Optimark 500 Mikromol/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Optimark 500 mikromol/ml injekční roztok v předplněné injekční stříkačce Optimark 500 mikromol/ml injekční roztok v injekční lahvičce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Předplněná injekční stříkačka
1 ml obsahuje 330,9 mg přípravku gadoversetamidum, odpovídající 500 mikromolům.
Jedna 10 ml injekční stříkačka obsahuje 3309 mg gadoversetamidu, odpovídající 5 milimolům. Jedna 15 ml injekční stříkačka obsahuje 4963,5 mg gadoversetamidu, odpovídající 7,5 milimolům. Jedna 20 ml injekční stříkačka obsahuje 6618 mg gadoversetamidu, odpovídající 10 milimolům. Jedna 30 ml injekční stříkačka obsahuje 9927 mg gadoversetamidu, odpovídající 15 milimolům.
Pomocná látka/Pomocné látky se známým účinkem:
20 ml roztoku obsahuje 28,75 mg sodíku.
30 ml roztoku obsahuje 43,13 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
Injekční lahvička
1 ml obsahuje 330,9 mg přípravku gadoversetamidum, odpovídající 500 mikromolům.
Jedna 10 ml injekční lahvička obsahuje 3309 mg gadoversetamidu, odpovídající 5 milimolům. Jedna 15 ml injekční lahvička obsahuje 4963,5 mg gadoversetamidu, odpovídající 7,5 milimolům. Jedna 20 ml injekční lahvička obsahuje 6618 mg gadoversetamidu, odpovídající 10 milimolům.
Pomocná látka/Pomocné látky se známým účinkem:
20 ml roztoku obsahuje 28,75 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Předplněná injekční stříkačka
Injekční roztok v předplněné injekční stříkačce.
Injekční lahvička
Injekční roztok v injekční lahvičce.
Čirý, bezbarvý až světle žlutý roztok. pH: 6,0 - 7,5
Osmolalita (37 °C): 1000 - 1200 mOsm/kg
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Tento přípravek je určen pouze k diagnostickým účelům.
Optimark je určen k použití pro zobrazování magnetickou rezonancí (MRI) centrálního nervového systému (CNS) a jater. Zajišťuje zesílení kontrastu a umožňuje vizualizaci a pomáhá při charakterizaci fekálních lézí a abnormálních struktur v CNS a játrech u dospělých pacientů a dětí ve věku od 2 let se známým nebo vysoce suspektním patologickým nálezem.
4.2 Dávkování a způsob podání
Přípravek Optimark může být podáván pouze lékaři, kteří mají klinickou praxi s metodou MRI.
Aby byla zajištěna v případě stavu nouze okamžitá pomoc, je nutné, aby byly připraveny všechny potřebné léčivé přípravky (např. epinefrin/adrenalin, theofylin, antihistaminika, kortikosteroidy a atropiny), endotracheální trubice a ventilátor.
Dávkování
Látka se musí podávat jako bolus periferní intravenózní injekcí v dávce 0,2 ml/kg (100 mikromolů/kg) tělesné hmotnosti. Po injekci by měl následovat proplach 5 ml roztoku chloridu sodného 9 mg/ml (0,9 %), aby bylo jisté, že se aplikovala celá injekce s kontrastní látkou. Procedura snímkování může být provedena během 1 hodiny po podání kontrastní látky.
Opakovaná dávka
U kraniální MRI, pokud přetrvává silné klinické podezření na existenci léze i přes normální nález z kontrastní MRI, nebo pokud přesnější informace o počtu, velikosti nebo rozsahu lézí může ovlivnit léčbu nebo způsob péče o pacienta, lze podat jedincům s normální funkcí ledvin druhou bolusovou injekci s dávkou 0,2 ml/kg (100 mikromolů/kg) tělesné hmotnosti během 30 minut po první injekci. Tímto způsobem se může zvýšit diagnostický přínos vyšetření. Bezpečnost opakovaných dávek není známa u dětí a dospívajících (ve věku od 2 let), u pacientů s poškozením ledvin nebo u starších jedinců. U těchto populací se opakovaná dávka nedoporučuje.
Omezená data o ostatních kontrastních látkách s obsahem gadolinia naznačují, že k vyloučení další kraniální metastázy u pacientů se známou ojedinělou metastázou odstranitelnou chirurgicky, může vést vyšetření MR s injekcí o dávce 300 mikromolů/kg tělesné hmotnosti přípravku Optimark k vyšší diagnostické jistotě.
Pediatrická populace
U dětí ve věku od 2 let není nutná žádná úprava dávky.
Přípravek Optimark je kontraindikován u novorozenců ve věku do 4 týdnů (viz bod 4.3).
Podávání přípravku Optimark dětem do 2 let věku se nedoporučuje, protože bezpečnost, účinnost a vliv na nezralé funkce ledvin nebyl u této věkové skupiny stanoven.
Starší pacienti (ve věku 65 let a starší)
Není nutná žádná úprava dávky. U starších pacientů je potřeba postupovat opatrně (viz bod 4.4). Porucha funkce ledvin a jater
Přípravek Optimark se nesmí podávat pacientům se závažnou poruchou funkce ledvin (GFR < 30 ml/min/1,73 m2) a/nebo akutním poškozením ledvin a pacientům s transplantovanými játry nebo pacientům v perioperačním období transplantace jater (viz bod 4.3). Pouze po pečlivém zvážení přínosu/rizika může být přípravek Optimark použit u pacientů se středně závažnou poruchou funkce ledvin (GFR 30-59 ml/min/1,73 m2), a to v dávce nepřekračující 100 mikromolů/kg tělesné hmotnosti (viz bod 4.4). Během jednoho vyšetření může být podána pouze jedna dávka. Vzhledem k nedostatku informací o opakovaném podání není možné injekce přípravku Optimark opakovat dříve, než interval mezi injekcemi dosáhne alespoň 7 dní.
Způsob podání
Přípravek se musí podávat jako bolus periferní intravenózní injekcí. Po injekci by měl následovat proplach 5 ml roztoku chloridu sodného 9 mg/ml (0,9 %), aby bylo jisté, že se aplikovala celá injekce s kontrastní látkou. Doporučuje se použít flexibilní zaváděcí žilní katétr, viz bod 4.4.
Optimark nesmí být podáván autoinjektorem u dětí ve věku od 2 do 11 let (viz bod 4.4).
Opatření, která je nutno učinit před zacházením s léčivým přípravkem nebo před jeho podáním Před použitím je třeba nádobu a roztok zkontrolovat podle popisu uvedeného v bodu 6.6.
4.3 Kontraindikace
Hypersenzitivita na gadoversetamid nebo další přípravky s obsahem gadolinia nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
Použití přípravku Optimark je kontraindikováno
• u pacientů se závažnou poruchou funkce ledvin (GFR <30ml/min/1,73 m2) a/nebo akutním poškozením ledvin,
• u pacientů s transplantovanými játry nebo
• u pacientů v perioperačním období transplantace jater a
• novorozenců ve věku do 4 týdnů (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Jako u každého paramagnetického kontrastního činidla, zesílení MRI gadoversetamidem může poškodit vizualizaci existujících lézí. Některé z těchto lézí lze vidět na nezesíleném nekontrastním MRI. Z tohoto důvodu je třeba při interpretaci zobrazení pomocí zesíleného kontrastu dávat pozor, pokud se neprovádí společně s nezesíleným MRI.
Před vyšetřením je třeba se postarat, aby byli pacienti dostatečně hydratováni.
Hypersenzitivita
Gadoversetamid může také způsobit alergoidní a další idiosynkratické reakce, které se mohou projevit ve formě kardiovaskulárních, respiračních a kožních reakcí (viz bod 4.8). Většina těchto reakcí se objeví do půl hodiny po podání kontrastní látky. Jako u všech ostatních kontrastních látek této třídy se mohou ve vzácných případech objevit pozdější reakce (po několika hodinách nebo dnech); avšak žádné takové nebyly hlášeny z již dokončených klinických zkoušek.
Jestliže se objeví hypersenzitivní reakce, musí se podání kontrastní látky okamžitě přerušit a je-li to zapotřebí, musí se začít s intravenózní léčbou.
Během vyšetření je nutný dohled lékaře a doporučuje se použít flexibilní zaváděcí katétr. Aby byla zajištěna v případě stavu nouze okamžitá pomoc, je nutné, aby byly připraveny všechny potřebné léčivé přípravky (např. epinefrin/adrenalin, theofylin, antihistaminika, kortikosteroidy a atropiny), endotracheální trubice a ventilátor.
Zvýšené riziko hypersenzitivních reakcí je v následujících případech:
- u pacientů s alergickou predispozicí
- u pacientů s bronchiálním astmatem; u těchto pacientů hrozí zejména riziko bronchospastických stavů, jež se tak zvyšuje
- u pacientů s anamnézou reakcí na kontrastní látky, včetně dřívější anamnézy reakce na kontrastní látky obsahující jod
Před podáním injekce s kontrastní látkou je třeba se zeptat pacienta, zda trpí nějakou alergií (např. alergie na mořské produkty nebo léčiva, senná rýma, kopřivka), zda je hypersenzitivní na kontrastní látku a zda má bronchiální astma. Je třeba zvážit premedikaci antihistaminiky nebo glukokortikoidy.
U pacientů užívajících beta-blokátory
Je třeba poznamenat, že pacienti užívající beta-blokátory nemusí mít odezvu na beta-agonisty, které se obvykle používají při léčbě hypersenzitivních reakcí.
U pacientů s kardiovaskulárním onemocněním
U této skupiny pacientů mohou být hypersenzitivní reakce závažného rázu. Zejména u pacientů se závažnými srdečními chorobami (např. závažné srdeční selhání, ischemická choroba srdeční) se kardiovaskulární reakce mohou zhoršit. Toto však při klinických zkouškách s přípravkem Optimark nebylo zjevné.
Onemocnění centrálního nervového systému
U pacientů trpících epilepsií nebo mozkovými lézemi se zvyšuje pravděpodobnost záchvatu během vyšetření. Je třeba dodržovat bezpečnostní opatření při vyšetřování těchto pacientů (např. monitorování pacientů) a musí být dostupné vybavení a léky pro potřeby rychlé léčby případného záchvatu.
Pacienti s poruchou funkce ledvin
Před aplikací přípravku Optimark musí být u všech pacientů proveden pomocí laboratorních testů skrínink zaměřený na poruchy funkce ledvin.
U pacientů s akutní nebo chronickou závažnou poruchou funkce ledvin (GFR < 30 ml/min/1,73 m2) a/nebo akutním poškozením ledvin byly v souvislosti s použitím přípravku Optimark a některých dalších kontrastních látek obsahujících gadolinium hlášeny případy nefrogenní systémové fibrózy (NSF). Přípravek Optimark je u těchto pacientů kontraindikován (viz bod 4.3). Ve zvýšené míře jsou ohroženi pacienti, kteří podstupují transplantaci jater, a to proto, že incidence akutního selhání ledvin je v této skupině vysoká. Proto se Optimark nesmí používat u pacientů s transplantovanými játry nebo pacientů v perioperačním období transplantace jater a u novorozenců (viz bod 4.3).
Riziko vzniku NSF u pacientů se středně závažnou poruchou funkce ledvin
2
(GFR 30-59 ml/min/1,73 m ) není známo. Proto se u pacientů se středně závažnou poruchou funkce ledvin může Optimark použít pouze po pečlivém zvážení rizika/prospěchu.
Gadoversetamid je dialyzovatelný. Hemodialýza krátce po podání přípravku Optimark může být vhodným postupem k odstranění Optimarku z těla. Neexistují důkazy na podporu zahájení hemodialýzy k prevenci nebo k léčbě NSF u pacientů, kteří hemodialýzu dosud nepodstupují.
U pacientů s výchozí poruchou funkce ledvin došlo při použití přípravku Optimark k akutnímu poranění ledvin vyžadujícímu dialýzu. Riziko akutního poranění ledvin se může zvýšit při zvýšené dávce kontrastní látky. Podávejte nejnižší možnou dávku pro odpovídající zobrazení.
