Palladone-Sr 2 Mg
sp. zn. sukls135744/2016
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
PALLADONE-SR 2 mg PALLADONE-SR 4 mg PALLADONE-SR 8 mg PALLADONE-SR 16 mg PALLADONE-SR 24 mg tvrdé tobolky s prodlouženým uvolňováním
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Palladone-SR 2 mg: jedna tobolka obsahuje hydromorphoni hydrochloridum 2mg (což odpovídá hydromorphonum 1,78 mg)
Palladone-SR 4 mg: jedna tobolka obsahuje hydromorphoni hydrochloridum 4mg (což odpovídá hydromorphonum 3,56 mg)
Palladone-SR 8 mg: jedna tobolka obsahuje hydromorphoni hydrochloridum 8 mg (což odpovídá hydromorphonum 7,12 mg)
Palladone-SR 16 mg: jedna tobolka obsahuje mg hydromorphoni hydrochloridum 16 (což odpovídá hydromorphonum 14,24 mg)
Palladone-SR 24 mg: jedna tobolka obsahuje hydromorphoni hydrochloridum 24 mg (což odpovídá hydromorphonum 21,36 mg)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka s prodlouženým uvolňováním
Palladone-SR 2 mg: tvrdé želatinové tobolky, černý potisk HCR 2, vrchní část žlutá, spodní část bílá, neprůhledná, uvnitř bílé až téměř bílé kulovité částice o průměru 0,85-1,50 mm Palladone-SR 4 mg: tvrdé želatinové tobolky, černý potisk HCR 4, vrchní část světle modrá, spodní část bezbarvá, průhledná, uvnitř bílé až téměř bílé kulovité částice o průměru 0,85-1,50 mm Palladone-SR 8 mg: tvrdé želatinové tobolky, černý potisk HCR 8, vrchní část růžová, spodní část bezbarvá, průhledná, uvnitř bílé až téměř bílé kulovité částice o průměru 0,85-1,50 mm Palladone-SR 16 mg: tvrdé želatinové tobolky, černý potisk HCR 16, vrchní část hnědá, spodní část bezbarvá, průhledná, uvnitř bílé až téměř bílé kulovité částice o průměru 0,85-1,50 mm Palladone-SR 24 mg: tvrdé želatinové tobolky, černý potisk HCR 24, vrchní část tmavě modrá, spodní část bezbarvá, průhledná, uvnitř bílé až téměř bílé kulovité částice o průměru 0,85-1,50 mm
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba středně silné až silné bolesti.
4.2 Dávkování a způsob podání
Dávkování
Dávkování přípravku Palladone-SR se musí přizpůsobit intenzitě bolesti a individuálním reakcím pacienta.
Interval mezi jednotlivými dávkami by neměl klesnout pod 12 hodin. Při léčbě chronické bolesti je lépe dodržovat při dávkování předem definovaný časový rozvrh.
Dávky by se měly zvyšovat postupně, dokud není dosaženo dostatečného analgetického účinku. Všeobecně by se měla podávat dostatečně vysoká dávka a v každém případě nejnižší možná analgeticky účinná dávka.
Doba užívání
Přípravek Palladone-SR by neměl být podáván déle, než je nezbytně nutné. Pokud typ a intenzita choroby vyžaduje dlouhodobou léčbu, je třeba pečlivými a pravidelnými prohlídkami zjistit, zda a do jaké míry je pokračování léčby nezbytné. Pokud již není léčba opiáty déle nutná, doporučuje se snižovat denní dávku postupně, aby se předešlo abstinenčním příznakům.
Dospělí a dospívající starší 12 let
Počáteční dávka přípravku Palladone-SR je 4 mg každých 12 hodin. Dávka může být opatrně zvyšována v závislosti na intenzitě bolesti a přijatelné míře nežádoucích účinků.
Pediatrická populace
Nedoporučuje se podávat přípravek Palladone-SR dětem mladším 12 let. Bezpečnost a účinnost přípravku Palladone-SR u dětí do 12 let nebyla stanovena. Nejsou dostupné žádné údaje.
Starší pacienti
U starších pacientů může k dosažení adekvátního analgetického účinku dostačovat nižší dávka než u jiných dospělých.
Pacienti s poruchou funkce ledvin a jater
U pacientů s poruchou funkce ledvin a jater může být potřeba upravit dávku a pacienti by měli být během léčby monitorováni. Dávky by měly být pacientům pečlivě titrovány pro dosažení klinického účinku (viz bod 5.2).
