Pemetrexed Accord 1000 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Pemetrexed Accord 100 mg prášek pro koncentrát pro infuzní roztok Pemetrexed Accord 500 mg prášek pro koncentrát pro infuzní roztok Pemetrexed Accord 1 000 mg prášek pro koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Pemetrexed Accord 100 mg, prášek pro koncentrát pro infuzní roztok
Jedna injekční lahvička obsahuje pemetrexedum 100 mg (jako pemetrexedum dinatricum
dihemihydricum).
Pomocná látka se známými účinky
Jedna injekční lahvička obsahuje přibližně 11 mg sodíku.
Pemetrexed Accord 500 mg, prášek pro koncentrát pro infuzní roztok
Jedna injekční lahvička obsahuje pemetrexedum 500 mg (jako pemetrexedum dinatricum
dihemihydricum).
Pomocná látka se známými účinky
Jedna injekční lahvička obsahuje přibližně 54 mg sodíku.
Pemetrexed Accord 1 000 mg, prášek pro koncentrát infuzní roztok
Jedna injekční lahvička obsahuje pemetrexedum 1 000 mg (jako pemetrexedum dinatricum dihemihydricum).
Pomocná látka se známými účinky
Jedna injekční lahvička obsahuje přibližně 108 mg sodíku.
Po rekonstituci (viz bod 6.6) obsahuje jedna injekční lahvička pemetrexedum o koncentraci 25 mg/ml.
3. LÉKOVÁ FORMA
Prášek pro koncentrát pro infuzní roztok.
Bílý až buď světle žlutý nebo zelenožlutý lyofilizovaný prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Maligní mezoteliom pleury
Přípravek Pemetrexed Accord je v kombinaci s cisplatinou indikován k léčbě pacientů bez předchozí chemoterapie s neresekovatelným maligním mezoteliomem pleury.
Nemalobuněčný karcinom plic
Přípravek Pemetrexed Accord je v kombinaci s cisplatinou indikován v první linii k léčbě pacientů s lokálně pokročilým nebo metastazujícím nemalobuněčným karcinomem plic jiného histologického typu, než predominantně z dlaždicových buněk (viz bod 5.1).
Přípravek Pemetrexed Accord je indikován jako monoterapie k udržovací léčbě lokálně pokročilého nebo metastazujícího nemalobuněčného karcinomu plic jiného histologického typu, než predominantně z dlaždicových buněk u pacientů, u kterých po chemoterapii založené na platině nedošlo k bezprostřední progresi onemocnění. (viz bod 5.1).
Přípravek Pemetrexed Accord je indikován ve druhé linii jako monoterapie k léčbě pacientů s lokálně pokročilým nebo metastazujícím nemalobuněčným karcinomem plic jiného histologického typu, než predominantně z dlaždicových buněk (viz bod 5.1).
4.2 Dávkování a způsob podání
Pemetrexed Accord se smí podávat pouze pod dohledem lékaře s kvalifikací pro podávání protinádorové chemoterapie.
Dávkování:
Pemetrexed Accord v kombinaci s cisplatinou
Doporučená dávka přípravku Pemetrexed Accord je 500 mg/m2 tělesného povrchu (body surface area -BSA) podávaná jako intravenózní infuze po dobu 10 minut první den každého 21denního cyklu. Doporučená dávka cisplatiny je 75 mg/m2 BSA, podaná infuzí během dvou hodin přibližně 30 minut po ukončení infuze pemetrexedu v první den každého 21denního cyklu. Pacienti musejí dostávat přiměřenou antiemetickou terapii a hydrataci před podáním cisplatiny, případně i po jejím podání (informace o dávkování cisplatiny - viz rovněž souhrn údajů o přípravku pro přípravky s obsahem cisplatiny).
Pemetrexed Accord v monoterapii
U pacientů léčených pro nemalobuněčný karcinom plic po předcházející chemoterapii je doporučená dávka přípravku Pemetrexed Accord 500 mg/m2 BSA podávaná jako intravenózní infuze po dobu 10 minut první den každého 21denního cyklu.
Režim premedikace
Ke snížení incidence a závažnosti kožních reakcí se podá kortikosteroid den před podáním pemetrexedu, v den jeho podání a v den po jeho podání. Kortikoid má být ekvivalentní 4 mg dexamethasonu podávanému perorálně 2x denně (viz bod 4.4).
Ke snížení toxicity musí pacienti léčení pemetrexedem dostávat rovněž vitaminovou suplementaci (viz bod 4.4). Pacienti musí denně užívat kyselinu listovou perorálně nebo multivitaminy s obsahem kyseliny listové (350-1 000 mikrogramů). Během sedmi dnů před první dávkou pemetrexedu se musí podat nejméně pět dávek kyseliny listové a její podávání musí pokračovat v průběhu celé léčby a po dobu 21 dní po poslední dávce pemetrexedu. Pacienti musejí rovněž dostat intramuskulární injekci vitaminu B12 (1000 mikrogramů) v týdnu před první dávkou pemetrexedu a poté jednou za každé tři cykly. Další injekce vitaminu B12 se mohou podávat ve stejný den jako pemetrexed.
Monitorování
Pacienti používající pemetrexed musejí mít před každou dávkou monitorovaný celý krevní obraz, včetně diferenciálního bílého krevního obrazu a počtu trombocytů. Před každým podáním chemoterapie musí být provedeno biochemické vyšetření za účelem vyhodnocení funkce ledvin a jater. Před zahájením každého cyklu chemoterapie je nutné, aby pacienti měli následující výsledky vyšetření: absolutní počet neutrofilů musí být > 1500 buněk/mm3 a počet trombocytů musí být > 100 000 buněk/mm3. Clearance kreatininu musí být > 45 ml/min.
Celkový bilirubin musí být < 1,5násobek horní hranice normálních hodnot. Alkalická fosfatáza (AP), aspartátaminotransferáza (AST) a alaninaminotransferáza (ALT) musejí být < 3násobek horní hranice normálních hodnot. V případě postižení jater tumorem jsou akceptovatelné hodnoty alkalické fosfatázy, AST a ALT < 5násobek horní hranice normálních hodnot.
Úprava dávek
Úprava dávky při zahájení následného cyklu se provede na základě krevního obrazu v době nejhlubšího poklesu nebo na základě maximální nehematologické toxicity zjištěné v předchozím cyklu terapie. Léčbu lze odložit, aby byl dostatek času k úpravě. Po úpravě se pacienti léčí podle pokynů uvedených v tabulce 1, 2 a 3, které se použijí v případě podávání přípravku Pemetrexed Accord v monoterapii nebo v kombinaci s cisplatinou.
|
Tabulka 1 -Úprava dávek pro pemetrexed (v monoterapii nebo v kombinaci) a cisplatinu -hematologické toxicity | |
|
Absolutní počet neutrofilů v době nejhlubšího poklesu < 500 /mm3 a počet trombocytů v době nejhlubšího poklesu >50 000 /mm3 |
75 % předchozí dávky (pemetrexedu i cisplatiny) |
|
Počet trombocytů v době nejhlubšího poklesu < 50 000 /mm3 bez ohledu na absolutní počet neutrofilů v době nejhlubšího poklesu |
75 % předchozí dávky (pemetrexedu i cisplatiny) |
|
Počet trombocytů v době nejhlubšího poklesu < 50 000 /mm3 s krváceníma bez ohledu na absolutní počet neutrofilů v době nejhlubšího poklesu |
50 % předchozí dávky (pemetrexedu i cisplatiny) |
a dle obecných kritérií toxicity (CTC) podle National Cancer Institute (CTC v2.0, NCI 1998) definice CTC krvácení > stupeň 2.
Pokud u pacientů dojde k rozvoji nehematologické toxicity > 3. stupně (s výjimkou neurotoxicity), musí se Pemetrexed Accord vysadit až do úpravy na hodnoty nižší nebo stejné, jako byly hodnoty před léčbou. Léčba se zahájí podle pokynů uvedených v tabulce 2.
|
Tabulka 2 - Úprava dávek pro pemetrexed (v monoterapii nebo v kombinaci ) a cisplatinu -nehematologické toxicity3’ b | ||
|
Dávka pemetrexedu (mg/m2) |
Dávka cisplatiny (mg/m2) | |
|
Jakákoli toxicita stupně 3 nebo 4 s výjimkou mukozitidy |
75 % předchozí dávky |
75 % předchozí dávky |
|
Jakýkoli průjem s nutností hospitalizace (bez ohledu na stupeň) nebo průjem stupně 3 nebo 4 |
75 % předchozí dávky |
75 % předchozí dávky |
|
Mukozitida stupně 3 nebo 4 |
50 % předchozí dávky |
100 % předchozí dávky |
aObecná kritéria toxicity podle National Cancer Institute (CTC v2.0; NCI 1998) bS výjimkou neurotoxicity
V případě neurotoxicity je doporučená úprava dávky pro přípravek Pemetrexed Accord a cisplatinu uvedena v tabulce 3. Pokud se vyskytnou projevy neurotoxicity stupně 3 nebo 4, musejí pacienti léčbu přerušit.
|
Tabulka 3 - Úprava dávek pro pemetrexed (v monoterapii nebo v kombinaci ) a cisplatinu -neurotoxicita | ||
|
Stupeň toxicity dle CTC a |
Dávka pemetrexedu (mg/m2) |
Dávka cisplatiny (mg/m2) |
|
0 - 1 |
100 % předchozí dávky |
100 % předchozí dávky |
|
2 |
100 % předchozí dávky |
50 % předchozí dávky |
a Obecná kritéria toxicity podle National Cancer Institute (CTC v2.0, NCI 1998)
Léčba přípravkem Pemetrexed Accord musí být přerušena, pokud se u pacienta vyskytnou jakékoli projevy hematologické nebo nehematologické toxicity stupně 3 nebo 4 po 2 sníženích dávky nebo ihned, pokud se vyskytnou projevy neurotoxicity stupně 3 nebo 4.
Starší _ pacienti
V klinických studiích nebyly žádné známky, že byse u pacientů ve věku 65 let nebo starších zvýšilo riziko nežádoucích účinků v porovnání s pacienty mladšími 65 let. Není nutné žádné snížení dávky, kromě případů, kdy je toto snížení nezbytné pro všechny pacienty.
Pediatrická _ populace
Použití přípravku Pemetrexed Accordu maligního mezoteliomu pleury a nemalobuněčného karcinomu plic není u pediatrické populace relevantní.
Pacienti s poruchou _ funkce ledvin (standardní Cockroftův a Gaultův vzorec nebo rychlost glomerulární filtrace měřená metodou clearance Tc99m-DPTA v séru):
Pemetrexed se primárně vylučuje v nezměněné formě ledvinami. V klinických studiích nebyla u pacientů s clearance kreatininu > 45 ml/min zapotřebí žádná úprava dávky, kromě úprav doporučovaných pro všechny pacienty. Údaje o použití pemetrexedu u pacientů s clearance kreatininu pod 45 ml/min jsou nedostatečné, a proto se u těchto pacientů používání pemetrexedu nedoporučuje (viz bod 4.4).
Pacienti s poruchou _ funkce _jater
Nebyl zjištěn žádný vztah mezi AST, ALT nebo celkovým bilirubinem a farmakokinetikou pemetrexedu. Nicméně, pacienti s poruchou funkce jater a bilirubinem > 1,5 x vyšším, než je horní hranice normální hodnoty nebo aminotransferázami > 3,0 x vyššími, než je horní hranice normálních hodnot (při absenci metastáz v játrech) nebo > 5,0 x vyššími, než je horní hranice normálních hodnot (při přítomnosti metastáz v játrech), nebyli specificky studováni.
Způsob podání:
Opatření, která je nutno učinit před zacházením s léčivým přípravkem Pemetrexed Accord nebo před jeho podáním, naleznete v bodě 6.6.
Pemetrexed Accord se podává jako intravenózní infuze po dobu 10 minut první den každého 21denního cyklu. Pokyny pro rekonstituci a naředění přípravku Pemetrexed Accord před jeho podáním naleznete v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Kojení (viz bod 4.6).
Současné podávání vakcíny proti žluté zimnici (viz bod 4.5).
4.4 Zvláštní upozornění a opatření pro použití
Pemetrexed může potlačit funkci kostní dřeně, která se manifestuje jako neutropenie, trombocytopenie a anémie (nebo pancytopenie) (viz bod 4.8). Myelosuprese představuje obvykle toxicitu, která limituje velikost použité dávky. Pacienti musejí být během léčby sledováni z hlediska myelosuprese a pemetrexed se nesmí podat do doby, než se absolutní počet neutrofilů nevrátí na hodnoty > 1500 buněk/mm3 a počet trombocytů se nevrátí na hodnoty > 100 000 buněk/mm3. Úprava dávek v následujících cyklech je dána hodnotami absolutního počtu neutrofilů v době nejhlubšího poklesu, počtu trombocytů a maximální nehematologickou toxicitou pozorovanými v předchozím cyklu (viz bod 4.2).
