Refacto Af 2000 Iu
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
ReFacto AF 250 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 500 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 1000 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 2000 IU prášek a rozpouštědlo pro injekční roztok
stříkačce
stříkačce
stříkačce
ReFacto AF 250 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce ReFacto AF 500 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce ReFacto AF 1000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční ReFacto AF 2000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční ReFacto AF 3000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
ReFacto AF 250 IU prášek a rozpouštědlo pro injekční roztok Jedna injekční lahvička obsahuje jmenovitě 250 m.j.* moroctocogum alfa1.
Po rozpuštění obsahuje 1 ml roztoku přibližně 62,5 m.j. moroctocogum alfa.
ReFacto AF 500 IU prášek a rozpouštědlo pro injekční roztok Jedna injekční lahvička obsahuje jmenovitě 500 m.j.* moroctocogum alfa1.
Po rozpuštění obsahuje 1 ml roztoku přibližně 125 m.j. moroctocogum alfa.
ReFacto AF 1000 IU prášek a rozpouštědlo pro injekční roztok Jedna injekční lahvička obsahuje jmenovitě 1000 m.j.* moroctocogum alfa1.
Po rozpuštění obsahuje 1 ml roztoku přibližně 250 m.j. moroctocogum alfa.
ReFacto AF 2000 IU prášek a rozpouštědlo pro injekční roztok Jedna injekční lahvička obsahuje jmenovitě 2000 m.j.* moroctocogum alfa1.
Po rozpuštění obsahuje 1 ml roztoku přibližně 500 m.j. moroctocogum alfa.
ReFacto AF 250 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje jmenovitě 250 m.j.* moroctocogum alfa1.
Po rozpuštění obsahuje 1 ml roztoku přibližně 62,5 m.j. moroctocogum alfa.
ReFacto AF 500 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje jmenovitě 500 m.j.* moroctocogum alfa1.
Po rozpuštění obsahuje 1 ml roztoku přibližně 125 m.j. moroctocogum alfa.
ReFacto AF 1000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje jmenovitě 1000 m.j.* moroctocogum alfa1.
Po rozpuštění obsahuje 1 ml roztoku přibližně 250 m.j. moroctocogum alfa.
ReFacto AF 2000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje jmenovitě 2000 m.j.* moroctocogum alfa1.
Po rozpuštění obsahuje 1 ml roztoku přibližně 500 m.j. moroctocogum alfa.
ReFacto AF 3000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje jmenovitě 3000 m.j.* moroctocogum alfa1.
Po rozpuštění obsahuje 1 ml roztoku přibližně 750 m.j. moroctocogum alfa.
* Účinnost (mezinárodní jednotky) je stanovena podle Evropského lékopisu chromogenní metodou. Specifická aktivita přípravku ReFacto AF je 7600-13800 m.j./mg bílkoviny.
aminokyselin srovnatelnou s 90 + 80 kDa formou faktoru VIII (tj. vymazání B-domény) a s posttranslačními modifikacemi podobnými modifikacím molekuly plasmatické.
Výrobní proces přípravku ReFacto byl modifikován tak, aby odstranil jakoukoli exogenní bílkovinu humánní nebo odvozenou od zvířat v procesu buněčné kultury purifikací, nebo finálního složení. Současně byl změněn smyšlený název přípravku na ReFacto AF.
Pomocná látka se známým účinkem:
Po rozpuštění obsahuje 1,23 mmol (29 mg) sodíku v jedné injekční lahvičce nebo předplněné injekční stříkačce.
Úplný seznam pomocných látek, viz bod 6.1.
3. LÉKOVÁ FORMA
ReFacto AF 250 IU, 500 IU, 1000 IU, 2000 IU prášek a rozpouštědlo pro injekční roztok Prášek a rozpouštědlo pro injekční roztok Bílý až téměř bílý koláč/prášek Čiré, bezbarvé rozpouštědlo
ReFacto AF 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce
Prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce
Bílý až téměř bílý koláč/prášek ve vrchním zásobníku předplněné injekční stříkačky
Čiré, bezbarvé rozpouštědlo ve spodním zásobníku předplněné injekční stříkačky
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a prevence krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII).
Přípravek ReFacto AF je vhodný k použití u dospělých pacientů a u dětí všech věkových skupin, včetně novorozenců.
Přípravek ReFacto AF neobsahuje von Willebrandův faktor, a proto není indikován u von Willebrandovy choroby.
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře se zkušenostmi v terapii hemofilie A.
Sledování léčby
Velikost podávané dávky a frekvence opakovaných infuzí musí být během léčby určovány podle hladin faktoru VIII. Každý pacient může na faktor VIII reagovat jinak, tj. s jinými poločasy a s jinou recovery. Pokud se dávka určuje podle tělesné hmotnosti, může být nutné ji u pacientů s nad- nebo podváhou příslušným způsobem upravit. Nepostradatelnou pomůckou je přesné sledování substituční léčby pomocí analýzy koagulace (plasmatické aktivity faktoru VIII), a to zejména v případě velkého chirurgického výkonu.
Při monitorování aktivity faktoru VIII u pacientů v průběhu léčby přípravkem ReFacto AF se doporučuje použití chromogenní metody. Používá-li se k určení aktivity faktoru VIII v krevních vzorcích pacienta jednofázová koagulační metoda založená na tromboplastinovém času (aPTT) in vitro, výsledky plasmatické aktivity faktoru VIII mohou být významně ovlivněny typem činidla aPTT a referenčním standardem použitými při dané metodě. Výsledky získané pomocí jednofázové koagulační metody aPTT se rovněž mohou významně lišit od výsledků získaných chromogenní metodou. Výsledky jednofázové koagulační metody jsou typicky o 20 - 50 % nižší než výsledky chromogenní metody. Ke korekci tohoto rozdílu se může použít laboratorní standard přípravku ReFacto AF (viz bod 5.2). Toto je důležité zejména tehdy, mění-li se laboratoř a/nebo použité reagencie.
Dávkování
Dávka a délka substituční terapie jsou závislé na závažnosti nedostatku faktoru VIII, místě a rozsahu krvácení a na klinickém stavu nemocného. Podané dávky musí být titrovány klinickou odpovědí pacienta. Přítomnost inhibitoru si může vyžádat vyšší dávky nebo vhodnou speciální léčbu.
Počet podaných jednotek faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které jsou vztaženy k aktuálnímu standardu WHO pro přípravky obsahující faktor VIII. Aktivita faktoru VIII v plasmě je vyjádřena buď v procentech (poměr k obsahu v normální lidské plasmě) nebo v IU (poměr k mezinárodnímu standardu obsahu faktoru VIII v plasmě). Jedna IU aktivity faktoru VIII odpovídá množství faktoru VIII v 1 ml normální lidské plasmy.
Jiný přípravek, obsahující moroktokog alfa a schválený pro používání mimo Evropu, má odlišnou sílu stanovenou výrobcem, jež byla kalibrována k Mezinárodnímu standardu WHO jednofázovou koagulační metodou; tento přípravek se nazývá XYNTHA. Vzhledem k použití odlišné metody ke stanovení účinnosti přípravků XYNTHA a ReFacto AF, je 1 IU přípravku XYNTHA (kalibrováno jednofázovou koagulační metodou) ekvivalentní s 1,38 IU přípravku ReFacto AF (kalibrováno chromogenní metodou). Je-li pacientovi, který je normálně léčen přípravkem XYNTHA, předepsán přípravek ReFacto AF, musí ošetřující lékař uvažovat o přizpůsobení doporučené dávky v závislosti na hodnotách dosažených hladin faktoru VIII.
Podle současných léčebných režimů se pacientům s hemofilií A doporučuje brát si s sebou na cestu dostatečnou zásobu faktoru VIII k předpokládané léčbě. Pacienti mají být upozorněni, aby se před cestováním poradili se svým poskytovatelem zdravotní péče.
Léčba dle potřeby
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že podání 1 IU faktoru VIII na kg tělesné hmotnosti zvýší plasmatickou aktivitu faktoru VIII o 2 IU/dl. Požadovaná dávka se stanoví podle následujícího vzorce:
Požadované jednotky (IU) = tělesná hmotnost (kg) x žádaný nárůst faktoru VIII (% nebo IU/dl) x 0,5 (IU/kg na IU/dl), kde 0,5 IU/kg na IU/dl odpovídá reciproční recovery, obecně pozorované po infuzi faktoru VIII.
Množství, které má být podáno a frekvence podání, mají v jednotlivých případech vždy vycházet z klinické účinnosti.
V případě následujících krvácivých příhod nemá aktivita faktoru VIII nikdy poklesnout pod danou plasmatickou hladinu (v % normálu nebo v IU/dl) v daném období. Následující tabulka může být použita jako průvodce dávkování při krvácivých příhodách a chirurgických výkonech:
|
Stupeň krvácení/ Typ chirurgického výkonu |
Požadovaná hladina faktoru VIII (% nebo IU/dl) |
Četnost dávek (hod.)/ trvání léčby (dny) |
|
Hemoragie Časný hemartros, svalové krvácení nebo krvácení do dutiny ústní |
O 1 o <N |
Opakovat každých 12 - 24 hod. Nejméně 1 den, až do zastavení krvácivé příhody, detekované bolestí, nebo do dosažení uzdravení. |
|
Rozsáhlejší hemartros, svalové krvácení nebo hematom |
30 -60 |
Opakované infuze každých 12 - 24 hod. po 3 - 4 dny nebo déle do ústupu bolesti a vyléčení akutní poruchy funkce. |
|
Život ohrožující hemoragie |
O O 1 O kO |
Opakované infuze každých 8 - 24 hod. až do vyřešení ohrožení. |
|
Chirurgické výkony Malé včetně extrakce zubů |
30 -60 |
Každých 24 hodin, nejméně 1 den, až do dosažení uzdravení. |
|
Velké |
80 - 100 (pre- a postoperační) |
Opakované infuze každých 8 až 24 hod. až do adekvátního zahojení rány, potom léčení nejméně dalších 7 dní k udržování aktivity faktoru VIII na 30 % - 60 % (IU/dl). |
Profylaxe
K dlouhodobé profylaxi krvácení pacientů se závažnou hemofilií A jsou obvyklé dávky 20 - 40 IU faktoru VIII na kg tělesné hmotnosti v rozmezí od 2 do 3 dnů. V některých případech, obzvláště u mladších pacientů, mohou být nezbytné kratší dávkové intervaly nebo vyšší dávky.
Pediatrická _ populace
Při léčbě mladších dětí (do 6 let věku) přípravkem ReFacto AF se předpokládá potřeba zvýšit dávky v porovnání s těmi, které se podávají starším dětem nebo dospělým. V klinických studiích s ReFacto prokázala farmakokinetická analýza nižší poločas vylučování a recovery u dětí pod 6 let věku než u starších dětí a dospělých (viz bod 5.2). V průběhu klinických studií dostávaly děti ve věku pod 6 let v profylaktickém režimu průměrnou dávku 50 IU/kg přípravku ReFacto AF a bylo u nich zjištěno průměrně 6,1 krvácivých příhod na rok. Starší děti a dospělí dostávali v profylaktickém režimu průměrnou dávku 27 IU/kg a bylo u nich zjištěno v průměru 10 krvácivých příhod na rok. V klinické studii stanovující střední dávku na infuzi přípravkem ReFacto AF na krvácivou epizodu u dětí ve věku pod 6 let, byla střední dávka vyšší než střední dávka pro starší děti a dospělé (51,3 IU/kg, respektive 29,3 IU/kg).
