Otezla 10 Mg+20 Mg+30 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Otezla 10 mg potahované tablety Otezla 20 mg potahované tablety Otezla 30 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje apremilastum 10 mg.
Jedna potahovaná tableta obsahuje apremilastum 20 mg.
Jedna potahovaná tableta obsahuje apremilastum 30 mg.
Pomocná látka/Pomocné látky se známým účinkem:
Jedna potahovaná tableta obsahuje 57 mg laktózy (ve formě monohydrátu laktózy). Jedna potahovaná tableta obsahuje 114 mg laktózy (ve formě monohydrátu laktózy). Jedna potahovaná tableta obsahuje 171 mg laktózy (ve formě monohydrátu laktózy).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Otezla 10 mg potahované tablety: Růžová 10mg potahovaná tableta ve tvaru kosočtverce, délka 8 mm, s vyrytým označením „APR“ na jedné straně a „10“ na druhé straně.
Otezla 20 mg potahované tablety: Hnědá 20mg potahovaná tableta ve tvaru kosočtverce, délka 10 mm, s vyrytým označením „APR“ na jedné straně a „20“ na druhé straně.
Otezla 30 mg potahované tablety: Béžová 30mg potahovaná tableta ve tvaru kosočtverce, délka 12 mm, s vyrytým označením „APR“ na jedné straně a „30“ na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Psoriatická artritida
Přípravek Otezla, samotný nebo v kombinaci s onemocnění modifikujícími antirevmatickými léky (DMARD), je indikován k léčbě aktivní psoriatické artritidy (PsA) u dospělých pacientů, kteří adekvátně neodpovídali nebo netolerovali předchozí léčbu DMARD (viz bod 5.1).
Psoriáza
Přípravek Otezla je indikován k léčbě středně těžké až těžké chronické ložiskové lupénky u dospělých pacientů, kteří neodpovídali nebo mají kontraindikovanou nebo netolerují jinou systémovou terapii, včetně cyklosporinu, methotrexátu nebo PUVA (kombinace psoralenu a UVA záření).
4.2 Dávkování a způsob podání
Léčba přípravkem Otezla má být zahájena odborným lékařem se zkušenostmi v diagnostice a léčbě psoriázy nebo psoriatické artritidy.
Dávkování
Doporučená dávka přípravku Otezla je 30 mg dvakrát denně perorálně, ráno a večer, v intervalu přibližně 12 hod, bez omezení přijmu potravin. Je nutné dodržovat plán úvodní titrace uvedený níže v Tabulce 1. Po úvodní titraci není nutná žádná další titrace.
Tabulka 1. Plán titrace dávek
|
1. den |
2. den |
3. den |
4. den |
5. den |
6. den a dále | |||||
|
DOP. |
DOP. |
ODP. |
DOP. |
ODP. |
DOP. |
ODP. |
DOP. |
ODP. |
DOP. |
ODP. |
|
10 mg |
10 mg |
10 mg |
10 mg |
20 mg |
20 mg |
20 mg |
20 mg |
30 mg |
30 mg |
30 mg |
Jestliže pacient zapomene užít dávku, měl by si ji vzít co nejdříve. Jestliže se však již blíží čas na další dávku, vynechaná dávka se nemá nahrazovat a má se užít další dávka v plánovaném čase.
V průběhu pivotních studií bylo největší zlepšení zaznamenáno během prvních 24 týdnů léčby.
U pacienta, který po 24 týdnech léčby nevykazuje žádné známky léčebného přínosu, je třeba léčbu přehodnotit. Odpověď pacienta na léčbu musí být pravidelně hodnocena. Klinické zkušenosti delší než 52 týdnů nejsou k dispozici (viz bod 5.1).
Zvláštní populace
Starší pacienti
U této skupiny pacientů není nutná úprava dávkování (viz body 4.8 a 5.2).
Pacienti s poruchou funkce ledvin
U pacientů s mírnou až středně závažnou poruchou funkce ledvin není nutná úprava dávkování.
U pacientů se závažnou poruchou funkce ledvin (clearance kreatininu méně než 30 ml/min dle Cockcroft-Gaultova vzorce) je třeba dávku apremilastu snížit na 30 mg jednou denně. Pro úvodní titraci dávky u této skupiny se doporučuje, aby byl přípravek Otezla titrován pouze s využitím dopoledních dávek uvedených v Tabulce 1 a aby byly odpolední dávky vynechány (viz bod 5.2).
Pacienti s poruchou funkce jater
U pacientů s poruchou funkce jater není nutná žádná úprava dávkování (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost apremilastu u dětí ve věku od 0 do 17 let nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Otezla je určen k perorálnímu podání. Potahované tablety se polykají celé a lze je užívat s jídlem nebo bez jídla.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku(y) nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Těhotenství (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Pacienti se vzácnými vrozenými poruchami, jako jsou intolerance galaktózy, vrozený deficit laktázy nebo malabsorpce glukózy-galaktózy, by neměli tento přípravek užívat.
U pacientů se závažnou poruchou funkce ledvin je třeba snížit dávku přípravku Otezla na 30 mg jednou denně (viz body 4.2 a 5 2).
U pacientů s podváhou na začátku léčby je třeba pravidelně kontrolovat tělesnou hmotnost. V případě nevysvětleného a klinicky významného váhového úbytku musí tyto pacienty vyšetřit lékař a je nutné zvážit ukončení léčby.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Souběžné podávání se silným induktorem enzymu cytochromu P450 3A4 (CYP3A4) rifampicinem vedlo ke snížení systémové expozice apremilastu, což může způsobit ztrátu účinnosti apremilastu. Proto se nedoporučuje současné užívání apremilastu se silnými induktory enzymu CYP3A4 (např. rifampicin, fenobarbital, karbamazepin, fenytoin a třezalka tečkovaná). Souběžné podávání apremilastu s opakovanými dávkami rifampicinu vedlo ke snížení plochy pod časovou křivkou plazmatické koncentrace apremilastu (AUC) přibližně o 72 % a ke snížení maximální koncentrace apremilastu v séru (Cmax) o 43 %. Expozice apremilastu se snižuje, pokud je přípravek podáván souběžně se silnými induktory CYP3A4 (např. rifampicinem), což může vést ke snížené klinické odpovědi.
V klinických studiích byl apremilast podáván souběžně s lokální léčbou (včetně kortikosteroidů, černouhelného dehtového šamponu a přípravků s kyselinou salicylovou určených k ošetření vlasové pokožky) a s léčbou UVB světlem.
Neobjevila se žádná klinicky významná léková interakce mezi ketokonazolem a apremilastem. Apremilast lze podávat souběžně s potentním inhibitorem CYP3A4, jakým je například ketokonazol.
U pacientů s psoriatickou artritidou nedošlo mezi apremilastem a methotrexátem k žádným farmakokinetickým lékovým interakcím. Apremilast lze podávat souběžně s methotrexátem.
Mezi apremilastem a perorální antikoncepcí obsahující ethinylestradiol a norgestimát nedošlo k žádným farmakokinetickým lékovým interakcím. Apremilast lze podávat souběžně s perorální antikoncepcí.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Před zahájením léčby je nezbytné vyloučit těhotenství. Ženy ve fertilním věku musí užívat účinnou metodu antikoncepce k zabránění otěhotnění po dobu léčby.
Údaje o podávání apremilastu těhotným ženám jsou omezené.
Apremilast je v těhotenství kontraindikován. U myší a opic byly pozorovány účinky apremilastu na těhotenství, a to včetně ztráty embrya/plodu a snížení hmotnosti plodu, dále opožděnou osifikaci u myší, při dávkách vyšších, než je současná doporučená maximální dávka u člověka. Žádné takové účinky nebyly pozorovány, byla-li expozice u zvířat 1,3krát vyšší než klinická expozice (viz bod 5.3).
Kojení
Apremilast byl zjištěn v mléce laktujících myších samic (viz bod 5.3). Není známo, zda se apremilast nebo jeho metabolity vylučují do lidského mateřského mléka. Riziko pro kojené děti nelze vyloučit, proto se podávání apremilastu během kojení nedoporučuje.
Fertilita
Údaje o fertilitě u lidí nejsou k dispozici. Při studiích na zvířatech nebyly pozorovány žádné nežádoucí účinky na fertilitu u myších samců při úrovni expozice odpovídající 3násobku klinické expozice a u samic při úrovni expozice odpovídající jednonásobku klinické expozice. Předklinické údaje o fertilitě viz bod 5.3.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Apremilast nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
V klinických studiích fáze III byly nejčastěji hlášenými nežádoucími účinky gastrointestinální poruchy včetně průjmu (15,7 %) a nevolnosti (13,9 %). Tyto gastrointestinální nežádoucí účinky byly většinou mírné až středně závažné, s 0,3 % průjmů a 0,3 % nevolností hlášených jako závažné. Tyto nežádoucí účinky se zpravidla vyskytovaly v prvních 2 týdnech léčby a obvykle vymizely do 4 týdnů. Další nejčastěji hlášené nežádoucí účinky zahrnovaly infekce horních cest dýchacích (8,4 %), bolest hlavy (7,9 %) a tenzní bolest hlavy (7,2 %). Většina nežádoucích účinků byla celkově považována za mírné až středně závažné.
K nejčastějším nežádoucím účinkům, které vedly k ukončení léčby v prvních 16 týdnech, patřil průjem (1,7 %) a nevolnost (1,5 %). Celková incidence závažných nežádoucích účinků byla nízká a nenaznačuje postižení specifického orgánového systému.
Reakce přecitlivělosti byly v klinických studiích apremilastu pozorovány méně často (viz bod 4.3). Seznam nežádoucích účinků v tabulce
Nežádoucí účinky pozorované u pacientů léčených apremilastem jsou uvedeny v tabulce níže podle třídy orgánových systémů a četnosti pro všechny nežádoucí účinky. V rámci každé třídy orgánových systémů a skupiny četnosti jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Nežádoucí účinky léku byly stanoveny na základě údajů získaných z programu klinického vývoje apremilastu. Uvedené četnosti nežádoucích účinků léku byly hlášeny ve větvích čtyř studií fáze III s apremilastem u PsA (n = 1945) nebo dvou studií fáze III s apremilastem u psoriázy (n = 1184) (nejvyšší četnost z obou souborů dat je uvedena v Tabulce 2).
Četnosti jsou definovány takto: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000).
Tabulka 2. Souhrn nežádoucích účinků u psoriatické artritidy (PsA) a/nebo psoriázy (PSOR)
|
Třída orgánového systému |
Četnost |
Nežádoucí účinek |
|
Infekce a infestace |
Časté |
Bronchitida |
|
Infekce horních cest dýchacích | ||
|
Nazofaryngitida* | ||
|
Poruchy imunitního systému |
Méně časté |
Přecitlivělost |
|
Třída orgánového systému |
Četnost |
Nežádoucí účinek |
|
Poruchy metabolismu a výživy |
Časté |
Snížená chuť k jídlu* |
|
Psychiatrické poruchy |
Časté |
Insomnie |
|
Poruchy nervového systému |
Časté |
Migréna* |
|
Tenzní bolesti hlavy* | ||
|
Bolest hlavy* | ||
|
Respirační, hrudní a mediastinální poruchy |
Časté | |
|
Gastrointestinální poruchy |
Velmi časté |
Průjem* |
|
Nevolnost* | ||
|
Časté |
Zvracení* | |
|
Časté vyprazdňování střev | ||
|
Bolest horní poloviny břicha* | ||
|
Gastroezofageální reflux | ||
|
Méně časté |
Gastrointestinální krvácení | |
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté |
Bolest zad* |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Únava |
|
Vyšetření |
Méně časté |
Snížení tělesné hmotnosti |
*Nejméně jeden z těchto nežádoucích účinků byl hlášen jako závažný.
Popis vybraných nežádoucích účinků
Pokles tělesné hmotnosti
V klinických studiích byla pravidelně zjišťována hmotnost pacientů. Průměrný váhový úbytek pozorovaný u pacientů léčených apremilastem po dobu až 52 týdnů byl 1,99 kg. Celkem u 14,3 % pacientů užívajících apremilast byl pozorován váhový úbytek 5 až 10 %, zatímco u 5,7 % pacientů užívajících apremilast byl pozorován váhový úbytek přesahující 10 %. Žádný z těchto pacientů neměl zjevné klinické následky váhového úbytku. Celkem u 0,1 % pacientů léčených apremilastem byla léčba přerušena v důsledku nežádoucího účinku snížení hmotnosti.
Viz dodatečné upozornění v bodě 4.4 pro pacienty, kteří trpí na začátku léčby podváhou.
Během placebem kontrolovaného období klinických studií fáze III u psoriázy byla u 1,2 % (14/1184) pacientů léčených apremilastem hlášena deprese ve srovnání s 0,5 % (2/418) pacientů léčených placebem. Žádné z těchto hlášení výskytu deprese nebylo závažné ani nevedlo k přerušení účasti ve studii.
Zvláštní populace
Starší pacienti
V klinických studiích nebyly celkově pozorovány žádné rozdíly v bezpečnostním profilu u starších pacientů ve věku > 65 let a mladších dospělých pacientů ve věku < 65 let.
Pacienti s poruchou funkce jater
Bezpečnost apremilastu v léčbě psoriatické artritidy a psoriázy u pacientů s poruchou funkce jater nebyla hodnocena.
Pacienti s poruchou funkce ledvin
V klinických studiích PsA nebo PSOR byl u pacientů s mírnou poruchou funkce ledvin pozorován bezpečnostní profil srovnatelný s pacienty s normální funkcí ledvin. Bezpečnost apremilastu
u pacientů s PsA nebo PSOR se středně závažnou nebo závažnou poruchou funkce ledvin nebyla v klinických studiích hodnocena.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Apremilast byl hodnocen u zdravých subjektů, jimž byla podávána maximální celková denní dávka 100 mg (podávaná jako 50 mg dvakrát denně) po dobu 4,5 dne, aniž by se prokázala dávku limitující toxicita. V případě předávkování se doporučuje u pacienta sledovat jakékoli známky nebo příznaky nežádoucích účinků a zahájit odpovídající symptomatickou léčbu. V případě předávkování se doporučují symptomatická a podpůrná léčebná opatření.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: imunosupresiva, selektivní imunosupresiva, ATC kód: L04AA32 Mechanismus účinku
Apremilast, perorální inhibitor fosfodiesterázy 4 (PDE4) s malou molekulou, působí intracelulárně a moduluje síť prozánětlivých a protizánětlivých mediátorů. PDE4 je PDE specifická pro cyklický adenosin monofosfát (cAMP) a dominantní PDE v zánětlivých buňkách. Inhibice PDE4 zvyšuje intracelulární hladiny cAMP, což omezuje zánětlivou odpověď díky modulaci exprese TNF-a, IL-23, IL-17 a jiných zánětlivých cytokinů. Cyklický AMP také moduluje hladiny protizánětlivých cytokinů jako například IL-10. Tyto prozánětlivé a protizánětlivé mediátory byly implikovány u psoriatické artritidy a psoriázy.
