Praxbind 2,5 G/50 Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Praxbind 2,5 g/50 ml injekční/infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml injekčního/infuzního roztoku obsahuje idarucizumabum 50 mg.
Jedna injekční lahvička obsahuje idarucizumabum 2,5 g v 50 ml.
Idarucizumab je vyráběný rekombinantní DNA technologií za použití ovariálních buněk čínského křečka.
Pomocné látky se známým účinkem
Jedna injekční lahvička o objemu 50 ml obsahuje 2 g sorbitolu a 25 mg sodíku (viz bod 4.4). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční/infuzní roztok
Čirý až mírně opalizující, bezbarvý až mírně nažloutlý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Praxbind je specifický přípravek k reverzi účinku dabigatranu a je indikován u dospělých pacientů léčených přípravkem Pradaxa (dabigatran-etexilát) v situacích, kdy je třeba urychleně zvrátit jeho antikoagulační účinky:
• při naléhavých chirurgických/urgentních výkonech
• při život ohrožujícím nebo nekontrolovaném krvácení.
4.2 Dávkování a způsob podání
Omezeno pouze pro použití v nemocnici.
Dávkování
Doporučená dávka přípravku Praxbind je 5 g (2 x 2,5 g/50 ml).
Po podání idarucizumabu podskupina pacientů znovu vykázala plazmatické koncentrace nevázaného dabigatranu a zároveň prodloužení testů srážlivosti do 24 hodin (viz bod 5.1).
Podání druhé 5 g dávky přípravku Praxbind lze zvážit v těchto situacích:
• rekurence klinicky významného krvácení spolu s prodloužením doby srážení krve;
• pokud by potenciální obnovení krvácení bylo život ohrožující a zjistí se prodloužená doba srážení krve;
• pokud pacienti vyžadují další naléhavý chirurgický/urgentní výkon a vykazují prodlouženou dobu srážení krve.
Relevantními koagulačními parametry jsou aktivovaný parciální tromboplastinový čas (aPTT), dilutovaný trombinový čas (dTT) nebo ekarinový koagulační čas (ECT) (viz bod 5.1).
Maximální denní dávka nebyla hodnocena.
Znovu zahájení antitrombotické terapie
Léčbu přípravkem Pradaxa (dabigatran-etexilát) lze znovu zahájit 24 hodin po podání přípravku Praxbind, pokud je pacient klinicky stabilní a bylo dosaženo odpovídající hemostázy.
Po podání přípravku Praxbind lze kdykoli zahájit jinou antitrombotickou terapii (např. nízkomolekulární heparin), pokud je pacient klinicky stabilní a bylo dosaženo odpovídající hemostázy.
Bez antitrombotické léčby jsou pacienti vystaveni riziku trombózy vzhledem k jejich základnímu onemocnění nebo zdravotnímu stavu.
Pacienti s poruchou funkce ledvin
Pacienti s poruchou funkce ledvin nevyžadují žádnou úpravu dávky. Porucha funkce ledvin neovlivnila reverzní účinek idarucizumabu.
Pacienti s poruchou funkce jater
Pacienti s poruchou funkce jater nevyžadují žádnou úpravu dávky (viz bod 5.2).
Starší pacienti
Starší pacienti ve věku 65 let a starší nevyžadují žádnou úpravu dávky (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Praxbind u dětí mladších 18 let nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Intravenózní podání.
Praxbind (2 x 2,5 g/50 ml) se podává intravenózně jako dvě po sobě následující infoze, každá v délce 5 až 10 minut, nebo jako bolusová injekce.
Další pokyny k použití a zacházení viz bod 6.6.
4.3 Kontraindikace
Žádné.
4.4 Zvláštní upozornění a opatření pro použití
Idarucizumab se specificky váže na dabigatran a zruší jeho antikoagulační účinek. Nezruší účinky jiných antikoagulancií (viz bod 5.1).
Léčbu přípravkem Praxbind lze kombinovat se standardními podpůrnými opatřeními, která jsou z medicínského hlediska považována za vhodná.
Hypersenzitivita
Riziko použití přípravku Praxbind u pacientů se známou hypersenzitivitou (např. anafylaktoidní reakcí) na idarucizumab nebo na kteroukoli pomocnou látku je třeba pečlivě zvážit oproti potenciálnímu přínosu této neodkladné léčby. Pokud dojde k anafylaktické reakci nebo jiné závažné alergické reakci, je třeba přípravek Praxbind okamžitě vysadit a zahájit vhodnou léčbu.
Hereditární intolerance fruktózy
Doporučená dávka přípravku Praxbind obsahuje 4 g sorbitolu jako pomocnou látku. U pacientů s hereditární intolerancí fruktózy je parenterální podávání sorbitolu spojováno s hlášeními hypoglykemie, hypofosfatemie, metabolické acidózy, zvýšené hladiny kyseliny močové, akutního jaterního selhání s narušením exkrečních a syntetických funkcí a úmrtí. Proto se u pacientů s hereditární intolerancí fruktózy musí riziko léčby přípravkem Praxbind zvážit oproti potenciálnímu přínosu takové neodkladné léčby. Pokud je přípravek Praxbind podáván těmto pacientům, je nutné zintenzivnit lékařskou péči během expozice přípravkem Praxbind a během 24 hodin po ní.
Tromboembolické příhody
Pacienti léčení dabigatranem mají základní onemocnění, která je predisponují k tromboembolickým příhodám. Zrušení účinků léčby dabigatranem vystavuje pacienty riziku trombózy vzhledem k jejich základnímu onemocnění. Aby se toto riziko snížilo, je třeba zvážit obnovení antikoagulační léčby, hned jak to bude z medicínského hlediska vhodné (viz bod 4.2).
Vyšetření bílkovin v moči
Praxbind způsobuje přechodnou proteinurii jako fyziologickou reakci na nadbytek bílkovin procházejících ledvinami po bolusové/krátkodobé intravenózní aplikaci 5 g idarucizumabu (viz bod 5.2). Při vyšetření moči je třeba vzít v úvahu, že přechodná proteinurie nesvědčí o poškození ledvin.
