Praluent 75 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Praluent 75 mg injekční roztok v předplněném peru Praluent 150 mg injekční roztok v předplněném peru Praluent 75 mg injekční roztok v předplněné injekční stříkačce Praluent 150 mg injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
75 mg injekční roztok
Jedno předplněné pero na jednorázové použití obsahuje alirocumabum 75 mg v 1 ml roztoku.
Jedna předplněná injekční stříkačka na jednorázové použití obsahuje alirocumabum 75 mg v 1 ml roztoku.
150 mg injekční roztok
Jedno předplněné pero na jednorázové použití obsahuje alirocumabum 150 mg v 1 ml roztoku.
Jedna předplněná injekční stříkačka na jednorázové použití obsahuje alirocumabum 150 mg v 1 ml roztoku.
Alirocumabum je lidská IgG1 monoklonální protilátka produkovaná rekombinantní DNA technologií v ovariálních buňkách čínského křečka.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce)
Čirý, bezbarvý až světle žlutý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Praluent je indikován k léčbě dospělých pacientů s primární hypercholesterolemií (heterozygotní familiární a nefamiliární) nebo se smíšenou dyslipidemií jako doplněk k dietním opatřením:
- v kombinaci se statinem nebo se statinem a jinou hypolipidemickou léčbou u pacientů, u kterých nelze dostáhnout cílových hodnot LDL cholesterolu maximální tolerovanou dávkou statinů, nebo
- samostatně nebo v kombinaci s jinou hypolipidemickou léčbou u pacientů, kteří netolerují statiny nebo u kterých je podávání statinů kontraindikováno.
Vliv přípravku Praluent na kardiovaskulární morbiditu a mortalitu nebyl dosud stanoven.
4.2 Dávkování a způsob podání
Dávkování
Před zahájením léčby přípravkem Praluent je třeba vyloučit sekundární příčiny hyperlipidemie nebo smíšené dyslipidemie (např. nefrotický syndrom, hypotyreózu).
Obvyklá počáteční dávka přípravku Praluent je 75 mg podaných subkutánně jednou za 2 týdny. U pacientů, u kterých je zapotřebí výraznější snížení LDL cholesterolu (> 60 %), může být počáteční dávka 150 mg podávaná jednou za 2 týdny.
Dávka přípravku Praluent může být u pacienta individuálně upravena, např. dle výchozí hodnoty LDL cholesterolu, cíle léčby nebo dle terapeutické odpovědi. Po 4 týdnech od zahájení léčby či úpravy dávky, kdy je obvykle dosažena stabilní hladina LDL cholesterolu, může být stanovena a zhodnocena hladina lipidů a následně upravena dávka (její zvýšení nebo snížení). Pacient má být léčen nejnižší možnou dávkou, které je zapotřebí, aby se dosáhlo požadovaného snížení hodnot LDL cholesterolu.
Pokud byla dávka vynechána, pacient si má podat dávku co nejdříve a poté pokračovat v podáních jednou za 2 týdny v původním schématu.
Zvláštní populace
Pediatrická populace
Bezpečnost a účinnost přípravku Praluent u dětí a dospívajících ve věku do 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Starší pacienti
U starších pacientů není nutná úprava dávky.
Porucha funkce jater
U pacientů s mírnou nebo se středně závažnou poruchou funkce jater není nutná úprava dávky. Nejsou dostupné žádné údaje o podávání pacientům se závažnou poruchou funkce jater.
Porucha funkce ledvin
U pacientů s mírnou nebo se středně závažnou poruchou funkce ledvin není nutná úprava dávky. O podávání pacientům se závažnou poruchou funkce ledvin jsou dostupné omezené údaje (viz bod 5.2).
Tělesná hmotnost
Není nutná úprava dávky dle tělesné hmotnosti pacienta.
Způsob podání Subkutánní podání.
Přípravek Praluent se podává injekčně subkutánně do stehna, břicha nebo horní části paže.
Je doporučeno pravidelné střídání místa injekční aplikace s každou injekcí.
Přípravek Praluent nemá být aplikován do míst s právě probíhající kožní nemocí nebo s úrazem, např. v místě popálenin, kožní vyrážky, zánětu nebo kožních infekcí.
Přípravek Praluent nesmí být podáván spolu s jinými přípravky podávanými injekčně do stejného místa aplikace.
Přípravek Praluent si může pacient aplikovat svépomocí nebo mu jej může aplikovat ošetřovatel(ka) po proškolení profesionálním zdravotníkem o správné technice podkožního podání.
Opatření, která je potřeba dodržovat před manipulací
Přípravek Praluent má být před použitím ponechán při pokojové teplotě, aby se vyrovnaly teplotní rozdíly (viz bod 6.6).
Každé předplněné pero nebo předplněná injekční stříkačka je pouze pro jednorázové použití.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Alergické reakce
V klinických studiích byly hlášeny případy obecných alergických reakcí, včetně svědění, ale i vzácných a někdy závažných alergických reakcí, např. hypersenzitivity, numulárního ekzému, kopřivky a hypersenzitivní vaskulitidy (viz bod 4.8). Pokud se vyskytnou známky a příznaky závažných alergických reakcí, léčba přípravkem Praluent musí být ukončena a musí být zahájena vhodná symptomatická léčba (viz bod 4.3).
Porucha funkce ledvin
V klinických studiích bylo omezené zastoupení pacientů se závažnou poruchou funkce ledvin (definovanou jako eGFR < 30 ml/min/1,73 m2) (viz bod 5.2). Přípravek Praluent má být u pacientů se závažnou poruchou funkce ledvin používán s opatrností.
Porucha funkce jater
Pacienti se závažnou poruchou funkce jater (Child-Pugh C) nebyli studováni (viz bod 5.2). Přípravek Praluent má být u pacientů se závažnou poruchou funkce jater používán s opatrností.
4.5 Interakce s jiným léčivými přípravky a jiné formy interakce
Vliv alirokumabu na jiné léčivé přípravky
Vzhledem k tomu, že je alirokumab biologický léčivý přípravek, nepředpokládá se jeho farmakokinetický vliv na jiné léčivé přípravky a na izoenzymy cytochromu P450.
Vliv jiných léčivých přípravků na alirokumab
Je známo, že statiny a jiná hypolipidemická terapie zvyšují produkci PCSK9, proteinu, na který cílí alirokumab. To vede ke zvýšení clearance touto cestou a ke snížení systémové expozice alirokumabu. Ve srovnání s monoterapií alirokumabem je expozice alirokumabu asi o 40 % nižší při současném užívání se statiny, o 15 % nižší při současném užívání s ezetimibem a o 35 % nižší při současném užívání s fenofibrátem. Avšak efekt na snížení LDL cholesterolu je v průběhu dávkovacího intervalu, kdy je alirokumab podáván jednou za 2 týdny, zachován.
4.6 Fertilita, těhotenství a kojení
Žádné údaje o podávání přípravku Praluent těhotným ženám nejsou k dispozici. Alirokumab je rekombinantní IgG1 protilátka, a proto se předpokládá jeho přechod přes placentární bariéru (viz bod 5.3). Studie na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky s ohledem na udržení těhotenství nebo vývoj embrya a plodu.V dávkách přesahujících humánní dávky byla mateřská toxicita zaznamenána u potkanů, ale ne u opic, a u potomků opic byla pozorována slabší sekundární imunitní odpověď na antigen (viz bod 5.3). Používání přípravku Praluent se nedoporučuje během těhotenství, pokud klinický stav ženy nevyžaduje léčbu alirokumabem.
Kojení
Není známo, zda se alirokumab vylučuje do lidského mateřského mléka. Lidský imunoglobulin G (IgG) je vylučován do lidského mateřského mléka, a zejména do mleziva; v tomto období kojení není přípravek Praluent doporučen. Po zbývající dobu kojení se očekává, že expozice touto cestou je nízká. Protože účinky alirokumabu na kojence nejsou známy, je nutné rozhodnout, zda v tomto období přerušit kojení nebo přerušit používání přípravku Praluent.
Fertilita
Ve studiích na zvířatech nebyly pozorovány žádné nežádoucí účinky na nepřímé ukazatele plodnosti (viz bod 5.3). Nejsou k dispozici žádné údaje o nepříznivých účincích na plodnost u lidí.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Praluent nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejčastějšími nežádoucími účinky byly lokální reakce v místě vpichu, příznaky onemocnění horních cest dýchacích a svědění. Nejčastějšími nežádoucími účinky, které vedly k přerušení léčby u pacientů léčených přípravkem Praluent, byly lokální reakce v místě vpichu.
Ve třetí fázi klinického programu nebyl pozorován žádný rozdíl v bezpečnostním profilu mezi užitými dvěma dávkami (75 mg a 150 mg).
Přehled nežádoucích účinků
Nežádoucí účinky jsou seřazeny podle třídy orgánových systémů. Četnost výskytu je definována následovně: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
Následující nežádoucí účinky byly hlášeny u pacientů léčených alirokumabem v souhrnných kontrolovaných studiích:
Tabulka 1: Nežádoucí účinky hlášené u pacientů léčených alirokumabem _v souhrnných kontrolovaných studiích_
|
Třídy orgánových systémů |
Časté |
Vzácné |
|
Poruchy imunitního systému |
Hypersenzitivita, hypersenzitivní vaskulitida | |
|
Respirační, hrudní a mediastinální poruchy |
Příznaky onemocnění horních cest dýchacích* | |
|
Poruchy kůže a podkožní tkáně |
Kopřivka, numulární ekzém | |
|
Celkové poruchy a reakce v místě aplikace |
Reakce v místě vpichu** |
* Zahrnující především orofaryngeální bolest, rinoreu, kýchání ** Zahrnující erytém/zčervenání, svědění, otoky, bolest/citlivost
Popis vybraných nežádoucích účinků
Lokální reakce v místě vpichu
Lokální reakce v místě vpichu, včetně erytému/zčervenání, svědění, otoků a bolesti/citlivosti, byly hlášeny u
6,1 % pacientů léčených alirokumabem oproti 4,1 % v kontrolní skupině (kterým byly podávány injekce placeba). Lokální reakce v místě vpichu byly nejčastěji přechodné a mírné intenzity. Četnost přerušení léčby v důsledku lokální reakce v místě vpichu byla mezi těmito dvěma skupinami srovnatelná (0,2 % ve skupině s alirokumabem oproti 0,3% v kontrolní skupině).
Celkové alergické reakce
Celkové alergické reakce byly ve skupině s alirokumabem hlášeny častěji (8,1 % pacientů) než v kontrolní skupině (7,0 % pacientů), zejména v důsledku rozdílu ve výskytu svědění. Typické pozorované případy svědění byly mírné a přechodné. Kromě toho byly v kontrolovaných klinických studiích hlášeny vzácné a někdy závažné alergické reakce, např. hypersenzitivita, numulární ekzém, kopřivka a hypersenzitivní vaskulitida (viz bod 4.4).
Zvláštní populace
Starší pacienti
Ačkoli u pacientů starších 75 let nebyly pozorovány žádné bezpečnostní problémy, údaje jsou u této věkové skupiny omezené.
V kontrolovaných studiích bylo 1158 pacientů (34,7 %) léčených přípravkem Praluent ve věku > 65 let a 241 pacientů (7,2 %) léčených přípravkem Praluent ve věku > 75 let. S narůstajícím věkem nebyly pozorovány žádné významné rozdíly v bezpečnosti a účinnosti.
Hodnoty LDL cholesterolu < 25 mg/dl (< 0,65 mmol/l)
V souhrnných kontrolovaných studiích mělo 796 z 3340 pacientů (23,8 %) léčených přípravkem Praluent dvě po sobě jdoucí hodnoty LDL cholesterolu < 25 mg/dl (< 0,65 mmol/l), včetně 288 pacientů (8,6 %) se dvěma po sobě následujícími hodnotami < 15 mg/dl (< 0,39 mmol/l). Toto většinou nastalo, když byla terapie zahájena a dále udržována na dávce 150 mg přípravku Praluent podávaného v intervalu 2 týdnů bez ohledu na počáteční hodnotu LDL cholesterolu nebo bez úpravy léčby. V souvislosti s těmito hodnotami LDL cholesterolu nebyl identifikován žádný nežádoucí účinek.
Imunogenicita/protilátky proti léčivu (Anti-drug-antibodies, ADA)
Ve 3. fázi klinického výzkumu se u 4,8 % pacientů léčených alirokumabem vyvinula ADA odpověď vyžadující léčbu ve srovnání s 0,6 % v kontrolní skupině (placebo nebo ezetimib). Většina z těchto pacientů vykazovala přechodně nízký pozitivní titr ADA s žádnou neutralizační aktivitou. Ve srovnání s pacienty, kteří byli ADA negativní, nevykazovali pacienti s pozitivním ADA žádný rozdíl v expozici alirokumabu, účinnosti nebo bezpečnosti, s výjimkou vyšší míry reakcí v místě vpichu. Pouze 1,2 % pacientů vykazovalo neutralizační protilátky (neutralising antibodies, NAB), všichni ve skupině alirokumabu. Většina z těchto pacientů měla jen jeden pozitivní neutralizační vzorek. Pouze 10 pacientů (0,3 %) mělo dva nebo více pozitivních vzorků NAB. Tyto údaje nenaznačují spojitost mezi přítomností NAB a účinností na snížení LDL cholesterolu nebo bezpečností. Údaje týkající se imunogenicity jsou vysoce závislé na senzitivitě a specifitě testu ADA.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V kontrolovaných klinických studiích nebyly identifikovány žádné bezpečnostní problémy při častějším dávkování, než při doporučeném dávkovacím schématu jednou za 2 týdny. Neexistuje žádná specifická léčba předávkování přípravkem Praluent. V případě předávkování má být pacient léčen symptomaticky a mají být podle potřeby zajištěna podpůrná opatření.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiné látky upravující hladinu lipidů, ATC kód: C10AX14.
Mechanismus účinku
Alirokumab je plně humánní monoklonální IgG1 protilátka, která má vysokou afinitu a specificitu k proprotein konvertáze subtilisin kexinu typu 9 (PCSK9). PCSK9 se váže na receptory pro lipoprotein s nízkou hustotou (LDLR) na povrchu hepatocytů, což podporuje degradaci LDLR v játrech. LDLR je hlavním receptorem, který vychytává cirkulující LDL cholesterol, proto pokles exprese LDLR pomocí PCSK9 má za následek vyšší hladiny LDL cholesterolu v krvi. Inhibicí navázání PCSK9 na LDLR zvyšuje alirokumab počet LDLR dostupných k vychytávání LDL, čímž se snižuje hladina LDL cholesterolu.