Subjekty starší 65 let nebyly zahrnuty do žádných klinických studií. Obecně platí, že dávku pro starší pacienty je třeba upravit dle konkrétních potřeb.
Porucha _ funkce _ jater nebo ledvin
Úprava dávkování u pacientů s poruchou funkce jater nebo ledvin nebyla v klinických hodnoceních studována.
Způsob podání
Intravenózní podání.
Přípravek ReFacto AF se podává intravenózní infuzí po dobu několika minut po rekonstituci lyofilizovaného prášku na injekci v roztoku chloridu sodného 9 mg/ml (0,9 %) na injekci (přiložen). Rychlost podání má být zvolena s ohledem na úroveň pohodlí nemocného.
Nezdravotnickým pracovníkům podávajícím přípravek se doporučuje absolvovat odpovídající školení. Návod k rekonstituci přípravku před podáním, viz bod 6.6.
4.3 Kontraindikace
Přecitlivělost na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Známá alergická reakce na křeččí bílkovinu.
4.4 Zvláštní upozornění a zvláštní opatření pro použití
Hypersenzitivita
U přípravku ReFacto AF byly pozorovány hypersenzitivní reakce alergického typu. Tento léčivý přípravek obsahuje stopy křeččích proteinů. Jestliže se objeví příznaky přecitlivělosti, pacienti mají být informováni, aby ihned přerušili užívání léčivého přípravku a kontaktovali svého lékaře. Pacienti mají být informováni o časných projevech přecitlivělosti, jako je vyrážka, generalizovaná kopřivka, tlak na hrudi, sípavé dýchání, hypotenze a anafylaxe.
V případě šoku mají být dodrženy léčebné standardy protišokové terapie.
Neutralizační protilátky (inhibitory)
Známou komplikací při léčbě nemocných s hemofilií A je tvorba neutralizačních protilátek (inhibitorů) faktoru VIII. Tyto inhibitory jsou obvykle IgG imunoglobuliny směrované proti prokoagulační aktivitě faktoru VIII, která je kvantifikována v Bethesda jednotkách (BU) v ml plasmy metodou Bethesda modifikovanou podle Nijmegena. Riziko rozvoje inhibitorů koreluje s vystavením faktoru VIII, toto riziko se zvyšuje v průběhu prvních 20 dnů expozice. Vzácně může dojít k rozvoji inhibitorů po prvních 100 dnech expozice.
Případy rekurentních inhibitorů (nízkého titru) byly pozorovány po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v anamnéze z jednoho přípravku faktoru VIII na jiný. Proto se doporučuje pečlivě monitorovat všechny pacienty na výskyt inhibitorů po každé změně přípravku.
V zásadě všichni pacienti léčení přípravky obsahující koagulační faktor VIII mají být pečlivě monitorováni na rozvoj inhibitorů vhodnými klinickými pozorováními a laboratorními testy. Jestliže se nedosáhne očekávané plasmatické hladiny faktoru VIII nebo nedojde-li ke zvládnutí krvácení
příslušnou dávkou, má být provedeno vyšetření, zda jsou přítomny inhibitory faktoru VIII. U pacientů s vysokým titrem inhibitorů nemusí být terapie faktorem VIII účinná a je třeba uvažovat o jiné terapeutické volbě. Léčba těchto pacientů má být řízena lékaři se zkušeností s léčbou pacientů s hemofilií a inhibitory faktoru VIII.
Hlášení o nedostatečné účinnosti
V klinických studiích a v postregistračním používání přípravku ReFacto byla podána hlášení o nedostatečné účinnosti, zejména v profylaxi pacientů. Hlášená nedostatečná účinnost přípravku ReFacto byla pacienty popisována jako krvácení do cílových kloubů, krvácení do nových kloubů nebo subjektivní pocit nového nástupu krvácení. Pro zajištění adekvátní terapeutické odpovědi při předepisování přípravku ReFacto AF je důležité individuální titrování a monitorování hladiny faktoru u každého pacienta (viz bod 4.8).
Důrazně se doporučuje po každém podání přípravku ReFacto AF pacientovi zaznamenat název přípravku a číslo šarže za účelem zachování vazby mezi pacientem a číslem šarže léčivého přípravku. Pacienti mohou dokumentovat číslo šarže nalepením štítku, který předtím odlepili z jedné injekční lahvičky nebo předplněné injekční stříkačky, do svého diáře pro hlášení možných nežádoucích účinků.
Kardiovaskulární příhody
Substituční léčba faktorem VIII může u pacientů s rizikovými faktory kardiovaskulárních poruch zvýšit riziko postižení kardiovaskulárního systému.
Komplikace spojené s katétrem
V případě nutnosti použití zařízení pro centrální venózní přístup (CVAD) je třeba zvážit riziko komplikací spojených s CVAD, včetně lokální infekce, bakteriemie a trombózy v místě katétru (viz bod 4.8).
Obsah sodíku
Po rekonstituci tento léčivý přípravek obsahuje 1,23 mmol (29 mg) sodíku v jedné injekční lahvičce nebo předplněné injekční stříkačce, což mají vzít v úvahu pacienti na řízené sodíkové dietě.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly hlášeny žádné interakce přípravků obsahujících rekombinantní koagulační faktor VIII a jiných léčivých přípravků.
4.6 Fertilita, těhotenství a kojení
Nebyly provedeny reprodukční studie podávání faktoru VIII zvířatům, a proto nejsou dostupné žádné údaje o fertilitě. Vzhledem ke vzácnému výskytu hemofilie A u žen nejsou k dispozici zkušenosti s podáváním faktoru VIII v období těhotenství a kojení. Proto má být faktor VIII podáván v průběhu těhotenství a kojení pouze tehdy, je-li jasně indikován.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek ReFacto AF nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nepříliš často byla po přípravku ReFacto pozorována hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, zimnici, nával horka, generalizovanou kopřivku, bolest hlavy, kopřivku, hypotenzi, letargii, nauzeu, neklid, tachykardii, napětí na hrudi, mravenčení, zvracení, sípání), které se mohou v některých případech rozvinout do závažné anafylaxe včetně šoku (viz bod 4.4).
V přípravku ReFacto AF mohou být přítomna stopová množství křeččích proteinů. Ve velmi vzácných případech byl pozorován rozvoj protilátek proti křeččím proteinům, avšak bez klinických následků.
V klinické studii s přípravkem ReFacto mělo 20 ze 113(18%) dříve léčených pacientů (PTP) zvýšený titr protilátek proti CHO, avšak bez zjevného klinického účinku.
Výskyt neutralizačních protilátek (inhibitorů) faktoru VIII je známou komplikací léčby nemocných s hemofilií A. Podobně jako při léčbě jakýmkoli přípravkem obsahujícím koagulační faktor VIII musí být pacienti pečlivě monitorováni na rozvoj inhibitorů, které musí být titrovány v Bethesda jednotkách (BU) při použití metody Bethesda upravené podle Nijmegena. Pokud se takové inhibitory vyskytnou, může se to projevit jako nedostatečná klinická odpověď na léčbu. V těchto případech se doporučuje vyhledat specializované pracoviště na léčbu hemofilie.
Přehled nežádoucích účinků v tabulce
Tabulka uvedená níže odpovídá klasifikaci orgánových systémů MedDRA (třídám orgánových systémů a úrovni preferovaných termínů). Frekvence byly hodnoceny na základě následujících konvencí: velmi časté (> 1/10); časté (> 1/100 až < 1/10) a méně časté (> 1/1000 až < 1/100). V této tabulce jsou uvedeny nežádoucí účinky, které byly hlášeny v klinických studiích s přípravkem ReFacto nebo ReFacto AF. Frekvence byly získány na základě nežádoucích účinků, které se vyskytly u všech léčených pacientů ve sloučených klinických studiích zahrnujících celkem 655 subjektů (554 PTP, 101 PUP).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Třídy orgánových systémů |
Frekvence výskytu na počet pacientů léčených přípravky ReFacto nebo ReFacto AF | ||
|
Velmi časté >1/10 |
Časté > 1/100 až < 1/10 |
Méně časté > 1/1000 až < 1/100 | |
|
Poruchy krve a lymfatického systému |
Inhibice faktoru VIII (PUP) |
Inhibice faktoru VIII (PTP) | |
|
Poruchy imunitního systému |
Anafylaktická reakce | ||
|
Poruchy metabolismu a výživy |
Snížená chuť k jídlu | ||
|
Poruchy nervového systému |
Závratě |
Periferní neuropatie; spavost; porucha chuti | |
|
Srdeční poruchy |
Angina pectoris; tachykardie; palpitace | ||
|
Cévní poruchy |
Hemoragie; hematom |
Hypotenze; tromboflebitida; návaly horka | |
|
Respirační, hrudní a mediastinální poruchy | |||
|
Gastrointestinální poruchy |
Průjem; zvracení; bolest břicha; nauzea | ||
|
Poruchy kůže a podkožní tkáně |
Kopřivka; vyrážka; svědění |
Hyperhidróza | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Artralgie |
Myalgie | |
|
Frekvence výskytu na počet pacientů léčených přípravky ReFacto nebo ReFacto AF | |||
|
Třídy orgánových systémů |
Velmi časté >1/10 |
Časté > 1/100 až < 1/10 |
Méně časté > 1/1000 až < 1/100 |
|
Celkové poruchy a |
Pyrexie |
Zimnice; lokální reakce |
Astenie; reakce v místě podání; |
|
reakce v místě aplikace |
spojená s katétrem |
bolest v místě podání; zánět v místě podání | |
|
Vyšetření |
Vyšetření protilátek pozitivní; vyšetření na protilátky proti faktoru VIII pozitivní |
Zvýšená hladina aspartátaminotransferázy; zvýšená hladina alaninaminotransferázy; zvýšená hladina bilirubinu v krvi; zvýšená hladina kreatininfosfokinázy v krvi | |
Popis vybraných nežádoucích účinků Inhibice faktoru VIII
V klinické studii s přípravkem ReFacto AF u dříve léčených pacientů (PTP) byla incidence inhibitorů faktoru VIII primárním bezpečnostním cílovým parametrem. Dva klinicky tiché přechodné inhibitory s nízkým titrem byly pozorovány u 94 pacientů s mediánem expozice 76 expozičních dnů (ED, rozmezí 1-92), odpovídající 2,2 % z 89 pacientů s minimálně 50 ED. V podpůrné studii s ReFacto AF byly pozorovány 1 de novo a 2 rekurentní inhibitory (všechny s nízkým titrem, stanovené v centrální laboratoři) u 110 pacientů; medián expozice 58 ED (rozmezí 5-140) a 98 pacientů mělo minimálně 50 ED s přípravkem ReFacto AF. Devadesát osm (98) z původních 110 pacientů pokračovalo v léčbě ve druhé podpůrné studii a mělo následnou rozšířenou expozici přípravku ReFacto AF s mediánem 169 ED navíc (rozmezí 9-425). Byl pozorován navíc jeden (1) de novo inhibitor s nízkým titrem. Četnost výskytu inhibitorů zjištěných v těchto studiích se pohybuje v očekávaném rozmezí.
V klinické studii u PTP s hemofilií A (faktor VIII:C < 2 %), kteří podstoupili velký chirurgický zákrok, byl pozorován 1 inhibitor u 30 pacientů léčených přípravkem ReFacto AF.
