Opdivo 10 Mg/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
OPDIVO 10 mg/ml koncentrát pro infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml koncentrátu obsahuje nivolumabum 10 mg.
Jedna 4ml lahvička obsahuje nivolumabum 40 mg.
Jedna 10ml lahvička obsahuje nivolumabum 100 mg.
Nivolumab je produkován v buňkách vaječníků čínských křečků rekombinantní DNA technologií. Pomocná látka se známým účinkem:
Jeden ml tohoto koncentrátu obsahuje 0,1 mmol sodíku (odpovídá 2,5 mg).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok (sterilní koncentrát).
Čirá až opalizující, bezbarvá až světle žlutá tekutina, která může obsahovat několik světlých částic. Roztok má přibližně pH 6,0 a osmolalitu přibližně 340 mosm/kg.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Melanom
Přípravek OPDIVO je indikován v monoterapii nebo v kombinaci s ipilimumabem k léčbě pokročilého (neresekovatelného nebo metastatického) melanomu u dospělých.
Zlepšení přežití bez progrese (PFS) u kombinace nivolumabu s ipilimumabem ve srovnání s monoterapií nivolumabem je potvrzeno jen u pacientů s nízkou úrovní nádorové exprese PD-L1 (viz body 4.4 a 5.1).
Nemalobuněčný karcinom plic (NSCLC)
Přípravek Opdivo je indikován k léčbě lokálně pokročilého nebo metastatického nemalobuněčného karcinomu plic (NSCLC) po předchozí chemoterapii u dospělých.
Renální karcinom (RCC)
Přípravek OPDIVO je indikován jako monoterapie k léčbě pokročilého renálního karcinomu po předchozí terapii u dospělých.
4.2 Dávkování a způsob podání
Léčbu musí zahájit a řídit lékař se zkušenostmi s léčbou nádorů. Dávkování
OPDIVO v monoterapiiDoporučená dávka přípravku OPDIVO je 3 mg/kg nivolumabu podávaného intravenózně po dobu 60 minut každé dva týdny.
OPDIVO v kombinaci s ipilimumabem
Doporučená dávka je 1 mg/kg nivolumabu podávaného intravenózní infuzí po dobu 60 minut každé tři týdny u prvních 4 dávek v kombinaci s ipilimumabem podávaným intravenózně v dávce 3mg/kg po dobu 90 minut.
Dále následuje druhá fáze, kde je podáváno 3 mg/kg nivolumabu intravenózně ve formě infuze po dobu 60 minut každé dva týdny.
Léčba přípravkem OPDIVO buď jako monoterapie nebo v kombinaci s ipilimumabem má pokračovat, dokud je pozorován klinický přínos nebo dokud ji pacient snáší.
Zvyšování nebo snižování dávky se nedoporučuje. V závislosti na individuální bezpečnosti a snášenlivosti může být nutné přerušení nebo vysazení dávky. Pokyny pro trvalé ukončení léčby nebo přerušení dávek jsou uvedeny v Tabulce 1. Podrobné pokyny pro řešení imunitně podmíněných nežádoucích účinků jsou popsány v bodě 4.4.
|
Tabulka 1: |
Doporučená úprava léčby přípravkem OPDIVO nebo přípravkem OPDIVO v kombinaci s ipilimumabem | |
|
Imunitně podmíněné nežádoucí účinky |
Závažnost |
Úprava léčby |
|
Imunitně podmíněná pneumonitida |
Pneumonitida 2. stupně Pneumonitida 3. nebo 4. stupně |
Vysaďte dávku(y), dokud symptomy neustoupí, nezlepší se rentgenové abnormality a není dokončena léčba kortikosteroidy. Trvale ukončete léčbu. |
|
Imunitně podmíněná kolitida |
Průjem nebo kolitida 2. stupně Průjem nebo kolitida 3. stupně - OPDIVO v monoterapii - OPDIVO + ipilimumab Průjem nebo kolitida 4. stupně |
Vysaďte dávku(y), dokud symptomy neustoupí a není dokončena léčba kortikosteroidy, je-li potřebná. Vysaďte dávku(y), dokud symptomy neustoupí a není dokončena léčba kortikosteroidy. Trvale ukončete léčbu. Trvale ukončete léčbu. |
|
Imunitně podmíněná hepatitida |
Zvýšení aspartátaminotransferázy (AST), alaninaminotransferázy (ALT), nebo celkového bilirubinu 2. stupně Zvýšení AST, ALT, nebo celkového bilirubinu 3. nebo 4. stupně |
Vysaďte dávku(y), dokud se laboratorní hodnoty nevrátí na výchozí hodnotu a není dokončena léčba kortikosteroidy, je-li potřebná. Trvale ukončete léčbu. |
|
Imunitně podmíněná nefritida a renální dysfunkce |
Zvýšení kreatininu 2. nebo 3. stupně Zvýšení kreatininu 4. stupně |
Vysaďte dávku(y), dokud se kreatinin nevrátí na výchozí hodnotu a není dokončena léčba kortikosteroidy. Trvale ukončete léčbu. |
|
Imunitně podmíněná endokrinopatie |
Symptomatická hypotyreóza 2. nebo 3. stupně, hypertyreóza, hypofyzitida, nedostatečnost nadledvin 2. stupně, diabetes mellitus 3. stupně Hypotyreóza 4. stupně Hypertyreóza 4. stupně Hypofyzitida 4. stupně Nedostatečnost nadledvin 3. nebo 4. stupně Diabetes mellitus 4. stupně |
Vysaďte dávku(y), dokud symptomy neustoupí a není dokončena léčba kortikosteroidy, je-li potřebná vzhledem k příznakům akutního zánětu. Léčba by měla pokračovat v případě hormonální substituční léčbya do doby, dokud nejsou přítomny žádné symptomy. Trvale ukončete léčbu. |
|
Imunitně podmíněná vyrážka |
Vysaďte dávku(y), dokud symptomy neustoupí a není dokončena léčba kortikosteroidy. Trvale ukončete léčbu. | |
|
Jiné nežádoucí účinky |
Stupeň 3 (první výskyt) Stupeň 4 nebo recidivující stupeň 3; přetrvávající stupeň 2 nebo 3 i přes úpravu léčby; nemožnost snížit dávku kortikosteroidu na 10 mg prednisonu nebo její ekvivalent denně |
Vysaďte dávku(y) Trvale ukončete léčbu. |
Poznámka: Stupně toxicity jsou v souladu s běžnými terminologickými kritérii nežádoucích účinků podle Národního institutu pro rakovinu (National Cancer Institute Common Terminology Criteria for Adverse Events), verze 4.0 (NCI-CTCAE v4).
Doporučení pro použití hormonální substituční léčby je uvedeno v bodu 4.4.
Pacienti léčení přípravkem OPDIVO musí dostat kartu pacienta a informace o rizicích přípravku OPDIVO (viz také příbalová informace).
Je-li přípravek OPDIVO podáván v kombinaci s ipilimumabem a je-li vysazen jeden z těchto přípravků, má se vysadit i druhý přípravek. Pokud se podávání přípravku po určité prodlevě obnoví, má se obnovit podle individuálního zhodnocení pacienta buď léčba v kombinaci nebo monoterapie přípravkem OPDIVO.
Zvláštní populace
Pediatrická populace
Bezpečnost a účinnost přípravku OPDIVO u dětí ve věku do 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Starší pacienti
U starších pacientů (> 65 let) není nutná žádná úprava dávkování (viz body 5.1 a 5.2).
Nemalobuněčný karcinom plic
Data získaná sledováním pacientů ve věku 75 let a více jsou příliš omezená na vyvození závěrů ohledně této populace.
Porucha funkce ledvin
Na základě výsledků populační farmakokinetiky (PK) není nutná žádná úprava dávky u pacientů s mírnou až středně závažnou poruchou funkce ledvin (viz bod 5.2). Data získaná sledováním pacientů se závažnou poruchou funkce ledvin jsou příliš omezená na vyvození závěrů ohledně této populace.
Porucha funkce jater
Na základě výsledků populační farmakokinetiky (PK) není nutná žádná úprava dávky u pacientů s mírnou poruchou funkce jater (viz bod 5.2). Data získaná sledováním pacientů se středně závažnou či závažnou poruchou funkce jater jsou příliš omezená na vyvození závěrů ohledně této populace. Přípravek OPDIVO musí být podáván s opatrností pacientům se středně závažnou poruchou funkce jater (celkový bilirubin > 1,5násobek až 3násobek horní hranice normálu [upper limit of normal - ULN] a jakékoli zvýšení AST) nebo u pacientů s závažnou poruchou funkce jater (celkový bilirubin > 3násobek ULN a jakékoli zvýšení AST).
Nemalobuněčný karcinom plic
Pacienti s ECOG skóre (Eastern Cooperative Oncology Group) fyzické aktivity > 2 byli z klinických studií s NSCLC vyloučeni (viz body 4.4 a 5.1).
Způsob podání
Přípravek OPDIVO je určen pouze k intravenóznímu podání. Podává se jako intravenózní infuze po dobu 60 minut. Infuze se musí podávat přes sterilní, nepyrogenní filtr s nízkou schopností vázat proteiny a s póry o velikosti 0,2-1,2 ^m.
Přípravek OPDIVO se nesmí podávat jako nitrožilní bolus nebo bolusová injekce.
Celková požadovaná dávka přípravku OPDIVO se může podávat v infuzi přímo jako roztok s koncentrací 10 mg/ml, nebo může být naředěna až na 1 mg/ml roztokem chloridu sodného 9 mg/ml (0,9%) pro injekce nebo roztokem glukózy 50 mg/ml (5%) pro injekce.
Pokud je podáván v kombinaci s ipilimumabem, má být přípravek OPDIVO podán jako první, poté má tentýž den následovat podání ipilimumabu. Je nutné použít samostatný infuzní vak a filtr pro každou infuzi.
Návod k zacházení s léčivým přípravkem před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Pokud je nivolumab podáván v kombinaci s ipilimumabem, je nutné se před zahájením léčby seznámit se Souhrnem údajů o přípravku ipilimumabu. Imunitně podmíněné nežádoucí účinky se objevovaly s vyšší frekvencí, pokud byl nivolumab podáván v kombinaci s ipilimumabem než při podávání nivolumabu v monoterapii. Většina imunitně podmíněných nežádoucích účinků se zlepšila či zcela ustoupila po použití vhodných léčebných opatření, jako je zahájení léčby kortikosteroidy či úpravy léčby (viz bod 4.2).
Během kombinované léčby byly také hlášeny kardiální nežádoucí účinky a plicní embolie.
Pacienti mají být nepřetržitě monitorováni vzhledem ke kardiálním a pulmonálním nežádoucím účinkům, a také s ohledem na klinické známky, příznaky a laboratorní odchylky ukazující na poruchy elektrolytů a dehydrataci, a to před léčbou a pravidelně v jejím průběhu. V případě život ohrožujících nebo recidivujících závažných kardiálních nebo pulmonálních nežádoucích účinků se má nivolumab v kombinaci s ipilimumabem vysadit.
Pacienti mají být průběžně sledováni (minimálně do 5 měsíců po poslední dávce), protože nežádoucí účinek nivolumabu nebo nivolumabu v kombinaci s ipilimumabem se může objevit kdykoli během podávání nebo po ukončení léčby.
U podezření na imunitně podmíněné nežádoucí účinky má být provedeno adekvátní hodnocení pro potvrzení etiologie nebo vyloučení jiných příčin. Podle závažnosti nežádoucího účinku má být nivolumab nebo nivolumab v kombinaci s ipilimumabem vysazen a mají se podat kortikosteroidy. Jestliže je pro léčení nežádoucícho účinku použita imunosuprese kortikosteroidy, musí být po zlepšení dávka snižována postupně po dobu nejméně jednoho měsíce. Rychlé snížení dávky by mohlo vést ke zhoršení nebo recidivě nežádoucího účinku. Jestliže dojde ke zhoršení nebo se nedostaví zlepšení navzdory použití kortikosteroidů, je třeba přidat jinou imunosupresivní léčbu než kortikosteroidy.
Nivolumab nebo nivolumab v kombinaci s ipilimumabem nemá být znovu nasazován, dokud pacient dostává imunosupresivní dávky kortikosteroidů nebo jinou imunosupresivní léčbu. K zabránění oportunních infekcí u pacientů dostávajících imunosupresivní terapii se má použít profylaktické podání antibiotik.
V případě závažných a opakujících se imunitně podmíněných nežádoucích účinků a jakýchkoli život ohrožujících imunitně podmíněných nežádoucích účinků musí být nivolumab nebo nivolumab v kombinaci s ipilimumabem trvale vysazen.
Používání nivolumabu u pacientů s melanomem, u kterých onemocnění rychle postupuje Lékaři mají zvážit před zahájením léčby pacientů s rychle postupujícím onemocněním opožděný nástup účinku nivolumabu (viz bod 5.1).
Používání nivolumabu u pacientů s neskvamózním NSCLC
Lékaři mají zvážit opožděný nástup účinku nivolumabu před zahájením léčby pacientů s horší prognózou nebo agresivním onemocněním. U neskvamózního NSCLC byl po dobu 3 měsíců pozorován zvýšený počet úmrtí ve srovnání s docetaxelem. Faktory, souvisejícími s časnými úmrtími, byly horší prognóza a/nebo agresivnější onemocnění kombinované s nízkou nebo žádnou expresí PD-L1 na nádorových buňkách (viz bod 5.1).
Imunitně podmíněná pneumonitida
Při léčbě monoterapií nivolumabem nebo nivolumabem v kombinaci s ipilimumabem byla pozorována závažná pneumonitida nebo intersticiální onemocnění plic, včetně fatálních případů (viz bod 4.8).
U pacientů mají být monitorovány známky a příznaky pneumonitidy, jako jsou rentgenové změny
(např. ohniskové opacity mléčného skla, ložiskové filtráty), dušnost a hypoxie. Infekční etiologii a etiologii spojenou se základním onemocněním je třeba vyloučit.
U pneumonitidy 3. nebo 4. stupně musíte ukončit podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem trvale a mají být nasazeny kortikosteroidy v dávce odpovídající 2-4 mg/kg metylprednisolonu denně.
U (symptomatické) pneumonitidy 2. stupně má být nivolumab nebo nivolumab v kombinaci s ipilimumabem vysazen a mají být nasazeny kortikosteroidy v dávce odpovídající 1 mg/kg metylprednisolonu denně. Po zlepšení lze znovu nasadit nivolumab nebo nivolumab v kombinaci s ipilimumabem po postupném utlumení kortikosteroidů. Jestliže dojde ke zhoršení nebo se nedostaví zlepšení přes použití kortikosteroidů, lze zvýšit na dávku odpovídající 2-4 mg/kg metylprednisolonu denně a musí být trvale ukončeno podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem.
Imunitně podmíněná kolitida
Při léčbě monoterapií nivolumabu nebo nivolumabem v kombinaci s ipilimumabem byly pozorovány závažné průjmy nebo kolitida (viz bod 4.8). Pacienti sprůjmem a dalšími symptomy kolitidy, jako je bolest břicha a hlen nebo krev ve stolici, mají být monitorováni. Infekční etiologie a etiologie spojená se základním onemocněním má být vyloučena.
U průjmu nebo kolitidy 4. stupně musí být trvale ukončeno podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem a mají být nasazeny kortikosteroidy v dávce odpovídající 1-2 mg/kg metylprednisolonu denně.
U průjmu nebo kolitidy 3. stupně má být monoterapie nivolumabem vysazena a mají být nasazeny kortikosteroidy v dávce odpovídající 1-2 mg/kg metylprednisolonu denně. Po zlepšení lze znovu nasadit monoterapii nivolumabem po postupném utlumení kortikosteroidů. Jestliže dojde ke zhoršení nebo se nedostaví zlepšení přes použití kortikosteroidů, podávání monoterapie nivolumabem musí být trvale ukončeno. Průjem nebo kolitida 3. stupně pozorované u léčby nivolumabem v kombinaci s ipilimumabem vyžadují také trvalé ukončení léčby a nasazení kortikosteroidů v dávce odpovídající 1-2 mg/kg metylprednisolonu denně.
U průjmu nebo kolitidy 2. stupně má být nivolumab nebo nivolumab v kombinaci s ipilimumabem vysazen. Jestliže potíže přetrvávají, řešte je kortikosteroidy v dávce odpovídající 0,5-1 mg/kg metylprednisolonu denně. Po zlepšení lze znovu nasadit nivolumab nebo nivolumab v kombinaci s ipilimumabem po postupném utlumení kortikosteroidů, je-li třeba. Jestliže dojde ke zhoršení nebo se nedostaví zlepšení přes použití kortikosteroidů, lze zvýšit na dávku odpovídající 1-2 mg/kg metylprednisolonu denně a podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem musí být trvale ukončeno.
Imunitně podmíněná hepatitida
Při léčbě monoterapií nivolumabem nebo nivolumabem v kombinaci s ipilimumabem byla pozorována závažná hepatitida (viz bod 4.8). Pacienti mají být monitorováni na známky a příznaky hepatitidy, jako je zvýšení transamináz a celkového bilirubinu. Infekční etiologie a etiologie spojená se základním onemocněním má být vyloučena.
Při zvýšení transamináz nebo celkového bilirubinu 3. a 4. stupně musí být podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem trvale ukončeno a mají být nasazeny kortikosteroidy v dávce odpovídající 1-2 mg/kg metylprednisolonu denně.
Při zvýšení transamináz nebo celkového bilirubinu 2. stupně má být nivolumab nebo nivolumab v kombinaci s ipilimumabem vysazen. Jestliže potíže přetrvávají, mají být nasazeny kortikosteroidy v dávce odpovídající 0,5-1 mg/kg metylprednisolonu denně. Po zlepšení lze znovu nasadit nivolumab nebo nivolumab v kombinaci s ipilimumabem po postupném utlumení kortikosteroidů, je-li třeba. Jestliže dojde ke zhoršení nebo se nedostaví zlepšení přes použití kortikosteroidů, lze zvýšit na dávku odpovídající 1-2 mg/kg metylprednisolonu denně a podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem musí být trvale ukončeno.
Imunitně podmíněná nefritida a renální dysfunkce
Při léčbě monoterapií nivolumabem nebo nivolumabem v kombinaci s ipilimumabem byla pozorována závažná nefritida a renální dysfunkce (viz bod 4.8). Pacienti mají být monitorováni na známky a příznaky nefritidy nebo renální dysfunkce. U většiny pacientů dochází k asymptomatickému nárůstu kreatininu v séru. Etiologie spojená se základním onemocněním má být vyloučena.
Při zvýšení kreatininu v séru 4. stupně musí být podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem trvale ukončeno a mají být nasazeny kortikosteroidy v dávce odpovídající 1-2 mg/kg metylprednisolonu denně.
Při zvýšení kreatininu v séru 2. nebo 3. stupně má být nivolumab nebo nivolumab v kombinaci s ipilimumabem vysazen a nasazeny kortikosteroidy v dávce odpovídající 0,5 až 1 mg/kg metylprednisolonu denně. Po zlepšení lze znovu nasadit nivolumab nebo nivolumab v kombinaci s ipilimumabem po postupném utlumení kortikosteroidů. Jestliže dojde ke zhoršení nebo se nedostaví zlepšení přes použití kortikosteroidů, má být dávka kortikosteroidů zvýšena na dávku odpovídající 1-2 mg/kg metylprednisolonu denně a podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem musí být trvale ukončeno.
Imunitně podmíněná endokrinopatie
Při léčbě monoterapií nivolumabem nebo nivolumabem v kombinaci s ipilimumabem byly pozorovány vážné endokrinopatie jako hypotyreóza, hypertyreóza, hypofyzitida, nedostatečnost nadledvin a diabetická ketoacidóza (viz bod 4.8).
Pacienti mají být sledováni kvůli klinickým příznakům a symptomům endokrinopatií a hyperglykémie a kvůli změnám funkce štítné žlázy (při zahájení léčby, pravidelně během léčby a podle potřeby na základě klinického zhodnocení). Pacienti mohou trpět únavou, bolestmi hlavy, změnami duševního stavu, bolestmi břicha, neobvyklými střevními projevy a hypotenzí, nebo nespecifickými symptomy, které se mohou podobat jiným příčinám jako mozkovým metastázám nebo základnímu onemocnění. Pokud není zjištěna jiná etiologie, mají být příznaky nebo symptomy endokrinopatií považovány za imunitně podmíněné.
U symptomatické hypotyreózy má být nivolumab nebo nivolumab v kombinaci s ipilimumabem vysazen a dle potřeby má být zahájena hormonální substituční léčba štítné žlázy. U symptomatické hypertyreózy má být nivolumab nebo nivolumab v kombinaci s ipilimumabem vysazen a dle potřeby má být zahájena antityreoidální léčba. Pokud existuje podezření na akutní zánět štítné žlázy, má být zváženo nasazení kortikosteroidů v dávce odpovídající 1-2 mg/kg metylprednisolonu denně.
Po zlepšení lze znovu nasadit nivolumab nebo nivolumab v kombinaci s ipilimumabem po postupném utlumení kortikosteroidů, je-li třeba. Monitorování funkce štítné žlázy má i nadále pokračovat, aby byla zajištěna odpovídající hormonální substituční léčba. U život ohrožující hypertyreózy nebo hypotyreózy musí být podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem trvale ukončeno.
