Omidria 10 Mg/Ml + 3 Mg/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
NÁZEV PŘÍPRAVKU
1.
Omidria 10 mg/ml + 3 mg/ml, koncentrát pro roztok k nitroočnímu výplachu
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Čtyři ml roztoku v injekční lahvičce obsahují phenylephrini hydrochloridum odpovídající phenylephrinum 40,6 mg (10,2 mg/ml) a ketorolacum trometamoli odpovídající ketorolacum 11,5 mg (2,88 mg/ml).
Ponaředění v 500 ml roztoku na výplach obsahuje 1 ml roztoku phenylephrinum 0,081 mg a ketorolacum 0,023 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro roztok k nitroočnímu výplachu.
Čirý bezbarvý až nažloutlý roztok s hodnotou pH 6,3 ± 0,3.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Omidria je indikován u dospělých k udržení intraoperační mydriázy, prevenci intraoperační miózy a snížení akutní pooperační bolesti očí při implantaci nitrooční čočky.
4.2 Dávkování a způsob podání
Přípravek Omidria musí být používán za kontrolovaných operačních podmínek kvalifikovaným očním chirurgem se zkušenostmi s implantací nitroočních čoček.
Dávkování
Doporučená dávka jsou 4,0 ml přípravku Omidria naředěné v 500 ml roztoku na výplach při použití k nitroočním výplachům postiženého oka během chirurgického výkonu.
Návod k naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
Zvláštní populace
Porucha funkce ledvin nebo jater
U pacientů s poruchou funkce ledvin nebo jater nebyly s přípravkem Omidria provedeny žádné oficiální studie. Nepředpokládá se žádná úprava dávky ani zvláštní upozornění u pacientů s poruchou funkce ledvin nebo jater (viz bod 5.2).
Starší lidé
V klinických studiích byla hodnocena i starší populace. Úprava dávkování není nutná.
Pediatrická populace
Bezpečnost a účinnost přípravku Omidria u dětí mladších 18 let nebyla zkoumána. Nej sou dostupné žádné údaje.
Způsob podání Nitrooční podání.
Pouze k jednorázovému použití.
Přípravek Omidria nebyl hodnocen bez použití standardních předoperačních mydriatických a anestetických přípravků. Podle rozhodnutí ošetřujícího oftalmologa mohou být předoperačně podány oční kapky obsahující antibiotika, anestetika, kortikosteroidy, mydriatika a nesteroidní protizánětlivá léčiva (NSAID).
Před podáním léčivého přípravku
Před použitím musí být přípravek Omidria naředěn v 500 ml roztoku na výplach. Pokyny k ředění viz bod 6.6.
Roztok na výplach obsahující přípravek Omidria je určen k použití během chirurgického výkonu stejným způsobem, jako se používá standardní roztok na výplach.
4.3 Kontraindikace
Hypersenzitivita na léčivé látky nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Pacienti s glaukomem s uzavřeným úhlem.
4.4 Zvláštní upozornění a opatření pro použití Tento přípravek musí být před nitroočním podáním zředěn.
Přípravek Omidria je indikován k přidání do roztoku na výplach používaného výhradně při implantaci nitrooční čočky.
Přípravek Omidria není indikován k použití bez ředění, k intravitreální injekci, obecnému topickému očnímu použití ani k systémovému použití mimo oko.
Bezpečnost a účinnost přípravku Omidria nebyly hodnoceny u pacientů s uveitidou, poraněním duhovky v anamnéze, ani při užívání alfa-adrenergních antagonistů.
Při používání přípravku Omidria je třeba vzít v úvahu následující zvláštní upozornění a opatření týkající se topického očního použití fenylefrinu a ketorolaku:
Kardiovaskulární reakce
U pacientů byly při očním použití fenylefrinu hlášeny závažné kardiovaskulární reakce, včetně komorových arytmií a infarktů myokardu. Tyto příhody, z nichž byly některé fatální, se obvykle objevily u pacientů s preexistujícími kardiovaskulárními chorobami.
