Orfadin 4Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Orfadin 2 mg tvrdé tobolky Orfadin 5 mg tvrdé tobolky Orfadin 10 mg tvrdé tobolky Orfadin 20 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tobolka obsahuje nitisinonum 2 mg.
Jedna tobolka obsahuje nitisinonum 5 mg.
Jedna tobolka obsahuje nitisinonum 10 mg.
Jedna tobolka obsahuje nitisinonum 20 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Bílé neprůsvitné tobolky (6x16 mm) černě označené „NTBC 2mg“ na jedné straně tobolky. Bílé neprůsvitné tobolky (6x16 mm) černě označené „NTBC 5mg“ na jedné straně tobolky. Bílé neprůsvitné tobolky (6x16 mm) černě označené „NTBC 10mg“ na jedné straně tobolky. Bílé neprůsvitné tobolky (6x16 mm) černě označené „NTBC 20mg“ na jedné straně tobolky. Tobolky obsahují bílý či téměr bílý prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých a pediatrických pacientů (jakéhokoli věkového rozmezí) s potvrzenou diagnózou hereditární tyrosinemie typu 1 (HT-1) kombinované s dietním omezením tyrosinu a fenylalaninu.
4.2 Dávkování a způsob podání
Léčbu nitisinonem má zahájit a kontrolovat lékař se zkušenostmi s léčbou pacientů s HT-1. Dávkování
Léčba všech genotypů nemoci se má zahájit co nejdříve, aby se zvýšila celková doba přežití a předešlo se komplikacím, jako je selhání jater, karcinom jater a onemocnění ledvin. Vedle léčby nitisinonem je nutno nasadit dietu s nízkým obsahem fenylalaninu a tyrosinu; kromě toho je třeba sledovat obsah aminokyselin v plazmě (viz body 4.4 a 4.8).
Doporučená úvodní dávka pro děti i dospělé je 1 mg/kg tělesné hmotnosti/den rozdělená do 2 dávek podávaných perorálně. Dávku nitisinonu je nutno jednotlivě upravit.
Úprava dávky
Při pravidelném sledování je třeba zjišťovat sukcinylaceton v moči, testovat funkci jater a měřit hladinu alfa-fetoproteinu (viz bod 4.4). Lze-li sukcinylaceton v moči zjistit i jeden měsíc po zahájení léčby nitisinonem, má se dávka nitisinonu zvýšit na 1,5 mg/kg tělesné hmotnosti/den, rozdělených do 2 dávek. Na základě zhodnocení všech biochemických parametrů je možné, že se má zvýšit na 2 mg/kg tělesné hmotnosti/den. U všech pacientů se však tato dávka má považovat za maximální dávku.
Je-li biochemická odezva uspokojivá, dávka se má upravit pouze podle zvýšení tělesné hmotnosti.
Během zavádění léčby nebo dojde-li ke zhoršení stavu, může však být kromě výše uvedených testů nutné také úzce sledovat všechny dostupné biochemické parametry (tj. sukcinylaceton v plazmě, 5-aminolevulinát v moči (ALA) a porfobilinogen v erytrocytech (činnost PBG-syntázy).
Zvláštní skupiny pacientů
Žádná zvláštní doporučení pro dávkování pacientům vyššího věku nebo pacientům s poruchou funkce ledvin či jater neexistují.
Pediatrická populace
Doporučené dávkování v mg/kg tělesné hmotnosti je u dětí stejné jako u dospělých.
Způsob podání
Tobolku lze otevřít a její obsah rozmíchat v malém množství vody nebo v předepsané dietě bezprostředně před požitím.
Přípravek Orfadin je také k dispozici ve formě perorální suspenze 4 mg/ml pro pediatrické pacienty, kteří mají problémy s polykáním tobolek.
Začne-li se proto při léčbě podávat nitisinon s jídlem, doporučuje se zachovat tento způsob podávání, viz bod 4.5.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoliv pomocnou látku uvedenou v bodě 6.1.
Matky užívající nitisinon nesmí kojit (viz body 4.6 a 5.3).
4.4 Zvláštní upozornění a opatření pro použití Monitorování hladiny tyrosinu v plazmě
Doporučuje se, aby před zahájením léčby nitisinonem byla provedena prohlídka očí pomocí štěrbinové lampy. Pacienta, u něhož se během léčby nitisinonem projeví poruchy zraku, má okamžitě vyšetřit oftalmolog. Je třeba ověřit, zda pacient dodržuje svůj dietní režim, a změřit koncentraci tyrosinu v plazmě. Je-li hladina tyrosinu v plazmě nad 500 mikromol/l, je třeba nasadit dietu s větším omezením tyrosinu a fenylalaninu. Nedoporučuje se snižovat koncentraci tyrosinu v plazmě snížením či vysazením nitisinonu, neboť vzhledem k metabolické poruše by se klinický stav pacienta mohl zhoršit.
Monitorování jater
Funkci jater je třeba pravidelně sledovat pomocí testů jaterní funkce a zobrazování jater. Doporučuje se také sledovat koncentrace alfa-fetoproteinu v séru. Zvýšení koncentrace alfa-fetoproteinu v séru může znamenat, že léčba je nedostatečná. Pacienty se zvyšující se hodnotou alfa-fetoproteinu nebo známkami uzlíků v játrech je třeba vždy vyšetřit vzhledem k možné hepatální malignitě.
Monitorování trombocytů a leukocytů
Doporučuje se pravidelně sledovat počet trombocytů a leukocytů, neboť při klinickém hodnocení bylo zjištěno několik případů reverzibilní trombocytopenie a leukopenie.
Monitorovací návštěva má probíhat každých 6 měsíců; kratší intervaly mezi návštěvami j sou doporučeny pouze v případě výskytu nežádoucích účinků.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí s jinými léčivými přípravky.
Nitisinon se metabolizuje in vitro pomocí CYP 3A4, takže při podávání nitisinonu společně s inhibitory nebo induktory tohoto enzymu je možné, že bude muset být upravena jeho dávka.
Na základě studií in vitro se neočekává, že by nitisinon inhiboval metabolismy zprostředkované CYP 1A2, 2C9, 2C19, 2D6, 2E1 nebo 3A4.
Žádné formální studie potravinových interakcí přípravku Orfadin tvrdé tobolky nebyly provedeny. Nitisinon byl však podáván s jídlem při získávání údajů o účinnosti a bezpečnosti. Začne-li se proto při léčbě přípravkem Orfadin tvrdé tobolky podávat nitisinon s jídlem, doporučuje se zachovat tento způsob podávání, viz bod 4.2.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání nitisinonu těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Potenciální riziko pro člověka není známé. Orfadin lze v těhotenství použít pouze tehdy, když klinický stav ženy vyžaduje léčbu nitisinonem.
Kojení
Není známo, zda se nitisinon vylučuje do mateřského mléka. Studie na zvířatech ukázaly nepříznivé postnatální účinky po vystavení nitisinonu v mléku. Matky užívající nitisinon proto nesmí kojit, neboť riziko pro kojené dítě nelze vyloučit (viz body 4.3 a 5.3).
Fertilita
Neexistují žádné údaje o tom, že by nitisinon ovlivňoval fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Orfadin má malý vliv na schopnost řídit nebo obsluhovat stroje. Oční nežádoucí účinky (viz bod 4.8) mohou nepříznivě působit na zrak a pokud je zrak ovlivněn, nemá pacient řídit nebo obsluhovat stroje, dokud účinek nevymizí.
4.8 Nežádoucí účinky
Přehled bezpečnostního profilu
Svým způsobem účinku nitisinon zvyšuje hladiny tyrosinu u všech pacientů léčených nitisinonem. Nežádoucí účinky související s očima, např. konjunktivitida, zákal rohovky, keratitida, fotofobie a bolest očí související se zvýšenými hodnotami tyrosinu jsou proto časté. Mezi jiné časté nežádoucí účinky patří trombocytopenie, leukopenie a granulocytopenie. Exfoliativní dermatitida se může objevit méně často.