Obsah sodíku
Jedna dávka tohoto léčivého přípravku obsahuje 2,2 mmol (50 mg) sodíku. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí přípravku Praxbind a jiných léčivých přípravků. Vzhledem k farmakokinetickým vlastnostem a vysoké specificitě vazby na dabigatran se klinicky relevantní interakce s jinými léčivými přípravky považují za nepravděpodobné.
Předklinický výzkum neprokázal u idarucizumabu žádné interakce s
• volumexpandéry;
• koncentráty koagulačního faktoru, např. koncentráty protrombinového komplexu (PCC, např. se 3 faktory a se 4 faktory), aktivované PCC (aPCC) a rekombinantní faktor VIIa;
• jinými antikoagulancii (např. inhibitory trombinu jinými než dabigatran, inhibitory faktoru Xa včetně nízkomolekulárního heparinu, antagonisty vitaminu K, heparinem). Idarucizumab tedy nezvrátí účinky jiných antikoagulancií.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání přípravku Praxbind těhotným ženám nejsou k dispozici. Studie reprodukční a vývojové toxicity nebyly vzhledem k charakteru a zamýšlenému klinickému použití léčivého přípravku provedeny. Přípravek Praxbind lze použít při těhotenství, pokud očekávaný klinický přínos převáží potenciální rizika.
Kojení
Není známo, zda se idarucizumab vylučuje do lidského mateřského mléka.
Fertilita
Údaje o účinku přípravku Praxbind na fertilitu nejsou k dispozici (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje Není relevantní.
4.8 Nežádoucí účinky
Bezpečnost přípravku Praxbind byla hodnocena u 224 zdravých dobrovolníků a u 123 pacientů v probíhajícím klinickém hodnocení fáze III, kteří nekontrolovaně krváceli nebo potřebovali neodkladný chirurgický výkon, a byli léčeni přípravkem Pradaxa (dabigatran-etexilát).
Nebyly zjištěny žádné nežádoucí účinky.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nejsou žádné klinické zkušenosti s předávkováním přípravkem Praxbind.
Nejvyšší jednotlivá dávka přípravku Praxbind studovaná u zdravých dobrovolníků byla 8 g. U této skupiny nebyly identifikovány žádné bezpečnostní signály.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: všechny jiné terapeutické přípravky, antidota, ATC kód: V03AB37 Mechanizmus účinku
Idarucizumab je specifická látka k reverzi účinku dabigatranu. Je to fragment humanizované monoklonální protilátky (Fab), který se s velmi vysokou afinitou váže na dabigatran; afinita jeho vazby je přibližně 300x vyšší než vazebná afinita dabigatranu na trombin. Komplex idarucizumab-dabigatran se vyznačuje rychlou asociací a extrémně pomalou disociací, proto je velmi stabilní. Idarucizumab se silně a specificky váže na dabigatran a jeho metabolity a neutralizuje jejich antikoagulační účinek.
Klinická účinnost a bezpečnost
Ke zhodnocení bezpečnosti, účinnosti, snášenlivosti, farmakokinetiky a farmakodynamiky idarucizumabu při podání samostatně nebo po podání dabigatran-etexilátu byly provedeny tři randomizované, dvojitě slepé, placebem kontrolované studie fáze I u 283 subjektů (224 léčených idarucizumabem). Hodnocená populace se sestávala ze zdravých dobrovolníků a subjektů se specifickými populačními parametry zahrnujícími věk, tělesnou hmotnost, rasu, pohlaví a poruchu funkce ledvin. Dávky idarucizumabu byly v těchto studiích v rozmezí od 20 mg do 8 g a délka infuze v rozmezí od 5 minut do 1 hodiny.
Reprezentativní hodnoty farmakokinetických a farmakodynamických parametrů byly stanoveny podle zdravých dobrovolníků ve věku 45-64 let, kteří dostali 5 g idarucizumabu (viz body 5.1 a 5.2).
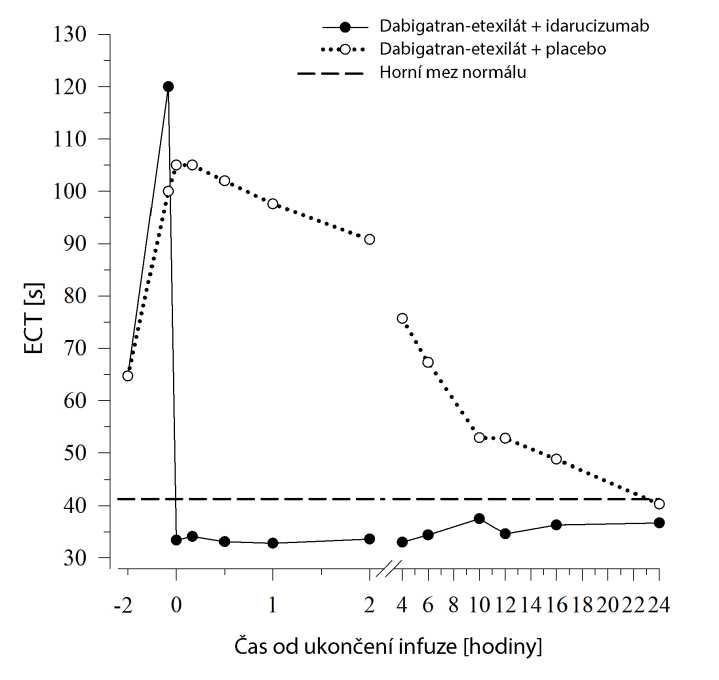
Aktuálně probíhá prospektivní, otevřená, nerandomizovaná, nekontrolovaná studie (RE-VERSE AD), kterou se hodnotí léčba dospělých pacientů, u kterých došlo k život ohrožujícímu nebo nekontrolovanému krvácení, které souvisí s dabigatranem (skupina A), nebo kteří vyžadovali neodkladný chirurgický nebo urgentní výkon (skupina B). Primárním cílovým parametrem byla maximální procentuální reverze antikoagulačního účinku dabigatranu během 4 hodin od podání idarucizumabu na základě dilutovaného trombinového času (dTT) nebo ekarinového koagulačního času (ECT) stanovených centrální laboratoří. Klíčovým sekundárním cílovým parametrem je obnovení hemostázy.