Farmakodynamické účinky
V klinických studiích u pacientů s psoriatickou artritidou apremilast významně moduloval, ale ne zcela inhiboval plazmatické hladiny proteinů IL-1a, IL-6, IL-8, MCP-1, MIP-1P, MMP-3 a TNF-a. Po 40 týdnech léčby apremilastem došlo ke snížení plazmatické hladiny proteinů IL-17 a IL-23 a zvýšení u IL-10. V klinických studiích u pacientů s psoriázou způsobil apremilast zmenšení tloušťky epidermu kůže poškozené lézí, infiltraci zánětlivých buněk a expresi prozánětlivých genů, včetně genů pro inducibilní syntázu oxidu dusnatého (iNOS), IL-12/IL-23p40, IL-17A, IL-22 a IL-8.
Apremilast podávaný v dávkách až do 50 mg dvakrát denně neměl vliv na délku QT intervalu u zdravých subjektů.
Psoriatická artritida
Účinnost a bezpečnost apremilastu byla hodnocena ve 3 multicentrických, randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích (studie PALACE 1, PALACE 2 a PALACE 3) s podobným uspořádáním, do nichž byli zařazeni dospělí pacienti s aktivní PsA (> 3 oteklé klouby a > 3 bolestivé klouby) i přes předchozí léčbu biologickými DMARD nebo DMARD s malou molekulou. Celkem 1493 pacientů bylo randomizováno a léčeno buď placebem, apremilastem 20 mg, nebo apremilastem 30 mg podávanými perorálně dvakrát denně.
Pacientům v těchto studiích byla PsA diagnostikována minimálně před 6 měsíci. Při zařazení do studie PALACE 3 byla navíc vyžadována jedna aktivní psoriatická kožní léze (alespoň 2 cm v průměru). Apremilast byl podáván v monoterapii (34,8 %) nebo v kombinaci se stabilními dávkami DMARD s malou molekulou (65,2 %). Pacienti dostávali apremilast v kombinaci s jednou látkou nebo několika z těchto látek: methotrexát (MTX, < 25 mg/týden, 54,5 %), sulfasalazin (SSZ, < 2 g/den, 9,0 %) a leflunomid (LEF, < 20 mg/den, 7,4 %). Souběžná léčba s biologickými DMARD včetně blokátorů TNF nebyla povolena. Tyto tři studie zahrnovaly pacienty se všemi podtypy PsA, včetně symetrické polyartritidy (62,0 %), asymetrické oligoartritidy (26,9 %), artritidy distálního interfalangeálního kloubu (6,2 %), mutilující artritidy (2,7 %) a predominující spondylitidy (2,1 %). Byli zahrnuti pacienti s preexistující entezopatií (63 %) nebo preexistující daktylitidou (42 %). Celkem 76,4 % pacientů bylo dříve léčeno pouze DMARD s malou molekulou a 22,4 % pacientů bylo dříve léčeno biologickými DMARD, což zahrnuje 7,8 % pacientů, u nichž byla předchozí léčba biologickým DMARD neúspěšná. Medián doby trvání onemocnění PsA byl 5 let.
Podle uspořádání studie byli pacienti, u nichž se počet bolestivých a oteklých kloubů nezlepšil alespoň o 20 % v 16. týdnu, považováni za pacienty nereagující na léčbu. U pacientů užívajících placebo, kteří byli považováni za pacienty nereagující na léčbu, byla provedena opakovaná zaslepená randomizace a v poměru 1: 1 byli zařazeni buď do skupiny léčené 20 mg apremilastu dvakrát denně, nebo do skupiny léčené 30 mg dvakrát denně. Ve 24. týdnu přešli všichni zbývající pacienti léčení placebem na léčbu buď 20 mg, nebo 30 mg apremilastu dvakrát denně.
Primárním cílovým parametrem účinnosti studie bylo procento pacientů dosahujících odpovědi ACR 20 (20% zlepšení dle kritérií American College of Rheumatology) v 16. týdnu.
Léčba apremilastem vedla ve srovnání s placebem k významnému zlepšení známek a příznaků PsA v 16. týdnu (dle hodnocení parametru odpovědi ACR 20). Podíly pacientů s ACR 20/50/70 (odpovědi ve studiích PALACE 1, PALACE 2 a PALACE 3 a souhrnné údaje ze studií PALACE 1, PALACE 2 a PALACE 3) u apremilastu 30 mg dvakrát denně v 16. týdnu jsou uvedeny v Tabulce 3. Odpovědi ACR 20/50/70 přetrvávaly v 24. týdnu.
Mezi pacienty, kteří byli původně randomizováni k léčbě apremilastem 30 mg dvakrát denně, byly četnosti odpovědí ACR 20/50/70 udrženy až do 52. týdne v souboru studií PALACE 1, PALACE 2 a PALACE 3 (Obrázek 1).
Tabulka 3. Podíl pacientů s ACR odpověďmi ve studiích PALACE 1, PALACE 2 a PALACE 3 a v souboru studií v 16. týdnu
|
PALACE1 |
PALACE 2 |
PALACE 3 |
SOUBOR STUDIÍ | |||||
|
Placebo |
Apremilast |
Placebo |
Apremilast |
Placebo |
Apremilast |
Placebo |
Apremilast | |
|
+/- |
30 mg BID |
+/- |
30 mg BID |
+/- |
30 mg BID |
+/- |
30 mg BID | |
|
DMARD |
+/- DMARD |
DMARD |
+/- DMARD |
DMARD |
+/- DMARD |
DMARD |
+/- DMARD | |
|
Na |
N = 168 |
N = 168 |
N = 159 |
N = 162 |
N = 169 |
N = 167 |
N = 496 |
N = 497 |
|
ACR 20a | ||||||||
|
16. týden |
19,0 % |
38,1 %** |
18,9 % |
32,1 %* |
18,3 % |
40,7 %** |
18,8 % |
37,0 %** |
|
ACR 50 | ||||||||
|
16. týden |
6,0 % |
16,1 %* |
5,0 % |
10,5 % |
8,3 % |
15,0 % |
6,5 % |
13,9 %** |
|
ACR 70 | ||||||||
|
16. týden |
1,2 % |
4,2 % |
0,6 % |
1,2 % |
2,4 % |
3,6 % |
1,4 % |
3,0 % |
*p < 0,01 pro apremilast vs. placebo.
**p < 0,001 pro apremilast vs. placebo. a N je počet randomizovaných a léčených pacientů.
Obrázek 1 Podíl pacientů s odpovědí ACR 20/50/70 až do 52. týdne v souhrnné analýze studií PALACE 1, PALACE 2 a PALACE 3 (NRI*)
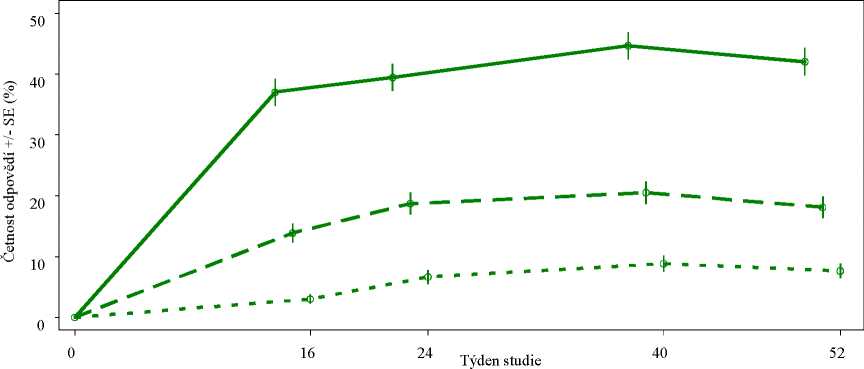
Cílový parametr
ACR 20 ACR 50 ACR 70
n/m (%) n/m (%)
184/497 (37,0) 196/497 (39,4)
69/497 (13,9) 93/497 (18,7)
15/497 ( 3,0) 33/497 ( 6,6)
n/m (%) 222/497 (44,7) 102/497 (20,5) 44/497 ( 8,9)
n/m (%) 209/497 (42,1) 90/497 (18,1) 38/497 ( 7,6)
Cílový parametr ACR 20 ■» ACR 50 e o o ACR 70
*NRI: imputace nereagujících subjektů. Za nereagující jsou považovány subjekty, které předčasně ukončily léčbu ještě před daným časovým bodem, a subjekty, u nichž nebyly v daném časovém bodě dostatečné údaje pro jednoznačné vyhodnocení odpovědi.
Z 497 pacientů původně randomizovaných do skupiny léčené 30 mg apremilastu dvakrát denně, bylo v 52. týdnů 375 (75 %) pacientů stále v léčení. Odpovědí ACR 20/50/70 bylo v 52. týdnu dosaženo u 57/25/11 % těchto pacientů.
Odpovědi pozorované u skupiny léčené apremilastem byly obdobné u pacientů, kteří souběžně užívali a neužívali DMARD, včetně methotrexátu. Pacienti dříve léčení DMARD nebo biologickými léky, kteří užívali apremilast, dosáhli v 16. týdnu častější odpovědi ACR 20 než pacienti užívající placebo.
Obdobné ACR odpovědi byly pozorovány u pacientů s různými podtypy PsA, včetně artritidy distálního interfalangeálního kloubu. Počet pacientů s mutilující artritidou a s podtypy predominující spondylitidy byl příliš malý, aby mohlo být provedeno smysluplné hodnocení.
Ve studiích PALACE 1, PALACE 2 a PALACE 3 bylo v 16. týdnu zaznamenáno významnější zlepšení hladiny C-reaktivního proteinu (CRP) dle stupnice aktivity onemocnění (DAS) 28 a podílu pacientů dosahujících modifikovaných kritérií léčebné odpovědi PsA (PsARC) u apremilastu oproti placebu (nominální p-hodnoty a p < 0,0004 a p-hodnota < 0,0017, v uvedeném pořadí). Tato zlepšení přetrvávala ve 24. týdnu. U pacientů, kteří pokračovali v léčbě apremilastem, k níž byli randomizováni na počátku studie, byly skóre DAS28 (CRP) a odpověď PsARC udrženy až do 52. týdne.
V 16. a 24. týdnu bylo u pacientů léčených apremilastem pozorováno zlepšení v parametrech periferních znaků aktivity psoriatické artritidy (jako je počet oteklých kloubů, počet bolestivých/citlivých kloubů, daktylitida a entezitida) a v kožních projevech psoriázy. U pacientů, kteří pokračovali v léčbě apremilastem, k níž byli randomizováni na počátku studie, byla tato zlepšení udržena až do 52. týdne.
Tělesné funkce a kvalita života související se zdravím
Ve studiích PALACE 1, PALACE 2 a PALACE 3 a souboru studií vykazovali pacienti léčení apremilastem v porovnání s placebem v 16. týdnu statisticky významné zlepšení tělesných funkcí hodnocené podle změny oproti výchozímu indexu postižení dle dotazníku pro posuzování zdravotního stavu (HAQ-DI). Zlepšení skóre HAQ-DI bylo udrženo v 24. týdnu.
Ve sdružené analýze otevřené fáze studií PALACE 1, PALACE 2 a PALACE 3 byla u pacientů, kteří byli původně randomizováni k léčbě 30 mg apremilastu dvakrát denně, v 52. týdnu změna skóre HAQ-DI oproti výchozí hodnotě -0,333 ve skupině léčené 30 mg apremilastu dvakrát denně.
Ve studiích PALACE 1, PALACE 2 a PALACE 3 bylo v 16. a 24. týdnu u pacientů léčených apremilastem ve srovnání s placebem zaznamenáno významné zlepšení kvality života související se zdravím měřené podle změny tělesných funkcí oproti výchozímu stavu, které byly hodnoceny pomocí krátkého dotazníku pro posuzování zdravotního stavu, verze 2 (SF-36v2), a změny skóre únavy dle dotazníku pro funkční hodnocení léčby chronického onemocnění (FACIT-f). U pacientů, kteří pokračovali v léčbě apremilastem, k níž byli randomizováni na počátku studie, byla zlepšení v parametru tělesných funkcí a skóre FACIT-f udržena až do 52. týdne.
Psoriáza
Bezpečnost a účinnost apremilastu byla hodnocena ve dvou multicentrických, randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích (studie ESTEEM 1 a ESTEEM 2). Tyto studie zahrnovaly celkem 1257 pacientů se středně těžkou až těžkou ložiskovou psoriázou na ploše > 10 % povrchu těla, se skóre oblasti psoriázy a indexu závažnosti (PASI ) > 12 a se statickým celkovým hodnocením lékařem (sPGA) > 3 (středně těžké nebo těžké), kteří byli kandidáty na léčbu světlem nebo systémovou terapii.
Tyto studie měly podobné uspořádání až do 32. týdne. V obou studiích byli pacienti randomizováni v poměru 2:1 do skupiny užívající 30 mg apremilastu dvakrát denně a do skupiny užívající placebo po dobu 16 týdnů (placebem kontrolovaná fáze), v 16.-32. týdnu užívali všichni pacienti 30 mg apremilastu dvakrát denně (udržovací fáze). Během randomizované fáze s vysazením léčby (32. až 52. týden) byli pacienti původně randomizovaní do skupiny užívající apremilast, kteří dosáhli alespoň 75% snížení skóre PASI (PASI-75) (ESTEEM 1) nebo 50% snížení skóre PASI (PASI-50)
(ESTEEM 2), znovu randomizováni v 32. týdnu buď do skupiny užívající placebo, nebo do skupiny užívající 30 mg apremilastu dvakrát denně. Pacienti, kteří byli znovu randomizováni do skupiny užívající placebo a kteří v 32. týdnu ztratili odpověď PASI-75 (ESTEEM 1) nebo ztratili 50% zlepšení skóre PASI oproti výchozím hodnotám (ESTEEM 2), byli převedeni na léčbu 30 mg apremilastu dvakrát denně. Pacienti, kteří nedosáhli stanovené odpovědi PASI do 32. týdne nebo kteří byli původně randomizováni do skupiny užívající placebo, užívali apremilast až do 52. týdne. Podávání slabých lokálních kortikosteroidů na tváři, v podpaží a tříslech, černouhelných dehtových šamponů a/nebo přípravků s kyselinou salicylovou určených k ošetření vlasové pokožky bylo po celou dobu studií povoleno. Ve 32. týdnu bylo subjektům, které nedosáhly odpovědi PASI-75 ve studii
ESTEEM 1 nebo odpovědi PASI-50 ve studii ESTEEM 2, kromě léčby 30 mg apremilastu dvakrát denně navíc povoleno používat lokální léčbu psoriázy a/nebo léčbu světlem.
