Ofev 100 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Ofev 100 mg měkké tobolky Ofev 150 mg měkké tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tobolka obsahuje nintedanibum 100 mg (ve formě nintedanibi esilas) Jedna tobolka obsahuje nintedanibum 150 mg (ve formě nintedanibi esilas)
Pomocné látky se známým účinkem:
Jedna tobolka obsahuje 1,2 mg sójového lecithinu.
Jedna tobolka obsahuje 1,8 mg sójového lecithinu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Měkká tobolka.
Ofev 100 mg měkké tobolky jsou neprůhledné, oválné, měkké želatinové tobolky broskvové barvy s černým potiskem na jedné straně - symbolem společnosti Boehringer Ingelheim a „100“.
Ofev 150 mg měkké tobolky jsou neprůhledné, oválné, měkké želatinové tobolky hnědé barvy s černým potiskem na jedné straně - symbolem společnosti Boehringer Ingelheim a „150“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Ofev je indikován k léčbě dospělých s idiopatickou plicní fibrózou (IPF).
4.2 Dávkování a způsob podání
Léčbu přípravkem Ofev mohou zahájit pouze lékaři, kteří mají zkušenosti s diagnózou a léčbou IPF. Dávkování
Doporučená dávka je 150 mg nintedanibu dvakrát denně podaných s odstupem přibližně 12 hodin. Dávku 100 mg dvakrát denně je doporučeno používat pouze u pacientů, kteří netolerují dávku 150 mg dvakrát denně.
Jestliže dojde k vynechání dávky, je třeba podávání obnovit podáním doporučené dávky v následující plánovaný termín. Jestliže dojde k vynechání dávky, pacient nesmí užít dodatečnou dávku. Doporučená maximální denní dávka 300 mg nesmí být překročena.
Úpravy dávky
Kromě případné symptomatické léčby by zvládání nežádoucích účinků přípravku Ofev (viz body 4.4 a 4.8) mohlo zahrnovat snížení dávky a dočasné přerušení do doby, než je příslušný nežádoucí účinek zvládnut do té míry, že lze pokračovat v léčbě. V léčbě přípravkem Ofev lze pokračovat podáváním plné dávky (150 mg dvakrát denně) nebo snížené dávky (100 mg dvakrát denně). Pokud pacient netoleruje dávku 100 mg dvakrát denně, léčbu přípravkem Ofev je třeba ukončit.
V případě přerušení z důvodu zvýšených hladin aspartátaminotransferázy (AST) nebo alaninaminotransferázy (ALT) > 3x horní limit normy (upper limit of normal, ULN) lze při návratu hladin transamináz na výchozí hodnotu obnovit léčbu přípravkem Ofev podáním snížené dávky (100 mg dvakrát denně) a dávku následně zvýšit na plnou výši (150 mg dvakrát denně) (viz body 4.4 a 4.8).
Zvláštní populace
Starší pacienti (> 65 let)
U starších pacientů nebyly zjištěny rozdíly v bezpečnosti a účinnosti. Dávku není třeba a priori upravovat na základě věku. U pacientů ve věku >75 let existuje větší pravděpodobnost, že ke zvládnutí nežádoucích účinků bude třeba dávku snížit (viz bod 5.2).
Porucha funkce ledvin
Ledvinami je vyloučeno méně než 1 % jednorázové dávky nintedanibu (viz bod 5.2). U pacientů s mírnou nebo středně těžkou poruchou funkce ledvin není třeba upravovat úvodní dávku. Bezpečnost, účinnost a farmakokinetika nintedanibu nebyla studována u pacientů s těžkou poruchou funkce ledvin (clearance kreatininu < 30 ml/min).
Porucha funkce jater
Nintedanib je vylučován převážně žlučí a stolicí (> 90 %). U pacientů s poruchou funkce jater se expozice zvýšila (Child Pugh A, Child Pugh B; viz bod 5.2). Na základě klinických údajů není u pacientů s lehkou poruchou funkce jater třeba upravovat úvodní dávku (Child Pugh A; viz bod 4.4). Bezpečnost a účinnost nintedanibu nebyla studována u pacientů s poruchou funkce jater klasifikovanou jako Child Pugh B a C. Léčba pacientů se středně těžkou (Child Pugh B) nebo těžkou (Child Pugh C) poruchou funkce jater přípravkem Ofev se nedoporučuje (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Ofev u dětí ve věku 0-18 let nebyla dosud stanovena. Nejsou k dispozici žádné údaje.
Způsob podání
Přípravek Ofev je určen k perorálnímu podání. Tobolky je třeba užívat s jídlem, spolknout celé s vodou a nežvýkat ani nedrtit.
4.3 Kontraindikace
Hypersenzitivita na nintedanib, arašídy nebo sóju nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Gastrointestinální poruchy
Průjem byl ve studiích INPULSIS (viz bod 5.1) nejčastějším gastrointestinálním nežádoucím účinkem a byl hlášen u 62,4 % pacientů léčených přípravkem Ofev oproti 18,4 % pacientů, jimž bylo podáváno placebo (viz bod 4.8). U většiny pacientů se jednalo o nežádoucí účinek mírné až střední intenzity a objevoval se během prvních 3 měsíců léčby. Průjem vedl ke snížení dávky u 10,7 % pacientů a k ukončení podávání nintedanibu u 4,4 % pacientů.
Průjem je třeba při prvních příznacích léčit adekvátní hydratací a léčivými přípravky proti průjmu, např. loperamidem, přičemž může být nutné přerušit léčbu. V léčbě přípravkem Ofev lze pokračovat podáváním snížené dávky (100 mg dvakrát denně) nebo plné dávky (150 mg dvakrát denně). Pokud těžký průjem přetrvává navzdory symptomatické léčbě, je třeba léčbu přípravkem Ofev ukončit.
Nauzea a zvracení byly často hlášenými gastrointestinálními nežádoucími účinky (viz bod 4.8).
U většiny pacientů byly nauzea a zvracení mírné až střední intenzity. Nauzea vedla k ukončení podávání nintedanibu u 2,0 % pacientů. Zvracení vedlo k ukončení podávání u 0,8 % pacientů.
Pokud potíže přetrvávají i přes odpovídající podpůrnou péči (včetně antiemetické léčby), může být nutné snížit dávku nebo léčbu přerušit. V léčbě lze pokračovat podáváním snížené dávky (100 mg dvakrát denně) nebo plné dávky (150 mg dvakrát denně). Pokud závažné příznaky přetrvávají, je třeba léčbu přípravkem Ofev ukončit.
Funkce jater
Bezpečnost a účinnost přípravku Ofev nebyla studována u pacientů se středně těžkou (Child Pugh B) nebo těžkou (Child Pugh C) poruchou funkce jater. Léčba přípravkem Ofev se proto u těchto pacientů nedoporučuje (viz bod 4.2). Vzhledem ke zvýšené expozici se u pacientů s lehkou poruchou funkce jater může zvýšit riziko nežádoucích účinků (Child Pugh A, viz body 4.2 a 5.2).
Podávání nintedanibu bylo spojeno se zvýšením hladin jaterních enzymů (ALT, AST, alkalická fosfatáza (ALP), gamma-glutamyl transferáza (GMT)) s potenciálně vyšším rizikem u pacientek. Zvýšení hladin transamináz bylo reverzibilní při snížení dávky nebo přerušení léčby. Podávání nintedanibu bylo též spojeno se zvýšením hladiny bilirubinu. Hladiny jaterních transamináz a bilirubinu je třeba vyšetřit před zahájením léčby přípravkem Ofev a poté v pravidelných intervalech (např. při každé kontrole pacienta) nebo dle klinické indikace. Jsou-li zjištěny zvýšené hladiny transamináz (AST nebo ALT) > 3x ULN, doporučuje se snížit dávku nebo přerušit léčbu přípravkem Ofev a pacienta je třeba pečlivě sledovat. Při návratu hladin transamináz na výchozí hodnotu lze v léčbě přípravkem Ofev pokračovat podáváním plné dávky (150 mg dvakrát denně) nebo léčbu obnovit podáváním snížené dávky (100 mg dvakrát denně), kterou lze následně zvýšit na plnou výši (viz bod 4.2). Pokud se kterékoli zvýšení hodnot jaterních testů pojí s klinickými příznaky poškození jater, např. žloutenkou, je léčbu přípravkem Ofev třeba trvale ukončit. Je třeba vyšetřit jiné možné příčiny zvýšení hladin jaterních enzymů.
Krvácení
Inhibice receptoru pro vaskulárni endoteliální růstový faktor (VEGFR) může být spojena se zvýšeným rizikem krvácení. Ve studiích INPULSIS byla četnost krvácivých nežádoucích účinků mírně vyšší u pacientů ve skupině léčené přípravkem Ofev (10,3 %) než ve skupině s placebem (7,8 %).
Nezávažná epistaxe byla nejčastější krvácivou příhodou. Četnost závažných krvácivých příhod byla u obou léčebných skupin nízká a vzájemně si podobná (placebo: 1,4 %; Ofev: 1,3 %).
Pacienti se známým rizikem krvácení, včetně pacientů s dědičnou predispozicí ke krvácení nebo pacientů, kterým byly podávány plné dávky antikoagulační léčby, nebyli do studií INPULSIS zařazeni. Po uvedení přípravku na trh byly hlášeny případy hemoragie (bez ohledu na to, zda pacienti užívali nebo neužívali antikoagulancia nebo jiné léčivé přípravky, které mohou způsobovat krvácení). Tyto pacienty lze proto přípravkem Ofev léčit pouze tehdy, jestliže předpokládaný přínos převažuje nad možným rizikem.