U symptomatické nedostatečnosti nadledvin 2. stupně má být nivolumab nebo nivolumab v kombinaci s ipilimumabem vysazen a dle potřeby má být zahájena léčba kortikosteroidy. U závažné (3. stupeň) nebo život ohrožující (4. stupeň) nedostatečnosti nadledvin musí být podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem trvale ukončeno. Monitorování funkce nadledvin a hormonální hladiny má i nadále pokračovat, aby byla zajištěna odpovídající léčba kortikosteroidy.
U symptomatické hypofyzitidy 2. nebo 3. stupně má být nivolumab nebo nivolumab v kombinaci s ipilimumabem vysazen a dle potřeby má být zahájena hormonální substituční léčba. Pokud existuje podezření na akutní zánět hypofýzy, má být zváženo nasazení kortikosteroidů v dávce odpovídající 1-2 mg/kg metylprednisolonu denně. Po zlepšení lze znovu nasadit nivolumab nebo nivolumab v kombinaci s ipilimumabem po postupném utlumení kortikosteroidů, je-li třeba. U život ohrožující (4. stupeň) hypofyzitidy musí být podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem trvale ukončeno. Monitorování funkce hypofýzy a hormonální hladiny má i nadále pokračovat, aby byla zajištěna odpovídající hormonální substituční léčba.
U symptomatického diabetu má být nivolumab nebo nivolumab v kombinaci s ipilimumabem vysazen a dle potřeby má být zahájena substituční léčba inzulinem. Monitorování krevního cukru má i nadále pokračovat, aby byla zajištěna odpovídající substituční léčba inzulinem. U život ohrožujícího diabetu musí být podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem trvale ukončeno.
Imunitně podmíněná vyrážka
Při léčbě nivolumabem v kombinaci s ipilimumabem a méně často při monoterapii nivolumabem byla pozorována závažná vyrážka. (viz bod 4.8). Podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem má být přerušeno při vyrážce 3. stupně a ukončeno při stupni 4. Závažná vyrážka má být léčena vysokými dávkami kortikosteroidů odpovídajícími dávce 1 až 2 mg/kg/den methylprednisolonu.
Vzácně byly pozorovány případy toxické epidermální nekrolýzy (TEN), některé z nich s fatálními následky. Pokud se objeví příznaky Steven-Johnsonova syndromu (SJS) nebo TEN, má být léčba nivolumabem přerušena a pacient má být vyšetřen a léčen na specializovaném oddělení. Pokud se během užívání nivolumabu u pacienta SJS nebo TEN vyvine, doporučuje se trvalé ukončení podávání přípravku.
Opatrnosti je třeba, pokud se zvažuje použití nivolumabu u pacientů, kteří prodělali závažné nebo život ohrožující kožní nežádoucí účinky na předchozí léčbu jinými imunostimulačními protinádorovými léky.
Jiné imunitně podmíněné nežádoucí účinky
Následující imunitně podmíněné nežádoucí účinky byly hlášeny u méně než 1 % pacientů léčených monoterapií nivolumabem v klinických studiích napříč různým dávkováním a typy nádorů: pankreatitida, uveitida, demyelinizace, autoimunitní neuropatie (včetně parézy n. facialis a n. abducens), syndrom Guillain-Barré, hypopituitarismus a myastenický syndrom.
Napříč klinickými studiemi zkoumajícími terapii nivolumabem v kombinaci s ipilimumabem byly u méně než 1 % pacientů hlášeny tyto další klinicky významné imunitně podmíněné nežádoucí účinky: gastritida, sarkoidóza a duodenitida.
U podezření na imunitně podmíněné nežádoucí účinky má být provedeno adekvátní vyhodnocení pro potvrzení etiologie nebo vyloučení jiných příčin. Na základě závažnosti nežádoucího účinku má být nivolumab nebo nivolumab v kombinaci s ipilimumabem vysazen a má být zahájena léčba kortikosteroidy. Po zlepšení lze znovu nasadit nivolumab nebo nivolumab v kombinaci s ipilimumabem po postupném utlumení kortikosteroidů. Podávání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem musí být trvale ukončeno v případě výskytu jakéhokoliv opakujícího se závažného imunitně podmíněného nežádoucího účinku nebo jakéhokoliv život ohrožujícího imunitně podmíněného nežádoucího účinku.
Reakce související s infuzí
V klinických studiích nivolumabem nebo nivolumabem v kombinaci s ipilimumabem byly hlášeny závažné reakce na infuzi (viz bod 4.8). V případě těžké nebo život ohrožující infuzní reakce musí být podání nivolumabu nebo nivolumabu v kombinaci s ipilimumabem ukončeno a zahájena odpovídající léčba. Pacienti s mírnou nebo středně těžkou infuzní reakcí mohou dostávat nivolumab nebo nivolumab v kombinaci s ipilimumabem za současného důkladného sledování a používání premedikace podle lokálních terapeutických postupů pro profylaxi reakcí na infuzi.
Opatření u jednotlivých onemocnění
Melanom
Pacienti s výchozím skóre fyzické aktivity > 2, s aktivními mozkovými metastázami, autoimunitním onemocněním a pacienti, kteří již užívali systémová imunosupresiva před vstupem do studie, byli z klinického hodnocení s nivolumabem nebo nivolumabem v kombinaci s ipilimumabem vyloučeni. Pacienti s okulárním/uveálním melanomem byli vyloučeni z klinických studií s melanomem. Kromě toho byli ze studie CA209037 vyloučeni pacienti, kteří měli nežádoucí účinek 4. stupně související s předchozí anti-CTLA-4 terapií (viz bod 5.1). Vzhledem k nedostatku údajů je třeba používat nivolumab u těchto pacientů s opatrností po pečlivém zvážení potenciálního individuálního rizika a prospěchu.
Zlepšení přežití bez progrese (PFS) u kombinace nivolumabu s ipilimumabem ve srovnání s monoterapií nivolumabem je potvrzeno jen u pacientů s nízkou úrovní nádorové exprese PD-L1. Před zahájením kombinované léčby je nutné provést pečlivé posouzení individuálních charakteristik pacienta a nádoru a vzít v úvahu zjištěný přínos a toxicitu kombinované léčby ve srovnání s monoterapií nivolumabem (viz body 4.8 a 5.1).
Nemalobuněčný karcinom plic
Pacienti s výchozím skóre fyzické aktivity > 2, s aktivními mozkovými metastázami nebo autoimunitním onemocněním, symptomatickým intersticiálním plicním onemocněním a pacienti, kteří již užívali systémová imunosupresiva před vstupem do studie, byli z klinických hodnocení NSCLC vyloučeni (viz body 4.5 a 5.1). Vzhledem k absenci údajů je třeba používat nivolumab u těchto pacientů s opatrností po pečlivém zvážení potenciálního individuálního rizika a prospěchu.
Renální karcinom
Pacienti s výskytem mozkových metastáz aktuálně nebo v anamnéze, s aktivním autoimunitním onemocněním či se zdravotním stavem vyžadujícím systémovou imunosupresi byli z pivotní studie RCC vyloučeni (viz body 4.5 a 5.1). Vzhledem k absenci údajů je třeba používat nivolumab u těchto pacientů s opatrností a po pečlivém zvážení potenciálního individuálního rizika a prospěchu.
Pacienti na dietě s nízkým obsahem sodíku
Jeden ml tohoto léčivého přípravku obsahuje 0,1 mmol (nebo 2,5 mg) sodíku. Toto je třeba vzít v úvahu při léčbě pacientů na dietě s omezeným příjmem sodíku.
Karta pacienta
Všichni lékaři předepisující přípravek OPDIVO se musí seznámit s Informací pro lékaře a Průvodcem zvládáním nežádoucích účinků. Předepisující lékař musí s pacientem prodiskutovat rizika léčby přípravkem OPDIVO. Pacientovi bude poskytnuta Karta pacienta vždy, když mu bude přípravek předepsán.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nivolumab je humánní monoklonální protilátka, studie farmakokinetických interakcí tedy nebyly provedeny. Protože monoklonální protilátky nejsou metabolizovány enzymy cytochromu P450 (CYP) nebo jinými enzymy metabolizujícími léky, nepředpokládá se, že inhibice nebo indukce těchto enzymů současně podávanými léčivými přípravky bude mít dopad na farmakokinetiku nivolumabu.
Jiné formy interakce
Systémová imunosuprese
Použití systémových kortikosteroidů a jiných imunosupresiv na počátku, před zahájením léčby nivolumabem, je třeba se vyhnout, protože může existovat jejich potenciální interference s farmakodynamickou aktivitou nivolumabu. Nicméně systémové kortikosteroidy a další imunosupresiva mohou být použita po spuštění léčby nivolumabem k léčbě imunitně podmíněných nežádoucích účinků. Předběžné výsledky nenaznačují, že by systémová imunosuprese po zahájení léčby nivolumabem vylučovala odpověď na nivolumab.
4.6 Fertilita, těhotenství a kojení
O použití nivolumabu u těhotných žen nejsou dostupné žádné údaje. Studie na zvířatech prokázaly embryofetální toxicitu (viz bod 5.3). O humánním IgG4 je známo, že prochází placentární bariérou; nivolumab je IgG4, proto je možné, že nivolumab přechází z matky na vyvíjející se plod. Nivolumab se nedoporučuje během těhotenství a také fertilním ženám, které nepoužívají účinnou antikoncepci, pokud klinický přínos nepřevyšuje možné riziko. Účinnou antikoncepci je třeba používat nejméně po dobu 5 měsíců od poslední dávky nivolumabu.
Kojení
Není známo, zda se nivolumab vylučuje do lidského mléka. Vhledem k tomu, že mnoho léčivých přípravků včetně protilátek může být vylučováno do mateřského mléka, riziko pro novorozence a kojence nelze vyloučit. Na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku je nutno rozhodnout, zda přerušit kojení nebo přerušit léčbu nivolumabem.
Fertilita
Studie hodnotící vliv nivolumabu na fertilitu nebyly provedeny. Proto není účinek nivolumabu na fertilitu mužů a žen znám.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Podle farmakodynamických vlastností nivolumabu není pravděpodobné, že by nivolumab ovlivňoval schopnost řídit a obsluhovat stroje. Pacienti musí být informováni, že by kvůli možným nežádoucím účinkům, jako je únava (viz bod 4.8), měli řídit nebo obsluhovat stroje s opatrností, dokud si nebudou jisti, že na ně nivolumab nemá nežádoucí vliv.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
V datovém souboru sloučeném ze studií zkoumajících nivolumab 3 mg/kg v monoterapii napříč typy tumorů (CA209066, CA209037, CA209067 (monoterapeutická skupina), CA209017, CA209057, CA209063 a CA209025) byly nejčastějšími nežádoucími účinky (> 10 %) únava (34 %), vyrážka (19 %), svědění (14 %), průjem (13 %), nauzea (13 %) a snížená chuť k jídlu (10 %).Většina nežádoucích účinků byla mírná až středně těžká (stupeň závažnosti 1 nebo 2).
V datovém souboru sloučeném ze studií s kombinací nivolumabu a ipilimumabu v léčbě melanomu (CA209067 (skupina s kombinovanou léčbou), CA209069 a CA209004-kohorta 8) byly nej častějšími nežádoucími účinky (> 10 %) vyrážka (51 %), únava (43 %), průjem (42 %), pruritus (35 %), nauzea (25 %), pyrexie (19 %), snížená chuť k jídlu (15 %), hypotyreóza (15 %), zvracení (14 %), kolitida (14 %), bolest břicha (13 %), artralgie (11 %) a bolest hlavy (11 %). Většina nežádoucích účinků byla mírná až středně těžká (stupeň závažnosti 1 nebo 2).
Ve skupině pacientů léčených nivolumabem v kombinaci s ipilimumabem ve studii CA209067 mělo 151/313 (48 %) pacientů první výskyt nežádoucího účinku 3. nebo 4. stupně závažnosti během počáteční fáze kombinované terapie.
Ze 147 pacientů této skupiny, kteří pokračovali v léčbě fází monoterapie, mělo 37 (25 %) zkušenost s alespoň jedním nežádoucím účinkem 3. nebo 4. stupně závažnosti během fáze monoterapie.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky hlášené ve sloučeném datovém souboru pacientů léčených nivolumabem v monoterapii (n=1728) a pacientů léčených nivolumabem v kombinaci s ipilimumabem (n=448) jsou prezentovány v Tabulce 2. Tyto nežádoucí účinky jsou prezentovány podle třídy orgánových systémů a frekvence. Frekvence jsou definovány jako: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně
časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 2: Nežádoucí účinky v klinických studiích
|
Nivolumab v monoterapii |
Nivolumab v kombinaci s ipilimumabem | |
|
Infekce a infestace | ||
|
Časté |
infekce horních cest dýchacích |
pneumoniea, infekce horních cest dýchacích |
|
Méně časté |
pneumoniea, bronchitida |
bronchitida |
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy) | ||
|
Vzácné |
histiocytární nekrotizující lymfadenitida (Kikuchiho lymfadenitida) | |
|
Poruchy krve a lymfatického systému | ||
|
Časté |
eozinofilie | |
|
Méně časté |
eozinofilie | |
|
Poruchy imunitního systému | ||
|
Časté |
reakce související s infuzíb, hypersensitivita |
reakce související s infuzíb, hypersensitivita |
|
Méně časté |
anafylaktická reakceb |
sarkoidóza |
|
Endokrinní poruchy | ||
|
Velmi časté |
hypotyreóza | |
|
Časté |
hypotyreóza, hypertyreóza, hyperglykémieb |
nedostatečnost nadledvin, hypopituitarismus, hypofyzitida, thyroiditis, hyperglykémieb |
|
Méně časté |
nedostatečnost nadledvin, hypopituitarismus, hypofyzitida, thyroiditis, diabetická ketoacidóza |
diabetická ketoacidóza, diabetes mellitus |
|
Vzácné |
diabetes mellitus | |
|
Poruchy metabolismu a výživy | ||
|
Velmi časté |
snížená chuť k jídlu |
snížená chuť k jídlu |
|
Časté |
dehydratace | |
|
Méně časté |
dehydratace, metabolická acidóza | |
|
Poruchy jater a žlučových cest | ||
|
Časté |
hepatitidab | |
|
Méně časté |
hepatitidab, hyperbilirubinémie | |
|
Vzácné |
cholestáza | |
|
Poruchy nervového systému | ||
|
Velmi časté | ||
|
Časté |
periferní neuropatie, bolest hlavy, závratě |
periferní neuropatie, závratě |
|
Méně časté |
polyneuropatie |
syndrom Guillain-Barré, polyneuropatie, neuritida, obrna nervus peroneus, autoimunní neuropatie (včetně parézy n. facialis a n. abducens) |
|
Vzácné |
syndrom Guillain-Barré, demyelinizace, myastenický syndrom, autoimunní neuropatie (včetně parézy n. facialis a n. abducens) | |
|
Poruchy oka | ||
|
Časté |
rozmazané vidění, suché oko |
uveitida, rozmazané vidění |
|
Méně časté |
uveitida | |
|
Srdeční poruchy | ||
|
Časté | ||
|
Méně časté |
arytmie (včetně ventrikulární arytmie)c, fibrilace síní | |
|
Vzácné |
arytmie (včetně ventrikulární arytmie)c, fibrilace síní | |
|
Cévní poruchy | ||
|
Časté |
hypertenze |
hypertenze |
|
Méne časté |
vaskulitida | |
|
Respirační, hrudní a mediastinální poruchy | ||
|
Časté |
neumonitidaa,b, plicní emboliea, dyspnoe, kašel | |
|
Méně časté |
pleurální výpotek |
pleurální výpotek |
|
Vzácné |
infiltrace plic | |
|
Gastrointestiná |
ní poruchy | |
|
Velmi časté |
kolitida, průjem, zvracení, nauzea, bolest břicha | |
|
Časté |
kolitida, stomatitida, zvracení, bolest břicha, zácpa, sucho v ústech |
stomatitida, gastritida, zácpa, sucho v ústech |
|
Méně časté |
pankreatitida |
pankreatitida, intestinální perforace, duodenitida |
|
Vzácné |
gastritida, duodenální vřed | |
|
Poruchy kůže a podkožní tkáně | ||
|
Velmi časté | ||
|
Časté |
vitiligo, suchá kůže, erytém, alopecie |
vitiligo, suchá kůže, erytém, alopecie, kopřivka |
|
Méně časté |
erythema multiforme, psoriáza, rosacea, kopřivka |
psoriáza |
|
Vzácné |
toxická epidermální nekrolýzaa,e |
toxická epidermální nekrolýzaa,e |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Velmi časté |
artralgie | |
|
Časté |
muskuloskeletální bolestf, artralgie |
muskuloskeletální bolestf |
|
Méně časté |
polymyalgia rheumatica, artritida |
spondyloartropatie, Sjogrenův syndrom, artritida, myopatie |
|
Vzácné |
myopatie | |
|
Poruchy ledvin a močových cest | ||
|
Časté |
selhání ledvina,b | |
|
Méně časté |
tubulointersticiální nefritida, selhání ledvinab |
tubulointersticiální nefritida |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Velmi časté | ||
|
Časté |
horečka, otok (včetně periferního otoku) |
otok (včetně periferního otoku), bolest |
|
Méně časté |
bolest, bolest na hrudi | |
|
Vyšetření8 | ||
|
Velmi časté |
zvýšená hladina AST, zvýšená hladina ALT, zvýšená alkalická fosfatáza, zvýšená lipáza, zvýšená amyláza, hypokalcémie, zvýšený kreatinin, lymfopenie, leukopenie, trombocytopenie, anemie, hyperkalcemie, hyperkalemie, hypokalemie, hypomagnezemie, hyponatremie |
zvýšená hladina AST, zvýšená hladina ALT, zvýšený celkový bilirubin, zvýšená alkalická fosfatáza, zvýšená lipáza, zvýšená amyláza, zvýšený kreatinin, lymfopenie, leukopenie, neutropenie, trombocytopenie, anemie, hypokalcemie, hyperkalemie, hypokalemie, hypomagnezemie, hyponatremie |
|
Časté |
zvýšení celkového bilirubinu, neutropenie, hypermagnezemie, hypernatremie, hmotnost snížená |
hyperkalcemie, hypermagnezemie, hypernatremie, snížená hmotnost |
a
b
Fatální případy byly hlášeny v dokončených nebo pokračujících klinických studiích.
Život ohrožující případy byly hlášeny v dokončených či pokračujících klinických studiích.
Frekvence nežádoucích účinků u třídy orgánových systémů srdeční poruchy byla bez ohledu na příčinu u populace s metastatickým melanomem, která již užívala CTLA4/BRAF inhibitor, vyšší ve skupině s nivolumabem než ve skupině s chemoterapií. Míra výskytu na 100 pacientoroků expozice činila 9,3 vs 0; závažné kardiální příhody byly hlášeny u 4,9 % pacientů ve skupině nivolumabu vs 0 ve skupině zvolené zkoušejícím. Frekvence kardiálních příhod byla u populace s metastatickým melanomem bez předchozí léčby nižší ve skupině nivolumabu než ve skupině dakarbazinu. Všechny byly zkoušejícími považovány za nesouvisející s nivolumabem kromě arytmie (atriální fibrilace, tachykardie a ventrikulární arytmie).
c
d
e
f
g
Vyrážka je souhrnný pojem, který zahrnuje makulopapulární vyrážku, erytematózní vyrážku, svědivou vyrážku, folikulární vyrážku, makulární vyrážku, morbiliformní vyrážku, papulární vyrážku, pustulární vyrážku, papuloskvamózní vyrážku, vezikulární vyrážku, vyrážku na kůži celého těla, dermatitidu, akneiformní dermatitidu, alergickou dermatitidu, atopickou dermatitidu, bulózní dermatitidu, exfoliativní dermatitidu, psoriatiformní dermatitidu a polékový kožní výsev.
Zaznamenáno ve studiích mimo sloučená data. Frekvence je založena na expozici v rámci celého programu.
Muskuloskeletální bolest je souhrnný pojem, který zahrnuje bolest zad, bolest kostí, muskuloskeletální bolest hrudníku, muskuloskeletální diskomfort, myalgii, bolest krku, bolest končetin a bolest páteře. Frekvence laboratorních údajů odrážejí procento pacientů, kteří měli zhoršení z výchozí hodnoty v laboratorních měřeních. Viz “Popis vybraných nežádoucích účinků; laboratorní abnormality” níže.
Popis vybraných nežádoucích účinků
Nivolumab nebo nivolumab v kombinaci s ipilimumabem je spojován s imunitně podmíněnými nežádoucími účinky. Pokud byla zvolena vhodná léčba, ve většině případů došlo k vyléčení těchto imunitně podmíněných nežádoucích účinků. Trvalé ukončení léčby si tyto účinky vyžádaly u více pacientů léčených kombinací nivolumabu s ipilimumabem než u pacientů léčených nivolumabem v monoterapii - u imunitně podmíněné kolitidy (16 % oproti 0,7 %), u imunitně podmíněné hepatitidy (9 % oproti 0,9 %) a u imunitně podmíněných endokrinopatií (2,5 % oproti 0,1 %). U pacientů s těmito nežádoucími účinky byly vysoké dávky kortikosteroidů (odpovídající alespoň 40 mg prednisonu denně) nutné u většího počtu pacientů užívajících kombinaci nivolumabu a ipilimumabu než u pacientů s nivolumabem v monoterapii - u imunitně podmíněné kolitidy (47 % oproti 14 %) a u imunitně podmíněné hepatitidy (46 % oproti 16 %). Pokyny pro řešení těchto nežádoucích účinků jsou popsány v bodě 4.4.
