Portrazza 800 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Portrazza 800 mg koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna 50ml injekční lahvička obsahuje necitumumabum 800 mg.
Jeden ml koncentrátu pro infuzní roztok obsahuje necitumumabum 16 mg.
Koncentrát je nutné před použitím zředit (viz bod 6.6).
Necitumumab je lidská IgG1 monoklonální protilátka produkovaná v myších (NS0) buňkách technologií rekombinantní DNA.
Pomocná látka se známým účinkem
Jedna 50ml injekční lahvička obsahuje přibližně 244,4 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok (sterilní koncentrát).
Čirá až mírně opalizující a bezbarvá až lehce nažloutlá tekutina s pH 6,0.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Portrazza je indikován v kombinaci s chemoterapií gemcitabinem a cisplatinou k léčbě dospělých pacientů s lokálně pokročilým nebo metastazujícím dlaždicobuněčným nemalobuněčným karcinomem plic s expresí receptoru pro epidermální růstový faktor (EGFR), kteří k léčbě tohoto onemocnění dosud neužívali chemoterapii.
4.2 Dávkování a způsob podání
Léčbu necitumumabem je třeba podávat pod dohledem lékaře kvalifikovaného k podávání protinádorové chemoterapie.
Během infuzí necitumumabu mají být k dispozici vhodné zdravotnické prostředky k léčbě závažných infuzních reakcí. Zajištěna musí být dostupnost resuscitačního vybavení.
Dávkování
Portrazza se přidává k chemoterapii založené na kombinaci gemcitabinu a cisplatiny po dobu až 6 cyklů léčby, následovaných u pacientů bez progrese choroby monoterapií přípravkem Portrazza až do progrese nebo nepřijatelné toxicity.
Doporučená dávka přípravku Portrazza je 800 mg (jednotná dávka) podávaná v intravenózní infuzi po dobu 60 minut 1. a 8. den každého 3týdenního cyklu. Pokud je indikována pomalejší rychlost infuze, neměla by délka infuze přesáhnout 2 hodiny.
U pacientů je během infuze třeba monitorovat známky reakcí souvisejících s infuzí (viz bod 4.4).
Premedikace
U pacientů, u nichž dříve došlo k přecitlivělosti stupně 1-2 nebo k reakci související s infuzí při podání přípravku Portrazza, se doporučuje kromě antihistaminika ještě premedikace kortikosteroidem a antipyretikem.
Před každou infuzí necitumumabu je nutné zvážit premedikaci z důvodu potenciálních kožních reakcí (viz bod 4.4).
Úprava dávkování
Doporučení k léčbě reakcí souvisejících s infuzí a kožních reakcí jsou uvedena v tabulkách 1 a 2.
Hypersenzitivita/Reakce související s infuzí
Tabulka 1 - Doporučení k léčbě hypersenzitivity/reakcí souvisejících s infuzí
|
Stupeň toxicitya |
Doporučení k léčbě (jakýkoliv výskyt) |
|
Stupeň 1 |
• Snižte rychlost infuze o 50 % po celou dobu trvání infuze.b • Monitorujte u pacienta případné zhoršení stavu. • Při dalších infuzích, viz bod premedikace. |
|
Stupeň 2 |
• Zastavte infuzi. Po zmírnění reakce na stupeň < 1 infuzi znovu zahajte rychlostí sníženou o 50 %b • Monitorujte u pacienta případné zhoršení stavu. • Při dalších infuzích, viz bod premedikace. |
|
Stupeň 3-4 |
• Léčbu necitumumabem okamžitě a trvale vysaďte. |
a Stupeň podle NCI-CTCAE, verze 3.0
b Pokud byla rychlost infuze snížena z důvodu hypersenzitivity/reakce související s infuzí stupně 1 nebo 2, doporučuje se použít nižší rychlost infuze i u všech následujících infuzí. Délka infuze nemá přesáhnout 2 hodiny.
Tabulka 2 - Doporučení k léčbě kožních reakcí
|
Stupeň toxicitya |
Doporučení k léčbě (jakýkoliv výskyt) |
|
Stupně 1 a 2 |
• Není nutná úprava dávky |
|
Stupeň 3 |
• Dočasně pozastavte podávání, nejvýše na 6 týdnů po 1. dni posledního terapeutického cyklu, až do zmírnění příznaků na stupeň < 2. Pokud po pozastavení podávání po dobu 2 po sobě následujících cyklů (6 týdnů) nedojde ke zmírnění příznaků na stupeň < 2, trvale vysaďte. • Po zmírnění na stupeň < 2 znovu zahajte podávání ve snížené dávce 400 mg. Pokud se příznaky při dávce 400 mg zhorší, trvale vysaďte. • Pokud se příznaky při dávce 400 mg nejméně po dobu 1 terapeutického cyklu nezhorší, lze dávku zvýšit na 600 mg. Pokud se příznaky při dávce 600 mg zhorší, dočasně pozastavte podávání, nejvýše na 6 týdnů po 1. dni posledního terapeutického cyklu, dokud se příznaky nezmírní na stupeň < 2. Po zmírnění na stupeň < 2 znovu zahajte podávání ve snížené dávce 400 mg. • Pokud se příznaky při dávce 600 mg během dalšího terapeutického cyklu nezhorší, lze dávku dále zvýšit na 800 mg. • U pacientů, u nichž dojde k induraci/fibróze kůže stupně 3, trvale vysaďte. |
|
Stupeň 4 |
• Léčbu necitumumabem okamžitě a trvale vysaďte. |
a Stupeň podle NCI-CTCAE, verze 3.0
Zvláštní populace
Pediatrická populace
Použití necitumumabu u pediatrické populace v indikaci nemalobuněčného karcinomu plic není relevantní.
Starší populace
Není třeba snižovat dávku kromě snížení doporučených pro všechny pacienty (viz body 4.4 a 5.1).
Porucha funkce ledvin
U pacientů s lehkou nebo středně těžkou poruchou funkce ledvin není potřeba nijak upravovat dávku (viz bod 5.2). Nejsou k dispozici žádné údaje o podávání necitumumabu u pacientů s těžkou poruchou funkce ledvin. Není doporučeno snížení dávky.
Porucha funkce jater
O podávání necitumumabu u pacientů se středně těžkou nebo těžkou poruchou funkce jater neexistují žádné údaje (viz bod 5.2). Není doporučeno snížení dávky.
Způsob podání
Přípravek Portrazza je určen výhradně k intravenóznímu podání. Podává se v intravenózní infuzi po dobu přibližně 60 minut pomocí infuzní pumpy. Přípravek Portrazza se nesmí podávat jako intravenózní bolus nebo jako injekce. V případě předchozí hypersenzitivity nebo reakce související s infuzí je třeba postupovat podle doporučení pro léčbu hypersenzitivity/reakcí souvisejících s infuzí uvedených v tabulce 1.
Jako ředicí roztok se musí použít výhradně injekční roztok chloridu sodného 9 mg/ml (0,9%). Infuze přípravku Portrazza se nesmí podávat ani míchat s roztoky glukózy.
Návod k naředění léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Pacienti s anamnézou závažné nebo život ohrožující hypersenzitivity na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Tromboembolické příhody
U necitumumabu v kombinaci s gemcitabinem a cisplatinou byly pozorovány žilní tromboembolické příhody (VTE) a arteriální tromboembolické příhody (ATE), včetně fatálních případů (viz také bod 4.8).
Podávání necitumumabu je třeba pečlivě zvážit u pacientů s předcházejícím výskytem tromboembolických příhod (jako je plicní embolie, hluboká žilní trombóza, infarkt myokardu, cévní mozková příhoda) nebo s preexistujícími rizikovými faktory tromboembolických příhod (jako je pokročilý věk, dlouhodobě imobilizovaní pacienti, pacienti se závažnou hypovolémií, pacienti se získanou nebo vrozenou trombofilií). U pacientů s VTE a ATE v anamnéze bylo relativní riziko VTE a ATE přibližně trojnásobně vyšší.
Necitumumab nemá být podáván pacientům s mnohočetnými rizikovými faktory tromboembolických příhod, pokud přínos u daného pacienta nepřeváží rizika.
Po pečlivém posouzení rizikových faktorů u daného pacienta (zahrnujících zvýšené riziko závažného krvácení u pacientů s kavitací nádoru nebo s nádorem obklopujícím velké centrální cévy) je třeba zvážit tromboprofylaxi.
