místě podání (např. píchání, pálení, bolest, zčervenání, edém)
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Zdraví dobrovolníci ve studii zaznamenali ortostatickou hypotenzi po dávce 0,64 mg/kg, podané jako intravenózní bolus.
V případě předávkování mají být známky a příznaky ortostatické hypotenze monitorovány a hlášeny lékaři. Léčení by mělo být zahájeno podle potřeby.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Laxativa, Antagonisté periferních opioidních receptorů, ATC kód: A06AH01
Mechanismus účinku
Methylnaltrexonium-bromid je selektivní antagonista vazby opioidů na mu-receptor. In vitro studie prokázaly, že methylnaltrexonium-bromid je antagonista opioidního mu-receptoru (inhibiční konstanta [Ki] = 28 nM) s 8 x nižším účinkem na kappa opioidní receptory (Ki = 230 nM) a mnohokrát nižší afinitou k opioidním delta receptorům.
Jako kvarterní amin má methylnaltrexonium-bromid omezenou schopnost přecházet přes hematoencefalickou bariéru. To umožňuje methylnaltrexonium-bromidu fungovat jako periferně účinkující mu-opioidní antagonista ve tkáních, jako je gastrointestinální trakt, aniž by ovlivnil analgetický účinek vyvolaný opioidy v centrálním nervovém systému.
Klinická účinnost a bezpečnost
Obstipace vyvolaná opioidy u dospělých pacientů s chronickou nenádorovou bolestí Účinnost a bezpečnost methylnaltrexonium-bromidu při léčbě obstipace vyvolané opioidy u pacientů s chronickou nenádorovou bolestí byla prokázána v randomizované, dvojitě zaslepené, placebem kontrolované studii (Studie 3356). V této studii byl medián věku pacientů 49 let (rozsah 23-83); 60 % bylo žen. Primární diagnózou většiny pacientů byla bolest zad.
Studie 3356 porovnávala 4-týdenní podávání 12 mg methylnaltrexonium-bromidu jedenkrát denně a 12 mg methylnaltrexonium-bromidu každý druhý den s placebem. Po 4-týdenním, dvojitě zaslepeném období následovala otevřená 8 týdenní fáze, ve které se methylnaltrexonium-bromid měl používat podle potřeby, ale ne víc než 1 dávka denně. Celkově bylo v dvojitě zaslepené fázi léčeno 460 pacientů (12 mg methylnaltrexonium-bromidu jedenkrát denně, n=150, 12 mg methylnaltrexonium-bromidu každý druhý den, n=148, placebo, n=162). Pacienti trpěli v minulosti chronickou nenádorovou bolestí a užívali stabilní dávku opioidu alespoň 50 mg perorálního ekvivalentu morfinu denně. Pacienti měli obstipaci vyvolanou opioidy (během období screeningu měli <3 stolice týdně bez podání záchranné léčby). Pacienti museli splnit požadavek, aby vysadili všechna dříve užívaná laxativa.
Prvním koprimámím ukazovatelem byl podíl pacientů s defekací bez záchranných laxativ (RFBM) během 4 hodin po podání první dávky a druhým byl percentuelní podíl aktivních injekcí, po kterých došlo k RFBM během 4 hodin v průběhu dvojitě zaslepené fáze. RFBM byla definována jako stolice, ke které došlo bez použití laxativ během předchozích 24 hodin.
Podíl pacientů, u kterých došlo k RFBM během 4 hodin po podání první dávky, byl 34,2 % v kombinované skupině s methylnaltrexonium-bromidem versus 9,9 % ve skupině s placebem (p<0,001). Průměrný percentuelní podíl podání methylnaltrexonium-bromidu, jehož důsledkem byla RFBM během 4 hodin, byl 28,9 % ve skupině s podáváním jedenkrát denně a 30,2 % ve skupině s podáváním každý druhý den v porovnání s 9,4 % a 9,3 % při podávání odpovídajícího režimu s placebem (p < 0,001).
Klíčový sekundární ukazatel upravené průměrné změny týdenních RFBM v porovnání s hodnotou při vstupu byl 3,1 v léčebné skupině s 12 mg methylnaltrexonium-bromidu jedenkrát denně, 2,1 v léčebné skupině s 12 mg methylnaltrexonium-bromidu každý druhý den a 1,5 v léčebné skupině s placebem během 4 týdenního dvojitě zaslepeného období. Rozdíl 1,6 RFBM za týden mezi 12 mg methylnaltrexonium-bromidu jedenkrát denně a placebem je statisticky významný (p < 0,001) a má význam i v klinické praxi.
Další sekundární ukazatel hodnotil podíl pacientů s >3 RFBM za týden během 4-týdenní dvojitě zaslepené fáze. Ten byl dosažen u 59 % pacientů ve skupině, která dostávala 12 mg methylnatrexonu denně (p<0,001 versus placebo), u 61 % pacientů, kteří ho dostávali každý druhý den (p<0,001 versus placebo), a u 38 % pacientů, kteří dostávali placebo. Doplňková analýza hodnotila percentuelní podíl pacientů, kteří dosáhli >3 úplných RFBM za týden a zvýšení o >1 úplných RFBM za týden alespoň ve 3 týdnech ze 4 týdnů léčby. Toto bylo dosaženo u 28,7 % pacientů ve skupině s podáváním 12 mg methylnatrexonium-bromidu denně (p<0,001 versus placebo), u 14,9 % pacientů, kteří ho dostávali každý druhý den (p=0,012 versus placebo), a u 6,2 % pacientů léčených placebem.
Nebyl získaný důkaz o rozdílech v účinnosti a bezpečnosti způsobených pohlavím. Vliv etnického původu nebylo možné analyzovat, protože populace byla především bělošská (90 %). Medián denní dávky opioidů se nijak významně neodlišoval od vstupních hodnot ani u pacientů užívajících methylnaltrexonium-bromid ani u pacientů dostávajících placebo.
Nezjistili se žádné klinicky významné změny v hodnocení bolesti v porovnání se vstupními hodnotami ani u pacientů užívajících methylnaltrexonium-bromid, ani u pacientů dostávajících placebo.
V klinických hodnoceních se nehodnotilo používání methylnaltrexonium-bromidu při léčbě obstipace vyvolané opioidy delší než 48 týdnů.
Obstipace vyvolaná opioidy u dospělých pacientů s pokročilým onemocněnímÚčinnost a bezpečnost methylnaltrexonium-bromidu při léčbě obstipace vyvolané opioidy u pacientů, kteří dostávají paliativní péči, byla prokázána ve dvou randomizovaných dvojitě zaslepených, placebem kontrolovaných studiích. V těchto studiích byl střední věk 68 let (rozmezí 21-100); 51 % byly ženy. Pacienti v obou studiích měli pokročilé terminální onemocnění s omezenou předpokládanou délkou přežití, ve většině případů s primární diagnózou nevyléčitelné rakoviny; ostatní primární diagnózy zahrnovaly konečné stádium CHOPN/emfyzém, onemocnění srdce/srdeční selhání, Alzheimerovu chorobu/demenci, HIV/AIDS a jiná onemocnění v pokročilém stádiu. Před screeningem měli pacienti opioidy vyvolanou obstipaci definovanou buď jako <3 stolice v předchozím týdnu, nebo žádná stolice po >2 dny.
Studie 301 porovnávala jednorázovou dvojitě zaslepenou subkutánní dávku methylnaltrexonium-bromidu 0,15 mg/kg, nebo 0,3mg/kg versus placebo. Po dvojitě zaslepené dávce následovala otevřená 4-týdenní perioda, během které mohl být podáván methylnaltrexonium-bromid podle potřeby, avšak ne častěji než 1 x za 24 hodin. Během obou period studie udržovali pacienti svůj obvyklý laxativní režim. V periodě dvojitě zaslepené studie bylo celkově zařazeno a léčeno 154 pacientů (47 dostalo methylnaltrexonium-bromid 0,15 mg/kg, 55 dostalo methylnaltrexonium-bromid 0,3 mg/kg, 52 dostalo placebo). Primárním cílovým parametrem byl podíl pacientů s defekací bez záchranné laxativní léčby do 4 hodin po podání dvojitě zaslepené dávky studovaného léčivého přípravku. Ve skupině léčené methylnaltrexonium-bromidem byl signifikantně vyšší podíl pacientů s defekací do 4 hodin po dvojitě zaslepené dávce (62 % u 0,15 mg/kg a 58 % u 0,3 mg/kg), než ve skupině léčené placebem (14 %); p<0,0001 pro každou dávku versus placebo.
Studie 302 porovnávala dvojitě zaslepené subkutánní dávky methylnaltrexonium-bromidu podané každý druhý den po 2 týdny proti placebu. V průběhu prvního týdne (dny 1., 3., 5., 7.) dostali pacienti buď 0,15 mg/kg methylnaltrexonium-bromidu, nebo placebo. Ve druhém týdnu mohla být dávka zvýšena na 0,30 mg/kg, pokud měl pacient do 8. dne bez použití záchranné dávky projímadla 2 nebo méně stolic. Předepsaná dávka mohla být pacientovi kdykoli snížena v závislosti na toleranci. Byly analyzovány údaje od 133 pacientů (62 užívalo methylnaltrexonium-bromidu, 71 užívalo placebo). Studie měla dva primární cíle: Podíl pacientů s defekací bez záchranné laxativní medikace do 4 hodin po první dávce léčivého přípravku a podíl pacientů s defekací bez záchranné laxativní medikace do 4 hodin po nejméně 2 z prvních 4 dávek léčivého přípravku. U pacientů léčených methylnaltrexonium-bromidem byl vyšší podíl defekací do 4 hodin po první dávce (48 %) než u pacientů léčených placebem (16 %); p<0,0001. Pacienti léčeni methylnaltrexonium-bromidem měli také signifikantně vyšší počet defekací do 4 hodin po podání alespoň 2 ze 4 dávek (52 %) než pacienti léčení placebem (9 %); p<0,0001. Konsistence stolice u pacientů, kteří měli zpočátku měkkou stolici, se významně nezlepšila.
V obou studiích nebyl prokázán rozdíl v účinnosti ani v bezpečnosti v závislosti na věku nebo pohlaví. Účinek nebylo možno analyzovat podle rasy, protože populace ve studii byla převážně bělošská
(88 %).
Trvání odpovědi bylo demonstrováno ve studii 302, v níž byl počet laxačních odpovědí konzistentní od dávky 1 až po dávku 7 v průběhu celé dvoutýdenní dvojitě zaslepené periody.
Účinnost a bezpečnost methylnaltrexonium-bromidu byly demonstrovány také v otevřené studii 301, při níž bylo léčivo podáváno od 2. dne po 4 týdny, a ve 2 otevřených rozšířených studiích (301EXT a 302EXT), v nichž byl methylnaltrexonium-bromid podáván podle potřeby po dobu až 4 měsíců (po tuto dobu pouze 8 pacientů). Celkově 136 pacientů ve studii 301, 21 pacientů ve studii 301EXT a 82 pacientů ve studii 302EXT dostalo nejméně jednu léčebnou dávku v otevřené studii. Přípravek Relistor byl podáván každých 3,2 dnů (medián dávkovacího intervalu s rozmezím 1-39 dnů).
Počet laxačních odpovědí byl udržován po dobu rozšířené studie u těch pacientů, kteří pokračovali v léčení.
V těchto studiích nebyl zjištěn žádný signifikantní vztah mezi úvodní dávkou opiátu a laxační odpovědí u pacientů léčených methylnaltrexonium-bromidem. Navíc střední dávka opiátu ve skupině pacientů léčených methylnaltrexonium-bromidem a pacientů léčených placebem se podstatně nelišila od úvodních dávek. Nedošlo ke klinicky relevantní změně skóre bolesti od úvodních hodnot ani ve skupině léčené methylnaltrexonium-bromidem, ani ve skupině léčené placebem.
Účinek na srdeční repolarizaci
Ve dvojitě zaslepené randomizované EKG studii s paralelní skupinou jednotlivá dávka methylnaltrexonium-bromidu (0,15; 0,30 a 0,50 mg/kg) u 207 zdravých dobrovolníků neprodloužila interval QT/QTc ani neovlivnila sekundární parametry EKG a tvar křivky byl srovnatelný s placebem a s pozitivní kontrolou (perorálně podaný moxifloxacin 400 mg).
5.2 Farmakokinetické vlastnosti
Absorpce
Methylnaltrexonium-bromid se po subkutánním podání rychle absorbuje a dosahuje maximální koncentrace (Cmax) přibližně po 0,5 hodině. Cmax a plocha pod křivkou koncentrace (AUC) se zvyšují úměrně se zvyšující se dávkou od 0,15 mg/kg do 0,5 mg/kg. Absolutní biologická dostupnost subkutánní dávky 0,30 mg/kg ve srovnání s intravenozní dávkou 0,30 mg/kg je 82 %.
Distribuce
Methylnaltrexonium-bromid podléhá středně silné distribuci do tkání. Distribuční objem v ustáleném stavu (Vss) je přibližně 1,1 l/kg. Methylnaltrexonium-bromid se minimálně váže na lidské plazmatické proteiny (11,0 % až 15,3 %), jak bylo zjištěno rovnovážnou dialýzou.
Biotransformace
Podle množství metabolitů získaných z exkretů je methylnaltrexonium-bromid u lidí metabolizován v malé míře. Zdá se, že primární metabolickou cestou je přeměna na isomery metyl-6-naltrexolu a na methylnatrexon-sulfát. Každý z izomerů metyl-6-naltrexolu má poněkud nižší antagonistický účinek než mateřská sloučenina a nižší expozici v plazmě ve výši přibližně 8 % z původního léčiva. Methylnaltrexon-sulfát je neúčinný metabolit a v plazmě je přítomný v hladině přibližně 25 % z původního léčiva. N-demetylace methylnaltrexonu na naltrexon není významná a činí asi 0,06 % podané dávky.