U pacientů se závažnou poruchou funkce jater je přípravek Palladone-SR kontraindikován (viz bod 4.3).
Způsob podání Perorální podání.
Tobolky se polykají celé s dostatečným množstvím tekutiny bez rozkousání. Obsah tobolky nesmí být rozdrcen nebo injekčně aplikován, protože to může vést k rychlému uvolnění a vstřebání potenciálně smrtelné dávky hydromorfonu.
4.3 Kontraindikace
- hypersenzitivita na hydromorfon nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
- závažná respirační deprese s hypoxií nebo zvýšenou hladinou oxidu uhličitého,
- těžká chronická obstrukční plicní nemoc,
- akutní nebo závažné astma bronchiale,
- koma,
- paralytický ileus a jiné náhlé příhody břišní,
- závažná porucha funkce j ater
- současné užívání inhibitorů monoaminooxidázy nebo přerušení jejich užívání v posledních dvou týdnech.
4.4 Zvláštní upozornění a opatření pro použití
Hlavním rizikem předávkování opiáty je respirační deprese. Se zvláštní opatrností by se měl přípravek podávat pacientům se závislostí na opiáty a u pacientů s poraněním hlavy (kvůli riziku zvýšení intrakraniálního tlaku), konvulzivním onemocněním, alkoholismem, deliriem tremens, toxickou psychózou, hypotenzí s hypovolemií, poruchami vědomí, chorobami žlučového ústrojí, s kolikami žlučových nebo močových cest, zánětem slinivky břišní, obstrukčním nebo zánětlivým onemocněním střev, hypertrofií prostaty, nedostatečnou funkcí kůry nadledvinek (např. Addisonova choroba), hypotyeroidismem, chronickou obstrukční plicní nemocí, bronchiálním astmatem, sníženou respirační rezervou; u oslabených starších pacientů nebo u pacientů s poruchou funkce ledvin nebo jater (viz bod 4.2). U pacientů, kteří vyžadují zvýšenou pozornost, se doporučuje snížené dávkování.
Při dlouhodobém užívání přípravku může u pacientů vzniknout tolerance a k zachování léčebných účinků je třeba progresivně zvyšovat dávky. Může docházet ke zkřížené toleranci s jinými opiáty. Dlouhodobé užívání tohoto přípravku může vést k fyzické závislosti a v případě náhlého ukončení léčby k abstinenčním příznakům. Pokud již pacient nevyžaduje léčbu hydromorfonem, doporučuje se snižovat dávky postupně, aby se předešlo abstinenčním příznakům.
Hydromorfon vykazuje podobný profil jako jiné silné opiáty. Hydromorfon mohou proto vyhledávat a zneužívat osoby se skrytým i zjevným návykem. Existuje riziko rozvoje psychické závislosti (návyku) na opiátová analgetika, včetně hydromorfonu. U pacientů, kteří v minulosti trpěli alkoholismem či drogovou závislostí, je třeba Palladone-SR používat se zvláštní opatrností.
Ojediněle se může při podávání vysokých dávek hydromorfonu vyskytnout hyperalgézie bez odezvy na zvýšenou dávku. V tomto případě je třeba snížit dávku hydromorfonu nebo změnit opiát.
Tobolky s prodlouženým uvolňováním nebo jejich obsah (granule/pelety) musí být polknuty vcelku a nesmí se lámat, žvýkat ani drtit. Užití rozlámaných, rozžvýkaných nebo rozdrcených granulí hydromorfonu může vést k rychlému uvolnění a absorpci potenciálně smrtelné dávky hydromorfonu. (viz bod 4.9).
Souběžné užívání alkoholu a přípravku Palladone-SR může zvýšit nežádoucí účinky přípravku; je třeba se vyvarovat jejich souběžného užívání.
Přípravek Palladone-SR je určen pro perorální užití. Zneužití perorální dávky pro jiný druh aplikace může mít za následek vážné zdravotní potíže, které mohou být i smrtelné.
Přípravek Palladone-SR by se neměl užívat, pokud existuje riziko vzniku paralytického ileu. Vyskytne-li se během užívání hydromorfonu paralytický ileus nebo podezření na něj, je třeba léčbu okamžitě přerušit.