LDLR také váže na triglyceridy bohaté VLDL remnantní lipoproteiny a lipoproteiny střední hustoty (IDL). Proto léčba alirokumabem může snížit hladinu těchto remnantních lipoproteinů, což se projeví snížením apolipoproteinu B (Apo B), non-HDL cholesterolu a triglyceridů (TG). Podání alirokumabu má také za následek snížení lipoproteinu (a) [Lp (a)], což je forma LDL cholesterolu, která se váže na apolipoprotein (a). Nicméně bylo prokázáno, že LDLR má k Lp (a) nízkou afinitu, a proto přesný mechanismus, kterým alirokumab snižuje Lp (a), není zcela objasněn.
V genetických studiích u lidí byly identifikovány varianty PCSK9 s mutacemi, při kterých došlo ke změně jejich funkce buď ve smyslu snížení (tzv. loss of function, ztráta funkce), nebo zvýšení (tzv. gain of function, získání funkce). Jedinci s jednou alelou PCSK9 s mutací loss of function mají nižší hladinu LDL cholesterolu, což koreluje s významně nižším výskytem ischemické choroby srdeční. Bylo hlášeno několik jedinců, kteří nesou dvě alely PCSK9 s mutací loss of function a kteří mají významně nízké hladiny LDL cholesterolu s normální hladinou HDL cholesterolu a TG. Naopak u pacientů se zvýšenou hladinou LDL cholesterolu a klinickou diagnózou familiární hypercholesterolemie byla v PCSK9 genu identifikována mutace gain of function.
V multicentrické, dvojitě zaslepené, placebem kontrolované, 14 týdnů trvající studii s 13 pacienty
s heterozygotní familiární hypercholesterolemií (HeFH) v důsledku mutace gain of function PCSK9 genu byli pacienti randomizováni buď do skupiny užívající alirokumab 150 mg jednou za 2 týdny, nebo do skupiny užívající placebo. Střední výchozí hodnota LDL cholesterolu byla 151,5 mg/dl (3,90 mmol/l). V 2. týdnu bylo průměrné snížení LDL cholesterolu oproti výchozím hodnotám 62,5 % ve skupině pacientů léčených alirokumabem ve srovnání s 8,8 % u pacientů užívajících placebo. V 8. týdnu bylo průměrné snížení LDL cholesterolu u pacientů léčených alirokumabem 72,4 % oproti výchozí hodnotě.
Farmakodynamické účinky
V in vitro testech neindukoval alirokumab aktivitu efektorových buněk prostřednictvím Fc receptoru (buňkami zprostředkovanou toxicitu závislou na protilátkách a cytotoxicitu závislou na komplementu), a to nezávisle na přítomnosti či nepřítomnosti PCSK9, a proti alirokumabu ve vazbě na PCSK9 nebyly zjištěny žádné rozpustné imunokomplexy schopné vázat proteiny komplementu.
Klinická účinnost a bezpečnost
Shrnutí studií třetí fáze klinického programu
Účinnost alirokumabu byla zkoumána v deseti studiích 3. fáze klinického výzkumu (pěti placebem kontrolovaných a pěti ezetimibem kontrolovaných studiích) zahrnujících 5296 pacientů randomizovaných do skupin s hypercholesterolemií (heterozygotní familiární a nefamiliární) nebo se smíšenou dyslipidemií, přičemž 3188 pacientů bylo randomizováno do skupiny užívající alirokumab. Ve třetí fázi studií mělo 31 % pacientů onemocnění diabetes mellitus 2. typu a 64 % pacientů mělo v anamnéze ischemickou chorobou srdeční. Tři z deseti studií byly provedeny výhradně u pacientů s heterozygotní familiární hypercholesterolemií (HeFH). Většina pacientů ve třetí fázi programu měla vysoké nebo velmi vysoké kardiovaskulární (KV) riziko a měla hypolipidemickou léčbu zahrnující maximální tolerované dávky statinů s další hypolipidemickou léčbou nebo bez ní. U pacientů, kteří nebyli současně léčeni statiny, byly provedeny dvě studie včetně jedné studie u pacientů s dokumentovanou statinovou intolerancí.
Celkem 2416 pacientů bylo zahrnuto ve dvou studiích (LONG TERM a HIGH FH), které byly provedeny pouze s dávkou 150 mg jednou za 2 týdny. Osm studií bylo provedeno s dávkou 75 mg jednou za 2 týdny s tím, že v případě nedosažení cílových hodnot LDL cholesterolu v 8. týdnu na základě předdefinovaných kritérií podle míry kardiovaskulárního rizika byla dávka ve 12. týdnu zvýšena na 150 mg jednou za 2 týdny.
Primárním kritériem účinnosti ve všech studiích fáze 3 bylo střední percentuální snížení výchozí hodnoty LDL cholesterolu ve 24. týdnu ve srovnání s placebem nebo ezetimibem. Ve všech studiích bylo primárního kritéria dosaženo. Obecně platí, že podávání alirokumabu také vede ke statisticky významně většímu percentuálnímu snížení hladiny celkového cholesterolu, non-HDL cholesterolu, apolipoproteinu B (Apo B) a lipoproteinu (a) [Lp (a )] ve srovnání s placebem/ezetimibem u pacientů, kteří byli či nebyli současně léčeni statiny. Alirokumab také snižuje triglyceridy (TG) a zvyšuje HDL cholesterol a apolipoprotein A-1 (Apo A-1) ve srovnání s placebem. Podrobné výsledky viz Tabulka 2 níže. Snížení LDL cholesterolu bylo pozorováno bez ohledu na věk, pohlaví, BMI, rasu, výchozí hladinu LDL cholesterolu, přítomnost či nepřítomnost HeFH, přítomnost smíšené dyslipidemie a diabetes. Ačkoli podobná účinnost byla pozorována u pacientů nad 75 let, údaje jsou u této věkové skupiny omezené. Snižování LDL cholesterolu bylo konzistentní bez ohledu na souběžnou léčbu statiny a jejich dávky. Významně vyšší podíl pacientů dosáhl hladiny LDL cholesterolu < 70 mg/dl (< 1,81 mmol/l) ve skupině užívající alirokumab ve srovnání se skupinou užívající placebo nebo ezetimib ve 12. týdnu a 24. týdnu. Ve studiích založených na režimu zvyšování dávek pomocí titrace většina pacientů dosáhla předem definované cílové hladiny LDL cholesterolu (založené na KV riziku) při dávce 75 mg jednou za 2 týdny a udržovací terapie u většiny pacientů zůstala na dávce 75 mg jednou za 2 týdny. Hypolipidemický účinek alirokumabu byl pozorován do 15 dnů po první dávce, maximum účinku bylo dosaženo přibližně po 4 týdnech. Při dlouhodobé léčbě účinnost přetrvávala na stejné úrovni po celou dobu trvání studií (až po dobu 2 let). Po vysazení alirokumabu nebyl pozorován tzv. rebound fenomén hladiny LDL cholesterolu, jehož hladina se k výchozím hodnotám vrátila postupně.
V předem specifikovaných analýzách před možným titrováním dávky ve 12. týdnu bylo v 8 studiích, ve kterých pacienti začali léčbu dávkou 75 mg jednou za 2 týdny, dosaženo průměrného snížení LDL cholesterolu v rozmezí od 44,5 % do 49,2 %. Ve 2 studiích, v nichž byla zahájena a udržována dávka 150 mg jednou za 2 týdny, bylo ve 12. týdnu dosaženo průměrného snížení LDL cholesterolu o 62,6%.
V souhrnných analýzách klinických studií třetí fáze, které umožnily titraci dávky, došlo v podskupině pacientů, kterým byla zvyšována dávka alirokumabu ze 75 mg jednou za 2 týdny na 150 mg jednou za
2 týdny ve 12. týdnu k dalšímu 14% průměrnému snížení hladiny LDL cholesterolu u pacientů současně užívající statin. U pacientů, kteří současně neužívali statin, vedla titrace dávky k dalšímu 3% průměrnému snížení hladiny LDL cholesterolu; největší účinek byl přitom pozorován u přibližně 25 % pacientů, kteří po zvýšení dávky dosáhli minimálně dalšího 10% snížení hladiny LDL cholesterolu. Pacienti, kterým byla titrována dávka na 150 mg jednou za 2 týdny, měli vyšší průměrnou výchozí hodnotu hladiny LDL cholesterolu.
Hodnocení kardiovaskulárních (KV) příhod
V současné době probíhá klinická studie, jejímž primárním cílem je sledování nežádoucích závažných kardiovaskulárních příhod (tzv. MACE - major adverse cardiovascular events, tj. úmrtí v následku ischemické choroby srdeční, infarkt myokardu, ischemická cévní mozková příhoda a nestabilní angina vyžadující hospitalizaci).
V předem stanovených souhrnných analýzách studií 3. fáze klinického výzkumu byly zaznamenány kardiovaskulární příhody vyžadující léčbu, které byly potvrzeny posouzením odborné komise a které sestávaly z úmrtí v následku ischemické choroby srdeční, infarktu myokardu, ischemické cévní mozkové příhody, nestabilní anginy pectoris vyžadující hospitalizaci, městnavého srdečního selhání vyžadujícího hospitalizaci a z revaskularizace, u 110 (3,5 %) pacientů ve skupině užívající alirokumab a u 53 (3,0 %) pacientů v kontrolní skupině (placebo nebo aktivní kontrola) s poměrem rizik HR = 1,08 (95% CI; 0,78 -1,50). MACE potvrzená posouzením odbornou komisí byly hlášeny u 52 z 3182 (1,6 %) pacientů ve skupině užívající alirokumab a u 33 z 1792 (1,8 %) pacientů v kontrolní skupině (placebo nebo aktivní kontrola);
HR = 0,81 (95% CI; 0,52 - 1,25).
V předem stanovené závěrečné analýze studie LONG TERM se vyskytly kardiovaskulární příhody potvrzené posouzením odborné komise a vyžadující léčbu u 72 z 1550 (4,6 %) pacientů ve skupině léčených alirokumabem a u 40 ze 788 (5,1 %) pacientů ve skupině užívající placebo; MACE potvrzená posouzením odbornou komisí byly hlášeny u 27 z 1550 (1,7 %) pacientů ve skupině užívající alirokumab a u 26 ze 788 (3,3 %) pacientů ve skupině užívající placebo. Poměry rizik (HR) byly vypočteny následně (post-hoc): pro všechny kardiovaskulární příhody bylo HR = 0,91 (95% CI; 0,62 - 1,34); pro mAcE bylo HR = 0,52 (95% CI; 0,31-0,90).
Celková mortalita
Celková mortalita ve studiích 3. fáze byla 0,6 % (20 z 3182 pacientů) ve skupině užívající alirokumab a 0,9 % (17 z 1792 pacientů) v kontrolní skupině. Hlavní příčinou úmrtí u většiny těchto pacientů byly KV příhody.
Kombinovaná terapie se statinem
Placebem kontrolované studie třetí fáze (se současně užívaným statinem) u pacientů s primární hypercholesterolemií nebo smíšenou dyslipidemií
Studie LONG TERM
Tato multicentrická, dvojitě zaslepená, placebem kontrolovaná, 18 měsíců trvající studie zahrnovala 2310 pacientů s primární hypercholesterolemií s vysokým nebo velmi vysokým KV rizikem, kterým byla podávána maximální tolerovaná dávka statinu s další hypolipidemickou terapií nebo bez ní. Pacienti užívali k jejich stávající hypolipidemické terapii buď alirokumab v dávce 150 mg jednou za 2 týdny nebo placebo. LONG TERM studie zahrnovala 17,7 % pacientů s HeFH, 34,6 % s onemocněním diabetes mellitus 2. typu a 68,6 % ischemickou chorobou srdeční v anamnéze. V týdnu 24 byl rozdíl mezi skupinami s alirokumabem a placebem v průměrném percentuálním snížení hladiny LDL cholesterolu proti výchozím hodnotám -61,9 % (95% CI: -64,3%, -59,4%, p < 0,0001). Podrobné výsledky viz Tabulka 2. Ve 12. týdnu dosáhlo 82,1 % pacientů ve skupině alirokumabu hodnoty LDL cholesterolu < 70 mg/dl (< 1,81 mmol/l) ve srovnání se
7,2 % u pacientů ve skupině s placebem. Rozdíl v porovnání s placebem byl statisticky signifikantní ve 24. týdnu pro všechny lipidy/lipoproteiny.
Studie COMBOI
Multicentrická, dvojitě zaslepená, placebem-kontrolovaná, 52 týdnů trvající studie zahrnovala 311 pacientů, kteří byli klasifikováni jako pacienti s velmi vysokým kardiovaskulárním rizikem a kteří nedosahovali předem definované cílové hladiny LDL cholesterolu navzdory maximálně tolerované dávce statinu, s další hypolipidemickou terapií nebo bez ní. Pacientům byl k jejich stávající hypolipidemické léčbě podáván buď alirokumab v dávce 75 mg jednou za 2 týdny, nebo placebo. Pacientům s hodnotou LDL cholesterolu > 70 mg/dl (> 1,81 mmol/l) byla dávka alirokumabu ve 12. týdnu titrována na dávku 150 mg jednou za 2 týdny. Ve 24. týdnu byl rozdíl při léčbě proti placebu v průměrném percentuálním snížení hodnoty LDL cholesterolu oproti výchozí hodnotě -45,9 % (95% CI: -52,5%, -39,3%, p < 0,0001). Podrobné výsledky viz Tabulka 2. Ve 12. týdnu (před titrací dávky) dosáhlo 76,0 % pacientů ve skupině užívající alirokumab hodnotu LDL cholesterolu < 70 mg/dl (< 1,81 mmol/l) ve srovnání s 11,3 % pacientů ve skupině užívající placebo. Dávka byla titrována na dávku 150 mg jednou za 2 týdny u 32 (16,8 %) pacientů léčených déle než 12 týdnů. V podskupině pacientů, kterým byla dávka zvýšena ve 12. týdnu, bylo dosaženo ve 24. týdnu dalšího 22,8% průměrného snížení hodnoty LDL cholesterolu. Rozdíl ve srovnání s placebem byl statisticky významný ve 24. týdnu pro všechny lipidy/lipoproteiny s výjimkou TG a Apo A-1.
