Novoeight 1500 Iu
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
NovoEight 250 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička s práškem obsahuje 250 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Po rozpuštění obsahuje přípravek NovoEight přibližně 62,5 IU/ml humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Účinnost (IU) se udává chromogenní metodou podle Evropského lékopisu. Specifická aktivita přípravku NovoEight je přibližně 8 300 IU/mg bílkoviny.
Turoktokog alfa (humánní koagulační faktor VIII (rDNA)) je čištěná bílkovina obsahující 1 445 aminokyselin s molekulovou hmotností asi 166 kDA. Je vyráběn rekombinantní DNA technologií z vaječníkových buněk čínského křečka (CHO buňky). Je vyroben bez přídavku jakékoliv bílkoviny lidského či zvířecího původu během kultivace buněk, čištění či konečné úpravy přípravku.
Turoktokog alfa je rekombinantní humánní koagulační faktor VIII se zkrácenou B-doménou (B-doména obsahuje 21 aminokyselin z přirozené B-domény) bez jakékoliv další změny v pořadí aminokyselin.
Pomocná látka se známým účinkem:
0,31 mmol sodíku (7 mg) v 1 ml vzniklého roztoku
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Bílý nebo lehce nažloutlý prášek či drobivá hmota.
Čirý a bezbarvý injekční roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a prevence krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). NovoEight lze používat ve všech věkových skupinách.
4.2 Dávkování a způsob podání
Léčba by měla být zahájena pod dohledem lékaře se zkušenostmi s léčbou hemofilie.
Doposud neléčení pacienti
Bezpečnost a účinnost přípravku NovoEight u dosud neléčených pacientů nebyly doposud stanoveny. Nejsou dostupné žádné údaje.
Dávkování
Dávkování a trvání substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a pacientově klinickém stavu.
Počet podávaných jednotek faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které odpovídají běžnému standardu WHO pro přípravky obsahující faktor VIII. Aktivita faktoru VIII v plazmě je vyjádřena buď v procentech (relativně k normální hladině v lidské plazmě) nebo v mezinárodních jednotkách (relativně k mezinárodnímu standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba v případě potřeby
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že jedna mezinárodní jednotka (IU) faktoru VIII na kg tělesné hmotnosti zvýší aktivitu faktoru VIII v plazmě asi o 2 IU/dl. Požadovaná dávka se vypočte podle následujícího vzorce:
Potřebný počet jednotek = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) (IU/dl) x 0,5 (IU/kg na IU/dl).
Množství, které má být podáno, a frekvence podání by měly být vždy přizpůsobeny klinické účinnosti v individuálním případě.
V případě následujících krvácivých příhod by v odpovídajícím období aktivita faktoru VIII neměla klesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může být použita jako návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Tabulka 1 Návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Stupeň krvácení/ Požadovaná hladina Četnost dávek (hodiny)/délka
Typ chirurgického výkonu FVIII (%) (IU/dl) trvání léčby (dny)
Krvácení
Časný hemartros, krvácení do svalů 20-40 Opakovat každých 12-24 hodin.
nebo do dutiny ústní Nejméně 1 den, dokud nedojde k
zástavě krvácení, indikované skončením bolestí, nebo ke zhojení.
Rozsáhlejší hemartros, krvácení do 30-60 svalů nebo hematom
Život ohrožující krvácení 60-100
Opakovat infuzi každých 12-24 hodin po dobu 3-4 dnů nebo déle dokud bolest a akutní porucha funkce neustoupí
Opakovat infuze každých 8-24 hodin dokud nepomine ohrožení života
Chirurgické zákroky
Každých 24 hodin, nejméně 1 den, až je dosaženo zhojení.
Menší operace včetně vytržení 30-60
zubu
|
Stupeň krvácení/ Typ chirurgického výkonu |
Požadovaná hladina FVIII (%) (IU/dl) |
Četnost dávek (hodiny)/délka trvání léčby (dny) |
|
Velké chirurgické výkony |
80-100 (před a po operaci) |
Opakovat infuzi každých 8-24 hodin až do adekvátního zhojení poranění, pak pokračovat v léčbě nejméně dalších 7 dní k udržení aktivity faktoru VIII na 30-60 % (IU/dl) |
Profylaxe
K dlouhodobé profylaxi krvácení u pacientů se závažnou hemofilií A. Obvyklé doporučené dávky jsou 20-40 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 20-50 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. V některých případech, zvláště u mladších pacientů, může být nutné podávat přípravek v kratších intervalech nebo ve vyšších dávkách.
Monitorování léčby
V průběhu léčby je doporučeno provádět příslušné stanovení hladin faktoru VIII za účelem získání vodítka pro velikost podávané dávky i četnost opakovaných aplikací. Zvláště v případě závažných chirurgických zákroků je nezbytné přesné monitorování substituční léčby, prováděné pomocí koagulační analýzy (aktivity faktoru VIII v plazmě). U jednotlivých pacientů se může jejich odezva na faktor VIII lišit dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním odlišných poločasů.
Chirurgický zákrok
U pediatrických pacientů neexistují žádné zkušenosti s chirurgickými zákroky.
Starší pacienti
U pacientů ve věku > 65 let neexistují žádné zkušenosti.
Pediatrická populace
K dlouhodobé profylaxi krvácení u pacientů do 12 let se doporučují dávky 25 - 50 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 25 - 60 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. Pro pediatrické pacienty od 12 let jsou doporučené dávky shodné s doporučeními pro dospělé pacienty.
Způsob podání Intravenózní podání
Doporučená rychlost infuze je u přípravku NovoEight 1-2 ml/min. Rychlost by měla být stanovena tak, aby vyhovovala pacientovi.
Instrukce pro rekonstituci léčivého přípravku před podáním naleznete v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Známá alergická reakce na křeččí bílkoviny.
4.4 Zvláštní upozornění a opatření pro použití Hypersenzitivita
Při používání přípravku NovoEight se mohou vyskytnout hypersenzitivní reakce alergického typu. Přípravek obsahuje stopy křeččích bílkovin, jež mohou u některých pacientů vyvolat alergické reakce. Obj eví-li se příznaky hypersenzitivity, musí být pacienti poučeni o tom, aby okamžitě přerušili léčbu tímto léčivým přípravkem a kontaktovali svého lékaře. Pacienti musí být informováni o časných příznacích hypersenzitivních reakcí včetně kopřivky, generalizované kopřivky, tlaku na hrudi, sípotu, hypotenze a anafylaxe.
V případě šoku je nutno nasadit standardní lékařskou léčbu šokového stavu.
Inhibitory
Známou komplikací léčby u individuálních případů hemofilie A je vznik neutralizačních protilátek (inhibitorů) proti faktoru VIII. Tyto inhibitory jsou obvykle IgG imunoglobuliny působící proti koagulační aktivitě faktoru VIII. Jsou kvantitativně udávané v Bethesda jednotkách (BU) na jeden ml plazmy a zjišťované pomocí modifikovaného testu. Riziko vzniku inhibitorů je ve vztahu k expozici organizmu faktorem VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Vzácně může dojít k tvorbě inhibitorů teprve po prvních 100 dnech expozice.
Při přechodu z jednoho přípravku obsahujícího faktor VIII na jiný byl u pacientů léčených již dříve, kteří měli v anamnéze dřívější výskyt inhibitoru, pozorován opětovný výskyt inhibitoru (nízký titr), a to i po více než 100 dnech expozice. Proto je při jakémkoliv převodu na jiný přípravek doporučeno vždy pečlivě monitorovat všechny pacienty s ohledem na vznik inhibitoru.
Obecně všichni pacienti léčení přípravky obsahujícími koagulační faktor VIII by měli být pečlivě sledováni z hlediska vzniku inhibitorů vhodnými klinickými vyšetřeními a laboratorními testy. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII nebo pokud není dosaženo kontroly krvácení příslušnou dávkou, musí být provedeny testy na přítomnost inhibitoru. U pacientů s vysokými hladinami inhibitorů, může být léčba faktorem VIII neúčinná, a je třeba zvážit jiné léčebné možnosti. Léčba takovýchto pacientů by měla být prováděna lékařem se zkušeností v péči o pacienty s hemofilií a s inhibitory proti faktoru VIII.
Důrazně se doporučuje, aby vždy při každém podání přípravku NovoEight pacientovi byl zaznamenán název a číslo šarže přípravku, aby bylo možno přiřadit číslo šarže přípravku k pacientovi.
Pomocné látky, které je nutno vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,31 mmol sodíku (7 mg) na 1 ml vzniklého roztoku. Tento fakt je nutno vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
Komplikace spojené s použitím katetru
Pokud je požadováno použití centrálního žilního katetru (CVAD), je nutno zvážit riziko komplikací spojené s jeho použitím včetně lokálních infekcí, bakteriémie a trombózy v místě katetru.
Pediatrická populace
Uvedená varování a preventivní opatření platí pro dospělé i děti.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce S přípravkem NovoEight nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
S přípravkem NovoEight nebyly prováděny žádné reprodukční studie na zvířatech. Na základě vzácného výskytu hemofilie A u žen nejsou zkušenosti týkající se použití faktoru VIII během těhotenství a kojení k dispozici. Z toho důvodu může být faktor VIII během těhotenství a kojení použit pouze, pokud je to jednoznačně indikováno.
4.7 Účinky na schopnost řídit a obsluhovat stroje
NovoEight nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Vzácně byly pozorovány hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a štípání v místě infuze, třesavku, zrudnutí, generalizovanou kopřivku, bolest hlavy, vyrážku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tlak na prsou, brnění, zvracení, sípot) a mohou v některých případech vyústit v těžkou anafylaxi (včetně šoku).
Velmi vzácně byl pozorován vznik protilátek proti křeččím proteinům se spojenou hypersenzitivitou.
U pacientů s hemofilií A může dojít ke vzniku neutralizačních protilátek (inhibitory) proti faktoru VIII. Jestliže dojde ke vzniku těchto inhibitorů, projeví se to jako nedostačující klinická odpověď. V takových případech se doporučuje vyhledat specializované centrum pro léčbu hemofilie.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky uvedené v tabulce níže jsou klasifikovány dle Tříd orgánových systémů podle databáze MedDRA (TOS a preferované termíny četností).
Frekvence výskytu jsou definovány podle následující konvence: Velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 2 Frekvence nežádoucích účinků v klinických studiích
|
Třída orgánových systémů |
Frekvence* |
Nežádoucí účinek |
|
Psychiatrické poruchy |
Méně časté | |
|
Poruchy nervového systému |
Méně časté |
Bolest hlavy, závratě |
|
Srdeční poruchy |
Méně časté |
Sinusová tachykardie |
|
Cévní poruchy |
Méně časté |
Hypertenze, lymfoedém |
|
Poruchy jater a žlučových cest |
Časté |
Zvýšení jaterních enzymů** |
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Méně časté |
Muskuloskeletální ztuhlost, artropatie, bolesti končetin, muskuloskeletální bolest |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Reakce v místě vpichu*** |
|
Méně časté |
Únava, pocit horka, periferní edém, pyrexie | |
|
Vyšetření |
Méně časté |
Zrychlená srdeční frekvence |
|
Poranění, otravy a procedurální komplikace |
Méně časté |
Kontuze |
*
**
Přepočteno na základě celkového počtu jednotlivých pacientů ve všech klinických studiích (214)
Zvýšení jaterních enzymů se týká alanin aminotransferázy, aspartát aminotransferázy, gama-glutamyltransferázy a bilirubinu
***
Reakce v místě vpichu zahrnují erytém v místě vpichu, extravazáty v místě vpichu a svědění v místě vpichu
Popis vybraných nežádoucích účinků
V průběhu všech klinických studií s přípravkem NovoEight bylo celkem hlášeno 30 nežádoucích účinků u 19 z 214 pacientů léčených přípravkem NovoEight. Nejčastěji hlášenými nežádoucími účinky byly reakce v místě vpichu a zvýšení jaterních enzymů. Z 30 nežádoucích účinků byly 2 hlášeny u jednoho z 31 pacientů do 6 let, žádný u pacientů ve věku 6-18 let a 28 bylo hlášeno u 18 ze 127 dospělých.
Pediatrická populace
V klinických studiích se 63 pediatrickými pacienty v rozmezí 0 až 12 let a s 24 dospívajícími
v rozmezí 12-18 let, kteří trpěli závažnou hemofilií A, nebyl nalezen žádný rozdíl v bezpečnostním profilu přípravku NovoEight mezi pediatrickými pacienty a dospělými.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika, krevní koagulační faktor VIII, ATC kód: B02BD02. Mechanismus účinku
NovoEight obsahuje turoktokog alfa - humánní koagulační faktor VIII (rDNA) se zkrácenou B-doménou. Tento glykoprotein má shodnou strukturu s humánním faktorem VIII, když je aktivován. Posttranslační modifikace jsou podobné modifikacím u molekul odvozených z plazmy. Sulfatační místo tyrosinu, jež je přítomno na Tyr1680 (přirozená plná délka) a jež je důležité pro vazbu na von Willebrandův faktor, je v molekule turoktokogu alfa plně sulfonované. Po podání infuze pacientovi s hemofilií se faktor VIII v krevním oběhu váže na endogenní von Willebrandův faktor. Komplex faktoru VIII s von Willebrandovým faktorem je tvořen 2 molekulami (faktor VIII a von Willebrandův faktor) s odlišnými fyziologickými funkcemi. Aktivovaný faktor VIII působí jako kofaktor pro aktivaci faktoru IX urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin pak konvertuje fibrinogen na fibrin a umožní tak tvorbu sraženiny. Hemofilie je na pohlaví závisející dědičná porucha krevní srážlivosti, jejíž příčinou je snížená hladina faktoru VIII:C. Výsledkem je silné krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánní nebo jako důsledek úrazu nebo chirurgického zákroku. Při substituční léčbě se hladiny plazmatického faktoru VIII zvýší, tím dojde k dočasné úpravě deficitu faktoru a tím také k úpravě sklonu ke krvácení.
Klinická účinnost
Byly provedeny tři multicentrické, otevřené, nekontrolované studie za účelem vyhodnotit bezpečnost a účinnost přípravku NovoEight v prevenci a léčbě krvácení u již dříve léčených pacientů s těžkou hemofilií A (aktivita FVIII <1 %). Do studie bylo zahrnuto 213 léčených pacientů; 150 dospívajících či dospělých pacientů bez inhibitorů ve věku od 12 let (>150 dní léčby) a 63 pediatrických pacientů bez inhibitorů do 12 let (>50 dní léčby). 187 z 213 pacientů pokračovalo v prodloužené studii bezpečnosti. Léčba přípravkem NovoEight byla prokázána jako bezpečná a měla předpokládaný hemostatický a preventivní účinek. Během akumulované léčby více než 54 000 dní (odpovídající 342 pacientoroků) nebyl ve fázi 3 a klinických studií u již dříve léčených pacientů pozorován rozvoj žádných inhibitorů proti faktoru VIII. Z 1 377 hlášených krvácivých příhod pozorovaných u 177 pacientů z 213, bylo 1 244 (90,3 %) krvácení zastaveno 1-2 infuzemi přípravku NovoEight.
Tabulka 3 Spotřeba turoktokogu alfa a celkový výskyt úspěšnosti
|
Mladší děti (0 - <6 let) |
Starší děti (6 - <12 let) |
Dospívající (12 -<18 let) |
Dospělí (>18 let) |
Celkem | |
|
Počet pacientů |
31 |
32 |
24 |
126 |
213 |
|
Dávka užitá k prevenci na pacienta (IU/kg TH) Průměr (SD) |
40,1 (8,5) |
36,6 (9,0) |
27,0 (7,6) |
26,9 (6,9) |
30,3 (9,2) |
|
Min ; Max |
26,5 ; 57,3 |
24,9 ; 57,9 |
20,5 ; 46,9 |
20,0 ; 50,8 |
20,0 ; 57,9 |
|
Dávka užitá k léčbě krvácení (IU/kg TH) Průměr (SD) Min ; Max |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Výskyt úspěšnosti* % |
92,9% |
88,9% |
79,7% |
85,6% |
85,9% |
TH: Tělesná hmotnost, SD: Směrodatná odchylka *Úspěšnost je definována buď jako „Výborná“ nebo „Dobrá“.
Celkem bylo provedeno 14 chirurgických zákroků u celkem 14 pacientů, z nichž 13 bylo závažných, a jeden byl lehký. Hemostáza byla úspěšná ve všech případech a nebylo hlášeno žádné selhání léčby.
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s turoktokogem alfa byly prováděny u pacientů trpících závažnou hemofilií A (aktivita FVIII <1 %), kteří již byli dříve léčeni. Analýza vzorků plazmy byla prováděna jak pomocí jednostupňového koagulačního testu, tak chromogenním testem.
V mezinárodní studii zahrnující 36 laboratoří byla testována aktivita přípravku NovoEight pomocí testu FVIII:C a porovnávána s na trhu dostupným přípravkem obsahujícím rekombinantní FVIII o plné délce. Studie prokázala srovnatelné a stabilní výsledky pro oba přípravky a rovněž to, že přípravek NovoEight může být v plazmě spolehlivě měřen, aniž by bylo zapotřebí speciálního standardu pro NovoEight.
Farmakokinetické parametry po jednorázové dávce přípravku NovoEight jsou shrnuty v Tabulce 4 pro test srážlivosti, v Tabulce 5 pro chromogenní test.
Tabulka 4 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), test srážlivosti_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((IU*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
t* (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (IU/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Průměrná doba setrvání v oběhu (hod.) |
9,63 (2,50) |
9,91 (2,57) |
14,19 5,08) |
Tabulka 5 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), chromogenní test_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((IU*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (IU/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Průměrná doba setrvání v oběhu (hod.) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Farmakokinetické parametry u pediatrických pacientů do 6 let a pediatrických pacientů ve věku 612 let byly srovnatelné. Byly pozorovány některé odchylky ve farmakokinetických parametrech přípravku NovoEight mezi pediatrickými a dospělými pacienty. U pediatrických pacientů byly nalezeny vyšší hodnoty CL a kratší ť/2 ve srovnání s dospělými pacienty s hemofilií A, což může být částečně způsobeno známým vyšším plazmatickým objemem na kilogram tělesné hmotnosti u mladších pacientů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po opakovaném podávání neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Chlorid sodný Histidin Sacharosa Polysorbát 80 Methionin
Dihydrát chloridu vápenatého Hydroxid sodný Kyselina chlorovodíková
Rozpouštědlo Chlorid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Před otevřením:
30 měsíců
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (< 30°C) po jedno nepřetržité období nepřesahující 9 měsíců. Jakmile byl přípravek jednou vyjmut z chladničky, nesmí tam již být vrácen zpět. Poznačte si prosím na krabičce datum, kdy jste přípravek začali uchovávat při pokojové teplotě. Injekční lahvičku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem.
Po rekonstituci:
Chemická a fyzikální stabilita po rekonstituci byla prokázána na dobu 24 hodin při 2°C - 8°C a 4 hodiny při uchovávání při pokojové teplotě (< 30°C).
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, jsou doba a podmínky uchovávání přípravku po otevření před použitím v odpovědnosti uživatele. Normálně by tato doba neměla být delší než 4 hodiny při uchovávání při pokojové teplotě (< 30°C) nebo 24 hodin při 2°C - 8°C, pokud rekonstituce neproběhla za kontrolovaných a validovaných aseptických podmínek.
Veškerý nepoužitý léčivý přípravek, který byl uchováván při pokojové teplotě déle než 4 hodiny, musí být zlikvidován.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem.
Uchovávání tohoto léčivého přípravku při pokojové teplotě a podmínky uchovávání po jeho rekonstituci viz bod 6.3.
6.5 Druh obalu a obsah balení
Jedno balení přípravku NovoEight 250 IU prášek a rozpouštědlo pro injekční roztok obsahuje:
- 1 skleněnou injekční lahvičku (sklo typu I) s práškem opatřenou chlorobutylovou pryžovou zátkou
- 1 sterilní adaptér injekční lahvičky k rozpuštění
- 1 předplněnou injekční stříkačku obsahující 4 ml rozpouštědla s polypropylenovým uzávěrem zpětného chodu, bromobutylovým pryžovým pístem a uzávěrem s bromobutylovou zátkou.
- 1 nástavec pístu (zhotovený z polypropylenu)
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
NovoEight je určen k intravenóznímu podání po rekonstituci prášku v rozpouštědle dodávaném v injekční stříkačce. Po rekonstituci je roztok čirý či lehce opalescentní. Roztok nepoužívejte, pokud je zakalený či obsahuje usazeniny.
Budete také potřebovat infuzní soupravu (infuzní set a jehlu s křidélky), sterilní alkoholové tampony, gázové polštářky a náplasti. Tyto pomůcky nejsou součástí balení přípravku NovoEight.
Vždy dodržujte aseptickou techniku.
Rekonstituce
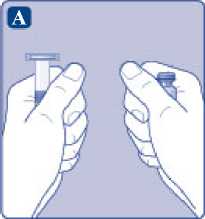
A)
Vyjměte injekční lahvičku, adaptér injekční lahvičky a předplněnou injekční stříkačku z krabičky. Nástavec pístu ponechte zatím v krabičce. Zahřejte injekční lahvičku a předplněnou injekční stříkačku na pokojovou teplotu. Můžete to udělat tak, že je podržíte v ruce, dokud nemají stejnou teplotu jako vaše dlaně. Jiné způsoby ohřátí injekční lahvičky a předplněné injekční stříkačky nepoužívejte.
B)
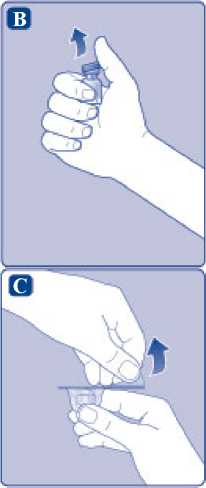