Užívání přípravku Palladone-SR se nedoporučuje v prvních 24 hodinách po operačním zákroku vzhledem k vysokému riziku neprůchodnosti střev v pooperační fázi ve srovnání s neoperovanými pacienty. Po uplynutí této doby, by se měl přípravek užívat opatrně zejména po operacích v oblasti břicha.
Pacienti před zákrokem za účelem dalšího potlačení bolesti (např. operace, blokáda plexu) by neměli užívat přípravek 12 hodin před tímto zákrokem. Pokud je indikována další léčba přípravkem Palladone-SR, dávkování by mělo být upraveno podle nových pooperačních požadavků.
Je třeba zdůraznit, že jakmile jsou pacienti jednou stabilizovaní na účinné dávce specifického opiátu, neměli by být převáděni na jiná opiátová analgetika bez provedení patřičných klinických testů a retitrace. Jinak není kontinuální analgetický účinek zajištěn.
Přípravky Palladone-SR 8 mg, 16mg a 24mg nejsou vhodné pro zahájení léčby opiáty. Tyto vyšší dávky přípravku Palladone-SR (8mg, 16mg nebo 24 mg) používejte pouze u pacientů, u nichž není v rámci léčby chronické bolesti dostatečná odezva na nižší dávky hydromorfonových preparátů (Palladone-SR 2 mg, Palladone-SR 4 mg) nebo jiných srovnatelně silných přípravků proti bolesti.
Při poruchách funkce kůry nadledvinek, by měla být sledována koncentrace kortisolu v plazmě a dle potřeby podávány kortikosteroidy.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Centrálně působící látky jako jsou trankvilizéry, anestetika, hypnotika a sedativa, neuroleptika, barbituráty, antidepresiva, antihistaminika/antiemetické látky a jiné opiáty mohou zvýšit tlumící účinek na CNS, který tyto látky mají, např. zklidnění, respirační deprese atd.
Alkohol může zesilovat farmakodynamické účinky přípravku Palladone-SR; je třeba se vyvarovat jejich souběžného užívání.
Je třeba se vyhnout užívání hydromorfonu současně s inhibitory monoaminooxidázy (IMAO) nebo v průběhu 14 dnů od jejich vysazení.
Nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
Nedoporučuje se podávat přípravek Palladone-SR v průběhu těhotenství nebo kojícím matkám. Těhotenství
Nejsou dostupné žádné klinické údaje o použití přípravku v těhotenství.
Nedoporučuje se podávat přípravek Palladone-SR během těhotenství a porodu. Může dojít k narušení kontraktility dělohy a riziku respirační deprese u novorozenců. Dlouhodobé užívání hydromorfonu během těhotenství může způsobit syndrom z vysazení léku u novorozenců.
Studie na zvířatech neprokázaly žádné teratogenní účinky při větších dávkách, než jsou očekávány u lidí (viz oddíl 5.3). Studie na zvířatech neprokázaly žádný vliv na plodnost a reprodukci při perorálně podávaných dávkách až ve výši 5 mg/kg/den. Perinatální toxicita byla zaznamenána u potkanů vystavených dávkám 2 a 5 mg/kg/den.
Kojení
Nejsou dostupné žádné klinické údaje o použití hydromorfonu během kojení. Kojící matky by neměly přípravek Palladone-SR užívat; je-li jeho užití nutné, doporučuje se kojení ukončit.
4.7 Vliv na řízení motorových vozidel a obsluhu strojů
Hydromorfon může ovlivnit schopnost řídit motorová vozidla a obsluhovat stroje. Toto je zejména pravděpodobné na začátku léčby hydromorfonem, při zvýšení dávky nebo změně léku a nebo pokud je hydromorfon kombinován s dalšími látkami ovlivňujícími CNS. Pacienti stabilizovaní na určité dávce, nemusí být již nadále omezováni. Pacienti by se proto měli poradit se svým lékařem, zda řízení motorových vozidel či obsluhu strojů povolí.
