Odomzo 200 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Odomzo 200 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje sonidegibum 200 mg (jako sonidegibi phosphas). Pomocná látka se známým účinkem:
Jedna tvrdá tobolka obsahuje 38,6 mg monohydrátu laktosy. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka (tobolka).
Neprůhledná růžová tvrdá tobolka, která obsahuje bílý až téměř bílý prášek s granulemi, s „NVR“ vytištěným černým inkoustem na víčku a „SONIDEGIB 200MG“ vytištěným černým inkoustem na těle tobolky.
Velikost tobolky je „Velikost #00“ (rozměry 23,3 x 8,53 mm).
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Odomzo je indikován k léčbě dospělých pacientů s lokálně pokročilým bazocelulárním karcinomem (BCC), který nelze léčit chirurgicky nebo radioterapií.
4.2 Dávkování a způsob podání
Přípravek Odomzo má být předepsán specializovaným lékařem nebo pod dohledem specializovaného lékaře, který má zkušenosti s léčbou dané indikace.
Dávkování
Doporučená dávka je 200 mg sonidegibu perorálně jednou denně, nejméně dvě hodiny po jídle a nejméně jednu hodinu před následujícím jídlem, každý den ve stejnou dobu.
Léčba by měla probíhat tak dlouho, dokud je pozorován klinický přínos nebo dokud se neprojeví nepřijatelná toxicita.
Změny v dávkování _při zvýšené hladině kreatinfosfokinázv (CK) a _při nežádoucích účincích týkajících se svalů
Pokud se objeví zvýšená hladina kreatinfosfokinázy (CK) nebo nežádoucí účinky týkající se svalů, může být potřeba přechodně přerušit dávkování a/nebo snížit dávky přípravku Odomzo.
Tabulka 1 shrnuje doporučení pro přerušení a/nebo snížení dávky přípravku Odomzo v managementu symptomatického zvýšení hladiny CK a nežádoucích účinků týkajících se svalů (jako jsou myalgie, myopatie a/nebo spasmus).
Tabulka 1 Doporučené změny dávkování a postup při symptomatickém zvýšení hladiny CK a při nežádoucích účincích týkajících se svalů.
|
Závažnost zvýšení hladiny CK |
Změny dávkování* a doporučení |
|
Stupeň 1 [CK zvýšení >ULN - 2,5 x ULN] |
• Pokračovat v léčbě se stejnou dávkou a sledovat hladiny CK týdně až do dosažení výchozí hladiny, a poté je sledovat měsíčně. Sledovat změny svalových symptomů až do dosažení výchozího stavu. • Pravidelně kontrolovat renální funkce (sérový kreatinin) a zajistit, že pacient je adekvátně hydratován. |
|
Stupeň 2 bez renálního poškození (sérový kreatinin < ULN) [CK zvýšení >2,5 x ULN - 5 x ULN] |
• Přerušit léčbu a sledovat hladiny CK týdně až do dosažení výchozí hladiny. • Sledovat změny svalových symptomů až do dosažení výchozího stavu. Po dosažení výchozího stavu/hladiny pokračovat v léčbě se stejnou dávkou, a poté měřit CK měsíčně. • Pravidelně kontrolovat renální funkce (sérový kreatinin) a zajistit, že pacient je adekvátně hydratován. • Pokud se symptomy vyskytnou znovu, přerušit léčbu až do dosažení výchozího stavu/hladiny. Znovu zahájit léčbu sonidegibem v dávce 200 mg obden a dodržovat stejná monitorovací doporučení. Pokud symptomy přetrvávají i přes dávkování obden, je nutné zvážit přerušení léčby. |
|
Stupeň 3 nebo 4 bez renálního poškození (sérový kreatinin < ULN) [stupeň 3 (CK zvýšení >5 x ULN - 10 x ULN)] [stupeň 4 (CK zvýšení >10 x ULN)] |
• Přerušit léčbu a sledovat hladiny CK týdně až do dosažení výchozí hladiny. Sledovat změny svalových symptomů až do dosažení výchozího stavu. • Pravidelně kontrolovat renální funkce (sérový kreatinin) a zajistit, že pacient je adekvátně hydratován. • Pokud není renální funkce poškozena a CK dosáhne výchozí hladiny, je možné zvážit obnovení léčby s dávkou 200 mg obden. Po obnovení podávání sonidegibu by měly být hladiny CK měřeny po dobu dvou měsíců každý týden, a poté jednou měsíčně. |
Stupeň 2, 3 nebo 4 s renálním poškozením (sérový kreatinin > ULN)
Pokud je renální funkce poškozena, je potřeba přerušit léčbu a zajistit, že pacient je adekvátně hydratován a posoudit jiné možné příčiny renálního poškození. Sledovat hladiny CK a sérového kreatininu týdně až do dosažení výchozí hladiny. Sledovat změny svalových symptomů až do dosažení výchozího stavu.
Pokud se hladina CK a sérového kreatininu vrátí na výchozí hladinu, je možné zvážit obnovení léčby s dávkou 200 mg obden a měřit hladiny CK po dobu dvou měsíců každý týden, a poté jednou měsíčně; jinak je třeba léčbu trvale ukončit._
* Výše uvedená doporučení pro změnu dávkování jsou založena na „Common Terminology
Criteria for Adverse Events (CTCAE) v4.03“, vyvinutá Národním ústavem pro rakovinu USA (the National Cancer Institute). CTCAE je standardizovaná klasifikace nežádoucích účinků, používaná v posuzování léčivých přípravků k léčbě rakoviny.
ULN: horní hranice normy
Jiné úpravy dávky
Postup při zvládnutí závažných nebo nepřijatelných nežádoucích účinků může vyžadovat dočasné přerušení léčby (s dalším snížením nebo bez dalšího snížení dávky) nebo ukončení léčby.
V případě nutného přerušení léčby a následného zmírnění nežádoucích účinků < stupeň 1, zvažte obnovení podávání přípravku Odomzo ve stejné dávce jako před přerušením léčby.
V případě nutného snížení dávky má být dávkování sníženo na 200 mg obden. Pokud se i po snížení dávky vyskytne stejný nežádoucí účinek a nedojde k jeho zlepšení, je nutné zvážit ukončení léčby přípravkem Odomzo.
Vzhledem k dlouhému poločasu sonidegibu se očekává objevení úplného účinku přerušení dávky nebo úpravy dávky sonidegibu u několika nežádoucích účinků obecně po několika týdnech (viz bod 5.2.).
Trvání léčby
V klinických studiích pokračovala léčba přípravkem Odomzo až do progrese onemocnění nebo do nepřijatelné toxicity. Přerušení léčby na dobu až 3 týdnů bylo povoleno vzhledem k individuální snášenlivosti.
Prospěch z pokračování léčby má být pravidelně hodnocen spolu s optimálním trváním léčby, které se mění u každého jednotlivého pacienta.
Zvláštní populace
Pacienti s poruchou funkce ledvin
Sonidegib nebyl hodnocen v samostatné farmakokinetické studii u pacientů s renálním poškozením.
Z dostupných údajů vyplývá, že eliminace sonidegibu ledvinami je zanedbatelná. Z populační farmakokinetické analýzy vyplývá, že lehké nebo středně těžké renální poškození nemělo významný vliv na zdánlivou clearance (CL/F) sonidegibu, což naznačuje, že úprava dávkování u pacientů s renálním poškozením není nutná (viz bod 5.2). U pacientů s těžkou poruchou funkce ledvin nejsou k dispozici žádné údaje o účinnosti a bezpečnosti.
Pacienti s poruchou funkce jater
U pacientů s poruchou funkce jater není nutná úprava dávky (viz bod 5.2).
Starší pacienti (>65 let)
Údaje o účinnosti a bezpečnosti u pacientů 65 let a starších nenaznačují nutnost úpravy dávkování u těchto pacientů (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Odomzo u dětí a dospívajících mladších 18 let s bazocelulárním karcinomem nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Odomzo se podává perorálně. Tobolky se musí polykat celé. Nesmí se kousat nebo drtit. Tobolky se nesmí otevírat kvůli riziku teratogenity (viz bod 5.3).
Přípravek Odomzo se musí užít nejméně dvě hodiny po jídle a nejméně jednu hodinu před následujícím jídlem. Pokud dojde během podávání ke zvracení, není povoleno opětovné podání dávky před další plánovanou dávkou.
Pokud se dávka vynechá, musí se užít hned, jak se to zjistí. Pokud však uběhlo víc než šest hodin od plánovaného podání dávky, dávka se neužije a je podána až následující plánovaná dávka.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 Těhotenství a kojení (viz body 4.4 a 4.6).
Ženy ve fertilním věku, které nesplňují požadavky Programu prevence početí pro pacienty/pacientky, užívající přípravek Odomzo (viz body 4.4 a 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Nežádoucí účinky týkající se svalů
V pivotní studii II. fáze byly pozorovány svalové spasmy, myalgie, myopatie a případy zvýšení hladiny CK. U většiny pacientů léčených přípravkem Odomzo 200 mg denně, kteří měli zvýšenou hladinu CK stupně 2 nebo vyšší, se objevily svalové symptomy dříve, než zvýšení hladiny CK.
U většiny pacientů byly svalové symptomy a zvýšená hladina CK vyřešeny vhodnými opatřeními.
Všichni pacienti, kteří začínají terapii přípravkem Odomzo, musí být informováni o rizicích nežádoucích účinků týkajících se svalů, včetně možnosti rabdomyolýzy. Pacienti musí být poučeni o tom, že mají ihned hlásit každou nevysvětlitelnou bolest svalů, citlivost nebo slabost, která se objeví během terapie přípravkem Odomzo nebo pokud symptomy přetrvávají i po přerušení léčby.