Pacienti i lékaři by měli znát projevy a příznaky tromboembolie. Pacienti mají být poučeni, aby v případě rozvoje příznaků, jako jsou dechová nedostatečnost, bolest na hrudi, otok paže či nohy, vyhledali lékařskou pomoc.
U pacientů, u nichž dojde k VTE nebo ATE, je třeba po pečlivém posouzení poměru rizik a přínosu léčby u daného pacienta zvážit vysazení necitumumabu.
V klinické studii u pokročilého nedlaždicobuněčného NSCLC měli pacienti v rameni s necitumumabem plus pemetrexedem a cisplatinou vyšší výskyt závažných tromboembolických příhod (včetně fatálních příhod) než pacienti v rameni s pemetrexedem a cisplatinou (viz také bod 4.8). Přidání necitumumabu u pokročilého nedlaždicobuněčného NSCLC nezlepšilo účinnost v porovnání se samotným pemetrexedem a cisplatinou.
Kardiorespirační onemocnění
U necitumumabu byl pozorován zvýšený výskyt kardiorespirační zástavy nebo náhlé smrti. Kardiorespirační zástava nebo náhlá smrt byla hlášena u 2,8 % (15/538) pacientů léčených necitumumabem v kombinaci s gemcitabinem a cisplatinou v porovnání s 0,6 % (3/541) pacientů léčených samotnou chemoterapií. U dvanácti z patnácti pacientů došlo k úmrtí během 30 dní po podání poslední dávky necitumumabu a tito pacienti měli v anamnéze přidružené onemocnění koronárních cév (n=3), hypomagnezémii (n=4), chronické obstrukční plicní onemocnění (n=7) a hypertenzi (n=5). U jedenácti z těchto 12 pacientů došlo k úmrtí beze svědků. Do pivotní studie nebyli zařazeni pacienti se závažným onemocněním koronárních cév, infarktem myokardu v posledních 6 měsících, nekontrolovanou hypertenzí a s nekontrolovaným městnavým srdečním selháním. Přírůstkové riziko kardiopulmonální zástavy nebo náhlé smrti u pacientů s anamnézou onemocnění koronárních cév, městnavého srdečního selhání nebo arytmií v porovnání s pacienty bez těchto přidružených onemocnění není známo.
Hypersenzitivita/reakce související s infuzí
U necitumumabu byly hlášeny reakce z přecitlivělosti/reakce související s infuzí (IRR). Ke vzniku příhody došlo obvykle po prvním nebo druhém podání necitumumabu. Během a po podání infuze u
pacientů monitorujte projevy hypersenzitivity a reakcí souvisejících s infuzí a mějte připravené k okamžitému použití resuscitační vybavení a příslušné zdravotnické prostředky. U pacientů, u nichž dříve došlo k hypersenzitivitě stupně 1-2 nebo k reakci související s infuzí při podání přípravku Portrazza, se doporučuje kromě antihistaminika premedikace kortikosteroidem a antipyretikem.
Řešení situace a úprava dávky viz bod 4.2.
Kožní reakce
U necitumumabu byly hlášeny kožní reakce (viz bod 4.8). K výskytu příhod docházelo hlavně během prvního cyklu léčby. Řešení situace a úprava dávky viz bod 4.2.
Pro řešení dermatologických reakcí může být dle klinické potřeby užitečné použití preventivního kožní ošetření zahrnujícího hydratační přípravky, ochranu před slunečním zářením, krém s topickými kortikosteroidy (1% hydrokortison) a perorální antibiotikum (např. doxycyklin). Pacientům lze doporučit, aby si nanášeli hydratační krém, opalovací krém a krém s topickými kortikosteroidy na obličej, ruce, nohy, krk, záda a hrudník.
Poruchy elektrolytů
Často (81,3 %) dochází v séru k progresivnímu poklesu hladiny hořčíku, což může vést k závažné hypomagnezémii (18,7%) (viz také bod 4.8). Po oddálení dávky může znovu dojít k hypomagnezémii stejného nebo závažnějšího stupně. U pacientů je třeba pečlivě monitorovat elektrolyty v séru včetně sérové hladiny hořčíku, draslíku a vápníku, a to před každým podáním necitumumabu a po dokončení léčby necitumumabem, až do dosažení normálních hodnot. V případě potřeby se doporučuje rychlé doplnění elektrolytů.
Starší populace
U pacientů starších 70 let nebyly mezi rameny pozorovány celkové rozdíly v účinnosti. Proto je u pacientů nad 70 let věku před zahájením léčby třeba pečlivě zhodnotit průvodní kardiovaskulární choroby, výkonnostní stav a pravděpodobnou snášenlivost chemoterapie s přidaným necitumumabem.
Ženy ve fertilním věku/antikoncepce u žen
Vzhledem ke svému mechanismu účinku a vzhledem ke zvířecím modelům, ve kterých je narušena exprese EGFR, může necitumumab způsobit poškození plodu nebo vývojové vady. Ženám ve fertilním věku je třeba doporučit, aby se po dobu užívání necitumumabu vyvarovaly otěhotnění. Během léčby necitumumabem a až 3 měsíce po posledním podání necitumumabu je nutné užívat účinnou antikoncepci. Doporučena jsou antikoncepční opatření nebo sexuální abstinence (viz bod 4.6).
Dieta s omezením sodíku
Tento léčivý přípravek obsahuje v jedné dávce 244 mg sodíku. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Mezi přípravkem Portrazza a gemcitabinem/cisplatinou nebyly pozorovány žádné lékové interakce. Farmakokinetika gemcitabinu/cisplatiny nebyla při současném podávání necitumumabu narušena a farmakokinetika necitumumabu nebyla narušena současným podáváním gemcitabinu/cisplatiny.
Žádné další formální studie interakcí s necitumumabem nebyly u lidí provedeny.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/antikoncepce u žen
Ženám ve fertilním věku je třeba doporučit, aby během užívání necitumumabu neotěhotněly, a je třeba je informovat o potenciálním riziku pro těhotenství a plod. Ženy ve fertilním věku musejí během léčby necitumumabem a až 3 měsíce po posledním podání necitumumabu užívat účinnou antikoncepci. Doporučuje se používání antikoncepčních metod nebo sexuální abstinence.
Nejsou k dispozici žádné údaje o podávání necitumumabu u těhotných žen. Nebyly provedeny reprodukční studie s necitumumabem u zvířat. Podle zvířecích modelů je receptor pro epidermální růstový faktor (EGFR) zapojen do prenatálního vývoje a může být nezbytný pro normální organogenezi, proliferaci a diferenciaci u vyvíjejícího se embrya. Přípravek Portrazza se nemá podávat během těhotenství ani u žen, které neužívají účinnou antikoncepci, pokud potenciální přínos nepřeváží potenciální riziko pro plod.
Kojení
Není známo, zda se necitumumab vylučuje do lidského mateřského mléka. Podle předpokladů jsou vylučování do mateřského mléka a perorální absorpce nízké. Riziko pro novorozence/kojence nelze vyloučit. Při léčbě přípravkem Portrazza a alespoň 4 měsíce po poslední dávce se má kojení přerušit.
Plodnost
Údaje o účinku necitumumabu na fertilitu u člověka nejsou k dispozici. Studie se zvířaty s přímým posouzením plodnosti nebyly provedeny (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Není známo, že by měl přípravek Portrazza vliv na schopnost řídit nebo obsluhovat stroje. U pacientů s takovými příznaky souvisejícími s léčbou, které narušují schopnost soustředění a reakce, se doporučuje, aby neřídili a neobsluhovali stroje, dokud tento účinek neodezní.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nejčastějšími závažnými nežádoucími reakcemi (stupně >3) pozorovanými u pacientů léčených necitumumabem jsou kožní reakce (6,3 %) a žilní tromboembolické příhody (4,3 %).
Nejčastějšími nežádoucími reakcemi byly kožní reakce, žilní tromboembolické příhody a abnormální laboratorní hodnoty (hypomagnezémie a albuminem korigovaná hypokalcémie).
