Nuwiq 500 Iu
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
V Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Nuwiq 250 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje nominálně 250 IU lidského koagulačního faktoru VIII (rDNA), simoctocog alfa.
Nuwiq obsahuje přibližně 100 IU/ml lidského koagulačního faktoru VIII (rDNA), simoctocog alfa po rekonstituci.
Účinnost (IU) se stanovuje podle Evropského lékopisu chromogenní analýzou. Specifická aktivita přípravku Nuwiq je přibližně 9500 IU/mg proteinu.
Simoctocog alfa (lidský koagulační faktor VIII (rDNA)) je čištěný protein, který obsahuje 1440 aminokyselin. Sekvence aminokyselin je srovnatelná s formou 90 + 80 kDa faktoru VIII lidské plazmy (tj. bez B-domény). Nuwiq se vyrábí rekombinantní DNA technologií v geneticky upravených lidských embryonálních renálních buňkách (HEK) 293F. V rámci výrobního procesu se k léčivému přípravku nepřidává žádný zvířecí ani lidský materiál.
Pomocná látka/Pomocné látky se známým účinkem:
1 ml rekonstituovaného roztoku obsahuje 7,35 mg sodíku (18,4 mg sodíku na 1 injekční lahvičku). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: bílý až téměř bílý drobivý prášek. Rozpouštědlo: voda na injekci, čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípraven Nuwiq lze použít pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má probíhat pod dohledem lékaře, který má zkušenosti s léčbou hemofilie.
Dosud neléčení pacienti
Bezpečnost a účinnost přípravku Nuwiq u pacientů bez předchozí léčby zatím nebyla stanovena. Dávkování
Dávka a doba trvání substituční léčby závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet podávaných jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (IU), které se vztahují k aktuálně platnému WHO standardu pro přípravky obsahující faktor VIII. Plazmatická aktivita faktoru VIII je vyjádřena buď v procentech (vztaženo k normální lidské plazmě), nebo v mezinárodních jednotkách (vztaženo k mezinárodnímu standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba dle potřeby
Výpočet požadované dávky faktoru VIII je založen na empirickém poznatku, že 1 mezinárodní jednotka (IU) faktoru VIII na 1 kilogram tělesné hmotnosti zvyšuje plazmatickou aktivitu faktoru VIII o přibližně 2 % normální aktivity nebo o 2 IU/dl. Požadovaná dávka se stanoví pomocí následujícího vzorce:
I. Požadované jednotky = tělesná hmotnost (kg) x požadované zvýšení faktoru VIII (%) (IU/dl) x 0,5 (IU/kg na 1 IU/dl)
II. Předpokládané zvýšení faktoru VIII (% z normální hodnoty) = podaná dávka vIU x 2
tělesná hmotnost (kg)
Podávané množství a četnost podávání by se měly vždy zaměřovat na klinickou efektivnost v každém jednotlivém případě.
V případě následného krvácení by aktivita faktoru VIII neměla poklesnout pod stanovenou hladinu plazmatické aktivity (v % normální hodnoty nebo IU/dl) ve sledovaném období. Následující tabulku lze použít jako návod pro dávkování při krvácivých příhodách nebo při chirurgickém zákroku.
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Frekvence dávkování (hodiny) / Délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny |
20-40 |
Opakujte každých 12 až 24 hodin. Po dobu nejméně jednoho dne, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne zhojení. |
|
Intenzivnější hemartróza, krvácení do svalu nebo tvorba hematomů |
30-60 |
Opakujte infúzi každých 12 až 24 hodin po dobu 3 až 4 dnů nebo déle, dokud bolest a akutní potíže neustoupí. |
|
Život ohrožující krvácení |
60-100 |
Opakujte infúzi každých 8 až 24 hodin, dokud není hrozba odvrácena. |
|
Chirurgický zákrok | ||
|
Drobný chirurgický zákrok, včetně trhání zubu |
30-60 |
Každých 24 hodin, po dobu nejméně 1 dne, dokud nedojde ke zhojení. |
|
Velký chirurgický zákrok |
80-100 (před operací a po ní) |
Opakujte infúzi každých 8-24 hodin, dokud nedojde ke zhojení rány, pak pokračujte v terapii po dobu nejméně dalších 7 dnů pro udržení aktivity faktoru VIII na úrovni 30 % až 60 % |
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Frekvence dávkování (hodiny) / Délka trvání léčby (dny) |
|
(IU/dl). | ||
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů s těžkou hemofilií A je obvyklé dávkování 20 až 40 IU faktoru VIII na 1 kilogram tělesné hmotnosti každé 2 až 3 dny. V některých případech, zejména u mladších pacientů, mohou být nutné kratší intervaly dávkování nebo vyšší dávky.
V průběhu léčby se doporučuje vhodné stanovení hladin faktoru VIII pro určení dávky, která má být podávána, a četnosti opakovaných infuzí. V případě velkých chirurgických zákroků je nezbytné přesné monitorování substituční léčby pomocí koagulační analýzy (aktivity faktoru VIII v plazmě). Jednotliví pacienti se mohou lišit ve své odpovědi na faktor VIII, mohou se lišit v poločasech eliminace a v hodnotách biologické dostupnosti.
Pediatrická populace
Dávkování je stejné u dospělých i dětských pacientů, ale u dětí mohou být nutné kratší intervaly mezi dávkami a vyšší dávky. V současnosti dostupné údaje jsou uvedeny v bodech 4.8, 5.1 a 5.2.
U dětí mladších 2 let nejsou dostupné žádné údaje.
Způsob podání
Intravenózní podání.
Doporučuje se podávat nejvýše 4 ml přípravku za minutu.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoliv pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Stejně jako u jiných intravenózně podávaných proteinových přípravků jsou možné hypersensitivní reakce alergického typu. Přípravek Nuwiq obsahuje stopová množství proteinů lidské hostitelské buňky jiných než faktor VIII. Vyskytnou-li se příznaky přecitlivělosti, pacienti mají být poučeni o neprodleném přerušení užívání léčivého přípravku a o návštěvě lékaře. Pacienti mají být poučeni o časných projevech přecitlivělosti, jako například vyrážka, generalizovaná kopřivka, tlak na hrudi, sípavé dýchání, hypotenze a anafylaxe.
V případě šoku je nutno dodržovat standardní lékařské postupy jeho léčby.
Inhibitory
Známou komplikací léčby u nemocných s hemofilií A je tvorba neutralizujících protilátek (inhibitorů) faktoru VIII. Tyto inhibitory jsou obvykle IgG imunoglobuliny směrované proti prokoagulační aktivitě faktoru VIII, a jsou kvantifikovány v Bethesda jednotkách (BU) na ml plazmy s použitím modifikovaného testu. Riziko rozvoje inhibitorů koreluje s expozicí faktoru VIII; toto riziko se zvyšuje v průběhu prvních 20 dnů expozice. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Z tohoto důvodu se doporučuje pečlivě monitorovat pacienty z hlediska možného vývoje inhibitorů po jakékoliv změně přípravku.
Obecně platí, že u všech pacientů léčených koagulačním faktorem VIII musí být pečlivě monitorován vznik inhibitorů vhodnými klinickými pozorováními a laboratorními testy. Pokud se nedosáhne očekávané hladiny aktivity plazmatického faktoru VIII nebo pokud krvácení není zastaveno odpovídající dávkou, měl by být proveden test za účelem určení přítomnosti inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční léčba faktorem VIII účinná a je třeba uvažovat o jiných možnostech léčby, jako je například navození imunitní tolerance (ITI). Léčba takových pacientů by měla být vedena lékaři se zkušenostmi s léčbou hemofilie a s inhibitory faktoru VIII.
Komplikace způsobené katétrem
Pokud je potřeba použít centrální žilní vstup (CVAD), má být zváženo riziko komplikací souvisejících s CVAD, včetně lokální infekce, bakteriémie a trombózy v místě vstupu katétru.
Důrazně se doporučuje, pokaždé, kdy je přípravek Nuwiq podán pacientovi, zaznamenat název a číslo šarže aplikovaného přípravku, aby bylo zachováno spojení mezi pacientem a číslem šarže léčivého přípravku.
Pediatrická populace
Uvedená upozornění a bezpečnostní pokyny se vztahují jak na dospělé, tak na dětské pacienty.
Pokyny vztahující se k pomocné látce (obsah sodíku)
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v 1 injekční lahvičce.
V závislosti na tělesné hmotnosti a dávkování by měl pacient obdržet více než jednu injekční lahvičku. To je třeba vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
U přípravku Nuwiq nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie na zvířatech nebyly s přípravkem Nuwiq prováděny.
Vzhledem k vzácnému výskytu hemofilie A u žen nejsou k dispozici žádné zkušenosti s používáním faktoru VIII během těhotenství a kojení. Z tohoto důvodu má být přípravek Nuwiq během těhotenství a kojení používán pouze, je-li to jasně indikováno. Nejsou k dispozici žádné údaje týkající se fertility.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Nuwiq nemá žádný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn informací o bezpečnostním profilu
U přípravků obsahujících faktor VIII byly vzácně pozorovány přecitlivělost nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, zimnici, zarudnutí, generalizovanou vyrážku, bolest hlavy, kopřivku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tlak na hrudi, mravenčení, zvracení, sípání) které se mohou v některých případech rozvinout do závažné anafylaxe (včetně šoku).
U pacientů s hemofilií A se mohou vytvořit neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory vyskytnou, mohou se projevit nedostatečnou klinickou odezvou na přípravek. V těchto případech se doporučuje vyhledat specializované pracoviště na léčení hemofilie.
Přehledný souhrn nežádoucích účinků
Během klinických studií přípravku Nuwiq provedených u již dříve léčených pediatrických pacientů (2 až 11 let, n = 58), dospívajících pacientů (12 až 17 let, n = 3) a dospělých pacientů (n = 74) s těžkou hemofilií A, byl celkový počet závažných nežádoucích účinků (ADR) 8 (6 u dospělých, 2 u dětí) hlášený u 5 pacientů (3 dospělých, 2 dětí).
Tabulka 1 uvedená níže je uspořádána v souladu s klasifikací tříd orgánových systémů MedDRa (SOC a preferované termíny četností).
Četnost je definována dle následující konvence: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000); velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 1. Frekvence výskytu nežádoucích účinků léčivého přípravku (ADR) na 1 pacienta
|
během klinických studií u 135 již dříve léčených pacientů s těžkou |
íemofilií A | |
|
Třída orgánových systémů podle databáze MedDRA |
Nežádoucí účinky |
Četnost* |
|
Poruchy nervové soustavy |
Parastézie Bolesti hlavy |
Méně časté |
|
Poruchy ucha a labyrintu |
Závratě |
Méně časté |
|
Gastrointestinální poruchy |
Sucho v ústech |
Méně časté |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolesti zad |
Méně časté |
|
Celkové poruchy a reakce v místě aplikace |
Zánět v místě vpichu Bolest v místě vpichu |
Méně časté |
|
Vyšetření |
Pozitivní protilátka faktoru VIII bez neutralizační aktivity |
Méně časté |
* Všechny tyto nežádoucí účinky se vyskytly pouze jednou. Vzhledem k tomu, že celkový počet studovaných pacientů je 135, četnost nemůže být jiná než „méně časté“, jakmile se závažné nežádoucí účinky objeví jednou.
Popis vybraných nežádoucích účinků
U jednoho dospělého pacienta byla zjištěna protilátka faktoru VIII bez neutralizační aktivity (viz Tabulka 1). Vzorek byl testován v centrální laboratoři v osmi zředěních. Výsledek byl pozitivní pouze při faktoru zředění 1 a titr protilátek byl velmi nízký. Inhibiční aktivita měřená pomocí modifikovaného Bethesda testu nebyla u tohoto pacienta zjištěna. Klinická účinnost a biologická dostupnost přípravku Nuwiq nebyly u tohoto pacienta narušeny.
Pediatrická populace
Četnost, typ a závažnost nežádoucích účinků u dětí se očekávají shodné jako u dospělých.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nejsou známy žádné případy předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika, krevní koagulační faktor VIII, ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. Po infuzi hemofilickému pacientovi se faktor VIII váže na von Willebrandův faktor přítomný v krevním oběhu pacienta. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin následně přeměňuje fibrinogen na fibrin a může dojít k vytvoření sraženiny. Hemofilie A je pohlavně vázaná dědičná porucha srážlivosti krve způsobená sníženou hladinou faktoru VIII:C, v důsledku které dochází k profuznímu krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánnímu, nebo jako následek úrazu při nehodě či chirurgickém zákroku. Substituční léčbou se hladiny faktoru VIII v plazmě zvýší, čímž je možná přechodná korekce nedostatku faktoru VIII a korekce sklonu ke krvácení.
Imunogenita přípravku Nuwiq byla hodnocena v rámci klinických zkoušek u 135 již dříve léčených pacientů s těžkou hemofilií A (74 dospělých a 61 dětských pacientů). U žádného z pacientů nedošlo k vývoji inhibitorů.
V rámci klinické studie provedené s 32 dospělými pacienty s těžkou hemofilií A byla střední hodnota spotřeby přípravku Nuwiq za účelem profylaxe 468,7 IU/kg/měsíc. Průměrná dávka při léčbě nečekaného krvácení byla 33,0 IU/kg u pacientů užívajících přípravek za účelem profylaxe. V rámci jiné studie bylo 22 dospělých pacientů léčeno na požádání. Celkem bylo 986 případů krvácení léčeno průměrnou dávkou 30,9 IU/kg. Obecně platí, že slabé krvácení vyžadovalo mírně nižší a silnější krvácení vyžadovalo až trojnásobně vyšší střední dávky.
Pediatrická populace
Data byla získána od 29 již dříve léčených dětí ve věku 2 až 5 let, 31 dětí ve věku 6 až 12 let a jednoho dospívajícího pacienta ve věku 14 let. Medián dávky na profylaktickou infuzi byl 37,8 IU/kg. Dvacet pacientů užívalo střední dávky vyšší než 45 IU/kg. Medián spotřeby přípravku Nuwiq za účelem profylaxe za měsíc byl 521,9 IU/kg. Vyšší medián dávky přípravku Nuwiq vyžadovala léčba krvácení u dětí (43,9 IU/kg) než u dospělých (33,0 IU/kg), a vyšší medián dávky vyžadovala léčba středního až silného krvácení v porovnání se slabým krvácením (78,2 IU/kg v porovnání s 41,7 IU/kg). Mladším dětem musel být obecně podáván vyšší medián dávky (6-12 let: 43,9 IU/kg; 2-5 let: 52,6 IU/kg).
Evropská agentura pro léčivé přípravky odložila závazek předložit výsledky studií přípravku Nuwiq u jednoho nebo více souborů pediatrické populace při léčbě hemofilie A (vrozeného nedostatku faktoru VIII) (viz bod 4.2, kde jsou uvedeny informace o použití pro pediatrické pacienty).
5.2 Farmakokinetické vlastnosti
Tabulka 2. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u dospělých již dříve léčených pacientů (věk 18-65 let) s těžkou hemofilií A (n = 20)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
22,6 ± 8,0 |
22,3 (8,4 - 38,1) |
|
T1/2 (hod) |
14,7 ± 10,4 |
12,5 (5,4 -55,6) |
|
IVR (%/IU/kg) |
2,5 ± 0,4 |
2,5 (1,7 -3,2) |
|
CL (ml/hod/kg) |
3,0 ± 1,2 |
2,7 (1,5-6,4) |
AUC = plocha pod křivkou (FVIII:C), Tm = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka
Tabulka 3. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u již dříve léčených dětských pacientů ve věku 6 až 12 let s těžkou hemofilií A (n = 12)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
13,2 ± 3,4 |
12,8 (7,8 - 19,1) |
|
T1/2 (hod) |
10,0 ± 1,9 |
9,9 (7,6 -14,1) |
|
IVR (%/IU/kg) |
1,9 ± 0,4 |
1,9 (1,2 -2,6) |
|
CL (ml/hod/kg) |
4,3 ± 1,2 |
4,2 (2,8 - 6,9) |
AUC = plocha pod křivkou (FVIII:C), T1/2 = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka
Tabulka 4. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u již dříve léčených dětských pacientů ve věku 2 až 5 let s těžkou hemofilií A (n = 13)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
11,7 ± 5,3 |
10,5 (4,9 -23,8) |
|
T1/2 (hod) |
9,5 ± 3,3 |
8,2 (4,3 - 17,3) |
|
IVR (%/IU/kg) |
1,9 ± 0,3 |
1,8 (1,5 -2,4) |
|
CL (ml/hod/kg) |
5,4 ± 2,4 |
5,1 (2,3 -10,9) |
AUC = plocha pod křivkou (FVIII:C), T1/2 = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka Pediatrická populace