Předběžná analýza RE-VERSE AD zahrnovala údaje od 123 pacientů: 66 pacientů se závažným krvácením (skupina A) a 57 pacientů, kteří vyžadovali naléhavý výkon (skupina B). Přibližně polovina pacientů v každé skupině byla mužského pohlaví. Medián věku byl 77 let a medián clearance kreatininu 61 ml/min. Přibližně 68 % pacientů ve skupině A a 63 % pacientů ve skupině B byl podáván dabigatran 110 mg dvakrát denně. Výsledky hodnocení centrální laboratoří byly k dispozici pro skupinu 90 pacientů (51 ve skupině A, 39 ve skupině B).
U většiny pacientů (> 89 %) ve skupině A i B bylo dosaženo úplné reverze antikoagulačního účinku dabigatranu (podle dTT nebo ECT) během prvních 4 hodin po podání 5 g idarucizumabu. Projevy reverze účinku byly patrné okamžitě po podání.
dTT Is]
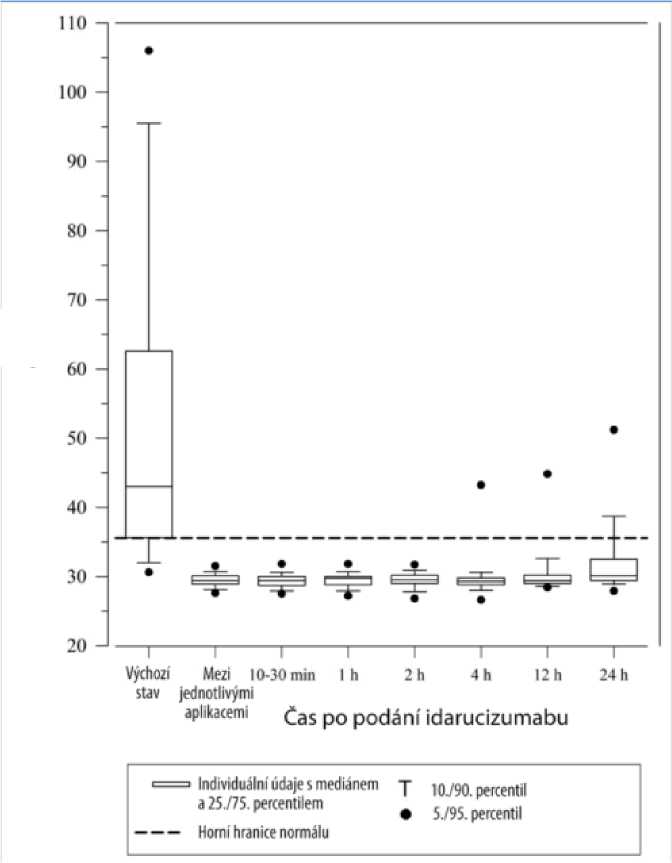
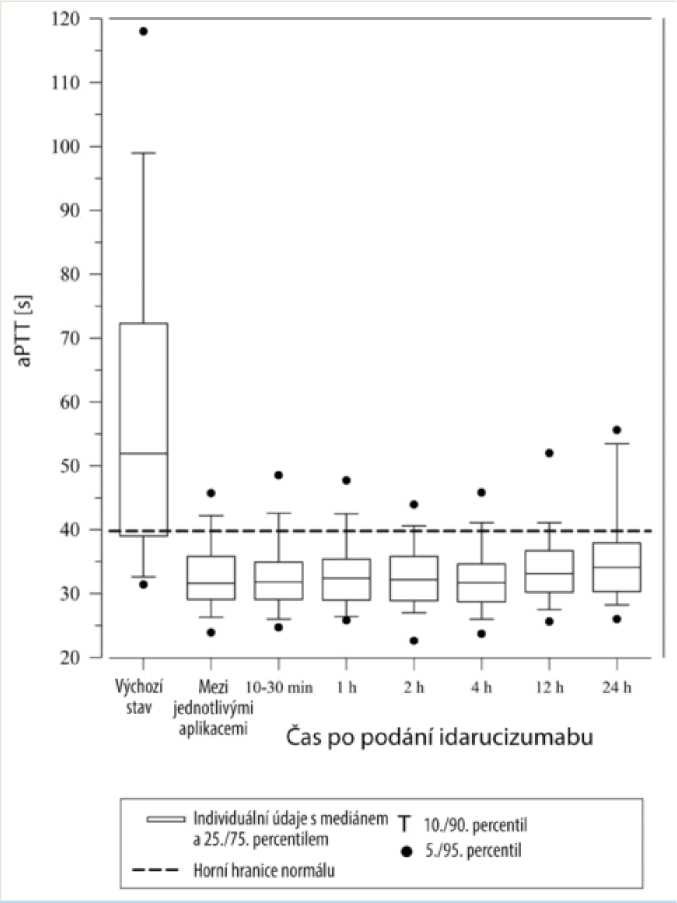
Obnovení hemostázy bylo dosaženo u 91 % hodnotitelných pacientů se závažným krvácením a normální hemostázu vykazovalo 92 % pacientů, u kterých bylo třeba provést naléhavý výkon.
Z celkového počtu 123 pacientů jich 26 zemřelo; všechna úmrtí souvisela buď s komplikacemi prvotní příhody nebo komorbiditami. Trombotické příhody byly hlášeny u 5 pacientů; žádnému z nich nebyla v okamžiku příhody podávána antitrombotická terapie a ve všech těchto případech lze trombotickou příhodu přiřadit základnímu onemocnění pacienta. Byly hlášeny mírné příznaky potenciální hypersenzitivity (pyrexie, bronchospasmus, hyperventilace, vyrážka nebo pruritus). Nebylo možné stanovit kauzální vztah s idarucizumabem. Dalšími nežádoucími účinky hlášenými u nejméně 5 % pacientů byly hypokalemie (9/123; 7 %), delirium (9/123; 7 %), obstipace (8/123; 7 %), pyrexie (7/123; 6 %) apneumonie (7/123; 6 %).