V obou studiích byl primárním cílovým parametrem účinnosti podíl pacientů, kteří v 16. týdnu dosáhli PASI-75. Hlavním sekundárním cílovým parametrem byl podíl pacientů, kteří v 16. týdnu dosáhli čistého (0) nebo minimálního (1) skóre sPGA.
Průměrné výchozí skóre PASI bylo 19,07 (medián 16,80), podíl pacientů s výchozím skóre sPGA 3 (středně těžké) a 4 (těžké) byl 70,0 %, resp. 29,8 % s průměrnou výchozí plochou povrchu těla 25,19 % (medián 21,0 %). Přibližně 30 % všech pacientů podstoupilo při léčbě psoriázy předchozí léčbu světlem a 54 % podstoupilo předchozí konvenční systémovou terapii a/nebo biologickou léčbu (včetně selhání léčby), přičemž 37 % předchozí konvenční systémovou terapii a 30 % předchozí biologickou léčbu. Přibližně jedna třetina pacientů nepodstoupila předchozí léčbu světlem, konvenční systémovou terapii ani biologickou léčbu. Celkem 18 % pacientů mělo psoriatickou artritidou v anamnéze.
Podíl pacientů dosahujících odpovědí PASI-50, -75 a -90 a čistého (0) nebo minimálního (1) skóre sPGA je uveden v Tabulce 4 níže. Léčba apremilastem vedla v porovnání s placebem k významnému zlepšení středně těžké až těžké ložiskové psoriázy, jak bylo prokázáno podílem pacientů s odpovědí PASI-75 v 16. týdnu. V 16. týdnu byla rovněž prokázána klinická zlepšení měřená podle odpovědí sPGA, PASI-50 a PASI-90. Přínos léčby apremilastem byl navíc prokázán na několika projevech psoriázy, včetně pruritu, onemocnění nehtu, postižení vlasové pokožky a měření kvality života.
Tabulka 4. Klinická odpověď v 16. týdnu studií ESTEEM 1 a ESTEEM 2 (FAS a LOCFb)
|
ESTEEM 1 |
ESTEEM 2 | |||
|
Placebo |
30 mg BID APR* |
Placebo |
30 mg BID APR* | |
|
N |
282 |
562 |
137 |
274 |
|
PASIc 75, n (%) |
15 (5,3) |
186 (33,1) |
8 (5,8) |
79 (28,8) |
|
sPGAd čisté nebo minimální, n (%) |
11 (3,9) |
122 (21,7) |
6 (4,4) |
56 (20,4) |
|
PASI 50, n (%) |
48 (17,0) |
330 (58,7) |
27 (19,7) |
152 (55,5) |
|
PASI 90, n (%) |
1 (0,4) |
55 (9,8) |
2 (1,5) |
24 (8,8) |
|
Procentuální změna BSAe (%) průměrná hodnota ± SD |
-6,9 |
-47,8 |
-6,1 |
-48,4 |
|
±38,95 |
±38,48 |
±47,57 |
±40,78 | |
|
Změna pruritu VASf(mm), |
-7,3 |
-31,5 |
-12,2 |
-33,5 |
|
průměrná hodnota ± SD |
±27,08 |
±32,43 |
±30,94 |
±35,46 |
|
Změna v DLQIg, průměrná |
-2,1 |
-6,6 |
-2,8 |
-6,7 |
|
hodnota ± SD |
±5,69 |
±6,66 |
±7,22 |
±6,95 |
|
Změna v SF-36 MCS h, |
-1,02 |
2,39 |
0,00 ± |
2,58 |
|
průměrná hodnota ± SD |
±9,161 |
±9,504 |
10,498 |
±10,129 |
* p < 0,0001 pro apremilast vs. placebo, s výjimkou PASI 90 ve studii ESTEEM 2 a změny v SF-36 MCS, kde p = 0,0042, resp. p = 0,0078. a FAS = celkový analyzovaný soubor b LOCF= poslední provedené sledování c PASI = skóre oblasti psoriázy a indexu závažnosti d sPGA = statické celkové hodnocení lékaře e BSA = plocha povrchu těla
f VAS = vizuální analogová stupnice; 0 = nejlepší, 100 = nejhorší g DLQI = index dermatologické kvality života; 0 = nejlepší, 30 = nejhorší
h SF-36 MCS = krátký 36položkový dotazník pro posuzování zdravotního stavu ve studii, souhrn otázek týkajících se duševního zdraví
Klinický přínos apremilastu byl prokázán na několika podskupinách definovaných na základě výchozích demografických údajů a výchozích charakteristik klinického onemocnění (včetně doby trvání psoriázy a pacientů s anamnézou psoriatické artritidy). Klinický přínos apremilastu byl také prokázán bez ohledu na předchozí medikaci k léčbě psoriázy a odpověď na předchozí léčbu psoriázy. Ve všech rozmezích hmotnosti byly pozorovány obdobné četnosti odpovědí.
Odpověď na apremilast v porovnání s placebem byla do 2. týdne rychlá, s významně většími zlepšeními známek a příznaků psoriázy, včetně PASI, kožních obtíží/bolesti a pruritu. Odpovědi PASI byly zpravidla dosaženy do 16. týdne a byly udrženy až do 32. týdne.
V obou studiích zůstalo průměrné procentuální zlepšení PASI oproti výchozím hodnotám stabilní během randomizované fáze s vysazením léčby u pacientů, kteří byli v 32. týdnu znovu randomizováni do skupiny léčené apremilastem (Tabulka 5).
Tabulka 5. Přetrvávání účinku u subjektů randomizovaných do APR 30 dvakrát denně na počátku studie a znovu randomizovaných do APR 30 dvakrát denně v 32.až 52.týdnu
|
Časový bod |
ESTEEM 1 |
ESTEEM 2 | |
|
Pacienti, kteří dosáhli PASI-75 v 32. týdnu |
Pacienti, kteří dosáhli PASI-50 v 32. týdnu | ||
|
Procentní změna PASI oproti výchozí hodnotě, průměrná hodnota (%) ± SDa |
16. týden |
-77,7 ± 20,30 |
-69,7 ± 24,23 |
|
32. týden |
-88 ± 8,30 |
-76,7 ± 13,42 | |
|
52. týden |
-80,5 ± 12,60 |
-74,4 ± 18,91 | |
|
Změna DLQI oproti výchozí hodnotě, průměrná hodnota (%)± SDa |
16. týden |
-8,3 ± 6,26 |
-7,8 ± 6,41 |
|
32. týden |
-8,9 ± 6,68 |
-7,7 ± 5,92 | |
|
52. týden |
-7,8 ± 5,75 |
-7,5 ± 6,27 | |
|
Podíl subjektů s psoriázou v oblasti vlasové pokožky PGA (ScPGA) 0 nebo 1, n/N (%)b |
16. týden |
40/48 (83,3) |
21/37 (56,8) |
|
32. týden |
39/48 (81,3) |
27/37 (73,0) | |
|
52. týden |
35/48 (72,9) |
20/37 (54,1) |
a Zahrnuje subjekty znovu randomizované do APR 30 dvakrát denně v 32. týdnu s výchozí hodnotou a následnou hodnotou v hodnoceném týdnu studie.
bN vychází ze subjektů se středně těžkou nebo těžší psoriázou v oblasti vlasové pokožky na počátku studie, které byly v 32. týdnu znovu randomizovány do APR 30 dvakrát denně. Subjekty s chybějícími údaji byly považovány za subjekty nereagující na léčbu.
V 52. týdnu studie ESTEEM 1 mělo odpověď PASI-75 přibližně 61 % pacientů, kteří byli v 32. týdnu znovu randomizovaní do skupiny léčené apremilastem. Z pacientů dosahujících alespoň odpovědi PASI-75, kteří byli v 32. týdnu znovu randomizováni do skupiny užívající placebo v průběhu randomizované fáze s vysazením léčby, jich dosáhlo v 52. týdnu 11,7 % odpovědi PASI-75. Střední doba do ztráty odpovědi PASI-75 mezi pacienty, kteří byli znovu randomizováni do skupiny užívající placebo, byl 5,1 týdne.
Ve studii ESTEEM 2 mělo v 52. týdnu odpověď PASI-50 přibližně 80,3 % pacientů, kteří byli v 32. týdnu znovu randomizováni do skupiny léčené apremilastem. Z pacientů dosahujících alespoň odpovědi PASI-50, kteří byli v 32. týdnu znovu randomizováni do skupiny užívající placebo, jich dosáhlo v 52. týdnu 24,2 % odpovědi PASI-50. Střední doba do ztráty 50 % zlepšení PASI v 32. týdnu byl 12,4 týdne.
Po randomizovaném vysazení léčby v 32. týdnu přibližně 70 % pacientů ve studii ESTEEM 1 a 65,6 % pacientů ve studii ESTEEM 2 znovu dosáhlo po opětovném zahájení léčby apremilastem odpovědí PASI-75 (ESTEEM 1) nebo PASI-50 (ESTEEM 2). Doba trvání opětovné léčby se kvůli uspořádání studií lišila a pohybovala se v rozmezí od 2,6 do 22,1 týdne.
Ve studii ESTEEM 1 bylo pacientům randomizovaným do skupiny léčené apremilastem na počátku studie, kteří nedosáhli odpovědi PASI-75 v 32. týdnu, povoleno užívat souběžnou lokální léčbu a/nebo léčbu UVB světlem mezi 32. a 52. týdnem. Z těchto pacientů dosáhlo 12 % odpovědi PASI-75 v 52. týdnu u léčby apremilastem v kombinaci s lokální léčbou a/nebo léčbou světlem.
Ve studiích ESTEEM 1 a ESTEEM 2 byla v 16. týdnu pozorována významná zlepšení (zmírnění) psoriázy nehtu, měřená průměrnou procentní změnou v indexu závažnosti psoriázy nehtu (NAPSI) oproti výchozí hodnotě u pacientů léčených apremilastem v porovnání s pacienty užívajícími placebo (p < 0,0001 a p = 0,0052, v uvedeném pořadí). Další zlepšení psoriázy nehtů byla pozorována v 32. týdnu u pacientů nepřetržitě léčených apremilastem .
V 16. týdnu studií ESTEEM 1 a ESTEEM 2 byla u pacientů léčených apremilastem oproti pacientům užívajícím placebo (p < 0,0001 v obou studiích) pozorována významná zlepšení psoriázy v oblasti vlasové pokožky s minimálně středně těžkou závažností (> 3), měřená dle podílu pacientů dosahujících čistého (0) nebo minimálního (1) celkového hodnocení psoriázy v oblasti vlasové pokožky lékařem (ScPGA). Zlepšení byla zpravidla udržena u subjektů, které byly v 32. týdnu znovu randomizovány do skupiny léčené přípravkem Otezla až do 52. týdne (Tabulka 5).
Ve studiích ESTEEM 1 a ESTEEM 2 byla u pacientů léčených apremilastem v porovnání s pacienty užívajícími placebo prokázána významná zlepšení v kvalitě života, měřená dle dermatologického indexu kvality života (DLQI) a SF-36v2MCS (Tabulka 4). Zlepšení v DLQI byla udržena až do 52. týdne u subjektů, které byly v 32. týdnu znovu randomizovány do skupiny léčené apremilastem (Tabulka 5). Ve studii ESTEEM 1 bylo navíc u pacientů léčených apremilastem v porovnání s placebem dosaženo významného zlepšení indexu v dotazníku pro posuzování pracovního omezení (WLQ-25).
5.2 Farmakokinetické vlastnosti
Absorpce
Apremilast se dobře vstřebává s absolutní biologickou dostupností po perorálním podání přibližně 73 % a s maximální plazmatickou koncentrací (Cmax) v mediánu doby (tmax) přibližně 2,5 hodiny. Farmakokinetické vlastnosti apremilastu jsou lineární, se zvýšením systémové expozice úměrně k dávce v dávkovém rozmezí 10 až 100 mg denně. Kumulace je minimální, je-li apremilast podáván jednou denně, a přibližně 53 % u zdravých subjektů a 68 % u pacientů s psoriázou, je-li podáván dvakrát denně. Podávání společně s jídlem nemění biologickou dostupnost, apremilast lze tedy podávat s jídlem nebo bez jídla.
Distribuce
Vazba apremilastu na plazmatické proteiny u člověka je přibližně 68 %. Střední zjevný distribuční objem (Vd) je 87 l, což svědčí o extravaskulární distribuci.
Biotransformace
Apremilast je ve velké míře metabolizován jak cestou zprostředkovanou CYP, tak i jinými (nezahrnujícími CYP) cestami, včetně oxidace, hydrolýzy a konjugace. To naznačuje, že inhibice jedné z cest ovlivňující clearance pravděpodobně nezpůsobí výrazné lékové interakce. Oxidační metabolismus apremilastu je zprostředkován primárně CYP3A4 s menším přispěním CYP1A2 a CYP2A6. Apremilast je hlavní cirkulující složkou po perorálním podání. Apremilast prochází rozsáhlou metabolizací, pouze 3 % podávané původní sloučeniny se vylučuje v moči a 7 % se vylučuje ve stolici. Hlavním cirkulujícím inaktivním metabolitem je glukuronidový konjugát O-demetylovaného apremilastu (M12). Jelikož je apremilast substrátem CYP3A4, snižuje se jeho expozice, je-li podáván souběžně s rifampicinem, silným induktorem CYP3A4.
In vitro není apremilast inhibitorem ani induktorem enzymů cytochromu P450. Proto je nepravděpodobné, že by apremilast podávaný souběžně se substráty enzymů CYP ovlivnil clearance a expozici léčivých látek, které jsou metabolizovány enzymy CYP.
In vitro je apremilast substrátem a slabým inhibitorem P-glykoproteinu (IC50 > 50pM), nepředpokládá se však, že by se objevily klinicky relevantní lékové interakce zprostředkované P-glykoproteinem (P-gp).
In vitro má apremilast malý nebo nemá žádný inhibiční účinek (IC50 > 10pM) na transportér organických aniontů (OAT) 1 a OAT3, transportér organických kationtů (OCT) 2, transportní polypeptid organických aniontů (OATP) 1B1 a OATP1B3 nebo na protein BCRP ((breast cancer resistant protein) a není substrátem těchto transportérů. Proto jsou klinicky relevantní lékové interakce nepravděpodobné, je-li apremilast podáván souběžně s léky, které jsou substráty nebo inhibitory těchto transportérů.