Eliminace
Methylnaltrexonium-bromid je vylučován převážně ve formě nezměněné léčivé látky. Přibližně polovina dávky je vyloučena močí a o něco méně stolicí. Konečný poločas vylučování^) je přibližně 8 hodin.
Zvláštní populace
Pacienti s jaterním poškozením
Celkový účinek methylnaltrexonium-bromidu u mírného a středně těžkého jaterního poškození byl studován u 8 pacientů s klasifikací Child-Pugh A a B a porovnáván se zdravými subjekty. Výsledky ukázaly, že jaterní poškození nemá podstatný vliv na AUC ani na Cmax methylnaltrexonium-bromidu. Vliv těžkého jaterního poškození na farmakokinetiku methylnaltrexonium-bromidu nebyl studován.
Pacienti s renálním poškozením
Ve studii s dobrovolníky s různým stupněm renálního poškození byla podávána jednotlivá dávka methylnaltrexonium-bromidu 0,30 mg/kg; renální poškození mělo značný účinek na renální vylučování methylnaltrexonium-bromidu. Renální clearance methylnaltrexonium-bromidu se snížila se zvýšenou závažností renálního poškození. Těžké renální poškození snížilo renální clearance methylnaltrexonium-bromidu 8 až 9-krát; to však vedlo pouze ke dvojnásobnému zvýšení celkové expozice (AUC) methylnaltrexonium-bromidu. Cmax se významně nezměnila. Nebyla provedena žádná studie u pacientů s renálním poškozením v terminálním stádiu vyžadujícím dialýzu.
Pediatrická populace
Nebyly provedeny žádné studie u pediatrické populace (viz bod 4.2).
Starší populace
Ve studii porovnávající farmakokinetické profily jednotlivé dávky a opakovaných dávek intravenozně podaného methylnaltrexonium-bromidu v dávce 24 mg zdravým mladým (věk 18-45 let, n=10) a starším (věk 65 let a více, n=10) subjektům byl shledán minimální vliv věku na účinky methylnatrexonium-bromidu. Střední Cmax a AUC v ustáleném stavu u starších subjektů byly 545 ng/ ml a 412 ng^hod/ml, tj. přibližně o 8,1 % resp. o 20 % vyšší než u zdravých subjektů. Proto se nedoporučuje upravovat dávkování v závislosti na věku.
Pohlaví
Nebyly pozorovány podstatné rozdíly v závislosti na pohlaví.
Hmotnost
Integrovaná analýza farmakokinetických údajů od zdravých subjektů prokázala, že expozice upravené dávky methylnaltrexonium-bromidu mg/kg se zvyšuje se zvyšující se hmotností. Průměrná expozice methylnaltrexonium-bromidu v dávce 0,15 mg/kg při hmotnosti v rozmezí od 38 do 114 kg byla 179 (rozmezí = 139-240) ng^hod/ml. Této expozice při dávce 0,15 mg/kg je možno dosáhnout úpravou dávky podle hmotnosti, použitím dávky 8 mg při tělesné hmotnosti od 38 do méně než 62 kg a dávky 12 mg při tělesné hmotnosti od 62 do 114 kg; průměrná dosažená expozice činí 187 (rozmezí = 148220) ng^hod/ml. Analýza údajů založená na rozdělení pacientů ve studii 301 a 302 podle hmotnosti dále ukázala, že dávka 8 mg pro tělesnou hmotnost od 38 do méně než 62 kg odpovídá průměrné dávce 0,16 (rozmezí = 0,21-0,13) mg/kg a dávka 12 mg pro tělesnou hmotnost od 62 do 114 kg odpovídá průměrné dávce 0,16 (rozmezí = 0,19-0,11) mg/kg.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a kancerogenního potenciálu neodhalily žádné zvláštní riziko pro člověka. V některých neklinických studiích na psech byly pozorovány účinky na srdce (prodloužení akčního potenciálu Purkyňových vláken nebo prodloužení QTc intervalu). Mechanizmus tohoto účinku není znám; zdá se však, že lidský srdeční kaliový iontový kanál (hERG) se na tom nepodílí.
Subkutánní injekce přípravku Relistor v dávce 150 mg/kg/den vedly ke snížení fertility u potkanů. Dávky až do 25 mg/kg/den (18-krát vyšší než expozice [AUC] u člověka po subkutánní dávce 0,3 mg/kg) neovlivňovaly fertilitu ani celkovou reprodukční schopnost.
Nebyla prokázána teratogenita u potkanů ani králíků. Subkutánní injekce přípravku Relistor v dávce 150/100 mg/kg/den u potkanů vedly ke snížení porodní hmotnosti mláďat; dávky až do 25 mg/kg/den (18-krát vyšší než expozice [AUC] u člověka po subkutánní dávce 0,3 mg/kg) neměly žádný vliv na průběh porodu, porod, přežívání ani na růst mláďat.
Methylnaltrexonium-bromid se vylučuje do mateřského mléka u potkanů.
Studie byly provedeny u mláďat potkanů a psů. Po intravenózní injekci methylnaltrexonium-bromidu bylo zjištěno, že mláďata potkanů jsou citlivější na toxicitu související s methylnaltrexonium-bromidem než dospělí potkani. U mláďat potkanů, kterým byl podáván intravenózně methylnaltrexonium-bromid po dobu 13 týdnů, se nežádoucí klinické příznaky (incidence křečí a namáhavý dech) objevily při dávkách (> 3 mg/kg/den) a expozicích (5,4 krát vyšší než expozice {AUC} u dospělého člověka při subkutánní dávce 0,15 mg/kg) nižších než byly ty, které způsobily podobnou toxicitu u dospělých potkanů (20 mg/kg/den). U mláďat potkanů při dávce 1 mg/kg/den nebo u dospělých potkanů při dávce 5 mg/kg/den se nevyskytly žádné nežádoucí účinky (1,6násobek resp. 7,8násobek expozice {AUC} u dospělého člověka při subkutánní dávce 0,15 mg/kg).
Po podávání intravenózních injekcí methylnaltrexonium-bromidu po dobu 13 týdnů byla u mladých i dospělých psů pozorována podobná toxicita související s methylnaltrexonium- bromidem. U dospělých a mladých psů, kteří dostávali methylnaltrexonium-bromid v dávce 20 mg/kg/den, byly pozorovány klinické příznaky CNS toxicity a prodloužení intervalu QTc. U mláďat ani u dospělých psů se při dávce 5 mg/kg/den (44násobek expozice {AUC} u dospělého člověka při subkutánní dávce 0,15 mg/kg) nevyskytly žádné nežádoucí účinky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
chlorid sodný natrium-kalcium-edetát glycin-hydrochlorid voda na injekci
kyselina chlorovodíková (k úpravě pH) hydroxid sodný (k úpravě pH)
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky.
6.3 Doba použitelnosti
18 měsíců
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
Jedna předplněná injekční stříkačka obsahuje 0,4 ml injekčního roztoku.
Předplněná injekční stříkačka z čirého skla typu I s jehlou z nerezové oceli, táhla z plastické hmoty a pevnou krytkou jehly z pryže.
Velikost balení: 4, 7, 8 nebo 10 předplněných inječkních stříkaček.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o.
Jankovcova 1569/2c 170 00, Praha 7 Česká republika
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/463/004
EU/1/08/463/005
EU/1/08/463/006
EU/1/08/463/007
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. července 2008
Datum posledního prodloužení registrace: 27. května 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Relistor 12 mg injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ"
Jedna předplněná injekční stříkačka 0,6 ml obsahuje 12 mg methylnaltrexonii bromidům. Jeden mililitr roztoku obsahuje 20 mg methylnaltrexonii bromidům.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce).
Čirý roztok, bezbarvý až světle žlutý, bez viditelných částic.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Relistor je určen k léčbě obstipace vyvolané opioidy, jestliže odpověď na léčbu laxativy není dostatečná u dospělých pacientů ve věku 18 let a starších,
4.2 Dávkování a způsob podání
Dávkování
Obstipace vyvolaná opioidy u dospělých pacientů s chronickou bolestí (kroměpacientů s pokročilým onemocněním, kteří dostávají paliativní péči)
Doporučená dávka methylnaltrexonium-bromidu je 12 mg (0,6 ml roztoku) subkutánně, podle potřeby, která je podávána jako minimálně 4 dávky týdně až maximálně jedenkrát denně (7 dávek za týden).
U těchto pacientů se má při zahájení léčby Relistorem ukončit léčba obvykle používanými laxativy (viz bod 5.1).
Obstipace vyvolaná opioidy u dospělých pacientů s pokročilým onemocněním (pacienti, kteří dostávají paliativní péči)
Doporučená dávka methylnaltrexonium-bromidu je 8 mg (0,4 ml roztoku) (pro pacienty o hmotnosti 38-61 kg) nebo 12 mg (0,6 ml roztoku) (pro pacienty o hmotnosti 62-114 kg).
Obvyklé dávkovací schéma je jedna samostatná dávka každý druhý den. Dávky mohou být podány také v delších intervalech, podle klinické potřeby.
Pouze pokud se nedostavila odpověď (defekace/zrychlení střevní peristaltiky) po dávce podané předcházející den, mohou pacienti dostat dvě následné, po sobě jdoucí dávky, mezi nimiž je interval 24 hodin.
Pacienti o hmotnosti nižší než 38 kg nebo vyšší než 114 kg by měli použít přípravek Relistor injekční lahvičky, protože doporučená dávka mg/kg nemůže být přesně podána předplněnou injekční stříkačkou.
U pacientů s paliativní péčí se Relistor přidá k obvyklé léčbě laxativy (viz bod 5.1).
Speciální populace
Starší populace
Není třeba úprava dávek v závislosti na věku (viz bod 5.2).
Pacienti s renálním poškozením
U pacientů s těžkým renálním poškozením (clearance kreatininu nižší než 30 ml/min.) má být snížena dávka methylnaltrexonium-bromidu z 12 mg na 8 mg (0,4 ml roztoku) u pacientů o hmotnosti 62 až 114 kg. U pacientů s těžkým renálním poškozením, jejichž hmotnost je mimo rozmezí od 62 do 114 kg (viz bod 5.2), musí být jejich dávka v mg/kg snížena o 50 %. Tito pacienti by měli používat přípravek Relistor v injekční lahvičce a nikoli v předplněné injekční stříkačce. Nejsou dostupné údaje o pacientech s renálním poškozením v terminálním stádiu na dialýze a podání methylnaltrexonium-bromidu těmto pacientům se nedoporučuje (viz bod 4.4).
Pacienti s jaterním poškozením
U pacientů s mírným až středně těžkým jaterním poškozením není třeba úprava dávek (viz bod 5.2).
Nejsou dostupné údaje o pacientech s těžkým jaterním poškozením (Child-Pugh C) a podání methylnaltrexonium-bromidu těmto pacientům se nedoporučuje (viz bod 4.4).
Pediatrická populace
Účinnost a bezpečnost methylnaltrexonium-bromidu u dětí ve věku do 18 et nebyla stanovena. Nejsou k dispozici žádné údaje.
Způsob podání
Přípravek Relistor se podává jako subkutánní injekce.
Doporučuje se pravidelně měnit místa podání injekce. Nedoporučuje se podávat injekci do oblastí, kde je jemná kůže, podlitiny, zarudnutí nebo tvrdá kůže. Vyhněte se místům s jizvami nebo s narušenou strukturou kůže.
Tři doporučené oblasti těla pro injekci přípravku Relistor jsou horní část dolních končetin, břicho a horní část paží.
Přípravek Relistor může být podáván bez ohledu na příjem potravy.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Podávání methylnaltrexonium-bromidu pacientům se známou nebo suspektní mechanickou gastrointestinální obstrukcí nebo po akutní břišní chirurgické operaci je kontraindikováno.
4.4 Zvláštní upozornění a opatření pro použití
Závažnost a zhoršení příznaků
Pacienti mají být poučeni, aby neprodleně hlásili závažné, přetrvávající a/nebo zhoršující se potíže.
Dojde-li v průběhu léčení k vážnému nebo přetrvávajícímu průjmu, pacientovi má být doporučeno, aby nepokračoval v terapii methylnaltrexonium-bromidema poradil se se svým lékařem.
Obstipace nesouvisející s používáním opioidů
Účinnost methylnaltrexonium-bromidu byla studována u pacientů s obstipací vyvolanou opioidy. Přípravek Relistor by neměl být proto používán u pacientů k léčbě obstipace, která s použitím opioidů nesouvisí.
Rychlý nástup defekace
Údaje z klinických studií svědčí o tom, že léčení methylnaltrexonium-bromidem může vést k rychlému nástupu (v průměru během 30 až 60 minut) defekace.
Trvání léčby
Obstipace vyvolaná opioidy u dospělých pacientů s pokročilým onemocněním Podávání methylnaltrexonium-bromidu nebylo u dospělých pacientů s pokročilým onemocněním v klinických studiích studováno déle než po dobu 4 měsíců, a proto by měl být podáván pouze po časově omezené období (viz bod 5.1
Pacienti s poškozením funkce jater nebo ledvin
Podávání methylnaltrexonium-bromidu pacientům s těžkou poruchou funkce jater nebo pacientům s poruchou funkce ledvin v terminálním stádiu vyžadujícím dialýzu se nedoporučuje (viz bod 4.2).
Gastrointestinální (GI) onemocnění a GI perforace
Methylnaltrexonium-bromid se má používat s opatrností u pacientů s potvrzenými lézemi GI traktu nebo s podezřením na ně.
Použití methylnaltrexonium-bromidu u pacientů s kolostomií, peritoneálním katetrem, aktivní divertikulární chorobou nebo s fekálním zaklíněním nebylo studováno. Proto má být přípravek Relistor podáván těmto pacientům s opatrností.
