Neorecormon 2000 Iu
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
NeoRecormon vícedávkový 50 000 IU lyofilizát a rozpouštědlo pro přípravu injekčního roztoku
2. KVALITATIVNÍ A KVANTITIATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje 50 000 mezinárodních jednotek (IU), což odpovídá 415 mikrogramům epoetinu beta* (rekombinantní lidský erytropoetin).
Jedna ampulka obsahuje 10 ml rozpouštědla (voda na injekce s benzylalkoholem a benzalkonium-chloridem jako konzervačními látkami).
Jeden ml připraveného roztoku obsahuje 5 000 IU epoetinu beta.
* Vyrobený technologií rekombinatní DNA v ovariálních buňkách čínských křečků.
Pomocné látky se známým účinkem:
Fenylalanin (až do 5,0 mg v injekční lahvičce)
Sodík (méně než 1 mmol v dávce)
Benzylalkohol (do 40 mg na ampulku vícedávkového rozpouštědla)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Lyofilizát a rozpouštědlo pro přípravu injekčního roztoku. Bílý lyofilizát a čiré, bezbarvé rozpouštědlo.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek NeoRecormon je indikován k léčbě:
- Léčba symptomatické anémie spojené s chronickým renálním selháním u dospělých a pediatrických pacientů.
- Léčba symptomatické anémie u dospělých pacientů s nemyeloidními malignitami, kteří jsou léčeni chemoterapií.
- Zvýšení počtu erytrocytů před odběrem krve k autologní transfuzi krve. Použití v této indikaci musí být zváženo vzhledem k udávanému zvýšení rizika vzniku tromboembolických příhod. Použití je indikováno pouze u pacientů s mírnou anémií (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], bez nedostatku železa), pokud postupy pro konzervování krve nejsou dostupné nebo účinné nebo plánovaný zvolený velký chirurgický zákrok vyžaduje velký objem krve (4 a více jednotek krve pro ženy nebo 5 a více jednotek pro muže). Viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba přípravkem NeoRecormon může být zahájena pouze lékařem se zkušenostmi s výše uvedenými
indikacemi přípravku. Protože byly vzácně zaznamenány anafylaktoidní reakce po podání přípravku,
první dávka má být aplikována pod dohledem zdravotnického personálu.
Dávkování
Léčba symptomatické anémie u dospělých a pediatrických pacientů s chronickým selháním ledvin Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta. Přípravek NeoRecormon má být podáván buď subkutánně nebo intravenózně, tak aby byla hladina hemoglobinu zvýšena maximálně na 12 g/dl (7,5 mmol/l). U pacientů, kteří nepodstupují dialýzu, se upřednostňuje subkutánní podání, kdy není zapotřebí vpichů do periferních žil. V případě intravenózního podání má být příp ravek podáván alespoň po dobu 2 minut, například přes arterio-venózní píštěl na konci dialýzy u pacientů na hemodialýze.
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu v rozsahu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Zvýšení hladiny hemoglobinu o více než 2 g/dl (1,25 mmol/l) během období čtyř týdnů není žádoucí. Pokud k němu dojde, má být dávka odpovídajícím způsobem upravena, jak je uvedeno dále. Je-li rychlost vzestupu hladiny hemoglobinu vyšší než 2 g/dl (1,25 mmol/l) za měsíc nebo pokud se hladina hemoglobinu zvýší až na hodnotu 12 g/dl (7,45 mmol/l) je třeba dávku snížit přibližně o 25 %. Pokud by se hladina hemoglobinu dále zvyšovala, má být léčba přerušena, dokud nezačne hladina opět klesat, a v tomto bodě má být znovu zahájena léčba dávkou přibližně o 25 % nižší, než byla předchozí podávaná dávka.
Pacienti mají být důkladně sledováni a je třeba ověřit, že byla použita nejnižší schválená účinná dávka přípravku NeoRecormon, která postačuje pro kontrolu symptomů anémie při zachování koncentrace hemoglobinu pod nebo na hodnotě 12 g/dl (7,45 mmol/l).
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin. U pacientů se slabou odpovědí hemoglobinu na přípravek NeoRecormon mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz bod 4.4 a 5.1).
V případě hypertenze nebo stávajících kardiovaskulárních, cerebrovaskulárních nebo periferních vaskulárních onemocnění má být týdenní zvýšení Hb a cílová hodnota Hb stanovena individuálně při zvážení klinického obrazu.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází.
1. Fáze korekční
- Subkutánní podání:
- Počáteční dávka je 3 x 20 IU/kg tělesné hmotnosti týdně. Dávka může být zvýšena každé
4 týdny o 3 x 20 IU/kg za týden, jestliže není dosaženo adekvátního vzestupu Hb (< 0,25 g/dl za týden).
- Týdenní dávka může být také rozdělena na denní dávky.
- Intravenózní podání:
Počáteční dávka je 3 x 40 IU/kg tělesné hmotnosti týdně. Dávka může být po 4 týdnech zvýšena na 80 IU/kg třikrát týdně a pokud je nutné další zvýšení dávky, má být o 20 IU/kg třikrát týdně v měsíčních intervalech.
Maximální dávka při obou způsobech podání nemá překročit 720 IU/kg za týden.
2. Fáze udržovací
Pro udržení Hb mezi 10 až 12 g/dl je dávka ze začátku snížena na polovinu předešlé dávky. Následně je u pacienta dávka nastavena v intervalech jednoho nebo dvou týdnů individuálně (udržovací dávka).
V případě podkožního podání může být týdenní dávka podána v jedné injekci týdně, nebo může být rozdělena do tří až sedmi dávek týdně. Pacienti, kteří jsou stabilní na režimu podávání jednou týdně, mohou být převedeni na dávkování jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Výsledky klinických studií u dětí prokázaly, že čím jsou pacienti mladší, tím v průměru vyšší dávky přípravku NeoRecormon vyžadují. Doporučený plán dávkování má však být dodržován, protože klinický účinek u jednotlivých pacientů nelze zcela předvídat.
Léčba přípravkem NeoRecormon je obvykle dlouhodobá. Může však být, pokud je to nutné, kdykoliv přerušena. Údaje týkající se dávkovacího schématu podání jednou týdně vycházejí z klinických studií s délkou léčby 24 týdnů.
Léčba symptomatické chemoterapií indukované anémie u pacientů s nádorovým onemocněním Přípravek NeoRecormon má být podáván subkutánně pacientům s anemií (např. koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l)). Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta.
Týdenní dávka může být podána v jedné injekci jednou týdně nebo může být rozdělena na 3 až 7 jednotlivých dávek.
Doporučená úvodní dávka je 30 000 IU týdně (to odpovídá přibližně 450 IU/kg tělesné hmotnosti týdně u pacienta s průměrnou hmotností).
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Pokud dojde po 4 týdnech léčby ke zvýšení hodnot hemoglobinu o minimálně 1 g/dl (0,62 mmol/l), má se v podávání této dávky dále pokračovat. Pokud hladina hemoglobinu nestoupne o alespoň 1 g/dl (0,62 mmol/l), má být zváženo podávání dvojnásobné týdenní dávky. Pokud hladina hemoglobinu nestoupne po 8 týdnech léčby o alespoň 1 g/dl (0,62 mmol/l), je odpověď nepravděpodobná a léčba má být ukončena.
Léčba má pokračovat až 4 týdny po ukončení chemoterapie.
Maximální dávka nemá překročit 60 000 IU týdně.
Jakmile je u jednotlivého pacienta dosaženo léčebného cíle, dávka má být snížena o 25 až 50 %, aby byla udržena hladina hemoglobinu na této úrovni. Je zapotřebí vzít v úvahu možnost titrace dávky.
Překročí-li hemoglobin hladinu 12 g/dl (7,5 mmol/l), má být dávka snížena zhruba o 25 až 50 %.
Pokud hladina hemoglobinu překročí 13 g/dl (8,1 mmol/l), má být léčba přípravkem NeoRecormon dočasně přerušena. Pokud hladina hemoglobinu klesne na 12 g/dl (7,5 mmol/l) nebo níže, má být léčba opět zahájena s dávkou přibližně o 25 % nižší, než byla předchozí dávka.
Pokud je nárůst hemoglobinu v průběhu 4 týdnů vyšší než 2 g/dl (1,3 mmol/l), dávka má být snížena o 25 až 50 %.
Pacienti mají být důkladně sledováni, přičemž je třeba ověřit, že byla použita nejnižší schválená dávka přípravku NeoRecormon postačující pro adekvátní kontrolu symptomů anémie.
Podávání pro zvýšení množství autologní krve
Rekonstituovaný roztok je aplikován intravenózně po dobu asi 2 minut nebo subkutánně.
Přípravek NeoRecormon je podáván dvakrát týdně po dobu 4 týdnů. V případě, kdy hematokrit pacienta umožňuje odběr krve, tj. hodnota hematokritu je > 33 %, je přípravek NeoRecormon podáván v okamžiku ukončení odběru.
V průběhu celé léčby nemá být překročena hodnota hematokritu 48 %.
Dávkování musí být stanoveno chirurgickým týmem zvlášť pro každého pacienta podle požadovaného množství krve pro autologní transfuzi a endogenní rezervy červených krvinek:
1. P ožadované množství krve pro autologní transfuzi závisí na předpokládané ztrátě krve, p opřípadě na použití postupů konzervování krve a na fyzické kondici pacienta.
Množství potřebné autologní krve má stačit k tomu, aby bylo možné se vyhnout transfuzi homologní krve.
Požadované množství předem odebrané krve je vyjádřeno v jednotkách, přičemž jedna jednotka v nomogramu je ekvivalentní 180 ml červených krvinek.
2. Možnost autologní transfuze krve závisí především na objemu pacientovy krve a na výchozí hodnotě hematokritu. Obě proměnné určují endogenní rezervu červených krvinek, kterou je možné vypočítat podle následujících vzorců:
Endogenní rezerva červených krvinek = objem krve [ml] x (hematokrit - 33)/100 Ženy: objem krve [ml] = 41 [ml/kg] x tělesná hmotnost [kg] + 1200 [ml]
Muži: objem krve [ml] = 44 [ml/kg] x tělesná hmotnost [kg] + 1600 [ml]
(tělesná hmotnost > 45 kg)
Indikace pro léčbu přípravkem NeoRecormon a jednotlivá dávka mají být stanoveny z požadovaného množství předem odebrané krve a endogenní rezervy červených krvinek podle následujících grafů.
Ženy
Požadované množství krve pro autologní transfuzi [jednotky]
Muži
Požadované množství krve pro autologní transfuzi [jednotky]
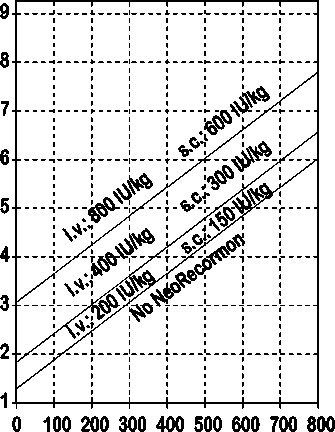
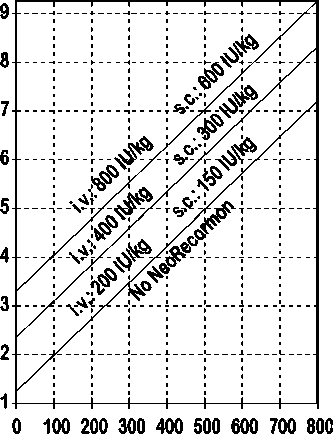
Endogenní rezerva červených krvinek [ml]
Endogenní rezerva červených krvinek [ml]
Takto stanovená jednotlivá dávka je podávána dvakrát týdně po dobu 4 týdnů. Maximální dávka nemá překročit 1600 IU/kg tělesné hmotnosti a týden při intravenózním podání nebo 1200 IU/kg tělesné hmotnosti a týden při subkutánním podávání.
Způsob podání
Přípravek ve formě „vícedávkový“ může být použit pro více pacientů. Aby bylo zabráněno riziku přenosu infekce, dodržujte vždy zásady aseptické aplikace a pro každé podání používejte vždy sterilní stříkačky a jehly. V jednom okamžiku musí být pro použití připravena jen jedna injekční lahvička přípravku NeoRecormon vícedávkový (tzn. je rozpuštěna).
Rekonstituovaný produkt je bezbarvý, čirý až lehce opalescentní roztok.
Pokyny pro rekonstituci léčivého přípravku před podáním viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku (uvedenou v bodě 6.1).
Špatně kontrolovatelná hypertenze.
V indikaci “podání pro zvýšení množství autologní krve”: infarkt myokardu nebo cévní mozková příhoda v průběhu jednoho měsíce před zákrokem, nestabilní angina pectoris, zvýšené riziko hluboké venózní trombózy jako např. po prodělaném tromboembolickém onemocnění.
Přípravek NeoRecormon vícedávkový obsahuje jako konzervační prostředek benzylalkohol a nesmí být proto podáván kojencům a dětem do 3 let věku.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek NeoRecormon má být užíván s opatrností u pacientů s refrakterní anémií s nadbytkem transformovaných blastů, při epilepsii, trombocytóze a chronickém selhání jater. Je nutné vyloučit nedostatek kyseliny listové a vitamínu BJ2, protože tyto stavy snižují účinek přípravku NeoRecormon.
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin, neboť vysoké kumulující se dávky epoetinu mohou mít spojitost se zvýšeným rizikem mortality, závažnými kardiovaskulárními a cerebrovaskulárními příhodami. U pacientů se slabou odpovědí hemoglobinu na epoetiny mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz body 4.2 a 5.1).
Je třeba zhodnotit u všech pacientů hladinu železa před a v průběhu léčby, aby byla zajištěna účinná erytropoéza. Může být nutná substituce železa prováděná v souladu s léčebnými doporučeními.
Závažné zatížení hliníkem, ke kterému během léčby renálního selhání může dojít, může rovněž účinek přípravku NeoRecormon snížit.
Indikace k léčbě přípravkem NeoRecormon u pacientů s nefrosklerózou, kteří ještě nejsou dialyzovaní, má být zvážena individuálně, protože u těchto pacientů nelze vyloučit urychlení postupu renálního selhání.
Čistá aplazie buněk červené krevní řady (PRCA)
PRCA způsobená neutralizujícími antierytropoetinovými protilátkami byla hlášena ve spojení s léčbou erytropoetiny, včetně přípravku NeoRecormon. Bylo prokázáno, že tyto protilátky zkříženě reagují se všemi erytropoetinovými proteiny, a proto pacienti, u kterých je podezření nebo je potvrzen výskyt neutralizujících protilátek proti erytropoetinu, nemají být převáděni na přípravek NeoRecormon (viz bod 4.8).
PRCA u pacientů s hepatitidou C
Paradoxní pokles hladiny hemoglobinu a rozvoj závažné anémie související s nízkým počtem retikulocytů má vést k okamžitému přerušení léčby epoetinem a provedení testů na protilátky proti erytropoetinu. Tyto případy byly hlášeny, pokud byly pacientům s hepatitidou C léčených interferonem a ribavirinem současně podávány epoetiny. Epoetiny nejsou schválené pro léčbu anémie související s hepatitidou C.
Monitorování krevního tlaku
Může dojít ke vzestupu krevního tlaku nebo ke zhoršení již existující hypertenze zejména v případě rychlého vzestupu hematokritu. Toto zvýšení krevního tlaku je možno upravit pomocí léků. Pokud zvýšení krevního tlaku nemůže být upraveno medikamentózně, je doporučeno krátkodobé přerušení léčby přípravkem NeoRecormon. Zvláště na začátku terapie se doporučuje pravidelné monitorování krevního tlaku, a to rovněž mezi dialýzami. Může dojít ke vzniku hypertenzní krize s příznaky napodobujícími encefalopatii, která vyžaduje okamžitou lékařskou intervenci a intenzivní péči.