Po nakapání topického očního fenylefrinu byly hlášeny případy významného zvýšení krevního tlaku. Předpokládaná systémová expozice je minimální a přechodná, nicméně při léčbě pacientů s nedostatečně kontrolovanou hypertenzí je třeba opatrnosti. Riziko zvýšení krevního tlaku může být vyšší u pacientů, u nichž je nutný delší chirurgický výkon.
Před chirurgickým výkonem je třeba řešit hypertyreózu a nestabilní kardiovaskulární onemocnění.
Zkřížená senzitivita.
Existuje potenciální zkřížená senzitivita s kyselinou acetylsalicylovou, deriváty kyseliny fenyloctové a dalšími nesteroidními protizánětlivými léčivy (NSAID). V souvislosti s použitím ketorolaku ve formě očního roztoku byly u pacientů, u nichž je buď známa hypersenzitivita na kyselinu acetylsalicylovou/NSAID, nebo kteří mají v předchozí anamnéze astma, hlášeny případy bronchospasmu nebo exacerbace astmatu. U osob, které dříve vykázaly citlivost na tyto léčivé látky, proto používejte přípravek Omidria s opatrností.
Použití přípravku Omidria během implantace nitrooční čočky může vést k dočasnému zhoršení zraku. (viz bod 4.7).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Nitrooční metabolické interakce jsou nepravděpodobné, protože fenylefrin a ketorolak jsou z přední komory odstraněny výplachy během chirurgického výkonu a normálním oběhem nitrooční tekutiny po chirurgickém výkonu. Rozsah mydriatického účinku přípravku Omidria může být narušen u pacientů, kteří současně užívají léčivé přípravky ovlivňující velikost pupily, jako jsou opioidy (miotika) nebo nesedativní antihistaminika (mydriatika).
Současné používání fenylefrinu a atropinu může u některých pacientů zvýšit presorické účinky a navodit tachykardii. Fenylefrin může potencovat kardiovaskulární depresorické účinky některých inhalačních anestetik. Ve studii, která hodnotila farmakokinetiku přípravku Omidria, byla systémová expozice fenylefrinu i ketorolaku minimální a přechodná. Proto se neočekávají žádné interakce.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku.
Přípravek Omidria není doporučen u žen, které mohou otěhotnět a nepoužívají antikoncepci. Těhotenství
Žádné údaje o podávání přípravku Omidria těhotným ženám nejsou k dispozici. Přípravek Omidria se nedoporučuje podávat během těhotenství.
Kojení
Není známo, zda se fenylefrin vylučuje do mateřského mléka. Ketorolak se po systémovém podání vylučuje do mateřského mléka. Riziko pro kojené dítě nelze vyloučit. Přípravek Omidria nemá být používán v těhotenství a během kojení.
Fertilita
Nejsou k dispozici žádné adekvátní údaje o účincích podávání fenylefrin-hydrochloridu nebo ketorolak-trometamolu na fertilitu u člověka.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Omidria může mít velký vliv na schopnost řídit nebo obsluhovat stroje. Vzhledem k tomu, že po implantaci nitrooční čočky může u pacientů, u nichž byl podán přípravek Omidria, dojít k dočasnému zhoršení zraku, je třeba pacienty poučit, že nemají řídit ani obsluhovat stroje, dokud nebudou vidět jasně. Další podrobnosti týkající se možných poruch zraku uvádí bod 4.8.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnostní profil přípravku Omidria vychází z údajů od 459 dospělých pacientů, které byly získány během klinického vývoje v randomizovaných kontrolovaných studiích. Nežádoucí účinky hlášené u pacientů, kteří používali přípravek Omidria, byly typické pro pooperační stav a většinou byly mírné až střední závažnosti a odezněly bez intervence nebo bez jakýchkoliv následků. Nejčastěji hlášenými nežádoucími účinky byly bolest oka (4,8 %), zánět přední komory (3,9 %), hyperemie spojivky (2,2 %), fotofobie (1,7 %), edém rohovky (1,3 %) a zánět (1,3 %). Každý z těchto účinků byl hlášen s podobnou frekvencí i u pacientů s placebem.