Farmakodynamické účinky
Farmakodynamika idarucizumabu po podání dabigatran-etexilátu byla hodnocena u 141 subjektů ve studiích fáze I a údaje jsou hlášeny pro reprezentativní podskupinu 6 zdravých dobrovolníků ve věku 45 až 64 let, kterým byla podána dávka 5 g formou intravenózní infuze. Medián vrcholové expozice u hodnocených zdravých dobrovolníků byl v rozmezí podávání 150 mg dabigatran-etexilátu dvakrát denně pacientům.
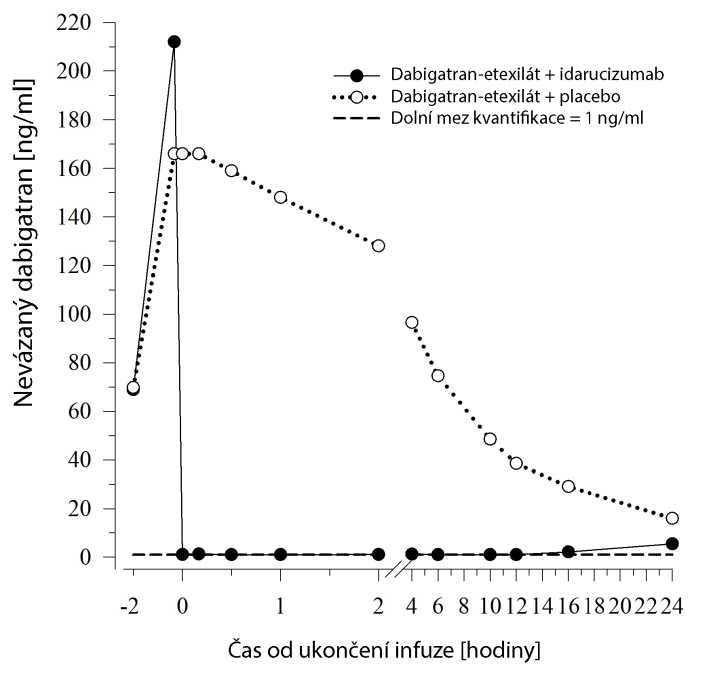
Okamžitě po podání idarucizumabu se plazmatická koncentrace nevázaného dabigatranu snížila o více než 99 %, čímž bylo dosaženo hladiny bez antikoagulační aktivity.
Většina pacientů vykázala reverzi koncentrace dabigatranu v plazmě přetrvávající až 12 hodin (> 90 %). U malého počtu pacientů byla zjištěna rekurence plazmatické hladiny nevázaného dabigatranu a souběžné zvýšení hodnot testů srážlivosti; možnou příčinou je redistribuce dabigatranu z periferie. Došlo k tomu během 2-24 hodin po podání idarucizumabu, a to zejména v časovém období >12 hodin.
Obrázek 3 - Hladina nevázaného dabigatranu v plazmě u reprezentativní skupiny zdravých dobrovolníků (podání idarucizumabu nebo placeba v 0 h)
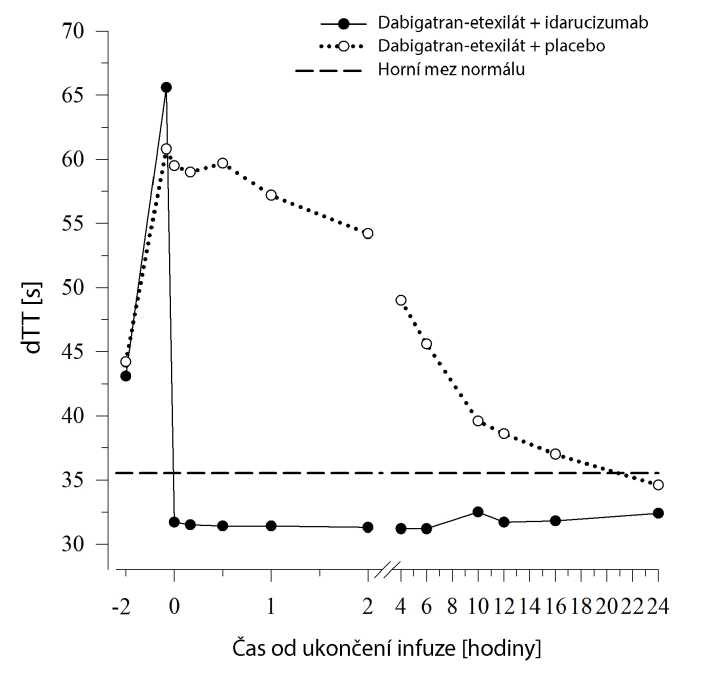
Dabigatran prodlužuje dobu srážení ukazatelů koagulace, jako je např. dilutovaný trombinový čas (dTT), trombinový čas (TT), aktivovaný parciální tromboplastinový čas (aPTT) a ekarinový koagulační čas (ECT), což dává přibližnou představu o intenzitě antikoagulace. Hodnota v normálním rozmezí po podání idarucizumabu udává, že pacient již není antikoagulován. Hodnota nad normálním rozmezím může značit zbytkový aktivní dabigatran nebo jiné klinické stavy, např. přítomnost jiných léků nebo transfuzní koagulopatii. Tyto testy byly použity k vyhodnocení antikoagulačního účinku dabigatranu. Úplná a trvalá reverze prodloužení doby srážení indukovaného dabigatranem byla zjištěna okamžitě po infuzi idarucizumabu a přetrvala po celou dobu sledování, tedy nejméně 24 h.
Obrázek 4 - Reverze prodloužení doby srážení indukovaného dabigatranem stanovené podle dTT u reprezentativní skupiny zdravých dobrovolníků (podání idarucizumabu nebo placeba v 0 li)
Obrázek 5 - Reverze prodloužení doby srážení indukovaného dabigatranem stanovené podle ECT u reprezentativní skupiny zdravých dobrovolníků (podání idarucizumabu nebo placeba v 0 h)
Parametry generace trombinu
Dabigatran výrazně ovlivňuje parametry endogenního potenciálu trombinu (ETP). Léčba idarucizumabem normalizovala jak poměr „lag time generace trombinu“ (čas od iniciace k první detekovatelné generaci trombinu), tak poměr času potřebného k dosažení maximální koncentrace vůči výchozím hodnotám při stanovení za 0,5 až 12 hodin po ukončení infuze idarucizumabu. Samotný idarucizumab nevykazoval žádný prokoagulační účinek měřený jako ETP. To naznačuje, že idarucizumab nemá žádný protrombotický účinek.
