Perjeta 420 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Perjeta 420 mg koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička se 14 ml koncentrátu obsahuje pertuzumabum 420 mg o koncentraci 30 mg/ml.
Po naředění obsahuje jeden mililitr roztoku přibližně 3,02 mg pertuzumabu pro úvodní dávku a přibližně 1,59 mg pertuzumabu pro udržovací dávku (viz bod 6.6).
Pertuzumabum je humanizovaná monoklonální protilátka IgG1 produkovaná savčími buňkami (ovariální buňky čínského křečka) technologií rekombinace DNA.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok.
Čirá až slabě opalescentní, bezbarvá až světle žlutá tekutina.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Metastazující karcinom prsu
Přípravek Perjeta je indikován k použití v kombinaci s trastuzumabem a docetaxelem u dospělých pacientů s HER2-pozitivním metastazujícím nebo lokálně rekurentním neresekovatelným karcinomem prsu, kteří dosud nebyli léčeni anti-HER2 léky nebo chemoterapií pro metastazující onemocnění.
Neoadjuvantní léčba karcinomu prsu
Přípravek Perjeta je indikován k neoadjuvantní léčbě v kombinaci s trastuzumabem a chemoterapií u dospělých pacientů s HER2-pozitivním, lokálně pokročilým, inflamatorním nebo časným karcinomem prsu s vysokým rizikem rekurence (viz bod 5.1).
4.2 Dávkování a způsob podání
Výdej přípravku Perjeta je vázán na lékařský předpis s omezením a léčba musí být zahájena pouze pod dohledem lékaře, který má zkušenosti s podáváním cytostatik. Přípravek Perjeta má být podáván zdravotnickým pracovníkem připraveným řešit anafylaxi a v zařízení, kde je okamžitě k dispozici kompletní vybavení pro resuscitaci.
Pacienti léčení přípravkem Perjeta musí mít HER2-pozitivní nádor s HER2 pozitivitou definovanou jako skóre 3+ při imunohistochemickém (IHC) stanovení a/nebo poměr > 2,0 při stanovení in situ hybridizací (ISH) validovaným testem.
Aby byla zajištěna přesnost a reprodukovatelnost výsledků, musí být hodnocení provedeno ve specializované laboratoři, ve které lze zaručit validaci postupů při testování. Úplný návod o provádění a hodnocení testu naleznete v příbalové informaci validované soupravy pro hodnocení HER2.
Dávkování
Doporučená úvodní dávka přípravku Perjeta je 840 mg podaná v intravenózní infuzi trvající 60 minut následovaná každé 3 týdny udržovací dávkou 420 mg podávanou po dobu 30 až 60 minut.
Při podávání s přípravkem Perjeta je doporučená úvodní dávka trastuzumabu 8 mg/kg tělesné hmotnosti podaná v intravenózní infuzi následovaná každé 3 týdny udržovací dávkou 6 mg/kg tělesné hmotnosti.
Doporučená úvodní dávka docetaxelu při podávání s přípravkem Perjeta je 75 mg/m2 s dalším podáváním v třítýdenních intervalech. Pokud je úvodní dávka dobře snášena, může být v následujících cyklech dávka docetaxelu zvýšena na 100 mg/m2 (dávka docetaxelu nemá být zvyšována při použití v kombinaci s karboplatinou, trastuzumabem nebo přípravkem Perjeta).
Léčivé přípravky mají být podávány postupně a nesmí být smíchány ve stejném infuzním vaku. Přípravek Perjeta a trastuzumab mohou být podány v libovolném pořadí. Pokud je pacient léčen docetaxelem, má být podán po přípravku Perjeta a trastuzumabu. Po každé infuzi přípravku Perjeta a před zahájením další infuze trastuzumabu nebo docetaxelu se doporučuje období 30 až 60 minut sledování (viz bod 4.4).
Metastazující karcinom prsu
Pacienti mají být léčeni přípravkem Perjeta a trastuzumabem do progrese nemoci nebo do nepřijatelné toxicity.
Neoadjuvantní léčba karcinomu prsu
Přípravek Perjeta má být podáván ve třech až šesti cyklech v kombinaci s neoadjuvantní léčbou trastuzumabem a chemoterapií, jako součást léčebného režimu časného karcinomu prsu.
Pacienti, následně po operaci, mají být léčeni adjuvantním trastuzumabem k dokončení jednoroční léčby (viz bod 5.1).
Opoždění nebo vynechání dávek
Pokud je doba mezi dvěma následnými infuzemi kratší než 6 týdnů, má být co nejdříve podán přípravek Perjeta v dávce 420 mg bez ohledu na termín příští plánované dávky.
Pokud je doba mezi dvěma následnými infuzemi 6 týdnů nebo déle, má být podán přípravek Perjeta znovu v úvodní dávce 840 mg v intravenózní infuzi trvající 60 minut, následované každé 3 týdny udržovací dávkou 420 mg podávanou po dobu 30 až 60 minut.
Úprava dávky
Snižování dávky přípravku Perjeta se nedoporučuje.
Pacienti mohou pokračovat v léčbě v době reverzibilní myelosuprese navozené chemoterapií, mají však být v této době pečlivě sledováni pro komplikace neutropenie. Úpravy dávky docetaxelu a jiné chemoterapie, viz příslušný Souhrn údajů o přípravku.
Snižování dávky trastuzumabu se nedoporučuje, viz Souhrn údajů o přípravku pro trastuzumab.
Pokud je ukončena léčba trastuzumabem, má být ukončeno i podávání přípravku Perjeta.
Pokud je ukončeno podávání docetaxelu, může u metastazujícího onemocnění léčba přípravkem Perjeta a trastuzumabem pokračovat až do progrese nemoci nebo nepřijatelné toxicity.
Dysfunkce levé srdeční komory
Podání přípravku Perjeta a trastuzumabu má být odloženo nejméně o 3 týdny při výskytu kteréhokoli z níže uvedených případů:
- známky a příznaky podezřelé z městnavého srdečního selhání (pokud je potvrzeno symptomatické městnavé srdeční selhání, má být podávání přípravku Perjeta ukončeno)
- pokles ejekční frakce levé srdeční komory na méně než 40 %
- hodnota ejekční frakce levé srdeční komory 40 až 45 % a současný pokles o > 10 procentních bodů pod hodnotu před léčbou.
Podávání přípravku Perjeta a trastuzumabu může být znovu zahájeno, pokud dojde k úpravě hodnoty ejekční frakce levé srdeční komory na > 45 % nebo je hodnota 40 až 45 % spojena s poklesem o < 10 procentních bodů od hodnoty před léčbou.
Pokud nedojde při vyšetření ejekční frakce levé srdeční komory opakovaném během přibližně 3 týdnů ke zlepšení nebo dojde k dalšímu poklesu hodnot, je nutno silně zvažovat ukončení léčby přípravkem Perjeta a trastuzumabem, pokud se nepředpokládá, že u daného pacienta prospěch nepřevažuje nad riziky (viz bod 4.4).
Reakce na infuzi
Pokud u pacienta vznikne reakce na infuzi, má být infuze zpomalena nebo přerušena (viz bod 4.8). Infuzi lze znovu zahájit po ústupu příznaků. Léčba zahrnující podání kyslíku, beta agonisty, antihistaminika, rychlé intravenózní podání tekutin a antipyretika může také napomoci ke zmírnění příznaků.
Hypersenzitivní reakce/anafylaxe
V případě reakce stupně 4 dle NCI-CTCAE (anafylaxe), bronchospasmu nebo syndromu akutní respirační tísně u pacienta je nutno infuzi okamžitě ukončit (viz bod 4.4).
Starší pacienti
Jsou k dispozici omezené údaje o bezpečnosti a účinnosti přípravku Perjeta u pacientů ve věku > 65 let. Nebyly pozorovány žádné významné rozdíly v bezpečnosti a účinnosti přípravku Perjeta mezi staršími pacienty ve věku 65 až 75 let a dospělými pacienty ve věku < 65 let. U starších pacientů ve věku > 65 let není nutná úprava dávky. Jsou k dispozici velmi omezené údaje o pacientech ve věku > 75 let.
Pacienti s poruchou renálních funkcí
U pacientů s mírnou nebo středně závažnou poruchou renálních funkcí není nutná úprava dávky přípravku Perjeta. U pacientů se závažnou poruchou renálních funkcí nelze stanovit žádná doporučení týkající se dávkování, protože jsou k dispozici jen omezené farmakokinetické údaje (viz bod 5.2).
Pacienti s poruchou jaterních funkcí
Bezpečnost a účinnost přípravku Perjeta nebyla u pacientů s poruchou jaterních funkcí hodnocena. Nelze stanovit žádná specifická doporučení týkající se dávkování.
Bezpečnost a účinnost přípravku Perjeta u dětí a dospívajících ve věku pod 18 let nebyla stanovena. Použití přípravku Perjeta u pediatrické populace v indikaci karcinom prsu není relevantní.
Způsob podání
Přípravek Perjeta se podává v intravenózní infuzi. Nemá se podávat jako intravenózní bolus. Návod k naředění přípravku Perjeta před jeho podáním je uveden v bodech 6.2 a 6.6.
Při úvodní dávce se doporučuje trvání infuze 60 minut. Pokud je první infuze dobře snášena, mohou být následné infuze podávány po dobu 30 minut až 60 minut (viz bod 4.4).
4.3 Kontraindikace
Hypersenzitivita na pertuzumab nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 tohoto přípravku.
4.4 Zvláštní upozornění a opatření pro použití
Z důvodu snadnější zpětné zjistitelnosti biologických léčivých přípravků má být obchodní název a číslo šarže podávaného přípravku zřetelně zaznamenáno (nebo vyznačeno) v pacientově dokumentaci.
Dysfunkce levé srdeční komory (včetně městnavého srdečního selhání)
Při použití léčivých přípravků blokujících aktivitu HER2, včetně přípravku Perjeta, byl hlášen pokles ejekční frakce levé srdeční komory. Riziko poklesu ejekční frakce levé srdeční komory může být vyšší u pacientů dříve léčených antracykliny nebo radioterapií na oblast hrudníku. V klíčové studii CLEOPATRA u pacientů s metastazujícím karcinomem prsu nebylo podávání přípravku Perjeta v kombinaci s trastuzumabem a docetaxelem spojeno s vyšší incidencí symptomatické systolické dysfunkce levé srdeční komory nebo poklesem ejekční frakce levé srdeční komory ve srovnání s pacienty s placebem, trastuzumabem a docetaxelem (viz bod 4.8).
Při neoadjuvantním podání (NEOSPHERE) byl výskyt systolické dysfunkce levé srdeční komory vyšší u skupiny léčené přípravkem Perjeta než u pacientů, kteří přípravkem Perjeta léčeni nebyli. Byl také pozorován zvýšený výskyt poklesu ejekční frakce levé srdeční komory u pacientů léčených přípravkem Perjeta v kombinaci s trastuzumabem a docetaxelem; ejekční frakce levé srdeční komory se u všech pacientů vrátila zpět na hodnotu > 50 %.
Přípravek Perjeta nebyl hodnocen u pacientů s hodnotou ejekční frakce levé srdeční komory < 50 % před léčbou, s anamnézou městnavého srdečního selhání, s poklesem ejekční frakce levé srdeční komory na < 50 % během předchozí adjuvantní léčby trastuzumabem nebo u pacientů se stavy, které mohou negativně ovlivnit činnost levé srdeční komory, jako jsou nekontrolovaná hypertenze, nedávný infarkt myokardu, závažná srdeční arytmie vyžadující léčbu nebo předchozí kumulativní expozice antracyklinu > 360 mg/m2 doxorubicinu nebo jeho ekvivalent.