Tabulkový seznam nežádoucích účinků
Frekvence nežádoucích účinku jsou definovány následovně: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Časté |
Méně časté |
|
Poruchy nervového systému | ||
|
Poruchy oka |
Bolest oka Zánět přední komory Hyperemie spojivky Edém rohovky Fotofobie |
Oční diskomfort Zánět oka Podráždění oka Edém spojivky Porucha rohovky Mydriáza Rozmazané vidění Snížená ostrost zraku Sklivcové zákaly Svědění očí Bolest očních víček Pocit cizího tělesa v oku Oslnění Zvýšený nitrooční tlak |
|
Gastrointestinální poruchy | ||
|
Celkové poruchy a reakce v místě aplikace |
Zánět |
Bolest |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě náhodné nitrokomorové injekce koncentrovaného roztoku je třeba přední komoru okamžitě vyprázdnit a vypláchnout standardním oftalmologickým roztokem na výplach.
Systémové předávkování fenylefrinem může vést k rychlému zvýšení krevního tlaku. Může také vyvolat bolest hlavy, úzkost, nauzeu a zvracení a komorové arytmie. V případě předávkování fenylefrinem je doporučeno okamžité podání injekce rychle působícího alfa-adrenergního blokátoru, jako je fentolamin.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: {dosud nepřidělena}, ATC kód: {dosud nepřidělen} Mechanismus účinku
Fenylefrin a ketorolak v přípravku Omidria působí odlišnými mechanismy. Udržují intraoperační mydriázu, předcházejí intraoperační mióze a snižují akutní pooperační bolest. Fenylefrin je agonista a1-adrenergních receptorů a působí jako mydriatikum kontrakce radiálního svalu duhovky a dilatací pupily, nanejvýš s malou nebo žádnou cykloplegií. V cirkulaci spojivky a dalších cévách oka nastává vazokonstrikce takového rozsahu, jaký odpovídá expozici léku.
Ketorolak je nesteroidní protizánětlivé léčivo (NSAID), které inhibuje oba enzymy cyklooxygenázy (COX1 a COX2), zmírňuje bolest a zánět snížením tkáňové koncentrace prostaglandinů v tkáni, která je důsledkem chirurgického traumatu. Ketorolak může také sekundárně přispívat k prevenci chirurgicky navozené miózy díky inhibici syntézy prostaglandinů v důsledku chirurgického poranění oka nebo přímé mechanické stimulace duhovky.
Klinická účinnost a bezpečnost
Účinnost a bezpečnost přípravku Omidria byla hodnocena ve dvou randomizovaných multicentrických placebem kontrolovaných dvojitě zaslepených klinických studiích fáze III u 808 dospělých pacientů podstupujících implantaci nitrooční čočky. Věk populace ve studiích se pohyboval od 26 do 90 let (59 % žen, 41 % mužů; 80 % osob bílé rasy, 12 % osob černé rasy a 8 % osob jiné rasy). Devatenáct procent případů katarakty bylo stupně 2 nebo 3 dle nukleárního gradingu LOCS II. 53 % pacientů mělo hnědé duhovky, 28 % modré duhovky a 19 % duhovky jiné barvy.
Pacienti byli randomizováni k používání přípravku Omidria nebo placeba (1:1). Všichni pacienti byli léčeni standardizovanými předoperačními topickými mydriatiky a anestetiky. Během celého chirurgického výkonu byl měřen průměr pupily. Pooperační bolest byla hodnocena pomocí samostatně vyplňované vizuální analogové škály (VAS) 0-100 mm.
Statistické hodnocení změny průměru pupily oproti vstupní hodnotě (mm) během chirurgického výkonu bylo provedeno pomocí Cochran-Mantel-Haenszelova (CMH) testu s úpravou na znáhodnění ve stratech. V 1. studii byl průměrný vážený rozdíl (Omidria - placebo) průměrné plochy pod křivkou (AUC) 0,58 mm [95% interval spolehlivosti: 0,48, 0,68] (P < 0,0001). Ve 2. studii byl průměrný vážený rozdíl (Omidria - placebo) průměrné AUC dle CMH 0,59 mm [95% interval spolehlivosti: 0,49, 0,69] (P < 0,0001).
