Ravicti 1,1 G/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
RAVICTI 1,1 g/ml perorální tekutina
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml tekutiny obsahuje glyceroli phenylbutyras 1,1 g. To odpovídá hustotě 1,1 g/ml. Léčivý přípravek neobsahuje žádné pomocné látky.
3. LÉKOVÁ FORMA
Perorální tekutina.
Čirá, bezbarvá až světle žlutá tekutina.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek RAVICTI je indikován k použití jako doplňující léčba při chronické léčbě dospělých a pediatrických pacientů > 2 měsíců věku s poruchami cyklu močoviny (urea cycle disorders, UCD), které zahrnují deficienci karbamoylfosfát syntázy I (CPS), ornithin transkarbamoylázy (OTC), argininosukcinát syntetázy (ASS), argininosukcinát lyázy (ASL), arginázy I (ARG) a deficienci ornithin translokázy u syndromu hyperornithinémie-hyperamonémie-homocitrulinémie (HHH), kteří nemohou být léčeni samotným omezením příjmu proteinů a/nebo suplementací aminokyselin.
Přípravek RAVICTI musí být požíván spolu s omezením příjmu proteinů a v některých případech s doplňky stravy (např. esenciální aminokyseliny, arginin, citrulin, kalorické doplňky neobsahující proteiny).
4.2 Dávkování a způsob podání
Přípravek RAVICTI má předepisovat lékař, který má zkušenosti s léčbou UCD.
Dávkování
Přípravek RAVICTI musí být požíván spolu s omezením příjmu proteinů a v některých případech s doplňky stravy (např. esenciální aminokyseliny, arginin, citrulin, kalorické doplňky neobsahující proteiny) v závislosti denním příjmu proteinů potřebných k podpoře růstu a vývoje.
Denní dávku je třeba individuálně přizpůsobit dle pacientovy proteinové tolerance a dle potřebného denního příjmu proteinů.
V terapii přípravkem RAVICTI může být nutné pokračovat celý život, pokud nedojde k ortotopické transplantaci jater.
Dospělí a děti ve věku > 2 měsíců do 18 let
Doporučené dávkování pro pacienty, kteří dosud nebyli kyselinou fenylbutyrovou léčeni, a pro pacienty přecházející z natrium-fenylbutyrátu na přípravek RAVICTI, je odlišné.
Doporučené celkové rozpětí denní dávky přípravku RAVICTI je 4,5 ml/m2/den až 11,2 ml/m2/den (5,3 g/m2/den až 12,4 g/m2/den) a je zapotřebí vzít v potaz následující:
Celkovou denní dávku je nutné rozdělit do stejných množství a podávat s každým jídlem nebo krmením (např. třikrát až šestkrát denně). Každou dávku je třeba zaokrouhlit na nejbližších 0,5 ml.
Doporučená výchozí dávka upacientů dosud neléčených fenylbutyrátem:
• 8,5 ml/m2/den (9,4 g/m2/den) u pacientů s tělesným povrchem (body surface area, BSA) < 1,3
m2
• 7 ml/m2/den (8 g/m2/den) u pacientů s BSA > 1,3 m2
Počáteční dávka u pacientů přecházejících z natrium-fenylbutyrátu na přípravek RAVICTI:
Pacienti přecházející z natrium-fenylbutyrátu na přípravek RAVICTI mají dostávat dávku přípravku RAVICTI, která obsahuje stejné množství kyseliny fenylbutyrové. Konverze se stanoví následovně:
• Celková denní dávka přípravku RAVICTI (ml) = celková denní dávka tablet natrium-fenylbutyrátu (g) x 0,86
• Celková denní dávka přípravku RAVICTI (ml) = celková denní dávka prášku natrium-fenylbutyrátu (g) x 0,81
Úprava dávky a sledování u dospělých a dětí ve věku >2 měsíce až 18 let:
Denní dávku je nutné individuálně upravit podle odhadované kapacity pacienta syntetizovat močovinu, pokud je nějaká, tolerance proteinů a denního příjmu proteinů ve stravě, který je potřebný na podporu růstu a vývoje. Proteiny ve stravě představují přibližně 16 % dusíku ve hmotnostním vyjádření. S ohledem na to, že přibližně 47 % dusíku ze stravy se vyloučí jako odpadní látka a přibližně 70 % dávky podané kyseliny 4-fenylbutyrové (PBA) bude přeměněno na fenylacetylglutamin, který se vyloučí močí (U-PAGN), je výchozí odhadovaná dávka glycerol-fenylbutyrátu na 24 hodin 0,6 ml glycerol-fenylbutyrátu na jeden gram proteinů ve stravě požité během 24hodinového období za předpokladu, že veškerý odpadní dusík bude pokryt glycerol-fenylbutyrátem a vyloučen jako fenylacetylglutamin (PAGN).
Úprava na základě hladiny amoniaku v plazmě
Dávku glycerol-fenylbutyrátu je nutné upravit, aby vedla k hladině amoniaku v plazmě nalačno, která je nižší než polovina horní hranice normálu (ULN) u pacientů ve věku 6 let a starších. U kojenců a menších dětí (obvykle mladších 6 let), kde je zjištění obsahu amoniaku nalačno problematické kvůli častému krmení, se první amoniak ráno má udržovat pod ULN.
Úprava na základě fenylacetylglutaminu v moči
Měření U-PAGN lze použít jako pomůcku při určení úpravy dávky glycerol-fenylbutyrátu a k hodnocení compliance. Každý gram U-PAGN vyloučený během 24 hodin obsahuje odpadní dusík vyprodukovaný ze 1,4 gramů proteinů v jídle. Pokud bude vylučování U-PAGN nedostatečné na pokrytí denního příjmu proteinů a amoniak nalačno bude vyšší než polovina doporučené ULN, je nutné dávku glycerol-fenylbutyrátu upravit směrem nahoru. Velikost úpravy dávky by měla vzít v úvahu množství proteinů ve stravě, které nebylo pokryto, jak to naznačuje 24hodinová hladina U-PAGN a odhadovaná dávka glycerol-fenylbutyrátu potřebná na jeden gram proteinů snědených v jídle.
Koncentrace U-PAGN v jednotlivém vzorku pod následujícími hladinami mohou signalizovat nesprávné podávání léčivého přípravku a/nebo nedostatečnou compliance:
• 9 000 mikrogramů (^g/ml) u pacientů mladších 2 let
• 7 000 mikrogramů (^g/ml) pro pacienty >2 let s BSA <1,3
• 5 000 mikrogramů (^g/ml) pro pacienty >2 let s BSA >1,3
Pokud koncentrace U-PAGN v jednotlivém vzorku poklesnou pod tyto hladiny, je třeba zhodnotit compliance s léčivým přípravkem a/nebo účinnost podávání léčivého přípravku (např. sondou) a zvážit zvýšení dávky glycerol-fenylbutyrátu u pacientů, kteří dodržují pokyny, k dosažení optimální kontroly amoniaku (v normálním rozmezí pro pacienty do 2 let věku a méně než polovina ULN u starších pacientů nalačno).
Úprava založená na fenylacetátu a fenylacetylglutaminu v plazmě
Příznaky jako zvracení, nauzea, bolest hlavy, somnolence, zmatenost nebo ospalost za nepřítomnosti vysoké hladiny amoniaku nebo komplikující nemoci mohou být známkami toxicity kyseliny fenyloctové (PAA) (viz bod 4.4, toxicita PAA). Proto může být měření plazmatických hladin PAA a PAGN užitečné jako vodítko pro dávkování. Ukázalo se, že poměr PAA vůči PAGN v plazmě (obě hodnoty měřené v pg/ml) je všeobecně nižší než 1 u pacientů bez akumulace PAA. U pacientů s poměrem PAA vůči PAGN nad 2,5 nemusí další zvýšení dávky glycerol-fenylbutyrátu zvýšit tvorbu PAGN, a to ani v případě, že jsou plazmatické koncentrace PAA zvýšené, kvůli saturaci konjugační reakce. V takových případech může zvýšená frekvence dávkování vést k nižší plazmatické koncentraci PAA a poměru PAA vůči PAGN. Při změně dávky glycerol-fenylbutyrátu se musí pečlivě sledovat hladiny amoniaku.
Deficit N-acetylglutamát syntázy (NAGS) a CITRINU (citrulinémie typu 2)
Bezpečnost a účinnost přípravku RAVICTI při léčbě pacientů s deficitem N-acetylglutamát syntázy (NAGS) a CITRINU (citrulinémie typu 2) nebyla stanovena.
Pediatrická populace
Pacienti ve věku od >2 měsíců do 2 let.
V současnosti dostupné údaje jsou uvedeny v bodě 5.2.
Pacienti od narození do <2 měsíců věku
Bezpečnost a účinnost přípravku RAVICTI pro tuto věkovou skupinu nebyla dosud stanovena.
Zvláštní populace
Starší pacienti (65 let a starší)
Klinické studie s přípravkem RAVICTI nezahrnovaly dostatečný počet subjektů > 65 let, aby bylo možné stanovit, zda reagují odlišně od mladších subjektů. Obecně má být výběr dávky pro staršího pacienta prováděn s opatrností, obvykle má začínat na dolním konci dávkového rozsahu, odrážet větší frekvenci snížené funkce jater, ledvin nebo srdce a souběžné onemocnění či terapii jinými léčivými přípravky.