Odstraňte plastové víčko z injekční lahvičky. Pokud je víčko uvolněné nebo chybí, injekční lahvičku nepoužívejte. Pryžovou zátku injekční lahvičky očistěte sterilním alkoholovým tamponem a nechte ji před použitím několik sekund na vzduchu oschnout.
C)
Sejměte ochranný papír z adaptéru injekční lahvičky. Pokud ochranný papír není zcela zatavený nebo je protržený, adaptér injekční lahvičky nepoužívejte. Nevyjímejte adaptér injekční lahvičky prsty z ochranného víčka.
D)
Otočte ochranné víčko a nasaďte adaptér na injekční lahvičku. Jakmile jste adaptér na injekční lahvičku nasadili, již ho z ní neodstraňujte.
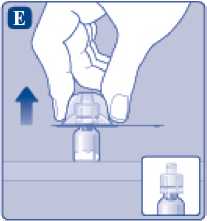
E)
Lehce stiskněte ochranné víčko mezi palcem a ukazováčkem, jak je patrné z obrázku. Sejměte ochranné víčko z adaptéru injekční lahvičky.
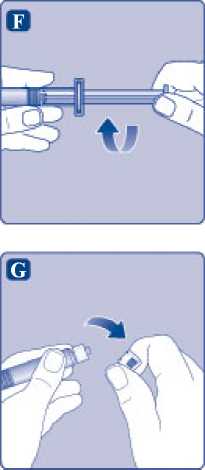
F)
Pevně uchopte nástavec pístu za širší konec a okamžitě ho našroubujte po směru hodinových ručiček na píst uvnitř předplněné injekční stříkačky, dokud nepocítíte odpor.
G)
Odstraňte ochranné víčko z předplněné injekční stříkačky ohnutím směrem dolů tak, aby se porušila perforace. Dbejte, abyste se nedotkli prsty hrotu injekční stříkačky pod jejím ochranným víčkem.
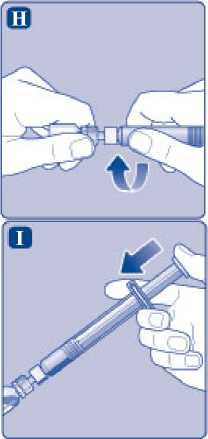
H)
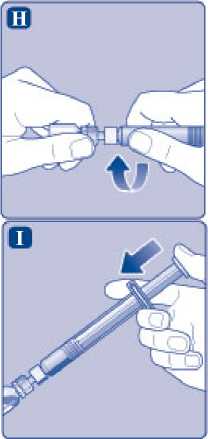
Našroubujte předplněnou injekční stříkačku bezpečně na adaptér injekční lahvičky, dokud nepocítíte odpor.
I)
Držte předplněnou injekční stříkačku lehce nakloněnou s injekční lahvičkou směřující dolů. Stisknutím nástavce pístu vstříkněte všechno rozpouštědlo do injekční lahvičky.
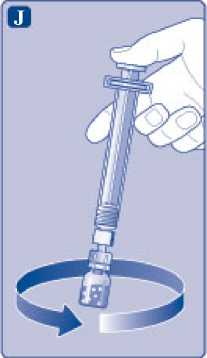
J)
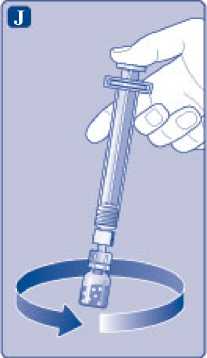
Nechte nástavec pístu zcela stlačený a jemným kroužením injekční lahvičkou rozpusťte všechen prášek. Injekční lahvičkou netřepejte, mohlo by to způsobit napěnění.
Doporučuje se použít NovoEight okamžitě po rekonstituci. Podmínky uchovávání rekonstituovaného léčivého přípravku viz bod 6.3.
Pokud je zapotřebí větší dávky, opakujte kroky A až J s dalšími injekčními lahvičkami, adaptéry injekčních lahviček a předplněnými injekčními stříkačkami.
Aplikace rekonstituovaného roztoku
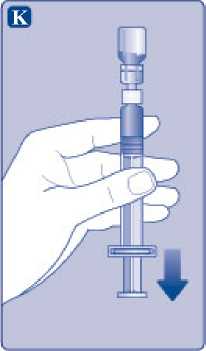
K)
Ponechte nástavec pístu zcela stlačený. Otočte injekční stříkačku tak, aby nasazená injekční lahvička byla dnem vzhůru. Uvolněte nástavec pístu a nechte ho samovolně vrátit se zpět. Tím se rekonstituovaný roztok natáhne do injekční stříkačky. Lehkým vytažením pístu směrem dolů pak zajistíte, že se do injekční stříkačky natáhne všechen roztok.
V případě, že potřebujete pouze část obsahu injekční lahvičky, použijte stupnici na injekční stříkačce, abyste si ověřili, že jste natáhli tolik vzniklého roztoku, kolik vám doporučil lékař či zdravotní sestra.
Stále držte injekční lahvičku dnem vzhůru a jemně poklepejte na injekční stříkačku, aby se vzduchové bubliny nashromáždily nahoře. Pomalu zatlačte na nástavec pístu, dokud všechny vzduchové bubliny neuniknou.
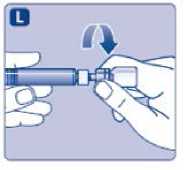
L)
Odšroubujte adaptér s injekční lahvičkou.
NovoEight je nyní připraven k aplikaci. Určete vhodné místo a pomalu aplikujte NovoEight do žíly v průběhu 2 až 5 minut.
Likvidace
Po aplikaci bezpečně zlikvidujte veškerý nepoužitý roztok přípravku NovoEight, injekční stříkačku s infuzní soupravou, injekční lahvičku s adaptérem a ostatní odpad dle doporučení lékárníka.
Nevhazujte do běžného domácího odpadu.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
8. REGISTRAČNÍ ČÍSLA
EU/1/13/888/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. listopad 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky (EMA) http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
NovoEight 500 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička s práškem obsahuje 500 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Po rozpuštění obsahuje přípravek NovoEight přibližně 125 IU/ml humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Účinnost (IU) se udává chromogenní metodou podle Evropského lékopisu. Specifická aktivita přípravku NovoEight je přibližně 8 300 IU/mg bílkoviny.
Turoktokog alfa (humánní koagulační faktor VIII (rDNA)) je čištěná bílkovina obsahující 1 445 aminokyselin s molekulovou hmotností asi 166 kDA. Je vyráběn rekombinantní DNA technologií z vaječníkových buněk čínského křečka (CHO buňky). Je vyroben bez přídavku jakékoliv bílkoviny lidského či zvířecího původu během kultivace buněk, čištění či konečné úpravy přípravku.
Turoktokog alfa je rekombinantní humánní koagulační faktor VIII se zkrácenou B-doménou (B-doména obsahuje 21 aminokyselin z přirozené B-domény) bez jakékoliv další změny v pořadí aminokyselin.
Pomocná látka se známým účinkem:
0,31 mmol sodíku (7 mg) v 1 ml vzniklého roztoku
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Bílý nebo lehce nažloutlý prášek či drobivá hmota.
Čirý a bezbarvý injekční roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a prevence krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). NovoEight lze používat ve všech věkových skupinách.
4.2 Dávkování a způsob podání
Léčba by měla být zahájena pod dohledem lékaře se zkušenostmi s léčbou hemofilie.
Doposud neléčení pacienti
Bezpečnost a účinnost přípravku NovoEight u dosud neléčených pacientů nebyly doposud stanoveny. Nejsou dostupné žádné údaje.
Dávkování
Dávkování a trvání substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a pacientově klinickém stavu.
Počet podávaných jednotek faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které odpovídají běžnému standardu WHO pro přípravky obsahující faktor VIII. Aktivita faktoru VIII v plazmě je vyjádřena buď v procentech (relativně k normální hladině v lidské plazmě) nebo v mezinárodních jednotkách (relativně k mezinárodnímu standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba v případě potřeby
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že jedna mezinárodní jednotka (IU) faktoru VIII na kg tělesné hmotnosti zvýší aktivitu faktoru VIII v plazmě asi o 2 IU/dl. Požadovaná dávka se vypočte podle následujícího vzorce:
Potřebný počet jednotek = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) (IU/dl) x 0,5 (IU/kg na IU/dl).
Množství, které má být podáno, a frekvence podání by měly být vždy přizpůsobeny klinické účinnosti v individuálním případě.
V případě následujících krvácivých příhod by v odpovídajícím období aktivita faktoru VIII neměla klesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může být použita jako návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Tabulka 1 Návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Stupeň krvácení/ Požadovaná hladina Četnost dávek (hodiny)/délka
Typ chirurgického výkonu FVIII (%) (IU/dl) trvání léčby (dny)
Krvácení
Časný hemartros, krvácení do svalů 20-40 Opakovat každých 12-24 hodin.
nebo do dutiny ústní Nejméně 1 den, dokud nedojde k
zástavě krvácení, indikované skončením bolestí, nebo ke zhojení.
Rozsáhlejší hemartros, krvácení do 30-60 svalů nebo hematom
Život ohrožující krvácení 60-100
Opakovat infuzi každých 12-24 hodin po dobu 3-4 dnů nebo déle dokud bolest a akutní porucha funkce neustoupí
Opakovat infuze každých 8-24 hodin dokud nepomine ohrožení života
Chirurgické zákroky
Každých 24 hodin, nejméně 1 den, až je dosaženo zhojení.
Menší operace včetně vytržení 30-60
zubu
|
Stupeň krvácení/ Typ chirurgického výkonu |
Požadovaná hladina FVIII (%) (IU/dl) |
Četnost dávek (hodiny)/délka trvání léčby (dny) |
|
Velké chirurgické výkony |
80-100 (před a po operaci) |
Opakovat infuzi každých 8-24 hodin až do adekvátního zhojení poranění, pak pokračovat v léčbě nejméně dalších 7 dní k udržení aktivity faktoru VIII na 30-60 % (IU/dl) |
Profylaxe
K dlouhodobé profylaxi krvácení u pacientů se závažnou hemofilií A. Obvyklé doporučené dávky jsou 20-40 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 20-50 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. V některých případech, zvláště u mladších pacientů, může být nutné podávat přípravek v kratších intervalech nebo ve vyšších dávkách.
Monitorování léčby
V průběhu léčby je doporučeno provádět příslušné stanovení hladin faktoru VIII za účelem získání vodítka pro velikost podávané dávky i četnost opakovaných aplikací. Zvláště v případě závažných chirurgických zákroků je nezbytné přesné monitorování substituční léčby, prováděné pomocí koagulační analýzy (aktivity faktoru VIII v plazmě). U jednotlivých pacientů se může jejich odezva na faktor VIII lišit dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním odlišných poločasů.
Chirurgický zákrok
U pediatrických pacientů neexistují žádné zkušenosti s chirurgickými zákroky.
Starší pacienti
U pacientů ve věku > 65 let neexistují žádné zkušenosti.
Pediatrická populace
K dlouhodobé profylaxi krvácení u pacientů do 12 let se doporučují dávky 25 - 50 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 25 - 60 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. Pro pediatrické pacienty od 12 let jsou doporučené dávky shodné s doporučeními pro dospělé pacienty.
Způsob podání Intravenózní podání
Doporučená rychlost infuze je u přípravku NovoEight 1-2 ml/min. Rychlost by měla být stanovena tak, aby vyhovovala pacientovi.
Instrukce pro rekonstituci léčivého přípravku před podáním naleznete v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Známá alergická reakce na křeččí bílkoviny.
4.4 Zvláštní upozornění a opatření pro použití Hypersenzitivita
Při používání přípravku NovoEight se mohou vyskytnout hypersenzitivní reakce alergického typu. Přípravek obsahuje stopy křeččích bílkovin, jež mohou u některých pacientů vyvolat alergické reakce. Obj eví-li se příznaky hypersenzitivity, musí být pacienti poučeni o tom, aby okamžitě přerušili léčbu tímto léčivým přípravkem a kontaktovali svého lékaře. Pacienti musí být informováni o časných příznacích hypersenzitivních reakcí včetně kopřivky, generalizované kopřivky, tlaku na hrudi, sípotu, hypotenze a anafylaxe.
V případě šoku je nutno nasadit standardní lékařskou léčbu šokového stavu.
Inhibitory
Známou komplikací léčby u individuálních případů hemofilie A je vznik neutralizačních protilátek (inhibitorů) proti faktoru VIII. Tyto inhibitory jsou obvykle IgG imunoglobuliny působící proti koagulační aktivitě faktoru VIII. Jsou kvantitativně udávané v Bethesda jednotkách (BU) na jeden ml plazmy a zjišťované pomocí modifikovaného testu. Riziko vzniku inhibitorů je ve vztahu k expozici organizmu faktorem VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Vzácně může dojít k tvorbě inhibitorů teprve po prvních 100 dnech expozice.
Při přechodu z jednoho přípravku obsahujícího faktor VIII na jiný byl u pacientů léčených již dříve, kteří měli v anamnéze dřívější výskyt inhibitoru, pozorován opětovný výskyt inhibitoru (nízký titr), a to i po více než 100 dnech expozice. Proto je při jakémkoliv převodu na jiný přípravek doporučeno vždy pečlivě monitorovat všechny pacienty s ohledem na vznik inhibitoru.
Obecně všichni pacienti léčení přípravky obsahujícími koagulační faktor VIII by měli být pečlivě sledováni z hlediska vzniku inhibitorů vhodnými klinickými vyšetřeními a laboratorními testy. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII nebo pokud není dosaženo kontroly krvácení příslušnou dávkou, musí být provedeny testy na přítomnost inhibitoru. U pacientů s vysokými hladinami inhibitorů, může být léčba faktorem VIII neúčinná, a je třeba zvážit jiné léčebné možnosti. Léčba takovýchto pacientů by měla být prováděna lékařem se zkušeností v péči o pacienty s hemofilií a s inhibitory proti faktoru VIII.
Důrazně se doporučuje, aby vždy při každém podání přípravku NovoEight pacientovi byl zaznamenán název a číslo šarže přípravku, aby bylo možno přiřadit číslo šarže přípravku k pacientovi.
Pomocné látky, které je nutno vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,31 mmol sodíku (7 mg) na 1 ml vzniklého roztoku. Tento fakt je nutno vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
Komplikace spojené s použitím katetru
Pokud je požadováno použití centrálního žilního katetru (CVAD), je nutno zvážit riziko komplikací spojené s jeho použitím včetně lokálních infekcí, bakteriémie a trombózy v místě katetru.
Pediatrická populace
Uvedená varování a preventivní opatření platí pro dospělé i děti.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce S přípravkem NovoEight nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
S přípravkem NovoEight nebyly prováděny žádné reprodukční studie na zvířatech. Na základě vzácného výskytu hemofilie A u žen nejsou zkušenosti týkající se použití faktoru VIII během těhotenství a kojení k dispozici. Z toho důvodu může být faktor VIII během těhotenství a kojení použit pouze, pokud je to jednoznačně indikováno.
4.7 Účinky na schopnost řídit a obsluhovat stroje
NovoEight nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Vzácně byly pozorovány hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a štípání v místě infuze, třesavku, zrudnutí, generalizovanou kopřivku, bolest hlavy, vyrážku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tlak na prsou, brnění, zvracení, sípot) a mohou v některých případech vyústit v těžkou anafylaxi (včetně šoku).
Velmi vzácně byl pozorován vznik protilátek proti křeččím proteinům se spojenou hypersenzitivitou.
U pacientů s hemofilií A může dojít ke vzniku neutralizačních protilátek (inhibitory) proti faktoru VIII. Jestliže dojde ke vzniku těchto inhibitorů, projeví se to jako nedostačující klinická odpověď. V takových případech se doporučuje vyhledat specializované centrum pro léčbu hemofilie.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky uvedené v tabulce níže jsou klasifikovány dle Tříd orgánových systémů podle databáze MedDRA (TOS a preferované termíny četností).
Frekvence výskytu jsou definovány podle následující konvence: Velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 2 Frekvence nežádoucích účinků v klinických studiích
|
Třída orgánových systémů |
Frekvence* |
Nežádoucí účinek |
|
Psychiatrické poruchy |
Méně časté | |
|
Poruchy nervového systému |
Méně časté |
Bolest hlavy, závratě |
|
Srdeční poruchy |
Méně časté |
Sinusová tachykardie |
|
Cévní poruchy |
Méně časté |
Hypertenze, lymfoedém |
|
Poruchy jater a žlučových cest |
Časté |
Zvýšení jaterních enzymů* * |
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Méně časté |
Muskuloskeletální ztuhlost, artropatie, bolesti končetin, muskuloskeletální bolest |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Reakce v místě vpichu* * * |
|
Méně časté |
Únava, pocit horka, periferní edém, pyrexie | |
|
Vyšetření |
Méně časté |
Zrychlená srdeční frekvence |
|
Poranění, otravy a procedurální komplikace |
Méně časté |
Kontuze |
*
**
Přepočteno na základě celkového počtu jednotlivých pacientů ve všech klinických studiích (214)
Zvýšení jaterních enzymů se týká alanin aminotransferázy, aspartát aminotransferázy, gama-glutamyltransferázy a bilirubinu
***
Reakce v místě vpichu zahrnují erytém v místě vpichu, extravazáty v místě vpichu a svědění v místě vpichu
Popis vybraných nežádoucích účinků
V průběhu všech klinických studií s přípravkem NovoEight bylo celkem hlášeno 30 nežádoucích účinků u 19 z 214 pacientů léčených přípravkem NovoEight. Nejčastěji hlášenými nežádoucími účinky byly reakce v místě vpichu a zvýšení jaterních enzymů. Z 30 nežádoucích účinků byly 2 hlášeny u jednoho z 31 pacientů do 6 let, žádný u pacientů ve věku 6-18 let a 28 bylo hlášeno u 18 ze 127 dospělých.
Pediatrická populace
V klinických studiích se 63 pediatrickými pacienty v rozmezí 0 až 12 let a s 24 dospívajícími
v rozmezí 12-18 let, kteří trpěli závažnou hemofilií A, nebyl nalezen žádný rozdíl v bezpečnostním profilu přípravku NovoEight mezi pediatrickými pacienty a dospělými.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika, krevní koagulační faktor VIII, ATC kód: B02BD02 Mechanismus účinku
NovoEight obsahuje turoktokog alfa - humánní koagulační faktor VIII (rDNA) se zkrácenou B-doménou. Tento glykoprotein má shodnou strukturu s humánním faktorem VIII, když je aktivován. Posttranslační modifikace jsou podobné modifikacím u molekul odvozených z plazmy. Sulfatační místo tyrosinu, jež je přítomno na Tyr1680 (přirozená plná délka) a jež je důležité pro vazbu na von Willebrandův faktor, je v molekule turoktokogu alfa plně sulfonované. Po podání infuze pacientovi s hemofilií se faktor VIII v krevním oběhu váže na endogenní von Willebrandův faktor. Komplex faktoru VIII s von Willebrandovým faktorem je tvořen 2 molekulami (faktor VIII a von Willebrandův faktor) s odlišnými fyziologickými funkcemi. Aktivovaný faktor VIII působí jako kofaktor pro aktivaci faktoru IX urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin pak konvertuje fibrinogen na fibrin a umožní tak tvorbu sraženiny. Hemofilie je na pohlaví závisející dědičná porucha krevní srážlivosti, jejíž příčinou je snížená hladina faktoru VIII:C. Výsledkem je silné krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánní nebo jako důsledek úrazu nebo chirurgického zákroku. Při substituční léčbě se hladiny plazmatického faktoru VIII zvýší, tím dojde k dočasné úpravě deficitu faktoru a tím také k úpravě sklonu ke krvácení.
Klinická účinnost
Byly provedeny tři multicentrické, otevřené, nekontrolované studie za účelem vyhodnotit bezpečnost a účinnost přípravku NovoEight v prevenci a léčbě krvácení u již dříve léčených pacientů s těžkou hemofilií A (aktivita FVIII <1 %). Do studie bylo zahrnuto 213 léčených pacientů; 150 dospívajících či dospělých pacientů bez inhibitorů ve věku od 12 let (>150 dní léčby) a 63 pediatrických pacientů bez inhibitorů do 12 let (>50 dní léčby). 187 z 213 pacientů pokračovalo v prodloužené studii bezpečnosti. Léčba přípravkem NovoEight byla prokázána jako bezpečná a měla předpokládaný hemostatický a preventivní účinek. Během akumulované léčby více než 54 000 dní (odpovídající 342 pacientoroků) nebyl ve fázi 3 a klinických studií u již dříve léčených pacientů pozorován rozvoj žádných inhibitorů proti faktoru VIII. Z 1 377 hlášených krvácivých příhod pozorovaných u 177 pacientů z 213, bylo 1 244 (90,3 %) krvácení zastaveno 1-2 infuzemi přípravku NovoEight.
Tabulka 3 Spotřeba turoktokogu alfa a celkový výskyt úspěšnosti
|
Mladší děti (0 - <6 let) |
Starší děti (6 - <12 let) |
Dospívající (12 -<18 let) |
Dospělí (>18 let) |
Celkem | |
|
Počet pacientů |
31 |
32 |
24 |
126 |
213 |
|
Dávka užitá k prevenci na pacienta (IU/kg TH) Průměr (SD) |
40,1 (8,5) |
36,6 (9,0) |
27,0 (7,6) |
26,9 (6,9) |
30,3 (9,2) |
|
Min ; Max |
26,5 ; 57,3 |
24,9 ; 57,9 |
20,5 ; 46,9 |
20,0 ; 50,8 |
20,0 ; 57,9 |
|
Dávka užitá k léčbě krvácení (IU/kg TH) Průměr (SD) Min ; Max |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Výskyt úspěšnosti* % |
92,9% |
88,9% |
79,7% |
85,6% |
85,9% |
TH: Tělesná hmotnost, SD: Směrodatná odchylka *Úspěšnost je definována buď jako „Výbomá“ nebo „Dobrá“.
Celkem bylo provedeno 14 chirurgických zákroků u celkem 14 pacientů, z nichž 13 bylo závažných, a jeden byl lehký. Hemostáza byla úspěšná ve všech případech a nebylo hlášeno žádné selhání léčby.
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s turoktokogem alfa byly prováděny u pacientů trpících závažnou hemofilií A (aktivita FVIII <1 %), kteří již byli dříve léčeni. Analýza vzorků plazmy byla prováděna jak pomocí jednostupňového koagulačního testu, tak chromogenním testem.
V mezinárodní studii zahrnující 36 laboratoří byla testována aktivita přípravku NovoEight pomocí testu FVIII:C a porovnávána s na trhu dostupným přípravkem obsahujícím rekombinantní FVIII o plné délce. Studie prokázala srovnatelné a stabilní výsledky pro oba přípravky a rovněž to, že přípravek NovoEight může být v plazmě spolehlivě měřen, aniž by bylo zapotřebí speciálního standardu pro NovoEight.
Farmakokinetické parametry po jednorázové dávce přípravku NovoEight jsou shrnuty v Tabulce 4 pro test srážlivosti, v Tabulce 5 pro chromogenní test.
Tabulka 4 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), test srážlivosti_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((IU*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
V (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (IU/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Průměrná doba setrvání v oběhu (hod.) |
9,63 (2,50) |
9,91 (2,57) |
14,19 5,08) |
Tabulka 5 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), chromogenní test_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((IU*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (IU/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Průměrná doba setrvání v oběhu (hod.) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Farmakokinetické parametry u pediatrických pacientů do 6 let a pediatrických pacientů ve věku 612 let byly srovnatelné. Byly pozorovány některé odchylky ve farmakokinetických parametrech přípravku NovoEight mezi pediatrickými a dospělými pacienty. U pediatrických pacientů byly nalezeny vyšší hodnoty CL a kratší ť/2 ve srovnání s dospělými pacienty s hemofilií A, což může být částečně způsobeno známým vyšším plazmatickým objemem na kilogram tělesné hmotnosti u mladších pacientů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po opakovaném podávání neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Chlorid sodný Histidin Sacharosa Polysorbát 80 Methionin
Dihydrát chloridu vápenatého Hydroxid sodný Kyselina chlorovodíková
Rozpouštědlo Chlorid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Před otevřením:
30 měsíců
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (< 30°C) po jedno nepřetržité období nepřesahující 9 měsíců. Jakmile byl přípravek jednou vyjmut z chladničky, nesmí tam již být vrácen zpět. Poznačte si prosím na krabičce datum, kdy jste přípravek začali uchovávat při pokojové teplotě. Injekční lahvičku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem.
Po rekonstituci:
Chemická a fyzikální stabilita po rekonstituci byla prokázána na dobu 24 hodin při 2°C - 8°C a 4 hodiny při uchovávání při pokojové teplotě (< 30°C).
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, jsou doba a podmínky uchovávání přípravku po otevření před použitím v odpovědnosti uživatele. Normálně by tato doba neměla být delší než 4 hodiny při uchovávání při pokojové teplotě (< 30°C) nebo 24 hodin při 2°C - 8°C, pokud rekonstituce neproběhla za kontrolovaných a validovaných aseptických podmínek.
Veškerý nepoužitý léčivý přípravek, který byl uchováván při pokojové teplotě déle než 4 hodiny, musí být zlikvidován.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem.
Uchovávání tohoto léčivého přípravku při pokojové teplotě a podmínky uchovávání po jeho rekonstituci viz bod 6.3.
6.5 Druh obalu a obsah balení
Jedno balení přípravku NovoEight 500 IU prášek a rozpouštědlo pro injekční roztok obsahuje:
- 1 skleněnou injekční lahvičku (sklo typu I) s práškem opatřenou chlorobutylovou pryžovou zátkou
- 1 sterilní adaptér injekční lahvičky k rozpuštění
- 1 předplněnou injekční stříkačku obsahující 4 ml rozpouštědla s polypropylenovým uzávěrem zpětného chodu, bromobutylovým pryžovým pístem a uzávěrem s bromobutylovou zátkou.
- 1 nástavec pístu (zhotovený z polypropylenu)
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
NovoEight je určen k intravenóznímu podání po rekonstituci prášku v rozpouštědle dodávaném v injekční stříkačce. Po rekonstituci je roztok čirý či lehce opalescentní. Roztok nepoužívejte, pokud je zakalený či obsahuje usazeniny.
Budete také potřebovat infuzní soupravu (infuzní set a jehlu s křidélky), sterilní alkoholové tampony, gázové polštářky a náplasti. Tyto pomůcky nejsou součástí balení přípravku NovoEight.
Vždy dodržujte aseptickou techniku.
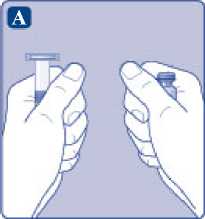
Rekonstituce
A)
Vyjměte injekční lahvičku, adaptér injekční lahvičky a předplněnou injekční stříkačku z krabičky. Nástavec pístu ponechte zatím v krabičce. Zahřejte injekční lahvičku a předplněnou injekční stříkačku na pokojovou teplotu. Můžete to udělat tak, že je podržíte v ruce, dokud nemají stejnou teplotu jako vaše dlaně. Jiné způsoby ohřátí injekční lahvičky a předplněné injekční stříkačky nepoužívejte.
B)