Imunitně _ podmíněná _ pneumonitida
U pacientů léčených nivolumabem v monoterapii byl výskyt pneumonitidy, včetně intersticiálního plicního onemocnění, hlášen u 3,2 % pacientů (56/1728). Většina těchto nežádoucích účinků byla 1. nebo 2. stupně závažnosti, pneumonitida byla hlášena u 0,7 % (12/1728) a intersticiální plicní onemocnění bylo hlášeno u 1,7 % (29/1728) pacientů. Nežádoucí účinky stupně 3 byly hlášeny u 0,8 % (14/1728) a stupně 4 u <0,1 % (1/1728) pacientů. Nežádoucí účinky 5. stupně závažnosti v těchto studiích hlášeny nebyly.Střední čas do nástupu byl 3,6 měsíce (rozpětí: 0,4-19,6). K vyléčení došlo u 47 pacientů (84 %) se střední dobou do vyléčení 5,3 týdnů (rozpětí: 0,6-53,1+); + vyjadřuje cenzurované sledování.
U pacientů léčených nivolumabem v kombinaci s ipilimumabem byl výskyt pneumonitidy, včetně intersticiálního plicního onemocnění, hlášen u 7,4 % pacientů (33/448). Nežádoucí účinky 2. stupně byly hlášeny u 4,5 % pacientů (20/448), 3. stupně u 1,1 % (5/448) a 4. stupně u 0,2 % pacientů (1/448). Jedna pneumonitida 3. stupně se zhoršila během 11 dní ve fatální. Střední čas do nástupu byl 2,3 měsíce (rozpětí: 0,7-6,7). K vyléčení došlo u 29 pacientů (87,9 %) se střední dobou do vyléčení
6,1 týdnů (rozpětí: 0,3-46,9+).
Imunitně podmíněná kolitida
U pacientů léčených nivolumabem v monoterapii byl výskyt průjmu či kolitidy hlášen u 13,6 % pacientů (235/1728). Většina nežádoucích účinků byla 1. či 2. stupně závažnosti, 1. stupeň závažnosti byl hlášen u 9,0 % (156/1728) a 2. stupeň byl hlášen u 3,0 % (52/1728) pacientů. Nežádoucí účinky 3. stupně závažnosti byly hlášeny u 1,6 % (27/1728) pacientů. V těchto studiích nebyly hlášeny žádné nežádoucí účinky 4. nebo 5. stupně závažnosti.
Střední čas do nástupu byl 1,8 měsíce (rozpětí: 0,0-20,9). K vyléčení došlo u 207 pacientů (89 %) se střední dobou do vyléčení 2,1 týdnů (rozpětí: 0,1-88,3+).
U pacientů léčených nivolumabem v kombinaci s ipilimumabem byl výskyt průjmu či kolitidy hlášen u 45,5 % pacientů (204/448). Nežádoucí účinky 2. stupně byly hlášeny u 13,2 % pacientů (59/448),
3. stupně u 15,4 % (69/448) a 4. stupně u 0,4 % pacientů (2/448). Nežádoucí účinky 5. stupně závažnosti v těchto studiích hlášeny nebyly. Střední čas do nástupu byl 1,1 měsíce (rozpětí: 0,0-10,4). K vyléčení došlo u 184 pacientů (90,6 %) se střední dobou do vyléčení 3,0 týdnů (rozpětí: 0,1-78,7+).
Imunitně podmíněná hepatitida
U pacientů léčených nivolumabem v monoterapii byl výskyt abnormálních testů funkce jater hlášen u 7,0 % pacientů (121/1728). Většina nežádoucích účinků byla 1. či 2. stupně závažnosti, 1. stupeň závažnosti byl hlášen u 3,9 % (68/1728) a 2. stupeň byl hlášen u 1,3 % (22/1728) pacientů. Nežádoucí účinky 3. a 4. stupně závažnosti byly hlášeny u 1,4 % (25/1728) a 0,3 % (6/1728) pacientů. V těchto studiích nebyly hlášeny žádné nežádoucí účinky 5. stupně závažnosti. Střední čas do nástupu byl
1,9 měsíců (rozpětí: 0,0-18,7). K vyléčení došlo u 95 pacientů (79 %) se střední dobou do vyléčení
5,1 týdnů (rozpětí: 0,1-82,6+).
U pacientů léčených nivolumabem v kombinaci s ipilimumabem byl výskyt abnormálních testů funkce jater hlášen u 27,9 % pacientů (125/448). Nežádoucí účinky 2. stupně byly hlášeny u 6,3 % pacientů (28/448), 3. stupně u 15,0 % (67/448) a 4. stupně u 1,8 % pacientů (8/448). Nežádoucí účinky
5. stupně závažnosti v těchto studiích hlášeny nebyly. Střední čas do nástupu byl 1,4 měsíce (rozpětí: 0,0-11,0). K vyléčení došlo u 116 pacientů (92,8 %) se střední dobou do vyléčení 5,0 týdnů (rozpětí: 0,1-53,1).
Imunitně podmíněná nefritida a renální dysfunkce
U pacientů léčených nivolumabem v monoterapii byl výskyt nefritidy a renální dysfunkce hlášen u 3,2 % pacientů (55/1728). Většina nežádoucích účinků byla 1. či 2. stupně závažnosti, 1. stupeň závažnosti byl hlášen u 1,9 % (32/1728) a 2. stupeň byl hlášen u 0,8 % (14/1728) pacientů. Nežádoucí účinky 3. a 4. stupně závažnosti byly hlášeny u 0,5 % (8/1728) a <0.1 % (1/1728) pacientů. V těchto studiích nebyla hlášena nefritida či renální dysfunkce 5. stupně závažnosti. Střední čas do nástupu byl 2,3 měsíce (rozpětí: 0,0-18,2). K vyléčení došlo u 33 pacientů (62 %) se střední dobou do vyléčení
11,1 týdnů (rozpětí: 0,1-77,1+).
U pacientů léčených nivolumabem v kombinaci s ipilimumabem byl výskyt nefritidy a renální dysfunkce hlášen u 4,2 % pacientů (19/448). Nežádoucí účinky 2. stupně byly hlášeny u 1,1 % pacientů (5/448), 3. stupně u 0,9 % (4/448) a 4. stupně u 0,7 % pacientů (3/448). Nežádoucí účinky 5. stupně závažnosti v těchto studiích hlášeny nebyly. Střední čas do nástupu byl 2,6 měsíce (rozpětí: 0,5-14,7). K vyléčení došlo u 17 pacientů (89,5 %) se střední dobou do vyléčení 1,9 týdnů (rozpětí: 0,4-42,6+).
Imunitně podmíněná endokrinopatie
U pacientů léčených nivolumabem v monoterapii byl výskyt poruch štítné žlázy, včetně hypotyreózy a hypertyreózy, hlášen u 8,6 % pacientů (149/1728). Většina nežádoucích účinků byla 1. či 2. stupně závažnosti, 1. stupeň závažnosti byl hlášen u 3,6 % (62/1728) a 2. stupeň byl hlášen u 4,9 % (85/1728) pacientů. Nežádoucí účinky 3. stupně závažnosti poruch štítné žlázy byly hlášeny u 0,1 % (2/1728) pacientů. Byla hlášena hypofýzitida (1 stupně 1; 1 stupně 2 a 3 stupně 3), nedostatečnost nadledvin (1 stupně 1; 5 stupně 2 a 4 stupně 3), diabetes mellitus (1 stupně 2) a diabetická ketoacidóza (2 stupně 3). V těchto studiích nebyly hlášeny žádné nežádoucí účinky stupně závažnosti 4 nebo 5. Střední čas do nástupu těchto endokrinopatií byl 2,8 měsíce (rozpětí: 0,4-14,0). K vyléčení došlo u 74 pacientů (45 %) se střední dobou do vyléčení 66,6 týdnů (0,4-96,1+).
U pacientů léčených nivolumabem v kombinaci s ipilimumabem byl výskyt poruch štítné žlázy hlášen u 23,7 % pacientů (106/448). Poruchy štítné žlázy 2. a 3. stupně byly hlášeny u 13,4 % (60/448), resp. 1,6 % pacientů (7/448). Hypolyzitida 2. a 3. stupně byla hlášena u 6,0 % (27/448), resp. 1,8 % (8/448) pacientů. Adrenální insuficience 2. a 3. stupně byla hlášena v obou případech u 1,1 % pacientů (5/448), stupeň 4 byl hlášen u 0,2 % pacientů (1/448). Diabetes mellitus stupně 1 a 2 a diabetická ketoacidóza byly vždy hlášeny u 0,2 % pacientů (1/448). Endokrinopatie 5. stupně závažnosti v těchto studiích hlášeny nebyly. Střední čas do nástupu těchto endokrinopatií byl 1,5 měsíce (rozpětí: 0,0-10,1). K vyléčení došlo u 59 pacientů (45,0 %). Střední doba do vyléčení se pohybovala v rozpětí 0,4 až 74,4+ týdnů.
Imunitně podmíněná vyrážka
U pacientů léčených nivolumabem v monoterapii byl výskyt vyrážky 28,0 % (484/1728). Většina nežádoucích účinků byla 1. stupně závažnosti, hlášeny byly u 21,9 % (378/1728) pacientů. Nežádoucí účinky 2. a 3. stupně závažnosti byly hlášeny u 5,2 % (89/1728), resp. 1,0 % (17/1728) pacientů.
V těchto studiích nebyly hlášeny žádné nežádoucí účinky stupně závažnosti 4 nebo 5. Střední čas do nástupu byl 1,4 měsíce (rozpětí: 0,0-17,2). K vyléčení došlo u 295 pacientů (62 %) se střední dobou do vyléčení 18,1 týdnů (0,1-113,7+).
U pacientů léčených nivolumabem v kombinaci s ipilimumabem byl výskyt vyrážky hlášen u 63,4 % pacientů (284/448). Nežádoucí účinky 2. stupně byly hlášeny u 19,2 % pacientů (86/448) a 3. stupně u 7,4 % pacientů (33/448). Nežádoucí účinky 4. a 5. stupně závažnosti v těchto studiích hlášeny nebyly. Střední čas do nástupu byl 0,5 měsíce (rozpětí: 0,0-9,7). K vyléčení došlo u 192 pacientů (67,6 %) se střední dobou do vyléčení 10,4 týdnů (rozpětí: 0,1-74,0+).
Reakce na infuzi
U pacientů léčených nivolumabem v monoterapii byl výskyt přecitlivělosti/reakce na infuzi 4,1 % (71/1728), včetně nežádoucího účinku stupně 3 u 3 pacientů a stupně 4 u 2 pacientů.
U pacientů léčených nivolumabem v kombinaci s ipilimumabem byl výskyt hypersenzitivity/reakce na infuzi hlášen u 3,8 % pacientů (17/448); všechny byly stupně závažnosti 1 nebo 2. Nežádoucí účinky 2. stupně byly hlášeny u 2,2 % pacientů (10/448). Nežádoucí účinky 3.-5. stupně závažnosti v těchto studiích hlášeny nebyly.
Laboratorní abnormality
U pacientů léčených nivolumabem v monoterapii bylo procento pacientů, u kterých došlo k posunu z výchozích hodnot na 3. nebo 4. stupeň laboratorní abnormality, následující: 4,4 % mělo anemii (všichni stupeň 3), 0,4 % trombocytopenii, 7,7 % lymfopenii, 0,5 % neutropenii, 1,7 % mělo zvýšenou alkalickou fosfatázu, 2,7 % zvýšené AST, 2,4 % zvýšené ALT, 1,0 % zvýšený celkový bilirubin, 0,8 % mělo zvýšený kreatinin, 2,4 % zvýšenou amylázu, 8,0 % zvýšenou lipázu, 5,9 % hypernatremii, 2,1 % hyperkalemii, 1,5 % hypokalemii, 1,3 % hyperkalcemii, 0,8 % hypermagnezemii, 0,5 % hypomagnezemii, 0,6 % hypokalcemii, 0,5 % leukopenii a 0,1 % hypernatremii.
U pacientů léčených nivolumabem v kombinaci s ipilimumabem bylo procento pacientů, u kterých došlo ke zhoršení z výchozích hodnot na 3. nebo 4. stupeň laboratorní abnormality, následující:2,8 % mělo anemii (všichni stupeň 3), 1,2 % trombocytopenii, 0,5 % leukopenii, 6,4 % lymfopenii, 0,7 % neutropenii, 4,1 % mělo zvýšenou alkalickou fosfatázu, 11,9 % zvýšené AST, 14,6 % zvýšené ALT, 0,9 % zvýšený celkový bilirubin, 2,4 % zvýšený kreatinin, 8,5 % zvýšenou amylázu, 18,2 % zvýšenou lipázu, 1,3 % hypokalcemii, 0,3 % mělo hyperkalcemii, hyperkalemii, hypermagnezemii nebo hypernatremii, 4,5 % hypokalemii a 9,2 % hyponatremii.
Imunogenicita
Z 1408 pacientů, kteří byli léčeni 3 mg/kg monoterapií nivolumabem každé dva týdny a u nichž bylo možno hodnotit přítomnost protilátky proti léku, mělo 155 pacientů (11,0 %) pozitivní test na protilátky proti léku vzniklé během léčby, přičemž test na neutralizující protilátky byl pozitivní u devíti pacientů (0,6 %).
Z 394 pacientů léčených nivolumabem v kombinaci s ipilimumabem a hodnotitelných na přítomnost protilátek proti nivolumabu bylo 149 pacientů (37,8 %) pozitivně testováno na přítomnost protilátky proti nivolumabu vzniklé během léčby, přičemž test na neutralizující protilátky byl pozitivní u 18 pacientů (4,6 %).
Přestože v případě, že byly přítomny protilátky proti nivolumabu, clearance nivolumabu vzrostla o 25 %, nebyla přítomnost protilátek spojena se snížením účinnosti či změnou profilu toxicity, jak vyplývá z analýzy farmakokinetiky a odpovědi na expozici, a to u monoterapie i kombinace.
Starší pacienti
Celkově nebyly hlášeny žádné rozdíly v bezpečnosti mezi strašími (> 65 let) a mladšími pacienty (< 65 let). Údaje od NSCLC pacientů ve věku 75 let a starších jsou příliš omezené, aby bylo možno pro tuto populaci vyvodit závěry (viz bod 5.1).
Porucha funkce jater nebo ledvin
Ve studii neskvamózního NSCLC (CA209057) byl bezpečnostní profil u pacientů s poškozením funkce jater a ledvin srovnatelný s celkovou populací. Tyto výsledky je třeba interpretovat s opatrností vzhledem k malé velikosti vzorku u jednotlivých podskupin.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V klinických studiích nebyly hlášeny žádné případy předávkování. V případě předávkování musí být pacienti pečlivě monitorováni s ohledem na známky nebo příznaky nežádoucích účinků a musí se okamžitě zahájit vhodná symptomatická léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antineoplastické látky, monoklonální protilátky. ATC kód: L01XC17. Mechanismus účinku
Nivolumab je humánní monoklonální protilátka (HuMAb) isotypu G4 (IgG4), která se váže na receptor označovaný jako PD-1 (receptor programované smrti) a blokuje jeho interakci s PD-L1 a PD-L2. Receptor PD-1 je negativním regulátorem aktivity T-buněk a bylo dokázáno, že se účastní kontroly imunitní odpovědi T-buněk. Vazba receptoru PD-1 na ligandy PD-L1 a PD-L2, které jsou exprimovány na antigen prezentujících buňkách nebo mohou být na nádorových či jiných buňkách v mikroprostředí nádoru, má za následek inhibici proliferace T-buněk a blokádu sekrece cytokinů. Nivolumab zesiluje odpověď T-buněk, včetně protinádorové odpovědi, blokádou vazby receptoru PD-1 na ligandy PD-L1 a PD-L2. Na syngenních myších modelech vedla blokační aktivita PD-1 k omezení růstu nádoru.
Kombinovaná terapie nivolumabem (anti-PD-1) a ipilimumabem (anti-CTLA-4) měla větší vliv na inhibiční funkci T buněk ve srovnání s efektem samostatného podání jednotlivých přípravků a prokázala tak vyšší protinádorovou účinnost v léčbě metastatického melanomu. Na syngenních myších modelech nádorů vedla dvojí blokační aktivita PD-1 a CTLA-4 k synergickému protinádorovému působení.
Klinická účinnost a bezpečnost
Melanom
Randomizovaná studie _ fáze 3 vs. dakarbazin (CA209066)
Bezpečnost a účinnost nivolumabu v dávce 3 mg/kg v monoterapii v léčbě pokročilého (neresekovatelného nebo metastatického) melanomu byly hodnoceny v randomizované, dvojitě zaslepené studii fáze 3 (CA209066). Studie zahrnovala dospělé pacienty (18leté a starší), s potvrzeným, dosud neléčeným, BRAF wild-type melanomem ve stadiu III nebo IV a se skóre fyzické aktivity podle ECOG 0 nebo 1. Ze studie byli vyloučeni pacienti s aktivním autoimunitním onemocněním, očním melanomem, nebo aktivními mozkovými či leptomeningeálními metastázami.
Celkem 418 pacientů bylo randomizováno, aby užívali buď nivolumab (n=210) podávaný intravenózně po dobu 60 minut v dávce 3 mg/kg každé dva týdny, nebo dakarbazin (n=208) v dávce 1000 mg/m2 každé 3 týdny. Randomizace byla stratifikována podle statusu nádorového PD-L1 a metastatického stadia (M0/M1a/M1b versus M1c). Léčba pokračovala tak dlouho, dokud byl pozorován klinický přínos, nebo do doby, než léčba přestala být tolerována. Léčba po progresi onemocnění byla povolena u pacientů, u kterých stále měla klinický přínos a nevyskytovaly se významné nežádoucí účinky studovaného léku (podle posouzení zkoušejícím). Vyhodnocení účinku léčby na tumor s pomocí Kritérií hodnocení odpovědi u solidních nádorů (RECIST 1.1) se provádělo po 9 týdnech od randomizace a opakovalo se každých 6 týdnů první rok a poté každých 12 týdnů. Primárním měřítkem účinnosti bylo celkové přežití (overall survival - OS). Klíčovým sekundárním měřítkem účinnosti byly zkoušejícím hodnocené PFS a objektivní míra odpovědi (objective response rate - ORR).
Charakteristiky při vstupu do studie byly mezi skupinami vyvážené. Medián věku byl 65 let (rozpětí: 18-87), 59 % byli muži a 99,5 % byli běloši. Většina pacientů měla ECOG skóre fyzické aktivity 0 (64 %) nebo 1 (34 %). 61 % pacientů mělo při vstupu do studie stadium M1c nemoci. Sedmdesát čtyři procent pacientů mělo kožní melanom a 11 % mělo slizniční melanom; 35 % pacientů mělo PD-L1 pozitivní melanom (exprese na >5 % buněčných membrán nádorových buněk). Šestnáct procent pacientů užívalo před tím adjuvantní léčbu; nejčastější adjuvantní léčbou bylo podávání interferonu (9 %). Čtyři procenta pacientů měla v anamnéze mozkové metastázy a 37 % pacientů mělo při vstupu do studie LDH větší než ULN.
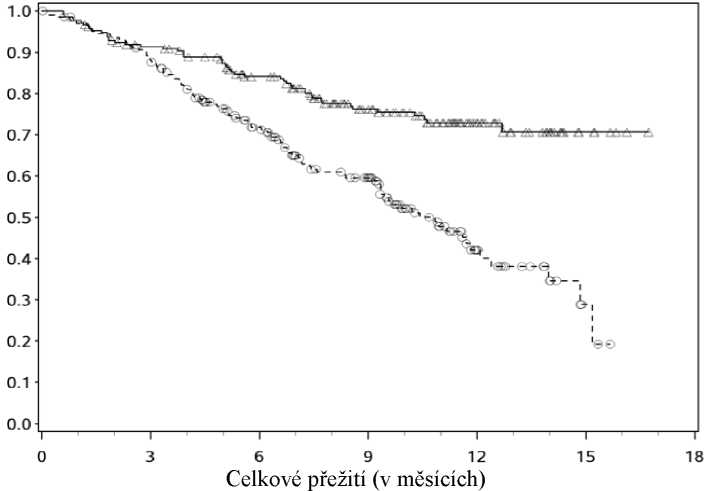
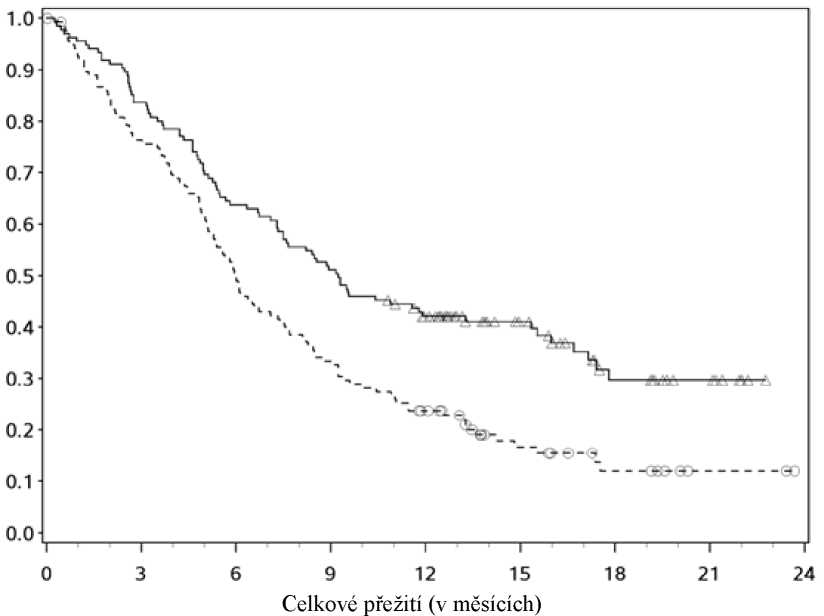
Kaplan-Meierovy křivky pro OS jsou znázorněny na Obrázku 1.