Po uvedení na trh byly u pacientů, kterým byl přípravek methylnaltrexonium-bromid podáván, hlášeny případy gastrointestinální perforace. Přestože onemocnění, pro která byli pacienti léčeni, mohla být spojena s lokalizovaným nebo difúzním snížením strukturální integrity stěny gastrointestinálního traktu (např. maligní onemocnění, peptický vřed, pseudoobstrukce), methylnaltrexonium-bromid mohl k těmto příhodám přispět.
Obsah sodíku
Tento lék obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce (tzn. je v podstatě sodíku prostý).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Methylnaltrexonium-bromid neovlivňuje farmakokinetiku léčivých přípravků metabolizovaných isoenzymy cytochromu P450 (CYP). Methylnaltrexonium-bromid je minimálně metabolizován isoenzymy CYP. Metabolické studie in vitro ukazují, že methylnaltrexonium-bromid neinhibuje aktivitu CYP1A2, CYP2E1, CYP2B6, CYP2A6, CYP2C9, CYP2C19 ani CYP3A4, zatímco je slabým inhibitorem metabolizmu modelového substrátu CYP2D6. V klinické studii interakcí u zdravých dospělých mužských subjektů neovlivnila dávka methylnaltrexonium-bromidu 0,3 mg/kg s.c. signifikantně metabolizmus substrátu CYP2D6 dextrometorfanu.
Interakční potenciál transportérů organických kationtů (OCT) vztahující se k transportérům OCT methylnaltrexonium-bromidu a OCT inhibitoru byl studován u 18 zdravých subjektů porovnáním farmakokinetických profilů po jednotlivé dávce methylnaltrexonium-bromidu před podáním a po podání vícenásobných dávek 400 mg cimetidinu. Renální clearance methylnaltrexonium-bromidu byla po podání vícenásobných dávek cimetidinu snížena (z 31 l/hod. na 18 l/hod.). To však vedlo jen k malému snížení celkové clearance (ze 107 l/hod. na 95 l/hod.). Návazně nebyla pozorována žádná významná změna AUC methylnaltrexonium-bromidu ani Cmax před podáním a po podání vícenásobných dávek cimetidinu.
4.6 Fertilita, těhotenství a kojení
Nejsou dostatečné údaje o podávání methylnaltrexonium-bromidu těhotným ženám. Studie na zvířatech prokázaly reprodukční toxicitu při podávání vysokých dávek (viz bod 5.3.). Potenciální riziko pro člověka není známo. Methylnaltrexonium-bromid nemá být podáván v průběhu těhotenství, pokud to není zcela nezbytné.
Kojení
Není známo, zda je methylnalteroxonium-bromid vylučován do mateřského mléka. Studie na zvířatech prokázaly vylučování methylnaltrexonium-bromidu do mateřského mléka. Je proto třeba rozhodnout, zda pokračovat/přerušit kojení, nebo pokračovat/přerušit podávání methylnaltrexonium-bromidu, a přitom vzít v úvahu přínos kojení pro dítě a přínos terapie methylnaltrexonium-bromidem pro matku.
Fertilita
Subkutánní injekce přípravku Relistor v dávce 150 mg/kg/den snížily fertilitu u potkanů. Dávky do 25 mg/kg/den (18ti násobek expozice [AUC] u lidí při subkutánní dávce 0,3 mg/kg) neovlivnily fertilitu nebo celkovou schopnost reprodukce.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Methylnaltrexonium-bromid ma malý vliv na schopnost řídit nebo obsluhovat stroje.
Může se vyskytnout závrať, a ta může ovlivnit schopnost řídit a obsluhovat stroje (viz bod 4.8).
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejčastějšími nežádoucími účinky u všech pacientů léčených methylnaltrexonium-bromidem v průběhu všech fází placebem kontrolovaných studií byly bolest břicha, nauzea, průjem a flatulence. Tyto účinky byly obecně mírné až středně silné.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky jsou klasifikovány jako: velmi časté (>1/10); časté (>1/100 až <1/10) ; méně časté (>1/1000 až <1/100); vzácné (>1/10 000 až <1/1000); velmi vzácné (<1/10 000) a frekvence neznámá (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti:
Poruchy nervového systému
Časté: Závrať
Časté: Mírné příznaky jako při náhlém přerušení léčby opioidy (kterýkoliv z uvedených příznaků: zimnice, třes, rhinorea, piloerekce, návaly horka, palpitace, hyperhidróza)
Gastrointestinální _ poruchy
Neznámé: Gastrointestinální perforace (viz bod 4.4)
Časté: Zvracení
Velmi časté: Bolest břicha, nauzea, flatulence, průjem
Poruchy kůže a podkožní tkáně
Časté: Reakce v místě podání (např. píchání, pálení, bolest, zčervenání, edém)
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Zdraví dobrovolníci ve studii zaznamenali ortostatickou hypotenzi po dávce 0,64 mg/kg, podané jako intravenózní bolus.
V případě předávkování mají být známky a příznaky ortostatické hypotenze monitorovány a hlášeny lékaři. Léčení by mělo být zahájeno podle potřeby.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Laxativa, Antagonisté periferních opioidních receptorů, ATC kód: A06AH01
Mechanismus účinku
Methylnaltrexonium-bromid je selektivní antagonista vazby opioidů na mu-receptor. In vitro studie prokázaly, že methylnaltrexonium-bromid je antagonista opioidního mu-receptoru (inhibiční konstanta [Ki] = 28 nM) s 8 x nižším účinkem na kappa opioidní receptory (Ki = 230 nM) a mnohokrát nižší afinitou k opioidním delta receptorům.
Jako kvarterní amin má methylnaltrexonium-bromid omezenou schopnost přecházet přes hematoencefalickou bariéru. To umožňuje methylnaltrexonium-bromidu fungovat jako periferně účinkující mu-opioidní antagonista ve tkáních, jako je gastrointestinální trakt, aniž by ovlivnil analgetický účinek vyvolaný opioidy v centrálním nervovém systému.
Klinická účinnost a bezpečnost
Obstipace vyvolaná opioidy u dospělých pacientů s chronickou nenádorovou bolestí Účinnost a bezpečnost methylnaltrexonium-bromidu při léčbě obstipace vyvolané opioidy u pacientů s chronickou nenádorovou bolestí byla prokázána v randomizované, dvojitě zaslepené, placebem kontrolované studii (Studie 3356). V této studii byl medián věku pacientů 49 let (rozsah 23-83); 60 % bylo žen. Primární diagnózou většiny pacientů byla bolest zad.
Studie 3356 porovnávala 4-týdenní podávání 12 mg methylnaltrexonium-bromidu jedenkrát denně a 12 mg methylnaltrexonium-bromidu každý druhý den s placebem. Po 4-týdenním, dvojitě zaslepeném období následovala otevřená 8 týdenní fáze, ve které se methylnaltrexonium-bromid měl používat podle potřeby, ale ne víc než 1 dávka denně. Celkově bylo v dvojitě zaslepené fázi léčeno 460 pacientů (12 mg methylnaltrexonium-bromidu jedenkrát denně, n=150, 12 mg methylnaltrexonium-bromidu každý druhý den, n=148, placebo, n=162). Pacienti trpěli v minulosti chronickou nenádorovou bolestí a užívali stabilní dávku opioidu alespoň 50 mg perorálního ekvivalentu morfinu denně. Pacienti měli obstipaci vyvolanou opioidy (během období screeningu měli <3 stolice týdně bez podání záchranné léčby). Pacienti museli splnit požadavek, aby vysadili všechna dříve užívaná laxativa.
Prvním koprimárním ukazovatelem byl podíl pacientů s defekací bez záchranných laxativ (RFBM) během 4 hodin po podání první dávky a druhým byl percentuelní podíl aktivních injekcí, po kterých došlo k RFBM během 4 hodin v průběhu dvojitě zaslepené fáze. RFBM byla definována jako stolice, ke které došlo bez použití laxativ během předchozích 24 hodin.
Podíl pacientů, u kterých došlo k RFBM během 4 hodin po podání první dávky, byl 34,2 % v kombinované skupině s methylnaltrexonium-bromidem versus 9,9 % ve skupině s placebem (p<0,001). Průměrný percentuelní podíl podání methylnaltrexonium-bromidu, jehož důsledkem byla RFBM během 4 hodin, byl 28,9 % ve skupině s podáváním jedenkrát denně a 30,2 % ve skupině s podáváním každý druhý den v porovnání s 9,4 % a 9,3 % při podávání odpovídajícího režimu s placebem (p < 0,001).
Klíčový sekundární ukazatel upravené průměrné změny týdenních RFBM v porovnání s hodnotou při vstupu byl 3,1 v léčebné skupině s 12 mg methylnaltrexonium-bromidu jedenkrát denně, 2,1 v léčebné skupině s 12 mg methylnaltrexonium-bromidu každý druhý den a 1,5 v léčebné skupině s placebem během 4 týdenního dvojitě zaslepeného období. Rozdíl 1,6 RFBM za týden mezi 12 mg methylnaltrexonium-bromidu jedenkrát denně a placebem je statisticky významný (p < 0,001) a má význam i v klinické praxi.
Další sekundární ukazatel hodnotil podíl pacientů s >3 RFBM za týden během 4-týdenní dvojitě zaslepené fáze. Ten byl dosažen u 59 % pacientů ve skupině, která dostávala 12 mg methylnatrexonu denně (p<0,001 versus placebo), u 61 % pacientů, kteří ho dostávali každý druhý den (p<0,001 versus placebo), a u 38 % pacientů, kteří dostávali placebo. Doplňková analýza hodnotila percentuelní podíl pacientů, kteří dosáhli >3 úplných RFBM za týden a zvýšení o >1 úplných RFBM za týden alespoň ve 3 týdnech ze 4 týdnů léčby. Toto bylo dosaženo u 28,7 % pacientů ve skupině s podáváním 12 mg methylnatrexonium-bromidu denně (p<0,001 versus placebo), u 14,9 % pacientů, kteří ho dostávali každý druhý den (p=0,012 versus placebo), a u 6,2 % pacientů léčených placebem.
Nebyl získaný důkaz o rozdílech v účinnosti a bezpečnosti způsobených pohlavím. Vliv etnického původu nebylo možné analyzovat, protože populace byla především bělošská (90 %). Medián denní dávky opioidů se nijak významně neodlišoval od vstupních hodnot ani u pacientů užívajících methylnaltrexonium-bromid ani u pacientů dostávajících placebo.
Nezjistili se žádné klinicky významné změny v hodnocení bolesti v porovnání se vstupními hodnotami ani u pacientů užívajících methylnaltrexonium-bromid, ani u pacientů dostávajících placebo.
V klinických hodnoceních se nehodnotilo používání methylnaltrexonium-bromidu při léčbě obstipace vyvolané opioidy delší než 48 týdnů.
Obstipace vyvolaná opioidy u dospělých pacientů s pokročilým onemocněním
Účinnost a bezpečnost methylnaltrexonium-bromidu při léčbě obstipace vyvolané opioidy u pacientů, kteří dostávají paliativní péči, byla prokázána ve dvou randomizovaných dvojitě zaslepených, placebem kontrolovaných studiích. V těchto studiích byl střední věk 68 let (rozmezí 21-100); 51 % byly ženy. Pacienti v obou studiích měli pokročilé terminální onemocnění s omezenou předpokládanou délkou přežití, ve většině případů s primární diagnózou nevyléčitelné rakoviny; ostatní primární diagnózy zahrnovaly konečné stádium CHOPN/emfyzém, onemocnění srdce/srdeční selhání, Alzheimerovu chorobu/demenci, HIV/AIDS a jiná onemocnění v pokročilém stádiu. Před screeningem měli pacienti opioidy vyvolanou obstipaci definovanou buď jako <3 stolice v předchozím týdnu, nebo žádná stolice po >2 dny.
Studie 301 porovnávala jednorázovou dvojitě zaslepenou subkutánní dávku methylnaltrexonium-bromidu 0,15 mg/kg, nebo 0,3 mg/kg versus placebo. Po dvojitě zaslepené dávce následovala otevřená 4-týdenní perioda, během které mohl být podáván methylnaltrexonium-bromid podle potřeby, avšak ne častěji než 1 x za 24 hodin. Během obou period studie udržovali pacienti svůj obvyklý laxativní režim. V periodě dvojitě zaslepené studie bylo celkově zařazeno a léčeno 154 pacientů (47 dostalo methylnaltrexonium-bromid 0,15 mg/kg, 55 dostalo methylnaltrexonium-bromid 0,3 mg/kg, 52 dostalo placebo). Primárním cílovým parametrem byl podíl pacientů s defekací bez záchranné laxativní léčby do 4 hodin po podání dvojitě zaslepené dávky studovaného léčivého přípravku. Ve skupině léčené methylnaltrexonium-bromidem byl signifikantně vyšší podíl pacientů s defekací do 4 hodin po dvojitě zaslepené dávce (62 % u 0,15 mg/kg a 58 % u 0,3 mg/kg), než ve skupině léčené placebem (14 %); p<0,0001 pro každou dávku versus placebo.