Arteriální tromboembolické příhody
Pacienti s nedávnou anamnézou infarktu myokardu nebo cévní mozkové příhody byli ze studií INPULSIS vyřazeni. Arteriální tromboembolické příhody byly hlášeny s nízkou četností: u 0,7 % pacientů ve skupině s placebem a u 2,5 % pacientů ve skupině s nintedanibem. Ačkoli nežádoucí příhody odrážející ischemickou chorobu srdeční byly mezi skupinami s nintedanibem a placebem vyrovnané, ve skupině s nintedanibem prodělalo infarkt myokardu vyšší procento pacientů (1,6 %) než ve skupině s placebem (0,5 %). Opatrnosti je zapotřebí při léčbě pacientů se zvýšeným kardiovaskulárním rizikem, včetně pacientů se známou koronární arteriální nemocí. U pacientů, u kterých dojde k rozvoji příznaků akutní ischemie myokardu, je třeba zvážit přerušení léčby.
Venózní tromboembolie
Ve studiích INPULSIS nebylo u pacientů léčených nintedanibem zjištěno žádné zvýšení rizika venózní tromboembolie. Vzhledem k mechanismu účinku nintedanibu se u pacientů může zvýšit riziko tromboembolických příhod.
Gastrointestinální perforace
Ve studiích INPULSIS nebylo u pacientů léčených nintedanibem zjištěno žádné zvýšení rizika gastrointestinální perforace. Vzhledem k mechanismu účinku nintedanibu mohou mít pacienti zvýšené riziko gastrointestinální perforace. Obzvláštní péče je zapotřebí při léčbě pacientů po předchozí operaci břicha. Léčbu přípravkem Ofev je možné zahájit nejdříve 4 týdny po operaci břicha.
U pacientů, u kterých dojde ke gastrointestinální perforaci, je třeba léčbu přípravkem Ofev trvale ukončit.
Hypertenze
Podávání přípravku Ofev může zvýšit krevní tlak. Systémový krevní tlak je třeba měřit pravidelně a dle klinické indikace.
Komplikace při hojení ran
Ve studiích INPULSIS nebyla zjištěna zvýšená frekvence narušeného hojení ran. Na základě mechanismu účinku může nintedanib narušit hojení ran. Nebyly provedeny žádné studie, které by zkoumaly specificky účinek nintedanibu na hojení ran. Léčbu přípravkem Ofev je proto možné zahájit nebo - v případě perioperativního přerušení - obnovit pouze na základě klinického zvážení adekvátního hojení ran.
Společné podávání s pirfenidonem
Souběžná léčba nintedanibem a pirfenidonem byla zkoumána u japonských pacientů s IPF ve studii s paralelním uspořádáním skupin. Dvacet čtyři pacientů bylo léčeno po dobu 28 dnů 150 mg nintedanibu dvakrát denně. U 13 pacientů byl nintedanib přidán k dlouhodobé léčbě standardními dávkami pirfenidonu. Jedenáct pacientů bylo léčeno monoterapií nintedanibem. Vzhledem ke krátkému trvání souběžné expozice a nízkému počtu pacientů nebyl poměr přínosu a rizika spojeného se společným podáváním s pirfenidonem stanoven.
Účinek na QT interval
V programu klinického hodnocení nebylo prokázáno prodloužení QT intervalu nintedanibem (bod 5.1). Vzhledem k tomu, že u některých jiných inhibitorů tyrozinkinázy byl prokázán účinek na QT, je při podávání nintedanibu pacientům, u kterých by mohlo dojít k prodloužení QTc, třeba opatrnosti.
Alergická reakce
Je známo, že potraviny obsahující sóju způsobují u osob s alergií na sóju alergické reakce, včetně závažné anafylaxe. U pacientů se známou alergií na bílkoviny obsažené v arašídech existuje zvýšené riziko závažných reakcí na přípravky obsahující sóju.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Glykoprotein P (P-gp)
Nintedanib je substrát P-gp (viz bod 5.2). Ve specifické lékové interakční studii došlo při společném podávání s potentním inhibitorem P-gp ketokonazolem ke zvýšení expozice nintedanibu 1,61krát vzhledem k AUC a 1,83krát vzhledem k Cmax. V lékové interakční studii s potentním induktorem P-gp rifampicinem došlo při společném podávání s rifampicinem v porovnání s podáváním samotného nintedanibu k poklesu expozice nintedanibu na 50,3 % vzhledem k AUC a na 60,3 % vzhledem k Cmax. Při společném podávání s přípravkem Ofev mohou silné inhibitory P-gp (např. ketokonazol, erythromycin nebo cyklosporin) zvýšit expozici nintedanibu. V takových případech je třeba pečlivě sledovat, zda pacienti nintedanib snášejí. Léčba nežádoucích účinků může vyžadovat přerušení léčby, snížení dávky nebo ukončení léčby přípravkem Ofev (viz bod 4.2).
Potentní induktory P-gp (např. rifampicin, karbamazepin, fenytoin a třezalka tečkovaná) mohou snižovat expozici nintedanibu. Je třeba zvážit výběr alternativního souběžně podávaného léčivého přípravku, který nemá žádný nebo má minimální potenciál indukovat P-gp.
Enzymy cytochromu (CYP)
CYP dráhy byly součástí biotransformace nintedanibu pouze v malé míře. V předklinických studiích nintedanib a jeho metabolity, volná kyselá frakce BIBF 1202 a její glukuronid BIBF 1202, neinhibovaly ani neindukovaly enzymy CYP (viz bod 5.2). Pravděpodobnost lékových interakcí s nintedanibem na základě CYP metabolismu je proto považována za nízkou.
Společné podávání s jinými léčivými přípravky
Potenciál pro interakce nintedanibu s hormonálními antikoncepčními přípravky nebyl zjišťován.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku / Antikoncepce
Nintedanib může poškozovat lidský plod (viz bod 5.3). Ženy ve fertilním věku je třeba poučit, aby se v době, kdy jsou léčeny přípravkem Ofev, vyhnuly otěhotnění. Je třeba je poučit, aby během léčby a ještě nejméně 3 měsíce po poslední dávce přípravku Ofev užívaly účinnou antikoncepci. Vzhledem k tomu, že nebyl zkoumán účinek nintedanibu na metabolismus a účinnost hormonálních antikoncepčních přípravků, je třeba jako další formu antikoncepce používat bariérové metody, aby nedošlo k otěhotnění.
Údaje o podávání přípravku Ofev těhotným ženám nejsou k dispozici, ale předklinické studie na zvířatech prokázaly reprodukční toxicitu této léčivé látky (viz bod 5.3). Vzhledem k tomu, že nintedanib může poškozovat také lidský plod, nesmí se během těhotenství použít.
Pacientky je třeba poučit, aby v případě, že během léčby přípravkem Ofev otěhotní, informovaly svého lékaře nebo lékárníka.
Jestliže pacientka v průběhu léčby přípravkem Ofev otěhotní, musí být informována o potenciálním nebezpečí pro plod. Je třeba zvážit ukončení léčby přípravkem Ofev.
Kojení
Informace o vylučování nintedanibu a jeho metabolitů do lidského mateřského mléka nejsou k dispozici.
Předklinické studie prokázaly, že se do mléka kojících samic potkanů vylučují malá množství nintedanibu a jeho metabolitů (< 0,5 % podané dávky). Riziko pro novorozence/kojence nelze vyloučit. Kojení má být během léčby přípravkem Ofev přerušeno.
Fertilita
Předklinické zkoumání nepotvrdilo negativní vliv na mužskou fertilitu (viz bod 5.3). Subchronické a chronické studie toxicity neposkytly při systémové expozici srovnatelné s maximální doporučenou dávkou pro člověka (maximum recommended human dose, MRHD) 150 mg dvakrát denně žádné důkazy o negativním vlivu na samičí fertilitu potkanů (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Ofev má mírný vliv na schopnost řídit nebo obsluhovat stroje. Pacienty je třeba poučit, aby byli v průběhu léčby přípravkem Ofev při řízení a obsluze strojů opatrní.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nintedanib byl testován v klinických hodnoceních s 1529 pacienty s IPF. Údaje o bezpečnosti poskytnuté níže vycházejí ze dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných
studií fáze III s 1061 pacienty srovnávajících léčbu nintedanibem 150 mg dvakrát denně s placebem po dobu 52 týdnů (INPULSIS-1 a INPULSIS-2).
Nejčastěji hlášenými nežádoucími účinky souvisejícími s použitím nintedanibu byly průjem, nauzea a zvracení, bolest břicha, snížená chuť k jídlu, úbytek tělesné hmotnosti a zvýšení hladin jaterních enzymů.
Informace k léčbě vybraných nežádoucích účinků jsou uvedeny také v bodě 4.4.
Tabulkový přehled nežádoucích účinků
Níže uvedená tabulka poskytuje souhrn nežádoucích účinků dle třídy orgánových systémů MedDRA a frekvenční kategorie.
Tabulka 1 shrnuje četnosti nežádoucích účinků hlášených ve skupině s nintedanibem (638 pacientů) kombinované z obou placebem kontrolovaných klinických hodnocení fáze III v trvání 52 týdnů.
Frekvenční kategorie jsou definovány takto:
velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000), velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit).
V rámci každé frekvence jsou nežádoucí účinky uvedeny v pořadí dle snižující se závažnosti.
Tabulka 1: Souhrn nežádoucích účinků dle _ frekvenční kategorie
|
Frekvence Třída orgánových systémů |
Velmi časté (> 1/10) |
Časté (> 1/100 < 1/10) |
Méně časté (> 1/1000 < 1/100) |
|
Poruchy metabolismu a výživy |
Snížená tělesná hmotnost, snížená chuť k jídlu | ||
|
Cévní poruchy |
Epistaxe |
Hypertenze | |
|
Gastrointestinální poruchy | |||
|
Poruchy jater a žlučových cest |
Zvýšené hladiny jaterních enzymů |
Zvýšené hladiny alaninaminotransferázy (ALT), zvýšené hladiny aspartátaminotransferázy (AST), zvýšené hladiny gamma-glutamyl transferázy (GMT) |
Hyperbilirubinemie, zvýšené hladiny alkalické fosfatázy (ALP) v krvi |
Popis vybraných nežádoucích účinků
Průjem byl hlášen u 62,4 % pacientů léčených nintedanibem. U 3,3 % pacientů léčených nintedanibem byl hlášen průjem závažné intenzity. Více než dvě třetiny pacientů s průjmem hlásily jeho první nástup již během prvních tří měsíců léčby. Průjem vedl k trvalému ukončení léčby u 4,4 % pacientů; v ostatních případech byl zvládán protiprůjmovou léčbou, snížením dávky nebo přerušením léčby (viz bod 4.4).