Odstraňte plastové víčko z injekční lahvičky. Pokud je víčko uvolněné nebo chybí, injekční lahvičku nepoužívejte. Pryžovou zátku injekční lahvičky očistěte sterilním alkoholovým tamponem a nechte ji před použitím několik sekund na vzduchu oschnout.
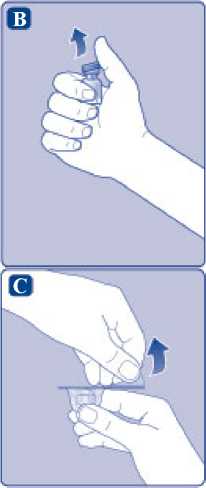
C)
Sejměte ochranný papír z adaptéru injekční lahvičky. Pokud ochranný papír není zcela zatavený nebo je protržený, adaptér injekční lahvičky nepoužívejte. Nevyjímejte adaptér injekční lahvičky prsty z ochranného víčka.
D)
Otočte ochranné víčko a nasaďte adaptér na injekční lahvičku. Jakmile jste adaptér na injekční lahvičku nasadili, již ho z ní neodstraňujte.
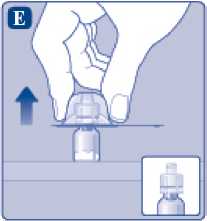
E)
Lehce stiskněte ochranné víčko mezi palcem a ukazováčkem, jak je patrné z obrázku. Sejměte ochranné víčko z adaptéru injekční lahvičky.
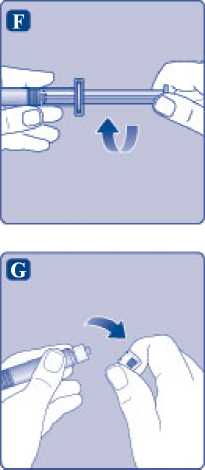
F)
Pevně uchopte nástavec pístu za širší konec a okamžitě ho našroubujte po směru hodinových ručiček na píst uvnitř předplněné injekční stříkačky, dokud nepocítíte odpor.
G)
Odstraňte ochranné víčko z předplněné injekční stříkačky ohnutím směrem dolů tak, aby se porušila perforace. Dbejte, abyste se nedotkli prsty hrotu injekční stříkačky pod jejím ochranným víčkem.
H)
Našroubujte předplněnou injekční stříkačku bezpečně na adaptér injekční lahvičky, dokud nepocítíte odpor.
I)
Držte předplněnou injekční stříkačku lehce nakloněnou s injekční lahvičkou směřující dolů. Stisknutím nástavce pístu vstříkněte všechno rozpouštědlo do injekční lahvičky.
J)
Nechte nástavec pístu zcela stlačený a jemným kroužením injekční lahvičkou rozpusťte všechen prášek. Injekční lahvičkou netřepejte, mohlo by to způsobit napěnění.
Doporučuje se použít NovoEight okamžitě po rekonstituci. Podmínky uchovávání rekonstituovaného léčivého přípravku viz bod 6.3.
Pokud je zapotřebí větší dávky, opakujte kroky A až J s dalšími injekčními lahvičkami, adaptéry injekčních lahviček a předplněnými injekčními stříkačkami.
Aplikace rekonstituovaného roztoku
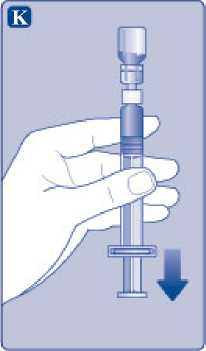
K)
Ponechte nástavec pístu zcela stlačený. Otočte injekční stříkačku tak, aby nasazená injekční lahvička byla dnem vzhůru. Uvolněte nástavec pístu a nechte ho samovolně vrátit se zpět. Tím se rekonstituovaný roztok natáhne do injekční stříkačky. Lehkým vytažením pístu směrem dolů pak zajistíte, že se do injekční stříkačky natáhne všechen roztok.
V případě, že potřebujete pouze část obsahu injekční lahvičky, použijte stupnici na injekční stříkačce, abyste si ověřili, že jste natáhli tolik vzniklého roztoku, kolik vám doporučil lékař či zdravotní sestra.
Stále držte injekční lahvičku dnem vzhůru a jemně poklepejte na injekční stříkačku, aby se vzduchové bubliny nashromáždily nahoře. Pomalu zatlačte na nástavec pístu, dokud všechny vzduchové bubliny neuniknou.
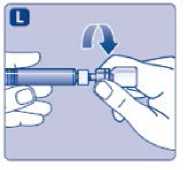
L)
Odšroubujte adaptér s injekční lahvičkou.
NovoEight je nyní připraven k aplikaci. Určete vhodné místo a pomalu aplikujte NovoEight do žíly v průběhu 2 až 5 minut.
Likvidace
Po aplikaci bezpečně zlikvidujte veškerý nepoužitý roztok přípravku NovoEight, injekční stříkačku s infuzní soupravou, injekční lahvičku s adaptérem a ostatní odpad dle doporučení lékárníka.
Nevhazujte do běžného domácího odpadu.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
8. REGISTRAČNÍ ČÍSLA
EU/1/13/888/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. listopad 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky (EMA) http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
NovoEight 1 000 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička s práškem obsahuje 1 000 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Po rozpuštění obsahuje přípravek NovoEight přibližně 250 IU/ml humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Účinnost (IU) se udává chromogenní metodou podle Evropského lékopisu. Specifická aktivita přípravku NovoEight je přibližně 8 300 IU/mg bílkoviny.
Turoktokog alfa (humánní koagulační faktor VIII (rDNA)) je čištěná bílkovina obsahující 1 445 aminokyselin s molekulovou hmotností asi 166 kDA. Je vyráběn rekombinantní DNA technologií z vaječníkových buněk čínského křečka (CHO buňky). Je vyroben bez přídavku jakékoliv bílkoviny lidského či zvířecího původu během kultivace buněk, čištění či konečné úpravy přípravku.
Turoktokog alfa je rekombinantní humánní koagulační faktor VIII se zkrácenou B-doménou (B-doména obsahuje 21 aminokyselin z přirozené B-domény) bez jakékoliv další změny v pořadí aminokyselin.
Pomocná látka se známým účinkem:
0,31 mmol sodíku (7 mg) v 1 ml vzniklého roztoku
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Bílý nebo lehce nažloutlý prášek či drobivá hmota.
Čirý a bezbarvý injekční roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a prevence krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). NovoEight lze používat ve všech věkových skupinách.
4.2 Dávkování a způsob podání
Léčba by měla být zahájena pod dohledem lékaře se zkušenostmi s léčbou hemofilie.
Doposud neléčení pacienti
Bezpečnost a účinnost přípravku NovoEight u dosud neléčených pacientů nebyly doposud stanoveny. Nejsou dostupné žádné údaje.
Dávkování
Dávkování a trvání substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a pacientově klinickém stavu.
Počet podávaných jednotek faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které odpovídají běžnému standardu WHO pro přípravky obsahující faktor VIII. Aktivita faktoru VIII v plazmě je vyjádřena buď v procentech (relativně k normální hladině v lidské plazmě) nebo v mezinárodních jednotkách (relativně k mezinárodnímu standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba v případě potřeby
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že jedna mezinárodní jednotka (IU) faktoru VIII na kg tělesné hmotnosti zvýší aktivitu faktoru VIII v plazmě asi o 2 IU/dl. Požadovaná dávka se vypočte podle následujícího vzorce:
Potřebný počet jednotek = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) (IU/dl) x 0,5 (IU/kg na IU/dl).
Množství, které má být podáno, a frekvence podání by měly být vždy přizpůsobeny klinické účinnosti v individuálním případě.
V případě následujících krvácivých příhod by v odpovídajícím období aktivita faktoru VIII neměla klesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může být použita jako návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Tabulka 1 Návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Stupeň krvácení/ Požadovaná hladina Četnost dávek (hodiny)/délka
Typ chirurgického výkonu FVIII (%) (IU/dl) trvání léčby (dny)
Krvácení
Časný hemartros, krvácení do svalů 20-40 Opakovat každých 12-24 hodin.
nebo do dutiny ústní Nejméně 1 den, dokud nedojde k
zástavě krvácení, indikované skončením bolestí, nebo ke zhojení.
Rozsáhlejší hemartros, krvácení do 30-60 svalů nebo hematom
Život ohrožující krvácení 60-100
Opakovat infuzi každých 12-24 hodin po dobu 3-4 dnů nebo déle dokud bolest a akutní porucha funkce neustoupí
Opakovat infuze každých 8-24 hodin dokud nepomine ohrožení života
Chirurgické zákroky
Každých 24 hodin, nejméně 1 den, až je dosaženo zhojení.
Menší operace včetně vytržení 30-60
zubu
|
Stupeň krvácení/ Typ chirurgického výkonu |
Požadovaná hladina FVIII (%) (IU/dl) |
Četnost dávek (hodiny)/délka trvání léčby (dny) |
|
Velké chirurgické výkony |
80-100 (před a po operaci) |
Opakovat infuzi každých 8-24 hodin až do adekvátního zhojení poranění, pak pokračovat v léčbě nejméně dalších 7 dní k udržení aktivity faktoru VIII na 30-60 % (IU/dl) |
Profylaxe
K dlouhodobé profylaxi krvácení u pacientů se závažnou hemofilií A. Obvyklé doporučené dávky jsou 20-40 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 20-50 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. V některých případech, zvláště u mladších pacientů, může být nutné podávat přípravek v kratších intervalech nebo ve vyšších dávkách.
Monitorování léčby
V průběhu léčby je doporučeno provádět příslušné stanovení hladin faktoru VIII za účelem získání vodítka pro velikost podávané dávky i četnost opakovaných aplikací. Zvláště v případě závažných chirurgických zákroků je nezbytné přesné monitorování substituční léčby, prováděné pomocí koagulační analýzy (aktivity faktoru VIII v plazmě). U jednotlivých pacientů se může jejich odezva na faktor VIII lišit dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním odlišných poločasů.
Chirurgický zákrok
U pediatrických pacientů neexistují žádné zkušenosti s chirurgickými zákroky.
Starší pacienti
U pacientů ve věku > 65 let neexistují žádné zkušenosti.
Pediatrická populace
K dlouhodobé profylaxi krvácení u pacientů do 12 let se doporučují dávky 25 - 50 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 25 - 60 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. Pro pediatrické pacienty od 12 let jsou doporučené dávky shodné s doporučeními pro dospělé pacienty.
Způsob podání Intravenózní podání
Doporučená rychlost infuze je u přípravku NovoEight 1-2 ml/min. Rychlost by měla být stanovena tak, aby vyhovovala pacientovi.
Instrukce pro rekonstituci léčivého přípravku před podáním naleznete v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Známá alergická reakce na křeččí bílkoviny.
4.4 Zvláštní upozornění a opatření pro použití Hypersenzitivita
Při používání přípravku NovoEight se mohou vyskytnout hypersenzitivní reakce alergického typu. Přípravek obsahuje stopy křeččích bílkovin, jež mohou u některých pacientů vyvolat alergické reakce. Obj eví-li se příznaky hypersenzitivity, musí být pacienti poučeni o tom, aby okamžitě přerušili léčbu tímto léčivým přípravkem a kontaktovali svého lékaře. Pacienti musí být informováni o časných příznacích hypersenzitivních reakcí včetně kopřivky, generalizované kopřivky, tlaku na hrudi, sípotu, hypotenze a anafylaxe.
V případě šoku je nutno nasadit standardní lékařskou léčbu šokového stavu.
Inhibitory
Známou komplikací léčby u individuálních případů hemofilie A je vznik neutralizačních protilátek (inhibitorů) proti faktoru VIII. Tyto inhibitory jsou obvykle IgG imunoglobuliny působící proti koagulační aktivitě faktoru VIII. Jsou kvantitativně udávané v Bethesda jednotkách (BU) na jeden ml plazmy a zjišťované pomocí modifikovaného testu. Riziko vzniku inhibitorů je ve vztahu k expozici organizmu faktorem VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Vzácně může dojít k tvorbě inhibitorů teprve po prvních 100 dnech expozice.
Při přechodu z jednoho přípravku obsahujícího faktor VIII na jiný byl u pacientů léčených již dříve, kteří měli v anamnéze dřívější výskyt inhibitoru, pozorován opětovný výskyt inhibitoru (nízký titr), a to i po více než 100 dnech expozice. Proto je při jakémkoliv převodu na jiný přípravek doporučeno vždy pečlivě monitorovat všechny pacienty s ohledem na vznik inhibitoru.
Obecně všichni pacienti léčení přípravky obsahujícími koagulační faktor VIII by měli být pečlivě sledováni z hlediska vzniku inhibitorů vhodnými klinickými vyšetřeními a laboratorními testy. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII nebo pokud není dosaženo kontroly krvácení příslušnou dávkou, musí být provedeny testy na přítomnost inhibitoru. U pacientů s vysokými hladinami inhibitorů, může být léčba faktorem VIII neúčinná, a je třeba zvážit jiné léčebné možnosti. Léčba takovýchto pacientů by měla být prováděna lékařem se zkušeností v péči o pacienty s hemofilií a s inhibitory proti faktoru VIII.
Důrazně se doporučuje, aby vždy při každém podání přípravku NovoEight pacientovi byl zaznamenán název a číslo šarže přípravku, aby bylo možno přiřadit číslo šarže přípravku k pacientovi.
Pomocné látky, které je nutno vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,31 mmol sodíku (7 mg) na 1 ml vzniklého roztoku. Tento fakt je nutno vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
Komplikace spojené s použitím katetru
Pokud je požadováno použití centrálního žilního katetru (CVAD), je nutno zvážit riziko komplikací spojené s jeho použitím včetně lokálních infekcí, bakteriémie a trombózy v místě katetru.
Pediatrická populace
Uvedená varování a preventivní opatření platí pro dospělé i děti.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce S přípravkem NovoEight nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
S přípravkem NovoEight nebyly prováděny žádné reprodukční studie na zvířatech. Na základě vzácného výskytu hemofilie A u žen nejsou zkušenosti týkající se použití faktoru VIII během těhotenství a kojení k dispozici. Z toho důvodu může být faktor VIII během těhotenství a kojení použit pouze, pokud je to jednoznačně indikováno.
4.7 Účinky na schopnost řídit a obsluhovat stroje
NovoEight nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Vzácně byly pozorovány hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a štípání v místě infuze, třesavku, zrudnutí, generalizovanou kopřivku, bolest hlavy, vyrážku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tlak na prsou, brnění, zvracení, sípot) a mohou v některých případech vyústit v těžkou anafylaxi (včetně šoku).
Velmi vzácně byl pozorován vznik protilátek proti křeččím proteinům se spojenou hypersenzitivitou.
U pacientů s hemofilií A může dojít ke vzniku neutralizačních protilátek (inhibitory) proti faktoru VIII. Jestliže dojde ke vzniku těchto inhibitorů, projeví se to jako nedostačující klinická odpověď. V takových případech se doporučuje vyhledat specializované centrum pro léčbu hemofilie.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky uvedené v tabulce níže jsou klasifikovány dle Tříd orgánových systémů podle databáze MedDRA (TOS a preferované termíny četností).
Frekvence výskytu jsou definovány podle následující konvence: Velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 2 Frekvence nežádoucích účinků v klinických studiích
|
Třída orgánových systémů |
Frekvence* |
Nežádoucí účinek |
|
Psychiatrické poruchy |
Méně časté | |
|
Poruchy nervového systému |
Méně časté |
Bolest hlavy, závratě |
|
Srdeční poruchy |
Méně časté |
Sinusová tachykardie |
|
Cévní poruchy |
Méně časté |
Hypertenze, lymfoedém |
|
Poruchy jater a žlučových cest |
Časté |
Zvýšení jaterních enzymů** |
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Méně časté |
Muskuloskeletální ztuhlost, artropatie, bolesti končetin, muskuloskeletální bolest |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Reakce v místě vpichu*** |
|
Méně časté |
Únava, pocit horka, periferní edém, pyrexie | |
|
Vyšetření |
Méně časté |
Zrychlená srdeční frekvence |
|
Poranění, otravy a procedurální komplikace |
Méně časté |
Kontuze |
*
**
Přepočteno na základě celkového počtu jednotlivých pacientů ve všech klinických studiích (214)
Zvýšení jaterních enzymů se týká alanin aminotransferázy, aspartát aminotransferázy, gama-glutamyltransferázy a bilirubinu
***
Reakce v místě vpichu zahrnují erytém v místě vpichu, extravazáty v místě vpichu a svědění v místě vpichu
Popis vybraných nežádoucích účinků
V průběhu všech klinických studií s přípravkem NovoEight bylo celkem hlášeno 30 nežádoucích účinků u 19 z 214 pacientů léčených přípravkem NovoEight. Nejčastěji hlášenými nežádoucími účinky byly reakce v místě vpichu a zvýšení jaterních enzymů. Z 30 nežádoucích účinků byly 2 hlášeny u jednoho z 31 pacientů do 6 let, žádný u pacientů ve věku 6-18 let a 28 bylo hlášeno u 18 ze 127 dospělých.
Pediatrická populace
V klinických studiích se 63 pediatrickými pacienty v rozmezí 0 až 12 let a s 24 dospívajícími
v rozmezí 12-18 let, kteří trpěli závažnou hemofilií A, nebyl nalezen žádný rozdíl v bezpečnostním profilu přípravku NovoEight mezi pediatrickými pacienty a dospělými.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika, krevní koagulační faktor VIII, ATC kód: B02BD02 Mechanismus účinku
NovoEight obsahuje turoktokog alfa - humánní koagulační faktor VIII (rDNA) se zkrácenou B-doménou. Tento glykoprotein má shodnou strukturu s humánním faktorem VIII, když je aktivován. Posttranslační modifikace jsou podobné modifikacím u molekul odvozených z plazmy. Sulfatační místo tyrosinu, jež je přítomno na Tyr1680 (přirozená plná délka) a jež je důležité pro vazbu na von Willebrandův faktor, je v molekule turoktokogu alfa plně sulfonované. Po podání infuze pacientovi s hemofilií se faktor VIII v krevním oběhu váže na endogenní von Willebrandův faktor. Komplex faktoru VIII s von Willebrandovým faktorem je tvořen 2 molekulami (faktor VIII a von Willebrandův faktor) s odlišnými fyziologickými funkcemi. Aktivovaný faktor VIII působí jako kofaktor pro aktivaci faktoru IX urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin pak konvertuje fibrinogen na fibrin a umožní tak tvorbu sraženiny. Hemofilie je na pohlaví závisející dědičná porucha krevní srážlivosti, jejíž příčinou je snížená hladina faktoru VIII:C. Výsledkem je silné krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánní nebo jako důsledek úrazu nebo chirurgického zákroku. Při substituční léčbě se hladiny plazmatického faktoru VIII zvýší, tím dojde k dočasné úpravě deficitu faktoru a tím také k úpravě sklonu ke krvácení.
Klinická účinnost
Byly provedeny tři multicentrické, otevřené, nekontrolované studie za účelem vyhodnotit bezpečnost a účinnost přípravku NovoEight v prevenci a léčbě krvácení u již dříve léčených pacientů s těžkou hemofilií A (aktivita FVIII <1 %). Do studie bylo zahrnuto 213 léčených pacientů; 150 dospívajících či dospělých pacientů bez inhibitorů ve věku od 12 let (>150 dní léčby) a 63 pediatrických pacientů bez inhibitorů do 12 let (>50 dní léčby). 187 z 213 pacientů pokračovalo v prodloužené studii bezpečnosti. Léčba přípravkem NovoEight byla prokázána jako bezpečná a měla předpokládaný hemostatický a preventivní účinek. Během akumulované léčby více než 54 000 dní (odpovídající 342 pacientoroků) nebyl ve fázi 3a klinických studií u již dříve léčených pacientů pozorován rozvoj žádných inhibitorů proti faktoru VIII. Z 1 377 hlášených krvácivých příhod pozorovaných u 177 pacientů z 213, bylo 1 244 (90,3 %) krvácení zastaveno 1-2 infuzemi přípravku NovoEight.
Tabulka 3 Spotřeba turoktokogu alfa a celkový výskyt úspěšnosti
|
Mladší děti (0 - <6 let) |
Starší děti (6 - <12 let) |
Dospívající (12 -<18 let) |
Dospělí (>18 let) |
Celkem | |
|
Počet pacientů |
31 |
32 |
24 |
126 |
213 |
|
Dávka užitá k prevenci na pacienta (IU/kg TH) Průměr (SD) |
40,1 (8,5) |
36,6 (9,0) |
27,0 (7,6) |
26,9 (6,9) |
30,3 (9,2) |
|
Min ; Max |
26,5 ; 57,3 |
24,9 ; 57,9 |
20,5 ; 46,9 |
20,0 ; 50,8 |
20,0 ; 57,9 |
|
Dávka užitá k léčbě krvácení (IU/kg TH) Průměr (SD) Min ; Max |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Výskyt úspěšnosti* % |
92,9% |
88,9% |
79,7% |
85,6% |
85,9% |
TH: Tělesná hmotnost, SD: Směrodatná odchylka *Úspěšnost je definována buď jako „Výbomá“ nebo „Dobrá“.
Celkem bylo provedeno 14 chirurgických zákroků u celkem 14 pacientů, z nichž 13 bylo závažných, a jeden byl lehký. Hemostáza byla úspěšná ve všech případech a nebylo hlášeno žádné selhání léčby.
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s turoktokogem alfa byly prováděny u pacientů trpících závažnou hemofilií A (aktivita FVIII <1 %), kteří již byli dříve léčeni. Analýza vzorků plazmy byla prováděna jak pomocí jednostupňového koagulačního testu, tak chromogenním testem.
V mezinárodní studii zahrnující 36 laboratoří byla testována aktivita přípravku NovoEight pomocí testu FVIII:C a porovnávána s na trhu dostupným přípravkem obsahujícím rekombinantní FVIII o plné délce. Studie prokázala srovnatelné a stabilní výsledky pro oba přípravky a rovněž to, že přípravek NovoEight může být v plazmě spolehlivě měřen, aniž by bylo zapotřebí speciálního standardu pro NovoEight.
Farmakokinetické parametry po jednorázové dávce přípravku NovoEight jsou shrnuty v Tabulce 4 pro test srážlivosti, v Tabulce 5 pro chromogenní test.
Tabulka 4 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), test srážlivosti_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((IU*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
t* (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (IU/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Průměrná doba setrvání v oběhu (hod.) |
9,63 (2,50) |
9,91 (2,57) |
14,19 5,08) |
Tabulka 5 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), chromogenní test_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((IU*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (IU/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Průměrná doba setrvání v oběhu (hod.) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Farmakokinetické parametry u pediatrických pacientů do 6 let a pediatrických pacientů ve věku 612 let byly srovnatelné. Byly pozorovány některé odchylky ve farmakokinetických parametrech přípravku NovoEight mezi pediatrickými a dospělými pacienty. U pediatrických pacientů byly nalezeny vyšší hodnoty CL a kratší ť/2 ve srovnání s dospělými pacienty s hemofilií A, což může být částečně způsobeno známým vyšším plazmatickým objemem na kilogram tělesné hmotnosti u mladších pacientů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po opakovaném podávání neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Chlorid sodný Histidin Sacharosa Polysorbát 80 Methionin
Dihydrát chloridu vápenatého Hydroxid sodný Kyselina chlorovodíková
Rozpouštědlo Chlorid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Před otevřením:
30 měsíců
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (< 30°C) po jedno nepřetržité období nepřesahující 9 měsíců. Jakmile byl přípravek jednou vyjmut z chladničky, nesmí tam již být vrácen zpět. Poznačte si prosím na krabičce datum, kdy jste přípravek začali uchovávat při pokojové teplotě. Injekční lahvičku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem.
Po rekonstituci:
Chemická a fyzikální stabilita po rekonstituci byla prokázána na dobu 24 hodin při 2°C - 8°C a 4 hodiny při uchovávání při pokojové teplotě (< 30°C).
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, jsou doba a podmínky uchovávání přípravku po otevření před použitím v odpovědnosti uživatele. Normálně by tato doba neměla být delší než 4 hodiny při uchovávání při pokojové teplotě (< 30°C) nebo 24 hodin při 2°C - 8°C, pokud rekonstituce neproběhla za kontrolovaných a validovaných aseptických podmínek.
Veškerý nepoužitý léčivý přípravek, který byl uchováván při pokojové teplotě déle než 4 hodiny, musí být zlikvidován.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem.
Uchovávání tohoto léčivého přípravku při pokojové teplotě a podmínky uchovávání po jeho rekonstituci viz bod 6.3.
6.5 Druh obalu a obsah balení
Jedno balení přípravku NovoEight 1 000 IU prášek a rozpouštědlo pro injekční roztok obsahuje:
- 1 skleněnou injekční lahvičku (sklo typu I) s práškem opatřenou chlorobutylovou pryžovou zátkou
- 1 sterilní adaptér injekční lahvičky k rozpuštění
- 1 předplněnou injekční stříkačku obsahující 4 ml rozpouštědla s polypropylenovým uzávěrem zpětného chodu, bromobutylovým pryžovým pístem a uzávěrem s bromobutylovou zátkou.
- 1 nástavec pístu (zhotovený z polypropylenu)
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
NovoEight je určen k intravenóznímu podání po rekonstituci prášku v rozpouštědle dodávaném v injekční stříkačce. Po rekonstituci je roztok čirý či lehce opalescentní. Roztok nepoužívejte, pokud je zakalený či obsahuje usazeniny.
Budete také potřebovat infuzní soupravu (infuzní set a jehlu s křidélky), sterilní alkoholové tampony, gázové polštářky a náplasti. Tyto pomůcky nejsou součástí balení přípravku NovoEight.
Vždy dodržujte aseptickou techniku.
Rekonstituce
A)
Vyjměte injekční lahvičku, adaptér injekční lahvičky a předplněnou injekční stříkačku z krabičky. Nástavec pístu ponechte zatím v krabičce. Zahřejte injekční lahvičku a předplněnou injekční stříkačku na pokojovou teplotu. Můžete to udělat tak, že je podržíte v ruce, dokud nemají stejnou teplotu jako vaše dlaně. Jiné způsoby ohřátí injekční lahvičky a předplněné injekční stříkačky nepoužívejte.
B)