Zvláštní pozoronost je třeba věnovat náhlým bodavým migrenosním bolestem hlavy jako možnému varovnému příznaku.
Chronické selhání ledvin
U pacientů s chronickým selháním ledvin, léčených přípravkem NeoRecormon, může dojít v průběhu léčby k mírnému, na dávce závislému nárůstu počtu krevních destiček v rozmezí normálních hodnot, zvláště pak v případě intravenózního podávání. K poklesu tohoto nárůstu dochází v dalším průběhu léčby. Je doporučeno pravidelně sledovat počet krevních destiček v průběhu prvních 8 týdnů léčby.
Koncentrace hemoglobinu
U pacientů s chronickým selháním ledvin nemá udržovací koncentrace hemoglobinu překročit horní limit cílové koncentrace hemoglobinu doporučované v bodě 4.2. V klinických studiích bylo pozorováno zvýšené riziko úmrtí a závažných kardiovaskulárních příhod nebo cerebrovaskulárních příhod, včetně cévní mozkové příhody, pokud byly používány přípravky stimulující erytropoézu (ESA) k dosažení cílových hodnot hemoglobinu vyšších než 12 g/dl (7,5 mmol/l).
V kontrolovaných klinických studiích se neprokázal žádný signifikantní přínos podávání epoetinů, pokud byla koncentrace hemoglobinu zvyšována nad hladinu nezbytně nutnou pro kontrolu symptomů anémie a předcházení krevní transfuze.
Účinek na růst nádorů
Epoetiny jsou růstové faktory, které primárně podporují tvorbu červených krvinek. Erytropoetinové receptory mohou být přítomny na povrchu různých nádorových buněk. Stejně jako u všech růstových faktorů je třeba mít na zřeteli, že i epoetiny mohou stimulovat rozvoj nádoru. V několika kontrolovaných klinických studiích nebylo ve spojení s epoetiny prokázáno zlepšení celkové doby přežití ani snížení rizika progrese nádorů u pacientů s anémií spojenou s nádorovým onemocněním.
V kontrolovaných klinických studiích zaměřených na použití přípravku NeoRecormon a ostatních erytropoézu stimulujících přípravků (ESA), bylo zjištěno:
- zkrácení doby do progrese nádoru u pacientů s pokročilým nádorovým onemocněním hlavy a krku, pokud byly dosahovány cílové hladiny hemoglobinu vyšší než 14 g/dl (8,7 mmol/l)
- zkrácení celkové doby přežití a zvýšení počtu úmrtí souvisejících s progresí onemocnění během 4 měsíců léčby u pacientů s metastazujícím karcinomem prsu léčených chemoterapií, pokud byly dosahovány cílové hladiny hemoglobinu 12-14 g/dl (7,5-8,7 mmol/l),
- zvýšení rizika úmrtí při dosahování cílové hladiny hemoglobinu 12 g/dl (7,5 mmol/l) u pacientů s aktivními maligními chorobami, kteří nedostávali ani chemoterapii ani ozařování. V této populaci pacientů není použití ESA indikováno.
Na základě výše uvedených skutečností má být za určitých klinických okolností při léčbě anémie u pacientů s nádorovým onemocněním upřednostněna transfuze krve. Rozhodnutí podat rekombinantní erytropoetin má být přijato na základě zvážení poměru přínosu a rizika a individuálního posouzení jednotlivého pacienta za daných specifických klinických podmínek. Další faktory, které mají být zváženy, je posouzení typu a stádia nádorového onemocnění; stupeň anémie; očekávaná délka přežití; podmínky, za kterých je pacient léčen; a vlastní volba pacienta (viz bod 5.1)
Může dojít ke vzestupu krevního tlaku, který lze farmakologicky léčit. Doporučuje se proto monitorovat krevní tlak, zejména v úvodní fázi léčby pacientů s nádorovým onemocněním.
U onkologických pacientů mají být také pravidelně sledovány počty krevních destiček a hladina hemoglobinu.
U pacientů v programu před autologní transfuzí krve může být zvýšení počtu krevních destiček, většinou v rozmezí normálních hodnot. Proto je doporučeno určovat počet krevních destiček u těchto pacientů alespoň jednou týdně. Pokud dojde ke zvýšení krevních destiček o více než 150 x 109/l nebo když nárůst přesáhne normální hodnoty, má být léčba přípravkem NeoRecormon přerušena.
U pacientů s chronickým selháním ledvin je v průběhu léčby přípravkem NeoRecormon při hemodialýze často nutné zvýšení dávky heparinu z důvodu zvýšeného hematokritu. Pokud není heparinizace optimální, může dojít k okluzi systému dialýzy.
U pacientů s chronickým selháním ledvin s rizikem trombózy cévního přístupu má být zvážena časná kontrola cévního přístupu a trombotická profylaxe kyselinou acetylsalicylovou.
V průběhu léčby přípravkem NeoRecormon má být pravidelně monitorována hladina sérového draslíku a fosfátů. Zvýšení kalémie bylo zaznamenáno u několika uremických pacientů léčených přípravkem NeoRecormon, i když příčinná souvislost nebyla ověřena. Pokud je pozorována zvýšená nebo stoupající hladina draslíku, pak má být zváženo přerušení podávání přípravku NeoRecormon dokud se kalémie neupraví.
Pro použití přípravku NeoRecormon v programu před autologní transfuzí krve musí být zváženy obecné zásady dárcovství krve, zvláště:
- dárci mají být pouze pacienti s hematokritem > 33 % (hemoglobin > 11g/dl [6,83 mmol/l];
- má být věnována zvláštní pozornost pacientům s hmotností nižší než 50 kg;
- jednotlivý čerpaný objem nemá překročit cca 12 % odhadnutého objemu krve pacienta.
Léčba má být vyhrazena pro pacienty, u kterých je zvlášť důležité vyhnout se transfuzi homologní krve a má být zvážena rizika a prospěch homologní transfuze.
Zneužití
Zneužití přípravku zdravými osobami může vést k nadměrnému zvýšení hematokritu. Toto zvýšení může být spojeno s život ohrožujícími kardiovaskulárními komplikacemi.
Pomocné látky
Přípravek NeoRecormon vícedávkový obsahuje až 5,0 mg fenylalaninu v jedné injekční lahvičce jako pomocnou látku. Tato skutečnost má být brána v úvahu při léčbě pacientů trpících vážnými formami fenylketonurie.
Přípravek NeoRecormon v rekonstituovaném vícedávkovém roztoku obsahuje benzylalkohol, který může způsobit toxické a anafylaktoidní reakce u kojenců a dětí do 3 let.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v dávce, tj. v podstatě je „bez sodíku“
Zpětná zjistitelnost přípravku NeoRecormon
Z důvodu snadnější zpětné zjistitelnosti látek stimulujících erytropoézu (ESAs) má být obchodní název předepsaného přípravku ESA zřetelně zaznamenán (nebo vyznačen) v pacientově dokumentaci.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Z dostupných klinických výsledků nejsou známy žádné interakce přípravku NeoRecormon s ostatními léčivými přípravky.
Pokusy na zvířatech prokázaly, že epoetin beta nezvyšuje myelotoxicitu cytostatických léčivých látek jako jsou etoposid, cisplatina, cyklofosfamid a fluorouracil.
4.6 Fertilita, těhotenství a kojení
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3).
Nejsou k dispozici žádné klinické údaje o účincích epoetinu beta během těhotenství.
Při předepisování těhotným ženám nutno postupovat opatrně.
Kojení
Není známo, zda se epoetin beta vylučuje do lidského mateřského mléka.
Rozhodnutí, zda pokračovat v kojení/přerušit kojení nebo pokračovat v léčbě/přerušit léčbu epoetinem beta, má být zváženo v závislosti na prospěšnosti kojení pro dítě a prospěšnosti léčby epoetinem beta u matky.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek NeoRecormon nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Na základě výsledků klinických studií s 1725 pacienty lze očekávat, že přibližně u 8 % pacientů léčených přípravkem NeoRecormon se mohou vyskytnout nežádoucí účinky.
Anemičtí pacienti s chronickým selháním ledvin
Nejčastějším nežádoucím účinkem v průběhu léčby přípravkem NeoRecormon je zvýšení krevního tlaku nebo zhoršení stávající hypertenze, zvláště pak v případech rychlého zvýšení hodnot hematokritu (viz bod 4.4). Hypertenzní krize s příznaky encefalopatie (např. bolesti hlavy a stavy zmatenosti, senzoricko-motorické poruchy jako například porucha řeči nebo zhoršení chůze až tonicko-klonické křeče) se mohou objevit také u jednotlivých pacientů s jinak normálním nebo nízkým krevním tlakem (viz bod 4.4).
Může dojít k trombóze žilního přístupu zejména u pacientů s tendencí k hypotenzi nebo s komplikacemi arteriovenózního zkratu (např. stenózy, aneuryzmata), viz bod 4.4. Ve většině případů je pozorován pokles hodnot sérového železa souběžně se vzestupem hematokritu (viz bod 4.4). Navíc byl v ojedinělých případech pozorován vzestup sérového draslíku a hladin fosfátů (viz bod 4.4).
V ojedinělých případech byla v souvislosti s léčbou přípravkem NeoRecormon hlášena čistá aplazie červených krvinek (PRCA) způsobená neutralizujícími antitrombopoetinovými protilátkami.
V případě diagnózy čisté aplazie červených krvinek (PRCA) způsobené neutralizujícími antitrombopoetinovými protilátkami musí být léčba přípravkem NeoRecormon ukončena a pacient nemá být převáděn na léčbu jinou erytropoetickou bílkovinou (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 1.
Pacienti s rakovinou
Bolesti hlavy související s léčbou epoetinem beta a hypertenze, kterou lze medikamentózně léčit, jsou časté (viz bod 4.4).
U některých pacientů je pozorován pokles sérového železa (viz bod 4.4).
V klinických studiích byl prokázán vyšší výskyt tromboembolických událostí u pacientů s karcinomem, léčených přípravkem NeoRecormon ve srovnání s neléčenými kontrolními skupinami nebo placebem. U pacientů léčených přípravkem NeoRecormon byla incidence 7 % ve srovnání s 4 % u kontrolních skupin; aniž by to bylo spojeno se zvýšenou mortalitou na tromboembolické události ve srovnání s kontrolními skupinami.
Nežádoucí účinky jsou uvedeny níže v tabulce 2.
Pacienti v programu autologního dárcovství krve
U pacientů v programu autologního dárcovství krve byla hlášena lehce zvýšená četnost tromboembolických událostí. Kauzální vztah k léčbě přípravkem NeoRecormon však nebyl prokázán.
Ve studiích kontrolovaných placebem byl více vyjádřen přechodný deficit železa u pacientů léčených přípravkem NeoRecormon než u kontrolní skupiny (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 3. Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů MedDRA a absolutní četnosti. Kategorie četnosti jsou definovány dle následující konvence:
velmi časté (> 1/10); časté (>1/100 to <1/10); méně časté (>1/1 000 to <1/100); vzácné (>1/10 000 to <1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
Tabulka 1: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů s chronickým selháním ledvin léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze Hypertenzní krize |
Časté Méně časté |
|
Poruchy nervového systému |
Časté | |
|
Poruchy krve a |
Trombóza cévního |
Vzácné |
|
lymfatického systému |
přístupu Trombocytóza |
Velmi vzácné |
Tabulka 2: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u onkologických pacientů léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze |
Časté |
|
Poruchy krve a lymfatického systému |
Tromboembolické účinky |
Časté |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Tabulka 3: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů v programu autologního dárcovství krve léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Popis vybraných nežádoucích účinků
Vzácně byly pozorovány kožní reakce jako jsou vyrážka, svědění, kopřivka nebo reakce v místě vpichu. Velmi vzácně byly popsány anafylaktoidní reakce související s léčbou epoetinem beta. V kontrolovaných klinických studiích však zvýšení výskytu hypersenzitivních reakcí nebylo popsáno.
Ve velmi vzácných případech, především na počátku léčby, byl zaznamenán výskyt příznaků podobných chřipce ("flu-like" symptomy) jako je horečka, zimnice, bolesti hlavy, bolest v končetinách, malátnost a/nebo bolest kostí. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Údaje z kontrolované klinické studie s epoetinem alfa nebo darbepoetinem alfa udávaly incidenci cévní mozkové příhody jako častou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Terapeutický rozsah přípravku NeoRecormon je velmi široký. Dokonce i při velmi vysokých hladinách v séru nebyly pozorovány žádné příznaky otravy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA01 Mechanismus účinku
Erytropoetin je glykoprotein, který stimuluje tvorbu erytrocytů z prekurzorů kmenových buněk.
Působí jako faktor stimulující mitózu kmenových buněk červené krevní řady a hormon působící na jejich diferenciaci.
Epoetin beta, léčivá látka přípravku NeoRecormon, je svým složením aminokyselin a cukrů identický s erytropoetinem, který byl izolován z moči anemických pacientů.
Biologický účinek epoetinu beta byl demonstrován po intravenózní a podkožní aplikaci u různých pokusných zvířat in vivo (normální a uremičtí potkani, polycytemické myši, psi). Po podání epoetinu beta se zvýší počet erytrocytů, hodnota Hb a retikulocytů stejně tak jako rychlost inkorporace 59Fe.
In vitro byla zjištěna zvýšená inkorporace 3H-thymidinu v erytroidních jaderných buňkách sleziny (kultura buněk sleziny myší) po inkubaci s epoetinem beta.
Pokusy na tkáňových kulturách buněk lidské kostní dřeně prokázaly, že epoetin beta stimuluje jen tvorbu červených krvinek a neovlivňuje tvorbu bílých krvinek. Nebyly zjištěny cytotoxické účinky epoetinu beta na kostní dřeň ani na lidské kožní buňky.
Po podání jednotlivé dávky epoetinu beta nebyly pozorovány žádné vlivy na chování nebo na pohybovou činnost u myší a oběhové nebo respirační funkce u psů.
Klinická účinnost a bezpečnost
V randomizované dvojitě zaslepené, placebem kontrolované studii, která hodnotila 4038 pacientů s chronickým selháním ledvin bez nutnosti dialýzy, s diabetem 2. typu a hladinami hemoglobinu < 11 g/dl, byli pacienti léčeni buď darbepoetinem alfa až do dosažení cílových hladin hemoglobinu 13 g/dl, nebo dostávali placebo (viz bod 4.4). Studie nesplnila žádný z primárních cílů, který by prokázal snížení rizika úmrtí ze všech příčin, kardiovaskulární morbidity nebo terminálního stádia onemocnění ledvin (ESRD). Analýza jednotlivých komponent složených cílových parametrů prokázala následující poměr rizik (95% CI): úmrtí 1,05 (0,92; 1,21), cévní mozková příhoda 1,92 (1,38; 2,68), městnavé srdeční selhání 0,89 (0,74; 1,08), infarkt myokardu 0,96 (0,75; 1,23), hospitalizace z důvodu ischemie myokardu 0,84 (0,55; 1,27), terminální stádium onemocnění ledvin 1,02 (0,87; 1,18).
Souhrnné post-hoc analýzy klinických studií s ESA byly provedeny u pacientů s chronickým selháním ledvin (u pacientů, kteří byli nebo nebyli na dialýze, u diabetiků a u pacientů bez diabetu). Byla pozorována tendence směrem ke zvýšení odhadovaného rizika mortality z jakýchkoli důvodů, rizika kardiovaskulárních a cerebrovaskulárních příhod spojených s vyššími kumulativními dávkami ESA, nezávisle na stavu diabetu nebo dialýze (viz body 4.2 a 4.4).