Zvýšené hladiny jaterních enzymů
Zvýšené hladiny jaterních enzymů (viz bod 4.4) byly hlášeny u 13,6 % pacientů léčených nintedanibem. Zvýšené hladiny jaterních enzymů byly reverzibilní a nesouvisely s klinicky prokázanou chorobou jater.
Další informace ke zvláštním populacím, doporučeným opatřením a úpravám dávek v případě průjmu a zvýšených hladin jaterních enzymů jsou uvedeny také v bodech 4.4, respektive 4.2.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Pro předávkování přípravkem Ofev neexistuje specifické antidotum ani léčba. U dvou pacientů v onkologickém programu došlo k předávkování maximální dávkou 600 mg dvakrát denně po dobu až osmi dnů. Zjištěné nežádoucí účinky byly v souladu se známým bezpečnostním profilem nintedanibu, tedy zvýšené hladiny jaterních enzymů a gastrointestinální příznaky. U obou pacientů tyto nežádoucí účinky odezněly. Ve studiích INPULSIS byl jeden pacient neúmyslně vystaven dávce 600 mg denně po celkovou dobu 21 dní. V době nesprávného dávkování se vyskytla a odezněla nezávažná nežádoucí příhoda (nazofaryngitida); žádné další příhody hlášeny nebyly. V případě předávkování je třeba přerušit léčbu a zahájit odpovídající obecná podpůrná opatření.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, inhibitory proteinkinázy, ATC kód: L01XE31 Mechanismus účinku
Nintedanib je malomolekulární inhibitor tyrozinkináz včetně receptorů destičkového růstového faktoru (PDGFR) a a B, receptoru fibroblastového růstového faktoru (FGFR) 1-3 a VEGFR 1-3. Nintedanib se kompetitivně váže na adenozintrifosfátovou (ATP) vazebnou kapsu těchto receptorů a blokuje intracelulární signalizaci. Nintedanib navíc inhibuje kinázy Flt-3 (tyrozinkináza podobná fms), Lck (tyrozinkináza specifická pro lymfocyty), Lyn (tyrozinkináza lyn) a Src (protoonkogen tyrozinkináza src).
Farmakodynamické účinky
Nintedanib inhibuje aktivaci signalizačních kaskád FGFR a PDGFR, které hrají kritickou roli v proliferaci, migraci a diferenciaci plicních fibroblastů/myofibroblastů, klíčových buněk z pohledu patologie idiopatické plicní fibrózy. Potenciální dopad inhibice VEGFR nintedanibem a antiangiogenní působení nintedanibu na patologii IPF nejsou zatím plně osvětleny. V předklinických modelech plicní fibrózy má nintedanib silně antifibrotický a protizánětlivý účinek. U humánních plicních fibroblastů pacientů s IPF nintedanib inhibuje proliferaci, migraci a transformaci na myofibroblasty.
Klinická účinnost a bezpečnost
Klinická účinnost nintedanibu byla hodnocena u pacientů s IPF ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III se stejným uspořádáním (INPULSIS-1 (1199.32) a INPULSIS-2 (1199.34)). Pacienti s výchozí FVC < 50 % předpokládané FVC nebo s výchozí difúzní kapacitou pro oxid uhelnatý (DLCO, korigovanou na koncentraci hemoglobinu)
< 30 % byly ze studií vyřazeni. Pacienti byli randomizováni v poměru 3:2 na léčbu přípravkem Ofev 150 mg nebo placebem dvakrát denně po dobu 52 týdnů.
Primárním cílovým parametrem byla roční míra poklesu usilovné vitální kapacity (forced vital capacity, FVC). Klíčovými sekundárními cílovými parametry byla změna kvality života oproti výchozímu celkovému skóre hodnocená v 52. týdnu pomocí dotazníku Saint George's Respiratory Questionnaire (SGRQ) a doba do první akutní exacerbace IPF.
Roční míra poklesu FVC
Roční míra poklesu FVC (v ml) byla významně snížena u pacientů, kteří byli léčeni nintedanibem, ve srovnání s pacienty, kteří dostávali placebo. Výsledný léčebný účinek byl v obou studiích konzistentní. Výsledky jednotlivých studií a souhrnné výsledky viz tabulka 2.
Tabulka 2: Roční míra poklesu FVC (ml) ve studiích INPULSIS-1 a INPULSIS-2 a souhrnné
_údaje - léčená skupina __
|
INPULSIS-1 |
INPU! |
lSIS-2 |
INPULSIS-1 a INPULSIS-2 souhrnné výsledky | |||
|
Placebo |
Ofev 150 mg dvakrát denně |
Placebo |
Ofev 150 mg dvakrát denně |
Placebo |
Ofev 150 mg dvakrát denně | |
|
Počet pacientů, jejichž data byla použita k analýze |
204 |
309 |
219 |
329 |
423 |
638 |
|
Míra1 (SE) poklesu FVC během 52 týdnů |
-239,9 (18,71) |
-114,7 (15,33) |
-207,3 (19,31) |
-113,6 (15,73) |
-223,5 (13,45) |
-113,6 (10,98) |
|
Porovnání s placebem | ||||||
|
Rozdíl1 |
125,3 |
93,7 |
109,9 | |||
|
95% CI |
(77,7, 172,8) |
(44,8, 142,7) |
(75,9, 144,0) | |||
|
Hodnota p |
<0,0001 |
0,0002 |
<0,0001 | |||
|
1 Odhad na základě modelu regrese náhodných koeficientů. CI: interval spolehlivosti | ||||||
Robustnost léčeného efektu nintedanibu při snižování roční míry poklesu FVC byla potvrzena ve všech předem specifikovaných analýzách citlivosti. U pacientů s chybějícími údaji primární analýza předpokládá, že pokles FVC po posledním měření, jehož výsledek je znám, je obdobný jako u jiných pacientů ve stejné léčebné skupině. V analýze citlivosti, která předpokládá, že u pacientů s chybějícími údaji v 52. týdnu je pokles FVC po posledním měření, jehož výsledek je znám, stejný jako u všech pacientů na placebu, byl upravený rozdíl v roční míře poklesu FVC mezi nintedanibem a placebem
113,9 ml/rok (95% CI 69,2; 158,5) ve studii INPULSIS-1 a 83,3 ml/rok (95% CI 37,6; 129,0) ve studii INPULSIS-2.
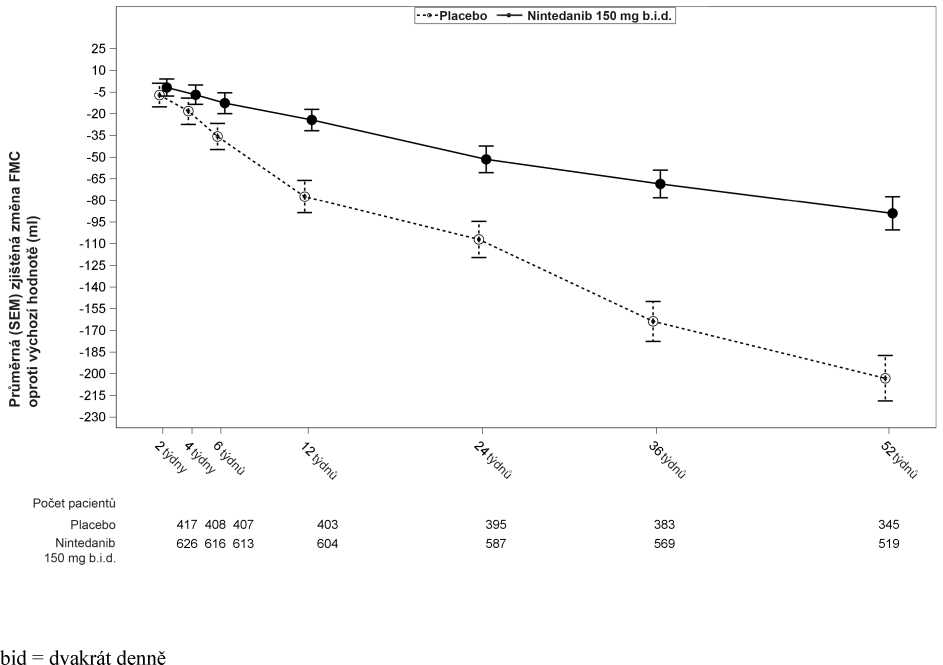
Podobné terapeutické účinky byly navíc pozorovány u ostatních cílových parametrů funkce plic, např. u změny výchozí hodnoty FVC v 52. týdnu a FVC analýz dat respondentů poskytujících další odůvodnění účinnosti nintedanibu na zpomalení progrese onemocnění. Vývoj změny oproti výchozí hodnotě v čase u obou léčebných skupin ve studii INPULSIS-1 a INPULSIS-2 (souhrnná analýza dat) je znázorněn na obr. 1.
Obrázek 1: Průměr (SEM) pozorované změny FVC oproti výchozí hodnotě (ml) v čase,
studie INPULSIS-1 a INPULSIS-2 souhrnně
Analýza dat pacientů reagujících na léčbu (hodnoceno pomocí parametru FVC)
V obou studiích INPULSIS byl podíl pacientů reagujících na léčbu z pohledu FVC, kteří jsou definováni jako pacienti s maximálním předpokládaným absolutním poklesem FVC do výše 5 % (prahová hodnota signalizující zvýšené riziko mortality u IPF), významně vyšší ve skupině s nintedanibem než s placebem. Podobné výsledky byly zjištěny v analýzách s využitím konzervativní prahové hodnoty 10 %. Výsledky jednotlivých studií a souhrnné výsledky viz tabulka 3.