Nežádoucí podmínky uvedené v tabulce
Nežádoucí účinky jsou uvedené níže podle třídy orgánových systémů MedDRA a absolutní frekvenci a jsou založeny na údajích z klinického hodnocení a postmarketingového použití. Frekvence se definuje jako velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nepříznivé nežádoucí účinky seřazeny podle klesající závažnosti.
|
Třída orgánového systému MedDRA |
F rekvence |
Nežádoucí účinek |
|
Poruchy krve a lymfatického systému |
Časté |
T rombocytopenie, leukopenie, granulocytopenie |
|
Méně časté |
Leukocytóza | |
|
Poruchy oka |
Časté |
Konjunktivitida, zákal oční rohovky, keratitida, fotofobie, bolest oka |
|
Méně časté |
Blefaritida | |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Exfoliativní dermatitida, erytematózní vyrážka, pruritus |
|
Vyšetření |
Velmi časté |
Zvýšené hodnoty tyrosinu |
Popis vybraných nežádoucích účinků
Léčba nitisinonem vede ke zvýšené hladině tyrosinu. Zvýšená hladina tyrosinu se spojuje s nežádoucími účinky spojenými s očima, např. zákalem rohovky a hyperkeratotickými lézemi. Omezením tyrosinu a fenylalaninu ve stravě by se měla omezit toxicita spojená s tímto druhem tyrosinemie pomocí snížení hladin tyrosinu (viz bod 4.4).
V klinických studiích byla granulocytopenie pouze méně často vážná (<0,5 x 109/l) a nebyla spojována s infekcemi. Nežádoucí účinky postihující třídu orgánových systémů MedDRA „Poruchy krve a lymfatického systému“ během pokračující léčby pomocí nitisinonu ustoupily.
Pediatrická populace
Bezpečností profil je založen zejména na pediatrické populaci, protože léčba nitisinonem má být zahájena, jakmile je stanovena diagnóza dědičné tyrosinemie typu 1 (HT-1). Klinická studie ani postmarketingové údaje nenasvědčují tomu, že bezpečnostní profil je jiný v různých dílčích skupinách pediatrické populace nebo jiný než bezpečnostní profil u dospělých pacientů.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Náhodné požití nitisinonu jedinci na normální dietě s neomezeným obsahem tyrosinu a fenylalaninu způsobí zvýšenou hladinu tyrosinu. Zvýšená hladina tyrosinu byla spojena s toxicitou pro oči, kůži a nervový systém. Omezením tyrosinu a fenylalaninu v dietě by se měla omezit toxicita spojená s tímto druhem tyrosinemie. Žádné informace o specifické léčbě předávkování nejsou k dispozici.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Trávicí trakt a metabolismus; Trávicí trakt a metabolismus, jiná léčiva, ATC kód: A16A X04.
Mechanismus účinku
Biochemická porucha u hereditámí tyrosinemie typu 1 (HT-1) je nedostatek hydrolázy fumarylacetoacetátu, která je konečným enzymem katabolické tyrosinové látkové výměny. Nitisinon je kompetitivní inhibitor dioxygenázy 4-hydroxyfenylpyruvátu, enzymu, který předchází hydrolázu fumarylacetoacetátu v katabolické tyrosinové látkové výměně. Tím, že inhibuje normální katabolismus tyrosinu u pacientů s HT-1, nitisinon brání akumulaci toxických meziproduktů maleylacetoacetátu a fumarylacetoacetátu. U pacientů s HT-1 se tyto meziprodukty přeměňují na toxické metabolity sukcinylaceton a sukcinylacetoacetát. Sukcinylaceton inhibuje látkovou výměnu syntézy porfyrinu, vedoucí k akumulaci 5-aminolevulinátu.
Farmakodynamické účinky
Léčba nitisinonem vede k normalizaci metabolismu porfyrinu s normální činností erytrocytové porfobilinogenové syntázy a močového 5-aminolevulinátu, sníženému vylučování sukcinylacetonu močí, zvýšené koncentraci tyrosinu v plazmě a zvýšenému vylučování fenolkyselin močí. Údaje dostupné z klinické studie indikují, že u více než 90 % pacientů byl sukcinylaceton v moči normalizován během prvního týdne léčby. Po správném upravení dávky nemá být sukcinylaceton v moči ani plazmě prokazatelný.
Klinická účinnost a bezpečnost
Ze srovnání údajů pro historické kontrolní pacienty vyplývá, že léčba nitisinonem spolu s dietním omezením má u všech fenotypů HT-1 za následek vyšší pravděpodobnost přežití. Je to zřejmé z následující tabulky:
|
Věk na začátku léčby nebo diagnózy |
Pravděpodo |
rnost přežití | ||
|
Léčba nitisinonu |
Kontrola diety | |||
|
5 let |
10 let |
5 let |
10 let | |
|
< 2 měsíce |
82 |
-- |
28 |
-- |
|
> 2-6 měsíců |
95 |
95 |
51 |
34 |
|
> 6 měsíců |
92 |
86 |
93 |
59 |
Bylo dále zjištěno, že ve srovnání s historickými údaji o léčbě pouhým dietním omezením měla léčba nitisinonem za následek i snížení rizika vývoje hepatocelulárního karcinomu (2,3 až 3,7krát). Bylo zjištěno, že včasným zahájení léčby se riziko vývoje hepatocelulárního karcinomu dále snížilo (13,5krát, byla-li léčba zahájena před dosažením věku 12 měsíců).
5.2 Farmakokinetické vlastnosti
Formální studie absorpce, distribuce, metabolismu a eliminace nitisinonu nebyly provedeny.
U 10 zdravých dobrovolníků mužského pohlaví po podání jediné dávky tobolek nitisinonu (1 mg/kg tělesné hmotnosti) byl terminální poločas (střední) nitisinonu v plazmě 54 hodin (v rozmezí od 39 do 86 hodin). Populační farmakokinetická analýza byla provedena na skupině 207 pacientů s HT-1. Pro clearance a poločas byly zjištěny hodnoty 0,0956 l/kg tělesné hmotnosti/day, respektive 52,1 hodin.
Studie in vitro s použitím mikrozomů lidských jater a enzymů P450 získaných z cDNA ukázaly omezený metabolismus zprostředkovaný enzymem CYP 3A4.
5.3 Předklinické údaje vztahující se k bezpečnosti
Při klinicky relevantních dávkách nitisinonu byla zjištěna embryofetální toxicita u myší a králíků.
U králíků nitisinon vyvolal s dávkou spojené zvýšení malformací (umbilikální hernie a gastrochíza), počínající při dávce 2,5krát vyšší, než je doporučená dávka pro člověka (2 mg/kg tělesné hmotnosti/den).
Studie prenatálního a postnatálního vývoje u myší ukázaly statisticky významné zkrácení přežívání a snížení růstu u mláďat po odstavení při dávkách 125 a 25krát vyšších, než je maximální doporučená dávka pro člověka. Tato tendence k negativnímu účinku na přežívání mláďat se začala projevovat při dávce od 5 mg/kg/den. U potkanů způsobilo vystavení mléku sníženou střední hmotnosti mláďat a komeální léze.
Ve studiích in vitro nebyly zpozorovány žádné mutagenní účinky, ale zjistil se slabý klastogenní účinek. Nebyly zjištěny žádné důkazy genotoxicity in vivo (analýza mikronukleí u myší a neplánovaná syntetická analýza DNA myších jater). Ve 26týdenní studii karcinogenity u transgenních myší (TgrasH2) se neprokázal karcinogenní potenciál nitisinonu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolek
Předbobtnalý kukuřičný škrob
Obal tobolky želatina
oxid titaničitý (E 171)
Potisk
černý oxid železitý (E 172), šelak,
propylenglykol hydroxid amonný.
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
18 měsíců.
Během doby použitelnosti může pacient uchovávat tobolky po jedno období 2 měsíce při teplotě nepřesahující 25°C, ale poté se přípravek musí zlikvidovat.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C-8°C).
6.5 Druh obalu a obsah balení
HDPE lahvička s LDPE víčkem odolným proti zneužití, obsahující 60 tobolek.
Každé balení obsahuje 1 lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Švédsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/303/001
EU/1/04/303/002
EU/1/04/303/003
EU/1/04/303/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21/02/2005
Datum posledního prodloužení registrace: 21/02/2010
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
1. NÁZEV PŘÍPRAVKU
Orfadin 4 mg/ml perorální suspenze
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje nitisinonum 4 mg.
Pomocné látky se známým účinkem:
Jeden ml obsahuje: sodík 0,7 mg (0,03 mmol) glycerol 500 mg natrium-benzoát 1 mg
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Perorální suspenze.