Erytropoetin je růstový faktor primárně podporující tvorbu červených krvinek. Receptory pro erytropoetin mohou být exprimovány na povrchu mnoha nádorových buněk.
Přežití a progrese nádoru byla zkoumána v pěti velkých kontrolovaných klinických studiích zahrnujících celkem 2833 pacientů; čtyři z těchto studií byly dvojitě zaslepené kontrolované placebem, jedna studie byla otevřená. Dvou studií se účastnili pacienti léčení chemoterapií. Cílová koncentrace hemoglobinu byla ve dvou studiích > 13 g/dl; v ostatních třech studiích 12-14 g/dl.
V otevřené studii nebyl z hlediska celkového přežití zjištěn žádný rozdíl mezi pacienty léčenými rekombinantním lidským erytropoetinem a mezi kontrolní skupinou. Ve čtyřech placebem kontrolovaných studiích se poměry rizik pro celkové přežití pohybovaly mezi 1,25 a 2,47 ve prospěch kontrolních skupin. Při srovnání s kontrolními skupinami ukázaly tyto studie konzistentní nevysvětlené statisticky významné zvýšení mortality u pacientů, kteří měli anémii spojenou s různými běžnými maligními nádory a kteří dostávali rekombinantní lidský erytropoetin. Výsledek celkového přežití zjištěný v klinických studiích nebylo možné uspokojivě vysvětlit rozdílem v incidenci trombózy a přidružených komplikací mezi skupinou dostávající rekombinantní lidský erytropoetin a mezi skupinou kontrolní.
Metaanalýza založená na údajích jednotlivých pacientů zahrnovala data ze všech 12 kontrolovaných klinických studií prováděných u pacientů s anémií a maligním nádorovým onemocněním, kteří byli léčeni přípravkem NeoRecormon (n=2301), ukázala celkový odhadovaný poměr rizik pro přežití 1,13 ve prospěch kontrol (95% CI 0,87; 1,46). U pacientů, kteří měli v baseline hladinu hemoglobinu <
10 g/dl (n=899), byl odhadovaný poměr rizik pro přežití 0,98 (95% CI 0,68 až 1,40). V celkové populaci bylo pozorováno zvýšené relativní riziko tromboembolických příhod (RR 1,62, 95% CI:
1,13; 2,31).
Byla provedena analýza dat na úrovni jednotlivých nemocných u více než 13 900 pacientů se zhoubným nádorem (léčených chemoterapií, radioterapií, chemoradioterapií nebo bez terapie) zařazených do 53 kontrolovaných klinických studií zahrnujících podávání několika epoetinů. Metaanalýza údajů celkového přežití prokázala, že poměr rizik (hazard ratio) je odhadem 1,06 ve prospěch kontrolních skupin (95% CI: 1,00, 1,12; 53 studií a 13 933 pacientů) a u pacientů se zhoubným nádorem podstupujících chemoterapii byl poměr rizik (hazard ratio) celkového přežití 1,04 (95% CI: 0,97, 1,11; 38 studií a 10 441 pacientů). Meta-analýzy zároveň konzistentně poukazují na významné zvýšení relativního rizika tromboembolických příhod u pacientů se zhoubným nádorem, kteří dostávají rekombinantní lidský erytropoetin (viz bod 4.4).
Ve velmi vzácných případech se v průběhu léčby rekombinantním lidským erytropoetinem objevily neutralizující protilátky proti erytropoetinu s nebo bez čisté aplazie červené krevní řady (PRCA).
5.2 Farmakokinetické vlastnosti
Farmakokinetické sledování u zdravých dobrovolníků a uremických pacientů ukázalo, že po intravenózním podání je poločas epoetinu beta mezi 4 - 12 hodinami a že distribuční objem odpovídá jedno- až dvojnásobku plazmatického objemu. Analogické výsledky byly nalezeny v pokusech na zvířatech u normálních i uremických potkanů.
Protrahovaná absorpce epoetinu beta po subkutánním podání uremickým pacientům vede ke vzniku koncentračního plató, přičemž maximální koncentrace je dosaženo v průměru za 12 - 28 hodin. Terminální poločas je delší než po intravenózním podání s průměrem 13 - 28 hodin.
Ve srovnání s intravenózní aplikací je biologická dostupnost epoetinu beta po podkožní aplikaci mezi 23 - 42 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
Studie kancerogenity s homologním erytropoetinem u myší neodhalily žádné známky proliferativního nebo kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 S eznam pomocných látek
Lyofilyzát:
Močovina,
Chlorid sodný,
Polysorbát 20,
Dihydrogenfosforečnan sodný,
Hydrogenfosforečnan sodný,
Chlorid vápenatý,
Glycin,
L-leucin,
L-isoleucin,
L-threonin,
L- kyselina glutamová,
L- fenylalanin.
Rozpouštědlo:
Benzylalkohol,
Benzalkonium chlorid,
Voda na injekci.
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6 (obsah přiložené ampulky s rozpouštědlem).
6.3 Doba použitelnosti
3 roky.
Chemická a fyzikální stabilita používaného roztoku byla prokázána jeden měsíc po rekonstituci, pokud byl roztok uchováván při 2 - 8 °C. Z mikrobiologického hlediska může být rekonstituovaný roztok po prvním otevření skladován nejvýše jeden měsíc při 2 - 8 °C. Používání roztoku při nedodržení doby a podmínek skladování je na zodpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte lahvičku v původním obalu, aby byl přípravek chráněn před světlem.
Pro usnadnění ambulantního použití může pacient vyjmout přípravek z chladničky a uchovávat jej jednorázově po dobu maximálně 5 dnů při pokojové teplotě (maximálně do 25 °C).
Chlazení již připraveného roztoku může být přerušeno na dobu nutnou pro podání přípravku.
Podmínky uchovávání rekonstituovaného léčivého přípravku viz bod 6.3.
6.5 Druh obalu a obsah balení
Lyofilizát: injekční lahvička (sklo typu I) se zátkou (pryž potažená teflonem).
Rozpouštědlo: 10 ml ampulka (sklo typu I)
Balení obsahuje 1 injekční lahvičku s 50 000 IU epoetinu beta, 1 ampulku obsahující 10 ml rozpouštědla, 1 rekonstituční zařízení pro manipulaci při rozpouštění a nabírání roztoku, 1 jehlu (21G2, nerezová ocel) a 1 jednorázovou injekční stříkačku (polypropylén a polyetylén).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek NeoRecormon vícedávkový je dodáván jako lyofilizát určený k přípravě injekčního roztoku v injekční lahvičce. Rozpouštědlo je v přiložené ampulce a rozpuštění se provede pomocí speciálního rekonstitučního zařízení pro manipulaci a nabírání roztoku dle pokynů viz níže. Podávány mohou být pouze takové roztoky, které jsou čiré nebo lehce opalescentní, bezbarvé a bez viditelných částic. Nepoužívejte skleněné injekční stříkačky, používejte pouze umělohmotné.
NeoRecormon vícedávkový je přípravek vtzv. mnohodávkové formě, tzn. že během 1 měsíce po rekonstituci lze z 1 lahvičky odebrat několik jednotlivých dávek pro několik pacientů. Abyste se vyhnuli riziku znečištění obsahu lahvičky, měli byste stále dodržovat aseptické podmínky (tj. při každém odběru používat sterilní jednorázové injekční stříkačky a jehly) a přísně dodržovat následující pokyny k manipulaci.
Před každým odběrem dezinfikujte gumovou zátku rekonstitučního zařízení na lahvičce alkoholem, aby nedošlo ke znečištění jejího obsahu opakovaným vpichem jehel.
Příprava roztoku přípravku NeoRecormon vícedávkový
(1) Vyjměte lahvičku se suchou substancí z obalu. Označte štítek datem rekonstituce a datem ukončení doby použitelnosti (expirace), což je přesně 1 měsíc po rekonstituci.
(2) Sejměte víčko z umělé hmoty z injekční lahvičky.
(3) Dezinfikujte gumovou zátku alkoholem.
(4) Vyjměte z obalu speciální rekonstituční zařízení pro rozpouštění a odběr (umožňuje sterilní výměnu vzduchu) a odstraňte ochrannou čepičku ze vpichovacího trnu.
(5) Nasaďte tento uzávěr na lahvičku a protlačte ho gumovou zátkou. Uzávěr musí zaskočit.
(6) Nasaďte zelenou jehlu na přibalenou stříkačku a odstraňte kryt jehly.
(7) Podržte ampulku modrou tečkou dopředu. Potřepte ampulkou nebo na ni zaklepejte, aby vytekla všechna kapalina ze zúženého místa dolů. Ulomte ampulku v místě jejího zúžení směrem dozadu. Nasajte všechnu kapalinu do stříkačky. Dezinfikujte gumovou zátku zařízení alkoholem.
(8) Propíchněte jehlu zátkou asi do hloubky 1 cm a vstříkněte pomalu všechno rozpouštědlo do injekční lahvičky. Poté odstraňte injekční stříkačku s jehlou.
(9) Lehce otáčejte lahvičkou,až se lyofilizát rozpustí. Netřepejte. Ujistěte se, že je roztok čirý, bezbarvý a bez jakýchkoli částic. Nasaďte ochrannou čepičku nahoru na rekonstituční zařízení.
(10) Před a po rozpouštění musí být lahvička s přípravkem NeoRecormon vícedávkový uchovávána v chladničce při +2°C až + 8° C.
Příprava jednotlivé dávky
(1) Dezinfikujte gumovou zátku rekonstitučního zařízení alkoholem před každým odběrem.
(2) Nasaďte jehlu 26G pevně na dodanou jednorázovou stříkačku (obsah 1 ml).
(3) Odstraňte kryt z jehly a propíchněte jehlu pryžovou zátkou rekonstitučního zařízení. Nasajte roztok přípravku NeoRecormon do stříkačky a vypusťte vzduch ze stříkačky do lahvičky a upravte množství roztoku přípravku NeoRecormon ve stříkačce na předepsanou dávku. Odstraňte injekční stříkačku (s jehlou) z rekonstitučního zařízení.
(4) Jehlu nahraďte novou jehlou (nová jehla má mít stejnou velikost jako jehla, kterou běžně používáte pro aplikaci injekce).
(5) Sejměte ochranný kryt z jehly a pečlivě odstraňte z jehly vzduch, tím, že podržíte stříkačku svisle vzhůru a budete lehce tlačit píst nahoru, až vystoupí kapka kapaliny ze špičky jehly.
Před podkožní injekcí očistěte kůži v místě vpichu tampónem s alkoholem. Pomocí palce a ukazováku utvořte kožní řasu. Uchopte stříkačku blízko jehly a rychle vpíchněte jehlu do kůže. Injikujte roztok s přípravkem NeoRecormon. Rychle vytáhněte jehlu a přitlačte na místo vpichu suchý sterilní tampón.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/031/019
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. července 1997
Datum posledního prodloužení registrace: 16. července 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
NeoRecormon 500 IU injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s 0,3 ml injekčního roztoku obsahuje 500 mezinárodních jednotek (IU), což odpovídá 4,15 mikrogramům epoetinu beta* (rekombinantní lidský erytropoetin).
Jeden ml injekčního roztoku obsahuje 1667 IU epoetinu beta.
* Vyrobený technologií rekombinatní DNA v ovariálních buňkách čínských křečků.
Pomocné látky se známým účinkem:
Fenylalanin (až do 0,3 mg v injekční stříkačce)
Sodík (méně než 1 mmol v injekční stříkačce)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Bezbarvý, čirý až slabě opalescentní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek NeoRecormon je indikován k léčbě:
- Léčba symptomatické anémie spojené s chronickým renálním selháním u dospělých a pediatrických pacientů.
- Prevence anémie u předčasně narozených dětí s porodní váhou v rozmezí 750-1500 g, které se narodily v době před 34. týdnem těhotenství.
- Léčba symptomatické anémie u dospělých pacientů s nemyeloidními malignitami, kteří jsou léčeni chemoterapií.
- Zvýšení počtu erytrocytů před odběrem krve k autologní transfuzi krve. Použití v této indikaci musí být zváženo vzhledem k udávanému zvýšení rizika vzniku tromboembolických příhod. Použití je indikováno pouze u pacientů s mírnou anémií (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], bez nedostatku železa), pokud postupy pro konzervování krve nejsou dostupné nebo účinné nebo plánovaný zvolený velký chirurgický zákrok vyžaduje velký objem krve (4 a více jednotek krve pro ženy nebo 5 a více jednotek pro muže). Viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba přípravkem NeoRecormon může být zahájena pouze lékařem se zkušenostmi s výše uvedenými
indikacemi přípravku. Protože byly vzácně zaznamenány anafylaktoidní reakce po podání přípravku,
první dávka má být aplikována pod dohledem zdravotnického personálu.
Dávkování
Léčba symptomatické anémie u dospělých a pediatrických pacientů s chronickým selháním ledvin Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta. Přípravek NeoRecormon má být podáván buď subkutánně nebo intravenózně, tak aby byla hladina hemoglobinu zvýšena maximálně na 12 g/dl (7,5 mmol/l). U pacientů, kteří nepodstupují dialýzu, se upřednostňuje subkutánní podání, kdy není zapotřebí vpichů do periferních žil. V případě intravenózního podání má být přípravek podáván alespoň po dobu 2 minut, například přes arterio-venózní píštěl na konci dialýzy u pacientů na hemodialýze.
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu v rozsahu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Zvýšení hladiny hemoglobinu o více než 2 g/dl (1,25 mmol/l) během období čtyř týdnů není žádoucí. Pokud k němu dojde, má být dávka odpovídajícím způsobem upravena, jak je uvedeno dále. Je-li rychlost vzestupu hladiny hemoglobinu vyšší než 2 g/dl (1,25 mmol/l) za měsíc nebo pokud se hladina hemoglobinu zvýší až na hodnotu 12 g/dl (7,45 mmol/l) je třeba dávku snížit přibližně o 25 %. Pokud by se hladina hemoglobinu dále zvyšovala, má být léčba přerušena, dokud nezačne hladina opět klesat, a v tomto bodě má být znovu zahájena léčba dávkou přibližně o 25 % nižší, než byla předchozí podávaná dávka.
Pacienti mají být důkladně sledováni a je třeba ověřit, že byla použita nejnižší schválená účinná dávka přípravku NeoRecormon, která postačuje pro kontrolu symptomů anémie při zachování koncentrace hemoglobinu pod nebo na hodnotě 12 g/dl (7,45 mmol/l).
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin. U pacientů se slabou odpovědí hemoglobinu na přípravek NeoRecormon mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz bod 4.4 a 5.1).
V případě hypertenze nebo stávajících kardiovaskulárních, cerebrovaskulárních nebo periferních vaskulárních onemocnění má být týdenní zvýšení Hb a cílová hodnota Hb stanovena individuálně při zvážení klinického obrazu.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází.
1. Fáze korekční
- Subkutánní podání:
Počáteční dávka je 3 x 20 IU/kg tělesné hmotnosti týdně. Dávka může být zvýšena každé 4 týdny o 3 x 20 IU/kg za týden, jestliže není dosaženo adekvátního vzestupu Hb (< 0,25 g/dl za týden).
Týdenní dávka může být také rozdělena na denní dávky.
- Intravenózní podání:
Počáteční dávka je 3 x 40 IU/kg tělesné hmotnosti týdně. Dávka může být po 4 týdnech zvýšena na 80 IU/kg třikrát týdně a pokud je nutné další zvýšení dávky, má být o 20 IU/kg třikrát týdně v měsíčních intervalech.
