Protelos 2 G
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
V Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky.
Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
PROTELOS 2 g granule pro perorální suspenzi
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden sáček obsahuje strontii ranelas 2 g.
Pomocné látky se známým účinkem:
Jeden sáček obsahuje též 20 mg aspartamu (E951).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Granule pro perorální suspenzi. Žluté granule.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba závažné osteoporózy:
- u žen po menopauze,
- u dospělých mužů,
s vysokým rizikem fraktury, u kterých léčba jinými léčivými přípravky schválenými pro léčbu osteoporózy není možná z důvodu například kontraindikací nebo intolerance. U žen po menopauze stroncium-ranelát snižuje riziko vertebrálních a kyčelních fraktur (viz bod 5.1).
Rozhodnutí o předepsání stroncium-ranelátu musí být založeno na posouzení celkového rizika jednotlivého pacienta (viz body 4.3 a 4.4).
4.2 Dávkování a způsob podání
Léčba musí být zahájena pouze lékařem se zkušenostmi s léčbou osteoporózy.
Dávkování
Doporučená dávka je jeden 2g sáček jednou denně perorálně.
Vzhledem k povaze léčeného onemocnění je stroncium-ranelát určen k dlouhodobému užívání.
Absorpce stroncium-ranelátu se snižuje jídlem, mlékem a mléčnými výrobky, proto by měl být PROTELOS užíván v době mezi jídly. Vzhledem k pomalé absorpci by měl být PROTELOS užíván před spaním, nejlépe alespoň dvě hodiny po jídle (viz body 4.5 a 5.2).
Pacienti léčení stroncium-ranelátem by měli užívat doplňky vitaminu D a vápníku, pokud je jejich příjem stravou nedostatečný.
Starší pacienti
Účinnost a bezpečnost stroncium-ranelátu byla stanovena na širokém věkovém spektru (až do věku 100 let při zařazení) dospělých mužů a žen po menopauze trpících osteoporózou. Není nutná úprava dávky ve vztahu k věku.
Porucha funkce ledvin
Stroncium-ranelát se nedoporučuje u pacientů s těžkou poruchou funkce ledvin (clearance kreatininu pod 30 ml/min) (viz body 4.4 a 5.2). U pacientů s mírnou až středně těžkou poruchou funkce ledvin (clearance kreatininu 30-70 ml/min) není nutná úprava dávky (viz body 4.4 a 5.2).
Porucha funkce jater
U pacientů s poruchou funkce jater není nutná úprava dávky (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku PROTELOS u dětí ve věku do 18 let nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání Perorální podání.
Granule v sáčcích se užívají jako suspenze ve sklenici obsahující minimálně 30 ml vody (přibližně jedna třetina standardní sklenice).
Ačkoli studie přípravku po rekonstituci (přípravě suspenze) před použitím prokázaly, že je stroncium-ranelát stabilní v suspenzi po dobu 24 hodin po přípravě, suspenze by měla být vypita bezprostředně po přípravě.
4.3 Kontraindikace
- Hypersensitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
- Současné nebo předchozí venózní tromboembolické příhody (VTE), zahrnující hlubokou venózní trombózu a plicní embolii.
- Dočasná nebo trvalá imobilizace následkem např. pooperační rekonvalescence nebo dlouhodobého klidu na lůžku.
- Prokázané v současné době nebo v anamnéze některé z následujících onemocnění: ischemická choroba srdeční, onemocnění periferních tepen a/nebo cerebrovaskulární onemocnění.
- Hypertenze neupravená léčbou.
4.4 Zvláštní upozornění a opatření pro použití
Srdeční ischemické příhody
Z nashromážděných randomizovaných placebem kontrolovaných studií u postmenopauzálních pacientek s osteoporózou bylo zjištěno signifikantně zvýšené riziko infarktu myokardu u pacientek léčených přípravkem PROTELOS ve srovnání s pacientkami léčenými placebem (viz bod 4.8).
Před zahájením léčby by měli být pacienti zhodnoceni s přihlédnutím k možnému kardiovaskulárnímu riziku.
Pacienti s významnými rizikovými faktory pro kardiovaskulární příhody (např. hypertenze, hyperlipidémie, diabetes mellitus, kouření) musí být stroncium-ranelátem léčeni pouze po pečlivém zvážení (viz body 4.3 a 4.8).
Během léčby přípravkem PROTELOS musí být pravidelně monitorováno kardiovaskulární riziko, zpravidla každých 6 až 12 měsíců.
Léčba musí být ukončena v případě, že u pacienta dojde k rozvoji ischemické choroby srdeční, onemocnění periferních tepen, cerebrovaskulárního onemocnění nebo je-li hypertenze neupravená léčbou (viz bod 4.3).
Venózní tromboembolismus
V placebem kontrolovaných studiích III. fáze byla léčba stroncium-ranelátem spojována se zvýšením ročního výskytu venózního tromboembolismu (VTE), včetně plicní embolie (viz bod 4.8). Příčina tohoto nálezu není známa. PROTELOS je kontraindikován u pacientů s anamnézou venózních tromboembolických příhod (viz bod 4.3) a s opatrností by měl být užíván u pacientů s rizikem VTE. Při léčbě pacientů starších 80 let s rizikem VTE by mělo být přehodnoceno pokračování v léčbě přípravkem PROTELOS.
Léčba přípravkem PROTELOS musí být přerušena v případě nemoci nebo stavu vedoucímu k imobilizaci (viz bod 4.3) a musí být provedena adekvátní preventivní opatření. Léčba nesmí být znovu zahájena, dokud se počáteční stav nevyřeší a pacient nebude plně mobilní. Pokud se objeví VTE, léčba přípravkem PROTELOS musí být ukončena.
Použití u pacientů s poruchou funkce ledvin
Vzhledem k nepřítomnosti údajů o bezpečnosti pro kosti u pacientů s těžkou poruchou funkce ledvin léčených stroncium-ranelátem se PROTELOS nedoporučuje u pacientů s clearance kreatininu nižší než 30 ml/min (viz bod 5.2). V souladu se správnou lékařskou praxí se u pacientů s chronickou poruchou funkce ledvin doporučuje pravidelné zhodnocení funkce ledvin. Pokračování léčby přípravkem PROTELOS u pacientů s rozvíjející se těžkou poruchou funkce ledvin by mělo být zváženo individuálně.
Kožní reakce
Při užívání přípravku PROTELOS byly hlášeny život ohrožující kožní reakce (Stevens-Johnsonův syndrom (SJS), toxická epidermální nekrolýza (TEN) a léková vyrážka s eosinofilií a systémovými symptomy (DRESS)).
Pacienti by měli být poučeni o příznacích a projevech a měli by být pečlivě sledováni kvůli kožním reakcím. Nejvyšší riziko výskytu SJS nebo TEN je během prvních týdnů léčby a syndromu DRESS obvykle kolem 3 - 6 týdnů.
Léčba přípravkem PROTELOS musí být okamžitě ukončena, pokud se objeví příznaky nebo projevy SJS nebo TEN (např. progresivní kožní vyrážka, často s puchýři nebo slizničními lézemi) nebo DRESS (např. vyrážka, horečka, eozinofilie) a systémové příznaky (např. adenopatie, hepatitida, intersticiální nefropatie, intersticiální plicní onemocnění).