Vyšetřete ejekční frakci levé srdeční komory před zahájením léčby přípravkem Perjeta a v průběhu léčby přípravkem Perjeta (každé 3 cykly u metastazujícího onemocnění a každé 2 cykly při neoadjuvantní léčbě), aby bylo zajištěno, že hodnota ejekční frakce levé srdeční komory je v mezích normálních hodnot daného zařízení. Při hodnotě ejekční frakce levé srdeční komory < 40 % nebo 40 % až 45 % ve spojení s poklesem o > 10 procentních bodů od hodnoty před léčbou má být léčba přípravkem Perjeta a trastuzumabem zastavena a vyšetření ejekční frakce levé srdeční komory zopakováno přibližně po 3 týdnech. Pokud nedojde ke zlepšení nebo dojde k dalšímu poklesu hodnot ejekční frakce levé srdeční komory, je nutno silně zvažovat ukončení léčby přípravkem Perjeta a trastuzumabem, pokud se nepředpokládá, že u daného pacienta prospěch nepřevažuje nad riziky (viz bod 4.2).
Před použitím přípravku Perjeta s antracykliny má být pečlivě zváženo a vyhodnoceno kardiální riziko v závislosti na potřebě léčby individuálního pacienta. K dispozici jsou omezené údaje o bezpečnosti ze studie TRYPHAENA, pokud jde o postupné nebo souběžné podávání přípravku Perjeta s epirubicinem jako součásti režimu FEC (viz body 4.8 a 5.1). Nejsou k dispozici žádné údaje o bezpečnosti týkající se podání přípravku Perjeta s doxorubicinem.
Vhledem k farmakologickému působení pertuzumabu a antracyklinů může být očekáváno zvýšené riziko kardiální toxicity při současném podání těchto přípravků v porovnání s postupným podáním, ačkoliv toto ve studii TRYPHAENA nebylo zaznamenáno. V této studii byli nízkou kumulativní dávkou epirubicinu, tj. až do výše 300 mg/m2 léčeni pouze pacienti bez předchozí chemoterapeutické léčby, kterým po operaci nebyla podávána další chemoterapeutická léčba.
Reakce na infuzi
Podání přípravku Perjeta bylo spojeno s reakcemi na infuzi (viz bod 4.8). Doporučuje se pečlivé sledování pacienta během podávání a po dobu 60 minut po první infuzi a během podávání a po dobu 30 - 60 minut po dalších infuzích přípravku Perjeta. Při významných projevech reakce na infuzi má být infuze zpomalena nebo přerušena a podána vhodná léčba. Pacienty je nutno vyšetřit a pečlivě sledovat až do úplného odeznění známek a příznaků. U pacientů se závažnými infuzními reakcemi má být zváženo trvalé přerušení. Klinické vyhodnocení má být provedeno na základě závažnosti probíhající reakce a odpovědi na podanou léčbu nežádoucího účinku (viz 4.2).
Hypersenzitivní reakce/anafylaxe
Pacienti mají být pečlivě sledováni pro případ výskytu hypersenzitivních reakcí. V klinických studiích přípravku Perjeta byla pozorována závažná hypersenzitivita, včetně anafylaxe (viz bod 4.8).
K dispozici pro okamžité použití mají být léky k léčbě těchto reakcí, stejně tak vybavení pro první pomoc. V případě hypersenzitivní reakce stupně 4 dle NCI-CTCAE (anafylaxe), bronchospasmu nebo syndromu akutní respirační tísně musí být léčba přípravkem Perjeta trvale ukončena (viz bod 4.2). Přípravek Perjeta je kontraindikován u pacientů se známou hypersenzitivitou na pertuzumab nebo na jakékoli jeho pomocné látky (viz bod 4.3).
Febrilní neutropenie
Pacienti léčení přípravkem Perjeta, trastuzumabem a docetaxelem mají vyšší riziko febrilní neutropenie než pacienti léčení placebem, trastuzumabem a docetaxelem, zejména v průběhu prvních 3 cyklů léčby (viz bod 4.8). Ve studii CLEOPATRA u metastazujícího karcinomu prsu, byly minimální hodnoty počtu neutrofilů podobné u pacientů léčených přípravkem Perjeta i u pacientů, kterým bylo podáváno placebo. Vyšší incidence febrilní neutropenie u pacientů léčených přípravkem Perjeta souvisela s vyšší incidencí mukozitidy a průjmů u těchto pacientů. Má být zvážena symptomatická léčba mukozitidy a průjmu. Nebyly hlášeny žádné příhody febrilní neutropenie po ukončení léčby docetaxelem.
Pertuzumab může vyvolat těžký průjem. V případě výskytu těžkého průjmu má být nasazena protiprůjmová léčba, a pokud nedojde ke zlepšení stavu, má být zváženo přerušení léčby pertuzumabem. V případě, že průjem je již pod kontrolou, může být léčba pertuzumabem obnovena.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
V sub-studii randomizované klíčové studie CLEOPATRA u metastazujícího karcinomu prsu nebyly u 37 pacientů pozorovány farmakokinetické interakce mezi pertuzumabem a trastuzumabem nebo mezi pertuzumabem a docetaxelem. Dále nebyly v populační farmakokinetické analýze pozorovány známky mezilékových interakcí mezi pertuzumabem a trastuzumabem nebo mezi pertuzumabem a docetaxelem. Tato nepřítomnost mezilékových interakcí byla potvrzena farmakokinetickými údaji z klinické studie NEOSPHERE při neoadjuvantním podání.
Ve čtyřech studiích byl hodnocen vliv pertuzumabu na farmakokinetiku souběžně podávaných cytotoxických přípravků, docetaxelu, gemcitabinu, erlotinibu a kapecitabinu. Nebyly zjištěny žádné známky jakékoli farmakokinetické interakce mezi pertuzumabem a kterýmkoli z těchto přípravků. Farmakokinetika pertuzumabu byla v těchto studiích srovnatelná s farmakokinetikou ve studiích s monoterapií.
4.6 Fertilita, těhotenství a kojení
Antikoncepce
Ženy ve fertilním věku musí během léčby přípravkem Peijeta a 6 měsíců po poslední dávce přípravku Perjeta používat účinnou antikoncepci.
O použití pertuzumabu u těhotných žen je k dispozici omezené množství údajů. Studie u zvířat prokázaly reprodukční toxicitu (viz bod 5.3).
Podávání přípravku Peijeta se nedoporučuje během těhotenství a ženám ve fertilním věku, které nepoužívají antikoncepci.
Kojení
Protože lidský IgG je vylučován do mateřského mléka a protože možnosti vstřebávání a škodlivosti pro dítě nejsou známy, má být rozhodnuto, zda ukončit kojení nebo ukončit léčbu při zvážení prospěchu kojení pro dítě a prospěchu léčby přípravkem Perjeta pro ženu (viz bod 5.2).
Fertilita
Nebyly provedeny studie specificky hodnotící vliv pertuzumabu na fertilitu zvířat. K dispozici jsou pouze velmi omezené údaje ze studií hodnotících toxicitu opakovaných dávek s ohledem na riziko nežádoucích účinků na samčí reprodukční systém. Nebyly pozorovány žádné nežádoucí účinky u sexuálně dospělých samic makaka jávského vystavených pertuzumabu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Na základě hlášených nežádoucích účinků se neočekává, že by přípravek Perjeta ovlivňoval schopnost řídit nebo obsluhovat stroje. Pacientům s reakcemi na infuzi má být doporučeno, aby neřídili a neobsluhovali stroje až do úplného odeznění příznaků.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Bezpečnost přípravku Perjeta byla hodnocena u více než 1 600 pacientů v randomizovaných studiích CLEOPATRA (n=808), NEOSPHERE (n=417) a TRYPHAENA (n=225) a ve studiích fáze I a fáze II prováděných u pacientů s různými zhoubnými nádory a léčenými převážně přípravkem Perjeta v kombinaci s dalšími cytostatiky. Bezpečnost přípravku Perjeta ve studiích fáze I a II byla obecně konzistentní s bezpečností pozorovanou ve studiích CLEOPATRA, NEOSPHERE a TRYPHAENA, ačkoliv incidence a nejčastější nežádoucí účinky se lišily v závislosti na tom, zda byl přípravek Perjeta podáván jako monoterapie nebo v kombinaci s jinými cytostatiky.
Metastazující karcinom prsu
V klíčové klinické studii CLEOPATRA byla 408 pacientům podána alespoň jedna dávka přípravku Perjeta v kombinaci s trastuzumabem a docetaxelem. Nejčastějšími nežádoucími účinky (> 50 %) pozorovanými u přípravku Perjeta v kombinaci s trastuzumabem a docetaxelem byly průjem, alopecie a neutropenie. Nejčastějšími nežádoucími účinky (> 10 %) stupně 3 až 4 dle NCI-CTCAE v.3 byly neutropenie, febrilní neutropenie a leukopenie a nejčastějšími závažnými nežádoucími příhodami byly febrilní neutropenie, neutropenie a průjem. K úmrtí v souvislosti s léčbou došlo u 1,2 % pacientů ve skupině léčené přípravkem Peijeta a u 1,5 % pacientů ve skupině s placebem. Nejčastější příčinou byly febrilní neutropenie a/nebo infekce.
V klíčové studii CLEOPATRA byly hlášeny nežádoucí účinky s nižší frekvencí po přerušení léčby docetaxelem. Po ukončení léčby docetaxelem se nežádoucí účinky ve skupině s přípravkem Peijeta a trastuzumabem vyskytly u < 10 % pacientů, s výjimkou průjmu (28,1 %), infekcí horních cest dýchacích (18,3 %), vyrážky (18,3 %), bolesti hlavy (17,0 %), únavy (13,4 %), zánětu nosohltanu (17,0 %), astenie (13,4 %), pruritu (13,7 %), artralgie (11,4 %), nauzey (12,7 %), bolesti v končetině (13,4 %), bolesti zad (12,1 %) a kašle (12,1 %).
Neoadjuvantní léčba karcinomu prsu
V klinické studii neoadjuvantní léčby NEOSPHERE byly nej častěji pozorovanými nežádoucími účinky (> 50 %) při podávání přípravku Peijeta v kombinaci s trastuzumabem a docetaxelem alopecie a neutropenie. Nejčastějším nežádoucím účinkem (> 10 %) stupně 3-4 dle NCI-CTCAE v.3 byla neutropenie.
V klinické studii neoadjuvantní léčby TRYPHAENA, kde byl přípravek Peijeta podáván v kombinaci s trastuzumabem a režimem FEC (5-fluorouracil, epirubicin, cyklofosfamid) ve 3 cyklech následovaných třemi cykly přípravku Peijeta, trastuzumabu a docetaxelu, byly nej častějšími nežádoucími účinky (> 50 %) neutropenie, průjem a nauzea. Nej častějšími nežádoucími účinky
(> 10 %) stupně 3-4 dle NCI-CTCAE v.3 byly neutropenie, febrilní neutropenie a leukopenie. Při podání přípravku Peijeta v kombinaci s trastuzumabem a docetaxelem ve 3 cyklech po 3 cyklech FEC (5-fluorouracil, epirubicin, cyklofosfamid), byly nej častějšími nežádoucími účinky (> 50 %) průjem, nauzea a alopecie. Nejčastějšími nežádoucími účinky (> 10 %) stupně 3-4 dle NCI-CTCAE v.3 byly neutropenie a leukopenie. Podobně, při podání přípravku Peijeta v kombinaci s režimem TCH (docetaxel, karboplatina a trastuzumab) v šesti cyklech, byly nejčastějšími nežádoucími účinky (> 50 %) průjem a alopecie. Nejčastějšími nežádoucími účinky (> 10 %) stupně 3-4 dle NCI-CTCAE v.3 byly neutropenie, febrilní neutropenie, anémie, leukopenie a průjem. Bezpečnost přípravku Peijeta podaného ve více než 6 cyklech při neoadjuvantní léčbě nebyla stanovena.