Děti a dospívající
Optimark nesmí být podáván autoinjektorem. Aby se zamezilo případnému nechtěnému předávkování, musí se dětem ve věku od 2 do 11 let požadovaná dávka podávat ručně.
U novorozenců a kojenců
Přípravek Optimark se nesmí používat u dětí mladších dvou let. Bezpečnost a účinnost přípravku u této věkové skupiny nebyla zjišťována.
Starší pacienti
U starších pacientů může být renální clearance gadoversetamidu zhoršená, proto je velmi důležité, aby byl u pacientů ve věku 65 let a starších proveden skrínink zaměřený na poruchy funkce ledvin.
Sodík
Tento přípravek obsahuje méně než 1 mmol sodíku (23 mg) v dávce až 17 ml, tj. v podstatě je „bez sodíku“.
10ml a 15ml injekční stříkačky obsahují méně než 1 mmol sodíku, tj. v podstatě jsou „bez sodíku“. Vyšší dávky obsahují 1 mmol nebo více sodíku, což je nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
Předplněná injekční stříkačka
20 ml roztoku obsahuje 28,75 mg sodíku.
30 ml roztoku obsahuje 43,13 mg sodíku.
Injekční lahvička
20 ml roztoku obsahuje 28,75 mg sodíku.
Hladina železa a zinku v séru
Je třeba dávat pozor, neboť v klinických zkouškách bylo pozorováno, že u některých osob dochází k přechodnému poklesu hladiny železa a zinku v krvi. Tyto změny však nemají klinický význam.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí.
Bylo prokázáno, že Optimark způsobuje interferenci při stanovení hladiny kalcia v séru použitím kolorometrické metody s o-kresolftalein komplexonem (OCP). Ale podávání gadoversetamidu nezpůsobuje skutečný pokles sérového kalcia. V přítomnosti gadoversetamidu poskytuje metoda OCP chybnou, nízkou hodnotu kalcia v plazmě. Závažnost tohoto artefaktu měření je úměrná koncentraci gadoversetamidu v krvi a u pacientů s normální clearancí ledvin lze přesné výsledky získat asi za 90 minut po podání injekce. U pacientů se sníženou funkcí ledvin se clearance gadoversetamidu zpomaluje a interference při stanovování kalcia metodou OCP se prodlužuje. Gadoversetamid neovlivňuje další metody stanovování hladin sérového kalcia, jako je například kolorimetrická metoda s činidlem arsenazo III, metoda atomové absorpční spektroskopie (AAS) a hmotnostní spektrometrie s indukčně vázaným plasmatem (ICP).
4.6 Fertilita, těhotenství a kojení
Údaje o podávání gadoversetamidu těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky (viz bod 5.3). Optimark lze v těhotenství použít pouze tehdy, když klinický stav ženy vyžaduje podání gadoversetamidu.
Kojení
Není známo, zda se gadoversetamid vylučuje do lidského mateřského mléka. Informace o vylučování gadoversetamidu do mateřského mléka u zvířat jsou nedostatečné. Riziko pro kojené děti nelze vyloučit. Kojení má být přerušeno minimálně po dobu 24 hodin od podání přípravku Optimark.
Fertilita
Neklinické údaje neodhalily žádné zvláštní riziko pro člověka na základě konvečních studií reprodukční toxicity. Klinické studie fertility nebyly provedeny.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Optimark nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Ambulantní pacienti by měli během řízení nebo obsluhování strojů vzít v úvahu, že je může v méně častých případech (>1/1 000 až <1/100) postihnout akutní závrať.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Většina nežádoucích účinků má mírnou nebo střední intenzitu a je přechodná. Nejčastějšími nežádoucími účinky byly dysgeuzie, pocity horka, bolest hlavy a závrať.
Většinu nežádoucích účinků pozorovaných po použití gadoversetamidu tvořily nežádoucí účinky na nervový systém, následované obecnými nežádoucími účinky, gastrointestinálními poruchami/poruchami kůže a podkožní tkáně.
Byly hlášeny závažné nežádoucí účinky zahrnující anafylaktické reakce, kardiovaskulární reakce a alergické respirační poruchy. Léčba má být symptomatická a pro případ výskytu závažné příhody je nutné mít okamžitě k dispozici potřebné léčivé přípravky a vybavení pro neodkladnou péči.
Seznam nežádoucích účinků v tabulkovém uspořádání
Z klinických zkoušek a post-marketingového použití byly hlášeny následující nežádoucí účinky v souvislosti s gadoversetamidem. U jednotlivých skupin vyjadřujících frekvence jsou nežádoucí účinky uváděny v pořadí snižující se závažnosti.
|
Třídy orgánových systémů podle databáze (MedDRA) |
časté (>1/100 až <1/10) |
méně časté (>1/1 000 až <1/100) |
vzácné (>1/10 000 až <1/1 000) |
velmi vzácné (<1/10 000) |
není známo |
|
Poruchy imunitního systému |
anafylaktické reakce | ||||
|
Poruchy metabolismu a výživy |
snížená chuť k jídlu | ||||
|
Psychiatrické poruchy |
pocity úzkosti, poruchy spánku, zmatenost a dezorientace | ||||
|
Poruchy nervového systému |
bolest hlavy, porucha chuti |
závrať, hypestézie, parestézie, porucha čichu |
křeče, tremor, somnolence, pocity pálení |
synkopa | |
|
Poruchy oka |
erytém víčka, bolest oka, rozmazané vidění, konjunktivitida, oční hyperémie | ||||
|
Poruchy ucha a labyrintu |
tinitus, vertigo | ||||
|
Srdeční poruchy |
palpitace, AV blok prvního stupně, extrasystoly, tachykardie, arytmie | ||||
|
Cévní poruchy |
zčervenání |
hypotenze, hypertenze | |||
|
Respirační, hrudní a mediastinální poruchy |
nazální kongesce, podráždění hrdla |
dyspnoe, dysfonie, rinorhea, pocit úzkosti v hrdle, bronchospasmus, kašel, hrtanový/hltanový edém, faryngitida, rinitida, kýchání | |||
|
Gastrointestinální poruchy |
nadměrné vylučování slin, bolest břicha, zácpa, sucho v ústech | ||||
|
Poruchy kůže a podkožní tkáně |
kopřivka, studený pot, erytém, hyperhidróza |
periorbitální edém |
nefrogenní systémová fibróza (NSF) |
|
Třídy orgánových systémů podle databáze (MedDRA) |
časté (>1/100 až <1/10) |
méně časté (>1/1 000 až <1/100) |
vzácné (>1/10 000 až <1/1 000) |
velmi vzácné (<1/10 000) |
není známo |
|
Poruchy ledvin a močových cest |
zvýšená hladina krevního kreatinu, hematurie | ||||
|
Celkové poruchy a reakce v místě aplikace |
pocity horka |
diskomfort na hrudi, bolest na hrudi, pocit chladu (včetně pocitu chladu akrálních částí těla), reakce v místě podání |
zimnice, bolest, edém tváře, astenické stavy včetně astenie, únavy a malátnosti, horečka, edém končetin, neobvyklé pocity | ||
|
Vyšetření |
abnormální hladina kalcia v krvi |
zvýšení ALT, abnormální výsledky rozboru moči, abnormální elektrolyty v moči, albumin v moči, zvýšené CPK, snížená hladina hemoglobinu |
prodloužení intervalu QT -elektrokardio-gram |
Podání je provázeno lokální reakcí a může vést k lokálnímu podráždění v místě vpichu.
V souvislosti s podáním přípravku Optimark byly hlášeny případy nefrogenní systémové fibrózy (NSF) (viz bod 4.4). U pacientů, kteří neměli jinak žádné příznaky nebo známky nefrogenní systémové fibrózy, byly u některých kontrastních látek s obsahem gadolinia hlášeny případy kožních ložisek souvisejících s gadoliniem s histologickým průkazem sklerotických útvarů.
Pediatrická populace
Přípravek Optimark byl studován u dětí ve věku 2 let a starších a byl zjištěn podobný bezpečnostní profil jako u dospělé populace.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Gadoversetamid byl testován u lidí v dávkách až 700 mikromolů/kg (sedmkrát více než standardní dávka). Klinické důsledky předávkování nebyly hlášeny. Příznaky akutní toxicity jsou u pacientů s normální funkcí ledvin velmi nepravděpodobné. Přípravek Optimark se dá odstranit z těla hemodialýzou. Neexistuje však žádný důkaz podporující předpoklad, že hemodialýza je vhodná k prevenci nefrogenní systémové fibrózy (NSF).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: kontrastní látka pro MRI, ATC kód: V08CA06
Gadoversetamid je chelát obsahující gadolinium - které má paramagnetické vlastnosti a je proto vhodné k zesílení kontrastního účinku MRI - a ligand versetamidu.
Cílem kontrastní látky pro MRI je indukovat změny intenzity signálu lézí, aby se docílilo jejich rozpoznání mezi dalšími normálními strukturami. Použití kontrastní látky tak sníží mezní hodnotu detekce lézí a jejich vizualizaci. Kontrastní látky pro MRI obsahující gadolinium (cheláty na bázi gadolinia) slouží k nepřímému působení na lokální magnetické prostředí pomocí změn protonu T1 (spin-souřadnice) a relaxačních časů T2 (spin-spin) a při obvyklé koncentraci 100 mikromolů/kg převládá zkrácení T1 a zkrácení T2 není signifikantní při použití T1-váhových sekvencí.
Gadoversetamid, extracelulární chelát gadolinia, se po intravenózním podání rychle vstřebává extracelulární tekutinou/prostorem a je vylučován zejména glomerulární filtrací.
Výsledkem těchto charakteristik je, že časování při získávání snímků po podání kontrastní látky je rozhodující při zobrazování jater. U metastází jater se rozdíl signálu mezi nádorem a okolní tkání výrazně zesiluje během prvních 90 sekund po podání extracelulární gadoliniové kontrastní látky. Proto rychlá zobrazovací sekvence musí začít 20 sekund po bolus injekci s kontrastní látkou, když je látka převážně v hepatických arteriích a potom opět po 60 sekundách po podání injekce během dominantní portálové venózní fáze. Jelikož hepatická arterie a portální venózní systém dodávají asi 20 %, resp.
80 % hepatické krve, včasné (hepatická arteriální fáze) zobrazení poskytuje optimální viditelnost lézí, co se týče hypervaskulárních lézí a snímky portální venózní fáze jsou užitečnou pomůckou pro hypovaskulární léze (většina metastatických lézí je poměrně hypovaskulární a jsou lépe prokazatelné během portální venózní fáze, zobrazující se jako oblasti s nižší intenzitou signálu v porovnání se zřetelně zesílenými játry). Viditelnost hypo- a hypervaskulárních lézí může být snížena, jestliže se zobrazování zpozdí o více než 3 minuty kvůli pronikání kontrastní látky do intersticiálního prostoru jak parenchymu jater, tak léze (např. metastázy), což má za následek, že léze má stejnou intenzitu signálu jako normální jatemí parenchym. Zpožděný kontrast nebo vyvážené snímky (> 5 minut po dávkování) napomáhají při charakterizaci lézí, např. střed metastázy shromáždí kontrast v intersticiálním prostoru léze a je oproti normálním játrům hyperintenzivní. Rozdíl ve vzoru zesílení signálu je užitečný při formulování různých diagnóz na základě charakterizace lézí a přispívá diagnostické jistotě.
Zesílení nádorů mozku pomocí kontrastní látky obsahující gadolinium (nebo jod) závisí na porušení krevní mozkové bariéry (BBB - bloodbrain barrier). Tyto látky jsou proto uváděny jako markery pro místa s abnormální poruchou BBB. Když je bariéra BBB porušena, molekuly gadoversetamidu pronikají do intersticiálních částí a produkují tak charakteristický paramagnetický efekt zkrácení T1 a T2. Obecně lze říci, že zesílení kontrastu při MRI, v standardní klinické dávce 100 mikromolů/kg, vede k výrazně lepší detekci lézí, senzitivitě a diagnostické přesnosti.