Včasná diagnóza a okamžité přerušení užívání přípravku se jeví jako nejlepší možnost, jak zvládat řešení SJS, TEN nebo DRESS. Včasné ukončení léčby je spojeno s lepší prognózou. Ve většině případů je výsledek DRESS příznivý po ukončení užívání přípravku PROTELOS a po zahájení léčby kortikosteroidy, pokud je to nutné. Uzdravení může být pomalé a v některých případech byla po ukončení léčby kortikosteroidy hlášena recidiva syndromu.
Pokud se u pacienta rozvinuly SJS, TEN nebo DRESS při užívání přípravku PROTELOS, nesmí být nikdy u tohoto pacienta znovu zahájena léčba přípravkem PROTELOS.
Byl hlášen vyšší výskyt, i když stále vzácný, hypersenzitivních reakcí včetně kožní vyrážky, SJS nebo TEN u pacientů asijského původu (viz bod 4.8).
V retrospektivní kontrolované farmakogenetické studii byly identifikovány alely HLA-A*33:03 a HLA-B*58:01 jako potenciální genetické rizikové faktory pro SJS/TEN spojené se stroncium-ranelátem u pacientů čínské etnické skupiny Chánové. U pacientů čínské etnické skupiny Chánové má být zvážen screening alel HLA-A*33:03 a HLA-B*58:01 před zahájením léčby přípravkem PROTELOS, pokud je to možné. Léčba přípravkem PROTELOS nemá být zahájena, pokud jsou testy pozitivní u jedné nebo obou alel. Přesto absence těchto alel v genotypu nevylučuje výskyt SJS/TEN.
Interakce s laboratorním vyšetřením
Stroncium interferuje s kolorimetrickými metodami ke stanovení koncentrací vápníku v krvi a moči. Proto by v lékařské praxi měla být k zajištění přesného stanovení koncentrací vápníku v krvi a moči používána indukční vazebná plazmatická atomová emisní spektrometrie nebo atomová absorpční spektrometrie.
Pomocné látky
PROTELOS obsahuje aspartam, zdroj fenylalaninu, což může být škodlivé pro osoby s fenylketonurií.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Jídlo, mléko a mléčné výrobky a léčivé přípravky obsahující vápník mohou snížit biologickou dostupnost stroncium-ranelátu přibližně o 60 -70 %. Proto by měl být udržován nejméně dvouhodinový odstup mezi podáním přípravku PROTELOS a těchto výrobků (viz body 4.2 a 5.2).
Jelikož dvojmocné kationty mohou na gastrointestinální úrovni vytvářet komplexy s perorálními tetracyklinovými (např. doxycyklin) a chinolonovými (např. ciprofloxacin) antibiotiky a tím snižovat jejich absorpci, nedoporučuje se současné podávání stroncium-ranelátu s těmito léčivými přípravky. Jako preventivní opatření by měla být léčba přípravkem PROTELOS přerušena během léčby perorálními tetracyklinovými nebo chinolonovými antibiotiky.
In vivo studie klinických interakcí ukázala, že podání hydroxidu hlinitého a hořečnatého buď dvě hodiny před nebo současně se stroncium-ranelátem způsobuje mírné snížení absorpce stroncium-ranelátu (20-25 % snížení AUC), kdežto absorpce byla prakticky neovlivněna, bylo-li antacidum podáno dvě hodiny po stroncium-ranelátu. Proto je vhodnější užívat antacida nejméně dvě hodiny po přípravku PROTELOS. Avšak je-li tento dávkovací režim nepraktický vzhledem k doporučenému podávání přípravku PROTELOS před spaním, je přijatelné i současné užívání.
Nebyly pozorovány žádné interakce s perorálními doplňky vitaminu D.
V klinických studiích nebyly zjištěny důkazy klinických interakcí nebo významného zvýšení krevních hladin stroncia s léčivými přípravky, u kterých se očekává, že budou běžně předepisovány současně
s přípravkem PROTELOS v cílové populaci. Tyto přípravky zahrnovaly: nesteroidní antiflogistika (včetně kyseliny acetylsalicylové), anilidy (jako paracetamol), H2 blokátory a inhibitory protonové pumpy, diuretika, digoxin a srdeční glykosidy, organické nitráty a jiná vazodilatancia k léčbě onemocnění srdce, blokátory kalciového kanálu, betablokátory, inhibitory ACE, antagonisty angiotenzinu II, selektivní agonisty beta-2 adrenoreceptorů, perorální antikoagulancia, inhibitory agregace trombocytů, statiny, fibráty a deriváty benzodiazepinu.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání stroncium-ranelátu těhotným ženám nejsou k dispozici.
Ve vysokých dávkách studie na zvířatech prokázaly reverzibilní účinky na kosti u potomků potkanů a králíků léčených během březosti (viz bod 5.3). Je-li PROTELOS omylem užit během těhotenství, léčba musí být ukončena.
Kojení
Fyzikálně-chemické údaje naznačují vylučování stroncium-ranelátu do lidského mateřského mléka. PROTELOS se nemá užívat během kojení.
Fertilita
Ve studiích na zvířatech nebyly pozorovány žádné účinky na fertilitu u samců a samic.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Stroncium-ranelát nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
PROTELOS byl studován v klinických studiích zahrnujících téměř 8000 účastnic. Dlouhodobá bezpečnost byla hodnocena u žen po menopauze s osteoporózou léčených až 60 měsíců stroncium-ranelátem 2 g/den (n=3352) nebo placebem (n=3317) ve studiích III. fáze. Průměrný věk byl 75 let při zařazení a 23 % zařazených pacientek bylo ve věku 80 až 100 let.
V nashromážděných analýzách z randomizovaných placebem kontrolovaných studií u postmenopauzálních pacientek s osteoporózou, byly nejčastějšími nežádoucími účinky nauzea a průjem, které byly obvykle zaznamenány na počátku léčby bez zaznamenatelného následného rozdílu mezi oběma skupinami. K vysazení léčby došlo zejména pro nauzeu. Nebyly zaznamenány rozdíly v povaze nežádoucích účinků mezi skupinami pacientek starších nebo mladších než 80 let při zařazení do studie.
Tabulkový přehled nežádoucích účinků
Následující nežádoucí účinky byly zaznamenány během klinických studií s přípravkem stroncium-ranelát a/nebo po uvedení přípravku na trh.
Nežádoucí účinky jsou uvedeny níže pomocí následujícího dělení: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000); velmi vzácné (<1/10000); není známo (z dostupných údajů nelze určit).