Hladiny CK se mají kontrolovat před začátkem léčby a pokud je klinicky indikováno, tak i potom, například v případě výskytu symptomů týkajících se svalů. Pokud je zaznamenána klinicky zvýšená hladina CK, je třeba zkontrolovat renální funkce (viz bod 4.2).
Při změně dávkování nebo přerušení je třeba dodržovat doporučení (viz bod 4.2). Postup při vysokém stupni zvýšení hladiny CK, kdy se používá podpůrná terapie včetně dostatečné hydratace, by měl být zvážen podle místních standardů lékařské praxe a léčebných doporučení.
Pacienti by měli být důkladně monitorováni pro symptomy týkající se svalů, pokud je přípravek Odomzo podáván v kombinaci s určitými léčivými přípravky, které mohou zvýšit potenciální riziko vzniku svalové toxicity (např. inhibitory CYP3A4, chlorochin, hydroxychlorochin, deriváty kyseliny fíbrové (fibráty), penicilamin, zidovudin, niacin a inhibitory HMG-CoA reduktázy) (viz bod 4.5).
Pacienti s neuromuskulámími poruchami (např. zánětlivé myopatie, svalová dystrofie, amyotrofická laterální skleróza, spinální muskulární atrofie) musí být důkladně monitorováni kvůli zvýšenému riziku svalové toxicity.
Embryofetální úmrtí nebo závažné vrozené vady
Přípravek Odomzo může způsobit embryofetální úmrtí nebo závažné vrozené vady při podávání v těhotenství. Na základě mechanismu účinku, ve studiích na zvířatech, sonidegib vykazuje teratogenní a fetotoxický účinek. Ženy, které užívají přípravek Odomzo, nesmí být těhotné nebo otěhotnět v průběhu léčby a 20 měsíců po ukončení léčby.
Kritéria definující ženy ve fertilním věku
V „Programu prevence početí pro pacienty/pacientky užívající přípravek Odomzo“ je žena ve fertilním věku definována jako pohlavně zralá žena, která
• menstruovala během předchozích 12 po sobě následujících měsíců,
• nepodstoupila hysterektomii nebo bilaterální ooforektomii, nebo která neměla lékařsky potvrzené předčasné ovariální selhání,
• nemá XY genotyp, Turnerův syndrom nebo děložní agenezi,
• se stane amenoreická po terapii rakoviny, včetně léčby přípravkem Odomzo.
Doporučení
Pro ženy ve fertilním věku
Přípravek Odomzo je kontraindikován u žen ve fertilním věku, které nesplňují podmínky Programu prevence početí pro pacienty/pacientky užívající přípravek Odomzo. Žena ve fertilním věku musí porozumět, že:
• Užíváním přípravku Odomzo vystavuje nenarozené dítě riziku teratogenity.
• Nesmí užívat přípravek Odomzo v těhotenství nebo pokud plánuje otěhotnět.
• Musí mít negativní těhotenský test provedený zdravotnickým pracovníkem v období 7 dnů před začátkem léčby přípravkem Odomzo.
• Během léčby musí mít negativní těhotenský test, který se provádí měsíčně, i kdyby se stala amenoreickou.
• Nesmí otěhotnět během léčby přípravkem Odomzo a 20 měsíců po poslední dávce.
• Musí dodržovat spolehlivá antikoncepční opatření.
• Musí používat dvě metody doporučené antikoncepce (viz bod „Antikoncepce“ uvedený níže a bod 4.6) během léčby přípravkem Odomzo, pokud nepotvrdí, že nemá pohlavní styk (abstinence).
• Musí oznámit svému poskytovateli zdravotní péče, pokud se objeví během léčby a během 20 měsíců po poslední dávce kterýkoli z následujících případů:
o otěhotní nebo se z jakékoli příčiny domnívá, že může být těhotná, o vynechání očekávané menstruace,
o přestane užívat antikoncepci (pokud nepotvrdí, že nemá pohlavní styk (abstinuje), o potřebuje změnit antikoncepci.
• Nesmí kojit během užívání přípravku Odomzo a 20 měsíců po poslední dávce.
Pro muže
Sonidegib může prostoupit do spermatu. Aby se předešlo případné fetální expozici během těhotenství, pacient musí porozumět, že:
• Užíváním přípravku Odomzo vystavuje nenarozené dítě riziku teratogenity, pokud provádí nechráněné sexuální aktivity s těhotnou ženou.
• Musí vždy použít doporučenou antikoncepci (viz bod „Antikoncepce“ uvedený níže a bod 4.6).
• Oznámí svému poskytovateli zdravotní péče, pokud jeho partnerka otěhotní během jeho užívání přípravku Odomzo nebo během 6 měsíců po poslední dávce.
Pro zdravotnické _pracovníky
Zdravotničtí pracovníci musí poučit pacienty, aby porozuměli a potvrdili všechny podmínky Programu prevence početí pro pacienty/pacientky užívající přípravek Odomzo.
Antikoncepce
Ženy ve fertilním věku
Ženy ve fertilním věku musí během užívání přípravku Odomzo a po dobu 20 měsíců po ukončení léčby používat dvě metody doporučené antikoncepce, včetně jedné vysoce spolehlivé metody a bariérové metody (viz bod 4.6).
Muži
Pacienti - muži, včetně těch, kteří podstoupili vasektomii, musí vždy používat kondom
(se spermicidem, pokud je dostupný) během pohlavního styku s partnerkou, a to během užívání
přípravku Odomzo a 6 měsíců po skončení terapie (viz body 4.6 a 5.3).
Těhotenský test
U žen ve fertilním věku musí být zdravotnickým pracovníkem proveden těhotenský test v průběhu 7 dnů před začátkem léčby přípravkem Odomzo a jednou měsíčně během léčby. Těhotenské testy musí mít minimální senzitivitu 25mIU/ml podle místní dostupnosti. V případě, že je pacientka těhotná, léčba nesmí být zahájena. Pokud otěhotní během léčby, podávání přípravku Odomzo musí být okamžitě ukončeno (viz bod 5.3). U pacientek, u kterých se vyskytne amenorea během terapie přípravkem Odomzo, se musí v průběhu léčby pokračovat v provádění těhotenského testu jednou měsíčně.
Omezení při předepisování a výdeji ženám ve fertilním věku
Přípravek Odomzo musí být předepsán a vydán do 7 dnů od provedení negativního těhotenského testu. Předepisuje se dávka na 30 dnů léčby, pokračování v léčbě vyžaduje novou preskripci.
Edukační materiál
Zdravotnickým pracovníkům a pacientům bude držitel rozhodnutí o registraci poskytovat edukační materiály (Program prevence početí pro pacienty/pacientky užívající přípravek Odomzo), jak se vyhnout embryonální a fetální expozici přípravkem Odomzo, aby se zdůraznila potenciální rizika spojená s užíváním tohoto léčivého přípravku.
Dárcovství krve
Pacienti by měli být poučení o tom, že nesmí darovat krev během užívání přípravku Odomzo a nejméně 20 měsíců po skončení léčby.
Dárcovství spermatu
Pacienti - muži nesmí darovat sperma během léčby přípravkem Odomzo a nejméně 6 měsíců po skončení léčby.
Interakce
Je třeba se vyvarovat současnému podávání se silnými CYP induktory (např. rifampicin, karbamazepin nebo fenytoin), protože nelze vyloučit riziko snížení plazmatických koncentrací a snížení účinnosti sonidegibu (viz také bod 4.5).
Kožní spinocelulámí karcinom (cutaneous squamous cell carcinoma. cuSCC)
Pacienti s pokročilým BCC mají zvýšené riziko vzniku cuSCC. Případy vzniku cuSCC byly zaznamenány u pacientů s pokročilým BCC léčených přípravkem Odomzo. Nebylo stanoveno. jestli je výskyt cuSCC spojen s podáváním přípravku Odomzo. Z tohoto důvodu by měli být všichni pacienti rutinně monitorováni v průběhu užívání přípravku Odomzo a cuSCC by měl být léčen podle standardů ošetřování.
Dodatečná opatření
Pacienti musí být poučeni, že nesmí nikdy dát tento léčivý přípravek jiné osobě. Veškeré tobolky, nevypotřebované na konci léčby. musí pacient zlikvidovat v souladu s místními požadavky (např. vrátit do lékárny nebo lékaři).
Pomocné látky
Tobolky přípravku Odomzo obsahují monohydrát laktosy. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Sonidegib je primárně metabolizován přes CYP3A4 a souběžné užívání silných inhibitorů nebo induktorů CYP3A4 může významně zvýšit nebo snížit koncentrace sonidegibu.
Látky, které mohou zvýšit plasmatickou koncentraci sonidegibu
Podávání 800 mg sonidegibu v jedné dávce s ketokonazolem (200 mg dvakrát denně po dobu 14 dní). který je silným inhibitorem CYP3A, zdravým subjektům, vyústilo v 2,25násobné a 1,49násobné zvýšení AUC, resp. Cmax sonidegibu, v porovnání s podáním sonidegibu samostatně. Podle simulace povede delší souběžné podávání silných inhibitorů CYP3A4 (např. déle než 14 dní) k několikanásobné změně v expozici sonidegibu. Pokud je nutné souběžné podání silného CYP3A inhibitoru, je nutné snížit dávku sonidegibu na 200 mg obden. Silné CYP3A inhibitory zahrnují (ale nejen) ritonavir, sachinavir, telithromycin, ketokonazol, itrakonazol, vorikonazol, posakonazol a nefazodon. Pacienti musí být pečlivě monitorováni z hlediska nežádoucích účinků, pokud je jedna z těchto látek podávána spolu se sonidegibem.