Bílá, mírně viskózní, neprůhledná suspenze.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých a pediatrických pacientů (jakéhokoli věkového rozmezí) s potvrzenou diagnózou hereditární tyrosinemie typu 1 (HT-1) kombinované s dietním omezením tyrosinu a fenylalaninu.
4.2 Dávkování a způsob podání
Léčbu nitisinonem má zahájit a kontrolovat lékař se zkušenostmi s léčbou pacientů s HT-1.
Dávkování
Léčba všech genotypů nemoci se má zahájit co nejdříve, aby se zvýšila celková doba přežití a předešlo se komplikacím, jako je selhání jater, karcinom jater a onemocnění ledvin. Vedle léčby nitisinonem je nutno nasadit dietu s nízkým obsahem fenylalaninu a tyrosinu; kromě toho je třeba sledovat obsah aminokyselin v plazmě (viz body 4.4 a 4.8).
Doporučená úvodní dávka pro děti i dospělé je 1 mg/kg tělesné hmotnosti/den rozdělená do 2 dávek podávaných perorálně. Dávku nitisinonu je nutno jednotlivě upravit.
Úprava dávky
Při pravidelném sledování je třeba zjišťovat sukcinylaceton v moči, testovat funkci jater a měřit hladinu alfa-fetoproteinu (viz bod 4.4). Lze-li sukcinylaceton v moči zjistit i jeden měsíc po zahájení léčby nitisinonem, má se dávka nitisinonu zvýšit na 1,5 mg/kg tělesné hmotnosti/den, rozdělených do 2 dávek. Na základě zhodnocení všech biochemických parametrů je možné, že semá zvýšit na 2 mg/kg tělesné hmotnosti/den. U všech pacientů se však tato dávka má považovat za maximální dávku.
Je-li biochemická odezva uspokojivá, dávka se má upravit pouze podle zvýšení tělesné hmotnosti.
Během zavádění léčby nebo dojde-li ke zhoršení stavu, může však být kromě výše uvedených testů nutné také úzce sledovat všechny dostupné biochemické parametry (tj. sukcinylaceton v plazmě, 5-aminolevulinát v moči (ALA) a porfobilinogen v erytrocytech (činnost PBG-syntázy).
Zvláštní skupiny pacientů
Žádná zvláštní doporučení pro dávkování pacientům vyššího věku nebo pacientům s poruchou funkce ledvin či jater neexistují.
Pediatrická populace
Doporučené dávkování v mg/kg tělesné hmotnosti je u dětí stejné jako u dospělých.
Způsob podání
Suspenze se podává pacientovi perorálně bez naředění pomocí stříkačky pro perorální podání. Stříkačky pro perorální podání o objemu 1 ml, 3 ml a 5 ml jsou přiloženy v balení k odměření přesné dávky v ml v souladu s předepsaným dávkováním. Stříkačky pro perorální podání mají stupnici po 0,01 ml; 0,1 ml a 0,2 ml.
Tabulka uvedená níže obsahuje převod dávek (mg/ml) pro tři velikosti stříkaček pro perorální podání: Tabulky převodu dávek pro jednotlivé tři velikosti stříkaček pro perorální podání:
|
1ml stříkačka pro |
Dá pnp Ori |
vka ravku adin |
|
perorální |
mg |
ml |
|
podání |
1,00 |
0,25 |
|
(stupnice |
1,25 |
0,31 |
|
p° 0,01 ml) |
1,50 |
0,38 |
|
1,75 |
0,44 | |
|
2,00 |
0,50 | |
|
2,25 |
0,56 | |
|
2,50 |
0,63 | |
|
2,75 |
0,69 | |
|
3,00 |
0,75 | |
|
3,25 |
0,81 | |
|
3,50 |
0,88 | |
|
3,75 |
0,94 | |
|
4,00 |
1,00 |
|
Dá pnp Ori |
vka ravku adin |
|
mg |
ml |
|
4,5 |
1,1 |
|
5,0 |
1,3 |
|
5,5 |
1,4 |
|
6,0 |
1,5 |
|
6,5 |
1,6 |
|
7,0 |
1,8 |
|
7,5 |
1,9 |
|
8,0 |
2,0 |
|
8,5 |
2,1 |
|
9,0 |
2,3 |
|
9,5 |
2,4 |
|
10,0 |
2,5 |
|
10,5 |
2,6 |
|
11,0 |
2,8 |
|
11,5 |
2,9 |
|
12,0 |
3,0 |
|
Dá pnp Ori |
vka ravku adin |
|
mg |
ml |
|
13,0 |
3,2 |
|
14, |
3,6 |
|
15,0 |
3,8 |
|
16,0 |
4,0 |
|
17,0 |
4,2 |
|
18,0 |
4,6 |
|
19,0 |
4,8 |
|
20,0 |
5,0 |
Důležité informace o návodu pro použití:
Před každým použitím se vyžaduje provedení redisperze důkladným protřepáním. Před redisperzí může léčivý přípravek vypadat jako kompaktní usazenina s mírně opalizujícím supernatantem. Dávka má být natažena a podána ihned po redisperzi. Je důležité pečlivě dodržovat instrukce pro přípravu a podání dávky uvedené v bodě 6.6, aby byla zajištěna přesnost dávkování.
Doporučuje se, aby zdravotnický pracovník poradil pacientovi nebo pečovateli, jak používat stříkačky pro perorální podání tak, aby se zajistilo podání správného objemu a aby byl předpis uveden v ml.
Orfadin je také dostupný ve 2mg, 5mg, 10mg a 20mg tobolkách, pokud jsou považovány za vhodnější pro pacienta.
Doporučuje se podávat perorální suspenzi s jídlem, viz bod 4.5.
Opatření, která je nutno učinit před zacházením s léčivým přípravkem nebo před jeho podáním K stříkačce pro perorální podání nemá být připojena žádná jehla, intravenózní linka nebo jiný prostředek pro parenterální podání.
Přípravek Orfadin je určen pouze pro perorální podání.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoliv pomocnou látku uvedenou v bodě 6.1.
Matky užívající nitisinon nesmí kojit (viz body 4.6 a 5.3).
4.4 Zvláštní upozornění a opatření pro použití Monitorování hladiny tvrosinu v plazmě
Doporučuje se, aby před zahájením léčby nitisinonem byla provedena prohlídka očí pomocí štěrbinové lampy. Pacienta, u něhož se během léčby nitisinonem projeví poruchy zraku, má okamžitě vyšetřit oftalmolog. Je třeba ověřit, zda pacient dodržuje svůj dietní režim, a změřit koncentraci tyrosinu v plazmě. Je-li hladina tyrosinu v plazmě nad 500 mikromol/l, je třeba nasadit dietu s větším omezením tyrosinu a fenylalaninu. Nedoporučuje se snižovat koncentraci tyrosinu v plazmě snížením či vysazením nitisinonu, neboť vzhledem k metabolické poruše by se klinický stav pacienta mohl zhoršit.
Monitorování jater
Funkci jater je třeba pravidelně sledovat pomocí testů jaterní funkce a zobrazování jater. Doporučuje se také sledovat koncentrace alfa-fetoproteinu v séru. Zvýšení koncentrace alfa-fetoproteinu v séru může znamenat, že léčba je nedostatečná. Pacienty se zvyšující se hodnotou alfa-fetoproteinu nebo známkami uzlíků v játrech je třeba vždy vyšetřit vzhledem k možné hepatální malignitě.
Monitorování trombocytů a leukocytů
Doporučuje se pravidelně sledovat počet trombocytů a leukocytů, neboť při klinickém hodnocení bylo zjištěno několik případů reverzibilní trombocytopenie a leukopenie.
Monitorovací návštěva má probíhat každých 6 měsíců; kratší intervaly mezi návštěvami j sou doporučeny pouze v případě výskytu nežádoucích účinků.
Pomocné látky se známým účinkem:
Glycerol
Jeden ml obsahuje 500 mg. Dávka 20 ml perorální suspenze (10 g glycerolu) nebo více může způsobit bolest hlavy, podráždění žaludku a průjem.
Sodík
Jeden ml obsahuje 0,7 mg (0,03 mmol).