|
Třídy orgánových systémů |
Frekvence |
Nežádoucí účinek |
|
Poruchy krve a lymfatického systému |
Méně časté |
Lymfadenopatie (ve spojení s kožními hypersenzitivními reakcemi) |
|
Vzácné |
Selhání kostní dřeně# | |
|
Eosinofilie (ve spojení s kožními hypersenzitivními reakcemi) | ||
|
Poruchy metabolismu a výživy |
Časté |
Hypercholesterolémie |
|
Psychiatrické poruchy |
Časté | |
|
Méně časté |
Stav zmatenosti | |
|
Poruchy nervového systému |
Časté | |
|
Poruchy vědomí | ||
|
Ztráta paměti | ||
|
Závrať | ||
|
Parestezie | ||
|
Méně časté | ||
|
Poruchy ucha a labyrintu |
Časté | |
|
Srdeční poruchy |
Časté |
Infarkt myokardu |
|
Cévní poruchy |
Časté |
Venózní tromboembolismus (VTE) |
|
Respirační, hrudní a mediastinální poruchy |
Časté |
Bronchiální hyperreaktivita |
|
Gastrointestinální poruchy |
Časté | |
|
Průjem a řídká stolice | ||
|
Abdominální bolest | ||
|
Gastrointestinální bolest | ||
|
Gastroesofageální reflux | ||
|
Zácpa | ||
|
Plynatost | ||
|
Méně časté |
Iritace orální sliznice (stomatitida a/nebo ulcerace v ústech) | |
|
Sucho v ústech | ||
|
Poruchy jater a žlučových cest |
Časté |
Hepatitida |
|
Méně časté |
Zvýšené koncentrace sérových transamináz (ve spojení s kožními hypersenzitivními reakcemi) | |
|
Poruchy kůže a podkožní tkáně |
Velmi časté |
Kožní hypersenzitivní reakce (vyrážka, svědění, kopřivka, angioedém)§ |
|
Časté |
Ekzém | |
|
Méně časté | ||
|
Alopecie | ||
|
Vzácné |
Léková vyrážka s eosinofilií a systémovými symptomy (DRESS) (viz bod 4.4)# | |
|
Velmi vzácné |
Závažné kožní nežádoucí účinky: Stevens- |
*V asijských zemích hlášeny jako vzácné.
|
Johnsonův syndrom a toxická epidermální nekrolýza* (viz bod 4.4)# | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Velmi časté |
Muskuloskeletální bolest (svalové křeče, myalgie, bolest kostí, artralgie a bolest končetin)§ |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Periferní otok |
|
Méně časté |
Pyrexie (ve spojení s kožními hypersenzitivními reakcemi) | |
|
Vyšetření |
Časté |
Zvýšení hladiny kreatinfosfokinázy (CPK)a |
|
§ Četnost v klinických studiích by |
a podobná v lékové a placebové skupině. | |
# Pro nežádoucí účinky, které nebyly zaznamenány v klinických studiích, není horní hranice 95% intervalu spolehlivosti vyšší než 3/X, přičemž X vyjadřuje celkový zhodnocený vzorek ze všech relevantních klinických sledování a studií.
a Muskuloskeletální frakce > 3násobek horní hranice normálního rozmezí. Ve Vtšině případů se tyto hodnoty spontánně vrátily k normálu beze změny léčby.
Popis vybraných nežádoucích účinků
Venózní tromboembolismus
Ve studiích III. fáze byla roční incidence venózního tromboembolismu (VTE) pozorována během 5 let přibližně 0,7%, s relativním rizikem 1,4 (95% CI = [1,0 ; 2,0]) u pacientek léčených stroncium-ranelátem v porovnání s placebem (viz bod 4.4).
Infarkt myokardu
Z nashromážděných randomizovaných placebem kontrolovaných studií u postmenopauzálních pacientek s osteoporózou bylo zjištěno signifikantně zvýšené riziko infarktu myokardu u pacientek léčených stroncium-ranelátem ve srovnání s pacientkami léčenými placebem (1,7 % versus 1,1 %) s relativním rizikem 1,6 (95% CI = (1,07; 2,38).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Symptomy
V klinické studii zkoumající opakované podání 4 g stroncium-ranelátu denně po dobu 25 dní u zdravých žen po menopauze byla prokázána dobrá snášenlivost. Jednorázové podání dávek až 11 g zdravým mladým dobrovolníkům (mužům) nevyvolalo žádné zvláštní symptomy.
Léčba předávkování
Při epizodách předávkování v klinických studiích (až 4 g/den po dobu maximálně 147 dní) nebyly pozorovány žádné klinicky relevantní příhody.
Podávání mléka nebo antacid může pomoci snížit absorpci léčivé látky. V případě závažného předávkování je možno zvážit vyvolání zvracení k odstranění nevstřebané léčivé látky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii nemocí kostí - Jiná léčiva ovlivňující stavbu a mineralizaci kostí, ATC kód: M05BX03.
Mechanismus účinku
In vitro, stroncium-ranelát:
- zvyšuje kostní formaci v kulturách kostní tkáně a replikaci prekurzorů osteoblastů a syntézu kolagenu v kulturách kostních buněk.
- snižuje resorpci kostí snížením diferenciace osteoklastů a resorpční aktivity.
Toto vede ke znovunastolení rovnováhy kostního obratu ve prospěch kostní formace.
Aktivita stroncium-ranelátu byla studována na různých neklinických modelech. Zvláště u zdravých potkanů zvyšuje stroncium-ranelát hmotu trámčité kosti, počet trámců a jejich tloušťku; to vede ke zvýšení pevnosti kostí.
V kostní tkáni léčených zvířat a lidí se stroncium adsorbuje zejména na povrch krystalů a jen mírně nahrazuje vápník v apatitových krystalech nově vytvořené kosti. Stroncium-ranelát nemění vlastnosti kostních krystalů. V biopsiích ze hřebenu kyčelní kosti odebraných po 60 měsících léčby stroncium-ranelátem 2 g/den ve studiích III. fáze nebyly pozorovány žádné škodlivé účinky na kvalitu nebo mineralizaci kostí.
Kombinované účinky distribuce stroncia v kosti (viz bod 5.2) a zvýšení RTG absorpce stroncia v porovnání s vápníkem vede ke zvýšení denzity kostního minerálu (BMD) měřené duální fotonovou rentgenovou absorpciometrií (DXA). Dostupné údaje ukazují, že tyto faktory zodpovídají za přibližně 50 % naměřené změny BMD během 3 let léčby přípravkem PROTELOS 2 g/den. To je třeba mít na paměti při interpretaci změn BMD při léčbě přípravkem PROTELOS. Ve studiích III. fáze, které prokázaly účinnost léčby přípravkem PROTELOS proti frakturám, se naměřená průměrná BMD v porovnání s počátečním stavem zvýšila s přípravkem PROTELOS o přibližně 4 % ročně na bederní páteři a 2 % ročně na krčku stehenní kosti, a po 3 letech se tyto hodnoty zvýšily na 13 až 15 % a 5 až 6 % v závislosti na studii.
Ve studiích III. fáze se biochemické markery kostní formace (kostní specifická alkalická fosfatáza a C-terminální propeptid prokolagenu typu I) ve srovnání s placebem zvyšovaly a markery kostní resorpce (příčné vazby sérového C-telopeptidu a močového N-telopeptidu) snižovaly od třetího měsíce léčby až do 3 let.
Sekundárně k farmakologickým účinkům stroncium-ranelátu bylo pozorováno mírné snížení sérových koncentrací vápníku a parathormonu (PTH), zvýšení krevních koncentrací fosforu a aktivity celkové alkalické fosfatázy, bez pozorovaných klinických důsledků.
Klinická, účinnost.
Osteoporóza je definována jako BMD na páteři či kyčli nižší o 2,5 SD nebo více než průměrná hodnota normální mladé populace. S postmenopauzální osteoporózou je spojováno mnoho rizikových faktorů zahrnujících nízkou hodnotu kostní hmoty, nízkou denzitu kostního minerálu, ranou menopauzu, anamnézu kouření a rodinnou anamnézu osteoporózy. Klinickým důsledkem osteoporózy je fraktura. Riziko faktur se zvyšuje s počtem rizikových faktorů.