Odstraňte plastové víčko z injekční lahvičky. Pokud je víčko uvolněné nebo chybí, injekční lahvičku nepoužívejte. Pryžovou zátku injekční lahvičky očistěte sterilním alkoholovým tamponem a nechte ji před použitím několik sekund na vzduchu oschnout.
C)
Sejměte ochranný papír z adaptéru injekční lahvičky. Pokud ochranný papír není zcela zatavený nebo je protržený, adaptér injekční lahvičky nepoužívejte. Nevyjímejte adaptér injekční lahvičky prsty z ochranného víčka.
D)
Otočte ochranné víčko a nasaďte adaptér na injekční lahvičku. Jakmile jste adaptér na injekční lahvičku nasadili, již ho z ní neodstraňujte.
E)
Lehce stiskněte ochranné víčko mezi palcem a ukazováčkem, jak je patrné z obrázku. Sejměte ochranné víčko z adaptéru injekční lahvičky.
F)
Pevně uchopte nástavec pístu za širší konec a okamžitě ho našroubujte po směru hodinových ručiček na píst uvnitř předplněné injekční stříkačky, dokud nepocítíte odpor.
G)
Odstraňte ochranné víčko z předplněné injekční stříkačky ohnutím směrem dolů tak, aby se porušila perforace. Dbejte, abyste se nedotkli prsty hrotu injekční stříkačky pod jejím ochranným víčkem.
H)
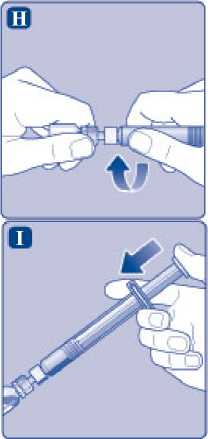
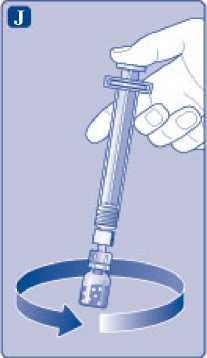
Našroubujte předplněnou injekční stříkačku bezpečně na adaptér injekční lahvičky, dokud nepocítíte odpor.
I)
Držte předplněnou injekční stříkačku lehce nakloněnou s injekční lahvičkou směřující dolů. Stisknutím nástavce pístu vstříkněte všechno rozpouštědlo do injekční lahvičky.
J)
Nechte nástavec pístu zcela stlačený a jemným kroužením injekční lahvičkou rozpusťte všechen prášek. Injekční lahvičkou netřepejte, mohlo by to způsobit napěnění.
Doporučuje se použít NovoEight okamžitě po rekonstituci. Podmínky uchovávání rekonstituovaného léčivého přípravku viz bod 6.3.
Pokud je zapotřebí větší dávky, opakujte kroky A až J s dalšími injekčními lahvičkami, adaptéry injekčních lahviček a předplněnými injekčními stříkačkami.
Aplikace rekonstituovaného roztoku
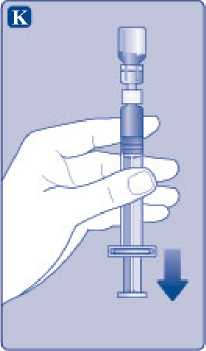
K)
Ponechte nástavec pístu zcela stlačený. Otočte injekční stříkačku tak, aby nasazená injekční lahvička byla dnem vzhůru. Uvolněte nástavec pístu a nechte ho samovolně vrátit se zpět. Tím se rekonstituovaný roztok natáhne do injekční stříkačky. Lehkým vytažením pístu směrem dolů pak zajistíte, že se do injekční stříkačky natáhne všechen roztok.
V případě, že potřebujete pouze část obsahu injekční lahvičky, použijte stupnici na injekční stříkačce, abyste si ověřili, že jste natáhli tolik vzniklého roztoku, kolik vám doporučil lékař či zdravotní sestra.
Stále držte injekční lahvičku dnem vzhůru a jemně poklepejte na injekční stříkačku, aby se vzduchové bubliny nashromáždily nahoře. Pomalu zatlačte na nástavec pístu, dokud všechny vzduchové bubliny neuniknou.
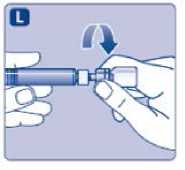
L)
Odšroubujte adaptér s injekční lahvičkou.
NovoEight je nyní připraven k aplikaci. Určete vhodné místo a pomalu aplikujte NovoEight do žíly v průběhu 2 až 5 minut.
Likvidace
Po aplikaci bezpečně zlikvidujte veškerý nepoužitý roztok přípravku NovoEight, injekční stříkačku s infuzní soupravou, injekční lahvičku s adaptérem a ostatní odpad dle doporučení lékárníka.
Nevhazujte do běžného domácího odpadu.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
8. REGISTRAČNÍ ČÍSLA
EU/1/13/888/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. listopad 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky (EMA) http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
NovoEight 1 500 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička s práškem obsahuje 1 500 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Po rozpuštění obsahuje přípravek NovoEight přibližně 375 IU/ml humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Účinnost (IU) se udává chromogenní metodou podle Evropského lékopisu. Specifická aktivita přípravku NovoEight je přibližně 8 300 IU/mg bílkoviny.
Turoktokog alfa (humánní koagulační faktor VIII (rDNA)) je čištěná bílkovina obsahující 1 445 aminokyselin s molekulovou hmotností asi 166 kDA. Je vyráběn rekombinantní DNA technologií z vaječníkových buněk čínského křečka (CHO buňky). Je vyroben bez přídavku jakékoliv bílkoviny lidského či zvířecího původu během kultivace buněk, čištění či konečné úpravy přípravku.
Turoktokog alfa je rekombinantní humánní koagulační faktor VIII se zkrácenou B-doménou (B-doména obsahuje 21 aminokyselin z přirozené B-domény) bez jakékoliv další změny v pořadí aminokyselin.
Pomocná látka se známým účinkem:
0,31 mmol sodíku (7 mg) v 1 ml vzniklého roztoku
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Bílý nebo lehce nažloutlý prášek či drobivá hmota.
Čirý a bezbarvý injekční roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a prevence krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). NovoEight lze používat ve všech věkových skupinách.
4.2 Dávkování a způsob podání
Léčba by měla být zahájena pod dohledem lékaře se zkušenostmi s léčbou hemofilie.
Doposud neléčení pacienti
Bezpečnost a účinnost přípravku NovoEight u dosud neléčených pacientů nebyly doposud stanoveny. Nejsou dostupné žádné údaje.
Dávkování
Dávkování a trvání substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a pacientově klinickém stavu.
Počet podávaných jednotek faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které odpovídají běžnému standardu WHO pro přípravky obsahující faktor VIII. Aktivita faktoru VIII v plazmě je vyjádřena buď v procentech (relativně k normální hladině v lidské plazmě) nebo v mezinárodních jednotkách (relativně k mezinárodnímu standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba v případě potřeby
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že jedna mezinárodní jednotka (IU) faktoru VIII na kg tělesné hmotnosti zvýší aktivitu faktoru VIII v plazmě asi o 2 IU/dl. Požadovaná dávka se vypočte podle následujícího vzorce:
Potřebný počet jednotek = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) (IU/dl) x 0,5 (IU/kg na IU/dl).
Množství, které má být podáno, a frekvence podání by měly být vždy přizpůsobeny klinické účinnosti v individuálním případě.
V případě následujících krvácivých příhod by v odpovídajícím období aktivita faktoru VIII neměla klesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může být použita jako návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Tabulka 1 Návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Stupeň krvácení/ Požadovaná hladina Četnost dávek (hodiny)/délka
Typ chirurgického výkonu FVIII (%) (IU/dl) trvání léčby (dny)
Krvácení
Časný hemartros, krvácení do svalů 20-40 Opakovat každých 12-24 hodin.
nebo do dutiny ústní Nejméně 1 den, dokud nedojde k
zástavě krvácení, indikované skončením bolestí, nebo ke zhojení.
Rozsáhlejší hemartros, krvácení do 30-60 svalů nebo hematom
Život ohrožující krvácení 60-100
Opakovat infuzi každých 12-24 hodin po dobu 3-4 dnů nebo déle dokud bolest a akutní porucha funkce neustoupí
Opakovat infuze každých 8-24 hodin dokud nepomine ohrožení života
Chirurgické zákroky
Každých 24 hodin, nejméně 1 den, až je dosaženo zhojení.
Menší operace včetně vytržení 30-60
zubu
|
Stupeň krvácení/ Typ chirurgického výkonu |
Požadovaná hladina FVIII (%) (IU/dl) |
Četnost dávek (hodiny)/délka trvání léčby (dny) |
|
Velké chirurgické výkony |
80-100 (před a po operaci) |
Opakovat infuzi každých 8-24 hodin až do adekvátního zhojení poranění, pak pokračovat v léčbě nejméně dalších 7 dní k udržení aktivity faktoru VIII na 30-60 % (IU/dl) |
Profylaxe
K dlouhodobé profylaxi krvácení u pacientů se závažnou hemofilií A. Obvyklé doporučené dávky jsou 20-40 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 20-50 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. V některých případech, zvláště u mladších pacientů, může být nutné podávat přípravek v kratších intervalech nebo ve vyšších dávkách.
Monitorování léčby
V průběhu léčby je doporučeno provádět příslušné stanovení hladin faktoru VIII za účelem získání vodítka pro velikost podávané dávky i četnost opakovaných aplikací. Zvláště v případě závažných chirurgických zákroků je nezbytné přesné monitorování substituční léčby, prováděné pomocí koagulační analýzy (aktivity faktoru VIII v plazmě). U jednotlivých pacientů se může jejich odezva na faktor VIII lišit dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním odlišných poločasů.
Chirurgický zákrok
U pediatrických pacientů neexistují žádné zkušenosti s chirurgickými zákroky.
Starší pacienti
U pacientů ve věku > 65 let neexistují žádné zkušenosti.
Pediatrická populace
K dlouhodobé profylaxi krvácení u pacientů do 12 let se doporučují dávky 25 - 50 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 25 - 60 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. Pro pediatrické pacienty od 12 let jsou doporučené dávky shodné s doporučeními pro dospělé pacienty.
Způsob podání Intravenózní podání
Doporučená rychlost infuze je u přípravku NovoEight 1-2 ml/min. Rychlost by měla být stanovena tak, aby vyhovovala pacientovi.
Instrukce pro rekonstituci léčivého přípravku před podáním naleznete v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Známá alergická reakce na křeččí bílkoviny.
4.4 Zvláštní upozornění a opatření pro použití Hypersenzitivita
Při používání přípravku NovoEight se mohou vyskytnout hypersenzitivní reakce alergického typu. Přípravek obsahuje stopy křeččích bílkovin, jež mohou u některých pacientů vyvolat alergické reakce. Obj eví-li se příznaky hypersenzitivity, musí být pacienti poučeni o tom, aby okamžitě přerušili léčbu tímto léčivým přípravkem a kontaktovali svého lékaře. Pacienti musí být informováni o časných příznacích hypersenzitivních reakcí včetně kopřivky, generalizované kopřivky, tlaku na hrudi, sípotu, hypotenze a anafylaxe.
V případě šoku je nutno nasadit standardní lékařskou léčbu šokového stavu.
Inhibitory
Známou komplikací léčby u individuálních případů hemofilie A je vznik neutralizačních protilátek (inhibitorů) proti faktoru VIII. Tyto inhibitory jsou obvykle IgG imunoglobuliny působící proti koagulační aktivitě faktoru VIII. Jsou kvantitativně udávané v Bethesda jednotkách (BU) na jeden ml plazmy a zjišťované pomocí modifikovaného testu. Riziko vzniku inhibitorů je ve vztahu k expozici organizmu faktorem VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Vzácně může dojít k tvorbě inhibitorů teprve po prvních 100 dnech expozice.
Při přechodu z jednoho přípravku obsahujícího faktor VIII na jiný byl u pacientů léčených již dříve, kteří měli v anamnéze dřívější výskyt inhibitoru, pozorován opětovný výskyt inhibitoru (nízký titr), a to i po více než 100 dnech expozice. Proto je při jakémkoliv převodu na jiný přípravek doporučeno vždy pečlivě monitorovat všechny pacienty s ohledem na vznik inhibitoru.
Obecně všichni pacienti léčení přípravky obsahujícími koagulační faktor VIII by měli být pečlivě sledováni z hlediska vzniku inhibitorů vhodnými klinickými vyšetřeními a laboratorními testy. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII nebo pokud není dosaženo kontroly krvácení příslušnou dávkou, musí být provedeny testy na přítomnost inhibitoru. U pacientů s vysokými hladinami inhibitorů, může být léčba faktorem VIII neúčinná, a je třeba zvážit jiné léčebné možnosti. Léčba takovýchto pacientů by měla být prováděna lékařem se zkušeností v péči o pacienty s hemofilií a s inhibitory proti faktoru VIII.
Důrazně se doporučuje, aby vždy při každém podání přípravku NovoEight pacientovi byl zaznamenán název a číslo šarže přípravku, aby bylo možno přiřadit číslo šarže přípravku k pacientovi.
Pomocné látky, které je nutno vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,31 mmol sodíku (7 mg) na 1 ml vzniklého roztoku. Tento fakt je nutno vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
Komplikace spojené s použitím katetru
Pokud je požadováno použití centrálního žilního katetru (CVAD), je nutno zvážit riziko komplikací spojené s jeho použitím včetně lokálních infekcí, bakteriémie a trombózy v místě katetru.
Pediatrická populace
Uvedená varování a preventivní opatření platí pro dospělé i děti.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce S přípravkem NovoEight nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
S přípravkem NovoEight nebyly prováděny žádné reprodukční studie na zvířatech. Na základě vzácného výskytu hemofilie A u žen nejsou zkušenosti týkající se použití faktoru VIII během těhotenství a kojení k dispozici. Z toho důvodu může být faktor VIII během těhotenství a kojení použit pouze, pokud je to jednoznačně indikováno.
4.7 Účinky na schopnost řídit a obsluhovat stroje
NovoEight nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Vzácně byly pozorovány hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a štípání v místě infuze, třesavku, zrudnutí, generalizovanou kopřivku, bolest hlavy, vyrážku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tlak na prsou, brnění, zvracení, sípot) a mohou v některých případech vyústit v těžkou anafylaxi (včetně šoku).
Velmi vzácně byl pozorován vznik protilátek proti křeččím proteinům se spojenou hypersenzitivitou.
U pacientů s hemofilií A může dojít ke vzniku neutralizačních protilátek (inhibitory) proti faktoru VIII. Jestliže dojde ke vzniku těchto inhibitorů, projeví se to jako nedostačující klinická odpověď. V takových případech se doporučuje vyhledat specializované centrum pro léčbu hemofilie.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky uvedené v tabulce níže jsou klasifikovány dle Tříd orgánových systémů podle databáze MedDRA (TOS a preferované termíny četností).
Frekvence výskytu jsou definovány podle následující konvence: Velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 2 Frekvence nežádoucích účinků v klinických studiích
|
Třída orgánových systémů |
Frekvence* |
Nežádoucí účinek |
|
Psychiatrické poruchy |
Méně časté | |
|
Poruchy nervového systému |
Méně časté |
Bolest hlavy, závratě |
|
Srdeční poruchy |
Méně časté |
Sinusová tachykardie |
|
Cévní poruchy |
Méně časté |
Hypertenze, lymfoedém |
|
Poruchy jater a žlučových cest |
Časté |
Zvýšení jaterních enzymů* * |
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Méně časté |
Muskuloskeletální ztuhlost, artropatie, bolesti končetin, muskuloskeletální bolest |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Reakce v místě vpichu* * * |
|
Méně časté |
Únava, pocit horka, periferní edém, pyrexie | |
|
Vyšetření |
Méně časté |
Zrychlená srdeční frekvence |
|
Poranění, otravy a procedurální komplikace |
Méně časté |
Kontuze |
*
**
Přepočteno na základě celkového počtu jednotlivých pacientů ve všech klinických studiích (214)
Zvýšení jaterních enzymů se týká alanin aminotransferázy, aspartát aminotransferázy, gama-glutamyltransferázy a bilirubinu
***
Reakce v místě vpichu zahrnují erytém v místě vpichu, extravazáty v místě vpichu a svědění v místě vpichu
Popis vybraných nežádoucích účinků
V průběhu všech klinických studií s přípravkem NovoEight bylo celkem hlášeno 30 nežádoucích účinků u 19 z 214 pacientů léčených přípravkem NovoEight. Nejčastěji hlášenými nežádoucími účinky byly reakce v místě vpichu a zvýšení jaterních enzymů. Z 30 nežádoucích účinků byly 2 hlášeny u jednoho z 31 pacientů do 6 let, žádný u pacientů ve věku 6-18 let a 28 bylo hlášeno u 18 ze 127 dospělých.
Pediatrická populace
V klinických studiích se 63 pediatrickými pacienty v rozmezí 0 až 12 let a s 24 dospívajícími
v rozmezí 12-18 let, kteří trpěli závažnou hemofilií A, nebyl nalezen žádný rozdíl v bezpečnostním profilu přípravku NovoEight mezi pediatrickými pacienty a dospělými.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika, krevní koagulační faktor VIII, ATC kód: B02BD02 Mechanismus účinku
NovoEight obsahuje turoktokog alfa - humánní koagulační faktor VIII (rDNA) se zkrácenou B-doménou. Tento glykoprotein má shodnou strukturu s humánním faktorem VIII, když je aktivován. Posttranslační modifikace jsou podobné modifikacím u molekul odvozených z plazmy. Sulfatační místo tyrosinu, jež je přítomno na Tyr1680 (přirozená plná délka) a jež je důležité pro vazbu na von Willebrandův faktor, je v molekule turoktokogu alfa plně sulfonované. Po podání infuze pacientovi s hemofilií se faktor VIII v krevním oběhu váže na endogenní von Willebrandův faktor. Komplex faktoru VIII s von Willebrandovým faktorem je tvořen 2 molekulami (faktor VIII a von Willebrandův faktor) s odlišnými fyziologickými funkcemi. Aktivovaný faktor VIII působí jako kofaktor pro aktivaci faktoru IX urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin pak konvertuje fibrinogen na fibrin a umožní tak tvorbu sraženiny. Hemofilie je na pohlaví závisející dědičná porucha krevní srážlivosti, jejíž příčinou je snížená hladina faktoru VIII:C. Výsledkem je silné krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánní nebo jako důsledek úrazu nebo chirurgického zákroku. Při substituční léčbě se hladiny plazmatického faktoru VIII zvýší, tím dojde k dočasné úpravě deficitu faktoru a tím také k úpravě sklonu ke krvácení.
Klinická účinnost
Byly provedeny tři multicentrické, otevřené, nekontrolované studie za účelem vyhodnotit bezpečnost a účinnost přípravku NovoEight v prevenci a léčbě krvácení u již dříve léčených pacientů s těžkou hemofilií A (aktivita FVIII <1 %). Do studie bylo zahrnuto 213 léčených pacientů; 150 dospívajících či dospělých pacientů bez inhibitorů ve věku od 12 let (>150 dní léčby) a 63 pediatrických pacientů bez inhibitorů do 12 let (>50 dní léčby). 187 z 213 pacientů pokračovalo v prodloužené studii bezpečnosti. Léčba přípravkem NovoEight byla prokázána jako bezpečná a měla předpokládaný hemostatický a preventivní účinek. Během akumulované léčby více než 54 000 dní (odpovídající 342 pacientoroků) nebyl ve fázi 3 a klinických studií u již dříve léčených pacientů pozorován rozvoj žádných inhibitorů proti faktoru VIII. Z 1 377 hlášených krvácivých příhod pozorovaných u 177 pacientů z 213, bylo 1 244 (90,3 %) krvácení zastaveno 1-2 infuzemi přípravku NovoEight.
Tabulka 3 Spotřeba turoktokogu alfa a celkový výskyt úspěšnosti
|
Mladší děti (0 - <6 let) |
Starší děti (6 - <12 let) |
Dospívající (12 -<18 let) |
Dospělí (>18 let) |
Celkem | |
|
Počet pacientů |
31 |
32 |
24 |
126 |
213 |
|
Dávka užitá k prevenci na pacienta (IU/kg TH) Průměr (SD) |
40,1 (8,5) |
36,6 (9,0) |
27,0 (7,6) |
26,9 (6,9) |
30,3 (9,2) |
|
Min ; Max |
26,5 ; 57,3 |
24,9 ; 57,9 |
20,5 ; 46,9 |
20,0 ; 50,8 |
20,0 ; 57,9 |
|
Dávka užitá k léčbě krvácení (IU/kg TH) Průměr (SD) Min ; Max |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Výskyt úspěšnosti* % |
92,9% |
88,9% |
79,7% |
85,6% |
85,9% |
TH: Tělesná hmotnost, SD: Směrodatná odchylka *Úspěšnost je definována buď jako „Výbomá“ nebo „Dobrá“.
Celkem bylo provedeno 14 chirurgických zákroků u celkem 14 pacientů, z nichž 13 bylo závažných, a jeden byl lehký. Hemostáza byla úspěšná ve všech případech a nebylo hlášeno žádné selhání léčby.
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s turoktokogem alfa byly prováděny u pacientů trpících závažnou hemofilií A (aktivita FVIII <1 %), kteří již byli dříve léčeni. Analýza vzorků plazmy byla prováděna jak pomocí jednostupňového koagulačního testu, tak chromogenním testem.
V mezinárodní studii zahrnující 36 laboratoří byla testována aktivita přípravku NovoEight pomocí testu FVIII:C a porovnávána s na trhu dostupným přípravkem obsahujícím rekombinantní FVIII o plné délce. Studie prokázala srovnatelné a stabilní výsledky pro oba přípravky a rovněž to, že přípravek NovoEight může být v plazmě spolehlivě měřen, aniž by bylo zapotřebí speciálního standardu pro NovoEight.
Farmakokinetické parametry po jednorázové dávce přípravku NovoEight jsou shrnuty v Tabulce 4 pro test srážlivosti, v Tabulce 5 pro chromogenní test.
Tabulka 4 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), test srážlivosti_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((IU*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
V (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (IU/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Průměrná doba setrvání v oběhu (hod.) |
9,63 (2,50) |
9,91 (2,57) |
14,19 5,08) |
Tabulka 5 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), chromogenní test_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((IU*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (IU/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Průměrná doba setrvání v oběhu (hod.) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Farmakokinetické parametry u pediatrických pacientů do 6 let a pediatrických pacientů ve věku 612 let byly srovnatelné. Byly pozorovány některé odchylky ve farmakokinetických parametrech přípravku NovoEight mezi pediatrickými a dospělými pacienty. U pediatrických pacientů byly nalezeny vyšší hodnoty CL a kratší ť/2 ve srovnání s dospělými pacienty s hemofilií A, což může být částečně způsobeno známým vyšším plazmatickým objemem na kilogram tělesné hmotnosti u mladších pacientů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po opakovaném podávání neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Chlorid sodný Histidin Sacharosa Polysorbát 80 Methionin
Dihydrát chloridu vápenatého Hydroxid sodný Kyselina chlorovodíková
Rozpouštědlo Chlorid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Před otevřením:
30 měsíců
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (< 30°C) po jedno nepřetržité období nepřesahující 9 měsíců. Jakmile byl přípravek jednou vyjmut z chladničky, nesmí tam již být vrácen zpět. Poznačte si prosím na krabičce datum, kdy jste přípravek začali uchovávat při pokojové teplotě. Injekční lahvičku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem.
Po rekonstituci:
Chemická a fyzikální stabilita po rekonstituci byla prokázána na dobu 24 hodin při 2°C - 8°C a 4 hodiny při uchovávání při pokojové teplotě (< 30°C).
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, jsou doba a podmínky uchovávání přípravku po otevření před použitím v odpovědnosti uživatele. Normálně by tato doba neměla být delší než 4 hodiny při uchovávání při pokojové teplotě (< 30°C) nebo 24 hodin při 2°C - 8°C, pokud rekonstituce neproběhla za kontrolovaných a validovaných aseptických podmínek.
Veškerý nepoužitý léčivý přípravek, který byl uchováván při pokojové teplotě déle než 4 hodiny, musí být zlikvidován.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem.
Uchovávání tohoto léčivého přípravku při pokojové teplotě a podmínky uchovávání po jeho rekonstituci viz bod 6.3.
6.5 Druh obalu a obsah balení
Jedno balení přípravku NovoEight 1 500 IU prášek a rozpouštědlo pro injekční roztok obsahuje:
- 1 skleněnou injekční lahvičku (sklo typu I) s práškem opatřenou chlorobutylovou pryžovou zátkou
- 1 sterilní adaptér injekční lahvičky k rozpuštění
- 1 předplněnou injekční stříkačku obsahující 4 ml rozpouštědla s polypropylenovým uzávěrem zpětného chodu, bromobutylovým pryžovým pístem a uzávěrem s bromobutylovou zátkou.
- 1 nástavec pístu (zhotovený z polypropylenu)
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
NovoEight je určen k intravenóznímu podání po rekonstituci prášku v rozpouštědle dodávaném v injekční stříkačce. Po rekonstituci je roztok čirý či lehce opalescentní. Roztok nepoužívejte, pokud je zakalený či obsahuje usazeniny.
Budete také potřebovat infuzní soupravu (infuzní set a jehlu s křidélky), sterilní alkoholové tampony, gázové polštářky a náplasti. Tyto pomůcky nejsou součástí balení přípravku NovoEight.
Vždy dodržujte aseptickou techniku.
Rekonstituce
A)
Vyjměte injekční lahvičku, adaptér injekční lahvičky a předplněnou injekční stříkačku z krabičky. Nástavec pístu ponechte zatím v krabičce. Zahřejte injekční lahvičku a předplněnou injekční stříkačku na pokojovou teplotu. Můžete to udělat tak, že je podržíte v ruce, dokud nemají stejnou teplotu jako vaše dlaně. Jiné způsoby ohřátí injekční lahvičky a předplněné injekční stříkačky nepoužívejte.
B)