Natrium-benzoát
Jeden ml obsahuje 1 mg. Zvýšení bilirubinu po jeho uvolnění z albuminu působením kyseliny benzoové a jejích solí může zvýšit žloutenku u předčasně narozených novorozenců a u novorozenců, narozených v termínu se žloutenkou, a může vést k jádrovému ikteru (depozita nekonjugovaného bilirubinu v mozkové tkáni). Proto má velký význam pečlivé monitorování plazmatických hladin bilirubinu u novorozenců. Před zahájením léčby je třeba stanovit hladinu bilirubinu: v případě významně zvýšených plazmatických hladin bilirubinu, zvláště u předčasně narozených pacientů s rizikovými faktory, jako je acidóza a nízká hladina albuminu, má být místo podání perorální suspenze zvážena léčba adekvátně odváženou částí tobolky přípravku Orfadin, dokud nedojde k normalizaci plazmatických hladin nekonjugovaného bilirubinu.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí s jinými léčivými přípravky.
Nitisinon se metabolizuje in vitro pomocí CYP 3A4, takže při podávání nitisinonu společně s inhibitory nebo induktory tohoto enzymu je možné, že bude muset být upravena jeho dávka.
Na základě studií in vitro se neočekává, že by nitisinon inhiboval metabolismy zprostředkované CYP 1A2, 2C9, 2C19, 2D6, 2E1 nebo 3A4.
Jídlo neovlivňuje biologickou dostupnost perorální suspenze nitisinonu, ale užití spolu s jídlem vede k nižší míře absorpce a následně k menšímu kolísání sérových koncentrací v rámci dávkovacího intervalu. Proto se doporučuje, aby byla perorální suspenze užívána s jídlem, viz bod 4.2.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání nitisinonu těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Potenciální riziko pro člověka není známé. Orfadin lze v těhotenství použít pouze tehdy, když klinický stav ženy vyžaduje léčbu nitisinonem.
Kojení
Není známo, zda se nitisinon vylučuje do mateřského mléka. Studie na zvířatech ukázaly nepříznivé postnatální účinky po vystavení nitisinonu v mléku. Matky užívající nitisinon proto nesmí kojit, neboť riziko pro kojené dítě nelze vyloučit (viz body 4.3 a 5.3).
Fertilita
Neexistují žádné údaje o tom, že by nitisinon ovlivňoval fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Orfadin má malý vliv na schopnost řídit nebo obsluhovat stroje. Oční nežádoucí účinky (viz bod 4.8) mohou nepříznivě působit na zrak a pokud je zrak ovlivněn, nemá pacient řídit nebo obsluhovat stroje, dokud účinek nevymizí.
4.8 Nežádoucí účinky
Přehled bezpečnostního profilu
Svým způsobem účinku nitisinon zvyšuje hladiny tyrosinu u všech pacientů léčených nitisinonem. Nežádoucí účinky související s očima, např. konjunktivitida, zákal rohovky, keratitida, fotofobie a bolest očí související se zvýšenými hodnotami tyrosinu jsou proto časté. Mezi jiné časté nežádoucí účinky patří trombocytopenie, leukopenie a granulocytopenie. Exfoliativní dermatitida se může objevit méně často.
Nežádoucí podmínky uvedené v tabulce
Nežádoucí účinky jsou uvedené níže podle třídy orgánových systémů MedDRA a absolutní frekvenci a jsou založeny na údajích z klinického hodnocení a postmarketingového použití. Frekvence se definuje jako velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nepříznivé nežádoucí účinky seřazeny podle klesající závažnosti.
|
Třída orgánového systému MedDRA |
F rekvence |
Nežádoucí účinek |
|
Poruchy krve a lymfatického systému |
Časté |
T rombocytopenie, leukopenie, granulocytopenie |
|
Méně časté |
Leukocytóza | |
|
Poruchy oka |
Časté |
Konjunktivitida, zákal oční rohovky, keratitida, fotofobie, bolest oka |
|
Méně časté |
Blefaritida | |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Exfoliativní dermatitida, erytematózní vyrážka, pruritus |
|
Vyšetření |
Velmi časté |
Zvýšené hodnoty tyrosinu |
Popis vybraných nežádoucích účinků
Léčba nitisinonem vede ke zvýšené hladině tyrosinu. Zvýšená hladina tyrosinu se spojuje s nežádoucími účinky spojenými s očima, např. zákalem rohovky a hyperkeratotickými lézemi. Omezením tyrosinu a fenylalaninu ve stravě by se měla omezit toxicita spojená s tímto druhem tyrosinemie pomocí snížení hladin tyrosinu (viz bod 4.4).
V klinických studiích byla granulocytopenie pouze méně často vážná (<0,5 x 109/l) a nebyla spojována s infekcemi. Nežádoucí účinky postihující třídu orgánových systémů MedDRA „Poruchy krve a lymfatického systému“ během pokračující léčby pomocí nitisinonu ustoupily.
Pediatrická populace
Bezpečností profil je založen zejména na pediatrické populaci, protože léčba nitisinonem má být zahájena, jakmile je stanovena diagnóza dědičné tyrosinemie typu 1 (HT-1). Klinická studie ani postmarketingové údaje nenasvědčují tomu, že bezpečnostní profil je jiný v různých dílčích skupinách pediatrické populace nebo jiný než bezpečnostní profil u dospělých pacientů.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Náhodné požití nitisinonu jedinci na normální dietě s neomezeným obsahem tyrosinu a fenylalaninu způsobí zvýšenou hladinu tyrosinu. Zvýšená hladina tyrosinu byla spojena s toxicitou pro oči, kůži a nervový systém. Omezením tyrosinu a fenylalaninu v dietě by se měla omezit toxicita spojená s tímto druhem tyrosinemie. Žádné informace o specifické léčbě předávkování nejsou k dispozici.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Trávicí trakt a metabolismus; Trávicí trakt a metabolismus, jiná léčiva, ATC kód: A16A X04.
Mechanismus účinku
Biochemická porucha u hereditámí tyrosinemie typu 1 (HT-1) je nedostatek hydrolázy fumarylacetoacetátu, která je konečným enzymem katabolické tyrosinové látkové výměny. Nitisinon je kompetitivní inhibitor dioxygenázy 4-hydroxyfenylpyruvátu, enzymu, který předchází hydrolázu fumarylacetoacetátu v katabolické tyrosinové látkové výměně. Tím, že inhibuje normální katabolismus tyrosinu u pacientů s HT-1, nitisinon brání akumulaci toxických meziproduktů maleylacetoacetátu a fumarylacetoacetátu. U pacientů s HT-1 se tyto meziprodukty přeměňují na toxické metabolity sukcinylaceton a sukcinylacetoacetát. Sukcinylaceton inhibuje látkovou výměnu syntézy porfyrinu, vedoucí k akumulaci 5-aminolevulinátu.
Farmakodynamické účinky
Léčba nitisinonem vede k normalizaci metabolismu porfyrinu s normální činností erytrocytové porfobilinogenové syntázy a močového 5-aminolevulinátu, sníženému vylučování sukcinylacetonu močí, zvýšené koncentraci tyrosinu v plazmě a zvýšenému vylučování fenolkyselin močí. Údaje dostupné z klinické studie indikují, že u více než 90 % pacientů byl sukcinylaceton v moči normalizován během prvního týdne léčby. Po správném upravení dávky nemá být sukcinylaceton v moči ani plazmě prokazatelný.
Klinická účinnost a bezpečnost
Ze srovnání údajů pro historické kontrolní pacienty vyplývá, že léčba nitisinonem spolu s dietním omezením má u všech fenotypů HT-1 za následek vyšší pravděpodobnost přežití. Je to zřejmé z následující tabulky:
|
Věk na začátku léčby nebo diagnózy |
Pravděpodo |
rnost přežití | ||
|
Léčba nitisinonu |
Kontrola diety | |||
|
5 let |
10 let |
5 let |
10 let | |
|
< 2 měsíce |
82 |
-- |
28 |
-- |
|
> 2-6 měsíců |
95 |
95 |
51 |
34 |
|
> 6 měsíců |
92 |
86 |
93 |
59 |
Bylo dále zjištěno, že ve srovnání s historickými údaji o léčbě pouhým dietním omezením měla léčba nitisinonem za následek i snížení rizika vývoje hepatocelulárního karcinomu (2,3 až -3,7krát). Bylo zjištěno, že včasným zahájení léčby se riziko vývoje hepatocelulárního karcinomu dále snížilo (13,5krát, byla-li léčba zahájena před dosažením věku 12 měsíců).