Počet subjektů s rizikem
|
Nivolumab 210 |
185 |
150 |
105 |
45 |
8 |
0 |
|
Dakarbazin 208 |
177 |
123 |
82 |
22 |
3 |
0 |
-A-Nivolumab (případů: 50/210), medián a 95% CI: NA
---O---Dakarbazin (případů: 96/208), medián a 95% CI: 10,84 (9,33; 12,09)
Pozorovaný přínos v celkovém přežití byl shodně prokázán napříč podskupinami pacientů, včetně podskupin podle výchozího ECOG skóre fyzické aktivity, M stadia, anamnézy metastáz mozku, či podle úrovně LDH na počátku léčby. Přínos v celkovém přežití byl pozorován bez ohledu na to, zda měli pacienti tumory typu PD-L1 negativní nebo PD-L1 pozitivní (nádorová membránová exprese tumoru nad nebo pod hraničními hodnotami 5 % nebo 10 %).
Dostupné údaje ukazují, že nástup účinku nivolumabu má takové zpoždění, že výhodnost léčby nivolumabem nad chemoterapií se může dostavit za 2 až 3 měsíce.
Výsledky účinnosti jsou zachyceny v Tabulce 3.
|
nivolumab (n = 210) |
dakarbazin (n = 208) | ||
|
Celkové přežití | |||
|
Případy |
50 (23,8) |
96 (46,2) | |
|
Poměr rizik 99,79% CI 95% CI p-hodnota Medián (95% CI) |
Nedosaženo |
0,42 (0,25; 0,73) (0,30; 0,60) < 0,0001 |
10,8 (9,33; 12,09) |
|
Výskyt (95% CI) | |||
|
v 6 měsících |
84,1 (78,3; 88,5) |
71,8 (64,9; 77,6) | |
|
ve 12 měsících |
72,9 (65,5; 78,9) |
42,1 (33,0; 50,9) | |
|
Přežití bez progrese | |||
|
Případy |
108 (51,4) |
163 (78,4) | |
|
Poměr rizik 95% CI p-hodnota Medián (95% CI) |
5,1 (3,48; 10,81) |
0,43 (0,34; 0,56) < 0,0001 |
2,2 (2,10; 2,40) |
|
Výskyt (95% CI) | |||
|
v 6 měsících |
48,0 (40,8; 54,9) |
18,5 (13,1; 24,6) | |
|
ve 12 měsících |
41,8 (34,0; 49,3) |
NA | |
|
Objektivní odpověď |
84 (40,0%) |
29 (13,9%) | |
|
(95% CI) |
(33,3; 47,0) |
(9,5; 19,4) | |
|
Míra relativního rizika (odds ratio) |
4,06 (2,52; 6,54) | ||
|
(95% CI) | |||
|
p-hodnota Kompletní odpověď (CR) |
16 (7,6%) |
< 0,0001 |
2 (1,0%) |
|
Částečná odpověď (PR) |
68 (32,4%) |
27 (13,0%) | |
|
Stabilní onemocnění (SD) |
35 (16,7%) |
46 (22,1%) | |
|
Medián doby trvání odpovědi | |||
|
Měsíce (rozpětí) |
Nedosažen (0+ - 12,5+) |
6,0 (1,1 - 10,0+) | |
|
Medián doby do nástupu odpovědi | |||
|
Měsíce (rozpětí) |
2,1 (1,2 - 7,6) |
2,1 (1,8 - 3,6) | |
“+” označuje cenzurované sledování.
Randomizovaná studie fáze 3 vs. chemoterapie (CA209037)
Bezpečnost a účinnost nivolumabu 3 mg/kg v monoterapii pro léčbu pokročilého (neresekovatelného nebo metastatického) melanomu byly hodnoceny v randomizované, otevřené studii fáze 3 (CA209037). Studie zahrnovala dospělé pacienty, kteří progredovali při nebo po léčbě ipilimumabem, a pokud byli pozitivní na mutaci BRAF V600, pak progredovali při léčbě inhibitorem BRAF kinázy nebo po ní. Ze studie byli vyloučeni pacienti s aktivním autoimunitním onemocněním, očním melanomem nebo se známými předchozími nežádoucími účinky vysokého stupně spojených s ipilimumabem (stupeň 4 podle CTCAE v4.0) v anamnéze, kromě zvládnuté nauzey, únavy, reakcí na infuzi nebo endokrinopatií.
Celkem 405 pacientů bylo randomizováno, aby užívali buď nivolumab (n=272) podávaný intravenózně po 60 minut v dávce 3 mg/kg každé dva týdny, nebo chemoterapii (n=133), která byla podávána dle volby zkoušejícího, a to buď dakarbazin (1 000 mg/m2 každé 3 týdny), nebo karboplatina (AUC 6 každé 3 týdny) a paklitaxel (175 mg/m2 každé 3 týdny). Randomizace byla stratifikována podle BRAF a nádorového PD-L1 stavu a míry nejlepší odpovědi na předchozí léčbu ipilimumabem.
Společným primárním měřítkem účinnosti byla potvrzená ORR u prvních 120 pacientů léčených nivolumabem, měřená nezávislou radiologickou kontrolní komisí (IRRC) s pomocí RECIST, verze 1.1, a srovnání OS u nivolumabu oproti chemoterapii. Další hodnocení účinnosti zahrnovalo nástup odpovědi a její trvání.
Medián věku byl 60 let (rozpětí: 23-88). 64 % pacientů byli muži a 98 % byli běloši. U 61 % pacientů bylo skóre fyzické aktivity ECOG 0 a u 39 % pacientů bylo 1. Většina (75 %) pacientů měla při vstupu do studie stadium M1c choroby. 37 % pacientů mělo kožní melanom a 10 % mělo mukózní melanom. Počet předchozích poskytnutých systémových terapií byl 1 u 27 % pacientů, 2 u 51 % pacientů, a > 2 u 21 % pacientů. 22 % pacientů mělo nádory, které byly dle testování pozitivní na BRAF mutaci a 50 % pacientů mělo nádory, které byly posouzeny jako PD-L1 pozitivní. 64 % pacientů nemělo žádný prospěch z předchozí léčby ipilimumabem (CR/PR nebo SD). Výchozí charakteristiky byly mezi skupinami vyrovnané, kromě podílu pacientů, kteří měli v anamnéze metastázy v mozku (19 % ve skupině nivolumabu a 13 % ve skupině chemoterapie), a pacientů s LDH na počátku větším než ULN (51 %, resp. 35 %).
V době této konečné analýzy ORR byly analyzovány výsledky od 120 pacientů léčených nivolumabem a 47 pacientů léčených chemoterapií, kteří měli za sebou minimálně 6 měsíců následného sledování. Výsledky účinnosti léčby jsou zachyceny v Tabulce 4.
Tabulka 4: Nejlepší celková míra odpovědi, nástup a doba trvání odpovědi (CA209037)_
nivolumab chemoterapie
(n = 120)_(n = 47)
|
Potvrzená objektivní odpověď (IRRC) |
38 (31,7%) |
5 |
(10,6%) |
|
(95% CI) |
(23,5, 40,8) |
(3,5; 23,1) | |
|
Kompletní odpověď (CR) |
4 (3,3%) |
0 | |
|
Částečná odpověď (PR) |
34 (28,3%) |
5 |
(10,6%) |
|
Stabilní onemocnění (SD) |
28 (23,3%) |
16 |
(34,0%) |
|
Medián doby trvání odpovědi | |||
|
Měsíce (rozpětí) |
Nedosažen |
3,6 |
(Údaj není dostupný) |
|
Medián doby do nástupu odpovědi | |||
|
Měsíce (rozpětí) |
2,1 (1,6-7,4) |
3,5 |
(2,1-6,1) |
Objektivní odpovědi na nivolumab byly pozorovány (podle definice koprimárního cílového parametru) u pacientů s pozitivitou na BRAF mutaci nebo bez ní. U pacientů, kteří dostávali nivolumab, byla ORR v podskupině (n=26) s BRAF pozitivní mutací 23 % (95% CI: 9,0; 43,6) a 34 % (95% CI: 24,6; 44,5) u pacientů, jejichž tumory byly BRAF negativní (n=94). Objektivní odpovědi na nivolumab byly pozorovány nezávisle na tom, zda měli pacienti tumory typu PD-L1 negativní nebo PD-L1 pozitivní (nádorová membránová exprese tumoru nad nebo pod hraničními hodnotami 5 % nebo 10 %). Nicméně role tohoto biomarkeru (exprese PD-L1 na nádorových buňkách) nebyla plně objasněna.
Údaje OS nebyly v době analýzy PFS konečné. V předběžné OS analýze, ve které nebyly zohledněny možné další účinky následné léčby, nebyly shledány žádné statisticky významné rozdíly mezi nivolumabem a chemoterapií. V rameni s chemoterapií užívalo následně léčbu anti-PD1 42 pacientů (31,6 %).
Dostupné údaje ukazují, že nástup účinku nivolumabu má takové zpoždění, že výhodnost léčby nivolumabem nad chemoterapií se může dostavit za 2 až 3 měsíce.
Zkoušejícím hodnocená, potvrzená ORR u všech léčených pacientů byla 25,7 % [95% CI: 20,6; 31,4] ve skupině s nivolumabem vs. 10,8 % [95% CI: 5,5; 18,5]) ve skupině s chemoterapií, s rozdílem ORR 15,0 % (95% CI: 6,0; 22,2). Zkoušejícím hodnocená, potvrzená ORR u pacientů s pozitivní BRAF mutací byla 19,3 % [95% CI: 10,0; 31,9] vs. 13,6 % [95% CI: 2,9; 34,9]), a u pacientů s negativní BRAF mutací byla 27,5 % [95% CI: 21,6; 34,0] vs. 10,0 % [95% CI: 4,4; 18,8]).
PFS numericky favorizoval skupinu s nivolumabem oproti skupině s chemoterapií u všech randomizovaných pacientů, u pacientů s BRAF pozitivní mutací i BRAF negativních pacientů (HR 0,74 [95% CI: 0,57; 0,97], resp. 0,98 [95% CI: 0,56; 1,70] a 0,63 [95% CI: 0,47; 0,85]).
Otevřená studie fáze 1 s eskalací dávky (MDX1106-03)
Bezpečnost a snášenlivost nivolumabu byly zkoumány v otevřené studii fáze 1 s eskalací dávky u různých typů nádorů, včetně maligního melanomu. Z 306 pacientů, kteří podstoupili předchozí léčbu, zahrnutých do studie mělo 107 pacientů melanom a dostávali nivolumab v dávce 0,1 mg/kg,
0,3 mg/kg, 1 mg/kg, 3 mg/kg nebo 10 mg/kg. V této pacientské populaci byla objektivní odpověď hlášena u 33 pacientů (31 %) se středním trváním odpovědi 22,9 měsíců (95% CI: 17,0, NR). Střední doba přežití bez progrese byla 3,7 měsíce (95% CI: 1,9; 9,3). Střední doba přežití byla 17,3 měsíců (95% CI: 12,5; 36,7) a odhadovaná míra celkového přežití byla 63 % (95% CI: 53; 71) v 1. roce, 48 % (95% CI: 38; 57) ve dvou letech a 41 % (95% CI: 31; 51) ve třech letech.
Randomizovaná studie fáze 3 hodnotící nivolumab v kombinaci s ipilimumabem nebo nivolumab v monoterapii ve srovnání s ipilimumabem v monoterapii (CA209067)
Bezpečnost a účinnost nivolumabu v kombinaci s ipilimumabem nebo nivolumabu v monoterapii ve srovnání s ipilimumabem v monoterapii k léčbě pokročilého (neresekovatelného nebo metastatického) melanomu byly hodnoceny v randomizované, dvojitě zaslepené studii fáze 3 (CA209067). Rozdíly mezi oběma skupinami obsahujícími nivolumab byly hodnoceny deskriptivně. Studie zahrnovala dospělé pacienty s potvrzeným neresekovatelným melanomem ve III. nebo IV. stádiu. Pacienti měli ECOG skóre fyzické aktivity 0 nebo 1. Byli zahrnuti pacienti, kteří neměli předchozí systémovou protinádorovou terapii neresekovatelného nebo metastatického melanomu. Předchozí adjuvantní nebo neoadjuvantní terapie byla povolena, pokud byla ukončena alespoň 6 měsíců před randomizací. Pacienti s aktivní autoimunitní chorobou, okulárním/uveálním melanomem nebo aktivními mozkovými nebo leptomeningeálními metastázami byli ze studie vyloučeni.
Celkem 945 pacientů bylo randomizováno k užívání nivolumabu v kombinaci s ipilimumabem (n = 314), nivolumabu v monoterapii (n = 316) nebo ipilimumabu v monoterapii (n = 315). Pacienti v rameni s kombinací užívali 1 mg/kg nivolumabu v 60minutové a 3 mg/kg ipilimumabu v 90minutové intravenózní infuzi každé 3 týdny u prvních 4 dávek a dále 3 mg/kg nivolumabu v monoterapii každé 2 týdny. Pacienti ve srovnávacím rameni dostávali 3 mg/kg ipilimumabu a placebo nivolumabu intravenózně každé 3 týdny u prvních 4 dávek a dále placebo každé 2 týdny. Randomizace byla stratifikována podle exprese PD-L1 (>5% vs. <5% exprese na nádorových buňkách), BRAF statusu a M stádia podle American Joint Committee on Cancer (AJCC) systému. Léčba pokračovala, dokud byl pozorován klinický přínos a dokud byla léčba tolerována. Vyhodnocení účinků léčby na tumor bylo provedeno 12 týdnů po randomizaci a dále každých 6 týdnů během prvního roku a potom každých 12 týdnů. Společným primárním měřítkem účinnosti bylo přežití bez progrese a OS. Byla rovněž hodnocena ORR a doba trvání odpovědi.
Základní charakteristiky na začátku studie byly ve všech třech léčebných skupinách vyvážené. Medián věku byl 61 let (rozpětí: 18 až 90 let), 65 % pacientů byli muži a 97 % byli běloši. ECOG skóre fyzické aktivity bylo 0 (73 %) nebo 1 (27 %). Většina pacientů měla chorobu stádia IV podle AJCC (93 %); 58 % mělo onemocnění stádia M1c při vstupu do studie. Dvacet dva procent pacientů mělo předchozí adjuvantní terapii. Třicet dva procent pacientů mělo melanom pozitivní na BRAF mutaci; 26,5 % pacientů mělo expresi PD-L1 na nádorových buňkách > 5 %. Čtyři procenta pacientů měla mozkové metastázy v anamnéze a 36 % mělo hladinu LDH při vstupu do studie vyšší než ULN. U pacientů s měřitelnou úrovní nádorové PD-L1 exprese byla jejich distribuce mezi léčebnými skupinami vyvážená. Úroveň exprese PD-L1 na nádorových buňkách byla stanovena pomocí PD-L1 IHC 28-8 pharmDx testu.
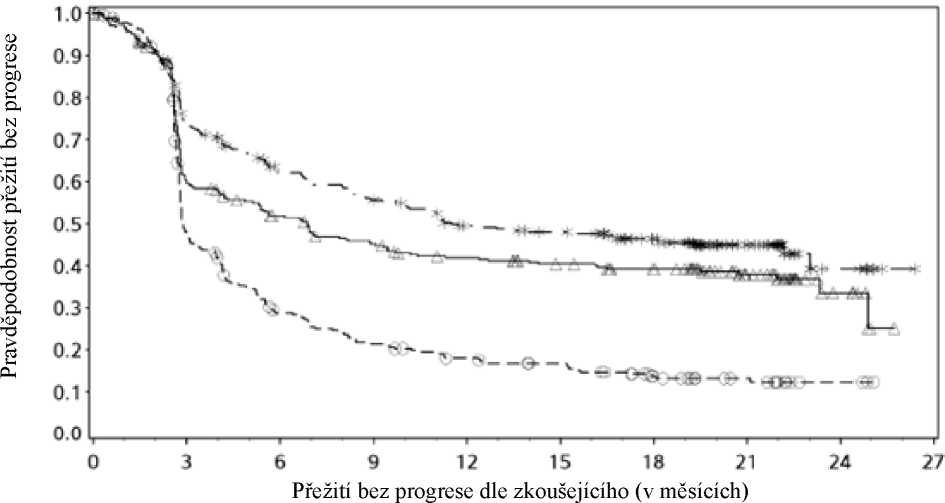
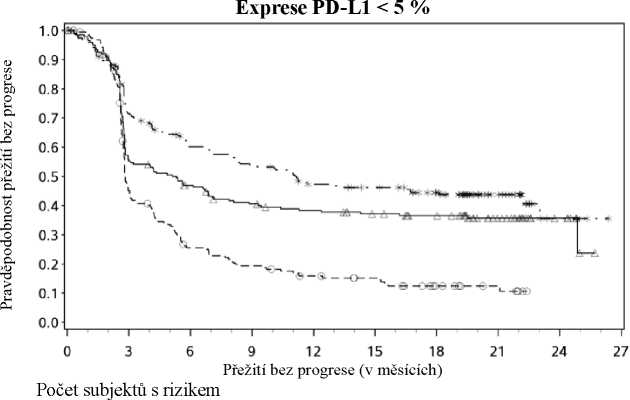
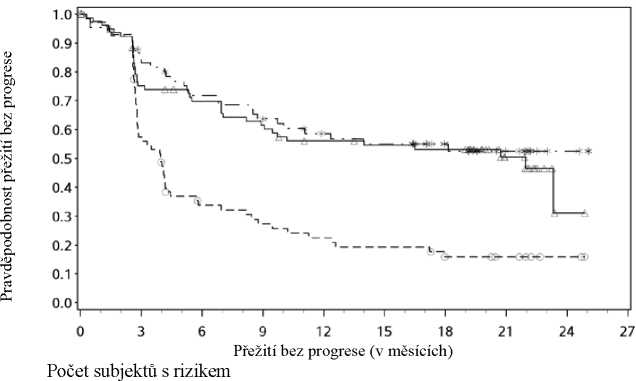
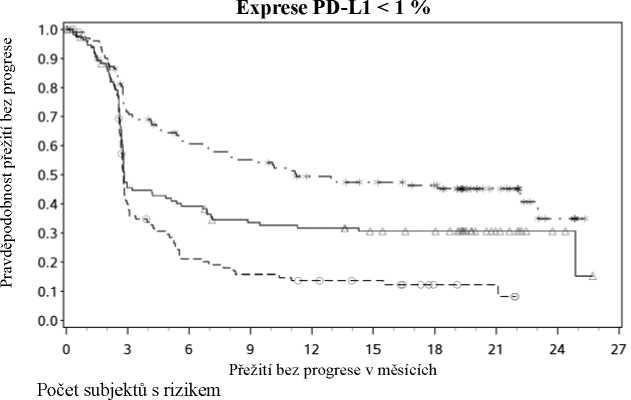
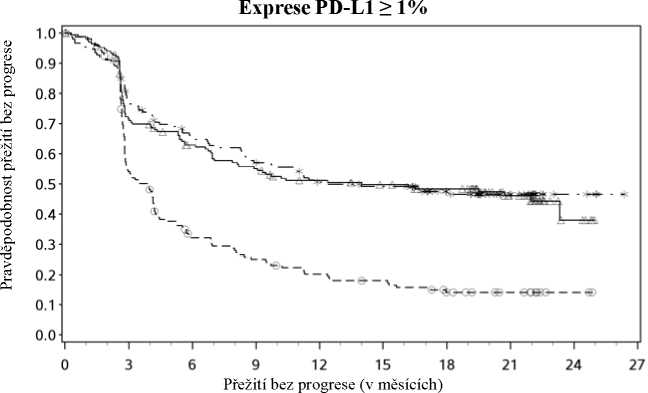
Následné sledování probíhalo minimálně 18 měsíců. Celkové přežití nebylo v okamžiku této analýzy ještě možno hodnotit. Výsledky PFS jsou uvedeny na obrázku 2 (veškerá randomizovaná populace), obrázku 3 (5% úroveň nádorové exprese PD-L1) a obrázku 4 (1% úroveň nádorové exprese PD-L1). Odpovědi jsou shrnuty v tabulce 5.