Pokud byla před léčbou podávána kyselina listová a vitamin B12, byla hlášena menší toxicita a snížení hematologické a nehematologické toxicity stupně 3 nebo 4, jako je neutropenie, febrilní neutropenie a infekce s neutropenií stupně 3 nebo 4. Proto musejí být všichni pacienti léčení pemetrexedem poučeni, aby užívali kyselinu listovou a vitamin B12 jako profylaktické opatření ke snížení toxicity související s léčbou (viz bod 4.2).
U pacientů, kteří nedostávali před léčbou kortikosteroid, byly popsány kožní reakce. Podávání dexamethasonu (nebo ekvivalentního kortikosteroidu) před léčbou pemetrexedem může snížit výskyt a závažnost kožních reakcí (viz bod 4.2).
Nebyl studován dostatečný počet pacientů s clearance kreatininu pod 45 ml/min. Proto se nedoporučuje používání pemetrexedu u pacientů s clearance kreatininu < 45 ml/min (viz bod 4.2).
Pacienti s lehkou až středně těžkou renální insuficiencí (clearance kreatininu 45-79 ml/min) se mají vyvarovat užívání nesteroidních protizánětlivých léků (NSAID), jako je ibuprofen a kyselina acetylsalicylová (> 1,3 g denně) dva dny před podáním pemetrexedu, v den jeho podání a dva dny po podání pemetrexedu (viz bod 4.5). U pacientů s lehkou až středně těžkou renální insuficiencí, u kterých je terapie pemetrexedem vhodná, má být přerušeno užívání NSAID s dlouhým eliminačním poločasem nejméně pět dnů před podáním pemetrexedu, v den jeho podání a nejméně dva dny po podání pemetrexedu (viz bod 4.5).
V souvislosti s podáváním pemetrexedu samotného nebo v kombinaci s jinými chemoterapeutiky byly hlášeny závažné renální příhody, včetně akutního selhání ledvin. U mnoha pacientů, u kterých
k renálním příhodám došlo, existovaly rizikové faktory pro rozvoj těchto příhod, včetně dehydratace, preexistující hypertenze nebo diabetu mellitu.
Efekt tekutiny ve třetím prostoru, jako je pleurální výpotek nebo ascites, na pemetrexed není zcela stanoven. Klinická studie fáze 2 s pemetrexedem u 31 pacienta se solidním tumorem a se stabilním výpotkem ve třetím prostoru neprokázala žádný rozdíl v plazmatických koncentracích normalizovaných podle dávky a v clearance pemetrexedu oproti pacientům bez přítomnosti tekutiny v třetím prostoru. Proto je vhodné před začátkem léčby pemetrexedem zvážit drenáž tekutiny z třetího prostoru , ale nemusí to být nutné.
V důsledku gastrointestinální toxicity pemetrexedu podávaného v kombinaci s cisplatinou byly popsány případy vážné dehydratace. Proto mají pacienti dostávat přiměřenou antiemetickou terapii a odpovídající hydrataci před podáním a případně i po podání medikace.
V průběhu klinických studií s pemetrexedem byly méně často hlášeny závažné kardiovaskulární příhody, zahrnující infarkt myokardu a cerebrovaskulární příhody obvykle, když byl pemetrexed podáván v kombinaci s dalšími cytostatiky. Většina pacientů, u kterých byly tyto příhody pozorovány, měla preexistující kardiovaskulární rizikové faktory (viz bod 4.8).
Pokles imunity je častým jevem u pacientů s onkologickým onemocněním. Proto současné používání živých oslabených vakcín není doporučeno (viz bod 4.3 a 4.5).
Pemetrexed může mít geneticky škodlivé účinky. Pohlavně zralým mužům se doporučuje, aby během léčby a až 6 měsíců po jejím ukončení nepočali dítě. Doporučuje se používání antikoncepčních metod nebo abstinence. Vzhledem k možnosti, že by pemetrexed způsobil ireverzibilní infertilitu, se mužům doporučuje, aby před zahájením léčby vyhledali konzultaci o možnosti uchování spermií.
Ženy ve fertilním věku musejí během léčby pemetrexedem používat účinnou antikoncepční metodu (viz bod 4.6).
U pacientů léčených ozařováním před léčbou, v průběhu nebo následovně po léčbě pemetrexedem byly hlášeny případy postradiační pneumonitidy. Těmto pacientům má být věnována zvýšená pozornost a opatrnost je také zapotřebí při použití dalších radiosenzibilizujících látek.
Byly hlášeny případy kožní reakce v místě předchozího ozařování (radiation recall) u pacientů ozařovaných před delší dobou - před týdny až roky.
Pemetrexed Accord 100 mg prášek pro koncentrát pro infuzní roztok
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) na jednu injekční lahvičku, tzn. je v podstatě ,bez obsahu sodíku‘.
Pemetrexed Accord 500 mg prášek pro koncentrát pro infiizní roztok
Jedna injekční lahvička tohoto léčivého přípravku obsahuje přibližně 54 mg sodíku. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku..
Pemetrexed Accord 1 000 mg prášek pro koncentrát pro infuzní roztok
Jedna injekční lahvička tohoto léčivého přípravku obsahuje přibližně 108 mg sodíku. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Pemetrexed se vylučuje hlavně ledvinami v nezměněné formě, a to tubulární sekrecí a v menším rozsahu glomerulární filtrací. Souběžné podávání nefrotoxických léků (např. aminoglykosidy, kličková diuretika, sloučeniny platiny, cyklosporin) může vést k opožděné clearance pemetrexedu. Tato kombinace se musí používat s opatrností. Pokud je to nutné, má být pečlivě monitorována clearance kreatininu.
Souběžné podávání látek, které se rovněž vylučují tubulární sekrecí (např. probenecid, penicilin), může vést k opožděné clearance pemetrexedu. Při kombinaci těchto látek je zapotřebí zvýšené opatrnosti. Pokud je to nutné, má být pečlivě monitorována clearance kreatininu.
U pacientů s normální renální funkcí (clearance kreatininu > 80 ml/min) mohou vysoké dávky nesteroidních protizánětlivých léků (NSAID, jako je ibuprofen > 1600 mg denně) a vysoké dávky kyseliny acetylsalicylové (> 1,3 g denně) snížit eliminaci pemetrexedu a tím zvýšit výskyt nežádoucích účinků. Proto je zapotřebí při současném podávání vyšších dávek NSAID nebo kyseliny acetylsalicylové společně s pemetrexedem u pacientů s normální renální funkcí (clearance kreatininu > 80 ml/min) zvýšené opatrnosti.
Pacienti s lehkou až středně těžkou renální insuficiencí (clearance kreatininu od 45 do 79 ml/min) se mají vyvarovat současného používání pemetrexedu s NSAID (např.ibuprofen) nebo s vyššími dávkami kyseliny acetylsalicylové 2 dny před podáním pemetrexedu, v den jeho podání a 2 dny po podání pemetrexedu ( viz bod 4.4).
Jelikož nejsou k dispozici údaje o potenciální interakci mezi pemetrexedem a NSAID s delším poločasem, jako je piroxikam a rofekoxib, je potřebné jejich podávání u pacientů s lehkou až středně těžkou renální insuficiencí přerušit nejméně 5 dní před podáním pemetrexedu, v den jeho podání a nejméně 2 dny po podání pemetrexedu (viz bod 4.4). Pokud je současné podávání NSAID nezbytné, mají být pacienti důkladně monitorováni z hlediska toxicity, zejména myelosuprese a gastrointestinální toxicity.
Pemetrexed prochází omezeným jaterním metabolismem. Výsledky studií in vitro s lidskými jaterními mikrozomy ukázaly, že nelze předpovědět, zda pemetrexed způsobí klinicky významnou inhibici metabolické clearance léků metabolizovaných CYP3A, CYP2D6, CYP2C9 a CYP1A2.
Interakce běžné u všech cytotoxických látek
Vzhledem ke zvýšenému riziku trombózy u pacientů s maligním onemocněním je časté používání antikoagulační léčby. Vysoká intraindividuální variabilita koagulačního stavu při těchto chorobách a možnost interakce mezi perorálními antikoagulancii a protinádorovou chemoterapií vyžaduje zvýšenou frekvenci monitorování INR (International Normalised Ratio), pokud se rozhodneme léčit pacienta perorálními antikoagulancii.
Kontraindikované souběžné používání: vakcína proti žluté zimnici: riziko fatálního generalizovaného vakcinačního onemocnění (viz bod 4.3/
Nedoporučené současné používání: živé oslabené vakcíny (vyjma žluté zimnice, kdy je současné používání kontraindikováno): riziko systémové reakce s možným fatálním průběhem. Toto riziko je zvýšeno u osob s již existujícím poklesem imunity způsobeným základním onemocněním. Kde je to možné, použijte inaktivované vakcíny (poliomyelitida) (viz bod 4.4).
4.6 Fertilita, těhotenství a kojení
Antikoncepce u mužů a u žen
Ženy ve fertilním věku musí v průběhu léčby pemetrexedem používat účinnou antikoncepci. Pemetrexed může mít geneticky škodlivé účinky. Pohlavně zralým mužům se doporučuje, aby během léčby a až 6 měsíců po jejím ukončení nepočali dítě. Doporučuje se používání antikoncepčních metod nebo abstinence.
Neexistují údaje o použití pemetrexedu u těhotných žen, avšak je podezření, že pemetrexed tak jako ostatní antimetabolity způsobuje vážné kongenitální vady, pokud je podáván v těhotenství. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Pemetrexed se nemá používat v těhotenství, pokud to není jednoznačně nezbytné a po pečlivém zvážení potřeby léčby u matky a rizika pro plod (viz bod 4.4).
Kojení
Není známo, zda se pemetrexed vylučuje do lidského mateřského mléka a nežádoucí účinky u kojeného dítěte nelze vyloučit. Při léčbě pemetrexedem musí být kojení přerušeno (viz bod 4.3).
Fertilita
Vzhledem k možnosti způsobit léčbou pemetrexedem ireverzibilní infertilitu se mužům doporučuje, aby před zahájením léčby vyhledali konzultaci o možnosti uchování spermií.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie hodnotící účinky na schopnost řídit a obsluhovat stroje nebyly provedeny. Bylo však popsáno, že pemetrexed může způsobovat únavu. Pacienti proto mají být upozorněni, aby neřídili nebo neobsluhovali stroje v případě, že se tyto nežádoucí účinky objeví.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nejčastěji hlášenými nežádoucími účinky v souvislosti s pemetrexedem používaným ať už v monoterapii nebo v kombinaci jsou myelosuprese, manifestující se jako anemie, neutropenie, leukopenie, trombocytopenie; a gastrointestinální toxicita, manifestující se jako anorexie, nauzea, zvracení, průjem, zácpa, faryngitida, mukozitida a stomatitida. Další nežádoucí účinky zahrnuj í renální toxicitu, zvýšení hladin aminotransferáz, alopecii, únavu, dehydrataci, vyrážku, infekci/sepsi a neuropatii. Mezi vzácné nežádoucí účinky patří Stevens-Johnsonův syndrom a toxická epidermální nekrolýza.
Tabulkový seznam nežádoucích účinků
V tabulce níže je uvedena frekvence a závažnost nežádoucích účinků, které byly hlášeny u více než 5 % ze 168 pacientů s mezoteliomem, kteří byli randomizováni k léčbě cisplatinou a pemetrexedem a u 163 pacientů s mezoteliomem, kteří byli randomizováni k léčbě cisplatinou v monoterapii. V obou léčebných ramenech byla pacientům, u kterých dosud nebyla prováděna chemoterapie, přidána kyselina listová a vitamin B12.