5.2 Farmakokinetické vlastnosti
Formální studie absorpce, distribuce, metabolismu a eliminace nitisinonu nebyly provedeny.
U 10 zdravých dobrovolníků mužského pohlaví po podání jediné dávky tobolek nitisinonu (1 mg/kg tělesné hmotnosti) byl terminální poločas (střední) nitisinonu v plazmě 54 hodin (v rozmezí od 39 do 86 hodin). Populační farmakokinetická analýza byla provedena na skupině 207 pacientů s HT-1. Pro clearance a poločas byly zjištěny hodnoty 0,0956 l/kg tělesné hmotnosti/day, respektive 52,1 hodin.
Studie in vitro s použitím mikrozomů lidských jater a enzymů P450 získaných z cDNA ukázaly omezený metabolismus zprostředkovaný enzymem CYP 3A4.
5.3 Předklinické údaje vztahující se k bezpečnosti
Při klinicky relevantních dávkách nitisinonu byla zjištěna embryofetální toxicita u myší a králíků.
U králíků nitisinon vyvolal s dávkou spojené zvýšení malformací (umbilikální hernie a gastrochíza), počínající při dávce 2,5krát vyšší, než je doporučená dávka pro člověka (2 mg/kg tělesné hmotnosti/den).
Studie prenatálního a postnatálního vývoje u myší ukázaly statisticky významné zkrácení přežívání a snížení růstu u mláďat po odstavení při dávkách 125 a 25krát vyšších, než je maximální doporučená dávka pro člověka. Tato tendence k negativnímu účinku na přežívání mláďat se začala projevovat při dávce od 5 mg/kg/den. U potkanů způsobilo vystavení mléku sníženou střední hmotnosti mláďat a komeální léze.
Ve studiích in vitro nebyly zpozorovány žádné mutagenní účinky, ale zjistil se slabý klastogenní účinek. Nebyly zjištěny žádné důkazy genotoxicity in vivo (analýza mikronukleí u myší a neplánovaná syntetická analýza DNA myších jater). Ve 26týdenní studii karcinogenity u transgenních myší (TgrasH2) se neprokázal karcinogenní potenciál nitisinonu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Hypromelóza Glycerol Polysorbát 80 Natrium-benzoát (E211)
Monohydrát kyseliny citronové
Natrium-citrát
Jahodové aroma (umělé)
Čištěná voda
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
Po prvním otevření je stabilita přípravku maximálně 2 měsíce při teplotě nepřesahující 25 °C, ale poté se musí zlikvidovat.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C-8 °C). Chraňte před mrazem.
Uchovávejte ve vzpřímené poloze.
Podmínky uchovávání tohoto léčivého přípravku po prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
100 ml hnědá skleněná lahvička (třídy III) s bílým HDPE dětským bezpečnostním šroubovacím uzávěrem a s kroužkem originality. Jedna lahvička obsahuje 90 ml perorální suspenze.
Jedno balení obsahuje jednu LDPE lahvičku, adaptér na lahvičku a 3 polypropylenové (PP) stříkačky pro perorální podání (1 ml, 3 ml a 5 ml).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Před každým použitím se vyžaduje provedení redisperze důkladným protřepáním. Před redisperzí může léčivý přípravek vypadat jako kompaktní usazenina s mírně opalizujícím supernatantem. Dávka má být natažena a podána ihned po redisperzi. Je důležité pečlivě
dodržovat níže uvedené instrukce pro přípravu a podání dávky, aby byla zajištěna přesnost dávkování.
Přiloženy jsou tři stříkačky pro perorální podání (1 ml, 3 ml a 5 ml) pro přesné odměření předepsané dávky. Doporučuje se, aby zdravotnický pracovník poradil pacientovi nebo ošetřovateli, jak používat stříkačku pro perorální podání tak, aby se zajistilo podání správného objemu.
Jak připravit novou lahvičku léku pro první použití:
Před užitím první dávky lahvičkou energicky zatřepejte, protože během dlouhodobého uchovávání částice vytvoří kompaktní usazeninu na dně lahvičky.
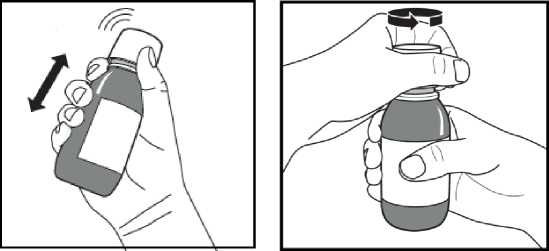
Obrázek A. Obrázek B.
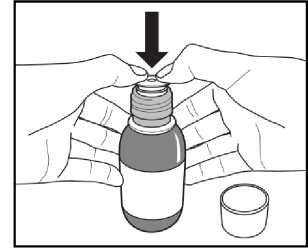
Obrázek C.
1. Lahvička se vyjme z chladničky. Na štítek na lahvičce se poznamená datum, kdy byla lahvička vytažena z chladničky.
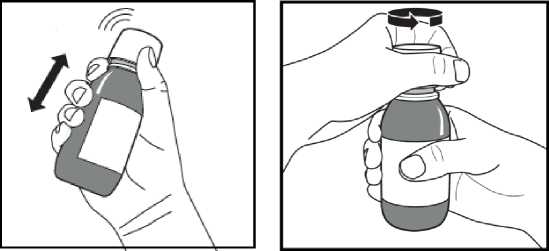
2. S lahvičkou se energicky třepe po dobu minimálně 20 sekund, dokud se kompaktní usazenina na dně lahvičky úplně nerozptýlí (Obrázek A).
3. Odjistí se dětský bezpečnostní šroubovací uzávěr tak, že se zatlačí silně dolů a otočí se jím proti směru hodinových ručiček (Obrázek B).
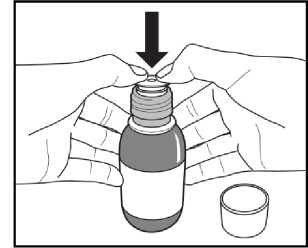
4. Otevřená lahvička se postaví na stůl ve vzpřímené poloze a plastový adaptér se silně zatlačí do hrdla lahvičky co možná nejhlouběji (Obrázek C). Lahvička se uzavře dětským bezpečnostním šroubovacím uzávěrem.
Pokyny k následnému dávkování jsou uvedeny níže v bodě ‘Jak připravit dávku léku’ Jak připravit dávku léku
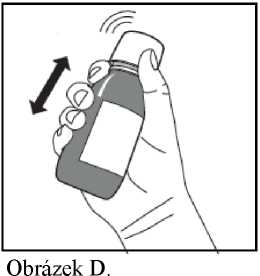
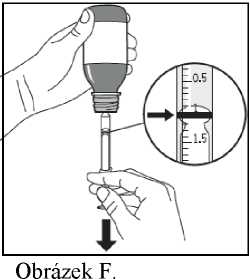
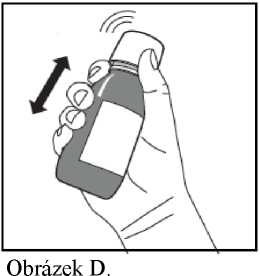
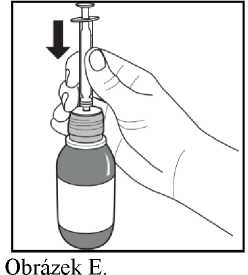
1. S lahvičkou se silně třepe po dobu minimálně 5 sekund (Obrázek D).
2. Okamžitě poté se má lahvička otevřít odjištěním dětského bezpečnostního šroubovacího uzávěru.
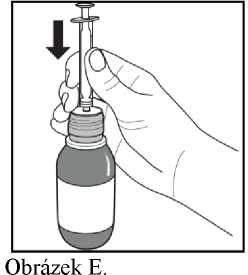
3. Píst ve stříkačce pro perorální podání se zatlačí úplně dolů.
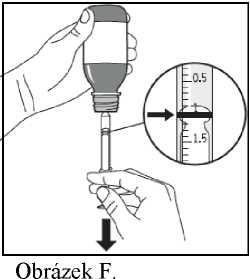
4. Lahvička se má držet ve vzpřímené poloze a stříkačka pro perorální podání se pevně zasune do otvoru adaptéru v horní části lahvičky (Obrázek E).