Tabulka 3: Podíl pacientů reagujících na léčbu (hodnoceno pomocí parametru FVC)
v 52. týdnu léčby ve studiích INPULSIS-1 a INPULSIS-2 a souhrnné údaje - léčená skupina___
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 a INPULSIS-2 souhrnné výsledky | ||||
|
Placebo |
Ofev 150 mg dvakrát denně |
Placebo |
Ofev 150 mg dvakrát denně |
Placebo |
Ofev 150 mg dvakrát denně | |
|
Počet pacientů, jejichž data byla použita k analýze |
204 |
309 |
219 |
329 |
423 |
638 |
|
5% práh | ||||||
|
Počet (%) pacientů s odpovědí na léčbu (hodnoceno pomocí FVC1) |
78 (38,2) |
163 (52,8) |
86 (39,3) |
175 (53,2) |
164 (38,8) |
338 (53,0) |
|
Porovnání s placebem | ||||||
|
Poměr šancí |
1,85 |
1,79 |
1,84 | |||
|
95% CI |
(1,28, 2,66) |
(1,26, 2,55) |
(1,43, 2,36) | |||
|
Hodnota p2 |
0,0010 |
0,0011 |
<0,0001 | |||
|
10% práh | ||||||
|
Počet (%) pacientů s odpovědí na léčbu (hodnoceno pomocí FVC1) |
116 (56,9) |
218 (70,6) |
140 (63,9) |
229 (69,6) |
256 (60,5) |
447 (70,1) |
|
Porovnání s placebem | ||||||
|
Poměr šancí |
1,91 |
1,29 |
1,58 | |||
|
95% CI |
(1,32, 2,79) |
(0,89, 1,86) |
(1,21, 2,05) | |||
|
Hodnota p2 |
0,0007 |
0,1833 |
0,0007 | |||
1 Pacienti reagující na léčbu jsou pacienti, u nichž nedošlo k absolutnímu předpokládanému poklesu FVC většímu než 5 % nebo 10 % v porovnání se vstupní hodnotou při vyhodnocení FVC v 52 týdnech.
2 Na základě logistické regrese.
Doba do progrese (absolutní předpokládaný pokles FVC > 10 % nebo úmrtí)
V obou studiích INPULSIS bylo riziko progrese statisticky významně nižší u pacientů léčených nintedanibem než u pacientů na placebu. V souhrnné analýze bylo HR 0,60, což představuje 40% snížení rizika progrese u pacientů léčených nintedanibem v porovnání s pacienty na placebu.
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 a INPULSIS-2 souhrnné výsledky | ||||
|
Placebo |
Ofev 150 mg dvakrát denně |
Placebo |
Ofev 150 mg dvakrát denně |
Placebo |
Ofev 150 mg dvakrát denně | |
|
Počet počet osob v riziku |
204 |
309 |
219 |
329 |
423 |
638 |
|
Pacienti s příhodami, n (%) |
83 (40,7) |
75 (24,3) |
92 (42,0) |
98 (29,8) |
175 (41,4) |
173 (27,1) |
|
Porovnání s placebem1 | ||||||
|
Hodnota p2 |
0,0001 |
0,0054 |
<0,0001 | |||
|
Poměr rizik3 |
0,53 |
0,67 |
0,60 | |||
|
95% CI |
(0,39; 0,72) |
(0,51; 0,89) |
(0,49; 0,74) | |||
|
1 Na základě údajů shromažďovaných po dobu až 372 dnů (52 týdnů + 7denní možný přesah). 2 Na základě log-rank testu. 3 Na základě Coxova regresního modelu. | ||||||
Změna celkového skóre SGRQ oproti výchozímu v 52. týdnu
Celkové skóre SGRQ, kterým se měří kvalita života související se zdravím (health-related quality of life, HRQoL), bylo analyzováno v 52. týdnu. Ve studii INPULSIS-2 vykázali pacienti, kteří dostávali placebo, větší nárůst oproti výchozímu celkovému skóre SGRQ než pacienti, kteří dostávali nintedanib 150 mg dvakrát denně. Pokles HRQoL byl menší ve skupině s nintedanibem; rozdíl mezi oběma léčebnými skupinami byl statisticky významný (-2,69; 95% CI: -4,95, -0,43; p=0,0197).
Ve studii INPULSIS-1 byl nárůst oproti výchozímu celkovému skóre SGRQ v 52. týdnu u nintedanibu a placeba podobný (rozdíl mezi léčebnými skupinami: -0,05; 95% CI:-2,50, 2,40; p=0,9657).
V souhrnné analýze obou studií INPULSIS byla odhadovaná průměrná změna celkového skóre SGRQ mezi výchozím stavem a 52. týdnem menší u skupiny s nintedanibem (3,53) než u skupiny s placebem (4,96) a rozdíl mezi léčebnými skupinami byl -1,43 (95% CI: -3,09, 0,23; p=0,0923). Celkově je účinek nintedanibu na kvalitu života související se zdravím měřenou celkovým skóre SGRQ mírný a naznačuje, že se kvalita života zhoršuje méně než u placeba.
Doba do první akutní exacerbace IPF
Ve studii INPULSIS-2 bylo u pacientů, kteří dostávali nintedanib, významně sníženo riziko první akutní exacerbace IPF v průběhu 52 týdnů ve srovnání s pacienty, kteří dostávali placebo; ve studii INPULSIS-1 nebyl mezi oběma léčebnými skupinami rozdíl. V souhrnné analýze obou studií INPULSIS bylo u pacientů, kteří dostávali nintedanib, zjištěno číselně nižší riziko první akutní exacerbace než u pacientů, kteří dostávali placebo. Výsledky jednotlivých studií a souhrnné výsledky viz tabulka 5.
Tabulka 5: Počet pacientů s akutními exacerbacemi IPF během 52 týdnů a analýza doby do
první exacerbace na základě příhod hlášených zkoušejícími ve studii
|
INP! |
JLSIS-1 |
INPULSIS-2 |
INPULSIS-1 a INPULSIS-2 souhrnné výsledky | |||
|
Placebo |
Ofev 150 mg dvakrát denně |
Placebo |
Ofev 150 mg dvakrát denně |
Placebo |
Ofev 150 mg dvakrát denně | |
|
Počet osob v riziku |
204 |
309 |
219 |
329 |
423 |
638 |
|
Pacienti s příhodami, n(%) |
11 (5,4) |
19 (6,1) |
21 (9,6) |
12 (3,6) |
32 (7,6) |
31 (4,9) |
|
Porovnání s placebem1 | ||||||
|
Hodnota p2 |
0,6728 |
0,0050 |
0,0823 | |||
|
Poměr rizik3 |
1,15 |
0,38 |
0,64 | |||
|
95% CI |
(0,54, 2,42) |
(0,19, 0,77) |
(0,39, 1,05) | |||
|
1 Na základě údajů shromažďovaných po dobu až 372 dnů (52 týdnů + 7denní možný přesah). 2 Na základě log-rank testu. 3 Na základě Coxova regresního modelu. | ||||||
Všechny nežádoucí příhody akutní exacerbace IPF hlášené zkoušejícím byly posouzeny zaslepenou posudkovou komisí. Souhrnné údaje byly podrobeny předem specifikované analýze citlivosti doby do prvního podezření nebo potvrzení akutní exacerbace IPF. Počet pacientů, u nichž v průběhu 52 týdnů došlo minimálně k 1 potvrzené exacerbaci, byla nižší u skupiny s nintedanibem (1,9 % pacientů) než u skupiny s placebem (5,7 % pacientů). Analýzou doby do výskytu sledované příhody s využitím souhrnných dat pro příhody potvrzené exacerbace byl zjištěn poměr rizik (hazard ratio, HR) ve výši 0,32 (95% CI 0,16, 0,65; p=0,0010). To znamená, že riziko první akutní exacerbace zjištěné IPF bylo v kterémkoli časovém bodě statisticky významně nižší u skupiny s nintedanibem než ve skupině s placebem.
Analýza přežívání
Předem specifikovanou souhrnnou analýzou údajů o přežívání ve studiích INPULSIS se zjistilo, že celková mortalita v průběhu 52 týdnů byla nižší u skupiny s nintedanibem (5,5 % pacientů) než u skupiny s placebem (7,8 % pacientů). Analýzou doby do úmrtí byl zjištěn HR ve výši 0,70 (95% CI 0,43, 1,12; p=0,1399). Výsledky všech cílových bodů přežití (např. mortalita v průběhu léčby a úmrtí v souvislosti s respiračním selháním) vykazovaly soustavné numerické rozdíly ve prospěch nintedanibu.
|
a INP1 |
ULSIS-2 a souhrnné údaje - léčená skupina | |||||
|
INP! |
JLSIS-1 |
INPULSIS-2 |
INPULSIS-1 a INPULSIS-2 souhrnné výsledky | |||
|
Placebo |
Ofev 150 mg dvakrát denně |
Placebo |
Ofev 150 mg dvakrát denně |
Placebo |
Ofev 150 mg dvakrát denně | |
|
Počet počet osob v riziku |
204 |
309 |
219 |
329 |
423 |
638 |
|
Pacienti s příhodami, n(%) |
13 (6,4) |
13 (4,2) |
20 (9,1) |
22 (6,7) |
33 (7,8) |
35 (5,5) |
|
Porovnání s placebem1 | ||||||
|
Hodnota p2 |
0,2880 |
0,2995 |
0,1399 | |||
|
Poměr rizik3 |
0,63 |
0,74 |
0,70 | |||
|
95% CI |
(0,29, 1,36) |
(0,40, 1,35) |
(0,43, 1,12) | |||
|
1 Na základě údajů shromažďovaných po dobu až 372 dnů (52 týdnů + 7denní možný přesah). 2 Na základě log-rank testu. 3 Na základě Coxova regresního modelu. | ||||||
Podpůrné důkazy z výsledků hodnocení fáze II (1199.30) Ofev 150 mg dvakrát denně Další důkaz účinnosti pochází z randomizovaného, dvojitě zaslepeného, placebem kontrolovaného klinického hodnocení pro určení dávek fáze II, které zahrnovalo skupinu s dávkováním nintedanibu 150 mg dvakrát denně.