Z literatury je známo, že biologická dostupnost a poločas byly nižší u dětí než u dospělých a clearance vyšší, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Podskupiny upravené podle hmotnosti
Tabulka 5. FK parametry pro přípravek Nuwiq upravené podle hmotnosti (Dávka: 50 IU/kg) u
|
dospělých již dříve |
éčených pacientů (věk 18-65 let) s těžkou hemofilií A (n = 20) | |||
|
FK parametr |
Všichni (n=20) |
Normální hmotnost (n=14) |
S nadváhou (n=4) |
Obézní (n=2) |
|
Chromogenní metoda Průměr ± SD | ||||
|
AUC (hod*IU/ml) |
22,6 ± 8,0 |
20,4 ± 6,9 |
24,9 ± 8,9 |
33,5 ± 6,5 |
|
T1/2 (hod) |
14,7 ± 10,4 |
14,7 ± 12,1 |
13,4 ± 5,9 |
17,2 ± 4,8 |
|
IVR (%/IU/kg) |
2,5 ± 0,4 |
2,4 ± 0,4 |
2,7 ± 0,4 |
2,8 ± 0,3 |
|
CL (ml/hod/kg) |
3,0 ± 1,2 |
3,2 ± 1,3 |
2,6 ± 1,0 |
1,8 ± 0,4 |
|
Chromogenní metoda Medián (rozsah) | ||||
|
AUC (hod*IU/ml) |
22,3 (8,4 -38,1) |
21,2 (8,4 -32,6) |
23,3 (17,4 - 35,5) |
33,5 (28,9 -38,1) |
|
T1/2 (hod) |
12,5 (5,4 - 55,6) |
12,3 (5,4 -55,6) |
11,2 (9,3 -22,0) |
17,2 (13,8 -20,6) |
|
IVR (%/IU/kg) |
2,5 (1,7 -3,2) |
2,4 (1,7 -3,1) |
2,8 (2,3 - 3,2) |
2,8 (2,6 -3,0) |
|
CL (ml/hod/kg) |
2,7 (1,5 -6,4) |
2,8 (1,7 -6,4) |
2,5 (1,6 -3,7) |
1,8 (1,5 -2,0) |
|
Normální hmotnost: |
BMI 18,5-25 kg/m2, |
Nadváha: BMI 25-30 kg/m2, Obezita: BM |
> 30 kg/m2, SD = | |
Směrodatná odchylka
5.3 Předklinické údaje vztahující se k bezpečnosti
V rámci předklinických studií byl přípravek Nuwiq bezpečně a úspěšně použit k obnově hemostázy u psů s hemofilií. Toxikologické studie ukázaly, že lokální intravenózní podání a systémová expozice byly dobře tolerovány u laboratorních zvířat (potkanů a opic makaka jávského).
Specifické studie reprodukční toxicity, chronické toxicity a kancerogenity s dlouhodobým opakovaným podáváním přípravku Nuwiq nebyly provedeny kvůli imunitní reakci na heterologní proteiny u všech nelidských druhů savců.
Nebyly provedeny žádné studie mutagenního potenciálu přípravku Nuwiq.
Ex vivo hodnocení při použití komerční testovací soupravy pro kvantifikaci odezvy T-buněk na proteinová terapeutika ukazují nízké riziko imunogenicity.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Sacharosa Chlorid sodný
Dihydrát chloridu vápenatého Arginin hydrochlorid Dihydrát citronanu sodného Poloxamer 188
Rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
Mají se používat pouze dodávané injekční soupravy, protože může dojít k selhání léčby v důsledku adsorpce lidského koagulačního faktoru VIII na vnitřní povrchy některých infuzních zařízení.
6.3 Doba použitelnosti
2 roky
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25°C) po dobu jednoho nepřetržitého období nepřesahujícího 1 měsíc. Jakmile byl přípravek jednou vyjmut z chladničky, nesmí tam být vrácen zpět. Označte si prosím na krabičce datum, kdy jste přípravek začali uchovávat při pokojové teplotě. Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Po rekonstituci je prokázána chemická a fyzikální stabilita po dobu 24 hodin při uchovávání při pokojové teplotě.
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, doba a podmínky uchovávání přípravku před použitím jsou v odpovědnosti uživatele. Uchovávejte rekonstituovaný přípravek při pokojové teplotě. Po rekonstituci chraňte před mrazem.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Uchovávání tohoto léčivého přípravku při pokojové teplotě a podmínky uchovávání po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Jedno balení přípravku Nuwiq 250 IU obsahuje:
- Prášek: 250 IU prášku v 8ml skleněné injekční lahvičce typu I, uzavřené potaženou bromobutylovou zátkou s pojistným hliníkovým uzávěrem
- Rozpouštědlo: 2,5 ml vody na injekci v předplněné injekční stříkačce z borosilikátového skla
- 1 sterilní adaptér injekční lahvičky pro rekonstituci s 1 motýlkovou jehlou a 2 tampóny napuštěnými alkoholem
Velikost balení po 1 kusu.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Prášek má být rekonstituován pouze v dodaném rozpouštědle (2,5 ml vody na injekce) za použití injekční soupravy, která je součástí dodávky. Při rozpouštění prášku má injekční lahvička vykonávat jemný krouživý pohyb, dokud se prášek nerozpustí. Po rekonstituci má být roztok natažen zpět do injekční stříkačky.
Rekonstituovaný léčivý přípravek má být před podáním vizuálně zkontrolován, zda neobsahuje cizorodé částice nebo zabarvení. Tento rekonstituovaný léčivý přípravek je čirý, bezbarvý roztok bez přítomnosti cizorodých částic s pH v rozmezí 6,5 až 7,5. Nepoužívejte roztok, je-li zakalený nebo obsahuje-li usazeniny.
Návod pro přípravu a podávání
1. Ponechte rozpouštědlo v injekční stříkačce (voda na injekci) a prášek v uzavřené injekční lahvičce dostatečnou dobu pro dosažení pokojové teploty. Můžete to udělat tak, že je podržíte v rukou, dokud nebudou stejně teplé jako vaše ruce. Na zahřívání lahvičky a předem naplněné stříkačky nepoužívejte jiné postupy. Tato teplota by měla být udržována během rekonstituce.
2. Odstraňte pojistné plastové víčko z injekční lahvičky s práškem, aby byla přístupná střední část gumové zátky. Neodstraňujte šedou zátku ani kovový kroužek kolem horní části injekční lahvičky.
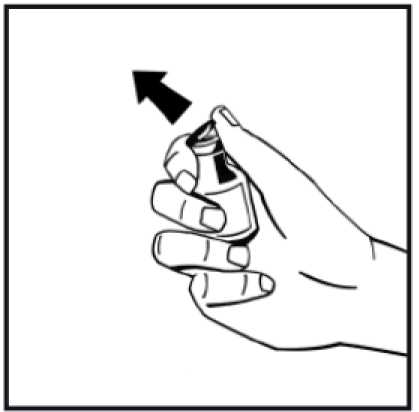
3. Otřete horní část injekční lahvičky tampónem napuštěným v alkoholu. Počkejte, dokud se alkohol nevypaří.
4. Sejměte papírový kryt z obalu adaptéru injekční lahvičky. Nevyndávejte adaptér z obalu.
Umístěte injekční lahvičku s práškem na rovný povrch a držte ji. Uchopte obal adaptéru a umístěte adaptér injekční lahvičky na střední část gumové zátky injekční lahvičky s práškem. Stlačte pevně obal adaptéru tak, aby hrot adaptéru prošel gumovou zátkou. Adaptér se přitom zacvakne na injekční lahvičku.
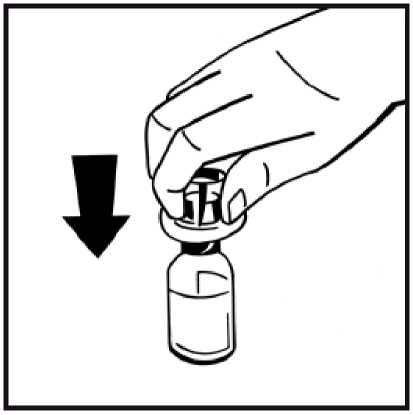
5.
6.
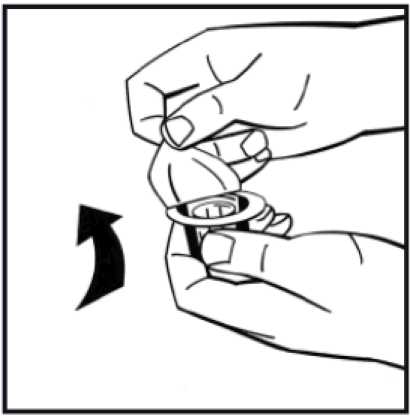
Sejměte papírový kryt z obalu předplněné injekční stříkačky. Uchopte píst za konec, a nedotýkejte se jeho střední části. Připojte konec pístu se závitem k injekční stříkačce s rozpouštědlem. Otáčejte pístem ve směru pohybu hodinových ručiček, dokud neucítíte slabý odpor.
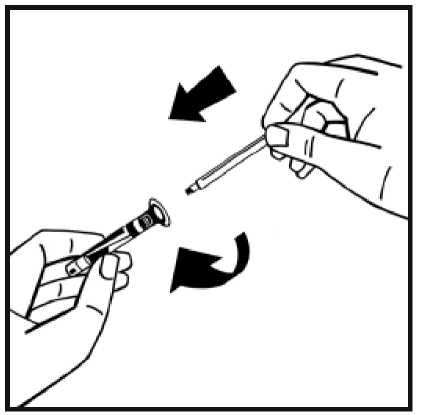
7. Odlomte ochranný plastový hrot z injekční stříkačky s rozpouštědlem v místě perforace víčka. Nedotýkejte se vnitřku víčka ani hrotu injekční stříkačky. Pokud roztok nebudete používat ihned, zavřete naplněnou stříkačku plastovou špičkou s odolností vůči nárazu pro uskladnění.
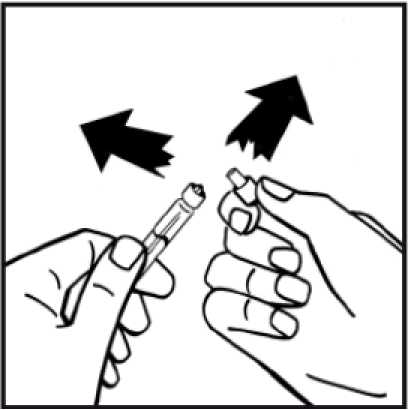
8. Sejměte obal adaptéru a vyhoďte jej.
9. Pevně připojte injekční stříkačku s rozpouštědlem k adaptéru injekční lahvičky otáčením ve směru pohybu hodinových ručiček, dokud neucítíte slabý odpor.
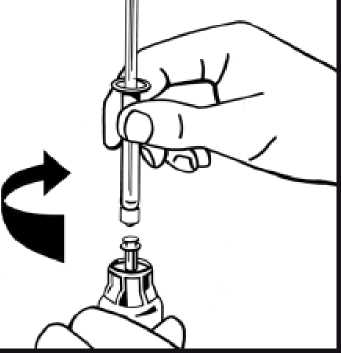
10. Pomalu vstřikujte veškeré rozpouštědlo do injekční lahvičky s práškem tlakem na píst.
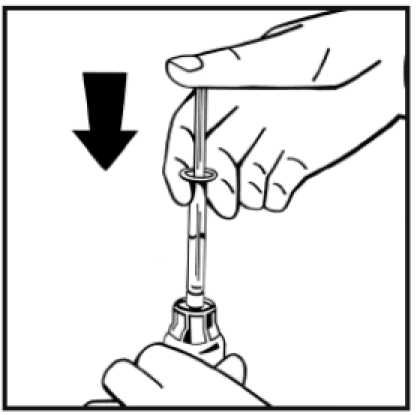
11. Aniž byste odstranili injekční stříkačku, rozpusťte prášek jemným pohybem nebo několika krouživými pohyby injekční lahvičky. Netřepejte. Počkejte, dokud se prášek zcela nerozpustí.
12. Před podáním zrakem zkontrolujte, že výsledný roztok neobsahuje žádné částice. Roztok má být čirý a bezbarvý, prakticky bez viditelných částic. Nepoužívejte roztok, je-li zakalený nebo obsahuje-li usazeniny.
13. Otočte injekční lahvičku připevněnou k injekční stříkačce dnem vzhůru a pomalu natáhněte výsledný roztok do stříkačky. Přesvědčte se, že nyní je celý obsah injekční lahvičky v injekční stříkačce.
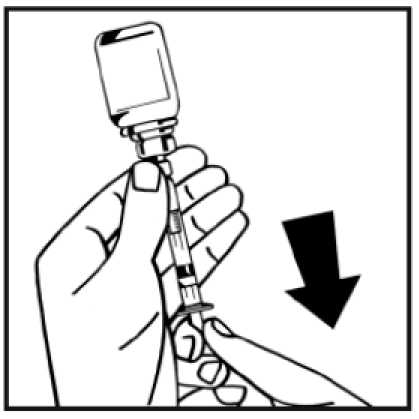
14. Odpojte naplněnou injekční stříkačku od adaptéru injekční lahvičky otáčením proti směru pohybu hodinových ručiček a prázdnou injekční lahvičku zlikvidujte.
15. Roztok je nyní připraven k okamžitému použití. Chraňte před chladem.
16. Očistěte zvolené místo vpichu jedním z dodaných tampónů napuštěných alkoholem.
17. Připojte k injekční stříkačce infuzní soupravu, která je součástí balení.
Zaveďte jehlu infuzní soupravy do zvolené žíly. Pokud jste použili škrtidlo pro zviditelnění žíly, má být toto škrtidlo uvolněno předtím, než začnete vstřikovat roztok.
Do injekční stříkačky se nesmí dostat žádná krev, aby nedošlo k tvorbě fibrinových sraženin.
18. Vstřikujte roztok do žíly pomalu, maximálně 4 ml za minutu.
Použijete-li více než jednu injekční lahvičku s práškem pro jednu léčbu, můžete použít stejnou injekční jehlu opakovaně. Adaptér injekční lahvičky a injekční stříkačka jsou určeny pouze na jedno použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Octapharma AB Lars Forssells gata 23 112 75 Stockholm Švédsko
8. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/936/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 22. července 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
V Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Nuwiq 500 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje nominálně 500 IU lidského koagulačního faktoru VIII (rDNA), simoctocog alfa.
Nuwiq obsahuje přibližně 200 IU/ml lidského koagulačního faktoru VIII (rDNA), simoctocog alfa po rekonstituci.
Účinnost (IU) se stanovuje podle Evropského lékopisu chromogenní analýzou. Specifická aktivita přípravku Nuwiq je přibližně 9500 IU/mg proteinu.
Simoctocog alfa (lidský koagulační faktor VIII (rDNA)) je čištěný protein, který obsahuje 1440 aminokyselin. Sekvence aminokyselin je srovnatelná s formou 90 + 80 kDa faktoru VIII lidské plazmy (tj. bez B-domény). Nuwiq se vyrábí rekombinantní DNA technologií v geneticky upravených lidských embryonálních renálních buňkách (HEK) 293F. V rámci výrobního procesu se k léčivému přípravku nepřidává žádný zvířecí ani lidský materiál.
Pomocná látka/Pomocné látky se známým účinkem:
1 ml rekonstituovaného roztoku obsahuje 7,35 mg sodíku (18,4 mg sodíku na 1 injekční lahvičku). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: bílý až téměř bílý drobivý prášek. Rozpouštědlo: voda na injekci, čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípraven Nuwiq lze použít pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má probíhat pod dohledem lékaře, který má zkušenosti s léčbou hemofilie.
Dosud neléčení pacienti
Bezpečnost a účinnost přípravku Nuwiq u pacientů bez předchozí léčby zatím nebyla stanovena. Dávkování
Dávka a doba trvání substituční léčby závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet podávaných jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (IU), které se vztahují k aktuálně platnému WHO standardu pro přípravky obsahující faktor VIII. Plazmatická aktivita faktoru VIII je vyjádřena buď v procentech (vztaženo k normální lidské plazmě), nebo v mezinárodních jednotkách (vztaženo k mezinárodnímu standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba dle potřeby
Výpočet požadované dávky faktoru VIII je založen na empirickém poznatku, že 1 mezinárodní jednotka (IU) faktoru VIII na 1 kilogram tělesné hmotnosti zvyšuje plazmatickou aktivitu faktoru VIII o přibližně 2 % normální aktivity nebo o 2 IU/dl. Požadovaná dávka se stanoví pomocí následujícího vzorce:
I. Požadované jednotky = tělesná hmotnost (kg) x požadované zvýšení faktoru VIII (%) (IU/dl) x 0,5 (IU/kg na 1 IU/dl)
II. Předpokládané zvýšení faktoru VIII (% z normální hodnoty) = podaná dávka vIU x 2
tělesná hmotnost (kg)
Podávané množství a četnost podávání by se měly vždy zaměřovat na klinickou efektivnost v každém jednotlivém případě.
V případě následného krvácení by aktivita faktoru VIII neměla poklesnout pod stanovenou hladinu plazmatické aktivity (v % normální hodnoty nebo IU/dl) ve sledovaném období. Následující tabulku lze použít jako návod pro dávkování při krvácivých příhodách nebo při chirurgickém zákroku.
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Frekvence dávkování (hodiny) / Délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny |
20-40 |
Opakujte každých 12 až 24 hodin. Po dobu nejméně jednoho dne, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne zhojení. |
|
Intenzivnější hemartróza, krvácení do svalu nebo tvorba hematomů |
30-60 |
Opakujte infúzi každých 12 až 24 hodin po dobu 3 až 4 dnů nebo déle, dokud bolest a akutní potíže neustoupí. |
|
Život ohrožující krvácení |
60-100 |
Opakujte infúzi každých 8 až 24 hodin, dokud není hrozba odvrácena. |
|
Chirurgický zákrok | ||
|
Drobný chirurgický zákrok, včetně trhání zubu |
30-60 |
Každých 24 hodin, po dobu nejméně 1 dne, dokud nedojde ke zhojení. |
|
Velký chirurgický zákrok |
80-100 (před operací a po ní) |
Opakujte infúzi každých 8-24 hodin, dokud nedojde ke zhojení rány, pak pokračujte v terapii po dobu nejméně dalších 7 dnů pro udržení aktivity faktoru VIII na úrovni 30 % až 60 % |
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Frekvence dávkování (hodiny) / Délka trvání léčby (dny) |
|
(IU/dl). | ||
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů s těžkou hemofilií A je obvyklé dávkování 20 až 40 IU faktoru VIII na 1 kilogram tělesné hmotnosti každé 2 až 3 dny. V některých případech, zejména u mladších pacientů, mohou být nutné kratší intervaly dávkování nebo vyšší dávky.
V průběhu léčby se doporučuje vhodné stanovení hladin faktoru VIII pro určení dávky, která má být podávána, a četnosti opakovaných infuzí. V případě velkých chirurgických zákroků je nezbytné přesné monitorování substituční léčby pomocí koagulační analýzy (aktivity faktoru VIII v plazmě). Jednotliví pacienti se mohou lišit ve své odpovědi na faktor VIII, mohou se lišit v poločasech eliminace a v hodnotách biologické dostupnosti.
Pediatrická populace
Dávkování je stejné u dospělých i dětských pacientů, ale u dětí mohou být nutné kratší intervaly mezi dávkami a vyšší dávky. V současnosti dostupné údaje jsou uvedeny v bodech 4.8, 5.1 a 5.2.
U dětí mladších 2 let nejsou dostupné žádné údaje.
Způsob podání
Intravenózní podání.
Doporučuje se podávat nejvýše 4 ml přípravku za minutu.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoliv pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Stejně jako u jiných intravenózně podávaných proteinových přípravků jsou možné hypersensitivní reakce alergického typu. Přípravek Nuwiq obsahuje stopová množství proteinů lidské hostitelské buňky jiných než faktor VIII. Vyskytnou-li se příznaky přecitlivělosti, pacienti mají být poučeni o neprodleném přerušení užívání léčivého přípravku a o návštěvě lékaře. Pacienti mají být poučeni o časných projevech přecitlivělosti, jako například vyrážka, generalizovaná kopřivka, tlak na hrudi, sípavé dýchání, hypotenze a anafylaxe.
V případě šoku je nutno dodržovat standardní lékařské postupy jeho léčby.
Inhibitory
Známou komplikací léčby u nemocných s hemofilií A je tvorba neutralizujících protilátek (inhibitorů) faktoru VIII. Tyto inhibitory jsou obvykle IgG imunoglobuliny směrované proti prokoagulační aktivitě faktoru VIII, a jsou kvantifikovány v Bethesda jednotkách (BU) na ml plazmy s použitím modifikovaného testu. Riziko rozvoje inhibitorů koreluje s expozicí faktoru VIII; toto riziko se zvyšuje v průběhu prvních 20 dnů expozice. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Z tohoto důvodu se doporučuje pečlivě monitorovat pacienty z hlediska možného vývoje inhibitorů po jakékoliv změně přípravku.
Obecně platí, že u všech pacientů léčených koagulačním faktorem VIII musí být pečlivě monitorován vznik inhibitorů vhodnými klinickými pozorováními a laboratorními testy. Pokud se nedosáhne očekávané hladiny aktivity plazmatického faktoru VIII nebo pokud krvácení není zastaveno odpovídající dávkou, měl by být proveden test za účelem určení přítomnosti inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční léčba faktorem VIII účinná a je třeba uvažovat o jiných možnostech léčby, jako je například navození imunitní tolerance (ITI). Léčba takových pacientů by měla být vedena lékaři se zkušenostmi s léčbou hemofilie a s inhibitory faktoru VIII.