5.2 Farmakokinetické vlastnosti
Distribuce v organizmu
Farmakokinetické vlastnosti gadoversetamidu odpovídají otevřenému modelu dvou kompartmentů. Při dávce 100 mikromolů/kg je průměrný distribuční poločas vypočtený metodou na základě zbytků u 12 normálních dobrovolníků 13,3 ± 6,8 min. Průměrný objem distribuce u dávky 100 mikromolů/kg u pacientů bez poškození ledvin (zahrnujících normální jedince a pacienty s CNS nebo jaterními patologickými strukturami) byl 158,7 ± 29 až 214,3 (rozpětí 116,4 až 295) ml/kg. Tento distribuční objem (asi 10-15 l pro tělesnou hmotnost 70 kg) je konzistentní s léčivým přípravkem rozšířeným do extracelulární tekutiny. Úroveň dávek nemá konzistentní účinek na distribuční objem v žádné studii. Gadoversetamid nebyl podroben proteinové vazbě in vitro.
Eliminace z organizmu
Poločas vylučování při dávce 100 mikromolů/kg kolísá mezi 1,49 ± 0,15 hod. u zdravých dobrovolníků a 2,11 ± 0,62 hod. u pacientů bez poškození ledvin (zahrnujících normální jedince a pacienty s CNS nebo jaterními patologickými strukturami).
Průměrná plasmatická clearance gadoversetamidu u zdravých jedinců
(111,0 ± 14,1 ml/min/1,73m2 BSA) se výrazně neliší od průměrné renální clearance. Podobné výsledky byly získány studiem normálních jedinců a pacientů s různými kombinacemi jaterních, CNS a renálních dysfunkcí, kdy renální clearance gadoversetamidu byla asi 95 % celkové plasmatické clearance. Tento výsledek (poměr renální clearance/celková plasmatická clearance blížící se 1) znamená, že gadoversetamid je zásadně vylučován ledvinami.
Nebyly zaznamenány žádné systematické odchylky u žádného kinetického parametru jako funkce úrovně dávkování (100 až 700 mikromolů/kg). Z tohoto důvodu se v tomto rozpětí dávek jeví kinetika gadoversetamidu jako lineární.
Metabolizmus
Vzhledem tomu, že podstatná část dávky byla v moči nalezena v intaktní formě, je pravděpodobné, že u člověka k významnému metabolizmu gadoversetamidu nedochází.
Zvláštní populace
Vliv pohlaví:
Dospělé ženy a muži byli zahrnuti do dvou farmakokinetických studií. Nebyly zaznamenány žádné významné odchylky ve farmakokinetice z důvodu pohlavní odlišnosti.
Vliv věku:
Při přepočtu podle tělesné hmotnosti je celková tělesná clearance gadoversetamidu větší ve věkové skupině 2 až 11 let (143 ± 27,9 ml/h/kg) než byla pozorována u věkové skupiny 12 až 18 let (117 ± 26,1 ml/h/kg) a než u dvou populací dospělých (82,1 ± 16,8 a 56,5 ± 9,7 ml/h/kg u věkové skupiny 19 až 64 let, resp. > 65 let).
Poločas vylučování u věkové skupiny 2 až 11 a 12 až 18 let (1,4 ± 0,3, resp. 1,6 ± 0,3 h"1) je kratší, než bylo pozorováno ve dvou populací dospělých (1,9 ± 0,5 a 2,5 ± 0,5 h"1 ve věkové skupině 19 až 64, resp.> 65 let). Počet starších pacientů, u kterých byla stanovována farmakokinetika, byl omezený (nad 65 let, N=3).
Vliv poškození ledvin
Plasmatické hladiny gadoversetamidu se zvyšují lineárně se snížením funkce ledvin; u pacientů se závažným poškozením ledvin (CrCi<30 ml/min) to dokonce vedlo k šestinásobnému snížení clearance gadoversetamidu a odpovídajícímu šestinásobnému zvětšení plochy expozice AUC a t/2. Protože gadoversetamid je podáván pouze jako jedna dávka, bude to mít za následek delší a vyšší expozici pro omezenou dobu trvání. Nicméně, po 72 hodinách i u pacientů se závažným poškozením ledvin je téměř celá dávka vyloučena v moči, zdravým jedincům byla podána dávka až 500 mikromolů/kg bez jakýchkoli problémů ohledně bezpečnosti. Avšak, vzhledem k hlášeným případům NSF, které mají zřejmě souvislost s renálním poškozením u dalších kontrastních látek obsahujících gadolinium a u gadoversetamidu, by neměl být přípravek Optimark u těchto pacientů používán.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, akutní toxicity, reprodukční toxicity, lokální tolerance, antigenicity a genotoxicity neodhalily žádné zvláštní riziko pro člověka. Studie karcinogenicity nebyly prováděny.
Studie toxicity při opakovaných dávkách na potkanech a psech odhalily tvoření tubulárních buněk ledvin, s přesvědčivým průkazem reverzibility účinku. Nebylo pozorováno žádné funkční poškození.
Eliminace přípravku Optimark u psů mladších než 3 měsíce byla výrazně opožděna z důvodu nevyvinutých ledvin, což mělo za následek vysokou systémovou expozici přípravku Optimark. Týdně opakované dávkování dvojnásobku až dvacetinásobku klinické dávky od jednoho týdne věku až do dospělosti vedlo k extenzivní mineralizaci tkání, které mělo za následek tvorbu lokalizovaných efektů, jako je například ulcerativní dermatitida, zhoršení cirkulace a dysfunkce jater.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Versetamid Hydroxid vápenatý Dihydrát chloridu vápenatého
Hydroxid sodný a/nebo kyselina chlorovodíková na úpravu pH Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být přípravek Optimark mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
3 roky.
Chemická a fyzikální stabilita po otevření byla prokázána na dobu 24 hodin při teplotě do 25 °C.
Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Předplněná injekční stříkačka
Injekční stříkačku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem.
Injekční lahvička
Injekční lahvičku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem.
Chraňte před chladem nebo mrazem.
Podmínky uchovávání tohoto léčivého přípravku po prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Předplněná injekční stříkačka
Optimark je plněn v injekční stříkačce vyrobené polypropylenu. Víčko špičky stříkačky a píst jsou vyrobeny z brombutylové pryže.
Velikosti balení:
1 x 10 ml 10 x 10 ml
1 x 15 ml 10 x 15 ml
1 x 20 ml 10 x 20 ml
1 x 30 ml 10 x 30 ml
Na trhu nemusí být k dispozici všechny velikosti balení.
Injekční lahvička
Optimark je plněn v injekční lahvičce vyrobené z bezbarvého, vysoce odolného borosilikátového skla (EP třídy I). Injekční lahvičky jsou uzavřeny uzávěry z brombutylové pryže, hliníkovými těsnícími uzávěry a plastovými víčky.
Velikosti balení:
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Optimark je určen k jednorázovému použití; nespotřebovaná část se musí zlikvidovat.
Roztok nepoužívejte, jestliže má změněnou barvu nebo obsahuje pevné částice. Jestliže se používá zařízení k opakovanému použití, je třeba úzkostlivě dbát na to, aby nebylo kontaminováno zbytky čisticích prostředků.
Předplněná injekční stříkačka
Předplněné injekční stříkačky:
Montáž a kontrola
Zkontrolujte stříkačku, zda nejeví známky netěsnosti. V případě netěsnosti nepoužívat.
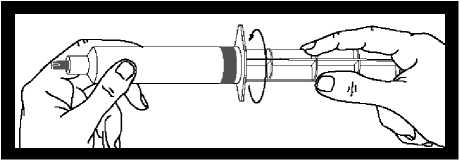
Po zašroubování dříku do pístu stříkačky je důležité ho pootočit o další V otáčky, aby se šedý píst volně otáčel.
Před použitím stříkačky odtrhněte šedý kryt koncovky a zlikvidujte ho. Stříkačka je nyní připravena k připojení jehly nebo infúzní hadičky.
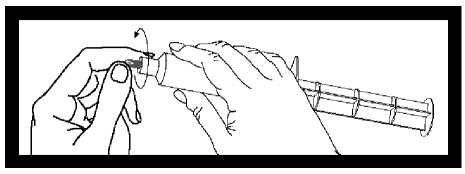
Po použití stříkačku a nespotřebovaný roztok zlikvidujte.
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. Oddělitelnou část štítku z předplněné injekční stříkačky je třeba vlepit do dokumentace pacienta, aby byl přesně zaznamenán použitý kontrastní přípravek s obsahem gadolinia. Též je nutno poznamenat podanou dávku.
Pokud se použijí elektronické záznamy pacientů, měly by být do záznamu pacienta uvedeny název přípravku, číslo šarže a dávka.
Injekční lahvička
Optimark je třeba natáhnout do injekční stříkačky a okamžitě použít.
Přípravek je třeba před použitím zkontrolovat, zda je celý obsah rozpuštěn a zda je nádoba a uzávěr v nepoškozeném stavu. Jsou-li přítomny pevné látky, injekční lahvičku zlikvidujte.
Po použití injekční stříkačku a nespotřebovaný roztok zlikvidujte.
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Oddělitelnou část štítku z injekční lahvičky je třeba vlepit do dokumentace pacienta, aby byl přesně zaznamenán použitý kontrastní přípravek s obsahem gadolinia. Též je nutno poznamenat podanou dávku. Pokud se použijí elektronické záznamy pacientů, měly by být do záznamu pacienta uvedeny název přípravku, číslo šarže a dávka.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Mallinckrodt Deutschland GmbH Josef-Dietzgen-Str. 1 53773 Hennef Německo
8. REGISTRAČNÍ ČÍSLO(A)
Předplněná injekční stříkačka 1 x 10 ml: EU/1/07/398/007 10 x 10 ml: EU/1/07/398/008 1 x 15 ml: EU/1/07/398/009 10 x 15 ml: EU/1/07/398/010 1 x 20 ml: EU/1/07/398/011 10 x 20 ml: EU/1/07/398/012 1 x 30 ml: EU/1/07/398/013 10 x 30 ml EU/1/07/398/014
Injekční lahvička 1 x 10 ml: EU/1/07/398/001 10 x 10 ml: EU/1/07/398/002 1 x 15 ml: EU/1/07/398/003 10 x 15 ml: EU/1/07/398/004 1 x 20 ml: EU/1/07/398/005 10 x 20 ml: EU/1/07/398/006
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 23. července 2007
Datum posledního prodloužení registrace: 15 června 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ/ VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/ VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Mallinckrodt Medical Imaging Ireland
Damastown
Mulhuddart
Dublin 15
Irsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ TOHOTO LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční činnosti v oblasti farmakovigilance podrobně uvedené v plánu farmakovigilance tak, jak byly schváleny v RMP uvedeném v modulu 1.8.2 schválené registrace, a dle případných následných aktualizací RMP schválených Výborem pro humánní léčivé přípravky (CHMP).
V souladu s pokynem Výboru pro humánní léčivé přípravky k systémům řízení rizik pro humánní léčivé přípravky má být aktualizovaný RMP předložen současně s příští periodicky aktualizovanou zprávou o bezpečnosti (PSUR).
Dále má být aktualizovaný RMP předložen:
• jestliže byly obdrženy nové informace, které mohou mít dopad na současné specifikace bezpečnosti, farmakovigilanční plán nebo na činnosti k minimalizaci rizik,
• do 60 dní po dosažení důležitého milníku (týkajícího se farmakovigilance nebo minimalizace rizik),
• na žádost Evropské agentury pro léčivé přípravky.
• Další opatření k minimalizaci rizik
Před uvedením na trh musí držitel rozhodnutí o registraci poskytnout všem potenciálním předepisujícím osobám kopii SPC (Souhrn údajů o přípravku) s průvodním dopisem, který obsahuje zvýrazněné bezpečnostní informace včetně bodů 4.3 a 4.4. Text musí být odsouhlasen s CHMP a musí obsahovat rovněž následující důležitou část:
• Podávání přípravku Optimark dětem ve věku do dvou let se nedoporučuje, protože bezpečnost, účinnost a dopad na funkci nevyvinutých ledvin nebyl u této věkové skupiny zkoumán.
Přípravek Optimark byl předmětem studie u dětí ve věku 2 let a starších s podobným profilem bezpečnosti, jako u starší populace.