Ve skupině léčené přípravkem Omidria přetrvávala mydriáza, zatímco ve skupině s placebem došlo k progresivní konstrikci pupily (viz obrázek).
Intraoperační změna průměru pupily (mm) oproti vstupní hodnotě
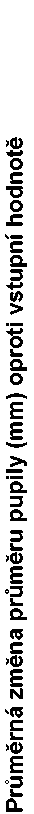
Minuty od iniciální chirurgické incize
Prevence miózy byla potvrzena v kategoriální analýze. V 1. studii měla v době odsátí hmot kortexu průměr pupily < 6 mm pouze 4 % pacientů ve skupině s přípravkem Omidria v porovnání s 23 % pacientů ve skupině s placebem a konstrikci pupily > 2,5 mm 3 % pacientů ve skupině s přípravkem Omidria v porovnání s 28 % pacientů ve skupině s placebem (p < 0,0001 v obou případech, chí-kvadrát test). Ve 2. studii měla při odsátí hmot kortexu průměr pupily < 6 mm pouze 4 % pacientů ve skupině s přípravkem Omidria v porovnání s 23 % pacientů ve skupině s placebem a konstrikci pupily > 2,5 mm 1 % pacientů ve skupině s přípravkem Omidria v porovnání s 27 % pacientů ve skupině s placebem (p < 0,0001, chí-kvadrát test).
|
Placebo |
Přípravek Omidria | |
|
1. studie |
N = 201 |
N = 201 |
|
Analyzovaný soubor (n) |
(n=180) |
(n=184) |
|
Změna AUC průměru pupily (mm) během chirurgického výkonu oproti vstupní hodnotě (ko-primární cílový parametr) [průměr (SD)] |
-0,5 (0,58) |
0,1 (0,41) |
|
Průměr < 6 mm kdykoliv |
85 (47 %) |
19(10%) |
|
Průměr < 6 mm při odsátí hmot kortexu |
41 (23 %) |
7 (4 %) |
|
Konstrikce pupily > 2,5 mm |
50 (28 %) |
6 (3 %) |
|
2. studie |
N = 204 |
N = 202 |
|
Analyzovaný soubor (n) |
(n=200) |
(n=195) |
|
Změna AUC průměru pupily (mm) během chirurgického výkonu oproti vstupní hodnotě (ko-primární cílový parametr) [průměr (SD)] |
-0,5 (0,57) |
0,1 (0,43) |
|
Průměr < 6 mm kdykoliv |
76 (38 %) |
18 (9 %) |
|
Průměr < 6 mm při odsátí hmot kortexu |
46 (23 %) |
8 (4 %) |
|
Konstrikce pupily > 2,5 mm |
53 (27 %) |
2 (1 %) |
Během prvních 10 až 12 hodin po chirurgickém výkonu bylo také prokázáno významné zmírnění bolesti oka. Statistické hodnocení bolesti určené podle 100mm VAS bylo provedeno pomocí CMH testu s úpravou na znáhodnění ve stratech. V 1. studii byl průměrný vážený rozdíl (Omidria - placebo) průměrné AUC dle CMH -5,20 mm [95% interval spolehlivosti: -7,31, -3,09] (P < 0,001). Ve 2. studii byl průměrný vážený rozdíl (Omidria - placebo) průměrné AUC dle CMH -4,58 mm [95% interval spolehlivosti: -6,92, -2,24] (P < 0,001).