Maximální dávka při obou způsobech podání nemá překročit 720 IU/kg za týden.
2. Fáze udržovací
Pro udržení Hb mezi 10 až 12 g/dl je dávka ze začátku snížena na polovinu předešlé dávky. Následně je u pacienta dávka nastavena v intervalech jednoho nebo dvou týdnů individuálně (udržovací dávka).
V případě podkožního podání může být týdenní dávka podána v jedné injekci týdně, nebo může být rozdělena do tří až sedmi dávek týdně. Pacienti, kteří jsou stabilní na režimu podávání jednou týdně, mohou být převedeni na dávkování jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Výsledky klinických studií u dětí prokázaly, že čím jsou pacienti mladší, tím v průměru vyšší dávky přípravku NeoRecormon vyžadují. Doporučený plán dávkování má však být dodržován, protože klinický účinek u jednotlivých pacientů nelze zcela předvídat.
Léčba přípravkem NeoRecormon v předplněné injekční stříkačce je obvykle dlouhodobá. Může však být, pokud je to nutné, kdykoliv přerušena. Údaje týkající se dávkovacího schématu podání jednou týdně vycházejí z klinických studií s délkou léčby 24 týdnů.
Prevence anémie u předčasně narozených dětí
Roztok je podáván subkutánně v dávce 3 x 250 IU/kg tělesné hmotnosti za týden. Předčasně narozené děti u kterých již byla provedena transfuze v okamžiku zahájení léčby přípravkem NeoRecormon nebudou pravděpodobně mít takový užitek z léčby jako předčasně narozené děti, u kterých transfuze provedena nebyla. Doporučená délka léčby je 6 týdnů.
Léčba symptomatické chemoterapií indukované anémie u pacientů s nádorovým onemocněním Přípravek NeoRecormon má být podáván subkutánně pacientům s anémií (např. koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l)). Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta.
Týdenní dávka může být podána v jedné injekci jednou týdně nebo může být rozdělena na 3 až 7 jednotlivých dávek.
Doporučená úvodní dávka je 30 000 IU týdně (to odpovídá přibližně 450 IU/kg tělesné hmotnosti týdně u pacienta s průměrnou hmotností).
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Pokud dojde po 4 týdnech léčby ke zvýšení hodnot hemoglobinu o minimálně 1 g/dl (0,62 mmol/l), má se v podávání této dávky dále pokračovat. Pokud hladina hemoglobinu nestoupne o alespoň 1 g/dl (0,62 mmol/l), má být zváženo podávání dvojnásobné týdenní dávky. Pokud hladina hemoglobinu nestoupne po 8 týdnech léčby o alespoň 1 g/dl (0,62 mmol/l), je odpověď nepravděpodobná a léčba má být ukončena.
Léčba má pokračovat až 4 týdny po ukončení chemoterapie.
Maximální dávka nemá překročit 60 000 IU týdně.
Jakmile je u jednotlivého pacienta dosaženo léčebného cíle, dávka má být snížena o 25 až 50 %, aby byla udržena hladina hemoglobinu na této úrovni. Je zapotřebí vzít v úvahu titrování dávky.
Překročí-li hemoglobin hladinu 12 g/dl (7,5 mmol/l), má být dávka snížena zhruba o 25 až 50 %.
Pokud hladina hemoglobinu překročí 13 g/dl (8,1 mmol/l), má být léčba přípravkem NeoRecormon dočasně přerušena. Pokud hladina hemoglobinu klesne na 12 g/dl (7,5 mmol/l) nebo níže, má být léčba opět zahájena s dávkou přibližně o 25 % nižší, než byla předchozí dávka.
Pokud je nárůst hemoglobinu v průběhu 4 týdnů vyšší než 2 g/dl (1,3 mmol/l), dávka má být snížena o 25 až 50 %.
Pacienti mají být důkladně sledováni, přičemž je třeba ověřit, že byla použita nejnižší schválená dávka přípravku NeoRecormon postačující pro adekvátní kontrolu symptomů anémie.
Podávání pro zvýšení množství autologní krve
Přípravek je aplikován intravenózně po dobu asi 2 minut nebo subkutánně.
Přípravek NeoRecormon je podáván dvakrát týdně po dobu 4 týdnů. V případě, kdy hematokrit pacienta umožňuje odběr krve, tj. hodnota hematokritu je > 33 %, je přípravek NeoRecormon podáván v okamžiku ukončení odběru.
V průběhu celé léčby nemá být překročena hodnota hematokritu 48 %.
Dávkování musí být stanoveno chirurgickým týmem zvlášť pro každého pacienta podle požadovaného množství krve pro autologní transfuzi a endogenní rezervy červených krvinek:
1. P ožadované množství krve pro autologní transfuzi závisí na předpokládané ztrátě krve, popřípadě na použití postupů konzervování krve a na fyzické kondici pacienta.
Množství potřebné autologní krve má stačit k tomu, aby bylo možné se vyhnout transfuzi homologní krve.
Požadované množství předem odebrané krve je vyjádřeno v jednotkách, přičemž jedna jednotka v nomogramu je ekvivalentní 180 ml červených krvinek.
2. Možnost autologní transfuze krve závisí především na objemu pacientovy krve a na výchozí hodnotě hematokritu. Obě proměnné určují endogenní rezervu červených krvinek, kterou je možné vypočítat podle následujících vzorců:
Endogenní rezerva červených krvinek = objem krve [ml] x (hematokrit - 33)/100 Ženy: objem krve [ml] = 41 [ml/kg] x tělesná hmotnost [kg] + 1200 [ml]
Muži: objem krve [ml] = 44 [ml/kg] x tělesná hmotnost [kg] + 1600 [ml]
(tělesná hmotnost > 45 kg)
Indikace pro léčbu přípravkem NeoRecormon a jednotlivá dávka mají být stanoveny z požadovaného množství předem odebrané krve a endogenní rezervy červených krvinek podle následujících grafů.
Ženy
Požadované množství krve pro autologní transfuzi [jednotky]
Muži
Požadované množství krve pro autologní transfuzi [jednotky]
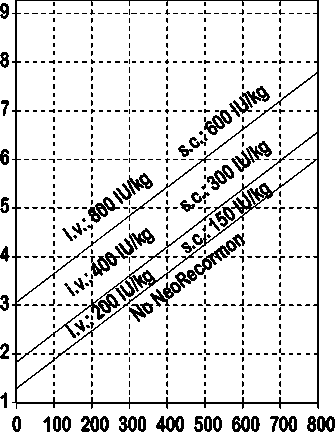
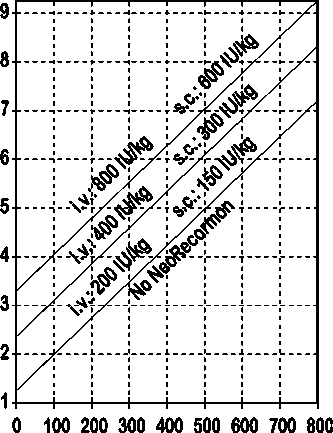
Endogenní rezerva červených krvinek [ml]
Endogenní rezerva červených krvinek [ml]
Takto stanovená jednotlivá dávka je podávána dvakrát týdně po dobu 4 týdnů. Maximální dávka nemá překročit 1600 IU/kg tělesné hmotnosti a týden při intravenózním podání nebo 1200 IU/kg tělesné hmotnosti a týden při subkutánním podávání.
Způsob podání
Přípravek NeoRecormon v předplněné injekční stříkačce je připraven k použití. Aplikován může být pouze roztok, který je čirý nebo lehce opalescentní, bezbarvý a bez viditelných částic.
Přípravek NeoRecormon v předplněné injekční stříkačce je sterilní, ale neobsahuje žádné konzervační látky. Za žádných okolností proto není možno aplikovat více než jednu dávku s jednou předplněnou injekční stříkačkou; léčivý přípravek je určen pouze k jednorázovému použití.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku (uvedenou v bodě 6.1).
Špatně kontrolovatelná hypertenze.
V indikaci “podání pro zvýšení množství autologní krve”: infarkt myokardu nebo cévní mozková příhoda v průběhu jednoho měsíce před zákrokem, nestabilní angina pectoris, zvýšené riziko hluboké venózní trombózy jako např. po prodělaném tromboembolickém onemocnění.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek NeoRecormon má být užíván s opatrností u pacientů s refrakterní anémií s nadbytkem transformovaných blastů, při epilepsii, trombocytóze a chronickém selhání jater. Je nutné vyloučit nedostatek kyseliny listové a vitamínu B12, protože tyto stavy snižují účinek přípravku NeoRecormon.
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin, neboť vysoké kumulující se dávky epoetinu mohou mít spojitost se zvýšeným rizikem mortality, závažnými kardiovaskulárními a cerebrovaskulárními příhodami. U pacientů se slabou odpovědí hemoglobinu na epoetiny mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz body 4.2 a 5.1).
Je třeba zhodnotit u všech pacientů hladinu železa před a v průběhu léčby, aby byla zajištěna účinná erytropoéza. Může být nutná substituce železa prováděná v souladu s léčebnými doporučeními.
Závažné zatížení hliníkem, ke kterému během léčby renálního selhání může dojít, může rovněž účinek přípravku NeoRecormon snížit.
Indikace k léčbě přípravkem NeoRecormon u pacientů s nefrosklerózou, kteří ještě nejsou dialyzovaní, má být zvážena individuálně, protože u těchto pacientů nelze vyloučit urychlení postupu renálního selhání.
Čistá aplazie buněk červené krevní řady (PRCA)
PRCA způsobená neutralizujícími antierytropoetinovými protilátkami byla hlášena ve spojení s léčbou erytropoetiny, včetně přípravku NeoRecormon. Bylo prokázáno, že tyto protilátky zkříženě reagují se všemi erytropoetinovými proteiny, a proto pacienti, u kterých je podezření nebo je potvrzen výskyt neutralizujících protilátek proti erytropoetinu, nemají být převáděni na přípravek NeoRecormon (viz bod 4.8).
PRCA u pacientů s hepatitidou C
Paradoxní pokles hladiny hemoglobinu a rozvoj závažné anémie související s nízkým počtem retikulocytů má vést k okamžitému přerušení léčby epoetinem a provedení testů na protilátky proti erytropoetinu. Tyto případy byly hlášeny, pokud byly pacientům s hepatitidou C léčených interferonem a ribavirinem současně podávány epoetiny. Epoetiny nejsou schválené pro léčbu anémie související s hepatitidou C.
Monitorování krevního tlaku
Může dojít ke vzestupu krevního tlaku nebo ke zhoršení již existující hypertenze zejména v případě rychlého vzestupu hematokritu. Toto zvýšení krevního tlaku je možno upravit pomocí léků. Pokud zvýšení krevního tlaku nemůže být upraveno medikamentózně, je doporučeno krátkodobé přerušení léčby přípravkem NeoRecormon. Zvláště na začátku terapie se doporučuje pravidelné monitorování krevního tlaku, a to rovněž mezi dialýzami. Může dojít ke vzniku hypertenzní krize s příznaky napodobujícími encefalopatii, která vyžaduje okamžitou lékařskou intervenci a intenzivní péči. Zvláštní pozoronost je třeba věnovat náhlým bodavým migrenosním bolestem hlavy jako možnému varovnému příznaku.
Chronické selhání ledvin
U pacientů 5 chronickým selháním ledvin, léčených přípravkem NeoRecormon, může dojít v průběhu léčby k mírnému, na dávce závislému nárůstu počtu krevních destiček v rozmezí normálních hodnot, zvláště pak v případě intravenózního podávání. K poklesu tohoto nárůstu dochází v dalším průběhu léčby. Je doporučeno pravidelně sledovat počet krevních destiček v průběhu prvních 8 týdnů léčby.
Koncentrace hemoglobinu
U pacientů s chronickým selháním ledvin nemá udržovací koncentrace hemoglobinu překročit horní limit cílové koncentrace hemoglobinu doporučovaný v bodě 4.2. V klinických studiích bylo pozorováno zvýšené riziko úmrtí a závažných kardiovaskulárních příhod nebo cerebrovaskulárních příhod, včetně cévní mozkové příhody, pokud byly používány přípravky stimulující erytropoézu (ESA) k dosažení cílových hodnot hemoglobinu vyšších než 12 g/dl (7,5 mmol/l).
V kontrolovaných klinických studiích se neprokázal žádný signifikantní přínos podávání epoetinů, pokud byla koncentrace hemoglobinu zvyšována nad hladinu nezbytně nutnou pro kontrolu symptomů anémie a předcházení krevní transfuze.
U předčasně narozených dětí může dojít k mírnému nárůstu počtu krevních destiček, především v období mezi 12. - 14. dnem života, proto má být počet krevních destiček sledován v pravidelných intervalech.
Účinek na růst nádorů
Epoetiny jsou růstové faktory, které primárně podporují tvorbu červených krvinek. Erytropoetinové receptory mohou být přítomny na povrchu různých nádorových buněk. Stejně jako u všech růstových faktorů je třeba mít na zřeteli, že i epoetiny mohou stimulovat rozvoj nádoru. V několika kontrolovaných klinických studiích nebylo ve spojení s epoetiny prokázáno zlepšení celkové doby přežití ani snížení rizika progrese nádorů u pacientů s anémií spojenou s nádorovým onemocněním.
V kontrolovaných klinických studiích zaměřených na použití přípravku NeoRecormon a ostatních erytropoézu stimulujících přípravků (ESA), bylo zjištěno:
- zkrácení doby do progrese nádoru u pacientů s pokročilým nádorovým onemocněním hlavy a krku, pokud byly dosahovány cílové hladiny hemoglobinu vyšší než 14 g/dl (8,7 mmol/l)
- zkrácení celkové doby přežití a zvýšení počtu úmrtí souvisejících s progresí onemocnění během 4 měsíců léčby u pacientů s metastazujícím karcinomem prsu léčených chemoterapií, pokud byly dosahovány cílové hladiny hemoglobinu 12-14 g/dl (7,5-8,7 mmol/l),
- zvýšení rizika úmrtí při dosahování cílové hladiny hemoglobinu 12 g/dl (7,5 mmol/l) u pacientů s aktivními maligními chorobami, kteří nedostávali ani chemoterapii ani ozařování. V této populaci pacientů není použití ESA indikováno.
Na základě výše uvedených skutečností má být za určitých klinických okolností při léčbě anémie u pacientů s nádorovým onemocněním upřednostněna transfuze krve. Rozhodnutí podat rekombinantní erytropoetin má být přijato na základě zvážení poměru přínosu a rizika a individuálního posouzení jednotlivého pacienta za daných specifických klinických podmínek. Další faktory, které mají být zváženy, je posouzení typu a stádia nádorového onemocnění; stupeň anémie; očekávaná délka přežití; podmínky, za kterých je pacient léčen; a vlastní volba pacienta (viz bod 5.1)
Může dojít ke vzestupu krevního tlaku, který lze farmakologicky léčit. Doporučuje se proto monitorovat krevní tlak, zejména v úvodní fázi léčby pacientů s nádorovým onemocněním.
U onkologických pacientů mají být také pravidelně sledovány počty krevních destiček a hladina hemoglobinu.
U pacientů v programu před autologní transfuzí krve může být zvýšení počtu krevních destiček, většinou v rozmezí normálních hodnot. Proto je doporučeno určovat počet krevních destiček u těchto pacientů alespoň jednou týdně. Pokud dojde ke zvýšení krevních destiček o více než 150 x 109/l nebo když nárůst přesáhne normální hodnoty, má být léčba přípravkem NeoRecormon přerušena.