5. Lahvička se opatrně otočí dnem vzhůru se vsazenou stříkačkou pro perorální podání (Obrázek
F).
6. Pro odebrání předepsané dávky (ml) se píst vytáhne pomalu dolů, dokud není horní okraj černého prstence přesně v rovině s čárou označující dávku (Obrázek F). Pokud jsou uvnitř naplněné stříkačky pro perorální podání vidět vzduchové bubliny, zatlačí se píst znovu vzhůru, dokud se vzduchové bubliny nevytlačí. Pak se vytáhne píst znovu dolů, dokud horní okraj černého prstence není přesně v rovině s čárou označující dávku.
7. Lahvička se znovu otočí do vzpřímené polohy. Stříkačka pro perorální podání se odpojí tak, že se opatrně vykroutí z lahvičky.
8. Dávka se má podat okamžitě perorálně (bez naředění), aby se předešlo tvorbě usazeniny ve stříkačce pro perorální podání. Stříkačka pro perorální podání se má vyprazdňovat pomalu, aby se usnadnilo polykání; rychlé vystříknutí léku může způsobit dušení.
9. Okamžitě po použití se nasadí dětský bezpečnostní šroubovací uzávěr. Adaptér lahvičky se ponechá na svém místě.
10. Lahvičku je možné uchovávat při pokojové teplotě do 25 °C nebo v chladničce.
Čištění
Stříkačka pro perorální podání se okamžitě vyčistí vodou. Oddělí se válec stříkačky a píst a obojí
se opláchne vodou. Nadbytečná voda se vytřepe a rozebraná stříkačka pro perorální podání se
nechá vysušit, dokud se nebude znovu skládat pro další dávkování.
Likvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Švédsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/303/005
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21/02/2005
Datum posledního prodloužení registrace: 21/02/2010
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců odpovědných za propouštění šarží
2 mg, 5 mg, 10 mg a 20 mg tvrdé tobolky:
Apotek Produktion & Laboratorier AB
Prismavagen2
SE-141 75 Kungens Kurva
Švédsko
4 mg/ml perorální suspenze:
Apotek Produktion & Laboratorier AB Celsiusgatan 43 SE-212 14 Malmo Švédsko
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2)
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Orfadin 2 mg tvrdé tobolky Orfadin 5 mg tvrdé tobolky Orfadin 10 mg tvrdé tobolky Orfadin 20 mg tvrdé tobolky Nitisinonum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje nitisinonum 2 mg Jedna tobolka obsahuje nitisinonum 5 mg Jedna tobolka obsahuje nitisinonum 10 mg Jedna tobolka obsahuje nitisinonum 20 mg
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
60 tvrdých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Sweden
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/04/303/001
EU/1/04/303/002
EU/1/04/303/003
EU/1/04/303/004
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Orfadin 2 mg Orfadin 5 mg Orfadin 10 mg Orfadin 20 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Orfadin 2 mg tvrdé tobolky Orfadin 5 mg tvrdé tobolky Orfadin 10 mg tvrdé tobolky Orfadin 20 mg tvrdé tobolky Nitisinonum Perorální podání
2. ZPŮSOB PODÁNÍ
3. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum International AB
4. POUŽITELNOST
EXP
5. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Přípravek může být uchováván maximálně 2 měsíce při teplotě nepřesahující 25 °C, ale poté se musí zlikvidovat.
Datum, kdy byl vyňat z chladničky:
6. ČÍSLO ŠARŽE
Lot
7. OBSAH UDANÝ JAKO POČET
60 tobolek
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Orfadin 4 mg/ml perorální suspenze Nitisinonum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje nitisinonum 4 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Perorální suspenze
1 lahvička 90 ml, 1 adaptér na lahvičku, 3 stříkačky pro perorální podání (1 ml, 3 ml, 5 ml).
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem. Uchovávejte ve vzpřímené poloze.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Sweden
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/04/303/005
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Orfadin 4 mg/ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Orfadin 4 mg/ml perorální suspenze Nitisinonum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje nitisinonum 4 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Perorální suspenze 90 ml
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem. Uchovávejte ve vzpřímené poloze.
Přípravek může být uchováván maximálně 2 měsíce při teplotě nepřesahující 25 °C, ale poté se musí zlikvidovat.
Datum, kdy byl vyňat z chladničky:
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Sweden
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/04/303/005
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
B. PŘÍBALOVÁ INFORMACE
Orfadin 2 mg tvrdé tobolky Orfadin 5 mg tvrdé tobolky Orfadin 10 mg tvrdé tobolky Orfadin 20 mg tvrdé tobolky
Nitisinonum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Orfadin a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Orfadin užívat
3. Jak se Orfadin užívá
4. Možné nežádoucí účinky
5 Jak Orfadin uchovávat
6. Obsah balení a další informace
1. Co je Orfadin a k čemu se používá
Orfadin obsahuje léčivou látku nitisinon. Tento přípravek se užívá k léčbě vzácné nemoci zvané dědičná tyrosinemie typu 1 u dospělých, dospívajících a dětí (jakéhokoli věkového rozmezí).
Při této nemoci není tělo schopno úplně rozložit aminokyselinu tyrosin (aminokyseliny staví naše proteiny) a dochází k tvorbě škodlivých látek. Tyto látky se hromadí ve Vašem těle. Orfadin blokuje rozklad tyrosinu, a brání tak tvorbě těchto škodlivých látek.
Jelikož tyrosin nadále zůstává ve vašem těle, musíte při užívání tohoto přípravku dodržovat speciální dietu. Tato speciální dieta je založena na nízkém obsahu tyrosinu a fenylalaninu (další aminokyselina).
2. Čemu musíte věnovat pozornost, než začnete Orfadin užívat Neužívejte Orfadin
- jestliže jste alergický(á) na nitisinon nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6).
Během užívání tohoto přípravku nekojte, viz bod „Těhotenství a kojení“.
Upozornění a opatření
Před užitím přípravku Orfadin se poraďte se svým lékařem nebo lékárníkem,
- jestliže Vám zrudnou oči nebo se objeví nějaký jiný příznak nepříznivého vlivu na oči. Okamžitě požádejte lékaře o vyšetření očí. Oční problémy, viz bod 4, mohou být příznakem nedostatečné dietní kontroly.
Během léčby Vám budou odebrány krevní vzorky, aby lékař mohl zjistit, zda je Vaše léčba vhodná a zda nedochází k žádným nežádoucím účinkům, které by mohly způsobit krevní onemocnění.
Pravidelně Vám budou kontrolovat játra, neboť na ně má onemocnění vliv.
Každých šest měsíců Vás má vyšetřit lékař. Pokud pociťujete nežádoucí účinky, mají vyšetření probíhat častěji.
Další léčivé přípravky a Orfadin
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Přípravek Orfadin s jídlem
Pokud začnete léčbu užíváním přípravku spolu s jídlem, doporučujeme přípravek užívat spolu s jídlem po celou dobu léčby.
Těhotenství a kojení
Bezpečnost přípravku tohoto přípravku nebyla u těhotných a kojících žen studována.
Jestliže plánujete těhotenství, sdělte to laskavě svému lékaři. Jestliže otěhotníte, musíte to okamžitě sdělit svému lékaři.
Během užívání tohoto přípravku nekojte, viz bod „Neužívejte Orfadin“.
Řízení dopravních prostředků a obsluha strojů
Tento přípravek má malý vliv na schopnost řídit nebo obsluhovat stroje. Pokud však na sobě pozorujete nežádoucí účinky, které nepříznivě působí na zrak, neřiďte ani neobsluhujte stroje, dokud není Váš zrak opět normální (viz bod 4 „Možné nežádoucí účinky“).
3. Jak se Orfadin užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Léčbu tímto přípravkem by měl zahájit a kontrolovat lékař se zkušenostmi s léčbou této nemoci (dědičné tyrosinemie typu 1).
Doporučená celková denní dávka přípravku je 1 mg/kg tělesné hmotnosti, rozdělená na 2 dávky. Váš lékař přizpůsobí dávku individuálně.
Máte-li problémy s polykáním tobolek, můžete tobolku otevřít a rozmíchat prášek v malém množství vody či předepsané diety těsně před požitím.