Eliminace
Plazmatická clearance apremilastu je v průměru asi 10 l/hod u zdravých subjektů s terminálním poločasem eliminace asi 9 hodin. Po perorálním podání radioaktivně značeného apremilastu bylo v moči zjištěno 58 % radioaktivity, resp. 39 % ve stolici, s přibližně 3 % radioaktivní dávky vyloučené ve formě apremilastu v moči, resp. 7 % ve stolici.
Starší pacienti
Apremilast byl hodnocen u mladých i starších zdravých subjektů. Expozice apremilastu u starších subjektů (65 až 85 let) je asi o 13 % vyšší u AUC a o 6 % vyšší u Cmax apremilastu než u mladších subjektů (18až 55 let). U subjektů starších 75 let jsou k dispozici jen omezené farmakokinetické údaje z klinických studií. U starších pacientů není nutná úprava dávkování.
Porucha funkce ledvin
Neexistuje žádný výrazný rozdíl ve farmakokinetice apremilastu mezi subjekty s mírnou nebo středně vážnou poruchou funkce ledvin a srovnatelnými zdravými subjekty (N = 8 každý). Z výsledků vyplývá, že u pacientů s mírnou až středně vážnou poruchou funkce ledvin není nutná žádná úprava dávkování. U pacientů se závažnou poruchou funkce ledvin (eGFR menší než 30 ml/min/1,73 m2 nebo clearance kreatininu < 30 ml/min) snižte dávku apremilastu na 30 mg jednou denně. U 8 subjektů se závažnou poruchou funkce ledvin, kterým byla podána jedna dávka 30 mg apremilastu, se AUC apremilastu zvýšila přibližně o 89 % a Cmax přibližně o 42 %.
Porucha funkce jater
Farmakokinetické vlastnosti apremilastu a jeho hlavního metabolitu M12 nejsou ovlivněny středně vážnou nebo vážnou poruchou funkce jater. U pacientů s poruchou funkce jater není nutná žádná úprava dávkování.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po opakovaném podávání neodhalily žádné zvláštní riziko pro člověka. Nebyl prokázán žádný potenciál k imunotoxicitě, podráždění kůže nebo fototoxicitě.
Fertilita a časný embryonální vývoj
Studie fertility myších samců neodhalila žádný účinek apremilastu na fertilitu samců po perorálním podání v dávkách 1, 10, 25 a 50 mg/kg/den; hladina NOAEL (žádného pozorovaného nežádoucího účinku) na fertilitu samců byla vyšší než při trojnásobné klinické expozici 50 mg/kg/den.
V kombinované studii fertility myších samic a embryonální/fetální vývojové toxicity s perorálními dávkami 10, 20, 40 a 80 mg/kg/den bylo u dávek 20 mg/kg/den a vyšších pozorováno prodloužení estrálních cyklů a doby do páření, i přesto se všechny myši pářily a počet březostí nebyl ovlivněn. Hladina žádného pozorovaného účinku (NOEL) na samičí fertilitu byla 10 mg/kg/den (jednonásobek klinické expozice).
Embryo-fetální vývoj
V kombinované studii fertility myších samic a embryonální/fetální vývojové toxicity s perorálními dávkami 10, 20, 40 a 80 mg/kg/den došlo při dávkách 20, 40 a 80 mg/kg/den ke zvýšení absolutní a/nebo relativní hmotnosti srdce matek. U dávek 20, 40 a 80 mg/kg/den byl pozorován zvýšený počet časných resorpcí a snížený počet osifikovaných zánártních kůstek. U dávek 40 a 80 mg/kg/den bylo pozorováno snížení hmotnosti plodu a zpoždění osifikace supraokcipitálního skeletu lebky. NOEL
u myších matek a plodů byla 10 mg/kg/den (1,3násobek klinické expozice).
Ve studii embryonální/fetální vývojové toxicity u opic měly perorální dávky 20, 50, 200 a 1000 mg/kg/den za následek s dávkou související nárůst prenatálních ztrát (potratů) při dávkách 50 mg/kg/den a vyšších; při dávkách 20 mg/kg/den (1,4násobek klinické expozice) nebyl pozorován žádný účinek testované látky na prenatální ztrátu.
Prenatální a postnatální vývoj
V prenatální a postnatální studii byl apremilast podáván perorálně březím myším samicím v dávkách 10, 80 a 300 mg/kg/den od 6. dne gestace do 20. dne laktace. Při dávkách 300 mg/kg/den byly pozorovány pokles a přírůstek tělesné hmotnosti matek a jedno úmrtí spojené s obtížemi při porodu mláďat. Byly rovněž pozorovány fyzické známky toxicity u matek spojované s porodem mláďat
u jedné myši pro každou dávku (80 a 300 mg/kg/den). Při dávkách > 80 mg/kg/den (> 4,0násobek klinické expozice) bylo během prvního týdne laktace pozorováno zvýšení perinatální a postnatální mortality mláďat a pokles tělesné hmotnosti mláďat. Nebyly zjištěny žádné účinky apremilastu na dobu trvání březosti, počet březích myší na konci gestačního období, počet myší, které porodily mláďata, ani žádný vliv na vývoj mláďat po 7. dni po porodu. Je pravděpodobné, že vliv na vývoj mláďat pozorovaný během prvního týdne postnatálního období souvisel s toxicitou pro mláďata spojenou s apremilastem (pokles hmotnosti a životaschopnosti mláďat) a/nebo nedostatkem mateřské péče (vyšší incidence mláďat bez mléka v žaludku). Veškeré vývojové účinky byly pozorovány v prvním týdnu postnatálního období, ve zbývajícím období před a po odstavení nebyly pozorovány žádné účinky spojené s apremilastem, včetně parametrů pohlavního dospívání, chování, páření, fertility a dělohy. NOEL u myší pro toxicitu pro matky a generaci F1 byla 10 mg/kg/den (1,3násobek klinické hodnoty AUC).
Studie kancerogenity
Studie kancerogenity u myší a potkanů neprokázaly žádnou kancerogenitu v souvislosti s léčbou apremilastem.
Studie genotoxicity
Apremilast není genotoxický. Apremilast nevyvolal mutace v Amesově testu ani chromozomální aberace u kultivovaných lidských lymfocytů z periferní krve s metabolickou aktivací nebo bez ní. Apremilast nebyl při dávkách do 2000 mg/kg/den klastogenní v in vivo mikronukleárním testu u myší.
Další studie
Nebyl prokázán žádný potenciál k imunotoxicitě, podráždění kůže nebo fototoxicitě.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety:
Mikrokrystalická celulóza Monohydrát laktózy Sodná sůl kroskarmelózy Magnesium-stearát
Potah tablety:
Polyvinylalkohol Oxid titaničitý (E171)
Makrogol 3350 Mastek
Oxid železitý červený (E172)
Tablety 20 mg obsahují navíc oxid železitý žlutý (E172).
Tablety 30 mg obsahují navíc oxid železitý žlutý (E172) a oxid železitý černý (E172).
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
24 měsíců.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30 °C.
6.5 Druh obalu a obsah balení
Balení pro úvodní léčbu obsahuje 27 potahovaných tablet (4x 10 mg, 4x 20 mg, 19x 30 mg). Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
CelgeneEurope Ltd.
1 LongwalkRoad Stockley Park Uxbridge UB11 1DB Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/981/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 15. ledna 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Otezla 30 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje apremilastum 30 mg.
Pomocná látka/Pomocné látky se známým účinkem:
Jedna potahovaná tableta obsahuje 171 mg laktózy (ve formě monohydrátu laktózy). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Otezla 30 mg potahované tablety: Béžová 30mg potahovaná tableta ve tvaru kosočtverce, délka 12 mm, s vyrytým označením „APR“ na jedné straně a „30“ na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Psoriatická artritida
Přípravek Otezla, samotný nebo v kombinaci s onemocnění modifikujícími antirevmatickými léky (DMARD), je indikován k léčbě aktivní psoriatické artritidy (PsA) u dospělých pacientů, kteří adekvátně neodpovídali nebo netolerovali předchozí léčbu DMARD (viz bod 5.1).
Psoriáza
Přípravek Otezla je indikován k léčbě středně těžké až těžké chronické ložiskové lupénky u dospělých pacientů, kteří neodpovídali nebo mají kontraindikovanou nebo netolerují jinou systémovou terapii, včetně cyklosporinu, methotrexátu nebo PUVA (kombinace psoralenu a UVA záření).
4.2 Dávkování a způsob podání
Léčba přípravkem Otezla má být zahájena odborným lékařem se zkušenostmi v diagnostice a léčbě psoriázy nebo psoriatické artritidy.
Dávkování
Doporučená dávka přípravku Otezla je 30 mg dvakrát denně perorálně, ráno a večer, v intervalu přibližně 12 hod, bez omezení přijmu potravin. Je nutné dodržovat plán úvodní titrace uvedený níže v Tabulce 1. Po úvodní titraci není nutná žádná další titrace.
Tabulka 1. Plán titrace dávek
|
1. den |
2. den |
3. den |
4. den |
5. den |
6. den a dále | |||||
|
DOP. |
DOP. |
ODP. |
DOP. |
ODP. |
DOP. |
ODP. |
DOP. |
ODP. |
DOP. |
ODP. |
|
10 mg |
10 mg |
10 mg |
10 mg |
20 mg |
20 mg |
20 mg |
20 mg |
30 mg |
30 mg |
30 mg |
Jestliže pacient zapomene užít dávku, měl by si ji vzít co nejdříve. Jestliže se však již blíží čas na další dávku, vynechaná dávka se nemá nahrazovat a má se užít další dávka v plánovaném čase.
V průběhu pivotních studií bylo největší zlepšení zaznamenáno během prvních 24 týdnů léčby.
U pacienta, který po 24 týdnech léčby nevykazuje žádné známky léčebného přínosu, je třeba léčbu přehodnotit. Odpověď pacienta na léčbu musí být pravidelně hodnocena. Klinické zkušenosti delší než 52 týdnů nejsou k dispozici (viz bod 5.1).
Zvláštní populace
Starší pacienti
U této skupiny pacientů není nutná úprava dávkování (viz body 4.8 a 5.2).
Pacienti s poruchou funkce ledvin
U pacientů s mírnou až středně závažnou poruchou funkce ledvin není nutná úprava dávkování.
U pacientů se závažnou poruchou funkce ledvin (clearance kreatininu méně než 30 ml/min dle Cockcroft-Gaultova vzorce) je třeba dávku apremilastu snížit na 30 mg jednou denně. Pro úvodní titraci dávky u této skupiny se doporučuje, aby byl přípravek Otezla titrován pouze s využitím dopoledních dávek uvedených v Tabulce 1 a aby byly odpolední dávky vynechány (viz bod 5.2).
Pacienti s poruchou funkce jater
U pacientů s poruchou funkce jater není nutná žádná úprava dávkování (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost apremilastu u dětí ve věku od 0 do 17 let nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Otezla je určen k perorálnímu podání. Potahované tablety se polykají celé a lze je užívat s jídlem nebo bez jídla.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku(y) nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Těhotenství (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Pacienti se vzácnými vrozenými poruchami, jako jsou intolerance galaktózy, vrozený deficit laktázy nebo malabsorpce glukózy-galaktózy, by neměli tento přípravek užívat.
U pacientů se závažnou poruchou funkce ledvin je třeba snížit dávku přípravku Otezla na 30 mg jednou denně (viz body 4.2 a 5 2).
U pacientů s podváhou na začátku léčby je třeba pravidelně kontrolovat tělesnou hmotnost. V případě nevysvětleného a klinicky významného váhového úbytku musí tyto pacienty vyšetřit lékař a je nutné zvážit ukončení léčby.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Souběžné podávání se silným induktorem enzymu cytochromu P450 3A4 (CYP3A4) rifampicinem vedlo ke snížení systémové expozice apremilastu, což může způsobit ztrátu účinnosti apremilastu. Proto se nedoporučuje současné užívání apremilastu se silnými induktory enzymu CYP3A4 (např. rifampicin, fenobarbital, karbamazepin, fenytoin a třezalka tečkovaná). Souběžné podávání apremilastu s opakovanými dávkami rifampicinu vedlo ke snížení plochy pod časovou křivkou plazmatické koncentrace apremilastu (AUC) přibližně o 72 % a ke snížení maximální koncentrace apremilastu v séru (Cmax) o 43 %. Expozice apremilastu se snižuje, pokud je přípravek podáván souběžně se silnými induktory CYP3A4 (např. rifampicinem), což může vést ke snížené klinické odpovědi.
V klinických studiích byl apremilast podáván souběžně s lokální léčbou (včetně kortikosteroidů, černouhelného dehtového šamponu a přípravků s kyselinou salicylovou určených k ošetření vlasové pokožky) a s léčbou UVB světlem.
Neobjevila se žádná klinicky významná léková interakce mezi ketokonazolem a apremilastem. Apremilast lze podávat souběžně s potentním inhibitorem CYP3A4, jakým je například ketokonazol.
U pacientů s psoriatickou artritidou nedošlo mezi apremilastem a methotrexátem k žádným farmakokinetickým lékovým interakcím. Apremilast lze podávat souběžně s methotrexátem.
Mezi apremilastem a perorální antikoncepcí obsahující ethinylestradiol a norgestimát nedošlo k žádným farmakokinetickým lékovým interakcím. Apremilast lze podávat souběžně s perorální antikoncepcí.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Před zahájením léčby je nezbytné vyloučit těhotenství. Ženy ve fertilním věku musí užívat účinnou metodu antikoncepce k zabránění otěhotnění po dobu léčby.
Údaje o podávání apremilastu těhotným ženám jsou omezené.
Apremilast je v těhotenství kontraindikován. U myší a opic byly pozorovány účinky apremilastu na těhotenství, a to včetně ztráty embrya/plodu a snížení hmotnosti plodu, dále opožděnou osifikaci u myší, při dávkách vyšších, než je současná doporučená maximální dávka u člověka. Žádné takové účinky nebyly pozorovány, byla-li expozice u zvířat 1,3krát vyšší než klinická expozice (viz bod 5.3).
Kojení
Apremilast byl zjištěn v mléce laktujících myších samic (viz bod 5.3). Není známo, zda se apremilast nebo jeho metabolity vylučují do lidského mateřského mléka. Riziko pro kojené děti nelze vyloučit, proto se podávání apremilastu během kojení nedoporučuje.