Látky, které mohou snížit plasmatickou koncentraci sonidegibu
Podávání 800 mg sonidegibu v jedné dávce s rifampicinem (600 mg denně po dobu 14 dní), který je silným induktorem CYP3A, zdravým subjektům, vyústilo v 72% a 54% snížení AUC, resp. Cmax sonidegibu, v porovnání s podáním sonidegibu samostatně. Souběžné podávání sonidegibu se silnými induktory CYP3A snižuje plasmatickou koncentraci sonidegibu. Souběžně nemají být podávány silné CYP3A induktory; včetně (ale nejen) karbamazepinu, fenobarbitalu, fenytoinu, rifabutinu, rifampicinu a třezalky tečkované (Hypericum perforatum). Pokud je nutné souběžně se sonidegibem podat silný induktor CYP3A4, je třeba zvážit zvýšení denní dávky sonidegibu na 400-800 mg. Na základě farmakokinetických dat se předpokládá se, že tato dávka sonidegibu upravuje AUC na běžnou hladinu pozorovanou bez podávání induktorů, pokud souběžné podávání s induktorem netrvá déle než 14 dní. Delší souběžné podávání s induktorem se nedoporučuje, protože se sníží expozice sonidegibu, což může ohrozit jeho účinnost. Po ukončení podávání silného induktoru se dávka sonidegibu sníží na původní.
Výsledky klinické studie u zdravých jedinců ukázaly změnu expozice sonidegibu (32% a 38% snížení AUC a Cmax) po souběžném podání jednotlivé dávky 200 mg přípravku Odomzo se 40 mg esomeprazolu (inhibitor protonové pumpy) denně po dobu 6 dní. Nepředpokládá se, že tato interakce je klinicky významná.
Účinky sonidegibu na jiné léčivé přípravky
Sonidegib je kompetitivní inhibitor CYP2B6 a CYP2C9 in vitro, potenciálně zvyšuje koncentrace látek, které jsou metabolizovány těmito enzymy. Sonidegib je také inhibitorem „breast cancer resistance proteinu“ (BCRP) (IC50 ~1,5 pM). Pacienti užívající souběžně substráty enzymů CYP2B6 a CYP2C9 nebo BCRP transportéry, musí být pečlivě monitorováni pro výskyt nežádoucích účinků. Substrátům enzymů CYP2B6 a CYP2C9 s úzkým terapeutickým rozmezím (např. warfarin, acenokumarol, efavirenz, methadon) nebo BCRP substrátům s úzkým terapeutickým rozmezím (např. methotrexát, mitoxantron, irinotekan, topotekan) je lépe se vyhnout.
Látky, které mohou zvýšit výskyt účinků týkajících se svalů
U pacientů užívajících přípravek Odomzo současně s jinými léčivými přípravky zvyšujícími riziko toxicity spojené se svaly, dochází k zvýšenému riziku vzniku nežádoucích účinků týkajících se svalů kvůli zkřížené toxicitě. Pokud se objeví svalové symptomy, pacienti musí být pečlivě monitorováni a je třeba zvážit úpravu dávkování.
V pivotní studii II. fáze užívalo 12 pacientů (15,2 %), léčených přípravkem Odomzo 200 mg, současně inhibitory HMG-CoA reduktázy (9 užívalo pravastatin, 3 užívali jiné inhibitory HMG-CoA reduktázy kromě pravastatinu, např. rosuvastatin a simvastatin). Z těchto pacientů mělo 7 (58,3 %) svalové symptomy stupně závažnosti až 1, zatímco u 43 pacientů (64,1 %), kteří neužívali inhibitory HMG-CoA reduktázy, se vyskytly symptomy stupně závažnosti až 3. U žádného z pacientů užívajících inhibitory HMG-CoA reduktázy se nevyskytl stupeň závažnosti 3/4 zvýšení hladiny CK, oproti 6 pacientům (9,0 %), kteří neužívali inhibitory HMG-CoA reduktázy.
Interakce s jídlem
Biologická dostupnost sonidegibu je zvýšena v přítomnosti jídla (viz bod 5.2). Přípravek Odomzo se musí užívat nejméně dvě hodiny po jídle a nejméně hodinu před následujícím jídlem.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Kvůli riziku embryofetálního úmrtí nebo závažných vrozených vad způsobených sonidegibem, nesmí být ženy užívající přípravek Odomzo těhotné nebo otěhotnět během léčby a po dobu 20 měsíců po jejím ukončení (viz bod 4.4).
Přípravek Odomzo je kontraindikován u žen ve fertilním věku, které nesplňují podmínky Programu prevence početí pro pacienty/pacientky užívající přípravek Odomzo (viz bod 4.3).
V případě otěhotnění nebo vynechání menstruace
Pokud pacientka otěhotní, vynechá jí menstruace nebo má z jakéhokoli důvodu podezření, že může být těhotná, musí to okamžitě oznámit svému ošetřujícímu lékaři.
Přetrvávající vynechání menstruace během léčby přípravkem Odomzo je nutné považovat za těhotenství až do vyšetření a potvrzení lékařem.
Antikoncepce u mužů a žen
Ženy ve _ fertilním věku
Ženy ve fertilním věku musí být schopny dodržovat vysoce spolehlivá antikoncepční opatření. Ženy musí používat dvě metody doporučené antikoncepce, včetně jedné vysoce spolehlivé metody a bariérové metody v průběhu terapie přípravkem Odomzo a 20 měsíců po poslední dávce. Ženy ve fertilním věku s nepravidelným menstruačním cyklem nebo bez menstruace, musí dodržovat všechna doporučení spolehlivé antikoncepce.
Muži
Není známo, jestli se sonidegib vyskytuje ve spermatu. Muži nesmí zplodit dítě nebo darovat sperma během užíváni přípravku Odomzo a nejméně 6 měsíců po ukončení terapie. Aby se předešlo potenciální fetální expozici během těhotenství, pacienti - muži, včetně těch, kteří podstoupili vasektomii, musí vždy používat kondom (se spermicidem, pokud je dostupný) při pohlavním styku s partnerkou během terapie přípravkem Odomzo a 6 měsíců po poslední dávce.
Doporučené _ formy vysoce spolehlivých metod antikoncepce:
• Tubální sterilizace
• Vasektomie
• Nitroděložní tělísko (IUD)
Doporučené bariérové metody:
• Jakýkoliv mužský kondom (se spermicidem, pokud je dostupný)
• Pesar (se spermicidem, pokud dostupný)
Údaje o podávání sonidegibu těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly teratogenitu a fetotoxicitu (viz bod 5.3). Přípravek Odomzo je v těhotenství kontraindikován.
Kojení
Není známo, zda se sonidegib vylučuje do lidského mateřského mléka. Vzhledem k možným závažným nežádoucím účinkům, jako jsou vážné vývojové vady u kojených novorozenců/kojenců, nesmí ženy během užívání přípravku Odomzo a 20 měsíců po ukončení léčby kojit (viz bod 5.3).
Fertilita
Údaje ze studií na potkanech a psech naznačují, že mužská a ženská fertilita, může být ireverzibilně ohrožena při užívání přípravku Odomzo (viz bod 5.3). U žen ve fertilním věku byla dodatečně v klinických studiích pozorována amenorea (viz bod 4.8). Před začátkem léčby přípravkem Odomzo by měly být diskutovány možnosti zachování plodnosti se ženami ve fertilním věku.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Odomzo nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Bezpečnost přípravku Odomzo byla hodnocena v pivotní studii II. fáze u celkově 229 dospělých pacientů s místně pokročilým nebo metastazujícím BCC. Pacienti byli léčeni přípravkem Odomzo 200 mg denně (n=79) nebo 800 mg denně (n=150). Střední doba trvání léčby byla 11,0 měsíců u pacientů léčených přípravkem Odomzo v doporučené dávce 200 mg (rozsah od 1,3 do 33,5 měsíců). Během léčby přípravkem Odomzo 200 mg denně nebo do 30 dnů od jejího ukončení bylo u pacientů s metastazujícím nebo lokálně pokročilým BCC zaznamenáno 1 úmrtí.
Nejčastějšími nežádoucími účinky vyskytujícími se u >10 % pacientů léčených přípravkem Odomzo 200 mg byly svalové spasmy, alopecie, porucha chuti, únava, nauzea, muskuloskeletální bolest, průjem, pokles tělesné hmotnosti, snížená chuť k jídlu, myalgie, abdominální bolest, bolest hlavy, bolest, zvracení a pruritus.
Nejčastějšími nežádoucími účinky stupně 3/4 vyskytujícími se u >2 % pacientů léčených přípravkem Odomzo 200 mg byly únava, pokles tělesné hmotnosti a svalové spasmy.
Četnější výskyt nežádoucích účinků (tabulka 2) byl hlášen u pacientů užívajících přípravek Odomzo 800 mg než u pacientů užívajících přípravek Odomzo 200 mg, kromě muskuloskeletální bolesti, průjmu, abdominální bolesti, bolesti hlavy a pruritu. To samé platilo u stupně 3/4 nežádoucích účinků, kromě únavy.
Tabulkový přehled nežádoucích účinků
Nežádoucí účinky u doporučené dávky z pivotní klinické studie II. fáze (tabulka 2) jsou uvedené podle MedDRA (Medical Dictionary for Regulatory Activities) verze 17.1 systémové orgánové klasifikace. Nežádoucí účinky jsou v rámci každé systémové orgánové klasifikace řazené dle četnosti, nejčastější nežádoucí účinky jsou uvedeny jako první. V rámci řazení do skupin podle četnosti jsou nežádoucí účinky uvedené podle klesající závažnosti. Navíc je odpovídající kategorie četnosti u každého nežádoucího účinku založena na následujících kritériích (CIOMS III): velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit).