Studie 302 porovnávala dvojitě zaslepené subkutánní dávky methylnaltrexonium-bromidu podané každý druhý den po 2 týdny proti placebu. V průběhu prvního týdne (dny 1., 3., 5., 7.) dostali pacienti buď 0,15 mg/kg methylnaltrexonium-bromidu, nebo placebo. Ve druhém týdnu mohla být dávka zvýšena na 0,30 mg/kg, pokud měl pacient do 8. dne bez použití záchranné dávky projímadla 2 nebo méně stolic. Předepsaná dávka mohla být pacientovi kdykoli snížena v závislosti na toleranci. Byly analyzovány údaje od 133 pacientů (62 užívalo methylnaltrexonium-bromidu, 71 užívalo placebo). Studie měla dva primární cíle: Podíl pacientů s defekací bez záchranné laxativní medikace do 4 hodin po první dávce léčivého přípravku a podíl pacientů s defekací bez záchranné laxativní medikace do 4 hodin po nejméně 2 z prvních 4 dávek léčivého přípravku. U pacientů léčených methylnaltrexonium-bromidem byl vyšší podíl defekací do 4 hodin po první dávce (48 %) než u pacientů léčených placebem (16 %); p<0,0001. Pacienti léčeni methylnaltrexonium-bromidem měli také signifikantně vyšší počet defekací do 4 hodin po podání alespoň 2 ze 4 dávek (52 %) než pacienti léčení placebem (9 %); p<0,0001. Konsistence stolice u pacientů, kteří měli zpočátku měkkou stolici, se významně nezlepšila.
V obou studiích nebyl prokázán rozdíl v účinnosti ani v bezpečnosti v závislosti na věku nebo pohlaví. Účinek nebylo možno analyzovat podle rasy, protože populace ve studii byla převážně bělošská
(88 %).
Trvání odpovědi bylo demonstrováno ve studii 302, v níž byl počet laxačních odpovědí konzistentní od dávky 1 až po dávku 7 v průběhu celé dvoutýdenní dvojitě zaslepené periody.
Účinnost a bezpečnost methylnaltrexonium-bromidu byly demonstrovány také v otevřené studii 301, při níž bylo léčivo podáváno od 2. dne po 4 týdny, a ve 2 otevřených rozšířených studiích (301EXT a 302EXT), v nichž byl methylnaltrexonium-bromid podáván podle potřeby po dobu až 4 měsíců (po tuto dobu pouze 8 pacientů). Celkově 136 pacientů ve studii 301, 21 pacientů ve studii 301EXT a 82 pacientů ve studii 302EXT dostalo nejméně jednu léčebnou dávku v otevřené studii. Přípravek Relistor byl podáván každých 3,2 dnů (medián dávkovacího intervalu s rozmezím 1-39 dnů).
Počet laxačních odpovědí byl udržován po dobu rozšířené studie u těch pacientů, kteří pokračovali v léčení.
V těchto studiích nebyl zjištěn žádný signifikantní vztah mezi úvodní dávkou opiátu a laxační odpovědí u pacientů léčených methylnaltrexonium-bromidem. Navíc střední dávka opiátu ve skupině pacientů léčených methylnaltrexonium-bromidem a pacientů léčených placebem se podstatně nelišila od úvodních dávek. Nedošlo ke klinicky relevantní změně skóre bolesti od úvodních hodnot ani ve skupině léčené methylnaltrexonium-bromidem, ani ve skupině léčené placebem.
Účinek na srdeční repolarizaci
Ve dvojitě zaslepené randomizované EKG studii s paralelní skupinou jednotlivá dávka methylnaltrexonium-bromidu (0,15; 0,30 a 0,50 mg/kg) u 207 zdravých dobrovolníků neprodloužila interval QT/QTc ani neovlivnila sekundární parametry EKG a tvar křivky byl srovnatelný s placebem a s pozitivní kontrolou (perorálně podaný moxifloxacin 400 mg).
5.2 Farmakokinetické vlastnosti
Absorpce
Methylnaltrexonium-bromid se po subkutánním podání rychle absorbuje a dosahuje maximální koncentrace (Cmax) přibližně po 0,5 hodině. Cmax a plocha pod křivkou koncentrace (AUC) se zvyšují úměrně se zvyšující se dávkou od 0,15 mg/kg do 0,5 mg/kg. Absolutní biologická dostupnost subkutánní dávky 0,30 mg/kg ve srovnání s intravenozní dávkou 0,30 mg/kg je 82 %.
Distribuce
Methylnaltrexonium-bromid podléhá středně silné distribuci do tkání. Distribuční objem v ustáleném stavu (Vss) je přibližně 1,1 l/kg. Methylnaltrexonium-bromid se minimálně váže na lidské plazmatické proteiny (11,0 % až 15,3 %), jak bylo zjištěno rovnovážnou dialýzou.
Biotransformace
Podle množství metabolitů získaných z exkretů je methylnaltrexonium-bromid u lidí metabolizován v malé míře. Zdá se, že primární metabolickou cestou je přeměna na isomery metyl-6-naltrexolu a na methylnatrexon-sulfát. Každý z izomerů metyl-6-naltrexolu má poněkud nižší antagonistický účinek než mateřská sloučenina a nižší expozici v plazmě ve výši přibližně 8 % z původního léčiva. Methylnaltrexon-sulfát je neúčinný metabolit a v plazmě je přítomný v hladině přibližně 25 % z původního léčiva. N-demetylace methylnaltrexonu na naltrexon není významná a činí asi 0,06 % podané dávky.
Eliminace
Methylnaltrexonium-bromid je vylučován převážně ve formě nezměněné léčivé látky. Přibližně polovina dávky je vyloučena močí a o něco méně stolicí. Konečný poločas vylučování1/2) je přibližně 8 hodin.
Zvláštní populace
Pacienti s jaterním poškozením
Celkový účinek methylnaltrexonium-bromidu u mírného a středně těžkého jaterního poškození byl studován u 8 pacientů s klasifikací Child-Pugh A a B a porovnáván se zdravými subjekty. Výsledky ukázaly, že jaterní poškození nemá podstatný vliv na AUC ani na Cmax methylnaltrexonium-bromidu. Vliv těžkého jaterního poškození na farmakokinetiku methylnaltrexonium-bromidu nebyl studován.
Pacienti s renálním poškozením
Ve studii s dobrovolníky s různým stupněm renálního poškození byla podávána jednotlivá dávka methylnaltrexonium-bromidu 0,30 mg/kg; renální poškození mělo značný účinek na renální vylučování methylnaltrexonium-bromidu. Renální clearance methylnaltrexonium-bromidu se snížila se zvýšenou závažností renálního poškození. Těžké renální poškození snížilo renální clearance methylnaltrexonium-bromidu 8 až 9-krát; to však vedlo pouze ke dvojnásobnému zvýšení celkové expozice (AUC) methylnaltrexonium-bromidu. Cmax se významně nezměnila. Nebyla provedena žádná studie u pacientů s renálním poškozením v terminálním stádiu vyžadujícím dialýzu.
Pediatrická populace
Nebyly provedeny žádné studie u pediatrické populace (viz bod 4.2).
Starší populace
Ve studii porovnávající farmakokinetické profily jednotlivé dávky a opakovaných dávek intravenozně podaného methylnaltrexonium-bromidu v dávce 24 mg zdravým mladým (věk 18-45 let, n=10) a starším (věk 65 let a více, n=10) subjektům byl shledán minimální vliv věku na účinky methylnatrexonium-bromidu. Střední Cmax a AUC v ustáleném stavu u starších subjektů byly 545 ng/ ml a 412 ng^hod/ml, tj. přibližně o 8,1 % resp. o 20 % vyšší než u zdravých subjektů. Proto se nedoporučuje upravovat dávkování v závislosti na věku.
Pohlaví
Nebyly pozorovány podstatné rozdíly v závislosti na pohlaví.
Hmotnost
Integrovaná analýza farmakokinetických údajů od zdravých subjektů prokázala, že expozice upravené dávky methylnaltrexonium-bromidu mg/kg se zvyšuje se zvyšující se hmotností. Průměrná expozice methylnaltrexonium-bromidu v dávce 0,15 mg/kg při hmotnosti v rozmezí od 38 do 114 kg byla 179(rozmezí = 139-240) ng^hod/ml. Této expozice při dávce 0,15 mg/kg je možno dosáhnout úpravou dávky podle hmotnosti, použitím dávky 8 mg při tělesné hmotnosti od 38 do méně než 62 kg a dávky 12 mg při tělesné hmotnosti od 62 do 114 kg; průměrná dosažená expozice činí 187 (rozmezí = 148220) ng^hod/ml. Analýza údajů založená na rozdělení pacientů ve studii 301 a 302 podle hmotnosti dále ukázala, že dávka 8 mg pro tělesnou hmotnost od 38 do méně než 62 kg odpovídá průměrné dávce 0,16 (rozmezí = 0,21-0,13) mg/kg a dávka 12 mg pro tělesnou hmotnost od 62 do 114 kg odpovídá průměrné dávce 0,16 (rozmezí = 0,19-0,11) mg/kg.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a kancerogenního potenciálu neodhalily žádné zvláštní riziko pro člověka. V některých neklinických studiích na psech byly pozorovány účinky na srdce (prodloužení akčního potenciálu Purkyňových vláken nebo prodloužení QTc intervalu). Mechanizmus tohoto účinku není znám; zdá se však, že lidský srdeční kaliový iontový kanál (hERG) se na tom nepodílí.
Subkutánní injekce přípravku Relistor v dávce 150 mg/kg/den vedly ke snížení fertility u potkanů. Dávky až do 25 mg/kg/den (18-krát vyšší než expozice [AUC] u člověka po subkutánní dávce 0,3 mg/kg) neovlivňovaly fertilitu ani celkovou reprodukční schopnost.
Nebyla prokázána teratogenita u potkanů ani králíků. Subkutánní injekce přípravku Relistor v dávce 150/100 mg/kg/den u potkanů vedly ke snížení porodní hmotnosti mláďat; dávky až do 25 mg/kg/den (18-krát vyšší než expozice [AUC] u člověka po subkutánní dávce 0,3 mg/kg) neměly žádný vliv na průběh porodu, porod, přežívání ani na růst mláďat.
Methylnaltrexonium-bromid se vylučuje do mateřského mléka u potkanů.
Studie byly provedeny u mláďat potkanů a psů. Po intravenózní injekci methylnaltrexonium-bromidu bylo zjištěno, že mláďata potkanů jsou citlivější na toxicitu související s methylnaltrexonium-bromidem než dospělí potkani. U mláďat potkanů, kterým byl podáván intravenózně methylnaltrexonium-bromid po dobu 13 týdnů, se nežádoucí klinické příznaky (incidence křečí a namáhavý dech) objevily při dávkách (> 3 mg/kg/den) a expozicích (5,4 krát vyšší než expozice {AUC} u dospělého člověka při subkutánní dávce 0,15 mg/kg) nižších než byly ty, které způsobily podobnou toxicitu u dospělých potkanů (20 mg/kg/den). U mláďat potkanů při dávce 1 mg/kg/den nebo u dospělých potkanů při dávce 5 mg/kg/den se nevyskytly žádné nežádoucí účinky (1,6násobek resp. 7,8násobek expozice {AUC} u dospělého člověka při subkutánní dávce 0,15 mg/kg).
Po podávání intravenózních injekcí methylnaltrexonium-bromidu po dobu 13 týdnů byla u mladých i dospělých psů pozorována podobná toxicita související s methylnaltrexonium- bromidem. U dospělých a mladých psů, kteří dostávali methylnaltrexonium-bromid v dávce 20 mg/kg/den, byly pozorovány klinické příznaky CNS toxicity a prodloužení intervalu QTc. U mláďat ani u dospělých psů se při dávce 5 mg/kg/den (44násobek expozice {AUC} u dospělého člověka při subkutánní dávce 0,15 mg/kg) nevyskytly žádné nežádoucí účinky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
chlorid sodný natrium-kalcium-edetát glycin-hydrochlorid voda na injekci
kyselina chlorovodíková (k úpravě pH) hydroxid sodný (k úpravě pH)
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky.
6.3 Doba použitelnosti
18 měsíců
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
Jedna předplněná injekční stříkačka obsahuje 0,6 ml injekčního roztoku.
Předplněná injekční stříkačka z čirého skla typu I s jehlou z nerezové oceli, táhla z plastické hmoty a pevnou krytkou jehly z pryže.
Velikost balení: 4, 7, 8 nebo 10 předplněných inječkních stříkaček.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o.
Jankovcova 1569/2c 170 00, Praha 7 Česká republika
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/463/008
EU/1/08/463/009
EU/1/08/463/010
EU/1/08/463/011
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. července 2008
Datum posledního prodloužení registrace: 27. května 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu
34
A. VÝROBCE/VÝROBCI ODPOVĚDNÝ/ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
VÝROBCE/VÝROBCI ODPOVĚDNÝ/ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
A.
Název a adresa výrobce odpovědného za propouštění šarží
Przedsi^biorstwo Farmaceutyczne Jelfa SA ul. Wincentego Pola 21 58-500 Jelenia Góra,
Polsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
Relistor 12 mg/0,6 ml injekční roztok methylnaltrexonii bromidům
Jedna injekční lahvička 0,6 ml obsahuje 12 mg methylnaltrexonii bromidům. Jeden mililitr roztoku obsahuje 20 mg methylnaltrexonii bromidům.
Chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Injekční roztok.
1 injekční lahvička 0,6 ml
Před použitím si přečtěte příbalovou informaci. Subkutánní podání
Uchovávejte mimo dohled a dosah dětí.
EXP
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
EU/1/08/463/001
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
RELISTOR 12 mg/0,6 ml
Relistor 12 mg/0,6 ml injekční roztok methylnaltrexonii bromidům
Jedna injekční lahvička 0,6 ml obsahuje 12 mg methylnaltrexonii bromidům. Jeden mililitr roztoku obsahuje 20 mg methylnaltrexonii bromidům.
Chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Injekční roztok.
2 injekční lahvičky po 0,6 ml
2 sterilní injekční stříkačky o objemu 1 ml se zasunovatelnou injekční jehlou 4 alkoholové tampony
Před použitím si přečtěte příbalovou informaci. Subkutánní podání
Uchovávejte mimo dohled a dosah dětí.
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/463/002
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Relistor 12 mg/0,6 ml injekční roztok methylnaltrexonii bromidům
Jedna injekční lahvička 0,6 ml obsahuje 12 mg methylnaltrexonii bromidům. Jeden mililitr roztoku obsahuje 20 mg methylnaltrexonii bromidům.
Chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Injekční roztok.
7 injekčních lahviček po 0,6 ml
7 sterilních injekčních stříkaček o objemu 1 ml se zasunovatelnou injekční jehlou 14 alkoholových tamponů
Před použitím si přečtěte příbalovou informaci. Subkutánní podání
Uchovávejte mimo dohled a dosah dětí.
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/463/003
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Relistor 8 mg injekční roztok v předplněné injekční stříkačce methylnaltrexonii bromidům
Jedna předplněná injekční stříkačka 0,4 ml obsahuje 8 mg methylnaltrexonii bromidům.
Chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Injekční roztok
4 předplněné injekční stříkačky
Před použitím si přečtěte příbalovou informaci.
Subkutánní podání
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte při teplotě do 30°C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11 NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/463/004
13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
RELISTOR 8 mg
Relistor 8 mg injekční roztok v předplněné injekční stříkačce methylnaltrexonii bromidům
Jedna předplněná injekční stříkačka 0,4 ml obsahuje 8 mg methylnaltrexonii bromidům.
Chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Injekční roztok
7 předplněných injekčních stříkaček
Před použitím si přečtěte příbalovou informaci. Subkutánní podání
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte při teplotě do 30°C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/463/005
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Relistor 8 mg injekční roztok v předplněné injekční stříkačce methylnaltrexonii bromidům
Jedna předplněná injekční stříkačka 0,4 ml obsahuje 8 mg methylnaltrexonii bromidům.
Chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Injekční roztok
8 předplněných injekčních stříkaček
Před použitím si přečtěte příbalovou informaci. Subkutánní podání
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte při teplotě do 30°C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/463/006
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Relistor 8 mg injekční roztok v předplněné injekční stříkačce methylnaltrexonii bromidům
Jedna předplněná injekční stříkačka 0,4 ml obsahuje 8 mg methylnaltrexonii bromidům.
Chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Injekční roztok
10 předplněných injekčních stříkaček
Před použitím si přečtěte příbalovou informaci. Subkutánní podání
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte při teplotě do 30°C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
12 REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/463/007
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Relistor 12 mg injekční roztok v předplněné injekční stříkačce methylnaltrexonii bromidům
Jedna předplněná injekční stříkačka 0,6 ml obsahuje 12 mg methylnaltrexonii bromidům.
Chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Injekční roztok
4 předplněné injekční stříkačky
Před použitím si přečtěte příbalovou informaci. Subkutánní podání
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte při teplotě do 30°C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
12 REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/463/008
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Relistor 12 mg injekční roztok v předplněné injekční stříkačce methylnaltrexonii bromidům
Jedna předplněná injekční stříkačka 0,6 ml obsahuje 12 mg methylnaltrexonii bromidům.
Chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Injekční roztok
7 předplněných injekčních stříkaček
Před použitím si přečtěte příbalovou informaci. Subkutánní podání
Uchovávejte mimo dohled a dosah.
Uchovávejte při teplotě do 30°C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/463/009
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Relistor 12 mg injekční roztok v předplněné injekční stříkačce methylnaltrexonii bromidům
Jedna předplněná injekční stříkačka 0,6 ml obsahuje 12 mg methylnaltrexonii bromidům.
Chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Injekční roztok
8 předplněných injekčních stříkaček
Před použitím si přečtěte příbalovou informaci. Subkutánní podání
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte při teplotě do 30°C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/463/010
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Relistor 12 mg injekční roztok v předplněné injekční stříkačce methylnaltrexonii bromidům
Jedna předplněná injekční stříkačka 0,6 ml obsahuje 12 mg methylnaltrexonii bromidům.
Chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Injekční roztok
10 předplněných injekčních stříkaček
Před použitím si přečtěte příbalovou informaci. Subkutánní podání
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte při teplotě do 30°C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/463/011
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU
Relistor 12 mg injekční roztok v předplněné injekční stříkačce methylnaltrexonii bromidům
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o.
3. POUŽITELNOST
EXP
4 ČÍSLO ŠARŽE
Lot
5. JINÉ
Subkutánní podání Uchovávejte při teplotě do 30°C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem. 0,6 ml injekčního roztoku (12 mg methylnaltrexonii bromidum)
Před použitím si přečtěte příbalovou informaci.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Relistor 8 mg injekce methylnaltrexonii bromidům
SC
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU
Relistor 8 mg injekční roztok v předplněné injekční stříkačce methylnaltrexonii bromidům
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o.
3. POUŽITELNOST
EXP
4 ČÍSLO ŠARŽE
Lot
5. JINÉ
Subkutánní podání Uchovávejte při teplotě do 30°C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem. 0,6 ml injekčního roztoku (12 mg methylnaltrexonii bromidum)
Před použitím si přečtěte příbalovou informaci.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Relistor 12 mg injekce methylnaltrexonii bromidům
SC
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU
Relistor 12 mg/0,6 ml injekční roztok methylnaltrexonii bromidům
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o.
3. POUŽITELNOST
EXP
4 ČÍSLO ŠARŽE
Lot
5. JINÉ
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem. Injekční jehla se po použití zatáhne
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Relistor 12 mg/0,6 ml injekční roztok methylnaltrexonii bromidům Subkutánní podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3 POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,6 ml injekčního roztoku (12 mg methylnaltrexonii bromidům)
6. JINÉ
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Relistor 12 mg/0,6 ml injekční roztok
Methylnaltrexonii bromidům
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo
lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Relistor a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Relistor používat
3. Jak se přípravek Relistor používá
4. Možné nežádoucí účinky
5 Jak přípravek Relistor uchovávat
6. Obsah balení a další informace
1. Co je přípravek Relistor a k čemu se používá
Přípravek Relistor obsahuje léčivou látku nazývanou methylnaltrexonium-bromid, která působí tak, že blokuje gastrointestinální účinky opioidních léků proti bolesti, které ovlivňují vyprazdňování
Léčí zácpu, která je způsobena léky k tišení středních až těžkých bolestí, nazývanými opioidy (např. morfin, kodein). Používá se u pacientů, kdy odpověď na léky na zácpu, tzv.laxativa, nebyla dostatečná. Opioidy Vám předepisuje lékař. Když začnete používat tento přípravek, lékař Vám řekne, jestli máte přestat užívat Vaše obvyklá laxativa nebo v jejich užívání pokračovat.
Tento přípravek se používá u dospělých (věk 18 let a více).
2. Čemu musíte věnovat pozornost, než začnete přípravek Relistor používat Nepoužívejte přípravek Relistor
- Jestliže jste alergický/á na methylnaltrexonium-bromid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- Jestliže Vy nebo Váš lékař víte, že máte neprůchodnost střeva, nebo že je nutný okamžitý chirurgický zákrok (diagnózu musí stanovit lékař).
Upozornění a opatření
Před použitím přípravku Relistor se poraďte se svým lékařem nebo lékarníkem
• Pokud máte závažné bolesti břicha, které nepřestávají nebo se zhoršují, informujte neprodleně svého lékaře, neboť by to mohly být příznaky tvořícího se otvoru ve střevní stěně (perforace střeva). Viz bod 4.
• Jestliže máte těžké onemocnění jater nebo ledvin.
• Jestliže máte těžký nebo přetrvávající průjem (častá vodnatá stolice), podávání přípravku přeruště a informujte neprodleně svého lékaře.
• Je důležité být poblíž toalety a mít k dispozici pomoc, je-li to nutné, protože účinek se může dostavit do 30 minut po podání injekce.
• Prosím, informujte lékaře, pokud máte bolest břicha, která nepřestává, nevolnost nebo zvracení, které nově nastoupilo, nebo se zhoršuje.
• Prosím, informujte také lékaře, pokud máte kolostomii (vývod tlustého střeva), peritoneální katetr (cévku do dutiny břišní), nebo máte divertikulární onemocnění (výchlipky v tlustém střevě) nebo zaklínění stolice (silná zácpa), protože tento lék musí být používán s opatrností při těchto stavech.
• Jestliže dostáváte podpůrnou péči při pokročilém onemocnění, budete tento lék používat jen omezenou dobu, která bude pravděpodobně kratší než 4 měsíce.
• Tento lék by neměl být podáván pacientům k léčbě zácpy, která s podáváním opioidů nesouvisí. Informujte, prosím, lékaře, pokud jste trpěl/a zácpou předtím, než jste užíval/a opioidy (pro léčbu bolesti).
Děti a dospívající
Nepodávejte tento lék dětem a dospívajícím mladším 18ti let, protože možná rizika a přínosy nejsou známy.
Další léčivé přípravky a přípravek Relistor
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, , které jste v nedávné době užíval(a) nebo které možná budete užívat.
Lékař Vám může doporučit současné užívání dalších léků včetně těch, které se používají při zácpě.
Těhotenství a kojení
Účinek methylnatrexonium- bromidu u těhotných žen není znám. Lékař rozhodne, jestli můžete užívat přípravek Relistor v případě, že jste těhotná.
Ženy užívající tento lék by neměly kojit, protože není známo, jestli methylnatrexonium-bromid přechází do mateřského mléka.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Závrať je častým nežádoucím účinkem tohoto léku. Může ovlivnit schopnost řízení dopravních prostředků a obsluhování strojů.
Důležité informace o některých složkách přípravku Relistor
Tento lék obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tzn. je v podstatě „bez sodíku“.
3. Jak se přípravek RELISTOR používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý/á, poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka pro pacienty s dlouhodobou bolestí (kromě pacientů dostávajících podpůrnou péči při pokročilém onemocnění) je 12 mg methylnaltrexonium-bromidu (0,6 ml roztoku) podávaného jako injekce pod kůži podle potřeby, ale minimálně 4-krát týdně až maximálně jedenkrát denně (7-krát týdně).
Doporučená dávka pro pacienty dostávající podpůrnou léčbu při pokročilém onemocnění je 8 mg methylnaltrexonium-bromidu (0,4 ml roztoku) pro pacienty o hmotnosti 38-61 kg nebo 12 mg (0,6 ml roztoku) pro pacienty o hmotnosti 62-114 kg. Dávka se podává každých 48 hodin (každý druhý den) jako injekce pod kůži.
Velikost dávky stanoví lékař.
Tento přípravek se podává injekcí pod kůži (podkožní injekce) buď (1) do horní části dolních končetin (stehna), nebo (2) do oblasti břicha nebo (3) do horní části paže (nejedná-li se o autoinjekci). (Viz NÁVOD K PŘÍPRAVĚ A PODÁNÍ INJEKCE PŘÍPRAVKU RELISTOR na konci této příbalové informace.)
Můžete dosáhnout pohybu střev do několika minut až hodin po podání injekce; proto se doporučuje být poblíž toalety nebo mít po ruce mísu.
Jestliže jste použil(a) více přípravku Relistor, než jste měl(a)
Jestliže jste užil/a větší množství tohoto přípravku, než jste měl/a (buď jednorázovým podáním většího množství, nebo podáním více injekcí v průběhu 24 hodin), můžete cítit závrať při vstávání, a proto neprodleně informujte svého lékaře nebo lékárníka. Vždy si vezměte vnější obal léku s sebou, a to i tehdy, je-li již prázdný.
Jestliže jste zapomněl(a) použít přípravek Relistor
Jestliže zapomenete užít dávku, co nejdříve informujte svého lékaře nebo lékárníka. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat přípravek Relistor
Jestliže chcete přestat užívat tento přípravek, měl/a byste informovat svého lékaře nebo lékárníka.
Máte-li jakékoli další otázky týkající se užívání tohoto léčivého přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento lék nežádoucí účinky, které se ale nemusí vyskytnout u každého.
U pacientů používajících přípravek Relistor byly hlášeny případy proděravění střevní stěny (gastrointestinální perforace). Z dostupných údajů není známo, jak často se to stává. Pokud začnete pociťovat bolest břicha, která je buď závažná nebo nepřestává, přestaňte užívat tento lék a okamžitě telefonicky informujte svého lékaře.
Následující nežádoucí účinky jsou velmi časté a mohou postihnout víc než 1 osobu z 10. Jestliže se u Vás vyskytne některý z těchto nežádoucích účinků a je buď závažný nebo nepřestává, měl/a byste kontaktovat svého lékaře:
• Bolest břicha (bolest žaludku)
• Nevolnost (žaludeční)
• Průjem (častá vodnatá stolice)
• Plynatost (odchod větrů)
Další časté nežádoucí účinky, které mohou postihnout až 1 osobu z 10, jsou:
• Závrať (pocit točení hlavy)
• Mírné příznaky jako při náhlém přerušení léčby opioidy (některý z uvedených příznaků: pocit chladu, třes, výtok z nosu, pocení, „husí kůže“, návaly horka, zrychlené bušení srdce)
• Reakce v místě podání (např. píchání, pálení, bolest, zčervenání, otok)
• Zvracení
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Relistor uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a injekční lahvičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní teplotní podmínky uchovávání.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Použijte tento přípravek pouze v případě, že je roztok čirý, bezbarvý až světle žlutý a pokud neobsahuje vločky ani mechanické částice.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebodomácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek obsahuje Relistor
• Léčivou látkou je methylnaltrexonii bromidum. Jedna injekční lahvička 0,6 ml obsahuje
12 mg methylnaltrexonii bromidum. Jeden mililitr roztoku obsahuje 20 mg methylnaltrexonii bromidum.
• Pomocnými látkami jsou: chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Jak přípravek Relistor vypadá a co obsahuje toto balení
Přípravek Relistor je injekční roztok. Je čirý, bezbarvý až světle žlutý a neobsahuje vločky ani pevné částice.
Jedna injekční lahvička obsahuje 0,6 ml roztoku.