V klinické studii s přípravkem ReFacto u PTP byl pozorován 1 inhibitor u 113 pacientů. Rovněž byly po uvedení přípravku na trh spontánně hlášeny inhibitory s vysokým titrem u PTP.
Klinické studie s přípravkem ReFacto AF probíhají u dosud neléčených pacientů (PUP, Previously Untreated Patients). V klinické studii s přípravkem ReFacto AF vyvinulo inhibitory 32 ze 101 (32 %) PUP (FVIII:C < 2 %). Ze 62 pacientů s FVIII:C < 1 % vyvinulo inhibitor 19 (31 %). Ze 32 případů s inhibitory z celé kohorty pacientů (n = 101) jich bylo 16 (16 %) klasifikováno jako s vysokým titrem (> 5 BU/ml) a 16 (16 %) jako s nízkým titrem (< 5 BU/ml). Střední počet expozičních dnů do vyvinutí inhibitorů u těchto 32 pacientů se rovnal 12 (rozmezí 3-49). 15 z 16 pacientů s vysokým titrem podstoupilo imunotoleranční léčbu (ITI). 10 pacientů z 16 s nízkým titrem zahájilo ITI.
Pediatrická populace
Byl hlášen jeden případ cysty u 11letého pacienta a jeden případ popsaný jako zmatenost u 13letého pacienta s možnou souvislostí s léčbou přípravkem ReFacto AF.
Bezpečnost přípravku ReFacto AF byla hodnocena u dříve léčených dětí a dospívajících (n=18, věk 12-16 let ve studii a n=49, věk 7-16 let v podpůrné studii). Třebaže bylo studováno omezené množství dětí, existuje tendence k vyšší frekvenci nežádoucích účinků u dětí ve věku 7-16 let v porovnání s dospělými. Probíhají klinické studie hodnotící podávání přípravku ReFacto AF u dětí ve věku pod 6 let.
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování přípravky obsahujícími rekombinantní koagulační faktor VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika, krevní koagulační faktor VIII;
ATC kód: B02BD02
Přípravek ReFacto AF obsahuje rekombinantní koagulační faktor VIII s odstraněnou B-doménou (moroctocogum alfa). Je to glykoprotein o přibližné molekulové hmotnosti 170 000 Da složený z 1438 aminokyselin. Přípravek ReFacto AF má funkční vlastnosti porovnatelné s vlastnostmi endogenního faktoru VIII. Aktivita faktoru VIII je u pacientů s hemofilií A značně snížena, a proto je nutná substituční terapie.
Po infuzi hemofilickému pacientovi se faktor VIII váže na von Willebrandův faktor přítomný v krevním oběhu nemocného.
Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který urychluje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin poté přemění fibrinogen na fibrin a vytvoří se sraženina. Hemofilie A je na pohlaví závislá dědičná porucha koagulace krve, která je důsledkem snížené hladiny faktoru VIII:C a vede k profuznímu krvácení do kloubů, svalů nebo vnitřních orgánů, a to buď spontánně nebo jako následek úrazu nebo chirurgického výkonu. Substituční terapií se zvýší hladina faktoru VIII v plasmě, což umožní dočasnou korekci nedostatku faktoru a úpravu sklonů ke krvácení.
Navození imunotolerance
Údaje týkající se imunotoleranční léčby (ITI, Immune Tolerance Induction) byly shromážděny od pacientů s hemofilií A, u kterých se rozvinuly inhibitory faktoru VIII. Údaje o ITI od 25 pacientů byly hodnoceny jako součást pivotní studie přípravku ReFacto u PUP (viz bod 4.8). Z těchto 25 pacientů mělo 20 pacientů pokles titrů inhibitoru na < 0,6 BU/ml, z nichž původně 11 z 15 mělo vysoký titr (>
5 BU/ml) a 9 z 10 pacientů mělo nízký titr. Z 6 pacientů, u nichž se rozvinuly inhibitory s nízkým titrem, avšak nebyla jim podána ITI, došlo u 5 k podobným poklesům titru. Nejsou k dispozici žádné dlouhodobé výsledky.
5.2 Farmakokinetické vlastnosti
Farmakokinetické parametry přípravku ReFacto odvozené ze zkřížené studie přípravku ReFacto a koncentrátu faktoru VIII vyrobeného z plasmy za použití chromogenní metody (viz bod 4.2) u 18 dříve léčených pacientů jsou seřazeny v následující tabulce.
|
Farmakokinetické parametry přípravku ReFacto u dříve léčených pacientů s hemofilií A | |||
|
PK parametr |
Průměr |
SD |
Medián |
|
AUCt (IUh/ml) |
19,9 |
4,9 |
19,9 |
|
t1/2 (h) |
14,8 |
5,6 |
12,7 |
|
CL (ml/hkg) |
2,4 |
0,75 |
2,3 |
|
MRT (h) |
20,2 |
7,4 |
18,0 |
|
Recovery (vzestup IU/dl v FVIII:C na IU/kg podaného FVIII) |
2,4 |
0,38 |
2,5 |
Zkratky: AUCt = plocha pod křivkou plasmatických koncentrací - od nuly až do poslední měřitelné koncentrace; t1/2 = poločas vylučování; CL = clearance; FVIII:C = aktivita FVIII; MRT = střední residenční doba
Ve studii, v níž byly měřeny aktivity přípravků ReFacto AF, ReFacto a faktoru VIII chromogenní metodou, byla prokázána bioekvivalence přípravku ReFacto AF a ReFacto. Poměry geometrických středních hodnot nejmenších čtverců přípravku ReFacto AF k přípravku ReFacto činily 100,6 % pro recovery, 99,5 % pro AUCt a 98,1 % pro AUC* (plocha pod křivkou plasmatické koncentrace od času nula do konce). Odpovídající 90 % interval spolehlivosti geometrických průměrů poměru přípravku ReFacto AF k přípravku ReFacto byl v bioekvivalenčním okně 80 % - 125 % a demonstroval bioekvivalenci přípravku ReFacto AF s přípravkem ReFacto.
Ve zkřížené farmakokinetické studii byly stanovovány farmakokinetické parametry přípravku ReFacto AF vzhledem k úvodním hodnotám a sledovány u 25 dříve léčených pacientů (> 12 let) po opakovaném podávání přípravku ReFacto AF po dobu šesti měsíců. Poměry geometrických středních hodnot nejmenších čtverců vzhledem k úvodním hodnotám v 6. měsíci činily 107 % pro recovery, 100 % pro AUCt a 104 % pro AUC*. Odpovídající 90% interval spolehlivosti poměrů v 6. měsíci vzhledem k úvodním hodnotám činil pro výše uvedené farmakokinetické parametry uvnitř ekvivalenčního okna 80 %-125 %. To indikuje změny ve farmakokinetických vlastnostech přípravku ReFacto AF, které nejsou závislé na čase.
Ve stejné studii u 30 dříve léčených pacientů (> 12 let) při použití standardního bioekvivalenčního přístupu, kdy byla stanovována účinnost přípravku ReFacto AF a nezkráceného rekombinantního faktoru VIII (FLrFVIII) a FVIII měřením plasmatických vzorků v centrální laboratoři jednofázovou koagulační metodou, byl shledán přípravek ReFacto AF farmakokineticky ekvivalentní s FLrFVIII.
U předtím neléčených pacientů (PUP) byly farmakokinetické parametry přípravku ReFacto stanoveny chromogenní metodou. Tito pacienti (n=59; průměrný věk 10 ± 8,3 měsíců) měli v týdnu 0 střední recovery 1,5 ± 0,6 IU/dl na IU/kg (rozmezí 0,2 až 2,8 IU/dl na IU/kg), která byla nižší než u dříve léčených pacientů (PTP) přípravkem ReFacto v týdnu 0 se střední recovery 2,4 ± 0,4 IU/dl na IU/kg (rozmezí 1,1 až 3,8 IU/dl na IU/kg). U PUP byla střední recovery stabilní v čase (5 návštěv v průběhu 2 let) a pohybovala se v rozmezí od 1,5 do 1,8 IU/dl na IU/kg. Farmakokinetické modelování populace s použitím údajů od 44 PUP vedlo ke střednímu očekávanému poločasu 8,0 ± 2,2 hodin.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Nebyly provedeny žádné studie karcinogenního potenciálu ani reprodukční toxicity.
FARMACEUTICKÉ ÚDAJE
6.
6.1 Seznam pomocných látek
Prášek
Sacharosa
Dihydrát chloridu vápenatého Histidin Polysorbát 80 Chlorid sodný
Rozpouštědlo Chlorid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky včetně jiných infuzních roztoků.
Smí se použít pouze dodaný infuzní set, neboť se může vyskytnout selhání léčby jako důsledek adsorpce lidského koagulačního faktoru VIII na vnitřní povrchy některého infuzního vybavení.
6.3 Doba použitelnosti
3 roky
Přípravek může být jednorázově vyjmut z chladničky a uložen při pokojové teplotě (do 25 °C), maximálně na dobu 3 měsíců. Na konci tohoto období nesmí být přípravek vrácen zpět do chladničky, ale musí být spotřebován nebo zlikvidován.
Po rekonstituci
Chemická a fyzikální stabilita po otevření před použitím byla doložena na dobu 3 hodin při teplotě do 25 °C.
ReFacto AF 250 IU, 500 IU, 1000 IU, 2000 IU prášek a rozpouštědlo pro injekční roztok Přípravek neobsahuje protimikrobní přísadu a rekonstituovaný přípravek musí být použitý ihned, nejpozději však do 3 hodin po rozpuštění. Jiné doby a podmínky uchovávání jsou v odpovědnosti uživatele.
ReFacto AF 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce
Přípravek neobsahuje protimikrobní přísadu a rekonstituovaný přípravek musí být použitý ihned, nejpozději však do 3 hodin po rozpuštění nebo po odstranění šedého víčka. Jiné doby a podmínky uchovávání jsou v odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
ReFacto AF 250 IU, 500 IU, 1000 IU, 2000 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce
Uchovávejte a převážejte chlazené (2oC - 8oC). Chraňte před mrazem.
Uchovávejte přípravek ve vnější krabičce, aby byl chráněn před světlem.
Podmínky uchovávání rekonstituovaného léčivého přípravku, viz bod 6.3.
6.5 Druh obalu a velikost balení
ReFacto AF 250 IU, 500 IU, 1000 IU, 2000 IU prášek a rozpouštědlo pro injekční roztok 250 IU, 500 IU, 1000 IU nebo 2000 IU prášku v 10 ml injekční lahvičce (sklo třídy I) se zátkou (butyl) a odtrhávacím uzávěrem (hliník) a 4 ml rozpouštědla v předplněné injekční stříkačce (sklo třídy I) s pístovou zátkou (butyl), krytem hrotu (butyl) a sterilním adaptérem lahvičky, sterilní infuzní souprava, alkoholem napuštěné tampony, náplast a gázový polštářek.
ReFacto AF 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce
250 IU, 500 IU, 1000 IU, 2000 IU nebo 3000 IU lyofilizovaného prášku ve vrchním zásobníku a 4 ml rozpouštědla ve spodním zásobníku předplněné injekční stříkačky (sklo třídy I) s butylovým pryžovým pístem a uzávěrem, jedno táhlo s pístem k sestavení, polypropylenové odvzdušněné sterilní víčko, sterilní infuzní souprava, alkoholem napuštěné tampony, náplast a gázový polštářek.