Tabulka 2 Nežádoucí účinky pozorované v pivotní studii II. fáze
|
Třídy primárních orgánových systémů Přednostní název |
Četnost všech stupňů 200 mg |
|
Poruchy metabolismu a výživy | |
|
Snížená chuť k jídlu |
Velmi časté |
|
Dehydratace |
Časté |
|
Poruchy nervového systému | |
|
Porucha chuti |
Velmi časté |
|
Velmi časté | |
|
Gastrointestinální poruchy | |
|
Velmi časté | |
|
Velmi časté | |
|
Abdominální bolest |
Velmi časté |
|
Velmi časté | |
|
Časté | |
|
Zácpa |
Časté |
|
Gastroezofageální reflux |
Časté |
|
Poruchy kůže a podkožní tkáně | |
|
Alopecie |
Velmi časté |
|
Pruritus |
Velmi časté |
|
Časté | |
|
Abnormální růst vlasů |
Časté |
|
Poruchy svalové a kosterní soustavy a pojivové t |
íáně |
|
Svalové spasmy |
Velmi časté |
|
Muskuloskeletální bolest |
Velmi časté |
|
Myalgie |
Velmi časté |
|
Myopatie [svalová únava a svalová slabost] |
Časté |
|
Reprodukční systém a choroby prsů | |
|
Amenorea* |
Velmi časté |
|
Celkové poruchy a reakce v místě aplikace | |
|
Únava |
Velmi časté |
|
Bolest |
Velmi časté |
|
Vyšetření | |
|
Snížení tělesné hmotnosti |
Velmi časté |
|
*Ze 79 pacientů užívajících přípravek Odomzo 200 mg bylo 5 žen ve fertilním věku. Mezi těmito ženami byla amenorea pozorována u 1 pacientky (20 %). | |
Klinicky významné laboratorní odchylky
Nejčastěji zaznamenanými laboratorními odchylkami stupně 3/4 s výskytem >5 % u pacientů léčených přípravkem Odomzo 200 mg byly zvýšení lipázy a zvýšení krevní CK (tabulka 3).
Tabulka 3 Laboratorní odchylky*
|
Laboratorní test |
Četnost všech stupňů 200 mg |
|
Hematologické parametry | |
|
Snížený hemoglobin |
Velmi časté |
|
Snížený počet lymfocytů |
Velmi časté |
|
Biochemické parametry | |
|
Zvýšený sérový kreatinin |
Velmi časté |
|
Zvýšená sérová kreatinfosfokináza (CK) |
Velmi časté |
|
Zvýšená krevní glukóza |
Velmi časté |
|
Zvýšená lipáza |
Velmi časté |
|
Zvýšená alaninaminotransamináza (ALT) |
Velmi časté |
|
Zvýšená aspartátaminotransamináza (AST) |
Velmi časté |
|
Zvýšená amyláza |
Velmi časté |
|
* Uvedeno podle nejhorší laboratorní hodnoty po léčbě bez ohledu na výchozí hodnoty, třídění podle CTCAE verze 4.03. | |
Popis vybraných nežádoucích účinků
Účinky týkající se svalů včetně zvýšení hladiny CK
Svalová toxicita je nejvíce klinicky relevantním nežádoucím účinkem hlášeným u pacientů léčených sonidegibem a předpokládá se, že se jedná o účinek třídy inhibitorů Hedgehog (Hh) signální dráhy. V pivotní studii II. fáze byly svalové spasmy nejčastějším nežádoucím účinkem týkajícím se svalů a byly zaznamenány u menšího počtu pacientů, kteří užívali přípravek Odomzo 200 mg (54 %), než u pacientů užívajících přípravek Odomzo 800 mg (69 %).
U 8 % pacientů užívajících přípravek Odomzo 200 mg bylo zaznamenáno zvýšení krevní hladiny CK stupně 3/4. U většiny pacientů, kteří měli zvýšení CK stupně 2 nebo víc, se objevily svalové symptomy před zvýšením hladiny CK. Medián času, kdy u těchto pacientů došlo ke zvýšení laboratorních hodnot CK stupně závažnosti 2 a vyšší, byl 12,9 týdne (rozsah 2 - 39 týdnů) po zahájení terapie přípravkem Odomzo a medián času, kdy došlo k vyřešení (k normalizaci nebo k stupni 1), byl 12 dní (95 % CI 8 až 14 dní).
Jeden pacient užívající přípravek Odomzo 200 mg zaznamenal svalové symptomy a zvýšení hladiny CK nad 10x ULN a vyžadoval podání tekutin intravenózně, v porovnání s 6 pacienty užívajícími přípravek Odomzo 800 mg.
V pivotní studii II. fáze nebyly potvrzeny žádné případy nahlášené rabdomyolýzy (definované jako hladiny CK >10 násobně nad úroveň před začátkem léčby nebo nad úroveň výchozí hladiny
nebo >10x ULN, pokud nebyla zaznamenána výchozí hladina plus 1,5 násobné zvýšení sérového kreatininu z hladiny před začátkem léčby nebo z výchozí hladiny). V nepivotní studii byl potvrzen jeden případ hlášený u pacienta, který užíval přípravek Odomzo 800 mg.
Amenorea
V pivotní studii II. fáze se u 2 (14,3 %) ze 14 žen s fertilním potenciálem nebo ve fertilním věku, které byly sterilizované podvázáním vejcovodů, vyskytla amenorea během léčby přípravkem Odomzo
200 mg nebo 800 mg jednou denně.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V studiích eskalace dávky byl přípravek Odomzo podáván v dávce do 3000 mg perorálně jednou denně. Pacienti mají být pečlivě monitorováni na nežádoucí účinky a ve všech případech předávkování by měla být přijata vhodná podpůrná opatření.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antineoplastické látky, jiné antineoplastické látky, ATC kód: L01XX48 Mechanismus účinku
Sonidegib je perorálně biologicky dostupný inhibitor Hh signální dráhy. Váže se k smoothened (Smo), což je molekula podobná receptoru spřaženému s G proteinem, která pozitivně reguluje Hh dráhu a nakonec aktivuje a uvolňuje gliom- asociované onkogenní (GLI) transkripční faktory, které indukují transkripci Hh cílových genů zapojených do proliferace, diferenciace a přežití. Chybná Hh signalizace je spojena s patogenezí několika typů karcinomu včetně bazocelulárního karcinomu (BCC). Sonidegib navázaný k Smo bude inhibovat Hh signalizaci a v důsledku toho blokovat signální transdukci.
Farmakodynamické účinky
Analýzy QTc intervalu v souvislosti s plazmatickou koncentrací sonidegibu naznačují, že horní mez jednostranného 95 % intervalu spolehlivosti pro zvýšení QTc byla pod 5 ms v ustáleném stavu Cmax u 800mg denní dávky, což způsobuje 2,3násobnou expozici v porovnání s doporučenou dávkou 200 mg. Z toho vyplývá, že terapeutické dávky přípravku Odomzo nezpůsobují klinicky signifikantní prodloužení QTc intervalu. Dále se zjistilo, že plasmatické koncentrace sonidegibu nad hladiny dosažené po podání terapeutické dávky nebyly spojené se život ohrožujícími arytmiemi nebo torsades de pointes.
Odpověď tumoru byla nezávislá na dávce přípravku Odomzo nebo plasmatické koncentraci v rozsahu dávky od 200 mg do 800 mg.
Klinická účinnost a bezpečnost
Randomizovaná dvojitě zaslepená studie II. fáze dvou různých dávek (200 mg nebo 800 mg jednou denně) přípravku Odomzo byla provedena u 230 pacientů s lokálně pokročilým bazocelulárním karcinomem (laBCC) (n=194) nebo s metastazujícím bazocelulárním karcinomem (mBCC=36).
Z 230 pacientů mělo 16 pacientů diagnózu Gorlinova syndromu (15 laBCC a 1 mBCC). Dospělí pacienti (>18 let) s laBCC nebo s mBCC, kteří nebyli kandidáty pro radioterapii, chirurgickou nebo jinou lokální terapii, byli randomizováni k užívání přípravku Odomzo v dávce buď 200 mg nebo 800 mg denně až do progrese nemoci nebo nepřijatelné toxicity.
Primárním cílovým parametrem účinnosti studie byla objektivní míra odpovědi podle modifikovaných Kritérií hodnocení odpovědi u solidních nádorů (modified Response Evaluation Criteria in Solid Tumours, mRECIST) u pacientů s laBCC a RECIST 1.1 u pacientů s mBCC stanovená podle centrálního hodnocení. Sekundární cílové parametry zahrnovaly trvání odpovědi, čas do odpovědi nádoru a přežívání bez progrese (PFS) podle mRECIST u pacientů s laBCC a RECIST 1.1 u pacientů s mBCC stanovených podle centrálního hodnocení.
Nezávislá posudková komise (IRC) posoudila u pacientů s laBCC celkovou odpověď, která byla integrována z centrálně hodnocených MRI snímků, digitálních klinických fotografií a histopatologie podle mRECIST. U laBCC byly odebrány vícenásobné punkční biopsie pokaždé, kdy posouzení odpovědi nebylo jednoznačné kvůli výskytu ulcerací, cyst a/nebo jizvení/fibrózy. MRI nádorová odpověď se hodnotila podle RECIST 1.1. Odpověď podle digitálních klinických fotografií byla hodnocena podle upravených kritérií Světové zdravotnické organizace (WHO) [částečná odpověď (PR): >50 % pokles v souhrnu svislých parametrů (SPD) léze; kompletní odpověď (CR): zmizení všech lézí; progrese nemoci: >25 % zvýšení v SPD lézí]. Pro stanovení úplné odpovědi musely všechny způsoby vyhodnocení prokázat nepřítomnost tumoru.