Komplikace způsobené katétrem
Pokud je potřeba použít centrální žilní vstup (CVAD), má být zváženo riziko komplikací souvisejících s CVAD, včetně lokální infekce, bakteriémie a trombózy v místě vstupu katétru.
Důrazně se doporučuje, pokaždé, kdy je přípravek Nuwiq podán pacientovi, zaznamenat název a číslo šarže aplikovaného přípravku, aby bylo zachováno spojení mezi pacientem a číslem šarže léčivého přípravku.
Pediatrická populace
Uvedená upozornění a bezpečnostní pokyny se vztahují jak na dospělé, tak na dětské pacienty.
Pokyny vztahující se k pomocné látce (obsah sodíku)
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v 1 injekční lahvičce.
V závislosti na tělesné hmotnosti a dávkování by měl pacient obdržet více než jednu injekční lahvičku. To je třeba vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
U přípravku Nuwiq nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie na zvířatech nebyly s přípravkem Nuwiq prováděny.
Vzhledem k vzácnému výskytu hemofilie A u žen nejsou k dispozici žádné zkušenosti s používáním faktoru VIII během těhotenství a kojení. Z tohoto důvodu má být přípravek Nuwiq během těhotenství a kojení používán pouze, je-li to jasně indikováno. Nejsou k dispozici žádné údaje týkající se fertility.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Nuwiq nemá žádný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn informací o bezpečnostním profilu
U přípravků obsahujících faktor VIII byly vzácně pozorovány přecitlivělost nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, zimnici, zarudnutí, generalizovanou vyrážku, bolest hlavy, kopřivku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tlak na hrudi, mravenčení, zvracení, sípání) které se mohou v některých případech rozvinout do závažné anafylaxe (včetně šoku).
U pacientů s hemofilií A se mohou vytvořit neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory vyskytnou, mohou se projevit nedostatečnou klinickou odezvou na přípravek. V těchto případech se doporučuje vyhledat specializované pracoviště na léčení hemofilie.
Přehledný souhrn nežádoucích účinků
Během klinických studií přípravku Nuwiq provedených u již dříve léčených pediatrických pacientů (2 až 11 let, n = 58), dospívajících pacientů (12 až 17 let, n = 3) a dospělých pacientů (n = 74) s těžkou hemofilií A, byl celkový počet závažných nežádoucích účinků (ADR) 8 (6 u dospělých, 2 u dětí) hlášený u 5 pacientů (3 dospělých, 2 dětí).
Tabulka 1 uvedená níže je uspořádána v souladu s klasifikací tříd orgánových systémů MedDRa (SOC a preferované termíny četností).
Četnost je definována dle následující konvence: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000); velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 1. Frekvence výskytu nežádoucích účinků léčivého přípravku (ADR) na 1 pacienta
|
během klinických studií u 135 již dříve léčených pacientů s těžkou |
íemofilií A | |
|
Třída orgánových systémů podle databáze MedDRA |
Nežádoucí účinky |
Četnost* |
|
Poruchy nervové soustavy |
Parastézie Bolesti hlavy |
Méně časté |
|
Poruchy ucha a labyrintu |
Závratě |
Méně časté |
|
Gastrointestinální poruchy |
Sucho v ústech |
Méně časté |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolesti zad |
Méně časté |
|
Celkové poruchy a reakce v místě aplikace |
Zánět v místě vpichu Bolest v místě vpichu |
Méně časté |
|
Vyšetření |
Pozitivní protilátka faktoru VIII bez neutralizační aktivity |
Méně časté |
* Všechny tyto nežádoucí účinky se vyskytly pouze jednou. Vzhledem k tomu, že celkový počet studovaných pacientů je 135, četnost nemůže být jiná než „méně časté“, jakmile se závažné nežádoucí účinky objeví jednou.
Popis vybraných nežádoucích účinků
U jednoho dospělého pacienta byla zjištěna protilátka faktoru VIII bez neutralizační aktivity (viz Tabulka 1). Vzorek byl testován v centrální laboratoři v osmi zředěních. Výsledek byl pozitivní pouze při faktoru zředění 1 a titr protilátek byl velmi nízký. Inhibiční aktivita měřená pomocí modifikovaného Bethesda testu nebyla u tohoto pacienta zjištěna. Klinická účinnost a biologická dostupnost přípravku Nuwiq nebyly u tohoto pacienta narušeny.
Pediatrická populace
Četnost, typ a závažnost nežádoucích účinků u dětí se očekávají shodné jako u dospělých.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nejsou známy žádné případy předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika, krevní koagulační faktor VIII, ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. Po infuzi hemofilickému pacientovi se faktor VIII váže na von Willebrandův faktor přítomný v krevním oběhu pacienta. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin následně přeměňuje fibrinogen na fibrin a může dojít k vytvoření sraženiny. Hemofilie A je pohlavně vázaná dědičná porucha srážlivosti krve způsobená sníženou hladinou faktoru VIII:C, v důsledku které dochází k profuznímu krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánnímu, nebo jako následek úrazu při nehodě či chirurgickém zákroku. Substituční léčbou se hladiny faktoru VIII v plazmě zvýší, čímž je možná přechodná korekce nedostatku faktoru VIII a korekce sklonu ke krvácení.
Imunogenita přípravku Nuwiq byla hodnocena v rámci klinických zkoušek u 135 již dříve léčených pacientů s těžkou hemofilií A (74 dospělých a 61 dětských pacientů). U žádného z pacientů nedošlo k vývoji inhibitorů.
V rámci klinické studie provedené s 32 dospělými pacienty s těžkou hemofilií A byla střední hodnota spotřeby přípravku Nuwiq za účelem profylaxe 468,7 IU/kg/měsíc. Průměrná dávka při léčbě nečekaného krvácení byla 33,0 IU/kg u pacientů užívajících přípravek za účelem profylaxe. V rámci jiné studie bylo 22 dospělých pacientů léčeno na požádání. Celkem bylo 986 případů krvácení léčeno průměrnou dávkou 30,9 IU/kg. Obecně platí, že slabé krvácení vyžadovalo mírně nižší a silnější krvácení vyžadovalo až trojnásobně vyšší střední dávky.
Pediatrická populace
Data byla získána od 29 již dříve léčených dětí ve věku 2 až 5 let, 31 dětí ve věku 6 až 12 let a jednoho dospívajícího pacienta ve věku 14 let. Medián dávky na profylaktickou infuzi byl 37,8 IU/kg. Dvacet pacientů užívalo střední dávky vyšší než 45 IU/kg. Medián spotřeby přípravku Nuwiq za účelem profylaxe za měsíc byl 521,9 IU/kg. Vyšší medián dávky přípravku Nuwiq vyžadovala léčba krvácení u dětí (43,9 IU/kg) než u dospělých (33,0 IU/kg), a vyšší medián dávky vyžadovala léčba středního až silného krvácení v porovnání se slabým krvácením (78,2 IU/kg v porovnání s 41,7 IU/kg). Mladším dětem musel být obecně podáván vyšší medián dávky (6-12 let: 43,9 IU/kg; 2-5 let: 52,6 IU/kg).
Evropská agentura pro léčivé přípravky odložila závazek předložit výsledky studií přípravku Nuwiq u jednoho nebo více souborů pediatrické populace při léčbě hemofilie A (vrozeného nedostatku faktoru VIII) (viz bod 4.2, kde jsou uvedeny informace o použití pro pediatrické pacienty).
5.2 Farmakokinetické vlastnosti
Tabulka 2. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u dospělých již dříve léčených pacientů (věk 18-65 let) s těžkou hemofilií A (n = 20)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
22,6 ± 8,0 |
22,3 (8,4 - 38,1) |
|
T1/2 (hod) |
14,7 ± 10,4 |
12,5 (5,4 -55,6) |
|
IVR (%/IU/kg) |
2,5 ± 0,4 |
2,5 (1,7 -3,2) |
|
CL (ml/hod/kg) |
3,0 ± 1,2 |
2,7 (1,5-6,4) |
AUC = plocha pod křivkou (FVIII:C), Tm = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka
Tabulka 3. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u již dříve léčených dětských pacientů ve věku 6 až 12 let s těžkou hemofilií A (n = 12)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
13,2 ± 3,4 |
12,8 (7,8 - 19,1) |
|
T1/2 (hod) |
10,0 ± 1,9 |
9,9 (7,6 -14,1) |
|
IVR (%/IU/kg) |
1,9 ± 0,4 |
1,9 (1,2 -2,6) |
|
CL (ml/hod/kg) |
4,3 ± 1,2 |
4,2 (2,8 - 6,9) |
AUC = plocha pod křivkou (FVIII:C), T1/2 = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka
Tabulka 4. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u již dříve léčených dětských pacientů ve věku 2 až 5 let s těžkou hemofilií A (n = 13)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
11,7 ± 5,3 |
10,5 (4,9 -23,8) |
|
T1/2 (hod) |
9,5 ± 3,3 |
8,2 (4,3 - 17,3) |
|
IVR (%/IU/kg) |
1,9 ± 0,3 |
1,8 (1,5 -2,4) |
|
CL (ml/hod/kg) |
5,4 ± 2,4 |
5,1 (2,3 -10,9) |
AUC = plocha pod křivkou (FVIII:C), T1/2 = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka Pediatrická populace
Z literatury je známo, že biologická dostupnost a poločas byly nižší u dětí než u dospělých a clearance vyšší, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Podskupiny upravené podle hmotnosti
Tabulka 5. FK parametry pro přípravek Nuwiq upravené podle hmotnosti (Dávka: 50 IU/kg) u
|
dospělých již dříve |
éčených pacientů (věk 18-65 let) s těžkou hemofilií A (n = 20) | |||
|
FK parametr |
Všichni (n=20) |
Normální hmotnost (n=14) |
S nadváhou (n=4) |
Obézní (n=2) |
|
Chromogenní metoda Průměr ± SD | ||||
|
AUC (hod*IU/ml) |
22,6 ± 8,0 |
20,4 ± 6,9 |
24,9 ± 8,9 |
33,5 ± 6,5 |
|
T1/2 (hod) |
14,7 ± 10,4 |
14,7 ± 12,1 |
13,4 ± 5,9 |
17,2 ± 4,8 |
|
IVR (%/IU/kg) |
2,5 ± 0,4 |
2,4 ± 0,4 |
2,7 ± 0,4 |
2,8 ± 0,3 |
|
CL (ml/hod/kg) |
3,0 ± 1,2 |
3,2 ± 1,3 |
2,6 ± 1,0 |
1,8 ± 0,4 |
|
Chromogenní metoda Medián (rozsah) | ||||
|
AUC (hod*IU/ml) |
22,3 (8,4 -38,1) |
21,2 (8,4 -32,6) |
23,3 (17,4 - 35,5) |
33,5 (28,9 -38,1) |
|
T1/2 (hod) |
12,5 (5,4 - 55,6) |
12,3 (5,4 -55,6) |
11,2 (9,3 -22,0) |
17,2 (13,8 -20,6) |
|
IVR (%/IU/kg) |
2,5 (1,7 -3,2) |
2,4 (1,7 -3,1) |
2,8 (2,3 - 3,2) |
2,8 (2,6 -3,0) |
|
CL (ml/hod/kg) |
2,7 (1,5 -6,4) |
2,8 (1,7 -6,4) |
2,5 (1,6 -3,7) |
1,8 (1,5 -2,0) |
|
Normální hmotnost: |
BMI 18,5-25 kg/m2, |
Nadváha: BMI 25-30 kg/m2, Obezita: BM |
> 30 kg/m2, SD = | |
Směrodatná odchylka
5.3 Předklinické údaje vztahující se k bezpečnosti
V rámci předklinických studií byl přípravek Nuwiq bezpečně a úspěšně použit k obnově hemostázy u psů s hemofilií. Toxikologické studie ukázaly, že lokální intravenózní podání a systémová expozice byly dobře tolerovány u laboratorních zvířat (potkanů a opic makaka jávského).
Specifické studie reprodukční toxicity, chronické toxicity a kancerogenity s dlouhodobým opakovaným podáváním přípravku Nuwiq nebyly provedeny kvůli imunitní reakci na heterologní proteiny u všech nelidských druhů savců.
Nebyly provedeny žádné studie mutagenního potenciálu přípravku Nuwiq.
Ex vivo hodnocení při použití komerční testovací soupravy pro kvantifikaci odezvy T-buněk na proteinová terapeutika ukazují nízké riziko imunogenicity.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Sacharosa Chlorid sodný
Dihydrát chloridu vápenatého Arginin hydrochlorid Dihydrát citronanu sodného Poloxamer 188
Rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
Mají se používat pouze dodávané injekční soupravy, protože může dojít k selhání léčby v důsledku adsorpce lidského koagulačního faktoru VIII na vnitřní povrchy některých infuzních zařízení.
6.3 Doba použitelnosti
2 roky
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25°C) po dobu jednoho nepřetržitého období nepřesahujícího 1 měsíc. Jakmile byl přípravek jednou vyjmut z chladničky, nesmí tam být vrácen zpět. Označte si prosím na krabičce datum, kdy jste přípravek začali uchovávat při pokojové teplotě. Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Po rekonstituci je prokázána chemická a fyzikální stabilita po dobu 24 hodin při uchovávání při pokojové teplotě.
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, doba a podmínky uchovávání přípravku před použitím jsou v odpovědnosti uživatele. Uchovávejte rekonstituovaný přípravek při pokojové teplotě. Po rekonstituci chraňte před mrazem.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Uchovávání tohoto léčivého přípravku při pokojové teplotě a podmínky uchovávání po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Jedno balení přípravku Nuwiq 500 IU obsahuje:
- Prášek: 500 IU prášku v 8ml skleněné injekční lahvičce typu I, uzavřené potaženou bromobutylovou zátkou s pojistným hliníkovým uzávěrem
- Rozpouštědlo: 2,5 ml vody na injekci v předplněné injekční stříkačce z borosilikátového skla
- 1 sterilní adaptér injekční lahvičky pro rekonstituci s 1 motýlkovou jehlou a 2 tampóny napuštěnými alkoholem
Velikost balení po 1 kusu.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Prášek má být rekonstituován pouze v dodaném rozpouštědle (2,5 ml vody na injekce) za použití injekční soupravy, která je součástí dodávky. Při rozpouštění prášku má injekční lahvička vykonávat jemný krouživý pohyb, dokud se prášek nerozpustí. Po rekonstituci má být roztok natažen zpět do injekční stříkačky.
Rekonstituovaný léčivý přípravek má být před podáním vizuálně zkontrolován, zda neobsahuje cizorodé částice nebo zabarvení. Tento rekonstituovaný léčivý přípravek je čirý, bezbarvý roztok bez přítomnosti cizorodých částic s pH v rozmezí 6,5 až 7,5. Nepoužívejte roztok, je-li zakalený nebo obsahuje-li usazeniny.
Návod pro přípravu a podávání
1. Ponechte rozpouštědlo v injekční stříkačce (voda na injekci) a prášek v uzavřené injekční lahvičce dostatečnou dobu pro dosažení pokojové teploty. Můžete to udělat tak, že je podržíte v rukou, dokud nebudou stejně teplé jako vaše ruce. Na zahřívání lahvičky a předem naplněné stříkačky nepoužívejte jiné postupy. Tato teplota by měla být udržována během rekonstituce.
2. Odstraňte pojistné plastové víčko z injekční lahvičky s práškem, aby byla přístupná střední část gumové zátky. Neodstraňujte šedou zátku ani kovový kroužek kolem horní části injekční lahvičky.
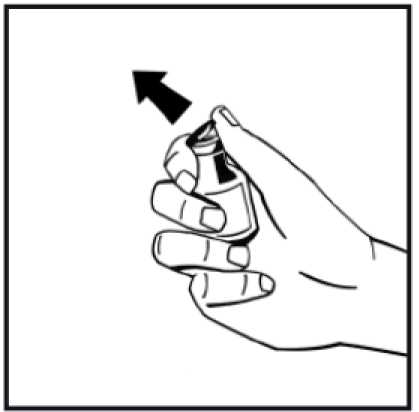
3. Otřete horní část injekční lahvičky tampónem napuštěným v alkoholu. Počkejte, dokud se alkohol nevypaří.
4. Sejměte papírový kryt z obalu adaptéru injekční lahvičky. Nevyndávejte adaptér z obalu.
Umístěte injekční lahvičku s práškem na rovný povrch a držte ji. Uchopte obal adaptéru a umístěte adaptér injekční lahvičky na střední část gumové zátky injekční lahvičky s práškem. Stlačte pevně obal adaptéru tak, aby hrot adaptéru prošel gumovou zátkou. Adaptér se přitom zacvakne na injekční lahvičku.
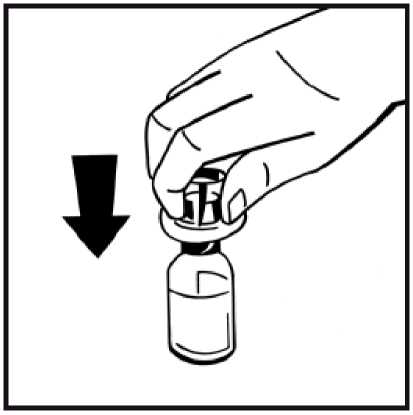
5.
6.
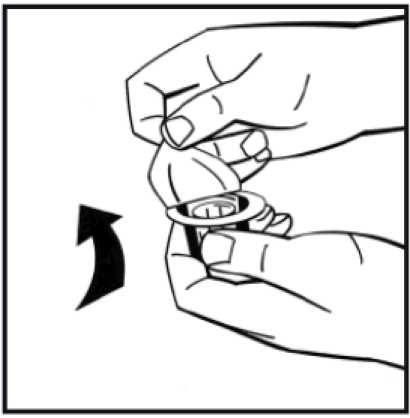
Sejměte papírový kryt z obalu předplněné injekční stříkačky. Uchopte píst za konec, a nedotýkejte se jeho střední části. Připojte konec pístu se závitem k injekční stříkačce s rozpouštědlem. Otáčejte pístem ve směru pohybu hodinových ručiček, dokud neucítíte slabý odpor.
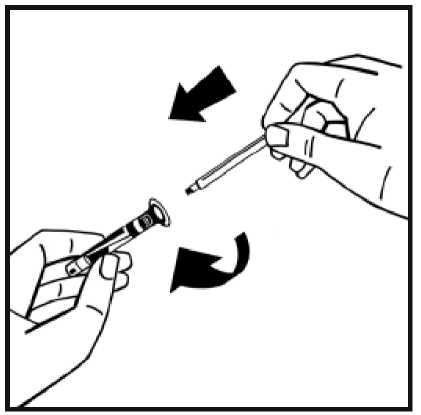
7. Odlomte ochranný plastový hrot z injekční stříkačky s rozpouštědlem v místě perforace víčka. Nedotýkejte se vnitřku víčka ani hrotu injekční stříkačky. Pokud roztok nebudete používat ihned, zavřete naplněnou stříkačku plastovou špičkou s odolností vůči nárazu pro uskladnění.
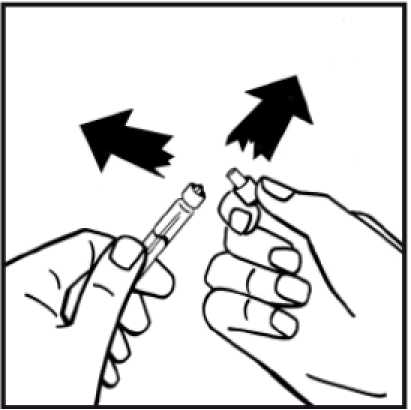
8. Sejměte obal adaptéru a vyhoďte jej.
9. Pevně připojte injekční stříkačku s rozpouštědlem k adaptéru injekční lahvičky otáčením ve směru pohybu hodinových ručiček, dokud neucítíte slabý odpor.
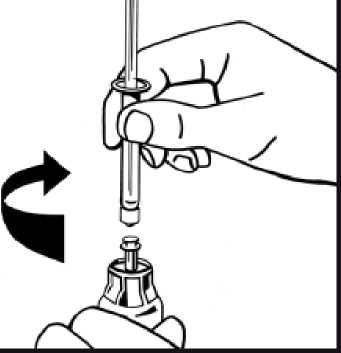
10. Pomalu vstřikujte veškeré rozpouštědlo do injekční lahvičky s práškem tlakem na píst.
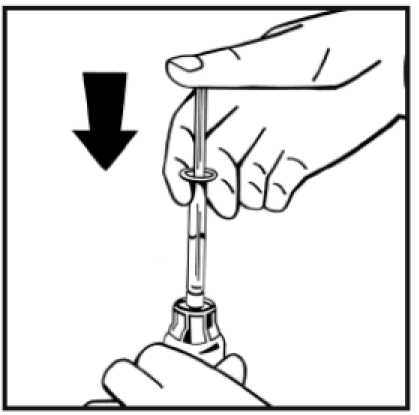
11. Aniž byste odstranili injekční stříkačku, rozpusťte prášek jemným pohybem nebo několika krouživými pohyby injekční lahvičky. Netřepejte. Počkejte, dokud se prášek zcela nerozpustí.