Velikost balení: 1 souprava.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
ReFacto AF 250 IU, 500 IU, 1000 IU, 2000 IU prášek a rozpouštědlo pro injekční roztok Lyofilizovaný prášek v injekční lahvičce se musí rekonstituovat přiloženým rozpouštědlem [roztok chloridu sodného 9 mg/ml (0,9 %)] z dodané předplněné injekční stříkačky za použití sterilního adaptéru lahvičky. Lahvičkou jemně otáčejte, až se veškerý prášek rozpustí. Další informace o rekonstituci a podání jsou, prosím, k dispozici v příbalové informaci, viz bod 3.
Po rozpuštění se roztok nasaje zpět do injekční stříkačky. Roztok je čirý nebo mírně opalescentní a bezbarvý. Pokud roztok obsahuje viditelné částice nebo je pozorováno zbarvení, musí být zlikvidován.
ReFacto AF 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce
Lyofilizovaný prášek ve vrchním zásobníku předplněné injekční stříkačky musí být rozpuštěn v rozpouštědle [roztok chloridu sodného 9 mg/ml (0,9 %)] ve spodním zásobníku předplněné injekční stříkačky. Předplněnou injekční stříkačkou jemně otáčejte, až se veškerý prášek rozpustí. Další informace o rekonstituci a podání jsou, prosím, k dispozici v příbalové informaci, viz bod 3.
Po rekonstituci bude roztok čirý nebo mírně opalescentní a bezbarvý. Pokud roztok obsahuje viditelné částice nebo je pozorováno zbarvení, musí být zlikvidován.
Přípravek po rozpuštění obsahuje polysorbát 80, o němž je známo, že zvyšuje míru vyluhování bis (2-ethylhexyl) ftalátu (DEHP) z polyvinylchloridu (PVC). Toto je třeba mít na mysli při přípravě a podávání přípravku, jakož i při délce jeho uchovávání v obalu z PVC po rekonstituci. Je důležité přesně dodržovat doporučení uvedená v bodě 6.3.
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/99/103/001
EU/1/99/103/002
EU/1/99/103/003
EU/1/99/103/004
EU/1/99/103/009
EU/1/99/103/006
EU/1/99/103/007
EU/1/99/103/008
EU/1/99/103/005
9. DATUM PRVNÍ REGISTRACE /PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. dubna 1999
Datum posledního prodloužení registrace: 15. dubna 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B . PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Swedish Orphan Biovitrum AB (publ)
Strandbergsgatan 49 SE-11276 Stockholm Švédsko
Název a adresa výrobce odpovědného za propouštění šarží
Wyeth Farma S.A
Autovia del Norte A-1 Km 23
Desvio Algete Km 1
28700 San Sebastian de los Reyes
Madrid
Španělsko
B . PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2)
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ TOHOTO LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
ReFacto AF 250 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 500 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 1000 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 2000 IU prášek a rozpouštědlo pro injekční roztok Moroctocogum alfa
(rekombinantní lidský koagulační faktor VIII)
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK)_
1 injekční lahvička: moroktokogum alfa 250 IU (přibližně 62,5 IU/ml po rozpuštění). 1 injekční lahvička: moroctocogum alfa 500 IU (přibližně 125 IU/ml po rozpuštění).
1 injekční lahvička: moroctocogum alfa 1000 IU (přibližně 250 IU/ml po rozpuštění). 1 injekční lahvička: moroctocogum alfa 2000 IU (přibližně 500 IU/ml po rozpuštění).
3. SEZNAM POMOCNÝCH LÁTEK
Sacharosa,
dihydrát chloridu vápenatého, histidin, polysorbát 80, chlorid sodný
Před použitím si přečtěte příbalovou informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok.
1 injekční lahvička s moroktokogem alfa 250 IU 1 injekční lahvička s moroctocogem alfa 500 IU 1 injekční lahvička s moroctocogem alfa 1000 IU 1 injekční lahvička s moroctocogem alfa 2000 IU
1 předplněná injekční stříkačka se 4 ml rozpouštědla 1 adaptér injekční lahvičky
1 sterilní infuzní set
2 alkoholové tampony 1 náplast
1 gáza
5. ZPŮSOB A CESTA PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Intravenózní podání po rozpuštění.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Nepoužívejte po uplynutí doby použitelnosti. Spotřebujte ihned nebo do 3 hodin po rozpuštění.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte a převážejte při 2 0C - 8 0C.
Chraňte před mrazem.
Uchovávejte injekční lahvičku v původním obalu, aby byla chráněna před světlem.
Přípravek ReFacto AF může být jednorázově uchováván až po dobu 3 měsíců při pokojové teplotě (do 25°C). Po uchovávání při pokojové teplotě nelze přípravek vrátit do chladničky.
Datum vyjmutí z chladničky:
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO OBALŮ Z NICH, POKUD JE TO VHODNÉ
Veškerý zbývající roztok zlikvidujte.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/99/103/001
EU/1/99/103/002
EU/1/99/103/003
EU/1/99/103/004
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE VBRAILLOVÉ PÍSMU
ReFacto AF 250 ReFacto AF 500 ReFacto AF 1000 ReFacto AF 2000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA (Y) PODÁNÍ
ReFacto AF 250 IU prášek pro injekční roztok ReFacto AF 500 IU prášek pro injekční roztok ReFacto AF 1000 IU prášek pro injekční roztok ReFacto AF 2000 IU prášek pro injekční roztok Moroctocogum alfa
(rekombinantní lidský koagulační faktor VIII) i.v. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
6. OSTATNÍ
Uchovávejte v chladničce.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA (Y) PODÁNÍ_
Rozpouštědlo k ReFacto AF
2. ZPŮSOB PODÁNÍ_
i.v. podání, po rozpuštění.
3. POUŽITELNOST_
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
Obsahuje 4 ml roztoku chloridu sodného 9 mg/ml (0,9%) na injekci
6. OSTATNÍ
Uchovávejte v chladničce.
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU VNĚJŠÍ KARTON
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
ReFacto AF 250 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce ReFacto AF 500 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce ReFacto AF 1000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce ReFacto AF 2000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce ReFacto AF 3000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce Moroctocogum alfa
(rekombinantní lidský koagulační faktor VIII)
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK)_
1 předplněná injekční stříkačka: moroktokogum alfa 250 IU (přibližně 62,5 IU/ml po rozpuštění). 1 předplněná injekční stříkačka: moroctocogum alfa 500 IU (přibližně 125 IU/ml po rozpuštění).
1 předplněná injekční stříkačka: moroctocogum alfa 1000 IU (přibližně 250 IU/ml po rozpuštění). 1 předplněná injekční stříkačka: moroctocogum alfa 2000 IU (přibližně 500 IU/ml po rozpuštění). 1 předplněná injekční stříkačka: moroctocogum alfa 3000 IU (přibližně 750 IU/ml po rozpuštění).
3. SEZNAM POMOCNÝCH LÁTEK
Pro další informace si přečtěte příbalovou informaci. Sacharosa,
dihydrát chloridu vápenatého, histidin, polysorbát 80, chlorid sodný
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce FuseNGo
1 předplněná injekční stříkačka (250 IU prášku ve vrchním zásobníku a 4 ml rozpouštědla ve spodním zásobníku)
1 předplněná injekční stříkačka (500 IU prášku ve vrchním zásobníku a 4 ml rozpouštědla ve spodním zásobníku)
1 předplněná injekční stříkačka (1000 IU prášku ve vrchním zásobníku a 4 ml rozpouštědla ve spodním zásobníku)
1 předplněná injekční stříkačka (2000 IU prášku ve vrchním zásobníku a 4 ml rozpouštědla ve spodním zásobníku)
1 předplněná injekční stříkačka (3000 IU prášku ve vrchním zásobníku a 4 ml rozpouštědla ve spodním zásobníku)
1 píst
1 sterilní infuzní set
2 alkoholové tampony 1 náplast
1 gáza
1 perforované sterilní víčko
5. ZPŮSOB A CESTA PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Intravenózní podání pouze pro jedno použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Použijte ihned nebo do 3 hodin po rekonstituci nebo po odstranění šedého pryžového víčka z injekční stříkačky.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte a transportujte v chladničce ( 2 0C - 8 0C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Přípravek ReFacto AF může být jednorázově uchováván až po dobu 3 měsíců při pokojové teplotě (do 25°C). Po uchovávání při pokojové teplotě nelze přípravek vrátit do chladničky.
Datum vyjmutí z chladničky:
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO OBALŮ Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road
Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/99/103/009
EU/1/99/103/006
EU/1/99/103/007
EU/1/99/103/008
EU/1/99/103/005
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE VBRAILLOVÉ PÍSMU
ReFacto AF 250 ReFacto AF 500 ReFacto AF 1000 ReFacto AF 2000 ReFacto AF 3000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA (Y) PODÁNÍ
ReFacto AF 250 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 500 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 1000 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 2000 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 3000 IU prášek a rozpouštědlo pro injekční roztok Moroctocogum alfa
(rekombinantní lidský koagulační faktor VIII) i.v. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
|
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK | |
|
Moroctocogum alfa 250 IU, i.v. podání pro jedno použití | |
|
Moroctocogum alfa 500 IU, i.v. podání pro jedno použití | |
|
Moroctocogum alfa 1000 IU, i.v. podání pro jedno použití | |
|
Moroctocogum alfa 2000 IU, i.v. podání pro jedno použití | |
|
Moroctocogum alfa 3000 IU, i.v. podání pro jedno použití | |
|
6. OSTATNÍ | |
|
Uchovávejte v chladničce. | |
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: Informace pro uživatele
ReFacto AF 250 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 500 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 1000 IU prášek a rozpouštědlo pro injekční roztok ReFacto AF 2000 IU prášek a rozpouštědlo pro injekční roztok
Moroctocogum alfa (rekombinantní lidský koagulační faktor VIII)
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci. Možná, že si ji budete potřebovat přečíst znovu.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek ReFacto AF a k čemu se používá.
2. Čemu musíte věnovat pozornost, než začnete přípravek ReFacto AF používat.
3. Jak se přípravek ReFacto AF používá.
4. Možné nežádoucí účinky.
5. Jak přípravek ReFacto AF uchovávat.
6. Obsah balení a další informace.
1. Co je přípravek ReFacto AF a k čemu se používá
Přípravek ReFacto AF obsahuje léčivou látku moroktokog alfa, lidský koagulační faktor VIII. Faktor VIII je nezbytný k tvorbě krevních sraženin a k zastavení krvácení. U pacientů s hemofilií A (vrozený nedostatek faktoru VIII) chybí nebo správně nefunguje.
Přípravek ReFacto AF se používá k léčbě nebo prevenci krvácení (profylaxe) u dospělých a dětí všech věkových skupin (včetně novorozenců) s hemofilií A.
2. Čemu musíte věnovat pozornost, než začnete přípravek ReFacto AF používat
Nepoužívejte ReFacto AF
- jestliže jste alergický(á) na moroctocog alfa nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže jste alergický(á) na křeččí proteiny.
Pokud si nejste jistý(a), zeptejte se svého lékaře.
Upozornění a opatření
Před použitím přípravku ReFacto AF se poraďte se svým lékařem nebo lékárníkem
- pokud dostanete alergickou reakci. Takovými příznaky alergické reakce jsou dechové potíže, dušnost, otok, kopřivka, svědění, tísnivý pocit na prsou, sípání a nízký krevní tlak. Anafylaxe je závažná alergická reakce, která může způsobit obtíže při polykání a/nebo dýchání, zrudnutí či otok tváře a/nebo rukou. Pokud se vyskytnou některé z těchto známek, zastavte ihned infuzi a spojte se s lékařem či vyhledejte pohotovostní lékařskou pomoc. V případě závažných alergických reakcí je třeba zvážit alternativní léčbu.