Léčba postmenopauzální osteoporózy:
Program studií proti frakturám s přípravkem PROTELOS byl vytvořen ze dvou placebem kontrolovaných studií III. fáze: studie SOTI a studie TROPOS. Do studie SOTI bylo zařazeno 1649 postmenopauzálních žen s diagnostikovanou osteoporózou (nízká bederní BMD a prodělaná vertebrální fraktura) a průměrným věkem 70 let. Do studie TROPOS bylo zařazeno 5091 postmenopauzálních žen s osteoporózou (nízká BMD krčku stehenní kosti a prodělaná fraktura u více než poloviny z nich) a průměrným věkem 77 let. Do studie SOTI a TROPOS bylo dohromady zařazeno 1556 pacientek starších 80 let při zařazení (23,1 % studované populace). V obou studiích pacientky dostávaly jako přídavek k léčbě (2 g/den stroncium-ranelát nebo placebo) upravené doplňky vápníku a vitaminu D.
PROTELOS snížil relativní riziko nové vertebrální fraktury o 41 % za 3 roky ve studii SOTI (tabulka 1). Účinek byl signifikantní od prvního roku. Podobné přínosy byly prokázány u žen s více frakturami
na počátku. Vzhledem ke klinickým vertebrálním frakturám (definovaných jako fraktury související s bolestí zad a/nebo poklesem tělesné výšky o nejméně 1 cm) bylo relativní riziko sníženo o 38 %. PROTELOS také snížil počet pacientek s poklesem tělesné výšky o nejméně 1 cm v porovnání s placebem. Hodnocení kvality života na specifické stupnici QUALIOST a skóre vnímání celkového zdravotního stavu obecné stupnice SF-36 ukázalo přínos přípravku PROTELOS ve srovnání s placebem.
Účinnost přípravku PROTELOS na snížení rizika nové vertebrální fraktury byla potvrzena ve studii TROPOS, včetně pacientek s osteoporózou bez fraktury z důvodu křehkosti kosti na počátku.
Tabulka 1: Výskyt pacientek s vertebrální frakturou a snížení relativního rizika
|
Studie |
Placebo |
PROTELOS |
Snížení relativního rizika vs. placebo (95% CI), hodnota p |
|
SOTI |
n=723 |
n=719 | |
|
Nová vertebrální fraktura během 3 let |
32,8 % |
20,9 % |
41 % (27-52), p<0,001 |
|
Nová vertebrální fraktura během 1. roku |
11,8 % |
6,1 % |
49 % (26-64), p<0,001 |
|
Nová klinická vertebrální fraktura během 3 let |
17,4 % |
11,3 % |
38 % (17-53), p<0,001 |
|
TROPOS |
n=1823 |
n=1817 | |
|
Nová vertebrální fraktura během 3 let |
20,0 % |
12,5 % |
39 % (27-49), p<0,001 |
U pacientek starších než 80 let při zařazení ukázala sdružená analýza studií SOTI a TROPOS, že PROTELOS snížil relativní riziko výskytu nové vertebrální fraktury o 32 % během 3 let (výskyt
19,1 % u stroncium-ranelátu vs. 26,5 % u placeba).
Při následné analýze pacientek ze sdružených studií SOTI a TROPOS s počáteční BMD bederní páteře a / nebo krčku stehenní kosti v osteopenickém rozmezí a bez prodělané fraktury, ale s nejméně jedním rizikovým faktorem pro frakturu navíc (n=176) PROTELOS snížil riziko první vertebrální fraktury o 72 % během 3 let (výskyt vertebrální fraktury 3,6 % u stroncium-ranelátu vs. 12,0 % u placeba).
Následná analýza byla provedena na podskupině pacientek ze studie TROPOS obzvláště důležité z medicínského hlediska a s vysokým rizikem fraktury [definovaným jako T-skóre BMD krčku stehenní kosti < -3 SD (rozmezí výrobce odpovídající -2,4 SD podle NHANES III) a ve věku > 74 let (n=1977, tj. 40 % populace studie TROPOS)]. V této skupině snížil PROTELOS během 3 let léčby riziko fraktury kyčle o 36 % v porovnání se skupinou léčenou placebem (tabulka 2).
|
Tabulka 2: Výskyt pacientek s frakturou kyčle a BMD < -2,4 SD (NHANES III) a věkem > 74 let |
snížení relativního |
rizika u pacientek s | |
|
Studie |
Placebo |
PROTELOS |
Snížení relativního rizika vs. placebo (95% CI), hodnota p |
|
TROPOS |
n=995 |
n=982 | |
|
Fraktura kyčle během 3 let |
6,4 % |
4,3 % |
36 % (0-59), p=0,046 |
Léčba osteoporózy u mužů:
Účinnost přípravku PROTELOS byla prokázána u mužů s osteoporózou ve dvouleté, dvojitě zaslepené, placebem kontrolované studii s hlavní analýzou po jednom roce léčby u 243 pacientů (populace intention to treat, 161 pacientů užívalo stroncium-ranelát) s vysokým rizikem zlomeniny (průměrný věk 72,7 let; průměrná hodnota BMD bederní páteře: T-skóre -2,6; prevalentní vertebrální fraktura u 28 % pacientů).
Všichni pacienti byli suplementováni vápníkem (1000 mg/den) a vitaminem D (800 IU/den).
Statisticky signifikantní nárůst BMD byl pozorován již 6 měsíců po zahájení léčby přípravkem PROTELOS versus placebo.
Po 12 měsících léčby byl pozorován statisticky signifikantní nárůst průměrné BMD bederní páteře, hlavního kritéria účinnosti (E (SE) = 5,32 % (0,75); 95%CI = (3,86; 6,79); p <0,001), podobně jak bylo pozorováno v pivotní studii III. fáze na snížení rizika fraktur, provedené u postmenopauzálních žen.
Po 12 měsících léčby byl pozorován statisticky signifikantní nárůst hodnoty BMD krčku stehenní kosti a BMD celkové oblasti proximálního femuru (total hip) (p<0,001).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem PROTELOS u všech podskupin pediatrické populace v indikaci osteoporózy (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Stroncium-ranelát je složen ze 2 atomů stabilního stroncia a 1 molekuly kyseliny ranelové, což je organická část umožňující nejlepší kompromis z hlediska molekulární hmotnosti, farmakokinetiky a snášenlivosti léčivého přípravku. Farmakokinetika stroncia a kyseliny ranelové byla hodnocena na zdravých mladých mužích a zdravých ženách po menopauze, a zároveň při dlouhodobé expozici u mužů s osteoporózou a u žen po menopauze s osteoporózou, včetně starých žen.
Absorpce, distribuce a vazba na plazmatické bílkoviny kyseliny ranelové je z důvodu její vysoké polarity nízká. Nedochází ke kumulaci kyseliny ranelové a nebyl prokázán její metabolismus u zvířat ani u člověka. Absorbovaná kyselina ranelová je rychle eliminována v nezměněné formě ledvinami.
Absorpce
Absolutní biologická dostupnost stroncia je přibližně 25 % (rozmezí 19-27 %) po perorální dávce 2 g stroncium-ranelátu. Maximálních plazmatických koncentrací je dosaženo za 3-5 hodin po jednorázové dávce 2 g. Rovnovážného stavu je dosaženo po 2 týdnech léčby. Užití stroncium-ranelátu společně s vápníkem nebo jídlem snižuje biologickou dostupnost stroncia přibližně o 60-70 % ve srovnání s podáním 3 hodiny po jídle. Vzhledem k relativně pomalé absorpci stroncia je třeba vyhnout se podání jídla a vápníku před a po podání přípravku PROTELOS. Perorální doplňky vitaminu D nemají vliv na expozici stronciu.