Primární cílový parametr, míra poklesu FVC v průběhu 52 týdnů, vykazoval nižší hodnotu v rameni s nintedanibem (-0,060 l/rok, n=84) než v rameni s placebem (-0,190 l/rok, n=83). Odhadovaný rozdíl mezi léčebnými skupinami byl 0,131 l/rok (95 % CI 0,027, 0,235). Rozdíl mezi léčebnými skupinami dosáhl nominální statistické významnosti (p=0,0136).
Odhadovaná průměrná změna celkového skóre SGRQ mezi výchozím stavem a 52. týdnem byla 5,46 pro placebo, což značí zhoršování kvality života související se zdravím, a -0,66 pro nintedanib, což značí stabilní kvalitu života související se zdravím. Odhadovaný průměrný rozdíl mezi nintedanibem a placebem byl -6,12 (95 % CI: -10,57, -1,67; p=0,0071).
Počet pacientů s akutními exacerbacemi IPF během 52 týdnů byl nižší u skupiny s nintedanibem (2,3 %, n=86) než u skupiny s placebem (13,8 %, n=87). Odhadovaný poměr rizik nintedanibu oproti placebu byl 0,16 (95 % CI 0,04, 0,71; p=0,0054).
QT interval
Ve specifické studii u pacientů s rakovinou ledvinových buněk bylo měřením QT/QTc zjištěno, že jednotlivá perorální dávka 200 mg nintedanibu ani opakované perorální dávky 200 mg nintedanibu podávané dvakrát denně po dobu 15 dnů neprodlužovaly QTcF interval.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Ofev u všech podskupin pediatrické populace s IPF (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Nintedanib dosáhl maximálních plazmatických koncentrací přibližně 2-4 hodiny po perorálním podání měkkých želatinových tobolek ve stavu sytosti (rozpětí 0,5-8 hodin). Absolutní biologická dostupnost 100mg dávky byla u zdravých dobrovolníků 4,69 % (90 % CI: 3,615-6,078). Absorpce a biologická dostupnost jsou sníženy účinky transportérů a významným předsystémovým metabolismem při prvním průchodu játry. Proporcionalita dávky byla prokázána zvýšením expozice nintedanibu (rozpětí dávek 50-450 mg jednou denně a 150-300 mg dvakrát denně. Rovnovážného stavu plazmatických koncentrací bylo dosaženo nejdéle do týdne od podání dávky.
Po požití stravy se expozice nintedanibu zvýšila o přibližně 20 % v porovnání s podáním nalačno (CI: 95,3-152,5 %) a absorpce byla zpožděna (medián tmax nalačno: 2,00 hod.; po jídle: 3,98 hod).
Distribuce
Nintedanib má nejméně dvoufázovou kinetiku. Po intravenózní infuzi byl zjištěn vysoký distribuční objem (Vss: 1 050 l, 45,0 %gCV).
Vazba nintedanibu na bílkoviny lidské krevní plazmy in vitro byla vysoká, vázaná frakce činila 97,8 %. Za hlavní vazebnou bílkovinu je považován sérový albumin. Nintedanib je distribuován především do plazmy, přičemž poměr krev ku plazmě je 0,869.
Biotransformace
Převažující metabolickou reakcí u nintedanibu je hydrolytické štěpení esterázami, které vede k tvorbě volné kyselé frakce BIBF 1202. BIBF 1202 je následně glukuronidována uridin-5-difosfát-glukuronosyltransferázami (UGT enzymy), především UGT 1A1, UGT 1A7,
UGT 1A8 a UGT 1A10 na BIBF 1202 glukuronid.
CYP dráhy byly součástí biotransformace nintedanibu pouze v malé míře, přičemž hlavním enzymem byl CYP 3A4. Ve studii absorpce, distribuce, metabolismu a eliminace (ADME) u člověka nebylo možné hlavní metabolit CYP dráhy v plazmě zjistit. In vitro představoval metabolismus závislý na CYP přibližně 5 % v porovnání s 25 % esterického štěpení. V předklinických studiích nintedanib, BIBF 1202 a glukuronid BIBF 1202 neinhibovaly ani neindukovaly enzymy CYP. Nepředpokládá se proto, že budou existovat lékové interakce mezi nintedanibem a substráty CYP, CYP inhibitory a CYP induktory.
Eliminace
Celková plazmatická clearance po intravenózní infuzi byla vysoká (CL: 1390 ^nl/^nin, 28,8 % gCV).
V podobě nezměněné léčivé látky bylo během 48 hodin močí vyloučeno přibližně 0,05 % perorálně podané dávky (31,5 % gCV) a přibližně 1,4 % intravenózně podané dávky (24,2 % gCV); renální clearance byla 20 ml/min (32,6 % gCV). Hlavní cestou eliminace lékové radioaktivity po perorálním podání [14C] nintedanibu bylo vylučování stolicí a žlučí (93,4 % dávky, 2,61 % gCV). Renální vylučování přispívalo k celkové clearanci jen v malé míře (0,649 % dávky, 26,3 % gCV). Celkové množství zjištěné látky (recovery) bylo považováno za úplné (více než 90 %) do 4 dnů po podání dávky. Terminální poločas nintedanibu byl mezi 10 a 15 hod. (gCV % přibližně 50 %).
Linearita/nelinearita
Farmakokinetiku (PK) nintedanibu lze považovat za lineární vzhledem k času (tedy údaje pro podání jednotlivých dávek lze extrapolovat na opakované dávky). Při opakovaném podání docházelo k akumulaci 1,04krát u Cmax a 1,38krát u AUCT. Minimální koncentrace nintedanibu zůstaly stabilní po dobu více než jednoho roku.
Transport
Nintedanib je substrát P-gp. Informace o interakčním potenciálu nintedanibu s tímto transportérem jsou uvedeny v bodě 4.5. Bylo prokázáno, že nintedanib není in vitro substrátem ani inhibitorem OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, ani MRP-2. Nintedanib rovněž nebyl substrátem BCRP.
In vitro byl pozorován pouze slabý inhibiční potenciál na OCT-1, BCRP a P-gp, což je považováno za klinicky málo významné. Totéž platí pro nintedanib jako substrát OCT-1.
Populační analýza farmakokinetiky u zvláštních populací
Farmakokinetické vlastnosti nintedanibu byly podobné u zdravých dobrovolníků, pacientů s IPF a onkologických pacientů. Podle populačních analýz farmakokinetiky (PopPK) u pacientů s IPF a nemalobuněčným karcinomem plic (non-small cell lung cancer, NSCLC) (n=1191) a deskriptivního výzkumu nebyla expozice nintedanibu ovlivněna pohlavím (korigováno dle tělesné hmotnosti), mírným či středním zhoršením funkce ledvin (odhadnuto na základě clearance kreatininu), požíváním alkoholu ani genotypem P-gp.
Populační analýzy PK naznačily středně významný vliv na expozici nintedanibu v závislosti na věku, tělesné hmotnosti a rase (viz níže). Vzhledem k velkým interindividuálním rozdílům v expozici nejsou zjištěné mírné účinky považovány za klinicky významné (viz bod 4.4).
Věk
Expozice nintedanibu se zvyšovala lineárně s věkem. AUCx,ss se u 45letého pacienta snížila o 16 % a u 76letého pacienta zvýšila o 13 % v porovnání s pacientem s mediánem věku 62 let. Věkové rozpětí pacientů v analýze bylo 29 až 85 let; přibližně 5 % populace bylo starších 75 let. Dle PopPK modelu měli pacienti ve věku > 75 let o přibližně 20 - 25 % vyšší expozici nintedanibu než pacienti mladší 65 let.
Studie u pediatrických populací nebyly provedeny.
Tělesná hmotnost
Byl zjištěn inverzní vztah mezi tělesnou hmotností a expozicí nintedanibu. AUCx,ss se zvýšila o 25 % u 50kg pacienta (5. percentil) a snížila o 19 % u 100kg pacienta (95. percentil) v porovnání s pacientem s mediánem hmotnosti 71,5 kg.
Rasa
Oproti bělochům byla geometrická průměrná expozice nintedanibu u pacientů z Číny, Tchaj-wanu a Indie o 33 % vyšší a u pacientů z Koreje o 22 % nižší (korigováno dle tělesné hmotnosti). Údaje od osob černé rasy byly velmi omezené ale ve stejném rozpětí jako u bělochů.
Porucha funkce jater
V dedikované studii fáze I s jednorázovou dávkou a v porovnání se zdravými jedinci byla expozice nintedanibu dle Cmax a AUC 2,2krát vyšší u dobrovolníků s lehkou poruchou funkce jater (Child Pugh A; 90% CI 1,3 - 3,7 u Cmax a 1,2 - 3,8 u AUC). U dobrovolníků se středně těžkou poruchou funkce jater (Child Pugh B) byla v porovnání se zdravými dobrovolníky expozice 7,6krát vyšší dle Cmax (90% CI 4,4 - 13,2) a 8,7krát vyšší dle AUC (90% CI 5,7 - 13,1). Pacienti s těžkou poruchou funkce jater (Child Pugh C) nebyli studováni.
Souběžná léčba pirfenidonem
V malé studii s paralelním uspořádáním skupin japonských pacientů s IPF (13 pacientů dostávalo nintedanib navíc k chronické léčbě standardními dávkami pirfenidonu; 11 pacientů dostávalo samotný nintedanib) poklesla při společném podávání s pirfenidonem expozice nintedanibu na 68,3 % vzhledem k AUC a na 59,2 % vzhledem k Cmax ve srovnání s podáváním samotného nintedanibu. Nintedanib neměl žádný účinek na farmakokinetiku pirfenidonu (viz bod 4.4).
5.3 Předklinické údaje vztahující se k bezpečnosti
Obecná toxikologie
Studie toxicity po podání jednorázové dávky potkanům a myším naznačily nízký potenciál nintedanibu k akutní toxicitě. V toxikologických studiích s opakovaným podáváním u potkanů byly nežádoucí účinky (např. ztluštění epifyzárních štěrbin, léze na řezácích) většinou spojené s mechanismem účinku nintedanibu (tedy inhibice VEGFR-2). Tyto změny jsou známy i u jiných VEGFR-2 inhibitorů a mohou být považovány za skupinový účinek.