|
Placebo |
Přípravek Omidria | |
|
1. studie |
N = 201 |
N = 201 |
|
Analyzovaný soubor (n) |
(n=201) |
(n=201) |
|
AUC 12hodinové bolest oka dle skóre VAS (ko-primární cílový parametr) [průměr±SD] |
9,2 ± 12,9 |
4,1 ± 8,07 |
|
Subjekty s VAS = 0 po celou dobu |
28 (14%) |
48 (24%) |
|
Subjekty s VAS > 40 kdykoliv |
30 (15%) |
13 (7%) |
|
2. studie |
N = 204 |
N = 202 |
|
Analyzovaný soubor (n) |
(n=202) |
(n=202) |
|
AUC 12hodinové bolest oka dle skóre VAS (ko-primární cílový parametr) [průměr±SD] |
8,9 ± 15,19 |
4,3 ± 8,75 |
|
Subjekty s VAS = 0 po celou dobu |
41 (20 %) |
56 (28 %) |
|
Subjekty s VAS > 40 kdykoliv |
27 (13 %) |
16 (8 %) |
Histologické vyšetření v neklinických toxikologických studiích neprokázalo žádné účinky na rohovku související s léčbou a v klinických studiích s přípravkem Omidria nebyly pozorovány žádné nepříznivé účinky na nejlépe korigovanou zrakovou ostrost (BCVA). V průběhu klinických studií nebyl zjišťován počet endoteliálních buněk.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Omidria u jedné nebo více podskupin pediatrické populace u terapeutických výkonů na čočce (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Ve studii, která hodnotila farmakokinetiku přípravku Omidria, byla systémová expozice fenylefrinu i ketorolaku minimální a přechodná.
Detekovatelná plazmatická koncentrace fenylefrinu byla zjištěna pouze u jednoho ze 14 pacientů. Maximální koncentrace pozorovaná u tohoto pacienta byla 1,7 ng/ml a objevila se po nakapání topických předoperačních kapek fenylefrinu ještě před expozicí přípravku Omidria.
Plazmatická koncentrace ketorolaku byla detekována u 11 ze 14 pacientů. Maximální zjištěná koncentrace ketorolaku byla 4,2 ng/ml.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje uváděné v literatuře týkající se jednotlivých složek přípravku Omidria získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity, kancerogenního potenciálu a reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka.
Toxikologická studie s jedinou dávkou byla provedena u afrických zelených opic s expozicí očním roztokům na výplach obsahujícím kombinaci fenylefrinu a ketorolaku použitým během implantace nitrooční čočky. Při podání kombinace fenylefrinu a ketorolaku v roztoku na výplach o koncentraci až 7 200 ^M fenylefrinu a 900 ^M ketorolaku nebyly pozorovány žádné nežádoucí příhody související s lékem ani patologické nálezy. Tyto koncentrace jsou více než 10krát vyšší než koncentrace každé z látek podávaných klinicky u pacientů léčených přípravkem Omidria.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Monohydrát kyseliny citronové, dihydrát natrium-citrátu, hydroxid sodný (k úpravě pH), kyselina chlorovodíková (k úpravě pH), voda na injekci.
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Neotevřené: 18 měsíců
Po otevření musí být přípravek okamžitě naředěn.
Po naředění byla chemická a fyzikální stabilita prokázána po dobu 6 hodin při 25 °C. Spotřebujte do 6 hodin po naředění. Z mikrobiologického hlediska by měl být přípravek použit okamžitě. Není-li použit okamžitě, jsou doba a podmínky uchovávání po otevření před použitím v odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 25 °C.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Po naředění: Uchovávejte při teplotě do 25 °C.
6.5 Druh obalu a obsah balení
Bezbarvá 5ml skleněná injekční lahvička třídy I se zátkou z butylové pryže a polypropylenovým odtrhovacím víčkem. Každá injekční lahvička je samostatně zabalená v krabičce.
Vícečetné balení (1 balení po 10 ks) obsahuje 10 injekčních lahviček k jednomu použití.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek Omidria se připravuje k nitroočnímu výplachu naředěním 4,0 ml koncentrátu v 500 ml standardního oftalmologického roztoku na výplach.
Je třeba dodržet následující pokyny:
- Injekční lahvičku je třeba vizuálně zkontrolovat, zda neobsahuje pevné částice. Použít lze pouze čirý, bezbarvý až nažloutlý roztok bez viditelných částic.
- Za aseptických podmínek odeberte 4,0 ml koncentrovaného roztoku pomocí vhodné sterilní jehly.
- 4,0 ml koncentrovaného roztoku je třeba vstříknout do 500ml vaku/lahve s roztokem na výplach.