Distribuce v organismu
Stroncium má distribuční objem přibližně 1 l/kg. Vazba stroncia na plazmatické bílkoviny u člověka je nízká (25 %) a stroncium má vysokou afinitu ke kostní tkáni. Měření koncentrace stroncia v biopsiích ze hřebenu kyčelní kosti u pacientek léčených stroncium-ranelátem 2 g/den až 60 měsíců ukazuje, že koncentrace stroncia v kosti mohou dosáhnout stabilní hladiny přibližně po 3 letech léčby. Neexistují údaje od pacientů, které by prokazovaly kinetiku eliminace stroncia z kostí po léčbě.
Biotransformace
Jakožto dvojmocný kation není stroncium metabolizováno. Stroncium-ranelát neinhibuje enzymy cytochromu P450.
Eliminace z organismu
Eliminace stroncia je nezávislá na čase a dávce. Skutečný poločas stroncia je přibližně 60 hodin.
K vylučování stroncia dochází ledvinami a gastrointestinálním traktem. Jeho plazmatická clearance je přibližně 12 ml/min (CV 22 %) a jeho renální clearance je přibližně 7 ml/min (CV 28 %).
Farmakokinetika ve zvláštních populacích
Starší pacienti
Populační farmakokinetické údaje neukázaly žádný vztah mezi věkem a domnělou clearance stroncia v cílové populaci.
Porucha funkce ledvin
U pacientů s mírnou až středně těžkou poruchou funkce ledvin (clearance kreatininu 30-70 ml/min) se clearance stroncia snižuje se snížením clearance kreatininu (přibližně 30% snížení v rozmezí clearance kreatininu 30 až 70 ml/min), což vede ke zvýšení plazmatických hladin stroncia. Ve studiích III. fáze mělo 85 % pacientek clearance kreatininu v rozmezí 30 až 70 ml/min a 6 % pod 30 ml/min při zařazení, a průměrná clearance kreatininu byla přibližně 50 ml/min. Proto není u pacientů s mírnou až středně těžkou poruchou funkce ledvin nutná úprava dávkování.
Neexistují farmakokinetické údaje u pacientů s těžkou poruchou funkce ledvin (clearance kreatininu pod 30 ml/min).
Porucha funkce jater
Neexistují farmakokinetické údaje u pacientů s poruchou funkce jater. Vzhledem k farmakokinetickým vlastnostem stroncia se neočekává žádný vliv.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, genotoxicity a hodnocení kancerogenního potenciálu neodhalily žádné zvláštní riziko pro člověka.
Chronické perorální podávání stroncium-ranelátu ve vysokých dávkách u hlodavců způsobilo abnormality kostí a zubů, spočívající zejména ve spontánních frakturách a zpožděné mineralizaci, které byly reverzibilní po ukončení léčby. Tyto účinky byly zaznamenány při hladinách stroncia v kostech, které byly 2-3krát vyšší než hladiny stroncia v kostech u lidí léčených po dobu až 3 let. Údaje ohledně akumulace stroncium-ranelátu ve skeletu při delší expozici jsou omezené.
Studie vývojové toxicity u potkanů a králíků vedly k abnormalitám kostí a zubů (např. ohnutí dlouhých kostí a zvlněná žebra) u potomků. U potkanů byly tyto účinky reverzibilní 8 týdnů po ukončení léčby.
Posouzení rizika pro životní prostředí (ERA)
Posouzení rizika stroncium-ranelátu pro životní prostředí bylo provedeno v souladu s Evropskými doporučeními ERA.
Stroncium-ranelát životní prostředí neohrožuje.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Aspartam (E951)
Maltodextrin Mannitol (E421)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
- 3 roky.
- Po rekonstituci přípravku ve vodě je suspenze stabilní po dobu 24 hodin. Nicméně, suspenze by měla být vypita bezprostředně po přípravě (viz bod 4.2).
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Papír/polyethylen/Al/polyethylenové sáčky.
Velikost balení
Krabičky obsahující 7, 14, 28, 56, 84 nebo 100 sáčků. Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
LES LABORATOIRES SERVIER 50, rue Carnot 92284 Suresnes cedex Francie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/288/001
EU/1/04/288/002
EU/1/04/288/003
EU/1/04/288/004
EU/1/04/288/005
EU/1/04/288/006
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. září 2004
Datum posledního prodloužení registrace: 22. května 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ /VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Les Laboratoires Servier Industrie 905, route de Saran, 45520 Gidy, Francie Przedsiebiorstwo Farmaceutyczne, ANPHARM S.A., ul Annopol 6B-03-236, Warszawa, Polsko
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Periodicky aktualizovaná zpráva o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Dále je třeba aktualizovaný RMP předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
Popis_
Neintervenční studie bezpečnosti k vyhodnocení efektivity aplikovaných opatření k minimalizaci rizika, zahrnující popis léčené populace pacientů v každodenní klinické praxi, metody použití a kardiovaskulární riziko.
Po schválení protokolu by měla být poskytována roční hlášení z této studie v rámci PSUR až do předložení závěrečné zprávy ze studie, která je očekávána do prosince 2017._
Další opatření k minimalizaci rizik
V každém členském státě, kde je PROTELOS na trhu, by se měl držitel rozhodnutí o registraci dohodnout s národním regulačním úřadem na finálním edukačním programu.
Po diskuzi a dohodě s národním regulačním úřadem v každém členském státě, kde je PROTELOS na trhu, držitel rozhodnutí o registraci zajistí, že bude všem lékařům, u kterých se předpokládá, že budou předepisovat PROTELOS, poskytnut tento edukační balíček:
• Souhrn údajů o přípravku
• Příbalová informace
• Příručka k předepisování a kontrolní seznam
• Karta s upozorněním pro pacienta
Příručka k předepisování a kontrolní seznam by měly obsahovat následující významná sdělení:
• PROTELOS je indikován pouze pro použití u pacientů se závažnou osteoporózou s vysokým rizikem fraktury, u kterých léčba jinými léčivými přípravky schválenými pro léčbu osteoporózy není možná z důvodu například kontraindikací nebo intolerance.
• Zahájení léčby přípravkem PROTELOS musí být založeno na zhodnocení celkového rizika každého pacienta.
• Všichni pacienti mají být informováni, že kardiovaskulární riziko musí být monitorováno
pravidelně každých 6 -12 měsíců.
• Každému pacientovi je třeba předat kartu s upozorněním pro pacienta.
• PROTELOS je kontraindikován a nesmí být užíván u pacientů:
o S prokázaným v současné době nebo v anamnéze některým z následujících onemocnění: ischemická choroba srdeční, onemocnění periferních tepen a/nebo cerebrovaskulární onemocnění.
o S hypertenzí neupravenou léčbou.
o S venózní tromboembolickou příhodou (VTE) v současné době nebo v anamnéze, zahrnující hlubokou žilní trombózu a plicní embolii.
o S dočasnou nebo trvalou imobilizací následkem např. pooperační rekonvalescence nebo dlouhodobého klidu na lůžku.
o S hypersenzitivitou na léčivou látku (stroncium-ranelát) nebo na kteroukoli pomocnou látku.