Porucha funkce jater
Protože k přeměně PAA na PAGN dochází v játrech, pacienti s těžkou poruchou funkce jater mohou mít zhoršenou konverzní schopnost a vyšší hladinu plazmatické PAA a poměr PAA vůči PAGN v plazmě. Proto má dávkování pro dospělé a pediatrické pacienty s lehkou, středně těžkou či těžkou poruchou funkce jater začínat na dolním konci doporučeného dávkového rozpětí (4,5 ml/m2/den) a je nutné ho udržovat na nejnižší dávce nezbytné ke kontrole hladin amoniaku u pacienta. Poměr PAA k PAGN v plazmě nad 2,5 může signalizovat saturaci konverzní kapacity z PAA na PAGN a potřebu snížit dávkování a/nebo zvýšit frekvenci dávkování. Poměr PAA k PAGN v plazmě může být užitečný pro sledování dávky. (viz bod 5.2).
Porucha funkce ledvin
U pacientů s UCD s poruchou funkce ledvin nebyly prováděny žádné studie; bezpečnost glycerol-fenylbutyrátu u pacientů s poruchou funkce ledvin není známa. U pacientů s těžkou poruchou funkce ledvin je nutné používat přípravek RAVICTI s opatrností. Takoví pacienti mají nejlépe začít na nejnižší dávce nezbytné ke kontrole hladin amoniaku v krvi a mají se na ní i udržovat.
Způsob podání
Perorální nebo gastroenterální podání.
Přípravek RAVICTI se užívá s jídlem a podává se přímo do úst perorální stříkačkou. Léčivý přípravek se nemá přidávat ani vmíchávat do většího objemu jiné tekutiny, protože glycerol-fenylbutyrát je těžší než voda a může tak dojít k neúplnému podání. Byly provedeny studie kompatibility (viz část 4.5). Přípravek RAVICTI lze přidávat do malého množství jablečného pyré, kečupu nebo kaše a musí se podat do 2 hodin, pokud se uchovává při pokojové teplotě (25 °C). Tento léčivý přípravek lze smíchat s léčivými preparáty (Cyclinex-1, Cyclinex-2, UCD-1, UCD-2, Polycose, Pro Phree a Citrulline) a použít do 2 hodin, pokud se uchovává při teplotě do 25 °C, nebo až do 24 hodin, pokud je v chladničce.
Při zahájení léčby poskytne lékárna balíček k zahájení léčby obsahující léčivý přípravek, opakovatelně uzavíratelný adaptér víčka lahve a 7 perorálních tříkaček s označením CE vhodné velikosti pro podávání správné dávky (viz bod 6.5). Po zahájení léčby lékárna poskytne standardní balíček obsahující léčivý přípravek a opakovatelně uzavíratelnýadaptér víčka lahve. Další perorální stříkačky s označením CE kompatibilní s opakovatelně uzavíratelným adaptérem víčka lahve lze získat v lékárně.
Lahev přípravku RAVICTI se otevře stlačením víčka směrem dolů a jeho otočením doleva. Opakovatelně uzavíratelný adaptér víčka lahve se našroubuje na lahev. Špička perorální stříkačky se zasune do opakovatelně uzavíratelného adaptéru víčka lahve. Lahev se otočí dnem vzhůru i se vsunutou perorální stříkačkou. Píst perorální stříkačky se postupně vytahuje směrem dolů, dokud se stříkačka nenaplní příslušným množstvím léčivého přípravku. Na perorální stříkačku se poklepe, aby se z ní odstranily všechny vzduchové bubliny a ověřilo se, že je naplněna správným množstvím tekutiny. Tekutina se polyká z perorální stříkačky nebo se perorální stříkačka připojí ke gastrostomi nebo nasogastrické sondě. Stejná perorální stříkačka se má používat pro všechny dávky užívané stejný den. Je důležité dbát na to, aby byla perorální
stříkačka v intervalech mezi dávkami udržována čistá a suchá. Opakovatelně uzavíratelný adaptér víčka lahve a perorální stříkačka se mezi denním dávkami neproplachují, protože přítomnost vody způsobuje degradaci glycerol-fenylbutyrátu. Po použití pevně nasaďte kryt na opakovatelně uzavíratelném adaptéru víčka lahve. Po poslední denní dávce se perorální stříkačku zlikviduje. Po vyprázdnění lahve nebo po 3 dnech od otevření, i když lahev není prázdná, je nutné opakovatelně uzavíratelný adaptér víčka lahve zlikvidovat. Pro každou novou lahev, která se otevře, je zapotřebí použít nový opakovatelně uzavíratelný adaptér víčka lahve.
Přípravek RAVICTI lze také podávat pomocí lékařské silikonové nasogastrické nebo gastrostomické sondy s označením CE u těch pacientů, kteří nemohou užívat léčivý přípravek perorálně.
Další informace ohledně způsobu podání a studiích kompatibility/stability po otevření před použitím naleznete v bodě 6.6.
Příprava pro _podání nasogastrickou sondou nebo gastrostomickou sondou:
Studie in vitro hodnotící procentuální množství zjištěné látky (recovery) z celkové dávky podané nasogastrickou, nasojejunální nebo gastrostomickou sondou prokázaly, že procentuální podíl zjištěné dávky byl > 99 % pro dávky > 1 ml a 70 % pro 0,5ml dávku. U pacientů, kteří mohou polykat tekutiny, se má přípravek RAVICTI podávat perorálně, a to i u pacientů s nasogastrickou a/nebo gastrostomickou sondou. U pacientů, kteří tekutiny polykat nemohou, se může k podání přípravku RAVICTI použít nasogastrická nebo gastrostomická sonda následujícím způsobem:
• K odběru předepsané dávky přípravku RAVICTI z lahve se použije perorální stříkačka.
• Špička perorální stříkačky se nasune na konec gastrostomické/nasogastrické sondy.
• Stlačením pístu perorální stříkačky se přípravek RAVICTI podá do sondy.
• Sonda se jednou propláchne 10 ml vody nebo léčivého preparátu a proplachovací roztok se nechá po podání vytéct.
S ohledem na nízké množství zjištěného léku (recovery) se dávku 0,5 ml nebo menší nedoporučuje podávat nasogastrickými, gastrostomickými nebo nasojejunálními sondami.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku.
• Léčba akutní hyperamonémie.
4.4 Zvláštní upozornění a opatření pro použití
I během léčby glycerol-fenylbutyrátem může u určitého procenta pacientů dojít k akutní hyperamonémii včetně hyperamonemické encefalopatie.
Snížená absorpce fenylbutyrátu při pankreatické insuficienci nebo intestinální malabsorpci Exokrinní pankreatické enzymy hydrolyzují glycerol-fenylbutyrát v tenkém střevě, kdy oddělují aktivní složku, fenylbutyrát, od glycerolu. Tento proces umožňuje absorpci fenylbutyrátu do oběhu. Nízká nebo nulová koncentrace pankreatických enzymů nebo střevní onemocnění s následnou malabsorpcí tuku mohou vést ke sníženému nebo nulovému trávení glycerol-fenylbutyrátu a/nebo absorpci fenylbutyrátu a snížené kontrole amoniaku v plazmě. Hladiny amoniaku je nutné pečlivě monitorovat u pacientů s pankreatickou insuficiencí nebo střevní malabsorpcí.
Neurotoxicita
Reverzibilní klinické projevy naznačující přítomnost neurotoxicity (např. nauzea, zvracení, somnolence) byly údajně spojeny s koncentracemi fenylacetátu v rozsahu 499-1285 pg/ml u pacientů s nádorovým onemocněním, kteří dostávali PAA intravenózně. Přestože toto nebylo pozorováno v klinických hodnoceních týkajících se pacientů s UCD, na vysoké hladiny PAA je nutné mít podezření u pacientů s nevysvětlenou somnolencí, zmateností, nauzeou a letargií, kteří mají normální nebo nízkou hladinu amoniaku.
Pokud příznaky jako zvracení, nauzea, bolest hlavy, somnolence, zmatenost nebo ospalost budou přítomny za absence vysoké koncentrace amoniaku nebo jiných komplikujících onemocnění, je třeba změřit plazmatickou PAA a poměr PAA vůči PAGN v plazmě a zvážit snížení dávky glycerol-fenylbutyrátu nebo zvýšení frekvence dávkování v případě, že koncentrace PAA překračuje 500 pg/l a poměr PAA k PAGN v plazmě překračuje 2,5.
Monitorování a laboratorní testy
Denní dávku je nutné individuálně upravit podle odhadované kapacity pacienta syntetizovat močovinu, pokud je nějaká, profilu aminokyselin, tolerance proteinů a denního příjmu proteinů ve stravě, který je potřebný na podporu růstu a vývoje. Doplňkové aminokyselinové preparáty budou možná nezbytné k zachování esenciálních aminokyselin a aminokyselin s rozvětveným řetězcem v normálním rozsahu. Další úpravy mohou být založeny na sledování amoniaku, glutaminu, U-PAGN a/nebo plazmatických PAA a PAGN a dále na poměru plazmatické PAA k PAGN (viz bod 4.2).