4.8 Nežádoucí účinky
Pro klasifikaci frekvence nežádoucích účinků se používá následující stupnice:
Velmi časté (> 1/10)
Časté (> 1/100 to < 1/10)
Méně časté (> 1/1,000 to < 1/100)
Vzácné (> 1/10,000 to < 1/1,000)
Velmi vzácné (<1/10,000)
Není známo (z dostupných údajů nelze určit)
Poruchy imunitního systému:
Není známo: anafylaktické reakce, reakce z přecitlivělosti (včetně orofaryngeálního otoku)
Poruchy metabolismu a výživy:
Časté: snížená chuť k jídlu
Psychiatrické poruchy:
Časté: úzkost, zmatenost, nespavost
Méně časté: agitovanost, deprese, euforická nálada, halucinace, noční děsy Není známo: léková závislost, dysforie
Poruchy nervového systému:
Velmi časté: závratě, ospalost Časté: bolest hlavy
Méně časté: třes, myoklonus, parestézie Vzácné: sedace, letargie
Není známo: konvulze, dyskineze, hyperalgezie (viz bod 4.4)
Poruchy oka:
Méně časté: poruchy zraku Není známo: mióza
Srdeční poruchy:
Vzácné: tachykardie, bradykardie, palpitace
Cévní poruchy Méně časté: hypotenze Není známo: zrudnutí
Respirační, hrudní a mediastinální poruchy:
Méně časté: dyspnoe
Vzácné: respirační deprese, bronchospasmus
Gastrointestinální poruchy:
Velmi časté: zácpa, nauzea Časté: bolest břicha, sucho v ústech, zvracení Méně časté: poruchy trávení, průjem, dysgeuzie Není známo: paralytický ileus
Poruchy jater a žlučových cest
Méně časté: zvýšená hladina jaterních enzymů
Vzácné: zvýšená hladina pankreatických enzymů
Poruchy kůže a podkožní tkáně:
Časté: pruritus, hyperhidróza Méně časté: vyrážka Není známo: urtikaria
Poruchy ledvin a močových cest:
Méně časté: zadržování moči a nucení na močení
Poruchy reprodukčního systému a prsu:
Méně časté: snížené libido, erektilní dysfunkce
Celkové poruchy a reakce v místě aplikace:
Časté: astenie
Méně časté: abstinenční syndrom*, únava, malátnost, periferní edém Není známo: léková tolerance, syndrom z vysazení léku u novorozenců
* Může se vyskytnout abstinenční syndrom, který zahrnuje symptomy jako neklid, úzkost, nervozitu, nespavost, hyperkinézii, třes a zažívací potíže.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www .sukl .cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Příznakem toxicity a předávkování hydromorfonem zahrnují zúžení zornic, bradykardii, respirační depresi, hypotenzi, ospalost vedoucí ke strnulosti a kómatu. V těžkých případech se může objevit cirkulační selhání končící i smrtí.
Čistý antagonista opioidů, jako je naloxon, je specifické antidotum proti příznakům předávkování opioidy. Doporučuje se podat nitrožilně 0,8 mg naloxonu. Pokud je třeba, opakovat ve 2-3 minutových intervalech nebo podat infúzi 2 mg naloxonu v 500 ml fyziologického roztoku nebo 5 % glukózy (0,004 mg/ml).
Rychlost infúze má korelovat s předcházejícím podáním bolusové dávky a má být v souladu s reakcí pacienta. Systém prodlouženého uvolňování může mít prodloužený účinek, který by měl být brán v úvahu.
Průchodnost dýchacích cest musí být zachována. Měly by se udržovat hladiny tekutin a elektrolytů.
V případě potřeby je třeba provést další podpůrná opatření.
Pacient by měl být pozorně sledován (alespoň 24 hodin), protože účinek opiátových antagonistů je kratší než účinek hydromorfonu, a proto lze očekávat opakované příznaky předávkování (např. respirační deprese).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: opioidní analgetika (anodyna), přírodní opiové alkaloidy ATC kód: N02A A03
Hydromorfon je p-selektivní, čistý opiátový agonista bez antagonistické aktivity. Hydromorfon a příbuzné opiáty mají největší účinek na centrální nervový systém a střevní trakt.
Účinky jsou zejména analgetické, anxiolytické, antitusické a sedativní. Dále se může vyskytnout ospalost, změny nálady, respirační deprese, snížení střevní motility, nevolnost, zvracení a změny v endokrinním a autonomním nervovém systému.
Endokrinní systém
Opiáty mohou ovlivnit osu hypotalamo-hypofyzo-adrenální nebo hypotalamo-hypofyzo-gonadální. Některé zřetelné změny zahrnují nárůst prolaktinu v séru a snížení kortisolu a testosteronu v plazmě. Klinické příznaky se mohou projevovat těmito hormonálními změnami.