Frekvence nežádoucích účinků je definována: velmi časté( >1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000), není známo (z dostupných údajů spontánních hlášení nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinek* |
pemetrexed/cisplatina |
cisplatina | ||
|
(n =168) |
(n =163) | |||||
|
Všechny stupně toxicity (%) |
Toxicita stupně 3-4 (%) |
Všechny stupně toxicity (%) |
Toxicita stupně 3-4 (%) | |||
|
Poruchy krve a lymfatického systému |
Velmi časté |
Snížení počtu neutrofilů/granul ocytů |
56,0 |
23,2 |
13,5 |
3,1 |
|
Snížení počtu leukocytů |
53,0 |
14,9 |
16,6 |
0,6 | ||
|
Snížení hladiny hemoglobinu |
26,2 |
4,2 |
10,4 |
0,0 | ||
|
Snížení počtu trombocytů |
23,2 |
5,4 |
8,6 |
0,0 | ||
|
Poruchy metabolismu a výživy |
Časté |
Dehydratace |
6,5 |
4,2 |
0,6 |
0,6 |
|
Poruchy nervového systému |
Velmi časté |
Senzorická neuropatie |
10,1 |
0,0 |
9,8 |
0,6 |
|
Časté |
Porucha chuti |
7,7 |
0,0*** |
6,1 |
0,0*** | |
|
Poruchy oka |
Časté |
Konjunktivitida |
5,4 |
0,0 |
0,6 |
0,0 |
|
Gastrointestinál ní poruchy |
Velmi časté |
16,7 |
3,6 |
8,0 |
0,0 | |
|
56,5 |
10,7 |
49,7 |
4,3 | |||
|
Stomatitida/ Faryngitida |
23,2 |
3,0 |
6,1 |
0,0 | ||
|
82,1 |
11,9 |
76,7 |
5,5 | |||
|
20,2 |
1,2 |
14,1 |
0,6 | |||
|
Zácpa |
11,9 |
0,6 |
7,4 |
0,6 | ||
|
Časté |
5,4 |
0,6 |
0,6 |
0,0 | ||
|
Poruchy kůže a podkožní tkáně |
Velmi časté |
16,1 |
0,6 |
4,9 |
0,0 | |
|
Alopecie |
11,3 |
0,0*** |
5,5 |
0,0*** | ||
|
Poruchy ledvin a močových cest |
Velmi časté |
Snížení hladiny reatininu |
10,7 |
0,6 |
9,8 |
1,2 |
|
Snížení clearance kreatininu** |
16,1 |
0,6 |
17,8 |
1,8 | ||
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Únava |
47,6 |
10,1 |
42,3 |
9,2 |
* Viz obecná kritéria toxicity podle National Cancer Institute, verze 2 pro každý stupeň toxicity s výjimkou termínu „snížení clearance kreatininu“
** který je odvozen z termínu „renální/urogenitální, jiné“
***V souladu s National Cancer Institute CTC (v2.0, NCI 1998), mají být případy poruchy chuti a alopecie hlášeny pouze jako stupeň 1 nebo 2.
Pro účely této tabulky byla použita hraniční hodnota 5 % pro zařazení všech příhod, kdy hlásící osoba považovala příhodu za možnou souvislost s léčbou pemetrexedem a cisplatinou.
Klinicky relevantní projevy toxicity (dle CTC), které byly hlášeny v > 1 % a < 5 % u pacientů náhodným výběrem přidělených k léčbě cisplatinou a pemetrexedem, jsou: renální selhání, infekce, horečka, febrilní neutropenie, zvýšení AST, ALT a GGT, kopřivka a bolest na hrudi.
Klinicky relevantní projevy toxicity (dle CTC), které byly hlášeny v < 1 % pacientů náhodným výběrem přidělených k léčbě cisplatinou a pemetrexedem, jsou: arytmie a motorická neuropatie.
V tabulce je uvedena frekvence a závažnost nežádoucích účinků, které byly hlášeny u více než 5 % z 265 pacientů, kteří byli randomizováni k léčbě pemetrexedem v monoterapii se suplementací kyselinou listovou a vitaminem B12 a u 276 pacientů, kteří byli randomizováni k léčbě docetaxelem v monoterapii. U všech pacientů byla stanovena diagnóza lokálně pokročilého nebo metastazujícího nemalobuněčného karcinomu plic a již byli léčeni chemoterapií.
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinek* |
pemetrexed n = 265 |
docetaxel n = 276 | ||
|
Všechny stupně toxicity (%) |
Toxicita stupně 3-4 (%) |
Všechny stupně toxicity (%) |
Toxicita stupně 3-4 (%) | |||
|
Poruchy krve a lymfatického systému |
Velmi časté |
Snížení počtu neutrofilů/ granulocytů |
10,9 |
5,3 |
45,3 |
40,2 |
|
Snížení počtu leukocytů |
12,1 |
4,2 |
34,1 |
27,2 | ||
|
Snížení hladiny hemoglobinu |
19,2 |
4,2 |
22,1 |
4,3 | ||
|
Časté |
Snížení počtu trombocytů |
8,3 |
1,9 |
1,1 |
0,4 | |
|
Gastrointestiná lní poruchy |
Velmi časté |
12,8 |
0,4 |
24,3 |
2,5 | |
|
16,2 |
1,5 |
12,0 |
1,1 | |||
|
Stomatitida/ faryngitida |
14,7 |
1,1 |
17,4 |
1,1 | ||
|
30,9 |
2,6 |
16,7 |
1,8 | |||
|
21,9 |
1,9 |
23,9 |
2,5 | |||
|
Časté |
Zácpa |
5,7 |
0,0 |
4,0 |
0,0 | |
|
Poruchy jater a žlučových cest |
Časté |
Zvýšení hladiny ALT |
7,9 |
1,9 |
1,4 |
0,0 |
|
Zvýšení hladiny AST |
6,8 |
1,1 |
0,7 |
0,0 | ||
|
Poruchy kůže a podkožní tkáně |
Velmi časté |
Vyrážka/ deskvamace |
14,0 |
0,0 |
6,2 |
0,0 |
|
Časté |
Pruritus |
6,8 |
0,4 |
1,8 |
0,0 | |
|
Alopecie |
6,4 |
0,4** |
37,7 |
2,2** | ||
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Únava |
34,0 |
5,3 |
35,9 |
5,4 |
|
Časté |
8,3 |
0,0 |
7,6 |
0,0 | ||
Viz obecná
kritéria toxicity podle National Cancer Institute, verze 2 pro každý stupeň toxicity.
** V souladu s National Cancer Institute CTC (v2.0, NCI 1998), mají být případy alopecie hlášeny pouze jako stupeň 1 nebo 2.
Pro účely této tabulky byla použita hraniční hodnota 5 % pro zařazení všech příhod, kdy hlásící osoba považovala příhodu za možnou souvislost s léčbou pemetrexedem.
Klinicky relevantní projevy toxicity (dle CTC), které byly hlášeny v > 1 % a <5 % u pacientů náhodným výběrem přidělených k léčbě pemetrexedem, jsou: infekce bez neutropenie, febrilní neutropenie, alergická reakce/hypersenzitivita, zvýšení kreatininu, motorická neuropatie, senzorická neuropatie, erythema multiforme a bolest břicha.
Klinicky relevantní projevy toxicity (dle CTC), které byly hlášeny v < 1 % pacientů náhodným výběrem přidělených k léčbě pemetrexedem, jsou: supraventrikulámí arytmie.
Klinicky relevantní laboratorní projevy toxicity stupně 3 a 4 byly obdobné při hodnocení integrovaných výsledků 3 studií fáze 2 sledujících monoterapii pemetrexedem (n = 164) a studie fáze 3 sledující monoterapii pemetrexedem (popsána výše), s výjimkou neutropenie (12,8 % oproti 5,3 %) a zvýšení alaninaminotransferázy (15,2 % oproti 1,9 %). Tyto rozdíly byly pravděpodobně dány rozdíly v populaci pacientů, protože do studií fáze 2 byli zařazeni pacienti dosud neléčení chemoterapií a pacientky s karcinomem prsu, předem intenzivně léčené s již existujícími metastázami do jater a/nebo s patologickými výchozími funkčními jaterními testy.
V tabulce níže je uvedena frekvence a závažnost nežádoucích účinků pravděpodobně souvisejících se studovanou medikací, které byly hlášeny u více než 5 % z 839 pacientů s NSCLC, kteří byli randomizováni k léčbě cisplatinou v kombinaci s pemetrexedem a z 830 pacientů s NSCLC, kteří byli randomizováni k léčbě cisplatinou v kombinaci s gemcitabinem. Všichni pacienti dostali studijní léčbu jako zahajovací léčbu lokálně pokročilého nebo metastazujícího nemalobuněčného karcinomu plic a pacienti v obou skupinách byli plně suplementováni kyselinou listovou a vitamnem B12. i
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinek** |
pemetrexed/cisplatina (n = 839) |
gem citabin/cisplatina (n= 830) | ||
|
Všechny stupně toxicity (%) |
Toxicita stupně 3-4 (%) |
Všechny stupně toxicity (%) |
Toxicita stupně 3-4 (%) | |||
|
Poruchy krve a lymfatického systému |
Velmi časté |
Snížení hladiny hemoglobinu |
33,0* |
5,6* |
45,7* |
9,9* |
|
Snížení počtu neutrofilů/ granulocytů |
29,0* |
15,1* |
38,4* |
26,7* | ||
|
Snížení počtu leukocytů |
17,8 |
4,8* |
20,6 |
7,6* | ||
|
Snížení počtu trombocytů |
10,1* |
4,1* |
26,6* |
12,7* | ||
|
Poruchy nervového systému |
Časté |
Senzorická europatie |
8,5* |
0,0* |
12,4* |
0,6* |
|
Porucha chuti |
8,1 |
0,0*** |
8,9 |
0,0*** | ||
|
Gastrointestinální poruchy |
Velmi časté |
56,1 |
7,2* |
53,4 |
3,9* | |
|
39,7 |
6,1 |
35,5 |
6,1 | |||
|
26,6 |
2,4* |
24,2 |
0,7* | |||
|
Zácpa |
21,0 |
0,8 |
19,5 |
0,4 | ||
|
Stomatitida/ faryngitida |
13,5 |
0,8 |
12,4 |
0,1 | ||
|
Průjem bez kolostomie |
12,4 |
1,3 |
12,8 |
1,6 | ||
|
Časté |
Dyspepsie/ pálení žáhy |
5,2 |
0,1 |
5,9 |
0,0 | |
|
Poruchy kůže a podkožní tkáně |
Velmi časté |
Alopecie |
11,9* |
0*** |
21,4* |
0,5*** |
|
Časté |
V yrážka/ deskvamace |
6,6 |
0,1 |
8,0 |
0,5 | |
|
Poruchy ledvin a močových cest |
Velmi časté |
Zvýšení hladiny kreatininu |
10,1* |
0,8 |
6,9* |
0,5 |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Únava |
42,7 |
6,7 |
44,9 |
4,9 |
* hodnota p <0,05 při srovnání kombinace pemetrexed/cisplatina a gemcitabin/cisplatina při použití Fisherova exaktního testu.
** Viz obecná kritéria toxicity podle National Cancer Institute, verze 2 pro každý stupeň toxicity.
*** V souladu s National Cancer Institute CTC (v2.0, NCI 1998), mají být případy poruchy chuti a alopecie hlášeny pouze jako stupeň 1 nebo 2.
Pro účely této tabulky byla použita hraniční hodnota 5 % pro zařazení všech příhod, kdy hlásící osoba považovala příhodu za eventuálně související s léčbou pemetrexedem a cisplatinou.
Klinicky relevantní toxicita, která byla hlášena u > 1 % a <5 % pacientů náhodným výběrem přidělených k léčbě pemetrexedem a cisplatinou, zahrnovala: zvýšení hodnot AST, zvýšení hodnot ALT, infekci, febrilní neutropenii, renální selhání, pyrexii, dehydrataci, konjunktivitidu a snížení clearance kreatininu.
Klinicky relevantní toxicita, která byla hlášena u < 1 % pacientů náhodným výběrem přidělených k léčbě pemetrexedem a cisplatinou, zahrnovala: zvýšení hodnot GGT, bolest na hrudi, arytmie a motorickou neuropatii.
Klinicky relevantní toxicita byla v celkové populaci u pacientů používajících pemetrexed v kombinaci s cisplatinou podobná bez ohledu na pohlaví.