12. Před podáním zrakem zkontrolujte, že výsledný roztok neobsahuje žádné částice. Roztok má být čirý a bezbarvý, prakticky bez viditelných částic. Nepoužívejte roztok, je-li zakalený nebo obsahuje-li usazeniny.
13. Otočte injekční lahvičku připevněnou k injekční stříkačce dnem vzhůru a pomalu natáhněte výsledný roztok do stříkačky. Přesvědčte se, že nyní je celý obsah injekční lahvičky v injekční stříkačce.
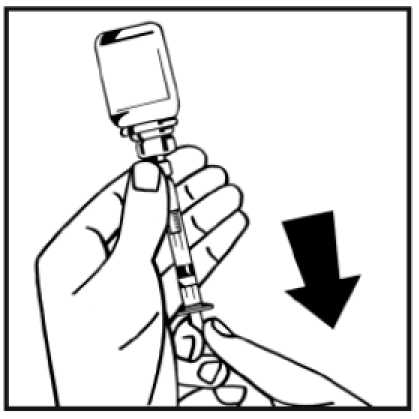
14. Odpojte naplněnou injekční stříkačku od adaptéru injekční lahvičky otáčením proti směru pohybu hodinových ručiček a prázdnou injekční lahvičku zlikvidujte.
15. Roztok je nyní připraven k okamžitému použití. Chraňte před chladem.
16. Očistěte zvolené místo vpichu jedním z dodaných tampónů napuštěných alkoholem.
17. Připojte k injekční stříkačce infuzní soupravu, která je součástí balení.
Zaveďte jehlu infuzní soupravy do zvolené žíly. Pokud jste použili škrtidlo pro zviditelnění žíly, má být toto škrtidlo uvolněno předtím, než začnete vstřikovat roztok.
Do injekční stříkačky se nesmí dostat žádná krev, aby nedošlo k tvorbě fibrinových sraženin.
18. Vstřikujte roztok do žíly pomalu, maximálně 4 ml za minutu.
Použijete-li více než jednu injekční lahvičku s práškem pro jednu léčbu, můžete použít stejnou injekční jehlu opakovaně. Adaptér injekční lahvičky a injekční stříkačka jsou určeny pouze na jedno použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Octapharma AB Lars Forssells gata 23 112 75 Stockholm Švédsko
8. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/936/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 22. července 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
V Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Nuwiq 1000 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje nominálně 1000 IU lidského koagulačního faktoru VIII (rDNA), simoctocog alfa.
Nuwiq obsahuje přibližně 400 IU/ml lidského koagulačního faktoru VIII (rDNA), simoctocog alfa po rekonstituci.
Účinnost (IU) se stanovuje podle Evropského lékopisu chromogenní analýzou. Specifická aktivita přípravku Nuwiq je přibližně 9500 IU/mg proteinu.
Simoctocog alfa (lidský koagulační faktor VIII (rDNA)) je čištěný protein, který obsahuje 1440 aminokyselin. Sekvence aminokyselin je srovnatelná s formou 90 + 80 kDa faktoru VIII lidské plazmy (tj. bez B-domény). Nuwiq se vyrábí rekombinantní DNA technologií v geneticky upravených lidských embryonálních renálních buňkách (HEK) 293F. V rámci výrobního procesu se k léčivému přípravku nepřidává žádný zvířecí ani lidský materiál.
Pomocná látka/Pomocné látky se známým účinkem:
1 ml rekonstituovaného roztoku obsahuje 7,35 mg sodíku (18,4 mg sodíku na 1 injekční lahvičku). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: bílý až téměř bílý drobivý prášek. Rozpouštědlo: voda na injekci, čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípraven Nuwiq lze použít pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má probíhat pod dohledem lékaře, který má zkušenosti s léčbou hemofilie.
Dosud neléčení pacienti
Bezpečnost a účinnost přípravku Nuwiq u pacientů bez předchozí léčby zatím nebyla stanovena. Dávkování
Dávka a doba trvání substituční léčby závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet podávaných jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (IU), které se vztahují k aktuálně platnému WHO standardu pro přípravky obsahující faktor VIII. Plazmatická aktivita faktoru VIII je vyjádřena buď v procentech (vztaženo k normální lidské plazmě), nebo v mezinárodních jednotkách (vztaženo k mezinárodnímu standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba dle potřeby
Výpočet požadované dávky faktoru VIII je založen na empirickém poznatku, že 1 mezinárodní jednotka (IU) faktoru VIII na 1 kilogram tělesné hmotnosti zvyšuje plazmatickou aktivitu faktoru VIII o přibližně 2 % normální aktivity nebo o 2 IU/dl. Požadovaná dávka se stanoví pomocí následujícího vzorce:
I. Požadované jednotky = tělesná hmotnost (kg) x požadované zvýšení faktoru VIII (%) (IU/dl) x 0,5 (IU/kg na 1 IU/dl)
II. Předpokládané zvýšení faktoru VIII (% z normální hodnoty) = podaná dávka vIU x 2
tělesná hmotnost (kg)
Podávané množství a četnost podávání by se měly vždy zaměřovat na klinickou efektivnost v každém jednotlivém případě.
V případě následného krvácení by aktivita faktoru VIII neměla poklesnout pod stanovenou hladinu plazmatické aktivity (v % normální hodnoty nebo IU/dl) ve sledovaném období. Následující tabulku lze použít jako návod pro dávkování při krvácivých příhodách nebo při chirurgickém zákroku.
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Frekvence dávkování (hodiny) / Délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny |
20-40 |
Opakujte každých 12 až 24 hodin. Po dobu nejméně jednoho dne, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne zhojení. |
|
Intenzivnější hemartróza, krvácení do svalu nebo tvorba hematomů |
30-60 |
Opakujte infúzi každých 12 až 24 hodin po dobu 3 až 4 dnů nebo déle, dokud bolest a akutní potíže neustoupí. |
|
Život ohrožující krvácení |
60-100 |
Opakujte infúzi každých 8 až 24 hodin, dokud není hrozba odvrácena. |
|
Chirurgický zákrok | ||
|
Drobný chirurgický zákrok, včetně trhání zubu |
30-60 |
Každých 24 hodin, po dobu nejméně 1 dne, dokud nedojde ke zhojení. |
|
Velký chirurgický zákrok |
80-100 (před operací a po ní) |
Opakujte infúzi každých 8-24 hodin, dokud nedojde ke zhojení rány, pak pokračujte v terapii po dobu nejméně dalších 7 dnů pro udržení aktivity faktoru VIII na úrovni 30 % až 60 % |
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Frekvence dávkování (hodiny) / Délka trvání léčby (dny) |
|
(IU/dl). | ||
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů s těžkou hemofilií A je obvyklé dávkování 20 až 40 IU faktoru VIII na 1 kilogram tělesné hmotnosti každé 2 až 3 dny. V některých případech, zejména u mladších pacientů, mohou být nutné kratší intervaly dávkování nebo vyšší dávky.
V průběhu léčby se doporučuje vhodné stanovení hladin faktoru VIII pro určení dávky, která má být podávána, a četnosti opakovaných infuzí. V případě velkých chirurgických zákroků je nezbytné přesné monitorování substituční léčby pomocí koagulační analýzy (aktivity faktoru VIII v plazmě). Jednotliví pacienti se mohou lišit ve své odpovědi na faktor VIII, mohou se lišit v poločasech eliminace a v hodnotách biologické dostupnosti.
Pediatrická populace
Dávkování je stejné u dospělých i dětských pacientů, ale u dětí mohou být nutné kratší intervaly mezi dávkami a vyšší dávky. V současnosti dostupné údaje jsou uvedeny v bodech 4.8, 5.1 a 5.2.
U dětí mladších 2 let nejsou dostupné žádné údaje.
Způsob podání
Intravenózní podání.
Doporučuje se podávat nejvýše 4 ml přípravku za minutu.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoliv pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Stejně jako u jiných intravenózně podávaných proteinových přípravků jsou možné hypersensitivní reakce alergického typu. Přípravek Nuwiq obsahuje stopová množství proteinů lidské hostitelské buňky jiných než faktor VIII. Vyskytnou-li se příznaky přecitlivělosti, pacienti mají být poučeni o neprodleném přerušení užívání léčivého přípravku a o návštěvě lékaře. Pacienti mají být poučeni o časných projevech přecitlivělosti, jako například vyrážka, generalizovaná kopřivka, tlak na hrudi, sípavé dýchání, hypotenze a anafylaxe.
V případě šoku je nutno dodržovat standardní lékařské postupy jeho léčby.
Inhibitory
Známou komplikací léčby u nemocných s hemofilií A je tvorba neutralizujících protilátek (inhibitorů) faktoru VIII. Tyto inhibitory jsou obvykle IgG imunoglobuliny směrované proti prokoagulační aktivitě faktoru VIII, a jsou kvantifikovány v Bethesda jednotkách (BU) na ml plazmy s použitím modifikovaného testu. Riziko rozvoje inhibitorů koreluje s expozicí faktoru VIII; toto riziko se zvyšuje v průběhu prvních 20 dnů expozice. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Z tohoto důvodu se doporučuje pečlivě monitorovat pacienty z hlediska možného vývoje inhibitorů po jakékoliv změně přípravku.
Obecně platí, že u všech pacientů léčených koagulačním faktorem VIII musí být pečlivě monitorován vznik inhibitorů vhodnými klinickými pozorováními a laboratorními testy. Pokud se nedosáhne očekávané hladiny aktivity plazmatického faktoru VIII nebo pokud krvácení není zastaveno odpovídající dávkou, měl by být proveden test za účelem určení přítomnosti inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční léčba faktorem VIII účinná a je třeba uvažovat o jiných možnostech léčby, jako je například navození imunitní tolerance (ITI). Léčba takových pacientů by měla být vedena lékaři se zkušenostmi s léčbou hemofilie a s inhibitory faktoru VIII.
Komplikace způsobené katétrem
Pokud je potřeba použít centrální žilní vstup (CVAD), má být zváženo riziko komplikací souvisejících s CVAD, včetně lokální infekce, bakteriémie a trombózy v místě vstupu katétru.
Důrazně se doporučuje, pokaždé, kdy je přípravek Nuwiq podán pacientovi, zaznamenat název a číslo šarže aplikovaného přípravku, aby bylo zachováno spojení mezi pacientem a číslem šarže léčivého přípravku.
Pediatrická populace
Uvedená upozornění a bezpečnostní pokyny se vztahují jak na dospělé, tak na dětské pacienty.
Pokyny vztahující se k pomocné látce (obsah sodíku)
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v 1 injekční lahvičce.
V závislosti na tělesné hmotnosti a dávkování by měl pacient obdržet více než jednu injekční lahvičku. To je třeba vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
U přípravku Nuwiq nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie na zvířatech nebyly s přípravkem Nuwiq prováděny.
Vzhledem k vzácnému výskytu hemofilie A u žen nejsou k dispozici žádné zkušenosti s používáním faktoru VIII během těhotenství a kojení. Z tohoto důvodu má být přípravek Nuwiq během těhotenství a kojení používán pouze, je-li to jasně indikováno. Nejsou k dispozici žádné údaje týkající se fertility.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Nuwiq nemá žádný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn informací o bezpečnostním profilu
U přípravků obsahujících faktor VIII byly vzácně pozorovány přecitlivělost nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, zimnici, zarudnutí, generalizovanou vyrážku, bolest hlavy, kopřivku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tlak na hrudi, mravenčení, zvracení, sípání) které se mohou v některých případech rozvinout do závažné anafylaxe (včetně šoku).
U pacientů s hemofilií A se mohou vytvořit neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory vyskytnou, mohou se projevit nedostatečnou klinickou odezvou na přípravek. V těchto případech se doporučuje vyhledat specializované pracoviště na léčení hemofilie.
Přehledný souhrn nežádoucích účinků
Během klinických studií přípravku Nuwiq provedených u již dříve léčených pediatrických pacientů (2 až 11 let, n = 58), dospívajících pacientů (12 až 17 let, n = 3) a dospělých pacientů (n = 74) s těžkou hemofilií A, byl celkový počet závažných nežádoucích účinků (ADR) 8 (6 u dospělých, 2 u dětí) hlášený u 5 pacientů (3 dospělých, 2 dětí).
Tabulka 1 uvedená níže je uspořádána v souladu s klasifikací tříd orgánových systémů MedDRa (SOC a preferované termíny četností).
Četnost je definována dle následující konvence: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000); velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 1. Frekvence výskytu nežádoucích účinků léčivého přípravku (ADR) na 1 pacienta
|
během klinických studií u 135 již dříve léčených pacientů s těžkou |
íemofilií A | |
|
Třída orgánových systémů podle databáze MedDRA |
Nežádoucí účinky |
Četnost* |
|
Poruchy nervové soustavy |
Parastézie Bolesti hlavy |
Méně časté |
|
Poruchy ucha a labyrintu |
Závratě |
Méně časté |
|
Gastrointestinální poruchy |
Sucho v ústech |
Méně časté |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolesti zad |
Méně časté |
|
Celkové poruchy a reakce v místě aplikace |
Zánět v místě vpichu Bolest v místě vpichu |
Méně časté |
|
Vyšetření |
Pozitivní protilátka faktoru VIII bez neutralizační aktivity |
Méně časté |
* Všechny tyto nežádoucí účinky se vyskytly pouze jednou. Vzhledem k tomu, že celkový počet studovaných pacientů je 135, četnost nemůže být jiná než „méně časté“, jakmile se závažné nežádoucí účinky objeví jednou.
Popis vybraných nežádoucích účinků
U jednoho dospělého pacienta byla zjištěna protilátka faktoru VIII bez neutralizační aktivity (viz Tabulka 1). Vzorek byl testován v centrální laboratoři v osmi zředěních. Výsledek byl pozitivní pouze při faktoru zředění 1 a titr protilátek byl velmi nízký. Inhibiční aktivita měřená pomocí modifikovaného Bethesda testu nebyla u tohoto pacienta zjištěna. Klinická účinnost a biologická dostupnost přípravku Nuwiq nebyly u tohoto pacienta narušeny.
Pediatrická populace
Četnost, typ a závažnost nežádoucích účinků u dětí se očekávají shodné jako u dospělých.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nejsou známy žádné případy předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika, krevní koagulační faktor VIII, ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. Po infuzi hemofilickému pacientovi se faktor VIII váže na von Willebrandův faktor přítomný v krevním oběhu pacienta. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin následně přeměňuje fibrinogen na fibrin a může dojít k vytvoření sraženiny. Hemofilie A je pohlavně vázaná dědičná porucha srážlivosti krve způsobená sníženou hladinou faktoru VIII:C, v důsledku které dochází k profuznímu krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánnímu, nebo jako následek úrazu při nehodě či chirurgickém zákroku. Substituční léčbou se hladiny faktoru VIII v plazmě zvýší, čímž je možná přechodná korekce nedostatku faktoru VIII a korekce sklonu ke krvácení.
Imunogenita přípravku Nuwiq byla hodnocena v rámci klinických zkoušek u 135 již dříve léčených pacientů s těžkou hemofilií A (74 dospělých a 61 dětských pacientů). U žádného z pacientů nedošlo k vývoji inhibitorů.
V rámci klinické studie provedené s 32 dospělými pacienty s těžkou hemofilií A byla střední hodnota spotřeby přípravku Nuwiq za účelem profylaxe 468,7 IU/kg/měsíc. Průměrná dávka při léčbě nečekaného krvácení byla 33,0 IU/kg u pacientů užívajících přípravek za účelem profylaxe. V rámci jiné studie bylo 22 dospělých pacientů léčeno na požádání. Celkem bylo 986 případů krvácení léčeno průměrnou dávkou 30,9 IU/kg. Obecně platí, že slabé krvácení vyžadovalo mírně nižší a silnější krvácení vyžadovalo až trojnásobně vyšší střední dávky.
Pediatrická populace
Data byla získána od 29 již dříve léčených dětí ve věku 2 až 5 let, 31 dětí ve věku 6 až 12 let a jednoho dospívajícího pacienta ve věku 14 let. Medián dávky na profylaktickou infuzi byl 37,8 IU/kg. Dvacet pacientů užívalo střední dávky vyšší než 45 IU/kg. Medián spotřeby přípravku Nuwiq za účelem profylaxe za měsíc byl 521,9 IU/kg. Vyšší medián dávky přípravku Nuwiq vyžadovala léčba krvácení u dětí (43,9 IU/kg) než u dospělých (33,0 IU/kg), a vyšší medián dávky vyžadovala léčba středního až silného krvácení v porovnání se slabým krvácením (78,2 IU/kg v porovnání s 41,7 IU/kg). Mladším dětem musel být obecně podáván vyšší medián dávky (6-12 let: 43,9 IU/kg; 2-5 let: 52,6 IU/kg).