Opakované podání dabigatran-etexilátu
Opakované podání dabigatran-etexilátu 24 hodin po infuzi idarucizumabu vedlo k očekávanému antikoagulačnímu účinku.
Imunogenita
Vzorky séra 283 subjektů (224 léčených idarucizumabem) byly před léčbou a po ní testovány na protilátky proti idarucizumabu.
Preexistující protilátky se zkříženou reaktivitou na idarucizumab byly zjištěny u přibližně 13 % (36/283) subjektů. U těchto subjektů nebyl pozorován žádný dopad na farmakokinetiku ani reverzní účinek idarucizumabu; nebyly pozorovány ani hypersenzitivní reakce.
U 4 % (9/224) subjektů byly zjištěny protilátky proti idarucizumabu vyplývající z léčby, což naznačuje nízký imunogenní potenciál idarucizumabu. U podskupiny 6 subjektů byl idarucizumab podán podruhé dva měsíce po prvním podání. U těchto subjektů nebyly před druhým podáním detekovány žádné protilátky proti idarucizumabu. U jednoho subjektu byly po druhém podání detekovány protilátky proti idarucizumabu vyplývající z léčby.
Předklinická farmakodynamika
Byl proveden model traumatu u prasat při použití tupého poranění jater po dávkování dabigatranu, kterým bylo dosaženo supraterapeutické koncentrace přibližně 10x přesahující hladinu v lidské plazmě. Idarucizumab účinně a rychle zvrátil život ohrožující krvácení do 15 minut po podání injekce. Všechna prasata přežila dávky idarucizumabu přibližně 2,5 a 5 g. Bez idarucizumabu byla mortalita u antikoagulované skupiny 100 %.
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Praxbind u jedné nebo více podskupin pediatrické populace při prevenci a léčbě krvácení souvisejícího s dabigatranem (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika idarucizumabu byla hodnocena u 224 subjektů ve studiích fáze I, přičemž prezentovány jsou údaje pro reprezentativní podskupinu 6 zdravých dobrovolníků ve věku 45 až 64 let, kterým byla podána dávka 5 g formou intravenózní infuze.
Distribuce
Idarucizumab vykazoval multifázickou kinetiku distribuce a eliminace a omezenou extravaskulární distribuci. Po intravenózní infuzi dávky 5 g byl geometrický průměr distribučního objemu v rovnovážném stavu (Vss) 8,9 l (geometrický koeficient variace (gCV) 24,8 %).
Biotransformace
Bylo popsáno několik cest, které se mohou podílet na metabolismu protilátek. Všechny tyto cesty zajišťují biodegradaci protilátky na menší molekuly, tj. malé peptidy nebo aminokyseliny, které jsou následně reabsorbovány a inkorporovány do obecné syntézy bílkovin.
Eliminace
Idarucizumab byl rychle eliminován při celkové clearance 47,0 ml/min (gCV 18,4 %), počátečním poločasu 47 minut (gCV 11,4 %) a terminálním poločasu 10,3 h (gCV 18,9 %). Po intravenózním podání dávky 5 g idarucizumabu bylo v moči nalezeno 32,1 % (gCV 60,0 %) dávky během odběrového období 6 hodin a méně než 1 % v následujících 18 hodinách. Předpokládá se, že zbývající část dávky se eliminuje prostřednictvím katabolismu bílkovin zejména v ledvinách.
Po léčbě idarucizumabem byla zjištěna proteinurie. Přechodná proteinurie je fyziologická reakce na nadbytek bílkovin procházejících ledvinami po bolusové/krátkodobé intravenózní aplikaci 5 g idarucizumabu. Přechodná proteinurie obvykle dosáhla maxima přibližně 4 hodiny po podání idarucizumabu a normalizovala se v průběhu 12-24 hodin. V jednotlivých případech proteinurie přetrvávala více než 24 hodin.
Pacienti s poruchou funkce ledvin
Ve studiích fáze I byl přípravek Praxbind hodnocen u subjektů s clearance kreatininu v rozmezí 44 až 213 ml/min. Subjekty s clearance kreatininu pod 44 ml/min nebyly ve fázi 1 hodnoceny.
V závislosti na stupni poruchy funkce ledvin byla celková clearance v porovnání se zdravými dobrovolníky snížena, což vedlo ke zvýšené expozici idarucizumabu.
Na základě farmakokinetických údajů od 68 pacientů s různým stupněm funkce ledvin (medián clearance kreatininu 19,2-126 ml/min) se odhaduje, že průměrná expozice idarucizumabu (AUC0-24h) se zvyšuje o 26 % u pacientů s lehkou poruchou funkce ledvin (clearance kreatininu 60-90 ml/min), o 78 % u pacientů se středně těžkou poruchou funkce ledvin (30-60 ml/min) a o 199 % u pacientů s těžkou poruchou funkce ledvin (0-30 ml/min). Jelikož dabigatran je také primárně vylučován ledvinami, lze vidět zvýšení expozice vůči dabigatranu také v závislosti na zhoršování funkce ledvin.
Na základě těchto údajů a míry reverze antikoagulačního účinku dabigatranu u pacientů zřejmě porucha funkce ledvin nemá vliv na reverzní účinek idarucizumabu, ačkoli tento závěr pro pacienty s těžkou poruchou funkce ledvin vychází jen z malé podskupiny pacientů.
Pacienti s poruchou funkce jater
Přípravek Praxbind nebyl hodnocen u pacientů s poruchou funkce jater. Je známo, že fragmenty protilátek jsou eliminovány zejména proteolytickým katabolismem v ledvinách. Neočekává se, že by porucha funkce jater měla mít vliv na farmakokinetiku idarucizumabu.