Jiné farmakologické účinky
Předklinické studie naznačují různé účinky opiátů na složky imunitního systému; klinický význam těchto poznatků není znám.
5.2 Farmakokinetické vlastnosti
Absorpce
Hydromorfon je absorbován z gastrointestinálního traktu a podléhá presystémové eliminaci. Výše absolutní biologické dostupnosti je 36,4 % (C.I. 90%: 32,7 - 40,5 %) u přípravku Palladone-SR a 32,3 % (C.I. 90%: 29,0 - 35,9 %) pro roztok hydromorfonu při podání ústy. Relativní biologická dostupnost přípravku Palladone-SR je srovnatelná s běžným uvolňováním hydromorfon hydrochloridu, avšak s nižší fluktuací plazmatické hladiny. Maximální plazmatické hladiny (Cmax=1,2 ± 1,24 ng/ml) po požití přípravku Palladone-SR je dosaženo po 2 až 5 hodinách (Tmax =3 (2 - 5), po nichž následuje rozsáhlá fáze, kdy se terapeutická plazmatická hladina udržuje na konstantní úrovni po nejméně dalších 12 hodin.
2 -i
Palladon® retard 4 mg
— ■ Palladon® 2,6 mg hard capsules (normal release)
0
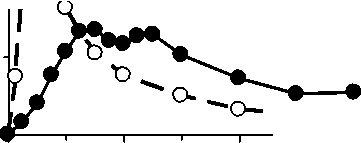
0 4 8 12 16 20 24
Distribuce
Vazba hydromorfonu na proteiny v plazmě je nízká (< 10 %) a zůstává konstantní počínaje nízkými plazmatickými koncentracemi od 2,46 ng/ml po velmi vysoké (81,99 ng/ml), jichž je dosaženo pouze při velmi vysokých dávkách hydromorfonu.
Hydromorfon hydrochlorid má relativně vysoký objem distribuce 1,22 + 0,23 l/kg (C.I.: 90%: 0,97 -1,60 l/kg, N=6 zdravých dobrovolníků mužského pohlaví). To svědčí o značné distribuci do tkání.
Z průběhu koncentrací jediné dávky hydromorfon hydrochloridu ve výši 2 mg i. v. nebo 4 mg perorálně podané nahodile 6 dobrovolníkům byl vypočten relativně krátký eliminační poločas rozpadu 2,64 ± 0,88 h (1,68 - 3,87 h).
Biotransformace
Hydromorfon je metabolizován přímou konjugací nebo redukcí ketonové funkce a následnou konjugací. Po absorpci je hydromorfon přeměněn zejména na hydromorfon-3-glukuronid, hydromorfon-3-glukosid a dihydroizomorfin-6-glukuronid. V menší míře jsou detekovány metabolity dihydroizomorfin-6-glukosid, dihydromorfin a dihydroizomorfin. Hydromorfon je metabolizován v játrech a vylučován ledvinami, v malém množství jako původní sloučenina.
Eliminace
Metabolity hydromorfonu byly detekovány v plazmě, moči a jaterními testy. Není potvrzeno, že je hydromorfon metabolizován in vivo prostřednictvím enzymatického systému Cytochrom P 450. In vitro je hydromorfon pouze v malé míře inhibován rekombinantními CYP-izoformami (IC50>50pM) včetně CYP1A2, 2A6, 2C8, 2D6 a 3A4. Proto se nepředpokládá, že hydromorfon inhibuje metabolismus jiných látek, které tyto CYP-izoformy metabolizují.
5.3 Předklinické údaje vztahující se k bezpečnosti
Reprodukční toxicita
U potkanů nebyly pozorovány žádné účinky na plodnost obou pohlaví nebo kvalitu spermií při perorálně podaných dávkách hydromorfonu až 5 mg/kg/den (30 mg/m2/den nebo 1,4 násobek očekávané dávky u lidí na základě specifického povrchu).
Hydromorfon nebyl teratogenní u březích samic potkanů ani králíků při podání perorálních dávek během hlavního období vývoje orgánů. Poruchy vývoje plodu byly pozorovány u králíků při 50 mg/kg (bez účinku na vývoj jsou dávky 25 mg/kg nebo 380mg/m2/den, AUC, přibližně 4 násobek dávky očekávané u lidí). Při perorálně podávaných dávkách hydromorfonu až 10 mg/kg (308 mg/m2/den, při AUC přibližně 1,8 násobku dávky očekávané u lidí) nebyla u potkanů a králíků pozorována toxicita pro plod. V literatuře byl zaznamenán průkaz teratogenních účinků u myší a křečků.