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
Držitel rozhodnutí o registraci musí předkládat každoroční kumulativní hlášení o případech nefrogenní systémové fibrózy (NSF). |
Červenec každého roku až do předložení výsledků studie u kostí. |
|
Držitel rozhodnutí o registraci musí provést studii hodnotící možné dlouhodobé hromadění gadolinia v kostech podle odsouhlaseného protokolu CHMP. |
Závěrečná zpráva o studii: Červen 2018 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
Optimark 500 mikromol/ml injekční roztok v předplněné injekční stříkačce Gadoversetamidum
1 ml obsahuje 330,9 mg přípravku gadoversetamidum, odpovídající 500 mikromolům.
Pomocné látky: versetamid, hydroxid vápenatý, dihydrát chloridu vápenatého, hydroxid sodný a/nebo kyselina chlorovodíková, voda na injekci.
Před použitím si přečtěte příbalovou informaci
Injekční roztok v předplněné injekční stříkačce 10 ml (1, 10 injekčních stříkačky)
15 ml (1, 10 injekčních stříkačky)
20 ml (1, 10 injekčních stříkačky)
30 ml (1, 10 injekčních stříkačky)
Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
Kontrastní látka pro zobrazování magnetickou rezonancí
Pro záznam: nalepte odlepovací štítek pro sledování do dokumentace pacienta. Pro elektronické záznamy: zadejte název produktu, č.š. a dávku.
EXP
Stříkačku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem. Chraňte před chladem nebo mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Pouze pro jednorázové použití. Nespotřebovaný roztok zlikvidujte.
Mallinckrodt Deutschland GmbH, Josef-Dietzgen-Str. 1, 53773 Hennef, Německo
EU/1/07/398/007 (1 x 10 ml) EU/1/07/398/008 (10 x 10 ml) EU/1/07/398/009 (1 x 15 ml) EU/1/07/398/010 (10 x 15 ml) EU/1/07/398/011 (1 x 20 ml) EU/1/07/398/012 (10 x 20 ml) EU/1/07/398/013 (1 x 30 ml) EU/1/07/398/014 (10 x 30 ml)
č.š.
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato
Optimark 500 mikromol/ml injekční roztok v předplněné injekční stříkačce Gadoversetamidum
1 ml obsahuje 330,9 mg přípravku gadoversetamidum, odpovídající 500 mikromolům.
Pomocné látky: versetamid, hydroxid vápenatý, dihydrát chloridu vápenatého, hydroxid sodný a/nebo kyselina chlorovodíková, voda na injekci.
Injekční roztok v předplněné injekční stříkačce
15 ml 20 ml 30 ml
Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
Tuto etiketu nalepte lepící stranou do dokumentace pacienta.
Pro elektronické záznamy: zadejte název produktu, č.š. a dávku.
EXP
Stříkačku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem. Chraňte před chladem nebo mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Pouze pro jednorázové použití. Nespotřebovaný roztok zlikvidujte.
Mallinckrodt Deutschland GmbH, Josef-Dietzgen-Str. 1, 53773 Hennef, Německo
EU/1/07/398/007 (1 x 10 ml) EU/1/07/398/008 (10 x 10 ml) EU/1/07/398/009 (1 x 15 ml) EU/1/07/398/010 (10 x 15 ml) EU/1/07/398/011 (1 x 20 ml) EU/1/07/398/012 (10 x 20 ml) EU/1/07/398/013 (1 x 30 ml) EU/1/07/398/014 (10 x 30 ml)
č.š.
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato
Optimark 500 mikromol/ml injekční roztok v předplněné injekční stříkačce
Gadoversetamidum
i.v. podání.
Před použitím si přečtěte příbalovou informaci.
EXP
Lot
10 ml
Stříkačku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem. Chraňte před chladem nebo mrazem.
Optimark 500 mikromol/ml injekční roztok v injekční lahvičce Gadoversetamidum
1 ml obsahuje 330,9 mg přípravku gadoversetamidum, odpovídající 500 mikromolům.
Pomocné látky: versetamid, hydroxid vápenatý, dihydrát chloridu vápenatého, hydroxid sodný a/nebo kyselina chlorovodíková, voda na injekci.
Injekční roztok v injekční lahvičce 10 ml (1, 10 injekčních lahviček) 15 ml (1, 10 injekčních lahviček) 20 ml (1, 10 injekčních lahviček)
Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
Kontrastní látka pro zobrazování magnetickou rezonancí
Pro záznam: nalepte odlepovací štítek pro sledování do dokumentace pacienta. Pro elektronické záznamy: zadejte název produktu, č.š. a dávku.
EXP
Lahvičku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem. Chraňte před chladem nebo mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Pouze pro jednorázové použití. Nespotřebovaný roztok zlikvidujte.
Mallinckrodt Deutschland GmbH, Josef-Dietzgen-Str. 1, 53773 Hennef, Německo
EU/1/07/398/001 (1 x 10 ml) EU/1/07/398/002 (10 x 10 ml) EU/1/07/398/003 (1 x 15 ml) EU/1/07/398/004 (10 x 15 ml) EU/1/07/398/005 (1 x 20 ml) EU/1/07/398/006 (10 x 20 ml)
č.š.
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato
Optimark 500 mikromol/ml injekční roztok v injekční lahvičce Gadoversetamidum
1 ml obsahuje 330,9 mg přípravku gadoversetamidum, odpovídající 500 mikromolům.
Pomocné látky: versetamid, hydroxid vápenatý, dihydrát chloridu vápenatého, hydroxid sodný a/nebo kyselina chlorovodíková, voda na injekci.
Injekční roztok v injekční lahvičce 15 ml 20 ml
Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
Tuto etiketu nalepte lepící stranou do dokumentace pacienta. Pro elektronické záznamy: zadejte název produktu, č.š. a dávku.
EXP
Lahvičku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem. Chraňte před chladem nebo mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Pouze pro jednorázové použití. Nespotřebovaný roztok zlikvidujte.
Mallinckrodt Deutschland GmbH, Josef-Dietzgen-Str. 1, 53773 Hennef, Německo
EU/1/07/398/001 (1 x 10 ml) EU/1/07/398/002 (10 x 10 ml) EU/1/07/398/003 (1 x 15 ml) EU/1/07/398/004 (10 x 15 ml) EU/1/07/398/005 (1 x 20 ml) EU/1/07/398/006 (10 x 20 ml)
č.š.
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato
Optimark 500 mikromol/ml injekční roztok v injekční lahvičce
Gadoversetamidum
i.v. podání.
Před použitím si přečtěte příbalovou informaci.
EXP
Lot
10 ml
Lahvičku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem. Chraňte před chladem nebo mrazem.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele Optimark 500 mikromol/ml injekční roztok v předplněné injekční stříkačce
Gadoversetamidum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než je Vám tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Optimark a k čemu se používá
2. Čemu musíte věnovat pozornost, než je Vám přípravek Optimark podán
3. Jak se přípravek Optimark podává
4. Možné nežádoucí účinky
5. Jak přípravek Optimark uchovávat
6. Obsah balení a další informace
1. Co je přípravek Optimark a k čemu se používá
Optimark obsahuje léčivou látku gadoversetamid. Gadoversetamid se používá jako ‘kontrastní látka’ při zobrazování magnetickou rezonancí.
Optimark je určen pouze k diagnostickým účelům. Používá se u dospělých pacientů a dětí ve věku od 2 let a starších, kteří podstupují zobrazování magnetickou rezonancí (MRI), typ zobrazování, kdy se pořizují snímky vnitřních orgánů. Optimark se používá k získání jasnějších snímků u pacientů, kteří mají, nebo u kterých se předpokládají abnormality mozku, páteře a jater.
2. Čemu musíte věnovat pozornost, než je Vám přípravek Optimark podán
Nepoužívejte přípravek Optimark
jestliže jste alergický/á
• na léčivou látku gadoversetamid nebo
• na kteroukoli další složku přípravku Optimark (viz bod 6) nebo
• na jiné kontrastní látky obsahující gadolinium
Přípravek Optimark Vám nesmí být podán, jestliže
• máte vážné a/nebo akutní problémy s ledvinami, nebo
• se chystáte podstoupit transplantaci jater, nebo jste v nedávné době transplantaci jater podstoupili, a to proto, že u pacientů v takovém stavu bylo po použití přípravku Optimark pozorováno onemocnění zvané nefrogenní systémová fibróza (NSF). NSF je onemocnění, při kterém dochází k tvrdnutí kůže a podkožních tkání. NSF může vést k vážnému znehybnění kloubů, svalové slabosti nebo může ovlivnit funkci vnitřních orgánů, to může vést až
k ohrožení života.
• Dále nesmí být přípravek Optimark podáván novorozencům ve věku do 4 týdnů.
Před podáním Přípravku Optimark musíte podstoupit krevní testy, aby se prověřila funkce ledvin.
Upozornění a opatření
Před použitím přípravku Optimark se poraďte se svým lékařem, jestliže:
• trpíte alergiemi (např. léčiva, mořské produkty, senná rýma, kopřivka) nebo astmatem
• jste měl(a) reakce na kontrastní látky, včetně dřívější anamnézy reakce na kontrastní látky obsahující jod
• Vaše ledviny nepracují správně
• jestliže jste podstoupil/a nebo v brzké době podstoupíte transplantaci jater
• máte pocit žízně nebo jste vypil(a) jenom malé nebo žádné množství tekutin před vyšetřením
• užíváte určitý druh léků k léčbě vysokého krevního tlaku, tj. beta-blokátor
• máte srdeční onemocnění
• trpíte epilepsií nebo mozkovými lézemi
• dodržujete dietu s kontrolovaným množstvím sodíku
Jestliže se Vás cokoli týká, Váš lékař rozhodne, zda je plánované vyšetření možné provést, či nikoli.
Děti a dospívající
Optimark se nedoporučuje pro děti mladší než dva roky.
Další léčivé přípravky a Optimark
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte nebo jste užíval(a) v nedávné době, a to i o lécích, které jsou dostupné bez lékařského předpisu.
Těhotenství a kojení
Přípravek Optimark se nemá podávat během těhotenství, jestliže to není zcela nezbytné.
Po té, co Vám bude Optimark podán, musí být kojení minimálně na 24 hodin přerušeno.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než je Vám tento přípravek podán.
Řízení dopravních prostředků a obsluha strojů
Jestliže jste ambulantní pacient a máte v úmyslu řídit nebo používat nástroje nebo stroje, mějte na zřeteli, že po podstoupení procedury zahrnující přípravek Optimark Vás mohou nenadále postihnout závratě.
Může se to týkat až 1 osoby ze 100.
Přípravek Optimark obsahuje sodík
Tento přípravek obsahuje méně než 1 mmol sodíku (23 mg) v dávce až 17 ml, tj. v podstatě je „bez sodíku“.
10 ml a 15 ml injekční stříkačky obsahují méně než 1 mmol sodíku, tj. v podstatě jsou „bez sodíku“. Vyšší dávky obsahují 1 mmol nebo více sodíku, což je nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
20 ml roztoku obsahuje 28,75 mg sodíku.
30 ml roztoku obsahuje 43,13 mg sodíku.
3. Jak se přípravek Optimark podává
Diagnostické procedury zahrnující použití kontrastních látek musí být prováděny pod dohledem lékaře, který má nezbytné školení a znalosti o postupu, který je prováděn.
Obvyklá dávka
Obvyklá dávka je 0,2 ml/kg tělesné hmotnosti a je stejná u dospělých i dětí ve věku 2 let a starších. Počítá se 14 ml pro jedince s hmotností 70 kg a toto množství se vstřikuje během asi 7-14 sekund do žíly, obvykle na ruce. Injekce je potom propláchnuta fyziologickým roztokem, aby se zajistilo, že nic nezůstalo v jehle nebo hadičce použité k injekci.
U dospělých lze podat druhou dávku během 30 minut od první injekce. Jestliže se hledají jisté abnormality mozku, může být nutné použít přípravek Optimark v trojnásobném množství obvyklé dávky u dospělých. Je na rozhodnutí lékaře, jakou dávku přípravku Optimark pro vyšetření potřebujete. Ihned informujte lékaře nebo zdravotní sestru nebo jiný personál, který Vám podal injekci, když cítíte bolest v místě vpichu.