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky na léky (ADR), které byly hlášeny u pacientů s pokročilým dlaždicobuněčným nemalobuněčným karcinomem plic, jsou uvedeny níže podle tříd orgánových systémů MedDRA, četnosti a stupně závažnosti. Pro klasifikaci četnosti výskytu je použito následující označení:
Velmi časté (> 1/10)
Časté (> 1/100 až < 1/10)
Méně časté (> 1/1000 až <1/100)
Vzácné (> 1/10000 až < 1/1 000)
Velmi vzácné (<1/10000)
V každé skupině četností jsou ADR uvedeny v pořadí podle klesající závažnosti.
Následující tabulka ukazuje četnost a závažnost ADR na základě výsledků studie SQUIRE, celosvětové multicentrické randomizované studie fáze 3 se dvěma rameny u dospělých pacientů se dlaždicobuněčným NSCLC, kteří byli randomizováni k léčbě necitumumabem v kombinaci s gemcitabinem/cisplatinou nebo k léčbě gemcitabinem/cisplatinou.
Tabulka 3. Nežádoucí účinky hlášené u > 1 % pacientů léčených necitumumabem ve studii SQUIRE
|
Třídy orgánových systémů |
Četnost |
Nežádoucí účinek |
Portrazza + GCb (N=538) |
GC (N=541) | ||
|
Jakýkoliv stupeň (%) |
Stupeň >3 (%) |
Jakýkoliv stupeň (%) |
Stupeň >3 (%) | |||
|
Infekce a infestace |
Časté |
Infekce močových cest |
4,1 |
0,2 |
1,7 |
0,2 |
|
Poruchy nervového systému |
Časté |
8,6 |
0 |
5,7 |
0,4 | |
|
Časté |
5,9 |
0,2 |
3,3 |
0 | ||
|
Poruchy oka |
Časté |
Konjunktivitida |
5,6 |
0 |
2,2 |
0 |
|
Cévní poruchy |
Časté |
Žilní tromboembolické příhody |
8,2 |
4,3 |
5,4 |
2,6 |
|
Časté |
Arteriální tromboembolické příhody |
4,3 |
3,0 |
3,9 |
2,0 | |
|
Časté |
Flebitida |
1,7 |
0 |
0,4 |
0 | |
|
Respirační, hrudní a mediastinální poruchy |
Časté |
Hemoptýza |
8,2 |
0,9 |
5,0 |
0,9 |
|
Časté |
Epistaxe |
7,1 |
0 |
3,1 |
0,2 | |
|
Časté |
Bolest v orofaryngu |
1,1 |
0 |
0,7 |
0 | |
|
Gastrointestinální poruchy |
Velmi časté |
28,8 |
2,8 |
25,0 |
0,9 | |
|
Velmi časté |
Stomatitida |
10,4 |
1,1 |
6,3 |
0,6 | |
|
Časté |
Dysfagie |
2,2 |
0,6 |
2,2 |
0,2 | |
|
Časté |
Vředy v ústech |
1,5 |
0 |
0,4 |
0 | |
|
Poruchy kůže a podkožní tkáně |
Velmi časté |
Kožní reakce |
77,9 |
6,3 |
11,8 |
0,6 |
|
Časté |
Hypersenzitivita/reakce související s infuzí |
1,5 |
0,4 |
2,0 |
0 | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté |
Svalové spasmy |
1,7 |
0 |
0,6 |
0 |
|
Poruchy ledvin a močových cest |
Časté |
2,4 |
0 |
0,9 |
0 | |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Pyrexie |
12,3 |
1,1 |
11,1 |
0,4 |
|
Vyšetření |
Velmi časté |
Hypomagnezémiec |
81,3 |
18,7 |
70,2 |
7,2 |
|
Velmi časté |
Albuminem korigovaná hypokalcémiec |
33,0 |
4,2 |
22,9 |
2,3 | |
|
Velmi časté |
Hypofosfatémiec |
28,9 |
6,3 |
22,7 |
5,7 | |
|
Velmi časté |
Hypokalémiec |
23,6 |
4,4 |
17,6 |
3,2 | |
|
Velmi časté |
Pokles tělesné hmotnosti |
12,1 |
0,6 |
6,3 |
0,6 |
Zkratky: GC = pouze gemcitabin a cisplatina; Portrazza+GC = necitumumab plus gemcitabin a
cisplatina; MedDRA = Medical Dictionary for Regulatory Activities. a Preferovaný MedDRA termín (verze 16).
b Tabulka ukazuje četnost ADR ve fázi chemoterapie hodnocené léčby, ve které byla kombinace Portrazza+GC přímo porovnávána s GC.
c Podle laboratorních vyšetření. Zařazeni jsou pouze pacienti s výsledky vstupní návštěvy a alespoň jedné návštěvy po vstupu do klinického hodnocení.
Popis vybraných nežádoucích účinků
Tromboembolické _ příhody
Žilní tromboembolické příhody (VTE) byly hlášeny přibližně u 8 % pacientů a převážně se jednalo o plicní embolii a hlubokou žilní trombózu. Závažné VTE byly hlášeny přibližně u 4 % pacientů.
Výskyt fatálních VTE byl mezi rameny podobný (0,2%).
Arteriální tromboembolické příhody (ATE) byly hlášeny přibližně u 4 % pacientů a převážně se jednalo o cévní mozkovou příhodu a infarkt myokardu. Závažné ATE byly hlášeny u 3 % pacientů. Výskyt fatálních ATE byl 0,6 % v experimentálním rameni versus 0,2% v kontrolním rameni (viz také bod 4.4).
V klinické studii u pokročilého nedlaždicobuněčného NSCLC byly žilní tromboembolické příhody (VTE) hlášeny přibližně u 11 % pacientů léčených necitumumabem v kombinaci s pemetrexedem a cisplatinou (v porovnání s 8 % v rameni se samotným pemetrexedem a cisplatinou) a převážně se jednalo o plicní embolii a hlubokou žilní trombózu. Závažné VTE byly hlášeny přibližně u 6 % pacientů léčených necitumumabem v kombinaci s pemetrexedem a cisplatinou (v porovnání se 4 % v rameni se samotným pemetrexedem a cisplatinou).
Arteriální tromboembolické příhody (ATE) byly hlášeny přibližně u 4 % pacientů léčených necitumumabem v kombinaci s pemetrexedem a cisplatinou (v porovnání se 6 % v rameni se samotným pemetrexedem a cisplatinou) a převážně se jednalo o cévní mozkovou příhodu a infarkt myokardu. Závažné ATE byly hlášeny přibližně u 3 % pacientů léčených necitumumabem v kombinaci s pemetrexedem a cisplatinou (v porovnání se 4 % v rameni se samotným pemetrexedem a cisplatinou).
Kožní reakce
Kožní reakce byly hlášeny přibližně u 78 % pacientů a jednalo se převážně o akneiformní vyrážku, akneiformní dermatitidu, suchou pokožku, pruritus, kožní fisury, paronychii a syndrom palmoplantární erytrodysestézie. Závažné kožní reakce byly hlášeny přibližně u 6 % pacientů, přičemž 1,7 % pacientů z důvodu kožních reakcí vysadilo léčbu. Většina kožních reakcí se rozvinula během prvního cyklu léčby a odezněla během 17 týdnů od nástupu (viz též bod 4.4).
Reakce související s infuzí
Reakce související s infuzí byly hlášeny u 1,5 % pacientů a jednalo se převážně o zimnici, horečku nebo dušnost. Závažné reakce související s infuzí byly hlášeny u 0,4 % pacientů. Většina reakcí souvisejících s infuzí se rozvinula po prvním nebo druhém podání necitumumabu.
Toxicita u starších pacientů a u pacientů s ECOG PS2
U pacientů léčených necitumumabem plus chemoterapií zahrnující gemcitabin a cisplatinu byla klinicky relevantní toxicita u starších pacientů a u pacientů se skóre 2 výkonnostního stavu dle ECOG (Eastern Cooperative Oncology Group), tj. ECOG PS2, podobná jako u celkové populace.