Placebem kontrolované studie 3. fáze u pacientů léčených statiny s heterozygotní familiární hypercholesterolemií (HeFH)
Studie FHI a FHII
Dvě multicentrické, placebem kontrolované, dvojitě zaslepené 18 měsíců trvající studie zahrnovaly 732 pacientů s HeFH, kterým byla podávána maximálně tolerovaná dávka statinu s další hypolipidemickou terapií nebo bez ní. Pacientům byl k jejich stávající hypolipidemické léčbě podáván buď alirokumab v dávce 75 mg jednou za 2 týdny nebo placebo. U pacientů s hodnotou LDL cholesterolu > 70 mg/dl (> 1,81 mmol/l) byla dávka alirokumabu ve 12. týdnu titrována na dávku 150 mg jednou za 2 týdny. Ve 24. týdnu byl rozdíl při léčbě oproti placebu v průměrném percentuálním snížení hodnoty LDL cholesterolu proti výchozí hodnotě -55,8% (95% CI: -60,0%, -51,6%, p < 0,0001). Podrobné výsledky viz Tabulka 2. Ve 12. týdnu (před titrací dávky) dosáhlo 50,2 % pacientů hodnotu LDL cholesterolu < 70 mg/dl (< 1,81 mmol/l) ve srovnání s 0,6 % pacientů ve skupině užívající placebo. V podskupině pacientů, kterým byla dávka zvýšena ve 12. týdnu, bylo dosaženo ve 24. týdnu dalšího 15,7% průměrného snížení hodnoty LDL cholesterolu. Rozdíl ve srovnání s placebem byl statisticky významný ve 24. týdnu pro všechny lipidy/lipoproteiny.
Studie HIGH FH
Třetí multicentrická, dvojitě zaslepená, placebem-kontrolovaná 18 měsíců trvající studie zahrnovala 106 pacientů s HeFH, kterým byla podávána maximálně tolerovaná dávka statinu s další hypolipidemickou terapií nebo bez ní a jejichž výchozí hodnota LDL cholesterolu byla > 160 mg/dl (> 4,14 mmol/l). Pacientům byl k jejich stávající hypolipidemické léčbě podáván buď alirokumab v dávce 150 mg jednou za 2 týdny nebo placebo. Ve 24. týdnu rozdíl při léčbě oproti placebu v průměrném percentuálním snížení hodnoty LDL cholesterolu proti výchozí hodnotě -39,1 % (95% CI: -51,1%, -27,1%, p < 0,0001). Podrobné výsledky viz Tabulka 2. Průměrné změny pro všechny ostatní lipidy/lipoproteiny byly podobné jako ve studiích FH I a FH II, nicméně nebylo dosaženo statisticky významného rozdílu v hodnotách TG, HDL cholesterolu a apo A-1.
Ezetimibem kontrolovaná studie 3. fáze (se současně užívanými statiny) u pacientů s primární hypercholesterolemií nebo smíšenou dyslipidemií
Studie COMBOII
V multicentrické, dvojitě zaslepené, ezetimibem kontrolované 2 roky trvající studii bylo zahrnuto 707 pacientů, kteří byli klasifikováni jako pacienti s velmi vysokým kardiovaskulárním rizikem a kteří nedosahovali předem definovanou cílovou hodnotu LDL cholesterolu navzdory maximálně tolerované dávce statinu. Pacientům byl k jejich stávající léčbě statinem podáván buď alirokumab v dávce 75 mg jednou za
2 týdny nebo ezetimib 10 mg jednou denně. Pacientům s hodnotou LDL cholesterolu > 70 mg/dl (> 1,81 mmol/l) byla dávka alirokumabu ve 12. týdnu titrována na dávku 150 mg jednou za 2 týdny. Ve 24. týdnu byl rozdíl při léčbě alirokumabem oproti ezetimibu v průměrném percentuálním snížení hodnoty LDL cholesterolu oproti výchozí hodnotě -29,8 % (95% CI: -34,4%, -25,3%, p < 0,0001). Podrobné výsledky viz Tabulka 2. Ve 12. týdnu (před titrací dávky) dosáhlo 77,2 % pacientů hodnotu LDL cholesterolu < 70 mg/dl (< 1,81 mmol/l) ve srovnání s 46,2 % pacientů ve skupině užívající ezetimib. V podskupině pacientů, kterým byla dávka zvýšena ve 12. týdnu, bylo dosaženo ve 24. týdnu dalšího 10,5% průměrného snížení hodnoty LDL cholesterolu. Rozdíl ve srovnání s ezetimibem byl statisticky významný ve 24. týdnu pro všechny lipidy/lipoproteiny s výjimkou TG a Apo A-1.
Monoterapie alirokumabem nebo doplněk k hypolipidemické terapii kromě statinů
Ezetimibem kontrolované studie 3. fáze u pacientů s primární hypercholesterolemií (bez současně užívaného statinu)
Studie ALTERNATIVE
Multicentrická, dvojitě zaslepená, ezetimibem kontrolovaná, 24 týdnů trvající studie zahrnovala 248 pacientů se zdokumentovanou statinovou intoleranci z důvodů příznaků souvisejících s kosterním svalstvem. Pacientům byl podáván buď alirokumab v dávce 75 mg jednou za 2 týdny nebo ezetimib 10 mg jednou denně, případně místo ezetimibu atorvastatin 20 mg jednou denně (jako opakovaný terapeutický pokus pro ověření intolerance). U pacientů s hodnotou LDL cholesterolu > 70 mg/dl (> 1,81 mmol/l) nebo > 100 mg/dl (> 2,59 mmol/l) v závislosti na míře KV rizika byla ve 12. týdnu dávka alirokumabu zvýšena na dávku 150 mg jednou za 2 týdny. Ve 24. týdnu byl rozdíl při léčbě alirokumabem oproti ezetimibu v průměrném percentuálním snížení hodnoty LDL cholesterolu proti výchozí hodnotě -30.4% (95% CI: -36.6%, -24.2%; p<0,0001). Podrobné výsledky viz Tabulka 2. Ve 12. týdnu (před titrací dávky) dosáhlo 34,9 % pacientů hodnoty LDL cholesterolu < 70 mg/dl (< 1,81 mmol/l) ve srovnání s 0 % pacientů ve skupině užívající ezetimib. V podskupině pacientů, kterým byla dávka zvýšena ve 12. týdnu, bylo dosaženo ve 24. týdnu dalšího průměrného snížení hodnoty LDL cholesterolu o 3,6%. Rozdíl ve srovnání s ezetimibem byl statisticky významný ve 24. týdnu pro LDL cholesterol, celkový cholesterol, non-HDL cholesterol, Apo B a Lp(a).
Tato studie hodnotila pacienty, kteří netolerovali alespoň dva statiny (z toho alespoň jeden v nejnižší schválené dávce). U těchto pacientů se nežádoucí účinky na kosterní svalstvo vyskytly s nižší četností ve skupině užívající alirokumab (32,5 %) ve srovnání se skupinou užívající atorvastatin (46,0 %) (HR = 0,61 [95% CI; 0,38-0,99]) a menší podíl pacientů přerušil léčbu ve skupině užívající alirokumab (15,9 %) v důsledku nežádoucích svalových účinků ve srovnání se skupinou užívající atorvastatin (22,2 %). V pěti placebem kontrolovaných studiích u pacientů užívajících maximální tolerované dávky statinů (n = 3752) byla četnost přerušení kvůli nežádoucím svalovým účinkům 0,4 % ve skupině užívající alirokumab a 0,5 % ve skupině užívající placebo.
Studie MONO
V multicentrické, dvojitě zaslepené, ezetimibem kontrolované, 24 týdnů trvající studii bylo zahrnuto
103 pacientů se středním KV rizikem, kteří neužívali statiny nebo jinou hypolipidemickou léčbu a jejichž
výchozí hodnota LDL cholesterolu byla v rozmezí od 100 mg/dl (2,59 mmol/l) až 190 mg/dl (4,91 mmol/l). Pacientům byl podáván buď alirokumab v dávce 75 mg jednou za 2 týdny nebo ezetimib 10 mg jednou denně. U pacientů s hodnotou LDL cholesterolu > 70mg/dl (> 1,81 mmol/l) byla ve 12. týdnu dávka alirokumabu zvýšena na dávku 150 mg jednou za 2 týdny. Ve 24. týdnu byl rozdíl při léčbě alirokumabem proti ezetimibu v průměrném percentuálním snížení hodnoty LDL cholesterolu oproti výchozí hodnotě -31,6 % (95% CI: -40,2%, -23,0%, p <0,0001). Podrobné výsledky viz Tabulka 2. Ve 12. týdnu (před titrací dávky) dosáhlo 57,7 % pacientů hodnotu LDL cholesterolu < 70 mg/dl (< 1,8 mmol/l) ve srovnání s 0 % pacientů ve skupině užívající ezetimib. Dávka byla titrována až na dávku 150 mg jednou za 2 týdny u 14 (30,4 %) pacientů léčených déle než 12 týdnů. V podskupině pacientů, kterým byla dávka titrována ve 12. týdnu, bylo dosaženo ve 24. týdnu dalšího 1,4% průměrného snížení hodnoty LDL cholesterolu. Rozdíl ve srovnání s ezetimibem byl statisticky významný ve 24. týdnu pro HDL cholesterol, celkový cholesterol, non-HDL cholesterol a Apo B.
Tabulka 2: Průměrná percentuální změna oproti výchozí hodnotě v hladinách LDL cholesterolu a jiných lipidů/lipoproteinů v placebem kontrolovaných a ezetimibem kontrolovaných studiích
|
Průměrná percentuální změna oproti výchozí hodnotě v placebem kontrolovaných studiích se současně užívaným statinem | ||||||||
|
LONG TERM (N=2310) |
FHI a FHII (N=732) |
High FH (N=106) |
COMBO I (N=311) | |||||
|
Placebo |
Alirokumab |
Placebo |
Alirokumab |
Placebo |
Alirokumab |
Placebo |
Alirokumab | |
|
Počet pacientů |
780 |
1530 |
244 |
488 |
35 |
71 |
106 |
205 |
|
Průměrná výchozí hodnota LDL cholesterolu v mg/dl (mmol/l) |
122,0 (3,16) |
122,8 (3,18) |
140,9 (3,65) |
141,3 (3,66) |
201,0 (5,21) |
196,3 (5,10) |
104,6 (2,71) |
100,3 (2,60) |
|
12. týden | ||||||||
|
LDL cholesterol (ITT)a |
1,5 |
-63,3 |
5,4 |
-43,6 |
-6,6 |
-46,9 |
1,1 |
-46,3 |
|
LDL cholesterol (On treatment)b |
1,4 |
-64,2 |
5,3 |
-44,0 |
-6,6 |
-46,9 |
1,7 |
-47,6 |
|
24. týden | ||||||||
|
LDL cholesterol (ITT)a |
0,8 |
-61,0c |
7,1 |
-48,8d |
-6,6 |
-45,7e |
-2,3 |
-48,2f |
|
LDL cholesterol (On treatment)b |
0,7 |
-62,8 |
6,8 |
-49,3 |
-6,6 |
-45,5 |
-0,8 |
-50,7 |
|
Non-HDL cholesterol |
0,7 |
-51,6 |
7,4 |
-42,8 |
-6,2 |
-41,9 |
-1,6 |
-39,1 |
|
Apo B |
1,2 |
-52,8 |
1,9 |
-41,7 |
-8,7 |
-39,0 |
-0,9 |
-36,7 |
|
Celkový cholesterol |
-0,3 |
-37,8 |
5,5 |
-31,2 |
-4,8 |
-33,2 |
-2,9 |
-27,9 |
|
Lp(a) |
-3,7 |
-29,3 |
-8,5 |
-26,9 |
-8,7 |
-23,5 |
-5,9 |
-20,5 |
|
TG |
1,8 |
-15,6 |
4,3 |
-9,8 |
-1,9 |
-10,5 |
-5,4 |
-6,0 |
|
HDL cholesterol |
-0,6 |
4,0 |
0,2 |
7,8 |
3,9 |
7,5 |
-3,8 |
3,5 |
|
Apo A-1 |
1,2 |
4,0 |
-0,4 |
4,2 |
2,0 |
5,6 |
-2,5 |
3,3 |
|
Průměrná % změna oproti výchozí |
hodnotě v ezetimibem-kontrolovaných studiích | |||||
|
Se současně užívaným statinem |
Bez současně užívaného statinu | |||||
|
COMBO II (N=707) |
ALTERNATIVE (N=248) |
MONO (N=103) | ||||
|
Ezetimib |
Alirokumab |
Ezetimib |
Alirokumab |
Ezetimib |
Alirokumab | |
|
Počet pacientů |
240 |
467 |
122 |
126 |
51 |
52 |
|
Střední výchozí hodnota LDL cholesterolu v mg/dl (mmol/l) |
104,5 (2,71) |
108,3 (2,81) |
194,2 (5,03) |
191,1 (5,0) |
138,3 (3,58) |
141,1 (3,65) |
|
12. týden | ||||||
|
LDL cholesterol (ITT )a |
-21,8 |
-51,2 |
-15,6 |
-47,0 |
-19,6 |
-48,1 |
|
LDL cholesterol (On treatment) |
-22,7 |
-52,4 |
-18,0 |
-51,2 |
-20,4 |
-53,2 |
|
24. týden | ||||||
|
LDL cholesterol (ITT)a |
-20,7 |
-50,6g |
-14,6 |
-45,0h |
-15,6 |
-47,2' |
|
LDL cholesterol (On treatment)b |
-21,8 |
-52,4 |
-17,1 |
-52,2 |
-17,2 |
-54,1 |
|
Non-HDL cholesterol |
-19,2 |
-42,1 |
-14,6 |
-40,2 |
-15,1 |
-40,6 |
|
Apo B |
-18,3 |
-40,7 |
-11,2 |
-36,3 |
-11,0 |
-36,7 |
|
Celkový cholesterol |
-14,6 |
-29,3 |
-10,9 |
-31,8 |
-10,9 |
-29,6 |
|
Lp(a) |
-6,1 |
-27,8 |
-7,3 |
-25,9 |
-12,3 |
-16,7 |
|
TG |
-12,8 |
-13,0 |
-3,6 |
-9,3 |
-10,8 |
-11,9 |
|
HDL cholesterol |
0,5 |
8,6 |
6,8 |
7,7 |
1,6 |
6,0 |
|
Apo A-1 |
-1,3 |
5,0 |
2,9 |
4,8 |
-0,6 |
4,7 |
a ITT analýza - intention-to-treat populace zahrnující všechna data o hladinách lipidů napříč trváním studie bez ohledu na adherenci jednotlivých pacientů ke studii.
b On treatment analýza - analýza omezená na pacienty, kteří byli skutečně léčeni.