Dávkování u zvláštní populace
Jestliže máte středně závažnou poruchu funkce ledvin, může Vám být během vyšetření podána pouze jedna dávka, další injekce může být podána nejdříve po 7 dnech.
Jestliže je Vám 65 let a více, není potřeba žádná úprava dávkování, budou však provedeny krevní testy ke zjištění funkce ledvin.
Jestliže jste použil(a) více přípravku Optimark než jste měl(a)
Jestliže obdržíte větší dávku přípravku Optimark s velkou pravděpodobností Vám to neuškodí, protože vyšší dávky přípravku nevedly k žádným problémům u lidí, kteří je obdrželi. Jestliže Vaše ledviny pracují normálně, je nepravděpodobné, že byste měli nějaké problémy. Optimark lze odstranit dialýzou. Jestliže si myslíte, že Vám bylo vstříknuto příliš mnoho přípravku Optimark, informujte lékaře nebo zdravotní sestru nebo jiný personál, který Vám podal injekci.
Máte-li jakékoli další otázky, týkající se užívání tohoto léku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Ihned informujte lékaře nebo zdravotní sestru/technologa o následujících příznacích, aby Vám byla poskytnuta okamžitá léčba, jelikož tyto příznaky mohou být nebo se mohou stát velmi závažné: nežádoucí účinky postihující srdce (mdloby, předčasné srdeční stahy (extrasystoly), bolest na hrudi) nebo dýchacího systému (dýchavičnost, zúžení dýchacích cest, otok hrdla nebo dráždění v hrdle, svědění v nose nebo výtok z nosu, kýchání).
Většina pozorovaných nežádoucích účinků po použití přípravku Optimark má mírnou nebo střední intenzitu a je přechodná. Nejčastějšími nežádoucími účinky byly pachuť v ústech, pocity horka, bolest hlavy a závrať.
Možné nežádoucí účinky jsou podrobněji popsány níže.
Skupiny četností uváděné níže a následující příznaky jsou na základě klinických zkoušek a zkušeností s používáním přípravku Optimark po jeho uvedení na trh:
|
Četnost |
Možné nežádoucí účinky |
|
Časté (mohou postihovat až 1 osobu z 10) |
bolest hlavy, pachuť v ústech, pocity horka |
|
Méně časté (mohou postihovat až 1 osobu ze 100) |
alergické reakce/reakce přecitlivělosti, závrať, brnění, snížená citlivost, snížení čichu, zarudnutí pokožky a teplo, překrvení nosní sliznice, podrážděné hrdlo, pocit na zvracení, průjem, svědění, vyrážka, nepříjemný pocit na hrudi, bolest na hrudi, pocit chladu včetně pocitu chladu v konečcích končetin, reakce v místě podání, změny hladin vápníku v krvi |
|
Četnost |
Možné nežádoucí účinky |
|
Vzácné (mohou postihovat až 1 osobu z 1000) |
snížená chuť k jídlu, pocit úzkosti, porucha spánku, ospalost, pocit pálení, pocit pohybu nebo točení, zvonění v uších, zarudlá oční víčka, bolest očí, rozmazané vidění, krví podlité oči, uvědomování si tlukotu srdce, nepravidelný srdeční tep, předčasné srdeční stahy, nízký krevní tlak, dýchavičnost, chrapot, výtok z nosu, stažené hrdlo, nadměrná tvorba slin, bolest břicha, zácpa, sucho v ústech, kopřivka, studený pot, červenání, zvýšené hladiny krevních parametrů (kreatin) normálně vylučované ledvinami, krev v moči, otok obličeje, slabost a podobné příznaky jako únava a celkový pocit nevolnosti, horečka, otoky končetin, zimnice, bolest, pocit chladu v konečcích končetin, zvýšená hladina enzymů v játrech, abnormální hodnoty rozboru moči, zvýšené hodnoty minerálů v moči, bílkovina v moči, zvýšené jaterní enzymy, zvýšení enzymů v srdci a svalech, snížená hladina hemoglobinu, pocit zmatení a dezorientace, třes, křeče, zarudlé oči, zrychlený srdeční tep, vysoký krevní tlak, zúžení dýchacích cest, otok hrdla nebo hlasivek, odřené hrdlo, kašel, svědění v nose, kýchání, pocení |
|
Velmi vzácné (mohou postihovat až 1 osobu z 10 000) |
otok kolem očí, abnormální EKG záznamy srdeční činnosti, mdloba, zvracení |
|
Není známo (z dostupných údajů nelze určit) |
ztvrdnutí kůže, které může postihovat také měkké tkáně a vnitřní orgány (nefrogenní systémová fibróza), pocit nevolnosti |
Byly zaznamenány případy nefrogenní systémové fibrózy (ta může způsobit tvrdnutí kůže a dále také může postihnout měkké tkáně a vnitřní orgány).
Pokud se přípravek Optimark použil u dětí ve věku dvou let a starších, měl podobné nežádoucí účinky jako u dospělých.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Optimark uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce a injekční lahvičce za EXP.
Injekční stříkačky uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem.
Chraňte před chladem nebo mrazem.
Přípravek má být použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
Roztok nepoužívejte, jestliže má změněnou barvu nebo obsahuje pevné částice.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
Obsah balení a další informace
6.
Co přípravek Optimark obsahuje
• Léčivou látkou je gadoversetamidum.
1 ml obsahuje 330,9 mg přípravku gadoversetamidum, odpovídající 500 mikromolům. Jedna 10 ml injekční lahvička obsahuje 3309 mg gadoversetamidu.
Jedna 15 ml injekční lahvička obsahuje 4963,5 mg gadoversetamidu.
Jedna 20 ml injekční lahvička obsahuje 6618 mg gadoversetamidu.
Jedna 30 ml injekční lahvička obsahuje 9927 mg gadoversetamidu.
• Dalšími pomocnými látkami jsou: versetamid, hydroxid vápenatý, dihydrát chloridu vápenatého, hydroxid sodný a/nebo kyselina chlorovodíková, voda na injekci.
Jak přípravek Optimark vypadá a co obsahuje toto balení
Injekční stříkačky Optimark obsahují čirý, bezbarvý až světle žlutý roztok.
Optimark je plněn v injekční stříkačce vyrobené polypropylenu. Kryt špičky stříkačky a píst jsou vyrobeny z brombutylové pryže.
Optimark předplněné injekční stříkačky jsou dodávané v následujících velikostech balení:
Na trhu nemusí být k dispozici všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci:
Mallinckrodt Deutschland GmbH Josef-Dietzgen-Str. 1 53773 Hennef Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci. Výrobce
Mallinckrodt Medical Imaging Ireland
Damastown
Mulhuddart, Dublin 15
Irsko
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky (EMA) na adrese: http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Terapeutické indikace
Optimark je určen k použití pro snímkování magnetickou rezonancí (MRI) centrálního nervového systému (CNS) a jater. Poskytuje zesílení kontrastu a umožňuje vizualizaci a pomáhá při charakterizaci fokálních lézí a abnormálních struktur v CNS a játrech u dospělých pacientů a dětí ve věku od 2 let, u nichž se předpokládá, že jsou s velkou pravděpodobností nemocní.
Kontraindikace
• Hypersensitivita na gadoversetamid nebo na jiné přípravky obsahující gadolinium nebo na kteroukoli pomocnou látku tohoto přípravku.
• Optimark je kontraindikován u pacientů se závažným poškozením ledvin (GFR <
30 ml/min/1,73 m2) a/nebo akutním poškozením ledvin,
• u pacientů s transplantovanými játry a
• u pacientů v předoperačním období pro transplantaci jater a
• u novorozenců do věku 4 týdnů.
Zvláštní upozornění a opatření pro použití
Jako u každého paramagnetického kontrastního činidla, zesílení MRI pomocí přípravku Optimark může zlepšit vizualizaci existujících lézí. Některé z těchto lézí lze vidět na nezesíleném, nekontrastním MRI. Z tohoto důvodu je třeba při interpretaci zobrazení pomocí zesíleného kontrastu dávat pozor, pokud se neprovádí společně s nezesíleným MRI.
Před vyšetřením je třeba se postarat, aby byli pacienti dostatečně hydratováni.
Hypersenzitivita
Gadoversetamid může také způsobit alergoidní a další idiosynkratické reakce, které se mohou projevit ve formě kardiovaskulárních, respiračních a kožních reakcí. Většina těchto reakcí se objeví do půl hodiny po podání kontrastní látky. Jako u všech ostatních kontrastních látek této třídy se mohou ve vzácných případech objevit pozdější reakce (po několika hodinách nebo dnech); avšak žádné takové nebyly hlášeny z již dokončených klinických zkoušek.
Jestliže se objeví hypersenzitivní reakce, musí se podání kontrastní látky okamžitě přerušit a je-li to zapotřebí, musí se začít s intravenózní léčbou.
Během vyšetření je nutný dohled lékaře a doporučuje se použít flexibilní zaváděcí katétr. Aby byla zajištěna v případě stavu nouze okamžitá pomoc, je nutné, aby byly připraveny všechny potřebné léčivé přípravky (např. epinefrin/adrenalin, theofylin, antihistaminiky, kortikosteroidy a atropiny), endotracheální trubice a ventilátor.
Zvýšené riziko hypersenzitivních reakcí je v následujících případech:
- u pacientů s alergickou predispozicí
- u pacientů s bronchiálním astmatem; u těchto pacientů hrozí zejména riziko bronchospastických stavů, jež se tak zvyšuje
- u pacientů s anamnézou reakcí na kontrastní látky, včetně dřívější anamnézy reakce na kontrastní látky obsahující jód
Před podáním injekce s kontrastní látkou je třeba se zeptat pacienta, zda trpí nějakou alergií (např. alergie na mořské produkty nebo léčiva, senná rýma, kopřivka), zda je hypersenzitivní na kontrastní látku a zda má bronchiální astma. Je třeba zvážit premedikaci antihistaminiky nebo glukokortikoidy.
Pacienti užívajících beta-blokátory
Je třeba poznamenat, že pacienti užívající beta-blokátory nemusí mít odezvu na beta-agonisty, které se obvykle používají při léčbě hypersenzitivních reakcí.
Pacienti s kardiovaskulárním onemocněním
U této skupiny pacientů mohou být hypersenzitivní reakce závažného rázu. Zejména u pacientů se závažnými srdečními chorobami (např. závažné srdeční selhání, ischemická choroba srdeční) se kardiovaskulární reakce mohou zhoršit. Toto však při klinických zkouškách s přípravkem Optimark nebylo zjevné.
Onemocnění centrálního nervového systému
U pacientů trpících epilepsií nebo mozkovými lézemi se zvyšuje pravděpodobnost záchvatu během vyšetření. Je třeba dodržovat bezpečnostní opatření při vyšetřování těchto pacientů (např. monitorování pacientů) a musí být dostupné vybavení a léky pro potřeby rychlé léčby případného záchvatu.
Pacienti s poruchou funkce ledvin
Před aplikací přípravku Optimark musí být u všech pacientů proveden pomocí laboratorních testů skrínink zaměřený na poruchy funkce ledvin
U pacientů s akutní nebo chronickou závažnou poruchou funkce ledvin (GFR < 30 ml/min/1,73 m2) a/nebo akutním poškozením ledvin byly v souvislosti s podáním přípravku Optimark a dalších kontrastních látek obsahujících gadolinium hlášeny případy nefrogenní systémové fibrózy (NSF). Přípravek Optimark je u těchto pacientů kontraindikován (viz bod Kontraindikace). Ve zvýšené míře jsou ohroženi pacienti, kteří podstoupili nebo podstupují transplantaci jater, a to proto, že incidence akutního selhání ledvin je v této skupině vysoká. Proto se Optimark nesmí podávat pacientům, kteří podstoupili nebo podstupují transplantaci jater a novorozencům.