Prenatální a postnatální studie u potkanů prokázaly, že došlo k nárůstu úmrtnosti mláďat a snížení tělesné hmotnosti v časném postnatálním období ve spojitosti s mateřskou toxicitou. Nebyly pozorovány žádné účinky na pokračující vývoj mláďat nebo reprodukční schopnosti.
Kancerogenita
Hydromorfon nebyl genotoxický při testu mutace bakterií, v in vitro testu aberací na lidských lymfocytech a v in vivo mikronukleárním testu u myší, ale byl pozitivní v testu na buňkách lymfomu s metabolickou aktivací u myší. Podobná zjištění byla hlášena i u jiných opioidních analgetik.
Dlouhodobé studie týkající se kancerogenity nebyly prováděny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Palladone-SR 2 mg:
Obsah tobolky
Mikrokrystalická celulosa, hypromelosa 2910/15.
Potahová vrstva
Ethylcelulosa, koloidní bezvodý oxid křemičitý, dibutyl-sebakát.
Vrchní část tobolky
Chinolinová žluť (E104), oxid titaničitý (E171), natrium-lauryl-sulfát, želatina.
Potisk tobolky
Černý inkoust: šelak, černý oxid železitý (E172), propylenglykol
Palladone-SR 4 mg:
Obsah tobolky
Mikrokrystalická celulosa, hypromelosa 2910/15.
Potahová vrstva
Ethylcelulosa, koloidní bezvodý oxid křemičitý, dibutyl-sebakát.
Vrchní část tobolky
Erythrosin (E127), indigokarmín (E132), oxid titaničitý (E171), natrium-lauryl-sulfát, želatina.
Spodní část tobolky Natrium-lauryl-sulfát, želatina.
Potisk tobolky
Černý inkoust: šelak, černý oxid železitý (E172), propylenglykol
Palladone-SR 8 mg:
Obsah tobolky
Mikrokrystalická celulosa, hypromelosa 2910/15.
Potahová vrstva
Ethylcelulosa, koloidní bezvodý oxid křemičitý, dibutyl-sebakát.
Vrchní část tobolky
Erythrosin (E127), oxid titaničitý (E171), natrium-lauryl-sulfát, želatina
Spodní část tobolky Natrium-lauryl-sulfát, želatina.
Potisk tobolky
Černý inkoust: šelak, černý oxid železitý (E172), propylenglykol
Palladone-SR 16 mg:
Obsah tobolky
Mikrokrystalická celulosa, hypromelosa 2910/15.
Potahová vrstva
Ethylcelulosa, koloidní bezvodý oxid křemičitý, dibutyl-sebakát.
Vrchní část tobolky
Červený oxid železitý (E172), žlutý oxid železitý (E172), černý oxid železitý (E172), oxid titaničitý (E171), natrium-lauryl-sulfát, želatina.
Potisk tobolky
Černý inkoust: šelak, černý oxid železitý (E172), propylenglykol
Palladone-SR 24 mg:
Obsah tobolky
Mikrokrystalická celulosa, hypromelosa 2910/15.
Potahová vrstva
Ethylcelulosa, koloidní bezvodý oxid křemičitý, dibutyl-sebakát.
Vrchní část tobolky
Indigokarmín (E132), oxid titaničitý (E171), natrium-lauryl-sulfát, želatina.
Spodní část tobolky Natrium-lauryl-sulfát, želatina.
Potisk tobolky
Černý inkoust: šelak, černý oxid železitý (E172), propylenglykol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním vnitřním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Čirý bezbarvý blistr Al/PVdC/PVC, krabička.
Velikost balení: 10, 20, 28, 30, 40, 50, 56 a 60 tobolek.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Mundipharma Gesellschaft m.b.H.
Apollogasse 16-18 A-1070 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLA
PALLADONE - SR 2 mg: 65/117/03-C PALLADONE - SR 4 mg: 65/118/03-C PALLADONE - SR 8 mg: 65/119/03-C PALLADONE - SR 16 mg: 65/120/03-C PALLADONE - SR 24 mg: 65/121/03-C
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
2. 4. 2003/11.3.2015
10. DATUM REVIZE TEXTU
18.7.2016
Strana 12 (celkem 12)