Percentuální snížení LDL cholesterolu ve 24. týdnu odpovídá střední absolutní změně:c -74,2 mg/dl (-1,92 mmol/l);d -
71.1 mg/dl (-1,84 mmol/l); e -90,8 mg/dl (-2,35 mmol/l); f -50,3 mg/dl (-1,30 mmol/l); g -55,4 mg/dl (1,44 mmol/l); h -
84.2 mg/dl (-2,18 mmol/l); 1 -66,9 mg/dl (-1,73 mmol/l).
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Praluent u jedné nebo více podskupin pediatrické populace v léčbě hypercholesterolemie (informace o použití u dětí viz bod 4.2).
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Praluent u všech podskupin pediatrické populace v léčbě smíšené dyslipidemie (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Po subkutánním podání 50 mg až 300 mg alirokumabu byl medián doby dosažení maximální koncentrace v séru (tmax) 3 až 7 dnů. Po jednorázovém subkutánním podání 75 mg do oblasti břicha, horní části paže nebo stehna byla farmakokinetika alirokumabu podobná. Populačními farmakokinetickými analýzami byla stanovena absolutní biologická dostupnost alirokumabu po subkutánním podání na přibližně 85 %. Rovnovážného stavu bylo dosaženo po 2-3 dávkách s asi dvojnásobným akumulačním poměrem.
Distribuce
Distribuční objem po intravenózním podání je v rozmezí od 0,04 do 0,05 l/kg, což naznačuje, že alirokumab je distribuován především v oběhovém systému.
Biotransformace
Specifické studie metabolismu nebyly provedeny, protože alirokumab je protein. Předpokládá se, že se alirokumab degraduje na malé peptidy a jednotlivé aminokyseliny.
Eliminace
Pro alirokumab byly pozorovány dvě eliminační fáze. Při nízkých koncentracích je eliminace zprostředkována převážně přes saturovatelnou vazbu na cílovou molekulu (PCSK9), zatímco při vyšších koncentracích je alirokumab eliminován do značné míry prostřednictvím nesaturovatelné proteolytické dráhy.
Na základě populační farmakokinetické analýzy byl střední zdánlivý poločas alirokumabu v rovnovážném stavu 17-20 dní u pacientů užívajících alirokumab v monoterapii v podkožních dávkách buď 75 mg jednou za 2 týdny nebo 150 mg jednou za 2 týdny. Při souběžném užívání se statinem byl střední zdánlivý poločas alirokumabu 12 dní.
Linearita/nelinearita
Při zdvojnásobení dávek ze 75 mg jednou za 2 týdny na 150 mg jednou za 2 týdny byl pozorován mírně vyšší nárůst koncentrace, než by odpovídalo 2x vyšší dávce, a to v rozmezí od 2,1 do 2,7násobku.
Zvláštní populace
Starší pacienti
Na základě populační farmakokinetické analýzy byl věk spojen s malým rozdílem v expozici alirokumabu v ustáleném stavu bez vlivu na účinnost nebo bezpečnost.
Pohlaví
Na základě populační farmakokinetické analýzy nemá pohlaví vliv na farmakokinetiku alirokumabu.
Rasa
Na základě populační farmakokinetické analýzy nemá rasa žádný vliv na farmakokinetiku alirokumabu.
Po jednotlivé dávce subkutánního podání 100 mg až 300 mg alirokumabu nebyl zjištěn žádný významný rozdíl v expozici mezi japonskou a bělošskou populací.
Tělesná hmotnost
Jako významná proměnná ovlivňující farmakokinetiku alirokumabu byla v konečném populačním farmakokinetickém modelu identifikována tělesná hmotnost. Expozice alirokumabu (AUC0-J4d) v rovnovážném stavu jak u dávkovacího režimu 75 mg, tak 150 mg jednou za 2 týdny byla snížena o 29 %, resp. 36 % u pacientů s hmotností více než 100 kg ve srovnání s pacienty s hmotností mezi 50 kg a 100 kg. Tento rozdíl však nevedl ke klinicky významnému rozdílu v účinku na snižování hladiny LDL cholesterolu.
Porucha funkce jater
Ve studii 1. fáze po podání jedné 75mg subkutánní dávky vykazoval alirokumab podobné farmakokinetické profily u subjektů s mírnou nebo středně závažnou poruchou funkce jater v porovnání se subjekty bez poruchy funkce jater. Nejsou k dispozici žádné údaje u pacientů se závažnou poruchou funkce jater.
Porucha funkce ledvin
Jelikož není známo, že by se monoklonální protilátky vylučovaly ledvinami, nepředpokládá se, že by na farmakokinetiku alirokumabu měla renální funkce vliv. Populační farmakokinetické analýzy ukázaly, že expozice alirokumabu (AUC0-J4d) v rovnovážném stavu dávkovacích režimů jak 75 mg, tak i 150 mg jednou za 2 týdny byla zvýšena o 22 %-35 % u pacientů s mírně závažnou poruchou funkce ledvin, resp. o 49 %-50 % u pacientů se středně závažnou poruchou funkce ledvin, ve srovnání s pacienty s normální funkcí ledvin. Rozložení tělesné hmotnosti a věku, dvou proměnných, které mají dopad na expozici alirokumabu, bylo mezi kategoriemi funkce ledvin odlišné a s největší pravděpodobností vysvětlují pozorované farmakokinetické rozdíly. U pacientů se závažnou poruchou funkce ledvin jsou k dispozici omezené údaje; u těchto pacientů je expozice alirokumabu přibližně 2krát vyšší v porovnání s pacienty s normální funkcí ledvin.
Vztah(y) mezi farmakokinetikou a farmakodynamikou
Farmakodynamický účinek alirokumabu na snižování LDL cholesterolu je nepřímý a zprostředkovaný vazbou na PCSK9. Bylo pozorováno na dávce závislé snížení koncentrace volného PCSK9 a LDL cholesterolu, dokud není úplného nasycení dosaženo. Další zvýšení koncentrace alirokumabu po nasycení vazby na PCSK9 nemá za následek další snížení LDL cholesterolu, avšak je pozorován déle trvající efekt na snížení LDL cholesterolu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje neodhalily žádné zvláštní riziko pro člověka na základě vyhodnocení farmakologie bezpečnosti a toxicity po opakovaném podání.
Studie reprodukční toxikologie u potkanů a opic naznačily, že alirokumab, stejně jako ostatní IgG protilátky, prochází přes placentární bariéru.
Nebyly zaznamenány žádné nežádoucí účinky na nepřímé ukazatele plodnosti u opic (např. estrální cyklus, testikulární objem, objem ejakulátu, pohyblivost spermií nebo celkový počet spermií v ejakulátu).
V toxikologických studiích nebyly zaznamenány ani žádné patologicko anatomické nebo histopatologické nálezy v reprodukčních tkáních potkanů a opic související s podáváním alirokumabu .
U potkanů a opic nebyly zaznamenány žádné nežádoucí účinky na růst a vývoj plodu. Mateřská toxicita nebyla patrná u březích opic při systémových expozicích, které byly 81krát vyšší než expozice u lidí při dávce 150 mg jednou za 2 týdny. Mateřská toxicita byla nicméně zaznamenána u březích samic potkanů při systémových expozicích, které jsou odhadovány na přibližně 5,3krát větší, než je expozice u člověka při dávce 150 mg jednou za 2 týdny (založené na expozici naměřené u samic potkanů, které nebyly březí, během 5týdenní toxikologické studie).
Potomci opic, kterým byly podávány vysoké dávky alirokumabu každý týden po celou dobu těhotenství, měli slabší sekundární imunitní odpověď na antigen než potomci kontrolních zvířat. U těchto potomků nebyl přítomen žádný jiný důkaz o imunitní dysfunkci související s alirokumabem .
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Histidin Sacharosa Polysorbát 20 Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C-8 °C). Chraňte před mrazem.
Přípravek Praluent může být uchováván mimo chladničku chráněn před světlem (do 25 °C) po dobu maximálně 30 dní. Po vyjmutí z chladničky musí být přípravek použit během 30 dní nebo musí být zlikvidován.
Uchovávejte pero nebo injekční stříkačku ve vnější krabičce, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
1 ml roztoku ve stříkačce ze silikonizovaného čirého skla typu 1, vybavená vsazenou jehlou z nerezové oceli, styren-butadienovým měkkým gumovým krytem jehly a ethylen tetrafluorethylenem potaženou brombutylovou pryžovou zátkou.
Předplněné pero 75 mg:
Komponenty stříkačky jsou sestaveny do předplněného pera na jednorázové použití s modrým víčkem a světle zeleným aktivačním tlačítkem.
Předplněné pero 150 mg:
Komponenty stříkačky jsou sestaveny do injekční stříkačky na jednorázové použití s modrým víčkem a tmavě šedým aktivačním tlačítkem.
Předplněná injekční stříkačka 75 mg:
Injekční stříkačka je vybavena světle zeleným polypropylenovým pístem.
Předplněná injekční stříkačka150 mg:
Injekční stříkačka je vybavena tmavě šedým polypropylenovým pístem.
Velikost balení:
1, 2 nebo 6 předplněných per.
1, 2 nebo 6 předplněných injekčních stříkaček.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Roztok má být čirý, bezbarvý až světle žlutý. Pokud je roztok jinak zabarven nebo pokud obsahuje viditelné částice, nemá být tento roztok používán.
Po použití umístěte předplněné pero/předplněnou injekční stříkačku do nepropíchnutelné nádoby a zlikvidujte v souladu s místními požadavky. Nerecyklujte tuto nádobu. Vždy uchovávejte tuto nádobu mimo dohled a dosah dětí. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Držitel rozhodnutí o registraci: sanofi-aventis groupe 54, rue La Boétie F - 75008 Paris Francie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1031/001
EU/1/15/1031/002
EU/1/15/1031/003
EU/1/15/1031/004
EU/1/15/1031/005
EU/1/15/1031/006 EU/1/15/1031/007 EU/1/15/1031/008 EU/1/15/1031/009 EU/1/15/1031/010 EU/1/15/1031/011 EU/1/15/1031/012
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY/BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY/BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Regeneron Pharmaceuticals, Inc.
81 Columbia Turnpike Rensselaer, NY 12144 USA
Název a adresa výrobce odpovědného za propouštění šarží
Pro předplněné injekční stříkačky
Sanofi Winthrop Industrie 1051 Boulevard Industriel,
76580 Le Trait Francie
Pro předplněná pera Sanofi-Aventis Deutschland GmbH Industriepark Hoechst BruningstraBe 50 65926 Frankfurt am Main Německo
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Praluent 75 mg injekční roztok v předplněném peru alirocumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedno předplněné pero obsahuje alirocumabum 75 mg (75 mg/ml) v 1 ml roztoku.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: histidin, sacharosa, polysorbát 20, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 předplněné pero
2 předplněná pera 6 předplněných per
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze na jednorázové použití.
Před použitím si přečtěte příbalovou informaci a podrobný návod k použití. Subkutánní podání Zde otevřít
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE TO POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem.
Přípravek může být uchováván mimo chladničku chráněn před světlem při teplotě do 25 °C po dobu maximálně 30 dní.
Uchovávejte pero ve vnější krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
sanofi-aventis groupe 54, rue La Boétie 75008 Paris Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1031/001 1 předplněné pero EU/1/15/1031/002 2 předplněná pera EU/1/15/1031/003 6 předplněných per
13. ČÍSLO ŠARŽE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
praluent 75 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK PERA - 75 mg_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Praluent 75 mg injekce alirocumabum Subkutánní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Č. šarže:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
75 mg/ml 1 ml
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Praluent 150 mg injekční roztok v předplněném peru alirocumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedno předplněné pero obsahuje alirocumabum 150 mg (150 mg/ml) v 1 ml roztoku.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: histidin, sacharosa, polysorbát 20, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 předplněné pero
2 předplněná pera 6 předplněných per
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze na jednorázové použití.
Před použitím si přečtěte příbalovou informaci a podrobný návod k použití. Subkutánní podání Zde otevřít
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE TO POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem.
Přípravek může být uchováván mimo chladničku chráněn před světlem při teplotě do 25 °C po dobu maximálně 30 dní.
Uchovávejte pero ve vnější krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
sanofi-aventis groupe 54, rue La Boétie 75008 Paris Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1031/007 1 předplněné pero EU/1/15/1031/008 2 předplněná pera EU/1/15/1031/009 6 předplněných per
13. ČÍSLO ŠARŽE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
praluent 150 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM UBALU ŠTÍTEK PERA - 150 mg_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Praluent 150 mg injekce alirocumabum Subkutánní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Č. šarže:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
150 mg/ml 1 ml
6. JINÉ
VNĚJŠÍ KRABIČKA - Předplněná injekční stříkačka 75 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Praluent 75 mg injekční roztok v předplněné injekční stříkačce alirocumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka obsahuje alirocumabum 75 mg (75 mg/ml) v 1 ml roztoku.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: histidin, sacharosa, polysorbát 20, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok v předplněné injekční stříkačce
1 předplněná injekční stříkačka
2 předplněné injekční stříkačky
6 předplněných injekčních stříkaček
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze na jednorázové použití.
Před použitím si přečtěte příbalovou informaci a podrobný návod k použití. Subkutánní podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE TO POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem.
Přípravek může být uchováván mimo chladničku chráněn před světlem při teplotě do 25 °C po dobu maximálně 30 dní.