Evropská agentura pro léčivé přípravky odložila závazek předložit výsledky studií přípravku Nuwiq u jednoho nebo více souborů pediatrické populace při léčbě hemofilie A (vrozeného nedostatku faktoru VIII) (viz bod 4.2, kde jsou uvedeny informace o použití pro pediatrické pacienty).
5.2 Farmakokinetické vlastnosti
Tabulka 2. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u dospělých již dříve léčených pacientů (věk 18-65 let) s těžkou hemofilií A (n = 20)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
22,6 ± 8,0 |
22,3 (8,4 - 38,1) |
|
T1/2 (hod) |
14,7 ± 10,4 |
12,5 (5,4 -55,6) |
|
IVR (%/IU/kg) |
2,5 ± 0,4 |
2,5 (1,7 -3,2) |
|
CL (ml/hod/kg) |
3,0 ± 1,2 |
2,7 (1,5-6,4) |
AUC = plocha pod křivkou (FVIII:C), Tm = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka
Tabulka 3. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u již dříve léčených dětských pacientů ve věku 6 až 12 let s těžkou hemofilií A (n = 12)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
13,2 ± 3,4 |
12,8 (7,8 - 19,1) |
|
T1/2 (hod) |
10,0 ± 1,9 |
9,9 (7,6 -14,1) |
|
IVR (%/IU/kg) |
1,9 ± 0,4 |
1,9 (1,2 -2,6) |
|
CL (ml/hod/kg) |
4,3 ± 1,2 |
4,2 (2,8 - 6,9) |
AUC = plocha pod křivkou (FVIII:C), T1/2 = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka
Tabulka 4. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u již dříve léčených dětských pacientů ve věku 2 až 5 let s těžkou hemofilií A (n = 13)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
11,7 ± 5,3 |
10,5 (4,9 -23,8) |
|
T1/2 (hod) |
9,5 ± 3,3 |
8,2 (4,3 - 17,3) |
|
IVR (%/IU/kg) |
1,9 ± 0,3 |
1,8 (1,5 -2,4) |
|
CL (ml/hod/kg) |
5,4 ± 2,4 |
5,1 (2,3 -10,9) |
AUC = plocha pod křivkou (FVIII:C), T1/2 = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka Pediatrická populace
Z literatury je známo, že biologická dostupnost a poločas byly nižší u dětí než u dospělých a clearance vyšší, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Podskupiny upravené podle hmotnosti
Tabulka 5. FK parametry pro přípravek Nuwiq upravené podle hmotnosti (Dávka: 50 IU/kg) u
|
dospělých již dříve |
éčených pacientů (věk 18-65 let) s těžkou hemofilií A (n = 20) | |||
|
FK parametr |
Všichni (n=20) |
Normální hmotnost (n=14) |
S nadváhou (n=4) |
Obézní (n=2) |
|
Chromogenní metoda Průměr ± SD | ||||
|
AUC (hod*IU/ml) |
22,6 ± 8,0 |
20,4 ± 6,9 |
24,9 ± 8,9 |
33,5 ± 6,5 |
|
T1/2 (hod) |
14,7 ± 10,4 |
14,7 ± 12,1 |
13,4 ± 5,9 |
17,2 ± 4,8 |
|
IVR (%/IU/kg) |
2,5 ± 0,4 |
2,4 ± 0,4 |
2,7 ± 0,4 |
2,8 ± 0,3 |
|
CL (ml/hod/kg) |
3,0 ± 1,2 |
3,2 ± 1,3 |
2,6 ± 1,0 |
1,8 ± 0,4 |
|
Chromogenní metoda Medián (rozsah) | ||||
|
AUC (hod*IU/ml) |
22,3 (8,4 -38,1) |
21,2 (8,4 -32,6) |
23,3 (17,4 - 35,5) |
33,5 (28,9 -38,1) |
|
T1/2 (hod) |
12,5 (5,4 - 55,6) |
12,3 (5,4 -55,6) |
11,2 (9,3 -22,0) |
17,2 (13,8 -20,6) |
|
IVR (%/IU/kg) |
2,5 (1,7 -3,2) |
2,4 (1,7 -3,1) |
2,8 (2,3 - 3,2) |
2,8 (2,6 -3,0) |
|
CL (ml/hod/kg) |
2,7 (1,5 -6,4) |
2,8 (1,7 -6,4) |
2,5 (1,6 -3,7) |
1,8 (1,5 -2,0) |
|
Normální hmotnost: |
BMI 18,5-25 kg/m2, |
Nadváha: BMI 25-30 kg/m2, Obezita: BM |
> 30 kg/m2, SD = | |
Směrodatná odchylka
5.3 Předklinické údaje vztahující se k bezpečnosti
V rámci předklinických studií byl přípravek Nuwiq bezpečně a úspěšně použit k obnově hemostázy u psů s hemofilií. Toxikologické studie ukázaly, že lokální intravenózní podání a systémová expozice byly dobře tolerovány u laboratorních zvířat (potkanů a opic makaka jávského).
Specifické studie reprodukční toxicity, chronické toxicity a kancerogenity s dlouhodobým opakovaným podáváním přípravku Nuwiq nebyly provedeny kvůli imunitní reakci na heterologní proteiny u všech nelidských druhů savců.
Nebyly provedeny žádné studie mutagenního potenciálu přípravku Nuwiq.
Ex vivo hodnocení při použití komerční testovací soupravy pro kvantifikaci odezvy T-buněk na proteinová terapeutika ukazují nízké riziko imunogenicity.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Sacharosa Chlorid sodný
Dihydrát chloridu vápenatého Arginin hydrochlorid Dihydrát citronanu sodného Poloxamer 188
Rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
Mají se používat pouze dodávané injekční soupravy, protože může dojít k selhání léčby v důsledku adsorpce lidského koagulačního faktoru VIII na vnitřní povrchy některých infuzních zařízení.
6.3 Doba použitelnosti
2 roky
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25°C) po dobu jednoho nepřetržitého období nepřesahujícího 1 měsíc. Jakmile byl přípravek jednou vyjmut z chladničky, nesmí tam být vrácen zpět. Označte si prosím na krabičce datum, kdy jste přípravek začali uchovávat při pokojové teplotě. Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Po rekonstituci je prokázána chemická a fyzikální stabilita po dobu 24 hodin při uchovávání při pokojové teplotě.
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, doba a podmínky uchovávání přípravku před použitím jsou v odpovědnosti uživatele. Uchovávejte rekonstituovaný přípravek při pokojové teplotě. Po rekonstituci chraňte před mrazem.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Uchovávání tohoto léčivého přípravku při pokojové teplotě a podmínky uchovávání po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Jedno balení přípravku Nuwiq 1000 IU obsahuje:
- Prášek: 1000 IU prášku v 8ml skleněné injekční lahvičce typu I, uzavřené potaženou bromobutylovou zátkou s pojistným hliníkovým uzávěrem
- Rozpouštědlo: 2,5 ml vody na injekci v předplněné injekční stříkačce z borosilikátového skla
- 1 sterilní adaptér injekční lahvičky pro rekonstituci s 1 motýlkovou jehlou a 2 tampóny napuštěnými alkoholem
Velikost balení po 1 kusu.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Prášek má být rekonstituován pouze v dodaném rozpouštědle (2,5 ml vody na injekce) za použití injekční soupravy, která je součástí dodávky. Při rozpouštění prášku má injekční lahvička vykonávat jemný krouživý pohyb, dokud se prášek nerozpustí. Po rekonstituci má být roztok natažen zpět do injekční stříkačky.
Rekonstituovaný léčivý přípravek má být před podáním vizuálně zkontrolován, zda neobsahuje cizorodé částice nebo zabarvení. Tento rekonstituovaný léčivý přípravek je čirý, bezbarvý roztok bez přítomnosti cizorodých částic s pH v rozmezí 6,5 až 7,5. Nepoužívejte roztok, je-li zakalený nebo obsahuje-li usazeniny.
Návod pro přípravu a podávání
1. Ponechte rozpouštědlo v injekční stříkačce (voda na injekci) a prášek v uzavřené injekční lahvičce dostatečnou dobu pro dosažení pokojové teploty. Můžete to udělat tak, že je podržíte v rukou, dokud nebudou stejně teplé jako vaše ruce. Na zahřívání lahvičky a předem naplněné stříkačky nepoužívejte jiné postupy. Tato teplota by měla být udržována během rekonstituce.
2. Odstraňte pojistné plastové víčko z injekční lahvičky s práškem, aby byla přístupná střední část gumové zátky. Neodstraňujte šedou zátku ani kovový kroužek kolem horní části injekční lahvičky.
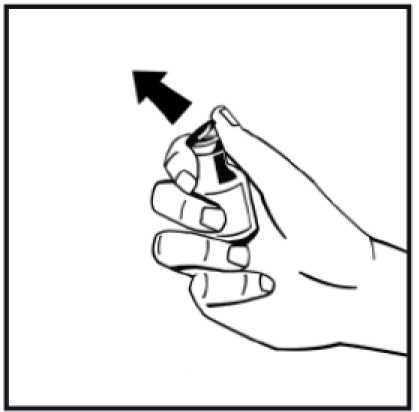
3. Otřete horní část injekční lahvičky tampónem napuštěným v alkoholu. Počkejte, dokud se alkohol nevypaří.
4. Sejměte papírový kryt z obalu adaptéru injekční lahvičky. Nevyndávejte adaptér z obalu.
Umístěte injekční lahvičku s práškem na rovný povrch a držte ji. Uchopte obal adaptéru a umístěte adaptér injekční lahvičky na střední část gumové zátky injekční lahvičky s práškem. Stlačte pevně obal adaptéru tak, aby hrot adaptéru prošel gumovou zátkou. Adaptér se přitom zacvakne na injekční lahvičku.
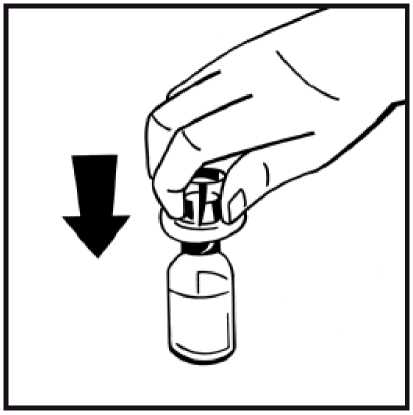
5.
6.
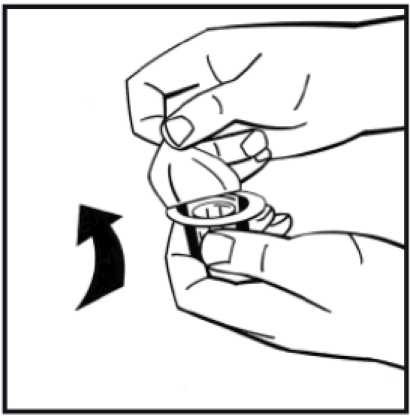
Sejměte papírový kryt z obalu předplněné injekční stříkačky. Uchopte píst za konec, a nedotýkejte se jeho střední části. Připojte konec pístu se závitem k injekční stříkačce s rozpouštědlem. Otáčejte pístem ve směru pohybu hodinových ručiček, dokud neucítíte slabý odpor.
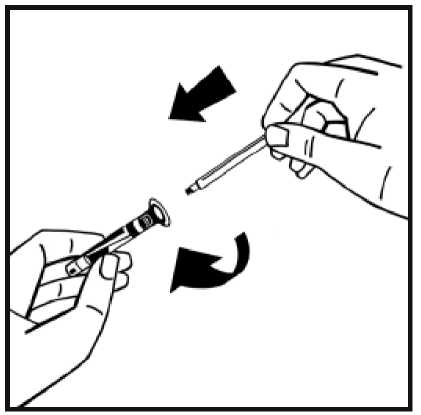
7. Odlomte ochranný plastový hrot z injekční stříkačky s rozpouštědlem v místě perforace víčka. Nedotýkejte se vnitřku víčka ani hrotu injekční stříkačky. Pokud roztok nebudete používat ihned, zavřete naplněnou stříkačku plastovou špičkou s odolností vůči nárazu pro uskladnění.
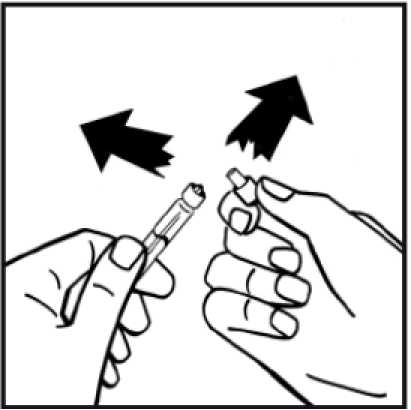
8. Sejměte obal adaptéru a vyhoďte jej.
9. Pevně připojte injekční stříkačku s rozpouštědlem k adaptéru injekční lahvičky otáčením ve směru pohybu hodinových ručiček, dokud neucítíte slabý odpor.
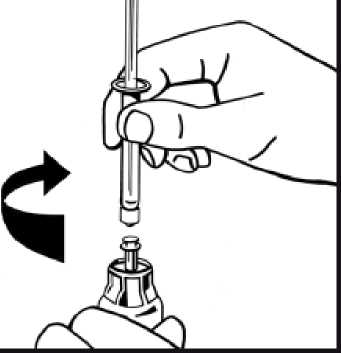
10. Pomalu vstřikujte veškeré rozpouštědlo do injekční lahvičky s práškem tlakem na píst.
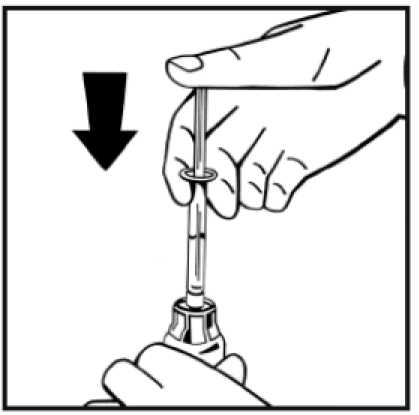
11. Aniž byste odstranili injekční stříkačku, rozpusťte prášek jemným pohybem nebo několika krouživými pohyby injekční lahvičky. Netřepejte. Počkejte, dokud se prášek zcela nerozpustí.
12. Před podáním zrakem zkontrolujte, že výsledný roztok neobsahuje žádné částice. Roztok má být čirý a bezbarvý, prakticky bez viditelných částic. Nepoužívejte roztok, je-li zakalený nebo obsahuje-li usazeniny.
13. Otočte injekční lahvičku připevněnou k injekční stříkačce dnem vzhůru a pomalu natáhněte výsledný roztok do stříkačky. Přesvědčte se, že nyní je celý obsah injekční lahvičky v injekční stříkačce.
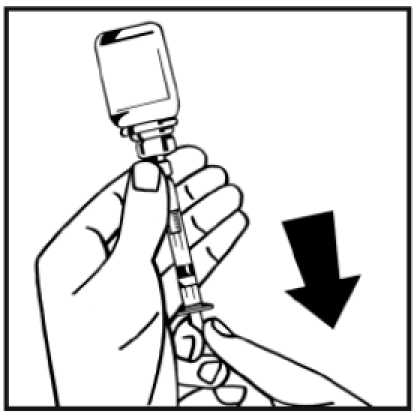
14. Odpojte naplněnou injekční stříkačku od adaptéru injekční lahvičky otáčením proti směru pohybu hodinových ručiček a prázdnou injekční lahvičku zlikvidujte.
15. Roztok je nyní připraven k okamžitému použití. Chraňte před chladem.
16. Očistěte zvolené místo vpichu jedním z dodaných tampónů napuštěných alkoholem.
17. Připojte k injekční stříkačce infuzní soupravu, která je součástí balení.
Zaveďte jehlu infuzní soupravy do zvolené žíly. Pokud jste použili škrtidlo pro zviditelnění žíly, má být toto škrtidlo uvolněno předtím, než začnete vstřikovat roztok.
Do injekční stříkačky se nesmí dostat žádná krev, aby nedošlo k tvorbě fibrinových sraženin.
18. Vstřikujte roztok do žíly pomalu, maximálně 4 ml za minutu.
Použijete-li více než jednu injekční lahvičku s práškem pro jednu léčbu, můžete použít stejnou injekční jehlu opakovaně. Adaptér injekční lahvičky a injekční stříkačka jsou určeny pouze na jedno použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Octapharma AB Lars Forssells gata 23 112 75 Stockholm Švédsko
8. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/936/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 22. července 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
V Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Nuwiq 2000 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje nominálně 2000 IU lidského koagulačního faktoru VIII (rDNA), simoctocog alfa.
Nuwiq obsahuje přibližně 800 IU/ml lidského koagulačního faktoru VIII (rDNA), simoctocog alfa po rekonstituci.
Účinnost (IU) se stanovuje podle Evropského lékopisu chromogenní analýzou. Specifická aktivita přípravku Nuwiq je přibližně 9500 IU/mg proteinu.
Simoctocog alfa (lidský koagulační faktor VIII (rDNA)) je čištěný protein, který obsahuje 1440 aminokyselin. Sekvence aminokyselin je srovnatelná s formou 90 + 80 kDa faktoru VIII lidské plazmy (tj. bez B-domény). Nuwiq se vyrábí rekombinantní DNA technologií v geneticky upravených lidských embryonálních renálních buňkách (HEK) 293F. V rámci výrobního procesu se k léčivému přípravku nepřidává žádný zvířecí ani lidský materiál.
Pomocná látka/Pomocné látky se známým účinkem:
1 ml rekonstituovaného roztoku obsahuje 7,35 mg sodíku (18,4 mg sodíku na 1 injekční lahvičku). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: bílý až téměř bílý drobivý prášek. Rozpouštědlo: voda na injekci, čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípraven Nuwiq lze použít pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má probíhat pod dohledem lékaře, který má zkušenosti s léčbou hemofilie.
Dosud neléčení pacienti
Bezpečnost a účinnost přípravku Nuwiq u pacientů bez předchozí léčby zatím nebyla stanovena. Dávkování
Dávka a doba trvání substituční léčby závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet podávaných jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (IU), které se vztahují k aktuálně platnému WHO standardu pro přípravky obsahující faktor VIII. Plazmatická aktivita faktoru VIII je vyjádřena buď v procentech (vztaženo k normální lidské plazmě), nebo v mezinárodních jednotkách (vztaženo k mezinárodnímu standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba dle potřeby
Výpočet požadované dávky faktoru VIII je založen na empirickém poznatku, že 1 mezinárodní jednotka (IU) faktoru VIII na 1 kilogram tělesné hmotnosti zvyšuje plazmatickou aktivitu faktoru VIII o přibližně 2 % normální aktivity nebo o 2 IU/dl. Požadovaná dávka se stanoví pomocí následujícího vzorce:
I. Požadované jednotky = tělesná hmotnost (kg) x požadované zvýšení faktoru VIII (%) (IU/dl) x 0,5 (IU/kg na 1 IU/dl)
II. Předpokládané zvýšení faktoru VIII (% z normální hodnoty) = podaná dávka vIU x 2
tělesná hmotnost (kg)
Podávané množství a četnost podávání by se měly vždy zaměřovat na klinickou efektivnost v každém jednotlivém případě.
V případě následného krvácení by aktivita faktoru VIII neměla poklesnout pod stanovenou hladinu plazmatické aktivity (v % normální hodnoty nebo IU/dl) ve sledovaném období. Následující tabulku lze použít jako návod pro dávkování při krvácivých příhodách nebo při chirurgickém zákroku.
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Frekvence dávkování (hodiny) / Délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny |
20-40 |
Opakujte každých 12 až 24 hodin. Po dobu nejméně jednoho dne, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne zhojení. |
|
Intenzivnější hemartróza, krvácení do svalu nebo tvorba hematomů |
30-60 |
Opakujte infúzi každých 12 až 24 hodin po dobu 3 až 4 dnů nebo déle, dokud bolest a akutní potíže neustoupí. |
|
Život ohrožující krvácení |
60-100 |
Opakujte infúzi každých 8 až 24 hodin, dokud není hrozba odvrácena. |
|
Chirurgický zákrok | ||
|
Drobný chirurgický zákrok, včetně trhání zubu |
30-60 |
Každých 24 hodin, po dobu nejméně 1 dne, dokud nedojde ke zhojení. |
|
Velký chirurgický zákrok |
80-100 (před operací a po ní) |
Opakujte infúzi každých 8-24 hodin, dokud nedojde ke zhojení rány, pak pokračujte v terapii po dobu nejméně dalších 7 dnů pro udržení aktivity faktoru VIII na úrovni 30 % až 60 % |
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Frekvence dávkování (hodiny) / Délka trvání léčby (dny) |
|
(IU/dl). | ||
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů s těžkou hemofilií A je obvyklé dávkování 20 až 40 IU faktoru VIII na 1 kilogram tělesné hmotnosti každé 2 až 3 dny. V některých případech, zejména u mladších pacientů, mohou být nutné kratší intervaly dávkování nebo vyšší dávky.
V průběhu léčby se doporučuje vhodné stanovení hladin faktoru VIII pro určení dávky, která má být podávána, a četnosti opakovaných infuzí. V případě velkých chirurgických zákroků je nezbytné přesné monitorování substituční léčby pomocí koagulační analýzy (aktivity faktoru VIII v plazmě). Jednotliví pacienti se mohou lišit ve své odpovědi na faktor VIII, mohou se lišit v poločasech eliminace a v hodnotách biologické dostupnosti.
Pediatrická populace
Dávkování je stejné u dospělých i dětských pacientů, ale u dětí mohou být nutné kratší intervaly mezi dávkami a vyšší dávky. V současnosti dostupné údaje jsou uvedeny v bodech 4.8, 5.1 a 5.2.
U dětí mladších 2 let nejsou dostupné žádné údaje.
Způsob podání
Intravenózní podání.