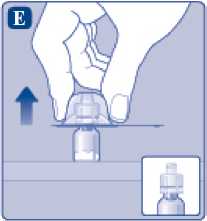
Odstraňte plastové víčko z injekční lahvičky. Pokud je víčko uvolněné nebo chybí, injekční lahvičku nepoužívejte. Pryžovou zátku injekční lahvičky očistěte sterilním alkoholovým tamponem a nechte ji před použitím několik sekund na vzduchu oschnout.
C)
Sejměte ochranný papír z adaptéru injekční lahvičky. Pokud ochranný papír není zcela zatavený nebo je protržený, adaptér injekční lahvičky nepoužívejte. Nevyjímejte adaptér injekční lahvičky prsty z ochranného víčka.
D)
Otočte ochranné víčko a nasaďte adaptér na injekční lahvičku. Jakmile jste adaptér na injekční lahvičku nasadili, již ho z ní neodstraňujte.
E)
Lehce stiskněte ochranné víčko mezi palcem a ukazováčkem, jak je patrné z obrázku. Sejměte ochranné víčko z adaptéru injekční lahvičky.
F)
Pevně uchopte nástavec pístu za širší konec a okamžitě ho našroubujte po směru hodinových ručiček na píst uvnitř předplněné injekční stříkačky, dokud nepocítíte odpor.
G)
Odstraňte ochranné víčko z předplněné injekční stříkačky ohnutím směrem dolů tak, aby se porušila perforace. Dbejte, abyste se nedotkli prsty hrotu injekční stříkačky pod jejím ochranným víčkem.
H)
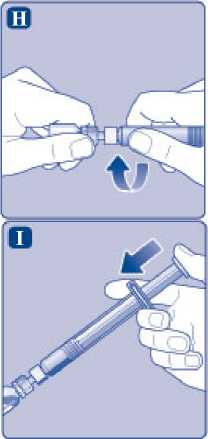
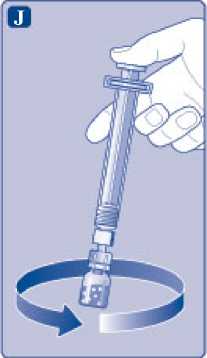
Našroubujte předplněnou injekční stříkačku bezpečně na adaptér injekční lahvičky, dokud nepocítíte odpor.
I)
Držte předplněnou injekční stříkačku lehce nakloněnou s injekční lahvičkou směřující dolů. Stisknutím nástavce pístu vstříkněte všechno rozpouštědlo do injekční lahvičky.
J)
Nechte nástavec pístu zcela stlačený a jemným kroužením injekční lahvičkou rozpusťte všechen prášek. Injekční lahvičkou netřepejte, mohlo by to způsobit napěnění.
Doporučuje se použít NovoEight okamžitě po rekonstituci. Podmínky uchovávání rekonstituovaného léčivého přípravku viz bod 6.3.
Pokud je zapotřebí větší dávky, opakujte kroky A až J s dalšími injekčními lahvičkami, adaptéry injekčních lahviček a předplněnými injekčními stříkačkami.
Aplikace rekonstituovaného roztoku
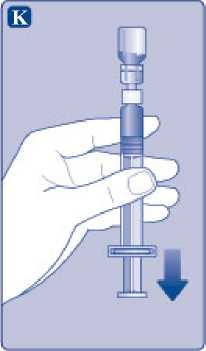
K)
Ponechte nástavec pístu zcela stlačený. Otočte injekční stříkačku tak, aby nasazená injekční lahvička byla dnem vzhůru. Uvolněte nástavec pístu a nechte ho samovolně vrátit se zpět. Tím se rekonstituovaný roztok natáhne do injekční stříkačky. Lehkým vytažením pístu směrem dolů pak zajistíte, že se do injekční stříkačky natáhne všechen roztok.
V případě, že potřebujete pouze část obsahu injekční lahvičky, použijte stupnici na injekční stříkačce, abyste si ověřili, že jste natáhli tolik vzniklého roztoku, kolik vám doporučil lékař či zdravotní sestra.
Stále držte injekční lahvičku dnem vzhůru a jemně poklepejte na injekční stříkačku, aby se vzduchové bubliny nashromáždily nahoře. Pomalu zatlačte na nástavec pístu, dokud všechny vzduchové bubliny neuniknou.
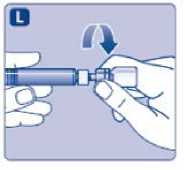
L)
Odšroubujte adaptér s injekční lahvičkou.
NovoEight je nyní připraven k aplikaci. Určete vhodné místo a pomalu aplikujte NovoEight do žíly v průběhu 2 až 5 minut.
Likvidace
Po aplikaci bezpečně zlikvidujte veškerý nepoužitý roztok přípravku NovoEight, injekční stříkačku s infuzní soupravou, injekční lahvičku s adaptérem a ostatní odpad dle doporučení lékárníka.
Nevhazujte do běžného domácího odpadu.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
8. REGISTRAČNÍ ČÍSLA
EU/1/13/888/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. listopad 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky (EMA) http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
NovoEight 2 000 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička s práškem obsahuje 2 000 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Po rozpuštění obsahuje přípravek NovoEight přibližně 500 IU/ml humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Účinnost (IU) se udává chromogenní metodou podle Evropského lékopisu. Specifická aktivita přípravku NovoEight je přibližně 8 300 IU/mg bílkoviny.
Turoktokog alfa (humánní koagulační faktor VIII (rDNA)) je čištěná bílkovina obsahující 1 445 aminokyselin s molekulovou hmotností asi 166 kDA. Je vyráběn rekombinantní DNA technologií z vaječníkových buněk čínského křečka (CHO buňky). Je vyroben bez přídavku jakékoliv bílkoviny lidského či zvířecího původu během kultivace buněk, čištění či konečné úpravy přípravku.
Turoktokog alfa je rekombinantní humánní koagulační faktor VIII se zkrácenou B-doménou (B-doména obsahuje 21 aminokyselin z přirozené B-domény) bez jakékoliv další změny v pořadí aminokyselin.
Pomocná látka se známým účinkem:
0,31 mmol sodíku (7 mg) v 1 ml vzniklého roztoku
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Bílý nebo lehce nažloutlý prášek či drobivá hmota.
Čirý a bezbarvý injekční roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a prevence krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). NovoEight lze používat ve všech věkových skupinách.
4.2 Dávkování a způsob podání
Léčba by měla být zahájena pod dohledem lékaře se zkušenostmi s léčbou hemofilie.
Doposud neléčení pacienti
Bezpečnost a účinnost přípravku NovoEight u dosud neléčených pacientů nebyly doposud stanoveny. Nejsou dostupné žádné údaje.
Dávkování
Dávkování a trvání substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a pacientově klinickém stavu.
Počet podávaných jednotek faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které odpovídají běžnému standardu WHO pro přípravky obsahující faktor VIII. Aktivita faktoru VIII v plazmě je vyjádřena buď v procentech (relativně k normální hladině v lidské plazmě) nebo v mezinárodních jednotkách (relativně k mezinárodnímu standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba v případě potřeby
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že jedna mezinárodní jednotka (IU) faktoru VIII na kg tělesné hmotnosti zvýší aktivitu faktoru VIII v plazmě asi o 2 IU/dl. Požadovaná dávka se vypočte podle následujícího vzorce:
Potřebný počet jednotek = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) (IU/dl) x 0,5 (IU/kg na IU/dl).
Množství, které má být podáno, a frekvence podání by měly být vždy přizpůsobeny klinické účinnosti v individuálním případě.
V případě následujících krvácivých příhod by v odpovídajícím období aktivita faktoru VIII neměla klesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může být použita jako návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Tabulka 1 Návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Stupeň krvácení/ Požadovaná hladina Četnost dávek (hodiny)/délka
Typ chirurgického výkonu FVIII (%) (IU/dl) trvání léčby (dny)
Krvácení
Časný hemartros, krvácení do svalů 20-40 Opakovat každých 12-24 hodin.
nebo do dutiny ústní Nejméně 1 den, dokud nedojde k
zástavě krvácení, indikované skončením bolestí, nebo ke zhojení.
Rozsáhlejší hemartros, krvácení do 30-60 svalů nebo hematom
Život ohrožující krvácení 60-100
Opakovat infuzi každých 12-24 hodin po dobu 3-4 dnů nebo déle dokud bolest a akutní porucha funkce neustoupí
Opakovat infuze každých 8-24 hodin dokud nepomine ohrožení života
Chirurgické zákroky
Každých 24 hodin, nejméně 1 den, až je dosaženo zhojení.
Menší operace včetně vytržení 30-60
zubu
|
Stupeň krvácení/ Typ chirurgického výkonu |
Požadovaná hladina FVIII (%) (IU/dl) |
Četnost dávek (hodiny)/délka trvání léčby (dny) |
|
Velké chirurgické výkony |
80-100 (před a po operaci) |
Opakovat infuzi každých 8-24 hodin až do adekvátního zhojení poranění, pak pokračovat v léčbě nejméně dalších 7 dní k udržení aktivity faktoru VIII na 30-60 % (IU/dl) |
Profylaxe
K dlouhodobé profylaxi krvácení u pacientů se závažnou hemofilií A. Obvyklé doporučené dávky jsou 20-40 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 20-50 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. V některých případech, zvláště u mladších pacientů, může být nutné podávat přípravek v kratších intervalech nebo ve vyšších dávkách.
Monitorování léčby
V průběhu léčby je doporučeno provádět příslušné stanovení hladin faktoru VIII za účelem získání vodítka pro velikost podávané dávky i četnost opakovaných aplikací. Zvláště v případě závažných chirurgických zákroků je nezbytné přesné monitorování substituční léčby, prováděné pomocí koagulační analýzy (aktivity faktoru VIII v plazmě). U jednotlivých pacientů se může jejich odezva na faktor VIII lišit dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním odlišných poločasů.
Chirurgický zákrok
U pediatrických pacientů neexistují žádné zkušenosti s chirurgickými zákroky.
Starší pacienti
U pacientů ve věku > 65 let neexistují žádné zkušenosti.
Pediatrická populace
K dlouhodobé profylaxi krvácení u pacientů do 12 let se doporučují dávky 25 - 50 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 25 - 60 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. Pro pediatrické pacienty od 12 let jsou doporučené dávky shodné s doporučeními pro dospělé pacienty.
Způsob podání Intravenózní podání
Doporučená rychlost infuze je u přípravku NovoEight 1-2 ml/min. Rychlost by měla být stanovena tak, aby vyhovovala pacientovi.
Instrukce pro rekonstituci léčivého přípravku před podáním naleznete v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Známá alergická reakce na křeččí bílkoviny.
4.4 Zvláštní upozornění a opatření pro použití Hypersenzitivita
Při používání přípravku NovoEight se mohou vyskytnout hypersenzitivní reakce alergického typu. Přípravek obsahuje stopy křeččích bílkovin, jež mohou u některých pacientů vyvolat alergické reakce. Obj eví-li se příznaky hypersenzitivity, musí být pacienti poučeni o tom, aby okamžitě přerušili léčbu tímto léčivým přípravkem a kontaktovali svého lékaře. Pacienti musí být informováni o časných příznacích hypersenzitivních reakcí včetně kopřivky, generalizované kopřivky, tlaku na hrudi, sípotu, hypotenze a anafylaxe.
V případě šoku je nutno nasadit standardní lékařskou léčbu šokového stavu.
Inhibitory
Známou komplikací léčby u individuálních případů hemofilie A je vznik neutralizačních protilátek (inhibitorů) proti faktoru VIII. Tyto inhibitory jsou obvykle IgG imunoglobuliny působící proti koagulační aktivitě faktoru VIII. Jsou kvantitativně udávané v Bethesda jednotkách (BU) na jeden ml plazmy a zjišťované pomocí modifikovaného testu. Riziko vzniku inhibitorů je ve vztahu k expozici organizmu faktorem VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Vzácně může dojít k tvorbě inhibitorů teprve po prvních 100 dnech expozice.
Při přechodu z jednoho přípravku obsahujícího faktor VIII na jiný byl u pacientů léčených již dříve, kteří měli v anamnéze dřívější výskyt inhibitoru, pozorován opětovný výskyt inhibitoru (nízký titr), a to i po více než 100 dnech expozice. Proto je při jakémkoliv převodu na jiný přípravek doporučeno vždy pečlivě monitorovat všechny pacienty s ohledem na vznik inhibitoru.
Obecně všichni pacienti léčení přípravky obsahujícími koagulační faktor VIII by měli být pečlivě sledováni z hlediska vzniku inhibitorů vhodnými klinickými vyšetřeními a laboratorními testy. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII nebo pokud není dosaženo kontroly krvácení příslušnou dávkou, musí být provedeny testy na přítomnost inhibitoru. U pacientů s vysokými hladinami inhibitorů, může být léčba faktorem VIII neúčinná, a je třeba zvážit jiné léčebné možnosti. Léčba takovýchto pacientů by měla být prováděna lékařem se zkušeností v péči o pacienty s hemofilií a s inhibitory proti faktoru VIII.
Důrazně se doporučuje, aby vždy při každém podání přípravku NovoEight pacientovi byl zaznamenán název a číslo šarže přípravku, aby bylo možno přiřadit číslo šarže přípravku k pacientovi.
Pomocné látky, které je nutno vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,31 mmol sodíku (7 mg) na 1 ml vzniklého roztoku. Tento fakt je nutno vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
Komplikace spojené s použitím katetru
Pokud je požadováno použití centrálního žilního katetru (CVAD), je nutno zvážit riziko komplikací spojené s jeho použitím včetně lokálních infekcí, bakteriémie a trombózy v místě katetru.
Pediatrická populace
Uvedená varování a preventivní opatření platí pro dospělé i děti.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce S přípravkem NovoEight nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
S přípravkem NovoEight nebyly prováděny žádné reprodukční studie na zvířatech. Na základě vzácného výskytu hemofilie A u žen nejsou zkušenosti týkající se použití faktoru VIII během těhotenství a kojení k dispozici. Z toho důvodu může být faktor VIII během těhotenství a kojení použit pouze, pokud je to jednoznačně indikováno.
4.7 Účinky na schopnost řídit a obsluhovat stroje
NovoEight nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Vzácně byly pozorovány hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a štípání v místě infuze, třesavku, zrudnutí, generalizovanou kopřivku, bolest hlavy, vyrážku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tlak na prsou, brnění, zvracení, sípot) a mohou v některých případech vyústit v těžkou anafylaxi (včetně šoku).
Velmi vzácně byl pozorován vznik protilátek proti křeččím proteinům se spojenou hypersenzitivitou.
U pacientů s hemofilií A může dojít ke vzniku neutralizačních protilátek (inhibitory) proti faktoru VIII. Jestliže dojde ke vzniku těchto inhibitorů, projeví se to jako nedostačující klinická odpověď. V takových případech se doporučuje vyhledat specializované centrum pro léčbu hemofilie.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky uvedené v tabulce níže jsou klasifikovány dle Tříd orgánových systémů podle databáze MedDRA (TOS a preferované termíny četností).
Frekvence výskytu jsou definovány podle následující konvence: Velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 2 Frekvence nežádoucích účinků v klinických studiích
|
Třída orgánových systémů |
Frekvence* |
Nežádoucí účinek |
|
Psychiatrické poruchy |
Méně časté | |
|
Poruchy nervového systému |
Méně časté |
Bolest hlavy, závratě |
|
Srdeční poruchy |
Méně časté |
Sinusová tachykardie |
|
Cévní poruchy |
Méně časté |
Hypertenze, lymfoedém |
|
Poruchy jater a žlučových cest |
Časté |
Zvýšení jaterních enzymů* * |
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Méně časté |
Muskuloskeletální ztuhlost, artropatie, bolesti končetin, muskuloskeletální bolest |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Reakce v místě vpichu* * * |
|
Méně časté |
Únava, pocit horka, periferní edém, pyrexie | |
|
Vyšetření |
Méně časté |
Zrychlená srdeční frekvence |
|
Poranění, otravy a procedurální komplikace |
Méně časté |
Kontuze |
*
**
Přepočteno na základě celkového počtu jednotlivých pacientů ve všech klinických studiích (214)
Zvýšení jaterních enzymů se týká alanin aminotransferázy, aspartát aminotransferázy, gama-glutamyltransferázy a bilirubinu
***
Reakce v místě vpichu zahrnují erytém v místě vpichu, extravazáty v místě vpichu a svědění v místě vpichu
Popis vybraných nežádoucích účinků
V průběhu všech klinických studií s přípravkem NovoEight bylo celkem hlášeno 30 nežádoucích účinků u 19 z 214 pacientů léčených přípravkem NovoEight. Nejčastěji hlášenými nežádoucími účinky byly reakce v místě vpichu a zvýšení jaterních enzymů. Z 30 nežádoucích účinků byly 2 hlášeny u jednoho z 31 pacientů do 6 let, žádný u pacientů ve věku 6-18 let a 28 bylo hlášeno u 18 ze 127 dospělých.
Pediatrická populace
V klinických studiích se 63 pediatrickými pacienty v rozmezí 0 až 12 let a s 24 dospívajícími
v rozmezí 12-18 let, kteří trpěli závažnou hemofilií A, nebyl nalezen žádný rozdíl v bezpečnostním profilu přípravku NovoEight mezi pediatrickými pacienty a dospělými.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika, krevní koagulační faktor VIII, ATC kód: B02BD02 Mechanismus účinku
NovoEight obsahuje turoktokog alfa - humánní koagulační faktor VIII (rDNA) se zkrácenou B-doménou. Tento glykoprotein má shodnou strukturu s humánním faktorem VIII, když je aktivován. Posttranslační modifikace jsou podobné modifikacím u molekul odvozených z plazmy. Sulfatační místo tyrosinu, jež je přítomno na Tyr1680 (přirozená plná délka) a jež je důležité pro vazbu na von Willebrandův faktor, je v molekule turoktokogu alfa plně sulfonované. Po podání infuze pacientovi s hemofilií se faktor VIII v krevním oběhu váže na endogenní von Willebrandův faktor. Komplex faktoru VIII s von Willebrandovým faktorem je tvořen 2 molekulami (faktor VIII a von Willebrandův faktor) s odlišnými fyziologickými funkcemi. Aktivovaný faktor VIII působí jako kofaktor pro aktivaci faktoru IX urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin pak konvertuje fibrinogen na fibrin a umožní tak tvorbu sraženiny. Hemofilie je na pohlaví závisející dědičná porucha krevní srážlivosti, jejíž příčinou je snížená hladina faktoru VIII:C. Výsledkem je silné krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánní nebo jako důsledek úrazu nebo chirurgického zákroku. Při substituční léčbě se hladiny plazmatického faktoru VIII zvýší, tím dojde k dočasné úpravě deficitu faktoru a tím také k úpravě sklonu ke krvácení.
Klinická účinnost
Byly provedeny tři multicentrické, otevřené, nekontrolované studie za účelem vyhodnotit bezpečnost a účinnost přípravku NovoEight v prevenci a léčbě krvácení u již dříve léčených pacientů s těžkou hemofilií A (aktivita FVIII <1 %). Do studie bylo zahrnuto 213 léčených pacientů; 150 dospívajících či dospělých pacientů bez inhibitorů ve věku od 12 let (>150 dní léčby) a 63 pediatrických pacientů bez inhibitorů do 12 let (>50 dní léčby). 187 z 213 pacientů pokračovalo v prodloužené studii bezpečnosti. Léčba přípravkem NovoEight byla prokázána jako bezpečná a měla předpokládaný hemostatický a preventivní účinek. Během akumulované léčby více než 54 000 dní (odpovídající 342 pacientoroků) nebyl ve fázi 3a klinických studií u již dříve léčených pacientů pozorován rozvoj žádných inhibitorů proti faktoru VIII. Z 1 377 hlášených krvácivých příhod pozorovaných u 177 pacientů z 213, bylo 1 244 (90,3 %) krvácení zastaveno 1-2 infuzemi přípravku NovoEight.
Tabulka 3 Spotřeba turoktokogu alfa a celkový výskyt úspěšnosti
|
Mladší děti (0 - <6 let) |
Starší děti (6 - <12 let) |
Dospívající (12 -<18 let) |
Dospělí (>18 let) |
Celkem | |
|
Počet pacientů |
31 |
32 |
24 |
126 |
213 |
|
Dávka užitá k prevenci na pacienta (IU/kg TH) Průměr (SD) |
40,1 (8,5) |
36,6 (9,0) |
27,0 (7,6) |
26,9 (6,9) |
30,3 (9,2) |
|
Min ; Max |
26,5 ; 57,3 |
24,9 ; 57,9 |
20,5 ; 46,9 |
20,0 ; 50,8 |
20,0 ; 57,9 |
|
Dávka užitá k léčbě krvácení (IU/kg TH) Průměr (SD) Min ; Max |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Výskyt úspěšnosti* % |
92,9% |
88,9% |
79,7% |
85,6% |
85,9% |
TH: Tělesná hmotnost, SD: Směrodatná odchylka *Úspěšnost je definována buď jako „Výbomá“ nebo „Dobrá“.
Celkem bylo provedeno 14 chirurgických zákroků u celkem 14 pacientů, z nichž 13 bylo závažných, a jeden byl lehký. Hemostáza byla úspěšná ve všech případech a nebylo hlášeno žádné selhání léčby.
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s turoktokogem alfa byly prováděny u pacientů trpících závažnou hemofilií A (aktivita FVIII <1 %), kteří již byli dříve léčeni. Analýza vzorků plazmy byla prováděna jak pomocí jednostupňového koagulačního testu, tak chromogenním testem.
V mezinárodní studii zahrnující 36 laboratoří byla testována aktivita přípravku NovoEight pomocí testu FVIII:C a porovnávána s na trhu dostupným přípravkem obsahujícím rekombinantní FVIII o plné délce. Studie prokázala srovnatelné a stabilní výsledky pro oba přípravky a rovněž to, že přípravek NovoEight může být v plazmě spolehlivě měřen, aniž by bylo zapotřebí speciálního standardu pro NovoEight.
Farmakokinetické parametry po jednorázové dávce přípravku NovoEight jsou shrnuty v Tabulce 4 pro test srážlivosti, v Tabulce 5 pro chromogenní test.
Tabulka 4 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), test srážlivosti_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((IU*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
t* (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (IU/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Průměrná doba setrvání v oběhu (hod.) |
9,63 (2,50) |
9,91 (2,57) |
14,19 5,08) |
Tabulka 5 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), chromogenní test_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((IU*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (IU/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Průměrná doba setrvání v oběhu (hod.) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Farmakokinetické parametry u pediatrických pacientů do 6 let a pediatrických pacientů ve věku 612 let byly srovnatelné. Byly pozorovány některé odchylky ve farmakokinetických parametrech přípravku NovoEight mezi pediatrickými a dospělými pacienty. U pediatrických pacientů byly nalezeny vyšší hodnoty CL a kratší ť/2 ve srovnání s dospělými pacienty s hemofilií A, což může být částečně způsobeno známým vyšším plazmatickým objemem na kilogram tělesné hmotnosti u mladších pacientů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po opakovaném podávání neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Chlorid sodný Histidin Sacharosa Polysorbát 80 Methionin
Dihydrát chloridu vápenatého Hydroxid sodný Kyselina chlorovodíková
Rozpouštědlo Chlorid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Před otevřením:
30 měsíců
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (< 30°C) po jedno nepřetržité období nepřesahující 9 měsíců. Jakmile byl přípravek jednou vyjmut z chladničky, nesmí tam již být vrácen zpět. Poznačte si prosím na krabičce datum, kdy jste přípravek začali uchovávat při pokojové teplotě. Injekční lahvičku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem.
Po rekonstituci:
Chemická a fyzikální stabilita po rekonstituci byla prokázána na dobu 24 hodin při 2°C - 8°C a 4 hodiny při uchovávání při pokojové teplotě (< 30°C).
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, jsou doba a podmínky uchovávání přípravku po otevření před použitím v odpovědnosti uživatele. Normálně by tato doba neměla být delší než 4 hodiny při uchovávání při pokojové teplotě (< 30°C) nebo 24 hodin při 2°C - 8°C, pokud rekonstituce neproběhla za kontrolovaných a validovaných aseptických podmínek.
Veškerý nepoužitý léčivý přípravek, který byl uchováván při pokojové teplotě déle než 4 hodiny, musí být zlikvidován.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem.
Uchovávání tohoto léčivého přípravku při pokojové teplotě a podmínky uchovávání po jeho rekonstituci viz bod 6.3.
6.5 Druh obalu a obsah balení
Jedno balení přípravku NovoEight 2 000 IU prášek a rozpouštědlo pro injekční roztok obsahuje:
- 1 skleněnou injekční lahvičku (sklo typu I) s práškem opatřenou chlorobutylovou pryžovou zátkou
- 1 sterilní adaptér injekční lahvičky k rozpuštění
- 1 předplněnou injekční stříkačku obsahující 4 ml rozpouštědla s polypropylenovým uzávěrem zpětného chodu, bromobutylovým pryžovým pístem a uzávěrem s bromobutylovou zátkou.
- 1 nástavec pístu (zhotovený z polypropylenu)
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
NovoEight je určen k intravenóznímu podání po rekonstituci prášku v rozpouštědle dodávaném v injekční stříkačce. Po rekonstituci je roztok čirý či lehce opalescentní. Roztok nepoužívejte, pokud je zakalený či obsahuje usazeniny.
Budete také potřebovat infuzní soupravu (infuzní set a jehlu s křidélky), sterilní alkoholové tampony, gázové polštářky a náplasti. Tyto pomůcky nejsou součástí balení přípravku NovoEight.
Vždy dodržujte aseptickou techniku.