• PROTELOS by měl být užíván pouze s opatrností u:
o Pacientů s významnými rizikovými faktory pro kardiovaskulární příhody, jako je hypertenze, hyperlipidémie, diabetes mellitus nebo kouření.
o Pacientů s rizikem VTE. Při léčbě pacientů starších 80 let s rizikem VTE by mělo být přehodnoceno pokračování v léčbě přípravkem PROTELOS.
• Léčba by měla být přerušena nebo ukončena v následujících situacích:
o Pokud u pacienta dojde k rozvoji ischemické choroby srdeční, onemocnění periferních tepen, cerebrovaskulárního onemocnění nebo je-li hypertenze neupravená léčbou, léčba musí být zastavena.
o Léčba musí být přerušena co nejdříve v případě nemoci nebo stavu vedoucímu k imobilizaci.
o Léčba přípravkem PROTELOS musí být okamžitě ukončena, pokud se objeví příznaky nebo projevy Stevens-Johnsonova syndromu (SJS), toxické epidermální nekrolýzy (TEN) nebo lékové vyrážky s eosinofilií a systémovými symptomy (DRESS) (např. vyrážka, horečka, eozinofilie a systémové příznaky např. adenopatie, hepatitida, intersticiální nefropatie, intersticiální plicní onemocnění). Pokud se u pacienta rozvinuly SJS, TEN nebo DRESS při užívání přípravku PROTELOS, nesmí být nikdy u tohoto pacienta znovu zahájena léčba přípravkem PROTELOS.
• Součástí příručky k předepisování bude kontrolní seznam, který bude předepisujícím lékařům připomínat kontraindikace, upozornění a opatření před předepsáním a podpoří pravidelné monitorování kardiovaskulárního rizika.
Karta s upozorněním pro pacienta by měla obsahovat následující významná sdělení:
• Důležitost ukázat kartu s upozorněním pro pacienta každému zdravotnickému odborníkovi, který tyto pacienty léčí.
• Kontraindikace pro léčbu přípravkem PROTELOS.
• Významné příznaky a projevy infarktu myokardu, VTE a závažných kožních reakcí.
• Kdy vyhledat urgentní lékařskou pomoc.
• Důležitost pravidelného monitorování kardiovaskulárního rizika.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
PROTELOS 2 g granule pro perorální suspenzi Strontii ranelas
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jeden sáček obsahuje strontii ranelas 2 g.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje též aspartam (E 951).
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Granule pro perorální suspenzi. 7 sáčků
5. ZPŮSOB A CESTA PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Pokud není užito okamžitě po rekonstituci, přípravek by měl být spotřebován do 24 hodin.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Les Laboratoires Servier 50, rue Carnot 92284 Suresnes cedex Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/04/288/001
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
PROTELOS 2 g granule pro perorální suspenzi Strontii ranelas
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jeden sáček obsahuje strontii ranelas 2 g.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje též aspartam (E 951).
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Granule pro perorální suspenzi. 14 sáčků
5. ZPŮSOB A CESTA PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.

Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Pokud není užito okamžitě po rekonstituci, přípravek by měl být spotřebován do 24 hodin.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Les Laboratoires Servier 50, rue Carnot 92284 Suresnes cedex Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/04/288/002
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
PROTELOS 2 g granule pro perorální suspenzi Strontii ranelas
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jeden sáček obsahuje strontii ranelas 2 g.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje též aspartam (E 951).
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Granule pro perorální suspenzi. 28 sáčků
5. ZPŮSOB A CESTA PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Pokud není užito okamžitě po rekonstituci, přípravek by měl být spotřebován do 24 hodin.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Les Laboratoires Servier 50, rue Carnot 92284 Suresnes cedex Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/04/288/003
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16 INFORMACE V BRAILLOVĚ PÍSMU
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
PROTELOS 2 g granule pro perorální suspenzi Strontii ranelas
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jeden sáček obsahuje strontii ranelas 2 g.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje též aspartam (E 951).
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Granule pro perorální suspenzi. 56 sáčků 84 sáčků 100 sáčků
5. ZPŮSOB A CESTA PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.

Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Pokud není užito okamžitě po rekonstituci, přípravek by měl být spotřebován do 24 hodin.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Les Laboratoires Servier 50, rue Carnot 92284 Suresnes cedex Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/04/288/004 56 sáčků EU/1/04/288/005 84 sáčků (3 balení po 28) EU/1/04/288/006 100 sáčků
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
PROTELOS 2 g
17. JEDINEČNÝ IDENFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU Sáček
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
PROTELOS 2 g granule pro perorální suspenzi Strontii ranelas
Perorální podání.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
c.s.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Před použitím si přeCtěte příbalovou informaci.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
PROTELOS 2 g - granule pro perorální suspenzi
Strontii ranelas
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je PROTELOS a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete PROTELOS užívat
3. Jak se PROTELOS užívá
4. Možné nežádoucí účinky
5 Jak PROTELOS uchovávat
6. Obsah balení a další informace
1. Co je PROTELOS a k čemu se používá
PROTELOS je přípravek užívaný k léčbě závažné osteoporózy:
- u žen po menopauze,
- u dospělých mužů,
s vysokým rizikem zlomeniny, u kterých jiná alternativní léčba není možná. U žen po menopauze stroncium-ranelát snižuje riziko zlomeniny páteře a kyčle.
O osteoporóze
Vaše tělo neustále rozkládá starou kost a vytváří novou kostní tkáň. Pokud máte osteoporózu, Vaše tělo rozkládá více kosti, než vytváří, takže postupně dochází k úbytku kosti a Vaše kosti se stanou řidšími a křehkými. Zvláště běžné je to u žen po menopauze.
Mnoho lidí s osteoporózou nemá žádné příznaky a dokonce ani nemusí vědět, že jí trpí. Osteoporóza však zvyšuje pravděpodobnost, že dojde k fraktuře (zlomenině kosti), zejména páteře, kyčle a zápěstí.
Jak PROTELOS působí
PROTELOS, který obsahuje léčivou látku stroncium-ranelát, patří do skupiny léků používaných k léčbě kostních onemocnění.
PROTELOS působí tak, že snižuje rozpad kosti a podporuje přestavbu kosti, čímž snižuje riziko zlomeniny. Nově vytvořená kost má normální kvalitu.
2. Čemu musíte věnovat pozornost, než začnete PROTELOS užívat Neužívejte PROTELOS
- jestliže jste alergický(á) na stroncium-ranelát nebo na kteroukoliv další složku přípravku PROTELOS (uvedenou v bodě 6).
- jestliže máte nebo jste měl(a) krevní sraženinu (např. v krevních cévách ve Vašich končetinách nebo v plicích).
- jestliže jste trvale nepohyblivý(á) nebo na nějaký čas vázán(a) na invalidní vozík, nebo upoután(a) na lůžko, nebo jestliže máte podstoupit operaci nebo se zotavujete po operaci. Riziko žilní trombózy (krevní sraženina v končetinách nebo v plicích) může být zvýšeno v případě delší nepohyblivosti.
- jestliže máte prokázanou ischemickou chorobu srdeční nebo cerebrovaskulární onemocnění, například byl u Vás diagnostikován srdeční infarkt, cévní mozková příhoda (mrtvice) nebo transitorní ischemická ataka (dočasné omezení krevního přítoku do mozku; také znám jako „malá cévní mozková příhoda“), angina pectoris nebo ucpání krevních cév do srdce nebo do mozku.
- jestliže máte nebo jste měl(a) problémy s krevním oběhem (onemocnění periferních tepen) nebo jestliže jste prodělal(a) chirurgický zákrok na tepnách nohou.