Riziko vzniku NSF u pacientů se středně závažnou poruchou funkce ledvin
(GFR 30-59 ml/min/1,73 m2) není známo. Proto se u pacientů se středně závažným poškozením
funkce ledvin může Optimark použít pouze po pečlivém zvážení rizika/prospěchu.
Gadoversetamid je dialyzovatelný. Hemodialýza krátce po podání přípravku Optimark může být vhodným postupem k odstranění Optimarku z těla. Neexistují důkazy na podporu zahájení hemodialýzy k prevenci nebo k léčbě NSF u pacientů, kteří hemodialýzu dosud nepodstupují.
U pacientů s výchozí poruchou funkce ledvin došlo při použití přípravku Optimark k akutnímu poranění ledvin vyžadujícímu dialýzu. Riziko akutního poranění ledvin se může zvýšit při zvýšené dávce kontrastní látky. Podávejte nejnižší možnou dávku pro odpovídající zobrazení.
Děti a dospívající
Optimark nesmí být podáván autoinjektorem. Aby se zamezilo případnému nechtěnému předávkování, musí se dětem ve věku od 2 do 11 let požadovaná dávka podávat ručně.
Novorozenci a kojenci
Přípravek Optimark se nesmí používat u dětí mladších dvou let. Bezpečnost a účinnost přípravku u této věkové skupiny nebyla zjišťována.
Starší pacienti
U starších pacientů může být renální clearance gadoversetamidu zhoršená, proto je velmi důležité, aby byl u pacientů ve věku 65 let a starších proveden skrínink zaměřený na poruchy funkce ledvin.
Sodík
Tento přípravek obsahuje méně než 1 mmol sodíku (23 mg) v dávce až 17 ml, tj. v podstatě je „bez sodíku“.
10 ml a 15 ml injekční stříkačky obsahují méně než 1 mmol sodíku, tj. v podstatě jsou „bez sodíku“. Vyšší dávky obsahují 1 mmol nebo více sodíku, což je nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
20 ml roztoku obsahuje 28,75 mg sodíku.
30 ml roztoku obsahuje 43,13 mg sodíku.
Hladina železa a zinku v séru
Je třeba dávat pozor, neboť v klinických zkouškách bylo pozorováno, že u některých osob dochází k přechodnému poklesu hladiny železa a zinku v krvi. Tyto změny však nemají klinický význam.
Fertilita, těhotenství a kojení
Údaje o podávání gadoversetamidu těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky. Optimark se nepodává během těhotenství, pokud klinický stav ženy nevyžaduje podání gadoversetamidu.
Kojení
Není známo, zda se gadoversetamid vylučuje do lidského mateřského mléka. Informace o vylučování gadoversetamidu do lidského mateřského mléka u zvířat jsou nedostatečné. Riziko pro kojené dítě nelze vyloučit. Kojení má být přerušeno minimálně po dobu 24 hodin od podání přípravku Optimark.
Fertilita
Neklinické údaje neodhalily žádné zvláštní riziko pro člověka na základě konvečních studií reprodukční toxicity. Klinické studie fertility nebyly provedeny.
Dávkování a způsob podání
Přípravek Optimark může být podáván pouze lékaři, kteří mají klinickou praxi s metodou MRI.
Aby byla zajištěna v případě stavu nouze okamžitá pomoc, je nutné, aby byly připraveny všechny potřebné léčivé přípravky (např. epinefrin/adrenalin, theofylin, antihistaminika, kortikosteroidy a atropiny), endotracheální trubice a ventilátor.
Dávkování
Látka se musí podávat jako bolus periferní intravenózní injekcí v dávce 0,2 ml/kg (100 mikromolů/kg) tělesné hmotnosti. Po injekci by měl následovat proplach 5 ml roztoku chloridu sodného 9 mg/ml (0,9 %), aby bylo jisté, že se aplikovala celá injekce s kontrastní látkou. Procedura snímkování může být provedena během 1 hodiny po podání kontrastní látky.
Opakovaná dávka
U kraniální MRI, pokud přetrvává silné klinické podezření na existenci léze i přes normální nález z kontrastní MRI, nebo pokud přesnější informace o počtu, velikosti nebo rozsahu lézí může ovlivnit léčbu nebo způsob péče o pacienta, lze podat jedincům s normální funkcí ledvin druhou bolusovou injekci s dávkou 0,2 ml/kg (100 mikromolů/kg) tělesné hmotnosti během 30 minut po první injekci. Tímto způsobem se může zvýšit diagnostický přínos vyšetření.
Bezpečnost opakovaných dávek není známa u dětí a dospívajících (ve věku od 2 let), u pacientů s poškozením ledvin nebo u starších jedinců. U těchto populací se opakovaná dávka nedoporučuje.
Omezená data o ostatních kontrastních látkách s obsahem gadolinia naznačují, že k vyloučení další kraniální metastáze u pacientů se známou ojedinělou metastází odstranitelnou chirurgicky, může vést vyšetření MR s injekcí o dávce 300 mikromolů/kg tělesné hmotnosti přípravku Optimark k vyšší diagnostické jistotě.
Pediatrická populace
U dětí ve věku od 2 let není nutná žádná úprava dávky.
Optimark je kontraindikován u novorozenců ve věku do 4 týdnů. Přípravek Optimark se nedoporučuje pro děti mladší 2 let, protože bezpečnost, účinnost a vliv na funkci nevyvinutých ledvin nebyl v této věkové skupině sledován.
Starší pacienti (ve věku 65 let a starší)
Není nutná žádná úprava dávkování. U starších pacientů je potřeba postupovat opatrně.
Porucha funkce ledvin a jater
Přípravek Optimark se nesmí podávat pacientům se závažnou poruchou funkce ledvin (GFR < 30 ml/min/1,73 m2) a/nebo akutním poškozením ledvin a pacientům s transplantovanými játry nebo pacientům v perioperačním období transplantace jater. Pouze po pečlivém zvážení přínosu/rizika může být přípravek Optimark použit u pacientů se středně závažnou poruchou funkce ledvin (GFR 30-59 ml/min/1,73 m2), a to v dávce nepřekračující 100 mikromolů/kg tělesné hmotnosti.
Během jednoho vyšetření může být podána pouze jedna dávka. Vzhledem k nedostatku informací o opakovaném podání není možné injekce přípravku Optimark opakovat dříve, než interval mezi injekcemi dosáhne alespoň 7 dní.
Způsob podání
Přípravek se musí podávat jako bolus periferní intravenózní injekcí. Po injekci by měl následovat proplach 5 ml roztoku chloridu sodného 9 mg/ml (0,9 %), aby bylo jisté, že se aplikovala celá injekce s kontrastní látkou. Doporučuje se použít flexibilní zaváděcí žilní katétr.
Optimark nesmí být podáván autoinjektorem u dětí ve věku od 2 do 11 let (viz bod 4.4).
Opatření, která je nutno učinit před zacházením s léčivým přípravkem nebo před jeho podáním Před použitím je třeba nádobu a roztok zkontrolovat.
Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí.
Bylo prokázáno, že gadoversetamid způsobuje interferenci při stanovení hladiny kalcia v séru použitím kolorimetrické metody s o-kresolftalein komplexonem (OCP). Ale podání gadoversetamidu nezpůsobuje skutečný pokles sérového kalcia. V přítomnosti gadoversetamidu poskytuje metoda OCP chybnou, nízkou hodnotu kalcia v plazmě. Závažnost tohoto artefaktu měření je úměrná koncentraci gadoversetamidu v krvi a u pacientů s normální clearancí ledvin lze přesné výsledky získat asi za 90 minut po podání injekce. U pacientů se sníženou funkcí ledvin se clearance gadoversetamidu zpomaluje a interference při stanovování kalcia metodou OCP se prodlužuje. Gadoversetamid neovlivňuje další metody stanovování hladin sérového kalcia, jako je například kolorimetrická metoda s činidlem arsenazo III, metoda atomové absorpční spektroskopie a hmotnostní spektrometrie s indukčně vázaným plazmatem.
Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Optimark je určen k jednorázovému použití; nespotřebovaná část se musí zlikvidovat.
Roztok nepoužívejte, jestliže má změněnou barvu nebo obsahuje pevné částice. Jestliže se používá zařízení k opakovanému použití, je třeba úzkostlivě dbát na to, aby nebylo kontaminováno zbytky čisticích prostředků.
Předplněné injekční stříkačky:
Montáž a kontrola
Zkontrolujte stříkačku, zda nejeví známky netěsnosti. V případě netěsnosti nepoužívat.
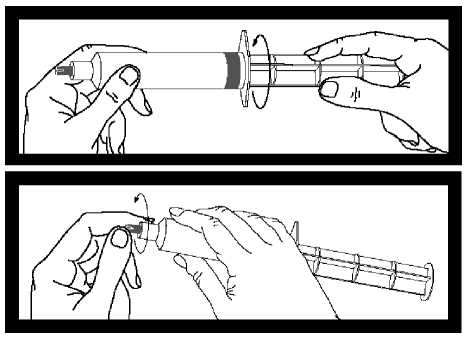
Po zašroubování dříku do pístu stříkačky je důležité ho pootočit o další V otáčky, aby se šedý píst volně otáčel.
Před použitím stříkačky odtrhněte šedý kryt koncovky a zlikvidujte ho. Stříkačka je nyní připravena k připojení jehly nebo infúzní hadičky.
Po použití stříkačku a nespotřebovaný roztok zlikvidujte.
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. Oddělitelnou část štítku z předplněné injekční stříkačky je třeba vlepit do dokumentace pacienta, aby byl přesně zaznamenán použitý kontrastní přípravek s obsahem gadolinia. Též je nutno poznamenat podanou dávku.
Pokud se použijí elektronické záznamy pacientů, měly by být do záznamu pacienta uvedeny název přípravku, číslo šarže a dávka.
Příbalová informace: informace pro uživatele Optimark 500 mikromol/ml injekční roztok v injekční lahvičce
Gadoversetamidum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než je Vám tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu,
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Optimark a k čemu se používá
2. Čemu musíte věnovat pozornost, než je Vám přípravek Optimark podán
3. Jak se přípravek Optimark podává
4. Možné nežádoucí účinky
5. Jak přípravek Optimark uchovávat
6. Obsah balení a další informace
1. Co je přípravek Optimark a k čemu se používá
Optimark obsahuje léčivou látku gadoversetamid. Gadoversetamid se používá jako ‘kontrastní látka’ při zobrazování magnetickou rezonancí.
Optimark je určen pouze k diagnostickým účelům. Používá se u dospělých pacientů a dětí ve věku od 2 let a starších, kteří podstupují zobrazování magnetickou rezonancí (MRI), typ zobrazování, kdy se pořizují snímky vnitřních orgánů. Optimark se používá k získání jasnějších snímků u pacientů, kteří mají, nebo u kterých se předpokládají abnormality mozku, páteře a játer.
2. Čemu musíte věnovat pozornost, než je Vám přípravek Optimark podán
Nepoužívejte přípravek Optimark
jestliže jste alergický/á
• na léčivou látku gadoversetamid nebo
• na kteroukoli další složku přípravku Optimark (viz bod 6) nebo
• na jiné kontrastní látky obsahující gadolinium
Přípravek Optimark Vám nesmí být podán, jestliže
• máte vážné a/nebo akutní problémy s ledvinami, nebo
• se chystáte podstoupit transplantaci jater, nebo jste v nedávné době transplantaci jater podstoupili, a to proto, že u pacientů v takovém stavu bylo po použití přípravku Optimark pozorováno onemocnění zvané nefrogenní systémová fibróza (NSF). NSF je onemocnění, při kterém dochází k tvrdnutí kůže a podkožních tkání. NSF může vést k vážnému znehybnění kloubů, svalové slabosti nebo může ovlivnit funkci vnitřních orgánů, to může vést až
k ohrožení života.
• Dále nesmí být přípravek Optimark podáván novorozencům ve věku do 4 týdnů.
Před podáním Přípravku Optimark musíte podstoupit krevní testy, aby se prověřila funkce ledvin.