Ve studiích toxicity s jinými živočichy než hlodavci byl pozorován průjem a zvracení spojené se snížením příjmu potravy a ztrátou tělesné hmotnosti.
U potkanů, psů a opic druhu Cynomolgus nebyl zjištěn nárůst hladin jaterních enzymů. Mírné zvýšení hladin jaterních enzymů, které nebylo spojeno se závažnými nežádoucími účinky, jako je průjem, bylo zjištěno pouze u opic druhu Rhesus.
Reprodukční toxicita
U potkanů byly embryofetální úmrtnost a teratogenní účinky pozorovány při hladinách expozice nižších než je expozice u člověka při maximální doporučené humánní dávce (MRHD) 150 mg dvakrát denně. Účinky na vývoj axiálního skeletu a na vývoj velkých cév byly rovněž zaznamenány při subterapeutických hladinách expozice.
U králíků byly embryofetální úmrtnost a teratogenní účinky pozorovány při expozici přibližně 3krát vyšší než u MRHD, ale nejednoznačné účinky na embryofetální vývoj axiálního skeletu a srdce byly zaznamenány již při expozici nižší než MRHD, tedy 150 mg dvakrát denně.
Ve studii pre- a postnatálního vývoje u potkanů byly účinky na pre-a postnatální vývoj pozorovány při expozici nižší než MRHD.
Studie samčí fertility a časného embryonálního vývoje až do nidace u potkanů neprokázala účinky na samčí reproduktivní soustavu a samčí fertilitu.
U potkanů byly malé dávky radioaktivně značeného nintedanibu nebo jeho metabolitů vyloučeny do mléka (< 0,5 % podané dávky).
Dvouleté studie kancerogenity u myší a potkanů nepřinesly žádné důkazy o kancerogenním potenciálu nintedanibu.
Studie genotoxicity nenaznačily žádný mutagenní potenciál nintedanibu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky
střední nasycené triacylglyceroly tvrdý tuk
sójový lecithin (E322)
Obal tobolky
želatina
glycerol 85%
oxid titaničitý (E171)
červený oxid železitý (E172)
žlutý oxid železitý (E172)
Potiskový inkoust šelak
černý oxid železitý (E172) propylenglykol (E1520)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Přípravek Ofev 100 mg měkké tobolky/přípravek Ofev 150 mg měkké tobolky je k dispozici v následujících baleních:
- 30x1 měkká tobolka v perforovaných jednodávkových Al/Al blistrech
- 60 x 1 měkká tobolka v perforovaných jednodávkových Al/Al blistrech
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH
Binger Strasse 173
55216 Ingelheim am Rhein
Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/979/001
EU/1/14/979/002
EU/1/14/979/003
EU/1/14/979/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 15. ledna 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Boehringer Ingelheim Pharma GmbH & Co. KG
Binger Strasse 173
55216 Ingelheim am Rhein
NĚMECKO
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky;
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Ofev 100 mg měkké tobolky nintedanibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje nintedanibum 100 mg (ve formě nintedanibi esilas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sójový lecithin. Další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
30 x 1 měkká tobolka 60 x 1 měkká tobolka
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/979/001
EU/1/14/979/002
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Ofev 100 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Ofev 150 mg měkké tobolky nintedanibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje nintedanibum 150 mg (ve formě nintedanibi esilas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sójový lecithin. Další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
30 x 1 měkká tobolka 60 x 1 měkká tobolka
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/979/003
EU/1/14/979/004
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Ofev 150 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
Ofev 100 mg tobolky nintedanibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI_
Boehringer Ingelheim (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE_
Lot
5. JINÉ_
Před použitím neotvírejte.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
Ofev 150 mg tobolky nintedanibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI_
Boehringer Ingelheim (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE_
Lot
5. JINÉ_
Před použitím neotvírejte.
B. PŘÍBALOVÁ INFORMACE
Ofev 100 mg měkké tobolky
Nintedanibum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Ofev a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Ofev užívat
3. Jak se přípravek Ofev užívá
4. Možné nežádoucí účinky
5. Jak přípravek Ofev uchovávat
6. Obsah balení a další informace
1. Co je přípravek Ofev a k čemu se používá
Přípravek Ofev obsahuje léčivou látku nintedanib a používá se k léčbě idiopatické plicní fibrózy (IPF).
IPF je stav, při kterém tkáň v plicích v průběhu času zbytňuje, tuhne a jizví se. Zjizvení následně omezuje schopnost přenášet kyslík z plic do krevního oběhu a začne být obtížné zhluboka dýchat. Přípravek Ofev pomáhá omezit jizvení a tuhnutí plic.
2. Čemu musíte věnovat pozornost, než začnete přípravek Ofev užívat
Neužívejte přípravek Ofev:
- jestliže jste alergický(á) na nintedanib, arašídy nebo sóju nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před užitím přípravku Ofev se poraďte se svým lékařem nebo lékárníkem,
- jestliže máte nebo jste někdy měl(a) problémy s játry,
- jestliže máte nebo jste někdy měl(a) problémy s krvácením,
- jestliže užíváte léčivé přípravky k ředění krve (jako je warfarin, fenprokumon nebo heparin) k prevenci tvorby krevních sraženin,
- jestliže máte nebo jste někdy měl(a) problémy se srdcem (například srdeční příhodu),
- jestliže jste v nedávné době podstoupil(a) operaci. Nintedanib může ovlivnit způsob, jak se hojí
rány. Léčba přípravkem Ofev proto obvykle bude na určitou dobu přerušena v případě, že máte být operován(a). Lékař rozhodne o tom, kdy bude léčba tímto přípravkem obnovena.
Na základě těchto informací může lékař provést některé krevní testy, například zkontrolovat funkci jater. Lékař s Vámi výsledky těchto testů prodiskutuje a rozhodne, zda můžete dostat přípravek Ofev.
Během užívání tohoto přípravku svého lékaře okamžitě informujte,
- jestliže dostanete průjem. Je důležité průjem včas léčit (viz bod 4);
- jestliže zvracíte nebo trpíte nevolností (pocit na zvracení);
- jestliže pociťujete silnou bolest břicha, máte horečku, zimnici, trpíte nevolností, zvracíte nebo máte napnuté břicho nebo se cítíte nafouklý(á), protože to by mohly být příznaky proděravění stěny trávicího traktu („gastrointestinální perforace“);
- jestliže pociťujete bolest v končetině, nebo ji máte oteklou, zarudlou či teplou, protože to by mohly být příznaky krevní sraženiny v jedné z žil (druh krevní cévy);
- jestliže pociťujete tlak nebo bolest na hrudi, obvykle na levé straně těla, bolest šíje, čelisti, ramene nebo paže, rychlý srdeční tep, dušnost, pocit na zvracení nebo zvracení, protože to by mohly být příznaky srdeční příhody (infarktu);
- jestliže máte velké krvácení.
Děti a dospívající
Děti a dospívající do 18 let věku nesmějí přípravek Ofev užívat.
Další léčivé přípravky a přípravek Ofev
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, včetně rostlinných přípravků a přípravků, které jste získal(a) bez lékařského předpisu.
Přípravek Ofev se může vzájemně ovlivňovat s některými jinými léčivými přípravky. Následující přípravky mohou zvyšovat hladiny nintedanibu v krvi a tak mohou zvyšovat riziko nežádoucích účinků (viz bod 4):
- léčivý přípravek používaný k léčbě plísňových infekcí (ketokonazol)
- léčivý přípravek používaný k léčbě bakteriálních infekcí (erythromycin)
- léčivý přípravek, který ovlivňuje imunitní systém (cyklosporin)
Následující léčivé přípravky mohou snižovat hladiny nintedanibu v krvi, a tak mohou snižovat účinnost přípravku Ofev:
- antibiotikum používané k léčbě tuberkulózy (rifampicin)
- léčivé přípravky používané k léčbě záchvatů křečí (karbamazepin, fenytoin)
- rostlinný přípravek k léčbě deprese (třezalka tečkovaná)
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Neužívejte tento přípravek během těhotenství, neboť může poškodit Vaše nenarozené dítě a způsobovat vrozené vady.
Ženy, které mohou otěhotnět, musí v době, kdy užívají přípravek Ofev, a nejméně 3 měsíce po ukončení léčby, používat účinnou kombinaci antikoncepčních metod, včetně bariérových metod jako druhé formy antikoncepce. O pro Vás nejvhodnějších metodách antikoncepce se poraďte se svým lékařem.
Okamžitě informujte svého lékaře nebo lékárníka v případě, že během léčby přípravkem Ofev otěhotníte.
Během léčby přípravkem Ofev nekojte, protože existuje určité riziko poškození kojeného dítěte.
Řízení dopravních prostředků a obsluha strojů
Přípravek Ofev může mít mírný vliv na schopnost řídit a obsluhovat stroje. Jestliže se necítíte dobře, neměl(a) byste řídit ani obsluhovat stroje.
Přípravek Ofev obsahuje sójový lecithin
Jestliže jste alergický(á) na sóju nebo arašídy, tento lék neužívejte (viz bod 2).
3. Jak se přípravek Ofev užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka je jedna 100mg tobolka dvakrát denně (celkem 200 mg denně). Tobolky užijte s odstupem 12 hodin každý den přibližně ve stejnou dobu, například jednu tobolku ráno a jednu tobolku večer. To zajistí, že se v krevním oběhu udržuje stálé množství nintedanibu. Tobolky spolkněte celé s vodou, tobolky nežvýkejte ani nedrťte. Doporučuje se, abyste tobolky užíval(a) s jídlem, tedy během nebo těsně před nebo po jídle.
Neužívejte víc než doporučenou dávku dvě 100mg tobolky přípravku Ofev denně.
Jestliže doporučenou dávku dvou 100mg tobolek přípravku Ofev denně netolerujete (viz možné nežádoucí účinky v bodě 4), lékař Vás může instruovat, abyste přípravek přestal(a) užívat. Sám (sama) dávku nesnižujte ani léčbu neukončujte bez předchozí konzultace s lékařem.