- Vak je třeba jemně převrátit, aby se roztok promíchal. Roztok je nutné podat do 6 hodin od přípravy.
- Vak je nutné vizuálně zkontrolovat, zda neobsahuje pevné částice. Použít lze pouze čirý, bezbarvý roztok bez viditelných částic.
- Do připraveného roztoku na výplach se nemají přidávat žádné jiné léčivé přípravky.
Použitou lahvičku a veškerý nespotřebovaný roztok na výplach je po jednom použití nutné zlikvidovat podle místních požadavků.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Omeros London Limited
Berkeley Square
London, W1J 6BD
Spojené království
Tel.: +44 (0) 20 7887 6296
Fax: +44 (0) 20 7887 6001
e-mail: regulatory@omeros.co.uk
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1018/001
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace: <datum rozhodnutíEU>
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ / VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ / VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného / výrobců odpovědných za propouštění šarží
Almac Pharma Services Limited Seagoe Industrial Estate, Craigavon, Co. Armagh BT63 5QD Severní Irsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Omidria 10 mg/ml + 3 mg/ml, koncentrát pro roztok k nitroočnímu výplachu phenylephrinum/ketorolacum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Čtyři ml roztoku v lahvičce obsahují phenylephrinum 40,6 mg (10,2 mg/ml) jako phenylephrini hydrochloridum a ketorolacum 11,5 mg (2,88 mg/ml) jako ketorolacum trometamoli.
Po naředění obsahuje 1 ml roztoku phenylephrinum 0,081 mg a ketorolacum'0,023 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: monohydrát kyseliny citronové, dihydrát natrium-citrátu, hydroxid sodný / kyselina chlorovodíková (pro úpravu pH), voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro roztok k nitroočmmu výplachu Vícečetné balení: 10 injekčních lahviček (1 balení po 10 ks).
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze k jednorázovému použití.
Před použitím si přečtěte příbalovou informaci. Nitrooční podání po naředění.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Spotřebujte bezprostředně po naředění.
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 25 °C.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Omeros London Limited London, W1J 6BD Spojené království
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1018/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Omidria 10 mg/ml + 3 mg/ml, koncentrát pro roztok k nitroočnímu výplachu phenylephrinum/ketorolacum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Čtyři ml roztoku v lahvičce obsahují phenylephrinum 40,6 mg (10,2 mg/ml) jako phenylephrini hydrochloridum a ketorolacum 11,5 mg (2,88 mg/ml) jako ketorolacum trometamoli.
Po naředění obsahuje 1 ml roztoku phenylephrinum 0,081 mg a ketorolacum 0,023 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: monohydrát kyseliny citronové, dihydrát natrium-citrátu, hydroxid sodný / kyselina chlorovodíková (pro úpravu pH), voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro roztok k nitroočnímu výplachu
1 injekční lahvička. Součást vícečetného balení, nelze prodávat samostatně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze k jednorázovému použití.
Před použitím si přečtěte příbalovou informaci. Nitrooční podání po naředění.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED a DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Spotřebujte bezprostředně po naředění.
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 25 °C.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Omeros London Limited London, W1J 6BD Spojené království
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1018/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Omidria 10 mg/ml + 3 mg/ml, koncentrát pro roztok k nitroočnímu výplachu phenylephrinum/ketorolacum
2. ZPŮSOB PODÁNÍ
Nitrooční podání po naředění.
Pouze k jednorázovému použití.
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Omidria 10 mg/ml + 3 mg/ml, koncentrát pro roztok k nitroočnímu výplachu
phenylephrinum/ketorolacum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Omidria a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Omidria používat
3. Jak se přípravek Omidria používá
4. Možné nežádoucí účinky
5. Jak přípravek Omidria uchovávat
6. Obsah balení a další informace
1. Co je přípravek Omidria a k čemu se používá
Omidria je léčivý přípravek používaný během chirurgického výkonu na oku. Obsahuje léčivé látky fenylefrin a ketorolak. Fenylefrin působí tak, že udržuje rozšířenou zornici. Ketorolak je lék proti bolesti, který patří do skupiny zvané nesteroidní protizánětlivá léčiva (NSAID); napomáhá také prevenci zúžení zornice.