Upředčasně narozených dětí nelze vyloučit možné riziko retinopatie způsobené erytropoetinem, proto je třeba zvýšené pozornosti a rozhodnutí o léčbě předčasně narozených dětí má být zvažováno na základě posouzení možného prospěchu a rizika této léčby a jiných dostupných možností.
U pacientů s chronickým selháním ledvin je v průběhu léčby přípravkem NeoRecormon při hemodialýze často nutné zvýšení dávky heparinu z důvodu zvýšeného hematokritu. Pokud není heparinizace optimální, může dojít k okluzi systému dialýzy.
U pacientů s chronickým selháním ledvin s rizikem trombózy cévního přístupu má být zvážena časná kontrola cévního přístupu a trombotická profylaxe kyselinou acetylsalicylovou.
V průběhu léčby přípravkem NeoRecormon má být pravidelně monitorována hladina sérového draslíku a fosfátů. Zvýšení kalémie bylo zaznamenáno u několika uremických pacientů léčených přípravkem NeoRecormon, i když příčinná souvislost nebyla ověřena. Pokud je pozorována zvýšená nebo stoupající hladina draslíku, pak má být zváženo přerušení podávání přípravku NeoRecormon dokud se kalémie neupraví.
Pro použití přípravku NeoRecormon v programu před autologní transfuzí krve musí být zváženy obecné zásady dárcovství krve, zvláště:
- dárci mají být pouze pacienti s hematokritem > 33 % (hemoglobin > 11g/dl [6,83 mmol/l];
- má být věnována zvláštní pozornost pacientům s hmotností nižší než 50 kg;
- jednotlivý čerpaný objem nemá překročit cca 12 % odhadnutého objemu krve pacienta.
Léčba má být vyhrazena pro pacienty, u kterých je zvlášť důležité vyhnout se transfuzi homologní krve a mají být zvážena rizika a prospěch homologní transfuze.
Zneužití
Zneužití přípravku zdravými osobami může vést k nadměrnému zvýšení hematokritu. Toto zvýšení může být spojeno s život ohrožujícími kardiovaskulárními komplikacemi.
Pomocné látky
Přípravek NeoRecormon v přeplněné injekční stříkačce obsahuje až 0,3 mg fenylalaninu v jedné injekční stříkačce jako pomocnou látku. Tato skutečnost má být brána v úvahu při léčbě pacientů trpících vážnými formami fenylketonurie.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v injekční stříkačce, tj. v podstatě je „bez sodíku“
Zpětná zjistitelnost přípravku NeoRecormon
Z důvodu snadnější zpětné zjistitelnosti látek stimulujících erytropoézu (ESAs) má být obchodní název předepsaného přípravku ESA zřetelně zaznamenán (nebo vyznačen) v pacientově dokumentaci.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Z dostupných klinických výsledků nejsou známy žádné interakce přípravku NeoRecormon s ostatními léčivými přípravky.
Pokusy na zvířatech prokázaly, že epoetin beta nezvyšuje myelotoxicitu cytostatických léčivých látekjako jsou etoposid, cisplatina, cyklofosfamid a fluorouracil.
4.6 Fertilita, těhotenství a kojení
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3).
Nejsou k dispozici žádné klinické údaje o účincích epoetinu beta během těhotenství.
Při předepisování těhotným ženám nutno postupovat opatrně.
Kojení
Není známo, zda se epoetin beta vylučuje do lidského mateřského mléka.
Rozhodnutí, zda pokračovat v kojení/přerušit kojení nebo pokračovat v léčbě/přerušit léčbu epoetinem beta, má být zvoleno v závislosti na prospěšnosti kojení pro dítě a prospěšnosti léčby epoetinem beta u matky.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek NeoRecormon nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Na základě výsledků klinických studií s 1725 pacienty lze očekávat, že přibližně u 8 % pacientů léčených přípravkem NeoRecormon se mohou vyskytnout nežádoucí účinky.
Anemičtí pacienti s chronickým selháním ledvin
Nejčastějším nežádoucím účinkem v průběhu léčby přípravkem NeoRecormon je zvýšení krevního tlaku nebo zhoršení stávající hypertenze, zvláště pak v případech rychlého zvýšení hodnot hematokritu (viz bod 4.4). Hypertenzní krize s příznaky encefalopatie (např. bolesti hlavy a stavy zmatenosti, senzoricko-motorické poruchy jako například porucha řeči nebo zhoršení chůze až tonicko-klonické křeče) se mohou objevit také u jednotlivých pacientů s jinak normálním nebo nízkým krevním tlakem (viz bod 4.4).
Může dojít k trombóze žilního přístupu zejména u pacientů s tendencí k hypotenzi nebo s komplikacemi arteriovenózního zkratu (např. stenózy, aneuryzmata), viz bod 4.4. Ve většině případů je pozorován pokles hodnot sérového železa souběžně se vzestupem hematokritu (viz bod 4.4). Navíc byl v ojedinělých případech pozorován vzestup sérového draslíku a hladin fosfátů (viz bod 4.4).
V ojedinělých případech byla v souvislosti s léčbou přípravkem NeoRecormon hlášena čistá aplazie červených krvinek (PRCA) způsobená neutralizujícími antitrombopoetinovými protilátkami.
V případě diagnózy čisté aplazie červených krvinek (PRCA) způsobené neutralizujícími antitrombopoetinovými protilátkami musí být léčba přípravkem NeoRecormon ukončena a pacient nemá být převáděn na léčbu jinou erytropoetickou bílkovinou (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 1.
Pacienti s rakovinou
Bolesti hlavy související s léčbou epoetinem beta a hypertenze, kterou lze medikamentózně léčit, jsou časté (viz bod 4.4).
U některých pacientů je pozorován pokles sérového železa (viz bod 4.4).
V klinických studiích byl prokázán vyšší výskyt tromboembolických událostí u pacientů s karcinomem léčených přípravkem NeoRecormon ve srovnání s neléčenými kontrolními skupinami nebo placebem. U pacientů léčených přípravkem NeoRecormon byla incidence 7 % ve srovnání s 4 % u kontrolních skupin; aniž by to bylo spojeno se zvýšenou mortalitou na tromboembolické události ve srovnání s kontrolními skupinami.
Nežádoucí účinky jsou uvedeny níže v tabulce 2.
Pacienti v programu autologního dárcovství krve
U pacientů v programu autologního dárcovství krve byla hlášena lehce zvýšená četnost tromboembolických událostí. Kauzální vztah k léčbě přípravkem NeoRecormon však nebyl prokázán.
Ve studiích kontrolovaných placebem byl více vyjádřen přechodný deficit železa u pacientů léčených přípravkem NeoRecormon než u kontrolní skupiny (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 3.
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů MedDRA a absolutní četnosti. Kategorie četnosti jsou definovány dle následující konvence:
velmi časté (> 1/10); časté (>1/100 to <1/10); méně časté (>1/1 000 to <1/100); vzácné (>1/10 000 to <1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
Tabulka 1: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů s chronickým selháním ledvin léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze Hypertenzní krize |
Časté Méně časté |
|
Poruchy nervového systému |
Časté | |
|
Poruchy krve a |
Trombóza cévního |
Vzácné |
|
lymfatického systému |
přístupu Trombocytóza |
Velmi vzácné |
Tabulka 2: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u onkologických pacientů léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze |
Časté |
|
poruchy krve a lymfatického systému |
Tromboembolické účinky |
Časté |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Tabulka 3: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů v programu autologního dárcovství krve léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Předčasně narozené děti
Pokles hodnot sérového ferritinu je velmi častý (viz bod 4.4).
Popis vybraných nežádoucích účinků
Vzácně byly pozorovány kožní reakce jako jsou vyrážka, svědění, kopřivka nebo reakce v místě vpichu. Velmi vzácně byly popsány anafylaktoidní reakce související s léčbou epoetinem beta. V kontrolovaných klinických studiích však zvýšení výskytu hypersenzitivních reakcí nebylo popsáno.
Ve velmi vzácných případech, především na počátku léčby, byl zaznamenán výskyt příznaků podobných chřipce ("flu-like" symptomy) jako je horečka, zimnice, bolesti hlavy, bolest v končetinách, malátnost a/nebo bolest kostí. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Údaje z kontrolované klinické studie s epoetinem alfa nebo darbepoetinem alfa udávaly incidenci cévní mozkové příhody jako častou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 P ředávkování
Terapeutický rozsah přípravku NeoRecormon je velmi široký. Dokonce i při velmi vysokých hladinách v séru nebyly pozorovány žádné příznaky otravy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA01 Mechanismus účinku
Erytropoetin je glykoprotein, který stimuluje tvorbu erytrocytů z prekurzorů kmenových buněk.
Působí jako faktor stimulující mitózu kmenových buněk červené krevní řady a hormon působící na jejich diferenciaci.
Epoetin beta, léčivá látka přípravku NeoRecormon, je svým složením aminokyselin a cukrů identický s erytropoetinem, který byl izolován z moči anemických pacientů.
Biologický účinek epoetinu beta byl demonstrován po intravenózní a podkožní aplikaci u různých pokusných zvířat in vivo (normální a uremičtí potkani, polycytemické myši, psi). Po podání epoetinu beta se zvýší počet erytrocytů, hodnota Hb a retikulocytů stejně tak jako rychlost inkorporace 59Fe.
In vitro byla zjištěna zvýšená inkorporace 3H-thymidinu v erytroidních jaderných buňkách sleziny (kultura buněk sleziny myší) po inkubaci s epoetinem beta.
Pokusy na tkáňových kulturách buněk lidské kostní dřeně prokázaly, že epoetin beta stimuluje jen tvorbu červených krvinek a neovlivňuje tvorbu bílých krvinek. Nebyly zjištěny cytotoxické účinky epoetinu beta na kostní dřeň ani na lidské kožní buňky.
Po podání jednotlivé dávky epoetinu beta nebyly pozorovány žádné vlivy na chování nebo na pohybovou činnost u myší a oběhové nebo respirační funkce u psů.
Klinická účinnost a bezpečnost
V randomizované dvojitě zaslepené, placebem kontrolované studii, která hodnotila 4038 pacientů s chronickým selháním ledvin bez nutnosti dialýzy, s diabetem 2. typu a hladinami hemoglobinu < 11 g/dl, byli pacienti léčeni buď darbepoetinem alfa až do dosažení cílových hladin hemoglobinu 13 g/dl, nebo dostávali placebo (viz bod 4.4). Studie nesplnila žádný z primárních cílů, který by prokázal snížení rizika úmrtí ze všech příčin, kardiovaskulární morbidity nebo terminálního stádia onemocnění ledvin (ESRD). Analýza jednotlivých komponent složených cílových parametrů prokázala následující poměr rizik (95% CI): úmrtí 1,05 (o,92; 1,21), cévní mozková příhoda 1,92 (1,38; 2,68), městnavé srdeční selhání 0,89 (0,74; 1,08), infarkt myokardu 0,96 (0,75; 1,23), hospitalizace z důvodu ischemie myokardu 0,84 (0,55; 1,27), terminální stádium onemocnění ledvin 1,02 (0,87; 1,18).
Souhrnné post-hoc analýzy klinických studií s ESA byly provedeny u pacientů s chronickým selháním ledvin (u pacientů, kteří byli nebo nebyli na dialýze, u diabetiků a u pacientů bez diabetu). Byla pozorována tendence směrem ke zvýšení odhadovaného rizika mortality z jakýchkoli důvodů, rizika kardiovaskulárních a cerebrovaskulárních příhod spojených s vyššími kumulativními dávkami ESA, nezávisle na stavu diabetu nebo dialýze (viz body 4.2 a 4.4).
Erytropoetin je růstový faktor primárně podporující tvorbu červených krvinek. Receptory pro erytropoetin mohou být exprimovány na povrchu mnoha nádorových buněk.
Přežití a progrese nádoru byla zkoumána v pěti velkých kontrolovaných klinických studiích zahrnujících celkem 2833 pacientů; čtyři z těchto studií byly dvojitě zaslepené kontrolované placebe m, jedna studie byla otevřená. Dvou studií se účastnili pacienti léčení chemoterapií. Cílová koncentrace hemoglobinu byla ve dvou studiích > 13 g/dl; v ostatních třech studiích 12-14 g/dl.
V otevřené studii nebyl z hlediska celkového přežití zjištěn žádný rozdíl mezi pacienty léčenými rekombinantním lidským erytropoetinem a mezi kontrolní skupinou. Ve čtyřech placebem kontrolovaných studiích se poměry rizik pro celkové přežití pohybovaly mezi 1,25 a 2,47 ve prospěch kontrolních skupin. Při srovnání s kontrolními skupinami ukázaly tyto studie konzistentní nevysvětlené statisticky významné zvýšení mortality u pacientů, kteří měli anémii spojenou s různými běžnými maligními nádory a kteří dostávali rekombinantní lidský erytropoetin. Výsledek celkového přežití zjištěný v klinických studiích nebylo možné uspokojivě vysvětlit rozdílem v incidenci trombózy a přidružených komplikací mezi skupinou dostávající rekombinantní lidský erytropoetin a mezi skupinou kontrolní.
Metaanalýza založené na údajích jednotlivých pacientů zahrnovala data ze všech 12 kontrolovaných klinických studií prováděných u pacientů s anémií a maligním nádorovým onemocněním, kteří byli léčeni přípravkem NeoRecormon (n=2301), ukázala celkový odhadovaný poměr rizik pro přežití 1,13 ve prospěch kontrol (95% CI 0,87; 1,46). U pacientů, kteří měli v baseline hladinu hemoglobinu <
10 g/dl (n=899), byl odhadovaný poměr rizik pro přežití 0,98 (95% CI 0,68 až 1,40). V celkové populaci bylo pozorováno zvýšené relativní riziko tromboembolických příhod (RR 1,62, 95% CI:
1,13; 2,31).
Byla provedena analýza dat na úrovni jednotlivých nemocných u více než 13 900 pacientů se zhoubným nádorem (léčených chemoterapií, radioterapií, chemoradioterapií nebo bez terapie) zařazených do 53 kontrolovaných klinických studií zahrnujících podávání několika epoetinů. Metaanalýza údajů celkového přežití prokázala, že poměr rizik (hazard ratio) je odhadem 1,06 ve prospěch kontrolních skupin (95% CI: 1,00, 1,12; 53 studií a 13 933 pacientů) a u pacientů se zhoubným nádorem podstupujících chemoterapii byl poměr rizik (hazard ratio) celkového přežití 1,04 (95% CI: 0,97, 1,11; 38 studií a 10 441 pacientů). Meta-analýzy zároveň konzistentně poukazují na významné zvýšení relativního rizika tromboembolických příhod u pacientů se zhoubným nádorem, kteří dostávají rekombinantní lidský erytropoetin (viz bod 4.4).
Ve velmi vzácných případech se v průběhu léčby rekombinantním lidským erytropoetinem objevily neutralizující protilátky proti erytropoetinu s nebo bez čisté aplazie červené krevní řady (PRCA).
5.2 Farmakokinetické vlastnosti
Farmakokinetické sledování u zdravých dobrovolníků a uremických pacientů ukázalo, že po intravenózním podání je poločas epoetinu beta mezi 4 - 12 hodinami a že distribuční objem odpovídá jedno- až dvojnásobku plazmatického objemu. Analogické výsledky byly nalezeny v pokusech na zvířatech u normálních i uremických potkanů.
Protrahovaná absorpce epoetinu beta po subkutánním podání uremickým pacientům vede ke vzniku koncentračního plató, přičemž maximální koncentrace je dosaženo v průměru za 12 - 28 hodin. Terminální poločas je delší než po intravenózním podání s průměrem 13 - 28 hodin.