Trichomegalie očních řas
U pacientů léčených necitumumabem byly hlášeny ojedinělé případy trichomegalie stupně 1.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
S předávkováním necitumumabem v klinických studiích u lidí jsou jen omezené zkušenosti. Nejvyšší klinicky hodnocenou dávkou necitumumabu ve studii fáze 1 s eskalací dávky u lidí je 1000 mg jednou týdně nebo jednou za dva týdny. Pozorované nežádoucí příhody zahrnovaly bolest hlavy, zvracení a nauzeu a odpovídaly bezpečnostnímu profilu u doporučené dávky. Pro předávkování necitumumabem není žádné známé antidotum.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, monoklonální protilátky, ATC kód: L01XC22 Mechanismus účinku
Necitumumab je rekombinantní lidská monoklonální IgG1 protilátka, která se s vysokou afinitou a specificitou váže na receptor 1 pro lidský epidermální růstový faktor (EGFR) a blokuje vazebné místo pro ligandy, čímž brání aktivaci všemi známými ligandy a in vitro inhibuje související biologické důsledky. Bylo zjištěno, že aktivace EGFR koreluje s progresí maligního onemocnění, navozením angiogeneze a inhibicí apoptózy nebo buněčné smrti. Kromě toho necitumumab in vitro navozuje internalizaci a odbourávání EGFR. Studie in vivo se štěpy odvozenými z buněčných linií modelů lidských nádorů (xenograft) včetně nemalobuněčného karcinomu plic prokázaly, že necitumumab má protinádorovou účinnost jak v monoterapii, tak v kombinaci s gemcitabinem a cisplatinou.
Imunogenita
Stejně jako všechny terapeutické proteiny je i tento potenciálně imunogenní.
Celkově byl u pacientů léčených necitumumabem zjištěn nízký výskyt protilátek proti léku a neutralizačních protilátek, bez korelace s výsledky týkajícími se bezpečnosti léčby u daných pacientů. Nebyl zjištěn vztah mezi imunogenitou a IRR ani nežádoucími příhodami při léčbě.
SQUIRE, celosvětová multicentrická randomizovaná studie se dvěma rameny s přípravkem Portrazza byla provedena u 1093 pacientů s dlaždicobuněčným NSCLC stadia IV (American Joint Committee on Cancer, verze 7) včetně pacientů s ECOG PS2, kteří dříve neužívali žádnou protinádorovou léčbu metastazujícího onemocnění. Pacienti byli randomizováni k užívání přípravku Portrazza 800 mg plus chemoterapie zahrnující gemcitabin 1250 mg/m2 a cisplatinu 75 mg/m2 (rameno Portrazza+GC) nebo samotné chemoterapie gemcitabinem a cisplatinou (rameno G+C) v první linii léčby. Portrazza a gemcitabin byly podávány 1. a 8. den každého 3týdenního terapeutického cyklu a cisplatina byla podávána 1. den každého 3týdenního terapeutického cyklu. Ve studii nebyla povinná premedikace před podáním přípravku Portrazza. Preventivní léčba kožních reakcí nebyla povolena do začátku druhého terapeutického cyklu. Pacienti dostali v každém rameni maximálně šest cyklů chemoterapie; pacienti v rameni s přípravkem Portrazza+GC, u nichž nedošlo k progresi, dále pokračovali v užívání přípravku Portrazza v monoterapii až do progrese choroby, nepřijatelné toxicity nebo odvolání souhlasu. Hlavním sledovaným parametrem účinnosti bylo celkové přežití (OS) a podpůrným sledovaným parametrem účinnosti bylo přežití bez progrese (PFS). Pacienti podstupovali každých 6 týdnů radiografické vyšetření stavu choroby, až do radiograficky doložené progrese onemocnění (PD).
Demografické a vstupní charakteristiky pacientů byly u obou terapeutických ramen vyvážené. Medián věku byl 62 (32-86), 83 % pacientů byli muži, 83,5 % bylo kavkazské rasy a 91 % byli kuřáci. ECOG PS 0 byl u 31,5 % pacientů; 1 u 59,7 %; a 2 u 9 %. Více než 50 % pacientů mělo metastatické onemocnění ve více než 2 lokalizacích. V rameni Portrazza+GC pokračovalo po dokončení chemoterapie 51 % pacientů v monoterapii přípravkem Portrazza. Užívání systémové léčby po studii bylo v obou ramenech podobné (47,3 % v rameni Portrazza+GC a 44,7 % v rameni GC).
Výsledky týkající se účinnosti jsou uvedeny v tabulce 4.
Tabulka 4. Souhrn údajů týkajících se účinnosti (ITT populace)
|
Rameno Portrazza+GC |
Rameno GC | |
|
N=545 |
N=548 | |
|
Celkové přežití | ||
|
Počet událostí (n) |
418 |
442 |
|
Medián - měsíce (95 % CIa) |
11,5 (10,4, 12,6) |
9,9 (8,9, 11,1) |
|
Poměr rizik (95 % CI)b c |
0,84 (0,74, 0,96) | |
|
Hodnota p dle dvoustranného |
0,012 | |
|
log-rank testuc | ||
|
1letá četnost celkového přežití (%) |
47,7 |
42,8 |
|
Přežití bez progrese | ||
|
Počet událostí (n) |
431 |
417 |
|
Medián - měsíce (95 % CI) |
5,7 (5,6, 6,0) |
5,5 (4,8, 5,6) |
|
Poměr rizik (95 % CI) b c |
0,85 (0,74, 0,98) | |
|
Hodnota p dle dvoustranného |
0,020 | |
|
log-rank testuc | ||
a Zkratky: CI = interval spolehlivosti
b Poměr rizik je vyjádřen jako léčba/kontrola a odhadován dle Coxova modelu
Stratifikováno podle randomizačních parametrů (ECOG PS [0-1 vs. 2] a zeměpisné oblasti (Severní Amerika, Evropa a Austrálie vs. Jižní Amerika, Jižní Afrika a Indie vs. Východní Asie])
c
Obr. 1. Kaplan-Meierův graf celkového přežití (ITT populace)
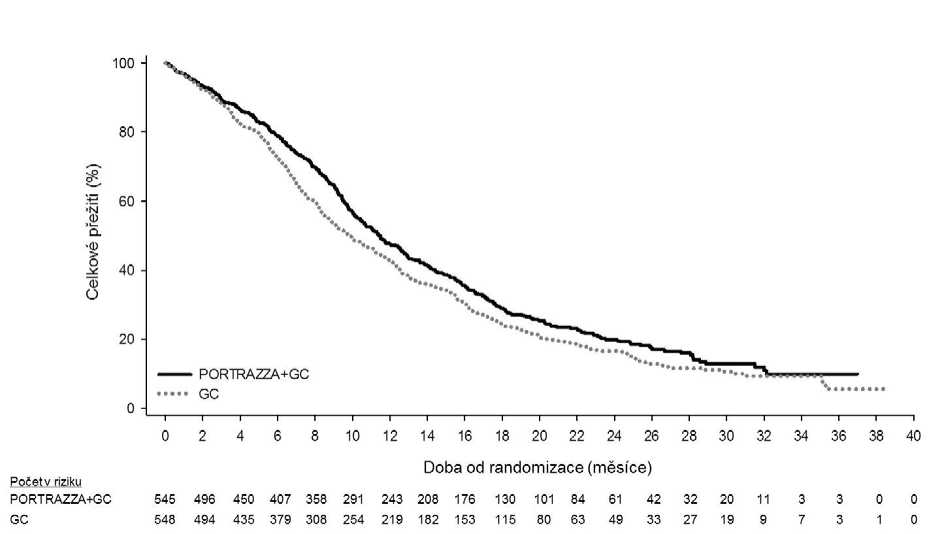
Zkratky: C = cisplatina, G = gemcitabin
Zlepšení u OS a PSF bylo pozorováno v podskupinách včetně předem definovaných stratifikačních faktorů [skóre ECOG PS [0-1 vs. 2] a zeměpisné oblasti (Severní Amerika, Evropa a Austrálie vs. Jižní Amerika, Jižní Afrika a Indie vs. východní Asie]); u pacientů ve věku 70 a více let byl poměr rizik pro celkové přežití 1,03 (0,75, 1,42) (viz obrázek 2).