Z 230 randomizovaných pacientů 79 užívalo přípravek Odomzo 200 mg. Z těchto 79 pacientů 66 (83,5 %) byli laBCC pacienti (37 [46,8 %] s agresivní histologií a 29 [36,7 %] s neagresivní histologií) a 13 (16,5 %) byli mBCC pacienti. Střední věk všech pacientů užívajících přípravek Odomzo 200 mg byl 67 let (59,5 % byli ve věku >65 let), 60,8 % byli muži a 89,9 % byli pacienti bělošské rasy.
Většina pacientů (laBCC 74 %, mBCC 92 %) podstoupila předchozí terapie včetně chirurgické terapie (laBCC 73 %, mBCC 85 %), radioterapie (laBCC 18 %, mBCC 54 %) a antineoplastické terapie (laBCC 23 %, mBCC 23 %).
Klíčové výsledky účinnosti podle centrálního hodnocení a podle hodnocení zkoušejícím jsou uvedeny v tabulce 4.
Tabulka 4 Přehled účinnosti podle centrálního hodnocení a podle hodnocení zkoušejícím FASa
|
Odomzo 200 mg Centrální Zkoušející laBCC laBCC N=66 N=66 | ||
|
Míra objektivní odpovědi, n (%) |
37 (56,1) |
47 (71,2) |
|
95% CI |
(43,3, 68,3) |
(58,7, 81,7) |
|
Nejlepší celková odpověď, n (%) Kompletní odpověď |
3 (4,5)b |
6 (9,1) |
|
Částečná odpověď |
34 (51,5) |
41 (62,1) |
|
Stabilizace nemoci |
23 (34,8) |
14 (21,2) |
|
Progrese nemoci |
1 (1,5) |
1 (1,5) |
|
Neznámá |
5 (7,6) |
4 (6,1) |
|
Čas do odpovědi nádoru (měsíce) Medián |
4,0 |
2,5 |
|
95% CI |
(3,8, 5,6) |
(1,9, 3,7) |
|
Trvání odpovědi Počet případů* |
10 |
21 |
|
Počet cenzurovaných |
27 |
26 |
|
Medián (měsíce) |
NE |
14,3 |
|
95% CI |
(NE) |
(12,0, 20,2) |
|
Pravděpodobnost bez událostí (%), (95% CI) 6 měsíců |
86,9 (68,6, 94,9) |
89,8 (74,8, 96,1) |
|
9 měsíců |
75,8 (55,7, 87,7) |
80,7 (63,5, 90,4) |
|
12 měsíců |
65,6 (43,2, 81,0) |
70,9 (52,2, 83,3) |
|
Přežívání bez progrese Počet případů* |
15 |
26 |
|
Počet cenzurovaných |
51 |
40 |
|
Medián (měsíce) |
22,1 |
19,4 |
|
95% CI |
(NE) |
(16,6, 22,6) |
|
Pravděpodobnost přežívání bez progrese (%), (95% CI) 6 měsíců |
94,8 (84,6, 98,3) |
94,8 (84,7, 98,3) |
|
12 měsíců |
82,2 (67,0, 90,8) |
76,0 (61,4, 85,7) |
|
a Set pro plnou analýzu zahrnoval všechny randomizované pacienty (zamýšlená populace). b Použita jenom negativní histologie na stanovení CR u pacientů, kteří mají alespoň PR od jiných způsobů | ||
|
vyhodnocení (MRI nebo fotografie), vyústila do míry CR 22,7%. *Událost odkazuje na progresi nemoci nebo úmrtí z jakékoliv příčiny. FAS: Set pro plnou analýzu CI: interval spolehlivosti NE: nevážený (medián nebyl ještě dosažen; celkový medián trvání odezvy 26,3 měsíce) | ||
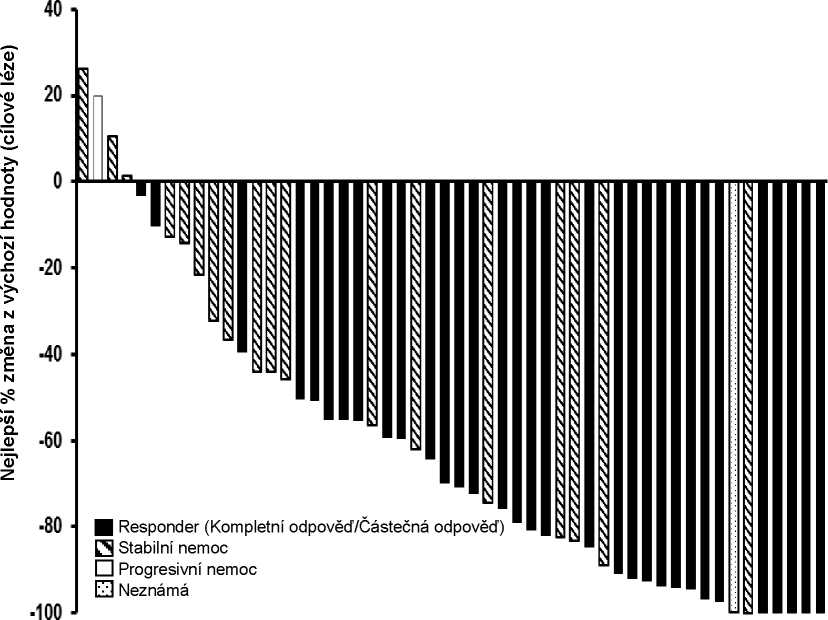
Obrázek 1 ukazuje nejlepší změnu velikosti cílové léze u každého pacienta s laBCC s dávkou 200 mg podle centrálního hodnocení.
Obrázek 1 Nejlepší změna z výchozí hodnoty cílové léze u pacientů s laBCC centrálním hodnocením FAS.
Pacienty hlášené výstupy byly hodnoceny jako výzkumný cílový parametr pomocí Dotazníku kvality života evropské organizace pro výzkum a léčbu rakoviny (EORTC QLQ-C30) a podle jeho modulu asociovaného s rakovinou krku a hlavy (H&N35).
U většiny pacientů se vyskytlo udržení a/nebo zlepšení symptomů spojených s nemocí, fungováním a zdravotním stavem. Čas do zhoršení v předem stanovených PRO-škálách (odpovídající >10bodovým zhoršením bez následného zlepšení) v podstatě odrážel odhadovaný PFS.
V pivotní studii 27,8% pacientů přerušilo léčbu kvůli nežádoucím účinkům, které byly většinou lehké nebo středně těžké (viz bod 4.8).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Odomzo u všech podskupin pediatrické populace s bazocelulárním karcinomem (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Po podání jednotlivé dávky přípravku Odomzo (100 mg až 3000 mg) bez jídla u pacientů s karcinomem, byl medián času k dosažení maximální koncentrace (Tmax) 2 až 4 hodiny. Sonidegib vykazoval v závislosti na dávce zvýšení AUC a Cmax nad dávkový rozsah od 100 mg do 400 mg, ale méně než u dávky nad 400 mg. Změny v clearance nebyly zaznamenány u opakovaného podání podle populační farmakokinetické analýzy a odhadovaná akumulace v rovnovážném stavu byla 19násobná
bez ohledu na dávku. Rovnovážný stav byl dosažen přibližně 4 měsíce po začátku užívání sonidegibu. Průměrný rovnovážný stav Ctrough u 200 mg byl 830 ng/ml (rozsah 200 až 2400 ng/ml) u pacientů s rakovinou. Ve srovnání se stavem nalačno se Cmax a AUC přípravku Odomzo 800 mg zvýšily 7,8 a 7,4krát při podání dávky s vysoce tučným jídlem.
Distribuce
Na základě populační farmakokinetické analýzy u 351 pacientů, kteří užívali perorálně dávky přípravku Odomzo v dávkovém rozsahu 100 mg až 3000 mg, ustálený stav zdánlivého distribučního objemu (Vss/F) byl 9 170 litrů. Hladina ustáleného stavu sonidegibu byla v kůži 6násobně vyšší než v plazmě.
Sonidegib byl vysoce vázán na lidské plasmatické proteiny (lidský sérový albumin a alfa-1 kyselý glykoprotein) in vitro (>97 %), a vazba nebyla závislá na koncentraci od 1 ng/ml do 2500 ng/ml.
Z in vitro údajů je zřejmé, že sonidegib není substrát P-gp, BCRP nebo multirezistentního proteinu 2 (MRP2). Sonidegib neinhiboval apikální efluxní transportéry, P-gp nebo MRP2, jaterní příjmové transportéry OATP1B1nebo OATP1B3, renální transportéry OAT1 a OAT3 přijímající organické anionty nebo transportéry OCT1 a OCT2 přijímající organické kationty v klinicky významných koncentracích.
Biotransformace
Sonidegib je primárně metabolizován přes CYP3A4. Nezměněný sonidegib představoval 36 % cirkulující radioaktivity a hlavním cirkulujícím metabolitem (45 % původní expozice) identifikovaným v plazmě je hydrolyzovaný produkt sonidegibu, který je farmakologicky inaktivní. Všechny metabolity byly považovány za 4 až 90krát méně silné než sonidegib.
Eliminace
Sonidegib a jeho metabolity jsou primárně eliminovány jaterní cestou, přičemž 93,4 % z podané dávky se objevilo ve stolici a 1,95 % v moči. Nezměněný sonidegib ve stolici představoval 88,7 % podané dávky a nebyl detekovatelný v moči. Poločas eliminace (tJ/2) sonidegibu odhadovaný z populačního farmakokinetického modelování byl přibližně 28 dní.