Jestliže jste užil(a) více Orfadinu, než jste měl(a)
Jestliže jste užil(a) více tohoto přípravku, než jste měl(a), co nejdříve kontaktujte svého lékaře nebo lékárníka.
Jestliže jste zapomněl(a) užít Orfadin
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Jestliže jste zapomněl(a) užít dávku, sdělte to svému lékaři nebo lékárníkovi.
Jestliže jste přestal(a) užívat přípravek Orfadin
Jestliže máte dojem, že přípravek má nedostatečný účinek, kontaktujte svého lékaře. Neměňte dávkování léku ani nezastavujte užívání Orfadinu, aniž byste to konzultovali s Vaším lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Možné nežádoucí účinky
4.
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud zaznamenáte jakékoli nežádoucí účinky, které se týkají očí, ihned si s Vaším lékařem domluvte oční prohlídku. Léčba pomocí nitisinonu vede k vyšším hladinám tyrosinu v krvi, což může vést k příznakům souvisejícím s očima. Mezi časté nežádoucí účinky související s očima (mohou se vyskytnout u více než 1 z 10 uživatelů) způsobené vyššími hladinami tyrosinu patří zánět oka (zánět spojivek), zákal a zánět rohovky (keratitida), citlivost na světlo (fotofobie) a bolest oka. Zánět očního víčka (blefaritida) je méně častým nežádoucím účinkem (může se vyskytnout až u 1 ze 100 uživatelů).
Jiné časté nežádoucí účinky
- Snížený počet krevních destiček (trombocytopenie) a bílých krvinek (leukopenie), nedostatek určitých bílých krvinek (granulocytopenie).
Jiné méně časté nežádoucí účinky
- zvýšený počet bílých krvinek (leukocytóza),
- svědění (pruritus), kožní zánět (exfoliativní dermatitida), vyrážka.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Orfadin uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na lahvičce a krabičce za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Přípravek může být uchováván maximálně 2 měsíce při teplotě nepřesahující 25 °C, ale poté se musí zlikvidovat.
Vyjímáte-li přípravek z chladničky, nezapomeňte na nálepce lahvičky vyznačit datum.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co Orfadin obsahuje
Léčivou látkou je nitisinonum.
Orfadin 2 mg: Jedna tobolka obsahuje nitisinonum 2 mg. Orfadin 5 mg: Jedna tobolka obsahuje nitisinonum 5 mg. Orfadin 10 mg: Jedna tobolka obsahuje nitisinonum 10 mg.
Orfadin 20 mg: Jedna tobolka obsahuje nitisinonum 20 mg.
Dalšími složkami jsou:
Obsah tobolek:
předbobtnalý kukuřičný škrob.
Obal tobolky: želatina
oxid titaničitý (E 171).
Potisk:
černý oxid železitý (E 172), šelak,
propylenglykol, hydroxid amonný.
Jak Orfadin vypadá a co obsahuje toto balení
Tvrdé tobolky jsou bílé, neprůsvitné, tvrdé a vyrobené z želatiny, potištěné černě nápisem „NTBC“ a označením síly „2 mg“, „5 mg“, „10 mg“ nebo „20 mg“. Tobolky obsahují bílý až téměř bílý prášek.
Tobolky se dodávají v plastových lahvičkách s víčkem odolným proti zneužití. Jedna lahvička obsahuje 60 tobolek.
Držitel rozhodnutí o registraci
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Švédsko
Výrobce
Apotek Produktion & Laboratorier AB Prismavagen 2 SE-141 75 Kungens Kurva Švédsko
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
Orfadin 4 mg/ml perorální suspenze
Nitisinonum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Orfadin a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Orfadin užívat
3. Jak se Orfadin užívá
4. Možné nežádoucí účinky
5 Jak Orfadin uchovávat
6. Obsah balení a další informace
1. Co je Orfadin a k čemu se používá
Orfadin obsahuje léčivou látku nitisinon. Tento přípravek se užívá k léčbě vzácné nemoci zvané dědičná tyrosinemie typu 1 u dospělých, dospívajících a dětí (jakéhokoli věkového rozmezí).
Při této nemoci není tělo schopno úplně rozložit aminokyselinu tyrosin (aminokyseliny staví naše proteiny) a dochází k tvorbě škodlivých látek. Tyto látky se hromadí ve Vašem těle. Orfadin blokuje rozklad tyrosinu, a brání tak tvorbě těchto škodlivých látek.
Jelikož tyrosin nadále zůstává ve vašem těle, musíte při užívání tohoto přípravku dodržovat speciální dietu. Tato speciální dieta je založena na nízkém obsahu tyrosinu a fenylalaninu (další aminokyselina).
2. Čemu musíte věnovat pozornost, než začnete Orfadin užívat Neužívejte Orfadin
- jestliže jste alergický(á) na nitisinon nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6).
Během užívání tohoto přípravku nekojte, viz bod „Těhotenství a kojení“.
Upozornění a opatření
Před užitím přípravku Orfadin se poraďte se svým lékařem nebo lékárníkem,
- jestliže Vám zrudnou oči nebo se objeví nějaký jiný příznak nepříznivého vlivu na oči. Okamžitě požádejte lékaře o vyšetření očí. Oční problémy, viz bod 4, mohou být příznakem nedostatečné dietní kontroly.
Během léčby Vám budou odebrány krevní vzorky, aby lékař mohl zjistit, zda je Vaše léčba vhodná a zda nedochází k žádným nežádoucím účinkům, které by mohly způsobit krevní onemocnění.
Pravidelně Vám budou kontrolovat játra, neboť na ně má onemocnění vliv.
Každých šest měsíců Vás má vyšetřit lékař. Pokud pociťujete nežádoucí účinky, mají vyšetření probíhat častěji.
Další léčivé přípravky a Orfadin
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Přípravek Orfadin s jídlem
Doporučuje se užívat perorální suspenzi s jídlem.
Těhotenství a kojení
Bezpečnost tohoto přípravku nebyla u těhotných a kojících žen studována.
Jestliže plánujete těhotenství, sdělte to laskavě svému lékaři. Jestliže otěhotníte, musíte to okamžitě sdělit svému lékaři.
Během užívání tohoto přípravku nekojte, viz bod „Neužívejte Orfadin“.
Řízení dopravních prostředků a obsluha strojů
Tento přípravek má malý vliv na schopnost řídit nebo obsluhovat stroje. Pokud však na sobě pozorujete nežádoucí účinky, které nepříznivě působí na zrak, neřiďte ani neobsluhujte stroje, dokud není Váš zrak opět normální (viz bod 4 „Možné nežádoucí účinky“).
Přípravek Orfadin obsahuje sodík, glycerol a natrium-benzoát.
Tento léčivý přípravek obsahuje 0,7 mg (0,03 mmol) sodíku v jednom ml.
Dávka 20 ml perorální suspenze (10 g glycerolu) nebo více může způsobit bolest hlavy, podráždění žaludku a průjem.
Natrium-benzoát může prohloubit žloutenku (zežloutnutí kůže a očí) u předčasně narozených novorozenců a u novorozenců narozených v termínu se žloutenkou, a může vést k jádrovému ikteru (poškození mozku v důsledku depozit nekonjugovaného bilirubinu). Hladina bilirubinu v krvi novorozence (látka, která ve velkém množství způsobuje zežloutnutí kůže) bude pečlivě sledována. Pokud je hladina výrazně vyšší, než by měla být, zvláště u předčasně narozených dětí s rizikovými faktory, jako je acidóza (nízké pH krve) a nízká hladina albuminu (bílkovina v krvi), je třeba zvážit léčbu přípravkem Orfadin tobolky místo podání perorální suspenze, dokud nedojde k normalizaci plazmatických hladin bilirubinu.
3. Jak se Orfadin užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Dodržujte pečlivě instrukce pro přípravu a podání dávky uvedené níže, aby bylo zajištěno podání správné dávky.
Léčbu tímto přípravkem by měl zahájit a kontrolovat lékař se zkušenostmi s léčbou této nemoci (dědičné tyrosinemie typu 1).
Doporučená celková denní dávka přípravku je 1 mg/kg tělesné hmotnosti, rozdělená na 2 dávky. Váš lékař přizpůsobí dávku individuálně.
Perorální suspenze se užívá pomocí stříkačky pro perorální podání přímo do úst a bez ředění. Přípravek Orfadin nesmí být podáván injekčně. Nenasazujte na stříkačku injekční jehlu.