Obr. 2. Graf "forest plot" pro analýzu celkového přežití u podskupin (populace ITT)
|
Kategorie |
Podskupina |
N Rameno |
|
Populace 111 |
PORTRAZZAi 545 | |
|
Věková . | ||
|
skupina |
< 65 let |
332 |
|
ž 65 let |
213 | |
|
< /Olei |
43/ | |
|
ž 70 let |
108 | |
|
Pohlaví |
Žena |
95 |
|
Muž |
450 | |
|
Rasa |
Bílá |
457 |
|
Jiná než bdá |
88 | |
|
Nikdv/bvvalv |
44 | |
|
Kurárky status | ||
|
Kuřák |
500 | |
|
FCOG P5 |
0-1 |
496 |
|
2 |
49 | |
|
Zeměpisné |
S Amerika, Evropa, Austrálie 4/2 | |
|
oblasti |
1 Amerika, 1 Afrika, |
Indie 30 |
|
Východní Asie |
43 | |
N
Rameno GC
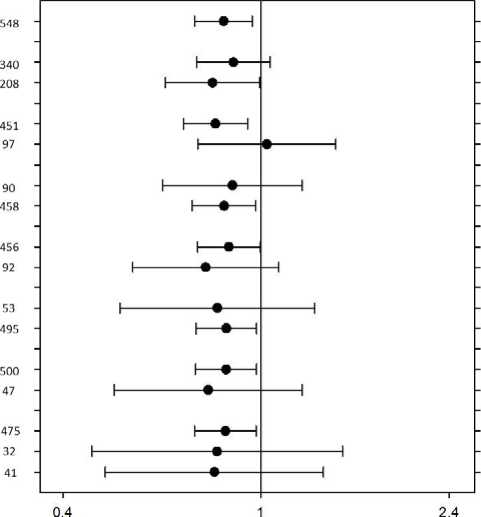
<-- -;->
Ve prospěch ramene PORTRAZZA+GC Ve prospěch ramene GC
HR
(95% Cl)
0,84(0,74,0,96)
0,88 (0,74,1,04) 0.80(0.64,0.99)
0,81(0,70,0,94) 1,03(0,75,1,42)
0,88(0,64,1,21) 0,84 (0./3,0,98)
0,8G (0,75,1,00) 0,78(0,55,1,09)
0,8? (0,57,1,79) 0,85(0,74,0,98)
0,85(0,74,0,98) 0,/8(0,bl, 1,21)
0,85(0,74,0,98) 0,82 (0,46,1,46) 0,81(0,49,1,34)
Zkratky: C = cisplatina, G = gemcitabin, ITT=intent-to-treat (populace se záměrem léčit).
Předem plánovaná exploratorní analýza, provedená po primární analýze, určila výsledky klinické účinnosti podle stupně exprese proteinu EGFR v nádoru.
V populaci ITT bylo u 982 (89,8 %) pacientů možné vyšetřit expresi EGRF imunohistochemickou metodou (IHC) pomocí soupravy Dako PharmDx Kit. Exprese EGFR v nádoru byla považována za pozitivní, když se podařilo identifikovat alespoň jednu obarvenou buňku. U velké většiny pacientů (95,2 % hodnotitelných pacientů, n — 935) vykázaly vzorky nádoru expresi EGFR; u 4,8 % (n = 47) nebyla exprese proteinu EGFR detekovatelná. Mezi podskupinou pacientů s detekovatelnou expresí proteinu EGFR a populací ITT nebyly relevantní rozdíly v rozložení demografických charakteristik, charakteristik onemocnění ani užívání systémové terapie po studii.
U pacientů s detekovatelnou expresí proteinu EFGR (indikovaná populace pacientů) bylo celkové přežití statisticky významně lepší v rameni Portrazza+GC v porovnání s ramenem GC s odhadovaným snížením rizika úmrtí o 21 % (poměr rizik [HR] — 0,79 [0,69-0,92], p = 0,002) a střední hodnotou OS 11,7 měsíce v rameni Portrazza+GC a 10,0 měsíce v rameni GC.
Bylo také pozorováno statisticky významné zlepšení přežití bez progrese (HR = 0,84 [0,72-0,97], p = 0,018) se střední hodnotou PFS 5,7 měsíce v rameni Portrazza+GC a 5,5 měsíce v rameni GC.
U pacientů s detekovatelnou expresí proteinu EFGR nebyl pozorován trend zvýšené účinnosti se zvyšujícím se stupněm exprese EGFR.
U pacientů bez detekovatelné exprese proteinu EGFR nebylo pozorováno žádné zlepšení celkového přežití (poměr rizik [HR] — 1,52 [0,74, 3,12]) ani přežití bez progrese (poměr rizik [HR] — 1,33 [0,65, 2,70]).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Portrazza u všech podskupin pediatrické populace s nemalobuněčným karcinomem plic (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Při dávkovacím schématu 800 mg necitumumabu 1. a 8. den v 21denním cyklu byl geometrický průměr Cmm necitumumabu v séru pacientů s dlaždicobuněčným NSCLC po pěti cyklech léčby v kombinaci s gemcitabinem a cisplatinou 98,5 pg/ml (variační koeficient 80 %).
Absorpce
Portrazza se podává v intravenózní infuzi. Nebyly provedeny žádné studie s jinou cestou podání. Distribuce
Distribuce přípravku Portrazza vykazuje dvoufázový pokles. Podle populačního farmakokinetického přístupu (PopPK) byl průměrný distribuční objem necitumumabu v ustáleném stavu (Vss) 6,97 l (CV 31 %).
Eliminace
Necitumumab vykazuje clearance závislou na koncentraci. Průměrná celková systémová clearance (CLtot) v ustáleném stavu při dávkování 800 mg 1. a 8. den 21denního cyklu byla 0,014 l/h (CV 39 %). To odpovídá poločasu přibližně 14 dní. Předpokládaná doba do dosažení ustáleného stavu byla přibližně 70 dní.
Zvláštní populace
Populační farmakokinetická analýza ukázala, že na farmakokinetiku necitumumabu nemá vliv věk, pohlaví ani rasa, zatímco CL a distribuční objem vykázaly méně než proporcionální pozitivní korelaci s tělesnou hmotností. I když výsledky modelování naznačují, že dispozice necitumumabu byla statisticky závislá na tělesné hmotnosti, simulace ukazují, že dávkování podle tělesné hmotnosti by variabilitu farmakokinetiky významně nesnížilo. U těchto subpopulací není nutná úprava dávkování.
Starší populace
Na základě výsledků populační farmakokinetické analýzy neměl věk vliv na expozici necitumumabu.
Porucha _ funkce ledvin
Nebyly provedeny žádné formální studie, které by hodnotily vliv poruchy funkce ledvin na farmakokinetiku necitumumabu. Na základě výsledků populační farmakokinetické analýzy neměly renální funkce hodnocené podle clearance kreatininu [CrCl] vliv na farmakokinetiku necitumumabu.
Porucha funkce jater
Nebyly provedeny žádné formální studie, které by hodnotily vliv poruchy funkce jater na farmakokinetiku necitumumabu. Na základě výsledků populační farmakokinetické analýzy neměl stav jaterních funkcí (hodnocených podle alaninaminotransferázy, aspartátaminotransferázy a celkového bilirubinu) významný vliv na farmakokinetiku necitumumabu.
5.3 Předklinické údaje vztahující se k bezpečnosti
V 26týdenní studii na opicích byla pozorována na dávce závislá reverzibilní kožní toxicita. Kožní účinky odpovídaly známým účinkům třídy inhibitorů EGFR.
Nebyly provedeny žádné speciální studie s necitumumabem u zvířat, které by hodnotily jeho kancerogenní potenciál nebo potenciál narušení plodnosti. Riziko narušení plodnosti není známo. Nicméně u opic, kterým byl podáván necitumumab po dobu 26 týdnů, nebyly pozorovány žádné nežádoucí účinky na samčí nebo samičí pohlavní orgány.
Je známo, že lidské IgG1 přecházejí přes placentu, proto může být necitumumab přenesen od matky na vyvíjející se plod. Nebyly provedeny žádné speciální studie, které by hodnotily účinek necitumumabu na rozmnožování a na vývoj plodu, ovšem vzhledem k jeho mechanismu účinku a vzhledem ke zvířecím modelům, ve kterých je narušena exprese EGFR, může necitumumab vést k poškození plodu nebo vývojovým vadám.
FARMACEUTICKÉ ÚDAJE
6.
6.1 Seznam pomocných látek
Dihydrát natrium-citrátu (E331)
Bezvodá kyselina citrónová (E330)
Chlorid sodný Glycin (E640)
Mannitol (E421)
Polysorbát 80 (E433)
Voda na injekci
6.2 Inkompatibility
Infuze přípravku Portrazza s nemá podávat ani míchat s roztoky glukózy. Tento léčivý přípravek se nesmí míchat s jinými léčivými přípravky, kromě přípravků uvedených v bodu 6.6.