Jestliže jste užil(a) více přípravku Ofev, než jste měl(a)
Okamžitě kontaktujte svého lékaře nebo lékárníka.
Jestliže jste zapomněl(a) užít přípravek Ofev
Jestliže jste zapomněl(a) užít předchozí dávku, neužívejte dvě tobolky najednou. Další 100mg dávku přípravku Ofev užijte dle plánu v další plánovanou dobu doporučenou lékařem nebo lékárníkem.
Jestliže jste přestal(a) užívat přípravek Ofev
Nepřerušujte užívání přípravku Ofev bez toho, aniž byste se nejprve poradil(a) s lékařem. Je důležité, abyste tento přípravek užíval(a) každý den po celou dobu, kdy Vám jej lékař předepisuje.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Musíte být obzvlášť obezřetný(á) v případě, že se u Vás v průběhu léčby přípravkem Ofev objeví následující nežádoucí účinky:
Průjem (velmi časté, může postihnout více než 1 z 10 lidí):
Průjem může vést k dehydrataci: ke ztrátě tekutin a důležitých solí (elektrolyty jako je sodík nebo draslík) z těla. Při prvních příznacích průjmu pijte velká množství vody a okamžitě kontaktujte svého lékaře. Co nejdříve začněte užívat vhodnou léčbu průjmu, například loperamid.
Během léčby tímto přípravkem byly pozorovány tyto další nežádoucí účinky:
Velmi časté nežádoucí účinky (mohou postihnout více než 1 z 10 lidí)
- Nevolnost (pocit na zvracení)
- Bolest ve spodní části těla (břicho)
- Abnormální výsledky jaterních testů.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 lidí)
- Zvracení
- Ztráta chuti k jídlu
- Pokles tělesné hmotnosti
- Krvácení z nosu
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 lidí)
- Vysoký krevní tlak (hypertenze)
- Žloutenka, tedy žlutá barva kůže a očního bělma způsobená vysokými hladinami bilirubinu Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Ofev uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a blistru. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte přípravek Ofev při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Nepoužívejte tento přípravek, pokud si všimnete, že blistr, ve kterém jsou tobolky umístěny, je otevřený nebo je tobolka poničená.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
Co přípravek Ofev obsahuje
- Léčivou látkou je nintedanibum. Jedna tobolka obsahuje nintedanibum 100 mg (ve formě nintedanibi esilas).
- Pomocnými látkami jsou:
Obsah tobolky: Střední nasycené triacylglyceroly, tvrdý tuk, sójový lecithin (E322)
Obal tobolky: Želatina, glycerol 85%, oxid titaničitý (E171), červený oxid železitý (E172),
žlutý oxid železitý (E172)
Potiskový inkoust: Šelak, černý oxid železitý (E172), propylenglykol (E1 520)
Jak přípravek Ofev vypadá a co obsahuje toto balení
Přípravek Ofev 100 mg tobolky jsou neprůhledné, oválné, měkké želatinové tobolky broskvové barvy s černým potiskem na jedné straně - symbolem společnosti Boehringer Ingelheim a číslicí „100“.
K dispozici jsou dvě velikosti balení přípravku Ofev 100 mg tobolky:
- 30x1 měkká tobolka v perforovaných jednodávkových hliníkových blistrech
- 60 x 1 měkká tobolka v perforovaných jednodávkových hliníkových blistrech
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Německo
Výrobce
Boehringer Ingelheim Pharma GmbH & Co. KG
Binger Strasse 173
D-55216 Ingelheim am Rhein
Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien SCS Boehringer Ingelheim Comm.V Tél/Tel: +32 2 773 33 11 |
Lietuva Boehringer Ingelheim RCV GmbH & Co KG Lietuvos filialas Tel: +370 37 473922 |
|
Btarapna Eboprarep HHrenxaňM P^B rMÓX h Ko. Kr - KnoH Ebnrapna Ten: +359 2 958 79 98 |
Luxembourg/Luxemburg SCS Boehringer Ingelheim Comm.V Tél/Tel: +32 2 773 33 11 |
|
Česká republika Boehringer Ingelheim spol. s r.o. Tel: +420 234 655 111 |
Magyarország Boehringer Ingelheim RCV GmbH & Co KG Magyarországi Fióktelepe Tel: +36 1 299 8900 |
|
Danmark Boehringer Ingelheim Danmark A/S Tlf: +45 39 15 88 88 |
Malta Boehringer Ingelheim Ltd. Tel: +44 1344 424 600 |
|
Deutschland Boehringer Ingelheim Pharma GmbH & Co. KG Tel: +49 (0) 800 77 90 900 |
Nederland Boehringer Ingelheim b.v. Tel: +31 (0) 800 22 55 889 |
|
Eesti Boehringer Ingelheim RCV GmbH & Co KG Eesti filiaal Tel: +372 612 8000 |
Norge Boehringer Ingelheim Norway KS Tlf: +47 66 76 13 00 |
|
EXXáSa Boehringer Ingelheim Ellas A.E. T^: +30 2 10 89 06 300 |
Osterreich Boehringer Ingelheim RCV GmbH & Co KG Tel: +43 1 80 105-0 |
|
Espaňa Boehringer Ingelheim Espana, S.A. Tel: +34 93 404 51 00 |
Polska Boehringer Ingelheim Sp. z o.o. Tel: +48 22 699 0 699 |
|
France Boehringer Ingelheim France S.A.S. Tél: +33 3 26 50 45 33 |
Portugal Boehringer Ingelheim, Unipessoal, Lda. Tel: +351 21 313 53 00 |
|
Hrvatska Boehringer Ingelheim Zagreb d.o.o. Tel: +385 1 2444 600 |
Románia Boehringer Ingelheim RCV GmbH & Co KG Viena - Sucursala Bucuresti Tel: +40 21 302 2800 |
|
Ireland Boehringer Ingelheim Ireland Ltd. Tel: +353 1 295 9620 |
Slovenija Boehringer Ingelheim RCV GmbH & Co KG Podružnica Ljubljana Tel: +386 1 586 40 00 |
Italia
Boehringer Ingelheim Italia S.p.A.
Tel: +39 02 5355 1
Kúnpoq
Boehringer Ingelheim Ellas A.E.
T^: +30 2 10 89 06 300
Latvija
Boehringer Ingelheim RCV GmbH & Co KG
Latvijas filiale
Tel: +371 67 240 011
Slovenská republika
Boehringer Ingelheim RCV GmbH & Co KG organizačná zložka Tel: +421 2 5810 1211
Suomi/Finland
Boehringer Ingelheim Finland Ky Puh/Tel: +358 10 3102 800
Sverige
Boehringer Ingelheim AB Tel: +46 8 721 21 00
United Kingdom
Boehringer Ingelheim Ltd.
Tel: +44 1344 424 600
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Ofev 150 mg měkké tobolky
Nintedanibum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Ofev a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Ofev užívat
3. Jak se přípravek Ofev užívá
4. Možné nežádoucí účinky
5. Jak přípravek Ofev uchovávat
6. Obsah balení a další informace
1. Co je přípravek Ofev a k čemu se používá
Přípravek Ofev obsahuje léčivou látku nintedanib a používá se k léčbě idiopatické plicní fibrózy (IPF).
IPF je stav, při kterém tkáň v plicích v průběhu času zbytňuje, tuhne a jizví se. Zjizvení následně omezuje schopnost přenášet kyslík z plic do krevního oběhu a začne být obtížné zhluboka dýchat. Přípravek Ofev pomáhá omezit jizvení a tuhnutí plic.
2. Čemu musíte věnovat pozornost, než začnete přípravek Ofev užívat
Neužívejte přípravek Ofev:
- jestliže jste alergický(á) na nintedanib, arašídy nebo sóju nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před užitím přípravku Ofev se poraďte se svým lékařem nebo lékárníkem,
- jestliže máte nebo jste někdy měl(a) problémy s játry,
- jestliže máte nebo jste někdy měl(a) problémy s krvácením,
- jestliže užíváte léčivé přípravky k ředění krve (jako je warfarin, fenprokumon nebo heparin) k prevenci tvorby krevních sraženin,
- jestliže máte nebo jste někdy měl(a) problémy se srdcem (například srdeční příhodu),
- jestliže jste v nedávné době podstoupil(a) operaci. Nintedanib může ovlivnit způsob, jak se hojí
rány. Léčba přípravkem Ofev proto obvykle bude na určitou dobu přerušena v případě, že máte být operován(a). Lékař rozhodne o tom, kdy bude léčba tímto přípravkem obnovena.
Na základě těchto informací může lékař provést některé krevní testy, například zkontrolovat funkci jater. Lékař s Vámi výsledky těchto testů prodiskutuje a rozhodne, zda můžete dostat přípravek Ofev.
Během užívání tohoto přípravku svého lékaře okamžitě informujte,
- jestliže dostanete průjem. Je důležité průjem včas léčit (viz bod 4);
- jestliže zvracíte nebo trpíte nevolností (pocit na zvracení);
- jestliže pociťujete silnou bolest břicha, máte horečku, zimnici, trpíte nevolností, zvracíte nebo máte napnuté břicho nebo se cítíte nafouklý(á), protože to by mohly být příznaky proděravění stěny trávicího traktu („gastrointestinální perforace“);
- jestliže pociťujete bolest v končetině, nebo ji máte oteklou, zarudlou či teplou, protože to by mohly být příznaky krevní sraženiny v jedné z žil (druh krevní cévy);
- jestliže pociťujete tlak nebo bolest na hrudi, obvykle na levé straně těla, bolest šíje, čelisti, ramene nebo paže, rychlý srdeční tep, dušnost, pocit na zvracení nebo zvracení, protože to by mohly být příznaky srdeční příhody (infarktu);
- jestliže máte velké krvácení.
Děti a dospívající
Děti a dospívající do 18 let věku nesmějí přípravek Ofev užívat.
Další léčivé přípravky a přípravek Ofev
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, včetně rostlinných přípravků a přípravků, které jste získal(a) bez lékařského předpisu.