Balení o více než jedné injekční lahvičce obsahuje vnitřní krabičky, přičemž v každé je jedna injekční lahvička, injekční stříkačka o objemu 1 ml se zasunovatelnou injekční jehlou a dva alkoholové tampony.
Dodávají se následující balení:
Samostatná injekční lahvička.
Balení obsahující 2 injekční lahvičky, 2 injekční stříkačky se zasunovatelnou injekční jehlou a 4 alkoholové tampony (tj. 2 vnitřní krabičky).
Balení obsahující 7 injekčních lahviček, 7 injekčních stříkaček se zasunovatelnou injekční jehlou a 14 alkoholových tamponů (tj. 7 vnitřních krabiček).
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
PharmaSwiss Česká republika s.r.o.
Jankovcova 1569/2c 170 00, Praha 7 Česká republika
Výrobce
Przedsi^biorstwo Farmaceutyczne Jelfa SA ul. Wincentego Pola 21 58-500 Jelenia Góra,
Polsko
u místního zástupce držitele rozhodnutí o registraci: Lietuva
PharmaSwiss UAB Tel. + 370 5 279 0762
Další informace o tomto přípravku získáte Belgie/Belgique/Belgien
Swedish Orphan Biovitrum BVBA Tél/Tel: + 32 2880 6119 e-mail: benelux@sobi.com
BunrapuH
PharmaSwiss EOOD Ten.: + 359 2 89 52 110
Česká republika
Swedish Orphan Biovitrum s.r.o. Tel: + 420 257 222 034 e-mail: mail.cz@sobi.com
Danmark
Swedish Orphan Biovitrum A/S Tlf: + 45 32 96 68 69 e-mail: mail.dk@sobi.com
Luxembourg/Luxemburg
Swedish Orphan Biovitrum BVBA Tél/Tel: +32 2880 6119 e-mail: benelux@sobi.com
Magyarország
Swedish Orphan Biovitrum s.r.o. Magyarországi Fióktelepe
Tel. +420 257 222 034 e-mail: mail.hu@sobi.com
Malta
Swedish Orphan Biovitrum S.r.l.
Tel: +39 0521 19 111 e-mail: mail.it@sobi.com
Deutschland
Swedish Orphan Biovitrum GmbH Tel: +49 89 55066760 e-mail: mail.de@sobi.com
Eesti
PharmaSwiss Eesti OU Tel: +372 6 827 400
Nederland
Swedish Orphan Biovitrum BVBA Tel: +32 288 06119 e-mail: benelux@sobi.com
Norge
Swedish Orphan Biovitrum AS Tlf: +47 66 82 34 00 e-mail: mail.no@sobi.com
EkXúba
Swedish Orphan Biovitrum S.r.l. TpA.: +39 0521 19 111
Osterreich
Swedish Orphan Biovitrum GmbH Tel: +49 89 55066760
e-mail: mail.it@sobi.com Espaňa
Swedish Orphan Biovitrum S.L Tel: + 34 913 91 35 80 e-mail: mail.es@sobi.com
France
Swedish Orphan Biovitrum SARL Tél: + 33 1 85 78 03 40 e-mail: mail.fr@sobi.com
Hrvatska
PharmaSwiss d.o.o.
Tel: +385 1 6311 833
Ireland
Swedish Orphan Biovitrum Ltd Tel: +44 1223 891854 e-mail: mail.uk@sobi.com
Island
Swedish Orphan Biovitrum A/S Tlf: + 45 32 96 68 69 e-mail: mail.dk@sobi.com
Italia
Swedish Orphan Biovitrum S.r.l. Tel: +39 0521 19 111 e-mail: mail.it@sobi.com
Kúnpoq
M.S. Jacovides & Co Ltd T@: + 357 22 0056200
Latvija
PharmaSwiss UAB Tel: + 371 67502185
e-mail: mail.de@sobi.com Polska
Valeant sp. z o.o. sp. j. Tel.: +48 17 865 51 00
Portugal
Swedish Orphan Biovitrum S.L Tel: +34 913 91 35 80 e-mail: mail.es@sobi.com
Románia
Valeant Pharma SRL Tel: +40 374 102 600
Slovenija
PharmaSwiss d.o.o.
Tel: + 386 1 2364 700
Slovenská republika
Swedish Orphan Biovitrum o.z.
Tel: +420 257 222 034 e-mail: mail.sk@sobi.com
Suomi/Finland
Oy Swedish Orphan Biovitrum Ab Puh./Tel: +358 201 558 840 e-mail: mail.fi@sobi.com
Sverige
Swedish Orphan Biovitrum AB (publ) Tel: +46 8 697 20 00 e-mail: mail.se@sobi.com
United Kingdom
Swedish Orphan Biovitrum Ltd Tel: +44 1223 891854 e-mail: mail.uk@sobi.com
Tato příbalová informace byla naposledy revidována.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu
SEZNAM OTÁZEK PRO PACIENTA
Tato část obsahuje důležité otázky, které si musíte zodpovědět předtím, než začnete užívat přípravek Relistor a během léčby přípravkem Relistor.
V případě, že v době, kdy je Vám podáván tento přípravek, odpovíte NE na některou z následujících otázek, kontaktujte, prosím, svého lékaře, zdravotní sestru nebo lékárníka.
Jsou Vám z důvodu Vašeho onemocnění podávány opioidy (například morfin nebo kodein)?
1. Je to již 48 hodin a více od Vaší poslední stolice?
2. Je Vám známa technika samopodávání injekce nebo jste ji diskutoval/a s Vaším lékařem (nebo zdravotní sestrou nebo lékárníkem)?
3. Jste natolik pohyblivý/á, abyste se dostal/a k toaletě, nebo máte asistenta, který se o Vás stará a může Vám pomoci?
4. Máte telefonní číslo na sestru nebo na zdravotní středisko?
NÁVOD K PŘÍPRAVÉ A PODÁVÁNÍ INJEKCE PŘÍPRAVKU RELISTOR
Tento bod je rozdělen do následujících podbodů:
Příprava před injekcí Příprava injekční stříkačky Výběr a příprava místa pro injekci
: Podání přípravku Relistor z balení obsahujícího injekční stříkačku se zasunovatelnou injekční jehlou
: Podání přípravku Relistor standardní injekční stříkačkou a injekční jehlou.
Likvidace zbylého odpadu
Úvod Krok 1:
Krok 2:
Krok 3:
Krok 4a
Krok 4b Krok 5:
Úvod
Tento návod vysvětluje, jak si připravit a aplikovat injekci přípravku Relistor. Pečlivě si návod přečtěte a postupujte podle něj krok za krokem. Lékař, zdravotní sestra nebo lékárník Vám vysvětlí správnou techniku podání injekce sobě samému. Nesnažte se podat si dávku léku, dokud si nejste jist/á, že rozumíte instrukci a že si umíte injekci připravit a podat. Tento lék nesmí být smíchán s žádným jiným lékem v jedné injekční stříkačce.
Můžete dostat buď balení s vnitřní krabičkou obsahující vše nezbytné pro podání injekce, nebo jen samostatnou injekční lahvičku. Když dostanete pouze samostatnou injekční lahvičku, budete ještě potřebovat alkoholové tampony a injekční stříkačku.
Krok 1: Příprava před injekcí
1. Vyberte si rovnou, čistou, dobře osvětlenou pracovní plochu, kde si můžete rozložit obsah krabičky přípravku Relistor. Ujistěte se, že máte dost času potřebného na celkovou aplikaci injekce.
2. Umyjte si ruce mýdlem a teplou vodou.
3. Připravte si vše, co budete k injekci potřebovat. To zahrnuje injekční lahvičku přípravku Relistor, 1 ml injekční stříkačku (se zasunovatelnou jehlou nebo bez ní),
2 alkoholové tampony a smotek vaty nebo gázy.
4. Ujistěte se, že roztok v injekční lahvičce je čirý a bezbarvý až světle žlutý a neobsahuje vločky ani viditelné mechanické částice. Pokud roztok obsahuje viditelné častice nebo má změněnou barvu, nepoužívejte jej. Pro pomoc kontaktujte svého lékárníka, sestru nebo lékaře.
Krok 2: Příprava injekční stříkačky
1. Sejměte ochranný plastový kryt z injekční lahvičky.
2. Otřete pryžovou zátku injekční lahvičky alkoholovým tamponem a položte lahvičku na rovnou pracovní plochu. Nedotýkejte se znovu pryžové zátky.
3. Vezměte z pracovní plochy injekční stříkačku. Držte tělo injekční stříkačky jednou rukou a rovně stáhněte kryt z injekční jehly. Položte kryt jehly zpět na pracovní plochu. Nedotýkejte se jehly a dávejte pozor, aby se jehla nedostala do kontaktu s žádným jiným povrchem.
Opatrně vytáhněte zpět píst stříkačky na značku 0,4 ml, pokud je Vaše dávka 8 mg přípravku Relistor, nebo na značku 0,6 ml, pokud je Vaše dávka 12 mg přípravku Relistor. Lékař, zdravotní sestra nebo lékárník Vám sdělí, jaká dávka Vám byla předepsána a jak často ji musíte užívat. Obvyklé dávky pro pacienty dostávající podpůrnou léčbu v pokročilém stádiu onemocnění jsou uvedeny níže v tabulce. Dávka se obvykle podává každých 48 hodin (každé dva dny) jako podkožní injekce.
Pacientova hmotnost v kg Méně než 38 kg 38-61 kg 62-114 kg Více než 114 kg
Naplňte injekční stříkačku do ml úrovně (dávky) 0,15 mg/kg 0,4 ml (8 mg)
0,6 ml (12 mg)
0,15 mg/kg
Pro pacienty s dlouhodobou bolestí (kromě pacientů dostávajících podpůrnou péči při pokročilém onemocnění) se injekční stříkačka naplní do úrovně 0,6 ml pro dávku 12 mg Relistoru.
Opatrně vytáhněte zpět píst stříkačky na správnou značku na stříkačce ( např. 0,4 ml, pokud je Vaše dávka 8 mg)
4. Vpíchněte jehlu kolmo středem pryžové zátky do injekční lahvičky. Vpich nesmí být
proveden pod úhlem, protože jehla by se mohla ohnout nebo zlomit. Držte lahvičku na pracovní ploše druhou rukou, aby Vám nevyklouzla. Při propichování pryžové zátky ucítíte slabý
odpor. Sledujte hrot jehly uvnitř injekční lahvičky.
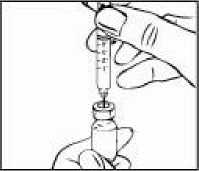
5. Abyste dostal/a ven vzduch, který je v injekční lahvičce přítomen, stlačte jemně píst a vytlačte
všechen vzduch do injekční lahvičky.
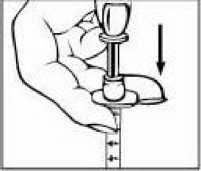
6. Pokud používáte dodanou injekční stříkačku se zasunovatelnou jehlou, NESTLAČUJTE PÍST AZ DO KONCE. Dbejte, aby se píst zastavil, jakmile ucítíte odpor. Kdybyste stlačil/a píst až do konce, uslyšel/a byste cvaknutí. To by znamenalo, že byl aktivován bezpečnostní mechanizmus a jehla by zmizela v injekční stříkačce. Pokud se to stane, zlikvidujte přípravek a začněte znovu s jinou injekční lahvičkou a stříkačkou.
Jehlu ponechte stále v injekční lahvičce a lahvičku obraťte dnem vzhůru. Držte stříkačku ve výši očí, abyste viděl/a dávkovací rysky a dávejte pozor, aby byl hrot jehly po celou dobu ponořen v tekutině. Pomalu stáhněte píst ke značce 0,4 ml nebo 0,6 ml na injekční stříkačce, nebo podle toho, jakou dávku Vám doporučil lékař, zdravotní sestra nebo lékárník. Když je injekční stříkačka správně naplněna, v injekční lahvičce může být vidět zbytek tekutiny nebo vzduchové bubliny. To je běžné.
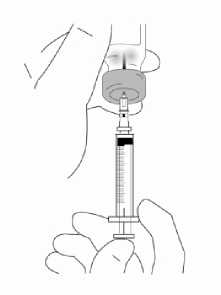
7 S jehlou stále v lahvičce dnem vzhůru zkontrolujte vzduchové bubliny v injekční stříkačce. Jemným poklepem na injekční stříkačku vyžeňte vzduchové bubliny k jejímu vrcholu; dbejte, abyste stále držel/a injekční lahvičku a injekční stříkačku ve svislé poloze. Mírně stlačte píst nahoru, abyste odstranil/a všechny vzduchové bubliny. Pokud přitom zpět do injekční lahvičky vytlačíte trochu roztoku, vytahujte pomalu opět píst stříkačky, abyste doplnil/a přesné množství roztoku zpět do stříkačky. Vzhledem k bezpečnostnímu provedení injekční stříkačky nemusí jít malé vzduchové bubliny odstranit. Nemusíte mít kvůli tomu obavy, protože to neovlivní přesnost dávky a nepředstavuje ani žádné riziko pro Vaše zdraví.
Poklepte na injekční stříkačku, držíte špičkou nahoru, a vytlačte pomocí pístu všechny bubliny
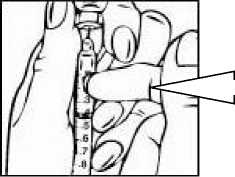
8. Vždy se ujistěte, že máte v injekční stříkačce odměřenou správnou dávku. Pokud si nejste čímkoli jist/a, poraďte se s lékařem, zdravotní sestrou nebo lékárníkem.