Ve srovnání s intravenózní aplikací je biologická dostupnost epoetinu beta po podkožní aplikaci mezi 23 - 42 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
Studie kancerogenity s homologním erytropoetinem u myší neodhalily žádné známky proliferativního nebo kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 S eznam pomocných látek
Močovina,
Chlorid sodný,
Polysorbát 20,
Dihydrogenfosforečnan sodný dihydrát,
Hydrogenfosforečnan sodný dodekahydrát,
Chlorid vápenatý dihydrát,
Glycin,
L-leucin,
L-isoleucin,
L-threonin,
L-kyselina glutamová,
L-fenylalanin.
Voda na injekci.
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C ).
Uchovávejte předplněnou injekční stříkačku v původním obalu, aby byl přípravek chráněn před světlem.
Pro usnadnění ambulantního použití může pacient vyjmout přípravek z chladničky a uchovávat jej jednorázově po dobu maximálně 3 dnů při pokojové teplotě (maximálně do 25 °C).
6.5 Druh obalu a obsah balení
Předplněná injekční stříkačka (sklo typu I) s víčkem a zátkou (pryž potažená teflonem) a s injekční jehlou (30G1/2). Jedna injekční stříkačka obsahuje 0,3 ml roztoku.
Velikost balení po 1 předplněné injekční stříkačce s 1 jehlou nebo 6 předplněných injekčních stříkačkách se 6 jehlami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Nejdříve si dobře umyjte ruce!
1. Vyjměte jednu předplněnou injekční stříkačku z balení a zkontrolujte, zda roztok ve stříkačce je čirý, bezbarvý a bez viditelných částic. Odstraňte víčko z injekční stříkačky.
2. Vyjměte jednu injekční jehlu z balení, nasaďte ji na injekční stříkačku a odstraňte z jehly ochranné víčko.
3. Vytlačte vzduch z injekční stříkačky tím, že stříkačku držíte ve vzpřímené poloze a jemně tlačíte píst vzhůru. Tlačte na píst tak dlouho, dokud množství přípravku NeoRecormon v injekční stříkačce neodpovídá množství předepsanému pro aplikaci.
4. Za použití tampónu namočeného v alkoholu si očistěte kůži v místě vpichu. Vytvořte záhyb na kůži tím, že ji sevřete mezi palec a ukazováček. Stiskněte tělo injekční stříkačky v části bližší k jehle a aplikujte jehlu do záhybu kůže rychlým a pevným pohybem. Vstříkněte roztok přípravku NeoRecormon. Jehlu rychle vytáhněte a zatlačte na místo vpichu suchým a sterilním tampónem.
Tento léčivý přípravek je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/031/025 -026
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. července 1997
Datum posledního prodloužení registrace: 16. července 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
NeoRecormon 2000 IU injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s 0,3 ml injekčního roztoku obsahuje 2000 mezinárodních jednotek (IU), což odpovídá 16,6 mikrogramům epoetinu beta* (rekombinantní lidský erytropoetin). Jeden ml injekčního roztoku obsahuje 6667 IU epoetinu beta.
* Vyrobený technologií rekombinatní DNA v ovariálních buňkách čínských křečků.
Pomocné látky se známým účinkem:
Fenylalanin (až do 0,3 mg v injekční stříkačce)
Sodík (méně než 1 mmol v injekční stříkačce)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Bezbarvý, čirý až lehce opalescentní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek NeoRecormon je indikován k léčbě:
- Léčba symptomatické anémie spojené s chronickým renálním selháním u dospělých a pediatrických pacientů.
- Prevence anémie u předčasně narozených dětí s porodní váhou v rozmezí 750-1500 g, které se narodily v době před 34. týdnem těhotenství.
- Léčba symptomatické anémie u dospělých pacientů s nemyeloidními malignitami, kteří jsou léčeni chemoterapií.
- Zvýšení počtu erytrocytů před odběrem krve k autologní transfuzi krve. Použití v této indikaci musí být zváženo vzhledem k udávanému zvýšení rizika vzniku tromboembolických příhod. Použití je indikováno pouze u pacientů s mírnou anémií (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], bez nedostatku železa), pokud postupy pro konzervování krve nejsou dostupné nebo účinné nebo plánovaný zvolený velký chirurgický zákrok vyžaduje velký objem krve (4 a více jednotek krve pro ženy nebo 5 a více jednotek pro muže). Viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba přípravkem NeoRecormon může být zahájena pouze lékařem se zkušenostmi s výše uvedenými
indikacemi přípravku. Protože byly vzácně zaznamenány anafylaktoidní reakce po podání přípravku,
první dávka má být aplikována pod dohledem zdravotnického personálu.
Dávkování
Léčba symptomatické anémie u dospělých a pediatrických pacientů s chronickým selháním ledvin Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta. Přípravek NeoRecormon má být podáván buď subkutánně nebo intravenózně, tak aby byla hladina hemoglobinu zvýšena maximálně na 12 g/dl (7,5 mmol/l). U pacientů, kteří nepodstupují dialýzu, se upřednostňuje subkutánní podání, kdy není zapotřebí vpichů do periferních žil. V případě intravenózního podání má být přípravek podáván alespoň po dobu 2 minut, například přes arterio-venózní píštěl na konci dialýzy u pacientů na hemodialýze.
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu v rozsahu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Zvýšení hladiny hemoglobinu o více než 2 g/dl (1,25 mmol/l) během období čtyř týdnů není žádoucí. Pokud k němu dojde, má být dávka odpovídajícím způsobem upravena, jak je uvedeno dále. Je-li rychlost vzestupu hladiny hemoglobinu vyšší než 2 g/dl (1,25 mmol/l) za měsíc nebo pokud se hladina hemoglobinu zvýší až na hodnotu 12 g/dl (7,45 mmol/l) je třeba dávku snížit přibližně o 25 %. Pokud by se hladina hemoglobinu dále zvyšovala, má být léčba přerušena, dokud nezačne hladina opět klesat, a v tomto bodě má být znovu zahájena léčba dávkou přibližně o 25 % nižší, než byla předchozí podávaná dávka.
Pacienti mají být důkladně sledováni a je třeba ověřit, že byla použita nejnižší schválená účinná dávka přípravku NeoRecormon, která postačuje pro kontrolu symptomů anémie při zachování koncentrace hemoglobinu pod nebo na hodnotě 12 g/dl (7,45 mmol/l).
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin. U pacientů se slabou odpovědí hemoglobinu na přípravek NeoRecormon mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz bod 4.4 a 5.1).
V případě hypertenze nebo stávajících kardiovaskulárních, cerebrovaskulárních nebo periferních vaskulárních onemocnění má být týdenní zvýšení Hb a cílová hodnota Hb stanovena individuálně při zvážení klinického obrazu.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází.
1. Fáze korekční
- Subkutánní podání:
Počáteční dávka je 3 x 20 IU/kg tělesné hmotnosti týdně. Dávka může být zvýšena každé 4 týdny o 3 x 20 IU/kg za týden, jestliže není dosaženo adekvátního vzestupu Hb (< 0,25 g/dl za týden).
Týdenní dávka může být také rozdělena na denní dávky.
- Intravenózní podání :
Počáteční dávka je 3 x 40 IU/kg tělesné hmotnosti týdně. Dávka může být po 4 týdnech zvýšena na 80 IU/kg třikrát týdně a pokud je nutné další zvýšení dávky, má být o 20 IU/kg třikrát týdně v měsíčních intervalech.
Maximální dávka při obou způsobech podání nemá překročit 720 IU/kg za týden.
2. Fáze udržovací
Pro udržení Hb mezi 10 až 12 g/dl je dávka ze začátku snížena na polovinu předešlé dávky. Následně je u pacienta dávka nastavena v intervalech jednoho nebo dvou týdnů individuálně (udržovací dávka).
V případě podkožního podání může být týdenní dávka podána v jedné injekci týdně, nebo může být rozdělena do tří až sedmi dávek týdně. Pacienti, kteří jsou stabilní na režimu podávání jednou týdně, mohou být převedeni na dávkování jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Výsledky klinických studií u dětí prokázaly, že čím jsou pacienti mladší, tím v průměru vyšší dávky přípravku NeoRecormon vyžadují. Doporučený plán dávkování má však být dodržován, protože klinický účinek u jednotlivých pacientů nelze zcela předvídat.
Léčba přípravkem NeoRecormon v předplněné injekční stříkačce je obvykle dlouhodobá. Může však být, pokud je to nutné, kdykoliv přerušena. Údaje týkající se dávkovacího schématu podání jednou týdně vycházejí z klinických studií s délkou léčby 24 týdnů.
Prevence anémie u předčasně narozených dětí
Roztok je podáván subkutánně v dávce 3 x 250 IU/kg tělesné hmotnosti za týden. Předčasně narozené děti u kterých již byla provedena transfuze v okamžiku zahájení léčby přípravkem NeoRecormon nebudou pravděpodobně mít takový užitek z léčby jako předčasně narozené děti, u kterých transfuze provedena nebyla. Doporučená délka léčby je 6 týdnů.
Léčba symptomatické chemoterapií indukované anémie u pacientů s nádorovým onemocněním Přípravek NeoRecormon má být podáván subkutánně pacientům s anémií (např. koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l)). Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta.
Týdenní dávka může být podána v jedné injekci jednou týdně nebo může být rozdělena na 3 až 7 jednotlivých dávek.
Doporučená úvodní dávka je 30 000 IU týdně (to odpovídá přibližně 450 IU/kg tělesné hmotnosti týdně u pacienta s průměrnou hmotností).
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Pokud dojde po 4 týdnech léčby ke zvýšení hodnot hemoglobinu o minimálně 1 g/dl (0,62 mmol/l), má se v podávání této dávky dále pokračovat. Pokud hladina hemoglobinu nestoupne o alespoň 1 g/dl (0,62 mmol/l), má být zváženo podávání dvojnásobné týdenní dávky. Pokud hladina hemoglobinu nestoupne po 8 týdnech léčby o alespoň 1 g/dl (0,62 mmol/l), je odpověď nepravděpodobná a léčba má být ukončena.
Léčba má pokračovat až 4 týdny po ukončení chemoterapie.
Maximální dávka nemá překročit 60 000 IU týdně.
Jakmile je u jednotlivého pacienta dosaženo léčebného cíle, dávka má být snížena o 25 až 50 %, aby byla udržena hladina hemoglobinu na této úrovni. Je zapotřebí vzít v úvahu titrování dávky.
Překročí-li hemoglobin hladinu 12 g/dl (7,5 mmol/l), má být dávka snížena zhruba o 25 až 50 %.
Pokud hladina hemoglobinu překročí 13 g/dl (8,1 mmol/l), má být léčba přípravkem NeoRecormon dočasně přerušena. Pokud hladina hemoglobinu klesne na 12 g/dl (7,5 mmol/l) nebo níže, má být léčba opět zahájena s dávkou přibližně o 25 % nižší, než byla předchozí dávka.
Pokud je nárůst hemoglobinu v průběhu 4 týdnů vyšší než 2 g/dl (1,3 mmol/l), dávka má být snížena o 25 až 50 %.
Pacienti mají být důkladně sledováni, přičemž je třeba ověřit, že byla použita nejnižší schválená dávka přípravku NeoRecormon postačující pro adekvátní kontrolu symptomů anémie.
Podávání pro zvýšení množství autologní krve
Přípravek je aplikován intravenózně po dobu asi 2 minut nebo subkutánně.
Přípravek NeoRecormon je podáván dvakrát týdně po dobu 4 týdnů. V případě, kdy hematokrit pacienta umožňuje odběr krve, tj. hodnota hematokritu je > 33 %, je přípravek NeoRecormon podáván v okamžiku ukončení odběru.
V průběhu celé léčby nemá být překročena hodnota hematokritu 48 %.
Dávkování musí být stanoveno chirurgickým týmem zvlášť pro každého pacienta podle požadovaného množství krve pro autologní transfuzi a endogenní rezervy červených krvinek:
1. P ožadované množství krve pro autologní transfuzi závisí na předpokládané ztrátě krve, popřípadě na použití postupů konzervování krve a na fyzické kondici pacienta.
Množství potřebné autologní krve má stačit k tomu, aby bylo možné se vyhnout transfuzi homologní krve.
Požadované množství předem odebrané krve je vyjádřeno v jednotkách, přičemž jedna jednotka v nomogramu je ekvivalentní 180 ml červených krvinek.
2. Možnost autologní transfuze krve závisí především na objemu pacientovy krve a na výchozí hodnotě hematokritu. Obě proměnné určují endogenní rezervu červených krvinek, kterou je možné vypočítat podle následujících vzorců:
Endogenní rezerva červených krvinek = objem krve [ml] x (hematokrit - 33)/100 Ženy: objem krve [ml] = 41 [ml/kg] x tělesná hmotnost [kg] + 1200 [ml]
Muži: objem krve [ml] = 44 [ml/kg] x tělesná hmotnost [kg] + 1600 [ml]
(tělesná hmotnost > 45 kg)
Indikace pro léčbu přípravkem NeoRecormon a jednotlivá dávka mají být stanoveny z požadovaného množství předem odebrané krve a endogenní rezervy červených krvinek podle následujících grafů.
Ženy
Požadované množství krve pro autologní transfuzi [jednotky]
Muži
Požadované množství krve pro autologní transfuzi [jednotky]
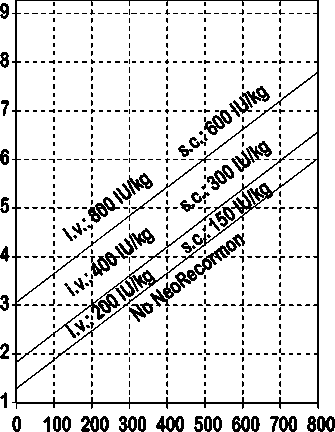
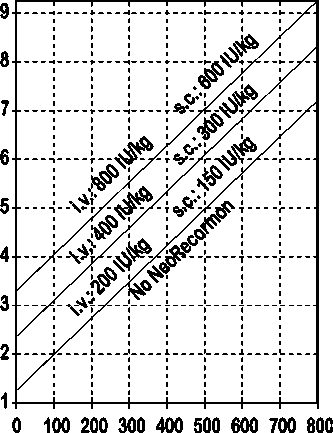
Endogenní rezerva červených krvinek [ml] Endogenní rezerva červených krvinek [ml]
Takto stanovená jednotlivá dávka je podávána dvakrát týdně po dobu 4 týdnů. Maximální dávka nemá překročit 1600 IU/kg tělesné hmotnosti a týden při intravenózním podání nebo 1200 IU/kg tělesné hmotnosti a týden při subkutánním podávání.
Způsob podání
Přípravek NeoRecormon v předplněné injekční stříkačce je připraven k použití. Aplikován může být pouze roztok, který je čirý nebo lehce opalescentní, bezbarvý a bez viditelných částic.
Přípravek NeoRecormon v předplněné injekční stříkačce je sterilní, ale neobsahuje žádné konzervační látky. Za žádných okolností proto není možno aplikovat více než jednu dávku s jednou předplněnou injekční stříkačkou; léčivý přípravek je určen pouze k jednorázovému použití.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku (uvedenou v bodě 6.1).
Špatně kontrolovatelná hypertenze.
V indikaci “podání pro zvýšení množství autologní krve”: infarkt myokardu nebo cévní mozková příhoda v průběhu jednoho měsíce před zákrokem, nestabilní angina pectoris, zvýšené riziko hluboké venózní trombózy jako např. po prodělaném tromboembolickém onemocnění.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek NeoRecormon má být užíván s opatrností u pacientů s refrakterní anémií s nadbytkem transformovaných blastů, při epilepsii, trombocytóze a chronickém selhání jater. Je nutné vyloučit nedostatek kyseliny listové a vitamínu BJ2, protože tyto stavy snižují účinek přípravku NeoRecormon.