Upozornění a opatření
Před použitím přípravku Optimark se poraďte se svým lékařem, jestliže:
• trpíte alergiemi (např. léčiva, mořské produkty, senná rýma, kopřivka) nebo astmatem
• jste měl(a) reakce na kontrastní látky, včetně dřívější anamnézy reakce na kontrastní látku obsahující jod
• Vaše ledviny nepracují správně
• jestliže jste podstoupil/a nebo v brzké době podstoupíte transplantaci jater
• máte pocit žízně nebo jste vypil(a) jenom malé nebo žádné množství tekutin před vyšetřením
• užíváte určitý druh léků k léčbě vysokého krevního tlaku, tj. beta-blokátor
• máte srdeční onemocnění
• trpíte epilepsií nebo mozkovými lézemi
• dodržujete dietu s kontrolovaným množstvím sodíku
Jestliže se Vás cokoli týká, Váš lékař rozhodne, zda je plánované vyšetření možné provést, či nikoli.
Děti a dospívající
Optimark se nedoporučuje pro dětí mladší než dva roky Další léčivé přípravky a Optimark
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte nebo jste užíval(a) v nedávné době, a to i o lécích, které jsou dostupné bez lékařského předpisu.
Těhotenství a kojení
Přípravek Optimark se nemá podávat během těhotenství, jestliže to není zcela nezbytné.
Po té, co Vám bude Optimark podán, musí být kojení minimálně na 24 hodin přerušeno.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než je Vám tento přípravek podán.
Řízení dopravních prostředků a obsluha strojů
Jestliže jste ambulantní pacient a máte v úmyslu řídit nebo používat nástroje nebo stroje, mějte na zřeteli, že po podstoupení procedury zahrnující přípravek Optimark Vás mohou nenadále postihnout závratě.
Může se to týkat až 1 osoby ze 100.
Přípravek Optimark obsahuje sodík
Tento přípravek obsahuje méně než 1 mmol sodíku (23 mg) v dávce až 17 ml, tj. v podstatě je „bez sodíku“.
10 ml a 15 ml injekční lahvičky obsahují méně než 1 mmol sodíku, tj. v podstatě jsou „bez sodíku“. Vyšší dávky obsahují 1 mmol nebo více sodíku, což je nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
20 ml roztoku obsahuje 28,75 mg sodíku.
3. Jak se přípravek Optimark podává
Diagnostické procedury zahrnující použití kontrastních látek musí být prováděny pod dohledem lékaře, který má nezbytné školení a znalosti o postupu, který je prováděn.
Obvyklá dávka
Obvyklá dávka je 0,2 ml/kg tělesné hmotnosti a je stejná u dospělých i dětí ve věku 2 let a starších. Počítá se 14 ml pro jedince s hmotností 70 kg a toto množství se vstřikuje během asi 7-14 sekund do žíly, obvykle na ruce. Injekce je potom propláchnuta fyziologickým roztokem, aby se zajistilo, že nic nezůstalo v jehle nebo hadičce použité k injekci.
U dospělých lze podat druhou dávku během 30 minut od první injekce. Jestliže se hledají jisté abnormality mozku, může být nutné použít přípravek Optimark v trojnásobném množství obvyklé dávky u dospělých. Je na rozhodnutí lékaře, jakou dávku přípravku Optimark pro vyšetření potřebujete. Ihned informujte lékaře nebo zdravotní sestru nebo jiný personál, který Vám podal injekci, když cítíte bolest v místě vpichu.
Dávkování u zvláštní populace
Jestliže máte středně závažnou poruchu funkce ledvin, může Vám být během vyšetření podána pouze jedna dávka, další injekce může být podána nejdříve po 7 dnech.
Jestliže je Vám 65 let a více, není potřeba žádná úprava dávkování, budou však provedeny krevní testy ke zjištění funkce ledvin.
Jestliže jste použil(a) více přípravku Optimark než jste měl(a)
Jestliže obdržíte větší dávku přípravku Optimark s velkou pravděpodobností Vám to neuškodí, protože vyšší dávky přípravku nevedly k žádným problémům u lidí, kteří je obdrželi. Jestliže Vaše ledviny pracují normálně, je nepravděpodobné, že byste měli nějaké problémy. Optimark lze odstranit dialýzou. Jestliže si myslíte, že Vám bylo vstříknuto příliš mnoho přípravku Optimark, informujte lékaře nebo zdravotní sestru nebo jiný personál, který Vám podal injekci.
Máte-li jakékoli další otázky, týkající se užívání tohoto léku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento lék nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Ihned informujte lékaře nebo zdravotní sestru/technologa o následujících příznacích, aby Vám byla poskytnuta okamžitá léčba, jelikož tyto příznaky mohou být nebo se mohou stát velmi závažné: nežádoucí účinky postihující srdce (mdloby, předčasné srdeční stahy (extrasystoly), bolest na hrudi) nebo dýchacího systému (dýchavičnost, zúžení dýchacích cest, otok hrdla nebo dráždění v hrdle, svědění v nose nebo výtok z nosu, kýchání).
Většina pozorovaných nežádoucích účinků po použití přípravku Optimark má mírnou nebo střední intenzitu a je přechodná. Nejčastějšími nežádoucími účinky byly pachuť v ústech, pocity horka, bolest hlavy a závrať.
Možné nežádoucí účinky jsou podrobněji popsány níže.
Skupiny četností uváděné níže a následující příznaky jsou na základě klinických zkoušek a zkušeností s používáním přípravku Optimark po jeho uvedení na trh:
|
Četnost |
Možné nežádoucí účinky |
|
Časté (mohou postihovat až 1 osobu z 10) |
bolest hlavy, pachuť v ústech, pocity horka |
|
Méně časté (mohou postihovat až 1 osobu ze 100) |
alergické reakce/reakce přecitlivělosti, závrať, brnění, snížená citlivost, snížení čichu, zarudnutí pokožky a teplo, překrvení nosní sliznice, podrážděné hrdlo, pocit na zvracení, průjem, svědění, vyrážka, nepříjemný pocit na hrudi, bolest na hrudi, pocit chladu včetně pocitu chladu v konečcích končetin, reakce v místě podání, změny hladin vápníku v krvi |
|
Četnost |
Možné nežádoucí účinky |
|
Vzácné (mohou postihovat až 1 osobu z 1000) |
snížená chuť k jídlu, pocit úzkosti, porucha spánku, ospalost, pocit pálení, pocit pohybu nebo točení, zvonění v uších, zarudlá oční víčka, bolest očí, rozmazané vidění, krví podlité oči, uvědomování si tlukotu srdce, nepravidelný srdeční tep, předčasné srdeční stahy, nízký krevní tlak, dýchavičnost, chrapot, výtok z nosu, stažené hrdlo, nadměrná tvorba slin, bolest břicha, zácpa, sucho v ústech, kopřivka, studený pot, červenání, zvýšené hladiny krevních parametrů (kreatin) normálně vylučované ledvinami, krev v moči, otok obličeje, slabost a podobné příznaky jako únava a celkový pocit nevolnosti, horečka, otoky končetin, zimnice, bolest, pocit chladu v konečcích končetin, zvýšená hladina enzymů v játrech, abnormální hodnoty rozboru moči, zvýšené hodnoty minerálů v moči, bílkovina v moči, zvýšené jaterní enzymy, zvýšení enzymů v srdci a svalech, snížená hladina hemoglobinu, pocit zmatení a dezorientace, třes, křeče, zarudlé oči, zrychlený srdeční tep, vysoký krevní tlak, zúžení dýchacích cest, otok hrdla nebo hlasivek, odřené hrdlo, kašel, svědění v nose, kýchání, pocení |
|
Velmi vzácné (mohou postihovat až 1 osobu z 10 000) |
otok kolem očí, abnormální EKG záznamy srdeční činnosti, mdloba, zvracení |
|
Není známo (z dostupných údajů nelze určit) |
ztvrdnutí kůže, které může postihovat také měkké tkáně a vnitřní orgány (nefrogenní systémová fibróza), pocit nevolnosti |
Byly zaznamenány případy nefrogenní systémové fibrózy (ta může způsobit tvrdnutí kůže a dále také může postihnout měkké tkáně a vnitřní orgány).
Pokud se přípravek Optimark použil u dětí ve věku dvou let a starších, měl podobné nežádoucí účinky jako u dospělých.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Optimark uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce a injekční lahvičce za EXP.
Injekční lahvičku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem.
Chraňte před chladem nebo mrazem.
Přípravek má být použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
Roztok nepoužívejte, jestliže má změněnou barvu nebo obsahuje pevné částice.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
Obsah balení a další informace
6.
Co přípravek Optimark obsahuje
• Léčivou látkou je gadoversetamidum.
1 ml obsahuje 330,9 mg přípravku gadoversetamidum, odpovídající 500 mikromolům.
Jedna 10 ml injekční lahvička obsahuje 3309 mg gadoversetamidu.
Jedna 15 ml injekční lahvička obsahuje 4963,5 mg gadoversetamidu.
Jedna 20 ml injekční lahvička obsahuje 6618 mg gadoversetamidu.
• Dalšími pomocnými látkami jsou: versetamid, hydroxid vápenatý, dihydrát chloridu vápenatého, hydroxid sodný a/nebo kyselina chlorovodíková, voda na injekci.
Jak přípravek Optimark vypadá a co obsahuje toto balení
Injekční lahvičky Optimark obsahují čirý, bezbarvý až světle žlutý roztok.
Optimark se dodává v injekčních lahvičkách s uzávěry z brombutylové pryže a hliníkovými těsnícími víčky.
Injekční lahvičky Optimark se dodávají v následujících velikostech obalů:
Na trhu nemusí být k dispozici všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Další informace o tomto přípravku získáte u držitele rozhodnutí o registraci:
Mallinckrodt Deutschland GmbH Josef-Dietzgen-Str. 1 53773 Hennef Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci. Výrobce
Mallinckrodt Medical Imaging Ireland
Damastown
Mulhuddart, Dublin 15
Irsko
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury (EMA) pro léčivé přípravky na adrese: http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Terapeutické indikace
Optimark je určen k použití pro snímkování magnetickou rezonancí (MRI) centrálního nervového systému (CNS) a jater. Poskytuje zesílení kontrastu a umožňuje vizualizaci a pomáhá při charakterizaci fokálních lézí a abnormálních struktur v CNS a játrech u dospělých pacientů a dětí ve věku od 2 let, u nichž se předpokládá, že jsou s velkou pravděpodobností nemocní.
Kontraindikace
• Hypersensitivita na gadoversetamid nebo na jiné přípravky obsahující gadolinium nebo na kteroukoli pomocnou látku tohoto přípravku.
• Optimark je kontraindikován u pacientů se závažným poškozením ledvin (GFR < 30 ml/min/1,73 m2) a/nebo akutním poškozením ledvin,
• u pacientů s transplantovanými játry a
• u pacientů v předoperačním období pro transplantaci jater a
• u novorozenců do věku 4 týdnů.
Zvláštní upozornění a opatření pro použití
Jako u každého paramagnetického kontrastního činidla, zesílení MRI pomocí přípravku Optimark může zlepšit vizualizaci existujících lézí. Některé z těchto lézí lze vidět na nezesíleném, nekontrastním MRI. Z tohoto důvodu je třeba při interpretaci zobrazení pomocí zesíleného kontrastu dávat pozor, pokud se neprovádí společně s nezesíleným MRI.
Před vyšetřením je třeba se postarat, aby byli pacienti dostatečně hydratováni.
Hypersenzitivita
Gadoversetamid může také způsobit alergoidní a další idiosynkratické reakce, které se mohou projevit ve formě kardiovaskulárních, respiračních a kožních reakcí. Většina těchto reakcí se objeví do půl hodiny po podání kontrastní látky. Jako u všech ostatních kontrastních látek této třídy se mohou ve vzácných případech objevit pozdější reakce (po několika hodinách nebo dnech); avšak žádné takové nebyly hlášeny z již dokončených klinických zkoušek.
Jestliže se objeví hypersenzitivní reakce, musí se podání kontrastní látky okamžitě přerušit a je-li to zapotřebí, musí se začít s intravenózní léčbou.