Starší pacienti/pohlaví/rasa
Podle populačních farmakokinetických analýz neměly pohlaví, věk ani rasa žádný klinicky významný vliv na farmakokinetiku idarucizumabu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Předklinické údaje získané na základě studií toxicity po opakovaném podávání po dobu až čtyř týdnů u potkanů a dvou týdnů u opic neodhalily žádné zvláštní riziko pro člověka. Bezpečnostní farmakologické studie neprokázaly žádné účinky na respirační centrální nervový ani kardiovaskulární systém.
Studie k vyhodnocení mutagenního a karcinogenního potenciálu idarucizumabu nebyly provedeny. Na základě mechanismu účinku a vlastností bílkovin se neočekávají žádné karcinogenní ani genotoxické účinky.
Studie k vyhodnocení potenciálních účinků idarucizumabu na reprodukci nebyly provedeny. Studie toxicity po opakovaném podávání po dobu až čtyř týdnů u potkanů a dvou týdnů u opic neodhalily v reprodukčních tkáních žádného z pohlaví žádné účinky související s léčbou. Navíc nebyla zjištěna žádná vazba idarucizumabu na lidské reprodukční tkáně ve studii zkřížené reaktivity tkání. Předklinické výsledky tedy nepoukazují na riziko v oblasti fertility ani embryo-fetálního vývoje.
Po intravenózním ani paravenózním podání idarucizumabu nebylo zjištěno žádné místní podráždění cévy. Struktura idarucizumabu nezpůsobovala hemolýzu lidské plné krve in vitro.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
trihydrát octanu sodného kyselina octová sorbitol polysorbát 20 voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
30 měsíců
Chemická a fyzikální stabilita idarucizumabu po otevření injekční lahvičky byla prokázána na dobu 1 hodiny při pokojové teplotě.
Z mikrobiologického hlediska je třeba přípravek použít okamžitě po otevření, pokud způsob otevření nezabraňuje riziku mikrobiální kontaminace. Pokud není použit okamžitě, odpovídá za délku a podmínky uchovávání otevřeného přípravku před použitím uživatel.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Neotevřenou injekční lahvičku lze před použitím uchovávat při pokojové teplotě (25 °C) po dobu až 48 hodin, pokud je uchovávána v původním obalu, chráněna před světlem, nebo až 6 hodin bez obalu na světle.
Podmínky uchovávání tohoto léčivého přípravku po prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
50 ml roztoku ve skleněné injekční lahvičce (sklo třídy I) s butylovou pryžovou zátkou a hliníkovým uzávěrem a štítkem s integrovaným držákem.
Velikost balení je 2 injekční lahvičky.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
U parenterálních léčivých přípravků, jako je přípravek Praxbind, je před podáním nutné vizuálně zkontrolovat přítomnost pevných částic a změnu zbarvení.
Přípravek Praxbind nesmí být mísen s jinými léčivými přípravky. K podání přípravku Praxbind lze použít stávající intravenózní linku. Linku je nutné před infuzí a po ní propláchnout 0,9% injekčním roztokem (9 mg/ml) chloridu sodného. Stejným intravenózním přístupem se nesmí současně podávat žádná jiná infuze.
Přípravek Praxbind je určen pouze pro jednorázové použití a neobsahuje konzervanty (viz bod 6.3).
Nebyly pozorovány inkompatibility mezi přípravkem Praxbind a polyvinylchloridovými, polyethylenovými ani polyuretanovými infuzními soupravami ani polypropylenovými injekčními stříkačkami.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH Binger Str. 173 D-55216 Ingelheim am Rhein Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1056/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 20. listopadu 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http: //www .ema. europa. eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA
PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Boehringer Ingelheim Pharma GmbH & Co. KG Birkendorfer Strasse 65 88397 Biberach an der Riss NĚMECKO
Název a adresa výrobce odpovědného za propouštění šarží
Boehringer Ingelheim Pharma GmbH & Co. KG Birkendorfer Strasse 65 88397 Biberach an der Riss NĚMECKO
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Praxbind 2,5 g/50 ml injekční/infuzní roztok idarucizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička o objemu 50 ml obsahuje idarucizumabum 2,5 g.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: trihydrát octanu sodného, kyselina octová, sorbitol, polysorbát 20, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
injekční/infuzní roztok 2 injekční lahvičky o objemu 50 ml
5. ZPŮSOB A CESTA/CESTY PODÁNÍ_
Intravenózní podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ_
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ_
Pouze pro jednorázové použití.
8. POUŽITELNOST_
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH Binger Str. 173 D-55216 Ingelheim am Rhein Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1056/001
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ_
15. NÁVOD K POUŽITÍ_
16. INFORMACE V BRAILLOVĚ PÍSMU_
Nevyžaduje se - odůvodnění přijato.
17. JINÉ - potisk na vnitřní straně
• Přiložená příbalová informace obsahuje další informace pro zdravotnické pracovníky.
• Doporučená dávka přípravku Praxbind je 5 g (2 x 2,5 g/50 ml).
• Intravenózní podání jako dvě po sobě následující infuze, každá v délce 5 až 10 minut, nebo jako bolusové injekce.
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU Štítek injekční lahvičky_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Praxbind 2,5 g/50 ml injekční/infuzní roztok idarucizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička o objemu 50 ml obsahuje idarucizumabum 2,5 g.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: trihydrát octanu sodného, kyselina octová, sorbitol, polysorbát 20, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
2 injekční lahvičky, každá o objemu 50 ml injekčního/infuzního roztoku
5. ZPŮSOB A CESTA/CESTY PODÁNÍ_
Intravenózní podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ_
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ_
Pouze pro jednorázové použití.