Seznam nežádoucích účinků v tabulce
Tabulka 1 shrnuje nežádoucí účinky v klíčové studii CLEOPATRA, ve které byl přípravek Peijeta podáván v kombinaci s docetaxelem a trastuzumabem pacientům s metastazujícím karcinomem prsu a v neoadjuvantních studiích NEOSPHERE a TRYPHAENA, ve kterých byl přípravek Perjeta podáván v kombinaci s trastuzumabem a chemoterapií pacientům s časným karcinomem prsu. Protože je přípravek Perjeta používán v kombinaci s trastuzumabem a chemoterapií, je obtížné stanovit kauzální souvislost nežádoucí příhody s jednotlivými léčivými přípravky.
Nežádoucí účinky jsou uvedeny níže podle MedDRA tříd orgánových systémů v kategoriích dle frekvence:
Velmi časté (> 1/10)
Časté (> 1/100 až < 1/10)
Méně časté (> 1/1000 až < 1/100)
Vzácné (> 1/10000 až < 1/1000)
Velmi vzácné (< 1/10000)
Není známo (z dostupných údajů nelze určit)
Nežádoucí účinky jsou v každé skupině frekvence a tříd orgánových systémů uvedeny v pořadí dle klesající závažnosti.
Tabulka 1 Souhrn nežádoucích účinků u pacientů léčených přípravkem Perjeta při metastazujícím onemocnění a neoadjuvantním podáníA
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
|
Infekce a infestace |
Infekce horních dýchacích cest Nazofaryngitida |
Paronychium | |
|
Poruchy krve a lymfatického systému |
Febrilní neutropenie* Neutropenie Leukopenie Anémie | ||
|
Poruchy imunitního systému |
Hypersenzitivní/ anafylaktická reakce° Reakce na infuzi/syndrom uvolňování cytokinů°° | ||
|
Poruchy metabolismu a výživy |
Snížená chuť k jídluf | ||
|
Psychiatrické poruchy |
Insomnie | ||
|
Poruchy nervového systému |
Periferní neuropatie Bolest hlavyf Dysgeuzie |
Periferní senzorická neuropatie Závratě | |
|
Poruchy oka |
Zvýšená tvorba slz | ||
|
Srdeční poruchy |
Dysfunkce levé srdeční komoryf (včetně městnavého srdečního selhání)** | ||
|
Respirační, hrudní a mediastinální poruchy |
Kašelf |
Pleurální výpotek Dušnostf |
Intersticiální plicní onemocnění |
|
Gastrointestinální poruchy |
Průjemf Zvraceníf Stomatitida Nauzeaf Zácpaf | ||
|
Poruchy kůže a podkožní tkáně |
Alopecie Vyrážkaf Onemocnění nehtů |
Pruritus Suchá kůže | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Myalgie Artralgie |
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
|
Celkové poruchy a reakce v místě aplikace |
Mukozitida/zánět sliznice Bolestf Edémf Pyrexie Únavaf Astenief |
ATabulka 1 ukazuje souhrnné údaje z celé doby léčby studie CLEOPATRA (data k 11. únoru 2014; medián počtu cyklů přípravku Perjeta byl 24); a z období neoadjuvantní léčby ve studií NEOSPHERE (medián počtu cyklů přípravku Perjeta byl 4, ve všech léčebných ramenech) a TRYPHAENA (medián počtu cyklů přípravku Perjeta byl 3-6 v jednotlivých léčebných ramenech).
* Včetně nežádoucích účinků s fatálním koncem.
** Pro celkovou dobu léčby ve všech třech studiích.
f Kromě febrilní neutropenie, neutropenie, leukopenie, zvýšené tvorby slz, intersticiálního plicního onemocnění, paronychia a alopecie byly všechny příhody uvedené v této tabulce hlášeny též u nejméně 1 % pacientů ve studiích s přípravkem Perjeta v monoterapii, ačkoli dle hodnocení řešitelů nemusely nutně souviset s přípravkem Perjeta. Velmi časté příhody (hlášené u > 10 % pacientů léčených přípravkem Perjeta v monoterapii) jsou v tabulce označeny f.
° Hypersenzitivní/anafylaktická reakce dle skupiny termínů.
00 Reakce na infuzi/syndrom uvolňování cytokinů zahrnuje škálu příhod ve stejném časovém období, viz níže „Popis vybraných nežádoucích účinků“.
Popis vybraných nežádoucích účinků
Dysfunkce levé srdeční komory
V klíčové studii CLEOPATRA metastazujícího karcinomu prsu, byl výskyt dysfunkce levé srdeční komory během léčby ve studii vyšší ve skupině léčené placebem (8,6 %) než ve skupině léčené přípravkem Perjeta (6,6 %). Také výskyt symptomatické dysfunkce levé srdeční komory byl nižší ve skupině léčené přípravkem Perjeta (1,8 % ve skupině léčené placebem versus 1,5 % ve skupině léčené přípravkem Perjeta) (viz bod 4.4).
V klinické studii neoadjuvantní léčby NEOSPHERE, ve které byl pacientům podáván přípravek Perjeta ve 4 cyklech jako neoadjuvantní léčba, byl výskyt dysfunkce levé srdeční komory (během celé doby léčby) vyšší u skupiny léčené přípravkem Perjeta, trastuzumabem a docetaxelem (7,5 %)
v porovnání se skupinou léčenou trastuzumabem a docetaxelem (1,9 %). Ve skupině léčené přípravkem Perjeta a trastuzumabem se vyskytl pouze jeden případ symptomatické dysfunkce levé srdeční komory.
V klinické studii neoadjuvantní léčby TRYPHAENA byl výskyt dysfunkce levé srdeční komory (během celé doby léčby) 8,3 % ve skupině léčené přípravkem Perjeta plus trastuzumab a FEC
(s následným podáním přípravku Perjeta plus trastuzumab a docetaxel); 9,3 % ve skupině léčené přípravkem Perjeta plus trastuzumab a docetaxel následně po režimu FEC; a 6,6 % ve skupině léčené přípravkem Perjeta v kombinaci s režimem TCH. Výskyt symptomatické dysfunkce levé srdeční komory (městnavé srdeční selhávání) byl 1,3 % ve skupině léčené přípravkem Perjeta plus trastuzumab a docetaxel následně po režimu FEC (tím byl vyloučen pacient, u kterého se vyskytla symptomatická dysfunkce levé srdeční komory v průběhu léčby režimem FEC před podáním přípravku Perjeta plus trastuzumab a docetaxel) a také 1,3 % ve skupině léčené přípravkem Perjeta v kombinaci s režimem TCH. U žádného pacienta ze skupiny léčené přípravkem Perjeta plus trastuzumab a FEC s následným podáním přípravku Perjeta plus trastuzumab a docetaxel se nevyskytla symptomatická dysfunkce levé srdeční komory.
Reakce na infuzi
V klíčové studii CLEOPATRA metastazujícího karcinomu prsu byla reakce na infuzi definována jako jakákoli příhoda hlášená jako hypersenzitivita, anafylaktická reakce, akutní reakce na infuzi nebo syndrom uvolňování cytokinů, která se projevila během infuze nebo téhož dne. V klíčové studii CLEOPATRA byla úvodní dávka přípravku Perjeta podána den před trastuzumabem a docetaxelem, aby byl o možno vyhodnotit reakce související s přípravkem Perjeta. První den, kdy byl podáván pouze přípravek Perjeta, byla celková frekvence reakcí na infuzi 9,8 % ve skupině s placebem a 13,2 % ve skupině léčené přípravkem Perjeta. Většina reakcí na infuzi byla mírného nebo středně závažného stupně. Nejčastějšími reakcemi na infuzi (> 1,0 %) ve skupině léčené přípravkem Perjeta byly pyrexie, třesavka, únava, bolest hlavy, astenie, hypersenzitivita a zvracení.
Ve druhém cyklu, v němž byly podány všechny léčivé přípravky ve stejný den, byly nej častější reakce na infuzi ve skupině léčené přípravkem Perjeta (> 1,0 %) únava, dysgeuzie, hypersenzitivita na léky, myalgie a zvracení (viz bod 4.4).
Ve studiích neoadjuvantní léčby NEOSPHERE a TRYPHAENA byl ve všech cyklech přípravek Perjeta podáván ve stejný den jako ostatní léky ve studii. Reakce na infuzi byly v souladu s reakcemi na infuzi pozorovanými ve studii CLEOPATRA v cyklech, kdy byl přípravek Perjeta podáván ve stejný den jako trastuzumab a docetaxel, a většina reakcí byla mírná až středně závažná.
Hypersenzitivní reakce/anafylaxe
V klíčové studii CLEOPATRA metastazujícího karcinomu prsu byla celková frekvence příhod hypersenzitivity/anafylaxe hlášených řešiteli během celého období léčby 9,3 % ve skupině s placebem a 11,3 % ve skupině léčené přípravkem Perjeta, z toho se ve 2,5 % respektive ve 2,0 % jednalo o příhody stupně 3 až 4 dle NCI-CTCAE. V souhrnu, příhody popisované řešiteli jako anafylaxe se projevily u 2 pacientů ve skupině s placebem a u 4 pacientů ve skupině léčené přípravkem Perjeta (viz bod 4.4).
V souhrnu byla většina hypersenzitivních reakcí mírného nebo středně závažného stupně a při léčbě tyto reakce ustoupily. Na základě modifikací léčby ve studii byla většina reakcí hodnocena jako související s infuzemi docetaxelu.
Ve studiích neoadjuvantní léčby NEOSPHERE a TRYPHAENA byly příhody hypersenzitivity/anafylaxe v souladu s příhodami pozorovanými ve studii CLEOPATRA. Ve studii NEOSPHERE se u dvou pacientů ve skupině léčené přípravkem Perjeta a docetaxelem vyskytla anafylaxe. Ve studii TRYPHAENA byla celková frekvence hypersenzitivity/anafylaxe vyšší u skupiny léčené přípravkem Perjeta a TCH (13,2 %), z toho 2,6 % bylo stupně 3-4 dle NCI-CTCAE v.3.
Febrilní neutropenie
V klíčové studii CLEOPATRA měla většina pacientů v obou léčebných skupinách nejméně jednu leukopenickou příhodu (63,0 % pacientů ve skupině léčené přípravkem Perjeta a 58,3 % pacientů ve skupině s placebem), z nichž většina byly neutropenické příhody. Febrilní neutropenie se vyskytla u 13,7 % pacientů léčených přípravkem Perjeta a u 7,6 % pacientů, kterým bylo podáváno placebo.
V obou léčebných skupinách byl počet pacientů s febrilní neutropenií nejvyšší v prvním cyklu léčby a následně postupně klesal. Zvýšená incidence febrilní neutropenie byla pozorována u asijských pacientů v obou léčebných skupinách ve srovnání s jinými rasami a pacienty z jiných zeměpisných oblastí. U asijských pacientů byla incidence febrilní neutropenie vyšší ve skupině léčené přípravkem Perjeta (25,8 %) než ve skupině s placebem (11,3 %).
Ve studii NEOSPHERE se u 8,4 % pacientů léčených neoadjuvantně přípravkem Perjeta, trastuzumab a docetaxel vyskytla febrilní neutropenie, v porovnání se 7,5 % pacientů léčených trastuzumabem a docetaxelem. Ve studii TRYPHAENA se febrilní neutropenie vyskytla u 17,1 % pacientů léčených neoadjuvantně přípravkem Perjeta + TCH a u 9,3 % pacientů léčených neoadjuvantně přípravkem Perjeta, trastuzumabem a docetaxelem následně po režimu FEC. Ve studii TRYPHAENA byl výskyt febrilní neutropenie vyšší u pacientů, kterým byl podán přípravek Perjeta v šesti cyklech v porovnání s pacienty, kterým byl přípravek Perjeta podán ve třech cyklech, nezávisle na podané chemoterapii. Stejně jako ve studii CLEOPATRA, byl v obou neoadjuvantních studiích pozorován vyšší výskyt neutropenie a febrilní neutropenie u asijských pacientů v porovnání s dalšími pacienty. Ve studii NEOSPHERE se febrilní neutropenie vyskytla u 8,3 % asijských pacientů léčených neoadjuvantně přípravkem Perjeta, trastuzumabem a docetaxelem, v porovnání se 4,0 % asijských pacientů léčených neoadjuvantně trastuzumabem a docetaxelem.