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin, neboť vysoké kumulující se dávky epoetinu mohou mít spojitost se zvýšeným rizikem mortality, závažnými kardiovaskulárními a cerebrovaskulárními příhodami. U pacientů se slabou odpovědí hemoglobinu na epoetiny mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz body 4.2 a 5.1).
Je třeba zhodnotit u všech pacientů hladinu železa před a v průběhu léčby, aby byla zajištěna účinná erytropoéza. Může být nutná substituce železa prováděná v souladu s léčebnými doporučeními.
Závažné zatížení hliníkem, ke kterému během léčby renálního selhání může dojít, může rovněž účinek přípravku NeoRecormon snížit.
Indikace k léčbě přípravkem NeoRecormon u pacientů s nefrosklerózou, kteří ještě nejsou dialyzovaní, má být zvážena individuálně, protože u těchto pacientů nelze vyloučit urychlení postupu renálního selhání.
Čistá aplazie buněk červené krevní řady (PRCA)
PRCA způsobená neutralizujícími antierytropoetinovými protilátkami byla hlášena ve spojení s léčbou erytropoetiny, včetně přípravku NeoRecormon. Bylo prokázáno, že tyto protilátky zkříženě reagují se všemi erytropoetinovými proteiny, a proto pacienti, u kterých je podezření nebo je potvrzen výskyt neutralizujících protilátek proti erytropoetinu, nemají být převáděni na přípravek NeoRecormon (viz bod 4.8).
PRCA u pacientů s hepatitidou C
Paradoxní pokles hladiny hemoglobinu a rozvoj závažné anémie související s nízkým počtem retikulocytů má vést k okamžitému přerušení léčby epoetinem a provedení testů na protilátky proti erytropoetinu. Tyto případy byly hlášeny, pokud byly pacientům s hepatitidou C léčených interferonem a ribavirinem současně podávány epoetiny. Epoetiny nejsou schválené pro léčbu anémie související s hepatitidou C.
Monitorování krevního tlaku
Může dojít ke vzestupu krevního tlaku nebo ke zhoršení již existující hypertenze zejména v případě rychlého vzestupu hematokritu. Toto zvýšení krevního tlaku je možno upravit pomocí léků. Pokud zvýšení krevního tlaku nemůže být upraveno medikamentózně, je doporučeno krátkodobé přerušení léčby přípravkem NeoRecormon. Zvláště na začátku terapie se doporučuje pravidelné monitorování krevního tlaku, a to rovněž mezi dialýzami. Může dojít ke vzniku hypertenzní krize s příznaky napodobujícími encefalopatii, která vyžaduje okamžitou lékařskou intervenci a intenzivní péči. Zvláštní pozoronost je třeba věnovat náhlým bodavým migrenosním bolestem hlavy jako možnému varovnému příznaku.
Chronické selhání ledvin
U pacientů 5 chronickým selháním ledvin, léčených přípravkem NeoRecormon, může dojít v průběhu léčby k mírnému, na dávce závislému, nárůstu počtu krevních destiček v rozmezí normálních hodnot, zvláště pak v případě intravenózního podávání. K poklesu tohoto nárůstu dochází v dalším průběhu léčby. Je doporučeno pravidelně sledovat počet krevních destiček v průběhu prvních 8 týdnů léčby.
Koncentrace hemoglobinu
U pacientů s chronickým selháním ledvin nemá udržovací koncentrace hemoglobinu překročit horní limit cílové koncentrace hemoglobinu doporučovaný v bodě 4.2. V klinických studiích bylo pozorováno zvýšené riziko úmrtí a závažných kardiovaskulárních příhod nebo cerebrovaskulárních příhod, včetně cévní mozkové příhody, pokud byly používány přípravky stimulující erytropoézu (ESA) k dosažení cílových hodnot hemoglobinu vyšších než 12 g/dl (7,5 mmol/l).
V kontrolovaných klinických studiích se neprokázal žádný signifikantní přínos podávání epoetinů, pokud byla koncentrace hemoglobinu zvyšována nad hladinu nezbytně nutnou pro kontrolu symptomů anémie a předcházení krevní transfuze.
U předčasně narozených dětí může dojít k mírnému nárůstu počtu krevních destiček, především v období mezi 12. - 14. dnem života, proto má být počet krevních destiček sledován v pravidelných intervalech.
Účinek na růst nádorů
Epoetiny jsou růstové faktory, které primárně podporují tvorbu červených krvinek. Erytropoetinové receptory mohou být přítomny na povrchu různých nádorových buněk. Stejně jako u všech růstových faktorů je třeba mít na zřeteli, že i epoetiny mohou stimulovat rozvoj nádoru. V několika kontrolovaných klinických studiích nebylo ve spojení s epoetiny prokázáno zlepšení celkové doby přežití ani snížení rizika progrese nádorů u pacientů s anémií spojenou s nádorovým onemocněním.
V kontrolovaných klinických studiích zaměřených na použití přípravku NeoRecormon a ostatních erytropoézu stimulujících přípravků (ESA), bylo zjištěno:
- zkrácení doby do progrese nádoru u pacientů s pokročilým nádorovým onemocněním hlavy a krku, pokud byly dosahovány cílové hladiny hemoglobinu vyšší než 14 g/dl (8,7 mmol/l)
- zkrácení celkové doby přežití a zvýšení počtu úmrtí souvisejících s progresí onemocnění během 4 měsíců léčby u pacientů s metastazujícím karcinomem prsu léčených chemoterapií, pokud byly dosahovány cílové hladiny hemoglobinu 12-14 g/dl (7,5-8,7 mmol/l),
- zvýšení rizika úmrtí při dosahování cílové hladiny hemoglobinu 12 g/dl (7,5 mmol/l) u pacientů s aktivními maligními chorobami, kteří nedostávali ani chemoterapii ani ozařování. V této populaci pacientů není použití ESA indikováno.
Na základě výše uvedených skutečností má být za určitých klinických okolností při léčbě anémie u pacientů s nádorovým onemocněním upřednostněna transfuze krve. Rozhodnutí podat rekombinantní erytropoetin má být přijato na základě zvážení poměru přínosu a rizika a individuálního posouzení jednotlivého pacienta za daných specifických klinických podmínek. Další faktory, které mají být zváženy, je posouzení typu a stádia nádorového onemocnění; stupeň anémie; očekávaná délka přežití; podmínky, za kterých je pacient léčen; a vlastní volba pacienta (viz bod 5.1)
Může dojít ke vzestupu krevního tlaku, který lze farmakologicky léčit. Doporučuje se proto monitorovat krevní tlak, zejména v úvodní fázi léčby pacientů s nádorovým onemocněním.
U onkologických pacientů mají být také pravidelně sledovány počty krevních destiček a hladina hemoglobinu.
U pacientů v programu před autologní transfuzí krve může být zvýšení počtu krevních destiček, většinou v rozmezí normálních hodnot. Proto je doporučeno určovat počet krevních destiček u těchto pacientů alespoň jednou týdně. Pokud dojde ke zvýšení krevních destiček o více než 150 x 109/l nebo když nárůst přesáhne normální hodnoty, má být léčba přípravkem NeoRecormon přerušena.
Upředčasně narozených dětí nelze vyloučit možné riziko retinopatie způsobené erytropoetinem, proto je třeba zvýšené pozornosti a rozhodnutí o léčbě předčasně narozených dětí má být zvažováno na základě posouzení možného prospěchu a rizika této léčby a jiných dostupných možností.
U pacientů s chronickým selháním ledvin je v průběhu léčby přípravkem NeoRecormon při hemodialýze často nutné zvýšení dávky heparinu z důvodu zvýšeného hematokritu. Pokud není heparinizace optimální, může dojít k okluzi systému dialýzy.
U pacientů s chronickým selháním ledvin s rizikem trombózy cévního přístupu má být zvážena časná kontrola cévního přístupu a trombotická profylaxe kyselinou acetylsalicylovou.
V průběhu léčby přípravkem NeoRecormon má být pravidelně monitorována hladina sérového draslíku a fosfátů. Zvýšení kalémie bylo zaznamenáno u několika uremických pacientů léčených přípravkem NeoRecormon, i když příčinná souvislost nebyla ověřena. Pokud je pozorována zvýšená nebo stoupající hladina draslíku, pak má být zváženo přerušení podávání přípravku NeoRecormon dokud se kalémie neupraví.
Pro použití přípravku NeoRecormon v programu před autologní transfuzí krve musí být zváženy obecné zásady dárcovství krve, zvláště:
- dárci mají být pouze pacienti s hematokritem > 33 % (hemoglobin > 11g/dl [6,83 mmol/l];
- má být věnována zvláštní pozornost pacientům s hmotností nižší než 50 kg;
- jednotlivý čerpaný objem nemá překročit cca 12 % odhadnutého objemu krve pacienta.
Léčba má být vyhrazena pro pacienty, u kterých je zvlášť důležité vyhnout se transfuzi homologní krve a mají být zvážena rizika a prospěch homologní transfuze.
Zneužití
Zneužití přípravku zdravými osobami může vést k nadměrnému zvýšení hematokritu. Toto zvýšení může být spojeno s život ohrožujícími kardiovaskulárními komplikacemi.
Pomocné látky
Přípravek NeoRecormon v přeplněné injekční stříkačce obsahuje až 0,3 mg fenylalaninu v jedné injekční stříkačce jako pomocnou látku. Tato skutečnost má být brána v úvahu při léčbě pacientů trpících vážnými formami fenylketonurie.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v injekční stříkačce, tj. v podstatě je „bez sodíku“
Zpětná zjistitelnost přípravku NeoRecormon
Z důvodu snadnější zpětné zjistitelnosti látek stimulujících erytropoézu (ESAs) má být obchodní název předepsaného přípravku ESA zřetelně zaznamenán (nebo vyznačen) v pacientově dokumentaci.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Z dostupných klinických výsledků nejsou známy žádné interakce přípravku NeoRecormon s ostatními léčivými přípravky.
Pokusy na zvířatech prokázaly, že epoetin beta nezvyšuje myelotoxicitu cytostatických léčivých látek jako jsou etoposid, cisplatina, cyklofosfamid a fluorouracil.
4.6 Fertilita, těhotenství a kojení
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3).
Nejsou k dispozici žádné klinické údaje o účincích epoetinu beta během těhotenství.
Při předepisování těhotným ženám nutno postupovat opatrně.
Kojení
Není známo, zda se epoetin beta vylučuje do lidského mateřského mléka.
Rozhodnutí, zda pokračovat v kojení/přerušit kojení nebo pokračovat v léčbě/přerušit léčbu epoetinem beta, má být zvoleno v závislosti na prospěšnosti kojení pro dítě a prospěšnosti léčby epoetinem beta u matky.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek NeoRecormon nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Na základě výsledků klinických studií s 1725 pacienty lze očekávat, že přibližně u 8 % pacientů léčených přípravkem NeoRecormon se mohou vyskytnout nežádoucí účinky.
Anemičtí pacienti s chronickým selháním ledvin
Nejčastějším nežádoucím účinkem v průběhu léčby přípravkem NeoRecormon je zvýšení krevního tlaku nebo zhoršení stávající hypertenze, zvláště pak v případech rychlého zvýšení hodnot hematokritu (viz bod 4.4). Hypertenzní krize s příznaky encefalopatie (např. bolesti hlavy a stavy zmatenosti, senzoricko-motorické poruchy jako například porucha řeči nebo zhoršení chůze až tonicko-klonické křeče) se mohou objevit také u jednotlivých pacientů s jinak normálním nebo nízkým krevním tlakem (viz bod 4.4).
Může dojít k trombóze žilního přístupu zejména u pacientů s tendencí k hypotenzi nebo s komplikacemi arteriovenózního zkratu (např. stenózy, aneuryzmata), viz bod 4.4. Ve většině případů je pozorován pokles hodnot sérového železa souběžně se vzestupem hematokritu (viz bod 4.4). Navíc byl v ojedinělých případech pozorován vzestup sérového draslíku a hladiny fosfátů (viz bod 4.4).
V ojedinělých případech byla v souvislosti s léčbou přípravkem NeoRecormon hlášena čistá aplazie červených krvinek (PRCA) způsobená neutralizujícími antitrombopoetinovými protilátkami.
V případě diagnózy čisté aplazie červených krvinek (PRCA) způsobené neutralizujícími antitrombopoetinovými protilátkami musí být léčba přípravkem NeoRecormon ukončena a pacient nemá být převáděn na léčbu jinou erytropoetickou bílkovinou (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 1.
Pacienti s rakovinou
Bolesti hlavy související s léčbou epoetinem beta a hypertenze, kterou lze medikamentózně léčit, jsou časté (viz bod 4.4).
U některých pacientů je pozorován pokles sérového železa (viz bod 4.4).
V klinických studiích byl prokázán vyšší výskyt tromboembolických událostí u pacientů s karcinomem léčených přípravkem NeoRecormon ve srovnání s neléčenými kontrolními skupinami nebo placebem. U pacientů léčených přípravkem NeoRecormon byla incidence 7 % ve srovnání s 4 % u kontrolních skupin; aniž by to bylo spojeno se zvýšenou mortalitou na tromboembolické události ve srovnání s kontrolními skupinami.
Nežádoucí účinky jsou uvedeny níže v tabulce 2.
Pacienti v programu autologního dárcovství krve
U pacientů v programu autologního dárcovství krve byla hlášena lehce zvýšená četnost tromboembolických událostí. Kauzální vztah k léčbě přípravkem NeoRecormon však nebyl prokázán.
Ve studiích kontrolovaných placebem byl více vyjádřen přechodný deficit železa u pacientů léčených přípravkem NeoRecormon než u kontrolní skupiny (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 3.
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů MedDRA a absolutní četnosti. Kategorie četnosti jsou definovány dle následující konvence:
velmi časté (> 1/10); časté (>1/100 to <1/10); méně časté (>1/1 000 to <1/100); vzácné (>1/10 000 to <1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
Tabulka 1: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů s chronickým selháním ledvin léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze Hypertenzní krize |
Časté Méně časté |
|
Poruchy nervového systému |
Časté | |
|
Poruchy krve a |
Trombóza cévního |
Vzácné |
|
lymfatického systému |
přístupu Trombocytóza |
Velmi vzácné |
Tabulka 2: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u onkologických pacientů léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze |
Časté |
|
Poruchy krve a lymfatického systému |
Tromboembolické účinky |
Časté |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Tabulka 3: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů v programu autologního dárcovství krve léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Předčasně narozené děti
Pokles hodnot sérového ferritinu je velmi častý (viz bod 4.4).
Popis vybraných nežádoucích účinků
Vzácně byly pozorovány kožní reakce jako jsou vyrážka, svědění, kopřivka nebo reakce v místě vpichu. Velmi vzácně byly popsány anafylaktoidní reakce související s léčbou epoetinem beta. V kontrolovaných klinických studiích však zvýšení výskytu hypersenzitivních reakcí nebylo popsáno.
Ve velmi vzácných případech, především na počátku léčby, byl zaznamenán výskyt příznaků podobných chřipce ("flu-like" symptomy) jako je horečka, zimnice, bolesti hlavy, bolest v končetinách, malátnost a/nebo bolest kostí. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Údaje z kontrolované klinické studie s epoetinem alfa nebo darbepoetinem alfa udávaly incidenci cévní mozkové příhody jako častou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 P ředávkování
Terapeutický rozsah přípravku NeoRecormon je velmi široký. Dokonce i při velmi vysokých hladinách v séru nebyly pozorovány žádné příznaky otravy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA01 Mechanismus účinku
Erytropoetin je glykoprotein, který stimuluje tvorbu erytrocytů z prekurzorů kmenových buněk.