V tabulce níže je uvedena frekvence a závažnost nežádoucích účinků pravděpodobně souvisejících se studijní medikací, které byly v klinických hodnoceních udržovací léčby pemetrexedem v monoterapii (JMEN: N=663) a pokračující udržovací léčby pemetrexedem (PARAMOUNT: n=539) hlášeny u více než 5 % z 800 pacientů, kteří byli randomizováni k léčbě samotným pemetrexedem a 402 pacientů, kteří byli randomizováni k léčbě placebem. U všech pacientů byl diagnostikován NSCLC stadia IIIB nebo IV a všichni podstoupili předchozí terapii založenou na platině. Pacienti v obou ramenech studie byli plně suplementováni kyselinou listovou a vitaminem B12.
|
Třída orgánových systémů |
Frekvence1 |
Nežádoucí ř V2 1 účinek |
pemetrexed3 (n = 800) |
placebo3 (n = 402) | ||
|
Všechny stupně toxicity (%) |
Toxicita stupně 3-4 (%) |
Všechny stupně toxicity (%) |
Toxicita stupně 3-4 (%) | |||
|
Poruchy krve a lymfatického systému |
Velmi časté |
Snížení hladiny hemoglobinu |
18,0 |
4,5 |
5,2 |
0,5 |
|
Časté |
Snížení počtu leukocytů |
5,8 |
1,9 |
0,7 |
0,2 | |
|
Snížení počtu neutrofilů |
8,4 |
4,4 |
0,2 |
0,0 | ||
|
Poruchy nervového systému |
Časté |
Senzorická neuropatie |
7,4 |
0,6 |
5,5 |
0,2 |
|
Gastrointestinální poruchy |
Velmi časté |
17,3 |
0,8 |
4,0 |
0,2 | |
|
12,8 |
1,1 |
3,2 |
0,0 | |||
|
Časté |
8,4 |
0,3 |
1,5 |
0,0 | ||
|
Mukozitida/ stomatitida |
6,8 |
0,8 |
1,7 |
0,0 | ||
|
Poruchy jater a žlučových cest |
Časté |
Zvýšení hladiny ALT |
6,5 |
0,1 |
2,2 |
0,0 |
|
Zvýšení hladiny AST |
5,9 |
0,0 |
1,7 |
0,0 | ||
|
Poruchy kůže a podkožní tkáně |
Časté |
Vyrážka/deskva mace |
8,1 |
0,1 |
3,7 |
0,0 |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Únava |
24,1 |
5,3 |
10,9 |
0,7 |
|
Časté |
Bolest |
7,6 |
0,9 |
4,5 |
0,0 | |
|
Edém |
5,6 |
0,0 |
1,5 |
0,0 | ||
|
Poruchy funkce ledvin a močových cest |
Časté |
Renální poruchy4 |
7,6 |
0,9 |
1,7 |
0,0 |
Zkratky: ALT = alaninaminotransferáza; AST = aspartátaminotrasferáza; CTCAE = obecná terminologická kritéria nežádoucích účinků (Common Terminology Criteria for Adverse Event); NCI = National Cancer Institute.
Klinicky relevantní projevy toxicity (dle CTC) jakéhokoli stupně, které byly hlášeny u > 1 % a <5 % pacientů náhodným výběrem přidělených k léčbě pemetrexedem zahrnují: febrilní neutropenii, infekci, snížení počtu trombocytů, průjem, zácpu, alopecii, pruritus/svědění, horečku (bez neutropenie), onemocnění očního povrchu (včetně konjunktivitidy), zvýšené slzení, závrať a motorickou neuropatii.
Klinicky relevantní projevy toxicity (dle CTC), které byly hlášeny u < 1 % pacientů náhodným výběrem přidělených k léčbě pemetrexedem, j sou: alergická reakce/hypersenzitivita, erythema multiforme, supraventrikulární arytmie a plicní embolie.
Bezpečnost byla hodnocena u pacientů, kteří byli randomizováni k léčbě pemetrexedem (n=800). Výskyt nežádoucích účinků byl hodnocen u pacientů, kteří dostali < 6 cyklů udržovací léčby pemetrexedem (n=519) a srovnán s výskytem u pacientů, kteří dostali > 6 cyklů léčby pemetrexedem (n=281). S déle trvající expozicí bylo pozorováno zvýšení výskytu nežádoucích účinků (všech stupňů). Významné zvýšení výskytu neutropenie stupně 3/4 potenciálně související s hodnoceným lékem, bylo pozorováno při delší expozici pemetrexedu (<6 cyklů: 3,3%, > 6 cyklů: 6,4%: p=0,046). U jednotlivých stupňů 3/4/5 dalších nežádoucích účinků, nebyly při delší expozici pozorovány statisticky významné rozdíly.
V průběhu klinických studií s pemetrexedem byly méně často hlášeny závažné kardiovaskulární a cerebrovaskulární příhody, zahrnující infarkt myokardu, anginu pectoris a tranzitorní ischemické ataky obvykle, když byl pemetrexed podáván v kombinaci s dalšími cytostatiky. Většina pacientů, u kterých byly tyto příhody pozorovány, měla preexistující kardiovaskulární rizikové faktory.
V průběhu klinických studií s pemetrexedem byly vzácně hlášeny potenciálně závažné případy hepatitidy.
Méně často byla u pacientů v průběhu klinických studií s pemetrexedem hlášena pancytopenie.
Méně často byly v klinických studiích u pacientů léčených pemetrexedem hlášeny případy kolitidy (včetně intestinálního a rektálního krvácení, někdy s fatálním průběhem, intestinální perforace, nekrózy a tyflitidy).
Méně často byly v klinických studiích u pacientů léčených pemetrexedem hlášeny případy intersticiální pneumonitidy s respirační nedostatečností, někdy s fatálním průběhem.
U pacientů léčených pemetrexedem byly méně často hlášeny případy edému.
Méně často byla u pacientů v průběhu klinických studií s pemetrexedem hlášena ezofagitida/radiační ezofagitida.
V průběhu klinických studií s pemetrexedem byly často hlášeny sepse, někdy s fatálním průběhem.
V průběhu postmarketingového sledování byly hlášeny u pacientů léčených pemetrexedem následující nežádoucí účinky:
V souvislosti s podáváním pemetrexedu samotného nebo v kombinaci s jinými chemoterapeutiky byly méně často hlášeny případy akutního selhání ledvin (viz bod 4.4).
U pacientů léčených ozařováním před léčbou, v průběhu nebo následně po léčbě pemetrexedem byly méně často hlášeny případy postradiační pneumonitidy (viz bod 4.4).
U pacientů, kteří podstoupili léčbu ozařováním, byly vzácně hlášeny případy kožní reakce v místě předchozího ozařování (radiation recall) (viz bod 4.4).
Méně často byly hlášeny případy periferní ischémie, které někdy vedly k nekróze končetiny.
Vzácně byly hlášeny případy tvorby puchýřů včetně Stevens-Johnsonova syndromu a toxické epidermální nekrolýzy, které byly v některých případech fatální.
Vzácně byla u pacientů léčených pemetrexedem hlášena hemolytická anemie, vyvolaná imunitní reakcí.
Vzácně byly hlášeny případy anafylaktického šoku.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
K popsaným symptomům předávkování patří neutropenie, anemie, trombocytopenie, mukozitida, senzorická polyneuropatie a vyrážka. K předpokládaným komplikacím předávkování patří myelosuprese, která se manifestuje neutropenií, trombocytopenií a anemií. Kromě toho lze pozorovat infekce s horečkou nebo bez ní, průjem a/nebo mukozitidu. V případě podezření na předávkování musí být u pacientů sledován krevní obraz a pacienti mají dostávat podle potřeby podpůrnou léčbu.
V léčbě předávkování pemetrexedem se má zvážit podávání kalcium-folinátu nebo kyseliny folinové.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatika, analogy kyseliny listové, ATC kód: L01BA04
Pemetrexed je tzv. „multi-targeted“ antifolikum, protinádorová látka působící narušení několika klíčových metabolických procesů závislých na kyselině listové, které jsou nezbytné pro replikaci buněk.
Studie in vitro prokázaly, že pemetrexed se chová jako „multi-targeted“ antifolikum tím, že inhibuje thymidylátsyntázu (TS), dihydrofolátreduktázu (DHFR) a glycinamid ribonukleotid formyltransferázu (GARFT), které jsou klíčové enzymy závislé na folátu pro biosyntézu thymidinu a purinových nukleotidů de novo. Pemetrexed je transportován do buněk redukovaným nosičem folátu a membránovým folátovým vazebným proteinovým transportním systémem. Jakmile je pemetrexed v buňce, přeměňuje se rychle a efektivně na polyglutamátové formy pomocí enzymu folylpolyglutamátsyntetáza. Polyglutamátové formy se zadržují v buňkách a jsou ještě silnějšími inhibitory TS a GARFT. Polyglutamace je proces závislý na čase a koncentraci, ke kterému dochází v nádorových buňkách a v menší míře i v normálních tkáních. Polyglutamátové metabolity mají zvýšený intracelulární poločas, což vede k protrahovanému účinku léku v maligních buňkách.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem pemetrexed u všech podskupin pediatrické populace ve schválených indikacích (viz bod 4.2).
Klinická účinnost:
Mezoteliom
Studie EMPHACIS byla multicentrická, randomizovaná, jednoduše zaslepená studie fáze 3 s přípravkem pemetrexed a cisplatinou oproti cisplatině u pacientů s maligním mezoteliomem pleury,
kteří dosud nepodstoupili chemoterapii. V této studii bylo prokázáno, že pacienti léčení přípravkem pemetrexed a cisplatinou měli klinicky významnou výhodu mediánu přežití, trvající 2,8 měsíce v porovnání s pacienty léčenými cisplatinou v monoterapii.
Během studie byla k léčbě zavedena dlouhodobá suplementace nízkými dávkami kyseliny listové a vitaminu B12 s cílem snížit toxicitu. Primární analýza této studie byla provedena na populaci všech pacientů randomizovaně přidělených do léčebné skupiny, kteří dostávali hodnocený lék (randomizovaní a léčení). Byla provedena analýza podskupin u pacientů, kteří dostávali suplementaci kyselinou listovou a vitaminem B12 v průběhu celé léčebné kúry hodnoceným lékem (úplná suplementace). Výsledky těchto analýz účinnosti jsou shrnuty v tabulce:
Účinnost přípravku pemetrexed v kombinaci s cisplatinou oproti cisplatině u pacientů s maligním _ mezoteliomem pleury__
|
Randomizovaní a léčení pacienti |
Plně suplementovaní pacienti | |||
|
Parametr účinnosti |
pemetrexed / |
cisplatina |
pemetrexed / |
cisplatina |
|
cisplatina |
cisplatina | |||
|
(n = 226) |
(n = 222) |
(n = 168) |
(n = 163) | |
|
Medián celkového přežití (měsíce) |
12,1 |
9,3 |
13,3 |
10,0 |
|
(95 % CI) |
(10,0 - 14,4) |
(7,8 - 10,7) |
(11,4 - 14,9) |
(8,4 - 11,9) |
|
Log Rank hodnota p* |
0,020 |
0,051 | ||
|
Medián doby do progrese tumoru |
5,7 |
3,9 |
6,1 |
3,9 |
|
(měsíce) | ||||
|
(95 % CI) |
(4,9 - 6,5) |
(2,8 - 4,4) |
(5,3 - 7,0) |
(2,8 - 4,5) |
|
Log Rank hodnota p* |
0,001 |
0,008 | ||
|
Doba do selhání léčby (měsíce) |
4,5 |
2,7 |
4,7 |
2,7 |
|
(95 % CI) |
(3,9 - 4,9) |
(2,1 - 2,9) |
(4,3 - 5,6) |
(2,2 - 3,1) |
|
Log Rank hodnota p* |
0,001 |
0,001 | ||
|
Výskyt celkové odpovědi* |
41,3 % |
16,7 % |
45,5 % |
19,6 % |
|
(95 % CI) |
(34,8 - 48,1) |
(12,0 - 22,2) |
(37,8 - 53,4) |
(13,8 - 26,6) |
|
Fisherova přesná hodnota p* |
< 0,001 |
< 0,001 | ||
Zkratky: CI = interval spolehlivosti * hodnota p se týká srovnání mezi rameny
** V rameni pemetrexed /cisplatina, randomizovaní a léčení (n = 225) a plně suplementovaní (n = 167) pacienti
Statisticky významné zlepšení klinicky významných symptomů (bolest a dušnost) vyskytujících se při maligním mezoteliomu pleury v rameni s pemetrexedem /cisplatinou (212 pacientů) oproti rameni s léčbou pouze cisplatinou (218 pacientů) bylo prokázáno pomocí škály symptomů karcinomu plic. Byly rovněž pozorovány statisticky významné rozdíly v plicních funkčních testech. Oddělení mezi léčebnými rameny bylo dosaženo zlepšením plicní funkce v rameni pemetrexed /cisplatina a zhoršením plicní funkce v čase u kontrolního ramene.
Existují omezené údaje u pacientů s maligním mezoteliomem pleury, léčených pemetrexedem v monoterapii. Pemetrexed v dávce 500 mg/m2 byl studován jako lék podávaný v monoterapii u 64 pacientů s maligním mezoteliomem pleury dosud neléčených chemoterapií. Celkový výskyt odpovědi na léčbu byl 14,1 %.
NSCLC, léčba v druhé linii
V multicentrické, randomizované, otevřené studii fáze 3 s pemetrexedem a docetaxelem u pacientů s lokálně pokročilým nebo metastatickým nemalobuněčným karcinomem plic po předchozí chemoterapii byl prokázán medián doby přežití 8,3 měsíce u pacientů léčených pemetrexed (ITT populace se záměrem léčit, n = 283) a 7,9 měsíců u pacientů léčených docetaxelem (ITT populace se záměrem léčit, n = 288). Předchozí chemoterapie nezahrnovala pemetrexed. Výsledky analýzy vlivu histologie NSCLC na celkové přežití svědčí ve prospěch pemetrexedu oproti docetaxelu u pacientů s NSCLC jiného histologického typu, než predominantně z dlaždicových buněk (n=399; 9,3 oproti 8,0 měsíců, adjustovaný HR=0,78; 95 CI=0,61-1,00, p=0,047) a ve prospěch docetaxelu u karcinomu s histologickou strukturou z dlaždicových buněk (n=172; 6,2 oproti 7,4 měsíců, adjustovaný HR=1,56; 95% CI =1,08-2,26; p = 0,018). V histologických podskupinách nebyly pozorovány žádné klinicky důležité rozdíly v bezpečnostním profilu pemetrexedu.