V klíčové studii CLEOPATRA metastazujícího karcinomu prsu se průjem vyskytl u 68,4 % pacientů léčených přípravkem Peijeta a u 48,7 % pacientů, kterým bylo podáváno placebo. Většina příhod byla mírného až středně závažného stupně a objevovala se v několika prvních cyklech léčby. Incidence průjmu stupně 3 až 4 dle NCI-CTCAE byla 9,3 % u pacientů léčených přípravkem Peijeta a 5,1 % u pacientů, kterým bylo podáváno placebo. Střední doba trvání nejdelší epizody bylo 18 dní u pacientů léčených přípravkem Peijeta a 8 dní u pacientů, kterým bylo podáváno placebo. Příhody průjmu dobře reagovaly na proaktivní opatření za použití protiprůjmových léků.
Ve studii NEOSPHERE se vyskytl průjem u 45,8 % pacientů léčených neoadjuvantně přípravkem Perjeta, trastuzumabem a docetaxelem, v porovnání s 33,6 % pacientů léčených trastuzumabem a docetaxelem. Ve studii TRYPHAENA se průjem vyskytl u 72,3 % pacientů léčených neoadjuvantně přípravkem Perjeta + TCH a u 61,4 % pacientů léčených neoadjuvantně přípravkem Perjeta, trastuzumabem a docetaxelem následně po režimu FEC. V obou studiích byly příhody mírné až středně závažné.
V klíčové studii CLEOPATRA u metastazujícího karcinomu prsu se vyrážka objevila u 51,7 % pacientů léčených přípravkem Perjeta ve srovnání s 38,9 % pacienty, kterým bylo podáváno placebo. Většina příhod byla stupně 1 nebo 2, objevily se v prvních 2 cyklech a odpověděly na standardní léčbu, jako je topická nebo perorální léčba akné.
Ve studii NEOSPHERE se vyrážka vyskytla u 40,2 % pacientů léčených neoadjuvantně přípravkem Perjeta, trastuzumabem a docetaxelem, v porovnání s 29,0 % pacientů léčených trastuzumabem a docetaxelem. Ve studii TRYPHAENA se vyrážka vyskytla u 36,8 % pacientů léčených neoadjuvantně přípravkem Perjeta + TCH a u 20,0 % pacientů léčených neoadjuvantně přípravkem Perjeta, trastuzumabem a docetaxelem následně po režimu FEC. Výskyt vyrážky byl vyšší u pacientů, kterým byl přípravek Perjeta podáván v šesti cyklech, v porovnání s pacienty, kterým byl přípravek Perjeta podáván ve třech cyklech, nezávisle na podané chemoterapii.
Laboratorní abnormality
V klíčové studii CLEOPATRA metastazujícího karcinomu prsu byla incidence neutropenie stupně 3 až 4 dle NCI-CTCAE v.3 ve dvou léčebných skupinách vyvážená (86,3 % u pacientů léčených přípravkem Perjeta a 86,6 % u pacientů, kterým bylo podáváno placebo, a to včetně 60,7 % respektive
64,8 % neutropenie stupně 4).
Ve studii NEOSPHERE byl výskyt neutropenie stupně 3-4 dle NCI-CTCAE v.3 74,5 % u pacientů léčených neoadjuvantně přípravkem Perjeta, trastuzumabem a docetaxelem, v porovnání s 84,5 % pacientů léčených trastuzumabem a docetaxelem, včetně 50,9 % respektive 60,2 % neutropenie stupně 4. Ve studii TRYPHAENA byl výskyt neutropenie stupně 3-4 dle NCI-CTCAE v.3 85,3 % u pacientů léčených neoadjuvantně přípravkem Perjeta + TCH a 77,0 % u pacientů léčených neoadjuvantně přípravkem Perjeta, trastuzumabem a docetaxelem následně po režimu FEC, včetně 66,7 % respektive 59,5 % neutropenie stupně 4.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Maximální tolerovaná dávka přípravku Perjeta nebyla stanovena. V klinických studiích nebyly testovány jednorázové dávky vyšší než 25 mg/kg (1727 mg).
V případě předávkování je nutné pacienty pečlivě sledovat na známky nebo příznaky nežádoucích účinků a zahájit příslušnou symptomatickou léčbu.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, monoklonální protilátky, ATC kód: L01XC13 Mechanismus účinku
Přípravek Perjeta je rekombinantní humanizovaná monoklonální protilátka specificky cílená na mimobuněčnou dimerizační doménu (subdoménu II) bílkoviny receptoru 2 lidského epidermálního růstového faktoru (HER2), která tím blokuje na ligandech závislou heterodimerizaci HER2 s ostatními členy skupiny HER včetně EGFR, HER3 a HER4. V důsledku toho přípravek Peijeta inhibuje na ligandech závislou nitrobuněčnou signalizaci dvěma hlavními signalizačními drahami: mitogenem aktivovanou proteinovou kinázou (MAP) a fosfoinositid 3-kinázou (PI3K). Inhibice těchto signálních drah může vést k zastavení buněčného růstu a apoptóze. Přípravek Peijeta kromě toho aktivuje na protilátkách závislou buněčnou cytotoxicitu.
Zatímco samotný přípravek Peijeta inhiboval proliferaci lidských nádorových buněk, kombinace přípravku Peijeta s trastuzumabem významně zvýšila protinádorovou aktivitu na modelu s xenoimplantáty nadměrně exprimujícími HER2.
Klinická účinnost a bezpečnost
Účinnost přípravku Perjeta u HER2-pozitivního karcinomu prsu je doložena randomizovanou srovnávací studií fáze III u metastazujícího karcinomu prsu a dvěma studiemi fáze II (jedna jednoramenná studie u metastazujícího karcinomu prsu a jedna randomizovaná srovnávací studie neoadjuvantního podání).
Metastazující karcinom _prsu
Přípravek Perjeta v kombinaci s trastuzumabem a docetaxelem
CLEOPATRA (WO20698) je multicentrická randomizovaná dvojitě zaslepená placebem kontrolovaná klinická studie fáze III provedená u 808 pacientů s HER2-pozitivním metastazujícím nebo lokálně rekurentním neresekovatelným karcinomem prsu. Pacienti s klinicky významnými kardiálními rizikovými faktory nebyli zařazeni (viz bod 4.4). Protože ze studie byli vyloučeni pacienti s mozkovými metastázami, nejsou k dispozici údaje o účinnosti přípravku Perjeta na mozkové metastázy. U pacientů s neresekovatelným lokálně rekurentním onemocněním jsou k dispozici velmi omezené údaje. Pacienti byli randomizováni v poměru 1:1 k léčbě placebem + trastuzumabem + docetaxelem nebo přípravkem Perjeta + trastuzumabem + docetaxelem.
Přípravek Perjeta a trastuzumab byly podávány ve standardních dávkách v třítýdenním režimu.
Pacienti byli léčeni přípravkem Perjeta a trastuzumabem do progrese nemoci, odvolání souhlasu nebo nepřijatelné toxicity. Docetaxel byl podáván v úvodní dávce 75 mg/m2 v intravenózní infuzi každé tři týdny v nejméně 6 cyklech. Pokud byla úvodní dávka dobře snášena, mohla být podle rozhodnutí řešitele dávka docetaxelu zvýšena na 100 mg/m2.
Primárním cílovým parametrem účinnosti této studie bylo přežití bez progrese (progression-free survival - PFS) podle nezávislého hodnocení, které bylo definováno jako doba od data randomizace do data progrese nemoci nebo úmrtí (z jakékoli příčiny), pokud k úmrtí došlo do 18 týdnů od posledního hodnocení nádoru. Sekundárními cílovými parametry účinnosti byly celkové přežití (overall survival - OS), přežití bez progrese (progression-free survival - PFS) (hodnocené řešitelem), četnost objektivních odpovědí (objective response rate - ORR), trvání odpovědi a čas progrese příznaků podle dotazníků kvality života FACT-B.
Přibližně polovina pacientů v každé léčebné skupině měla onemocnění s pozitivitou hormonálních receptorů (definovanou jako pozitivita estrogenních (ER) a/nebo progesteronových (PgR) receptorů) a přibližně polovina pacientů v každé léčebné skupině dostala předchozí adjuvantní nebo neoadjuvantní léčbu. Většina z těchto pacientů dostala předchozí léčbu antracyklinem a 11 % všech pacientů podstoupilo předchozí léčbu trastuzumabem. Celkem 43 % pacientů v obou léčebných skupinách bylo léčeno předchozí radioterapii. Střední vstupní hodnota ejekční frakce levé srdeční komory u pacientů byla 65,0 % (rozptyl 50 % až 88 %) v obou skupinách.
Výsledky účinnosti ve studii CLEOPATRA jsou shrnuty v tabulce 2. Bylo dosaženo statisticky významného zlepšení nezávisle hodnoceného přežití bez progrese ve skupině léčené přípravkem Perjeta ve srovnání se skupinou s placebem. Výsledky řešiteli hodnoceného přežití bez progrese byly podobné jako nezávisle hodnocené přežití bez progrese.
Tabulka 2 Souhrn údajů o účinnosti ve studii CLEOPATRA
|
Parametr |
Placebo+ trastuzumab + docetaxel n=406 |
Perjeta+ trastuzumab + docetaxel n=402 |
Poměr rizik (HR) (95% interval spolehlivosti) |
Hodnota p |
|
Přežití bez progrese (nezávislé hodnocení) - primární cílový parametr účinnosti* Počet pacientů s příhodou Medián, měsíce |
242 (59 %) 12,4 |
191 (47,5 %) 18,5 |
0,62 [0,51 - 0,75] |
<0,0001 |
|
Celkové přežití - sekundární cílový parametr účinnosti** Počet pacientů s příhodou Medián, měsíce |
221 (54,4 %) 40,8 |
168 (41,8 %) 56,5 |
0,68 [0,56 - 0,84] |
0,0002 |
|
Četnost objektivních odpovědí (ORR)A - sekundární cílový parametr účinnosti Počet pacientů s měřitelným onemocněním Respondenti*** 95% interval spolehlivosti pro ORR Úplná odpověď (CR) Částečná odpověď (PR) Stabilizace nemoci (SD) Progrese nemoci (PD) |
336 233 (69,3 %) [64,1 - 74,2] 14 (4,2 %) 219 (65,2 %) 70 (20,8 %) 28 (8,3 %) |
343 275 (80,2 %) [75,6 - 84,3] 19 (5,5 %) 256 (74,6 %) 50 (14,6 %) 13 (3,8 %) |
Rozdíl ORR: 10,8 % [4,2 - 17,5] % |
0,0011 |
|
Trvání odpovědi fa n= Medián, týdny 95% interval spolehlivosti pro medián |
233 54,1 [46 - 64] |
275 87,6 [71 - 106] |
* Primární analýza celkového přežití bez progrese, ukončení sběru údajů dne 13. května 2011.
**Konečná analýza celkového přežití, ukončení sběru údajů dne 11. února 2014.
*** Pacienti s nejlepší celkovou odpovědí hodnocenou jako potvrzená úplná nebo částečná odpověď podle
RECIST.
f Hodnoceno u pacientů s nejlepší celkovou odpovědí - úplnou nebo částečnou odpovědí.
A Četnost objektivních odpovědí a trvání odpovědi je na základě údajů nezávislého hodnocení nádoru.