- jestliže máte vysoký krevní tlak neupravený léčbou.
Upozornění a opatření
Před použitím přípravku PROTELOS se poraďte se svým lékařem nebo lékárníkem:
- jestliže jste ohrožen(a) srdečním onemocněním, tj. máte vysoký krevní tlak, vysokou hladinu cholesterolu, cukrovku, kouříte,
- jestliže jste ohrožen(a) krevními sraženinami,
- jestliže trpíte závažným onemocněním ledvin.
Váš lékař bude pravidelně hodnotit stav Vašeho srdce a krevních cév, zpravidla každých 6 až 12 měsíců, po dobu, kdy budete užívat přípravek PROTELOS.
Jestliže se během léčby objeví alergická reakce (jako je otok obličeje, jazyka nebo hrdla, obtíže s dýcháním nebo s polykáním, kožní vyrážka), musíte ihned přestat užívat PROTELOS a vyhledat lékařskou pomoc (viz bod 4).
Při užívání přípravku PROTELOS byly hlášeny potenciálně život ohrožující kožní vyrážky (Stevens-Johnsonův syndrom, toxická epidermální nekrolýza a závažné reakce přecitlivělosti (DRESS)). Nejvyšší riziko výskytu závažných kožních reakcí Stevens-Johnsonova syndromu a toxické epidermální nekrolýzy je během prvních týdnů léčby a syndromu DRESS obvykle kolem 3 - 6 týdnů. Pokud se u Vás rozvine vyrážka nebo závažné kožní příznaky (viz bod 4), přestaňte užívat PROTELOS, vyhledejte okamžitě lékaře a řekněte mu, že užíváte tento lék.
Pokud se u Vás rozvinuly Stevens-Johnsonův syndrom nebo toxická epidermální nekrolýza nebo DRESS při užívání přípravku PROTELOS, nesmíte nikdy znovu zahájit léčbu přípravkem PROTELOS.
Jestliže máte asijský původ, můžete mít vyšší riziko výskytu kožních reakcí.
Riziko těchto kožních reakcí lze předpokládat u pacientů asijského původu, zejména u čínské etnické skupiny Chánové. U pacientů s geny HLA-A*33:03 a/nebo HLA-B*58:01 dojde pravděpodobněji ke vzniku závažné kožní reakce než u těch, kteří tyto geny nemají.
Lékař má zvážit, zda provést krevní testy před užitím přípravku PROTELOS.
Děti a dospívající
PROTELOS není určen pro použití u dětí a dospívajících (do 18 let).
Další léčivé přípravky a přípravek PROTELOS
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Pokud musíte užívat perorální tetracykliny jako doxycyklin nebo chinolony jako je ciprofloxacin (dva typy antibiotik), měl(a) byste ukončit užívání přípravku PROTELOS. Jakmile ukončíte užívání těchto antibiotik, můžete opět začít užívat PROTELOS. Pokud si nejste jistý(á), zeptejte se svého lékaře nebo lékárníka.
Pokud užíváte léky obsahující vápník, měl(a) byste počkat alespoň 2 hodiny, než si vezmete PROTELOS.
Pokud užíváte antacida (léky ke zmírnění pálení žáhy), měl(a) byste je užívat nejméně 2 hodiny po přípravku PROTELOS. Není-li to možné, je přijatelné užít tyto dva léky najednou.
Pokud potřebujete provedení krevních testů nebo zkoušku moči kvůli kontrole hladiny vápníku, měl(a) byste oznámit laboratoři, že užíváte PROTELOS, protože užívání může zasahovat do některých testovacích metod.
Přípravek PROTELOS s jídlem a pitím
Jídlo, mléko a mléčné výrobky snižují vstřebání stroncium-ranelátu. Doporučuje se, abyste užíval(a) PROTELOS mezi jídly, nejlépe před spaním nejméně dvě hodiny po jídle, mléce, mléčných výrobcích nebo doplňcích vápníku.
Těhotenství a kojení
Neužívejte PROTELOS v době těhotenství nebo pokud kojíte. Pokud jste jej nedopatřením užila během těhotenství nebo kojení, jeho užívání okamžitě ukončete a poraďte se s lékařem.
Řízení dopravních prostředků a obsluha strojů
Je nepravděpodobné, že by PROTELOS ovlivňoval schopnost řídit nebo obsluhovat stroje. PROTELOS obsahuje aspartam (E951)
Pokud trpíte fenylketonurií (vzácné, dědičné onemocnění látkové přeměny), poraďte se před zahájením léčby tímto přípravkem s lékařem.
3. Jak se PROTELOS užívá
Léčba musí být zahájena pouze lékařem se zkušenostmi s léčbou osteoporózy.
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
PROTELOS je určen k perorálnímu podání (užití ústy).
Doporučená dávka je jeden 2g sáček denně.
Doporučuje se, abyste užíval(a) PROTELOS před spaním, nejlépe nejméně 2 hodiny po večeři. Pokud chcete, tak si po užití přípravku PROTELOS můžete hned lehnout.
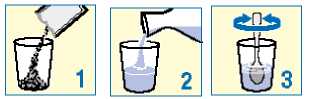
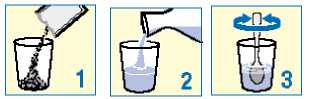
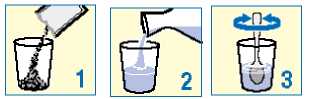
Granule obsažené v sáčcích užívejte jako suspenzi ve sklenici obsahující minimálně 30 ml (přibližně jednu třetinu běžné sklenice) vody. Viz návod uvedený níže. PROTELOS může reagovat s mlékem a mléčnými výrobky, je proto důležité, abyste PROTELOS k zajištění správného účinku míchal(a) jenom s vodou.


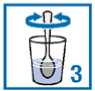
Vysypte granule ze sáčku do sklenice;
Přidejte vodu;
Míchejte, dokud se všechny granule nerozptýlí ve vodě.
Poté hned vypijte. Před vypitím nesmíte suspenzi nechat stát déle než 24 hodin. Pokud z nějakého důvodu nemůžete lék vypít ihned, musíte jej před vypitím znovu promíchat.
Váš lékař Vám může poradit, abyste společně s přípravkem PROTELOS užíval(a) doplňky vápníku a vitaminu D. Neužívejte doplňky vápníku před spaním, ve stejné době jako PROTELOS.
Váš lékař Vám řekne, jak dlouho máte pokračovat v léčbě přípravkem PROTELOS. Léčba osteoporózy obvykle vyžaduje dlouhou dobu. Je důležité, abyste v užívání přípravku PROTELOS pokračoval(a) tak dlouho, jak Vám lékař předepsal.
Jestliže jste užil(a) více přípravku PROTELOS, než jste měl(a)
Pokud užijete více sáčků přípravku PROTELOS, než je doporučeno Vaším lékařem, informujte svého lékaře nebo lékárníka. Ti Vám mohou doporučit, abyste vypil(a) mléko nebo užila nějaké antacidum ke snížení vstřebávání léčivé látky.
Jestliže jste zapomněl(a) užít PROTELOS
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Jednoduše pokračujte s další dávkou v obvyklém čase.