Fertilita
Údaje o fertilitě u lidí nejsou k dispozici. Při studiích na zvířatech nebyly pozorovány žádné nežádoucí účinky na fertilitu u myších samců při úrovni expozice odpovídající 3násobku klinické expozice a u samic při úrovni expozice odpovídající jednonásobku klinické expozice. Předklinické údaje o fertilitě viz bod 5.3.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Apremilast nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
V klinických studiích fáze III byly nejčastěji hlášenými nežádoucími účinky gastrointestinální poruchy včetně průjmu (15,7 %) a nevolnosti (13,9 %). Tyto gastrointestinální nežádoucí účinky byly většinou mírné až středně závažné, s 0,3 % průjmů a 0,3 % nevolností hlášených jako závažné. Tyto nežádoucí účinky se zpravidla vyskytovaly v prvních 2 týdnech léčby a obvykle vymizely do 4 týdnů. Další nejčastěji hlášené nežádoucí účinky zahrnovaly infekce horních cest dýchacích (8,4 %), bolest hlavy (7,9 %) a tenzní bolest hlavy (7,2 %). Většina nežádoucích účinků byla celkově považována za mírné až středně závažné.
K nejčastějším nežádoucím účinkům, které vedly k ukončení léčby v prvních 16 týdnech, patřil průjem (1,7 %) a nevolnost (1,5 %). Celková incidence závažných nežádoucích účinků byla nízká a nenaznačuje postižení specifického orgánového systému.
Reakce přecitlivělosti byly v klinických studiích apremilastu pozorovány méně často (viz bod 4.3). Seznam nežádoucích účinků v tabulce
Nežádoucí účinky pozorované u pacientů léčených apremilastem jsou uvedeny v tabulce níže podle třídy orgánových systémů a četnosti pro všechny nežádoucí účinky. V rámci každé třídy orgánových systémů a skupiny četnosti jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Nežádoucí účinky léku byly stanoveny na základě údajů získaných z programu klinického vývoje apremilastu. Uvedené četnosti nežádoucích účinků léku byly hlášeny ve větvích čtyř studií fáze III s apremilastem u PsA (n = 1945) nebo dvou studií fáze III s apremilastem u psoriázy (n = 1184) (nejvyšší četnost z obou souborů dat je uvedena v Tabulce 2).
Četnosti jsou definovány takto: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000).
Tabulka 2. Souhrn nežádoucích účinků u psoriatické artritidy (PsA) a/nebo psoriázy (PSOR)
|
Třída orgánového systému |
Četnost |
Nežádoucí účinek |
|
Infekce a infestace |
Časté |
Bronchitida |
|
Infekce horních cest dýchacích | ||
|
Nazofaryngitida* | ||
|
Poruchy imunitního systému |
Méně časté |
Přecitlivělost |
|
Poruchy metabolismu a výživy |
Časté |
Snížená chuť k jídlu* |
|
Psychiatrické poruchy |
Časté |
Insomnie |
|
Poruchy nervového systému |
Časté |
Migréna* |
|
Tenzní bolesti hlavy* | ||
|
Bolest hlavy* | ||
|
Respirační, hrudní a mediastinální poruchy |
Časté |
|
Třída orgánového systému |
Četnost |
Nežádoucí účinek |
|
Gastrointestinální poruchy |
Velmi časté |
Průjem* |
|
Nevolnost* | ||
|
Časté |
Zvracení* | |
|
Časté vyprazdňování střev | ||
|
Bolest horní poloviny břicha* | ||
|
Gastroezofageální reflux | ||
|
Méně časté |
Gastrointestinální krvácení | |
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté |
Bolest zad* |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Únava |
|
Vyšetření |
Méně časté |
Snížení tělesné hmotnosti |
*Nejméně jeden z těchto nežádoucích účinků byl hlášen jako závažný.
Popis vybraných nežádoucích účinků
Pokles tělesné hmotnosti
V klinických studiích byla pravidelně zjišťována hmotnost pacientů. Průměrný váhový úbytek pozorovaný u pacientů léčených apremilastem po dobu až 52 týdnů byl 1,99 kg. Celkem u 14,3 % pacientů užívajících apremilast byl pozorován váhový úbytek 5 až 10 %, zatímco u 5,7 % pacientů užívajících apremilast byl pozorován váhový úbytek přesahující 10 %. Žádný z těchto pacientů neměl zjevné klinické následky váhového úbytku. Celkem u 0,1 % pacientů léčených apremilastem byla léčba přerušena v důsledku nežádoucího účinku snížení hmotnosti.
Viz dodatečné upozornění v bodě 4.4 pro pacienty, kteří trpí na začátku léčby podváhou.
Během placebem kontrolovaného období klinických studií fáze III u psoriázy byla u 1,2 % (14/1184) pacientů léčených apremilastem hlášena deprese ve srovnání s 0,5 % (2/418) pacientů léčených placebem. Žádné z těchto hlášení výskytu deprese nebylo závažné ani nevedlo k přerušení účasti ve studii.
Zvláštní populace
Starší pacienti
V klinických studiích nebyly celkově pozorovány žádné rozdíly v bezpečnostním profilu u starších pacientů ve věku > 65 let a mladších dospělých pacientů ve věku < 65 let.
Pacienti s poruchou funkce jater
Bezpečnost apremilastu v léčbě psoriatické artritidy a psoriázy u pacientů s poruchou funkce jater nebyla hodnocena.
Pacienti s poruchou funkce ledvin
V klinických studiích PsA nebo PSOR byl u pacientů s mírnou poruchou funkce ledvin pozorován bezpečnostní profil srovnatelný s pacienty s normální funkcí ledvin. Bezpečnost apremilastu u pacientů s PsA nebo PSOR se středně závažnou nebo závažnou poruchou funkce ledvin nebyla v klinických studiích hodnocena.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Apremilast byl hodnocen u zdravých subjektů, jimž byla podávána maximální celková denní dávka 100 mg (podávaná jako 50 mg dvakrát denně) po dobu 4,5 dne, aniž by se prokázala dávku limitující toxicita. V případě předávkování se doporučuje u pacienta sledovat jakékoli známky nebo příznaky nežádoucích účinků a zahájit odpovídající symptomatickou léčbu. V případě předávkování se doporučují symptomatická a podpůrná léčebná opatření.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: imunosupresiva, selektivní imunosupresiva, ATC kód: L04AA32 Mechanismus účinku
Apremilast, perorální inhibitor fosfodiesterázy 4 (PDE4) s malou molekulou, působí intracelulárně a moduluje síť prozánětlivých a protizánětlivých mediátorů. PDE4 je PDE specifická pro cyklický adenosin monofosfát (cAMP) a dominantní PDE v zánětlivých buňkách. Inhibice PDE4 zvyšuje intracelulární hladiny cAMP, což omezuje zánětlivou odpověď díky modulaci exprese TNF-a, IL-23, IL-17 a jiných zánětlivých cytokinů. Cyklický AMP také moduluje hladiny protizánětlivých cytokinů jako například IL-10. Tyto prozánětlivé a protizánětlivé mediátory byly implikovány u psoriatické artritidy a psoriázy.
Farmakodynamické účinky
V klinických studiích u pacientů s psoriatickou artritidou apremilast významně moduloval, ale ne zcela inhiboval plazmatické hladiny proteinů IL-1a, IL-6, IL-8, MCP-1, MIP-ip, MMP-3 a TNF-a. Po 40 týdnech léčby apremilastem došlo ke snížení plazmatické hladiny proteinů IL-17 a IL-23 a zvýšení u IL-10. V klinických studiích u pacientů s psoriázou způsobil apremilast zmenšení tloušťky epidermu kůže poškozené lézí, infiltraci zánětlivých buněk a expresi prozánětlivých genů, včetně genů pro inducibilní syntázu oxidu dusnatého (iNOS), IL-12/IL-23p40, IL-17A, IL-22 a IL-8.
Apremilast podávaný v dávkách až do 50 mg dvakrát denně neměl vliv na délku QT intervalu u zdravých subjektů.
Zkušenosti z klinických studií
Psoriatická artritida
Účinnost a bezpečnost apremilastu byla hodnocena ve 3 multicentrických, randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích (studie PALACE 1, PALACE 2 a PALACE 3) s podobným uspořádáním, do nichž byli zařazeni dospělí pacienti s aktivní PsA (> 3 oteklé klouby a > 3 bolestivé klouby) i přes předchozí léčbu biologickými DMARD nebo DMARD s malou molekulou. Celkem 1493 pacientů bylo randomizováno a léčeno buď placebem, apremilastem 20 mg, nebo apremilastem 30 mg podávanými perorálně dvakrát denně.
Pacientům v těchto studiích byla PsA diagnostikována minimálně před 6 měsíci. Při zařazení do studie PALACE 3 byla navíc vyžadována jedna aktivní psoriatická kožní léze (alespoň 2 cm v průměru). Apremilast byl podáván v monoterapii (34,8 %) nebo v kombinaci se stabilními dávkami DMARD s malou molekulou (65,2 %). Pacienti dostávali apremilast v kombinaci s jednou látkou nebo několika z těchto látek: methotrexát (MTX, < 25 mg/týden, 54,5 %), sulfasalazin (SSZ, < 2 g/den, 9,0 %) a leflunomid (LEF, < 20 mg/den, 7,4 %). Souběžná léčba s biologickými DMARD včetně blokátorů TNF nebyla povolena. Tyto tři studie zahrnovaly pacienty se všemi podtypy PsA, včetně symetrické polyartritidy (62,0 %), asymetrické oligoartritidy (26,9 %), artritidy distálního interfalangeálního kloubu (6,2 %), mutilující artritidy (2,7 %) a predominující spondylitidy (2,1 %). Byli zahrnuti pacienti s preexistující entezopatií (63 %) nebo preexistující daktylitidou (42 %). Celkem 76,4 % pacientů bylo dříve léčeno pouze DMARD s malou molekulou a 22,4 % pacientů bylo dříve léčeno biologickými DMARD, což zahrnuje 7,8 % pacientů, u nichž byla předchozí léčba biologickým DMARD neúspěšná. Medián doby trvání onemocnění PsA byl 5 let.
Podle uspořádání studie byli pacienti, u nichž se počet bolestivých a oteklých kloubů nezlepšil alespoň o 20 % v 16. týdnu, považováni za pacienty nereagující na léčbu. U pacientů užívajících placebo, kteří byli považováni za pacienty nereagující na léčbu, byla provedena opakovaná zaslepená randomizace a v poměru 1: 1 byli zařazeni buď do skupiny léčené 20 mg apremilastu dvakrát denně, nebo do skupiny léčené 30 mg dvakrát denně. Ve 24. týdnu přešli všichni zbývající pacienti léčení placebem na léčbu buď 20 mg, nebo 30 mg apremilastu dvakrát denně.
Primárním cílovým parametrem účinnosti studie bylo procento pacientů dosahujících odpovědi ACR 20 (20% zlepšení dle kritérií American College of Rheumatology) v 16. týdnu.
Léčba apremilastem vedla ve srovnání s placebem k významnému zlepšení známek a příznaků PsA v 16. týdnu (dle hodnocení parametru odpovědi ACR 20). Podíly pacientů s ACR 20/50/70 (odpovědi ve studiích PALACE 1, PALACE 2 a PALACE 3 a souhrnné údaje ze studií PALACE 1, PALACE 2 a PALACE 3) u apremilastu 30 mg dvakrát denně v 16. týdnu jsou uvedeny v Tabulce 3. Odpovědi ACR 20/50/70 přetrvávaly v 24. týdnu.
Mezi pacienty, kteří byli původně randomizováni k léčbě apremilastem 30 mg dvakrát denně, byly četnosti odpovědí ACR 20/50/70 udrženy až do 52. týdne v souboru studií PALACE 1, PALACE 2 a PALACE 3 (Obrázek 1).
Tabulka 3. Podíl pacientů s ACR odpověďmi ve studiích PALACE 1, PALACE 2 a PALACE 3 a v souboru studií v 16. týdnu
|
PALACE1 |
PALACE 2 |
PALACE 3 |
SOUBOR STUDIÍ | |||||
|
Placebo |
Apremilast |
Placebo |
Apremilast |
Placebo |
Apremilast |
Placebo |
Apremilast | |
|
+/- |
30 mg BID |
+/- |
30 mg BID |
+/- |
30 mg BID |
+/- |
30 mg BID | |
|
DMARD |
+/- DMARD |
DMARD |
+/- DMARD |
DMARD |
+/- DMARD |
DMARD |
+/- DMARD | |
|
Na |
N = 168 |
N = 168 |
N = 159 |
N = 162 |
N = 169 |
N = 167 |
N = 496 |
N = 497 |
|
ACR 20a | ||||||||
|
16. týden |
19,0 % |
38,1 %** |
18,9 % |
32,1 %* |
18,3 % |
40,7 %** |
18,8 % |
37,0 %** |
|
ACR 50 | ||||||||
|
16. týden |
6,0 % |
16,1 %* |
5,0 % |
10,5 % |
8,3 % |
15,0 % |
6,5 % |
13,9 %** |
|
ACR 70 | ||||||||
|
16. týden |
1,2 % |
4,2 % |
0,6 % |
1,2 % |
2,4 % |
3,6 % |
1,4 % |
3,0 % |
*p < 0,01 pro apremilast vs. placebo.
**p < 0,001 pro apremilast vs. placebo. a N je počet randomizovaných a léčených pacientů.
Obrázek 1 Podíl pacientů s odpovědí ACR 20/50/70 až do 52. týdne v souhrnné analýze studií PALACE 1, PALACE 2 a PALACE 3 (NRI*)
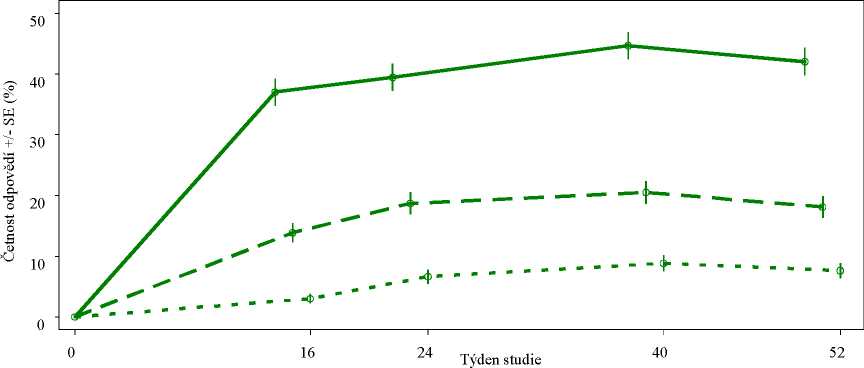
Cílový parametr
ACR 20 ACR 50 ACR 70
n/m (%) n/m (%)
184/497 (37,0) 196/497 (39,4)
69/497 (13,9) 93/497 (18,7)
15/497 ( 3,0) 33/497 ( 6,6)
n/m (%) 222/497 (44,7) 102/497 (20,5) 44/497 ( 8,9)
n/m (%) 209/497 (42,1) 90/497 (18,1) 38/497 ( 7,6)
Cílový parametr ACR 20 0-0 ■» ACR 50 e « o ACR 70
*NRI: imputace nereagujících subjektů. Za nereagující jsou považovány subjekty, které předčasně ukončily léčbu ještě před daným časovým bodem, a subjekty, u nichž nebyly v daném časovém bodě dostatečné údaje pro jednoznačné vyhodnocení odpovědi.