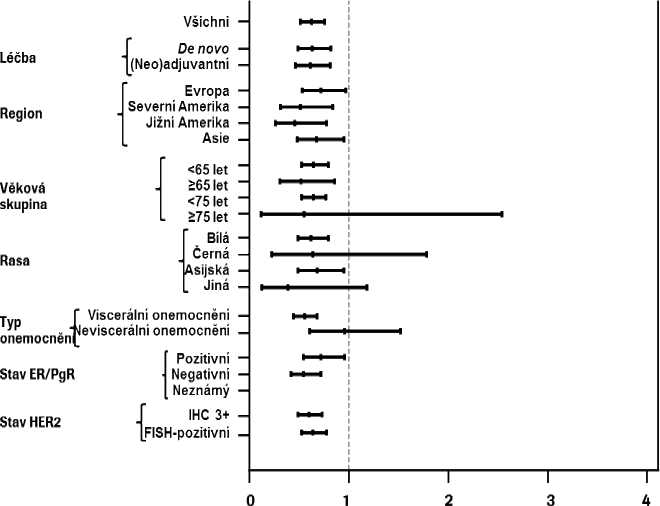
Byly pozorovány konzistentní výsledky u předem definovaných podskupin pacientů včetně podskupin definovaných stratifikačními faktory zeměpisné oblasti a předchozí adjuvantní/neoadjuvantní léčby nebo de novo metastazujícího karcinomu prsu (viz obrázek 1). Následná explorativní analýza ukázala, že u pacientů dříve léčených trastuzumabem (n = 88) byl poměr rizik pro nezávisle hodnocené přežití
bez progrese 0,62 (95% interval spolehlivosti 0,35 až 1,07) ve srovnání s hodnotou 0,60 (95% interval spolehlivosti 0,43 až 0,83) u pacientů, kteří měli jinou předchozí léčbu než trastuzumab (n = 288).
Obrázek 1 Nezávisle hodnocené přežití bez progrese v podskupinách pacientů
|
Dolni hranice |
Horni hranice | ||
|
N |
intervalu |
Odhad |
intervalu |
|
spolehlivosti |
spolehlivosti | ||
|
808 |
0,52 |
0,63 |
0,76 |
|
432 |
0,49 |
0,63 |
0,82 |
|
376 |
0,46 |
0,61 |
0,81 |
|
306 |
0,53 |
0,72 |
0,97 |
|
135 |
0,31 |
0,51 |
0,84 |
|
114 |
0,27 |
0,46 |
0,78 |
|
253 |
0,48 |
0,68 |
0,95 |
|
681 |
0,53 |
0,65 |
0,80 |
|
127 |
0,31 |
0,52 |
0,86 |
|
789 |
0,53 |
0,64 |
0,78 |
|
19 |
0,12 |
0,55 |
2,54 |
|
480 |
0,49 |
0,62 |
0,80 |
|
30 |
0,23 |
0,64 |
1,79 |
|
261 |
0,49 |
0,68 |
0,95 |
|
37 |
0,13 |
0,39 |
1,18 |
|
630 |
0,45 |
0,55 |
0,68 |
|
178 |
0,61 |
0,96 |
1,52 |
|
388 |
0,55 |
0,72 |
0,95 |
|
408 |
0,42 |
0,55 |
0,72 |
|
12 |
■ |
- |
■ |
|
721 |
0,49 |
0,60 |
0,74 |
|
767 |
0,53 |
0,64 |
0,78 |
Poměr rizik
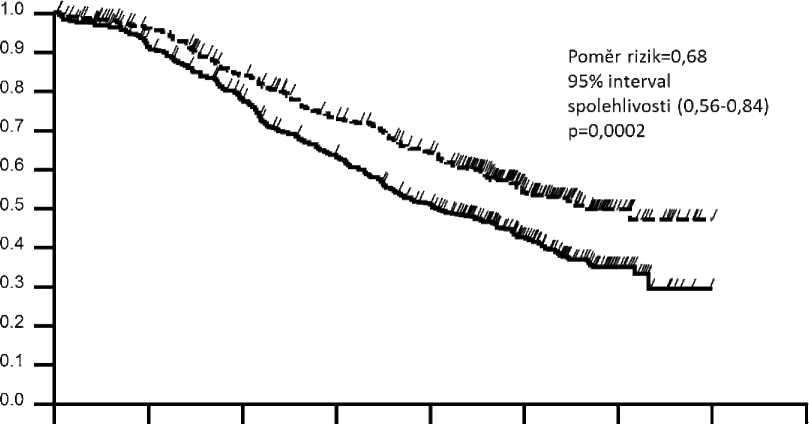
Konečná analýza celkového přežití byla provedena po úmrtí 389 pacientů (221 ve skupině léčené placebem a 168 ve skupině léčené přípravkem Peijeta). Byl zachován statisticky významný přínos celkového přežití ve prospěch skupiny léčené přípravkem Peijeta, pozorovaný dříve při průběžné analýze celkového přežití (provedené jeden rok po primární analýze) (poměr rizik 0,68, p = 0,0002 log-rank test). Střední doba do úmrtí byla 40,8 měsíců ve skupině léčené placebem a 56,5 měsíců ve skupině léčené přípravkem Peijeta (viz tabulka 2, obrázek 2).
Placebo + T + D
>N
<D
V)
O
e
n
o
■o
o
Q.
><1>
>
0
10 20
30 40
50
60
70 80
počet v riziku
Pertuzumab + T + D 402 Placebo + T + D 406
Měsíce
|
371 |
318 |
268 |
226 |
104 |
|
350 |
289 |
230 |
179 |
91 |
Pertuzumab + T + D
28
23
1
0
0
0
Randomizovaná léčba
Pla = placebo; Ptz = pertuzumab (Perjeta); T = trastuzumab (Herceptin); D = docetaxel
Mezi dvěma léčebnými skupinami nebyly zjištěny statisticky významné rozdíly v kvalitě života související se zdravím, která byla hodnocena skórem dle FACT-B TOI-PFB.
Další informace z podpůrných klinických studií
BO17929 - jednoramenná studie u metastazujícího karcinomu prsu
BO17929 byla nerandomizovaná studie fáze II u pacientů s metastazujícím karcinomem prsu, jejichž nádory progredovaly v průběhu léčby trastuzumabem. Léčba přípravkem Perjeta a trastuzumabem vyústila v četnost odpovědí 24,2 % s dalšími 25,8 % pacientů, kteří zaznamenali stabilizaci nemoci trvající alespoň 6 měsíců, což ukazuje, že přípravek Perjeta je aktivní po progresi na trastuzumabu.
Neoadjuvantní léčba karcinomu prsu
Při neoadjuvantní léčbě jsou lokálně pokročilé nebo inflamatorní karcinomy prsu považovány za vysoce rizikové bez ohledu na stav hormonálních receptorů. Při hodnocení rizika u časného karcinomu prsu je nutno brát v úvahu velikost nádoru, stupeň, stav hormonálních receptorů a metastázy v mízních uzlinách.
Indikace neoadjuvantní léčby karcinomu prsu je podložena důkazy o zvýšení četnosti patologické úplné odpovědi a trendem k delšímu přežití bez nemoci, na což však nenavazují benefity ve smyslu dlouhodobých výsledků, například celkové přežití nebo přežití bez nemoci.
NEOSPHERE (WO20697)
NEOSPHERE (WO20697) je multicentrická mezinárodní randomizovaná kontrolovaná studie fáze II s přípravkem Perjeta, která byla provedena u 417 dospělých pacientek s nově zjištěným časným, inflamatorním nebo lokálně pokročilým HER2-pozitivním karcinomem prsu (T2-4d; primární nádor > 2 cm v průměru), které dosud nebyly léčeny trastuzumabem, chemoterapií nebo radioterapií.
Pacientky s metastázami, bilaterálním karcinomem prsu, klinicky významnými kardiálními rizikovými
faktory (viz bod 4.4) nebo ejekční frakcí levé komory < 55 % nebyly zařazeny. Většina pacientek byla mladší 65 let.
Pacientky byly randomizovány k předoperační léčbě 4 cykly jedním z následujících neoadjuvantních režimů:
• Trastuzumab plus docetaxel
• Přípravek Perjeta plus trastuzumab a docetaxel
• Přípravek Perjeta plus trastuzumab
• Přípravek Perjeta plus docetaxel.
Randomizace byla stratifikována podle typu karcinomu prsu (operabilní, lokálně pokročilý nebo inflamatorní) a dle ER nebo PgR pozitivity.
Přípravek Perjeta byl podáván intravenózně v úvodní dávce 840 mg, následně 420 mg každé tři týdny. Trastuzumab byl podáván intravenózně v úvodní dávce 8 mg/kg, následně 6 mg/kg každé tři týdny. Docetaxel byl podáván intravenózně v úvodní dávce 75 mg/m2, následně 75 mg/m2 nebo 100 mg/m2 (pokud byl tolerován) každé tři týdny. Po operaci všechny pacientky dostávaly intravenózně 3 cykly 5-fluorouracilu (600 mg/m2), epirubicinu (90 mg/m2), cyklofosfamidu (600 mg/m2) (FEC) každé tři týdny, a trastuzumab byl podáván intravenózně každé tři týdny až do dokončení jednoroční léčby. Pacientkám, které dostávaly před operací pouze přípravek Perjeta plus trastuzumab, byly následně po operaci podány FEC i docetaxel.
Primárním cílovým parametrem účinnosti ve studii bylo hodnocení četnosti úplné patologické odpovědi (pCR) v prsu (ypT0/is). Sekundárními cílovými parametry účinnosti byly četnost klinických odpovědí, četnost konzervativních operací prsu (jen při nádoru T2-3), přežití bez nemoci (DFS) a přežití bez progrese (PFS). Další explorativní hodnocení četnosti pCR zahrnovalo stav lymfatických uzlin (ypT0/isN0 a ypT0N0).
Demografické hodnoty byly dobře vyváženy (stření věk byl 49-50 let, většina byla bílé rasy (71 %)) a vše byly pacientky - ženy. Celkem 7 % pacientek mělo inflamatorní karcinom prsu, 32 % lokálně pokročilý karcinom prsu a 61 % operabilní karcinom prsu. Přibližně polovina pacientek v každé léčebné skupině měla hormonálně pozitivní onemocnění (definované jako ER pozitivní a/nebo PgR pozitivní)
Výsledky účinnosti jsou uvedeny v tabulce 3. U pacientek léčených přípravkem Perjeta plus trastuzumabem + docetaxelem bylo pozorováno statisticky významné zlepšení četnosti pCR (ypT0/is) ve srovnání s pacientkami, které dostávaly trastuzumab a docetaxel (45,8 % versus 29,0 %, p = 0,0141). Byly pozorovány podobné výsledky nezávisle na definici pCR. Předpokládá se, že se tento rozdíl v četnosti pCR promítne do klinicky významných rozdílů při hodnocení dlouhodobé účinnosti, což je podporováno pozitivním vývojem přežití bez progrese (HR 0,69, 95% interval spolehlivosti 0,34; 1,40) a přežití bez nemoci (HR 0,60, 95% interval spolehlivosti 0,28; 1,27).
Četnost pCR a velikost benefitu při léčbě přípravkem Perjeta (Perjeta plus trastuzumab a docetaxel v porovnání s pacienty užívajícími trastuzumab a docetaxel) byly nižší v podskupině pacientů s hormonálně pozitivními nádory (rozdíl 6 % pCR v prsu) než u pacientek s nádory hormonálně negativními (rozdíl 26,4 % pCR v prsu). Četnost pCR byla podobná u pacientek s operabilním onemocněním ve srovnání s onemocněním lokálně pokročilým. Pacientek s inflamatorním karcinomem bylo příliš málo na to, aby bylo možno učinit jasné závěry, avšak četnost pCR byla vyšší u pacientek, které dostaly přípravek Perjeta plus trastuzumab a docetaxel.
TRYPHAENA (BO22280)
TRYPHAENA je multicentrická randomizovaná klinická studie fáze II provedená u 225 dospělých pacientek (žen) s HER2-pozitivním lokálně pokročilým, operabilním nebo inflamatorním karcinomem prsu (T2-4d; primární nádor >2 cm v průměru), které dosud nebyly léčeny trastuzumabem, chemoterapií nebo radioterapií. Nebyly zařazeny pacientky s metastázami, bilaterálním karcinomem prsu, klinicky významnými kardiálními rizikovými faktory (viz bod 4.4) nebo ejekční frakcí levé komory (LVEF) <55 %. Většina pacientek byla mladší než 65 let. Pacientky byly randomizovány k předoperační léčbě jedním ze tří neoadjuvantních režimů:
• 3 cykly režimu FEC následované 3 cykly docetaxelu, vše souběžně s přípravkem Perjeta a
trastuzumabem
• 3 cykly režimu FEC samotného následované 3 cykly docetaxelu podaného souběžně s
přípravkem Perjeta a trastuzumabem
• 6 cyklů režimu TCH v kombinaci s přípravkem Perjeta.