Rekonstituce
A)
Vyjměte injekční lahvičku, adaptér injekční lahvičky a předplněnou injekční stříkačku z krabičky. Nástavec pístu ponechte zatím v krabičce. Zahřejte injekční lahvičku a předplněnou injekční stříkačku na pokojovou teplotu. Můžete to udělat tak, že je podržíte v ruce, dokud nemají stejnou teplotu jako vaše dlaně. Jiné způsoby ohřátí injekční lahvičky a předplněné injekční stříkačky nepoužívejte.
B)




Odstraňte plastové víčko z injekční lahvičky. Pokud je víčko uvolněné nebo chybí, injekční lahvičku nepoužívejte. Pryžovou zátku injekční lahvičky očistěte sterilním alkoholovým tamponem a nechte ji před použitím několik sekund na vzduchu oschnout.
C)
Sejměte ochranný papír z adaptéru injekční lahvičky. Pokud ochranný papír není zcela zatavený nebo je protržený, adaptér injekční lahvičky nepoužívejte. Nevyjímejte adaptér injekční lahvičky prsty z ochranného víčka.
D)
Otočte ochranné víčko a nasaďte adaptér na injekční lahvičku. Jakmile jste adaptér na injekční lahvičku nasadili, již ho z ní neodstraňujte.
E)
Lehce stiskněte ochranné víčko mezi palcem a ukazováčkem, jak je patrné z obrázku. Sejměte ochranné víčko z adaptéru injekční lahvičky.
F)
Pevně uchopte nástavec pístu za širší konec a okamžitě ho našroubujte po směru hodinových ručiček na píst uvnitř předplněné injekční stříkačky, dokud nepocítíte odpor.
G)
Odstraňte ochranné víčko z předplněné injekční stříkačky ohnutím směrem dolů tak, aby se porušila perforace. Dbejte, abyste se nedotkli prsty hrotu injekční stříkačky pod jejím ochranným víčkem.
H)


Našroubujte předplněnou injekční stříkačku bezpečně na adaptér injekční lahvičky, dokud nepocítíte odpor.
I)
Držte předplněnou injekční stříkačku lehce nakloněnou s injekční lahvičkou směřující dolů. Stisknutím nástavce pístu vstříkněte všechno rozpouštědlo do injekční lahvičky.
J)
Nechte nástavec pístu zcela stlačený a jemným kroužením injekční lahvičkou rozpusťte všechen prášek. Injekční lahvičkou netřepejte, mohlo by to způsobit napěnění.
Doporučuje se použít NovoEight okamžitě po rekonstituci. Podmínky uchovávání rekonstituovaného léčivého přípravku viz bod 6.3.
Pokud je zapotřebí větší dávky, opakujte kroky A až J s dalšími injekčními lahvičkami, adaptéry injekčních lahviček a předplněnými injekčními stříkačkami.
Aplikace rekonstituovaného roztoku

K)
Ponechte nástavec pístu zcela stlačený. Otočte injekční stříkačku tak, aby nasazená injekční lahvička byla dnem vzhůru. Uvolněte nástavec pístu a nechte ho samovolně vrátit se zpět. Tím se rekonstituovaný roztok natáhne do injekční stříkačky. Lehkým vytažením pístu směrem dolů pak zajistíte, že se do injekční stříkačky natáhne všechen roztok.
V případě, že potřebujete pouze část obsahu injekční lahvičky, použijte stupnici na injekční stříkačce, abyste si ověřili, že jste natáhli tolik vzniklého roztoku, kolik vám doporučil lékař či zdravotní sestra.
Stále držte injekční lahvičku dnem vzhůru a jemně poklepejte na injekční stříkačku, aby se vzduchové bubliny nashromáždily nahoře. Pomalu zatlačte na nástavec pístu, dokud všechny vzduchové bubliny neuniknou.