Jestliže přestanete užívat PROTELOS
Je důležité, že užíváte PROTELOS po dobu, jakou Vám předepsal Váš lékař. PROTELOS může léčit Vaši závažnou osteoporózu, pouze pokud pokračujete v užívání přípravku.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Pokud se u Vás objeví následující nežádoucí účinky, přestaňte užívat PROTELOS a kontaktujte
okamžitě svého lékaře:
Časté (mohou postihnout až 1 z 10 lidí):
- Srdeční infarkt: náhlé tlakové bolesti na hrudi, které mohou vystřelovat do levé paže, čelisti, žaludku, zad a/nebo lopatek. Další symptomy mohou být pocit na zvracení/zvracení, pocení, dušnost, bušení srdce, (extrémní) únava a/nebo závrať. U pacientů s vysokým rizikem srdečního onemocnění může dojít k srdečnímu infarktu. Váš lékař Vám nepředepíše PROTELOS, pokud máte takovéto riziko.
- Krevní sraženiny v žilách: bolest, zarudnutí, otok na končetině, náhlá bolest na hrudi nebo obtíže při dýchání.
Vzácné (mohou postihnout až 1 z 1000 lidí):
- Známky závažné reakce přecitlivělosti (DRESS): zpočátku jako příznaky podobné chřipce a vyrážka na obličeji, poté rozšířená vyrážka s vysokou teplotou (méněčasté), zvýšené hladiny jaterních enzymů v krevních testech (méně časté), zvýšení počtu určitého druhu bílých krvinek (eozinofilie) (vzácné) a zvětšené lymfatické (mízní) uzliny (méně časté).
Velmi vzácné (mohou postihnout až 1 z 10000 lidí):
- Známky potenciálně život ohrožující vyrážky na kůži (Stevens-Johnsonův syndrom, toxická epidermální nekrolýza): zpočátku jako načervenalé terčovité skvrny nebo kruhové skvrny často s centrálně lokalizovanými puchýři na trupu. Přídatné znaky mohou zahrnovat vředy v ústech, v krku,v nose, na genitáliích a zánět spojivek (červené a napuchlé oči). Tyto potenciálně život ohrožující kožní vyrážky jsou často doprovázeny příznaky podobnými chřipce. Vyrážka může vést k rozšíření puchýřů nebo odlupování kůže.
Další možné nežádoucí účinky
Velmi časté (mohou postihnout více než 1 z 10 lidí):
Svědění, kopřivka, kožní vyrážka, angioedem (jako oteklý obličej, jazyk nebo hrdlo, obtíže s dýcháním nebo polykáním), bolest kostí, končetin, svalů a/nebo kloubů, svalové křeče.
Časté:
Zvracení, bolest břicha, reflux (obsah žaludku vracející se do jícnu), porucha trávení, zácpa, plynatost, potíže se spaním, zánět jater (hepatitida), otok končetin, bronchiální hyperreaktivita (symptomy zahrnují sípot a dušnost a kašel), zvýšená hladina svalového enzymu (kreatinin fosfokináza), zvýšené hladiny cholesterolu.
Pocit na zvracení, průjem, bolest hlavy, ekzém, problémy s pamětí, mdloba, mravenčení, točení hlavy, závrať. Tyto účinky však byly mírné a krátkodobé a obvykle si nevyžádaly ukončení užívání tohoto léku. Promluvte si s lékařem, pokud začnou být některé účinky nepříjemné nebo pokud přetrvávají.
Méně časté (mohou postihnout až 1 ze 100 lidí):
Záchvaty, podráždění úst (jako vředy v ústech a záněty dásní), úbytek vlasů, pocit zmatenosti, pocit nevolnosti, sucho v ústech, podráždění pokožky.
Vzácné:
Snížení tvorby krevních buněk v kostní dřeni.
Neužívejte PROTELOS znovu, jestliže jste ukončil(a) léčbu z důvodu reakcí přecitlivělosti.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak PROTELOS uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti vyznačené na krabičce a na sáčku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Po rozpuštění přípravku ve vodě je vzniklá suspenze stabilní po dobu 24 hodin. Nicméně, suspenze by měla být vypita bezprostředně po přípravě (viz bod 3).
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte.Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co PROTELOS obsahuje
- Léčivou látkou je strontii ranelas. Jeden sáček obsahuje strontii ranelas 2 g.
- Pomocné látky jsou aspartam (E 951), maltodextrin, mannitol (E 421).
Jak PROTELOS vypadá a co obsahuje toto balení
PROTELOS je dostupný v sáčcích obsahujících žluté granule pro perorální suspenzi. PROTELOS je dodáván v krabičkách po 7, 14, 28, 56, 84 nebo 100 sáčcích.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci
Les Laboratoires Servier 50, rue Carnot 92284 Suresnes cedex Francie
Výrobce
Les Laboratoires Servier Industrie 905, route de Saran 45520 Gidy Francie
Anpharm Przedsiebiorstwo Farmaceutyczne S.A.
03-236 Warszawa Ul. Annopol 6B Polsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien S.A. Servier Benelux N.V. Tel: +32 (0)2 529 43 11 |
Lietuva UAB "SERVIER PHARMA" Tel: +370 (5) 2 63 86 28 |
|
Btarapnu CepBue MeguKaa EOOfl Tea.: +359 2 921 57 00 |
Luxembourg/Luxemburg S.A. Servier Benelux N.V. Tel: +32 (0)2 529 43 11 |
|
Česká republika Servier s.r.o. Tel: +420 222 118 111 |
Magyarország Servier Hungaria Kft. Tel: +36 1 238 7799 |
|
Danmark Servier Danmark A/S Tlf: +45 36 44 22 60 |
Malta Galepharma Ltd Tel: +(356) 21 247 082 |
|
Deutschland Servier Deutschland GmbH Tel: +49 (0)89 57095 01 |
Nederland Servier Nederland Farma B.V. Tel: +31 (0)71 5246700 |
|
Eesti Servier Laboratoires OU Tel: + 372 664 5040 |
Norge Servier Danmark A/S Tlf: +45 36 44 22 60 |
|
ELLáSa IEPBIE EAAAI OAPMAKEYTIKH EnE TqL +30 210 939 1000 |
Osterreich Servier Austria GmbH Tel: +43 (1) 524 39 99 |
|
Espaňa Laboratorios Servier S.L. Tel: +34 91 748 96 30 |
Polska Servier Polska Sp. z o.o. Tel: +48 (0) 22 594 90 00 |
|
France Les Laboratoires Servier Tel: +33 (0)1 55 72 60 00 |
Portugal Servier Portugal, Lda Tel.: +351 21 312 20 00 |
|
Hrvatska Servier Pharma, d. o. o. Tel.: +385 (0)1 3016 222 |
Románia Servier Pharma SRL Tel: +40 21 528 52 80 |
|
Ireland Servier Laboratories (Ireland) Ltd. Tel: +353 (0)1 6638110 |
Slovenija Servier Pharma d.o.o. Tel.: +386 (0)1 563 48 11 |
|
Island Servier Laboratories c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Servier Slovensko spol. s r.o Tel.:+421 (0)2 5920 41 11 |
|
Italia Servier Italia S.p.A. Tel: +39 06 669081 |
Suomi/Finland Servier Finland Oy P./Tel: +358 (0)9 279 80 80 |
|
Kúnpoq C.A. Papaellinas Ltd. TpL +357 22741741 |
Sverige Servier Sverige AB Tel: +46 (0)8 522 508 00 |
|
Latvija SIA Servier Latvia Tel. +371 67502039 |
United Kingdom Servier Laboratories Ltd Tel: +44 (0)1753 666409 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
38