Přípravek Ofev se může vzájemně ovlivňovat s některými jinými léčivými přípravky. Následující přípravky mohou zvyšovat hladiny nintedanibu v krvi a tak mohou zvyšovat riziko nežádoucích účinků (viz bod 4):
- léčivý přípravek používaný k léčbě plísňových infekcí (ketokonazol)
- léčivý přípravek používaný k léčbě bakteriálních infekcí (erytromycin)
- léčivý přípravek, který ovlivňuje imunitní systém (cyklosporin)
Následující léčivé přípravky mohou snižovat hladiny nintedanibu v krvi, a tak mohou snižovat účinnost přípravku Ofev:
- antibiotikum používané k léčbě tuberkulózy (rifampicin)
- léčivé přípravky používané k léčbě záchvatů křečí (karbamazepin, fenytoin)
- rostlinný přípravek k léčbě deprese (třezalka tečkovaná)
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Neužívejte tento přípravek během těhotenství, neboť může poškodit ještě nenarozené dítě a způsobovat vrozené vady.
Ženy, které mohou otěhotnět, musí v době, kdy užívají přípravek Ofev, a nejméně 3 měsíce po ukončení léčby, používat účinnou kombinaci antikoncepčních metod, včetně bariérových metod jako druhé formy antikoncepce. O pro Vás nejvhodnějších metodách antikoncepce se poraďte se svým lékařem.
Okamžitě informujte svého lékaře nebo lékárníka v případě, že během léčby přípravkem Ofev otěhotníte.
Během léčby přípravkem Ofev nekojte, protože existuje určité riziko poškození kojeného dítěte.
Řízení dopravních prostředků a obsluha strojů
Přípravek Ofev může mít mírný vliv na schopnost řídit a obsluhovat stroje. Jestliže se necítíte dobře, neměl(a) byste řídit ani obsluhovat stroje.
Přípravek Ofev obsahuje sójový lecithin
Jestliže jste alergický(á) na sóju nebo arašídy, tento lék neužívejte (viz bod 2).
3. Jak se přípravek Ofev užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka je jedna 150mg tobolka dvakrát denně (celkem 300 mg denně). Tobolky užijte dvakrát denně s odstupem 12 hodin každý den přibližně ve stejnou dobu, například jednu tobolku ráno a jednu tobolku večer. To zajistí, že se v krevním oběhu udržuje stálé množství nintedanibu. Tobolky spolkněte celé s vodou, tobolky nežvýkejte ani nedrťte. Doporučuje se, abyste tobolky užíval(a) s jídlem, tedy během nebo těsně před nebo po jídle.
Neužívejte víc než doporučenou dávku dvě 150mg tobolky přípravku Ofev denně.
Jestliže doporučenou dávku dvou 150mg tobolek přípravku Ofev denně netolerujete (viz možné nežádoucí účinky v bodě 4), lékař Vám může denní dávku přípravku Ofev snížit. Sám (sama) dávku nesnižujte ani léčbu neukončujte bez předchozí konzultace s lékařem.
Lékař Vám může doporučenou dávku snížit na dvakrát 100 mg denně (celkem 200 mg denně).
V takovém případě Vám lékař k léčbě předepíše přípravek Ofev 100 mg tobolky. Neužívejte víc než doporučenou dávku dvou 100 mg tobolek přípravku Ofev denně, pokud Vám byla denní dávka snížena na 200 mg denně.
Jestliže jste užil(a) více přípravku Ofev, než jste měl(a)
Okamžitě kontaktujte svého lékaře nebo lékárníka.
Jestliže jste zapomněl(a) užít přípravek Ofev
Jestliže jste zapomněl(a) užít předchozí dávku, neužívejte dvě tobolky najednou. Další 150mg dávku přípravku Ofev užijte dle plánu v další plánovanou dobu doporučenou lékařem nebo lékárníkem.
Jestliže jste přestal(a) užívat přípravek Ofev
Nepřerušujte užívání přípravku Ofev bez toho, aniž byste se nejprve poradil(a) s lékařem. Je důležité, abyste tento přípravek užíval(a) každý den po celou dobu, kdy Vám jej lékař předepisuje.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Musíte být obzvlášť obezřetný(á) v případě, že se u Vás v průběhu léčby přípravkem Ofev objeví následující nežádoucí účinky:
Průjem (velmi časté, může postihnout více než 1 z 10 lidí):
Průjem může vést k dehydrataci: ke ztrátě tekutin a důležitých solí (elektrolyty jako je sodík nebo draslík) z těla. Při prvních příznacích průjmu pijte velká množství vody a okamžitě kontaktujte svého lékaře. Co nejdříve začněte užívat vhodnou léčbu průjmu, například loperamid.
Během léčby tímto přípravkem byly pozorovány tyto další nežádoucí účinky:
Velmi časté nežádoucí účinky (mohou postihnout více než 1 z 10 lidí)
- Nevolnost (pocit na zvracení)
- Bolest ve spodní části těla (břicho)
- Abnormální výsledky jaterních testů.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 lidí)
- Zvracení
- Ztráta chuti k j ídlu
- Pokles tělesné hmotnosti
- Krvácení z nosu
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 lidí)
- Vysoký krevní tlak (hypertenze)
- Žloutenka, tedy žlutá barva kůže a očního bělma způsobená vysokými hladinami bilirubinu Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Ofev uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a blistru. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte přípravek Ofev při teplotě do 25 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Nepoužívejte tento přípravek, pokud si všimnete, že blistr, ve kterém jsou tobolky umístěny, je otevřený nebo je tobolka poničená.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
Co přípravek Ofev obsahuje
- Léčivou látkou je nintedanibum. Jedna tobolka obsahuje nintedanibum 150 mg (ve formě nintedanibi esilas).
- Pomocnými látkami jsou:
Obsah tobolky: Střední nasycené triacylglyceroly, tvrdý tuk, sójový lecithin (E322)
Obal tobolky: Želatina, glycerol 85%, oxid titaničitý (E171), červený oxid železitý (E172),
žlutý oxid železitý (E172)
Potiskový inkoust: Šelak, černý oxid železitý (E172), propylenglykol (E1 520)
Jak přípravek Ofev vypadá a co obsahuje toto balení
Přípravek Ofev 150 mg tobolky jsou neprůhledné, oválné, měkké želatinové tobolky hnědé barvy s černým potiskem na jedné straně - symbolem společnosti Boehringer Ingelheim a číslicí „150“.
K dispozici jsou dvě velikosti balení přípravku Ofev 150 mg tobolky:
- 30x1 měkká tobolka v perforovaných jednodávkových hliníkových blistrech
- 60 x 1 měkká tobolka v perforovaných jednodávkových hliníkových blistrech
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Německo
Výrobce
Boehringer Ingelheim Pharma GmbH & Co. KG
Binger Strasse 173
D-55216 Ingelheim am Rhein
Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien SCS Boehringer Ingelheim Comm.V Tél/Tel: +32 2 773 33 11 |
Lietuva Boehringer Ingelheim RCV GmbH & Co KG Lietuvos filialas Tel: +370 37 473922 |
|
Btarapna Eboprarep HHrenxaňM P^B rMÓX h Ko. Kr - KnoH Ebnrapna Ten: +359 2 958 79 98 |
Luxembourg/Luxemburg SCS Boehringer Ingelheim Comm.V Tél/Tel: +32 2 773 33 11 |
|
Česká republika Boehringer Ingelheim spol. s r.o. Tel: +420 234 655 111 |
Magyarország Boehringer Ingelheim RCV GmbH & Co KG Magyarországi Fióktelepe Tel: +36 1 299 8900 |
|
Danmark Boehringer Ingelheim Danmark A/S Tlf: +45 39 15 88 88 |
Malta Boehringer Ingelheim Ltd. Tel: +44 1344 424 600 |
|
Deutschland Boehringer Ingelheim Pharma GmbH & Co. KG Tel: +49 (0) 800 77 90 900 |
Nederland Boehringer Ingelheim b.v. Tel: +31 (0) 800 22 55 889 |
|
Eesti Boehringer Ingelheim RCV GmbH & Co KG Eesti filiaal Tel: +372 612 8000 |
Norge Boehringer Ingelheim Norway KS Tlf: +47 66 76 13 00 |
|
EXXáSa Boehringer Ingelheim Ellas A.E. T^: +30 2 10 89 06 300 |
Osterreich Boehringer Ingelheim RCV GmbH & Co KG Tel: +43 1 80 105-0 |
|
Espaňa Boehringer Ingelheim Espana, S.A. Tel: +34 93 404 51 00 |
Polska Boehringer Ingelheim Sp. z o.o. Tel: +48 22 699 0 699 |
|
France Boehringer Ingelheim France S.A.S. Tél: +33 3 26 50 45 33 |
Portugal Boehringer Ingelheim, Unipessoal, Lda. Tel: +351 21 313 53 00 |
|
Hrvatska Boehringer Ingelheim Zagreb d.o.o. Tel: +385 1 2444 600 |
Románia Boehringer Ingelheim RCV GmbH & Co KG Viena - Sucursala Bucuresti Tel: +40 21 302 2800 |
|
Ireland Boehringer Ingelheim Ireland Ltd. Tel: +353 1 295 9620 |
Slovenija Boehringer Ingelheim RCV GmbH & Co KG Podružnica Ljubljana Tel: +386 1 586 40 00 |
Italia
Boehringer Ingelheim Italia S.p.A.
Tel: +39 02 5355 1
Kúnpoq
Boehringer Ingelheim Ellas A.E.
T^: +30 2 10 89 06 300
Latvija
Boehringer Ingelheim RCV GmbH & Co KG
Latvijas filiale
Tel: +371 67 240 011
Slovenská republika
Boehringer Ingelheim RCV GmbH & Co KG organizačná zložka Tel: +421 2 5810 1211
Suomi/Finland
Boehringer Ingelheim Finland Ky Puh/Tel: +358 10 3102 800
Sverige
Boehringer Ingelheim AB Tel: +46 8 721 21 00
United Kingdom
Boehringer Ingelheim Ltd.
Tel: +44 1344 424 600
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
43