Během vyšetření je nutný dohled lékaře a doporučuje se použít flexibilní zaváděcí katétr. Aby byla zajištěna v případě stavu nouze okamžitá pomoc, je nutné, aby byly připraveny všechny potřebné léčivé přípravky (např. epinefrin/adrenalin, theofylin, antihistaminiky, kortikosteroidy a atropiny), endotracheální trubice a ventilátor.
Zvýšené riziko hypersenzitivních reakcí je v následujících případech:
- u pacientů s alergickou predispozicí
- u pacientů s bronchiálním astmatem; u těchto pacientů hrozí zejména riziko bronchospastických stavů, jež se tak zvyšuje
- u pacientů s anamnézou reakcí na kontrastní látky, včetně dřívější anamnézy reakce na kontrastní látky obsahující jód
Před podáním injekce s kontrastní látkou je třeba se zeptat pacienta, zda trpí nějakou alergií (např. alergie na mořské produkty nebo léčiva, senná rýma, kopřivka), zda je hypersenzitivní na kontrastní látku a zda má bronchiální astma. Je třeba zvážit premedikaci antihistaminiky nebo glukokortikoidy.
Pacienti užívajících beta-blokátory
Je třeba poznamenat, že pacienti užívající beta-blokátory nemusí mít odezvu na beta-agonisty, které se obvykle používají při léčbě hypersenzitivních reakcí.
Pacienti s kardiovaskulárním onemocněním
U této skupiny pacientů mohou být hypersenzitivní reakce závažného rázu. Zejména u pacientů se závažnými srdečními chorobami (např. závažné srdeční selhání, ischemická choroba srdeční) se kardiovaskulární reakce mohou zhoršit. Toto však při klinických zkouškách s přípravkem Optimark nebylo zjevné.
Onemocnění centrálního nervového systému
U pacientů trpících epilepsií nebo mozkovými lézemi se zvyšuje pravděpodobnost záchvatu během vyšetření. Je třeba dodržovat bezpečnostní opatření při vyšetřování těchto pacientů (např. monitorování pacientů) a musí být dostupné vybavení a léky pro potřeby rychlé léčby případného záchvatu.
Pacienti s poruchou funkce ledvin
Před aplikací přípravku Optimark musí být u všech pacientů proveden pomocí laboratorních testů skrínink zaměřený na poruchy funkce ledvin
U pacientů s akutní nebo chronickou závažnou poruchou funkce ledvin (GFR < 30 ml/min/1,73 m2) a/nebo akutním poškozením ledvin byly v souvislosti s podáním přípravku Optimark a dalších kontrastních látek obsahujících gadolinium hlášeny případy nefrogenní systémové fibrózy (NSF).
Přípravek Optimark je u těchto pacientů kontraindikován (viz bod Kontraindikace). Ve zvýšené míře jsou ohroženi pacienti, kteří podstoupili nebo podstupují transplantaci jater, a to proto, že incidence akutního selhání ledvin je v této skupině vysoká. Proto se Optimark nesmí podávat pacientům, kteří podstoupili nebo podstupují transplantaci jater a novorozencům.
Riziko vzniku NSF u pacientů se středně závažnou poruchou funkce ledvin
2
(GFR 30-59 ml/min/1,73 m ) není známo. Proto se u pacientů se středně závažným poškozením funkce ledvin může Optimark použít pouze po pečlivém zvážení rizika/prospěchu.
Gadoversetamid je dialyzovatelný. Hemodialýza krátce po podání přípravku Optimark může být vhodným postupem k odstranění Optimarku z těla. Neexistují důkazy na podporu zahájení hemodialýzy k prevenci nebo k léčbě NSF u pacientů, kteří hemodialýzu dosud nepodstupují.
U pacientů s výchozí poruchou funkce ledvin došlo při použití přípravku Optimark k akutnímu poranění ledvin vyžadujícímu dialýzu. Riziko akutního poranění ledvin se může zvýšit při zvýšené dávce kontrastní látky. Podávejte nejnižší možnou dávku pro odpovídající zobrazení.
Děti a dospívající
Optimark nesmí být podáván autoinjektorem. Aby se zamezilo případnému nechtěnému předávkování, musí se dětem ve věku od 2 do 11 let požadovaná dávka podávat ručně.
Novorozenci a kojenci
Přípravek Optimark se nesmí používat u dětí mladších dvou let. Bezpečnost a účinnost přípravku u této věkové skupiny nebyla zjišťována.
Starší pacienti
U starších pacientů může být renální clearance gadoversetamidu zhoršená, proto je velmi důležité, aby byl u pacientů ve věku 65 let a starších proveden skrínink zaměřený na poruchy funkce ledvin.
Sodík
Tento přípravek obsahuje méně než 1 mmol sodíku (23 mg) v dávce až 17 ml, tj. v podstatě je „bez sodíku“.
10 ml a 15 ml injekční lahvičky obsahují méně než 1 mmol sodíku, tj. v podstatě jsou „bez sodíku“.
Vyšší dávky obsahují 1 mmol nebo více sodíku, což je nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
20 ml roztoku obsahuje 28,75 mg sodíku.
30 ml roztoku obsahuje 43,13 mg sodíku.
Hladina železa a zinku v séru
Je třeba dávat pozor, neboť v klinických zkouškách bylo pozorováno, že u některých osob dochází k přechodnému poklesu hladiny železa a zinku v krvi. Tyto změny však nemají klinický význam.
Fertilita, těhotenství a kojení
Údaje o podávání gadoversetamidu těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky. Optimark se nepodává během těhotenství, pokud klinický stav ženy nevyžaduje podání gadoversetamidu.
Kojení
Není známo, zda se gadoversetamid vylučuje do lidského mateřského mléka. Informace o vylučování gadoversetamidu do lidského mateřského mléka u zvířat jsou nedostatečné. Riziko pro kojené dítě nelze vyloučit. Kojení má být přerušeno minimálně po dobu 24 hodin od podání přípravku Optimark.
Fertilita
Neklinické údaje neodhalily žádné zvláštní riziko pro člověka na základě konvečních studií reprodukční toxicity. Klinické studie fertility nebyly provedeny.
Dávkování a způsob podání
Přípravek Optimark může být podáván pouze lékaři, kteří mají klinickou praxi s metodou MRI.
Aby byla zajištěna v případě stavu nouze okamžitá pomoc, je nutné, aby byly připraveny všechny potřebné léčivé přípravky (např. epinefrin/adrenalin, theofylin, antihistaminika, kortikosteroidy a atropiny), endotracheální trubice a ventilátor.
Dávkování
Látka se musí podávat jako bolus periferní intravenózní injekcí v dávce 0,2 ml/kg (0,1 mikromolů/kg) tělesné hmotnosti. Po injekci by měl následovat proplach 5 ml roztoku chloridu sodného 9 mg/ml (0,9 %), aby bylo jisté, že se aplikovala celá injekce s kontrastní látkou. Procedura snímkování může být provedena během 1 hodiny po podání kontrastní látky.
Opakovaná dávka
U kraniální MRI, pokud přetrvává silné klinické podezření na existenci léze i přes normální nález z kontrastní MRI, nebo pokud přesnější informace o počtu, velikosti nebo rozsahu lézí může ovlivnit léčbu nebo způsob péče o pacienta, lze podat jedincům s normální funkcí ledvin druhou bolusovou injekci s dávkou 0,2 ml/kg (0,1 mikromolů/kg) tělesné hmotnosti během 30 minut po první injekci. Tímto způsobem se může zvýšit diagnostický přínos vyšetření.
Bezpečnost opakovaných dávek není známa u dětí a dospívajících (ve věku od 2 let), u pacientů s poškozením ledvin nebo u starších jedinců. U těchto populací se opakovaná dávka nedoporučuje.
Omezená data o ostatních kontrastních látkách s obsahem gadolinia naznačují, že k vyloučení další kraniální metastáze u pacientů se známou ojedinělou metastází odstranitelnou chirurgicky, může vést vyšetření MR s injekcí o dávce 0,3 mikromolů/kg tělesné hmotnosti přípravku Optimark k vyšší diagnostické jistotě.
Pediatrická populace
U dětí ve věku od 2 let není nutná žádná úprava dávky.
Optimark je kontraindikován u novorozenců ve věku do 4 týdnů. Přípravek Optimark se nedoporučuje pro děti mladší 2 let, protože bezpečnost, účinnost a vliv na funkci nevyvinutých ledvin nebyl v této věkové skupině sledován.
Starší pacienti (ve věku 65 let a starší)
Není nutná žádná úprava dávkování. U starších pacientů je potřeba postupovat opatrně.
Porucha funkce ledvin a jater
Přípravek Optimark se nesmí podávat pacientům se závažnou poruchou funkce ledvin (GFR < 30 ml/min/1,73 m2) a/nebo akutním poškozením ledvin a pacientům s transplantovanými játry nebo pacientům v perioperačním období transplantace jater. Pouze po pečlivém zvážení přínosu/rizika může být přípravek Optimark použit u pacientů se středně závažnou poruchou funkce ledvin (GFR 30-59 ml/min/1,73 m2), a to v dávce nepřekračující 100 mikromolů/kg tělesné hmotnosti.
Během jednoho vyšetření může být podána pouze jedna dávka. Vzhledem k nedostatku informací o opakovaném podání není možné injekce přípravku Optimark opakovat dříve, než interval mezi injekcemi dosáhne alespoň 7 dní.
Způsob podání
Přípravek se musí podávat jako bolus periferní intravenózní injekcí. Po injekci by měl následovat proplach 5 ml roztoku chloridu sodného 9 mg/ml (0,9 %), aby bylo jisté, že se aplikovala celá injekce s kontrastní látkou. Doporučuje se použít flexibilní zaváděcí žilní katétr.
Optimark nesmí být podáván autoinjektorem u dětí ve věku od 2 do 11 let (viz bod 4.4).
Opatření, která je nutno učinit před zacházením s léčivým přípravkem nebo před jeho podáním Před použitím je třeba nádobu a roztok zkontrolovat.
Interakce s jinými léčivými přípravky a jiné formy interakce
Žádné formální studie interakcí nebyly provedeny.
Bylo prokázáno, že gadoversetamid způsobuje interferenci při stanovení hladiny kalcia v séru použitím kolorimetrické metody s o-kresolftalein komplexonem (OCP). Ale podání gadoversetamidu nezpůsobuje skutečný pokles sérového kalcia. V přítomnosti gadoversetamidu poskytuje metoda OCP chybnou, nízkou hodnotu kalcia v plazmě. Závažnost tohoto artefaktu měření je úměrná koncentraci gadoversetamidu v krvi a u pacientů s normální clearancí ledvin lze přesné výsledky získat asi za 90 minut po podání injekce. U pacientů se sníženou funkcí ledvin se clearance gadoversetamidu zpomaluje a interference při stanovování kalcia metodou OCP se prodlužuje. Gadoversetamid neovlivňuje další metody stanovování hladin sérového kalcia, jako je například kolorimetrická metoda s činidlem arsenazo III, metoda atomové absorpční spektroskopie a hmotnostní spektrometrie s indukčně vázaným plazmatem.
Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Optimark je určen k jednorázovému použití; nespotřebovaná část se musí zlikvidovat.
Optimark je třeba natáhnout do injekční stříkačky a okamžitě použít.
Roztok nepoužívejte, jestliže má změněnou barvu nebo obsahuje pevné částice. Jestliže se používá zařízení k opakovanému použití, je třeba úzkostlivě dbát na to, aby nebylo kontaminováno zbytky čisticích prostředků.
Přípravek je třeba před použitím zkontrolovat, zda je celý obsah rozpuštěn a zda je nádoba a uzávěr v nepoškozeném stavu. Jsou-li přítomny pevné látky, injekční lahvičku zlikvidujte.
Po použití injekční stříkačku a nespotřebovaný roztok zlikvidujte.
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. Oddělitelnou část štítku z injekční lahvičky je třeba vlepit do dokumentace pacienta, aby byl přesně zaznamenán použitý kontrastní přípravek s obsahem gadolinia. Též je nutno poznamenat podanou dávku. Pokud se použijí elektronické záznamy pacientů, měly by být do záznamu pacienta uvedeny název přípravku, číslo šarže a dávka.
49