Potenciál ostatních léčivých přípravků k ovlivnění amoniaku Kortikosteroidy
Použití kortikosteroidů může způsobit rozklad proteinů v těle a zvýšit hladiny amoniaku v plazmě. Hladiny amoniaku je třeba pečlivě monitorovat v případě, že se současně používají kortikosteroidy a glycerol-fenylbutyrát.
Kyselina valproová a haloperidol
Haloperidol a kyselina valproová mohou vyvolat hyperamonémii. Je třeba pečlivě sledovat koncentrace amoniaku v případě, že je nezbytné použít kyselinu valproovou nebo haloperidol u pacientů s UCD.
Probenecid
Probenecid může inhibovat vylučování metabolitů glycerol-fenylbutyrátu včetně PAGN ledvinami.
Ženy ve _ fertilním věku/antikoncepce u mužů a u žen
Ženy ve fertilním věku musí používat účinnou antikoncepci (viz bod 4.6).
Přípravek RAVICTI lze v těhotenství a u žen v reprodukčním věku, které nepoužívají antikoncepci, pouze tehdy, když klinický stav ženy vyžaduje léčbu glycerol-fenylbutyrátem, viz bod 4.6.
Glycerol
Glycerol-fenylbutyrát podávaný v maximální doporučené dávce obsahuje méně než 0,5 g glycerolu v jedné dávce.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Při současném používání léčivých přípravků, o nichž je známo, že inhibují lipázu, je třeba postupovat opatrně s ohledem na to, že glycerol-fenylbutyrát je hydrolyzován trávicí lipázou na kyselinu fenylbutyrovou a glycerol. To může být spojeno se zvýšeným rizikem interakcí léčivého přípravku s inhibitory lipázy a s lipázou obsaženou v substitučních terapiích pankreatickými enzymy.
Nelze vyloučit potenciální vliv na izoenzym CYP2D6 a u pacientů, kteří užívají léčivé přípravky, jež jsou substráty CYP2D6, je zapotřebí opatrnosti.
Prokázalo se, že glycerol-fenylbutyrát a/nebo jeho metabolity, PAA a PBA, jsou slabými induktory enzymu CYP3A4 in vivo. In vivo expozice glycerol-fenylbutyrátu vedla ke snížené systémové expozici midazolamu o přibližně 32 % a zvýšené expozici 1-hydroxy metabolitu midazolamu, což naznačuje, že dávkování glycerol-fenylbutyrátu v ustáleném stavu způsobuje indukci CYP3A4. Může existovat potenciál pro interakce glycerol-fenylbutyrátu jakožto induktoru CYP3A4 a těch přípravků, které jsou převážně metabolizovány pomocí dráhy CYP3A4. Proto mohou být sníženy terapeutické účinky a/nebo hladiny metabolitu léčivých přípravků, včetně některých perorálních kontraceptiv, které jsou substráty tohoto enzymu, a jejich plné účinky nelze po společném podání s glycerol-fenylbutyrátem zaručit.
Další léčivé přípravky jako kortikosteroidy, kyselina valproová, haloperidol a probenecid mohou mít potenciál ovlivnit hladiny amoniaku, viz bod 4.4.
Účinky glycerol-fenylbutyrátu na izoenzymy cytochromu P450 (CYP) 2C9 a jeho možné reakce s celecoxibem byly ověřovány u lidí; žádné známky interakce nebyly zjištěny.
Účinky glycerol-fenylbutyrátu na ostatní izoenzymy CYP nebyly u člověka studovány a nelze je vyloučit.
Studie kompatibility prokázaly chemickou a fyzikální stabilitu glycerol-fenylbutyrátu po otevření před použitím při podání s následujícími potravinami a potravinovými doplňky: jablečné pyré, kečup, kaše a pět léčivých preparátů (Cyclinex-1, Cyclinex-2, UCD-1, UCD-2, Polycose, Pro Phree a Citrulline), které obvykle dostávají pacienti s UCD (viz bod 4.2).
4.6 Fertilita, těhotenství a kojení
Ženy v reprodukčním věku/antikoncepce u mužů a žen
Použití přípravku RAVICTI u žen v reprodukčním věku musí být doprovázeno použitím účinné antikoncepce (viz část 4.4).
Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Údaje o podávání glycerol-fenylbutyrátu těhotným ženám jsou omezené.
Podávání glycerol-fenylbutyrátu se v těhotenství a u žen v reprodukčním věku, které nepoužívají antikoncepci, nedoporučuje.
Kojení
Není známo, zda se glycerol-fenylbutyrát nebo jeho metabolity vylučují do lidského mateřského mléka. Riziko pro kojené novorozence/děti nelze vyloučit. Na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku je nutno rozhodnout, zda přerušit kojení, nebo ukončit/přerušit podávání glycerol-fenylbutyrátu.
Fertilita
Glycerol-fenylbutyrát neměl žádný účinek na fertilitu nebo reprodukční funkci u samců a samic potkanů (viz bod 5.3). Pro fertilitu u člověka nejsou k dispozici žádné údaje.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek RAVICTI může mít výrazný vliv na schopnost řídit a obsluhovat stroje, neboť léčba pomocí glycerol-fenylbutyrátu může způsobovat závratě nebo bolest hlavy (viz bod 4.8). Pacienti nemají řídit ani obsluhovat stroje, když pociťují tyto nežádoucí účinky.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Hodnocení nežádoucích účinků bylo založeno na expozici u 114 pacientů s UCD (65 dospělých a 49 dětí v rozmezí věku od 2 měsíců do 17 let) s deficity CPS, OTC, ASS, ASL, ARG nebo HHH napříč 4 krátkodobými a 3 dlouhodobými klinickými studiemi, v nichž 90 pacientů dokončilo dobu trvání 12 měsíců (medián expozice = 51 týdnů).
Na počátku léčby se může vyskytnout bolest břicha, nauzea, průjem a/nebo bolest hlavy; tyto účinky obvykle vymizí během několika dnů, i když se v léčbě pokračuje. Nej častěji hlášenými nežádoucími účinky (>5 %) během léčby glycerol-fenylbutyrátem byly průjem, flatulence a bolest hlavy (8,8 % každý), snížená chuť k jídlu (7,0 %), zvracení (6,1 %) a únava, nauzea a abnormální zápach kůže (5,3 % každý).
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky jsou uvedeny níže podle třídy orgánových systémů a podle frekvence. Frekvence je vyjádřena jako velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10000 až <1/1000), velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Každý nežádoucí účinek hlášený u jednoho pacienta splňoval kritéria jako méně častý. Díky vzácnosti populace s UCD a malé velikosti databáze bezpečnostní populace léčivého přípravku (N=114) není známa frekvence nežádoucích účinků pro vzácné a velmi vzácné účinky.
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinek |
|
Infekce a infestace |
Méně časté |
Gastrointestinální virová infekce |
|
Endokrinní poruchy |
Méně časté |
Hypothyroidismus |
|
Poruchy metabolismu a výživy |
Časté |
Snížená chuť k jídlu, zvýšená chuť k jídlu |
|
Méně časté |
Hypoalbuminémie, hypokalémie | |
|
Psychiatrické poruchy |
Časté |
Averze vůči jídlu |
|
Poruchy nervového systému |
Časté |
Závratě, bolest hlavy, třes |
|
Méně časté |
Dysgeuzie, letargie, parestézie, psychomotorická hyperaktivita, somnolence, porucha řeči | |
|
Méně časté |
Stav zmatenosti, depresivní nálada | |
|
Srdeční poruchy |
Méně časté |
Ventrikulární arytmie |
|
Cévní poruchy |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté |
Dysfonie, epistaxe, nazální kongesce, orofaryngeální bolest, podráždění hrdla |
|
Gastrointestinální poruchy |
Časté |
Flatulence, průjem, zvracení, nauzea, bolest břicha, dyspepsie, abdominální distenze, zácpa, orální diskomfort, dávení |
|
Méně časté |
Abdominální diskomfort, abnormální stolice, sucho v ústech, eruktace, nutkání k defekaci, bolest v horní a/nebo dolní části břicha, bolestivá defekace, steatorhoea, stomatitida | |
|
Poruchy jater a žlučových cest |
Méně časté |
Bolest žlučníku |
|
Poruchy kůže a podkožní tkáně |
Časté |
Abnormální zápach kůže, akné |
|
Méně časté |
Alopécie, hyperhidróza, svědivá vyrážka | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Méně časté |
Bolest zad, otoky kloubů, svalové křeče, bolest končetin, plantární fasciitida |
|
Poruchy ledvin a močových cest |
Méně časté | |
|
Poruchy reprodukčního systému a prsu |
Časté |
Metroragie |
|
Méně časté |
Amenorea, nepravidelná menstruace | |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Únava, periferní edém |
|
Méně časté |
Hlad, pyrexie | |
|
Vyšetření |
Časté |
Zvýšený obsah aspartátaminotransferázy, zvýšený obsah alaninaminotransferázy, zvýšená aniontová mezera, snížený počet lymfocytů, snížený obsah vitamínu D |
|
Méně časté |
Zvýšený obsah draslíku v krvi, zvýšený obsah triglyceridů v krvi, abnormální elektrokardiogram, zvýšený obsah lipoproteinu s nízkou hustotou, prolongovaný protrombinový čas, zvýšený počet bílých krvinek, zvýšení tělesné hmotnosti, snížení tělesné hmotnosti |
Pediatrická populace
Nežádoucí účinky hlášené u více pediatrických než dospělých pacientů během dlouhodobé léčby glycerol-fenybutyrátem zahrnovaly bolest v horní části břicha (3 ze 49 pediatrických (6,1 %) v porovnání s 1 z 51 (2,0 %) dospělých) a zvýšenou aniontovou mezeru (2 ze 49 pediatrických (4,1 %) v porovnání s 0 z 51 (0,0 %) dospělých).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
PAA, aktivní metabolit glycerol-fenylbutyrátu, je spojována se známkami a příznaky neurotoxicity (viz bod 4.4) a mohla by se hromadit v těle pacientů, u nichž dojde k předávkování. V případě předávkování je třeba léčivý přípravek vysadit a pacienta sledovat, zda se u něj neprojeví jakékoliv známky či příznaky nežádoucích účinků.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Trávicí trakt a metabolismus, jiná léčiva, Trávicí trakt a metabolismus, různá léčiva, ATC kód: A16AX09
Mechanismus účinku
Glycerol-fenylbutyrát je léčivý přípravek vážící dusík. Je to triglycerid obsahující 3 molekuly PBA navázané na glycerolovou kostru.