8. POUŽITELNOST_
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH Binger Str. 173 D-55216 Ingelheim am Rhein Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1056/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Praxbind 2,5 g/50 ml injekční/infuzní roztok
idarucizumabum
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci, protože obsahuje pro Vás důležité údaje. Vezměte prosím na vědomí, že tento léčivý přípravek se používá primárně v naléhavých situacích a že o tom, zda ho potřebujete, rozhodne lékař.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Praxbind a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Praxbind používat
3. Jak se přípravek Praxbind používá
4. Možné nežádoucí účinky
5. Jak přípravek Praxbind uchovávat
6. Obsah balení a další informace
1. Co je přípravek Praxbind a k čemu se používá Co je přípravek Praxbind
Přípravek Praxbind je specifickým přípravkem ke zvrácení účinku dabigatranu (Pradaxa), léčivého přípravku na ředění krve, který v těle blokuje látku, která se podílí na srážení krve. Praxbind slouží k rychlému vychytání dabigatranu s cílem neutralizovat jeho účinek.
Přípravek Praxbind obsahuje léčivou látku idarucizumab.
K čemu se přípravek Praxbind používá
Praxbind se používá u dospělých pacientů v naléhavých situacích, když lékař usoudí, že je třeba rychle neutralizovat účinek přípravku Pradaxa
- při naléhavých chirurgických/urgentních výkonech
- při život ohrožujícím nebo nekontrolovaném krvácení.
2. Čemu musíte věnovat pozornost, než začnete přípravek Praxbind používat Upozornění a opatření
Informujte svého lékaře nebo zdravotní sestru
- jestliže jste alergický(á) na idarucizumab nebo na kteroukoli další složku uvedenou v bodě 6.
- jestliže trpíte genetickým onemocněním s názvem vrozená nesnášenlivost fruktózy. V takovém případě může sorbitol, který je složkou tohoto léčivého přípravku, způsobit závažné nežádoucí účinky.
Na tyto informace bude brán zřetel, než u Vás bude zahájena léčba přípravkem Praxbind.
Tento léčivý přípravek odstraní z organismu pouze dabigatran. Neodstraní jiné léčivé přípravky, které zabraňují srážení krve.
Po odstranění dabigatranu z těla nebudete chráněn(a) proti tvorbě krevních sraženin. Jakmile to Váš zdravotní stav umožní, lékař znovu nasadí léčivé přípravky, které zabraňují srážení krve.
Děti a dospívající
Nejsou dostupné žádné informace o použití přípravku Praxbind u dětí.
Další léčivé přípravky a přípravek Praxbind
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a), nebo které možná budete užívat.
Tento léčivý přípravek byl navržen tak, aby se vázal pouze na dabigatran. Není pravděpodobné, že by přípravek Praxbind ovlivnil účinek jiných léčivých přípravků, ani že by na přípravek Praxbind měly vliv jiné léčivé přípravky.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem.
Nejsou dostupné žádné informace o účincích tohoto léčivého přípravku u těhotných nebo kojících žen. Praxbind nemá vliv na žádné tělesné funkce jako takové, a lékař se proto může rozhodnout Vám tento léčivý přípravek podat, pokud očekávaný přínos převáží potenciální rizika.
Praxbind obsahuje sodík
Jedna dávka tohoto léčivého přípravku obsahuje 50 mg sodíku. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
3. Jak se přípravek Praxbind používá
Tento léčivý přípravek je určen pouze pro nemocniční podání.
Doporučená dávka je 5 g (2 injekční lahvičky o objemu 50 ml).
Ve vzácných případech se může stát, že po první dávce přípravku Praxbind budete mít v krvi ještě příliš mnoho dabigatranu, a lékař se ve specifických situacích může rozhodnout podat Vám druhou 5g dávku.
Lékař nebo zdravotní sestra Vám tento přípravek podá injekcí nebo infuzí do žíly.
Po podání přípravku Praxbind lékař rozhodne o pokračování Vaší léčby pro prevenci srážení krve. Přípravek Pradaxa lze znovu podat za 24 hodin po podání přípravku Praxbind.
Podrobné pokyny k podání přípravku Praxbind pro lékaře nebo zdravotní sestru jsou uvedeny na konci této příbalové informace (viz „Pokyny k zacházení s přípravkem“).
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře.
Možné nežádoucí účinky
4.
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Dosud nebyly zjištěny žádné nežádoucí účinky.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Praxbind uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za „Použitelné do:“ a na injekční lahvičce za „EXP“ Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2° C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Přípravek Praxbind je po otevření určen k okamžitému použití.
6. Obsah balení a další informace
Co přípravek Praxbind obsahuje
- Léčivou látkou je idarucizumabum.
- Pomocnými látkami jsou trihydrát octanu sodného, kyselina octová, sorbitol, polysorbát 20 a voda na injekci.
Jak přípravek Praxbind vypadá a co obsahuje toto balení
Praxbind injekční/infuzní roztok je čirý až mírně opalizující, bezbarvý až mírně nažloutlý roztok, který se dodává ve skleněné injekční lahvičce uzavřené butylovou pryžovou zátkou a hliníkovým uzávěrem.
Jedno balení obsahuje dvě injekční lahvičky.