Speciální populace
Pacienti s poruchou funkce jater
Farmakokinetika sonidegibu byla sledována u pacientů s lehkou (Child-Pugh třída A; n=8), středně těžkou (Child-Pugh třída B; n=8) nebo těžkou (Child-Pugh třída C; n=9) poruchou funkce jater a u 8 zdravých jedinců s normální funkcí jater. Cmax sonidegibu po jednotlivé perorální 800mg dávce byla o 20 % a 21 % a 60 % nižší u lehkého, středně těžkého a těžkého jaterního poškození, v porovnání s normální jaterní funkcí. AUCinf sonidegibu byla o 40 %, 22 % a 8 % nižší, v uvedeném pořadí. AUClast byla o 35 % nižší u lehkého jaterního poškození, o 14 % vyšší u středně těžkého jaterního poškození a o 23 % nižší u těžkého jaterního poškození. U pacientů s jaterním poškozením není nutná úprava dávkování.
Pacienti s poruchou funkce ledvin
Vliv renálního poškození na systémovou expozici sonidegibu nebyl studován. Protože sonidegib není vylučován ledvinami, předpokládá se, že u pacientů s renálním poškozením nenastane změna v systémové expozici. Populační farmakokinetická analýza nepotvrdila signifikantní vliv renální funkce (clearance kreatininu >27 ml/min) na zdánlivou clearance (CL/F) sonidegibu, což naznačuje, že úprava dávkování u pacientů s renálním poškozením není nutná.
Vliv věku, tělesné hmotnosti a pohlaví
Populační farmakokinetická analýza ukázala, že neexistuje žádný klinicky významný vliv věku (testovaný rozsah 20-93 let, průměr 61 let), tělesné hmotnosti (testovaný rozsah 42-181 kg, průměr
77 kg), pohlaví nebo clearance kreatininu (testovaný rozsah 27,3-290 ml/min, průměr 92,9 ml/min.) na systémovou expozici sonidegibu.
Vliv etnika
Cmax a AUCinf sonidegibu u zdravé japonské populace byly 1,56 a 1,68krát vyšší než u zdravé západní populace v jednotlivé dávce 200 mg.
5.3 Předklinické údaje vztahující se k bezpečnosti
Sonidegib byl hodnocen na potkanech a psech.
Obecná toxikologie
Většina nežádoucích účinků sonidegibu může být připsána jeho farmakologickému mechanismu účinku na vývojové dráhy a účinky byly podobné u potkanů i psů. Většina účinků se vyskytla těsně k plánovaným lidským expozicím. Tyto účinky pozorované u klinicky závažných expozic zahrnují uzavření kostních růstových štěrbin, účinek na růst zubů, účinek na mužský a ženský reprodukční systém, atrofii vlasových folikulů s alopecií, gastrointestinální toxicita s poklesem tělesné hmotnosti a vlivy na lymfatické uzliny. U expozic vysoko nad klinickou expozicí byla dodatečným cílovým orgánem ledvina.
Kancerogeneze a mutageneze
Studie kancerogenity nebyly u sonidegibu provedeny, ale sonidegib neměl genotoxický účinek ve studiích prováděných in vitro a in vivo.
Reprodukční a vývojová toxicita
U sonidegibu byla prokázána fetotoxicita u králíků, jak bylo dokázáno potratem a/nebo kompletní resorpcí plodů a teratogenitou, která vyústila do závažných malformací u velmi nízké expozice. Teratogenní účinky zahrnovaly vertebrální malformace a malformace distálních končetin a prstů, závažné kraniofaciální malformace a jiné závažné vady střední čáry. Fetotoxicita u králíků byla také pozorována ve velmi nízké mateřské expozici. U potkaních samic byla snížena fertilita v nízké expozici. U potkaních samců, léčených sonidegibem, expozice přibližně 2násobkem klinické expozice neovlivnila plodnost samců.
6. FARMACEUTICKÉ ÚDAJE 6.1 Seznam pomocných látek
Obsah tobolky
Krospovidon typ A Monohydrát laktosy Magnesium-stearát Poloxamer 188
Koloidní bezvodý oxid křemičitý Natrium-lauryl-sulfát
Obal tobolky
Želatina
Červený oxid železitý (E172) Oxid titaničitý (E171)
Potiskový inkoust
Černý oxid železitý (E172)
Propylenglykol (E1520)
Šelak
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
10 x 1 tvrdá tobolka v PCTFE/PVC/Al jednodávkových perforovaných blistrech.
Balení obsahuje 10 nebo 30 tvrdých tobolek.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1030/001
EU/1/15/1030/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
14. srpna 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Novartis Pharma GmbH Roonstrasse 25 90429 Norimberk Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ
LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Před uvedením přípravku na trh v každém členském státě si musí držitel rozhodnutí o registraci (MAH) nechat schválit následující příslušným národním úřadem:
• Národní část informačních dopisů pro zdravotnické pracovníky (DHPC)
• Metodologii sběru informací o užívání přípravku Odomzo a compliance s farmakovigilančním programem v těhotenství a jeho účinnosti
• Formát a obsah příručky pro zdravotnické pracovníky a pacienty
MAH musí rozeslat DHPC při uvedení přípravku na trh, který musí obsahovat následující:
• Hlavní text odsouhlasen CHMP
• Národní specifické požadavky odsouhlasené národní kompetentní autoritou týkající se:
- Distribuce přípravku
- Opatření k zajištění, že všechna příslušná opatření byla provedena před předepsáním a dispenzací přípravku Odomzo.
MAH musí nepřetržitě kontrolovat, že všichni lékaři předepisující přípravek Odomzo, poskytují
následující:
• Informaci o přípravku
• Edukační materiál pro zdravotnické pracovníky
• Připomínkovou kartu zdravotnického pracovníka
• Edukační materiál pro pacienty
• Připomínkovou kartu pacienta
Edukační materiál k přípravku Odomzo pro zdravotnické pracovníky musí obsahovat následující
důležité součásti:
• Stručný popis přípravku Odomzo, jeho povolenou indikaci a dávkování
• Požadavky k informování pacientů o teratogenním riziku spojeném s přípravkem Odomzo a potřebou vyhnout se fetální expozici
• Popis programu prevence otěhotnění a kategorizaci pacientů založenou na pohlaví a schopnosti otěhotnět
• Informaci o doporučených formách antikoncepce u žen a mužů
• Povinnosti zdravotnických pracovníků v souvislosti s předepisováním přípravku Odomzo
• Bezpečnostní doporučení pro ženy ve fertilním věku
• Bezpečnostní doporučení pro muže
• Požadavky v případě otěhotnění
• Informujte pacienty, že nesmí darovat krev v průběhu terapie přípravkem Odomzo a nejméně 20 měsíců po poslední dávce
• Kontrolní seznam pro zdravotnické pracovníky k ujištění, že pacienti dostávají náležité poradenství
• Zajistit, že všichni pacienti vyplní a podepíšou Formulář ověření poskytnutého poradenství při užívání přípravku Odomzo, který se nachází v edukačních materiálech pro zdravotnické pracovníky
• Hlášení nežádoucích účinků
Edukační materiál k přípravku Odomzo pro pacienty musí obsahovat následující důležité součásti:
• Informace pro pacienty o rizicích teratogenity spojených s přípravkem Odomzo a nutnosti vyhnout se fetální expozici
• Nutnost používat vhodnou antikoncepci a definici vhodné antikoncepce.
• Národní nebo jiné vhodné specifické požadavky na preskripci přípravku Odomzo a jeho následnou dispenzaci
• Informaci o tom, že se přípravek Odomzo nesmí dávat žádné jiné osobě stejně jako informaci o likvidaci nepotřebného léku a nutnosti mít přípravek Odomzo mimo dohled a dosah dětí.
• Pacient nesmí darovat krev v průběhu terapie a nejméně 20 měsíců po poslední dávce
• Pacientka nesmí kojit v průběhu terapie a nejméně 20 měsíců po poslední dávce
• Pacient musí říct zdravotnickému pracovníkovi o kterémkoli nežádoucím účinku
• Informaci pro ženy ve fertilním věku
• Informace pro muže
Připomínková karta pro zdravotnické pracovníky musí obsahovat následující důležité části:
• Informaci pro ženy ve fertilním věku
• Informaci pro muže
• Nutnost říct pacientům, aby ihned oznámili ošetřujícímu zdravotnickému pracovníkovi, pokud se domnívají, že je pacientka těhotná nebo je těhotná partnerka pacienta.
• Připomenout pacientům vrátit nepoužité tobolky na konci léčby (likvidace bude záviset na místních požadavcích)
• Připomenout pacientům, že nesmí darovat krev v průběhu léčby a nejméně 20 měsíců po poslední dávce.
Připomínková karta pacienta musí obsahovat následující důležité části:
• Informaci pro pacienty o teratogenním riziku spojeném s přípravkem Odomzo a nutnosti vyhnout se fetální expozici
• Zákaz darování krve v průběhu léčby a nejméně 20 měsíců po poslední dávce
• Informaci pro ženy ve fertilním věku
• Informaci pro muže
• Nepoužité tobolky vrátit na konci léčby (likvidace bude záviset na místních požadavcích)
• Kontaktní telefonní čísla pro případ nouze
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Poregistrační studie účinnosti (PAES): MAH musí předložit analýzu studie CLDE225A2201 spolu s: • aktualizovanými analýzami účinnosti a bezpečnosti • korelativní analýzou odpovědi na léčbu a hladiny Gli 1 pro celou populaci studie pivotní studie v různých časových úsecích (např. výchozí hladina, čas odpovědi, čas progrese a j.) • aktualizovanými analýzami výsledků agresivními vs non-agresivními histologickými subtypy. |
30/10/2016 |
|
Poregistrační studie účinnosti (PAES): MAH musí předložit konečný CSR pro studii CLDE225A2201, včetně aktualizovaných analýz výsledků agresivními vs non-agresivními histologickými subtypy. |
30/10/2017 |
|
Poregistrační studie účinnosti (PAES): MAH musí předložit molekulární analýzu nádorového materiálu, který je stále dostupný, od léčených pacientů ze studie CLDE225A2201 s výskytem progrese na zkoumání mechanismů rezistence spojených s bodovou mutací v SMO, která může vést k reaktivaci Hh signální dráhy a znovunarůstání nádoru. |
30/10/2016 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
VNĚJŠÍ OBAL JEDNOTLIVÉHO BALENÍ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Odomzo 200 mg tvrdé tobolky sonidegibum
2. OBSAH LÉČIVÉ LÁTKY
Jedna tvrdá tobolka obsahuje sonidegibum 200 mg (jako sonidebigi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdé tobolky
10 x 1 tvrdá tobolka 30 x 1 tvrdá tobolka
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci. Tobolku nedrťte, neotevírejte a nežvýkejte.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Riziko vzniku závažných vrozených vad.