Randomizace byla stratifikována dle typu nádoru prsu (operabilní, lokálně pokročilý nebo inflamatorní) a pozitivity ER a/nebo PgR.
Přípravek Perjeta byl podáván intravenózně v úvodní dávce 840 mg, následně 420 mg každé 3 týdny. Trastuzumab byl podáván v úvodní dávce 8 kg/kg, následně 6 mg/kg každé 3 týdny. Režim FEC (5-fluorouracil [500 mg/m2], epirubicin [100 mg/m2], cyklofosfamid [600 mg/m2]) byl podáván intravenózně každé 3 týdny, 3 cykly. Docetaxel byl podáván v úvodní dávce 75 mg/m2 v intravenózní infuzi s možností zvýšení dávky na 100 mg/m2 dle rozhodnutí řešitele, pokud byla úvodní dávka dobře snášena. Avšak ve skupině léčené přípravkem Perjeta v kombinaci s režimem TCH byly podávány docetaxel nitrožilně v dávce 75 mg/m2 (zvýšení dávky nebylo povoleno) a karboplatina (AUC 6) nitrožilně každé 3 týdny. Po operaci dostávaly všechny pacientky trastuzumab do celkové doby 1 rok léčby.
Primárním cílovým parametrem bylo hodnocení kardiální bezpečnosti v neoadjuvantní části studie. Sekundárními cílovými parametry byla četnost úplné patologické odpovědi (pCR) v prsu (ypT0/is), přežití bez nemoci (DFS), přežití bez progrese (PFS) a celkové přežití (OS)
Demografické hodnoty byly mezi rameny dobře vyváženy (střední věk 49-50 let, většina byla bílé rasy [77 %]) a vše byly pacientky - ženy. Celkem 6 % pacientek mělo inflamatorní karcinom prsu, 25 % lokálně pokročilý karcinom prsu a 69 % operabilní karcinom prsu. Přibližně polovina pacientek v každé léčebné skupině měla hormonálně pozitivní onemocnění.
Ve srovnání s publikovanými daty pro podobné režimy bez přípravku Perjeta byly ve všech třech ramenech pozorovány vysoké četnosti pCR (viz tabulka 3). Byly pozorovány podobné výsledky nezávisle na použité definici pCR. Četnosti pCR byly nižší u pacientek s nádorem hormonálně pozitivním (rozptyl 46,2 až 50,0 %) než u pacientek s nádorem hormonálně negativním (rozptyl 65,0 až 83,8 %).
Četnost pCR byla podobná u pacientek s operabilním a lokálně pokročilým onemocněním. Pacientek s inflamatorním karcinomem bylo příliš málo na to, aby bylo možno učinit jasné závěry.
Tabulka 3 NEOSPHERE (WO20697) a TRYPHAENA (BO22280): Přehled účinnosti (všechny zařazené pacientky)__
|
NEOSPHERE (WO20697) |
TRYPHAENA (BO22280) | ||||||
|
Parametr |
trastuzuma b + docetaxel N=107 |
Perjeta + trastuzuma b + docetaxel N=107 |
Perjeta + trastuzumab N=107 |
Perjeta + docetaxel N=96 |
Perjeta + trastuzumab + FEC^ Perjeta + trastuzumab + docetaxel N=73 |
FEC^ Perjeta + trastuzumab+ docetaxel N=75 |
Perjeta + TCH N=77 |
|
Hodnota četnosti pCR v prsu (ypT0/is) n (%) [95% interval spolehlivos ti]1 |
31 (29,0 %) [20,6; 38,5] |
49 (45,8 %) [36,1; 55,7] |
18 (16,8 %) [10,3; 25,3] |
23 (24,0 %) [15,8; 33,7] |
45 (61,6 %) [49,5; 72,8] |
43 (57,3 %) [45,4; 68,7] |
51 (66,2 %) [54,6; 76,6] |
|
Rozdíl četností pCR2 [95% interval spolehlivos ti]3 |
+16,8 % [3,5; 30,1] |
-12,2 % [-23,8; -0,5] |
-21,8 % [-35,1; -8,5] |
NA |
NA |
NA | |
|
p-hodnota (se Simesovou korekcí pro CMH test)4 |
0,0141 (vs. trastuzumab + docetaxel) |
0,0198 (vs. trastuzumab + docetaxel) |
0,0030 (vs Perjeta + trastuzumab + docetaxel) |
NA |
NA |
NA | |
|
Četnost pCR v prsu a lymfatické uzlině (ypT0/is N0) n (%) [95% interval spolehlivos ti] |
23 (21,5 %) [14,1; 30,5] |
42 (39,3 %) [30,3; 49,2] |
12 (11,2 %) [5,9; 18,8] |
17 (17,7 %) [10,7; 26,8] |
41 (56,2 %) [44,1; 67,8] |
41 (54,7 %) [42,7; 66,2] |
49 (63,6 %) [51,9; 74,3] |
|
ypT0 N0 n (%) [95% interval spolehlivos ti] |
13 (12,1 %) [6,6; 19,9] |
35 (32,7 %) [24,0; 42,5] |
6 (5,6 %) [2,1; 11,8] |
13 (13,2 %) [7,4; 22,0] |
37 (50,7 %) [38,7; 62,6] |
34 (45,3 %) [33,8; 57,3] |
40 (51,9 %) [40,3; 63,5] |
|
NEOSPHERE (WO20697) |
TRYPHAENA (BO22280) | ||||||
|
Parametr |
trastuzuma b + docetaxel N=107 |
Perjeta + trastuzuma b + docetaxel N=107 |
Perjeta + trastuzumab N=107 |
Perjeta + docetaxel N=96 |
Perjeta + trastuzumab + FEC^ Perjeta + trastuzumab + docetaxel N=73 |
FEC^ Perjeta + trastuzumab+ docetaxel N=75 |
Perjeta + TCH N=77 |
|
Klinická odpověď5 |
79 (79,8 %) |
89 (88,1 %) |
69 (67,6 %) |
65 (71,4 %) |
67 (91,8 %) |
71 (94,7 %) |
69 (89,6 %) |
FEC: 5-fluorouracil, epirubicin, cyklofosfamid; TCH: docetaxel, karboplatina a trastuzumab, CMH: Cochran-Mantel-Haenszel
1. 95% interval spolehlivosti pro jeden binomiální vzorek za použití metody Pearson-Cloppera.
2. Léčba přípravkem Perjeta + trastuzumab + docetaxel a přípravek Perjeta + trastuzumab je porovnávána s režimem trastuzumab + docetaxel, zatímco režim Perjeta + docetaxel je porovnáván s režimem Perjeta + trastuzumab + docetaxel.
3. Přibližný 95% interval spolehlivosti pro rozdíl dvou četností odpovědí za použití metody dle Hauck-Andersona.
4. Hodnota p dle Cochran-Mantel-Haenszel testu se Simesovou úpravou na multiplicitu.
5. Klinická odpověď reprezentuje pacienty s nejlepší celkovou odpovědí CR nebo PR v průběhu neoadjuvantního období (primární prsní léze).
Imunogenicita
U pacientů v klíčové studii CLEOPATRA byly v několika časových intervalech provedeny testy na přítomnost protilátek proti léčebné protilátce Perjeta. Test na přítomnost protilátek proti léčebné protilátce byl pozitivní přibližně u 2,8 % (11/386 pacientů) pacientů léčených přípravkem Perjeta a u
6.2 % (23/372 pacientů) pacientů léčených placebem. U žádného z těchto 34 pacientů nedošlo
k závažné (stupně 4 dle NCI-CTCAE) reakci na infuzi nebo hypersenzitivní reakci (anafylaxi), která by jasně souvisela s protilátkami proti léčebné protilátce. U 2 ze 366 (0,5 %) pacientů léčených přípravkem Perjeta ve studiích fáze I a II se však projevila hypersenzitivní reakce stupně 3 v souvislosti s detekovatelnými protilátkami proti léčebné protilátce. V současné době není k dispozici dostatek údajů, aby bylo možno zhodnotit vliv protilátek proti léčebné protilátce na účinnost přípravku Perjeta v kombinaci s trastuzumabem a docetaxelem.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Perjeta u všech podskupin pediatrické populace v indikaci karcinom prsu (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Analýza populační farmakokinetiky byla provedena s údaji 481 pacientů s různými typy pokročilých maligních tumorů zařazených do různých klinických studií (fáze I, II a III), kteří dostali přípravek Perjeta v monoterapii nebo v kombinaci v dávkách pohybujících se od 2 do 25 mg/kg podávaných každé 3 týdny v intravenózní infuzi trvající 30 až 60 minut.
Absorpce
Přípravek Perjeta se podává v intravenózní infuzi. Studie s jinou cestou podání nebyly prováděny. Distribuce v organismu
V různých klinických studiích byly u typického pacienta distribuční objemy centrálního kompartmentu (V ) 3,11 litru a periferního kompartmentu (V ) 2,46 litru.
c p
Biotransformace
Metabolismus přípravku Perjeta nebyl přímo studován. Protilátky jsou odstraňovány hlavně katabolicky.
Eliminace z organismu
Střední clearance přípravku Perjeta byla 0,235 litru/den a střední poločas byl 18 dní.
Linearita/nelinearita
Přípravek Perjeta vykázal v rozsahu doporučených dávek lineární farmakokinetiku.
Starší pacienti
Na základě analýzy populační farmakokinetiky nebyl pozorován významný rozdíl farmakokinetiky přípravku Perjeta mezi pacienty ve věku < 65 let (n=306) a pacienty ve věku > 65 let (n=175).
Pacienti s poruchou renálních funkcí
S přípravkem Perjeta nebyla provedena žádná studie při poruše renálních funkcí. Na základě výsledků analýzy populační farmakokinetiky byla expozice přípravku Perjeta podobná u pacientů s mírnou (clearance kreatininu 60 až 90 ml/min, n=200) a středně závažnou poruchou renálních funkcí (clearance kreatininu 30 až 60 ml/min, n=71) jako u pacientů s normální renální funkcí (clearance kreatininu vyšší než 90 ml/min, n=200). Nebyl pozorován žádný vztah mezi clearance kreatininu a expozicí přípravku Perjeta při rozsahu pozorovaných hodnot clearance kreatininu (27 až 244 ml/min).
Jiné zvláštní populace
Na základě analýzy populační farmakokinetiky se nepředpokládají žádné rozdíly ve farmakokinetice v závislosti na věku, pohlaví a etniku (Japonci versus ne-Japonci). Nejvýznamnějšími proměnnými ovlivňujícími clearance byly hladina albuminu před léčbou a tělesná hmotnost bez tuku. Clearance klesala u pacientů s vyšší koncentrací albuminu před léčbou a stoupala u pacientů s vyšší tělesnou hmotností bez tuku. Analýza senzitivity provedená při doporučeném dávkování a režimu pro přípravek Perjeta a při extrémních hodnotách těchto dvou proměnných však neprokázala jejich významný vliv na možnost dosažení cílové koncentrace v ustáleném stavu, jak byla definována v preklinických modelech s xenoimplantáty. Není tedy nutné upravovat dávkování přípravku Perjeta s ohledem na tyto proměnné.
Farmakokinetické výsledky pertuzumabu ve studii NEOSPHERE jsou ve shodě s prognózami z předchozího farmakokinetického populačního modelu.