Počet subjektů s rizikem Nivolumab + Ipilimumab
|
314 219 Nivolumab |
174 |
156 |
133 |
126 |
103 |
48 |
8 |
0 |
|
316 177 Ipilimumab |
148 |
127 |
114 |
104 |
94 |
46 |
8 |
0 |
|
315 137 |
78 |
58 |
46 |
40 |
25 |
15 |
3 |
0 |
---*----Nivolumab+ipilimumab (případy: 161/314), medián a 95% CI: 11,50 (8,90; 22,18)
Výskyt PFS ve 12 měsících a 95% CI: 49 % (44; 55)
-A- Nivolumab (případy: 183/316), medián a 95% CI: 6,87 (4,34; 9,46)
Výskyt PFS ve 12 měsících a 95% CI: 42 % (36; 47)
—O— Ipilimumab (případy: 245/315), medián a 95% CI: 2,89 (2,79; 3,42)
Výskyt PFS ve 12 měsících a 95% CI: 18 % (14; 23)
Nivolumab+ipilimumab vs ipilimumab (primární analýza) - HR (99,5% CI): 0,42 (0,32, 0,56); p-hodnota: <0.0001
Nivolumab vs ipilimumab (primární analýza) - HR (99,5% CI): 0,55 (0,42; 0,73); p- hodnota: <0.0001 Nivolumab+ipilimumab vs nivolumab (deskriptivní analýza) - HR (95% CI): 0,76 (0,62; 0,95)
Nivolumab + Ipilimumab
|
210 142 Nivolumab |
113 |
101 |
86 |
81 |
69 |
31 |
5 |
0 |
|
208 108 Ipilimumab |
89 |
75 |
69 |
62 |
55 |
29 |
7 |
0 |
|
202 82 |
45 |
34 |
26 |
22 |
12 |
7 |
0 |
0 |
---*---- Nivolumab+Ipilimumab (případy: 111/210 medián a 95% CI: 11,10 (7,98; 22,18)
—A- Nivolumab (případy: 125/208), medián a 95% CI: 5,32 (2,83; 7,06)
- - -O- - - Ipilimumab (případy: 159/202), medián a 95% CI: 2,.83 (2,7; 3,09)
Nivolumab+Ipilimumab vs. Ipilimumab - poměr rizik: 0,42 (0,33; 0,54) Nivolumab vs. Ipilimumab - poměr rizik: 0,57 (0,45; 0,72) Nivolumab+Ipilimumab vs. Nivolumab - poměr rizik: 0,74 (0,58; 0,96)
Exprese PD-L1 > 5%
Nivolumab + Ipilimumab
|
68 53 Nivolumab |
44 |
39 |
33 |
31 |
22 |
13 |
3 |
0 |
|
80 57 Ipilimumab |
51 |
45 |
39 |
37 |
36 |
16 |
1 |
0 |
|
75 40 |
21 |
17 |
14 |
12 |
8 |
6 |
2 |
0 |
---*---- Nivolumab+Ipilimumab (případy: 29/68), medián a 95% CI: N.A. (9,72; N.A.)
—A- Nivolumab (případy: 38/80), medián a 95% CI: 21,95 (8,90; N.A.)
- - -O- - - Ipilimumab (případy: 57/75), medián a 95% CI: 3,94 (2,79; 4,21)
Nivolumab+Ipilimumab vs. Ipilimumab - poměr rizik: 0,35 (0,22; 0,55) Nivolumab vs. Ipilimumab - poměr rizik: 0,41 (0,27; 0,62) Nivolumab+Ipilimumab vs. Nivolumab - poměr rizik: 0,87 (0,54; 1,41)
Nivolumab + Ipilimumab
|
123 82 Nivolumab |
65 |
59 |
50 |
46 |
41 |
18 |
4 |
0 |
|
117 50 Ipilimumab |
43 |
35 |
33 |
29 |
27 |
11 |
3 |
0 |
|
113 39 |
20 |
15 |
12 |
10 |
4 |
3 |
0 |
0 |
---*---- Nivolumab+Ipilimumab (případy: 63/123), medián a 95% CI: 11,24 (6,93; 23,03)
—A- Nivolumab (případy: 77/117), medián a 95% CI: 2,83 (2,76; 5,13)
---O--- Ipilimumab (případy: 87/113), medián a 95% CI: 2,79 (2,66; 2,96)
Nivolumab+Ipilimumab vs. Ipilimumab - poměr rizik: 0,39 (0,28; 0,54) Nivolumab vs. Ipilimumab - poměr rizik: 0,65 (0,48; 0,88) Nivolumab+Ipilimumab vs. Nivolumab - poměr rizik: 0,60 (0,43; 0,84)
Počet subjektů s rizikem Nivolumab + Ipilimumab
|
155 113 Nivolumab |
92 |
81 |
69 |
66 |
50 |
26 |
4 |
0 |
|
171 115 Ipilimumab |
97 |
85 |
75 |
70 |
64 |
34 |
5 |
0 |
|
164 83 |
46 |
36 |
28 |
24 |
16 |
10 |
2 |
0 |
---*---- Nivolumab+Ipilimumab (případy: 77/155), medián a 95% CI: 12,35 (8,74; N.A.)
—A- Nivolumab (případy: 86/171), medián a 95% CI: 14,00 (7,03; N.A.)
- - -O- - - Ipilimumab (případy: 129/164), medián a 95% CI: 3,91 (2,83; 4,17)
Nivolumab+Ipilimumab vs. Ipilimumab - poměr rizik: 0,42 (0,31; 0,55) Nivolumab vs. Ipilimumab - poměr rizik: 0,44 (0,34; 0,58) Nivolumab+Ipilimumab vs. Nivolumab - poměr rizik: 0,94 (0,69; 1,28)
|
nivolumab + ipilimumab (n=314) |
nivolumab (n=316) |
ipilimumab (n=315) | |
|
Objektivní odpověď |
181 (58 %) |
138 (44 %) |
60 (19 %) |
|
(95% CI) |
(52,0; 63,2) |
(38,1; 49,3) |
(14,9; 23,8) |
|
Míra relativního rizika (odds ratio) (vs. ipilimumab) |
6,09 |
3,40 | |
|
(99,5% CI) |
(3,59; 10,33) |
(2,02; 5,72) | |
|
p-hodnota |
p<0,0001 |
p<0,0001 | |
|
Kompletní odpověď (CR) |
38 (12 %) |
31 (10%) |
7 (2 %) |
|
Částečná odpověď (PR) |
143 (46 %) |
107 (34 %) |
53 (17 %) |
|
Stabilní onemocnění (SD) |
41(13 %) |
33 (10 %) |
69 (22 %) |
|
Medián trvání odpovědi | |||
|
Měsíce (rozmezí) |
nedosaženo (0+ - 24+) |
22,3 (0+ - 23+) |
14,4 (1,4 - 22,3+) |
|
ORR (95% CI) podle úrovně nádorové exprese PD-L1 | |||
|
<5% |
55 % (47,8; 61,6) n=210 |
41 % (34,6; 48,4) n=208 |
18 % (12,8; 23,8) n=202 |
|
>5% |
72 % (59,9; 82,3) n=68 |
58 % (45,9; 68,5) n=80 |
21 % (12,7; 32,3) n=75 |
|
<1% |
52 % (42,8; 61,1) n=123 |
33 % (24,9; 42,6) n=117 |
19 % (11,9; 27,0) n=113 |
|
>1% |
65 % (56,4; 72,0) n=155 |
54 % (46,6; 62,0) n=171 |
19 % (13,2; 25,7) n=164 |
V obou ramenech obsahujících nivolumab byl prokázán významný přínos v PFS a vyšší ORR ve srovnání se samotným ipilimumabem a zjištěné výsledky PFS i ORR ve 12 měsících následného sledování byly konzistentně potvrzeny u všech podskupin pacientů včetně těch členěných podle ECOG skóre, BRAF statusu, M stádia, věku, mozkových metastáz v anamnéze a hladiny LDH na počátku léčby.
U 128 pacientů, kteří přerušili užívání nivolumabu v kombinaci s ipilimumabem kvůli nežádoucím účinkům dosahoval medián PFS 16,7 měsíců (95% CI: 10,2; NA) a ORR byla 69 % (88/128) přičemž 15 % (19/128) pacientů dosáhlo kompletní odpovědi.
U obou ramen obsahujících nivolumab byl prokázán vyšší výskyt objektivní odpovědi než u ipilimumabu bez ohledu na úroveň exprese PD-L1. ORR byl vyšší u kombinace nivolumabu s ipilimumabem než u monoterapie nivolumabem napříč úrovněmi exprese PD-L1 (tabulka 5). Medián trvání odpovědi u pacientů s úrovní exprese PD-L1 > 5% nebyl v rameni s kombinací dosažen (rozmezí: 0+-22,3+); v rameni s monoterapií nivolumabem činil 20,8 měsíců (rozmezí 2,8-20,8) a v rameni s ipilimumabem nebyl dosažen (rozmezí 1,4-19,9+). U nádorové exprese PD-L1 expression <5 % nebyl medián trvání odpovědi dosažen (rozmezí: 0+-24+) v rameni s kombinací a činil 22,3 měsíců (rozmezí: 0+-23+) v rameni s monoterapií nivolumabem a 18,2 měsíců (rozmezí: 1,4-19,8+) v rameni s monoterapií ipilimumabem.
S ohledem na relevantní kritéria nádorové odpovědi a PFS nelze spolehlivě stanovit žádnou jasnou hranici pro expresi PD-L1. Výsledky následné komplexní analýzy ukazují, že ke klinickému výsledku mohou přispívat další charakteristiky pacienta a nádoru (např. ECOG skóre fyzické aktivity,
M stádium, AJCC stádium, pohlaví, oblast a LDH na počátku studie).
Účinnost dle BRAF statusu: pacienti s pozitivní BRAF[V600] mutací i BRAF wild-type pacienti randomizovaní k nivolumabu v kombinaci s ipilimumabem dosáhli mediánu PFS 15,5 měsíců (95% CI: 8,0; NA) resp. 11,3 měsíců (95% CI: 8,3; 22,2) a ORR 66,7 % (95% CI: 56,6; 75,7; n=102)
resp. 53,3 % (95% CI: 46,3; 60,2; n=212), zatímco pacienti randomizovaní k nivolumabu v monoterapii měli medián PFS 5,6 měsíců (95% CI: 2,8; 9,3) resp. 7,1 měsíců (95% CI:4,9; 14,3) a ORR 36,7 % (95% CI: 27,2; 47,1; n=98) resp. 46,8 % (95% CI: 40,0; 53.6; n=218).
Randomizovaná studie fáze 2 s nivolumabem v kombinaci s ipilimumabem a ipilimumabem (CA209069)
Studie CA209069 byla randomizovaná dvojitě zaslepená studie fáze 2 hodnotící bezpečnost a účinnost
nivolumabu v kombinaci s ipilimumabem ve srovnání se samotným ipilimumabem u 142 pacientů
s pokročilým (neresekovatelným nebo metastatickým) melanomem, se vstupními kritérii podobnými
jako ve studii CA209067 a primární analýzou pacientů s BRAF wild-type melanomem
(77 % pacientů). Zkoušejícím hodnocená ORR byla 61 % (95% CI: 48,9; 72,4) v rameni s kombinací
(n=72) vs 11% (95% CI: 3,0; 25,4) v rameni s ipilimumabem (n=37). Odhadovaná míra OS po
12 a 18 měsících byla 79 % (95% CI: 67; 87), resp. 73 % (95% CI: 61; 82) u kombinace a 62 %
(95% CI: 44; 75), resp. 56 % (95% CI: 39; 70) u ipilimumabu.
Nemalobuněčný karcinom plic
Skvamózní NSCLC
Randomizovaná studie fáze 3 vs. docetaxel (CA209017)
Bezpečnost a účinnost nivolumabu v dávce 3 mg/kg v monoterapii v léčbě pokročilého nebo metastatického skvamózního NSCLC byly hodnoceny v randomizované, otevřené studii fáze 3 (CA209017). Studie zahrnovala dospělé (18leté a starší), kteří měli progresi onemocnění během nebo po jedné předchozí chemoterapii platinovým dubletem a s ECOG skóre fyzické aktivity 0 nebo 1. Pacienti byli zařazeni bez ohledu na jejich stav nádorového PD-L1. Ze studie byli vyřazeni pacienti s aktivním autoimunitním onemocněním, symptomatickou intersticiální plicní chorobou nebo neléčenými metastázami mozku. Pacienti s léčenými metastázami mozku byli vhodní, pokud se neurologicky vrátili na výchozí hodnotu nejméně 2 týdny před zařazením, a byli buď bez léčby kortikosteroidy, nebo byli na stabilní nebo snižující se dávce odpovídající <10 mg prednisonu denně.
Celkem 272 pacientů bylo randomizováno buď k nivolumabu podávanému intravenózně po dobu 60 minut v dávce 3 mg/kg každé dva týdny (n = 135) nebo k docetaxelu v dávce 75 mg/m2 každé 3 týdny (n = 137). Léčba pokračovala tak dlouho, dokud byl pozorován klinický přínos, nebo do doby, než léčba přestala být tolerována. Vyhodnocení účinku léčby na tumor pomocí RECIST 1.1 se provádělo po 9 týdnech od randomizace a poté pokračovalo každých 6 týdnů. Primárním měřítkem účinnosti bylo OS. Klíčovým sekundárním měřítkem účinnosti byly zkoušejícím hodnocený ORR a PFS). Navíc bylo hodnoceno zlepšení příznaků pomocí škály (Lung Cancer Symptom Score - LCSS) průměrného indexu symptomové zátěže a celkový zdravotní stav pomocí skóre EQ-5D a vizuální analogovou škálou (EQ-VAS).
Charakteristiky při vstupu do studie byly mezi skupinami vyvážené. Medián věku byl 63 let (rozpětí: 39-85), 44 % >65 let a 11 % >75 let. Většina pacientů byli běloši (93 %) a muži (76 %). Třicet jedna procent mělo progresi onemocnění hlášenou jako nejlepší odpověď na jejich poslední předchozí režim a 45 % pacientů dostalo nivolumab do 3 měsíců od ukončení svého posledního předchozího režimu. Počáteční skóre fyzické aktivity podle škály ECOG bylo 0 (24 %) nebo 1 (76 %).
Kaplan-Meierovy křivky pro OS jsou znázorněny na obrázku 5.
Obrázek 5: Kaplan Meierovy křivky OS (CA209017)
Pravděpodobnost přežití
Počet subjektů s rizikem Nivolumab 3 mg/kg
|
135 |
113 |
86 |
69 |
52 |
31 |
15 |
7 |
0 |
|
Docetaxel 137 |
103 |
68 |
45 |
30 |
14 |
7 |
2 |
0 |
-A-Nivolumab 3 mg/kg (případů: 86/135), medián a 95% CI : 9,23 (7,33, 13,27)
---O— Docetaxel (případů: 113/137), medián a 95% CI : 6,01 (5,13, 7,33)
Pozorovaný přínos v celkovém přežití byl shodně prokázán napříč podskupinami pacientů. Přínos v přežití byl pozorován bez ohledu na to, zda pacienti měli nádory, které byly označeny jako PD-L1 negativní nebo PD-L1 pozitivní (membránová exprese tumoru s hraniční hodnotou 1 %, 5 % nebo 10 %). Nicméně role tohoto biomarkeru (exprese PD-L1 na nádorových buňkách) nebyla plně objasněna.
Součástí studie CA209017 byl i omezený počet pacientů > 75 let (11 ve skupině nivolumabu a 18 ve skupině docetaxelu). Nivolumab vykázal numericky menší vliv na OS (HR 1,85; 95% CI: 0,76, 4,51), PFS (HR = 1,76; 95% CI: 0,77, 4,05) a ORR (9,1 % vs. 16,7 %). Vzhledem k malé velikosti vzorku nelze z těchto údajů vyvodit žádné definitivní závěry.
Výsledky účinnosti jsou zachyceny v tabulce 6.
|
nivolumab (n = 135) |
docetaxel (n = 137) | |
|
Celkové přežití | ||
|
Případy |
86 (63,7) |
113 (82,5) |
|
Poměr rizik |
0,59 | |
|
96.85% CI |
(0,43; 0,81) | |
|
p-hodnota |
0,0002 | |
|
Medián (95% CI) měsíce |
9,23 (7,33; 13,27) |
6,01 (5,13, 7.33) |
|
Výskyt (95% CI) ve |
42,1 (33,7; 50,3) |
23,7 (16,9; 31,1) |
|
12 měsících | ||
|
Potvrzená objektivní odpověď |
27 (20,0 %) |
12 (8,8 %) |
|
(95% CI) |
(13,6; 27,7) |
(4,6; 14,8) |
|
Míra relativního rizika (odds |
2,64 (1,27; 5,49) | |
|
ratio) (95% CI) | ||
|
p-hodnota |
0,0083 | |
|
Kompletní odpověď (CR) |
1 (0,7 %) |
0 |
|
Částečná odpověď (PR) |
26 (19,3 %) |
12 (8,8 %) |
|
Stabilní onemocnění (SD) |
39 (28,9 %) |
47 (34,3 %) |
|
Medián doby trvání odpovědi | ||
|
Měsíce (rozpětí) |
Nedosažen (2,9 - 20,5+) |
8,4 (1,4+ - 15,2+) |
|
Medián doby do nástupu odpovědi | ||
|
Měsíce (rozpětí) |
2,2 (1,6 - 11,8) |
2,1 (1,8 - 9,5) |
|
Přežití bez progrese | ||
|
Případy |
105 (77,8) |
122 (89,1) |
|
Poměr rizik |
0,62 | |
|
95% CI |
(0,47; 0,81) | |
|
p-hodnota |
< 0,0004 | |
|
Medián (95% CI) (měsíce) |
3,48 (2,14; 4,86) |
2,83 (2,10; 3,52) |
|
Výskyt (95% CI) ve |
20,8 (14,0; 28,4) |
6,4 (2,9; 11,8) |
|
12 měsících | ||
Výskyt zlepšení příznaků souvisejících s onemocněním, měřeno pomocí LCSS, byl podobný mezi skupinami s nivolumabem (18,5 %) a docetaxelem (21,2 %). Průměrná hodnota EQ-VAS se v průběhu času zvýšila u obou léčebných skupin, což naznačuje lepší celkový zdravotní stav pacientů, kteří zůstali na léčbě.
Jednoramenná studie fáze 2(CA209063)
Studie CA 209063 byla jednoramenná, otevřená studie hodnotící 117 pacientů s lokálně pokročilým nebo metastatickým skvamózním NSCLC po dvou nebo více terapeutických režimech; kromě toho byla použita podobná vstupní kritéria jako ve studii CA209017. Nivolumab v dávce 3 mg/kg vykázal stejný výskyt celkové odpovědi 14,5 % (95% CI: 8,7-22,2 %), medián OS byl 8,21 měsíců (95% CI: 6,05-10,9) a medián PFS 1,87 měsíce (95% CI: 1,77-3,15 měsíců). PFS byl měřen pomocí RECIST, verze 1.1. Odhadovaný výskyt 1ročního přežití byl 41 %.
Neskvamózní NSCLC
Randomizovaná studie fáze 3 proti docetaxelu (CA209057)
Bezpečnost a účinnost samotného nivolumabu v dávce 3 mg/kg v léčbě pokročilého nebo metastatického neskvamózního NSCLC byla hodnocena v otevřené randomizované studii fáze 3 (CA209057). Studie zahrnovala pacienty (18 let a starší), u kterých došlo k progresi onemocnění během nebo po podání jedné terapie založené na platinovém dubletu, která mohla zahrnovat udržovací
terapii, a kteří měli hodnoty ECOG skóre 0 nebo 1. Další linie TKI léčby byla povolena pacientům se známou mutací EGFR nebo translokací ALK. Pacienti byli zahrnuti bez ohledu na jejich PD-L1 stav. Pacienti s aktivním autoimunitním onemocněním, symptomatickým intersticiálním onemocněním plic nebo neléčenými mozkovými metastázami byli ze studie vyloučeni. Pacienty s léčenými mozkovými metastázami bylo možné zařadit, pokud se neurologický obraz vrátil k počátečnímu stavu alespoň 2 týdny před vstupem do studie a to bez kortikosteroidů nebo s jejich stabilní nebo klesající dávkou odpovídající <10 mg prednisonu denně.
Celkem bylo randomizováno 582 pacientů buď k nivolumabu v dávce 3 mg/kg podávané 60 minut intravenózně každé 2 týdny (n = 292) nebo k docetaxelu v dávce 75 mg/m2 každé 3 týdny (n = 290). Léčba probíhala, dokud byl pozorován klinický přínos nebo dokud nepřestala být tolerována. Hodnocení nádoru podle RECIST. Primárním cílem v hodnocení účinnosti bylo OS. Hlavními sekundárními cíly v hodnocení účinnosti byly ORR a PFS hodnocené zkoušejícím. Byly provedeny další předem specifikované analýzy podskupin k posouzení účinnosti u exprese PD-L1 na nádorových buňkách na předdefinované úrovni 1 %, 5 % a 10 % nádorových buněk. Posouzení podle intervalů PD-L1 exprese nebylo do předem specifikovaných analýz zahrnuto vzhledem k malé velikosti vzorků v jednotlivých intervalech.
Před randomizací byly systematicky shromažďovány vzorky nádorové tkáně odebrané před vstupem do studie, aby bylo možno provést předem plánované analýzy účinnosti podle úrovně exprese PD-L1 na nádorových buňkách. Úroveň exprese PD-L1 na nádorových buňkách byla stanovena pomocí PD-L1 IHC 28-8 pharmDx testu.
Medián věku byl 62 let (rozpětí: 21 až 85), přičemž 34 % pacientů bylo > 65 let a 7 % > 75 let. Většina pacientů byli běloši (92 %) a muži (55 %). Počáteční ECOG skóre bylo 0 (31 %) nebo 1 (69 %). Sedmdesát devět procent pacientů byli bývalí nebo současní kuřáci.
Kaplan-Meierovy křivky pro OS jsou znázorněny na obrázku 6.