Omezené klinické údaje z jiného randomizovaného kontrolovaného klinického hodnocení fáze 3 naznačují, že údaje o účinnosti pemetrexedu (Overall survival OS - celková doba přežití, Progression free survival PFS - doba přežití bez progrese) jsou podobné pro skupinu pacientů s předchozí léčbou docetaxelem (n=41) a pacientů bez předchozí léčby docetaxelem (n = 540).
Účinnost pemetrexedu oproti docetaxelu u pacientů s nemalobuněčným karcinomem plic _- ITT populace__
|
pemetrexed |
docetaxel | |
|
Doba přežití (měsíce) • Medián (m) • 95 % CI pro medián • HR • 95 % CI pro HR • Hodnota p pro neinferioritu (HR) |
(n = 283) (n = 288) 8,3 7,9 (7,0 - 9,4) (6,3 - 9,2) 0,99 (0,82 - 1,20) 0,226 | |
|
Doba přežití bez progrese (měsíce) • Medián • HR (95 % CI) |
(n = 283) (n = 288) 2,9 2,9 0,97 (,82 - 1,16) | |
|
Doba do selhání léčby (TTTF - měsíce) • Medián • HR (95 % CI) |
(n = 283) (n = 288) 2,3 2,1 0,84 (,71 - ,997) | |
|
Odpověď (n: kvalifikovaní pro odpověď) • Výskyt odpovědi (%) (95 % CI) • Stabilní onemocnění (%) |
(n = 264) 9,1 (5,9 - 13,2) 45,8 |
(n = 274) 8,8 (5,7 - 12,8) 46,4 |
Zkratky: CI = interval spolehlivosti; HR = poměr rizik; ITT = záměr léčit; n = celková velikost
populace
NSCLC, léčba v _ první linii
Multicentrická, randomizovaná, otevřená studie fáze 3 s přípravkem pemetrexed a cisplatinou oproti gemcitabinu s cisplatinou u pacientů bez předchozí chemoterapie s lokálně pokročilým nebo metastazujícím (stadium IIIB nebo IV) nemalobuněčným karcinomem plic (NSCLC) prokázala, že přípravek pemetrexed v kombinaci s cisplatinou (populace ITT Intent-to-treat), n =862) dosáhl primárního cílového parametru a prokázal podobný klinický účinek jako gemcitabin v kombinaci s cisplatinou (ITT n =863) na celkové přežití (adjustovaný poměr rizik 0,94; 95% CI 0,84-1,05). Všichni pacienti účastnící se této studie měli ECOG výkonnostní stav 0 nebo 1.
Primární analýza účinnosti byla založena na ITT populaci. Analýzy citlivosti pro hlavní cílové parametry studie byly také vyhodnoceny u populace pacientů splňujících vstupní kritéria protokolu (Protocol Qualified -PQ). Výsledky analýz účinnosti u populace PQ jsou v souladu s analýzami populace ITT a podporují noninferioritu kombinace AC oproti GC.
Doba přežití bez progrese (Progression free survival - PFS) a výskyt celkové odpovědi byly podobné v obou ramenech léčby: medián PFS byl 4,8 měsíců pro pemetrexed v kombinaci s cisplatinou oproti
5,1 měsíců pro gemcitabin v kombinaci s cisplatinou (adjustovaný poměr rizik 1,04; 95% CI 0,941,15) a četnost celkové odpovědi byla 30,6% (95% CI 27,3- 33,9) pro pemetrexed s cisplatinou oproti 28,2% (95% CI 25,0-31,4) pro gemcitabin s cisplatinou. Údaje o PFS byly částečně potvrzeny nezávislým přezkoumáním (pro přezkoumání bylo náhodně vybráno 400/1725 pacientů).
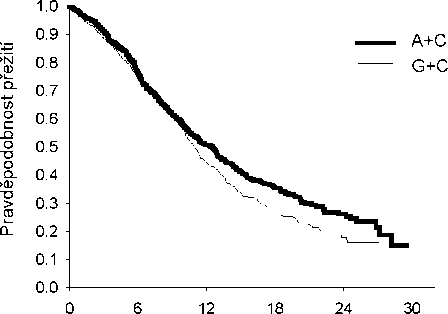
Analýza vlivu histologického původu NSCLC na celkovou dobu přežití prokázala klinicky významné rozdíly mezi jednotlivými histologickými typy, viz níže uvedená tabulka.
Účinnost kombinace pemetrexed + cisplatina oproti kombinaci gemcitabin + cisplatina v první linii nemalobuněčného karcinomu plic - ITT populace a histologické podskupiny.
|
ITT populace a histologické podskupiny |
Medián celkové doby přežití v měsících (95% CI) |
Adjustovaný poměr rizik (HR) (95% CI) |
Superiorita hodnota p | |||
|
pemetrexed + cisplatina |
gemcitabin + cisplatina | |||||
|
ITT populace (n= 1725) |
10,3 (9,8 - 11,2) |
n=862 |
10,3 (9,6 - 10,9) |
n=863 |
0,94a (0,84 - 1,05) |
0,259 |
|
Adenokarcinom (n=847) |
12,6 (10,7 - 13,6) |
n=436 |
10,9 (10,2 - 11,9) |
n=411 |
0,84 (0,71-0,99) |
0,033 |
|
Velkobuněčný (n=153) |
10,4 (8,6 - 14,1) |
n=76 |
6,7 (5,5 - 9,0) |
n=77 |
0,67 (0,48-0,96) |
0,027 |
|
Jiný (n=252) |
8,6 (6,8 - 10,2) |
n=106 |
9,2 (8,1 - 10,6) |
n=146 |
1,08 (0,81-1,45) |
0,586 |
|
Dlaždicobuněčný (n=473) |
9,4 (8,4 - 10,2) |
n=244 |
10,8 (9,5 - 12,1) |
n=229 |
1,23 (1,00-1,51) |
0,050 |
Zkratky: CI = interval spolehlivosti; ITT = záměr léčit; n = celková velikost souboru.
a Statisticky významné pro noninferioritu, s celkovým intervalem spolehlivosti pro HR dostatečně pod hranicí noninferiority 1,17645 (p <0,001).
Kaplan - Meierova křivka celkové doby přežití podle histologického typu
Adenokarcinom
Doba přežití (měsíce)
Velkobuněčný karcinom
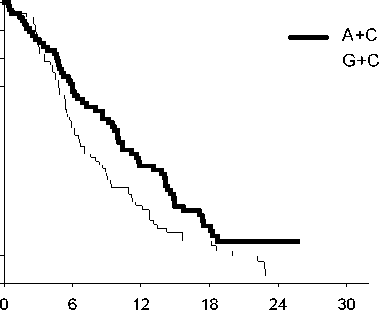
Doba přežití (měsíce)
V histologických podskupinách nebyly pozorovány žádné klinicky významné rozdíly v bezpečnostním profilu pemetrexedu v kombinaci s cisplatinou.
U pacientů léčených pemetrexedem v kombinaci s cisplatinou byl zapotřebí menší počet transfúzí (16,4% oproti 28,9 %, p<0,001), transfuzí erytrocytů (16,1 % oproti 27,3 %, p<0,001) a transfuzí trombocytů (1,8 % oproti 4,5%, p=0,002). Rovněž byl zapotřebí menší počet podání erytropoetinu/darbopoetinu (10,4 % oproti 18,1 %, p<0,001), G-CSF/GM-CSF (3,1 % oproti 6,1 %, p=0,004) a přípravků obsahujících železo (4,3 % oproti 7,0 %, p=0,021).
NSCLC, udržovací léčba
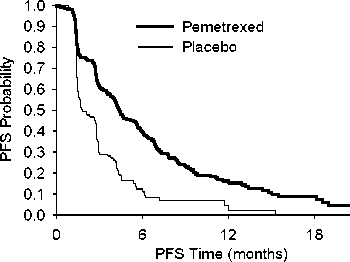
JMEN
Multicentrická, randomizovaná, dvojitě zaslepená studie fáze 3 kontrolovaná placebem (JMEN), srovnávala účinnost a bezpečnost udržovací léčby pemetrexedem spolu s nejlepší možnou podpůrnou léčbou (BSC) (n=441) a podávání placeba spolu s BSC (n=222) u pacientů s lokálně pokročilým (stadium IIIB) nebo metastazujícím (stadium IV) nemalobuněčným karcinomem plic (NSCLC), u kterých nedošlo k progresi po 4 cyklech terapie první linie dvojkombinací obsahující cisplatinu nebo karboplatinu v kombinaci s gemcitabinem, paklitaxelem nebo docetaxelem. Kombinovaná léčba obsahující v první linii v dvojkombinaci pemetrexed nebyla zahrnuta. Všichni pacienti účastnící se této studie měli ECOG výkonnostní stav 0 nebo 1. Udržovací léčba byla pacientům podávána do doby progrese nemoci. Účinnost a bezpečnost byly měřeny od doby randomizace po dokončení (indukční) terapie první linie. Střední hodnota počtu cyklů podaných pacientům byla 5 cyklů udržovací léčby pemetrexedem a 3,5 cyklů podávání placeba. Celkem 213 pacientů (48,3 %) dokončilo > 6 cyklů 103 pacientů (23,4 %) > 10 cyklů léčby pemetrexedem.
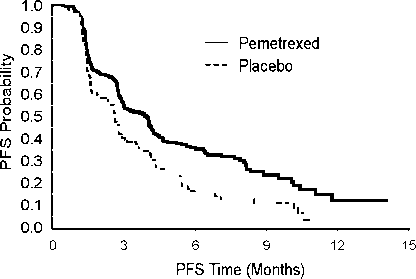
Studie dosáhla primárního cílového parametru a prokázala statisticky významné zlepšení PFS ve skupině pemetrexedu oproti skupině placeba (n = 581, nezávisle hodnocená populace, medián 4,0 měsíce, resp. 2,0 měsíce) (poměr rizika = 0,60, 95% CI: 0.49-0.73, p < 0.00001). Nezávislé hodnocení pacientských skenů potvrdilo závěry hodnocení PFS ze strany zkoušejících. Střední hodnota celkové doby přežití (OS) pro celkovou populaci (n=663) byla 13,4 měsíců ve skupině pemetrexedu a 10,6 měsíců ve skupině placeba, poměr rizik = 0,79 (95% CI: 0,65 až 0,95; p = 0,01192).
V souladu s dalšími studiemi pemetrexedu byl ve studii JMEN pozorován rozdíl v účinnosti s ohledem na histologii NSCLC. U pacientů s NSCLC jiného histologického typu, než predominantně
z dlaždicových buněk (n=430, nezávisle hodnocená populace) byla střední doba přežití bez progrese (PFS) 4,4 měsíce u pemetrexedu a 1,8 měsíců u skupiny placeba, poměr rizik = 0,47, 95% CI: 0,370,60, p= 0,00001. Střední hodnota celkové doby přežití (OS) u pacientů s NSCLC jiného histologického typu, než predominantně z dlaždicových buněk (n=481) byla 15,5 měsíců ve skupině pemetrexedu a 10,3 měsíce ve skupině placeba (poměr rizik = 0,70, 95% CI: 0,56-0,88, p=0,002). Střední doba OS včetně indukční fáze byla u pacientů s NSCLC jiného histologického typu, než predominantně z dlaždicových buněk 18,6 měsíců ve skupině pemetrexedu a 13,6 měsíců ve skupině placeba (poměr rizik =0,71, 95% CI: 0,56-0,88, p=0,002).
U pacientů s karcinomem histologického typu z dlaždicových buněk nenaznačují výsledky PFS a OS výhodu léčby pemetrexedem oproti placebu.
V histologických podskupinách nebyly pozorovány žádné klinicky důležité rozdíly v bezpečnostním profilu pemetrexedu.