UCD jsou vrozené deficity enzymů nebo transportérů nezbytných pro syntézu močoviny z amoniaku (NH3, NH4+). Nepřítomnost těchto enzymů nebo transportérů vede k akumulaci toxických hladin amoniaku v krvi a mozku dotčených pacientů. Glycerol-fenylbutyrát je hydrolyzován pankreatickými lipázami za vzniku PBA, která je přeměněna beta-oxidací na PAA, což je aktivní složka glycerol-fenylbutyrátu. PAA se konjuguje s glutaminem (který obsahuje 2 molekuly dusíku) acetylací v játrech a ledvinách za vzniku PAGN, který je vylučován ledvinami. V molárními vyjádření PAGN podobně jako močovina obsahuje 2 moly dusíku a slouží jako alternativní prostředek pro vylučování odpadního dusíku.
Farmakodynamické účinky
Farmakologické účinky
Ve sdružené analýze studií, v nichž pacienti přešli z natrium-fenylbutyrátu na glycerol-fenylbutyrát, byla AUC0_24h amoniaku 774,11 a 991,19 [(^mol/l)*hod.] během léčby glycerol-fenylbutyrátem a natrium-fenylbutyrátem v uvedeném pořadí (n = 80, poměr geometrických průměrů 0,84, 95% intervaly spolehlivosti 0,740 a 0,949)
Srdeční elektrofyziologie
V randomizované, placebem a aktivní léčbou (moxifloxacin 400 mg) kontrolované studii se čtyřmi rameny a překřížením byl u 57 zdravých subjektů hodnocen účinek opakovaných dávek glycerol-fenylbutyrátu 13,2 g/den a 19,8 g/den (přibližně 69 % a 104 % maximální doporučení denní dávky) na interval QTc. Horní rozsah jednostranného 95% IS pro největší QTc upravený podle placeba a korigovaný dle výchozí hodnoty, na základě individuální korekční metody (QTcI) pro glycerol-fenylbutyrát, byl nižší než 10 ms, což prokazuje, že glycerol-fenylbutyrát neměl žádný prolongační účinek na QT/QTc. Citlivost metody byla potvrzena významnou prolongací QTc u pozitivní kontroly, moxifloxacinu.
Klinická účinnost a bezpečnost
Klinické studie u dospělých pacientů s UCD
Aktivní léčbou kontrolovaná, 4týdenní, zaslepená zkřížená studie noninferiority (Studie 1) Randomizovaná, dvojitě zaslepená, aktivní léčbou kontrolovaná zkřížená studie noninferiority (Studie 1) porovnávala ekvivalentní dávky glycerol-fenylbutyrátu a natrium-fenylbutyrátu pomocí vyhodnocení 24hodinových koncentrací amoniaku v žilní krvi u pacientů s UCD, kteří užívali natrium-fenylbutyrát ke kontrole UCD před zařazením do studie. Bylo nutné, aby pacienti měli diagnostikovanou UCD zahrnující deficity CPS, OTC nebo ASS potvrzenou enzymatickými, biochemickými nebo genetickými testy. U pacientů při zařazení nesměla být klinicky prokázána hyperamonémie a nesměli dostávat léčivé přípravky, o nichž je známo, že zvyšují koncentrace amoniaku (např. valproát), zvyšují katabolismu proteinů (např. kortikosteroidy) nebo významně ovlivňují renální clearance (např. probenecid).
Glycerol-fenylbutyrát byl noninferiorní vůči natrium-fenylbutyrátu, pokud jde o 24hodinovou AUC pro amoniak. V této analýze bylo hodnoceno čtyřicet čtyři pacientů. Průměrné 24hodinové AUC pro amoniak v žilní krvi během dávkování v ustáleném stavu byly 866 ^mol/l*hod. u glycerol-fenylbutyrátu a 977 ^mol/l*hod. u natrium-fenylbutyrátu (n = 44, poměr geometrických průměrů 0,91; 95% intervaly spolehlivosti 0,799, 1,034).
V souladu s plazmatickým amoniakem byly koncentrace glutaminu v krvi nižší během léčby glycerol-fenylbutyrátem v porovnání s natrium-fenylbutyrátem v každém rameni zkřížené studie (pokles o 44,3 ± 154,43 ^mol/l po glycerol-fenylbutyrátu v porovnání s NaPBA, p = 0,064; párový t-test; p = 0,048, Wilcoxonův znaménkový test).
Otevřená nekontrolovaná rozšířená studie u dospělých
K vyhodnocení měsíční kontroly amoniaku a hyperamonémické krize za období 12 měsíců byla provedena dlouhodobá (12měsíční), nekontrolovaná, otevřená studie (Studie 2). Do studie bylo zařazeno celkem 51 dospělých pacientů s deficity CPS, OTC, ASS, ASL, ARG a HHH a všichni až na 6 byli převedeni z natrium-fenylbutyrátu na ekvivalentní dávky glycerol-fenylbutyrátu. Koncentrace amoniaku v žilní krvi byly sledovány měsíčně. Průměrné hodnoty amoniaku v žilní krvi nalačno u dospělých ve studii 2 byly v normálních mezích během dlouhodobé léčby glycerol-fenylbutyrátem (rozsah: 6-30 pmol/l). Z 51 dospělých pacientů účastnících se studie 2 udávalo 7 pacientů (14 %) celkem 10 hyperamonémických krizí během léčby glycerol-fenylbutyrátem v porovnání s 9 pacienty (18 %), kteří udávali celkem 15 krizí během 12 měsíců před vstupem do studie, zatímco byli léčení natrium-fenylbutyrátem.
Pediatrická populace
Klinické studie u pediatrických pacientů s UCD
Účinnost glycerol-fenylbutyrátu u pediatrických pacientů ve věku od 2 měsíců do 17 let s deficity OTC, ASS, ASL a ARG byla hodnocena ve 2 otevřených studiích s fixní sekvencí s převedením z natrium-fenylbutyrátu na ekvivalentní dávkování glycerol-fenylbutyrátu (studie 3 a 4). Studie 3 trvala 14 dnů a studie 4 trvala 10 dnů.
Zjistilo se, že glycerol-fenylbutyrát je noninferiorní vůči natrium-fenylbutyrátu, pokud jde o kontrolu amoniaku v obou těchto pediatrických studiích. Ve sdružené analýze krátkodobých studií u dětí (studie 3 a studie 4) byla koncentrace amoniaku v plazmě významně nižší po přechodu na glycerol-fenylbutyrát, AUC0-24hod. amoniaku byla 626,79 a 871,72 (pmol/l)*hod. během léčby glycerol-fenylbutyrátem a natrium-fenylbutyrátem v uvedeném pořadí (n = 26, poměr geometrických průměrů 0,79, 95% intervaly spolehlivosti 0,647 a 0,955).
Průměrné hladiny glutaminu v krvi byly rovněž nevýznamně nižší po léčbě glycerol-fenylbutyrátem v porovnání s léčbou natrium-fenylbutyrátem o -45,2 ± 142,94 pmol/l (p = 0,135, párový t test; p = 0,114, Wilcoxonův znaménkový test).
Otevřené, nekontrolované, rozšířené studie u pediatrických pacientů
Dlouhodobé (12měsíční), nekontrolované, otevřené studie byly prováděny s cílem vyhodnotit měsíční kontroly amoniaku a hyperamonémické krize za období 12 měsíců ve třech studiích (studie 2, která rovněž zařazovala dospělé, a rozšíření studií 3 a 4). Bylo zařazeno celkem 49 dětí ve věku od 2 měsíců do 17 let s deficity OTC, ASS, ASL a ARG a všechny až na 1 byly převedeny z natrium-fenylbutyrátu na glycerol-fenylbutyrát. Průměrné hodnoty amoniaku v žilní krvi nalačno byly v normálních mezích během dlouhodobé léčby glycerol-fenylbutyrátem (rozsah: 17-25 pmol/l). Ze 49 pediatrických pacientů účastnících se těchto rozšířených studií udávalo 12 pacientů (25 %) celkem 17 hyperamonémických krizí během léčby glycerol-fenylbutyrátem v porovnání s 21 pacienty (43 %), kteří udávali 38 krizí během předchozích 12 měsíců před vstupem do studie, zatímco byli léčení natrium-fenylbutyrátem.