Uchovávejte injekční stříkačku ve vnější krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
sanofi-aventis groupe 54, rue La Boétie 75008 Paris Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1031/004 1 předplněná injekční stříkačka EU/1/15/1031/005 2 předplněné injekční stříkačky EU/1/15/1031/006 6 předplněných injekčních stříkaček
13. ČÍSLO ŠARŽE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
praluent 75 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
Praluent 75 mg injekční roztok v předplněné injekční stříkačce alirocumabum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI_
sanofi-aventis groupe
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE_
Č. šarže:
5. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK INJEKČNÍ STŘÍKAČKY - 75 mg_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Praluent 75 mg injekce
alirocumabum
s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Č. šarže:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 ml
6. JINÉ
VNĚJŠÍ KRABIČKA - Předplněná injekční stříkačka 150 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Praluent 150 mg injekční roztok v předplněné injekční stříkačce alirocumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka obsahuje alirocumabum 150 mg (150 mg/ml) v 1 ml roztoku.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: histidin, sacharosa, polysorbát 20, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 předplněná injekční stříkačka
2 předplněné injekční stříkačky
6 předplněných injekčních stříkaček
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze na jednorázové použití.
Před použitím si přečtěte příbalovou informaci a podrobný návod k použití. Subkutánní podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE TO POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem.
Přípravek může být uchováván mimo chladničku chráněn před světlem při teplotě do 25 °C po dobu maximálně 30 dní.
Uchovávejte injekční stříkačku ve vnější krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
sanofi-aventis groupe 54, rue La Boétie 75008 Paris Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1031/010 1 předplněná injekční stříkačka EU/1/15/1031/011 2 předplněné injekční stříkačky EU/1/15/1031/012 6 předplněných injekčních stříkaček
13. ČÍSLO ŠARŽE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
praluent 150 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
Praluent 150 mg injekční roztok v předplněné injekční stříkačce alirocumabum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI_
sanofi-aventis groupe
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE_
Č. šarže:
5. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK INJEKČNÍ STŘÍKAČKY - 150 mg_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Praluent 150 mg injekce
alirocumabum
s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Č. šarže:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Praluent 75 mg injekční roztok v předplněném peru Praluent 150 mg injekční roztok v předplněném peru
alirocumabum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti.
Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí
účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože
obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Praluent a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Praluent používat
3. Jak se přípravek Praluent používá
4. Možné nežádoucí účinky
5. Jak přípravek Praluent uchovávat
6. Obsah balení a další informace
1. Co je přípravek Praluent a k čemu se používá
Co je přípravek Praluent
• Přípravek Praluent obahuje účinnou látku alirokumab.
• Alirokumab je tzv. monoklonální protilátka (speciální druh bílkoviny, která je schopna se napojit na jinou cílovou látku v těle). Monoklonální protilátky jsou bílkoviny, které rozpoznávají jiné specifické bílkoviny a váží se na ně. Alirokumab se váže na PCSK9.
Jak funguje přípravek Praluent
Přípravek Praluent pomáhá snížit hladinu „špatného“ cholesterolu (také nazývaného „LDL cholesterol“).
Přípravek Praluent blokuje bílkovinu nazývanou PCSK9.
• PCSK9 je bílkovina vylučovaná j aterními buňkami.
• „Špatný“ cholesterol je za normálních okolností odstraňován z krve vazbou na specifické „receptory“ (jakési „přístavy pro cholesterol“) v játrech.
• PCSK9 snižuje počet těchto receptorů v játrech, což má za následek, že je hladina „špatného“ cholesterolu vyšší, než by měla.
• Přípravek Praluent zvyšuje počet receptorů dostupných k odstraňování „špatného“ cholesterolu tím, že blokuje PCSK9, což má za následek snížení „špatného“ cholesterolu.
Na co se přípravek Praluent používá
• U dospělých s vysokou hladinou cholesterolu v krvi [hypercholesterolemie (heterozygotní familiární a nefamiliární) nebo smíšená dyslipidemie]. Používá se:
- současně se statinem (běžně používaný léčivý přípravek ke snížení vysokého cholesterolu) nebo s jinými léčivými přípravky snižujícími hladinu cholesterolu v případě, pokud maximální dávka statinu nesnižuje hladinu cholesterolu dostatečně, nebo
- samostatně nebo současně s jinými léky snižujícími hladiny cholesterolu v případě, když nejsou statiny tolerovány nebo nemohou být použity.
• Při používání tohoto přípravku je třeba pokračovat v dietě snižující cholesterol.
2. Čemu musíte věnovat pozornost, než začnete přípravek Praluent používat Nepoužívejte přípravek Praluent
• jestliže j ste alergický(á) na alirokumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Praluent se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou o svém zdravotním stavu včetně alergií.
Pokud se u Vás vyskytne závažná alergická reakce, přestaňte používat přípravek Praluent a ihned se poraďte se svým lékařem. V klinických studiích byly zaznamenány někdy závažné alergické reakce, jako jsou reakce z přecitlivělosti (ztížené dýchání), numulární ekzém (načervenalé skvrny na kůži někdy s puchýři) a zánět cév z přecitlivělosti (tzv. hypersenzitivní vaskulitida, specifická forma reakce přecitlivělosti s příznaky, např. s průjmy, s vyrážkou nebo s purpurově zbarvenými skvrnami na kůži). Pro popis alergických reakcí, které se mohou vyskytnout při užívání přípravku Praluent, viz bod 4.
Pokud trpíte onemocněním ledvin nebo jater, informujte před použitím tohoto léčivého přípravku svého lékaře, protože přípravek Praluent byl zkoumán pouze u několika pacientů se závažným onemocněním ledvin a u pacientů se závažným onemocněním jater studován nebyl.
Děti a dospívající
Přípravek Praluent není doporučen pro použití u dětí a dospívajících mladších 18 let, protože v těchto věkových skupinách neexistují s použitím tohoto léčivého přípravku žádné zkušenosti.
Další léčivé přípravky a přípravek Praluent
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Přípravek Praluent není doporučen v období těhotenství nebo v období kojení.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Řízení dopravních prostředků a obsluha strojů
Nepředpokládá se, že má přípravek Praluent vliv na schopnost řídit dopravní prostředky nebo obsluhovat stroje.
3. Jak se přípravek Praluent používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře, lékárníka nebo zdravotní sestry. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Kolik podat
Lékař Vám sdělí, jaká dávka je pro Vás vhodná (75 mg nebo 150 mg). Lékař Vám zkontroluje hladinu cholesterolu a v průběhu léčby Vám může upravit dávku (směrem nahoru nebo dolů).
Vždy zkontrolujte štítek na peru, abyste se ujistil(a), že máte správný léčivý přípravek a správnou sílu.
Kdy podat
Přípravek Praluent se injekčně podává jednou za 2 týdny.
Před podáním
Před podáním přípravku Praluent si pečlivě přečtěte návod k použití.
Kam podat
Pro informaci, kam podat přípravek Praluent, si pečlivě přečtěte detailní návod k použití.
Naučte se používat předplněné pero
Než použijete pero poprvé, Váš lékař, lékárník nebo zdravotní sestra Vám ukáže, jak přípravek Praluent podávat.
• Vždy si přečtěte „Návod k použití“ uvedený na krabičce.
• Vždy používejte pero dle pokynů popsaných v „Návodu k použití“.
Jestliže jste použil(a) více přípravku Praluent, než jste měl(a)
Jestliže jste použil(a) více přípravku Praluent, než jste měl(a), poraďte se se svým lékařem nebo lékárníkem. Jestliže jste zapomněl(a) použít přípravek Praluent
Jestliže jste zapomněl(a) podat dávku přípravku Praluent, podejte zapomenutou dávku co nejdříve, jak je to možné. Poté podejte další dávku za dva týdny ode dne, kdy jste dávku zapomněl(a). Např. pokud jste normálně podával(a) dávku každé druhé úterý, podávejte nadále dávku každé druhé úterý. Takto dodržujte pravidelný dávkovací režim. Pokud si nejste jistý(á), kdy podávat přípravek Praluent, kontaktujte svého lékaře, lékárníka nebo zdravotní sestru.
Jestliže jste přestal(a) používat přípravek Praluent
Nepřestávejte používat přípravek Praluent, aniž byste se poradil(a) se svým lékařem. Pokud přestanete používat přípravek Praluent, může se Vám zvýšit hladina cholesterolu.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravodní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se u Vás vyskytne závažná alergická reakce, přestaňte používat přípravek Praluent a ihned se poraďte se svým lékařem. Byly zaznamenány někdy i závažné alergické reakce, jako jsou reakce s přecitlivělosti (ztížené dýchání), numulární ekzém (načervenalé skvrny na kůži někdy s puchýři) a zánět cév z přecitlivělosti (specifická forma reakce přecitlivělosti s příznaky, např. s průjmy, s vyrážkou nebo s purpurově zbarvenými skvrnami na kůži) (mohou postihnout až 1 z 1000 osob).
Další nežádoucí účinky jsou:
Časté (mohou se vyskytnout až u 1 z 10 osob)
• zčervenání, svědění, otok, bolest/citlivost v místě podání léčivého přípravku (lokální reakce v místě podání injekce)
• příznaky onemocnění horních cest dýchacích, např. bolest v krku, rýma, kýchání
• svědění (pruritus).
Vzácné (mohou se vyskytnout až u 1 z 1000 osob)
• červené a svědivé zvýšené hrbolky nebo vyrážka (kopřivka).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V*. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Praluent uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C až 8 °C). Chraňte před mrazem.
Uchovávejte pero ve vnější krabičce, aby byl přípravek chráněn před světlem.
Pokud je to zapotřebí, jednotlivá předplněná pera mohou být uchovávána mimo chladničku při teplotě do 25 °C po dobu maximálně 30 dní. Chraňte před světlem. Po vyjmutí z chladničky musí být přípravek Praluent použit během 30 dní nebo musí být zlikvidován.
Nepoužívejte tento přípravek, pokud je nepřirozeně zabarvený nebo zakalený či pokud obsahuje viditelné vločky nebo částice.
Po použití umístěte předplněné pero do nepropíchnutelné nádoby. Tuto nádobu vždy uchovávejte mimo dohled a dosah dětí. Zeptejte se svého lékaře, lékárníka nebo zdravotní sestry, jak tuto nádobu zlikvidovat. Nerecyklujte tuto nádobu.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Praluent obsahuje
• Léčivou látkou je alirocumabum. Jedno pero na jednorázové použití obsahuje alirocumabum buď 75 miligramů (75 miligramů na 1 ml roztoku) nebo 150 miligramů (150 miligramů na 1 ml roztoku).
• Dalšími složkami jsou histidin, sacharosa, polysorbát 20 a voda na injekci.
Jak přípravek Praluent vypadá a co obsahuje toto balení
Přípravek Praluent je čirý, bezbarvý až světle žlutý injekční roztok v předplněném peru.
Jedno předplněné pero se zeleným tlačítkem obsahuje 1 ml roztoku s jednorázovou dávkou 75 miligramů. Je k dispozici ve velikostech balení po 1, 2 nebo 6 předplněných perech.
Jedno předplněné pero s šedým tlačítkem obsahuje 1 ml roztoku s jednorázovou dávkou 150 miligramů.
Je k dispozici ve velikostech balení po 1, 2 nebo 6 předplněných perech.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
sanofi-aventis groupe 54, rue La Boétie F - 75008 Paris Francie
Výrobce
Sanofi-Aventis Deutschland GmbH Industriepark Hoechst
BruningstraBe 50
65926 Frankfurt am Main
Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Sanofi Belgium Tél/Tel: +32 (0)2 710 54 00 |
Lietuva UAB “SANOFI-AVENTIS LIETUVA” Tel: +370 5 2755224 |
|
Btarapna sanofi-aventis Bulgaria EOOD Ten.: +359 (0)2 970 53 00 |
Luxembourg/Luxemburg Sanofi Belgium Tél/Tel: +32 (0)2 710 54 00 (Belgique/Belgien) |
|
Česká republika sanofi-aventis, s.r.o. Tel: +420 233 086 111 |
Magyarország SANOFI-AVENTIS Zrt. Tel.: +36 1 505 0050 |
|
Danmark sanofi-aventis Denmark A/S Tlf: +45 45 16 70 00 |
Malta Sanofi Malta Ltd. Tel: +356 21493022 |
|
Deutschland Sanofi-Aventis Deutschland GmbH Tel: +49 (0)180 2 222010 |
Nederland sanofi-aventis Netherlands B.V. Tel: +31 (0)182 557 755 |
|
Eesti sanofi-aventis Estonia OU Tel: +372 627 34 88 |
Norge sanofi-aventis Norge AS Tlf: +47 67 10 71 00 |
|
EXXáSa sanofi-aventis AEBE Tr|k: +30 210 900 16 00 |
Osterreich sanofi-aventis GmbH Tel: +43 1 80 185 - 0 |
|
Espaňa sanofi-aventis, S.A Tel: +34 93 485 94 00 |
Polska sanofi-aventis Sp. z o.o. Tel.: +48 22 280 00 00 |
|
France sanofi-aventis France Tél: 0 800 222 555 Appel depuis l’étranger : +33 1 57 63 23 23 |
Portugal Sanofi - Produtos Farmaceuticos, Lda. Tel: +351 21 35 89 400 |
|
Hrvatska sanofi-aventis Croatia d.o.o. Tel: +385 1 600 34 00 |
Románia Sanofi Romania SRL Tel: +40 (0) 21 317 31 36 |
|
Ireland sanofi-aventis Ireland Ltd. T/A SANOFI Tel: +353 (0) 1 403 56 00 |
Slovenija sanofi-aventis d.o.o. Tel: +386 1 560 48 00 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika sanofi-aventis Pharma Slovakia s.r.o. Tel: +421 2 33 100 100 |
Italia Suomi/Finland
Sanofi S.p.A. Sanofi Oy
Tel: 800 131212 (domande di tipo tecnico) Puh/Tel: +358 (0) 201 200 300
800 536389 (altre domande)
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu .
Příbalová informace: informace pro uživatele
Praluent 75 mg injekční roztok v předplněné injekční stříkačce Praluent 150 mg injekční roztok v předplněné injekční stříkačce
alirocumabum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti.
Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí
účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože
obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Praluent a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Praluent používat
3. Jak se přípravek Praluent používá
4. Možné nežádoucí účinky
5. Jak přípravek Praluent uchovávat
6. Obsah balení a další informace
1. Co je přípravek Praluent a k čemu se používá
Co je přípravek Praluent
• Přípravek Praluent obahuje účinnou látku alirokumab.