Neužívat v průběhu těhotenství a kojení.
Musíte dodržovat Program prevence početí pro pacienty/pacientky užívající přípravek Odomzo.
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 30 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Nepoužité tobolky se musí na konci léčby vrátit.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1030/001 10 tvrdých tobolek
EU/1/15/1030/002 30 tvrdých tobolek
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Odomzo 200 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTRY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Odomzo 200 mg tobolky sonidegibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
B. PŘÍBALOVÁ INFORMACE
Odomzo 200 mg tvrdá tobolka
sonidegibum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Odomzo může způsobit závažné vrozené vady. To může vést k úmrtí dítěte před narozením nebo krátce po narození. V průběhu užívání tohoto přípravku nesmíte otěhotnět. Musíte dodržovat antikoncepční opatření popsaná v této příbalové informaci.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Odomzo a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Odomzo užívat
3. Jak se přípravek Odomzo užívá
4. Možné nežádoucí účinky
5 Jak přípravek Odomzo uchovávat
6. Obsah balení a další informace
1. Co je přípravek Odomzo a k čemu se používá Co je přípravek Odomzo
Přípravek Odomzo obsahuje léčivou látku sonidegib. Je to přípravek určený k léčbě rakoviny.
K čemu se přípravek Odomzo používá
Přípravek Odomzo se používá k léčbě dospělých s rakovinou kůže zvané bazocelulární karcinom, a to v případech, kdy se rakovina rozšířila do okolí a nemůže být léčena chirurgicky nebo radiací (ozařováním).
Jak přípravek Odomzo působí
Normální růst buněk je kontrolován různými chemickými signály. U pacientů s bazocelulárním karcinomem (basaliomem) se změny vyskytují u genů kontrolujících část tohoto procesu, známého jako „hedgehog dráha“. To zapíná signály, které způsobují nekontrolovaný růst nádorových buněk. Přípravek Odomzo způsobuje blokování tohoto procesu, zastavení růstu rakovinných buněk a zastavení tvorby nových buněk.
2. Čemu musíte věnovat pozornost, než začnete přípravek Odomzo užívat
Přečtěte si zvláštní pokyny, které jste obdržel(a) od svého lékaře, zvláště působení přípravku Odomzo na nenarozené děti.
Čtěte pozorně a dodržujte doporučení obsažená v pacientské příručce a připomínkové kartě, kterou jste obdržel(a) od svého lékaře.
Neužívejte přípravek Odomzo:
• jestliže jste alergický(á) na sonidegib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
• jestliže jste těhotná nebo si myslíte, že můžete být těhotná. To je proto, že přípravek Odomzo může způsobit poškození nebo úmrtí Vašeho nenarozeného dítěte (viz bod „Těhotenství“).
• jestliže kojíte. To je proto, že není známo, jestli přípravek Odomzo může prostupovat do mateřského mléka a působit škodlivě na Vaše dítě (viz bod „Kojení“).
• jestliže jste schopná otěhotnět, ale nejste schopná nebo nejste ochotná dodržovat nezbytná opatření prevence početí uvedená v Programu prevence početí pro pacienty/pacientky užívající přípravek Odomzo.
Neužívejte přípravek Odomzo, pokud se Vás týká kterýkoli bod uvedený výše. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem před tím, než začnete užívat přípravek Odomzo.
Více informací k výše uvedeným bodům naleznete v odstavcích „Těhotenství“, „Kojení“ a „Plodnost“ a „Antikoncepce pro ženy a muže“.
Upozornění a opatření
- Přípravek Odomzo může způsobit svalové problémy. Řekněte svému lékaři před tím, než začnete přípravek Odomzo užívat, pokud jste někdy měl(a) svalové křeče nebo slabost nebo pokud užíváte ještě jiné léky. Některé léky (např. léky k léčbě vysokého cholesterolu) mohou zvýšit riziko svalových potíží. Okamžitě řekněte svému lékaři nebo lékárníkovi, pokud Vás budou bolet svaly nebo budete mít bezdůvodně svalové křeče nebo slabost v průběhu léčby přípravkem Odomzo. Váš lékař může změnit dávku nebo zastavit léčbu dočasně nebo úplně.
- V průběhu léčby přípravkem Odomzo a 20 měsíců po jejím ukončení nesmíte darovat krev.
- Jestliže jste pacient - muž, nesmíte darovat sperma během léčby přípravkem Odomzo a 6 měsíců po poslední dávce.
- Lékař bude pravidelně kontrolovat Vaši kůži pro další případ rakoviny zvané kožní spinocelulární karcinom (spinaliom, SCC). Není známo, jestli může být SCC spojen s léčbou přípravkem Odomzo. Tento typ rakoviny se obvykle objevuje na kůži poškozené sluncem, nerozšiřuje se a lze ho léčit. Oznamte lékaři, pokud zaznamenáte jakékoli změny na své kůži.
- Nikdy nedávejte tento lék žádné další osobě. Na konci léčby vraťte nepoužité tobolky. Zeptejte se svého lékaře nebo lékárníka, kam tobolky vrátit.
Krevní testy v průběhu terapie přípravkem Odomzo
Váš lékař bude provádět krevní testy před léčbou a možná také v průběhu léčby. Tyto testy budou kontrolovat zdravotní stav svalů měřením hladiny enzymu, který se jmenuje kreatinfosfokináza.
Děti a dospívající (do 18 let)
Přípravek Odomzo se nesmí podávat dětem a dospívajícím do 18 let. To je proto, že není známo, jestli je v této věkové skupině bezpečný nebo účinný. Ve studiích na zvířatech s tímto lékem byly zaznamenány problémy s růstem zubů a kostí.
Další léčivé přípravky a přípravek Odomzo
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. To zahrnuje i léky obdržené bez lékařského předpisu a rostlinné přípravky. To je proto, že přípravek Odomzo může ovlivnit působení některých léčiv. Některá jiná léčiva mohou též ovlivnit působení přípravku Odomzo nebo mohou víc přispět k tomu, že se u Vás objeví nežádoucí účinky.
Zejména řekněte svému lékaři nebo lékárníkovi, pokud užíváte kterýkoli z následujících přípravků:
• léky jako statiny a fibráty užívané k léčbě vysokého cholesterolu a lipidů (tuků)
• vitamin B3, známý též jako niacin
• léky jako methotrexát, mitoxantron, irinotekan nebo topotekan, užívané k léčbě určitých typů rakoviny nebo jiných onemocnění, jako jsou závažné kloubní problémy (revmatoidní artritida) a psoriáza (lupénka)
• léky jako warfarin nebo acenokumarol užívané k prevenci vzniku krevních sraženin
• léky jako telithromycin, rifampicin a rifabutin užívané k léčbě bakteriálních infekcí
• léky jako ketokonazol (kromě šamponů a krémů), itrakonazol, posakonazol nebo vorikonazol užívané k léčbě plísňových infekcí
• léky jako chlorochin a hydroxychlorochin užívané k léčbě parazitárních infekcí stejně jako jiných nemocí, jako např. revmatoidní artritida nebo lupus erythematodes
• léky jako ritonavir, sachinavir, efavirenz nebo zidovidin užívané k léčbě AIDS nebo HIV
• léky jako karbamazepin, fenytoin nebo fenobarbital užívané k léčbě akutních záchvatů (křečí)
• lék zvaný nefazodon užívaný k léčbě deprese
• lék zvaný penicilamin užívaný k léčbě revmatoidní artritidy
• rostlinný přípravek zvaný třezalka tečkovaná (též známá jako Hypericum perforatum) užívaná k léčbě deprese.
Pokud se na Vás vztahuje kterýkoli z výše uvedených, nebo si nejste jistý(á), řekněte to svému lékaři nebo lékárníkovi před zahájením užívání přípravku Odomzo.
Tyto přípravky by se měly užívat s opatrností nebo by se v průběhu léčby přípravkem Odomzo užívat neměly. Pokud užíváte jakýkoli z nich, lékař Vám může předepsat náhradní lék.
V průběhu léčby přípravkem Odomzo musíte svému lékaři nebo lékárníkovi sdělit, pokud Vám je předepsán jiný lék, který jste dosud neužíval(a).
Těhotenství
Neužívejte přípravek Odomzo, pokud jste těhotná nebo se domníváte, že můžete být těhotná, nebo plánujete otěhotnět v průběhu léčby přípravkem Odomzo nebo v průběhu 20 měsíců po jejím ukončení. Jakmile otěhotníte nebo se domníváte, že můžete být těhotná, musíte přestat užívat přípravek Odomzo a sdělit to svému lékaři. Přípravek Odomzo může způsobit závažné vrozené vady u dítěte nebo vést k úmrtí nenarozeného dítěte. Přesná opatření (Programu prevence početí pro pacienty/pacientky užívající přípravek Odomzo), které jste obdržel(a) od svého lékaře, obsahují zvláště informace o účincích přípravku Odomzo na nenarozené děti.