L)
Odšroubujte adaptér s injekční lahvičkou.
NovoEight je nyní připraven k aplikaci. Určete vhodné místo a pomalu aplikujte NovoEight do žíly v průběhu 2 až 5 minut.
Likvidace
Po aplikaci bezpečně zlikvidujte veškerý nepoužitý roztok přípravku NovoEight, injekční stříkačku s infuzní soupravou, injekční lahvičku s adaptérem a ostatní odpad dle doporučení lékárníka.
Nevhazujte do běžného domácího odpadu.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
8. REGISTRAČNÍ ČÍSLA
EU/1/13/888/005
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. listopad 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky (EMA) http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
NovoEight 3 000 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička s práškem obsahuje 3 000 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Po rozpuštění obsahuje přípravek NovoEight přibližně 750 IU/ml humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Účinnost (IU) se udává chromogenní metodou podle Evropského. Specifická aktivita přípravku NovoEight je přibližně 8 300 IU/mg bílkoviny.
Turoktokog alfa (humánní koagulační faktor VIII (rDNA)) je čištěná bílkovina obsahující 1 445 aminokyselin s molekulovou hmotností asi 166 kDA. Je vyráběn rekombinantní DNA technologií z vaječníkových buněk čínského křečka (CHO buňky). Je vyroben bez přídavku jakékoliv bílkoviny lidského či zvířecího původu během kultivace buněk, čištění či konečné úpravy přípravku.
Turoktokog alfa je rekombinantní humánní koagulační faktor VIII se zkrácenou B-doménou (B-doména obsahuje 21 aminokyselin z přirozené B-domény) bez jakékoliv další změny v pořadí aminokyselin.
Pomocná látka se známým účinkem:
0,31 mmol sodíku (7 mg) v 1 ml vzniklého roztoku
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Bílý nebo lehce nažloutlý prášek či drobivá hmota.
Čirý a bezbarvý injekční roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a prevence krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). NovoEight lze používat ve všech věkových skupinách.
4.2 Dávkování a způsob podání
Léčba by měla být zahájena pod dohledem lékaře se zkušenostmi s léčbou hemofilie.
Doposud neléčení pacienti
Bezpečnost a účinnost přípravku NovoEight u dosud neléčených pacientů nebyly doposud stanoveny. Nejsou dostupné žádné údaje.
Dávkování
Dávkování a trvání substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a pacientově klinickém stavu.
Počet podávaných jednotek faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které odpovídají běžnému standardu WHO pro přípravky obsahující faktor VIII. Aktivita faktoru VIII v plazmě je vyjádřena buď v procentech (relativně k normální hladině v lidské plazmě) nebo v mezinárodních jednotkách (relativně k mezinárodnímu standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba v případě potřeby
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že jedna mezinárodní jednotka (IU) faktoru VIII na kg tělesné hmotnosti zvýší aktivitu faktoru VIII v plazmě asi o 2 IU/dl. Požadovaná dávka se vypočte podle následujícího vzorce:
Potřebný počet jednotek = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) (IU/dl) x 0,5 (IU/kg na IU/dl).
Množství, které má být podáno, a frekvence podání by měly být vždy přizpůsobeny klinické účinnosti v individuálním případě.
V případě následujících krvácivých příhod by v odpovídajícím období aktivita faktoru VIII neměla klesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může být použita jako návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Tabulka 1 Návod pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Stupeň krvácení/ Požadovaná hladina Četnost dávek (hodiny)/délka
Typ chirurgického výkonu FVIII (%) (IU/dl) trvání léčby (dny)
Krvácení
Časný hemartros, krvácení do svalů 20-40 Opakovat každých 12-24 hodin.
nebo do dutiny ústní Nejméně 1 den, dokud nedojde k
zástavě krvácení, indikované skončením bolestí, nebo ke zhojení.
Rozsáhlejší hemartros, krvácení do 30-60 svalů nebo hematom
Život ohrožující krvácení 60-100
Opakovat infuzi každých 12-24 hodin po dobu 3-4 dnů nebo déle dokud bolest a akutní porucha funkce neustoupí
Opakovat infuze každých 8-24 hodin dokud nepomine ohrožení života
Chirurgické zákroky
Každých 24 hodin, nejméně 1 den, až je dosaženo zhojení.
Menší operace včetně vytržení 30-60
zubu
|
Stupeň krvácení/ Typ chirurgického výkonu |
Požadovaná hladina FVIII (%) (IU/dl) |
Četnost dávek (hodiny)/délka trvání léčby (dny) |
|
Velké chirurgické výkony |
80-100 (před a po operaci) |
Opakovat infuzi každých 8-24 hodin až do adekvátního zhojení poranění, pak pokračovat v léčbě nejméně dalších 7 dní k udržení aktivity faktoru VIII na 30-60 % (IU/dl) |
Profylaxe
K dlouhodobé profylaxi krvácení u pacientů se závažnou hemofilií A. Obvyklé doporučené dávky jsou 20-40 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 20-50 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. V některých případech, zvláště u mladších pacientů, může být nutné podávat přípravek v kratších intervalech nebo ve vyšších dávkách.
Monitorování léčby
V průběhu léčby je doporučeno provádět příslušné stanovení hladin faktoru VIII za účelem získání vodítka pro velikost podávané dávky i četnost opakovaných aplikací. Zvláště v případě závažných chirurgických zákroků je nezbytné přesné monitorování substituční léčby, prováděné pomocí koagulační analýzy (aktivity faktoru VIII v plazmě). U jednotlivých pacientů se může jejich odezva na faktor VIII lišit dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním odlišných poločasů.
Chirurgický zákrok
U pediatrických pacientů neexistují žádné zkušenosti s chirurgickými zákroky.
Starší pacienti
U pacientů ve věku > 65 let neexistují žádné zkušenosti.
Pediatrická populace
K dlouhodobé profylaxi krvácení u pacientů do 12 let se doporučují dávky 25 - 50 IU faktoru VIII na kg tělesné hmotnosti každý druhý den nebo 25 - 60 IU faktoru VIII na kg tělesné hmotnosti 3krát týdně. Pro pediatrické pacienty od 12 let jsou doporučené dávky shodné s doporučeními pro dospělé pacienty.
Způsob podání Intravenózní podání
Doporučená rychlost infuze je u přípravku NovoEight 1-2 ml/min. Rychlost by měla být stanovena tak, aby vyhovovala pacientovi.
Instrukce pro rekonstituci léčivého přípravku před podáním naleznete v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Známá alergická reakce na křeččí bílkoviny.
4.4 Zvláštní upozornění a opatření pro použití Hypersenzitivita
Při používání přípravku NovoEight se mohou vyskytnout hypersenzitivní reakce alergického typu. Přípravek obsahuje stopy křeččích bílkovin, jež mohou u některých pacientů vyvolat alergické reakce. Obj eví-li se příznaky hypersenzitivity, musí být pacienti poučeni o tom, aby okamžitě přerušili léčbu
tímto léčivým přípravkem a kontaktovali svého lékaře. Pacienti musí být informováni o časných příznacích hypersenzitivních reakcí včetně kopřivky, generalizované kopřivky, tlaku na hrudi, sípotu, hypotenze a anafylaxe.
V případě šoku je nutno nasadit standardní lékařskou léčbu šokového stavu.
Inhibitory
Známou komplikací léčby u individuálních případů hemofilie A je vznik neutralizačních protilátek (inhibitorů) proti faktoru VIII. Tyto inhibitory jsou obvykle IgG imunoglobuliny působící proti koagulační aktivitě faktoru VIII. Jsou kvantitativně udávané v Bethesda jednotkách (BU) na jeden ml plazmy a zjišťované pomocí modifikovaného testu. Riziko vzniku inhibitorů je ve vztahu k expozici organizmu faktorem VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Vzácně může dojít k tvorbě inhibitorů teprve po prvních 100 dnech expozice.
Při přechodu z jednoho přípravku obsahujícího faktor VIII na jiný byl u pacientů léčených již dříve, kteří měli v anamnéze dřívější výskyt inhibitoru, pozorován opětovný výskyt inhibitoru (nízký titr), a to i po více než 100 dnech expozice. Proto je při jakémkoliv převodu na jiný přípravek doporučeno vždy pečlivě monitorovat všechny pacienty s ohledem na vznik inhibitoru.
Obecně všichni pacienti léčení přípravky obsahujícími koagulační faktor VIII by měli být pečlivě sledováni z hlediska vzniku inhibitorů vhodnými klinickými vyšetřeními a laboratorními testy. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII nebo pokud není dosaženo kontroly krvácení příslušnou dávkou, musí být provedeny testy na přítomnost inhibitoru. U pacientů s vysokými hladinami inhibitorů, může být léčba faktorem VIII neúčinná, a je třeba zvážit jiné léčebné možnosti. Léčba takovýchto pacientů by měla být prováděna lékařem se zkušeností v péči o pacienty s hemofilií a s inhibitory proti faktoru VIII.
Důrazně se doporučuje, aby vždy při každém podání přípravku NovoEight pacientovi byl zaznamenán název a číslo šarže přípravku, aby bylo možno přiřadit číslo šarže přípravku k pacientovi.
Pomocné látky, které je nutno vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,31 mmol sodíku (7 mg) na 1 ml vzniklého roztoku. Tento fakt je nutno vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
Komplikace spojené s použitím katetru
Pokud je požadováno použití centrálního žilního katetru (CVAD), je nutno zvážit riziko komplikací spojené s jeho použitím včetně lokálních infekcí, bakteriémie a trombózy v místě katetru.
Pediatrická populace
Uvedená varování a preventivní opatření platí pro dospělé i děti.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce S přípravkem NovoEight nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
S přípravkem NovoEight nebyly prováděny žádné reprodukční studie na zvířatech. Na základě vzácného výskytu hemofilie A u žen nejsou zkušenosti týkající se použití faktoru VIII během těhotenství a kojení k dispozici. Z toho důvodu může být faktor VIII během těhotenství a kojení použit pouze, pokud je to jednoznačně indikováno.
4.7 Účinky na schopnost řídit a obsluhovat stroje
NovoEight nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Vzácně byly pozorovány hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a štípání v místě infuze, třesavku, zrudnutí, generalizovanou kopřivku, bolest hlavy, vyrážku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tlak na prsou, brnění, zvracení, sípot) a mohou v některých případech vyústit v těžkou anafylaxi (včetně šoku).
Velmi vzácně byl pozorován vznik protilátek proti křeččím proteinům se spojenou hypersenzitivitou.
U pacientů s hemofilií A může dojít ke vzniku neutralizačních protilátek (inhibitory) proti faktoru VIII. Jestliže dojde ke vzniku těchto inhibitorů, projeví se to jako nedostačující klinická odpověď. V takových případech se doporučuje vyhledat specializované centrum pro léčbu hemofilie.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky uvedené v tabulce níže jsou klasifikovány dle Tříd orgánových systémů podle databáze MedDRA (TOS a preferované termíny četností).
Frekvence výskytu jsou definovány podle následující konvence: Velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 2 Frekvence nežádoucích účinků v klinických studiích
|
Třída orgánových systémů |
Frekvence* |
Nežádoucí účinek |
|
Psychiatrické poruchy |
Méně časté | |
|
Poruchy nervového systému |
Méně časté |
Bolest hlavy, závratě |
|
Srdeční poruchy |
Méně časté |
Sinusová tachykardie |
|
Cévní poruchy |
Méně časté |
Hypertenze, lymfoedém |
|
Poruchy jater a žlučových cest |
Časté |
Zvýšení jaterních enzymů* * |
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Méně časté |
Muskuloskeletální ztuhlost, artropatie, bolesti končetin, muskuloskeletální bolest |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Reakce v místě vpichu* * * |
|
Méně časté |
Únava, pocit horka, periferní edém, pyrexie | |
|
Vyšetření |
Méně časté |
Zrychlená srdeční frekvence |
|
Poranění, otravy a procedurální komplikace |
Méně časté |
Kontuze |
*
**
Přepočteno na základě celkového počtu jednotlivých pacientů ve všech klinických studiích (214)
Zvýšení jaterních enzymů se týká alanin aminotransferázy, aspartát aminotransferázy, gama-glutamyltransferázy a bilirubinu
***
Reakce v místě vpichu zahrnují erytém v místě vpichu, extravazáty v místě vpichu a svědění v místě vpichu
Popis vybraných nežádoucích účinků
V průběhu všech klinických studií s přípravkem NovoEight bylo celkem hlášeno 30 nežádoucích účinků u 19 z 214 pacientů léčených přípravkem NovoEight. Nejčastěji hlášenými nežádoucími účinky byly reakce v místě vpichu a zvýšení jaterních enzymů. Z 30 nežádoucích účinků byly 2 hlášeny u jednoho z 31 pacientů do 6 let, žádný u pacientů ve věku 6-18 let a 28 bylo hlášeno u 18 ze 127 dospělých.
Pediatrická populace
V klinických studiích se 63 pediatrickými pacienty v rozmezí 0 až 12 let a s 24 dospívajícími
v rozmezí 12-18 let, kteří trpěli závažnou hemofilií A, nebyl nalezen žádný rozdíl v bezpečnostním profilu přípravku NovoEight mezi pediatrickými pacienty a dospělými.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika, krevní koagulační faktor VIII, ATC kód: B02BD02 Mechanismus účinku
NovoEight obsahuje turoktokog alfa - humánní koagulační faktor VIII (rDNA) se zkrácenou B-doménou. Tento glykoprotein má shodnou strukturu s humánním faktorem VIII, když je aktivován. Posttranslační modifikace jsou podobné modifikacím u molekul odvozených z plazmy. Sulfatační místo tyrosinu, jež je přítomno na Tyr1680 (přirozená plná délka) a jež je důležité pro vazbu na von Willebrandův faktor, je v molekule turoktokogu alfa plně sulfonované. Po podání infuze pacientovi s hemofilií se faktor VIII v krevním oběhu váže na endogenní von Willebrandův faktor. Komplex faktoru VIII s von Willebrandovým faktorem je tvořen 2 molekulami (faktor VIII a von Willebrandův faktor) s odlišnými fyziologickými funkcemi. Aktivovaný faktor VIII působí jako kofaktor pro aktivaci faktoru IX urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin pak konvertuje fibrinogen na fibrin a umožní tak tvorbu sraženiny. Hemofilie je na pohlaví závisející dědičná porucha krevní srážlivosti, jejíž příčinou je snížená hladina faktoru VIII:C. Výsledkem je silné krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánní nebo jako důsledek úrazu nebo chirurgického zákroku. Při substituční léčbě se hladiny plazmatického faktoru VIII zvýší, tím dojde k dočasné úpravě deficitu faktoru a tím také k úpravě sklonu ke krvácení.
Klinická účinnost
Byly provedeny tři multicentrické, otevřené, nekontrolované studie za účelem vyhodnotit bezpečnost a účinnost přípravku NovoEight v prevenci a léčbě krvácení u již dříve léčených pacientů s těžkou hemofilií A (aktivita FVIII <1 %). Do studie bylo zahrnuto 213 léčených pacientů; 150 dospívajících či dospělých pacientů bez inhibitorů ve věku od 12 let (>150 dní léčby) a 63 pediatrických pacientů bez inhibitorů do 12 let (>50 dní léčby). 187 z 213 pacientů pokračovalo v prodloužené studii bezpečnosti. Léčba přípravkem NovoEight byla prokázána jako bezpečná a měla předpokládaný hemostatický a preventivní účinek. Během akumulované léčby více než 54 000 dní (odpovídající 342 pacientoroků) nebyl ve fázi 3 a klinických studií u již dříve léčených pacientů pozorován rozvoj žádných inhibitorů proti faktoru VIII. Z 1 377 hlášených krvácivých příhod pozorovaných u 177 pacientů z 213, bylo 1 244 (90,3 %) krvácení zastaveno 1-2 infuzemi přípravku NovoEight.
Tabulka 3 Spotřeba turoktokogu alfa a celkový výskyt úspěšnosti
|
Mladší děti (0 - <6 let) |
Starší děti (6 - <12 let) |
Dospívající (12 -<18 let) |
Dospělí (>18 let) |
Celkem | |
|
Počet pacientů |
31 |
32 |
24 |
126 |
213 |
|
Dávka užitá k prevenci na pacienta (IU/kg TH) Průměr (SD) |
40,1 (8,5) |
36,6 (9,0) |
27,0 (7,6) |
26,9 (6,9) |
30,3 (9,2) |
|
Min ; Max |
26,5 ; 57,3 |
24,9 ; 57,9 |
20,5 ; 46,9 |
20,0 ; 50,8 |
20,0 ; 57,9 |
|
Dávka užitá k léčbě krvácení (IU/kg TH) Průměr (SD) Min ; Max |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Výskyt úspěšnosti* % |
92,9% |
88,9% |
79,7% |
85,6% |
85,9% |
TH: Tělesná hmotnost, SD: Směrodatná odchylka *Úspěšnost je definována buď jako „Výbomá“ nebo „Dobrá“.
Celkem bylo provedeno 14 chirurgických zákroků u celkem 14 pacientů, z nichž 13 bylo závažných, a jeden byl lehký. Hemostáza byla úspěšná ve všech případech a nebylo hlášeno žádné selhání léčby.
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s turoktokogem alfa byly prováděny u pacientů trpících závažnou hemofilií A (aktivita FVIII <1 %), kteří již byli dříve léčeni. Analýza vzorků plazmy byla prováděna jak pomocí jednostupňového koagulačního testu, tak chromogenním testem.
V mezinárodní studii zahrnující 36 laboratoří byla testována aktivita přípravku NovoEight pomocí testu FVIII:C a porovnávána s na trhu dostupným přípravkem obsahujícím rekombinantní FVIII o plné délce. Studie prokázala srovnatelné a stabilní výsledky pro oba přípravky a rovněž to, že přípravek NovoEight může být v plazmě spolehlivě měřen, aniž by bylo zapotřebí speciálního standardu pro NovoEight.
Farmakokinetické parametry po jednorázové dávce přípravku NovoEight jsou shrnuty v Tabulce 4 pro test srážlivosti, v Tabulce 5 pro chromogenní test.
Tabulka 4 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), test srážlivosti_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((IU*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
t* (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (IU/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Průměrná doba setrvání v oběhu (hod.) |
9,63 (2,50) |
9,91 (2,57) |
14,19 5,08) |
Tabulka 5 Farmakokinetika po podání jednorázové dávky turoktokogu alfa pacientům se závažnou hemofilií A (FVIII <1%), chromogenní test_
|
Parametr |
0 - <6 let |
6 - <12 let |
>12 let |
|
n=14 |
n=14 |
n=33 | |
|
Průměr (SD) |
Průměr (SD) |
Průměr (SD) | |
|
Přírůstkové recovery (IU/ml)/(IU/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((IU*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (IU/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Průměrná doba setrvání v oběhu (hod.) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Farmakokinetické parametry u pediatrických pacientů do 6 let a pediatrických pacientů ve věku 612 let byly srovnatelné. Byly pozorovány některé odchylky ve farmakokinetických parametrech přípravku NovoEight mezi pediatrickými a dospělými pacienty. U pediatrických pacientů byly nalezeny vyšší hodnoty CL a kratší ť/2 ve srovnání s dospělými pacienty s hemofilií A, což může být částečně způsobeno známým vyšším plazmatickým objemem na kilogram tělesné hmotnosti u mladších pacientů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po opakovaném podávání neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Chlorid sodný Histidin Sacharosa Polysorbát 80 Methionin
Dihydrát chloridu vápenatého Hydroxid sodný Kyselina chlorovodíková
Rozpouštědlo Chlorid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Před otevřením:
30 měsíců
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (< 30°C) po jedno nepřetržité období nepřesahující 9 měsíců. Jakmile byl přípravek jednou vyjmut z chladničky, nesmí tam již být vrácen zpět. Poznačte si prosím na krabičce datum, kdy jste přípravek začali uchovávat při pokojové teplotě. Injekční lahvičku uchovávejte ve vnějším obalu, aby byl přípravek chráněn před světlem.
Po rekonstituci:
Chemická a fyzikální stabilita po rekonstituci byla prokázána na dobu 24 hodin při 2°C - 8°C a 4 hodiny při uchovávání při pokojové teplotě (< 30°C).
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, jsou doba a podmínky uchovávání přípravku po otevření před použitím v odpovědnosti uživatele. Normálně by tato doba neměla být delší než 4 hodiny při uchovávání při pokojové teplotě (< 30°C) nebo 24 hodin při 2°C - 8°C, pokud rekonstituce neproběhla za kontrolovaných a validovaných aseptických podmínek.
Veškerý nepoužitý léčivý přípravek, který byl uchováván při pokojové teplotě déle než 4 hodiny, musí být zlikvidován.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem.
Uchovávání tohoto léčivého přípravku při pokojové teplotě a podmínky uchovávání po jeho rekonstituci viz bod 6.3.
6.5 Druh obalu a obsah balení
Jedno balení přípravku NovoEight 3 000 IU prášek a rozpouštědlo pro injekční roztok obsahuje:
- 1 skleněnou injekční lahvičku (sklo typu I) s práškem opatřenou chlorobutylovou pryžovou zátkou
- 1 sterilní adaptér injekční lahvičky k rozpuštění
- 1 předplněnou injekční stříkačku obsahující 4 ml rozpouštědla s polypropylenovým uzávěrem zpětného chodu, bromobutylovým pryžovým pístem a uzávěrem s bromobutylovou zátkou.
- 1 nástavec pístu (zhotovený z polypropylenu)
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
NovoEight je určen k intravenóznímu podání po rekonstituci prášku v rozpouštědle dodávaném v injekční stříkačce. Po rekonstituci je roztok čirý či lehce opalescentní. Roztok nepoužívejte, pokud je zakalený či obsahuje usazeniny.
Budete také potřebovat infuzní soupravu (infuzní set a jehlu s křidélky), sterilní alkoholové tampony, gázové polštářky a náplasti. Tyto pomůcky nejsou součástí balení přípravku NovoEight.
Vždy dodržujte aseptickou techniku.

Rekonstituce
A)
Vyjměte injekční lahvičku, adaptér injekční lahvičky a předplněnou injekční stříkačku z krabičky. Nástavec pístu ponechte zatím v krabičce. Zahřejte injekční lahvičku a předplněnou injekční stříkačku na pokojovou teplotu. Můžete to udělat tak, že je podržíte v ruce, dokud nemají stejnou teplotu jako vaše dlaně. Jiné způsoby ohřátí injekční lahvičky a předplněné injekční stříkačky nepoužívejte.
B)




Odstraňte plastové víčko z injekční lahvičky. Pokud je víčko uvolněné nebo chybí, injekční lahvičku nepoužívejte. Pryžovou zátku injekční lahvičky očistěte sterilním alkoholovým tamponem a nechte ji před použitím několik sekund na vzduchu oschnout.
C)
Sejměte ochranný papír z adaptéru injekční lahvičky. Pokud ochranný papír není zcela zatavený nebo je protržený, adaptér injekční lahvičky nepoužívejte. Nevyjímejte adaptér injekční lahvičky prsty z ochranného víčka.
D)
Otočte ochranné víčko a nasaďte adaptér na injekční lahvičku. Jakmile jste adaptér na injekční lahvičku nasadili, již ho z ní neodstraňujte.
E)
Lehce stiskněte ochranné víčko mezi palcem a ukazováčkem, jak je patrné z obrázku. Sejměte ochranné víčko z adaptéru injekční lahvičky.
F)
Pevně uchopte nástavec pístu za širší konec a okamžitě ho našroubujte po směru hodinových ručiček na píst uvnitř předplněné injekční stříkačky, dokud nepocítíte odpor.
G)
Odstraňte ochranné víčko z předplněné injekční stříkačky ohnutím směrem dolů tak, aby se porušila perforace. Dbejte, abyste se nedotkli prsty hrotu injekční stříkačky pod jejím ochranným víčkem.
H)


Našroubujte předplněnou injekční stříkačku bezpečně na adaptér injekční lahvičky, dokud nepocítíte odpor.
I)
Držte předplněnou injekční stříkačku lehce nakloněnou s injekční lahvičkou směřující dolů. Stisknutím nástavce pístu vstříkněte všechno rozpouštědlo do injekční lahvičky.
J)
Nechte nástavec pístu zcela stlačený a jemným kroužením injekční lahvičkou rozpusťte všechen prášek. Injekční lahvičkou netřepejte, mohlo by to způsobit napěnění.
Doporučuje se použít NovoEight okamžitě po rekonstituci. Podmínky uchovávání rekonstituovaného léčivého přípravku viz bod 6.3.
Pokud je zapotřebí větší dávky, opakujte kroky A až J s dalšími injekčními lahvičkami, adaptéry injekčních lahviček a předplněnými injekčními stříkačkami.
Aplikace rekonstituovaného roztoku

K)
Ponechte nástavec pístu zcela stlačený. Otočte injekční stříkačku tak, aby nasazená injekční lahvička byla dnem vzhůru. Uvolněte nástavec pístu a nechte ho samovolně vrátit se zpět. Tím se rekonstituovaný roztok natáhne do injekční stříkačky. Lehkým vytažením pístu směrem dolů pak zajistíte, že se do injekční stříkačky natáhne všechen roztok.
V případě, že potřebujete pouze část obsahu injekční lahvičky, použijte stupnici na injekční stříkačce, abyste si ověřili, že jste natáhli tolik vzniklého roztoku, kolik vám doporučil lékař či zdravotní sestra.
Stále držte injekční lahvičku dnem vzhůru a jemně poklepejte na injekční stříkačku, aby se vzduchové bubliny nashromáždily nahoře. Pomalu zatlačte na nástavec pístu, dokud všechny vzduchové bubliny neuniknou.