Doporučuje se podávat nejvýše 4 ml přípravku za minutu.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoliv pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Stejně jako u jiných intravenózně podávaných proteinových přípravků jsou možné hypersensitivní reakce alergického typu. Přípravek Nuwiq obsahuje stopová množství proteinů lidské hostitelské buňky jiných než faktor VIII. Vyskytnou-li se příznaky přecitlivělosti, pacienti mají být poučeni o neprodleném přerušení užívání léčivého přípravku a o návštěvě lékaře. Pacienti mají být poučeni o časných projevech přecitlivělosti, jako například vyrážka, generalizovaná kopřivka, tlak na hrudi, sípavé dýchání, hypotenze a anafylaxe.
V případě šoku je nutno dodržovat standardní lékařské postupy jeho léčby.
Inhibitory
Známou komplikací léčby u nemocných s hemofilií A je tvorba neutralizujících protilátek (inhibitorů) faktoru VIII. Tyto inhibitory jsou obvykle IgG imunoglobuliny směrované proti prokoagulační aktivitě faktoru VIII, a jsou kvantifikovány v Bethesda jednotkách (BU) na ml plazmy s použitím modifikovaného testu. Riziko rozvoje inhibitorů koreluje s expozicí faktoru VIII; toto riziko se zvyšuje v průběhu prvních 20 dnů expozice. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Z tohoto důvodu se doporučuje pečlivě monitorovat pacienty z hlediska možného vývoje inhibitorů po jakékoliv změně přípravku.
Obecně platí, že u všech pacientů léčených koagulačním faktorem VIII musí být pečlivě monitorován vznik inhibitorů vhodnými klinickými pozorováními a laboratorními testy. Pokud se nedosáhne očekávané hladiny aktivity plazmatického faktoru VIII nebo pokud krvácení není zastaveno odpovídající dávkou, měl by být proveden test za účelem určení přítomnosti inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční léčba faktorem VIII účinná a je třeba uvažovat o jiných možnostech léčby, jako je například navození imunitní tolerance (ITI). Léčba takových pacientů by měla být vedena lékaři se zkušenostmi s léčbou hemofilie a s inhibitory faktoru VIII.
Komplikace způsobené katétrem
Pokud je potřeba použít centrální žilní vstup (CVAD), má být zváženo riziko komplikací souvisejících s CVAD, včetně lokální infekce, bakteriémie a trombózy v místě vstupu katétru.
Důrazně se doporučuje, pokaždé, kdy je přípravek Nuwiq podán pacientovi, zaznamenat název a číslo šarže aplikovaného přípravku, aby bylo zachováno spojení mezi pacientem a číslem šarže léčivého přípravku.
Pediatrická populace
Uvedená upozornění a bezpečnostní pokyny se vztahují jak na dospělé, tak na dětské pacienty.
Pokyny vztahující se k pomocné látce (obsah sodíku)
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v 1 injekční lahvičce.
V závislosti na tělesné hmotnosti a dávkování by měl pacient obdržet více než jednu injekční lahvičku. To je třeba vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
U přípravku Nuwiq nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie na zvířatech nebyly s přípravkem Nuwiq prováděny.
Vzhledem k vzácnému výskytu hemofilie A u žen nejsou k dispozici žádné zkušenosti s používáním faktoru VIII během těhotenství a kojení. Z tohoto důvodu má být přípravek Nuwiq během těhotenství a kojení používán pouze, je-li to jasně indikováno. Nejsou k dispozici žádné údaje týkající se fertility.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Nuwiq nemá žádný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn informací o bezpečnostním profilu
U přípravků obsahujících faktor VIII byly vzácně pozorovány přecitlivělost nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, zimnici, zarudnutí, generalizovanou vyrážku, bolest hlavy, kopřivku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tlak na hrudi, mravenčení, zvracení, sípání) které se mohou v některých případech rozvinout do závažné anafylaxe (včetně šoku).
U pacientů s hemofilií A se mohou vytvořit neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory vyskytnou, mohou se projevit nedostatečnou klinickou odezvou na přípravek. V těchto případech se doporučuje vyhledat specializované pracoviště na léčení hemofilie.
Přehledný souhrn nežádoucích účinků
Během klinických studií přípravku Nuwiq provedených u již dříve léčených pediatrických pacientů (2 až 11 let, n = 58), dospívajících pacientů (12 až 17 let, n = 3) a dospělých pacientů (n = 74) s těžkou hemofilií A, byl celkový počet závažných nežádoucích účinků (ADR) 8 (6 u dospělých, 2 u dětí) hlášený u 5 pacientů (3 dospělých, 2 dětí).
Tabulka 1 uvedená níže je uspořádána v souladu s klasifikací tříd orgánových systémů MedDRa (SOC a preferované termíny četností).
Četnost je definována dle následující konvence: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000); velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 1. Frekvence výskytu nežádoucích účinků léčivého přípravku (ADR) na 1 pacienta
|
během klinických studií u 135 již dříve léčených pacientů s těžkou |
íemofilií A | |
|
Třída orgánových systémů podle databáze MedDRA |
Nežádoucí účinky |
Četnost* |
|
Poruchy nervové soustavy |
Parastézie Bolesti hlavy |
Méně časté |
|
Poruchy ucha a labyrintu |
Závratě |
Méně časté |
|
Gastrointestinální poruchy |
Sucho v ústech |
Méně časté |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolesti zad |
Méně časté |
|
Celkové poruchy a reakce v místě aplikace |
Zánět v místě vpichu Bolest v místě vpichu |
Méně časté |
|
Vyšetření |
Pozitivní protilátka faktoru VIII bez neutralizační aktivity |
Méně časté |
* Všechny tyto nežádoucí účinky se vyskytly pouze jednou. Vzhledem k tomu, že celkový počet studovaných pacientů je 135, četnost nemůže být jiná než „méně časté“, jakmile se závažné nežádoucí účinky objeví jednou.
Popis vybraných nežádoucích účinků
U jednoho dospělého pacienta byla zjištěna protilátka faktoru VIII bez neutralizační aktivity (viz Tabulka 1). Vzorek byl testován v centrální laboratoři v osmi zředěních. Výsledek byl pozitivní pouze při faktoru zředění 1 a titr protilátek byl velmi nízký. Inhibiční aktivita měřená pomocí modifikovaného Bethesda testu nebyla u tohoto pacienta zjištěna. Klinická účinnost a biologická dostupnost přípravku Nuwiq nebyly u tohoto pacienta narušeny.
Pediatrická populace
Četnost, typ a závažnost nežádoucích účinků u dětí se očekávají shodné jako u dospělých.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nejsou známy žádné případy předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika, krevní koagulační faktor VIII, ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. Po infuzi hemofilickému pacientovi se faktor VIII váže na von Willebrandův faktor přítomný v krevním oběhu pacienta. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin následně přeměňuje fibrinogen na fibrin a může dojít k vytvoření sraženiny. Hemofilie A je pohlavně vázaná dědičná porucha srážlivosti krve způsobená sníženou hladinou faktoru VIII:C, v důsledku které dochází k profuznímu krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánnímu, nebo jako následek úrazu při nehodě či chirurgickém zákroku. Substituční léčbou se hladiny faktoru VIII v plazmě zvýší, čímž je možná přechodná korekce nedostatku faktoru VIII a korekce sklonu ke krvácení.
Imunogenita přípravku Nuwiq byla hodnocena v rámci klinických zkoušek u 135 již dříve léčených pacientů s těžkou hemofilií A (74 dospělých a 61 dětských pacientů). U žádného z pacientů nedošlo k vývoji inhibitorů.
V rámci klinické studie provedené s 32 dospělými pacienty s těžkou hemofilií A byla střední hodnota spotřeby přípravku Nuwiq za účelem profylaxe 468,7 IU/kg/měsíc. Průměrná dávka při léčbě nečekaného krvácení byla 33,0 IU/kg u pacientů užívajících přípravek za účelem profylaxe. V rámci jiné studie bylo 22 dospělých pacientů léčeno na požádání. Celkem bylo 986 případů krvácení léčeno průměrnou dávkou 30,9 IU/kg. Obecně platí, že slabé krvácení vyžadovalo mírně nižší a silnější krvácení vyžadovalo až trojnásobně vyšší střední dávky.
Pediatrická populace
Data byla získána od 29 již dříve léčených dětí ve věku 2 až 5 let, 31 dětí ve věku 6 až 12 let a jednoho dospívajícího pacienta ve věku 14 let. Medián dávky na profylaktickou infuzi byl 37,8 IU/kg. Dvacet pacientů užívalo střední dávky vyšší než 45 IU/kg. Medián spotřeby přípravku Nuwiq za účelem profylaxe za měsíc byl 521,9 IU/kg. Vyšší medián dávky přípravku Nuwiq vyžadovala léčba krvácení u dětí (43,9 IU/kg) než u dospělých (33,0 IU/kg), a vyšší medián dávky vyžadovala léčba středního až silného krvácení v porovnání se slabým krvácením (78,2 IU/kg v porovnání s 41,7 IU/kg). Mladším dětem musel být obecně podáván vyšší medián dávky (6-12 let: 43,9 IU/kg; 2-5 let: 52,6 IU/kg).
Evropská agentura pro léčivé přípravky odložila závazek předložit výsledky studií přípravku Nuwiq u jednoho nebo více souborů pediatrické populace při léčbě hemofilie A (vrozeného nedostatku faktoru VIII) (viz bod 4.2, kde jsou uvedeny informace o použití pro pediatrické pacienty).
5.2 Farmakokinetické vlastnosti
Tabulka 2. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u dospělých již dříve léčených pacientů (věk 18-65 let) s těžkou hemofilií A (n = 20)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
22,6 ± 8,0 |
22,3 (8,4 - 38,1) |
|
T1/2 (hod) |
14,7 ± 10,4 |
12,5 (5,4 -55,6) |
|
IVR (%/IU/kg) |
2,5 ± 0,4 |
2,5 (1,7 -3,2) |
|
CL (ml/hod/kg) |
3,0 ± 1,2 |
2,7 (1,5-6,4) |
AUC = plocha pod křivkou (FVIII:C), Tm = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka
Tabulka 3. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u již dříve léčených dětských pacientů ve věku 6 až 12 let s těžkou hemofilií A (n = 12)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
13,2 ± 3,4 |
12,8 (7,8 - 19,1) |
|
T1/2 (hod) |
10,0 ± 1,9 |
9,9 (7,6 -14,1) |
|
IVR (%/IU/kg) |
1,9 ± 0,4 |
1,9 (1,2 -2,6) |
|
CL (ml/hod/kg) |
4,3 ± 1,2 |
4,2 (2,8 - 6,9) |
AUC = plocha pod křivkou (FVIII:C), T1/2 = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka
Tabulka 4. FK parametry pro přípravek Nuwiq (Dávka: 50 IU/kg) u již dříve léčených dětských pacientů ve věku 2 až 5 let s těžkou hemofilií A (n = 13)_
|
FK parametr |
Chromogenní metoda | |
|
Průměr ± SD |
Medián (rozsah) | |
|
AUC (hod*IU/ml) |
11,7 ± 5,3 |
10,5 (4,9 -23,8) |
|
T1/2 (hod) |
9,5 ± 3,3 |
8,2 (4,3 - 17,3) |
|
IVR (%/IU/kg) |
1,9 ± 0,3 |
1,8 (1,5 -2,4) |
|
CL (ml/hod/kg) |
5,4 ± 2,4 |
5,1 (2,3 -10,9) |
AUC = plocha pod křivkou (FVIII:C), T1/2 = terminální poločas,
IVR = inkrementální biologická dostupnost in vivo, CL = clearance, SD = Směrodatná odchylka Pediatrická populace
Z literatury je známo, že biologická dostupnost a poločas byly nižší u dětí než u dospělých a clearance vyšší, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Podskupiny upravené podle hmotnosti
Tabulka 5. FK parametry pro přípravek Nuwiq upravené podle hmotnosti (Dávka: 50 IU/kg) u
|
dospělých již dříve |
éčených pacientů (věk 18-65 let) s těžkou hemofilií A (n = 20) | |||
|
FK parametr |
Všichni (n=20) |
Normální hmotnost (n=14) |
S nadváhou (n=4) |
Obézní (n=2) |
|
Chromogenní metoda Průměr ± SD | ||||
|
AUC (hod*IU/ml) |
22,6 ± 8,0 |
20,4 ± 6,9 |
24,9 ± 8,9 |
33,5 ± 6,5 |
|
T1/2 (hod) |
14,7 ± 10,4 |
14,7 ± 12,1 |
13,4 ± 5,9 |
17,2 ± 4,8 |
|
IVR (%/IU/kg) |
2,5 ± 0,4 |
2,4 ± 0,4 |
2,7 ± 0,4 |
2,8 ± 0,3 |
|
CL (ml/hod/kg) |
3,0 ± 1,2 |
3,2 ± 1,3 |
2,6 ± 1,0 |
1,8 ± 0,4 |
|
Chromogenní metoda Medián (rozsah) | ||||
|
AUC (hod*IU/ml) |
22,3 (8,4 -38,1) |
21,2 (8,4 -32,6) |
23,3 (17,4 - 35,5) |
33,5 (28,9 -38,1) |
|
T1/2 (hod) |
12,5 (5,4 - 55,6) |
12,3 (5,4 -55,6) |
11,2 (9,3 -22,0) |
17,2 (13,8 -20,6) |
|
IVR (%/IU/kg) |
2,5 (1,7 -3,2) |
2,4 (1,7 -3,1) |
2,8 (2,3 - 3,2) |
2,8 (2,6 -3,0) |
|
CL (ml/hod/kg) |
2,7 (1,5 -6,4) |
2,8 (1,7 -6,4) |
2,5 (1,6 -3,7) |
1,8 (1,5 -2,0) |
|
Normální hmotnost: |
BMI 18,5-25 kg/m2, |
Nadváha: BMI 25-30 kg/m2, Obezita: BM |
> 30 kg/m2, SD = | |
Směrodatná odchylka
5.3 Předklinické údaje vztahující se k bezpečnosti
V rámci předklinických studií byl přípravek Nuwiq bezpečně a úspěšně použit k obnově hemostázy u psů s hemofilií. Toxikologické studie ukázaly, že lokální intravenózní podání a systémová expozice byly dobře tolerovány u laboratorních zvířat (potkanů a opic makaka jávského).
Specifické studie reprodukční toxicity, chronické toxicity a kancerogenity s dlouhodobým opakovaným podáváním přípravku Nuwiq nebyly provedeny kvůli imunitní reakci na heterologní proteiny u všech nelidských druhů savců.
Nebyly provedeny žádné studie mutagenního potenciálu přípravku Nuwiq.
Ex vivo hodnocení při použití komerční testovací soupravy pro kvantifikaci odezvy T-buněk na proteinová terapeutika ukazují nízké riziko imunogenicity.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Sacharosa Chlorid sodný
Dihydrát chloridu vápenatého Arginin hydrochlorid Dihydrát citronanu sodného Poloxamer 188
Rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
Mají se používat pouze dodávané injekční soupravy, protože může dojít k selhání léčby v důsledku adsorpce lidského koagulačního faktoru VIII na vnitřní povrchy některých infuzních zařízení.
6.3 Doba použitelnosti
2 roky
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25°C) po dobu jednoho nepřetržitého období nepřesahujícího 1 měsíc. Jakmile byl přípravek jednou vyjmut z chladničky, nesmí tam být vrácen zpět. Označte si prosím na krabičce datum, kdy jste přípravek začali uchovávat při pokojové teplotě. Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Po rekonstituci je prokázána chemická a fyzikální stabilita po dobu 24 hodin při uchovávání při pokojové teplotě.
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, doba a podmínky uchovávání přípravku před použitím jsou v odpovědnosti uživatele. Uchovávejte rekonstituovaný přípravek při pokojové teplotě. Po rekonstituci chraňte před mrazem.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Uchovávání tohoto léčivého přípravku při pokojové teplotě a podmínky uchovávání po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Jedno balení přípravku Nuwiq 2000 IU obsahuje:
- Prášek: 2000 IU prášku v 8ml skleněné injekční lahvičce typu I, uzavřené potaženou bromobutylovou zátkou s pojistným hliníkovým uzávěrem
- Rozpouštědlo: 2,5 ml vody na injekci v předplněné injekční stříkačce z borosilikátového skla
- 1 sterilní adaptér injekční lahvičky pro rekonstituci s 1 motýlkovou jehlou a 2 tampóny napuštěnými alkoholem
Velikost balení po 1 kusu.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Prášek má být rekonstituován pouze v dodaném rozpouštědle (2,5 ml vody na injekce) za použití injekční soupravy, která je součástí dodávky. Při rozpouštění prášku má injekční lahvička vykonávat jemný krouživý pohyb, dokud se prášek nerozpustí. Po rekonstituci má být roztok natažen zpět do injekční stříkačky.
Rekonstituovaný léčivý přípravek má být před podáním vizuálně zkontrolován, zda neobsahuje cizorodé částice nebo zabarvení. Tento rekonstituovaný léčivý přípravek je čirý, bezbarvý roztok bez přítomnosti cizorodých částic s pH v rozmezí 6,5 až 7,5. Nepoužívejte roztok, je-li zakalený nebo obsahuje-li usazeniny.
Návod pro přípravu a podávání
1. Ponechte rozpouštědlo v injekční stříkačce (voda na injekci) a prášek v uzavřené injekční lahvičce dostatečnou dobu pro dosažení pokojové teploty. Můžete to udělat tak, že je podržíte v rukou, dokud nebudou stejně teplé jako vaše ruce. Na zahřívání lahvičky a předem naplněné stříkačky nepoužívejte jiné postupy. Tato teplota by měla být udržována během rekonstituce.
2. Odstraňte pojistné plastové víčko z injekční lahvičky s práškem, aby byla přístupná střední část gumové zátky. Neodstraňujte šedou zátku ani kovový kroužek kolem horní části injekční lahvičky.