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s glycerol-fenylbutyrátem u jedné nebo více podskupin pediatrické populace ve schválené indikaci léčby poruch cyklu močoviny (informace o použití u dětí viz bod 4.2).
Po léčbě je nepravděpodobný zvrat již dříve existující poruchy funkce nervového systému a neurologické zhoršování může u některých pacientů pokračovat.
5.2 Farmakokinetické vlastnosti
Absorpce
Přípravek RAVICTI je proléčivo PBA. Po perorálním podání se PBA uvolňuje z glycerolové kostry v gastrointestinálním traktu pomocí pankreatické lipázy. PBA odvozená z glycerol-fenylbutyrátu je dále přeměňována P-oxidací na PAA.
U zdravých dospělých subjektů, které nalačno dostaly jednorázovou perorální dávku 2,9 ml/m2 glycerol-fenylbutyrátu, nastaly vrcholové hladiny PBA, PAA a PAGN v plazmě po 2, 4 a 4 hod. v uvedeném pořadí. Po jednorázovém podání glycerol-fenylbutyrátu byly plazmatické koncentrace PBA kvantifikovatelné u 15 z 22 účastníků při první době odběru vzorku po dávce (0,25 hod.). Průměrná maximální koncentrace (Cmax) pro PBA byla 37,0 pg/ml, pro PAA byla 14,9 pg/ml a pro PAGN byla
30,2 pg/ml. U zdravých subjektů nebyl v plazmě detekován intaktní glycerol-fenylbutyrát.
U zdravých subjektů se zvyšovala systémová expozice PAA, PBA a PAGN v závislosti na dávce. Po podávání 4 ml glycerol-fenylbutyrátu po dobu 3 dnů (3krát denně) byla průměrná Cmax 66 pg/ml a AUC byla 930 pg^hod./ml v případě PBA, zatímco pro PAA to bylo 28 pg/ml a 942 pg^hod./ml v uvedeném pořadí. V téže studii po podávání 6 ml glycerol-fenylbutyrátu po dobu 3 dnů (3x denně) byla průměrná Cmax 100 pg/ml a AUC byla 1 400 pg^hod/ml v případě PBA, zatímco pro PAA to bylo 65 pg/ml a 2 064 pg^hod/ml v uvedeném pořadí.
U dospělých pacientů s UCD, kteří dostávali opakované dávky glycerol-fenylbutyrátu, se maximální plazmatické koncentrace v ustáleném stavu (Cmaxss) PBA, PAA a PAGN objevovaly za 8, 12 a 10 hod. po první dávce v daný den, v uvedeném pořadí. Intaktní glycerol-fenylbutyrát nebyl detekovatelný v plazmě u pacientů s UCD.
Farmakokinetické modelování populace a simulace dávkování naznačují, že PBA vstupuje do oběhu přibližně o 70-75 % pomaleji, když je podávána perorálně jako glycerol-fenylbutyrát, v porovnání s natrium-fenylbutyrátem a dále ukazují, že plocha povrchu těla je nejvýznamnější kovariátou vysvětlující variabilitu clearance PAA.
Distribuce
In vitro byl rozsah vazby na lidské plazmatické proteiny u 14C-značených metabolitů 80,6 % až 98,0 % pro PBA (rozsah 1-250 pg/ml), a 37,1 % až 65,6 % pro PAA (rozsah 5-500 pg/ml). Vazba PAGN na proteiny byla 7 % až 12 % a nebyly zaznamenány žádné účinky koncentrace.
Biotransformace
Po perorální podání pankreatické lipázy hydrolyzují glycerol-fenylbutyrát a uvolňují PBA. PBA prochází p-oxidací na PAA, která se konjuguje s glutaminem v jádrech a v ledvinách prostřednictvím enzymu fenylacetyl-CoA: L-glutamin-N-acetyltransferázy za vzniku PAGN. PAGN se následně eliminuje močí.
Saturace konjugace PAA a glutaminu za vzniku PAGN byla naznačena zvýšením poměru plazmatické PAA vůči PAGN s rostoucí dávkou a s rostoucí závažností poruchy funkce jater.
U zdravých subjektů po podávání 4 ml, 6 ml a 9 ml 3krát denně po dobu 3 dnů byl poměr průměrné AUC0-23 hod. PAA vůči PAGN 1, 1,25 a 1,6 v uvedeném pořadí. V samostatné studii u pacientů s poruchou funkce jater (Child-Pugh B a C) se poměry průměrných hodnot pro PAA vůči PAGN mezi všemi pacienty, kteří dostávali dávku 6 ml a 9 ml dvakrát denně, pohybovaly od 0,96 do 1,28 a u pacientů s dávkou 9 ml dvakrát denně bylo toto rozpětí 1,18 až 3,19.
Ve studiích in vitro byla zaznamenána specifická aktivita lipáz pro glycerol-fenylbutyrát v následujícím sestupném pořadí: pankreatická triglyceridová lipáza, karbonyl-ester lipáza a pankreatický s lipázou související protein 2. Navíc byl pomocí esteráz v lidské plazmě hydrolyzován glycerol-fenylbutyrát in vitro. V těchto studiích in vitro úplné vymizení glycerol-fenylbutyrátu nevyprodukovalo molárně ekvivalentní PBA, což naznačuje tvorbu mono- nebo bis-esterových metabolitů. Ovšem tvorba mono- nebo bis-esterů nebyla u člověka studována.
Eliminace
Průměrný (SD) procentuální podíl podané PBA eliminované jako PAGN byl přibližně 68,9 % (17,2) u dospělých a 66,4 % (23,9) u dětských pacientů s UCD v ustáleném stavu. PAA a PBA zastupovaly minoritní močové metabolity, každý představující <1 % podané dávky PBA.
Zvláštní populace
Porucha funkce jater
Ve studii s pacienty s klinicky dekompenzovanou cirhózou a hepatickou encefalopatií (Child-Pugh B a C) byla průměrná Cmax PAA 144 pg/ml (rozsah: 14-358 pg/ml) po denní dávce 6 ml glycerol-fenylbutyrátu dvakrát denně, zatímco průměrná Cmax PAA byla 292 pg/ml (rozsah: 57-655 pg/ml) po denní dávce 9 ml glycerol-fenylbutyrátu dvakrát denně. Poměr průměrných hodnot pro PAA vůči PAGN mezi všemi pacienty, kteří dostali dávku 6 ml dvakrát denně, se pohyboval od 0,96 do 1,28 a u pacientů, kteří dostali dávku 9 ml dvakrát denně, se pohyboval v rozsahu 1,18 až 3,19. Po opakovaných dávkách byla koncentrace PAA >200 pg/l spojena s vyšším poměrem plazmatických koncentrací PAA vůči PAGN než 2,5.
Tyto nálezy společně signalizují, že konverze PAA na PAGN může být zhoršená u pacientů se závažnou poruchou funkce jater a že poměr PAA vůči PAGN >2,5 v plazmě odhaluje pacienty s rizikem zvýšených hladin PAA.
Porucha funkce ledvin
Farmakokinetika glycerol-fenylbutyrátu u pacientů s poruchou funkce ledvin, včetně pacientů s terminálním stádiem renálního onemocnění (ESRD) nebo pacientů na hemodialýze, nebyla hodnocena.
Pohlaví
U zdravých dospělých dobrovolníků byl zjištěn vliv pohlaví u všech metabolitů, kdy u žen byly obecně vyšší plazmatické koncentrace všech metabolitů než u mužů při dané hladině dávky. U zdravých dobrovolnic-žen byla průměrná Cmax pro PAA o 51 % a 120 % vyšší než u dobrovolníků-mužů po podávání 4 ml a 6 ml 3krát denně po dobu 3 dnů v uvedeném pořadí. Dávkově normalizovaná průměrná AUC0-23 hod. pro PAA byla o 108 % vyšší u žen než u mužů. Dávkování u pacientů s UCD však musí být individuální na základě specifických metabolických potřeb a zbytkové enzymové kapacity pacienta bez ohledu na pohlaví.
Pediatrická populace
Farmakokinetické modelování a simulace dávkování u jednotlivých populací naznačují, že nejvýznamnější kovariátou vysvětlující variabilitu clearance PAA je plocha tělesného povrchu. Clearance PAA byla 7,1 l/hod., 10,9 l/hod., 16,4 l/hod. a 24,4 l/hod. u pacientů s UCD ve věku < 2, 3 až 5, 6 až 11 a 12 až 17 v jednotlivých případech. U 7 pediatrických pacientů mladších 2 let, kteří dostávali přípravek RAVICTI až po dobu 12 měsíců, nevzrostly koncentrace PAA, PBA a PAGN během doby léčby a celkové střední koncentrace PAA, PBA a PAGN u těchto pacientů byly podobné koncentracím podobným u starších věkových pediatrických skupin.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Kancerogeneze
Ve studii s potkany glycerol-fenylbutyrát způsobil statisticky významné zvýšení incidence pankreatického adenomu z acinárních buněk, karcinomu a kombinovaného adenomu nebo karcinomu u samců a samic v dávce 4,7- a 8,4krát vyšší, než jsou dávky u dospělých pacientů (6,87 ml/m2/den na základě kombinované AUC pro PBA a PAA). U samic potkanů rovněž vzrostla incidence následujících tumorů: thyroidální adenom z folikulárních buněk, karcinom a kombinovaný adenom nebo karcinom, adrenální kortikální kombinovaný adenom nebo karcinom, cervikální schwannom, uterinní endometriální stromální polyp a kombinovaný polyp nebo sarkom.