Obrázek 6: Kaplan-Meierovy křivky pro OS (CA209057)
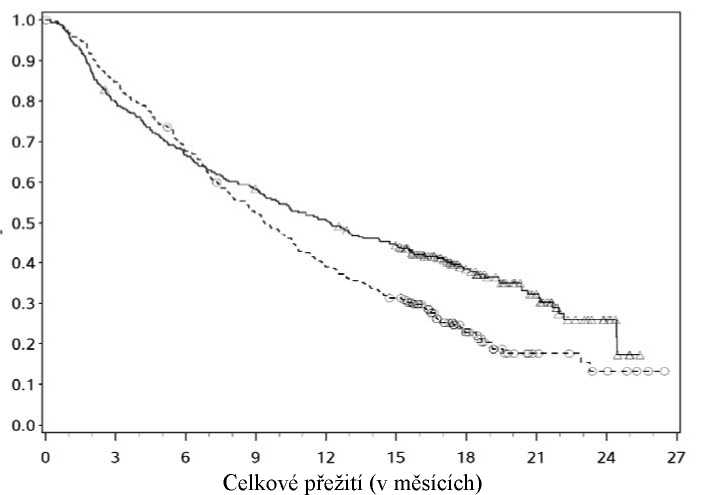
Počet subjektů s rizikem Nivolumab 3 mg/kg
|
292 |
232 |
194 |
169 |
146 |
123 |
62 |
32 |
9 |
0 |
|
Docetaxel 290 |
244 |
194 |
150 |
111 |
88 |
34 |
10 |
5 |
0 |
-A-Nivolumab 3 mg/kg (případy: 190/292), medián and 95% CI: 12.19 (9,66; 14,98)
---O---Docetaxel (případy: 223/290), medián and 95% CI: 9,36 (8,05; 10,68)
Studie prokázala statisticky významné zlepšení OS u pacientů randomizovaných k užívání nivolumabu ve srovnání s docetaxelem v předdefinované interim analýze, ve které bylo pozorováno 413 případů (93 % počtu případů plánovaných pro finální analýzu). Výsledky účinnosti jsou uvedeny v tabulce 7.
Tabulka 7: Výsledky účinnosti (CA209057)
|
nivolumab (n = 292) |
docetaxel (n = 290) | ||
|
Předdefinovaná interim analýza Celkové přežití | |||
|
Případy (%) |
190 (65,1%) |
223 (76,9%) | |
|
Poměr rizika (95,92% CI) p-hodnotab Medián (95% CI) měsíce |
12,19 (9,66; 14,98) |
0,73 (0,59; 0,89) 0,0015 |
9,36 (8,05; 10,68) |
|
Výskyt (95% CI) ve 12 měsících |
50,5 (44,6; 56,1) |
39,0 (33,3; 44,6) | |
|
Potvrzená objektivní odpověď |
56 (19,2%) |
36 (12,4%) | |
|
(95% CI) |
(14,8; 24,2) |
(8,8; 16,8) | |
|
Míra relativního rizika (odds ratio) |
1,68 (1,07; 2,64) | ||
|
(95% CI) | |||
|
p-hodnota Kompletní odpověď (CR) |
4 (1,4%) |
0,0246 |
1 (0,3%) |
|
Částečná odpověď (PR) |
52 (17,8%) |
35 (12,1%) | |
|
Stabilní onemocnění (SD) |
74 (25,3%) |
122 (42,1%) | |
|
Medián doby trvání odpovědi | |||
|
Měsíce (rozpětí) |
17,15 (1,8; 22,6+) |
5,55 (1,2+; 15,2+) | |
|
Medián doby do nástupu odpovědi | |||
|
Měsíce (rozpětí) |
2,10 (1,2; 8,6) |
2,61 (1,4; 6,3) | |
|
Přežití bez progrese | |||
|
Případy |
234 (80,1%) |
245 (84,5%) | |
|
Poměr rizik 95% CI p-hodnota Medián (95% CI) (měsíce) |
2,33 (2,17; 3,32) |
0,92 (0,77; 1,11) 0,3932 |
4,21 (3,45; 4,86) |
|
Výskyt (95% CI) ve 12 měsících |
18,5 (14,1; 23,4) |
8,1 (5,1; 12,0) | |
a
b
Odvozeno z modelu stratifikovaných proporčních rizik.
P-hodnota je odvozena z log-rank testu stratifikovaného podle předchozí udržovací terapie a linie terapie; odpovídající hladina významnosti hranice účinnosti podle O’Brien-Fleminga je 0,0408. označuje cenzurované sledování.
Kvantitativní exprese nádorového PD-L1 byla měřena u 79 % pacientů ve skupině přípravku OPDIVO a u 77 % pacientů ve skupině docetaxelu. Výskyt exprese nádorového PD-L1 u obou léčebných skupin (nivolumab vs. docetaxel) byl vyvážený u všech předdefinovaných úrovní exprese nádorového PD-L1: > 1 % (53 % vs. 55 %), > 5 % (41 % vs. 38 %) a > 10 % (37 % vs. 35 %).
Pacienti se všemi předdefinovanými úrovněmi exprese PD-L1 na nádorových buňkách prokázali vyšší pravděpodobnost zlepšení přežití ve srovnání s docetaxelem, zatímco u pacientů s nízkou nebo žádnou expresí nádorového PD-L1 bylo přežití podobné jako u docetaxelu. Pokud jde o ORR, zvýšená exprese PD-L1 byla spojena s vyšším ORR. Ve srovnání s celkovou populací se medián trvání odpovědi zvýšil u nivolumabu proti docetaxelu u pacientů bez PD-L1 exprese (18,3 měsíců vs.
5,6 měsíců) a u pacientů s PD-L1 expresí (16,0 měsíců vs. 5,6 měsíců).
Tabulka 8 shrnuje výsledky ORR a OS podle exprese PD-L1 na nádorových buňkách.
|
PD-L1 exprese |
nivolumab |
docetaxel | |
|
ORR podle exprese PD-L1 na nádorových buňkách | |||
|
Míra relativního rizika | |||
|
(odds ratio) (95% CI) | |||
|
<1 % |
10/108 (9,3 %) |
15/101 (14,9 %) |
0,59 (0,22; 1,48) |
|
95% CI: 4,5; 16,4 |
95% CI: 8,6; 23,3 | ||
|
>1 % |
38/123 (30,9 %) |
15/123 (12,2 %) |
3,22 (1,60; 6,71) |
|
95% CI: 22,9; 39,9 |
95% CI: 7,0; 19,3 | ||
|
>1 % to <10 %a |
6/37 (16,2 %) |
5/ 44 (11,4 %) |
1,51 (0,35; 6,85) |
|
95% CI: 6,2; 32,0 |
95% CI: 3,8; 24,6 | ||
|
>10 % to <50 % |
1 5/20 (25,0 %) |
7/33 (21,2 %) |
1,24 (0,26; 5,48) |
|
95% CI: 8,7; 49,1 |
95% CI: 9,0; 38,9 | ||
|
>50%a |
27/66 (40,9 %) |
3/46 (6,5 %) |
9,92 (2,68; 54,09) |
|
95% CI: 29,0; 53,7 |
95% CI: 1,4; 17,9 | ||
|
Celkové přežití podle exprese PD-L1 na nádorových buňkách | |||
|
Počet případů (počet pacientů) |
Nestratifikovaný poměr | ||
|
rizik (95% CI) | |||
|
<1 % |
77 (108) |
75 (101) |
0,90 (0,66; 1,24) |
|
>1 %* |
68 (123) |
93 (123) |
0,59 (0,43; 0,82) |
|
>1% to <10%a |
27 (37) |
30 (44) |
1,33 (0,79; 2,24) |
|
>10% to <50%a |
11 (20) |
26 (33) |
0,61 (0,30; 1,23) |
|
>50%a |
30 (66) |
37 (46) |
0,32 (0,20; 0,53) |
Následná analýza; výsledky je třeba interpretovat s opatrností vzhledem k malé velikosti vzorků a ke skutečnosti, že test PD-L1 IHC 28-8 pharmDx nebyl pro expresi na úrovni 10 % a 50 % buněk analyticky validován.
Podíl úmrtí během prvních 3 měsíců u pacientů v rameni nivolumabu byl vyšší (59/292, 20,2 %) než v rameni docetaxelu (44/290, 15,2 %). Výsledky následné komplexní analýzy ukazují, že pacienti s horší prognózou nebo agresivním onemocněním léčení nivolumabem mohou mít vyšší riziko úmrtí během prvních 3 měsíců, pokud mají zároveň nižší (tj. < 50 %) nebo žádnou expresi PD-L1 na nádorových buňkách.
V analýze podskupin pacientů, kteří nikdy nekouřili nebo jejichž nádory obsahovaly mutace aktivující EGFR, nebylo prokázáno lepší přežití; nicméně vzhledem k malým počtům pacientů nelze z těchto údajů učinit definitivní závěry.
Renální karcinom
Bezpečnost a účinnost nivolumabu v dávce 3 mg/kg v monoterapii v léčbě pokročilého RCC s komponentou světlých buněk byla hodnocena v randomizované otevřené studii (CA209025) 3. fáze. Ve studii byli zahrnuti pacienti (18 let a starší), u kterých došlo k progresi onemocnění během nebo po 1 nebo 2 předcházejících anti-angiogenních terapiích a po ne více než celkem 3 předchozích systémových léčebných režimech. Pacienti museli mít skóre podle Karnofského (KPS) > 70 %. Tato studie zahrnovala pacienty bez ohledu na status nádorového PD-L1. Pacienti s výskytem mozkových metastáz aktuálně nebo v anamnéze, s předchozí léčbou inhibitorem savčího rapamycinového cíle (mTOR), aktivním autoimunitním onemocněním nebo stavy vyžadujícími systémovou imunosupresi byli ze studie vyřazeni.
Celkem 821 pacientů bylo randomizováno k užívání buď nivolumabu v dávce 3 mg/kg podávanému intravenózně 60 minut každé 2 týdny (n=410) nebo everolimu v dávce 10 mg denně perorálně (n=411). Léčba pokračovala, pokud byl pozorován klinický přínos a dokud byla léčba snášena. První hodnocení nádorové odpovědi byla provedena 8 týdnů po randomizaci a dále každých 8 týdnů během prvního roku a potom každých 12 týdnů do progrese onemocnění nebo ukončení léčby, podle toho, co nastalo později. U pacientů, kteří léčbu ukončili z jiných důvodů než kvůli progresi, pokračovalo hodnocení nádorové odpovědi i po ukončení léčby. Pokračování léčby po progresi zhodnocené zkoušejícím podle definice RECIST, verze 1.1, bylo dovoleno, pokud měla léčba podle hodnocení zkoušejícího pro pacienta klinický přínos a byla jím snášena. Primárním cílem účinnosti bylo celkové přežití (OS). Sekundární hodnocení účinnosti zahrnovala ORR a PFS podle posouzení zkoušejícího. Počáteční charakteristiky byly mezi oběma skupinami vyvážené. Medián věku činil 62 let (rozpětí: 18-88) přičemž 40 % pacientů bylo > 65 let a 9 % > 75 let. Většina pacientů byli muži (75 %) a běloši (88 %), všechny rizikové skupiny Memorial Sloan Kettering Cancer Center (MSKCC) byly zastoupeny a 34 % resp. 66 % mělo počáteční hodnotu KPS 70 resp. 80 % a 90 resp. 100 %. Většina pacientů (72 %) prošla jednou předchozí anti-angiogenní léčbou. Střední doba od prvotní diagnózy do randomizace činila 2,6 roku jak ve skupině nivolumabu, tak everolimu. Střední doba trvání léčby byla
5,5 měsíců (rozpětí: 0-29,6+ měsíců) u pacientů léčených nivolumabem a 3,7 měsíců (rozpětí:
6 dní-25,7+ měsíců) u pacientů léčených everolimem.
U 44 % pacientů podávání nivolumabu pokračovalo i po progresi onemocnění.
Kaplan-Meierovy křivky pro OS jsou znázorněny na obrázku 7.
Obrázek 7: Kaplan-Meierovy křivky OS (CA209025)
Pravděpodobnost přežití
Počet subjektů s rizikem
|
Nivolumab 410 |
389 |
359 |
337 |
305 |
275 |
213 |
139 |
73 |
29 |
3 |
0 |
|
Everolimus 411 |
366 |
324 |
287 |
265 |
241 |
187 |
115 |
61 |
20 |
2 |
0 |
Nivolumab 3 mg/kg (případů: 183/410), medián a 95% Cl: 25,00 (21,75, N.A.) -■ - - Everolimus 10 mg (případů: 215/411), medián a 95% CI: 19,55 (17,64; 23,06)
Studie prokázala statisticky významné zlepšení celkového přežití u pacientů randomizovaných k užívání nivolumabu ve srovnání s everolimem v předem určené interim analýze, která hodnotila 398 případů (70 % z plánovaného počtu případů pro konečnou analýzu) (tabulka 9 a obrázek 7). Přínos v OS byl pozorován bez ohledu na úroveň exprese PD-L1 na nádorových buňkách. Výsledky účinnosti jsou zachyceny v tabulce 9.
Tabulka 9: Výsledky účinnosti (CA209025)
|
nivolumab (n = 410) |
everolimus (n = 411) | ||
|
Overall survival | |||
|
Případy Poměr rizik 98,52% CI p-hodnota |
183 (45) |
0,73 (0,57; 0,93) 0,0018 |
215 (52) |
|
Medián (95% CI) |
25,0 (21,7; NE) |
19,6 (17,6; 23,1) | |
|
Výskyt (95% CI) | |||
|
v 6 měsících |
89,2 (85,7; 91,8) |
81,2 (77,0; 84,7) | |
|
ve 12 měsících |
76,0 (71,5; 79,9) |
66,7 (61,8; 71,0) | |
|
Objektivní odpověď |
103 (25,1%) |
22 (5,4%) | |
|
(95% CI) Míra relativního rizika (odds ratio) |
(21,0; 29,6) |
5,98 (3,68; 9,72) |
(3,4; 8,0) |
|
(95% CI) | |||
|
p-hodnota |
< 0,0001 | ||
|
Kompletní odpověď (CR) |
4 (1,0%) |
2 (0,5%) | |
|
Částečná odpověď (PR) |
99 (24,1%) |
20 (4,9%) | |
|
Stabilní onemocnění (SD) |
141 (34,4%) |
227 (55,2%) | |
|
Medián doby trvání odpovědi | |||
|
Měsíce (rozpětí) |
11,99 (0,0-27,6+) |
11,99 (0,0+-22,2+) | |
|
Medián doby do nástupu odpovědi | |||
|
Měsíce (rozpětí) |
3,5 (1,4-24,8) |
3,7 (1,5-11,2) | |
|
Přežití bez progrese | |||
|
Případy Poměr rizik 95% CI p-hodnota |
318 (77,6) |
0,88 (0,75; 1,03) 0,1135 |
322 (78,3) |
|
Medián (95% CI) |
4,6 (3,71; 5,39) |
4,4 (3,71; 5,52) | |
označuje cenzurované sledování.
Medián doby do nástupu objektivní odpovědi byl 3,5 měsíce (rozpětí: 1,4-24,8 měsíce) po zahájení terapie nivolumabem. U čtyřiceti devíti (47,6 %) odpovídajících byla odpověď setrvalá po dobu v rozmezí 0,0-27,6+ měsíců.
Celkové přežití mohlo být spojeno se zlepšením příznaků souvisejících s onemocněním i v nespecifické kvalitě života (QoL), což bylo hodnoceno podle validních a spolehlivých škál Functional Assessment of Cancer Therapy-Kidney Symptom Index-Disease Related Symptoms (FKSI-DRS) a EuroQoL EQ-5D. Zjevně významné zlepšení příznaků (změna hodnoty MID=2 ve skóre FKSI-DRS; p<0,001) a doby do zlepšení (HR = 1,66 (1,33;2,08), p<0,001) bylo významně lepší u pacientů v rameni nivolumabu. Protože obě ramena ve studii užívala aktivní léčbu, mají být QoL data interpretována v kontextu otevřeného designu studie a tedy přijímána s opatrností.
Bezpečnost a účinnost u starších _pacientů
Celkově nebyly zjištěny rozdíly v bezpečnosti či účinnosti mezi staršími (> 65 let) a mladšími pacienty (< 65 let). Data získaná sledováním pacientů s NSCLC ve věku 75 let a více jsou příliš omezená na vyvození závěrů ohledně této populace.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s nivolumabem u všech podskupin pediatrické populace v léčbě maligních solidních tumorů (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika (PK) nivolumabu je lineární v rozpětí dávek 0,1 až 10 mg/kg. Na základě populační PK analýzy údajů činily geometrický průměr clearance (CL), terminální biologický poločas a míra průměrné expozice v ustáleném stavu při dávce 3 mg/kg nivolumabu každé dva týdny 9,5 ml/h, resp. 26,7 dní a 75,3 pg/ml.
CL nivolumabu se zvyšuje s rostoucí tělesnou hmotností. Dávkování normalizované dle tělesné hmotnosti udržuje přibližně stejnou minimální koncentraci látky v ustáleném stavu v širokém rozmezí tělesné hmotnosti (34-162 kg).
Metabolická cesta nivolumabu nebyla plně charakterizována. U nivolumabu se očekává, že jeho degradace na malé peptidy a aminokyseliny skrze katabolické dráhy probíhá stejným způsobem jako u endogenní IgG.
OPDIVO v kombinaci s ipilimumabem: Geometrický průměr CL, Vss a terminální poločas nivolumabu byly 9,83 ml/h, resp. 7,62 l a 24,1 dny. CL nivolumabu se zvýšila o 35 %, pokud byl podáván v kombinaci; CL ipilimumabu nijak ovlivněna nebyla.
V přítomnosti protilátek proti nivolumabu se CL nivolumabu zvýšila o 25 %, pokud byl podáván v kombinaci; CL ipilimumabu nijak přítomnost protilátek neovlivnila.
Zvláštní populace
Populační PK analýza ukázala, že není rozdíl v CL nivolumabu v závislosti na věku, pohlaví, rase, typu tumoru a velikosti nádoru a poruše funkce jater. Ačkoli skóre ECOG, výchozí míra glomerulární filtrace (GFR), albumin, tělesná hmotnost a mírná porucha funkce jater měly vliv na CL nivolumabu, tento účinek nebyl klinicky významný.
Porucha _ funkce ledvin
Vliv poruchy ledvin na CL nivolumabu byl v populační PK analýze hodnocen u pacientů s mírnou (GFR < 90 a > 60 ml/min/1,73 m2; n = 379), střední (GFR < 60 a > 30 ml/min/1,73 m2; n = 179), nebo závážnou (GFR < 30 a > 15 ml/min/1,73 m2; n = 2) poruchou funkce ledvin oproti pacientům s normální funkcí ledvin (GFR > 90 ml/min/1,73 m2; n = 342). Nebyly zjištěny žádné klinicky důležité rozdíly v CL nivolumabu mezi pacienty s mírnou či středně těžkou poruchou funkce ledvin a pacienty s normální funkcí ledvin. Údaje od pacientů se závažnou poruchou ledvin jsou příliš omezené a neumožňují učinit pro tuto populaci nějaké závěry (viz bod 4.2).
Porucha funkce jater
Účinek poruchy funkce jater na CL nivolumabu byl v populační PK analýze hodnocen u pacientů s mírnou poruchou funkce jater (celkový bilirubin 1,0 x až 1,5 x ULN nebo AST > ULN podle definice jaterní dysfunkce Národního onkologického institutu; n = 92) oproti pacientům s normální funkcí jater (celkový bilirubin a AST < ULN; n = 804). Nebyly zjištěny žádné klinicky důležité rozdíly v CL nivolumabu mezi pacienty s mírnou poruchou funkce jater a normální funkcí jater. Nivolumab nebyl studován u pacientů se středně závažnou (celkový bilirubin > 1,5 x až 3 x ULN a jakékoli AST) nebo závážnou poruchou funkce jater (celkový bilirubin > 3 x ULN a jakékoli AST) (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
Blokáda signalizace PD-L1 prokázala u myších modelů narušení této tolerance matky k plodu a tudíž zvýšení počtu potratů. Účinky nivolumabu na prenatální a postnatální vývoj byly hodnoceny u opic, které dostávaly nivolumab dvakrát týdně od počátku organogeneze v prvním trimestru až do porodu, při expozici osmkrát nebo 35krát vyšší, než jaká je pozorována při klinické dávce 3 mg/kg nivolumabu (podle AUC). Na počátku třetího trimestru byl pozorován nárůst potratů v závislosti na dávce a zvýšená novorozenecká úmrtnost.
Zbývající potomstvo nivolumabem léčených samic přežilo do plánovaného ukončení studie, bez klinických příznaků spojených s lékem, změn v normálním vývoji, účinků na hmotnost orgánů nebo makro- a mikroskopických patologických změn. Výsledky indexů růstu, stejně jako teratogenních, neurobehaviorálních, imunologických a klinických patologických parametrů během 6měsíčního postnatálního období byly srovnatelné s výsledky kontrolní skupiny. Nicméně vzhledem k mechanismu účinku nivolumabu, fetální expozice nivolumabu může zvýšit riziko vzniku chorob souvisejících s imunitním systémem nebo ovlivnit normální imunitní odpověď; choroby související s imunitním systémem byly pozorovány u myší s odstraněným genem pro receptor PD-1.
Studie fertility nebyly s nivolumabem provedeny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát natrium-citrátu Chlorid sodný Mannitol (E421)
Kyselina pentetová Polysorbát 80
Hydroxid sodný (k úpravě pH)
Kyselina chlorovodíková (k úpravě pH)
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky. Přípravek OPDIVO se nesmí podávat jako infuze současně stejnou intravenózní linkou jako jiné léčivé přípravky.
6.3 Doba použitelnosti
Neotevřená injekční lahvička 2 roky.