5.3 Předklinické údaje vztahující se k bezpečnosti
U zvířat nebyly provedeny studie specificky hodnotící vliv pertuzumabu na fertilitu. Nelze učinit definitivní závěry o nežádoucích účincích na samčí reprodukční orgány u makaků jávských ve studiích toxicity opakovaných dávek.
Studie reprodukční toxikologie byly provedeny u březích samic makaků jávských (v 19. až 50. dnu březosti) při úvodní dávce 30 až 150 mg/kg následované dávkou 10 až 100 mg/kg každé 2 týdny. Toto dávkování vedlo ke klinicky relevantní expozici dle Cmax 2,5 až 20krát vyšší než při doporučeném dávkování pro člověka. Intravenózní podávání pertuzumabu od 19. do 50. dne březosti (období organogeneze) bylo embryotoxické, incidence úmrtí embrya/plodu mezi 25. a 70. dnem březosti se zvyšovala v závislosti na dávce. Incidence ztráty embrya/plodu byla 33, 50 a 85 % u gravidních opičích samic, kterým byl podáván každé 2 týdny pertuzumab v dávce 10, 30 a 100 mg/kg (dle Cmax dávky 2,5 až 20krát vyšší než je doporučené dávkování pro člověka). Při císařském řezu 100. den březosti byly nalezeny ve všech skupinách dle dávky pertuzumabu oligohydramnion, nižší relativní hmotnost plic a ledvin a mikroskopické známky hypoplazie ledvin odpovídající opožděnému vývoji ledvin. Kromě toho byly rovněž zaznamenány v souvislosti s omezením růstu plodu při oligohydramnionu hypoplazie plic (1 ze 6 při dávce 30 mg/kg a 1 ze 2 při dávce 100 mg/kg), defekty přepážky srdečních komor (1 ze 6 při dávce 30 mg/kg), zeslabení stěny srdečních komor (1 ze 2 při dávce 100 mg/kg) a menší defekty skeletu (externí - 3 ze 6 při dávce 30 mg/kg). Ve všech léčených skupinách byla 100. den březosti hlášena expozice pertuzumabu u potomků na úrovni 29 až 40 % hladiny v séru matky.
Makakové jávští celkově dobře snášeli týdenní intravenózní podávání pertuzumabu v dávkách až 150 mg/kg. Při dávkách 15 mg/kg a vyšších byl zaznamenán občasný mírný průjem související s léčbou. U podskupiny opic vedlo chronické dávkování (7 až 26 týdenních dávek) k epizodám závažného průjmu. Průjem byl zvládán (s výjimkou eutanazie u jednoho zvířete při dávkování 50 mg/kg) podpůrnou léčbou včetně intravenózního doplňování tekutin.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Kyselina octová 98%
Histidin Sacharóza Polysorbát 20 Voda na injekci
6.2 Inkompatibility
Nebyly pozorovány žádné inkompatibility mezi přípravkem Peijeta a vaky z polyvinylchloridu (PVC) nebo non-PVC polyolefinu včetně polyetylenu. K naředění přípravku Peijeta nemá být používán roztok glukózy (5%), protože přípravek Perjeta je v takovém roztoku chemicky a fyzikálně nestabilní.
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Neotevřená injekční lahvička 2 roky.
Naředěný roztok
Byla prokázána chemická a fyzikální stabilita během používání po dobu 24 hodin při teplotě 30 °C.
Z mikrobiologického hlediska má být přípravek použit okamžitě. Pokud není použit okamžitě, je za další použití a uchovávání před použitím odpovědný uživatel. Uchovávání by nemělo přesáhnout 24 hodin při teplotě 2 °C až 8 °C, pokud naředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička (sklo třídy I) s uzávěrem (butylová pryž) obsahující 14 ml roztoku.
Balení obsahuje 1 injekční lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek Perjeta neobsahuje žádné antimikrobiální látky. Při přípravě infuzního roztoku proto musí být zaji štěna sterilita a přípravu musí provádět zdravotnický pracovník.
Přípravek Perjeta je určen pouze pro jednorázové použití a je podáván v intravenózní infuzi.
Injekční lahvička se nesmí protřepávat. 14 ml přípravku Perjeta koncentrát pro infuzní roztok má být nataženo z injekční lahvičky a naředěno v infuzním vaku z PVC nebo non-PVC polyolefinu o obsahu 250 ml infuzního roztoku chloridu sodného o koncentraci 9 mg/ml (0,9 %). Po naředění má jeden ml roztoku obsahovat přibližně 3,02 mg pertuzumabu (840 mg/278 ml) pro úvodní dávku, ke které jsou potřeba dvě injekční lahvičky, a přibližně 1,59 mg pertuzumabu (420 mg/264 ml) pro udržovací dávku, ke které je potřeba jedna injekční lahvička.
Pro mísení roztoku má být vak zvolna převracen, aby se zabránilo napěnění.
Léčivé přípravky pro parenterální použití je nutno před podáním vizuálně zkontrolovat na přítomnost částic a změnu barvy. Pokud j sou pozorovány částice nebo změna barvy, nemá se roztok použít. Jakmile je infuze připravena, má být podána okamžitě (viz bod 6.3).
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/813/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 4. března 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky/biologických léčivých látek
Genentech, Inc.
1000 New Horizons Way Vacaville, CA 95688-9431 USA
Název a adresa výrobce odpovědného za propouštění šarží
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Whylen Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP se předkládá každoročně až do prodloužení registrace.
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je třeba je předložit současně.
Dále je třeba aktualizovaný RMP předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření
|
Popis |
Termín splnění |
|
MO22324 (PHEREXA) Multicentrická randomizovaná studie fáze III, která porovnává kombinaci trastuzumabu a kapecitabinu, s/nebo bez pertuzumabu u pacientů s HER2-pozitivní metastazující rakovinou prsu, u kterých došlo k progresi po jedné linii léčby založené na trastuzumabu při metastazujícím onemocnění. |
Červenec 2016 |
|
MO28047 (PERUSE) Multicentrická, otevřená, jednoramenná studie pertuzumabu v kombinaci s trastuzumabem a taxany v první linii léčby u pacientů s HER2- pozitivní rakovinou prsu v pokročilém stádiu (metastazující nebo lokálně rekurentní). |
Září 2020 |
|
Poregistrační studie účinnosti (PAES): Z důvodu poskytnutí dlouhodobých údajů o účinnosti týkající se přežití bez nemoci (DFS) a celkového přežití (OS), má držitel rozhodnutí o registraci předložit výsledky studie BO25126 (APHINITY), randomizované, multicentrické, dvojitě zaslepené, placebem kontrolované porovnání chemoterapie plus trastuzumab plus placebo versus chemoterapie plus trastuzumab plus pertuzumab jako adjuvatní léčba u pacientů s operovatelným HER2-pozitivním primárním karcinomem prsu. |
Květen 2017 |
|
Poregistrační studie bezpečnosti (PASS): Z důvodu vyhodnocení kardiální bezpečnosti a poskytnutí dalších údajů o účinnosti při neoadjuvantní léčbě, má držitel rozhodnutí o registraci předložit výsledky studie WO29217 (BERENICE), multicentrická, multinárodnostní studie fáze II k vyhodnocení pertuzumabu v kombinaci s trastuzumabem a standardním neoadjuvantním antracyklinem, v závislosti na chemoterapii u pacientů s HER2-pozitivním, lokálně pokročilým, inflamatorním nebo časným karcinomem prsu. |
Květen 2017 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Perjeta 420 mg koncentrát pro infuzní roztok pertuzumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička se 14 ml obsahuje pertuzumabum 420 mg o koncentraci 30 mg/ml.
3. SEZNAM POMOCNÝCH LÁTEK
Kyselina octová 98%, histidin, sacharóza a polysorbát 20. Voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro infuzní roztok 420 mg/14 ml 1 x 14 ml
5 ZPŮSOB A CESTA/CESTY PODÁNÍ
Intravenózní podání po naředění Neprotřepávejte
Před použitím si přečtěte příbalovou informaci
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9 ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce (2 °C - 8 °C)
Chraňte před mrazem
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/813/001
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU NÁLEPKA NA INJEKČNÍ LAHVIČKU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Perjeta 420 mg koncentrát pro infuzní roztok
pertuzumabum
i.v.
2. ZPŮSOB PODÁNÍ
Intravenózní podání po naředění
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
420 mg/14 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Perjeta 420 mg koncentrát pro infuzní roztok
pertuzumabum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotní sestry.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Peij eta a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Perjeta používat
3. Jak se přípravek Perjeta používá
4. Možné nežádoucí účinky
5. Jak přípravek Peij eta uchovávat
6. Obsah balení a další informace
1. Co je přípravek Perjeta a k čemu se používá
Přípravek Perjeta obsahuje léčivou látku pertuzumab a používá se k léčbě dospělých pacientů s rakovinou prsu, pokud:
• bylo zjištěno, že se jedná o „HER2-pozitivní“ formu rakoviny prsu - Váš lékař Vám na to provede test.
• se nádor rozšířil (metastazoval) do jiných částí těla a nebyl dříve léčen protinádorovými léky (chemoterapií) nebo jinými léky, které se vážou na HER2, nebo se po předchozí léčbě nádor v prsu znovu objevil.
• nádor není rozšířen do jiných částí těla a léčba je podána před operací (léčba před operací se nazývá neoadjuvantní léčba).
Kromě přípravku Perjeta budete léčen(a) rovněž protinádorovými léky trastuzumabem a docetaxelem. Pokud užíváte přípravek Perjeta před operací, může Vám být podána další chemoterapie jako součást celkové léčby. Informace o těchto lécích jsou obsaženy v samostatných příbalových informacích. Požádejte svého lékaře nebo zdravotní sestru o informace o těchto dalších lécích.
Jak přípravek Perjeta působí
Přípravek Perjeta je typ léku, který se nazývá „monoklonální protilátka“, která se váže na specifické cíle ve Vašem těle a na buňky nádoru.
Přípravek Perjeta rozpoznává a váže se na cíl nazývaný „receptor 2 lidského epidermálního růstového faktoru“ (HER2). HER2 je ve větším množství přítomen na povrchu některých nádorových buněk, kde jejich růst podporuje. Pokud se přípravek Perjeta naváže na HER2 nádorových buněk, může zpomalit nebo zastavit jejich růst nebo je zahubit.
2. Čemu musíte věnovat pozornost, než začnete přípravek Perjeta používat Přípravkem Perjeta nesmíte být léčen(a)
• jestliže j ste alergický(á) na pertuzumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Pokud si nejste jistý(á), poraďte se před zahájením léčby přípravkem Perjeta se svým lékařem nebo zdravotní sestrou.
Upozornění a opatření
Před použitím přípravku Perjeta se poraďte se svým lékařem nebo zdravotní sestrou, jestliže:
• jste kdykoli měl(a) problémy se srdcem (jako jsou srdeční selhání, léčba pro závažné nepravidelnosti srdeční akce, nekontrolovaný vysoký krevní tlak, nedávno prodělaný srdeční záchvat) - Váš lékař provede vyšetření, zda Vaše srdce pracuje správně.
• jste kdykoli měl(a) problémy se srdcem v průběhu předchozí léčby trastuzumabem.
• jste byl(a) kdykoli dříve léčen(a) protinádorovým lékem ze skupiny nazývané antracykliny, např. doxorubicin nebo epirubicin - tyto léky mohou poškodit srdeční sval a zvýšit tak riziko problémů se srdcem při léčbě přípravkem Perjeta.
Pokud se Vás týká cokoli z výše uvedeného (nebo pokud si nejste jistý(á)), poraďte se před zahájením léčby přípravkem Perjeta se svým lékařem nebo zdravotní sestrou.
Reakce na infuzi
Může dojít k reakci na infuzi, alergické nebo anafylaktické (závažnější alergické) reakci. Váš lékař nebo zdravotní sestra Vás budou sledovat pro možnost výskytu nežádoucích účinků během infuze a 30 až 60 minut po jejím ukončení. V případě jakékoli závažné reakce může Váš lékař léčbu přípravkem Perjeta ukončit. Další informace o reakcích na infuzi, které je nutno sledovat během infuze a po jejím ukončení, jsou uvedeny v bodě 4 „Závažné nežádoucí účinky“.