V 26týdenní studii u myší nebyl glycerol-fenylbutyrát tumorogenní v dávkách až do 1 000 mg/kg/den.
Glycerol-fenylbutyrát byl hodnocen v řadě studií genotoxicity in vitro a in vivo a neprokázal žádnou genotoxickou aktivitu.
Porucha _ fertility
Glycerol-fenylbutyrát neměl žádný účinek na fertilitu nebo reprodukční funkci u samců a samic potkanů při hladinách klinické expozice, ovšem v perorálních dávkách až do přibližně 7násobku dávky u dospělých pacientů byla pozorována maternální toxicita společně toxicitou u samců a zvýšil se počet neživotaschopných embryí.
Vývojové studie
Perorální podávání glycerol-fenylbutyrátu v průběhu organogeneze u potkanů a králíků nemělo žádné účinky na embryofetální vývoj při dávkách 2,7- a 1,9násobku dávky pro dospělé pacienty v uvedeném pořadí. Ve studii s potkany však byla pozorována maternální toxicita a nežádoucí účinky na embryofetální vývoj včetně snížených hmotností plodu a cervikálních žeber při dávkách odpovídajících přibližně 6násobku dávky u dospělých pacientů na základě kombinované AUC pro PBA a PAA. Během organogeneze a laktace u potkanů nebyly pozorovány žádné vývojové abnormality do 92. dne po vrhu při perorálním podání u březích samic potkana.
Studie na juvenilních zvířatech
Ve studii u juvenilních potkanů s denní perorální dávkou podanou 2. den postpartum po dobu páření a březosti po dosažení dospělosti se terminální tělesná hmotnost snížila v závislosti na dávce u samců a samic až o 16 % a 12 % v uvedeném pořadí. Fertilita (počet březích potkaních samic) poklesla až o 25 % při dávce představující 2,6násobek dávky u dospělých pacientů. Rovněž byla zaznamenána toxicita pro embryo (zvýšené resorpce) a snížená velikost při vrhu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Žádné.
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
1 rok.
Po první otevření lahve musí být léčivý přípravek spotřebován do 3 dnů a lahev a její obsah je třeba zlikvidovat, i pokud není prázdná.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Lahev ze skla třídy III s dětským bezpečnostním uzávěrem z polyethylenu s vysokou hustotou (HDPE).
Jedna lahev obsahuje 25 ml tekutiny.
Velikost balení k zahájení léčby:
• 1 balíček k zahájení léčby obsahující 1 lahev, 1 zavírací adaptér víčka lahve a sedm (7) perorálních stříkaček s označením CE o obsahu 1 ml v papírové krabičce
• 1 balíček k zahájení léčby obsahující 1 lahev, 1 zavírací adaptér víčka lahve a sedm (7) perorálních stříkaček s označením CE o obsahu 3 ml v papírové krabičce
• 1 balíček k zahájení léčby obsahující 1 lahev, 1 zavírací adaptér víčka lahve a sedm (7)
perorálních stříkaček s označením CE o obsahu 5 ml v papírové krabičce a speciální předpis.
Tato velikost stříkaček je vhodná pro možný interval úvodních dávek a množství bude dostatečné na týdenní léčbu. Další stříkačky se poskytují v případě, že je denně potřeba více než jedna stříkačka. Zbývající stříkačky je třeba odložit pro použití s další lahví. Každou lahev je třeba po 3 dnech zlikvidovat. Po úvodní léčbě lze v lékárně získat další perorální stříkačky s označením CE kompatibilní se zavíracím adaptérem víčka lahve.
Velikost standardního balení: 1 lahev a 1 zavírací adaptér víčka lahve v papírové krabičce.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Jedna perorální stříkačka se používá jeden den. Neoplachujte opakovatelně uzavíratelný adaptér víčka lahve ani perorální stříkačku mezi denními dávkami, protože voda způsobuje degradaci glycerol-fenylbutyrátu. Každý den po poslední dávce perorální stříkačku zlikvidujte. Stejný opakovatelně uzavíratelný adaptér víčka lahve lze používat, dokud lahev nebude prázdná. Pro každou novou lahev, která se otevře, je zapotřebí použít nový opakovatelně uzavíratelný adaptér víčka lahve.
Byla prokázána chemická kompatibilita glycerol-fenylbutyrátu s lékařskými silikonovými nasogastrickými, gastrostomickými a nasojejunálními sondami. Studie in vitro vyhodnocující procentuální znovuzískání z celkové dávky dodané nasogastrickou nebo gastrostomickou sondou prokázaly, že procentuální podíl znovuzískané dávky byl > 99 % pro dávky > 1 ml a přibližně 70 % pro dávku 0,5 ml. Proto se doporučuje, aby se k podávání dávek > 1 ml používaly pouze nasogastrické, nasojejunální nebo gastrostomické sondy. Pokud bude zapotřebí podat takovými nasogastrickými, gastrostomickými nebo nasojejunálními sondami dávku 0,5 ml nebo menší, je třeba vzít v úvahu nízké znovuzískání léčivého přípravku.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Horizon Pharma Ireland Limited Connaught House, 1st Floor,
1 Burlington Road,
Dublin 4, D04C5Y6 Irsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1062/001
EU/1/15/1062/002
EU/1/15/1062/003
EU/1/15/1062/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 27. listopadu 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
AndersonBrecon (UK) Limited Units 2-7
Wye Valley Business Park
Brecon Road
Hay-on-Wye
Hereford
Herefordshire
HR3 5PG
Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky na předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán na řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit.
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedené opatření:
|
Popis |
Termín splnění |
|
Neintervenční poregistrační studie bezpečnosti (PASS): Multicentrická prospektivní neintervenční registrační studie u pacientů s poruchami cyklu močoviny při léčbě glycerol-fenylbutyrátem určená k charakterizaci demografických údajů pacientů a dokumentaci dlouhodobé bezpečnosti a klinických výsledků. |
Závěrečná zpráva ze studie se očekává do konce července 2030. |
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU VNĚJŠÍ KRABICE (STANDARDNÍ BALENÍ)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
RAVICTI 1,1 g/ml perorální tekutina Glyceroli phenylbutyras
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje glyceroli phenylbutyras 1,1 g.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Perorální tekutina.
1 x 25 ml lahev.
1 zavírací adaptér víčka lahve.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální nebo gastroenterální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Použijte do 3 dnů od otevření.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Horizon Pharma Ireland Limited Connaught House, 1st Floor,
1 Burlington Road,
Dublin 4, D04C5Y6 Irsko
EU/1/15/1062/001
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
RAVICTI 1,1 g/ml
RAVICTI 1,1 g/ml perorální tekutina Glyceroli phenylbutyras
Jeden ml obsahuje glyceroli phenylbutyras 1,1 g.
Perorální tekutina.
1ml balíček k zahájení léčby 1 x 25ml lahev.
1 zavírací adaptér víčka lahve. 7 x 1ml perorální stříkačka.
Před použitím si přečtěte příbalovou informaci. Perorální nebo gastroenterální podání. Používejte pouze poskytnutou 1ml stříkačku.
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Horizon Pharma Ireland Limited Connaught House, 1st Floor,
1 Burlington Road,
Dublin 4, D04C5Y6 Irsko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1062/002
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
RAVICTI 1,1 g/ml
RAVICTI 1,1 g/ml perorální tekutina Glyceroli phenylbutyras
Jeden ml obsahuje glyceroli phenylbutyras 1,1 g.
Perorální tekutina.
3ml balíček k zahájení léčby 1 x 25ml lahev.
1 zavírací adaptér víčka lahve. 7 x 3ml perorální stříkačka.
Před použitím si přečtěte příbalovou informaci. Perorální nebo gastroenterální podání. Používejte pouze poskytnutou 3ml stříkačku.
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Horizon Pharma Ireland Limited Connaught House, 1st Floor,
1 Burlington Road,
Dublin 4, D04C5Y6 Irsko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1062/003
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
RAVICTI 1,1 g/ml
RAVICTI 1,1 g/ml perorální tekutina Glyceroli phenylbutyras
Jeden ml obsahuje glyceroli phenylbutyras 1,1 g.
Perorální tekutina.
5ml balíček k zahájení léčby 1 x 25ml lahev.
1 zavírací adaptér víčka lahve. 7 x 5ml perorální stříkačka.
Před použitím si přečtěte příbalovou informaci. Perorální nebo gastroenterální podání. Používejte pouze poskytnutou 5ml stříkačku.