- nedojde-li k zastavení krvácení podle očekávání, spojte se s lékařem či vyhledejte lékařskou pohotovostní službu.
- jestliže se nepodaří zastavit krvácení obvyklou dávkou. U pacientů, kteří dostávají přípravky obsahující faktor VIII, se mohou někdy vyvinout protilátky proti faktoru VIII (též známé jako inhibitory faktoru VIII), které mohou zabránit správnému účinku přípravků obsahujících faktor VIII. Pokud jste léčen(a) faktorem VIII, měl(a) byste být sledován(a) na rozvoj inhibitorů faktoru VIII.
Další léčivé přípravky a přípravek ReFacto AF
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte nebo jste užíval(a) v nedávné
době nebo které možná budete užívat.
Těhotenství, kojení a plodnost
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte
se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Řízení dopravních prostředků a obsluha strojů
Přípravek ReFacto AF nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
Přípravek ReFacto AF obsahuje sodík.
Přípravek ReFacto AF obsahuje 1,23 mmol (nebo 29 mg) sodíku v jedné injekční lahvičce
rozpuštěného prášku. Informujte svého lékaře, pokud jste na řízené sodíkové dietě.
3. Jak se přípravek ReFacto AF používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Léčbu přípravkem ReFacto AF má zahajovat lékař se zkušeností s léčbou pacientů s hemofilií A. Váš lékař Vám stanoví dávku přípravku ReFacto AF, kterou dostanete. Tato dávka a trvání léčby bude záviset na Vaší individuální potřebě náhrady faktoru VIII. Přípravek ReFacto AF se podává injekcí do žíly po dobu několika minut. Pacienti nebo jejich pečovatelé mohou podávat injekce přípravku ReFacto AF, pokud byli řádně proškoleni.
V průběhu léčby může lékař rozhodnout o změně dávky přípravku ReFacto AF, kterou dostáváte.
Před cestováním se, prosím, poraďte se svým poskytovatelem zdravotní péče. Na cestu byste si měl(a) opatřit dostatečnou zásobu přípravku obsahující faktor VIII ke své předpokládané léčbě.
Doporučuje se, kdykoli použijete přípravek ReFacto AF, zaznamenat si z obalu název přípravku a číslo šarže. Pro účely hlášení nežádoucích účinků můžete dokumentovat číslo šarže odlepením štítku z jedné injekční lahvičky a jeho nalepením do svého diáře.
Rekonstituce (rozpuštění) a podání
Níže popsaný postup slouží jako obecný návod pro rekonstituci a podávání přípravku ReFacto AF. Pacienti by měli sledovat specifický postup rekonstituce a podávání přípravku lékařem.
K rekonstituci použijte pouze předplněnou injekční stříkačku dodávanou v krabičce. K podání přípravku může být použita jiná sterilní injekční stříkačka pro jednorázové použití.
Přípravek ReFacto AF se podává intravenózní (i.v.) infuzí po rekonstituci lyofilizovaného prášku na injekci přiloženým rozpouštědlem [roztok chloridu sodného 9 mg/ml (0,9%)] v injekční stříkačce. Přípravek ReFacto AF nemá být mísen s jinými infuzními roztoky.
Před rekonstitucí a podáním léku si vždy umyjte ruce. Během rekonstituce musí být dodrženy zásady aseptické přípravy (tj. čisté a bezmikrobiální).
Rekonstituce:
1. Nechejte injekční lahvičku s lyofilizovaným přípravkem ReFacto AF a rozpouštědlo v předplněné injekční stříkačce zahřát na pokojovou teplotu.
2. Odstraňte plastové víčko z injekční lahvičky ReFacto AF, aby se odkryla střední část gumového uzávěru.
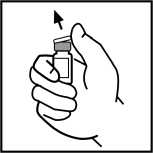
3. Otřete vršek lahvičky přiloženým tampónem s alkoholem nebo použijte jiný antiseptický roztok a nechejte oschnout. Po očištění se už nedotýkejte rukama gumové zátky a nedovolte, aby se zátka něčeho dotkla.
4. Odloupněte víčko z čirého plastového obalu adaptéru injekční lahvičky. Nevyjímejte adaptér z obalu.
5. Položte injekční lahvičku na plochý povrch. Vezměte obal s adaptérem, položte adaptér na injekční lahvičku a přitlačte obal silně dolů, až adaptér pronikne shora na vrchol injekční lahvičky a jehla adaptéru pronikne gumovou zátkou.
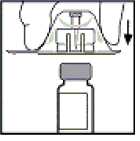
6. Sejměte obal z adaptéru a vyhoďte jej.
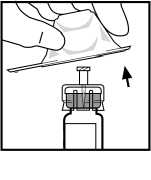
Připevněte táhlo s pístem k injekční stříkačce s rozpouštědlem vložením táhla do otvoru v zátce injekční stříkačky za stálého tlaku a otáčení, až bezpečně sedí v zátce.
Odlomte bezpečnostní kryt hrotu injekční stříkačky rozpouštědla ulomením perforace krytky. To se provede střídavým ohýbáním krytu nahoru a dolů dokud se perforace neulomí. Nedotýkejte se vnitřní části krytu ani hrotu stříkačky. Možná že kryt bude muset být znovu nasazen (jestliže se rekonstituovaný přípravek ReFacto AF nepodává bezprostředně), proto jej umístěte stranou a postavte na jeho vrchol.
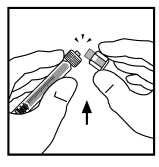
9.
Postavte injekční lahvičku na rovnou plochu. Vložením hrotu do otvoru adaptéru spojte injekční stříkačku rozpouštědla s adaptérem injekční lahvičky za stálého tlaku a otáčení stříkačky ve směru hodinových ručiček až do zajištění spojení.
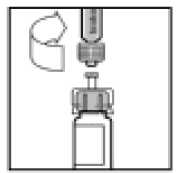
10.
Pomalým tlakem na táhlo pístu vstříkněte veškeré rozpouštědlo do injekční lahvičky ReFacto AF.
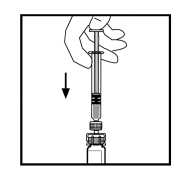
11.
Zatímco injekční stříkačka je stále spojena s adaptérem, jemně otáčejte injekční lahvičkou až do rozpuštění prášku.
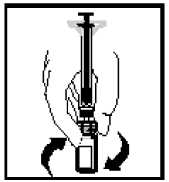
Připravený roztok musí být před podáním vizuálně zkontrolován, aby neobsahoval mechanické částice. Roztok musí být čirý až mírně opalescentní a bezbarvý.
Poznámka: Jestliže použijete na infuzi více než jednu injekční lahvičku přípravku ReFacto AF, obsah každé injekční lahvičky má být rozpuštěn podle předchozího návodu. Injekční stříkačka rozpouštědla se má odstranit, přičemž adaptér injekční lahvičky se ponechá na místě a ke stažení rozpuštěného obsahu jednotlivých injekčních lahviček může být použita samostatná velká injekční stříkačka s otvorem luer.
Přesvědčte se, že táhlo pístu je stále úplně stlačené a převraťte injekční lahvičku dnem vzhůru. Pomalu stáhněte všechen roztok přes adaptér lahvičky zpět do injekční stříkačky.
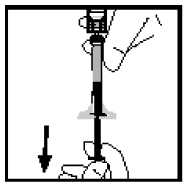
14. Odpojte injekční stříkačku od adaptéru injekční lahvičky jemným tahem a otáčením injekční stříkačky proti směru hodinových ručiček. Zlikvidujte injekční lahvičku s připojeným adaptérem.
Poznámka: Jestliže roztok nemá být podán bezprostředně, na stříkačku se musí opatrně nasadit kryt. Nedotýkejte se hrotu stříkačky ani vnitřní strany krytu.
Přípravek ReFacto AF musí být použit nejpozději do 3 hodin po rekonstituci. Připravený roztok může být před podáním uchováván při pokojové teplotě.
Podání (intravenózní infuze):
Přípravek ReFacto AF má být podán přiloženou infuzní soupravou a předplněnou injekční stříkačkou s rozpouštědlem nebo samostatnou sterilní injekční stříkačkou z plastické hmoty k jednorázovému použití s otvorem luer.
1. Nasaďte injekční stříkačku na konec luer hadičky infuzního setu.
2. Přiložte škrtidlo a připravte si injekční místo otřením kůže alkoholovým tampónem, dodaným v soupravě.
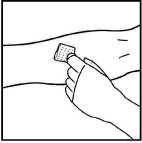
3. Napíchněte žílu jehlou s infuzní soupravou podle pokynů lékaře a sejměte škrtidlo. Odstraňte veškerý vzduch z infuzní soupravy tak, že natáhnete roztok zpět do injekční stříkačky. Rozpuštěný přípravek musí být podán intravenózně během několika minut. Lékař může změnit rychlost infuze tak, aby to bylo pro Vás pohodlnější.
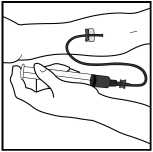
Veškerý nespotřebovaný roztok, prázdné injekční lahvičky a použité jehly a stříkačky vyhoďte do vhodného kontejneru mimo zdravotnický odpad, protože tyto materiály by mohly při neopatrné likvidaci někoho poranit.
Jestliže jste užil(a) více přípravku ReFacto AF než jste měl(a)
Poraďte se s lékařem nebo lékárníkem.
Jestliže jste přestal(a) užívat ReFacto AF
Nepřerušujte podávání přípravku ReFacto AF, aniž byste se předem poradil(a) se svým lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Alergické reakce
Pokud se náhle vyskytnou závažné alergické (anafylaktické) reakce, musí se infuze ihned přerušit.
Pokud máte některý z následujících časných příznaků alergických reakcí, musíte ihned informovat svého lékaře:
• vyrážka, kopřivka, místní otok kůže, celkové svědění
• otok rtů a jazyka
• problémy s dýcháním, sípot, tíha na hrudi
• celkový pocit nemoci
• závratě a ztráta vědomí
Závažné příznaky zahrnující problémy s dýcháním a (téměř) mdloby vyžadují neodkladnou léčbu. Závažné náhlé alergické (anafylaktické) reakce jsou méně časté (postihují až 1 ze 100 pacientů).
Vytvoření inhibitorů
U pacientů s hemofilií A se mohou vyvinout neutralizační protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory vyskytnou, mohou se projevit zvýšeným množstvím přípravku ReFacto AF typicky potřebným k léčbě krvácení a/nebo pokračujícím krvácením po léčbě. V takových případech se doporučuje kontaktovat specializované centrum pro hemofiliky. Lékař může požadovat sledování možného vyvinutí inhibitoru. V klinických studiích došlo k vyvinutí inhibitorů přibližně u 2 % pacientů léčených přípravkem ReFacto AF.