• Alirokumab je tzv. monoklonální protilátka (speciální druh bílkoviny, která je schopna se napojit na jinou cílovou látku v těle). Monoklonální protilátky jsou bílkoviny, které rozpoznávají jiné specifické bílkoviny a váží se na ně. Alirokumab se váže na PCSK9.
Jak funguje přípravek Praluent
Přípravek Praluent pomáhá snížit hladinu „špatného“ cholesterolu (také nazývaného „LDL cholesterol“).
Přípravek Praluent blokuje bílkovinu nazývanou PCSK9.
• PCSK9 je bílkovina vylučovaná j aterními buňkami.
• „Špatný“ cholesterol je za normálních okolností odstraňován z krve vazbou na specifické „receptory“ (jakési „přístavy pro cholesterol“) v játrech.
• PCSK9 snižuje počet těchto receptorů v játrech, což má za následek, že je hladina „špatného“ cholesterolu vyšší, než by měla.
• Přípravek Praluent zvyšuje počet receptorů dostupných k odstraňování „špatného“ cholesterolu tím, že blokuje PCSK9, což má za následek snížení „špatného“ cholesterolu.
Na co se přípravek Praluent používá
• U dospělých s vysokou hladinou cholesterolu v krvi [hypercholesterolemie (heterozygotní familiární a nefamiliární) nebo smíšená dyslipidemie]. Používá se:
- současně se statinem (běžně používaný léčivý přípravek ke snížení vysokého cholesterolu) nebo s jinými léčivými přípravky snižujícími hladinu cholesterolu v případě, pokud maximální dávka statinu nesnižuje hladinu cholesterolu dostatečně, nebo
- samostatně nebo současně s jinými léky snižujícími hladiny cholesterolu v případě, když nejsou statiny tolerovány nebo nemohou být použity.
• Při používání tohoto přípravku je třeba pokračovat v dietě snižující cholesterol.
2. Čemu musíte věnovat pozornost, než začnete přípravek Praluent používat Nepoužívejte přípravek Praluent
• jestliže j ste alergický(á) na alirokumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Praluent se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou o svém zdravotním stavu včetně alergií.
Pokud se u Vás vyskytne závažná alergická reakce, přestaňte používat přípravek Praluent a ihned se poraďte se svým lékařem. V klinických studiích byly zaznamenány někdy závažné alergické reakce, jako jsou reakce z přecitlivělosti (ztížené dýchání), numulární ekzém (načervenalé skvrny na kůži někdy s puchýři) a zánět cév z přecitlivělosti (tzv. hypersenzitivní vaskulitida, specifická forma reakce přecitlivělosti s příznaky, např. s průjmy, s vyrážkou nebo s purpurově zbarvenými skvrnami na kůži). Pro popis alergických reakcí, které se mohou vyskytnout při užívání přípravku Praluent, viz bod 4.
Pokud trpíte onemocněním ledvin nebo jater, informujte před použitím tohoto léčivého přípravku svého lékaře, protože přípravek Praluent byl zkoumán pouze u několika pacientů se závažným onemocněním ledvin a u pacientů se závažným onemocněním jater studován nebyl.
Děti a dospívající
Přípravek Praluent není doporučen pro použití u dětí a dospívajících mladších 18 let, protože v těchto věkových skupinách neexistují s použitím tohoto léčivého přípravku žádné zkušenosti.
Další léčivé přípravky a přípravek Praluent
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Přípravek Praluent není doporučen v období těhotenství nebo v období kojení.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Řízení dopravních prostředků a obsluha strojů
Nepředpokládá se, že má přípravek Praluent vliv na schopnost řídit dopravní prostředky nebo obsluhovat stroje.
3. Jak se přípravek Praluent používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře, lékárníka nebo zdravotní sestry. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Kolik podat
Lékař Vám sdělí, jaká dávka je pro Vás vhodná (75 mg nebo 150 mg). Lékař Vám zkontroluje hladinu cholesterolu a v průběhu léčby Vám může upravit dávku (směrem nahoru nebo dolů).
Vždy zkontrolujte štítek na injekční stříkačce, abyste se ujistil(a), že máte správný léčivý přípravek a správnou sílu.
Kdy podat
Přípravek Praluent se injekčně podává jednou za 2 týdny.
Před podáním
Před podáním přípravku Praluent si pečlivě přečtěte návod k použití.
Kam podat
Pro informaci, kam podat přípravek Praluent, si pečlivě přečtěte detailní návod k použití.
Naučte se používat předplněnou injekční stříkačku
Než použijete injekční stříkačku poprvé, Váš lékař, lékárník nebo zdravotní sestra Vám ukáže, jak přípravek Praluent podávat.
• Vždy si přečtěte „Návod k použití“ uvedený na krabičce.
• Vždy používejte injekční stříkačku dle pokynů popsaných v „Návodu k použití“.
Jestliže jste použil(a) více přípravku Praluent, než jste měl(a)
Jestliže jste použil(a) více přípravku Praluent, než jste měl(a), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Jestliže jste zapomněl(a) použít přípravek Praluent
Jestliže jste zapomněl(a) podat dávku přípravku Praluent, podejte zapomenutou dávku co nejdříve, jak je to možné. Poté podejte další dávku za dva týdny ode dne, kdy jste dávku zapomněl(a). Např. pokud jste normálně podával(a) dávku každé druhé úterý, podávejte nadále dávku každé druhé úterý. Takto dodržujte pravidelný dávkovací režim. Pokud si nejste jistý(á), kdy podávat přípravek Praluent, kontaktujte svého lékaře, lékárníka nebo zdravotní sestru.
Jestliže jste přestal(a) používat přípravek Praluent
Nepřestávejte používat přípravek Praluent, aniž byste se poradil(a) se svým lékařem. Pokud přestanete používat přípravek Praluent, může se Vám zvýšit hladina cholesterolu.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se u Vás vyskytne závažná alergická reakce, přestaňte používat přípravek Praluent a ihned se poraďte se svým lékařem. Byly zaznamenány někdy i závažné alergické reakce, jako jsou reakce s přecitlivělosti (ztížené dýchání), numulární ekzém (načervenalé skvrny na kůži někdy s puchýři) a zánět cév z přecitlivělosti (specifická forma reakce přecitlivělosti s příznaky, např. s průjmy, s vyrážkou nebo s purpurově zbarvenými skvrnami na kůži) (mohou postihnout až 1 z 1000 osob).
Další nežádoucí účinky jsou:
Časté (mohou se vyskytnout až u 1 z 10 osob)
• zčervenání, svědění, otok, bolest/citlivost v místě podání léčivého přípravku (lokální reakce v místě podání injekce)
• příznaky onemocnění horních cest dýchacích, např. bolest v krku, rýma, kýchání
• svědění (pruritus).
Vzácné (mohou se vyskytnout až u 1 z 1000 osob)
• červené a svědivé zvýšené hrbolky nebo vyrážka (kopřivka).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V*. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Praluent uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C až 8 °C). Chraňte před mrazem.
Uchovávejte injekční stříkačku ve vnější krabičce, aby byl přípravek chráněn před světlem.
Pokud je to zapotřebí, jednotlivé předplněné stříkačky mohou být uchovávána mimo chladničku při teplotě do 25 °C po dobu maximálně 30 dní. Chraňte před světlem. Po vyjmutí z chladničky musí být přípravek Praluent použit během 30 dní nebo musí být zlikvidován.
Nepoužívejte tento přípravek, pokud je nepřirozeně zabarvený nebo zakalený či pokud obsahuje viditelné vločky nebo částice.
Po použití umístěte předplněnou injekční stříkačku do nepropíchnutelné nádoby. Tuto nádobu vždy uchovávejte mimo dohled a dosah dětí. Zeptejte se svého lékaře, lékárníka nebo zdravotní sestry, jak tuto nádobu zlikvidovat. Nerecyklujte tuto nádobu.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Praluent obsahuje
• Léčivou látkou je alirocumabum. Jedna injekční stříkačka na jednorázové použití obsahuje alirocumabum buď 75 miligramů (75 miligramů na 1 ml roztoku) nebo 150 miligramů (150 miligramů na 1 ml roztoku).
• Dalšími složkami jsou histidin, sacharosa, polysorbát 20 a voda na injekci.
Jak přípravek Praluent vypadá a co obsahuje toto balení
Přípravek Praluent je čirý, bezbarvý až světle žlutý injekční roztok v předplněné injekční stříkačce.
Jedna předplněná injekčka stříkačka se zeleným pístem obsahuje 1 ml roztoku s jednorázovou dávkou 75 miligramů.
Je k dispozici ve velikostech balení po 1, 2 nebo 6 předplněných injekčních stříkačkách.
Jedna předplněná injekční stříkačka s šedým pístem obsahuje 1 ml roztoku s jednorázovou dávkou 150 miligramů.
Je k dispozici ve velikostech balení po 1, 2 nebo 6 předplněných injekčních stříkačkách.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
sanofi-aventis groupe 54, rue La Boétie F - 75008 Paris Francie
Manufacturer
Sanofi Winthrop Industrie 1051 Boulevard Industriel 76580 Le Trait Francie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Sanofi Belgium Tél/Tel: +32 (0)2 710 54 00 |
Lietuva UAB “SANOFI-AVENTIS LIETUVA” Tel: +370 5 2755224 |
|
Btarapna sanofi-aventis Bulgaria EOOD Ten.: +359 (0)2 970 53 00 |
Luxembourg/Luxemburg Sanofi Belgium Tél/Tel: +32 (0)2 710 54 00 (Belgique/Belgien) |
|
Česká republika sanofi-aventis, s.r.o. Tel: +420 233 086 111 |
Magyarország SANOFI-AVENTIS Zrt. Tel.: +36 1 505 0050 |
|
Danmark sanofi-aventis Denmark A/S Tlf: +45 45 16 70 00 |
Malta Sanofi Malta Ltd. Tel: +356 21493022 |
|
Deutschland Sanofi-Aventis Deutschland GmbH Tel: +49 (0)180 2 222010 |
Nederland sanofi-aventis Netherlands B.V. Tel: +31 (0)182 557 755 |
|
Eesti sanofi-aventis Estonia OU Tel: +372 627 34 88 |
Norge sanofi-aventis Norge AS Tlf: +47 67 10 71 00 |
|
EXXáSa sanofi-aventis AEBE Tr^: +30 210 900 16 00 |
Osterreich sanofi-aventis GmbH Tel: +43 1 80 185 - 0 |
|
Espaňa sanofi-aventis, S.A Tel: +34 93 485 94 00 |
Polska sanofi-aventis Sp. z o.o. Tel.: +48 22 280 00 00 |
|
France sanofi-aventis France Tél: 0 800 222 555 Appel depuis l’étranger : +33 1 57 63 23 23 |
Portugal Sanofi - Produtos Farmaceuticos, Lda. Tel: +351 21 35 89 400 |
|
Hrvatska sanofi-aventis Croatia d.o.o. Tel: +385 1 600 34 00 |
Románia Sanofi Romania SRL Tel: +40 (0) 21 317 31 36 |
Ireland
sanofi-aventis Ireland Ltd. T/A SANOFI Tel: +353 (0) 1 403 56 00
Ísland
Vistor hf.
Sími: +354 535 7000 Italia
Sanofi S.p.A.
Tel: 800 131212 (domande di tipo tecnico) 800 536389 (altre domande)
Knrcpog
sanofi-aventis Cyprus Ltd.
Tr^: +357 22 871600
Latvija
sanofi-aventis Latvia SIA Tel: +371 67 33 24 51
Slovenija
sanofi-aventis d.o.o.
Tel: +386 1 560 48 00
Slovenská republika
sanofi-aventis Pharma Slovakia s.r.o. Tel: +421 2 33 100 100
Suomi/Finland
Sanofi Oy
Puh/Tel: +358 (0) 201 200 300
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu .
Praluent 75 mg injekční roztok v předplněném peru
alirokumab
Návod k použití
Součásti pera přípravku Praluent jsou znázorněny na tomto obrázku.
Zelené tlačítko
Tělo
Okénko
Žlutá bezpečnostní krytka ukrývající jehlu
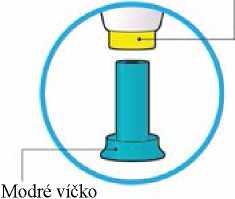
Pouze pro jednorázové použití
Důležité informace
• Tento prostředek je předplněné pero na jednorázové použití. Obsahuje 75 mg přípravku Praluent (alirokumab) v 1 ml.
• Tento léčivých přípravek se podává injekčně pod kůži a můžete si jej podat sami nebo Vám jej může podat někdo jiný (ošetřovatel(ka)).
• Toto pero může být použito pouze pro jednorázové podání injekce a poté musí být vyhozeno.
Co ano
V Uchovávejte pero přípravku Praluent mimo dohled a dosah dětí.
V Před použitím pera přípravku Praluent si pečlivě přečtěte celý návod.
V Při každém použití pera přípravku Praluent postupujte podle návodu.
V Uchovávejte nepoužívaná pera v chladničce při teplotě 2 °C až 8 °C. Pro podrobné podmínky uchovávání viz samostatná příbalová informace přípravku Praluent.
Co ne
x Nedotýkejte se žluté bezpečnostní krytky.
x Nepoužívejte pero, pokud Vám upadlo nebo pokud je poničené.
x Nepoužívejte pero, pokud modré víčko chybí nebo pokud není bezpečně připevněno.
x Nepoužívejte pero vícekrát než jednou. x Netřepejte s perem. x Nevystavujte pero mrazu. x Nevystavujte pero přímému slunečnímu světlu.
Ponechte si tento leták. Pokud máte nějaké otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry nebo zavolejte na telefonní číslo sanofi-aventis, které je uvedené v příbalové informaci.
KROK A: Příprava na podání injekce
Předtím než začnete, budete potřebovat:
• pero přípravku Praluent
• tampón namočený v alkoholu
• vatový tampón nebo gázu
• nepropíchnutelnou nádobu (viz krok B, 8).
© Zkontrolujte štítek na peru.
• Zkontrolujte, že máte správný produkt a správnou dávku.
• Zkontrolujte datum expirace a uplynulo-li již toto datum, pero nepoužívejte.

® Zkontrolujte okénko.
• Zkontrolujte, zda je tekutina čirá, bezbarvá až světle žlutá a neobsahuje částice; pokud toto nesplňuje, pero nepoužívejte (viz obrázek A).