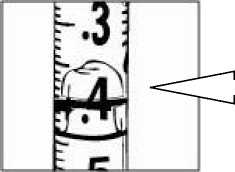
Vždy se ujistěte, že máte v injekční stříkačce odměřenou správnou dávku (např. 0,4 ml, pokud je Vaše dávka 8 mg)
9. Vyjměte jehlu s injekční stříkačkou z injekční lahvičky. Ponechejte injekční jehlu připojenou k injekční stříkačce. Nedotýkejte se jehly a dávejte pozor, aby se jehla nedotkla žádného jiného povrchu. Jakmile je lék natažený do stříkačky, musí se do 24 hodin použít, protože přípravek Relistor je ovlivněn světlem a nemusel by dobře účinkovat, pokud ve stříkačce zůstane déle než 24 hodin.
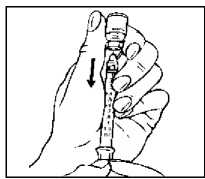
Krok 3: Výběr a příprava místa pro injekci
1. Pro podání injekce přípravku Relistor jsou doporučené tři oblasti těla: (1) horní část dolních končetin (stehna), (2) břicho ,a (3) horní část paže (pokud Vám přípravek podává jiná osoba).
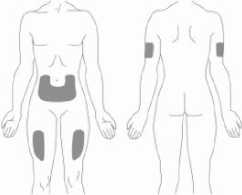
2. Doporučuje se pro každou injekci vybrat vždy odlišné místo podání. Vyhněte se podání injekce přesně do předchozího místa podání. Nepodávejte injekci do oblasti, kde je kůže bolestivá, pohmožděná, zčervenalá nebo tvrdá. Vyhněte se oblastem s jizvami nebo s narušenou strukturou kůže (strie).
3. Místo na kůži, kam má být podána injekce přípravku Relistor, potřete alkoholovým amponem. AŽ DO PODÁNÍ INJEKCE SE TOHOTO MÍSTA NEDOTÝKEJTE. Před podáním injekce nechejte místo injekce oschnout.
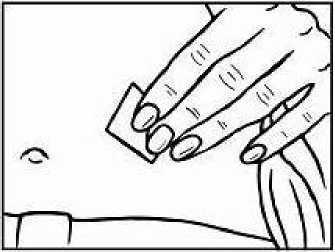
Krok 4a: Podání přípravku Relistor z balení obsahujícího injekční stříkačku se zasunovatelnou injekční jehlou
1. Naplněnou injekční stříkačku držte svisle s jehlou směrem nahoru a zkontrolujte, zda v ní nej sou vzduchové bubliny. Pokud tam jsou vzduchové bubliny, vyžeňte j e nahoru j emným poklepem prstu na stříkačku, až vzduchové bubliny stoupnou k jejímu vrcholu. Pomalu tlačte píst nahoru
a vzduchové bubliny z injekční stříkačky vytlačte ven.
2. Držte injekční stříkačku jednou rukou jako tužku. Druhou rukou na očištěném místě kůže vytvořte kožní řasu a pevně ji držte.
3. Krátkým rychlým pohybem zatlačte celou délku jehly do kůže pod malým úhlem (45 stupňů).
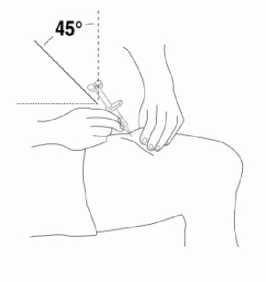
4. Jakmile je jehla vpíchnutá, pusťte kůži a pomalu stlačujte píst směrem dolů, dokud není stříkačka prázdná a neuslyšíte cvaknutí.
5. Když uslyšíte cvaknutí, znamená to, že byl podán celý obsah. Jehla automaticky vyjede z kůže ven a zakryje se. V místě vpichu může dojít ke slabému krvácení. Na místo vpichu můžete přitisknout bavlněný tampon nebo gázu. Netřete si toto místo. Podle potřeby je možné místo vpichu překrýt náplastí.
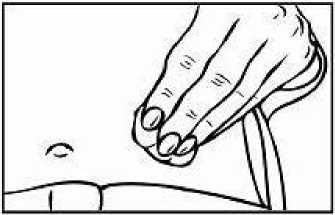
Krok 4b: Podání přípravku Relistor standardní injekční stříkačkou a injekční jehlou.
1. Naplněnou injekční stříkačku držte svisle s jehlou směrem nahoru a zkontrolujte, zda v ní nejsou vzduchové bubliny. Pokud tam jsou vzduchové bubliny, vyžeňte je nahoru jemným poklepem prstu na stříkačku, až vzduchové bubliny stoupnou k jejímu vrcholu. Pomalu tlačte píst nahoru
a vzduchové bubliny z injekční stříkačky vytlačte ven.
2. Držte injekční stříkačku jednou rukou jako tužku. Druhou rukou na očištěném rmístě kůže vytvořte kožní řasu a pevně ji držte.
3. Krátkým rychlým pohybem zatlačte celou délku jehly do kůže pod malým úhlem (45 stupňů).
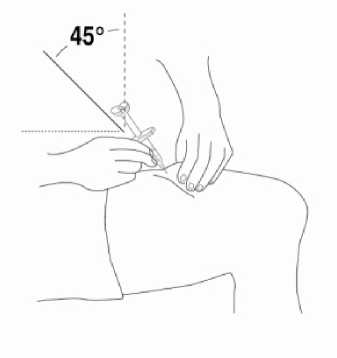
4. Jakmile je jehla vpíchnutá, pusťte kůži a pomalu stlačujte píst směrem dolů a podejte přípravek Relistor.
5. Jakmile je injekční stříkačka prázdná, rychle vytáhněte jehlu z kůže. Držte přitom jehlu stále pod stejným úhlem jako při vpichu. V místě vpichu může dojít ke slabému krvácení. Na místo vpichu můžete přitisknout bavlněný tampon nebo gázu. Netřete si toto místo. Podle potřeby je možné místo vpichu překrýt náplastí.
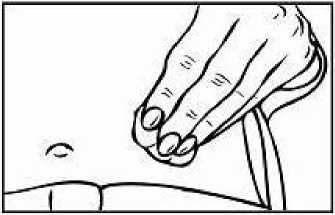
Krok 5: Likvidace zbylého odpadu
Zakrytá injekční stříkačka nebo injekční stříkačka a jehla se NIKDY nesmí znovu použít. NIKDY znovu nezakrývejte jehlu krytem. Použité injekční jehly a stříkačky odkládejte do uzavíratelného kontejneru odolného proti propíchnutí podle pokynů lékaře, sestry nebo lékárníka.
PŘÍBALOVÁ INFORMACE: INFORMACE PRO UŽIVATELE
Relistor 8 mg injekční roztok v předplněné injekční stříkačce Relistor 12 mg injekční roztok v předplněné injekční stříkačce
Methylnaltrexonii bromidům
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné p známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo
lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Relistor a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Relistor používat
3. Jak se přípravek Relistor používá
4. Možné nežádoucí účinky
5 Jak přípravek Relistor uchovávat
6. Obsah balení a další informace
1. Co je přípravek Relistor a k čemu se používá
Přípravek Relistor obsahuje léčivou látku nazývanou methylnaltrexonium-bromid, která působí tak, že blokuje gastrointestinální účinky opioidních léků proti bolesti, které ovlivňují vyprazdňování.
Léčí zácpu, která je způsobena léky k tišení středních až těžkých bolestí, nazývanými opioidy (např. morfin, kodein). Používá se u pacientů, kdy odpověď na léky na zácpu, tzv. laxativa, nebyla dostatečná. Opioidy Vám předepisuje lékař. Když začnete užívat tento přípravek, lékař Vám řekne, jestli máte přestat užívat Vaše obvyklá laxativa nebo v jejich užívání pokračovat.
Tento přípravek se používá u dospělých (věk 18 let a více).
2. Čemu musíte věnovat pozornost, než začnete přípravek Relistor používat Nepoužívejte přípravek Relistor
- Jestliže jste alergický/á na methylnaltrexonium-bromid nebo na kteroukoli další složku tohoto
přípravku (uvedenou v bodě 6.
- Jestliže Vy nebo Váš lékař víte, že máte neprůchodnost střeva, nebo že je nutnýí okamžitý
chirurgický zákrok (diagnózu musí stanovit lékař).
Upozornění a opatření
Před použitím přípravku Relistor se poraďte se svým lékařem nebo lékarníkem
• Pokud máte závažné bolesti břicha, které neřestávají nebo se zhoršují, informujte neprodleně svého lékaře, neboť by to mohly být příznaky tvořícího se otvoru ve střevní stěně (perforace střeva). Viz bod 4.
• Jestliže máte těžké onemocnění jater nebo ledvin.
• Jestliže máte těžký nebo přetrvávající průjem (častá vodnatá stolice), podávání přípravku přeruště a informujte neprodleně svého lékaře.
• Je důležité být poblíž toalety a mít k dispozici pomoc, je-li to nutné, protože účinek se může dostavit do 30 minut po podání injekce.
• Prosím, informujte lékaře, pokud máte bolest břicha, která nepřestává, nevolnost nebo zvracení, které nově nastoupilo, nebo se zhoršuje.
• Prosím, informujte také lékaře, pokud máte kolostomii (vývod tlustého střeva), peritoneální katetr (cévku do dutiny břišní), nebo máte divertikulární onemocnění (výchlipky v tlustém střevě) nebo zaklínění stolice (silná zácpa), protože tento lék musí být používán s opatrností při těchto stavech.
• Jestliže dostáváte podpůrnou péči při pokročilém onemocnění, budete tento lék užívat jen omezenou dobu, která bude pravděpodobně kratší než 4 měsíce.
• Tento lék by neměl být podáván pacientům k léčbě zácpy, která s podáváním opioidů nesouvisí. Informujte, prosím, lékaře, pokud jste trpěl/a zácpou předtím, než jste užíval/a opioidy (pro léčbu bolesti).
Děti a dospívající
Nepodávejte tento lék dětem a dospívajícím mladším 18ti let, protože možná rizika a přínosy nejsou známy.
Další léčivé přípravky a přípravek Relistor
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Lékař Vám může doporučit současné užívání dalších léků včetně těch, které se používají při zácpě. Těhotenství a kojení
Účinek methylnatrexonium-bromidu u těhotných žen není znám. Lékař rozhodne, jestli můžete užívat přípravek Relistor v případě, že jste těhotná.
Ženy užívající tento lék by neměly kojit, protože není známo, jestli methylnatrexonium-bromidu přechází do mateřského mléka.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Závrať je častým nežádoucím účinkem tohoto léku. Může ovlivnit schopnost řízení dopravních prostředků a obsluhování strojů.
Důležité informace o některých složkách přípravku Relistor
Tento lék obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tzn. je v podstatě „bez sodíku“.
3. Jak se přípravek Relistor používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý/á, poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka pro pacienty s dlouhodobou bolestí (kromě pacientů dostávajících podpůrnou péči při pokročilém onemocnění) je 12 mg methylnaltrexonium-bromidu (0,6 ml roztoku) podávaného jako injekce pod kůži podle potřeby, ale minimálně 4-krát týdně až maximálně jedenkrát denně (7-krát týdně).
8mg předplněná injekční stříkačka Relistor se má použít jen na léčbu těchto pacientů s nenádorovou bolestí, u kterých je potřebné snížit dávku z důvodu jiného zdravotního problému.
Doporučená dávka pro pacienty dostávající podpůrnou léčbu při pokročilém onemocnění je 8 mg methylnaltrexonium-bromidu (0,4 ml roztoku) pro pacienty o hmotnosti 38-61 kg nebo 12 mg (0,6 ml roztoku) pro pacienty o hmotnosti 62-114 kg. Dávka se podává každých 48 hodin (každý druhý den) jako injekce pod kůži.
Velikost dávky stanoví lékař.
Pokud je Vaše hmotnost menší než 38 kg nebo vyšší než 114 kg měl/a byste použít přípravek Relistor injekční lahvičky, protože správná dávka nemůže být přesně podána předplněnou injekční stříkačkou.
Tento přípravek se podává injekcí pod kůži (podkožní injekce) buď (1) do horní části dolních končetin (stehna), nebo (2) do oblasti břicha, nebo (3) do horní části paže (nejedná-li se o autoinjekci). (Viz NÁVOD K PŘÍPRAVĚ A PODÁNÍ INJEKCE PŘÍPRAVKU RELISTOR na konci této příbalové informace.)
Můžete dosáhnout pohybu střev do několika minut až hodin po podání injekce; proto se doporučuje být poblíž toalety nebo mít po ruce mísu.
Jestliže jste použil(a) více přípravku Relistor, než jste měl(a)
Jestliže jste užil/a větší množství tohoto přípravku, než jste měl/a (buď jednorázovým podáním většího množství, nebo podáním více injekcí v průběhu 24 hodin), můžete cítit závrať při vstávání, a proto neprodleně informujte svého lékaře nebo lékárníka. Vždy si vezměte vnější obal léku s sebou, a to i tehdy, je-li již prázdný.
Jestliže jste zapomněl(a) použít přípravek Relistor
Jestliže zapomenete užít dávku, co nejdříve informujte svého lékaře nebo lékárníka. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat přípravek Relistor
Jestliže chcete přestat užívat tento přípravek, měl/a byste informovat svého lékaře nebo lékárníka.
Máte-li jakékoli další otázky týkající se užívání tohoto léčivého přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento lék nežádoucí účinky, které se ale nemusí vyskytnout u každého.