Velmi časté nežádoucí účinky (postihují více než 1 z 10 pacientů)
• vývoj inhibitorů u pacientů, kteří nikdy předtím nebyli léčeni přípravky obsahujícími faktor VIII
• bolest hlavy
• kašel
• bolest kloubů
• horečka
Časté nežádoucí účinky (postihují až 1 z 10 pacientů)
• krvácení
• vývoj inhibitorů u pacientů, kteří již byli léčeni přípravky obsahujícími faktor VIII
• závratě
• snížená chuť k jídlu, průjem, zvracení, bolest břicha, pocit na zvracení
• bolest svalů
• zimnice, reakce v místě katétru
• některé testy mohou ukázat zvýšení protilátek proti faktoru VIII
Méně časté nežádoucí účinky (postihují až 1 ze 100 pacientů)
• závažné alergické reakce
• necitlivost, spavost, změny chuti
• bolest na hrudi, zrychlený srdeční rytmus, palpitace
• nízký krevní tlak, bolest a zarudnutí žil související s krevní sraženinou, zrudnutí
• dušnost
• nadměrné pocení
• slabost, reakce v místě injekce včetně bolesti
• mírné zvýšení hladin srdečních enzymů
• zvýšení hladin jaterních enzymů, zvýšení hladiny bilirubinu
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek ReFacto AF uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku lahvičky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte a přepravujte chlazené (2°C - 8°C). Chraňte před mrazem, aby se předešlo poškození předplněné injekční stříkačky s rozpouštědlem.
Pro Vaše pohodlí je možno léčivý přípravek vyjmout z chladničky na jediné období trvající maximálně 3 měsíce a uchovávat při pokojové teplotě (do 25 °C). Po uplynutí tohoto období uchovávání při pokojové teplotě se přípravek nesmí vrátit do chladničky, ale musí se spotřebovat, či zlikvidovat. Na krabičce si zaznamenejte datum, kdy byl přípravek ReFacto AF vyjmut z chladničky a uchováván při pokojové teplotě (do 25 °C). Uchovávejte lahvičku ve vnější krabičce, aby byl přípravek chráněn před světlem.
Rozpuštěný přípravek použijte nejpozději do 3 hodin od rozpuštění.
Přípravek je bezbarvý a čirý až mírně opalescentní. Nepoužívejte tento přípravek, pokud zjistíte, že je roztok zakalený nebo obsahuje viditelné částice.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo do domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek ReFacto AF obsahuje
- Léčivou látkou je moroctocogum alfa (rekombinantní koagulační faktor VIII). Jedna injekční lahvička ReFacto AF obsahuje moroctocogum alfa 250, 500, 1000 nebo 2000 IU.
- Pomocnými látkami jsou sacharosa, dihydrát chloridu vápenatého, histidin, polysorbát 80 a chlorid sodný. Přiloženo je také rozpouštědlo k rozpuštění [roztok chloridu sodného 9mg/ml (0,9%) na injekci].
- Po rozpuštění dodaným rozpouštědlem [roztok chloridu sodného 9mg/ml (0,9%)] jedna injekční lahvička připraveného injekčního roztoku obsahuje 62,5; 125; 250 nebo 500 IU moroctocogum alfa (podle síly moroctocogum alfa, tj. 250, 500, 1000, nebo 2000 IU).
Jak přípravek ReFacto AF vypadá a co obsahuje toto balení
Přípravek ReFacto AF se dodává jako prášek na injekci ve skleněné injekční lahvičce a s rozpouštědlem v předplněné injekční stříkačce.
Obsahem balení je:
- jedna injekční lahvička s práškem moroctocogum alfa 250, 500, 1000, nebo 2000 IU
- jedna předplněná injekční stříkačka s rozpouštědlem, 4 ml sterilního roztoku chloridu sodného 9 mg/ml (0,9%) k rozpuštění, s táhlem a pístem
- jeden sterilní adaptér lahvičky
- jedna sterilní infuzní souprava
- dva alkoholové tampony
- jedna náplast
- jeden gázový polštářek
Držitel rozhodnutí o registraci
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
Výrobce
Wyeth Farma S.A
Autovia del Norte A-1 Km 23
Desvio Algete Km 1
28700 San Sebastian de los Reyes
Madrid
Španělsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
|
Belgie /Belgique / Belgien Pfizer S.A./N.V. Tél/Tel: +32 (0)2 554 62 11 |
Lietuva Pfizer Luxembourg SARL filialas Lietuvoje Tel. +3705 2514000 |
|
Btarapna n^aň3ep HroKceMÓypr CAPH, KnoH EtnrapHa Ten.: +359 2 970 4333 |
Luxembourg/Luxemburg Pfizer S.A. Tél/Tel: +32 (0)2 554 62 11 |
|
Česká republika Pfizer PFE, spol. s.r.o. Tel: +420 283 004 111 |
Magyarország Pfizer Kft. Tel.: + 36 1 488 37 00 |
|
Danmark Pfizer ApS Tlf: +45 44 20 11 00 |
Malta Vivian Corporation Ltd. Tel: +35621 344610 |
|
Deutschland Pfizer Pharma GmbH Tel: +49 (0)30 550055 51000 |
Nederland Pfizer bv Tel: +31 (0)10 406 43 01 |
|
Eesti Pfizer Luxembourg SARL Eesti filiaal Tel: +372 666 7500 |
Norge Pfizer Norge AS Tlf: +47 67 526 100 |
|
EXXáSa PFIZER EAAAI A.E TpA.: +30 210 678 5800 |
Osterreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0 |
|
Espaňa Pfizer S.L. Tel: +34 91 490 99 00 |
Polska Pfizer Polska Sp. z o.o., Tel.: +48 22 335 61 00 |
|
France Pfizer Tél: +33 (0)1 58 07 34 40 |
Portugal Pfizer Biofarmaceutica, Sociedade Unipessoal Lda Tel: +351 21 423 5500 |
|
Hrvatska Pfizer Croatia d.o.o. Tel: + 385 1 3908 777 |
Románia Pfizer Románia S.R.L. Tel: +40 21 207 28 00 |
|
Ireland Pfizer Healthcare Ireland Tel: 1800 633 363 (toll free) +44 (0)1304 616161 |
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana Tel: + 386 (0) 1 52 11 400 |
|
Ísland Icepharma hf. Sími: + 354 540 8000 |
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel: +421-2-3355 5500 |
Italia
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 43 00 40 Sverige
Pfizer Innovations AB Tel: + 46 (0)8 550 520 00
United Kingdom
Pfizer Limited
Tel: +44 (0)1304 616161
Pfizer S.r.l.
Tel: +39 06 33 18 21
Kúrcpog
PFIZER EAAAI A.E. (CYPRUS BRANCH)
T^: +357 22 817690
Latvija
Pfizer Luxembourg SARL filiale Latvija
Tel: +371 670 35 775
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
Příbalová informace: Informace pro uživatele
ReFacto AF 250 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce ReFacto AF 500 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce ReFacto AF 1000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce
ReFacto AF 2000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce
ReFacto AF 3000 IU prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce
Moroctocogum alfa (rekombinantní lidský koagulační faktor VIII)
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci. Možná, že si ji budete potřebovat přečíst znovu.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek ReFacto AF a k čemu se používá.
2. Čemu musíte věnovat pozornost, než začnete přípravek ReFacto AF používat.
3. Jak se přípravek ReFacto AF používá.
4. Možné nežádoucí účinky.
5. Jak přípravek ReFacto AF uchovávat.
6. Obsah balení a další informace.
1. Co je přípravek ReFacto AF a k čemu se používá
Přípravek ReFacto AF obsahuje léčivou látku moroktokog alfa, lidský koagulační faktor VIII. Faktor VIII je nezbytný k tvorbě krevních sraženin a k zastavení krvácení. U pacientů s hemofilií A (vrozený nedostatek faktoru VIII) chybí nebo správně nefunguje.
Přípravek ReFacto AF se používá k léčbě nebo prevenci krvácení (profylaxe) u dospělých a dětí všech věkových skupin (včetně novorozenců) s hemofilií A.
2. Čemu musíte věnovat pozornost, než začnete přípravek ReFacto AF používat Nepoužívejte přípravek ReFacto AF
- jestliže jste alergický(á) (přecitlivělý(á)) na moroctocog alfa nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže jste alergický(á) na křeččí proteiny.
Pokud si nejste jistý(a), zeptejte se svého lékaře.
Upozornění a opatření
Před použitím přípravku ReFacto AF se poraďte se svým lékařem nebo lékárníkem
- pokud dostanete alergickou reakci. Takovými příznaky alergické reakce jsou dechové potíže, dušnost, otok, kopřivka, svědění, tísnivý pocit na prsou, dýchavičnost a nízký krevní tlak. Anafylaxe je závažná alergická reakce, která může způsobit obtíže při polykání a/nebo dýchání, zrudnutí či otok tváře a/nebo rukou. Pokud se vyskytnou některé z těchto známek, zastavte ihned infuzi a spojte se s lékařem či vyhledejte pohotovostní lékařskou pomoc. V případě závažných alergických reakcí je třeba zvážit alternativní léčbu.
- nedojde-li k zastavení krvácení podle očekávání, spojte se s lékařem či vyhledejte lékařskou pohotovostní službu.
- jestliže se nepodaří zastavit krvácení obvyklou dávkou. U pacientů, kteří dostávají přípravky obsahující faktor VIII, se mohou někdy vyvinout protilátky proti faktoru VIII (též známé jako inhibitory faktoru VIII), které mohou zabránit správnému účinku přípravků obsahujících faktor VIII. Pokud jste léčen(a) faktorem VIII, měl(a) byste být sledován(a) na rozvoj inhibitorů faktoru VIII.
Další léčivé přípravky a přípravek ReFacto AF
Prosím, informujte svého lékaře nebo lékárníka o všech lécích, které užíváte nebo jste užíval(a)
v nedávné době nebo které možná budete užívat.
Těhotenství, kojení a plodnost
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte
se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Řízení dopravních prostředků a obsluha strojů
Přípravek ReFacto AF nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
Přípravek ReFacto AF obsahuje sodík.
Přípravek ReFacto AF obsahuje 1,23 mmol (nebo 29 mg) sodíku v jedné předplněné injekční stříkačce
rozpuštěného prášku. Informujte svého lékaře, pokud jste na řízené sodíkové dietě.
3. Jak se přípravek ReFacto AF užívá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Léčbu přípravkem ReFacto AF má zahajovat lékař se zkušeností s léčbou pacientů s hemofilií A. Váš lékař Vám stanoví dávku přípravku ReFacto AF, kterou dostanete. Tato dávka a trvání léčby bude záviset na Vaší individuální potřebě náhrady faktoru VIII. Přípravek ReFacto AF se podává injekcí do žíly po dobu několika minut. Pacienti nebo jejich pečovatelé mohou podávat injekce přípravku ReFacto AF, pokud byli řádně proškoleni.
V průběhu léčby může lékař rozhodnout o změně dávky přípravku ReFacto AF, kterou dostáváte.
Před cestováním se prosím poraďte se svým poskytovatelem zdravotní péče. Na cestu byste si měl(a) opatřit dostatečnou zásobu přípravku obsahující faktor VIII ke své předpokládané léčbě.
Doporučuje se, kdykoli použijete přípravek ReFacto AF, zaznamenat si z obalu název přípravku a číslo šarže. Pro účely hlášení nežádoucích účinků můžete dokumentovat číslo šarže odlepením štítku z jedné předplněné injekční stříkačky a jeho nalepením do svého diáře.
Rekonstituce (rozpuštění) a podání
Níže popsaný postup slouží jako obecný návod pro rekonstituci a podávání přípravku ReFacto AF dodávaného v předplněné injekční stříkačce. Pacienti by měli sledovat specifický postup rekonstituce a podávání přípravku lékařem.