3. Otřete horní část injekční lahvičky tampónem napuštěným v alkoholu. Počkejte, dokud se alkohol nevypaří.
4. Sejměte papírový kryt z obalu adaptéru injekční lahvičky. Nevyndávejte adaptér z obalu.
Umístěte injekční lahvičku s práškem na rovný povrch a držte ji. Uchopte obal adaptéru a umístěte adaptér injekční lahvičky na střední část gumové zátky injekční lahvičky s práškem. Stlačte pevně obal adaptéru tak, aby hrot adaptéru prošel gumovou zátkou. Adaptér se přitom zacvakne na injekční lahvičku.

5.
6.

Sejměte papírový kryt z obalu předplněné injekční stříkačky. Uchopte píst za konec, a nedotýkejte se jeho střední části. Připojte konec pístu se závitem k injekční stříkačce s rozpouštědlem. Otáčejte pístem ve směru pohybu hodinových ručiček, dokud neucítíte slabý odpor.

7. Odlomte ochranný plastový hrot z injekční stříkačky s rozpouštědlem v místě perforace víčka. Nedotýkejte se vnitřku víčka ani hrotu injekční stříkačky. Pokud roztok nebudete používat ihned, zavřete naplněnou stříkačku plastovou špičkou s odolností vůči nárazu pro uskladnění.