Po otevření
Z mikrobiologického hlediska je třeba, aby byl léčivý přípravek po otevření okamžitě aplikován v infuzi, nebo naředěn a podán v infuzi.
Po přípravě infuze
Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, je chemická a fyzikální stabilita přípravku OPDIVO prokázána po dobu 24 hodin při teplotě 2 °C až 8 °C a při ochraně před světlem, při teplotě 20 °C až 25 °C a pokojovém světle maximálně 4 hodiny (z celkových 24 hodin se má do této 4hodinové doby započítat doba podávání přípravku).
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Podmínky pro uchovávání po přípravě infuze viz bod 6.3.
6.5 Druh obalu a obsah balení
4 ml koncentrátu v 10ml injekční lahvičce (sklo třídy I) s uzávěrem (potaženým butylovou gumou) a tmavě modrým odklápěcím uzávěrem (hliník). Velikost balení 1 injekční lahvička.
10 ml koncentrátu v 10ml injekční lahvičce (sklo třídy I) s uzávěrem (potaženým butylovou gumou) a šedým odklápěcím uzávěrem (hliník). Velikost balení 1 injekční lahvička.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravu musí provádět vyškolený personál v souladu s pravidly správné praxe, zejména s ohledem na asepsi.
Příprava a podání
Výpočet dávky
Předepsaná dávka pro pacienta se udává v mg/kg. Podle této předepsané dávky vypočítejte celkovou dávku, která se má podat. Pro přípravu celé dávky pro pacienta může být zapotřebí více než jedna lahvička koncentrátu OPDIVO.
■ Celková dávka nivolumabu v mg = tělesná hmotnost pacienta v kg * předepsaná dávka v mg/kg.
■ Objem koncentrátu OPDIVO k přípravě dávky (ml) = celková dávka v mg, dělená deseti (síla koncentrátu OPDIVO je 10 mg/ml).
Příprava infuze
Když připravujete infuzi, věnujte pozornost zajištění aseptické manipulace. Infuze se má připravovat v digestoři s laminárním prouděním nebo v bezpečnostním boxu pomocí standardních opatřeních pro bezpečné zacházení s intravenózními látkami.
Přípravek OPDIVO lze použít pro intravenózní podání buď:
■ neředěný po přenosu do infuzní nádoby pomocí vhodné sterilní stříkačky; nebo
■ po naředění v koncentraci až do 1 mg/ml. Konečná koncentrace infuze by se měla pohybovat v rozpětí mezi 1 a 10 mg/ml. Koncentrát přípravku OPDIVO se může ředit buď:
■ roztokem chloridu sodného 9 mg/ml (0,9%) pro injekce; nebo
■ roztokem glukózy 50 mg/ml (5%) pro injekce.
KROK 1
■ Prohlédněte koncentrát přípravku OPDIVO, zda neobsahuje částice nebo se nezměnila barva. Lahvičku neprotřepávejte. Koncentrát přípravku OPDIVO je čirá až opalizující, bezbarvá až světle žlutá tekutina. Pokud je roztok zakalený, změnil barvu či pokud obsahuje jiné částice než několik průsvitných až bílých částic, lahvičku zlikvidujte.
■ Odeberte požadovaný objem koncentrátu OPDIVO pomocí vhodné sterilní stříkačky.
KROK 2
■ Vstříkněte koncentrát do sterilní, prázdné skleněné lahvičky nebo intravenózní nádobky (PVC nebo polyolefin).
■ Podle potřeby nařeďte roztokem chloridu sodného 9 mg/ml (0,9%) pro injekce nebo roztokem glukózy 50 mg/ml (5%) pro injekce. Jemně promíchejte infuzi otáčením v ruce. Neprotřepávejte.
Infuze přípravku OPDIVO se nesmí podávat jako intravenózní bolus nebo bolusová injekce. Podávejte infuzi přípravku OPDIVO intravenózně po dobu 60 minut.
Přípravek OPDIVO se nesmí podávat jako infuze současně ve stejné intravenózní lince s jinými látkami. Pro infuzi používejte oddělenou infuzní linku.
Používejte infuzní set a sériový, sterilní, apyrogenní filtr s nízkou schopností vázat proteiny (velikost pórů 0,2-1,2 pm).
Infuze přípravku OPDIVO je kompatibilní s:
PVC a polyolefinovými nádobami, skleněnými lahvemi, infuzními sety z PVC a sériovými filtry s polyétersulfonovou membránou o velikostí pórů 0,2 pm až 1,2 pm.
Po podání dávky nivolumabu vypláchněte linku roztokem chloridu sodného 9 mg/ml (0,9%) pro injekce nebo roztokem glukózy 50 mg/ml (5%) pro injekce.
Likvidace
Neuchovávejte nepoužitou část infuzního roztoku pro další použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bristol-Myers Squibb Pharma EEIG Uxbridge Business Park Sanderson Road Uxbridge UB8 1DH Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1014/001-002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
19. června 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA
PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců biologické léčivé látky
Bristol-Myers Squibb Company 6000 Thompson Road East Syracuse, New York 13057 USA
Lonza Biologics, Inc.
101 International Drive Portsmouth, New Hampshire 03801 USA
Samsung Biologics Co. Ltd.
125, Cheomdan-daero Yeonsu-gu, Incheon, 21987 Korea
Název a adresa výrobce odpovědného za propouštění šarží
Bristol-Myers Squibb S.r.l.
Loc. Fontana del Ceraso 03012 Anagni (FR)
Itálie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky;
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
V každém členském státě musí držitel rozhodnutí o registraci (MAH) před uvedením přípravku OPDIVO na trh získat souhlas národní regulační autority s obsahem a formátem edukačního programu včetně způsobů komunikace a distribuce, i všech ostatních aspektů programu.
Edukační materiály jsou zaměřeny na zvýšení povědomí o možném riziku imunitně podmíněných nežádoucích příhod v souvislosti s podání přípravku OPDIVO a poskytnutí pokynů, jak je zvládat, a rovněž zvýšit povědomí osob pečujících o pacienty o známkách a příznacích těchto nežádoucích příhod v jejich časném stádiu.
Držitel rozhodnutí o registraci zajistí, že zdravotničtí pracovníci a pacienti/pečující osoby, u kterých se předpokládá, že budou předepisovat nebo užívat přípravek OPDIVO, budou mít přístup nebo jim bude poskytnut následující edukační balíček:
• Edukační materiál pro lékaře
• Karta pacienta
Edukační materiál pro lékaře má obsahovat:
• Souhrn údajů o přípravku
• Průvodce zvládáním nežádoucích účinků
Pokyny ke zvládnutí nežádoucích účinků mají obsahovat tyto klíčové informace:
• Relevantní informace (tj. závažnost, frekvence, rychlost nástupu, reverzibilita nežádoucího účinku) u následujících bezpečnostních problémů:
o imunitně podmíněná pneumonitida
o imunitně podmíněná kolitida
o imunitně podmíněná hepatitida
o imunitně podmíněná nefritida a renální dysfunkce
o imunitně podmíněné endokrinopatie
o imunitně podmíněná vyrážka
o ostatní imunitně podmíněné nežádoucí účinky.
• Podrobnosti o tom, jak minimalizovat bezpečnostní problémy vhodným monitorováním a jinými opatřeními
• Karta pacienta má obsahovat tato klíčová sdělení:
• Léčba přípravkem OPDIVO může zvýšit riziko:
o imunitně podmíněné pneumonitidy
o imunitně podmíněné kolitidy
o imunitně podmíněné hepatitidy
o imunitně podmíněné nefritidy a renální dysfunkce
o imunitně podmíněných endokrinopatií
o imunitně podmíněné vyrážky
o ostatních imunitně podmíněných nežádoucích účinků.
• Známky a příznaky bezpečnostního problému a kdy se poradit se zdravotnickým pracovníkem
• Kontaktní údaje na lékaře, který přípravek OPDIVO předepsal
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření
|
Popis |
Termín splnění |
|
1. Poregistrační studii účinnosti (PAES): MAH předloží závěrečnou zprávu ze studie CA209037: Randomizovaná otevřená studie fáze 3 hodnotící nivolumab ve srovnání s léčbou dle výběru zkoušejícího u pacientů s pokročilým (neresekovatelným nebo metastatickým) melanomem progredujícím po anti-CTLA-4 terapii. |
Závěrečná zpráva ze studie má být předložena do 31. října 2016 |
|
2. Poregistrační studii účinnosti (PAES): MAH předloží závěrečnou zprávu ze studie CA209067: Randomizovaná dvojitě zaslepená studie u pacientů léčených nivolumabem v monoterapii, ipilimumabem v monoterapii a nivolumabem v kombinaci s ipilimumabem. |
Závěrečná zpráva ze studie má být předložena do 31. března 2017 |
|
3. Význam biomarkerů pro predikci účinnosti nivolumabu a/nebo kombinované terapie nivolumab + ipilimumab má být dále zkoumán, zvláště: 1. Pokračovat ve výzkumu optimální hranice pro PD-L1 pozitivitu na základě nyní používané metody stanovení, aby se dále objasnil její význam pro predikci účinnosti nivolumabu. Tyto analýzy budou prováděny na celé populaci studie CA209037 u pacientů s pokročilým melanomem. Tyto analýzy mohou být předloženy jako dodatek k závěrečné zprávě z klinické studie CA209037. 2. Pro predikci účinnosti nivolumabu a/nebo kombinované terapie nivolumab + ipilimumab imunohistochemickými metodami dále zkoumat výpovědní hodnotu jiných biomarkerů než jen stav membránové exprese PD-L1 na nádorové buňce (tj. jiné genotypové metody/testy a příslušné hranice, které se mohou ukázat jako citlivější a specifičtější v predikci odpovědi na léčbu než jsou PD-L1, PD-L2, tumor infiltrující lymfocyty s měřením hustoty CD8+ T-buněk, měření RNA podpisu, exprese složek komplexů prezentujících antigen a /nebo dalších inhibičních kontrolních receptorů/ligandů nádoru apod). Toto bude předloženo pro všechny schválené indikace: |
31. říjen 2016 |
|
- Melanom - monoterapie: studie CA209038 a CA209066 - Melanom - kombinace (s ipilimumabem): studie CA209038, CA209067 nebo CA209069 |
30. září 2017 31. březen 2019 |
|
- NSCLC: studie CA209017, CA209057 a CA209026 - RCC: studie CA209025 a CA209009 |
31. březen 2018 31. březen 2018 |
|
Dále budou ve studii CA209038 zkoumány hladiny myeloidních supresorových buněk. 3. Dále zkoumat vztah mezi expresí PDL-1 a PDL-2 ve studiích fáze 1 (CA209009, CA209038 and CA209064). |
31. březen 2017 |
|
4. Dále zkoumat asociativní analýzy mezi expresí PDL-1 a PDL-2 prováděné ve studiích CA209-066, CA209057 a CA209025. |
30. červen 2018 |
|
5. Dále zkoumat možnou změnu v PD-L1 stavu nádoru během léčby a/nebo jeho progrese ve studiích CA209-009, CA209-038 and CA209-064. |
30. září 2017 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
OPDIVO 10 mg/ml koncentrát pro infuzní roztok nivolumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml koncentrátu obsahuje nivolumabum 10 mg.
Jedna injekční lahvička 4 ml obsahuje nivolumabum 40 mg. Jedna injekční lahvička 10 ml obsahuje nivolumabum 100 mg.
3. SEZNAM POMOCNÝCH LÁTEK_
Pomocné látky: dihydrát natrium-citrátu, chlorid sodný, mannitol (E421), kyselina pentetová, polysorbát 80, hydroxid sodný, kyselina chlorovodíková, voda na injekci.
Další informace naleznete v příbalové informaci
4. LÉKOVÁ FORMA A OBSAH BALENÍ_
Koncentrát pro infuzní roztok.
40 mg/4 ml 100 mg/10 ml
1 injekční lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Pouze pro jednorázové použití.
8. POUŽITELNOST
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bristol-Myers Squibb Pharma EEIG Uxbridge Business Park Sanderson Road Uxbridge UB8 1DH Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1014/001 40 mg injekční lahvička EU/1/15/1014/002 100 mg injekční lahvička
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato.
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK NA LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
OPDIVO 10 mg/ml sterilní koncentrát
nivolumabum
i.v. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
40 mg/4 ml 100 mg/10 ml
6. JINÉ
Pouze pro jednorázové použití.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
OPDIVO 10 mg/ml koncentrát pro infuzní roztok
nivolumabum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Je důležité, abyste po dobu léčby u sebe stále nosil(a) Kartu pacienta.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek OPDIVO a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek OPDIVO používat
3. Jak se přípravek OPDIVO používá
4. Možné nežádoucí účinky
5. Jak přípravek OPDIVO uchovávat
6. Obsah balení a další informace
1. Co je přípravek OPDIVO a k čemu se používá
Přípravek OPDIVO se používá k léčbě:
■ pokročilého melanomu (typ rakoviny kůže) u dospělých
■ pokročilého nemalobuněčného karcinomu plic (typ rakoviny plic) u dospělých
■ pokročilého renálního karcinomu (pokročilé rakoviny ledvin) u dospělých.
Obsahuje léčivou látku nivolumab, což je monoklonální protilátka, druh proteinu, který rozpoznává a váže se na specifickou cílovou látku v těle.
Nivolumab se váže na cílový protein, který se nazývá receptor programovaného zániku nádorové buňky (PD-1), který může vypnout činnost T-buněk (druh bílých krvinek, které tvoří součást imunitního systému, přirozené obrany organismu). Navázáním na PD-1 blokuje nivolumab tuto činnost a zabraňuje tak vypnutí T-buněk. To pomáhá zvýšit jejich aktivitu proti buňkám melanomu, nádoru plic nebo ledvin.
Přípravek OPDIVO může být podáván v kombinaci s ipilimumabem. Je důležité si také přečíst příbalovou informaci tohoto přípravku. Pokud máte k ipilimumabu jakékoliv dotazy, obraťte se na svého lékaře.
2. Čemu musíte věnovat pozornost, než začnete přípravek OPDIVO používat Nepoužívejte přípravek OPDIVO
■ jestliže jste alergický(á) na nivolumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6 "Obsah balení a další informace"). Jestliže si nejste jistý(á), poraďte se se svým lékařem.
Upozornění a opatření
Před použitím přípravku OPDIVO se poraďte se svým lékařem. Přípravek OPDIVO může způsobit:
■ plicní potíže, jako je obtížné dýchání nebo kašel. Tyto potíže mohou znamenat zánět plic (pneumonitidu nebo intersticiální onemocnění plic).
■ průjem (vodnatou, řídkou nebo měkkou stolici) nebo příznaky zánětu střev (kolitidy), jako je bolest břicha a hlen nebo krev ve stolici.
■ zánět jater (hepatitidu). Známky a příznaky hepatitidy mohou zahrnovat abnormální testy funkce jater, žloutnutí očí nebo kůže (žloutenku), bolest na pravé straně břicha nebo únavu.
■ zánět ledvin nebo ledvinové potíže. Známky a příznaky mohou zahrnovat abnormální testy funkce ledvin nebo snížené množství moči.
■ problémy se žlázami produkujícími hormony (včetně hypofýzy (podvěsku mozkového), štítné žlázy a nadledvinek), které mohou mít dopad na funkci těchto žláz. Známky a příznaky signalizující, že žlázy nefungují řádně, mohou zahrnovat vyčerpanost (extrémní únavu), změnu tělesné hmotnosti nebo bolest hlavy a poruchy zraku.
■ diabetes (cukrovka) (příznaky zahrnují nadměrnou žízeň, značně zvýšené množství moči, zvýšenou chuť k jídlu spojenou se ztrátou tělesné hmotnosti, pocit únavy, ospalosti, malátnosti, sklíčený, podrážděný pocit, kdy se celkově necítíte dobře) nebo diabetickou ketoacidózu (tj. kyselina v krvi v důsledku diabetu).
■ zánět kůže, který může vést k vyrážce a svědění.
Informujte okamžitě svého lékaře, jestliže máte nebo zhoršují-li se kterékoli z těchto známek nebo příznaků. Nepokoušejte se sám (sama) léčit příznaky jinými léky. Váš lékař Vám může
■ dát jiné léky, aby zabránil komplikacím a omezil příznaky,
■ vysadit příští dávku přípravku OPDIVO,
■ nebo léčbu přípravkem OPDIVO zcela ukončit.
Prosím povšimněte si, že tyto známky a příznaky se mohou někdy objevit se zpožděním, a mohou se rozvinout týdny nebo měsíce po poslední dávce. Před léčbou lékař zkontroluje Váš celkový zdravotní stav. Během léčby budou také prováděny krevní testy.
Než dostanete přípravek OPDIVO, poraďte se s lékařem nebo zdravotní sestrou, jestliže:
■ trpíte autoimunitním onemocněním (stav, kdy tělo napadá své vlastní buňky);
■ máte oční melanom;
■ Vám byl dříve podáván ipilimumab, jiný lék k léčbě melanomu, a měl(a) jste kvůli tomuto léku vážné nežádoucí účinky;
■ jste byl(a) informována, že se nádorové onemocnění rozšířilo do mozku;
■ máte zánět plic v anamnéze;
■ Vám byly dříve podávány léky k potlačení imunitního systému.
Děti a dospívající
Přípravek OPDIVO by se neměl používat u dětí a dospívajících do věku 18 let.
Další léčivé přípravky a přípravek OPDIVO
Předtím, než Vám bude podán přípravkek OPDIVO, informujte svého lékaře, pokud užíváte některé léky, které potlačují funkci imunitního systému, jako jsou kortikosteroidy, protože tyto léky mohou ovlivňovat účinek přípravku OPDIVO. Nicméně jakmile jste léčen(a) přípravkem OPDIVO, Váš lékař Vám může předepsat kortikosteroidy, aby se zmírnily projevy nežádoucích účinků, které se mohou objevit během léčby, a nebude to mít vliv na účinek léčiva.
Informujte svého lékaře o všech lécích, které užíváte nebo které jste v nedávné době užíval(a). Neužívejte během své léčby žádné jiné léky, aniž byste se nejdříve poradil(a) s lékařem.
Těhotenství a kojení
Informujte svého lékaře, jestliže jste těhotná, domníváte se, že můžete být těhotná, plánujete otěhotnění nebo kojíte.
Nepoužívejte přípravek OPDIVO, jste-li těhotná, pokud Vám to lékař výslovně neřekne. Účinky přípravku OPDIVO u těhotných žen nejsou známy, ale je možné, že léčivá látka nivolumab by mohla poškodit nenarozené dítě.
■ Jste-li žena, která by mohla otěhotnět, musíte po dobu léčby přípravkem OPDIVO a ještě alespoň 5 měsíců po podání poslední dávky přípravku OPDIVO používat účinnou antikoncepci.
■ Jestliže otěhotníte během používání přípravku OPDIVO, informujte svého lékaře.
Není známo, zda se nivolumab vylučuje do lidského mateřského mléka. Riziko pro kojence nelze vyloučit. Zeptejte se svého lékaře, zda můžete kojit během nebo po léčbě přípravkem OPDIVO.
Řízení dopravních prostředků a obsluha strojů
Není pravděpodobné, že by nivolumab měl dopad na schopnost řídit a obsluhovat stroje; provádějte však tyto činnosti s opatrností, dokud si nebudete jistý(a), že na Vás nivolumab nemá nežádoucí účinky.
Přípravek OPDIVO obsahuje sodík
Pokud držíte dietu s nízkým obsahem sodíku (s nízkým obsahem soli), informujte svého lékaře, než Vám bude přípravek OPDIVO podán. Tento přípravek obsahuje 2,5 mg sodíku na jeden ml koncentrátu.
Tyto informace naleznete rovněž v Kartě pacienta, kterou jste dostal(a) od lékaře. Je důležité, abyste měl(a) tuto kartu u sebe a ukázal(a) ji svému partnerovi nebo pečovateli.
3. Jak se přípravek OPDIVO používá Kolik přípravku OPDIVO dostanete
Množství přípravku OPDIVO, které budete dostávat, se vypočítá podle Vaší tělesné hmotnosti. Doporučená dávka je 3 mg nivolumabu na kilogram tělesné hmotnosti.
V závislosti na Vaší dávce bude patřičné množství přípravku OPDIVO před použitím naředěno roztokem chloridu sodného 9 mg/ml (0,9 %) pro injekce nebo roztokem glukózy 50 mg/ml (5 %) pro injekce. Pro dosažení požadované dávky se může použít i více než jedna lahvička přípravku OPDIVO.
Pokud je přípravek OPDIVO podáván v kombinaci s ipilimumabem, je doporučená dávka přípravku OPDIVO 1 mg nivolumabu na kilogram tělesné hmotnosti pro první 4 dávky (fáze kombinované léčby). Dále je doporučená dávka přípravku OPDIVO 3 mg nivolumabu na kilogram tělesné hmotnosti (fáze léčby jedním přípravkem).
Jak se přípravek OPDIVO podává
Léčbu přípravkem OPDIVO budete dostávat v nemocnici nebo na klinice pod dohledem zkušeného lékaře.
Přípravek OPDIVO budete dostávat jako infuzi (kapačku) do žíly (intravenózně, nitrožilně) po dobu 60 minut každé dva týdny. Váš lékař Vám bude podávat přípravek OPDIVO tak dlouho, dokud z něho budete mít prospěch nebo dokud budete léčbu snášet.
Pokud je přípravek OPDIVO podáván v kombinaci s ipilimumabem, budete dostávat infuzi po dobu 60 minut každé 3 týdny u prvních 4 dávek (fáze kombinované léčby). Poté budete dostávat infuzi po dobu 60 minut každé 2 týdny (fáze léčby jedním přípravkem).