Kojení
V průběhu léčby nebo v průběhu 20 měsíců po jejím ukončení nekojte. Není známo, zda přípravek Odomzo prochází do mateřského mléka a poškozuje dítě.
Plodnost
Přípravek Odomzo může mít vliv na plodnost u mužů a žen. Pokud plánujete mít v budoucnu děti, poraďte se se svým lékařem.
Antikoncepce pro ženy a muže
Ženy
Před začátkem léčby přípravkem Odomzo se zeptejte svého lékaře, jestli byste mohla otěhotnět, dokonce i v případě, pokud už nemáte menstruaci (menopauza). Je důležité zkontrolovat se svým lékařem, jestli existuje riziko, že byste mohla otěhotnět.
Pokud jste schopna otěhotnět:
• musíte přijmout opatření, která zabrání otěhotnění v průběhu léčby přípravkem Odomzo,
• musíte v průběhu užívání přípravku Odomzo používat 2 metody antikoncepce, jednu vysoce spolehlivou a jednu bariérovou metodu (viz příklady níže),
• tuto antikoncepci musíte užívat ještě 20 měsíců po ukončení užívání přípravku Odomzo, protože zbytky léku zůstávají v těle dlouhou dobu.
Lékař s Vámi probere nejlepší metodu antikoncepce pro Vás.
Musíte použít jednu vysoce spolehlivou metodu, jako:
• nitroděložní tělísko (IUD)
• chirurgickou sterilizaci.
Musíte též použít jednu bariérovou metodu, jako:
• kondom (se spermicidem, pokud je dostupný)
• diafragma neboli pesar (se spermicidem, pokud je dostupný).
Lékař bude provádět těhotenské testy:
• alespoň 7 dní před začátkem léčby - na ujištění, že už nejste těhotná
• každý měsíc v průběhu léčby.
V průběhu léčby a 20 měsíců po jejím ukončení řekněte svému lékaři okamžitě, pokud:
• se domníváte, že antikoncepce z jakéhokoliv důvodu nefunguje
• j ste přestala menstruovat
• jste přestala užívat antikoncepci
• potřebujete změnit antikoncepci
Muži
V průběhu léčby přípravkem Odomzo vždy používejte kondom (se spermicidem, pokud je to možné) při pohlavním styku s partnerkou, i kdybyste měl vazektomii (podvázané chámovody). Toto musíte dodržovat ještě 6 měsíců po ukončení léčby.
Řekněte svému lékaři ihned, jakmile Vaše partnerka otěhotní v průběhu Vaší léčby přípravkem Odomzo a 6 měsíců po jejím ukončení.
V průběhu léčby přípravkem Odomzo a 6 měsíců po jejím ukončení nesmíte darovat sperma.
Řízení dopravních prostředků a obsluha strojů
Není pravděpodobné, že Odomzo ovlivňuje schopnost řídit nebo používání nářadí nebo stroje. Řekněte svému lékaři, pokud si nejste jistý(á).
Přípravek Odomzo obsahuje laktózu
Přípravek Odomzo obsahuje laktózu (mléčný cukr). Pokud Vám lékař řekl, že nesnášíte některé cukry, poraďte se s lékařem, než začnete tento léčivý přípravek užívat.
3. Jak se přípravek Odomzo užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Užívání tohoto přípravku
Doporučená dávka přípravku je 200 mg (1 tobolka) denně.
• Nejezte 2 hodiny před užitím přípravku Odomzo a 1 hodinu poté.
• Užívejte tobolku každý den přibližně ve stejnou dobu. To Vám pomůže si zapamatovat, kdy máte lék brát.
• Tobolku spolkněte celou. Neotevírejte ji, nežvýkejte ani nedrťte.
Neměňte svou dávku bez porady s lékařem. Nepřekračujte doporučenou dávku předepsanou lékařem. Pokud se po spolknutí tobolky vyzvracíte, neužívejte další tobolku až do následující plánované dávky.
Jak dlouho se přípravek Odomzo užívá
Přípravek Odomzo užívejte tak dlouho, jak Vám řekl lékař. Pokud máte dotazy k tomu, jak dlouho máte přípravek Odomzo užívat, zeptejte se svého lékaře nebo lékárníka.
Jestliže jste užil(a) více přípravku Odomzo, než jste měl(a)
Jestliže jste užil(a) více přípravku Odomzo než jste měl(a) nebo jestliže někdo jiný náhodně užil Váš lék, řekněte to svému lékaři nebo okamžitě jděte do nemocnice. Vezměte s sebou lék, jeho obal
Jestliže jste zapomněl(a) užít přípravek Odomzo
Jestliže jste zapoměl(a) užít přípravek Odomzo, užijte ho ihned poté, jakmile si to uvědomíte. Pokud uběhlo více než šest hodin od zapomenuté dávky, vynechejte tuto dávku, užijte až následující dávku v obvyklou dobu. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Odomzo
Nepřestávejte užívat přípravek Odomzo bez předchozí porady se svým lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Přípravek Odomzo může způsobit závažné vrozené vady. V průběhu užívání tohoto přípravku nesmíte otěhotnět (viz „Těhotenství“, „Kojení“ a „Plodnost“ a „Antikoncepce pro ženy a muže“ v bodě 2 pro více informací).
Přestaňte užívat přípravek Odomzo a lékaři okamžitě oznamte, pokud zaznamenáte nějakou z následujících známek, což mohou být příznaky alergické reakce:
• ztížené dýchání nebo polykání
• otok obličeje, rtů, jazyka nebo krku
• těžké svědění kůže s červenou nebo vystouplou vyrážkou.
Některé nežádoucí účinky mohou být závažné
Oznamte okamžitě svému lékaři nebo lékárníkovi, pokud zaznamenáte nějakou z následujících známek:
• těžké svalové křeče, bolest svalů nebo svalová slabost. To mohou být příznaky tzv. rabdomyolýzy (rozpad svalových vláken kosterních svalů), která zahrnuje porušení svalové tkáně.
• tmavá moč, snížené vylučování moči nebo žádné vylučování moči. To by mohlo být známkou rozpadu svalových vláken, který následně poškozuje ledviny.
Další možné nežádoucí účinky
Jestliže se jakýkoli z následujících nežádoucích účinků stane závažným, oznamte to svému lékaři nebo lékárníkovi.
Velmi časté: mohou postihovat více jak 1 z 10 lidí
• svalové křeče, bolest svalů, bolest v kostech, vazech a šlachách
• vynechání menstruace
• průjem a pálení žáhy
• snížená chuť k j ídlu
• bolest hlavy
• porušení chuti nebo zvláštní chuť v ústech
• bolest břicha
• nevolnost
• zvracení
• svědění
• ztráta vlasů
• únava
• bolest
• pokles tělesné hmotnosti.
Časté: mohou postihovat až 1 z 10 lidí
• žaludeční nevolnost a poruchy trávení
• zácpa
• vyrážka
• abnormální růst vlasů
• žízeň, zadržování moče, pokles tělesné hmotnosti, suchá zarudlá kůže, podrážděnost (možné příznaky nízké hladiny tekutin v těle, známé jako dehydratace).
V průběhu léčby přípravkem Odomzo se mohou vyskytnout abnormální výsledky krevních testů. Ty mohou upozornit Vašeho lékaře na možné změny v některých částech Vašeho těla, například:
• vysoké hladiny následujících enzymů: kreatinfosfokináza (svalová funkce), lipáza a/nebo amyláza (funkce slinivky břišní), alaninaminotransferáza (ALT) a/nebo aspartátaminotransferáza (AST) (jaterní funkce)
• vysoká hladina kreatininu (funkce ledvin)
• vysoká hladina krevního cukru (známá j ako hyperglykemie)
• nízká hladina hemoglobinu (nutný k přenosu kyslíku v krvi)
• nízká hladina bílých krvinek.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Odomzo uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a blistru za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Neuchovávejte při teplotě nad 30 °C.
• Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
• Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se
svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Odomzo obsahuje
• Léčivou látkou je sonidegibum (jako sonidegibi phosphas). Jedna tobolka obsahuje sonidegibum 200 mg.
• Dalšími složkami jsou:
o Obsah tobolky: krospovidon, monohydrát laktosy, magnesium-stearát, poloxamer 188, koloidní bezvodý oxid křemičitý, natrium-lauryl-sulfát. o Obal tobolky: želatina, červený oxid železitý (E 172), oxid titaničitý (E171). o Potiskový inkoust: černý oxid železitý (E172), propylenglykol (E1520), šelak.
Jak přípravek Odomzo vypadá a co obsahuje toto balení
Tobolky přípravku Odomzo 200 mg jsou růžové a neprůhledné. Na jejich povrchu je vytištěno „SONIDEGIB 200 mg“ a „NVR“.
Přípravek Odomzo je dostupný v jednodávkových perforovaných blistrech obsahujících
10 x 1 tobolku. Je k dispozici v baleních po 10 a 30 tobolkách. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Bt^rapnn Novartis Pharma Services Inc. Tem +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Česká republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 555 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EkXába Novartis (Hellas) A.E.B.E. TpA.: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
Románia
Hrvatska
Novartis Hrvatska d.o.o. Tel. +385 1 6274 220
Ireland
Novartis Ireland Limited Tel: +353 1 260 12 55
Novartis Pharma Services Romania SRL Tel: +40 21 31299 01
Slovenija
Novartis Pharma Services Inc. Tel: +386 1 300 75 50
Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Kúrcpog
Novartis Pharma Services Inc. T^: +357 22 690 690
Suomi/Finland
Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Jako součást Programu prevence početí pro pacienty/pacientky užívající přípravek Odomzo všichni pacienti obdrží:
• Pacientskou příručku
• Připomínkovou kartu pacienta
Pro získání dalších informací se podívejte na tyto dokumenty.
37