6.3 Doba použitelnosti
Neotevřená injekční lavička 2 roky.
Po naředění
Při přípravě podle pokynů neobsahuje roztok přípravku Portrazza žádné antimikrobiální konzervační látky.
Pro minimalizaci rizika mikrobiální kontaminace se doporučuje se, aby byla připravená dávka roztoku podána okamžitě. Pokud se připravená dávka roztoku necitumumabu nepoužije okamžitě, musí být uchována při 2 °C až 8 °C po dobu nepřesahující 24 hodin nebo ji lze uchovat při 9 °C až 25 °C po dobu až 4 hodin. Uchovávejte chráněné před světlem. Krátká expozice okolnímu světlu během přípravy a podání je přijatelná.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v lednici (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
50 ml roztoku v injekční lahvičce (sklo třídy I) s chlorbutylovou elastomerovou zátkou, hliníkovým uzávěrem a polypropylenovým víčkem.
Balení s 1 injekční lahvičkou.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Připravte infuzní roztok za použití aseptické techniky, aby byla zajištěna sterilita připraveného roztoku.
Injekční lahvička je určena pouze pro jedno použití. Zkontrolujte obsah injekčních lahviček, zda neobsahují drobné částice nebo nedošlo ke změně barvy. Koncentrát pro přípravu infuzního roztoku musí být před naředěním čirý až mírně opalizující a bezbarvý až lehce nažloutlý. Pokud zjistíte přítomnost částic nebo zabarvení, injekční lahvičku nepoužívejte.
Injekční lahvička obsahuje 800 mg necitumumabu, tj. 16 mg/ml roztoku; jedna 50ml injekční lahvička obsahuje celou dávku. Jako ředicí roztok použijte výhradně injekční roztok chloridu sodného 9 mg/ml (0,9%).
Podání pomocí předplněných balení k intravenózní infuzi
Asepticky odeberte 50 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%) z předplněného 250ml balení pro intravenózní aplikaci a pak přeneste do balení 50 ml léčivého přípravku necitumumab, aby bylo opět dosaženo konečného objemu 250 ml v balení. Kontejner jemně převraťte, aby se obsah promíchal. Infuzní roztok NEZMRAZUJTE ani NEPROTŘEPÁVEJTE. NEŘEĎTE jinými roztoky ani nepodávejte spolu s infuzí s jinými elektrolyty nebo léčivými přípravky.
Podání pomocí prázdného obalu pro intravenózní infuzi
Asepticky přeneste 50 ml léčivého přípravku necitumumab do prázdného obalu pro intravenózní infuzi a do obalu přidejte 200 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), aby bylo dosaženo konečného objemu 250 ml. Obal jemně převraťte, aby se obsah promíchal. Infuzní roztok NEZMRAZUJTE ani NEPROTŘEPÁVEJTE. NEŘEĎTE jinými roztoky ani nepodávejte spolu s infuzí s jinými elektrolyty nebo léčivými přípravky.
Podejte pomocí infuzní pumpy. K infuzi je nutné použít samostatnou infuzní linku a na konci inluze je nutné linku vypláchnout injekčním roztokem chloridu sodného 9 mg/ml (0,9 %).
Parenterální léčivé přípravky je třeba před podáním vizuálně zkontrolovat, zda neobsahují částice. Pokud zjistíte přítomnost částic, infuzní roztok nepoužívejte.
Nepoužívejte zbývající objem necitumumabu, který zůstane v injekční lahvičce, protože přípravek neobsahuje antimikrobiální konzervační látky.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Eli Lilly Nederland B.V.
Papendorpseweg 83 3528 BJ Utrecht Nizozemsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1084/001
9. DATUM REGISTRACE/ PRODLOUŽENÍ REGISTRACE
Datum první registrace: 15. únor 2016
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY /BIOLOGICKÝCH LÉČIVÝCH LÁTEK A> VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY /BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa vvrobce/vvrobců biologické léčivé látky/biologických léčivých látek
ImClone Systems LLC
33 ImClone Drive
Branchburg
New Jersey
NJ 08876
USA
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží Lilly, S.A.
Avda. de la Industria, 30 Alcobendas Madrid 28108 Španělsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl.
107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Před uvedením přípravku Portrazza (necitumumab) na trh v každém členském státě musí držitel rozhodnutí o registraci získat souhlas odpovídající národní autority s obsahem a formátem edukačních materiálů, včetně způsobu komunikace, distribučního postupu a jakýchkoliv dalších aspektů programu.
Držitel rozhodnutí o registraci musí zajistit, aby v každém členském státě, ve kterém je přípravek Portrazza na trhu, byli všichni lékaři (tzn. onkologové) seznámeni s klíčovými podmínkami pro bezpečné používání necitumumabu. Tyto materiály budou informovat o rizicích týkajících se arteriálních / žilních tromboembolických příhod a kardiorespiračních poruch.
Klíčové prvky edukačních materiálů pro lékaře:
o Důležitost posouzení rizik před zahájením léčby necitumumabem
o Popis tromboembolických příhod včetně četnosti jejich výskytu z klinických studií
o Doporučení, aby si pacienti a lékaři byli vědomi příznaků a projevů tromboembolie. Pacienti mají být poučeni, aby vyhledali lékařskou pomoc, pokud se u nich vyvinou příznaky tromboembolie, jako jsou obtížné dýchání, bolest na hrudi, otok paže či nohy
o Nutnost pečlivě zvážit použití necitumumabu u pacientů s tromboembolickými příhodami v anamnéze nebo s preexistujícími rizikovými faktory tromboembolických příhod.
o Informaci o relativním riziku VTE nebo ATE u pacientů s VTE nebo ATE v anamnéze
o Doporučení, že necitumumab nemá být podáván pacientům s vícečetnými rizikovými faktory tromboembolických příhod, pokud u nich přínosy nepřeváží rizika
o Nutnost zvážení tromboprofylaxe po pečlivém zhodnocení rizikových faktorů pacienta
o Potřeba zvážit po pečlivém posouzení poměru rizik a přínosu léčby u daného pacienta vysazení necitumumabu u pacientů, u nichž dojde k VTE nebo ATE.
o Popis kardiorespiračních příhod včetně četnosti jejich výskytu z klinických studií
o Informace o tom, že není známo přírůstkové riziko kardiopulmonální zástavy nebo náhlé smrti u pacientů s anamnézou onemocnění koronárních cév, městnavého srdečního selhání nebo arytmií v porovnání s pacienty bez těchto přidružených onemocnění
o Instrukce zdravotnickým pracovníkům, aby si přečetli tyto materiály spolu se souhrnem informací o přípravku (SPC)
Edukační materiál pro lékaře musí také obsahovat: o Souhrn informací o přípravku o Příbalovou informaci pro pacienta
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Portrazza 800 mg koncentrát pro infuzní roztok necitumumabum
2. OBSAH LÉČIVÉ LÁTKY
Jedna 50ml injekční lahvička obsahuje necitumumabum 800 mg (16 mg/ml).
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: dihydrát natrium-citrátu, bezvodá kyselina citronová, chlorid sodný, glycin, mannitol, polysorbát 80 a voda na injekci. Další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro infuzní roztok
800 mg/50 ml 1 injekční lahvička
5. ZPŮSOB A CESTA
Intravenózní podání po naředění.
Pouze pro jednorázové podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Neprotřepávejte.
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Eli Lilly Nederland B.V. Papendorpseweg 83 3528 BJ Utrecht Nizozemsko
12 REGISTRAČNÍ ČÍSLO
EU/1/15/1084/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU štítek
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
Portrazza 800 mg sterilní koncentrát
necitumumabum
i.v. podání po naředění
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
800 mg/50 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: Informace pro uživatele Portrazza 800 mg koncentrát pro infuzní roztok
necitumumabum
'VTento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně tuto příbalovou informaci dříve, než Vám bude tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou v této příbalové informaci uvedeny. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Portrazza a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Portrazza podán
3. Jak Vám bude přípravek Portrazza podáván
4. Možné nežádoucí účinky
5. Jak přípravek Portrazza uchovávat
6. Obsah balení a další informace
1. Co je Portrazza a k čemu se používá
Portrazza obsahuje léčivou látku necitumumab, která patří do skupiny látek zvaných monoklonální protilátky.