Držitel rozhodnutí o registraci
Boehringer Ingelheim International GmbH Binger Str. 173 D-55216 Ingelheim am Rhein Německo
Výrobce
Boehringer Ingelheim Pharma GmbH & Co. KG Birkendorfer Strasse 65 D-88397 Biberach an der Riss Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien SCS Boehringer Ingelheim Comm.V Tél/Tel: +32 2 773 33 11 |
Lietuva Boehringer Ingelheim RCV GmbH & Co KG Lietuvos filialas Tel: +370 37 473922 |
|
Btarapna Eboprarep HHrenxaňM P^B rMÓX h Ko. Kr - KnoH Ebnrapna Ten: +359 2 958 79 98 |
Luxembourg/Luxemburg SCS Boehringer Ingelheim Comm.V Tél/Tel: +32 2 773 33 11 |
|
Česká republika Boehringer Ingelheim spol. s r.o. Tel: +420 234 655 111 |
Magyarország Boehringer Ingelheim RCV GmbH & Co KG Magyarországi Fióktelepe Tel: +36 1 299 8900 |
|
Danmark Boehringer Ingelheim Danmark A/S Tlf: +45 39 15 88 88 |
Malta Boehringer Ingelheim Ltd. Tel: +44 1344 424 600 |
|
Deutschland Boehringer Ingelheim Pharma GmbH & Co. KG Tel: +49 (0) 800 77 90 900 |
Nederland Boehringer Ingelheim b.v. Tel: +31 (0) 800 22 55 889 |
|
Eesti Boehringer Ingelheim RCV GmbH & Co KG Eesti filiaal Tel: +372 612 8000 |
Norge Boehringer Ingelheim Norway KS Tlf: +47 66 76 13 00 |
|
EXXáSa Boehringer Ingelheim Ellas A.E. T^: +30 2 10 89 06 300 |
Osterreich Boehringer Ingelheim RCV GmbH & Co KG Tel: +43 1 80 105-0 |
|
Espaňa Boehringer Ingelheim Espana, S.A. Tel: +34 93 404 51 00 |
Polska Boehringer Ingelheim Sp. zo.o. Tel: +48 22 699 0 699 |
|
France Boehringer Ingelheim France S.A.S. Tél: +33 3 26 50 45 33 |
Portugal Boehringer Ingelheim, Unipessoal, Lda. Tel: +351 21 313 53 00 |
|
Hrvatska Boehringer Ingelheim Zagreb d.o.o. Tel: +385 1 2444 600 |
Románia Boehringer Ingelheim RCV GmbH & Co KG Viena-Sucursala Bucuresti Tel: +40 21 302 2800 |
|
Ireland Boehringer Ingelheim Ireland Ltd. Tel: +353 1 295 9620 |
Slovenija Boehringer Ingelheim RCV GmbH & Co KG Podružnica Ljubljana Tel: +386 1 586 40 00 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Boehringer Ingelheim RCV GmbH & Co KG organizačná zložka Tel: +421 2 5810 1211 |
Sverige
Boehringer Ingelheim AB
Tel: +46 8 721 21 00
Puh/Tel: +358 10 3102 800
Kúnpoq
Boehringer Ingelheim Ellas A.E. T^: +30 2 10 89 06 300
Latvija United Kingdom
Boehringer Ingelheim RCV GmbH & Co KG Boehringer Ingelheim Ltd. Latvijas filiale Tel: +44 1344 424 600
Tel: +371 67 240 011
Tato příbalová informace byla naposledy revidována MM/RRRR.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Praxbind se specificky váže na dabigatran a zruší jeho antikoagulační účinek. Nezruší účinky jiných antikoagulancií.
Léčbu přípravkem Praxbind lze kombinovat se standardními podpůrnými opatřeními, která jsou považována z medicínského hlediska za vhodná.
Doporučená dávka přípravku Praxbind obsahuje 4 g sorbitolu jako pomocnou látku. U pacientů s hereditární intolerancí fruktózy existuje riziko závažných nežádoucích účinků, které se musí zvážit oproti potenciálnímu přínosu neodkladné léčby přípravkem Praxbind. Pokud je přípravek Praxbind podáván těmto pacientům, je nutné zintenzivnit lékařskou péči během expozice přípravkem Praxbind a během 24 hodin po ní.
Dávkování a podání:
Doporučená dávka přípravku Praxbind je 5 g (2 x 2,5 g/50 ml).
Podání druhé 5 g dávky přípravku Praxbind lze zvážit v těchto situacích:
• rekurence klinicky významného krvácení spolu s prodloužením doby srážení krve;
• pokud by potenciální obnovení krvácení bylo život ohrožující a zjistí se prodloužená doba srážení krve;
• pokud pacienti vyžadují další naléhavý chirurgický/urgentní výkon a vykazují prodlouženou dobu srážení krve.
Relevantními koagulačními parametry jsou aktivovaný parciální tromboplastinový čas (aPTT), dilutovaný trombinový čas (dTT) nebo ekarinový koagulační čas (ECT).
Maximální denní dávka nebyla hodnocena.
Praxbind (2 x 2,5 g/50 ml) se podává intravenózně jako dvě po sobě následující infuze, každá v délce 5 až 10 minut, nebo jako bolusová injekce.
Pacienti léčení dabigatranem mají základní onemocnění, která je predisponují k tromboembolickým příhodám. Zrušení účinků léčby dabigatranem vystavuje pacienty riziku trombózy vzhledem k jejich základnímu onemocnění. Aby se toto riziko snížilo, je třeba zvážit obnovení antikoagulační léčby, hned jak to bude z medicínského hlediska vhodné.
Léčbu přípravkem Pradaxa (dabigatran-etexilát) lze znovu zahájit 24 hodin po podání přípravku Praxbind, pokud je pacient klinicky stabilní a bylo dosaženo odpovídající hemostázy.
Po podání přípravku Praxbind lze kdykoli zahájit jinou antitrombotickou terapii (např. nízkomolekulární heparin), pokud je pacient klinicky stabilní a bylo dosaženo odpovídající hemostázy.
Pokyny k zacházení s přípravkem:
Přípravek Praxbind nesmí být mísen s jinými léčivými přípravky. K podání přípravku Praxbind lze použít stávající intravenózní linku. Linku je nutné před infuzí a po ní propláchnout 0,9% injekčním roztokem (9 mg/ml) chloridu sodného. Stejným intravenózním přístupem se nesmí současně podávat žádná jiná infuze.
Přípravek Praxbind je určen pouze pro jednorázové použití a neobsahuje konzervanty.
Neotevřenou injekční lahvičku lze před použitím uchovávat při pokojové teplotě (25 °C) po dobu až 48 hodin, pokud je uchovávána v původním obalu, chráněna před světlem, nebo až 6 hodin bez obalu na světle. Chemická a fyzikální stabilita idarucizumabu po otevření injekční lahvičky byla prokázána na dobu 1 hodiny při pokojové teplotě.
Z mikrobiologického hlediska je třeba přípravek použít okamžitě po otevření, pokud způsob otevření nezabraňuje riziku mikrobiální kontaminace. Pokud není použit okamžitě, odpovídá za délku a podmínky uchovávání otevřeného přípravku před použitím uživatel.
Nebyly pozorovány inkompatibility mezi přípravkem Praxbind a polyvinylchloridovými, polyethylenovými ani polyuretanovými infuzními soupravami ani polypropylenovými injekčními stříkačkami.
31