Přípravek ReFacto AF se podává intravenózní (i.v.) infuzí po rekonstituci. Předplněná injekční stříkačka se skládá ze dvou zásobníků, jeden zásobník obsahuje lyofilizovaný prášek přípravku ReFacto AF a druhý zásobník obsahuje rozpouštědlo [roztok chloridu sodného 9mg/ml (0,9%)]. Pro potřeby této příbalové informace bude toto zařízení popisováno jako předplněná injekční stříkačka.
K rekonstituci (rozpuštění) používejte pouze předplněnou injekční stříkačku dodávanou v krabičce.
K podání tohoto léčivého přípravku mohou být použity jiné sterilní injekční stříkačky, určené k jednorázovému použití.
Přípravek ReFacto AF nesmí být mísen s jinými infuzními roztoky.
Poznámka: Jestliže potřebujete použít více než jednu předplněnou injekční stříkačku přípravku ReFacto AF, jednotlivé předplněné injekční stříkačky by měly být rekonstituovány podle těchto specifických instrukcí. Samostatná 10ml nebo větší uzavřená injekční stříkačka (není součástí této soupravy) může být použita k natažení rekonstituovaných obsahů jednotlivých stříkaček (viz Další pokyny).
Příprava
1. Umyjte si vždy ruce před provedením následujících postupů.
2. Během rekonstituce musí být dodrženy zásady aseptické techniky (tj. čisté a bezmikrobiální).
3. Všechny komponenty používané při rekonstituci a podávání tohoto přípravku by měly být použity co nejdříve po otevření jejich sterilních obalů, aby se minimalizovalo jejich vystavení vzduchu.
Rekonstituce (rozpouštění)
1. Vyjměte předplněnou injekční stříkačku z chladničky a ponechejte ji, aby dosáhla pokojové teploty.
2. Vyjměte obsah soupravy přípravku ReFacto AF předplněná injekční stříkačka a umístěte jej na čistý povrch, ujistěte se, že máte vše, co budete potřebovat.
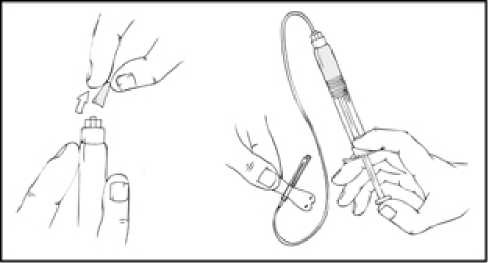
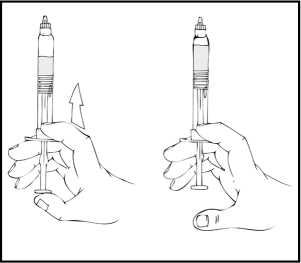
3. Pevně uchopte píst tak, jak je znázorněno na následujícím obrázku. Našroubujte píst pevně do uzávěru předplněné injekční stříkačky přípravku ReFacto AF a otáčejte po směru hodinových ručiček, až ucítíte odpor (přibližně 2 otočení).
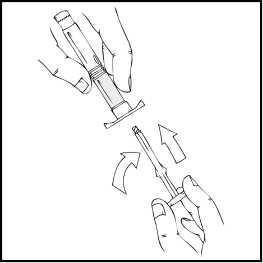
Během procesu rekonstituce je důležité držet předplněnou injekční stříkačku přípravku ReFacto AF svisle (s bílým práškem nad čirým roztokem), aby se zabránilo možnému prosakování.
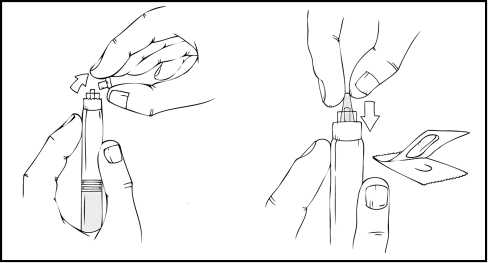
4. Držte předplněnou injekční stříkačku svisle, odstraňte bílý uzávěr ohnutím zprava doleva (nebo lehkým kolébavým pohybem), aby se rozlomila perforace víčka a objevilo se šedé pryžové víčko předplněné injekční stříkačky přípravku ReFacto AF.
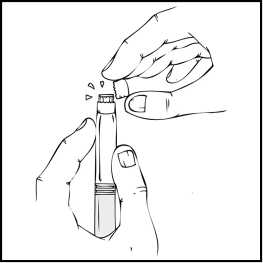
5. Sejměte modré ochranné víčko z obalu přípravku ReFacto AF.
Zatímco stále držíte předplněnou injekční stříkačku ReFacto AF ve svislé poloze, odstraňte šedé pryžové víčko a nahraďte je modrým perforovaným ochranným víčkem. Toto ochranné víčko je perforované, což dovoluje unikat vzduchu, a tím se zabraňuje zvýšení tlaku uvnitř předplněné injekční stříkačky. Nedotýkejte se otevřeného konce injekční stříkačky ani modrého ochranného víčka.
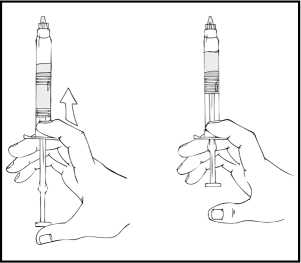
6. Zlehka a pomalu zatlačujte píst, dokud se dva písty uvnitř předplněné injekční stříkačky nepotkají a dokud se rozpouštědlo nepřemístí do vrchního zásobníku, který obsahuje prášek přípravku ReFacto AF.
Poznámka: Aby se zabránilo úniku tekutiny ze stříkačky, netlačte na píst příliš velkou silou.
7. Stále držte předplněnou injekční stříkačku ReFacto AF svisle, jemně s ní několikrát zakružte, dokud se prášek nerozpustí.
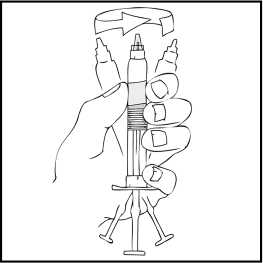
Podívejte se na výsledný roztok a zkontrolujte, zdali se v něm nevyskytují viditelné částečky prášku a zdali je bezbarvý. Roztok by měl být čirý až slabě opalescentní a bezbarvý, Pokud se v roztoku vyskytnou viditelné částice nebo je roztok zbarvený, předplněnou stříkačku zlikvidujte.
8. Stále držte předplněnou injekční stříkačku ReFacto AF ve svislé poloze a pomalým zatlačováním pístu odstraňte většinu vzduchu (ale ne všechen) z vrchního zásobníku.
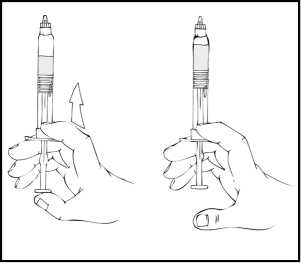
Přípravek ReFacto AF by měl být podán do tří hodin po rekonstituci nebo po odstranění šedého víčka z předplněné injekční stříkačky.
Pokud nepoužijete přípravek ReFacto AF okamžitě, měl(a) byste injekční stříkačku skladovat ve svislé poloze s modrým ochranným víčkem na předplněné injekční stříkačce až do doby, kdy budete chtít přípravek ReFacto AF použít. Rekonstituovaný roztok může být uchováván při pokojové teplotě po dobu až 3 hodin. Pokud nepoužijete přípravek do 3 hodin, zlikvidujte ho.
Podání (intravenózní infuze)
Váš lékař nebo jiný zdravotnický pracovník by Vám měl vysvětlit, jak si podat infuzi přípravku ReFacto AF. Jakmile se naučíte, jak si podat infuzi, můžete postupovat dle pokynů v této Příbalové informaci.
Přípravek ReFacto AF se podává intravenózní (i.v.) infuzí („kapačkou“ do žíly) po rekonstituci prášku v rozpouštědle (0,9% roztok chloridu sodného). Jakmile dojde k rekonstituci, přípravek ReFacto AF by měl být před podáním zkontrolován, zdali se v něm nevyskytují viditelné částečky a zdali je bezbarvý.
Přípravek ReFacto AF by měl být podáván použitím infuzního setu obsaženého v soupravě, jestliže Vám lékař nebo jiný zdravotnický pracovník neporadí jinak.
1. Odstraňte modré ochranné perforované víčko a pevně připojte intravenózní infuzní set k předplněné injekční stříkačce ReFacto AF.
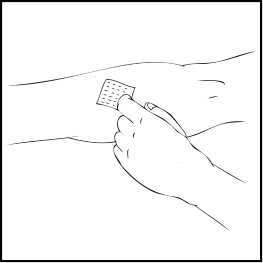
2. Použijte škrtidlo a připravte místo, kam budete přípravek podávat. Kůži dobře otřete alkoholovým tamponem dodávaným v soupravě přípravku ReFacto AF.
3. Odstraňte ochranné víčko jehly a zasuňte jehlu infuzního setu do žíly tak, jak Vás poučil Váš lékař nebo jiný zdravotnický pracovník. Odstraňte škrtidlo. Rekonstituovaný přípravek ReFacto AF se podává intravenózně a podání by mělo trvat několik minut. Váš lékař může změnit rychlost infuze, aby pro Vás byla infuze příjemnější. Poraďte se o podání intravenózní infuze se svým lékařem nebo s jiným zdravotnickým pracovníkem. Neaplikujte infuzi sami, dokud jste nebyl(a) řádně poučen(a).
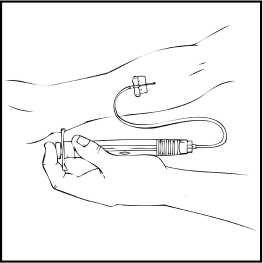
Rekonstituovaný přípravek ReFacto AF nesmí být podáván stejným setem nebo ve stejném obalu s jiným léčivým přípravkem.
4. Po infuzi přípravku ReFacto AF infuzní set odstraňte a zlikvidujte. Zbylé množství léčivého přípravku v infuzním setu neovlivní Vaši léčbu.
Poznámka: Prosím, zlikvidujte všechen nepoužitý roztok, prázdnou předplněnou injekční stříkačku a použité pomůcky do vhodného obalu pro likvidaci zdravotnického odpadu, jelikož tyto materiály mohou někoho zranit, jestliže nejsou zlikvidovány náležitým způsobem.
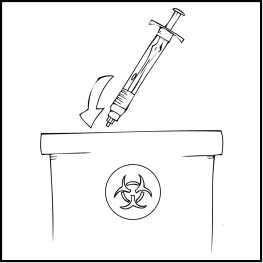
Doporučuje se zaznamenat si číslo šarže přípravku („Lot“), které je uvedeno na štítku předplněné stříkačky ReFacto AF, pokaždé když tento přípravek použijete. K zaznamenání čísla šarže můžete použít samolepku na přípravku ReFacto AF předplněná injekční stříkačka.
Další pokyny:
Vícenásobná rekonstituce přípravku ReFacto AF předplněná injekční stříkačka do 10ml nebo větší injekční stříkačky (Luer Lock Syringe) (10ml nebo větší injekční stříkačka není součástí této soupravy)
Pokyny níže jsou pro použití více souprav přípravku ReFacto AF předplněná injekční stříkačka, spolu s 10ml (nebo větší) injekční stříkačkou.