JMEN: Kaplan - Meierova křivka doby přežití bez progrese (PFS) a celkové doby přežití u pacientů s NSCLC jiného histologického typu, než predominantně z dlaždicových buněk, užívajících pemetrexed nebo placebo
Doba přežití bez progrese Celková doba přežití
0 6 12 18 24 30 36 42
Survival Time (months)
1.0
0.9
Ě 0.8
Í5 0.7 ro
2 0.6
í 0.5 g 0.4 | 0.3
W 0.2 0.1 0.0
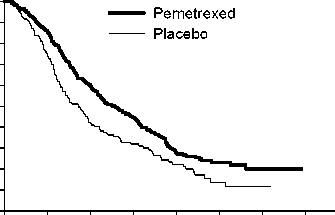
PARAMOUNT
Mutlicentrická, randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie fáze 3 (PARAMOUNT) porovnávala účinnost a bezpečnost pokračující udržovací léčby pemetrexedem plus BSC_(n = 359) s léčbou placebem plus BSC (n = 180) u pacientů s lokálně pokročilým (stadium IIIB) nebo metastazujícím (stadium IV) NSCLC jiného histologického typu, než predominantně z dlaždicových buněk, u kterých nedošlo k progresi onemocnění po 4 cyklech první linie léčby dvojkombinací pemetrexedu a cisplatiny. Z celkového počtu 939 pacientů léčených indukcí ppemetrexedem s cisplatinou, bylo 539 pacientů randomizováno na udržovací léčbu pemetrexedem nebo placebem. Z randomizovaných pacientů mělo 44,9 % úplnou/částečnou odpověď a u 51,9 % došlo ke stabilizaci onemocnění po indukci pemetrexedem s cisplatinou. Pacienti, kteří byli randomizováni k udržovací léčbě, museli mít výkonnostní stav ECOG 0 nebo 1. Medián doby od začátku indukční léčby pemetrexedem s cisplatinou do začátku udržovací léčby byl 2,96 měsíce jak v rameni s pemetrexedem, tak v rameni s placebem. Randomizovaní pacienti dostávali udržovací léčbu do doby progrese onemocnění. Účinnost a bezpečnost byly měřeny od doby randomizace po ukončení prvoliniové (indukční) léčby. Střední hodnoty počtu cyklů podaných pacientům byly 4 cykly léčby pemetrexedem a 4 cykly placeba. Celkem dokončilo >6 cyklů udržovací léčby pemetrexedem 169 pacientů (47,1 %), což představovalo nejméně 10 cyklů pemetrexedu celkem.
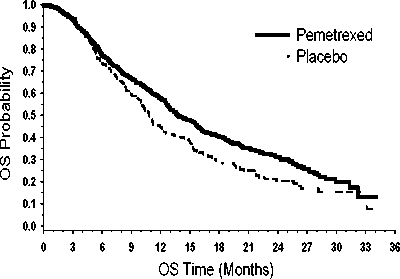
Studie splnila svůj primární cílový parametr a ukázala statisticky významné zlepšení PFS v rameni s pemetrexedem ve srovnání s placebovým ramenem (n=472, nezávisle hodnocená populace, medián
3,9 měsíců a 2,6 měsíců v tomto pořadí) (poměr rizik = 0,64, 95% CI = 0,51-0,81, p = 0,0002). Nezávislé posouzení skenů pacientů potvrdilo nálezy z hodnocení PFS zkoušejícími. Pro randomizované pacienty byl, měřeno od zahájení první linie indukční léčby pemetrexedem s cisplatinou, medián PFS stanovený zkoušejícím 6,9 měsíců v rameni s pemetrexedem a 5,6 měsíce v rameni s placebem (poměr rizik = 0,59 95% CI = 0,47-0,74).
Po indukci pemetrexedem s cisplatinou (4 cykly), byla léčba pemetrexedem statisticky lepší než placebo z hlediska celkového přežití (medián 13,9 měsíce versus 11,0 měsíců , poměr rizik = 0,78, 95% CI = 0,64-0,96, p = 0,0195). V době, kdy byla tato finální analýza doby přežití provedena, bylo v rameni s pemetrexedem 28,7% pacientů naživu nebo byl kontakt s nimi ztracen oproti 21,7 % pacientům v rameni s placebem. Relativní léčebný účinek pemetrexedu byl napříč podskupinami (včetně stadia nemoci, odpovědi na indukci, ECOG PS, kuřáckého statusu, pohlaví, histologie a věku) vnitřně konzistentní a podobný tomu, který byl pozorován v neadjustovaných analýzách OS a PFS. Jednoletá a 2letá četnost přežití pacientů s pemetrexedem byla 58 % a 32 % dle uvedeného pořadí, ve srovnání s 45 % a 21 % u pacientů s placebem. Od začátku indukční léčby pemetrexedem s cisplatinou v první linii byl medián OS pacientů 16,9 měsíce u ramene s pemetrexedem a 14,0 měsíců u ramene s placebem (poměr rizik= 0,78, 95% CI= 0,64-0,96). Procento pacientů, kteří dostali postudijní léčbu bylo 64,3 % u pemetrexedu a 71,7 % u placeba.
PARAMOUNT: Kaplan - Meierova křivka doby přežití bez progrese (PFS) a celkové doby přežití (OS) u pacientů s NSCLC jiného histologického typu, než predominantně z dlaždicových buněk, pokračujících v udržovací léčbě ppemetrexedem nebo placebem (měřeno od randomizace)
Doba přežití bez progrese (PFS)
Celková doba přežití (OS)
Bezpečnostní profily udržovací léčby pemetrexedem ze dvou klinických hodnocení JMEN a PARAMOUNT byly podobné.
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti pemetrexedu po jeho podání v monoterapii byly hodnoceny u 426 pacientů s různými maligními solidními tumory, kterým byl lék podáván v dávkách od 0,2 do 838 mg/m5 infuzí po dobu 10 minut. Distribuční objem pemetrexedu v ustáleném stavu činil 9 l/m5. Studie in vitro ukazují, že pemetrexed se přibližně z 81 % váže na plazmatické proteiny. Různý stupeň poruchy funkce ledvin nevede k významnému ovlivnění této vazby. Pemetrexed podstupuje v omezené míře metabolismus v játrech. Pemetrexed se primárně vylučuje močí, přičemž 70-90 % podané dávky se odstraní močí v nezměněné formě během prvních 24 hodin po jeho podání. Studie in vitro naznačují, že pemetrexed je aktivně vylučován pomocí OAT3 (přenašeč organických aniontů). Celková systémová clearance pemetrexedu je 91,8 ml/min a eliminační poločas plazmy je 3,5 hodin u pacientů s normální funkcí ledvin (clearance kreatininu 90 ml/min). Variabilita clearance mezi pacienty je střední, a to 19,3 %. Celková systémová expozice pemetrexedu (AUC) a maximální plazmatická koncentrace rostou proporcionálně s dávkou. Farmakokinetika pemetrexedu je stejná i při více léčebných cyklech.
Farmakokinetické vlastnosti pemetrexedu nejsou ovlivněny souběžně podanou cisplatinou. Suplementace kyselinou listovou perorálně a vitaminem B12 intramuskulárně neovlivňuje farmakokinetiku pemetrexedu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Podání pemetrexedu březím myším vedlo ke snížení viability plodů, ke snížení hmotnosti plodů, neúplné osifikaci některých kosterních struktur a rozštěpu patra.
Podání pemetrexedu samcům myší vedlo k reprodukční toxicitě charakterizované snížením fertility a testikulární atrofií. V devítiměsíční studii provedené na bíglech s použitím intravenózní bolusové injekce byl pozorován nález na varlatech (degenerace/nekróza seminiferní výstelky). To naznačuje, že pemetrexed může poškodit mužskou fertilitu. Fertilita žen nebyla studována.
Pemetrexed nebyl mutagenní in vitro ani u testu chromozomální aberace na buňkách ovarií čínských křečků ani u Amesova testu. V mikronukleárním testu in vivo u myši bylo prokázáno, že pemetrexed je klastogenní.
Studie hodnotící kancerogenní potenciál pemetrexedu nebyly provedeny. 6
6.1 Seznam pomocných látek
Mannitol (E421)
Kyselina chlorovodíková (E507) (k úpravě pH)
Hydroxid sodný (E524) (k úpravě pH)
6.2 Inkompatibility
Pemetrexed je fyzikálně inkompatibilní s diluenty obsahujícími kalcium, jako je laktátový Ringerův roztok a Ringerův roztok. Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Neotevřená lahvička 3 roky.
Rekonstituované a infuzní roztoky
Chemická a fyzikální stabilita po otevření před použitím rekonstituovaného a infuzního roztoku pemetrexedu byla prokázána na dobu 24 hodin při teplotě 25 °C. Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím j sou v odpovědnosti uživatele a normálně nemá být doba delší než 24 hodin při teplotě 2 °C -8 °C, pokud rekonstituce/ředění neproběhly za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Pemetrexed Accord 100 mg, prášek pro koncentrát pro infuzní roztok
Injekční skleněná lahvička (třídy I) s šedou brombutylovou pryžovou zátkou s fialovým odtrhávacím uzávěrem obsahující 100 mg pemetrexedu Balení po linjekční lahvičce.
Pemetrexed Accord 500 mg, prášek pro koncentrát pro infuzní roztok
Injekční skleněná lahvička ( třída I) s šedou brombutylovou pryžovou zátkou s modrým odtrhovacím víčkem obsahující 500 mg pemetrexedu.
Balení po 1 injekční lahvičce.
Pemetrexed Accord 1 000 mg, prášek pro koncentrát pro infuzní roztok
Injekční skleněná lahvička (střída I) s šedou brombutylovou pryžovou zátkou s modrým (královská modř) odtrhovacím víčkem obsahující 1000 mg pemetrexedu.
Balení po 1 injekční lahvičce.
6.6 Zvláštní opatření pro likvidaci přípravku a zacházení s ním
1. Při rekonstituci a dalším ředění pemetrexedu k podání intravenózní infuze používejte aseptickou techniku. 5
3. Rekonstituujte obsah 100mg injekční lahvičky se 4,2 ml 0,9% injekčního roztoku chloridu sodného (9 mg/ml)bez konzervačních látek, čímž vznikne roztok s koncentrací pemetrexedu 25 mg/ml.
Rekonstituujte obsah 500mg injekční lahvičky s 20 ml 0,9% injekčního roztoku chloridu sodného (9 mg/ml) bez konzervačních látek, čímž vznikne roztok s koncentrací pemetrexedu 25 mg/ml.
Rekonstituujte obsah 1000mg injekční lahvičky se 40 ml 0,9% injekčního roztoku chloridu sodného (9 mg/ml)bez konzervačních látek, čímž vznikne roztok s koncentrací pemetrexedu 25 mg/ml.
Pohybujte jemným krouživým pohybem každou injekční lahvičkou, dokud se prášek zcela nerozpustí. Výsledný roztok je čirý a jeho barva kolísá od bezbarvé po žlutou nebo žlutozelenou, aniž by byla narušena jeho kvalita. pH rekonstituovaného roztoku se pohybuje mezi 6,6 a 7,8. Je potřebné další ředění.
4. Náležitý objem rekonstituovaného roztoku pemetrexedu musí být dále naředěn na 100 ml 0,9% injekčním roztokem chloridu sodného (9 mg/m)bez konzervačních látek a podá se intravenózní infuzí po dobu 10 minut.
5. Infuzní roztoky pemetrexedu, připravené podle návodu, jsou kompatibilní
s polyvinylchloridovými a polyolefinovými infuzními sety a infuzními vaky.
6. Léčivé přípravky pro parenterální použití se musí před podáním vizuálně zkontrolovat, zda neobsahují pevné částice a nedošlo ke změně barvy. Jestliže zpozorujete pevné částice, přípravek nepodávejte.
7. Roztok pemetrexedu je určen pouze na jedno použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Bezpečnostní opatření při přípravě a podání přípravku :
Tak jako i u jiných potenciálně toxických cytostatických látek je nutné udržovat pozornost při zacházení s infuzním roztokem pemetrexedu a při jeho přípravě. Doporučuje se používat ochranné rukavice. Pokud dojde ke kontaktu roztoku pemetrexedu s kůží, umyjte ihned a důkladně kůži mýdlem a vodou. Pokud dojde ke kontaktu roztoku pemetrexedu se sliznicemi, opláchněte je důkladně vodou. Pemetrexed není puchýřotvorná látka. V případě podání extravazálně neexistuje specifické antidotum. Bylo popsáno několik případů podání pemetrexedu mimo žílu, které hodnotící lékař nepovažoval za závažné. Únik pemetrexedu extravazálně se léčí místními standardními postupy jako u jiných nepuchýřotvorných látek.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4HF Velká Británie
8. REGISTRAČNÍ ČÍSLO
EU/1/15/1071/001
EU/1/15/1071/002
EU/1/15/1071/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců odpovědných za propouštění šarží
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow HA1 4HF Velká Británie
Wessling Hungary Kft.
Foti Ú56
Budapest
1047
Maďarsko
V příbalové informaci k léčivému přípravku musí být uvedený název a adresa výrobců zodpovědných za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Požadavky na předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst 7 směrnice 2001/83/ES a jakékoli následné změnyjsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Neuplatňuje se.