Z 497 pacientů původně randomizovaných do skupiny léčené 30 mg apremilastu dvakrát denně, bylo v 52. týdnů 375 (75 %) pacientů stále v léčení. Odpovědí ACR 20/50/70 bylo v 52. týdnu dosaženo u 57/25/11 % těchto pacientů.
Odpovědi pozorované u skupiny léčené apremilastem byly obdobné u pacientů, kteří souběžně užívali a neužívali DMARD, včetně methotrexátu. Pacienti dříve léčení DMARD nebo biologickými léky, kteří užívali apremilast, dosáhli v 16. týdnu častější odpovědi ACR 20 než pacienti užívající placebo.
Obdobné ACR odpovědi byly pozorovány u pacientů s různými podtypy PsA, včetně artritidy distálního interfalangeálního kloubu. Počet pacientů s mutilující artritidou a s podtypy predominující spondylitidy byl příliš malý, aby mohlo být provedeno smysluplné hodnocení.
Ve studiích PALACE 1, PALACE 2 a PALACE 3 bylo v 16. týdnu zaznamenáno významnější zlepšení hladiny C-reaktivního proteinu (CRP) dle stupnice aktivity onemocnění (DAS) 28 a podílu pacientů dosahujících modifikovaných kritérií léčebné odpovědi PsA (PsARC) u apremilastu oproti placebu (nominální p-hodnot a p < 0,0004 a p-hodnota < 0,0017, v uvedeném pořadí). Tato zlepšení přetrvávala ve 24. týdnu. U pacientů, kteří pokračovali v léčbě apremilastem, k níž byli randomizováni na počátku studie, byly skóre DAS28 (CRP) a odpověď PsARC udrženy až do 52. týdne.
V 16. a 24. týdnu bylo u pacientů léčených apremilastem pozorováno zlepšení v parametrech periferních znaků aktivity psoriatické artritidy (jako je počet oteklých kloubů, počet bolestivých/citlivých kloubů, daktylitida a entezitida) a v kožních projevech psoriázy. U pacientů, kteří pokračovali v léčbě apremilastem, k níž byli randomizováni na počátku studie, byla tato zlepšení udržena až do 52. týdne.
Tělesné funkce a kvalita života související se zdravím
Ve studiích PALACE 1, PALACE 2 a PALACE 3 a souboru studií vykazovali pacienti léčení apremilastem v porovnání s placebem v 16. týdnu statisticky významné zlepšení tělesných funkcí hodnocené podle změny oproti výchozímu indexu postižení dle dotazníku pro posuzování zdravotního stavu (HAQ-DI). Zlepšení skóre HAQ-DI bylo udrženo v 24. týdnu.
Ve sdružené analýze otevřené fáze studií PALACE 1, PALACE 2 a PALACE 3 byla u pacientů, kteří byli původně randomizováni k léčbě 30 mg apremilastu dvakrát denně, v 52. týdnu změna skóre HAQ-DI oproti výchozí hodnotě -0,333 ve skupině léčené 30 mg apremilastu dvakrát denně.
Ve studiích PALACE 1, PALACE 2 a PALACE 3 bylo v 16. a 24. týdnu u pacientů léčených apremilastem ve srovnání s placebem zaznamenáno významné zlepšení kvality života související se zdravím měřené podle změny tělesných funkcí oproti výchozímu stavu, které byly hodnoceny pomocí krátkého dotazníku pro posuzování zdravotního stavu, verze 2 (SF-36v2), a změny skóre únavy dle dotazníku pro funkční hodnocení léčby chronického onemocnění (FACIT-f). U pacientů, kteří pokračovali v léčbě apremilastem, k níž byli randomizováni na počátku studie, byla zlepšení v parametru tělesných funkcí a skóre FACIT-f udržena až do 52. týdne.
Psoriáza
Bezpečnost a účinnost apremilastu byla hodnocena ve dvou multicentrických, randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích (studie ESTEEM 1 a ESTEEM 2). Tyto studie zahrnovaly celkem 1257 pacientů se středně těžkou až těžkou ložiskovou psoriázou na ploše > 10 % povrchu těla, se skóre oblasti psoriázy a indexu závažnosti (PASI ) > 12 a se statickým celkovým hodnocením lékařem (sPGA) > 3 (středně těžké nebo těžké), kteří byli kandidáty na léčbu světlem nebo systémovou terapii.
Tyto studie měly podobné uspořádání až do 32. týdne. V obou studiích byli pacienti randomizováni v poměru 2:1 do skupiny užívající 30 mg apremilastu dvakrát denně a do skupiny užívající placebo po dobu 16 týdnů (placebem kontrolovaná fáze), v 16.-32. týdnu užívali všichni pacienti 30 mg apremilastu dvakrát denně (udržovací fáze). Během randomizované fáze s vysazením léčby (32. až 52. týden) byli pacienti původně randomizovaní do skupiny užívající apremilast, kteří dosáhli alespoň 75% snížení skóre PASI (PASI-75) (ESTEEM 1) nebo 50% snížení skóre PASI (PASI-50)
(ESTEEM 2), znovu randomizováni v 32. týdnu buď do skupiny užívající placebo, nebo do skupiny užívající 30 mg apremilastu dvakrát denně. Pacienti, kteří byli znovu randomizováni do skupiny užívající placebo a kteří v 32. týdnu ztratili odpověď PASI-75 (ESTEEM 1) nebo ztratili 50% zlepšení skóre PASI oproti výchozím hodnotám (ESTEEM 2), byli převedeni na léčbu 30 mg apremilastu dvakrát denně. Pacienti, kteří nedosáhli stanovené odpovědi PASI do 32. týdne nebo kteří byli původně randomizováni do skupiny užívající placebo, užívali apremilast až do 52. týdne. Podávání slabých lokálních kortikosteroidů na tváři, v podpaží a tříslech, černouhelných dehtových šamponů a/nebo přípravků s kyselinou salicylovou určených k ošetření vlasové pokožky bylo po celou dobu studií povoleno. Ve 32. týdnu bylo subjektům, které nedosáhly odpovědi PASI-75 ve studii ESTEEM 1 nebo odpovědi PASI-50 ve studii ESTEEM 2, kromě léčby 30 mg apremilastu dvakrát denně navíc povoleno používat lokální léčbu psoriázy a/nebo léčbu světlem.
V obou studiích byl primárním cílovým parametrem účinnosti podíl pacientů, kteří v 16. týdnu dosáhli PASI-75. Hlavním sekundárním cílovým parametrem byl podíl pacientů, kteří v 16. týdnu dosáhli čistého (0) nebo minimálního (1) skóre sPGA.
Průměrné výchozí skóre PASI bylo 19,07 (medián 16,80), podíl pacientů s výchozím skóre sPGA 3 (středně těžké) a 4 (těžké) byl 70,0 %, resp. 29,8 % s průměrnou výchozí plochou povrchu těla 25,19 % (medián 21,0 %). Přibližně 30 % všech pacientů podstoupilo při léčbě psoriázy předchozí léčbu světlem a 54 % podstoupilo předchozí konvenční systémovou terapii a/nebo biologickou léčbu (včetně selhání léčby), přičemž 37 % předchozí konvenční systémovou terapii a 30 % předchozí biologickou léčbu. Přibližně jedna třetina pacientů nepodstoupila předchozí léčbu světlem, konvenční systémovou terapii ani biologickou léčbu. Celkem 18 % pacientů mělo psoriatickou artritidou v anamnéze.
Podíl pacientů dosahujících odpovědí PASI-50, -75 a -90 a čistého (0) nebo minimálního (1) skóre sPGA je uveden v Tabulce 4 níže. Léčba apremilastem vedla v porovnání s placebem k významnému zlepšení středně těžké až těžké ložiskové psoriázy, jak bylo prokázáno podílem pacientů s odpovědí PASI-75 v 16. týdnu. V 16. týdnu byla rovněž prokázána klinická zlepšení měřená podle odpovědí sPGA, PASI-50 a PASI-90. Přínos léčby apremilastem byl navíc prokázán na několika projevech psoriázy, včetně pruritu, onemocnění nehtu, postižení vlasové pokožky a měření kvality života.
Tabulka 4. Klinická odpověď v 16. týdnu studií ESTEEM 1 a ESTEEM 2 (FAS a LOCFb)
|
ESTEEM 1 |
ESTEEM 2 | |||
|
Placebo |
30 mg BID APR* |
Placebo |
30 mg BID APR* | |
|
N |
282 |
562 |
137 |
274 |
|
PASIc 75, n (%) |
15 (5,3) |
186 (33,1) |
8 (5,8) |
79 (28,8) |
|
sPGAd čisté nebo minimální, n (%) |
11 (3,9) |
122 (21,7) |
6 (4,4) |
56 (20,4) |
|
PASI 50, n (%) |
48 (17,0) |
330 (58,7) |
27 (19,7) |
152 (55,5) |
|
PASI 90, n (%) |
1 (0,4) |
55 (9,8) |
2 (1,5) |
24 (8,8) |
|
Procentuální změna BSAe (%) průměrná hodnota ± SD |
-6,9 |
-47,8 |
-6,1 |
-48,4 |
|
±38,95 |
±38,48 |
±47,57 |
±40,78 | |
|
Změna pruritu VASf(mm), |
-7,3 |
-31,5 |
-12,2 |
-33,5 |
|
průměrná hodnota ± SD |
±27,08 |
±32,43 |
±30,94 |
±35,46 |
|
Změna v DLQIg, průměrná |
-2,1 |
-6,6 |
-2,8 |
-6,7 |
|
hodnota ± SD |
±5,69 |
±6,66 |
±7,22 |
±6,95 |
|
Změna v SF-36 MCS h, |
-1,02 |
2,39 |
0,00 ± |
2,58 |
|
průměrná hodnota ± SD |
±9,161 |
±9,504 |
10,498 |
±10,129 |
* p < 0,0001 pro apremilast vs. placebo, s výjimkou PASI 90 ve studii ESTEEM 2 a změny v SF-36 MCS, kde p = 0,0042, resp. p = 0,0078. a FAS = celkový analyzovaný soubor b LOCF= poslední provedené sledování c PASI = skóre oblasti psoriázy a indexu závažnosti d sPGA = statické celkové hodnocení lékaře e BSA = plocha povrchu těla
f VAS = vizuální analogová stupnice; 0 = nejlepší, 100 = nejhorší g DLQI = index dermatologické kvality života; 0 = nejlepší, 30 = nejhorší
h SF-36 MCS = krátký 36položkový dotazník pro posuzování zdravotního stavu ve studii, souhrn otázek týkajících se duševního zdraví
Klinický přínos apremilastu byl prokázán na několika podskupinách definovaných na základě výchozích demografických údajů a výchozích charakteristik klinického onemocnění (včetně doby trvání psoriázy a pacientů s anamnézou psoriatické artritidy). Klinický přínos apremilastu byl také prokázán bez ohledu na předchozí medikaci k léčbě psoriázy a odpověď na předchozí léčbu psoriázy. Ve všech rozmezích hmotnosti byly pozorovány obdobné četnosti odpovědí.
Odpověď na apremilast v porovnání s placebem byla do 2. týdne rychlá, s významně většími zlepšeními známek a příznaků psoriázy, včetně PASI, kožních obtíží/bolesti a pruritu. Odpovědi PASI byly zpravidla dosaženy do 16. týdne a byly udrženy až do 32. týdne.
V obou studiích zůstalo průměrné procentuální zlepšení PASI oproti výchozím hodnotám stabilní během randomizované fáze s vysazením léčby u pacientů, kteří byli v 32. týdnu znovu randomizováni do skupiny léčené apremilastem (Tabulka 5).
Tabulka 5. Přetrvávání účinku u subjektů randomizovaných do APR 30 dvakrát denně na počátku studie a znovu randomizovaných do APR 30 dvakrát denně v 32.až 52.týdnu
|
Časový bod |
ESTEEM 1 |
ESTEEM 2 | |
|
Pacienti, kteří dosáhli PASI-75 v 32. týdnu |
Pacienti, kteří dosáhli PASI-50 v 32. týdnu | ||
|
Procentní změna PASI oproti výchozí hodnotě, průměrná hodnota (%) ± SDa |
16. týden |
-77,7 ± 20,30 |
-69,7 ± 24,23 |
|
32. týden |
-88 ± 8,30 |
-76,7 ± 13,42 | |
|
52. týden |
-80,5 ± 12,60 |
-74,4 ± 18,91 | |
|
Změna DLQI oproti výchozí hodnotě, průměrná hodnota (%)± SDa |
16. týden |
-8,3 ± 6,26 |
-7,8 ± 6,41 |
|
32. týden |
-8,9 ± 6,68 |
-7,7 ± 5,92 | |
|
52. týden |
-7,8 ± 5,75 |
-7,5 ± 6,27 | |
|
Podíl subjektů s psoriázou v oblasti vlasové pokožky PGA (ScPGA) 0 nebo 1, n/N (%)b |
16. týden |
40/48 (83,3) |
21/37 (56,8) |
|
32. týden |
39/48 (81,3) |
27/37 (73,0) | |
|
52. týden |
35/48 (72,9) |
20/37 (54,1) |
a Zahrnuje subjekty znovu randomizované do APR 30 dvakrát denně v 32. týdnu s výchozí hodnotou a následnou hodnotou v hodnoceném týdnu studie.
bN vychází ze subjektů se středně těžkou nebo těžší psoriázou v oblasti vlasové pokožky na počátku studie, které byly v 32. týdnu znovu randomizovány do APR 30 dvakrát denně. Subjekty s chybějícími údaji byly považovány za subjekty nereagující na léčbu.
V 52. týdnu studie ESTEEM 1 mělo odpověď PASI-75 přibližně 61 % pacientů, kteří byli v 32. týdnu znovu randomizovaní do skupiny léčené apremilastem. Z pacientů dosahujících alespoň odpovědi PASI-75, kteří byli v 32. týdnu znovu randomizováni do skupiny užívající placebo v průběhu randomizované fáze s vysazením léčby, jich dosáhlo v 52. týdnu 11,7 % odpovědi PASI-75. Střední doba do ztráty odpovědi PASI-75 mezi pacienty, kteří byli znovu randomizováni do skupiny užívající placebo, byl 5,1 týdne.