• Můžete pozorovat vzduchové bublinky. To je normální.
• Nepoužívejte v případě, že je okénko zabarveno výrazně žlutou barvou (viz obrázek B).
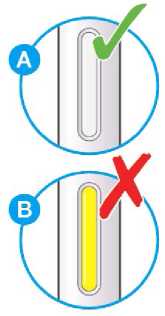
® Ponechte pero při pokojové teplotě po dobu 30 až 40 minut.
• Nezahřívejte pero, nechejte jej při pokojové teplotě, aby se teplotní rozdíly vyrovnaly samy.
• Nevkládejte pero zpět do chladničky.
© Připravte si místo podání injekce.
• Umyjte si ruce mýdlem a vodou a osušte ručníkem.
• Injekci můžete podat do:
o stehna
o břicha (kromě oblasti 5 cm kolem pupku) o horní části paže (Viz obrázek).
• Při podávání injekce můžete stát nebo sedět.
• Ošetřete místo podání injekce tampónem namočeným v alkoholu.
• Nepodávejte do kůže, která je citlivá, tvrdá, červená nebo horká.
• Nepodávejte do místa v blízkosti viditelné žíly.
• Zvolte jiné místo pro každé podání.
• Nepodávejte přípravek Praluent do stejného místa, kam podáváte jiné injekční léčivé přípravky.
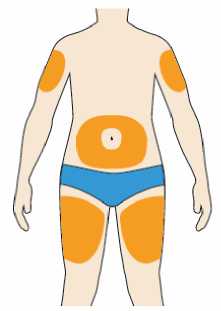
KROK B: Jak podat injekci
© Po dokončení všech kroků v „Kroku A: Příprava na podání injekce“ sundejte modré víčko.
• Nesundávejte modré víčko, dokud nejste připraven(á) podat injekci.
• Nenasazujte zpátky modré víčko.
Modré víčko
© Držte pero přípravku Praluent takto.
• Nedotýkejte se žluté bezpečnostní krytky.
• Ujistěte se, že vidíte okénko.
© Přitiskněte žlutou bezpečnostní krytku na kůži v přibližně v 90stupňovém úhlu.
• Přitiskněte a pevně držte oproti svému tělu, až přestane být žlutá bezpečnostní krytka viditelná. Pero nebude správně fungovat, pokud není žlutá bezpečnostní krytka dostatečně přitisknutá.
• V případě potřeby zmáčkněte kůži, abyste se ujistil(a), že místo vpichu je pevné.
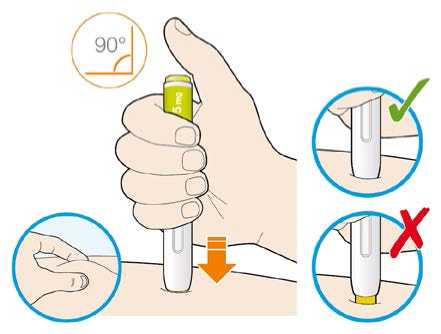
© Stiskněte a ihned uvolněte zelené tlačítko pomocí palce.
• Uslyšíte kliknutí. Tímto začalo podání injekce.
• Okénko se bude postupně zabarvovat do žluta.
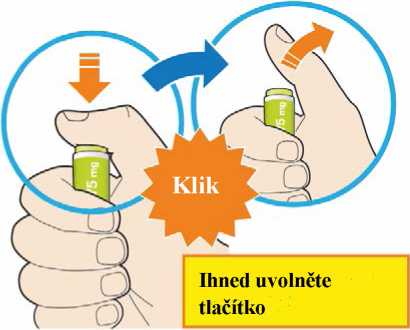
© Po uvolnění tlačítka stále držte pero oproti své kůži.
• Podání injekce trvá přibližně 20 vteřin.
© Než odejmete pero z kůže, zkontrolujte, zda se okénko zbarvilo do žluta.
• Nevyjímejte pero, dokud se celé okénko nezabarví do žluta.
• Podání injekce je kompletní v případě, že se celé okénko zabarví do žluta, a poté uslyšíte druhé kliknutí.
• Pokud se okénko celé nezabarví do žluta, zavolejte pro pomoc na linku sanofi-aventis. Nepodávejte druhou dávku, aniž byste se poradili se svým lékařem, lékárníkem nebo zdravotní sestrou.

® Odejměte pero z kůže.
• Po podání injekce nemasírujte kůži.
• Pokud se objeví krev, přitiskněte na místo vpichu vatový tampón nebo gázu, dokud se krvácení nezastaví.
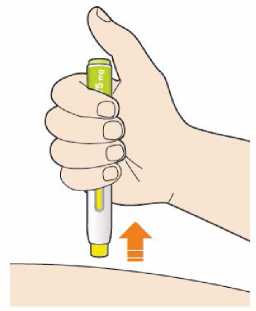
® Vyhoďte pero a víčko.
• Nenasazujte modré víčko zpět.
• Ihned po použití vyhoďte pero a víčko do nepropíchnutelné nádoby.
• Zeptejte se svého lékaře, lékárníka nebo zdravotní sestry jak tuto nádobu zlikvidovat.
• Vždy uchovávejte tuto nádobu mimo dohled a dosah dětí.
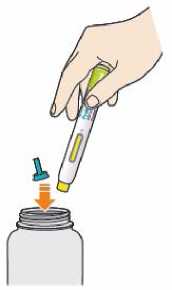
Praluent 150 mg injekční roztok v předplněném peru
alirokumab
Návod k použití
Součásti pera přípravku Praluent jsou znázorněny na tomto obrázku.

Okénko
Žlutá bezpečnostní krytka ukývající jehlu
Šedé tlačítko
Tělo
Modré víčko
Pouze pro je dnorázové použití
Důležité informace
• Tento prostředek je předplněné pero na jednorázové použití. Obsahuje 75 mg přípravku Praluent (alirokumab) v 1 ml.
• Tento léčivých přípravek se podává injekčně pod kůži a můžete si jej podat sami nebo Vám jej může podat někdo jiný (ošetřovatel(ka)).
• Toto pero může být použito pouze pro jednorázové podání injekce a poté musí být vyhozeno.
Co ano
V Uchovávejte pero přípravku Praluent mimo dohled a dosah dětí.
V Před použitím pera přípravku Praluent si pečlivě přečtěte celý návod.
V Při každém použití pera přípravku Praluent postupujte podle návodu.
V Uchovávejte nepoužívaná pera v chladničce při teplotě 2 °C až 8 °C. Pro podrobné podmínky uchovávání viz samostatná příbalová informace přípravku Praluent.
Co ne
x Nedotýkejte se žluté bezpečnostní krytky.
x Nepoužívejte pero, pokud Vám upadlo nebo pokud je poničené.
x Nepoužívejte pero, pokud modré víčko chybí nebo pokud není bezpečně připevněno.
x Nepoužívejte pero vícekrát než jednou.
x Netřepejte s perem.
x Nevystavujte pero mrazu.
x Nevystavujte pero přímému slunečnímu světlu.
Ponechte si tento leták. Pokud máte nějaké otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry nebo zavolejte na telefonní číslo sanofi-aventis, které je uvedené v příbalové informaci.
KROK A: Příprava na podání injekce
Předtím než začnete, budete potřebovat:
• pero přípravku Praluent
• tampón namočený v alkoholu
• vatový tampón nebo gázu
• nepropíchnutelnou nádobu (viz krok B, 8).
© Zkontrolujte štítek na peru.
• Zkontrolujte, že máte správný produkt a správnou dávku.
• Zkontrolujte datum expirace a uplynulo-li již toto datum, pero nepoužívejte.

© Zkontrolujte okénko.
• Zkontrolujte, zda je tekutina čirá, bezbarvá až světle žlutá a neobsahuje částice; pokud toto nesplňuje, pero nepoužívejte (viz obrázek A).
• Můžete pozorovat vzduchové bublinky. To je normální.
• Nepoužívejte v případě, že je okénko zabarveno výrazně žlutou barvou (viz obrázek B).
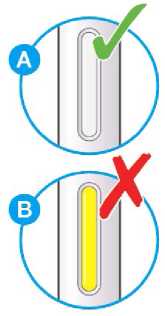
® Ponechte pero při pokojové teplotě po dobu 30 až 40 minut.
• Nezahřívejte pero, nechejte jej při pokojové teplotě, aby se teplotní rozdíly vyrovnaly samy.
• Nevkládejte pero zpět do chladničky.
© Připravte si místo podání injekce.
• Umyjte si ruce mýdlem a vodou a osušte ručníkem.
• Injekci můžete podat do:
o stehna
o břicha (kromě oblasti 5 cm kolem pupku) o horní části paže (Viz obrázek).
• Při podávání injekce můžete stát nebo sedět.
• Očistěte místo podání injekce tampónem namočeným v alkoholu.
• Nepodávejte do kůže, která je citlivá, tvrdá, červená nebo horká.
• Nepodávejte do místa v blízkosti viditelné žíly.
• Zvolte jiné místo pro každé podání.
• Nepodávejte přípravek Praluent do stejného místa, kam podáváte jiné injekční léčivé přípravky.
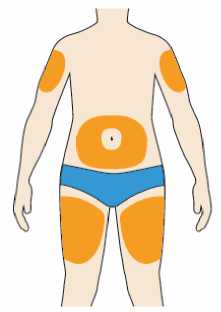
KROK B: Jak podat injekci
© Po dokončení všech kroků v „Kroku A: Příprava na podání injekce“ sundejte modré víčko.
• Nesundávejte modré víčko, dokud nejste připraven(á) podat injekci.
• Nenasazujte zpátky modré víčko.
Modré víčko
© Držte pero přípravku Praluent takto.
• Nedotýkejte se žluté bezpečnostní krytky.
• Ujistěte se, že vidíte okénko.
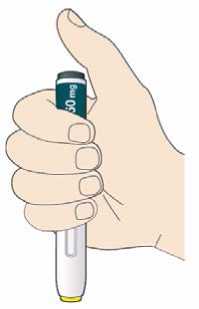
® Přitiskněte žlutou bezpečnostní krytku na kůži v přibližně v 90stupňovém úhlu.
• Přitiskněte a pevně držte oproti svému tělu, až přestane být žlutá bezpečnostní krytka viditelná. Pero nebude správně fungovat, pokud není žlutá bezpečnostní krytka dostatečně přitisknutá.
• V případě potřeby zmáčkněte kůži, abyste se ujistil (a), že místo vpichu je pevné.
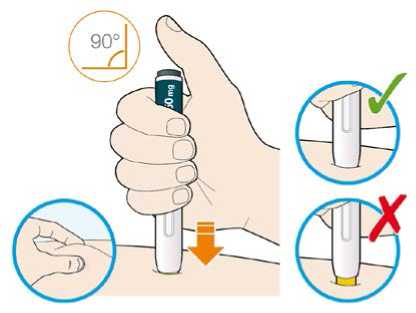
© Stiskněte a ihned uvolněte šedé tlačítko pomocí palce.
• Uslyšíte kliknutí. Tímto začalo podání injekce.
• Okénko se bude postupně zabarvovat do žluta.
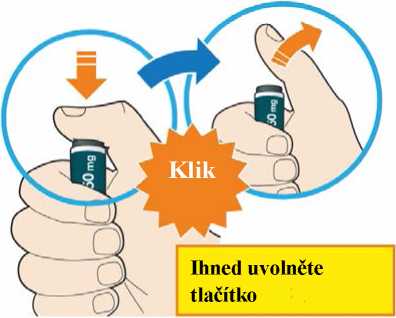
© Po uvolnění tlačítka stále držte pero oproti své kůži.
• Podání injekce trvá přibližně 20 vteřin.
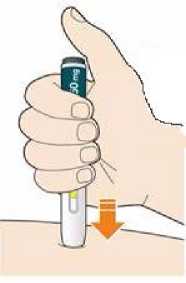
© Než odejmete pero z kůže, zkontrolujte, zda se okénko zbarvilo do žluta.
• Nevyjímejte pero, dokud se celé okénko nezabarví do žluta.
• Podání injekce je kompletní v případě, že se celé okénko zabarví do žluta, a poté uslyšíte druhé kliknutí.
• Pokud se okénko celé nezabarví do žluta, zavolejte pro pomoc na linku sanofi-aventis. Nepodávejte druhou dávku, aniž byste se poradili se svým lékařem lékárníkem nebo zdravotní sestrou.

© Odejměte pero z kůže.
• Po podání injekce nemasírujte kůži.
• Pokud se objeví krev, přitiskněte na místo vpichu vatový tampón nebo gázu, dokud se krvácení nezastaví.
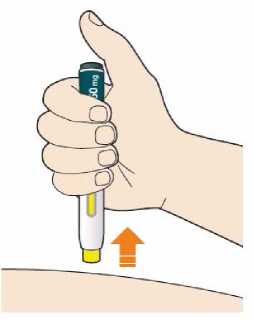
® Vyhoďte pero a víčko.
• Nenasazujte modré víčko zpět.
• Ihned po použití vyhoďte pero a víčko do nepropíchnutelné nádoby.
• Zeptejte se svého lékaře, lékárníka nebo zdravotní sestry jak tuto nádobu zlikvidovat.
• Vždy uchovávejte tuto nádobu mimo dohled a dosah dětí.
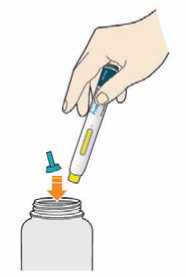
Praluent 75 mg injekční roztok v předplněné injekční stříkačce
alirokumab
Návod k použití
Součásti stříkačky přípravku Praluent jsou znázorněny na tomto obrázku.

Zelený píst

Tělo stříkačky
Důležité informace
• Tento prostředek je předplněná injekční stříkačka na jednorázové použití. Obsahuje 75 mg přípravku Praluent (alirokumab) v 1 ml.
• Tento léčivých přípravek se podává injekčně pod kůži a můžete si jej podat sami nebo Vám jej může podat někdo jiný (ošetřovatel(ka)).
• Tato injekční stříkačka může být použita pouze pro jednorázové podání injekce a poté musí být vyhozena.
Co ano
V Uchovávejte injekční stříkačku přípravku Praluent mimo dohled a dosah dětí.
V Před použitím injekční stříkačky přípravku Praluent si pečlivě přečtěte celý návod.