L)
Odšroubujte adaptér s injekční lahvičkou.
NovoEight je nyní připraven k aplikaci. Určete vhodné místo a pomalu aplikujte NovoEight do žíly v průběhu 2 až 5 minut.
Likvidace
Po aplikaci bezpečně zlikvidujte veškerý nepoužitý roztok přípravku NovoEight, injekční stříkačku s infuzní soupravou, injekční lahvičku s adaptérem a ostatní odpad dle doporučení lékárníka.
Nevhazujte do běžného domácího odpadu.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
8. REGISTRAČNÍ ČÍSLA
EU/1/13/888/006
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. listopad 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky (EMA) http://www.ema.europa.eu.
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců biologické léčivé látky
BioReliance Ltd
Todd Campus, West of Scotland Science Park,
Glasgow, G20 0XA Velká Británie
Novo Nordisk A/S Brennum Park DK-3400 Hillerad Dánsko
Název a adresa výrobce odpovědného za propouštění šarží
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
B PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2)
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 8 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
NovoEight 250 IU prášek a rozpouštědlo pro injekční roztok turoctocogum alfa (humánní koagulační faktor VIII (rDNA))
Po rozpuštění obsahuje 1 ml přípravku NovoEight přibližně 62,5 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Prášek: chlorid sodný, histidin, sacharosa, polysorbát 80, methionin, dihydrát chloridu vápenatého, hydroxid sodný, kyselina chlorovodíková Rozpouštědlo: chlorid sodný
Prášek a rozpouštědlo pro injekční roztok
Balení obsahuje: 1 injekční lahvičku s práškem, 4 ml rozpouštědla v předplněné injekční stříkačce, nástavec pístu a adaptér injekční lahvičky
Intravenózní podání
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Může být uchováváno při pokojové teplotě < 30°C po jedno nepřetržité období nepřesahující 9 měsíců.
Z chladničky vyjmuto:_
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
EU/1/13/888/001
č.š.:
Výdej léčivého přípravku vázán na lékařský předpis
NovoEight 250 IU
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
NovoEight 250 IU prášek pro injekční roztok turoctocogum alfa Intravenózní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO SARZE
č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
250 IU
6. OTHER
NovoEight 500 IU prášek a rozpouštědlo pro injekční roztok turoctocogum alfa (humánní koagulační faktor VIII (rDNA))
Po rozpuštění obsahuje 1 ml přípravku NovoEight přibližně 125 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Prášek: chlorid sodný, histidin, sacharosa, polysorbát 80, methionin, dihydrát chloridu vápenatého, hydroxid sodný, kyselina chlorovodíková Rozpouštědlo: chlorid sodný
Prášek a rozpouštědlo pro injekční roztok
Balení obsahuje: 1 injekční lahvičku s práškem, 4 ml rozpouštědla v předplněné injekční stříkačce, nástavec pístu a adaptér injekční lahvičky
Intravenózní podání
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Může být uchováváno při pokojové teplotě < 30°C po jedno nepřetržité období nepřesahující 9 měsíců.
Z chladničky vyjmuto:_
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
EU/1/13/888/002
č.š.:
Výdej léčivého přípravku vázán na lékařský předpis
NovoEight 500 IU
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
NovoEight 500 IU prášek pro injekční roztok turoctocogum alfa Intravenózní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO SARZE
č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
500 IU
6. OTHER
NovoEight 1 000 IU prášek a rozpouštědlo pro injekční roztok turoctocogum alfa (humánní koagulační faktor VIII (rDNA))
Po rozpuštění obsahuje 1 ml přípravku NovoEight přibližně 250 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Prášek: chlorid sodný, histidin, sacharosa, polysorbát 80, methionin, dihydrát chloridu vápenatého, hydroxid sodný, kyselina chlorovodíková Rozpouštědlo: chlorid sodný
Prášek a rozpouštědlo pro injekční roztok
Balení obsahuje: 1 injekční lahvičku s práškem, 4 ml rozpouštědla v předplněné injekční stříkačce, nástavec pístu a adaptér injekční lahvičky
Intravenózní podání
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Může být uchováváno při pokojové teplotě < 30°C po jedno nepřetržité období nepřesahující 9 měsíců.
Z chladničky vyjmuto:_
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
12. REGISTRAČNÍ ČÍSLO
EU/1/13/888/003
13 ČÍSLO ŠARŽE č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
NovoEight 1 000 IU
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
NovoEight 1 000 IU prášek pro injekční roztok turoctocogum alfa Intravenózní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO SARZE
č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 000IU
6. OTHER
NovoEight 1 500 IU prášek a rozpouštědlo pro injekční roztok turoctocogum alfa (humánní koagulační faktor VIII (rDNA))
Po rozpuštění obsahuje 1 ml přípravku NovoEight přibližně 375 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Prášek: chlorid sodný, histidin, sacharosa, polysorbát 80, methionin, dihydrát chloridu vápenatého, hydroxid sodný, kyselina chlorovodíková Rozpouštědlo: chlorid sodný
Prášek a rozpouštědlo pro injekční roztok
Balení obsahuje: 1 injekční lahvičku s práškem, 4 ml rozpouštědla v předplněné injekční stříkačce, nástavec pístu a adaptér injekční lahvičky
Intravenózní podání
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Může být uchováváno při pokojové teplotě < 30°C po jedno nepřetržité období nepřesahující 9 měsíců.
Z chladničky vyjmuto:_
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
12. REGISTRAČNÍ ČÍSLO
EU/1/13/888/004
13 ČÍSLO ŠARŽE č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
NovoEight 1 500 IU
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
NovoEight 1 500 IU prášek pro injekční roztok turoctocogum alfa Intravenózní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO SARZE
č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 500IU
6. OTHER
NovoEight 2 000 IU prášek a rozpouštědlo pro injekční roztok turoctocogum alfa (humánní koagulační faktor VIII (rDNA))
Po rozpuštění obsahuje 1 ml přípravku NovoEight přibližně 500 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Prášek: chlorid sodný, histidin, sacharosa, polysorbát 80, methionin, dihydrát chloridu vápenatého, hydroxid sodný, kyselina chlorovodíková Rozpouštědlo: chlorid sodný
Prášek a rozpouštědlo pro injekční roztok
Balení obsahuje: 1 injekční lahvičku s práškem, 4 ml rozpouštědla v předplněné injekční stříkačce, nástavec pístu a adaptér injekční lahvičky
Intravenózní podání
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Může být uchováváno při pokojové teplotě < 30°C po jedno nepřetržité období nepřesahující 9 měsíců.
Z chladničky vyjmuto:_
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
12. REGISTRAČNÍ ČÍSLO
EU/1/13/888/005
13 ČÍSLO ŠARŽE č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
NovoEight 2 000 IU
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
NovoEight 2 000 IU prášek pro injekční roztok turoctocogum alfa Intravenózní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO SARZE
č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2 000 IU
6. OTHER
NovoEight 3 000 IU prášek a rozpouštědlo pro injekční roztok turoctocogum alfa (humánní koagulační faktor VIII (rDNA))
Po rozpuštění obsahuje 1 ml přípravku NovoEight přibližně 750 IU humánního koagulačního faktoru VIII (rDNA) turoctocogum alfa.
Prášek: chlorid sodný, histidin, sacharosa, polysorbát 80, methionin, dihydrát chloridu vápenatého, hydroxid sodný, kyselina chlorovodíková Rozpouštědlo: chlorid sodný
Prášek a rozpouštědlo pro injekční roztok
Balení obsahuje: 1 injekční lahvičku s práškem, 4 ml rozpouštědla v předplněné injekční stříkačce, nástavec pístu a adaptér injekční lahvičky
Intravenózní podání
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Může být uchováváno při pokojové teplotě < 30°C po jedno nepřetržité období nepřesahující 9 měsíců.
Z chladničky vyjmuto:_
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
12. REGISTRAČNÍ ČÍSLO
EU/1/13/888/006
13 ČÍSLO ŠARŽE č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
NovoEight 3 000 IU
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
NovoEight 3 000 IU prášek pro injekční roztok turoctocogum alfa Intravenózní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO SARZE
č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
3 000 IU
6. OTHER
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
Rozpouštědlo pro NovoEight Chlorid sodný 9 mg/ml
|
2. |
ZPŮSOB PODÁNÍ |
|
3. |
POUŽITELNOST |
|
Použitelné do: | |
|
ČÍSLO ŠARŽE | |
|
4. | |
|
č.š.: | |
|
5. |
OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET |
|
4 ml | |
|
6. |
OTHER |
Novo Nordisk A/S
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
NovoEight 250 IU prášek a rozpouštědlo pro injekční roztok NovoEight 500 IU prášek a rozpouštědlo pro injekční roztok NovoEight 1 000 IU prášek a rozpouštědlo pro injekční roztok NovoEight 1 500 IU prášek a rozpouštědlo pro injekční roztok NovoEight 2 000 IU prášek a rozpouštědlo pro injekční roztok NovoEight 3 000 IU prášek a rozpouštědlo pro injekční roztok
turoctocogum alfa (humánní koagulační faktor VIII (rDNA))
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je NovoEight a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete NovoEight používat
3. Jak se NovoEight používá
4. Možné nežádoucí účinky
5. Jak přípravek NovoEight uchovávat
6. Obsah balení a další informace
1. Co je NovoEight a k čemu se používá
NovoEight obsahuje léčivou látku turoktokog alfa, lidský koagulační faktor VIII. Faktor VIII je bílkovina, která je přirozeně obsažena v krvi a pomáhá vytvářet krevní sraženinu.
NovoEight se používá k léčbě a k prevenci krvácivých příhod u pacientů s hemofilií A (vrozený nedostatek faktoru VIII) a lze ho používat ve všech věkových skupinách.
U pacientů s hemofilií A faktor VIII chybí nebo nefunguje dostatečně. NovoEight nahrazuje tento defekt nebo chybějící faktor VIII a pomáhá tělu vytvářet sraženiny v místě krvácení.
2. Čemu musíte věnovat pozornost, než začnete NovoEight používat Nepoužívejte NovoEight:
• jste-li alergický(á) na léčivou látku nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
• jste-li alergický(á) na křeččí bílkoviny.
Pokud platí něco z výše uvedeného, NovoEight nepoužívejte. Nejste-li si jistý(á), se poraďte před použitím tohoto přípravku s lékařem.
Upozornění a opatření
Před použitím přípravku NovoEight se poraďte se svým lékařem.
Existuje určité riziko, že se u vás vzácně vyskytne anafylaktická reakce (náhlá závažná alergická reakce) na přípravek NovoEight. Časné příznaky alergických reakcí jsou vyrážka, kopřivka, podlitiny, celkové svědění, otoky rtů a jazyka, dýchací potíže, sípot, tlak na prsou, celkový pocit nemoci a závratě.
Pokud se tyto příznaky projeví, okamžitě ukončete aplikaci a kontaktujte lékaře.
Poraďte se s lékařem, pokud se vám zdá, že při podání současné dávky není krvácení pod kontrolou. Může k tomu být několik důvodů. U některých osob používajících tento léčivý přípravek může dojít ke vzniku protilátek proti faktoru VIII (také známých jako inhibitory faktoru VIII). Inhibitory faktoru VIII činní přípravek NovoEight méně účinný při prevenci nebo kontrole krvácení. Pokud k tomu dojde, budete možná potřebovat vyšší dávku přípravku NovoEight nebo může být nutno přejít na jiný přípravek, aby došlo ke kontrole krvácení. Abyste měl(a) krvácení pod kontrolou, nezvyšujte celkovou dávku přípravku NovoEight bez porady s ošetřujícím lékařem. Pokud jste byl(a) dříve léčen(a) přípravky obsahujícími faktor VIII, měl(a) byste to oznámit lékaři. Zvláště, pokud u vás došlo ke vzniku inhibitorů, protože může existovat vyšší riziko jejich opětovného vzniku.
Další léčivé přípravky a přípravek NovoEight
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se s lékařem před použitím tohoto přípravku.
Řízení dopravních prostředků a obsluha strojů
NovoEight nemá vliv na vaši schopnost řídit nebo obsluhovat stroje.
NovoEight obsahuje sodík
Tento léčivý přípravek po rekonstituci (rozpuštění) obsahuje 28 mg sodíku (7 mg/ml).
Oznamte lékaři, pokud dodržujete dietu s kontrolovaným příjmem sodíku.
3. Jak se NovoEight používá
Léčbu přípravkem NovoEight vám předepíše lékař, který má zkušenosti s péčí o pacienty s hemofilií A. Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Lékař vám vypočte dávku. Ta bude záviset na vaší tělesné hmotnosti a důvodu, proč tento přípravek používáte.
Prevence krvácení
Obvyklá dávka je 20-50 mezinárodních jednotek (IU) na kilogram tělesné hmotnosti. Injekce se aplikuje každé 2-3 dny. V některých případech, zvlášť u mladších pacientů, mohou být zapotřebí častější podání nebo vyšší dávky.
Léčba krvácení
Dávka přípravku NovoEight je vypočtena v závislosti na vaší tělesné hmotnosti a hladinách faktoru VIII, kterých je nutno dosáhnout. Cílové hladiny faktoru VIII budou záviset na závažnosti a místě, kde došlo ke krvácení.
Použití u dětí a dospívajících
NovoEight je možno podávat dětem bez ohledu na věk. U dětí (do 12 let) může být zapotřebí vyšších dávek nebo častějších aplikací. Děti (od 12 let) a dospívající mohou používat stejné dávky jako dospělí.
Jak přípravek NovoEight podávat
NovoEight se aplikuje jako injekce do žíly. Více informací naleznete v části „Instrukce pro použití přípravku NovoEight“.
Jestliže si aplikujete více NovoEight, než jste měl(a)
Jestliže jste si aplikoval(a) více NovoEight, než jste měl(a), oznamte to hned lékaři nebo navštivte nemocnici.
Jestliže jste zapomněl(a) použít přípravek NovoEight
Jestliže jste vynechal(a) dávku a nevíte, jak ji nahradit, poraďte se s lékařem.
Jestliže jste přestal(a) používat přípravek NovoEight
Pokud přestanete přípravek NovoEight používat, nejste chráněn(a) proti krvácení či při již probíhajícím krvácení nemusí dojít k jeho zástavě. Nepřestávejte NovoEight používat bez porady s lékařem.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Při používání tohoto přípravku může dojít k níže uvedeným nežádoucím účinkům.
Pokud (velmi vzácně) dojde k náhlým závažným alergickým reakcím (anafylaktickým reakcím), musí být aplikace okamžitě ukončena. Pokud máte některý z následujících časných příznaků, musíte okamžitě kontaktovat lékaře:
• dýchací obtíže, dušnost nebo sípot
• tlak na prsou
• otok rtů a jazyka
• vyrážka, kopřivka, podlitiny nebo generalizovaná vyrážka
• pocit závratě nebo ztráta vědomí
• nízký krevní tlak (bledá chladná pokožka, zrychlená srdeční činnost)
Závažné příznaky jako potíže s polykáním či dýchací obtíže a zarudlý či oteklý obličej nebo ruce vyžadují urychlenou naléhavou léčbu.
Pokud u vás dojde k závažným alergickým reakcím, lékař vám může změnit lék.
Časté nežádoucí účinky (mohou se projevit až u 1 pacienta z 10)
• krevní testy ukazují na změny ve funkci jater
• reakce okolo místa aplikace léku (zarudnutí a svědění)
Méně časté nežádoucí účinky (mohou se projevit až u 1 pacienta ze 100)
• pocit únavy
• bolest hlavy
• pocit závratí
• potíže se spaním (nespavost)
• zrychlený srdeční tep
• zvýšení krevního tlaku
• vyrážka
• horečka
• návaly horka
• svalová ztuhlost
• bolest svalů
• bolest v nohou a pažích
• otoky dolních končetin a chodidel
• onemocnění kloubů
• modřiny.
Další nežádoucí účinky u dětí a dospívajících
Nežádoucí účinky pozorované u dětí a dospívajících jsou shodné s nežádoucími účinky pozorovanými u dospělých.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek NovoEight uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce, na štítku injekční lahvičky a štítku předplněné injekční stříkačky za „Použitelné do“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem.
Před rozpuštěním může být NovoEight uchováván při pokojové teplotě (do 30°C) po jedno nepřetržité období nepřesahující 9 měsíců. Zaznamenejte si prosím na vnější krabičku datum, od kdy uchováváte NovoEight při pokojové teplotě. Poté, co jste začal (a) NovoEight uchovávat při pokojové teplotě, nevracejte jej již nikdy zpět do chladničky.
Jakmile byl NovoEight rozpuštěn, měl by být použit okamžitě. Nemůžete-li ho použít okamžitě po rekonstituci, měl(a) byste ho použít během 4 hodin, pokud byl uchováván při pokojové teplotě (do 30°C) a během 24 hodin, pokud byl uchováván při teplotě 2°C - 8°C. Rekonstituovaný přípravek uchovávejte v injekční lahvičce. Nepoužije-li se přípravek okamžitě, nemusí být již sterilní a může způsobit infekci. Roztok neuchovávejte bez předchozí porady s lékařem.
Prášek v injekční lahvičce je bílý nebo lehce nažloutlý. Prášek nepoužívejte, pokud má jinou barvu.
Připravený roztok je čirý až lehce opalescentní. Nepoužívejte tento přípravek, pokud si všimnete, že je zakalený nebo obsahuje viditelné částice.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co NovoEight obsahuje
- Léčivou látkou je turoctocogum alfa (humánní koagulační faktor VIII (rDNA)). Jedna injekční lahvička s přípravkem NovoEight obsahuje 250, 500, 1 000, 1 500, 2 000 nebo 3 000 IU turoktokogu alfa.
- Pomocnými látkami jsou histidin, sacharosa, polysorbát 80, chlorid sodný, methionin, dihydrát chloridu vápenatého, hydroxid sodný a kyselina chlorovodíková.
- Pomocnými látkami v rozpouštědle je chlorid sodný 9 mg/ml.
Po rekonstituci v přiloženém rozpouštědle (roztok chloridu sodného 9 mg/ml (0,9%) pro injekce) obsahuje připravený injekční roztok 62,5 IU, 125 IU, 250 IU, 375 IU, 500 IU nebo 750 IU turoktokogu alfa v ml (v závislosti na síle turoktokogu alfa tj. 250, 500, 1 000, 1 500, 2 000 nebo 3 000 IU).
Jak NovoEight vypadá a co obsahuje toto balení
Přípravek NovoEight je dostupný v baleních obsahujících 250, 500, 1 000, 1 500, 2 000 nebo 3 000 IU.
Jedno balení přípravku NovoEight obsahuje injekční lahvičku s bílým nebo lehce nažloutlým práškem, předplněnou injekční stříkačku o objemu 4 ml s čirým bezbarvým roztokem, nástavec pístu a adaptér injekční lahvičky.
Držitel rozhodnutí o registraci a výrobce
Novo Nordisk A/S Novo Allé
DK-2880 Bagsv^rd, Dánsko
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Instrukce pro použití přípravku NovoEight
PŘED POUŽITÍM PŘÍPRAVKU NOVOEIGHT SI PEČLIVĚ PŘEČTĚTE TYTO INSTUKCE
NovoEight je dodáván jako prášek. Před podáním (aplikací) musí být rekonstituován v rozpouštědle, které je dodáváno v injekční stříkačce. Rozpouštědlo je roztok chloridu sodného o koncentraci 9 mg/ml (0,9%). Rekonstituovaný NovoEight musí být aplikován do žíly (intravenózní injekce). Toto balení obsahuje potřebné vybavení, které poslouží k rozpuštění a aplikaci přípravku NovoEight.
Budete také potřebovat infuzní soupravu (infuzní set a jehlu s křidélky), sterilní alkoholové tampony, kousky gázy a náplasti. Tyto pomůcky nejsou součástí balení přípravku NovoEight.
Toto vybavení nepoužívejte bez příslušného proškolení lékařem nebo zdravotní sestrou.
Vždy si umyjte ruce a ujistěte se, že pracovní plocha je čistá.
Při přípravě injekce a její aplikaci přímo do žíly je důležité používat čistou a mikrobů prostou (aseptickou) techniku. Nesprávná technika může zanést do krve infekci.
Krabičku s vybavením neotevírejte, dokud nejste připraven(a) ho použít.
Vybavení nepoužívejte, pokud upadlo nebo pokud je poškozené. Použijte místo něj nové balení.
Vybavení nepoužívejte po uplynutí doby použitelnosti. Použijte místo něj nové balení. Doba použitelnosti je vyznačena na krabičce, injekční lahvičce s práškem, adaptéru injekční lahvičky a na předplněné injekční stříkačce za „Použitelné do“.
Vybavení nepoužívejte, pokud máte podezření, že je kontaminováno. Použijte místo něj nové balení.
Žádnou ze součástí vybavení nevyhazujte, dokud nedokončíte aplikaci injekčního roztoku. Vybavení je určeno pouze pro jednorázové použití.
Obsah
Balení obsahuje:
• 1 injekční lahvičku s práškem NovoEight
• 1 adaptér injekční lahvičky
• 1 předplněnou injekční stříkačku s rozpouštědlem
• 1 nástavec pístu (umístěný pod injekční stříkačkou)
Přehled
Injekční lahvička s práškem NovoEight
Plastové víčko
Adaptér injekční lahvičky
Pryžová zátka (pod plastovým víčkem)
Ochranné víčko
Tm Ochranný
(pod ochranným papírem) papír
Předplněná injekční stříkačka s rozpouštědlem
Hrot injekční
stříkačky Píst
(pod víčkem
■ ■ , - , .w, N Stupnice injekční stříkačky) ť
Nástavec pístu
Závit
Horní široký konec
Z
Ochranné víčko injekční stříkačky
1. Příprava injekční lahvičky a injekční stříkačky

Vezměte si potřebný počet balení přípravku NovoEight.
Ověřte si datum doby použitelnosti.
Ověřte název, sílu a barvu balení, abyste se ujistil(a), že obsahuje správný přípravek.
Umyjte si ruce a pečlivě je osušte čistým ručníkem či elektrickým vysoušečem.
Vyjměte z krabičky injekční lahvičku, adaptér injekční lahvičky a předplněnou injekční stříkačku. Nástavec pístu zatím ponechte v krabičce.
Zahřejte injekční lahvičku a předplněnou injekční stříkačku na pokojovou teplotu. Můžete to udělat tak, že je podržíte v ruce, dokud nemají stejnou teplotu jako vaše dlaně.
Jiné způsoby ohřátí injekční lahvičky a předplněné injekční stříkačky nepoužívejte.
Odstraňte plastové víčko z injekční lahvičky. Pokud je víčko uvolněné nebo chybí, injekční lahvičku nepoužívejte.
Pryžovou zátku očistěte sterilním alkoholovým tamponem a nechte ji před použitím několik sekund na vzduchu oschnout, abyste zajistil(a) co možná nejvyšší sterilitu.
Pryžové zátky se prsty nedotýkejte, abyste nezanesl(a) infekci.
2. Připojení adaptéru injekční lahvičky
Sejměte ochranný papír z adaptéru injekční lahvičky.
Pokud ochranný papír není zcela zatavený nebo je protržený, adaptér injekční lahvičky nepoužívejte.
Adaptér injekční lahvičky nevyndávejte prsty z ochranného víčka. Pokud byste se dotkl(a) tmu adaptéru injekční lahvičky, mohla by se z vašich prstů přenést infekce.
Položte injekční lahvičku na hladký a pevný povrch.
Otočte ochranné víčko a nasaďte adaptér na injekční lahvičku.
Jakmile je jednou adaptér nasazen, neodstraňujte jej z injekční lahvičky.
Lehce stiskněte ochranné víčko mezi palcem a ukazováčkem, jak je znázorněno na obrázku.
Sejměte ochranné víčko z adaptéru injekční lahvičky.
Při odstraňování ochranného víčka dejte pozor, abyste nenadzvedl(a) adaptér injekční lahvičky.
3. Nasazení nástavce pístu na injekční stříkačku
• Pevně uchopte nástavec pístu za širší konec a vyjměte ho z krabičky. Nedotýkejte se boků nebo závitu na nástavci pístu.
Pokud byste se dotkl(a) boků nebo závitu, mohla by se z vašich prstů přenést infekce.




|
• Okamžitě nástavec pístu našroubujte po směru hodinových ručiček na píst uvnitř předplněné injekční stříkačky, dokud nepocítíte odpor. |
e_iÉ_. | |
|
• Odstraňte ochranné víčko z předplněné injekční stříkačky ohnutím směrem dolů tak, aby se porušila perforace. Dbejte, abyste se nedotkl(a) hrotu injekční stříkačky pod ochranným víčkem. Pokud byste se dotkl(a) hrotu injekční stříkačky, mohla by se z vašich prstů přenést infekce. Pokud je ochranné víčko uvolněné nebo chybí, předplněnou injekční stříkačku nepoužívejte. |
© Wmy fv | |
|
• Našroubujte předplněnou injekční stříkačku bezpečně na adaptér injekční lahvičky, dokud nepocítíte odpor. |
© | |
|
4. Rekonstituce prášku v rozpouštědle • Držte předplněnou injekční stříkačku lehce nakloněnou s injekční lahvičkou směřující dolů. • Stisknutím nástavce pístu vstříkněte všechno rozpouštědlo do injekční lahvičky. |
jj?' . _ j |
Nechte nástavec pístu zcela stlačený a jemným kroužením injekční lahvičkou rozpusťte všechen prášek.

Injekční lahvičkou netřepejte, mohlo by to způsobit napěnění.
Připravený roztok zkontrolujte. Musí být čirý až lehce opalescentní (mírně zakalený). Pokud obsahuje částečky hmoty nebo má jinou barvu, nepoužívejte ho. Použijte místo toho nové balení.
Doporučuje se použít NovoEight okamžitě po rekonstituci. Je to z toho důvodu, že uchovávaný přípravek už nemusí být sterilní a může způsobit infekci.
Pokud rekonstituovaný roztok přípravku NovoEight nemůžete použít ihned, je třeba ho použít během 4 hodin, pokud byl uchováván při pokojové teplotě (do 30 C), a během 24 hodin, pokud byl uchováván při teplotě 2 °C - 8 °C. Rekonstituovaný přípravek uchovávejte v injekční lahvičce.
Rekonstituovaný roztok nezmrazujte ani ho neuchovávejte v injekční stříkačce.
Bez doporučení lékaře roztok neuchovávejte.
Rekonstituovaný roztok chraňte před přímým slunečním světlem.
CD
Pokud pro svou dávku potřebujete více než jednu injekční lahvičku, opakujte kroky A až J s dalšími injekčními lahvičkami, adaptéry injekčních lahviček a předplněnými injekčními stříkačkami, dokud nedosáhnete požadované dávky.

Ponechte nástavec pístu zcela stlačený.
Otočte injekční stříkačku tak, aby nasazená injekční lahvička byla dnem vzhůru.
Uvolněte nástavec pístu a nechte ho samovolně vrátit se zpět. Tím se
připravený roztok natáhne do injekční stříkačky.
Lehkým vytažením nástavce pístu směrem dolů pak zajistíte, že se do injekční stříkačky natáhne všechen připravený roztok.
V případě, že potřebujete pouze část obsahu injekční lahvičky, použijte stupnici na injekční stříkačce, abyste si ověřil(a), že jste natáhl(a) tolik vzniklého
roztoku, kolik vám doporučil lékař či zdravotní sestra.
Pokud se kdykoliv vyskytne v injekční stříkačce příliš vzduchu, vytlačte ho zpět do injekční lahvičky.
Zatímco držíte injekční lahvičku dnem vzhůru, jemně poklepejte na injekční stříkačku, aby se vzduchové bubliny nashromáždily nahoře.
Pomalu zatlačte na nástavec pístu, dokud všechny vzduchové bubliny neuniknou.

Odšroubujte adaptér s injekční lahvičkou.
Dbejte, abyste se nedotkl(a) hrotu injekční stříkačky. Pokud byste se dotkl(a) hrotu injekční stříkačky, mohla by se z vašich prstů přenést infekce.
5. Aplikace připraveného roztoku
NovoEight je nyní připraven k aplikaci do žíly.
• Aplikujte připravený roztok dle instrukcí lékaře či zdravotní sestry.
• Aplikujte pomalu v průběhu 2 až 5 minut.
• Nemíchejte NovoEight se žádnými dalšími intravenózními infuzemi či přípravky.
Aplikace NovoEight za použití bezjehlových spojek k intravenózním (i.v.) katetrům
Upozornění: předplněná injekční stříkačka je zhotovena ze skla a je kompatibilní (slučitelná) se standardními spoji luer-lock. Některé bezjehlové spojky s vnitřním hrotem jsou s předplněnými injekčními stříkačkami nekompatibilní. Tato nekompatibilita může zabránit aplikaci léku a/nebo může způsobit poškození bezjehlové spojky.
Aplikace roztoku centrálním venózním přístupovým zařízením (CVAD) jako jsou například centrální žilní katetr nebo podkožní port:
• Vždy pracujte čistě a použijte choroboplodných zárodků prostou (aseptickou) techniku. Dodržujte instrukce pro správné používání spojky a CVAD, které vám poskytl lékař nebo zdravotní sestra.
• Aplikace pomocí CVAD může vyžadovat použití sterilní 10ml plastové injekční stříkačky pro natažení rekonstituovaného roztoku. To by mělo být provedeno ihned po kroku J.
• Pokud hadičky CVAD potřebují před aplikací přípravku NovoEight nebo po ní propláchnout, použijte injekční roztok chloridu sodného 9 mg/ml.
Zlikvidování odpadu
• Po aplikaci bezpečně zlikvidujte veškerý _nepoužitý roztok přípravku NovoEight,
|
injekční stříkačku s infuzní soupravou, injekční lahvičku s adaptérem a ostatní odpad dle instrukcí lékárníka. Nevyhazujte do běžného domácího odpadu. |
(33 0 | |
|
Před likvidací použité vybavení nerozebírejte. Použité vybavení znovu nepoužívejte. | ||
1
109