1. Rekonstituujte všechny předplněné injekční stříkačky přípravku ReFacto AF podle pokynů popsaných výše (v kapitole Rekonstituce a podání).
Držte ReFacto AF předplněnou injekční stříkačku ve svislé poloze a pomalým zatlačováním pístu odstraňte většinu vzduchu (ale ne všechen) ze zásobníku s léčivým přípravkem.
2. Vyjměte kuželovitý spojovací díl injekční stříkačky z obalu (kuželovitý spojovací díl není součástí této soupravy).
3. Spojte sterilní 10ml (nebo větší) injekční stříkačku s předplněnou injekční stříkačkou přípravku ReFacto AF pomocí spojovacího dílu.
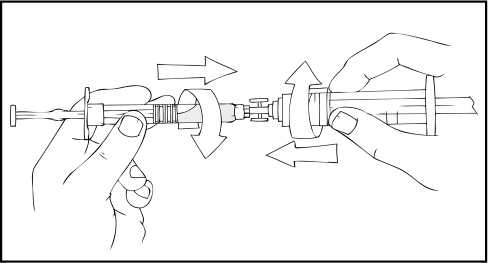
S předplněnou injekční stříkačkou přípravku ReFacto AF nahoře pomalu stlačujte píst, až se všechen obsah přemístí do 10ml (nebo větší) injekční stříkačky.
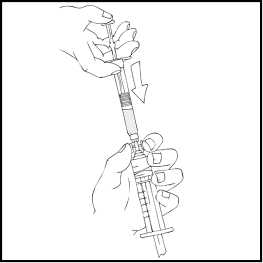
5. Sejměte prázdnou předplněnou injekční stříkačku přípravku ReFacto AF a zopakujte pokyny 3 a 4 popsané výše s rekonstituovanými obsahy dalších předplněných injekčních stříkaček.
6. Sejměte kuželovitý spojovací díl z 10ml (nebo větší) injekční stříkačky a připojte infuzní set tak, jak je popsáno výše v pokynech pro podání předplněné injekční stříkačky [viz kapitola Podání (intravenózní infuze)].
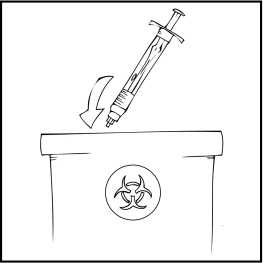
Poznámka: Prosím, odstraňte všechen nepoužitý roztok, prázdnou předplněnou injekční stříkačku a použité pomůcky do vhodného obalu pro likvidaci zdravotnického odpadu, jelikož tyto materiály mohou někoho zranit, jestliže nejsou zlikvidovány náležitým způsobem.
Jestliže si podáte větší množství přípravku ReFacto AF, než byste měl(a)
Poraďte se se svým lékařem nebo lékárníkem.
Jestliže přestanete přípravek ReFacto AF používat
Nepřestávejte používat přípravek ReFacto AF bez porady s lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Alergické reakce
Pokud se náhle vyskytnou závažné alergické (anafylaktické) reakce, musí se infuze ihned přerušit.
Pokud máte některý z následujících časných příznaků alergických reakcí, musíte ihned informovat svého lékaře:
• vyrážka, kopřivka, místní otok kůže, celkové svědění
• otok rtů a jazyka
• problémy s dýcháním, sípot, tíha na hrudi
• celkový pocit nemoci
• závratě a ztráta vědomí
Závažné příznaky zahrnující problémy s dýcháním a (téměř) mdloby vyžadují neodkladnou léčbu. Závažné náhlé alergické reakce (anafylaktické) jsou méně časté (postihují až 1 ze 100 pacientů).
Vytvoření inhibitorů
U pacientů s hemofilií A se mohou vyvinout neutralizační protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory vyskytnou, mohou se projevit zvýšeným množstvím přípravku ReFacto AF, typicky potřebným k léčbě krvácení a/nebo pokračujícím krvácením po léčbě. V takových případech se doporučuje kontaktovat specializované centrum pro hemofiliky. Lékař může požadovat sledování možného vyvinutí inhibitoru. V klinických studiích došlo k vyvinutí inhibitorů přibližně u 2% pacientů léčených přípravkem ReFacto AF.
Velmi časté nežádoucí účinky (postihují více než 1 z 10 pacientů)
• vývoj inhibitorů u pacientů, kteří nikdy předtím nebyli léčeni přípravky obsahujícími faktor VIII
• bolest hlavy
• kašel
• bolest kloubů
• horečka
Časté nežádoucí účinky (postihují až 1 z 10 pacientů)
• krvácení
• vývoj inhibitorů u pacientů, kteří již byli léčeni přípravky obsahujícími faktor VIII
• závratě
• snížená chuť k jídlu, průjem, zvracení, bolest břicha, pocit na zvracení
• bolest svalů
• zimnice, reakce v místě katétru
• některé testy mohou ukázat zvýšení protilátek proti faktoru VIII Méně časté nežádoucí účinky (postihují až 1 ze 100 pacientů)
• závažné alergické reakce
• necitlivost, spavost, změny chuti
• bolest na hrudi, zrychlený srdeční rytmus, palpitace
• nízký krevní tlak, bolest a zarudnutí žil související s krevní sraženinou, zrudnutí
• dušnost
• nadměrné pocení
• slabost, reakce v místě injekce včetně bolesti
• mírné zvýšení hladin srdečních enzymů
• zvýšení hladin jaterních enzymů, zvýšení hladiny bilirubinu
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek ReFacto AF uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku předplněné injekční stříkačky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte a přepravujte chlazené (2°C - 8°C). Chraňte před mrazem, aby se předešlo poškození předplněné injekční stříkačky.
Pro Vaše pohodlí je možno léčivý přípravek vyjmout z chladničky na jediné období trvající maximálně 3 měsíce a uchovávat při pokojové teplotě (do 25 °C). Po uplynutí tohoto období uchovávání při pokojové teplotě se přípravek nesmí vrátit do chladničky, ale musí se spotřebovat, či zlikvidovat. Na krabičku si zaznamenejte datum, kdy byl přípravek ReFacto AF předplněná injekční stříkačka vyjmut z chladničky a uchováván při pokojové teplotě (do 25 °C).
Uchovávejte předplněnou injekční stříkačku ve vnější krabičce, aby byl přípravek chráněn před světlem.
Rozpuštěný přípravek použijte nejpozději do 3 hodin od rozpuštění nebo po odstranění šedé pryžového víčka.
Přípravek je bezbarvý a čirý až mírně opalescentní. Nepoužívejte tento přípravek, pokud zjistíte, že je roztok zakalený nebo obsahuje viditelné částice.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo do domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek ReFacto AF obsahuje
- Léčivou látkou je moroctocogum alfa (rekombinantní koagulační faktor VIII). Jedna předplněná injekční stříkačka ReFacto AF obsahuje moroctocogum alfa 250, 500, 1000, 2000 nebo 3000 IU.
Rozpouštědlo [chlorid sodný 9 mg/ml (0.9%) injekční roztok] je obsažen v balení přípravku ReFacto AF předplněná injekční stříkačka pro rekonstituci léčivé látky moroctocog alfa.
- Pomocnými látkami jsou sacharosa, dihydrát chloridu vápenatého, histidin, polysorbát 80 a chlorid sodný.
- Po rozpuštění dodaným rozpouštědlem [roztok chloridu sodného 9mg/ml (0,9%)] připravený injekční roztok obsahuje buď 62,5; 125; 250; 500 nebo 750 IU moroctocogum alfa v ml, respektive (podle síly moroctocogum alfa, tj. 250, 500, 1000, 2000 nebo 3000 IU).
Jak přípravek ReFacto AF vypadá a co obsahuje toto balení
Přípravek ReFacto AF se dodává jako prášek a rozpouštědlo pro injekční roztok v předplněné injekční stříkačce, která obsahuje ReFacto AF prášek ve vrchním zásobníku a rozpouštědlo (chlorid sodný 9mg/ml, (0,9% roztok)) ve spodním zásobníku.
Obsahem balení je:
- jedna předplněná injekční stříkačka obsahující prášek moroctocogum alfa 250, 500, 1000, 2000 nebo 3000 IU a rozpouštědlo, 4 ml sterilního chloridu sodného 9 mg/ml (0,9%) injekční roztok pro naředění
- jeden píst
- jedno modré ochranné sterilní víčko
- jedna sterilní infuzní souprava
- dva alkoholové tampony
- jedna náplast
- jeden gázový polštářek
Držitel rozhodnutí o registraci
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
Výrobce
Wyeth Farma S.A
Autovia del Norte A-1 Km 23
Desvio Algete Km 1
8700 San Sebastian de los Reyes
Madrid
Španělsko
Další informace o tomto léčivém přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie /Belgique / Belgien
Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje
Tel. +3705 2514000
Btnrapuu
n$aň3ep HroKceMÓypr CAPH, KnoH Etnrapna Ten.: +359 2 970 4333
Česká republika
Pfizer PFE, spol. s.r.o.
Tel: +420 283 004 111
Luxembourg/Luxemburg
Pfizer S.A.
Tél/Tel: +32 (0)2 554 62 11
Magyarország
Pfizer Kft.
Tel.: + 36 1 488 37 00
|
Danmark Pfizer ApS Tlf: +45 44 20 11 00 |
Malta Vivian Corporation Ltd. Tel: +35621 344610 |
|
Deutschland Pfizer Pharma GmbH Tel: +49 (0)30 550055 51000 |
Nederland Pfizer bv Tel: +31 (0)10 406 43 01 |
|
Eesti Pfizer Luxembourg SARL Eesti filiaal Tel: +372 666 7500 |
Norge Pfizer Norge AS Tlf: +47 67 526 100 |
|
EHá8a Pfizer EAAAI A.E. Tni: +30 210 678 5800 |
Osterreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0 |
|
Espaňa Pfizer S.L. Tel: +34 91 490 99 00 |
Polska Pfizer Polska Sp. z o.o., Tel.: +48 22 335 61 00 |
|
France Pfizer Tél: +33 (0)1 58 07 34 40 |
Portugal Pfizer Biofarmaceutica, Sociedade Unipessoal Lda Tel: +351 21 423 5500 |
|
Hrvatska Pfizer Croatia d.o.o. Tel: + 385 1 3908 777 |
Románia Pfizer Románia S.R.L. Tel: +40 21 207 28 00 |
|
Ireland Pfizer Healthcare Ireland Tel: 1800 633 363 (toll free) +44 (0)1304 616161 |
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana Tel: + 386 (0) 1 52 11 400 |
|
Ísland Icepharma hf. Sími: + 354 540 8000 |
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel: +421-2-3355 5500 |
|
Italia Pfizer S.r.l. Tel: +39 06 33 18 21 |
Suomi/Finland Pfizer Oy Puh/Tel: +358 (0)9 43 00 40 |
|
Kúnpoq PFIZER EAAAI A.E. (CYPRUS BRANCH) Tni: +357 22 817690 |
Sverige Pfizer Innovations AB Tel: + 46 (0)8 550 520 00 |
|
Latvija Pfizer Luxembourg SARL filiale Latvija Tel: +371 670 35 775 |
United Kingdom Pfizer Limited Tel: +44 (0)1304 616161 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
51
Lidský koagulační faktor VIII produkovaný rekombinantní technologií DNA v ovariálních buňkách čínského křečka (CHO). Moroktokog alfa je glykoprotein obsahující 1438 aminokyselin se sekvencí