Necitumumab rozpozná a specificky se naváže na bílkovinu na povrchu některých nádorových buněk. Tato bílkovina se nazývá receptor pro epidermální růstový faktor (EGFR). Na EGRF se mohou navázat jiné tělesné bílkoviny (zvané růstové faktory) a podporovat růst a dělení nádorových buněk. Necitumumab brání jiným bílkovinám, aby se navázaly na EGFR a tím brání růstu a dělení nádorových buněk.
Portrazza se používá v kombinaci s dalšími protinádorovými léčivými přípravky u dospělých k léčbě určitého typu rakoviny plic v pokročilém stadiu (dlaždicobuněčného nemalobuněčného karcinomu plic), jehož buňky mají na svém povrchu bílkovinu EGFR. Protinádorové léčivé přípravky, se kterými se kombinuje, jsou gemcitabin a cisplatina.
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Portrazza podán Přípravek Portrazza nesmíte dostat
- jestliže j ste někdy měl(a) závažnou alergickou reakci na necitumumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Ihned lékaři nebo zdravotní sestře sdělte, pokud se u vás během léčby přípravkem Portrazza nebo po léčbě objeví některý z následujících stavů (i když si tím nebudete jistý/á):
- Krevní sraženiny v tepnách nebo žílách
Portrazza může způsobit vznik krevních sraženin v tepnách nebo žílách. Příznaky mohou zahrnovat otok, bolest a citlivost končetiny, obtížné dýchání, bolest na hrudi nebo abnormální srdeční tep a nepříjemné pocity. Lékař s vámi probere, zda nepotřebujete nějaká preventivní opatření. Příznaky vzniku krevních sraženin najdete také v bodu 4.
- Poruchy týkající se srdce a dýchacího systému
U pacientů léčených přípravkem Portrazza v kombinaci s gemcitabinem a cisplatinou i u pacientů léčených samotným gemcitabinem a cisplatinou byly pozorovány případy poruch týkajících se srdce a dýchacího systému a nevysvětlená úmrtí. Příčiny těchto úmrtí a jejich souvislost s léčbou nebyly vždy známy. Portrazza může toto riziko zvyšovat. Lékař s vámi tuto záležitost probere.
- Reakce související s infuzí
Při léčbě přípravkem Portrazza může dojít k reakcím souvisejícím s infuzí. Tyto reakce mohou být alergie. Lékař s vámi probere, zda nepotřebujete nějaká preventivní opatření nebo časnou léčbu. Během infuze vás bude lékař nebo zdravotní sestra kvůli možnému vzniku nežádoucích účinků kontrolovat. Pokud u vás dojde k závažné reakci související s infuzí, může vám lékař doporučit úpravu dávky přípravku Portrazza nebo léčbu přípravkem Portrazza ukončit. Další podrobnosti o reakcích souvisejících s infuzí, ke kterým může dojít při infuzi nebo po ní, najdete v bodu 4.
- Kožní reakce
Portrazza může vyvolávat nežádoucí účinky postihující kůži. Lékař s vámi probere, zda nepotřebujete nějaká preventivní opatření nebo časnou léčbu. Pokud u vás dojde k závažné kožní reakci, může vám lékař doporučit úpravu dávky přípravku Portrazza nebo léčbu přípravkem Portrazza ukončit. Další podrobnosti o kožních reakcích najdete v bodu 4.
- Hladina hořčíku, vápníku, draslíku a fosfátů v krvi
Během léčby vám bude lékař kontrolovat pravidelně hladinu některých látek, jako je hořčík, vápník, draslík a fosfáty v krvi. Pokud bude jejich hladina příliš nízká, může vám lékař předepsat přípravky na jejich doplnění.
Děti a dospívající
Přípravek Portrazza se nemá podávat pacientům do 18 let věku, protože neexistují žádné informace o tom, jak u této věkové skupiny působí.
Další léčivé přípravky a Portrazza
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Týká se to i léčivých přípravků, které jsou dostupné bez lékařského předpisu, a rostlinných přípravků.
Těhotenství a kojení
Před zahájením léčby musíte informovat svého lékaře, pokud jste těhotná nebo kojíte, myslíte si, že byste mohla být těhotná nebo plánujete počít dítě.
Během užívání tohoto léčivého přípravku a nejméně 3 měsíce po poslední dávce přípravku Portrazza se vyhněte otěhotnění, protože tento léčivý přípravek by mohl poškodit vaše nenarozené dítě. Promluvte si s lékařem o tom, jaká antikoncepce je pro vás nejvhodnější.
Během léčby přípravkem Portrazza a nejméně 4 měsíce po užití poslední dávky nekojte, protože tento léčivý přípravek by mohl poškodit růst a vývoj vašeho dítěte.
Řízení dopravních prostředků a obsluha strojů
Pokud se u vás objeví příznaky, které narušují schopnost soustředění a reakce, neřiďte ani neobsluhujte stroje, dokud tento účinek neodezní.
Portrazza obsahuje sodík
Tento léčivý přípravek obsahuje v jedné dávce 244 mg sodíku. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
3. Jak budete přípravek Portrazza dostávat
Na vaši léčbu přípravkem Portrazza bude dohlížet lékař se zkušenostmi s podáváním protinádorových léčivých přípravků.
Premedikace
Před podáním přípravku Portrazza můžete dostat léčivé přípravky na snížení rizika reakce spojené s infuzí a kožní reakce.
Dávka a podávání
Doporučená dávka přípravku Portrazza je 800 mg 1. a 8. den každého 3týdenního cyklu. Portrazza se podává v kombinaci s léčivými přípravky gemcitabinem a cisplatinou až po dobu 6 cyklů a poté se podává samostatně. Počet infuzí, které dostanete, bude záviset na tom, jakým způsobem a jak dlouho budete na léčbu přípravkem Portrazza reagovat. Lékař to s vámi probere.
Tento léčivý přípravek se podává intravenózní infuzí (kapáním do žíly). Infuze trvá přibližně 60 minut.
Na konci této příbalové informace je podrobný návod pro lékaře nebo zdravotní sestru, jak infuzi přípravku Portrazza připravit (viz "Pokyny pro manipulaci").
Úpravy dávkování
Během každé infuze u vás bude lékař nebo zdravotní sestra kontrolovat nežádoucí účinky. Pokud se u vás během léčby objeví reakce v souvislosti s infuzí, rychlost infuze bude zpomalena a všechny další infuze budou také podávány pomaleji. Délka infuze by neměla přesáhnout 2 hodiny. Viz také bod 2 "Upozornění a opatření".
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Významnými nežádoucími účinky přípravku Portrazza jsou kožní reakce a tvorba krevních sraženin v žílách.
Pokud se u vás objeví jakýkoliv z následujících stavů, vyhledejte okamžitě lékařskou pomoc: Krevní sraženiny v žílách
Pravděpodobnost vzniku krevních sraženin v žílách je přibližně u 8 ze 100 pacientů. Pravděpodobnost, že tyto nežádoucí účinky budou závažné, je přibližně u 4 ze 100 pacientů. Mohou vést k ucpání krevní cévy v dolní končetině. Příznaky mohou zahrnovat otok, bolest a citlivost končetiny. Krevní sraženiny mohou také vest k ucpání krevních cév v plicích. Mezi příznaky může patřit obtížné dýchání, bolest na hrudi nebo abnormální srdeční tep a obtíže.
Kožní reakce
Kožní reakce se mohou vyskytnout přibližně u 80 ze 100 pacientů, kteří užívají přípravek Portrazza, a tyto reakce jsou obvykle mírné až středně závažné. Pravděpodobnost závažných kožních reakcí je přibližně u 5 ze 100 pacientů. Mezi příznaky závažné kožní reakce mohou patřit projevy na kůži připomínající akné a kožní vyrážka. Kožní vyrážka obvykle připomíná akné a často postihuje obličej, horní část hrudníku a zad, ale může se objevit na kterékoliv části těla. Většina těchto nežádoucích účinků obvykle po ukončení léčby přípravkem Portrazza časem vymizí.