U pacientů používajících přípravek Relistor byly hlášeny případy proděravění střevní stěny (gastrointestinální perforace). Z dostupných údajů není známo, jak často se to stává. Pokud začnete pociťovat bolest břicha, která je buď závažná nebo nepřestává, přestaňte užívat tento lék a okamžitě telefonicky informujte svého lékaře
Následující nežádoucí účinky jsou velmi časté a mohou postihnout víc než 1 osobu z 10. Jestliže se u Vás vyskytne některý z těchto nežádoucích účinků a je buď závažný nebo nepřestává, měl/a byste kontaktovat svého lékaře:
• Bolest břicha (bolest žaludku)
• Nevolnost (žaludeční)
• Průjem (častá vodnatá stolice)
• Plynatost (odchod větrů)
Další časté nežádoucí účinky, které mohou postihnout až 1 osobu z 10, jsou:
• Závrať (pocit točení hlavy)
• Mírné příznaky jako při náhlém přerušení léčby opioidy (některý z uvedených příznaků: pocit chladu, třes, výtok z nosu, pocení, „husí kůže“, návaly tepla, bušení srdce)
• Reakce v místě podání (např. píchání, pálení, bolest, zčervenání, otok)
• Zvracení
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Relistor uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce, víku podnosu a štítku injekční stříkačky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 30°C.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
Použijte tento přípravek pouze v případě, že je roztok čirý, bezbarvý až světle žlutý a pokud neobsahuje vločky ani mechanické částice.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte.. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Relistor obsahuje
- Léčivou látkou je methylnaltrexonii bromidum. Jedna injekční stříkačka 0,4 ml obsahuje 8 mg methylnaltrexonii bromidum. Jedna injekční stříkačka 0,6 ml obsahuje 12 mg methylnaltrexonii bromidum. Jeden mililitr roztoku obsahuje 20 mg methylnaltrexonii bromidum.
- Pomocnými látkami jsou: chlorid sodný, natrium-kalcium-edetát, glycin-hydrochlorid, voda na injekci, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH).
Jak přípravek Relistor vypadá a co obsahuje toto balení
Přípravek Relistor je injekční roztok. Je čirý, bezbarvý až světle žlutý a neobsahuje vločky ani pevné
částice.
Dodávají se následující balení:
Balení obsahující 4, 7, 8 nebo 10 předplněných injekčních stříkaček s krytem jehly.
Na trhu nemusí být všechny velikosti balení. Držitel rozhodnutí o registraci
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00, Praha 7 Česká republika
Výrobce
Przedsi^biorstwo Farmaceutyczne Jelfa SA ul. Wincentego Pola 21 58-500 Jelenia Góra,
Polsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Swedish Orphan Biovitrum BVBA Tél/Tel: + 32 2880 6119 e-mail: benelux@sobi.com
Lietuva
PharmaSwiss UAB Tel. + 370 5 279 0762
BtarapHH
PharmaSwiss EOOD Tea.: + 359 2 89 52 110
Luxembourg/Luxemburg
Swedish Orphan Biovitrum BVBA Tél/Tel: +32 2880 6119 e-mail: benelux@sobi.com
Česká republika
Swedish Orphan Biovitrum s.r.o. Tel: + 420 257 222 034 e-mail: mail.cz@sobi.com
Magyarország
Swedish Orphan Biovitrum s.r.o. Magyarországi Fióktelepe
Tel. +420 257 222 034 e-mail: mail.hu@sobi.com
Danmark
Swedish Orphan Biovitrum A/S Tlf: + 45 32 96 68 69 e-mail: mail.dk@sobi.com
Deutschland
Swedish Orphan Biovitrum GmbH Tel: +49 89 55066760 e-mail: mail.de@sobi.com
Eesti
PharmaSwiss Eesti OU Tel: +372 6 827 400
EkXába
Swedish Orphan Biovitrum S.r.l. TpT,: +39 0521 19 111 e-mail: mail.it@sobi.com
Espaňa
Swedish Orphan Biovitrum S.L Tel: + 34 913 91 35 80 e-mail: mail.es@sobi.com
Malta
Swedish Orphan Biovitrum S.r.l. Tel: +39 0521 19 111 e-mail: mail.it@sobi.com
Nederland
Swedish Orphan Biovitrum BVBA Tel: +32 288 06119 e-mail: benelux@sobi.com
Norge
Swedish Orphan Biovitrum AS Tlf: +47 66 82 34 00 e-mail: mail.no@sobi.com
Osterreich
Swedish Orphan Biovitrum GmbH
Tel: +49 89 55066760 e-mail: mail.de@sobi.com
Polska
Valeant sp. z o.o. sp. j.
Tel.: +48 17 865 51 00
France
Swedish Orphan Biovitrum SARL Tél: + 33 1 85 78 03 40 e-mail: mail.fr@sobi.com
Hrvatska
PharmaSwiss d.o.o.
Tel: +385 1 6311 833
Ireland
Swedish Orphan Biovitrum Ltd Tel: +44 1223 891854 e-mail: mail.uk@sobi.com
Island
Swedish Orphan Biovitrum A/S Tlf: + 45 32 96 68 69 e-mail: mail.dk@sobi.com
Italia
Swedish Orphan Biovitrum S.r.l. Tel: +39 0521 19 111 e-mail: mail.it@sobi.com
Kórcpog
M.S. Jacovides & Co Ltd Tnk: + 357 22 0056200
Latvija
PharmaSwiss UAB Tel: + 371 67502185
Portugal
Swedish Orphan Biovitrum S.L Tel: +34 913 91 35 80 e-mail: mail.es@sobi.com
Románia
Valeant Pharma SRL Tel: +40 374 102 600
Slovenija
PharmaSwiss d.o.o.
Tel: + 386 1 2364 700
Slovenská republika
Swedish Orphan Biovitrum o.z.
Tel: +420 257 222 034 e-mail: mail.sk@sobi.com
Suomi/Finland
Oy Swedish Orphan Biovitrum Ab Puh./Tel: +358 201 558 840 e-mail: mail.fi@sobi.com
Sverige
Swedish Orphan Biovitrum AB (publ) Tel: +46 8 697 20 00 e-mail: mail.se@sobi.com
United Kingdom
Swedish Orphan Biovitrum Ltd Tel: +44 1223 891854 e-mail: mail.uk@sobi.com
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu
SEZNAM OTÁZEK PRO PACIENTA
Tato část obsahuje důležité otázky, které si musíte zodpovědět předtím, než začnete užívat přípravek Relistor a během léčby přípravkem Relistor.
V případě, že v době, kdy je Vám podáván tento přípravek, odpovíte NE na některou z následujících otázek, kontaktujte, prosím, svého lékaře, zdravotní sestru nebo lékárníka.
• Jsou Vám z důvodu Vašeho onemocnění podávány opioidy (například morfin nebo kodein)?
• Je to již 48 hodin a více od Vaší poslední stolice?
• Je Vám známa technika samopodávání injekce nebo jste ji diskutoval/a s Vaším lékařem (nebo zdravotní sestrou nebo lékárníkem)?
• Jste natolik pohyblivý/á, abyste se dostal/a k toaletě, nebo máte asistenta, který se o Vás stará a může Vám pomoci?
• Máte telefonní číslo na sestru nebo na zdravotní středisko?
NÁVOD K PŘÍPRAVÉ A PODÁVÁNÍ INJEKCE PŘÍPRAVKU RELISTOR
Tento bod je rozdělen do následujících podbodů:
Úvod
Krok 1: Příprava před injekcí
Krok 2: Výběr a příprava místa pro injekci
Krok 3: Podání přípravku Relistoru předplněnou injekční stříkačkou Krok 4: Likvidace zbylého odpadu
Úvod
Tento návod Vám vysvětlí, jak si připravit a aplikovat injekci přípravku Relistor při použití předplněné injekční stříkačky. Pečlivě si návod přečtěte a postupujte podle něj krok za krokem. Váš lékař, zdravotní sestra nebo lékárník Vám vysvětlí správnou techniku podání injekce sobě samému. Nesnažte se podat si dávku léku, dokud si nejste jist/á, že rozumíte instrukci a že si umíte injekci připravit a podat.
Důležitá upozornění:
• Nepoužívejte předplněnou injekční stříkačku přípravku Relistor více než jednou, i když ještě v injekční stříkačce zbývá lék.
• Po použití (krok 4) bezpečně vyhoďte předplněnou injekční stříkačku přípravku Relistor.
• Abyste se vyhnul/a poranění jehlou, použité jehly znovu nezakrývejte.
Připravte si vše, co budete k injekci potřebovat:
1. Relistor předplněná injekční stříkačka
2. alkoholové tampony
3. bavlněný tampon nebo gáza
4. náplast
Krok 1: Příprava před injekcí
1. Vyberte si rovnou, čistou, dobře osvětlenou pracovní plochu, kde si můžete rozložit obsah krabičky přípravku Relistor. Ujistěte se, že máte dost času potřebného na celkovou aplikaci
injekce.
2. Umyjte si ruce mýdlem a teplou vodou.
3. Podívejte se na předplněnou injekční stříkačku. Ujistěte se, že dávka předepsaná Vám Vaším lékařem odpovídá dávce na štítku předplněné injekční stříkačky.
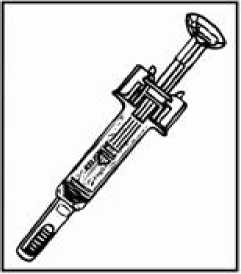
4. Ujistěte se, že roztok v předplněné injekční stříkačce je čirý a bezbarvý až světle žlutý a neobsahuje vločky ani viditelné mechanické částice. Pokud tomu tak není, nepoužívejte předplněnou injekční stříkačku a pro pomoc kontaktujte Vaši zdravotní sestru, lékaře nebo lékárníka.
5. Držte pevně tělo předplněné injekční stříkačky a rovně stáhněte kryt z injekční jehly. Nedotýkejte se jehly a dávejte pozor, aby se jehla nedotkla žádného jiného povrchu.
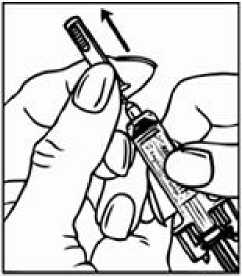
Krok 2: Výběr a příprava místa pro injekci
1. Pro podání injekce přípravku Relistor jsou doporučené tři oblasti těla: (1) horní část dolních končetin (stehna), (2) břicho, a (3) horní část paže (pokud Vám přípravek podává jiná osoba).
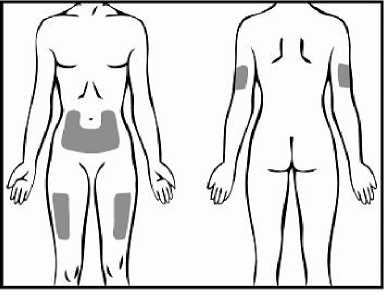
2. Doporučuje se pro každou injekci vybrat vždy odlišné místo podání. Vyhněte se podání
injekce přesně do předchozího místa podání. Nepodávejte injekci do oblasti, kde je kůže bolestivá, pohmožděná, zčervenalá nebo tvrdá. Vyhněte se oblastem s jizvami nebo s narušenou
strukturou kůže (strie).
3. Místo na kůži, kam má být podána injekce, očistěte alkoholovým tamponem a nechte oschnout. Až do podání injekce se tohoto místa nedotýkejte.
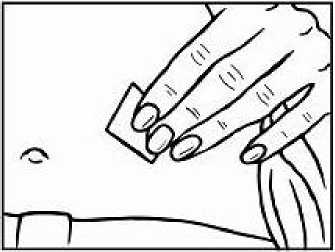
Krok 3: Podání přípravku Relistor předplněnou injekční stříkačkou
1. Držte injekční stříkačku jednou rukou jako tužku. Druhou rukou na očištěném místě kůže vytvořte kožní řasu a pevně ji držte.
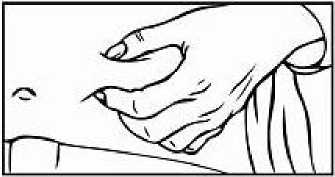
2.
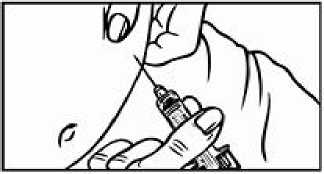
Krátkým rychlým pohybem zatlačte celou délku jehly do kůže pod malým úhlem (45 stupňů).
3. Jakmile je jehla vpíchnutá, pusťte kůži a pomalu stlačujte píst směrem dolů, dokud není předplněná injekční stříkačka prázdná.
4. Rychle vytáhněte jehlu z kůže, držte přitom jehlu stále pod stejným úhlem jako při vpichu. Vytáhněte Váš palec z pístu, abyste umožnil/a zakrytí jehly ochranným obalem. V místě vpichu může dojít ke slabému krvácení.
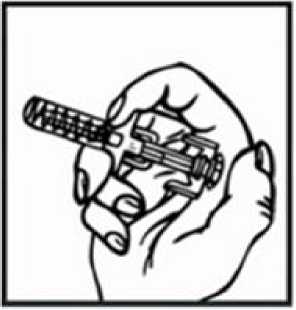
5. Na místo vpichu můžete přitisknout bavlněný tampon nebo gázu. Netřete si toto místo. Podle potřeby je možné místo vpichu překrýt náplastí.
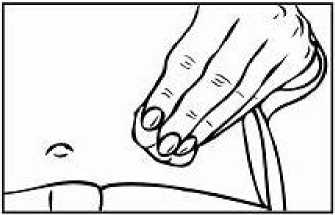
Krok 4: Likvidace zbylého odpadu
Předplněná injekční stříkačka se NIKDY nesmí znovu použít. NIKDY znovu nezakrývejte jehlu krytem. Zlikvidujte předplněnou injekční stříkačku podle pokynů lékaře, sestry nebo lékárníka.
Použitou předplněnou injekční stříkačku odkládejte do uzavíratelného, propíchnutí odolného kontejneru. Můžete použít kontejner k likvidaci ostrých předmětů (jako např. žlutý kontejner pro nebezpečný biologický odpad). Zeptejte se svého lékaře, sestry nebo lékárníka, jak správně zlikvidovat kontejner. Mohou existovat místní předpisy o tom, jak správně zlikvidovat použité injekční jehly a injekční stříkačky.
98