Ve studii ESTEEM 2 mělo v 52. týdnu odpověď PASI-50 přibližně 80,3 % pacientů, kteří byli v 32. týdnu znovu randomizováni do skupiny léčené apremilastem. Z pacientů dosahujících alespoň odpovědi PASI-50, kteří byli v 32. týdnu znovu randomizováni do skupiny užívající placebo, jich dosáhlo v 52. týdnu 24,2 % odpovědi PASI-50. Střední doba do ztráty 50 % zlepšení PASI v 32. týdnu byl 12,4 týdne.
Po randomizovaném vysazení léčby v 32. týdnu přibližně 70 % pacientů ve studii ESTEEM 1 a 65,6 % pacientů ve studii ESTEEM 2 znovu dosáhlo po opětovném zahájení léčby apremilastem odpovědí PASI-75 (ESTEEM 1) nebo PASI-50 (ESTEEM 2). Doba trvání opětovné léčby se kvůli uspořádání studií lišila a pohybovala se v rozmezí od 2,6 do 22,1 týdne.
Ve studii ESTEEM 1 bylo pacientům randomizovaným do skupiny léčené apremilastem na počátku studie, kteří nedosáhli odpovědi PASI-75 v 32. týdnu, povoleno užívat souběžnou lokální léčbu a/nebo léčbu UVB světlem mezi 32. a 52. týdnem. Z těchto pacientů dosáhlo 12 % odpovědi PASI-75 v 52. týdnu u léčby apremilastem v kombinaci s lokální léčbou a/nebo léčbou světlem.
Ve studiích ESTEEM 1 a ESTEEM 2 byla v 16. týdnu pozorována významná zlepšení (zmírnění) psoriázy nehtu, měřená průměrnou procentní změnou v indexu závažnosti psoriázy nehtu (NAPSI) oproti výchozí hodnotě u pacientů léčených apremilastem v porovnání s pacienty užívajícími placebo (p < 0,0001 a p = 0,0052, v uvedeném pořadí). Další zlepšení psoriázy nehtů byla pozorována v 32. týdnu u pacientů nepřetržitě léčených apremilastem .
V 16. týdnu studií ESTEEM 1 a ESTEEM 2 byla u pacientů léčených apremilastem oproti pacientům užívajícím placebo (p < 0,0001 v obou studiích) pozorována významná zlepšení psoriázy v oblasti vlasové pokožky s minimálně středně těžkou závažností (> 3), měřená dle podílu pacientů dosahujících čistého (0) nebo minimálního (1) celkového hodnocení psoriázy v oblasti vlasové pokožky lékařem (ScPGA). Zlepšení byla zpravidla udržena u subjektů, které byly v 32. týdnu znovu randomizovány do skupiny léčené přípravkem Otezla až do 52. týdne (Tabulka 5).
Ve studiích ESTEEM 1 a ESTEEM 2 byla u pacientů léčených apremilastem v porovnání s pacienty užívajícími placebo prokázána významná zlepšení v kvalitě života, měřená dle dermatologického indexu kvality života (DLQI) a SF-36v2MCS (Tabulka 4). Zlepšení v DLQI byla udržena až do 52. týdne u subjektů, které byly v 32. týdnu znovu randomizovány do skupiny léčené apremilastem (Tabulka 5). Ve studii ESTEEM 1 bylo navíc u pacientů léčených apremilastem v porovnání s placebem dosaženo významného zlepšení indexu v dotazníku pro posuzování pracovního omezení (WLQ-25).
5.2 Farmakokinetické vlastnosti
Absorpce
Apremilast se dobře vstřebává s absolutní biologickou dostupností po perorálním podání přibližně 73 % a s maximální plazmatickou koncentrací (Cmax) v mediánu doby (tmax) přibližně 2,5 hodiny. Farmakokinetické vlastnosti apremilastu jsou lineární, se zvýšením systémové expozice úměrně k dávce v dávkovém rozmezí 10 až 100 mg denně. Kumulace je minimální, je-li apremilast podáván jednou denně, a přibližně 53 % u zdravých subjektů a 68 % u pacientů s psoriázou, je-li podáván dvakrát denně. Podávání společně s jídlem nemění biologickou dostupnost, apremilast lze tedy podávat s jídlem nebo bez jídla.
Distribuce
Vazba apremilastu na plazmatické proteiny u člověka je přibližně 68 %. Střední zjevný distribuční objem (Vd) je 87 l, což svědčí o extravaskulární distribuci.
Biotransformace
Apremilast je ve velké míře metabolizován jak cestou zprostředkovanou CYP, tak i jinými (nezahrnujícími CYP) cestami, včetně oxidace, hydrolýzy a konjugace. To naznačuje, že inhibice jedné z cest ovlivňující clearance pravděpodobně nezpůsobí výrazné lékové interakce. Oxidační metabolismus apremilastu je zprostředkován primárně CYP3A4 s menším přispěním CYP1A2 a CYP2A6. Apremilast je hlavní cirkulující složkou po perorálním podání. Apremilast prochází rozsáhlou metabolizací, pouze 3 % podávané původní sloučeniny se vylučuje v moči a 7 % se vylučuje ve stolici. Hlavním cirkulujícím inaktivním metabolitem je glukuronidový konjugát O-demetylovaného apremilastu (M12). Jelikož je apremilast substrátem CYP3A4, snižuje se jeho expozice, je-li podáván souběžně s rifampicinem, silným induktorem CYP3A4.
In vitro není apremilast inhibitorem ani induktorem enzymů cytochromu P450. Proto je nepravděpodobné, že by apremilast podávaný souběžně se substráty enzymů CYP ovlivnil clearance a expozici léčivých látek, které jsou metabolizovány enzymy CYP.
In vitro je apremilast substrátem a slabým inhibitorem P-glykoproteinu (IC50 > 50pM), nepředpokládá se však, že by se objevily klinicky relevantní lékové interakce zprostředkované P-glykoproteinem (P-gp).
In vitro má apremilast malý nebo nemá žádný inhibiční účinek (IC50 > 10pM) na transportér organických aniontů (OAT) 1 a OAT3, transportér organických kationtů (OCT) 2, transportní polypeptid organických aniontů (OATP) 1B1 a OATP1B3 nebo na protein BCRP ((breast cancer resistant protein) a není substrátem těchto transportérů. Proto jsou klinicky relevantní lékové interakce nepravděpodobné, je-li apremilast podáván souběžně s léky, které jsou substráty nebo inhibitory těchto transportérů.
Eliminace
Plazmatická clearance apremilastu je v průměru asi 10 l/hod u zdravých subjektů s terminálním poločasem eliminace asi 9 hodin. Po perorálním podání radioaktivně značeného apremilastu bylo v moči zjištěno 58 % radioaktivity, resp. 39 % ve stolici, s přibližně 3 % radioaktivní dávky vyloučené ve formě apremilastu v moči, resp. 7 % ve stolici.
Starší pacienti
Apremilast byl hodnocen u mladých i starších zdravých subjektů. Expozice apremilastu u starších subjektů (65 až 85 let) je asi o 13 % vyšší u AUC a o 6 % vyšší u Cmax apremilastu než u mladších subjektů (18až 55 let). U subjektů starších 75 let jsou k dispozici jen omezené farmakokinetické údaje z klinických studií. U starších pacientů není nutná úprava dávkování.
Porucha funkce ledvin
Neexistuje žádný výrazný rozdíl ve farmakokinetice apremilastu mezi subjekty s mírnou nebo středně vážnou poruchou funkce ledvin a srovnatelnými zdravými subjekty (N = 8 každý). Z výsledků vyplývá, že u pacientů s mírnou až středně vážnou poruchou funkce ledvin není nutná žádná úprava dávkování. U pacientů se závažnou poruchou funkce ledvin (eGFR menší než 30 ml/min/1,73 m2 nebo clearance kreatininu < 30 ml/min) snižte dávku apremilastu na 30 mg jednou denně. U 8 subjektů se závažnou poruchou funkce ledvin, kterým byla podána jedna dávka 30 mg apremilastu, se AUC apremilastu zvýšila přibližně o 89 % a Cmax přibližně o 42 %.
Porucha funkce jater
Farmakokinetické vlastnosti apremilastu a jeho hlavního metabolitu M12 nejsou ovlivněny středně vážnou nebo vážnou poruchou funkce jater. U pacientů s poruchou funkce jater není nutná žádná úprava dávkování.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po opakovaném podávání neodhalily žádné zvláštní riziko pro člověka. Nebyl prokázán žádný potenciál k imunotoxicitě, podráždění kůže nebo fototoxicitě.
Fertilita a časný embryonální vývoj
Studie fertility myších samců neodhalila žádný účinek apremilastu na fertilitu samců po perorálním podání v dávkách 1, 10, 25 a 50 mg/kg/den; hladina NOAEL (žádného pozorovaného nežádoucího účinku) na fertilitu samců byla vyšší než při trojnásobné klinické expozici 50 mg/kg/den.
V kombinované studii fertility myších samic a embryonální/fetální vývojové toxicity s perorálními dávkami 10, 20, 40 a 80 mg/kg/den bylo u dávek 20 mg/kg/den a vyšších pozorováno prodloužení estrálních cyklů a doby do páření, i přesto se všechny myši pářily a počet březostí nebyl ovlivněn. Hladina žádného pozorovaného účinku (NOEL) na samičí fertilitu byla 10 mg/kg/den (jednonásobek klinické expozice).
Embrvo-fetální vývoj
V kombinované studii fertility myších samic a embryonální/fetální vývojové toxicity s perorálními dávkami 10, 20, 40 a 80 mg/kg/den došlo při dávkách 20, 40 a 80 mg/kg/den ke zvýšení absolutní a/nebo relativní hmotnosti srdce matek. U dávek 20, 40 a 80 mg/kg/den byl pozorován zvýšený počet časných resorpcí a snížený počet osifikovaných zánártních kůstek. U dávek 40 a 80 mg/kg/den bylo pozorováno snížení hmotnosti plodu a zpoždění osifikace supraokcipitálního skeletu lebky. NOEL
u myších matek a plodů byla 10 mg/kg/den (1,3násobek klinické expozice).
Ve studii embryonální/fetální vývojové toxicity u opic měly perorální dávky 20, 50, 200 a 1000 mg/kg/den za následek s dávkou související nárůst prenatálních ztrát (potratů) při dávkách 50 mg/kg/den a vyšších; při dávkách 20 mg/kg/den (1,4násobek klinické expozice) nebyl pozorován žádný účinek testované látky na prenatální ztrátu.
Prenatální a postnatální vývoj
V prenatální a postnatální studii byl apremilast podáván perorálně březím myším samicím v dávkách 10, 80 a 300 mg/kg/den od 6. dne gestace do 20. dne laktace. Při dávkách 300 mg/kg/den byly pozorovány pokles a přírůstek tělesné hmotnosti matek a jedno úmrtí spojené s obtížemi při porodu mláďat. Byly rovněž pozorovány fyzické známky toxicity u matek spojované s porodem mláďat
u jedné myši pro každou dávku (80 a 300 mg/kg/den). Při dávkách > 80 mg/kg/den (> 4,0násobek klinické expozice) bylo během prvního týdne laktace pozorováno zvýšení perinatální a postnatální mortality mláďat a pokles tělesné hmotnosti mláďat. Nebyly zjištěny žádné účinky apremilastu na dobu trvání březosti, počet březích myší na konci gestačního období, počet myší, které porodily mláďata, ani žádný vliv na vývoj mláďat po 7. dni po porodu. Je pravděpodobné, že vliv na vývoj mláďat pozorovaný během prvního týdne postnatálního období souvisel s toxicitou pro mláďata spojenou s apremilastem (pokles hmotnosti a životaschopnosti mláďat) a/nebo nedostatkem mateřské péče (vyšší incidence mláďat bez mléka v žaludku). Veškeré vývojové účinky byly pozorovány v prvním týdnu postnatálního období, ve zbývajícím období před a po odstavení nebyly pozorovány žádné účinky spojené s apremilastem, včetně parametrů pohlavního dospívání, chování, páření, fertility a dělohy. NOEL u myší pro toxicitu pro matky a generaci F1 byla 10 mg/kg/den (1,3násobek klinické hodnoty AUC).
Studie kancerogenity
Studie kancerogenity u myší a potkanů neprokázaly žádnou kancerogenitu v souvislosti s léčbou apremilastem.
Studie genotoxicity
Apremilast není genotoxický. Apremilast nevyvolal mutace v Amesově testu ani chromozomální aberace u kultivovaných lidských lymfocytů z periferní krve s metabolickou aktivací nebo bez ní. Apremilast nebyl při dávkách do 2000 mg/kg/den klastogenní v in vivo mikronukleárním testu u myší.
Další studie
Nebyl prokázán žádný potenciál k imunotoxicitě, podráždění kůže nebo fototoxicitě.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety:
Mikrokrystalická celulóza Monohydrát laktózy Sodná sůl kroskarmelózy Magnesium-stearát
Potah tablety: Polyvinylalkohol Oxid titaničitý (E171) Makrogol 3350 Mastek
Oxid železitý červený (E172)
Tablety 30 mg obsahují navíc oxid železitý žlutý (E172) a oxid železitý černý (E172).
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
24 měsíců.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30 °C.
6.5 Druh obalu a obsah balení
Blistry z PVC/Al fólie obsahující14 potahovaných tablet, v balení s 56 tabletami (30 mg) a 168 tabletami (30 mg).
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
CelgeneEurope Ltd.
1 LongwalkRoad Stockley Park Uxbridge UB11 1DB Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/981/002
EU/1/14/981/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 15. ledna 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Celgene Europe Limited
1 Longwalk Road
Stockley Park
Uxbridge
Middlesex UB11 1DB Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
Otezla 10 mg, potahované tablety Otezla 20 mg, potahované tablety Otezla 30 mg, potahované tablety apremilastum
Jedna potahovaná tableta obsahuje apremilastum 10 mg, 20 mg nebo 30 mg.
Obsahuje laktózu. Podrobnější informace naleznete v příbalové informaci.
Potahované tablety Balení pro úvodní léčbu 4 potahované tablety 10 mg 4 potahované tablety 20 mg 19 potahovaných tablet 30 mg
Před použitím si přečtěte příbalovou informaci. Perorální podání.
Uchovávejte mimo dohled a dosah dětí.
EXP
Uchovávejte při teplotě do 30 °C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
CelgeneEurope Ltd. 1 LongwalkRoad Stockley Park Uxbridge UB11 1DB Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/981/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Otezla 10 mg Otezla 20 mg Otezla 30 mg
Blistr (údaje vytištěné přímo na obalové pouzdro, v němž je nepotištěný blistr s přípravkem zapečetěn)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Otezla 10 mg tablety Otezla 20 mg tablety Otezla 30 mg tablety
apremilastum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Celgene
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Otezla 30 mg potahované tablety apremilastum
Jedna potahovaná tableta obsahuje apremilastum 30 mg.