Mezi další nežádoucí účinky patří:
Velmi časté (mohou postihnout více než 1 osobu z 10):
- svědění, suchá kůže, šupinatění kůže, poruchy nehtů (kožní reakce)
- zvracení
- horečka nebo zvýšená teplota (pyrexie)
- pokles tělesné hmotnosti
- vředy v ústech a opary (stomatitida)
Časté (mohou postihnout až 1 z 10 osob)
- bolest hlavy
- vykašlávání krve (hemoptýza)
- krvácení z nosu (epistaxe)
- divná chuť v ústech, kovová chuť v ústech (dysgeuzie)
- zánět očí (konjunktivitida)
- krevní sraženiny v tepnách
- infekce močových cest (močového měchýře a/nebo ledvin)
- bolest při močení (dysurie)
- obtížné polykání (dysfagie)
- svalové spasmy
- zánět žil dolních končetin (flebitida)
- alergické reakce
- bolest v ústech a hrdle (orofaryngeální bolest)
Portrazza může také vést ke změnám výsledků krevních testů. Zahrnují nízkou hladinu hořčíku, vápníku, draslíku nebo fosfátů v krvi.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou v této příbalové informaci uvedeny. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak uchovávat přípravek Portrazza
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a na štítku na injekční lahvičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Infuzní roztok: Po naředění a přípravě je nutné léčivý přípravek okamžitě použít. Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 při 2 °C až 8 °C nebo 4 hodiny při teplotě 9 °C až 25 °C. Infuzní roztok nezmrazujte a netřepejte s ním. Nepodávejte roztok, pokud si v něm všimnete jakýchkoliv částic nebo zabarvení.
Tento léčivý přípravek je určen pouze k jednorázovému použití.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Portrazza obsahuje
- Léčivou látkou je necitumumabum. Jeden mililitr koncentrátu pro infuzní roztok obsahuje necitumumabum 16 mg.
Jedna 50ml injekční lahvička obsahuje necitumumabum 800 mg.
- Dalším složkami j sou dihydrát natrium-citrátu (E331), bezvodá kyselina citronová (E330), chlorid sodný (viz bod 2 “Portrazza obsahuje sodík”), glycin (E640), mannitol (E421), polysorbát 80 (E433) a voda na injekci.
Jak Portrazza vypadá a co obsahuje toto balení
Portrazza 800 mg, koncentrát pro infuzní roztok (sterilní koncentrát), je čirá až mírně opalizující a bezbarvá až lehce nažloutlá tekutina ve skleněné injekční lahvičce s pryžovou zátkou.
Je k dispozici v baleních:
- 1 injekční lahvička s 50 ml
Držitel rozhodnutí o registraci
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht, Nizozemsko Výrobce
Lilly, S.A., Avda de la Industria, 30, Alcobendas, 28108 Madrid, 28108, Španělsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Lietuva
Eli Lilly Holdings Limited atstovybé Tel. +370 (5) 2649600
Luxembourg/Luxemburg
Eli Lilly Benelux S.A./N.V.
Tél/Tel: + 32-(0)2 548 84 84
Magyarország
Lilly Hungária Kft.
Tel: + 36 1 328 5100
Malta
Charles de Giorgio Ltd.
Tel: + 356 25600 500
Nederland
Eli Lilly Nederland B.V.
Tel: + 31-(0) 30 60 25 800
Norge
Eli Lilly Norge A.S.
Tlf: + 47 22 88 18 00
Belgique/Belgie/Belgien
Eli Lilly Benelux S.A./N.V.
Tél/Tel: + 32-(0)2 548 84 84
Eunrapnu
Tn "Enn Hunn HegepnaHg" E.B. - Eunrapna Ten. + 359 2 491 41 40
Česká republika ELI LILLY ČR, s.r.o.
Tel: + 420 234 664 111
Danmark
Eli Lilly Danmark A/S Tlf: +45 45 26 60 00
Deutschland
Lilly Deutschland GmbH Tel. + 49-(0) 6172 273 2222
Eesti
Eli Lilly Holdings Limited Eesti filiaal Tel: +372 6 817 280
EXXáSa Osterreich
OAPMAIEPB-AIAAY A.E.B.E. Eli Lilly Ges.m.b.H.
Tn^: +30 210 629 4600 Tel: + 43-(0) 1 711 780
Espaňa Lilly S.A.
Tel: + 34-91 663 50 00 France
Lilly France SAS
Tél: +33-(0) 1 55 49 34 34
Hrvatska
Eli Lilly Hrvatska d.o.o.
Tel: +385 1 2350 999
Ireland
Eli Lilly and Company (Ireland) Limited Tel: + 353-(0) 1 661 4377
Island
Icepharma hf.
Sími + 354 540 8000
Italia
Eli Lilly Italia S.p.A.
Tel: + 39- 055 42571
Kúrcpog
Phadisco Ltd Tn^: +357 22 715000
Latvija
Eli Lilly Holdings Limited parstavnieciba Latvija Tel: +371 67364000
Polska
Eli Lilly Polska Sp. z o.o.
Tel: +48 22 440 33 00
Portugal
Lilly Portugal Produtos Farmaceuticos, Lda Tel: + 351-21-4126600
Románia
Eli Lilly Románia S.R.L.
Tel: + 40 21 4023000
Slovenija
Eli Lilly farmacevtska družba, d.o.o.
Tel: +386 (0)1 580 00 10
Slovenská republika
Eli Lilly Slovakia, s.r.o.
Tel: + 421 220 663 111
Suomi/Finland
Oy Eli Lilly Finland Ab Puh/Tel: + 358-(0) 9 85 45 250
Sverige
Eli Lilly Sweden AB Tel: + 46-(0) 8 7378800
United Kingdom
Eli Lilly and Company Limited Tel: + 44-(0) 1256 315000
Tato příbalová informace byla naposledy revidována (měsíc RRRR).
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Pokyny pro zacházení s přípravkem Portrazza 800 mg koncentrát pro infuzní roztok necitumumab
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Připravte infuzní roztok za použití aseptické metody, aby byla zajištěna sterilita připraveného roztoku.
Injekční lahvička je určena pouze pro jedno použití. Zkontrolujte obsah injekčních lahviček, zda neobsahují drobné částice nebo nedošlo ke změně barvy. Koncentrát pro přípravu infuzního roztoku musí být před naředěním čirý až mírně opalizující a bezbarvý až lehce nažloutlý. Pokud zjistíte přítomnost částic nebo zabarvení, injekční lahvičku nepoužívejte.
Injekční lahvička obsahuje 800 mg necitumumabu, tj. 16 mg/ml roztoku, přičemž jedna 50ml injekční lahvička obsahuje celou dávku. Jako ředicí roztok použijte výhradně injekční roztok chloridu sodného 9 mg/ml (0,9%).
Podání pomocí předplněných balení k intravenózní infuzi
Asepticky odeberte 50 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%) z předplněného 250ml balení pro intravenózní aplikaci a pak přeneste do balení 50 ml léčivého přípravku necitumumab, aby bylo opět dosaženo konečného objemu 250 ml v balení. Kontejner jemně převraťte, aby se obsah promíchal. Infuzní roztok NEZMRAZUJTE ani NEPROTŘEPÁVEJTE. NEŘEĎTE jinými roztoky ani nepodávejte spolu s infuzí s jinými elektrolyty nebo léčivými přípravky.
Podání pomocí prázdného obalu pro intravenózní infuzi
Asepticky přeneste 50 ml léčivého přípravku necitumumab do prázdného obalu pro intravenózní infuzi a do obalu přidejte 200 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), aby bylo dosaženo konečného objemu 250 ml. Obal jemně převraťte, aby se obsah promíchal. Infuzní roztok NEZMRAZUJTE ani NEPROTŘEPÁVEJTE. NEŘEĎTE jinými roztoky ani nepodávejte spolu s infuzí s jinými elektrolyty nebo léčivými přípravky.
Podejte pomocí infuzní pumpy. K infuzi je nutné použít samostatnou infuzní linku a na konci infuze je nutné linku vypláchnout injekčním roztokem chloridu sodného 9 mg/ml (0,9 %).
Parenterální léčivé přípravky je třeba před podáním vizuálně zkontrolovat, zda neobsahují částice. Pokud zjistíte přítomnost částic, infuzní roztok nepoužívejte.
Nepoužívejte žádný zbývající objem necitumumabu, který zůstane v injekční lahvičce, protože přípravek neobsahuje antimikrobiální konzervační látky.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
34