Jak připravit dávku k podání
Dávka, kterou Vám lékař předepíše, má být uvedena v ml suspenze a ne v mg. To proto, že stříkačka pro perorální podání, která se používá k odebrání správné dávky z lahvičky, bude označena v ml. Pokud je Váš předpis v mg, poraďte se se svým lékárníkem nebo lékařem.
Balení obsahuje lahvičku léku s uzávěrem, adaptér na lahvičku a tři stříkačky pro perorální podání (1 ml, 3 ml a 5 ml). Vždy používejte k odebrání léku jednu ze tří přiložených stříkaček pro perorální podání.
• 1 ml stříkačka pro perorální podání (nejmenší stříkačka pro perorální podání) je značena od 0,1 ml do 1 ml s dílky stupnice po 0,01 ml. Používá se k odměřování dávek do 1 ml.
• 3 ml stříkačka pro perorální podání (středně velká stříkačka pro perorální podání) je značena od 1 ml do 3 ml s dílky stupnice po 0,1 ml. Používá se k odměřování dávek od 1 ml do 3 ml.
• 5 ml stříkačka pro perorální podání (největší stříkačka pro perorální podání) je značena od 1 ml do 5 ml s dílky stupnice po 0,2 ml. Používá se k odměřování dávek větších než 3 ml.
Při dávkování léku je důležité, abyste používal(a) správnou stříkačku pro perorální podání. Váš lékař, lékárník nebo zdravotní sestra Vám poradí, kterou stříkačku pro perorální podání máte použít, a to v závislosti na dávce, která Vám byla předepsána.
Jak připravit novou lahvičku léku pro první použití:
Před užitím první dávky silně lahvičkou zatřepejte, protože během dlouhodobého uchovávání částice vytvoří kompaktní usazeninu na dně lahvičky. Dodržujte pokyny uvedené níže:
Obrázek A. Obrázek B.
Obrázek C.
1. Vyjměte lahvičku z chladničky. Na štítek na lahvičce poznamenejte datum, kdy jste ji vytáhli z chladničky.
2. Energicky lahvičkou třepejte po dobu minimálně 20 sekund, dokud se kompaktní usazenina na dně lahvičky úplně nerozpustí (Obrázek A).
3. Odjistěte dětský bezpečnostní šroubovací uzávěr tak, že ho zatlačíte silně dolů a otočíte jej proti směru hodinových ručiček (Obrázek B).
4. Postavte otevřenou lahvičku na stůl ve vzpřímené poloze. Zatlačte plastový adaptér silně do hrdla lahvičky co možná nejhlouběji (Obrázek C) a uzavřete lahvičku dětským bezpečnostním šroubovacím uzávěrem.
Pokyny k následnému dávkování jsou uvedeny níže v bodě ‘Jak připravit dávku léku’
Jak připravit dávku léku
1. Lahvičkou energicky třepejte po dobu minimálně 5 sekund (Obrázek D).
2. Okamžitě poté lahvičku otevřete odstraněním dětského bezpečnostního šroubovacího uzávěru.
3. Zatlačte píst uvnitř stříkačky pro perorální podání úplně dolů.
4. Držte lahvičku ve vzpřímené poloze a stříkačku pro perorální podání pevně zasuňte do otvoru adaptéru v horní části lahvičky (Obrázek E).
5. Opatrně otočte lahvičku dnem vzhůru se vsazenou stříkačkou pro perorální podání (Obrázek
F).
6. Pro odebrání předepsané dávky (ml) zatáhněte píst pomalu dolů, dokud není horní okraj černého prstence přesně v rovině s čárou označující dávku (Obrázek F). Pokud uvnitř naplněné stříkačky pro perorální podání pozorujete vzduchové bubliny, zatlačte píst znovu vzhůru, dokud vzduchové bubliny nevytlačíte. Pak zatlačte píst znovu dolů, dokud horní okraj černého prstence není přesně v rovině s čárou označující dávku.
7. Znovu lahvičku otočte do vzpřímené polohy. Odpojte stříkačku pro perorální podání tak, že ji opatrně vykroutíte z lahvičky.
8. Dávka se má podat okamžitě do úst (bez naředění), aby se předešlo tvorbě usazeniny ve stříkačce pro perorální podání. Stříkačka pro perorální podání se má vyprazdňovat pomalu, aby se usnadnilo polykání; rychlé vystříknutí léku může způsobit dušení.
9. Okamžitě po použití nasaďte dětský bezpečnostní šroubovací uzávěr. Adaptér lahvičky nechejte na svém místě.
10. Lahvičku je možné uchovávat při pokojové teplotě (neuchovávejte při teplotě nad 25 °C).
Čištění:
Stříkačku pro perorální podání okamžitě vypláchněte vodou. Oddělte válec stříkačky a píst a obojí
opláchněte vodou. Vytřepejte nadbytečnou vodu a nechejte rozebranou stříkačku pro perorální
podání vysušit, dokud ji nebudete znovu skládat pro další dávkování.
Jestliže jste užil(a) více Orfadinu, než jste měl(a)
Jestliže jste užil(a) více tohoto přípravku, než jste měl(a), co nejdříve kontaktujte svého lékaře nebo lékárníka.
Jestliže jste zapomněl(a) užít Orfadin
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Jestliže jste zapomněl(a) užít dávku, sdělte to svému lékaři nebo lékárníkovi.
Jestliže jste přestal(a) užívat přípravek Orfadin
Jestliže máte dojem, že přípravek má nedostatečný účinek, kontaktujte svého lékaře. Neměňte dávkování léku ani nezastavujte užívání Orfadinu, aniž byste to konzultovali s Vaším lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Možné nežádoucí účinky
4.
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud zaznamenáte jakékoli nežádoucí účinky, které se týkají očí, ihned si s Vaším lékařem domluvte oční prohlídku. Léčba pomocí nitisinonu vede k vyšším hladinám tyrosinu v krvi, což může vést k příznakům souvisejícím s očima. Mezi časté nežádoucí účinky související s očima (mohou se vyskytnout u více než 1 z 10 uživatelů) způsobené vyššími hladinami tyrosinu patří zánět oka (zánět spojivek), zákal a zánět rohovky (keratitida), citlivost na světlo (fotofobie) a bolest oka. Zánět očního víčka (blefaritida) je méně častým nežádoucím účinkem (může se vyskytnout až u 1 ze 100 uživatelů).
Jiné časté nežádoucí účinky
- Snížený počet krevních destiček (trombocytopenie) a bílých krvinek (leukopenie), nedostatek určitých bílých krvinek (granulocytopenie).
Jiné méně časté nežádoucí účinky
- zvýšený počet bílých krvinek (leukocytóza),
- svědění (pruritus), kožní zánět (exfoliativní dermatitida), vyrážka.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Orfadin uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na lahvičce a krabičce za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte lahvičku ve vzpřímené poloze.
Po prvním otevření lze přípravek uchovávat maximálně 2 měsíce při teplotě nepřevyšující 25 °C, poté se přípravek musí zlikvidovat.
Vyjímáte-li přípravek z chladničky, nezapomeňte na nálepce lahvičky vyznačit datum.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Orfadin obsahuje
- Léčivou látkou je nitisinonum. Jeden ml obsahuje nitisinonum 4 mg.
- Dalšími složkami jsou hypromelóza, glycerol (viz bod 2), polysorbát 80, natrium-benzoát (E211) (viz bod 2), monohydrát kyseliny citronové, natrium-citrát (viz bod 2), jahodové aroma (umělé) a čištěná voda.
Jak Orfadin vypadá a co obsahuje toto balení
Perorální suspenze je bílá, hustší, neprůhledná suspenze. Před zatřepáním lahvičkou může vypadat jako kompaktní usazenina na dně a mírně opalizující tekutina.
Dodává se ve 100 ml hnědé skleněné lahvičce s bílým dětským bezpečnostním šroubovacím uzávěrem. Jedna lahvička obsahuje 90 ml supenze.
Jedno balení obsahuje jednu lahvičku, jeden adaptér na lahvičku a tři stříkačky pro perorální podání.
Držitel rozhodnutí o registraci
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Švédsko
Výrobce
Apotek Produktion & Laboratorier AB Celsiusgatan 43 SE-212 14 Malmo Švédsko
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
39