Jestliže vynecháte dávku přípravku OPDIVO
Je velmi důležité, abyste dodržoval(a) všechny termíny k podávání přípravku OPDIVO. Jestliže některý termín nestihnete, zeptejte se svého lékaře, na kdy se má naplánovat další dávka.
Jestliže jste přestal(a) užívat přípravek OPDIVO
Ukončení léčby může zastavit účinek léku. Nepřerušujte léčbu přípravkem OPDIVO, dokud toto neproberete se svým lékařem.
Máte-li jakékoli další otázky týkající se Vaší léčby nebo používání tohoto přípravku, zeptejte se svého lékaře.
Pokud je přípravek OPDIVO podáván v kombinaci s ipilimumabem, dostanete nejprve OPDIVO a potom ipilimumab.
Seznamte se s příbalovou informací pro ipilimumab, abyste porozuměli užívání tohoto přípravku. Pokud máte jakékoliv dotazy týkající se tohoto přípravku, obraťte se prosím, na svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Váš lékař s Vámi tyto účinky prodiskutuje a vysvětlí Vám rizika a přínosy léčby.
Sledujte důležité příznaky zánětu. Přípravek OPDIVO působí na Váš imunitní systém a může způsobit zánět v určitých částech těla. Zánět může způsobit závažné poškození Vašeho těla a některé zánětlivé stavy mohou být život ohrožující a potřebovat léčbu nebo vysazení nivolumabu.
V klinických studiích se samotným nivolumabem byly hlášeny tyto nežádoucí účinky:
Velmi časté (mohou postihnout více než 1 pacienta z 10)
■ snížená chuť k jídlu
■ průjem (vodnatá, řídká nebo měkká stolice), pocit na zvracení
■ vyrážka na kůži, svědění
■ pocity únavy nebo slabosti
Časté (mohou postihnout až 1 pacienta z 10)
■ infekce horních cest dýchacích
■ alergická reakce, reakce spojené s infuzí léku
■ nedostatečná funkce štítné žlázy, což může způsobit únavu nebo zvyšování tělesné hmotnosti, nebo nadměrná činnost štítné žlázy, což může vést ke zrychlenému pulzu, pocení a ztrátě tělesné hmotnosti
■ vysoká hladina cukru v krvi (hyperglykémie)
■ zánět nervů způsobující necitlivost, slabost, štípavou nebo pálivou bolest paží a nohou; bolest hlavy, závratě
■ rozmazané vidění, suché oči
■ vysoký krevní tlak (hypertenze)
■ zánět plic (pneumonitida), charakterizovaný kašlem a obtížným dýcháním, dušnost (dyspnoe), kašel
■ zánět střev (kolitida), vřídky a ranky v ústech (stomatitida), zvracení, bolest břicha, zácpa, sucho v ústech
■ skvrny se změněnou barvou kůže (vitiligo), suchá kůže, začervenání kůže, neobvyklá ztráta nebo řídnutí vlasů
■ bolest svalů, kostí a kloubů
■ horečka, edém (otok)
Méně časté (mohou postihnout až 1 pacienta ze 100)
■ závažná plicní infekce (zápal plic), zánět průdušek (bronchitida)
■ zvýšení počtu některých bílých krvinek
■ snížené vylučování hormonů produkovaných nadledvinkami (žlázy uložené nad ledvinami), nedostatečná funkce (hypopituitarismus) nebo zánět (hypofyzitida) žlázy uložené na bázi lebky (hypofýze, podvěsek mozkový), otok štítné žlázy, kyselina v krvi zapříčiněná diabetem (diabetická ketoacidóza), diabetes
■ dehydratace, zvýšená kyselost krve
■ zánět jater (hepatitida)
■ žloutnutí kůže a/nebo očí (žloutenka)
■ poškození nervů způsobující necitlivost a slabost (polyneuropatie)
■ zánět oka, který způsobuje bolest a začervenání, problémy s viděním nebo rozmazané vidění
■ zvýšená tepová frekvence
■ zánětlivé postižení žil
■ tekutina kolem plic
■ zánět pankreatu (slinivky břišní)
■ závažný stav kůže, který způsobuje červené, často svědivé skvrny, podobně jako vyrážka při spalničkách, která začíná na končetinách a někdy na obličeji a zbytku těla (erythema multiforme); kožní onemocnění se zesílenými červenými skvrnami na kůži, často se stříbřitými šupinami (psoriáza, lupénka); stav kůže na obličeji, kdy jsou nos a tváře neobvykle červené (rosacea, růžovka), kopřivka (svědivá, vystouplá vyrážka)
■ zánět svalů způsobující bolest nebo ztuhlost, bolest v kloubech
■ zánět ledvin, selhání ledvin
■ bolest, bolest na hrudi
Vzácné (mohou postihnout až 1 pacienta z 1000)
■ onemocnění způsobující zánět nebo zvětšení lymfatických (mízních) uzlin (Kikuchiho lymfadenitida)
■ cukrovka (diabetes mellitus)
■ upání žlučovodů
■ přechodný zánět nervů, který způsobuje bolest, slabost a paralýzu končetin (Guillainův-Barrého syndrom); ztráta ochranného pouzdra nervových vláken (demyelinizace); stav, kdy dochází ke svalové slabosti a snadnému vyčerpání (myastenický syndrom); záněty nervů způsobené tím, že tělo tvoří látky, poškozující vlastní orgány, které způsobují necitlivost, slabost, brnění nebo palčivou bolest,
■ změny srdečního rytmu nebo frekvence, zrychlený puls, abnormální srdeční rytmus
■ tekutina v plicích
■ gastritida (zánět žaludku), vřed tenkého střeva
■ závažné odlupování kůže (toxická epidermální nekrolýza), které může vést k úmrtí
■ myopatie (svalová bolest, svalová citlivost a slabost nezpůsobené cvičením)
Informujte okamžitě svého lékaře, pokud se u Vás vyskytne kterýkoli z nežádoucích účinků uvedených výše. Nepokoušejte se sám (sama) léčit své příznaky jinými léky.
Změny ve výsledcích testů
Přípravek OPDIVO může způsobit změny ve výsledcích testů prováděných Vaším lékařem. Mezi ně patří:
■ abnormální testy funkce jater (zvýšené množství jaterních enzymů aspartátaminotransferázy, alaninaminotransferázy nebo alkalické fosfatázy v krvi, vyšší hladina odpadní látky bilirubinu v krvi)
■ abnormální testy funkce ledvin (zvýšené množství kreatininu v krvi)
■ snížený počet červených krvinek (které přenášejí kyslík), bílých krvinek (které jsou důležité pro boj s infekcí), nebo krevních destiček (buněk napomáhajících srážení krve)
■ zvýšená hladina enzymu, který štěpí tuky, a enzymu, který štěpí škrob
■ zvýšené nebo snížené množství vápníku nebo draslíku
■ zvýšené nebo snížené hladiny hořčíku nebo sodíku
■ snížení hmotnosti
V klinických studiích s nivolumabem v kombinaci s ipilimumabem byly hlášeny tyto nežádoucí účinky:
Velmi časté (mohou postihnout více než 1 pacienta z 10)
■ nedostatečná funkce štítné žlázy, což může způsobit únavu nebo zvyšování tělesné hmotnosti
■ snížená chuť k jídlu
■ bolest hlavy
■ zánět střev (kolitida), průjem (vodnatá, řídká nebo měkká stolice), zvracení, pocit na zvracení, bolest žaludku
■ vyrážka na kůži, svědění
■ bolest kloubů
■ pocity únavy nebo slabosti, horečka
Časté (mohou postihnout až 1 pacienta z 10)
■ infekce horních cest dýchacích, závažná plicní infekce (zápal plic)
■ zvýšení počtu některých bílých krvinek
■ alergická reakce, reakce spojené s infuzí léku
■ snížené vylučování hormonů produkovaných nadledvinkami (žlázy uložené nad ledvinami), nedostatečná funkce (hypopituitarismus) nebo zánět (hypofyzitida) žlázy uložené na bázi lebky (hypofýza, podvěsek mozkový), nadměrná činnost štítné žlázy, což může vést ke zrychlenému pulzu, pocení a ztrátě tělesné hmotnosti, zánět štítné žlázy, otok štítné žlázy, vysoká hladina cukru v krvi (hyperglykémie)
■ dehydratace
■ zánět jater (hepatitida)
■ zánět nervů způsobující necitlivost, slabost, štípavou nebo pálivou bolest paží a nohou; závratě
■ zánět oka, který způsobuje bolest a začervenání, problémy s viděním nebo rozmazané vidění
■ zvýšená tepová frekvence
■ vysoký krevní tlak (hypertenze)
■ zánět plic (pneumonitida), charakterizovaný kašlem a obtížným dýcháním, krevní sraženiny, dušnost (dyspnoe), kašel
■ vřídky a ranky v ústech (stomatitida), gastritida (zánět žaludku), zácpa, sucho v ústech
■ skvrny se změněnou barvou kůže (vitiligo), suchá kůže, začervenání kůže, neobvyklá ztráta nebo řídnutí vlasů, kopřivka (svědivá vyrážka)
■ bolest svalů a kostí
■ selhání ledvin
■ edém (otok), bolest
Méně časté (mohou postihnout až 1 pacienta ze 100)
■ zánět průdušek (bronchitida)
■ chronické onemocnění spojené s tvorbou zánětu v různých orgánech a tkáních, zvláště v plicích (sarkoidóza)
■ kyselina v krvi zapříčiněná diabetem (diabetická ketoacidóza), diabetes
■ přechodný zánět nervů, který způsobuje bolest, slabost a paralýzu končetin (Guillainův-Barrého syndrom); poškození nervů způsobující necitlivost a slabost (polyneuropatie), zánět nervů, obrna lýtkového nervu, záněty nervů způsobené tím, že tělo tvoří látky, poškozující vlastní orgány, které způsobují necitlivost, slabost, brnění nebo palčivou bolest
■ změny srdečního rytmu nebo frekvence, abnormální srdeční rytmus
■ tekutina v plicích
■ zánět pankreatu (slinivky břišní), proděravění střeva, zánět dvanáctníku
■ kožní onemocnění se zesílenými červenými skvrnami na kůži, často se stříbřitými šupinami (psoriáza, lupénka);
■ chronické onemocnění kloubů (spondyloartropatie); onemocnění, kde imunitní systém napadá žlázy produkující tělní tekutiny jako jsou slzy nebo sliny (Sjoegrenův syndrom), bolestivé klouby, myopatie (svalová bolest, svalová citlivost a slabost nezpůsobené cvičením)
■ zánět ledvin,
■ bolest na hrudi
Vzácné (mohou postihnout až 1 pacienta z 1000)
■ závažné odlupování kůže (toxická epidermální nekrolýza), které může vést k úmrtí
Informujte okamžitě svého lékaře, pokud se u Vás vyskytne kterýkoli z nežádoucích účinků
uvedených výše. Nepokoušejte se sám (sama) léčit své příznaky jinými léky.
Změny ve výsledcích testů
Přípravek OPDIVO v kombinaci s ipilimumabem může způsobit změny ve výsledcích testů prováděných Vaším lékařem. Mezi ně patří:
■ abnormální testy funkce jater (zvýšené množství jaterních enzymů aspartátaminotransferázy, alaninaminotransferázy nebo alkalické fosfatázy v krvi, vyšší hladina odpadní látky bilirubinu v krvi)
■ abnormální testy funkce ledvin (zvýšené množství kreatininu v krvi)
■ snížený počet červených krvinek (které přenášejí kyslík), bílých krvinek (které jsou důležité pro boj s infekcí), nebo krevních destiček (buněk napomáhajících srážení krve)
■ zvýšená hladina enzymu, který štěpí tuky, a enzymu, který štěpí škrob
■ zvýšené nebo snížené množství vápníku nebo draslíku
■ zvýšené nebo snížené hladiny hořčíku nebo sodíku
■ snížení hmotnosti
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek OPDIVO uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku lahvičky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C až 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Neuchovávejte žádnou nespotřebovanou část infuzního roztoku pro další použití. Veškerý nepoužitý léčivý přípravek nebo odpadní materiál musí být zlikvidován v souladu s místními požadavky.
6. Obsah balení a další informace
Co přípravek OPDIVO obsahuje
■ Léčivou látkou je nivolumabum.
Jeden ml koncentrátu pro infuzní roztok obsahuje nivolumabum 10 mg.
Jedna lahvička obsahuje nivolumabum buď 40 mg (v 4 ml) nebo 100 mg (v 10 ml).
■ Dalšími složkami jsou dihydrát natrium-citrátu, chlorid sodný (viz bod 2 "OPDIVO obsahuje sodík"), mannitol (E421), kyselina pentetová, polysorbát 80, hydroxid sodný, kyselina chlorovodíková a voda na injekci.
Jak přípravek OPDIVO vypadá a co obsahuje toto balení
OPDIVO koncentrát pro infuzní roztok (sterilní koncentrát) je a čirá až opalizující, bezbarvá až světle žlutá tekutina, která může obsahovat několik světlých částic.
Dodává se v baleních obsahujících jednu lahvičku se 4 ml nebo jednu lahvičku s10 ml.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Bristol-Myers Squibb Pharma EEIG Uxbridge Business Park Sanderson Road Uxbridge UB8 1DH Velká Británie
Výrobce
Bristol-Myers Squibb S.r.l.
Loc. Fontana del Ceraso 03012 Anagni (FR)
Itálie
Pro jakékoli informace o tomto léku kontaktujte prosím místního zástupce držitele rozhodnutí o registraci.
|
Belgique/Belgie/Belgien N.V. Bristol-Myers Squibb Belgium S.A. Tél/Tel: + 32 2 352 76 11 |
Lietuva Bristol-Myers Squibb Gyógyszerkereskedelmi Kft. Tel: + 370 52 369140 |
Etarapnu Luxembourg/Luxemburg
Bristol-Myers Squibb Gyógyszerkereskedelmi Kft. N.V. Bristol-Myers Squibb Belgium S.A Tea.: + 359 800 12 400 Tél/Tel: + 32 2 352 76 11
|
Česká republika Bristol-Myers Squibb spol. s r.o. Tel: + 420 221 016 111 |
Magyarország Bristol-Myers Squibb Gyógyszerkereskedelmi Kft. Tel.: + 36 1 301 9700 |
|
Danmark Bristol-Myers Squibb Tlf: + 45 45 93 05 06 |
Malta Bristol-Myers Squibb S.r.l. Tel: + 39 06 50 39 61 |
|
Deutschland Bristol-Myers Squibb GmbH & Co. KGaA Tel: + 49 89 121 42-0 |
Nederland Bristol-Myers Squibb B.V. Tel: + 31 (0)30 300 2222 |
Eesti Norge
Bristol-Myers Squibb Gyógyszerkereskedelmi Kft. Bristol-Myers Squibb Norway Ltd Tel: + 372 640 1030 Tlf: + 47 67 55 53 50
|
Ekkába Bristol-Myers Squibb A.E. Tr(k: + 30 210 6074300 |
Osterreich Bristol-Myers Squibb GesmbH Tel: + 43 1 60 14 30 |
|
Espaňa Bristol-Myers Squibb, S.A. Tel: + 34 91 456 53 00 |
Polska Bristol-Myers Squibb Polska Sp. z o.o. Tel.: + 48 22 5796666 |
|
France Bristol-Myers Squibb SARL Tél: + 33 (0)1 58 83 84 96 |
Portugal Bristol-Myers Squibb Farmaceutica Portuguesa, S.A. Tel: + 351 21 440 70 00 |
|
Hrvatska Bristol-Myers Squibb spol. s r.o. TEL: +385 1 7100 030 |
Románia Bristol-Myers Squibb Gyógyszerkereskedelmi Kft. Tel: + 40 (0)21 272 16 00 |
Ireland Slovenija
Bristol-Myers Squibb Pharmaceuticals Ltd Bristol-Myers Squibb spol. s r.o.
Tel: + 353 (1 800) 749 749 Tel: + 386 1 2355 100
Slovenská republika
Bristol-Myers Squibb spol. s r.o.
Tel: + 421 2 59298411
Italia
Bristol-Myers Squibb S.r.l.
Tel: + 39 06 50 39 61
Kúrcpog
Bristol-Myers Squibb A.E.
Tnh + 357 800 92666
Latvija
Bristol-Myers Squibb Gyógyszerkereskedelmi Tel: + 371 67708347
Suomi/Finland
Oy Bristol-Myers Squibb (Finland) Ab Puh/Tel: + 358 9 251 21 230
Sverige
Bristol-Myers Squibb AB Tel: + 46 8 704 71 00
United Kingdom
Kft. Bristol-Myers Squibb Pharmaceuticals Ltd Tel: + 44 (0800) 731 1736
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Příprava a podání přípravku OPDIVO
Přípravu musí provádět školený personál v souladu s pravidly správné praxe, zejména s ohledem na aseptickou manipulaci.
Výpočet dávky
Předepsaná dávka pro pacienta se udává v mg/kg. Podle této předepsané dávky vypočítejte celkovou dávku, která se má podat. Pro přípravu celé dávky pro pacienta může být zapotřebí více než jedna lahvička koncentrátu přípravku OPDIVO.
■ Celková dávka nivolumabu v mg = tělesná hmotnost pacienta v kg * předepsaná dávka v mg/kg.
■ Objem koncentrátu přípravku OPDIVO k přípravě dávky (ml) = celková dávka v mg dělená deseti (síla koncentrátu OPDIVO je 10 mg/ml).
Příprava infuze
Když připravujete infuzi, věnujte pozornost zajištění aseptické manipulace. Infuze se má připravovat v digestoři s laminárním prouděním nebo v bezpečnostním boxu pomocí standardních opatřeních pro bezpečné zacházení s intravenózními látkami.
Přípravek OPDIVO lze použít pro intravenózní podání buď:
■ neředěný po přenosu do infuzní nádobky pomocí vhodné sterilní stříkačky; nebo
■ po naředění v koncentraci až do 1 mg/ml. Konečná koncentrace infuze by se měla pohybovat v rozpětí mezi 1 a 10 mg/ml. Koncentrát přípravku OPDIVO se může ředit buď:
■ roztokem chloridu sodného 9 mg/ml (0,9%) pro injekce; nebo
■ roztokem glukózy 50 mg/ml (5%) pro injekce.
KROK 1
■ Prohlédněte koncentrát přípravku OPDIVO, zda neobsahuje částice nebo se nezměnila barva. Lahvičku neprotřepávejte. Koncentrát přípravku OPDIVO je čirá až opalizující, bezbarvá až světle žlutá tekutina. Pokud je roztok zakalený, změnil barvu či pokud obsahuje jiné částice než několik průsvitných až bílých částic, lahvičku zlikvidujte.
■ Odeberte požadovaný objem koncentrátu OPDIVO pomocí vhodné sterilní stříkačky.
KROK 2
■ Vstříkněte koncentrát do sterilní, prázdné skleněné lahvičky nebo intravenózní nádobky (PVC nebo polyolefin).
■ Podle potřeby nařeďte roztokem chloridu sodného 9 mg/ml (0,9%) pro injekce nebo roztokem glukózy 50 mg/ml (5%) pro injekce. Jemně promíchejte infuzi otáčením v ruce. Neprotřepávejte.
Podávání:
Infuze přípravku OPDIVO se nesmí podávat jako intravenózní bolus nebo bolusová injekce.
Podávejte infuzi přípravku OPDIVO intravenózně po dobu 60 minut.
Přípravek OPDIVO se nesmí podávat jako infuze současně ve stejné intravenózní lince s jinými látkami. Pro infuzi použijte oddělenou infuzní linku.
Používejte infuzní set a sériový, sterilní, nepyrogenní filtr s nízkou schopností vázat proteiny (velikost pórů 0,2-1,2 pm).
Infuze přípravku OPDIVO je kompatibilní s:
■ PVC nádobami
■ polyolefmovými nádobami
■ skleněnými lahvemi
■ infuzními sety z PVC
■ sériovými filtry s polyétersulfonovou membránou o velikostí pórů 0,2 pm až 1,2 pm.
Po podání dávky nivolumabu vypláchněte linku roztokem chloridu sodného 9 mg/ml (0,9%) pro injekce nebo roztokem glukózy 50 mg/ml (5%) pro injekce.
Podmínky pro uchovávání a doba použitelnosti:
Neotevřená lahvička
Přípravek OPDIVO musí být uchováván v chladničce (2 °C až 8 °C). Lahvičky musí být uchovávány v původním obalu, aby byl přípravek chráněn před světlem. Přípravek OPDIVO musí být chráněn před mrazem.
Nepoužívejte přípravek OPDIVO po uplynutí doby použitelnosti uvedené na krabičce a štítku lahvičky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Infuze přípravku OPDIVO
Podávání infuze přípravku OPDIVO musí být dokončeno do 24 hodin po přípravě. Není-li roztok použit okamžitě, může být uchován v chladničce v teplotě 2 °C - 8 °C a ochráněn před světlem až 24 hodin (z celkových 24 hodin smí být maximálně 4 hodiny v pokojové teplotě (20 °C - 25 °C) a osvětlené místnosti). Další doba a podmínky skladování po otevření jsou v odpovědnosti uživatele.
Likvidace
Neuchovávejte žádnou nepoužitou část infuzního roztoku pro další použití. Veškerý nepoužitý léčivý přípravek nebo odpadní materiál musí být zlikvidován v souladu s místními požadavky.
59