8. Sejměte obal adaptéru a vyhoďte jej.
9. Pevně připojte injekční stříkačku s rozpouštědlem k adaptéru injekční lahvičky otáčením ve směru pohybu hodinových ručiček, dokud neucítíte slabý odpor.

10. Pomalu vstřikujte veškeré rozpouštědlo do injekční lahvičky s práškem tlakem na píst.

11. Aniž byste odstranili injekční stříkačku, rozpusťte prášek jemným pohybem nebo několika krouživými pohyby injekční lahvičky. Netřepejte. Počkejte, dokud se prášek zcela nerozpustí.
12. Před podáním zrakem zkontrolujte, že výsledný roztok neobsahuje žádné částice. Roztok má být čirý a bezbarvý, prakticky bez viditelných částic. Nepoužívejte roztok, je-li zakalený nebo obsahuje-li usazeniny.
13. Otočte injekční lahvičku připevněnou k injekční stříkačce dnem vzhůru a pomalu natáhněte výsledný roztok do stříkačky. Přesvědčte se, že nyní je celý obsah injekční lahvičky v injekční stříkačce.

14. Odpojte naplněnou injekční stříkačku od adaptéru injekční lahvičky otáčením proti směru pohybu hodinových ručiček a prázdnou injekční lahvičku zlikvidujte.
15. Roztok je nyní připraven k okamžitému použití. Chraňte před chladem.
16. Očistěte zvolené místo vpichu jedním z dodaných tampónů napuštěných alkoholem.
17. Připojte k injekční stříkačce infuzní soupravu, která je součástí balení.
Zaveďte jehlu infuzní soupravy do zvolené žíly. Pokud jste použili škrtidlo pro zviditelnění žíly, má být toto škrtidlo uvolněno předtím, než začnete vstřikovat roztok.
Do injekční stříkačky se nesmí dostat žádná krev, aby nedošlo k tvorbě fibrinových sraženin.
18. Vstřikujte roztok do žíly pomalu, maximálně 4 ml za minutu.
Použijete-li více než jednu injekční lahvičku s práškem pro jednu léčbu, můžete použít stejnou injekční jehlu opakovaně. Adaptér injekční lahvičky a injekční stříkačka jsou určeny pouze na jedno použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Octapharma AB Lars Forssells gata 23 112 75 Stockholm Švédsko
8. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/936/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 22. července 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY /BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY /BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa vvrobce/vvrobců biologické léčivé látky/biologických léčivých látek
Octapharma AB Lars Forssells gata 23 112 75 Stockholm Švédsko
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Octapharma AB Lars Forssells gata 23 112 75 Stockholm Švédsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
Nuwiq 250 mezinárodních jednotek
Prášek a rozpouštědlo pro injekční roztok
simoctocog alfa (rekombinantní lidský koagulační faktor VIII)
Jedna injekční lahvička obsahuje simoctocog alfa 250 mezinárodních jednotek (100 IU/ml po rekonstituci)
Pomocné látky: sacharosa, chlorid sodný, dihydrát chloridu vápenatého, arginin hydrochlorid, dihydrát citronanu sodného, poloxamer 188 Rozpouštědlo: Voda na injekci
Viz příbalová informace, kde jsou uvedeny další údaje.
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s práškem, 1 předplněná injekční stříkačka, 1 adaptér pro injekční lahvičku, 1 motýlková jehla, 2 tampony napuštěné alkoholem
1 injekční lahvička s práškem obsahuje 250 mezinárodních jednotek simoctocog alfa.
1 předplněná injekční stříkačka obsahuje 2,5 ml vody na injekci.
Před použitím si přečtěte příbalovou informaci. Intravenózní podání po rekonstituci.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte v chladničce. Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po dobu jednoho nepřetržitého období nepřesahujícího 1 měsíc.
Z chladničky vyjmuto:_
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Octapharma AB Lars Forssells gata 23 112 75 Stockholm Švédsko
EU/1/14/936/001
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
Nuwiq 250
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Nuwiq 250 mezinárodních jednotek, prášek pro injekční roztok, simoctocog alfa (rekombinantní lidský koagulační faktor VIII).
Intravenózní podání po rekonstituci.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
250 IU/injekční lahvička
6. JINÉ
Octapharma-Logo
Nuwiq 500 mezinárodních jednotek
Prášek a rozpouštědlo pro injekční roztok
simoctocog alfa (rekombinantní lidský koagulační faktor VIII)
Jedna injekční lahvička obsahuje simoctocog alfa 500 mezinárodních jednotek (200 IU/ml po rekonstituci)
Pomocné látky: sacharosa, chlorid sodný, dihydrát chloridu vápenatého, arginin hydrochlorid, dihydrát citronanu sodného, poloxamer 188 Rozpouštědlo: Voda na injekci
Viz příbalová informace, kde jsou uvedeny další údaje.
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s práškem, 1 předplněná injekční stříkačka, 1 adaptér pro injekční lahvičku, 1 motýlková jehla, 2 tampony napuštěné alkoholem
1 injekční lahvička s práškem obsahuje 500 mezinárodních jednotek simoctocog alfa.
1 předplněná injekční stříkačka obsahuje 2,5 ml vody na injekci.
Před použitím si přečtěte příbalovou informaci. Intravenózní podání po rekonstituci.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte v chladničce. Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po dobu jednoho nepřetržitého období nepřesahujícího 1 měsíc.
Z chladničky vyjmuto:_
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Octapharma AB Lars Forssells gata 23 112 75 Stockholm Švédsko
EU/1/14/936/002
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
Nuwiq 500
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Nuwiq 500 mezinárodních jednotek, prášek pro injekční roztok, simoctocog alfa (rekombinantní lidský koagulační faktor VIII).
Intravenózní podání po rekonstituci.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
500 IU/injekční lahvička
6. JINÉ
Octapharma-Logo
Nuwiq 1000 mezinárodních jednotek
Prášek a rozpouštědlo pro injekční roztok
simoctocog alfa (rekombinantní lidský koagulační faktor VIII)
Jedna injekční lahvička obsahuje simoctocog alfa 1000 mezinárodních jednotek (400 IU/ml po rekonstituci)
Pomocné látky: sacharosa, chlorid sodný, dihydrát chloridu vápenatého, arginin hydrochlorid, dihydrát citronanu sodného, poloxamer 188 Rozpouštědlo: Voda na injekci
Viz příbalová informace, kde jsou uvedeny další údaje.
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s práškem, 1 předplněná injekční stříkačka, 1 adaptér pro injekční lahvičku, 1 motýlková jehla, 2 tampony napuštěné alkoholem
1 injekční lahvička s práškem obsahuje 1000 mezinárodních jednotek simoctocog alfa.
1 předplněná injekční stříkačka obsahuje 2,5 ml vody na injekci.
Před použitím si přečtěte příbalovou informaci. Intravenózní podání po rekonstituci.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte v chladničce. Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po dobu jednoho nepřetržitého období nepřesahujícího 1 měsíc.
Z chladničky vyjmuto:_
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Octapharma AB Lars Forssells gata 23 112 75 Stockholm Švédsko
EU/1/14/936/003
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
Nuwiq 1000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Nuwiq 1000 mezinárodních jednotek, prášek pro injekční roztok, simoctocog alfa (rekombinantní lidský koagulační faktor VIII).
Intravenózní podání po rekonstituci.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1000 injekční lahvička
6. JINÉ
Octapharma-Logo
Nuwiq 2000 mezinárodních jednotek
Prášek a rozpouštědlo pro injekční roztok
simoctocog alfa (rekombinantní lidský koagulační faktor VIII)
Jedna injekční lahvička obsahuje simoctocog alfa 2000 mezinárodních jednotek (800 IU/ml po rekonstituci)
Pomocné látky: sacharosa, chlorid sodný, dihydrát chloridu vápenatého, arginin hydrochlorid, dihydrát citronanu sodného, poloxamer 188 Rozpouštědlo: Voda na injekci
Viz příbalová informace, kde jsou uvedeny další údaje.
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s práškem, 1 předplněná injekční stříkačka, 1 adaptér pro injekční lahvičku, 1 motýlková jehla, 2 tampony napuštěné alkoholem
1 injekční lahvička s práškem obsahuje 2000 mezinárodních jednotek simoctocog alfa.
1 předplněná injekční stříkačka obsahuje 2,5 ml vody na injekci.
Před použitím si přečtěte příbalovou informaci. Intravenózní podání po rekonstituci.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte v chladničce. Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po dobu jednoho nepřetržitého období nepřesahujícího 1 měsíc.
Z chladničky vyjmuto:_
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Octapharma AB Lars Forssells gata 23 112 75 Stockholm Švédsko
EU/1/14/936/004
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
Nuwiq 2000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Nuwiq 2000 mezinárodních jednotek, prášek pro injekční roztok, simoctocog alfa (rekombinantní lidský koagulační faktor VIII).
Intravenózní podání po rekonstituci.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2000 IU/injekční lahvička
6. JINÉ
Octapharma-Logo
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Rozpouštědlo pro přípravek Nuwiq Voda na injekci
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2,5 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Nuwiq 250 IU prášek a rozpouštědlo pro injekční roztok Nuwiq 500 IU prášek a rozpouštědlo pro injekční roztok Nuwiq 1000 IU prášek a rozpouštědlo pro injekční roztok Nuwiq 2000 IU prášek a rozpouštědlo pro injekční roztok
Simoctocog alfa (rekombinantní lidský koagulační faktor VIII)
V Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Nuwiq a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Nuwiq používat
3. Jak používat přípravek Nuwiq
4. Možné vedlejší účinky
5. Jak uchovávat přípravek Nuwiq
6. Obsah balení a další informace
1. Co je přípravek Nuwiq a k čemu se používá
Přípravek Nuwiq obsahuje léčivou látku lidský rekombinantní koagulační faktor VIII (simoctocog alfa). Faktor VIII je potřebný pro tvorbu krevních sraženin a zastavení krvácení. U pacientů s hemofilií A (vrozený nedostatek faktoru VIII) faktor VIII chybí nebo nefunguje správně.
Přípravek Nuwiq nahrazuje faktor VIII a používá se k léčbě a prevenci krvácení u pacientů s hemofilií A a je vhodný pro použití u všech věkových skupin.
2. Čemu musíte věnovat pozornost, než začnete přípravek Nuwiq používat Nepoužívejte přípravek Nuwiq:
• jestliže jste alergický(á) na léčivou látku simoctocog alfa nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Pokud si nejste jisti, zeptejte se svého lékaře.
Upozornění a opatření
Před použitím přípravku Nuwiq se poraďte se svým lékařem nebo lékárníkem.
Existuje vzácná možnost, že dojde k anafylaktické reakci (závažná, náhlá alergická reakce) na přípravek Nuwiq. Máte být poučeni o časných příznacích alergických reakcí, jak jsou uvedeny v bodu 4 „Alergické reakce“.
Pokud nastane jakákoliv z těchto reakcí, okamžitě přerušte podávání injekce a obraťte se na svého lékaře.
Pokud se krvácení nezastaví používáním přípravku Nuwiq, informujte neprodleně svého lékaře. Pravděpodobně u vás došlo k vytvoření inhibitorů faktoru VIII a Váš lékař bude chtít provést testy, aby toto potvrdil. Inhibitory faktoru VIII jsou protilátky v krvi, které blokují faktor VIII, který užíváte. Způsobují, že faktor VIII je méně účinný pro zastavení krvácení. Pokud jste byli dříve léčeni produkty faktoru VIII, zejména pak, pokud se u vás vyvinuli inhibitory, měli byste o tom informovat svého lékaře, protože existuje zvýšené riziko, že k tomu dojde opakovaně.
Komplikace způsobené katétrem
Pokud je pro podání přípravku potřeba použít centrální žilní vstup (CVAD), lékař zváží riziko možných komplikací souvisejících s CVAD, včetně lokální infekce, bakteriémie (přítomnost bakterií v krvi) a trombózy (krevní sraženina) v místě vstupu katétru.
Důrazně se doporučuje, pokaždé, kdy je přípravek Nuwiq podán pacientovi, zaznamenat název a číslo šarže aplikovaného přípravku z důvodů zachování spojení mezi pacientem a číslem šarže léčivého přípravku.
Další léčivé přípravky a přípravek Nuwiq
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Přípravek Nuwiq nemá žádný vliv na Vaši schopnost řídit a obsluhovat stroje.
Přípravek Nuwiq obsahuje sodík
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v 1 injekční lahvičce.
Nicméně v závislosti na tělesné hmotnosti a dávkování by měl pacient obdržet více než jednu injekční lahvičku. To je třeba vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
3. Jak používat přípravek Nuwiq
Léčba přípravkem Nuwiq bude zahájena lékařem, který má zkušenosti s péčí o pacienty s hemofilií typu A. Vždy používejte tento přípravek přesně podle pokynů svého lékaře nebo sestry. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo sestrou.
Přípravek Nuwiq je obvykle podáván injekcí do žíly (intravenózně) Váším lékařem nebo sestrou, kteří mají zkušenosti s péčí o pacienty s hemofilií A. Vy nebo jiná osoba můžete podávat přípravek Nuwiq injekčně také, ale až po řádném zaškolení.
Váš lékař vypočte dávku přípravku Nuwiq (v mezinárodních jednotkách = IU) podle Vašeho zdravotního stavu a tělesné hmotnosti a s ohledem na to, zda je přípravek použit za účelem prevence nebo léčby krvácení. Jak často bude nutno injekci podávat, bude záviset na tom, jak bude u Vás přípravek Nuwiq účinkovat. Léčba přípravkem proti hemofílii A je obvykle celoživotní.
Prevence krvácení
Obvyklá dávka přípravku Nuwiq alfa je 20 až 40 IU na kilogram tělesné hmotnosti, podávaná každé 2 až 3 dny. V některých případech, zejména u mladších pacientů, však mohou být nutné kratší intervaly mezi jednotlivými injekcemi nebo vyšší dávky.
Léčba krvácení
Dávka přípravku Nuwiq se vypočte podle Vaší tělesné hmotnosti a hladiny faktoru VIII, která má být dosažena. Cílové hodnoty faktoru VIII závisejí na závažnosti a místě krvácení.
Máte-li pocit, že účinek přípravku Nuwiq není dostatečný, poraďte se se svým lékařem. Váš lékař provede příslušné laboratorní testy, aby se přesvědčil, že Vaše hladiny faktoru VIII jsou přiměřené. Uvedené je důležité zejména v případě, že máte podstoupit velký chirurgický zákrok.
Pacienti s vyvinutými inhibitory proti faktoru VIII
Nedosahuje-li Váš plazmatický faktor VIII očekávaných hladin při léčbě přípravkem Nuwiq nebo nedojde-li k zastavení krvácení, může to být proto, že jste si vytvořili inhibitory faktoru VIII. Toto zkontroluje Váš lékař. K zastavení krvácení budete možná potřebovat vyšší dávky přípravku Nuwiq nebo jiný přípravek. Nezvyšujte celkovou dávku přípravku Nuwiq pro zastavení krvácení bez konzultace se svým lékařem.
Použití u dětí
Způsob, jakým se používá přípravek Nuwiq u dětí se neliší od způsobu podávání u dospělých. Protože přípravky s faktorem VIII mohou být častěji podávány dětem, může být nezbytné použít centrální žilní vstup (CVAD) s externím konektorem umožňujícím přístup do krevního řečiště před katétr bez injekce pokožkou.
Jestliže jste použil(a) více přípravku Nuwiq než jste měl(a)
Nejsou známy žádné příznaky při předávkování. Pokud jste užil(a) více přípravku Nuwiq než jste měl(a), informujte prosím svého lékaře.
Jestliže jste zapomněl(a) použít přípravek Nuwiq
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Pokračujte okamžitě s další dávkou a dále pokračujte v pravidelných intervalech dle pokynů svého lékaře nebo lékárníka.
Jestliže jste přestal(a) používat přípravek Nuwiq
Nepřestávejte používat přípravek Nuwiq bez konzultace se svým lékařem nebo lékárníkem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Alergické reakce
Máte být informováni o časných známkách alergických reakcí. Dojde-li k závažným, náhlým alergickým reakcím (anafylaktickému šoku) (velmi vzácně, až 1 případ z 10 000), infuze musí být okamžitě zastavena. Musíte se neprodleně obrátit na svého lékaře, objeví-li se u Vás následující příznaky:
- vyrážka, kopřivka, podlitiny, celkové svědění,
- otok rtů nebo jazyka,
- dýchací obtíže, sípání, tlak na hrudi,
- celková nevolnost,
- mdloby a ztráta vědomí.
Tyto příznaky mohou být počátečními příznaky anafylaktického šoku. Objeví-li se kterékoli z těchto příznaků, okamžitě zastavte infúzi a přivolejte svého lékaře. Závažné příznaky vyžadují neodkladnou léčbu.
Méně časté nežádoucí účinky, mohou se týkat až 1 osoby ze 100
Brnění nebo necitlivost (parestézie), bolest hlavy, zánět v místě vpichu, bolest v místě vpichu, bolest zad, závratě, sucho v ústech.
Nežádoucí účinky v souvislosti s podáním do centrální žíly (CVAD):
infekce související s katétrem, celková (systémová) infekce a lokální krevní sraženina v místě zavedení katétru.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak uchovávat přípravek Nuwiq
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku za zkratkou EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem. Uchovávejte injekční lahvičku v uzavřeném krabičce, aby byl přípravek chráněn před světlem.
Před rozpuštěním může být Nuwiq uchováván při pokojové teplotě (do 25°C) po dobu jednoho nepřetržitého období nepřesahujícího 1 měsíc. Zaznamenejte si prosím na krabičku datum, od kdy uchováváte Nuwiq při pokojové teplotě. Poté, co jste začal(a) Nuwiq uchovávat při pokojové teplotě, nevracejte jej zpět do chladničky.
Rekonstituovaný roztok použijte okamžitě po rekonstituci.
Upozornění na určité viditelné známky snížené kvality přípravku
Nepoužívejte přípravek, pokud vidíte známky snížené kvality v důsledku manipulace s balením, zejména se stříkačkou a/nebo injekční lahvičkou.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domovního odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Nuwiq obsahuje
Prášek:
- Léčivá látka je rekombinantní lidský koagulační faktor VIII (simoctocog alfa).
Jedn injekční lahvička s práškem obsahuje 250, 500, 1000 nebo 2000 IU látky simoctocog alfa.
- Další látky jsou sacharosa, chlorid sodný, dihydrát chloridu vápenatého, arginin hydrochlorid, dihydrát citronanu sodného a poloxamer 188
Rozpouštědlo:
Voda na injekci
Jak přípravek Nuwiq vypadá a co obsahuje toto balení
Přípravek Nuwiq se dodává jako prášek a rozpouštědlo pro injekční roztok a jedná se o bílý až téměř bílý drobivý prášek ve skleněné ampuli. Rozpouštědlem je voda na injekci a dodává se v předplněné skleněné injekční stříkačce.
Po rekonstituci je roztok čirý, bezbarvý a bez cizorodých částic.
Každé balení přípravku Nuwiq obsahuje:
- 1 injekční lahvičku s práškem s 250, 500, 1000 nebo 2000 IU látky simoctocog alfa
- 1 předplněnou injekční stříkačku s 2,5 ml vody na injekci
- 1 adaptér pro injekční lahvičku
- 1 motýlkovou jehlu
- 2 tampóny napuštěné alkoholem
Držitel rozhodnutí o registraci a výrobce
Octapharma AB, Lars Forssells gata 23, 112 75 Stockholm, Švédsko
Další informace o tomto léčivém přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien Octapharma Benelux (Belgium) Tél/Tel: +32 2 3730890 |
Ireland Octapharma Limited (UK) Tel: +44 161 8373770 |
Norge Octapharma AS Tlf: +47 63988860 |
|
BtnrapHH Octapharma Nordic AB (Sweden) Ten.: +46 8 56643000 |
Ísland Octapharma AS (Norway) Sími: +47 63988860 |
Osterreich Octapharma Handelsgesellschaft m.b.H Tel: +43 1 610321222 |
|
Česká republika Octapharma CZ s.r.o. Tel: +420 266 793 510 |
Italia Kedrion S.p.A. Tel: +39 0583 1969603 |
Polska Octapharma Poland Sp. z o.o. Tel: +48 22 2082734 |
|
Danmark Octapharma Nordic AB (Sweden) Tlf: +46 8 56643000 |
Kúnpoq Octapharma Nordic AB (Sweden) Tn^: +46 8 56643000 |
Portugal Octapharma Produtos Farmaceuticos Tel: +351 21 8160820 |
|
Deutschland Octapharma GmbH Tel: +49 2173 9170 |
Latvija Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
Románia Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
|
Eesti Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
Lietuva Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
Slovenija Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
|
EXXáSa Octapharma Hellas SA Tn^: +30 210 8986500 |
Luxembourg/Luxemburg Octapharma Benelux (Belgium) Tél/Tel: +32 2 3730890 |
Slovenská republika Octapharma AG, o.z.z.o. Tel: +421 2 54646701 |
|
Espaňa Octapharma S.A. Tel: +34 91 6487298 |
Magyarország Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
Suomi/Finland Octapharma Nordic AB Puh/Tel: +358 9 85202710 |
|
France Octapharma France Tél: +33 1 41318000 |
Malta Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
Sverige Octapharma Nordic AB Tel: +46 8 56643000 |
|
Hrvatska Octapharma Nordic AB |
Nederland Octapharma Benelux |
United Kingdom Octapharma Limited |
(Sweden)
Tel: +46 8 56643000
(Belgium)
Tel: +32 2 3730890
Tel: +44 161 8373770
Tato příbalová informace byla naposledy revidována v
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Léčba dle potřeby
Podávané množství a četnost podávání by se měly vždy zaměřovat na klinickou efektivnost v každém jednotlivém případě.
V případě následného krvácení by aktivita faktoru VIII neměla poklesnout pod stanovenou hladinu plazmatické aktivity (v % normální hodnoty nebo IU/dl) ve sledovaném období. Následující tabulku lze použít jako návod pro dávkování při krvácivých příhodách nebo při chirurgickém zákroku.
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Frekvence dávkování (hodiny) / Délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, do svalů nebo ústní dutiny |
20-40 |
Opakujte každých 12 až 24 hodin. Po dobu nejméně jednoho dne, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne zhojení. |
|
Intenzivnější hemartróza, krvácení do svalu nebo tvorba hematomů |
30-60 |
Opakujte infúzi každých 12 až 24 hodin po dobu 3 až 4 dnů nebo déle, dokud bolest a akutní potíže neustoupí. |
|
Život ohrožující krvácení |
60-100 |
Opakujte infúzi každých 8 až 24 hodin, dokud není hrozba odvrácena. |
|
Chirurgický zákrok | ||
|
Drobný chirurgický zákrok, včetně trhání zubu |
30-60 |
Každých 24 hodin, po dobu nejméně 1 dne, dokud nedojde ke zhojení. |
|
Velký chirurgický zákrok |
80-100 (před operací a po ní) |
Opakujte infúzi každých 8-24 hodin, dokud nedojde ke zhojení rány, pak pokračujte v terapii po dobu nejméně dalších 7 dnů pro udržení aktivity faktoru VIII na úrovni 30 % až 60 % (IU/dl). |
NÁVOD PRO PŘÍPRAVU A PODÁVÁNÍ
1. Ponechte rozpouštědlo v injekční stříkačce (voda na injekci) a prášek v uzavřené injekční lahvičce dostatečnou dobu pro dosažení pokojové teploty. Můžete to udělat tak, že je podržíte v rukou, dokud se nebudou zdát stejně teplé jako vaše ruce. Na zahřívání lahvičky a předem naplněné stříkačky nepoužívejte jiné postupy. Tato teplota by měla být udržována během rekonstituce.
2. Odstraňte pojistné plastové víčko z injekční lahvičky s práškem, aby byla přístupná střední část gumové zátky. Neodstraňujte šedou zátku ani kovový kroužek kolem horní části injekční
lahvičky.

3. Otřete horní část injekční lahvičky tampónem napuštěným v alkoholu. Počkejte, dokud se alkohol nevypaří.
4. Sejměte papírový kryt z obalu adaptéru injekční lahvičky. Nevyndávejte adaptér z obalu.

5. Umístěte injekční lahvičku s práškem na rovný povrch a držte ji. Uchopte obal adaptéru a umístěte adaptér injekční lahvičky na střední část gumové zátky injekční lahvičky s práškem. Stlačte pevně obal adaptéru tak, aby hrot adaptéru prošel gumovou zátkou. Adaptér se přitom zacvakne na injekční lahvičku.
6.

Sejměte papírový kryt z obalu předplněné injekční stříkačky. Uchopte píst za konec, a nedotýkejte se jeho střední části. Připojte konec pístu se závitem k injekční stříkačce s rozpouštědlem. Otáčejte pístem ve směru pohybu hodinových ručiček, dokud neucítíte slabý odpor.

Odlomte ochranný plastový hrot z injekční stříkačky s rozpouštědlem v místě perforace víčka. Nedotýkejte se vnitřku víčka ani hrotu injekční stříkačky. Pokud roztok nebudete používat ihned, zavřete naplněnou stříkačku plastovou špičkou s odolností vůči nárazu pro uskladnění.

Sejměte obal adaptéru a vyhoďte jej.
Pevně připojte injekční stříkačku s rozpouštědlem k adaptéru injekční lahvičky otáčením ve směru pohybu hodinových ručiček, dokud neucítíte slabý odpor.

10. Pomalu vstřikujte veškeré rozpouštědlo do injekční lahvičky s práškem tlakem na píst.

11. Aniž byste odstranili injekční stříkačku, rozpusťte prášek jemným pohybem nebo několika krouživými pohyby injekční lahvičky. Netřepejte. Počkejte, dokud se prášek zcela nerozpustí.
12. Před podáním zrakem zkontrolujte, že výsledný roztok neobsahuje žádné částice. Roztok má být čirý a bezbarvý, prakticky bez viditelných částic. Nepoužívejte roztok, je-li zakalený nebo obsahuje-li usazeniny.
13. Otočte injekční lahvičku připevněnou k injekční stříkačce dnem vzhůru a pomalu natáhněte výsledný roztok do stříkačky. Přesvědčte se, že nyní je celý obsah injekční lahvičky v injekční stříkačce.

14. Odpojte naplněnou injekční stříkačku od adaptéru injekční lahvičky otáčením proti směru pohybu hodinových ručiček a prázdnou injekční lahvičku zlikvidujte.
15. Roztok je nyní připraven k okamžitému použití. Chraňte před chladem.
16. Očistěte zvolené místo vpichu jedním z dodaných tampónů napuštěných alkoholem.
17. Připojte k injekční stříkačce infuzní soupravu, která je součástí balení.
Zaveďte jehlu infuzní soupravy do zvolené žíly. Pokud jste použili škrtidlo pro zviditelnění žíly, má být toto škrtidlo uvolněno předtím, než začnete vstřikovat roztok.
Do injekční stříkačky se nesmí dostat žádná krev, aby nedošlo k tvorbě fibrinových sraženin.
18. Vstřikujte roztok do žíly pomalu, maximálně 4 ml za minutu.
Použijete-li více než jednu injekční lahvičku s práškem pro jednu léčbu, můžete použít stejnou injekční jehlu opakovaně. Adaptér injekční lahvičky a injekční stříkačka jsou určeny pouze na jedno použití.
82