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Horizon Pharma Ireland Limited Connaught House, 1st Floor,
1 Burlington Road,
Dublin 4, D04C5Y6 Irsko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1062/004
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
RAVICTI 1,1 g/ml
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU ŠTÍTEK 25ml LAHVE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
RAVICTI 1,1 g/ml perorální tekutina Glyceroli phenylbutyras
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje glyceroli phenylbutyras 1,1 g.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Perorální tekutina. 1 x 25ml lahev.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální nebo gastroenterální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Použijte do 3 dnů od otevření.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Horizon Pharma Ireland Limited Connaught House, 1st Floor,
1 Burlington Road,
Dublin 4, D04C5Y6 Irsko
EU/1/15/1062/001
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
Příbalová informace: informace pro pacienta
RAVICTI 1,1 g/ml perorální tekutina
Glyceroli phenylbutyras
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli otázky, zeptejte se svého lékaře nebo lékárníka.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek RAVICTI a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek RAVICTI užívat
3. Jak se přípravek RAVICTI užívá
4. Možné nežádoucí účinky
5. Jak přípravek RAVICTI uchovávat
6. Obsah balení a další informace
1. Co je přípravek RAVICTI a k čemu se používá
PŘÍPRAVEK RAVICTI obsahuje léčivou látku „glycerol-fenylbutyrát“, která se používá k léčbě šesti známých „poruch cyklu močoviny“ (UCD) u dospělých a dětí ve věku 2 měsíců a starších. UCD zahrnuje deficity (nedostatečnosti) karbamoylfosfát syntázy I (CPS), ornithin transkarbamoylázy (OTC), argininosukcinát syntetázy (ASS), argininosukcinát lyázy (ASL), arginázy I (ARG) a deficit ornithin translokázy u syndromu hyperornithinémie-hyperamonémie-homocitrulinémie (HHH).
O poruchách cyklu močoviny
• Při poruchách cyklu močoviny tělo nemůže odstranit dusík z bílkovin, které jíme.
• Normálně tělo mění nadbytečný dusík v bílkovině do odpadní sloučeniny nazývané „amoniak“. Játra pak odstraňují amoniak z těla cyklem, který se nazývá „cyklus močoviny“.
• Při poruchách cyklu močoviny tělo není schopno vytvořit dostatek jaterních enzymů k odstranění nadbytečného dusíku.
• To znamená, že se v organismu hromadí amoniak. Pokud není amoniak z těla odstraňován, může poškodit mozek a vést k poruchám vědomí nebo kómatu.
• Poruchy cyklu močoviny j sou vzácné.
Jak přípravek RAVICTI působí
Přípravek RAVICTI pomáhá tělu eliminovat odpadní dusík. Tím se snižuje množství amoniaku v těle.
2. Čemu musíte věnovat pozornost, než začnete přípravek RAVICTI užívat
Neužívejte přípravek RAVICTI:
• jestliže j ste alergický(á) na glycerol-fenylbutyrát,
• jestliže máte akutní hyperamonémii, která vyžaduje rychlejší zásah.
Pokud si nejste jist(á), zda se kterákoliv ze shora uvedených podmínek vztahuje i na Vás, poraďte se svým lékařem nebo lékárníkem dříve, než budete přípravek RAVICTI užívat.
Upozornění a opatření
Před užitím přípravku RAVICTI se poraďte se svým lékařem nebo lékákrníkem:
• jestliže máte obtíže s ledvinami nebo játry - je to proto, že se glycerol-fenylbutyrát odstraňuje z těla ledvinami a játry,
• jestliže máte obtíže se slinivkou břišní, žaludkem nebo střevy - tyto orgány odpovídají za vstřebávání glycerol-fenylbutyrátu do těla.
Pokud se kterákoliv ze shora uvedených podmínek na Vás nevztahuje (nebo si nejste jist(á)), poraďte se svým lékařem nebo lékárníkem dříve, než budete přípravek RAVICTI užívat.
V některých případech, jako je infekce nebo stav po chirurgickém výkonu, se může množství amoniaku zvýšit navzdory léčbě tímto lékem a může poškodit mozek (hyperamonemická encefalopatie).
V jiných případech množství amoniaku v krvi rychle vzrůstá. V tomto případě glycerol-fenylbutyrát nezastaví závažné zvyšování koncentrace amoniaku v krvi.
Vysoké koncentrace amoniaku vedou k nevolnosti (pocitu na zvracení), zvracení nebo pocitu zmatenosti. Informujte svého lékaře nebo ihned jděte do nemocnice, pokud si povšimnete jakéhokoliv z těchto příznaků.
Přípravek RAVICTI se musí používat se speciální dietou s nízkým obsahem bílkovin.
• Tuto dietu vám navrhne lékař nebo dietní sestra.
• Tuto dietu musíte pečlivě dodržovat.
• Léčbu a dietu budete muset dodržovat po celý život, pokud nedojde k úspěšné transplantaci jater.
Dítě
Přípravek RAVICTI se nedoporučuje používat u dětí mladších 2 měsíců.
Další léčivé přípravky a přípravek RAVICTI
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Zvláště informujte svého lékaře nebo lékárníka, pokud užíváte jakýkoliv z následujících léků, které mohou být méně účinné, když se používají s glycerol-fenylbutyrátem. Pokud budete tyto léky užívat, možná budete potřebovat pravidelné krevní testy:
• midazolam a barbituráty - používané ke zklidnění, při obtížích se spánkem nebo k léčbě epilepsie
• antikoncepce
Rovněž informujte svého lékaře nebo lékárníka, pokud užíváte jakýkoliv z následujících léků, protože mohou zvyšovat množství amoniaku v těle nebo měnit způsob, jak glycerol-fenylbutyrát působí:
• kortikosteroidy - používané k léčbě zánětlivých oblastí těla
• valproát - lék k léčbě epilepsie
• haloperidol - používá se k léčbě některých duševních obtíží
• probenecid - k léčbě vysokých hladin kyseliny močové v krvi, které by mohly způsobit dnu („hyperurikémie“).
• inhibitory lipázy - používané k léčbě obezity
• lipáza v pankreatických substitučních terapiích
Pokud se kterákoliv ze shora uvedených podmínek na Vás vztahuje (nebo si nejste jist(á)), poraďte se svým lékařem dříve, než budete přípravek RAVICTI užívat.
Těhotenství, antikoncepce a kojení
• Pokud jste těhotná, poraďte se svým lékařem dříve, než začnete přípravek RAVICTI užívat. Pokud otěhotníte během užívání přípravku RAVICTI, poraďte se svým lékařem. Je to proto, že léčba přípravkem RAVICTI může poškodit Vaše nenarozené dítě.
• Pokud jste žena, která by mohla otěhotnět, musíte používat účinnou metodu antikoncepce během léčby přípravkem RAVICTI. Poraďte se svým lékařem o nejlepší metodě antikoncepce.
• Neužívejte přípravek RAVICTI, pokud kojíte. Je to proto, že může přecházet do mateřského mléka a může Vašemu dítěti uškodit.
Řízení dopravních prostředků a obsluha strojů
RAVICTI může mít výrazný vliv na schopnost řídit a obsluhovat stroje. Při užívání glycerol-
fenylbutyrátu můžete pociťovat závratě nebo mít bolesti hlavy. Pokud se objeví tyto nežádoucí účinky,
neřiďte dopravní prostředky ani neobsluhujte stroje.
3. Jak se přípravek RAVICTI užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Během léčby glycerol-fenylbutyrát musíte dodržovat speciální dietu s nízkým obsahem bílkovin.
Jaké množství mám užívat
Lékař Vám řekne, kolik přípravku RAVICTI byste měl(a) každý den užívat.
• Vaše denní dávka bude záviset na Vaší velikosti a tělesné hmotnosti, množství bílkovin ve stravě a celkovém stavu poruchy cyklu močoviny.
• Pokud máte obtíže s ledvinami nebo játry, lékař Vám může stanovit nižší dávku.
• Budete muset podstoupit pravidelné krevní testy, aby lékař mohl stanovit správnou dávku.
• Lékař Vám může doporučit, abyste přípravek RAVICTI užíval(a) více než 3krát denně. U malých dětí to může být 4krát až 6krát denně. Mezi každou dávkou musí uplynout nejméně 3 hodiny.
Jak se tento přípravek užívá
Lékař vám oznámí, jak budete užívat přípravek RAVICTI perorální tekutina. Lze jej užívat následujícími způsoby:
• ústy,
• hadičkou, která prochází břichem do žaludku - nazývá se „gastrostomická sonda“,
• hadičkou, která prochází nosem do žaludku - nazývá se „nasogastrická sonda“.
Užívejte přípravek RAVICTI ústy, pokud Vám lékař neurčil jinak.
Přípravek RAVICTI s jídlem
Užívejte přípravek RAVICTI s jídlem nebo bezprostředně po něm. Malým dětem se lék podává během krmení nebo bezprostředně po něm.
Měření dávky
K odměření dávky používejte perorální stříkačku.
• Měl(a) byste mít lék s opakovatelně uzavíratelným adaptérem víčka lahve společně s perorální stříkačkou pro podání správné dávky glycerol-fenylbutyrátu.
1. Stlačením víčka směrem dolů a jeho otočením doleva otevřete lahev přípravku RAVICTI.
2. Našroubujte opakovatelně uzavíratelný adaptér víčka lahve na láhev.
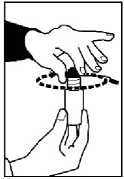
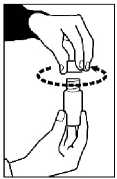
3. Špičku perorální stříkačky vsuňte do opakovatelně uzavíratelného adaptéru víčka lahve.