Povinnost uskutečnit poregistrační opatření
Neuplatňuje se.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU
Pemetrexed Accord 100 mg prášek pro koncentrát pro infuzní roztok pemetrexedum
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jedna lahvička obsahuje pemetrexedum 100 mg (jako pemetrexedum dinatricum dihemihydricum) Po rekonstituci (viz příbalová informace) obsahuje jedna injekční lahvička pemetrexedum o koncentraci 25 mg/ml.
3. SEZNAM POMOCNÝCH LÁTEK
Mannitol, kyselina chlorovodíková, hydroxid sodný (další informace viz příbalová informace)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro koncentrát pro infuzní roztok. 1 injekční lahvička
5. ZPŮSOB A CESTA PODÁNÍ
Pouze pro jednorázové podání.
Intravenózní podání po rekonstituci a naředění. Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
CYTOTOXICKÁ LÁTKA
8 POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU
NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4HF Velká Británie
12. REGISTRAČNÍ CÍSLO(A)
EU/1/15/1071/001
13. ČÍSLO SARZE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato..
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Pemetrexed Accord 100 mg prášek pro koncentrát pro infuzní roztok
pemetrexedum
IV podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4 ČÍSLO ŠARŽE
Lot:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK 100 mg
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU
Pemetrexed Accord 500 mg prášek pro koncentrát pro infuzní roztok pemetrexedum
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jedna lahvička obsahuje pemetrexedum 500 mg (jako pemetrexedum dinatricum dihemihydricum) Po rekonstituci (viz příbalová informace) obsahuje jedna injekční lahvička pemetrexedum o koncentraci 25 mg/ml.
3. SEZNAM POMOCNÝCH LÁTEK
Mannitol, kyselina chlorovodíková, hydroxid sodný (viz příbalová informace)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro koncentrát pro infuzní roztok. 1 lahvička
5. ZPŮSOB A CESTA PODÁNÍ
Pouze pro jednorázové podání.
Intravenózní podání po rekonstituci a naředění. Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
CYTOTOXICKÁ LÁTKA
8 POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU
NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4HF Velká Británie
12. REGISTRAČNÍ CÍSLO(A)
EU/1/15/1071/002
13. ČÍSLO SARZE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato.
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Pemetrexed Accord 500 mg prášek pro koncentrát pro infuzní roztok pemetrexedum
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
5. ZPŮSOB A CESTA PODÁNÍ
Intravenózní podání po rekonstituci a naředění.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
CYTOTOXICKÁ LÁTKA
8. POUŽITELNOST
EXP:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Accord
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1071/002
Lot:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato.
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU
Pemetrexed Accord 1000 mg prášek pro koncentrát pro infuzní roztok pemetrexedum
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jedna lahvička obsahuje pemetrexedum 1000 mg (jako pemetrexedum dinatricum dihemihydricum) Po rekonstituci (viz příbalová informace) obsahuje jedna injekční lahvička pemetrexedum o koncentraci 25 mg/ml.
3. SEZNAM POMOCNÝCH LÁTEK
Mannitol, kyselina chlorovodíková, hydroxid sodný (viz příbalová informace)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro koncentrát pro infuzní roztok. 1 lahvička
5. ZPŮSOB A CESTA PODÁNÍ
Pouze pro jednorázové podání.
Intravenózní podání po rekonstituci a naředění. Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
CYTOTOXICKÁ LÁTKA
8 POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ
NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4HF Velká Británie
12. REGISTRAČNÍ CÍSLO(A)
EU/1/15/1071/003
13. ČÍSLO SARZE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato.
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Pemetrexed Accord 1000 mg prášek pro koncentrát pro infuzní roztok pemetrexedum
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
5. ZPŮSOB A CESTA PODÁNÍ
Intravenózní podání po rekonstituci a naředění.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
CYTOTOXICKÁ LÁTKA
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10 ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Accord
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1071/003
Lot:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16 INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Pemetrexed Accord 100 mg prášek pro koncentrát pro infuzní roztok Pemetrexed Accord 500 mg prášek pro koncentrát pro infuzní roztok Pemetrexed Accord 1000 mg prášek pro koncentrát pro infuzní roztok
pemetrexedum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu..
- Máte-li jakékoliv další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento lék byl předepsán pouze pro vás. Nepřenechávejte ho nikomu jinému. Mohl by mu uškodit, i když příznaky onemocnění mohou být stejné.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků , sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Pemetrexed Accord a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Pemetrexed Accord používat
3. Jak se přípravek Pemetrexed Accord používá
4. Možné nežádoucí účinky
5. Jak přípravek Pemetrexed Accord uchovávat
6. Obsah balení s další informace
1. Co je přípravek Pemetrexed Accord a k čemu se používá
Pemetrexed Accord je léčivý přípravek používaný k léčbě zhoubných nádorů.
Přípravek Pemetrexed Accord se podává pacientům bez předchozí chemoterapie v kombinaci s dalším protinádorovým lékem cisplatinou k léčbě maligního mezoteliomu pleury, což je forma zhoubného nádoru postihujícího výstelku dutiny hrudní a pokrývající plíce.
Přípravek Pemetrexed Accord se v kombinaci s cisplatinou podává také jako počáteční léčba u pacientů s pokročilými stadiemi rakoviny plic.
Přípravek Pemetrexed Accord Vám může být předepsán i pokud máte rakovinu plic v pokročilém stadiu a pokud Vaše onemocnění příznivě reagovalo na léčbu nebo zůstalo po počáteční chemoterapii převážně nezměněno.
Pemetrexed Accord je rovněž určena k léčbě pacientů s pokročilými stadiem rakoviny plic, u kterých došlo k dalšímu rozvoji onemocnění poté, co byla použita jiná úvodní chemoterapie.
2. Čemu musíte věnovat pozornost, než začnete přípravek Permetrexed Accord používat Nepoužívejte přípravek Pemetrexed Accord:
- jestliže j ste alergický(á) na pemetrexed nebo na kteroukoli další složku přípravku Pemetrexed Accord (uvedenou v bodě 6).
- pokud kojíte, musíte během léčby přípravkem Pemetrexed Accord přestat kojit.
- pokud jste nedávno byl(a) nebo máte být očkován(a) vakcínou proti žluté zimnici.
Upozornění a opatření
Před použitím přípravku Pemetrexed Accord se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
Pokud máte nebo j ste měl(a) problémy s ledvinami, oznamte to svému lékaři nebo nemocničnímu lékárníkovi, protože by nemuselo být vhodné, abyste dostával(a) přípravek Pemetrexed Accord.
Před každou infuzí Vám bude odebrána krev k vyšetření, zda máte v pořádku funkci ledvin a jater a ke kontrole, zda máte dostatečný počet krvinek, abyste mohl(a) dostat přípravek Pemetrexed Accord. Váš lékař se může rozhodnout změnit dávku nebo odložit léčbu v závislosti na Vašem celkovém zdravotním stavu a v případě, že máte příliš nízký počet krvinek. Pokud je Vám podána rovněž cisplatina, Váš lékař zkontroluje, zda jste dostatečně hydratován(a) a před léčbou cisplatinou a po ní dostanete vhodné léky, které zabrání zvracení.
Oznamte svému lékaři, pokud j ste podstoupil(a) nebo máte podstoupit léčbu ozařováním, protože může dojít k časné nebo opožděné reakci na ozařování při používání přípravku Pemetrexed Accord .
Oznamte svému lékaři, jestliže jste byl(a) nedávno očkován(a), protože pak může při používání přípravku Pemetrexed Accord dojít nežádoucím účinkům.
Oznamte svému lékaři, že máte nebo jste měl(a) srdeční onemocnění.
Pokud u Vás došlo k nahromadění tekutiny okolo plic, může se lékař rozhodnout před podáním přípravku Pemetrexed Accord tuto tekutinu odstranit.
Děti a dospívající
Použití přípravku Pemetrexed Accord u dětí a dospívajících není relevantní.
Další léčivé přípravky a přípravek Pemetrexed Accord
Informujte svého lékaře nebo nemocničního lékárníka o všech lécích, které užíváte nebo které jste v nedávné době užíval(a), jako jsou přípravky proti bolesti nebo zánětu (otokům), jako jsou tzv. nesteroidní protizánětlivé léky (NSA), včetně léků, které jsou volně prodejné bez lékařského předpisu (jako je např. ibuprofen). Existuje mnoho druhů NSA s různou dobou účinnosti. Na základě plánovaného data infuze přípravku Pemetrexed Accord a stavu funkce ledvin Vám lékař doporučí, které léky smíte užívat a kdy je smíte užívat. Pokud si nejste jistý(á), zeptejte se svého lékaře nebo lékárníka, zda některý z Vašich léků není NSA.
Stejně jako u jiných chemoterapeutik, není doporučeno kombinování přípravku Pemetrexed Accord s živými oslabenými vakcínami. Pokud možno mají být použity inaktivní vakcíny.
Těhotenství
Pokud jste těhotná, domníváte, že můžete být těhotná nebo plánujete otěhotnět oznamte to svému lékaři nebo lékárníkovi, než je Vám podán tento léčivý přípravek. Přípravku Pemetrexed Accord je třeba se v těhotenství vyvarovat. Váš lékař s Vámi probere možná rizika používání přípravku Pemetrexed Accord během těhotenství. Během léčby přípravkem Pemetrexed Accord musejí ženy používat vhodnou antikoncepci.
Kojení
Pokud kojíte, oznamte to svému lékaři.
Během léčby přípravkem Pemetrexed Accord se musí kojení přerušit.
Plodnost
Muži nemají počít dítě během léčby a po dobu 6 měsíců po skončení léčby přípravkem Pemetrexed Accord a z těchto důvodů mají během léčby přípravkem Pemetrexed Accord a po dobu 6 měsíců po jejím skončení používat účinnou antikoncepci. Pokud během léčby tímto přípravkem nebo během 6 měsíců po ukončení této léčby chcete počít dítě, poraďte se se svým lékařem nebo lékárníkem. Můžete vyhledat konzultaci ohledně uchování spermií před zahájením léčby.
Řízení dopravních prostředků a obsluha strojů
Přípravek Pemetrexed Accord může vyvolat únavu. Při řízení dopravních prostředků a obsluze strojů buďte opatrný(á).
Přípravek Pemetrexed Accord obsahuje sodík
Přípravek Pemetrexed Accord 100 mg obsahuje méně než 1 mmol sodíku (23 mg) na jednu injekční lahvičku, takže je v podstatě bez obsahu sodíku‘.
Přípravek Pemetrexed Accord 500 mg obsahuje přibližně 54 mg sodíku v jedné injekční lahvičce. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
Přípravek Pemetrexed Accord 1000 mg obsahuje přibližně 108 mg sodíku v jedné injekční lahvičce. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
3. Jak se přípravek Pemetrexed Accord používá
Přípravek Pemetrexed Accord používejte přesně podle pokynů svého lékaře nebo lékárníka. Nejste-li si jisti, poraďte se s lékařem nebo lékárníkem.
Doporučená dávka Pemetrexed Accord je 500 mg na jeden čtvereční metr plochy povrchu těla. Tato plocha se vypočítá z Vaší výšky a tělesné hmotnosti. Lékař poté z tohoto údaje vypočte potřebnou dávku. Tato dávka může být upravena, případně léčba může být oddálena v závislosti na počtu krvinek a celkovém zdravotním stavu. Než Vám bude přípravek podán, nemocniční lékárník, zdravotní sestra nebo lékař smísí prášek přípravku Pemetrexed Accord s injekčním roztokem chloridu sodného o koncentraci 0,9 mg/ml (0,9%).
Přípravek Pemetrexed Accord dostanete vždy v infuzi do žíly. Tato infuze bude trvat přibližně 10 minut.
Pokud dostanete přípravek Pemetrexed Accord s cisplatinou:
Lékař nebo nemocniční lékárník vypočítá potřebnou dávku na základě Vaší výšky a tělesné hmotnosti. Cisplatina se podává rovněž do žíly a podává se přibližně 30 minut po ukončení infuze přípravku Pemetrexed Accord. Infuze cisplatiny bude trvat přibližně 2 hodiny.
Infuze budete obvykle dostávat 1x za 3 týdny.
Další léky:
Kortikosteroidy: Váš lékař Vám předepíše tablety se steroidem (v dávce odpovídající 4 mg dexamethasonu dvakrát denně), které budete užívat den před léčbou přípravkem Pemetrexed Accord, v den jeho podání a následující den po jeho podání. Tento lék budete dostávat ke snížení frekvence a závažnosti kožních reakcí, které lze předpokládat během protinádorové léčby.