V Při každém použití injekční stříkačky přípravku Praluent postupujte podle návodu.
V Uchovávejte nepoužívané injekční stříkačky v chladničce při teplotě 2 °C až 8 °C. Pro podrobné podmínky uchovávání viz samostatná příbalová informace přípravku Praluent.
Co ne
x Nedotýkejte se jehly.
x Nepoužívejte injekční stříkačku, pokud Vám upadla nebo pokud je poničená.
x Nepoužívejte injekční stříkačku, pokud šedá krytka jehly chybí nebo pokud není bezpečně připevněna. x Nepoužívejte injekční stříkačku vícekrát než jednou. x Netřepejte s injekční stříkačkou. x Nevystavujte injekční stříkačku mrazu. x Nevystavujte injekční stříkačku přímému slunečnímu světlu.
Ponechte si tento leták. Pokud máte nějaké otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry nebo zavolejte na telefonní číslo sanofi-aventis, které je uvedené v příbalové informaci.
KROK A: Příprava na podání injekce
Předtím, než začnete, budete potřebovat:
• injekční stříkačku přípravku Praluent
• tampón namočený v alkoholu
• vatový tampón nebo gázu
• nepropíchnutelnou nádobu (viz krok B, 6).
© Než začnete.
• Vyjměte injekční stříkačku z balení a uchopte ji za tělo stříkačky.
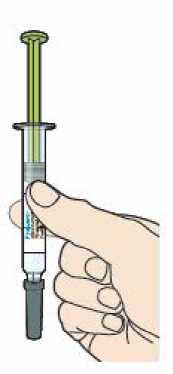
© Zkontrolujte štítek na injekční stříkačce.
• Zkontrolujte, že máte správný produkt a správnou dávku (zelený píst pro dávku 75 mg/ml).
• Zkontrolujte datum expirace a uplynulo-li již toto datum, injekční stříkačku nepoužívejte.
• Zkontrolujte, zda je tekutina čirá, bezbarvá až světle žlutá a neobsahuje částice; pokud toto nesplňuje, tuto injekční stříkačku nepoužívejte.
• Zkontrolujte, zda není injekční stříkačka otevřená nebo poničená.
® Ponechte injekční stříkačku při pokojové teplotě po dobu 30 až 40 minut.
• Nezahřívejte injekční stříkačku, nechejte ji při pokojové teplotě, aby se teplotní rozdíly vyrovnaly samy.
• Nevkládejte injekční stříkačku zpět do chladničky.
© Připravte si místo podání injekce.
• Umyjte si ruce mýdlem a vodou a osušte je ručníkem.
• Injekci můžete podat do:
o stehna
o břicha (kromě oblasti 5 cm kolem pupku) o horní části paže (Viz obrázek).
• Při podávání injekce můžete stát nebo sedět.
• Ošetřete místo podání injekce tampónem namočeným v alkoholu.
• Nepodávejte do kůže, která je citlivá, tvrdá, červená nebo horká.
• Nepodávejte do místa v blízkosti viditelné žíly.
• Zvolte jiné místo pro každé podání.
• Nepodávejte přípravek Praluent do stejného místa, kam podáváte jiné injekční léčivé přípravky.
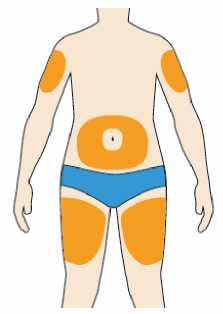
KROK B: Jak podat injekci
© Po dokončení všech kroků v „Kroku A: Příprava na podání injekce“ sundejte krytku jehly.
• Nesundávejte krytku jehly, dokud nejste připraven(á) podat injekci.
• Držte injekční stříkačku uprostřed a jehlou směřujte od těla.
• Nedotýkejte se pístu.
• Můžete pozorovat vzduchové bublinky. To je normální. Nezbavujte se vzduchových bublinek v injekční stříkačce před podáním injekce.
• Nenasazujte zpátky šedou krytku jehly.
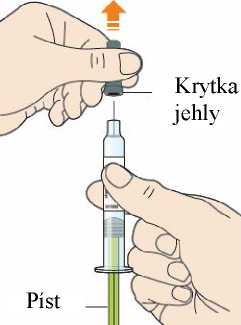
® V případě potřeby zmáčkněte kůži.
• Pomocí palce a ukazováku vytvořte v místě podání injekce kožní řasu.
• Držte kůži po celou dobu podání injekce.
® Vpíchněte jehlu do kožní řasy rychlým pohybem připomínajícím zapíchnutí šipky.
• Pokud vytvoříte 5cm kožní řasu, zvolte úhel 90°.
• Pokud vytvoříte 2cm kožní řasu, zvolte úhel 45°.
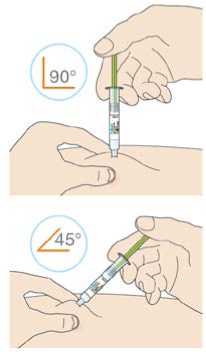
© Stiskněte píst směrem dolů.
• Podejte všechen injekční roztok plynulým stlačením pístu směrem dolů.
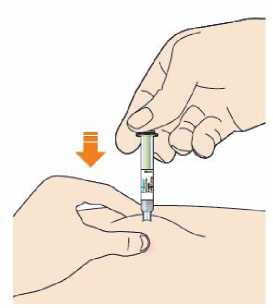
© Než vyjmete jehlu z kůže, zkontrolujte, zda je injekční stříkačka prázdná.
• Nevyjímejte jehlu stříkačky, pokud není úplně prázdná.
• Vyjměte jehlu z kůže ve stejném směru, v jakém jste ji vpíchli.
• Po podání injekce nemasírujte kůži.
• Pokud se objeví krev, přitiskněte na místo vpichu vatový tampón nebo gázu, dokud se krvácení nezastaví.
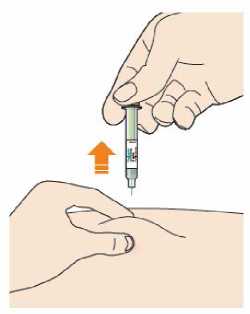
© Vyhoďte injekční stříkačku a krytku
• Nenasazujte šedou krytku jehly zpět.
• Nepoužívejte injekční stříkačku vícekrát než jednou.
• Ihned po použití vyhoďte injekční stříkačku do nepropíchnutelné nádoby.
Zeptejte se svého lékaře, lékárníka nebo zdravotní sestry, jak tuto nádobu zlikvidovat. Vždy uchovávejte tuto nádobu mimo dohled a dosah dětí,
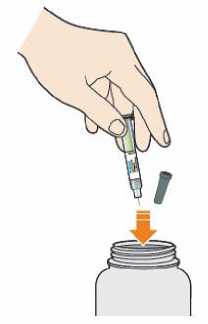
Praluent 150 mg injekční roztok v předplněné injekční stříkačce
alirokumab
Návod k použití
Součásti stříkačky přípravku Praluent jsou znázorněny na tomto obrázku.
Šedý píst
i
Tělo stříkačky
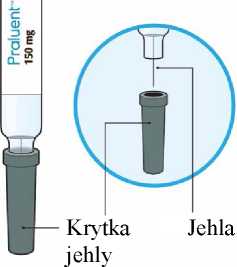
Důležité informace
• Tento prostředek je předplněná injekční stříkačka na jednorázové použití. Obsahuje 75 mg přípravku Praluent (alirokumab) v 1 ml.
• Tento léčivých přípravek se podává injekčně pod kůži a můžete si jej podat sami nebo Vám jej může podat někdo jiný (ošetřovatel(ka)).
• Tato injekční stříkačka může být použita pouze pro jednorázové podání injekce a poté musí být vyhozena.
Co ano
V Uchovávejte injekční stříkačku přípravku Praluent mimo dohled a dosah dětí.
V Před použitím injekční stříkačky přípravku Praluent si pečlivě přečtěte celý návod.
V Při každém použití injekční stříkačky přípravku Praluent postupujte podle návodu.
V Uchovávejte nepoužívané injekční stříkačky v chladničce při teplotě 2 °C až 8 °C. Pro podrobné podmínky uchovávání viz samostatná příbalová informace přípravku Praluent.
Co ne
x Nedotýkejte se jehly.
x Nepoužívejte injekční stříkačku, pokud Vám upadla nebo pokud je poničená.
x Nepoužívejte injekční stříkačku, pokud šedá krytka jehly chybí nebo pokud není bezpečně připevněna. x Nepoužívejte injekční stříkačku vícekrát než jednou. x Netřepejte s injekční stříkačkou. x Nevystavujte injekční stříkačku mrazu. x Nevystavujte injekční stříkačku přímému slunečnímu světlu.
Ponechte si tento leták. Pokud máte nějaké otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry nebo zavolejte na telefonní číslo sanofi-aventis, které je uvedené v příbalové informaci.
KROK A: Příprava na podání injekce
Předtím, než začnete, budete potřebovat:
• injekční stříkačku přípravku Praluent
• tampón namočený v alkoholu
• vatový tampón nebo gázu
• nepropíchnutelnou nádobu (viz krok B, 6).
© Než začnete.
• Vyjměte injekční stříkačku z balení a uchopte ji za tělo stříkačky.
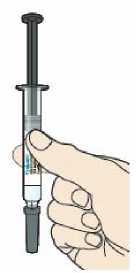
© Zkontrolujte štítek na injekční stříkačce.
• Zkontrolujte, že máte správný produkt a správnou dávku (šedý píst pro dávku150 mg/ml).
• Zkontrolujte datum expirace a uplynulo-li již toto datum, injekční stříkačku nepoužívejte.
• Zkontrolujte, zda je tekutina čirá, bezbarvá až světle žlutá a neobsahuje částice; pokud toto nesplňuje, tuto injekční stříkačku nepoužívejte.
• Zkontrolujte, zda není injekční stříkačka otevřená nebo poničená.
® Ponechte injekční stříkačku při pokojové teplotě po dobu 30 až 40 minut.
• Nezahřívejte injekční stříkačku, nechejte ji při pokojové teplotě, aby se teplotní rozdíly vyrovnaly samy.
• Nevkládejte injekční stříkačku zpět do chladničky.
© Připravte si místo podání injekce.
• Umyjte si ruce mýdlem a vodou a osušte je ručníkem.
• Injekci můžete podat do:
o stehna
o břicha (kromě oblasti 5 cm kolem pupku) o horní části paže (Viz obrázek).
• Při podávání injekce můžete stát nebo sedět.
• Ošetřete místo podání injekce tampónem namočeným v alkoholu.
• Nepodávejte do kůže, která je citlivá, tvrdá, červená nebo horká.
• Nepodávejte do místa v blízkosti viditelné žíly.
• Zvolte jiné místo pro každé podání.
• Nepodávejte přípravek Praluent do stejného místa, kam podáváte jiné injekční léčivé přípravky.
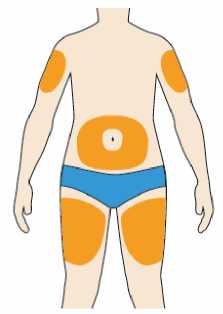
KROK B: Jak podat injekci
© Po dokončení všech kroků v „Kroku A: Příprava na podání injekce“ sundejte krytku jehly.
• Nesundávejte krytku jehly, dokud nejste připraven(á) podat injekci.
• Držte injekční stříkačku uprostřed a jehlou směřujte od těla.
• Nedotýkejte se pístu.
• Můžete pozorovat vzduchové bublinky. To je normální. Nezbavujte se vzduchových bublinek v injekční stříkačce před podáním injekce.
• Nenasazujte zpět šedou krytku jehly.
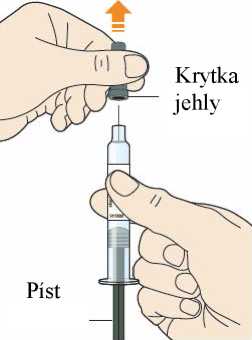
® V případě potřeby zmáčkněte kůži.
• Pomocí palce a ukazováku vytvořte v místě podání injekce kožní řasu.
• Držte kůži po celou dobu podání injekce.
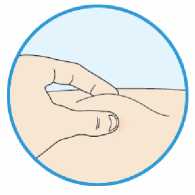
® Vpíchněte jehlu do kožní řasy rychlým pohybem připomínajícím zapíchnutí šipky.
• Pokud vytvoříte 5cm kožní řasu, zvolte úhel 90°.
• Pokud vytvoříte 2cm kožní řasu, zvolte úhel 45°.
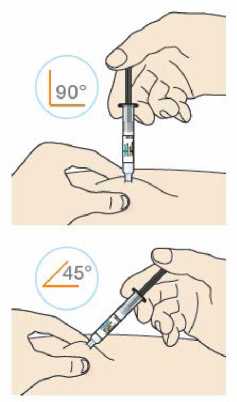
© Stiskněte píst směrem dolů.
• Podejte všechen injekční roztok plynulým stlačením pístu směrem dolů.
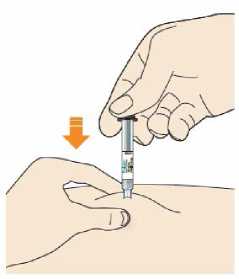
© Než vyjmete jehlu z kůže, zkontrolujte, zda je injekční stříkačka prázdná.
• Nevyjímejte jehlu stříkačky, pokud není úplně prázdná.
• Vyjměte jehlu z kůže ve stejném směru, v jakém jste ji vpíchli.
• Po podání injekce nemasírujte kůži.
• Pokud se objeví krev, přitiskněte na místo vpichu vatový tampón nebo gázu, dokud se krvácení nezastaví.
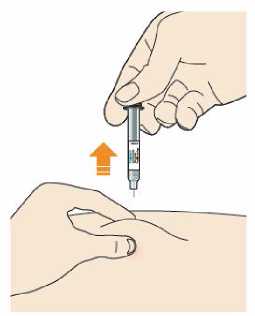
© Vyhoďte injekční stříkačku a krytku
• Nenasazujte šedou krytku jehly zpět.
• Nepoužívejte injekční stříkačku vícekrát než jednou.
Ihned po použití vyhoďte injekční stříkačku do nepropíchnutelné nádoby.
Zeptejte se svého lékaře, lékárníka nebo zdravotní sestry, jak tuto nádobu zlikvidovat. Vždy uchovávejte tuto nádobu mimo dohled a dosah dětí,
70