Přípravek Omidria se používá u dospělých k výplachu oka během chirurgické implantace nové čočky (části oka, která zaostřuje světlo procházející zornicí a umožňující ostré vidění). Tento výkon se nazývá implantace nitrooční čočky. Léčivý přípravek se používá k udržení rozšířené zornice během chirurgického výkonu a ke zmírnění bolesti oka po výkonu.
2. Čemu musíte věnovat pozornost, než začnete přípravek Omidria používat Přípravek Omidria se nesmí použít:
- jestliže j ste alergický(á) na fenylefrin nebo ketorolak nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6);
- jestliže máte onemocnění zvané glaukom s uzavřeným úhlem.
Upozornění a opatření
Před použitím přípravku Omidria se poraďte se svým lékařem nebo zdravotní sestrou, jestliže:
- máte onemocnění srdce,
- máte zvýšený krevní tlak,
- máte zvýšenou činnost štítné žlázy (hypertyreózu),
- máte alergii na kyselinu acetylsalicylovou nebo jiné léky proti bolesti zvané nesteroidní protizánětlivá léčiva (NSAID),
- máte astma.
Pokud se vás týká cokoliv z výše uvedeného, oznamte to prosím svému lékaři. Váš lékař rozhodne, zda je pro vás přípravek Omidria vhodný.
Děti a dospívající
Přípravek Omidria se nemá používat u dětí a dospívajících do 18 let věku, protože nebyl u těchto skupin populace hodnocen.
Další léčivé přípravky a přípravek Omidria
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
- Zejména sdělte lékaři, pokud používáte atropin, léčivý přípravek používaný k rozšíření zornice. Použití atropinu spolu s přípravkem Omidria může u některých pacientů zvýšit krevní tlak
a zrychlit činnost srdce.
- Jedna z léčivých látek v přípravku Omidria může reagovat s několika typy anestetik (znecitlivujících přípravků). Váš lékař o tom bude vědět. Pokud budete při očním chirurgickém výkonu v celkové anestezii, promluvte si o tom se svým lékařem.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete vám je tento přípravek podán.
Přípravek Omidria se nesmí používat v těhotenství. Pokud můžete otěhotnět, je třeba, abyste před použitím přípravku Omidria používala vhodnou antikoncepci.
Přípravek Omidria nemá být používán v těhotenství a během kojení.
Řízení dopravních prostředků a obsluha strojů
Tento přípravek může mít velký vliv na schopnost řídit nebo obsluhovat stroje. Vzhledem k tomu, že u vás může dojít ke zhoršení zraku, neřiďte ani neobsluhujte stroje, dokud nezačnete vidět ostře.
3. Jak se přípravek Omidria používá
Přípravek Omidria vám podá v nemocnici nebo na klinice kvalifikovaný lékař nebo operatér, který se specializuje na oční chirurgii.
Přípravek Omidria se používá jako roztok k výplachu očí během chirurgického výkonu, při kterém se nahrazuje čočka.
Jestliže jste dostal(a) více přípravku Omidria, než jste měl(a)
Fenylefrin, jedna z léčivých látek přípravku Omidria, může vyvolat rychlé zvýšení krevního tlaku, jestliže je podán v příliš velkém množství a pronikne do krve v dostatečném množství na to, aby ovlivnil i jiné části těla. Může rovněž vyvolat bolest hlavy, úzkost, pocit na zvracení, zvracení a neobvykle rychlou srdeční frekvenci.
Lékař u vás bude sledovat všechny známky a příznaky nežádoucích účinků a bude je v případě potřeby léčit.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Níže uvedené nežádoucí účinky jsou obvykle mírné až středně závažné intenzity a většinou samy odezní bez dlouhodobých následků.
Nežádoucí účinky postihující oko:
Časté nežádoucí účinky (mohou postihnout až 1 osobu z 10):
- bolest oka,
- zánět přední části oka,
- zarudnutí oka,
- otok rohovky (průhledné vrstvy překrývající přední část oka),
- citlivost na světlo.