Doplňování léčby o vitaminy: během léčby přípravkem Pemetrexed Accord Vám Váš lékař předepíše užívat kyselinu listovou (vitamin) nebo multivitamin s obsahem kyseliny listové (350 až 1000 mikrogramů), který musíte užívat 1x denně. Během sedmi dní před první dávkou přípravku Pemetrexed Accord si musíte vzít nejméně 5 dávek kyseliny listové. Po poslední dávce přípravku Pemetrexed Accord musíte pokračovat 21 dní v užívání kyseliny listové. Dostanete rovněž injekci vitaminu B12 (1000 mikrogramů), a to v týdnu před podáním přípravku Pemetrexed Accord a dále přibližně každých 9 týdnů (což odpovídá 3 cyklům léčby přípravkem Pemetrexed Accord). Vitamin B12 a kyselinu listovou dostanete ke snížení možných toxických účinků protinádorové léčby.
Máte-li jakékoli další otázky, týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Možné nežádoucí účinky
4.
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Jestliže zaznamenáte jakýkoliv z níže uvedených nežádoucích účinků, musíte ihned kontaktovat svého lékaře:
• Horečka nebo infekce (časté): pokud máte teplotu 38 °C nebo vyšší, pocení nebo jiné známky infekce (protože můžete mít méně bílých krvinek než je obvyklé, což je velmi časté). Infekce (sepse) může mít závažný průběh a může vést k úmrtí.
• Pokud začnete pociťovat bolest na hrudi (časté) nebo máte rychlou srdeční frekvenci (méně časté).
• Pokud máte bolest, zarudnutí, otok nebo vřídky v ústech (velmi časté).
• Alergické reakce: pokud se vyvine kožní vyrážka (velmi časté), pocity pálení nebo brnění (časté), případně horečka (časté). Ve vzácných případech může být kožní reakce závažná a může vést k úmrtí. Obraťte se na svého lékaře, pokud se u Vás vyskytne závažná vyrážka nebo svědění anebo puchýře (Stevens-Johnsonův syndrom nebo toxická epidermální nekrolýza).
• Pokud pozorujete únavu, slabost, snadnější zadýchávání nebo jste bledý(á) (protože můžete mít méně krevního barviva hemoglobinu, než je obvyklé, což je velmi časté).
• Pokud pozorujete krvácení z dásní, nosu nebo úst, případně jiné krvácení, které se obtížně zastavuje, načervenalou nebo narůžovělou moč, neočekávanou tvorbu modřin (protože můžete mít nižší počet krevních destiček, než je obvyklé, což je velmi časté).
• Pokud pozorujete náhlou dušnost, intenzivní bolest na hrudi nebo kašel s vykašláváním krve (méně časté) (může to znamenat přítomnost krevní sraženiny v plicních cévách).
Nežádoucí účinky mohou zahrnovat:
Velmi časté (mohou se vyskytnout u více než 1 z 10 pacientů)
Snížení počtu bílých krvinek Nízké hladiny hemoglobinu (anemie)
Snížení počtu krevních destiček
Bolest, zarudnutí, otok nebo vřídky v ústech Pocit na zvracení Ztráta chuti k jídlu Únava
Kožní vyrážka Vypadávání vlasů Zácpa
Ztráta hmatového citu
Ledviny: abnormální nálezy při vyšetření krve
Časté (mohou se vyskytnout až u 1 z 10 pacientů)
Alergické reakce: kožní vyrážka/pocity pálení nebo brnění
Infekce včetně sepse
Dehydratace
Selhání ledvin
Podráždění kůže a svědění
Svalová slabost
Zánět spojivek
Žaludeční nevolnost
Změny vnímání chuti
Játra: abnormální nálezy při vyšetření krve
Zvýšené slzení
Méně časté (mohou se vyskytnout až u 1 ze 100 pacientů)
Náhlé selhání ledvin Zrychlení srdečního tepu
Zánět sliznice jícnu byl zaznamenán při kombinaci pemetrexedu a ozařování.
Kolitida (zánět tlustého střeva, který může být doprovázen střevním krvácením nebo krvácením z konečníku)
Intersticiální pneumonitida (zjizvení plicních sklípků).
Otoky (hromadění tekutin v tělesných tkáních vedoucím k otokům).
U některých pacientů se při léčbě pemetrexedem vyskytla srdeční příhoda, cévní mozková příhoda nebo malá mozková příhoda, obvykle při používání v kombinaci s další protinádorovou terapií. Pancytopenie-kombinované nízké počty bílých krvinek, červených krvinek a destiček U některých pacientů, kteří podstoupili před léčbou, v průběhu nebo po léčbě pemetrexedem ozařování, se může vyskytnout postradiační pneumonitida (zjizvení plicních sklípků v souvislosti s léčbou ozařováním).
Byly hlášeny případy bolesti, snížené teploty a změny zbarvení končetin.
Krevní sraženiny v plicních cévách (plicní embolie)
Vzácné (mohou se vyskytnout až u 1 z 1 000 pacientů)
Reakce zvaná „radiation recall“ (kožní vyrážka podobná výraznému spálení od sluníčka), která se objeví na kůži, která byla před delší dobou vystavena záření -dny až roky.
Výskyt puchýřů (zpuchýřující onemocnění kůže) - včetně Stevens-Johnsonova syndromu a toxické epidermální nekrolýzy
Hemolytická anemie vyvolaná imunitní reakcí (chudokrevnost způsobená rozpadem červených krvinek vyvolaným protilátkami)
Hepatitida (zánět jater)
Anafylaktický šok (těžká alergická reakce)
Může se u Vás vyskytnout jakýkoliv z těchto příznaků a/nebo onemocnění. Pokud zpozorujete některý z těchto nežádoucích účinků, musíte to oznámit svému lékaři, jakmile to bude možné.
Pokud se obáváte některých nežádoucích účinků, řekněte to svému lékaři.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Pemetrexed Accord uchovávat
Uchovávejte tento lék mimo dohled a dosah dětí.
Tento léčivý přípravek nepoužívejte po uplynutí doby použitelnosti vyznačené na obalu za zkratkou EXP.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Rekonstituovaný a infuzní roztok: Přípravek musí být použit okamžitě. Chemická a fyzikální stabilita přípravku po otevření před použitím byla prokázána na dobu 24 hodin při uchovávání v chladničce, pokud byl přípravek připraven dle návodu.
Tento léčivý přípravek nepoužívejte, jestliže si povšimnete jakýchkoli známek zhoršení kvality.
Tento léčivý přípravek je určen pouze pro jednorázové podání. Žádné léčivé přípravky nelikvidujte v odpadních vodách ani s domovním odpadem. Zeptejte se svého lékárníka, jak zlikvidovat léčivé přípravky, které již nepoužíváte. Tato opatření pomohou chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Pemetrexed Accord obsahuje
Léčivou látkou je pemetrexedum.
Pemetrexed Accord 100 mg: Jedna injekční lahvička obsahuje pemetrexedum 100 mg (jako pemetrexedum dinatricum dihemihydricum).
Pemetrexed Accord 500 mg: Jedna injekční lahvička obsahuje pemetrexedum 500 mg (jako pemetrexedum dinatricum dihemihydricum).
Pemetrexed Accord 1 000 mg: Jedna injekční lahvička obsahuje pemetrexedum 1 000 mg (jako pemetrexedum dinatricum dihemihydricum).
Po rekonstituci obsahuje roztok pemetrexed o koncentraci 25 mg/ml. Před podáním je nutné další naředění, které provede zdravotnický pracovník.
Pomocné látky jsou mannitol, kyselina chlorovodíková a hydroxid sodný (viz bod 2).
Jak přípravek Pemetrexed Accord vypadá a co obsahuje toto balení
Pemetrexed Accord je prášek pro koncentrát pro infuzní roztok v injekční lahvičce. Je to bílý až buď světle žlutý nebo zelenožlutý lyofilizovaný prášek.
Jedno balení přípravku Pemetrexed Accord obsahuje 1 injekční lahvičku.
Držitel rozhodnutí o registraci
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4HF Velká Británie
Výrobce
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4HF Velká Británie
Wessling Hungary Kft.
Foti út 56., Budapest, 1047,
Maďarsko
Tato příbalová informace byla naposledy revidována {MM/RRRR}.
Další zdroje informací
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Návod k použití přípravku, zacházení s ním a k jeho likvidaci
1. Při rekonstituci a dalším ředění pemetrexedu k podání intravenózní infuze používejte aseptickou techniku.
2. Vypočtěte dávku a počet potřebných injekčních lahviček přípravku Pemetrexed Accord.
Injekční lahvička obsahuje větší množství pemetrexedu k usnadnění přenosu označeného množství.
3. Pemetrexed Accord 100 mg:
Rekonstituujte obsah 100mg injekční lahvičky se 4,2 ml 0,9 % injekčního roztoku chloridu sodného (9 mg/m)l (bez konzervačních látek, čímž vznikne roztok obsahující pemetrexed o koncentraci 25 mg/ml.
Pemetrexed Accord 500 mg:
Rekonstituujte obsah 500mg injekční lahvičky s 20 ml 0,9 % injekčního roztoku chloridu sodného (9 mg/ml) bez konzervačních látek, čímž vznikne roztok obsahující pemetrexed o koncentraci 25 mg/ml.
Pemetrexed Accord 1 000 mg:
Rekonstituujte obsah 1 000mg injekční lahvičky s 40 ml 0,9 % injekčního roztoku chloridu sodného (9 mg/ml) bez konzervačních látek, čímž vznikne roztok obsahující pemetrexed o koncentraci 25 mg/ml.
Pohybujte jemným krouživým pohybem každou injekční lahvičkou, dokud se prášek zcela nerozpustí. Výsledný roztok je čirý a jeho barva kolísá od bezbarvé po žlutou nebo žlutozelenou, aniž by byla narušena jeho kvalita. pH rekonstituovaného roztoku se pohybuje mezi 6,6 a 7,8. Potřebné je další ředění.
4. Náležitý objem rekonstituovaného roztoku pemetrexedu musí být dále naředěn na 100 ml 0,9 % injekčním roztokem chloridu sodného (9 mg/ml)bez konzervačních látek a podá se intravenózní infuzí po dobu 10 minut.
5. Infuzní roztoky pemetrexedu. připravené podle návodu, j sou kompatibilní
s polyvinylchloridovými a polyolefinovými infuzními sety a infuzními vaky. Pemetrexed je inkompatibilní s rozpouštědly obsahujícími kalcium, jako je laktátový Ringerův roztok a Ringerův roztok.
6. Léčivé přípravky pro parenterální použití se musí před podáním vizuálně zkontrolovat, zda neobsahují částečky a nedošlo ke změně barvy. Jestliže zpozorujete pevné částice, přípravek nepodávejte.
7. Roztok pemetrexedu je určen pouze na jedno použití. Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Uchovávání
Chemická a fyzikální stabilita po otevření před použitím rekonstituovaného a infuzního roztoku pemetrexedu byla prokázána na dobu 24 hodin při teplotě 25 °C. Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně nemá být doba delší než 24 hodin při teplotě 2 °C -8 °C, pokud rekonstituce/ředění neproběhly za kontrolovaných a validovaných aseptických podmínek.
Bezpečnostní opatření při přípravě a podání přípravku Pemetrexed Accord: Tak jako i u jiných potenciálně toxických cytostatických látek je nutné udržovat pozornost při zacházení s infuzním roztokem pemetrexedu a při jeho přípravě. Doporučuje se používat ochranné rukavice. Pokud dojde ke kontaktu roztoku pemetrexedu s kůží, umyjte ihned a důkladně kůži mýdlem a vodou. Pokud dojde ke kontaktu roztoku pemetrexedu se sliznicemi, opláchněte je důkladně vodou. Pemetrexed není puchýřotvorná látka. V případě podání paravazálně neexistuje specifické antidotum. Bylo popsáno několik případů podání pemetrexedu paravazálně, které hodnotící lékař nepovažoval za závažné. Únik pemetrexedu mimo žílu se léčí podle místních standardních postupů jako u jiných nepuchýřotvorných ch látek.
49
Definice frekvence nežádoucích účinků: velmi časté - > 10 %; časté - > 5 % a < 10 %. Pro účely této tabulky byla použita hraniční hodnota 5 % pro zařazení všech příhod, kdy hlásící osoba považovala příhodu za možnou souvislost s léčbou pemetrexedem.
Viz NCI CTCAE kritéria (verze 3.0; NCI 2003) pro každý stupeň toxicity. Výskyt toxicit uveden podle CTCAE verze 3.0
V integrované tabulce nežádoucích účinků jsou kombinovány výsledky klinických hodnocení udržovací léčby pemetrexedem JMEN (n=663) a pokračující udržovací léčby pemetrexedem PARAMOUNT (n=539).
Souhrnné označení zahrnuje zvýšení kreatininu v séru/krvi, pokles glomerulární filtrace, renální selhání a renální/urogenitální- jiné.
Vypočtěte dávku a počet potřebných injekčních lahviček přípravku Pemetrexed Accord.
Injekční lahvička obsahuje větší množství pemetrexedu k usnadnění přenosu označeného množství.
FARMACEUTICKÉ ÚDAJE