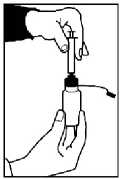
4. Lahev otočte dnem vzhuru i se vsunutou perorální stříkačkou.
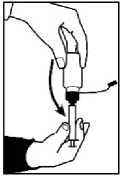
5. Naplňte do perorální stříkačky takové množství tekutiny glycerol-fenylbutyrátu, jaké Vám lékař řekl, že máte užívat, a to tak tak, že budete vytahovat píst, dokud se stříkačka tímto množstvím nenaplní.
- Poznámka: Pokud to bude možné, použijte velikost perorální stříkačky v ml, která je co nejbližší (ale ne menší než) doporučená dávka (například když bude dávka 0,8 ml, použijte 1ml perorální stříkačku).
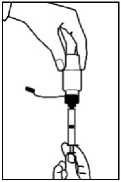
6. Poklepáním na perorální stříkačku odstraňte vzduchové bubliny, čímž zajistíte, že bude naplněna správným množstvím tekutiny.
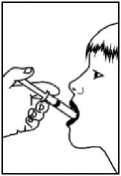
7. Spolkněte tekutinu z perorální stříkačky nebo připevněte perorální stříkačku ke gastrostomické nebo nasogastrické sondě.
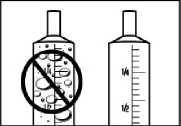
8. Nepřidávejte ani nemíchejte přípravek RAVICTI do větších objemů tekutiny, jako je voda nebo šťáva. Pokud se přípravek RAVICTI přidá do vody, tekutina se zakalí a možná nedostanete plnou dávku.
9. Přípravek RAVICTI lze přidat do malého množství měkkých potravin, jako je kečup, lékařské preparáty, jablečné pyré nebo kaše.
10. Pokud bude objem perorální stříkačky menší než předepsaná dávka, musíte tyto kroky opakovat, abyste dostal(a) celou dávku. Použijte jednu perorální stříkačku pro všechny dávky užívané jeden den.
11. Po užití plné dávky se napijte vody, aby bylo jisté, že Vám v ústech nezůstane žádný lék, nebo propláchněte gastrostomickou nebo nasogastrickou sondu 10 ml vody pomocí nové perorální stříkačky.
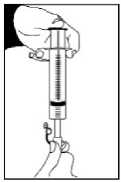
12. Zavřete kryt na opakovatelně uzavíratelném adaptéru víčka lahve.
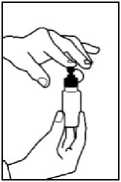
13. Neoplachujte opakovatelně uzavíratelný adaptér víčka lahve mezi jednotlivými použitími během dne, protože voda způsobí zakalený vzhled glycerol-fenylbutyrátu. Lahev a perorální stříkačku mezi dávkami uchovávejte na čistém, suchém místě.
14. Po poslední denní dávce perorální stříkačku zlikvidujte. Opakovatelně uzavíratelný adaptér víčka lahve lze používat, dokud lahev nebude prázdná. Když se otevře nová lahev, je zapotřebí použít nový opakovatelně uzavíratelnýadaptér víčka lahve.
15. Další stříkačky jsou součástí balíčku k zahájení léčby, pokud potřebujete denně více než jednu stříkačku. Zbývající stříkačky je třeba uchovat pro použití s další lahví. Každou lahev je třeba zlikvidovat po 3 dnech.
Jestliže jste užil(a) více přípravku RAVICTI, než jste měl(a)
Jestliže jste užil(a) více tohoto léku, než jste měl(a), informujte svého lékaře.
Jestliže si povšimnete kterékoliv z následujících známek, oznamte to svému lékaři nebo odejděte ihned
do nemocnice, protože se může jednat o známky předávkování nebo vysoké hladiny amoniaku:
• pocit ospalosti, únava, točení hlavy nebo někdy zmatenost
• bolest hlavy
• změny chuti
• obtíže se slyšením
• pocit ztráty orientace
• menší schopnost si zapamatovat věci
• stávající neurologická onemocnění se mohou zhoršit
Jestliže jste zapomněl(a) užít přípravek RAVICTI
Jestliže jste zapomněl užít dávku, vezměte si vynechanou dávku, co nejdříve si na to vzpomenete. Když však další dávka u dospělých bude za méně než 2 hodiny, pak ji přeskočte a vezměte si svoji další dávku jako obvykle.
Pro děti: pokud další dávka bude za méně než 30 minut, pak ji přeskočte a podejte další dávku jako obvykle.
• Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Jestliže jste přestal(a) užívat přípravek RAVICTI
Budete muset užít tento lék a dodržovat speciální dietu s nízkým obsahem bílkovin po celý svůj život. Nepřestávejte užívat glycerol-fenylbutyrát, aniž byste si o tom promluvit(a) se svým lékařem.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. U tohoto léku se mohou objevit následující nežádoucí účinky:
Závažné nežádoucí účinky
Neprodleně informujte svého lékaře, pokud:
• zvracíte, což je častý nežádoucí účinek, více než jedenkrát a zvracení neustává.
Jiné nežádoucí účinky
Informujte svého lékaře či lékárníka, pokud si povšimnete kteréhokoliv z následujících nežádoucích účinků:
Časté: mohou postihnout až 1 pacienta z 10
• nafouknuté nebo bolestivé břicho, zácpa, průjem, pálení žáhy, plynatost, nevolnost (pocit na zvracení), bolest v ústech, říhání
• otok rukou a nohou
• pocit únavy, pocit závratí, bolest hlavy nebo třes
• snížená nebo zvýšená chuť k j ídlu, nechuť j íst určité potraviny
• krvácení v období mezi menstruacemi, akné nenormální zápach kůže
• testy vykazují zvýšené hladiny jaterních enzymů, nerovnováha solí v krvi, nízké hladiny typu bílých krvinek („lymfocyty“) nebo nízké hladiny vitamínu D
Méně časté: mohou postihnout až 1 pacienta ze 100
• sucho v ústech, změna chuti k jídlu
• říhání, bolest břicha, změny stolice, například je olejovitá, naléhavá potřeba vyprázdnění střev, bolestivé střevní pohyby
• pocit hlavu, ztráta tělesné hmotnosti nebo nárůst tělesné hmotnosti
• zvýšená teplota, návaly horka
• bolest žlučníku, močového měchýře nebo zad
• otok kloubů, svalové spasmy, bolest rukou a nohou, bolest paty
• virová infekce ve střevech
• pocit mravenčení, pocit velkého neklidu, obtíže při probouzení se, pocit ospalosti, problémy s řečí, pocit zmatenosti, deprese
• zastavení menstruace nebo nepravidelné menstruace, hrubý hlas, krvácení z nosu, ucpaný nos, zanícené nebo bolavé hrdlo, ztráta vlasů, větší pocení než obvykle, svědivá vyrážka, nepravidelný srdeční tep, snížená funkce štítné žlázy
• testy ukazující vyšší nebo nižší hladinu draslíku v krvi
• testy ukazující vyšší obsah triglyceridů, lipoproteinů s nízkou hustotou nebo bílých krvinek v krvi
• testy ukazující abnormální EKG (elektrokardiogram)
• testy ukazující delší protrombinový čas
• testy ukazující nižší hodnotu albuminu v krvi
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek RAVICTI uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte přípravek RAVICTI po uplynutí doby použitelnosti uvedené na krabičce a štítku na lahvi za písmeny EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání. Jakmile bude lahev otevřená, musíte svůj lék použít do 3 dnů od otevření. Lahev je třeba zlikvidovat, i pokud není prázdná.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek RAVICTI obsahuje
• Léčivou látkou je glyceroli phenylbutyras.
• Jeden ml tekutiny obsahuje glyceroli phenylbutyras 1,1 g. To odpovídá hustotě 1,1 g/ml.
• Neobsahuje žádné další složky.
Jak přípravek RAVICTI vypadá a co obsahuje toto balení
Tekutina se plní do 25ml čiré skleněné lahve a je uzavřena plastovým dětským bezpečnostním víčkem.
Při zahájení léčby poskytne lékárna balíček k zahájení léčby obsahující 1 lahev přípravku RAVICTI, zavírací adaptér víčka lahve a 7 kusů perorálních stříkaček vhodné velikosti. Velikost í stříkačky (1 ml, 3 ml, 5 ml) bude uvedena ve Vašem přepisu. Pokud potřebujete více než jednu stříkačku denně, lékárna Vám vydá další stříkačky. Zbývající stříkačky je třeba uchovat pro použití s další lahví. Každou lahev je třeba zlikvidovat po 3 dnech. Po zahájení léčby lékárna poskytne standardní balíček obsahující 1 lahev přípravku RAVICTI a zavírací adaptér víčka lahve. Další stříkačky lze získat v lékárně.
Držitel rozhodnutí o registraci
Horizon Pharma Ireland Limited Connaught House, 1st Floor,
1 Burlington Road,
Dublin 4, D04C5Y6 Irsko
Výrobce
AndersonBrecon (UK) Limited Units 2-7,
Wye Valley Business Park,
Brecon Road,
Hay-on-Wye,
Hereford, Herefordshire, HR3 5PG Velká Británie
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
40