Obsahuje laktózu. Podrobnější informace naleznete v příbalové informaci.
Potahované tablety 56 potahovaných tablet 168 potahovaných tablet
Před použitím si přečtěte příbalovou informaci. Perorální podání.
Uchovávejte mimo dohled a dosah dětí.
EXP
Uchovávejte při teplotě do 30 °C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
CelgeneEurope Ltd. 1 LongwalkRoad Stockley Park Uxbridge UB11 1DB Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/981/002
EU/1/14/981/003
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Otezla 30 mg
BLISTR_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
Otezla 30 mg tablety apremilastum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Celgene
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Otezla 10 mg potahované tablety Otezla 20 mg potahované tablety Otezla 30 mg potahované tablety
Apremilastum
^FtciUo přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Otezla a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Otezla užívat
3. Jak se přípravek Otezla užívá
4. Možné nežádoucí účinky
5. Jak přípravek Otezla uchovávat
6. Obsah balení a další informace
1. Co je přípravek Otezla a k čemu se používá
Co je přípravek Otezla
Přípravek Otezla obsahuje léčivou látku apremilast. Ta patří do skupiny léků nazývaných inhibitory fosfodiesterázy typu 4, které pomáhají zmírňovat zánět.
K čemu se přípravek Otezla používá
Přípravek Otezla se používá k léčbě dospělých s následující onemocněními:
• Psoriatická artritida - pokud nemůžete používat jiný typ léku nazývaných „chorobu-modifikující antirevmatické léky“ (DMARD) nebo jste některý z těchto léků vyzkoušel(a) a nepůsobil na Vás.
• Středně těžká až těžká ložisková psoriáza- jestliže nemůžete používat jednu z následujících terapií nebo jste některou z těchto terapií vyzkoušel(a) a nepůsobila na Vás:
- léčba světlem - léčba, při níž jsou určité oblasti kůže vystaveny ultrafialovému světlu;
- systémová terapie - léčba, která ovlivňuje celé tělo spíš než jen jednu oblast, např. léky
cyklosporin nebo methotrexát.
Co je psoriatická artritida
Psoriatická artritida je zánětlivé onemocnění kloubů, obvykle doprovázené psoriázou (lupénka), zánětlivým onemocněním kůže.
Co je ložisková psoriáza
Psoriáza je zánětlivé onemocnění kůže, které může způsobovat červená, šupinatá, ztluštělá, svědivá a bolestivá místa na kůži a může také postihovat vlasovou pokožku a nehty.
Jak přípravek Otezla působí
Psoriatická artritida a psoriáza jsou obvykle celoživotní onemocnění a v současné době neexistuje vyléčení. Přípravek Otezla působí tak, že snižuje aktivitu enzymu v lidském těle, který se nazývá „fosfodiesteráza 4“ a je zapojen do zánětlivého procesu. Snížením aktivity tohoto enzymu může přípravek Otezla pomoci kontrolovat zánět spojený s psoriatickou artritidou a psoriázou, a tak omezit známky a příznaky těchto onemocnění.
U psoriatické artritidy vede léčba přípravkem Otezla ke zlepšení oteklých a bolestivých kloubů a může zlepšit Vaši celkovou tělesnou funkci.
U psoriázy vede léčba přípravkem Otezla ke snížení kožních ložisek psoriázy a dalších známek a příznaků onemocnění.
Bylo také prokázáno, že přípravek Otezla zlepšuje kvalitu života pacientů s psoriázou nebo psoriatickou artritidou. To znamená, že dopad Vašeho zdravotního stavu na každodenní činnosti, vztahy a další faktory by měl být menší než dříve.
2. Čemu musíte věnovat pozornost, než začnete přípravek Otezla užívat
Neužívejte přípravek Otezla:
• jestliže jste alergický(á) na apremilast nebo kteroukoli další složku tohoto přípravku (uvedenou v bodě 6);
• jestliže jste těhotná nebo se domníváte, že můžete být těhotná.
Upozornění a opatření
Před užitím přípravku Otezla se poraďte se svým lékařem nebo lékárníkem.
Jestliže Váš lékař usoudí, že máte podváhu, a Vy zaznamenáte neúmyslný pokles tělesné hmotnosti v průběhu léčby přípravkem Otezla, sdělte to svému lékaři.
Jestliže máte těžké onemocnění ledvin, pak je doporučená dávka přípravku Otezla 30 mg jednou denně (ranní dávka). Váš lékař Vám poradí, jak máte zvyšovat dávku, pokud poprvé začínáte užívat přípravek Otezla.
Děti a dospívající
Přípravek Otezla nebyl hodnocen u dětí a dospívajících, a proto se jeho použití u dětí a dospívajících ve věku do 17 let nedoporučuje.
Další léčivé přípravky a přípravek Otezla
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. To platí i pro léky, které jsou dostupné bez lékařského předpisu, a bylinné přípravky, neboť přípravek Otezla může ovlivnit způsob, jakým některé jiné léky působí. Také některé jiné léky mohou ovlivnit působení přípravku Otezla.
Především informujte svého lékaře nebo lékárníka dříve, než začnete užívat přípravek Otezla, jestliže užíváte kterýkoli z následujících léků:
• rifampicin - antibiotikum používané k léčbě tuberkulózy;
• fenytoin, fenobarbital a karbamazepin - léky používané k léčbě záchvatů nebo epilepsie;
• třezalka tečkovaná - bylinný přípravek používaný k léčbě mírné úzkosti a deprese.
Těhotenství a kojení
O účincích přípravku Otezla na těhotenství je k dispozici málo informací. Během užívání tohoto léčivého přípravku byste neměla otěhotnět a musíte používat účinnou metodu antikoncepce. Není známo, zda se tento lék vylučuje do lidského mateřského mléka. Přípravek Otezla se během kojení nemá používat.
Sdělte svému lékaři, pokud se domníváte, že můžete být těhotná, nebo plánujete otěhotnět, nebo pokud kojíte nebo chcete kojit.
Řízení dopravních prostředků a obsluha strojů
Přípravek Otezla nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
Přípravek Otezla obsahuje laktózu
Přípravek Otezla obsahuje laktózu (typ cukru). Jestliže Vám lékař sdělil, že netolerujete nebo nemůžete trávit některé cukry, poraďte se se svým lékařem dříve, než začnete tento přípravek používat.
3. Jak se přípravek Otezla užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se
svým lékařem nebo lékárníkem.
Kolik přípravku Otezla se užívá
• Když poprvé začínáte užívat přípravek Otezla, dostanete „balení pro úvodní léčbu“, které obsahuje všechny dávky tak, jak jsou uvedeny v tabulce níže.
• „Balení pro úvodní léčbu“ je jasně označeno, aby bylo zajištěno, že užijete správnou tabletu ve správnou dobu.
• Vaše léčba začne nejnižší dávkou, která se bude během prvních 6 dní léčby postupně zvyšovat.
• „Balení pro úvodní léčbu“ rovněž obsahuje dostatečné množství tablet v doporučené dávce na dalších 8 dní (7. až 14. den).
• Doporučená dávka přípravku Otezla je 30 mg dvakrát denně po dokončení fáze postupného zvyšování dávky - jedna 30mg dávka ráno a jedna 30mg dávka večer, v intervalu přibližně 12 hod, s nebo bez jídla.
• To představuje celkovou denní dávku 60 mg. Této doporučené dávky dosáhnete na konci 6. dne.
• Po dosažení doporučené dávky dostanete v předepsaném balení pouze tablety se silou 30 mg. Touto fází postupného zvyšování dávky budete muset projít pouze jednou, a to i v případě, že budete léčbu zahajovat znovu.
|
Den |
Ranní dávka |
Večerní dávka |
Celková denní dávka |
|
1. den |
10 mg (růžová) |
Neužívejte dávku |
10 mg |
|
2. den |
10 mg (růžová) |
10 mg (růžová) |
20 mg |
|
3. den |
10 mg (růžová) |
20 mg (hnědá) |
30 mg |
|
4. den |
20 mg (hnědá) |
20 mg (hnědá) |
40 mg |
|
5. den |
20 mg (hnědá) |
30 mg (béžová) |
50 mg |
|
6. den a dále |
30 mg (béžová) |
30 mg (béžová) |
60 mg |
Osoby s onemocněním ledvin
Jestliže máte vážné onemocnění ledvin, pak je doporučená dávka přípravku Otezla 30 mg jednou denně (ranní dávka). Váš lékař Vám poradí, jak máte zvyšovat dávku, pokud poprvé začínáte užívat přípravek Otezla.
Jak a kdy se přípravek Otezla užívá
• Tablety polykejte celé, nejlépe s vodou.
• Tablety můžete užívat s jídlem nebo bez jídla.
• Užívejte přípravek Otezla každý den přibližně ve stejnou dobu, jednu tabletu ráno a jednu tabletu večer.
• Pokud se Váš stav po šesti měsících léčby nezlepší, sdělte to svému lékaři.
Jestliže jste užil(a) více přípravku Otezla, než jste měl(a)
Jestliže jste užil(a) více přípravku Otezla, než jste měl(a), sdělte to svému lékaři nebo navštivte přímo nemocnici. Vezměte s sebou obal léku a tuto příbalovou informaci.
Jestliže jste zapomněl(a) užít přípravek Otezla
• Jestliže jste vynechal(a) dávku přípravku Otezla, užijte ji, jakmile si to uvědomíte. Jestliže se již blíží doba pro další dávku, vynechanou dávku neužívejte. Pokračujte další dávkou v obvyklou dobu.
• Neužívejte dvě dávky zároveň.
Jestliže jste přestal(a) užívat přípravek Otezla
• Přípravek Otezla užívejte, dokud Vám lékař neřekne, že máte léčbu ukončit.
• Nepřestávejte přípravek Otezla užívat bez předchozí konzultace s lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Velmi časté nežádoucí účinky (mohou postihnout více než 1 z 10 osob)
• průjem
• nevolnost
Časté nežádoucí účinky (mohou postihnout až 1 z 10 osob)
• kašel
• bolest zad
• zvracení
• pocit únavy
• bolest v žaludku
• ztráta chuti k jídlu
• časté vyprazdňování střev
• problémy se spaním (insomnie)
• porucha trávení nebo pálení žáhy
• bolest hlavy, migrény nebo tenzní bolesti hlavy
• infekce horních cest dýchacích, jako jsou nachlazení, příznaky rýmy, zánět paranazální dutiny
• zánět a otok průdušek (bronchitida)
• běžné nachlazení (zánět nosohltanu, nazofaryngitida)
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 osob)
• vyrážka
• váhový úbytek
• alergická reakce
• krvácení ve střevech nebo v žaludku
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Otezla uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na blistru a krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Uchovávejte při teplotě do 30 °C.
• Nepoužívejte přípravek Otezla, pokud si všimnete poškození obalu nebo známek manipulace s balením přípravku.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Otezla obsahuje
• Léčivou látkou je apremilastum.
• Jedna potahovaná tableta obsahuje apremilastum 10 mg.
• Jedna potahovaná tableta obsahuje apremilastum 20 mg.
• Jedna potahovaná tableta obsahuje apremilastum 30 mg.
• Dalšími složkami jádra tablety jsou mikrokrystalická celulóza, monohydrát laktózy, sodná sůl kroskarmelózy a magnesium-stearát.
• Potah tablety obsahuje polyvinylalkohol, oxid titaničitý (E171), makrogol, mastek, oxid železitý červený (E172).
• Potahované tablety 20 mg obsahují navíc oxid železitý žlutý (E172).
• Potahované tablety 30 mg obsahují navíc oxid železitý žlutý (E172) a oxid železitý černý
(E172).
Jak přípravek Otezla vypadá a co obsahuje toto balení
Přípravek Otezla 10 mg potahované tablety jsou růžové, ve tvaru kosočtverce, s vyrytým označením „APR“ na jedné straně a „10“ na druhé straně.
Přípravek Otezla 20 mg potahované tablety jsou hnědé, ve tvaru kosočtverce, s vyrytým označením „APR“ na jedné straně a „20“ na druhé straně.
Přípravek Otezla 30 mg potahované tablety jsou béžové, ve tvaru kosočtverce, s vyrytým označením „APR“ na jedné straně a „30“ na druhé straně.
Velikosti balení
• Balení pro úvodní léčbu je přehnuté pouzdro obsahující 27 tablet: 4 x 10mg tablety, 4 x 20mg tablety a 19 x 30mg tablety.
• Standardní balení na jeden měsíc obsahuje 56 x 30mg tablety.
• Standardní balení na tři měsíce obsahuje 168 x 30mg tablety.
Držitel rozhodnutí o registraci a výrobce
CelgeneEurope Ltd.
1 LongwalkRoad Stockley Park Uxbridge UB11 1DB Velká Británie
Tato příbalová informace byla naposledy revidována
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY V REGISTRACI
Vědecké závěry
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizované zprávy / aktualizovaných zpráv o bezpečnosti (PSUR) apremilastu dospěl výbor CHMP k těmto vědeckým závěrům:
Na základě analýzy případů gastrointestinálního krvácení, hlášených po uvedení přípravku na trh a v klinických hodnoceních, byl nalezen důkaz, který naznačuje příčinnou souvislost mezi gastrointestinálním krvácením a použitím apremilastu. Tato souvislost byla dále podpořena přezkoumáním hlášení ze systému Eudravigilance shromážděných na základě následujících parametrů: odpovídající časová souvislost; pozitivní dechallenge, ve všech případech se spontánním zotavením po ukončení podávání apremilastu a bez matoucích faktorů (souběžná medikace, zdravotní stav). V průběhu placebem kontrolovaných studií byl navíc zaznamenán vyšší počet případů gastrointestinálního krvácení u pacientů užívajících apremilast než u pacientů užívajících placebo, i když celkové počty byly nízké.
Vzhledem k dostupným údajům týkajícím se gastrointestinálního krvácení při použití apremilastu výbor PRAC považuje změny v údajích o přípravku za odůvodněné.
Výbor CHMP souhlasí s vědeckými závěry výboru PRAC.
Zdůvodnění změny v registraci
Na základě vědeckých závěrů týkajících se apremilastu výbor CHMP zastává stanovisko, že poměr přínosů a rizik léčivého přípravku obsahujícího / léčivých přípravků osahujících apremilast zůstává nezměněný, a to pod podmínkou, že v informacích o přípravku budou provedeny navrhované změny.
Výbor CHMP doporučuje změnu v registraci.
52