Méně časté nežádoucí účinky (mohou postihnout až 1 osobu ze 100):
- nepříj emný pocit v oku,
- zánět oka,
- podráždění oka,
- zarudnutí oka,
- potíže s rohovkou,
- rozšíření zornice,
- rozmazané vidění,
- snížení ostrosti zraku,
- malé tmavé stíny pohybující se v zorném poli,
- svědění oka,
- bolest očních víček,
- pocit cizího tělíska v oku,
- oslnění,
- zvýšený nitrooční tlak.
Nežádoucí účinky postihující celý organismus:
Časté nežádoucí účinky:
- zánět.
Méně časté nežádoucí účinky:
- pocit na zvracení,
- bolest,
- bolest hlavy.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Omidria uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku lahvičky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Neuchovávejte při teplotě nad 25 °C. Uchovávejte injekční lahvičku v původním obalu, aby byl přípravek chráněn před světlem.
Nepoužívejte, pokud je roztok zakalený nebo pokud obsahuje částice.
Naředěný roztok se má použít do 6 hodin po naředění.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Omidria obsahuje
Léčivými látkami jsou phenylephrinum jako phenylephrini hydrochloridum a ketorolacum jako ketorolacum trometamoli.
Čtyři ml roztoku v lahvičce obsahují phenylephrinum 40,6 mg (10,2 mg/ml) a ketorolacum 11,5 mg (2,88 mg/ml).
Dalšími složkami jsou:
- monohydrát kyseliny citronové,
- dihydrát natrium-citrátu,
- hydroxid sodný (k úpravě pH),
- kyselina chlorovodíková (k úpravě pH),
- voda na injekci.
Jak přípravek Omidria vypadá a co obsahuje toto balení
Čirý bezbarvý až nažloutlý sterilní koncentrát pro roztok k nitroočnímu výplachu.
Dodává se v injekčních lahvičkách k jednomu použití a je určen k podání 4,0 ml koncentrátu naředěného v 500 ml roztoku na výplach pro oční podání. Bezbarvá 5ml skleněná lahvička třídy I se zátkou z butylové pryže a polypropylenovým odtrhovacím víčkem.
Vícečetné balení obsahuje 10 krabiček, jedna krabička obsahuje jednu injekční lahvičku k jednomu použití.
Držitel rozhodnutí o registraci
Omeros London Limited
Berkeley Square
London, W1J 6BD
Spojené království
Tel.: +44 (0) 20 7887 6296
Fax: +44 (0) 20 7887 6001
E-mail: regulatorv@,omeros.co.uk
Výrobce
Almac Pharma Services Limited Seagoe Industrial Estate, Craigavon, Co. Armagh BT63 5QD Severní Irsko
Další informace o tomto přípravku získáte u držitele rozhodnutí o registraci.
Tato příbalová informace byla naposledy revidována <datum rozhodnutíKomise>.
Přípravek Omidria se připravuje k nitroočnímu výplachu naředěním 4,0 ml koncentrátu přípravku
Omidria v 500 ml standardního oftalmologického roztoku na výplach.
Je třeba dodržet následující pokyny:
- Injekční lahvičku je třeba vizuálně zkontrolovat, zda neobsahuje pevné částice. Použít lze pouze čirý, bezbarvý až nažloutlý roztok bez viditelných částic.
- Za aseptických podmínek odeberte 4,0 ml koncentrovaného roztoku pomocí vhodné sterilní jehly.
- 4,0 ml koncentrovaného roztoku je třeba injikovat do 500ml vaku/lahve s roztokem na výplach.
- Vak je třeba jemně převrátit, aby se roztok promíchal. Roztok je nutné podat do 6 hodin od přípravy.
- Vak je nutné vizuálně zkontrolovat, zda neobsahuje pevné částice. Použít lze pouze čirý, bezbarvý roztok bez viditelných částic.
- Do připraveného roztoku na výplach se nemají přidávat žádné jiné léčivé přípravky.
Použitou injekční lahvičku a veškerý nespotřebovaný roztok na výplach je po jednom použití nutné
zlikvidovat podle místních požadavků.