Problémy se srdcem
Léčba přípravkem Perjeta může ovlivnit činnost srdce. Před léčbou přípravkem Perjeta a v jejím průběhu bude proto činnost srdce kontrolována. Další informace o známkách problémů se srdcem, které je nutno sledovat, jsou uvedeny v bodě 4 „Závažné nežádoucí účinky“.
Febrilní neutropenie (nízký počet bílých krvinek a horečka)
Pokud je přípravek Perjeta podáván s jinými protinádorovými léky (trastuzumab a docetaxel), může dojít k poklesu počtu bílých krvinek a vzniku horečky (vzestupu teploty). Pokud máte zánět trávicího ústrojí (například zánět sliznice dutiny ústní nebo průjem), může být vznik tohoto nežádoucího účinku pravděpodobnější.
Průjem
Léčba přípravkem Perjeta může způsobit těžký průjem. Průjem je stav, kdy Vaše tělo produkuje více vodnaté stolice, než je obvyklé. Pokud se u Vás během užívání protinádorové léčby vyskytne těžký průjem, může Váš lékař zahájit léčbu proti průjmu a přerušit léčbu přípravkem Perjeta, dokud nebude průjem pod kontrolou.
Použití u dětí a dospívajících
Přípravek Perjeta se nemá podávat pacientům mladším než 18 let, protože nejsou k dispozici žádné informace, jak přípravek účinkuje u této věkové skupiny.
Další léčivé přípravky a přípravek Perjeta
Informujte svého lékaře nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. To zahrnuje i léky, které jsou dostupné bez lékařského předpisu, a rostlinná léčiva.
Těhotenství a kojení
Pokud j ste těhotná nebo kojíte, nebo pokud se domníváte, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo zdravotní sestrou dříve, než začnete tento přípravek používat. Budou Vás informovat o přínosech a rizicích pro Vás a Vaše dítě, pokud byste byla léčena přípravkem Peijeta během těhotenství.
• Ihned informujte svého lékaře, pokud byste otěhotněla během léčby přípravkem Peijeta nebo do 6 měsíců po ukončení léčby.
• Zeptejte se svého lékaře, zda můžete kojit během léčby přípravkem Perjeta nebo po jejím ukončení.
Přípravek Perjeta může poškodit nenarozené dítě. Během léčby přípravkem Perjeta a 6 měsíců po jejím ukončení je třeba používat účinnou antikoncepci. Poraďte se se svým lékařem, jaká antikoncepce je pro Vás nejvhodnější.
Řízení dopravních prostředků a obsluha strojů
Je nepravděpodobné, že by přípravek Perjeta ovlivňoval Vaši schopnost řídit nebo obsluhovat stroje. Jestliže se však u Vás projeví jakákoli reakce na infuzi, alergická nebo anafylaktická reakce, vyčkejte až do jejího odeznění, než budete řídit nebo obsluhovat stroje.
3. Jak se přípravek Perjeta používá Způsob podání
Přípravek Perjeta Vám lékař nebo zdravotní sestra podá v nemocnici nebo v ambulanci.
• Podává se formou kapací infuze do žíly (intravenózní infuze) jednou za tři týdny.
• Množství podaného léku a trvání infuze je různé při prvním podání a při dalším podání.
• Počet infuzí, které dostanete, závisí na tom, jak budete reagovat na léčbu a zda Vám je léčba podávána před operací (neoadjuvantní léčba) nebo k léčbě onemocnění, které se rozšířilo.
• Přípravek Perjeta se podává v kombinaci s dalšími protinádorovými léky (trastuzumab a docetaxel).
Při první infuzi:
• Dostanete 840 mg přípravku Perjeta po dobu 60 minut. Váš lékař nebo zdravotní sestra Vás budou sledovat pro případ nežádoucích účinků během infuze a 60 minut po jejím ukončení.
• Dostanete rovněž trastuzumab a docetaxel.
Při všech dalších infuzích, pokud první infuze byla dobře snášena:
• Dostanete 420 mg přípravku Perjeta po dobu 30 až 60 minut. Váš lékař nebo zdravotní sestra Vás budou sledovat pro případ nežádoucích účinků během infuze a 30 až 60 minut po jejím ukončení.
• Dostanete rovněž trastuzumab a docetaxel.
Prosím, přečtete si další informace o dávkování trastuzumabu a docetaxelu (rovněž jejich podání může způsobit nežádoucí účinky) v příbalové informaci těchto přípravků, abyste dobře rozuměli, jak se tyto přípravky používají. Máte-li otázky týkající se těchto přípravků, zeptejte se, prosím, svého lékaře nebo zdravotní sestry.
Jestliže jste zapomněl(a) použít přípravek Perjeta
Pokud zapomenete nebo vynecháte návštěvu lékaře, abyste dostal(a) přípravek Perjeta, domluvte si co nejdříve nový termín. Pokud uběhlo od Vaší poslední návštěvy 6 týdnů nebo více, bude Vám podána vyšší dávka 840 mg přípravku Perjeta.
Jestliže jste přestal(a) používat přípravek Perjeta
Neukončujte léčbu tímto přípravkem bez předchozí domluvy s lékařem. Je důležité, abyste dostal(a) všechny doporučené infuze.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Závažné nežádoucí účinky
Informujte okamžitě lékaře nebo zdravotní sestru, pokud zaznamenáte jakýkoli z níže uvedených nežádoucích účinků:
• Nej častějšími nežádoucími účinky, které se mohou vyskytnout u přibližně 2 pacientů ze 3, jsou průjem, vypadávání vlasů a snížení počtu bílých krvinek (zjišťuje se vyšetřením krve) s horečkou nebo bez horečky.
• U přibližně 13 pacientů ze 100 se mohou projevit rekce na infuzi, které mohou zahrnovat pocit nevolnosti (pocit na zvracení), horečku, třesavku, pocit únavy, bolest hlavy, ztrátu chuti k jídlu. Alergické a anafylaktické (závažnější alergické) reakce se mohou projevit u přibližně 1 pacienta z 10. Mohou zahrnovat otok obličeje a krku s dýchacími obtížemi.
• Příznaky problémů se srdcem (srdeční selhání) byly pozorovány u přibližně 5 pacientů ze 100 a mohou zahrnovat kašel, dušnost při spánku vleže a otoky (zadržování tekutin) nohou nebo rukou.
Informujte okamžitě lékaře nebo zdravotní sestru, pokud zaznamenáte jakýkoli z výše uvedených nežádoucích účinků.
Další nežádoucí účinky zahrnují:
Velmi časté (mohou postihnout více než 1 z 10 osob):
• Horečka
• Nespavost
• Pokles počtu červených krvinek - zjišťuje se vyšetřením krve
• Bolest v krku, zarudnutí, bolavý nos nebo příznaky rýmy, příznaky podobné chřipce a horečka
• Pocity slabosti, necitlivosti, brnění nebo svědění postihující především chodidla a nohy
• Změny nehtů
• Ztráta nebo změna vnímání chuti
• Pocit nevolnosti nebo nevolnost
• Menší chuť k jídlu
• Zácpa
• Vyrážka
• Bolest kloubů nebo svalů, svalová slabost
• Bolest (bolest kostí, krku, hrudníku, břicha)
• Zánět zažívacího ústrojí (např. zánět sliznice dutiny ústní)
• Otoky kotníků nebo jiných částí těla v důsledku zadržování nadměrného množství vody v těle
Časté (mohou postihnout až 1 z 10 osob):
• Pocity závratě
• Dušnost
• Zvýšené slzení
• Suchá nebo svědící kůže, vyrážka podobná akné
• Tekutina na plicích způsobující obtíže s dýcháním
• Zánět nehtového lůžka v místě spojení nehtu a kůže
• Stavy, při kterých správně nepracuje levá srdeční komora, s příznaky nebo bez příznaků Méně časté (mohou postihnout až 1 ze 100 osob):
• Hrudní příznaky, jako je suchý kašel nebo dušnost (možné známky intersticiálního plicního onemocnění, stavu, kdy jsou poškozeny tkáně kolem plicních sklípků)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Pokud zaznamenáte jakýkoli z výše uvedených příznaků po ukončení léčby přípravkem Perjeta, promluvte si ihned se svým lékařem a informujte jej, že jste byl(a) dříve léčen(a) přípravkem Peijeta.
Některé nežádoucí účinky, které budete pozorovat, mohou souviset s rakovinou prsu. Pokud jste léčen(a) přípravkem Perjeta v kombinaci s trastuzumabem a docetaxelem, mohou některé nežádoucí účinky souviset s těmito přípravky.
5. Jak přípravek Perjeta uchovávat
Přípravek Perjeta je uchováván zdravotnickými pracovníky v nemocnici nebo v ambulanci. Pokyny pro uchovávání jsou následující:
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za „Použitelné do:“ Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Uchovávejte v chladničce (2 °C - 8 °C).
• Chraňte před mrazem.
• Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
• Nepoužívejte tento přípravek, pokud si všimnete pevných částic v tekutině nebo má tekutina nesprávnou barvu (podívejte se, prosím, do bodu 6).
• Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Perjeta obsahuje
• Léčivou látkou je pertuzumabum. Jedna injekční lahvička obsahuje celkem 420 mg pertuzumabu o koncentraci 30 mg/ml.
• Dalšími složkami jsou kyselina octová 98%, histidin, sacharóza, polysorbát 20 a voda na injekci.
Jak přípravek Perjeta vypadá a co obsahuje toto balení
Přípravek Perjeta je koncentrát pro infuzní roztok. Je to čirá až slabě opalescentní, bezbarvá až světle žlutá tekutina. Přípravek je dodáván ve skleněné injekční lahvičce obsahující 14 ml koncentrátu.
Jedno balení obsahuje jednu injekční lahvičku.
Držitel rozhodnutí o registraci
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
Výrobce
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien N.V. Roche S.A. Tél/Tel: +32 (0) 2 525 82 11 |
Lietuva UAB “Roche Lietuva” Tel: +370 5 2546799 |
|
Bt^rapnn Pom Etarapua EOOfl Tea: +359 2 818 44 44 |
Luxembourg/Luxemburg (Voir/siehe Belgique/Belgien) |
|
Česká republika Roche s. r. o. Tel: +420 - 2 20382111 |
Magyarország Roche (Magyarország) Kft. Tel: +36 - 23 446 800 |
|
Danmark Roche a/s Tlf: +45 - 36 39 99 99 |
Malta (See United Kingdom) |
|
Deutschland Roche Pharma AG Tel: +49 (0) 7624 140 |
Nederland Roche Nederland B.V. Tel: +31 (0) 348 438050 |
|
Eesti Roche Eesti OU Tel: + 372 - 6 177 380 |
Norge Roche Norge AS Tlf: +47 - 22 78 90 00 |
|
ELXáSa Roche (Hellas) A.E. Tr|k: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
Espaňa Roche Farma S.A. Tel: +34 - 91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 - 22 345 18 88 |
|
France Roche Tél: +33 (0)1 47 61 40 00 |
Portugal Roche Farmaceutica Química, Lda Tel: +351 - 21 425 70 00 |
|
Hrvatska Roche d.o.o. Tel: + 385 1 47 22 333 |
Románia Roche Románia S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska družba d.o.o Tel: +386 - 1 360 26 00 |
|
Ísland Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 - 2 52638201 |
|
Italia Roche S.p.A. Tel: +39 - 039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
|
Kónpoq r.A.Exapáxn? & Eia AxS. Tr|k: +357 - 22 76 62 76 |
Sverige Roche AB Tel: +46 (0) 8 726 1200 |
|
Latvija Roche Latvija SIA Tel: +371 - 6 7039831 |
United Kingdom Roche Products Ltd. Tel: +44 (0) 1707 366000 |
Tato příbalová informace byla naposledy revidována {MM/RRRR}.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
39