Působí jako faktor stimulující mitózu kmenových buněk červené krevní řady a hormon působící na jejich diferenciaci.
Epoetin beta, léčivá látka přípravku NeoRecormon, je svým složením aminokyselin a cukrů identický s erytropoetinem, který byl izolován z moči anemických pacientů.
Biologický účinek epoetinu beta byl demonstrován po intravenózní a podkožní aplikaci u různých pokusných zvířat in vivo (normální a uremičtí potkani, polycytemické myši, psi). Po podání epoetinu beta se zvýší počet erytrocytů, hodnota Hb a retikulocytů stejně tak jako rychlost inkorporace 59Fe.
In vitro byla zjištěna zvýšená inkorporace 3H-thymidinu v erytroidních jaderných buňkách sleziny (kultura buněk sleziny myší) po inkubaci s epoetinem beta.
Pokusy na tkáňových kulturách buněk lidské kostní dřeně prokázaly, že epoetin beta stimuluje jen tvorbu červených krvinek a neovlivňuje tvorbu bílých krvinek. Nebyly zjištěny cytotoxické účinky epoetinu beta na kostní dřeň ani na lidské kožní buňky
Po podání jednotlivé dávky epoetinu beta nebyly pozorovány žádné vlivy na chování nebo na pohybovou činnost u myší a oběhové nebo respirační funkce u psů.
Klinická účinnost a bezpečnost
V randomizované dvojitě zaslepené, placebem kontrolované studii, která hodnotila 4038 pacientů s chronickým selháním ledvin bez nutnosti dialýzy, s diabetem 2. typu a hladinami hemoglobinu < 11 g/dl, byli pacienti léčeni buď darbepoetinem alfa až do dosažení cílových hladin hemoglobinu 13 g/dl, nebo dostávali placebo (viz bod 4.4). Studie nesplnila žádný z primárních cílů, který by prokázal snížení rizika úmrtí ze všech příčin, kardiovaskulární morbidity nebo terminálního stádia onemocnění ledvin (ESRD). Analýza jednotlivých komponent složených cílových parametrů prokázala následující poměr rizik (95% CI): úmrtí 1,05 (0,92; 1,21), cévní mozková příhoda 1,92 (1,38; 2,68), městnavé srdeční selhání 0,89 (0,74; 1,08), infarkt myokardu 0,96 (0,75; 1,23), hospitalizace z důvodu ischemie myokardu 0,84 (0,55; 1,27), terminální stádium onemocnění ledvin 1,02 (0,87; 1,18).
Souhrnné post-hoc analýzy klinických studií s ESA byly provedeny u pacientů s chronickým selháním ledvin (u pacientů, kteří byli nebo nebyli na dialýze, u diabetiků a u pacientů bez diabetu). Byla pozorována tendence směrem ke zvýšení odhadovaného rizika mortality z jakýchkoli důvodů, rizika kardiovaskulárních a cerebrovaskulárních příhod spojených s vyššími kumulativními dávkami ESA, nezávisle na stavu diabetu nebo dialýze (viz body 4.2 a 4.4).
Erytropoetin je růstový faktor primárně podporující tvorbu červených krvinek. Receptory pro erytropoetin mohou být exprimovány na povrchu mnoha nádorových buněk.
Přežití a progrese nádoru byla zkoumána v pěti velkých kontrolovaných klinických studiích zahrnujících celkem 2833 pacientů; čtyři z těchto studií byly dvojitě zaslepené kontrolované placebe m, jedna studie byla otevřená. Dvou studií se účastnili pacienti léčení chemoterapií. Cílová koncentrace hemoglobinu byla ve dvou studiích >13 g/dl; v ostatních třech studiích 12-14 g/dl.
V otevřené studii nebyl z hlediska celkového přežití zjištěn žádný rozdíl mezi pacienty léčenými rekomb inantním lidským erytropoetinem a mezi kontrolní skupinou. Ve čtyřech placebem kontrolovaných studiích se poměry rizik pro celkové přežití pohybovaly mezi 1,25 a 2,47 ve prospěch kontrolních skupin. Při srovnání s kontrolními skupinami ukázaly tyto studie konzistentní nevysvětlené statisticky významné zvýšení mortality u pacientů, kteří měli anémii spojenou s různými běžnými maligními nádory a kteří dostávali rekombinantní lidský erytropoetin. Výsledek celkového přežití zjištěný v klinických studiích nebylo možné uspokojivě vysvětlit rozdílem v incidenci trombózy a přidružených komplikací mezi skupinou dostávající rekombinantní lidský erytropoetin a mezi skupinou kontrolní.
Metaanalýza založená na údajích jednotlivých pacientů zahrnovala data ze všech 12 kontrolovaných klinických studií prováděných u pacientů s anémií a maligním nádorovým onemocněním, kteří byli léčeni přípravkem NeoRecormon (n=2301), ukázala celkový odhadovaný poměr rizik pro přežití 1,13 ve prospěch kontrol (95% CI 0,87; 1,46). U pacientů, kteří měli v baseline hladinu hemoglobinu < 10 g/dl (n=899), byl odhadovaný poměr rizik pro přežití 0,98 (95% CI 0,68 až 1,40). V celkové populaci bylo pozorováno zvýšené relativní riziko tromboembolických příhod (RR 1,62, 95% CI: 1,13; 2,31).
Byla provedena analýza dat na úrovni jednotlivých nemocných u více než 13 900 pacientů se zhoubným nádorem (léčených chemoterapií, radioterapií, chemoradioterapií nebo bez terapie) zařazených do 53 kontrolovaných klinických studií zahrnujících podávání několika epoetinů. Metaanalýza údajů celkového přežití prokázala, že poměr rizik (hazard ratio) je odhadem 1,06 ve prospěch kontrolních skupin (95% CI: 1,00, 1,12; 53 studií a 13 933 pacientů) a u pacientů se zhoubným nádorem podstupujících chemoterapii byl poměr rizik (hazard ratio) celkového přežití 1,04 (95% CI: 0,97, 1,11; 38 studií a 10 441 pacientů). Meta-analýzy zároveň konzistentně poukazují na významné zvýšení relativního rizika tromboembolických příhod u pacientů se zhoubným nádorem, kteří dostávají rekombinantní lidský erytropoetin (viz bod 4.4).
Ve velmi vzácných případech se v průběhu léčby rekombinantním lidským erytropoetinem objevily neutralizující protilátky proti erytropoetinu s nebo bez čisté aplazie červené krevní řady (PRCA).
5.2 Farmakokinetické vlastnosti
Farmakokinetické sledování u zdravých dobrovolníků a uremických pacientů ukázalo, že po intravenózním podání je poločas epoetinu beta mezi 4 - 12 hodinami a že distribuční objem odpovídá jedno- až dvojnásobku plazmatického objemu. Analogické výsledky byly nalezeny v pokusech na zvířatech u normálních i uremických potkanů.
Protrahovaná absorpce epoetinu beta po subkutánním podání uremickým pacientům vede ke vzniku koncentračního plató, přičemž maximální koncentrace je dosaženo v průměru za 12 - 28 hodin. Terminální poločas je delší než po intravenózním podání s průměrem 13 - 28 hodin.
Ve srovnání s intravenózní aplikací je biologická dostupnost epoetinu beta po podkožní aplikaci mezi 23 - 42 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
Studie kancerogenity s homologním erytropoetinem u myší neodhalily žádné známky proliferativního nebo kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 S eznam pomocných látek
Močovina,
Chlorid sodný,
Polysorbát 20,
Dihydrogenfosforečnan sodný dihydrát,
Hydrogenfosforečnan sodný dodekahydrát,
Chlorid vápenat dihydrát,
Glycin,
L-leucin,
L-isoleucin,
L-threonin,
L-kyselina glutamová,
L-fenylalanin.
Voda na injekci.
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku v původním obalu, aby byl přípravek chráněn před světlem.
Pro usnadnění ambulantního použití může pacient vyjmout přípravek z chladničky a uchovávat jej jednorázově po dobu maximálně 3 dnů při pokojové teplotě (maximálně do 25 °C).
6.5 Druh obalu a obsah balení
Předplněná injekční stříkačka (sklo typu I) s víčkem a zátkou (pryž potažená teflonem) a s injekční jehlou (27G1/2). Jedna injekční stříkačka obsahuje 0,3 ml roztoku.
Velikost balení po 1 předplněné injekční stříkačce s 1 jehlou nebo 6 předplněných injekčních stříkačkách se 6 jehlami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Nejdříve si dobře umyjte ruce!
1. Vyjměte jednu předplněnou injekční stříkačku z balení a zkontrolujte, zda roztok ve stříkačce je čirý, bezbarvý a bez viditelných částic. Odstraňte víčko z injekční stříkačky.
2. Vyjměte jednu injekční jehlu z balení, nasaďte ji na injekční stříkačku a odstraňte z jehly ochranné víčko.
3. Vytlačte vzduch z injekční stříkačky tím, že stříkačku držíte ve vzpřímené poloze a jemně tlačíte píst vzhůru. Tlačte na píst tak dlouho, dokud množství přípravku NeoRecormon v injekční stříkačce neodpovídá množství předepsanému pro aplikaci.
4. Za použití tampónu namočeného v alkoholu si očistěte kůži v místě vpichu. Vytvořte záhyb na kůži tím, že ji sevřete mezi palec a ukazováček. Stiskněte tělo injekční stříkačky v části bližší k jehle a aplikujte jehlu do záhybu kůže rychlým a pevným pohybem. Vstříkněte roztok přípravku NeoRecormon. Jehlu rychle vytáhněte a zatlačte na místo vpichu suchým a sterilním tampónem.
Tento léčivý přípravek je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/031/029 -030
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. července 1997
Datum posledního prodloužení registrace: 16. července 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
NeoRecormon 3000 IU injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s 0,3 ml injekčního roztoku obsahuje 3000 mezinárodních jednotek (IU), což odpovídá 24,9 mikrogramům epoetinu beta* (rekombinantní lidský erytropoetin). Jeden ml injekčního roztoku obsahuje 10 000 IU epoetinu beta.
* Vyrobený technologií rekombinatní DNA v ovariálních buňkách čínských křečků.
Pomocné látky se známým účinkem:
Fenylalanin (až do 0,3 mg v injekční stříkačce)
Sodík (méně než 1 mmol v injekční stříkačce)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Bezbarvý, čirý až slabě opalescentní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek NeoRecormon je indikován k léčbě:
- Léčba symptomatické anémie spojené s chronickým renálním selháním u dospělých a pediatrických pacientů.
- Prevence anémie u předčasně narozených dětí s porodní váhou v rozmezí 750-1500 g, které se narodily v době před 34. týdnem těhotenství.
- Léčba symptomatické anémie u dospělých pacientů s nemyeloidními malignitami, kteří jsou léčeni chemoterapií.
- Zvýšení počtu erytrocytů před odběrem krve k autologní transfuzi krve. Použití v této indikaci musí být zváženo vzhledem k udávanému zvýšení rizika vzniku tromboembolických příhod. Použití je indikováno pouze u pacientů s mírnou anémií (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], bez nedostatku železa), pokud postupy pro konzervování krve nejsou dostupné nebo účinné nebo plánovaný zvolený velký chirurgický zákrok vyžaduje velký objem krve (4 a více jednotek krve pro ženy nebo 5 a více jednotek pro muže). Viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba přípravkem NeoRecormon může být zahájena pouze lékařem se zkušenostmi s výše uvedenými
indikacemi přípravku. Protože byly vzácně zaznamenány anafylaktoidní reakce po podání přípravku,
první dávka má být aplikována pod dohledem zdravotnického personálu.
Dávkování
Léčba symptomatické anémie u dospělých a pediatrických pacientů s chronickým selháním ledvin Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta. Přípravek NeoRecormon má být podáván buď subkutánně nebo intravenózně, tak aby byla hladina hemoglobinu zvýšena maximálně na 12 g/dl (7,5 mmol/l). U pacientů, kteří nepodstupují dialýzu, se upřednostňuje subkutánní podání, kdy není zapotřebí vpichů do periferních žil. V případě intravenózního podání má být přípravek podáván alespoň po dobu 2 minut, například přes arterio-venózní píštěl na konci dialýzy u pacientů na hemodialýze.
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu v rozsahu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Zvýšení hladiny hemoglobinu o více než 2 g/dl (1,25 mmol/l) během období čtyř týdnů není žádoucí. Pokud k němu dojde, má být dávka odpovídajícím způsobem upravena, jak je uvedeno dále. Je-li rychlost vzestupu hladiny hemoglobinu vyšší než 2 g/dl (1,25 mmol/l) za měsíc nebo pokud se hladina hemoglobinu zvýší až na hodnotu 12 g/dl (7,45 mmol/l) je třeba dávku snížit přibližně o 25 %. Pokud by se hladina hemoglobinu dále zvyšovala, má být léčba přerušena, dokud nezačne hladina opět klesat, a v tomto bodě má být znovu zahájena léčba dávkou přibližně o 25 % nižší, než byla předchozí podávaná dávka.
Pacienti mají být důkladně sledováni a je třeba ověřit, že byla použita nejnižší schválená účinná dávka přípravku NeoRecormon, která postačuje pro kontrolu symptomů anémie při zachování koncentrace hemoglobinu pod nebo na hodnotě 12 g/dl (7,45 mmol/l).
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin. U pacientů se slabou odpovědí hemoglobinu na přípravek NeoRecormon mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz bod 4.4 a 5.1).
V případě hypertenze nebo stávajících kardiovaskulárních, cerebrovaskulárních nebo periferních vaskulárních onemocnění má být týdenní zvýšení Hb a cílová hodnota Hb stanovena individuálně při zvážení klinického obrazu.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází.
1. Fáze korekční
- Subkutánní podání:
Počáteční dávka je 3 x 20 IU/kg tělesné hmotnosti týdně. Dávka může být zvýšena každé 4 týdny o 3 x 20 IU/kg za týden, jestliže není dosaženo adekvátního vzestupu Hb (< 0,25 g/dl za týden).
Týdenní dávka může být také rozdělena na denní dávky.
- Intravenózní podání:
Počáteční dávka je 3 x 40 IU/kg tělesné hmotnosti týdně. Dávka může být po 4 týdnech zvýšena na 80 IU/kg třikrát týdně a pokud je nutné další zvýšení dávky, má být o 20 IU/kg třikrát týdně v měsíčních intervalech.
Maximální dávka při obou způsobech podání nemá překročit 720 IU/kg za týden.
2. Fáze udržovací
Pro udržení Hb mezi 10 až 12 g/dl je dávka ze začátku snížena na polovinu předešlé dávky. Následně je u pacienta dávka nastavena v intervalech jednoho nebo dvou týdnů individuálně (udržovací dávka).
V případě podkožního podání může být týdenní dávka podána v jedné injekci týdně, nebo může být rozdělena do tří až sedmi dávek týdně. Pacienti, kteří jsou stabilní na režimu podávání jednou týdně, mohou být převedeni na dávkování jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Výsledky klinických studií u dětí prokázaly, že čím jsou pacienti mladší, tím v průměru vyšší dávky přípravku NeoRecormon vyžadují. Doporučený plán dávkování má však být dodržován, protože klinický účinek u jednotlivých pacientů nelze zcela předvídat.
Léčba přípravkem NeoRecormon v předplněné injekční stříkačce je obvykle dlouhodobá. Může však být, pokud je to nutné, kdykoliv přerušena. Údaje týkající se dávkovacího schématu podání jednou týdně vycházejí z klinických studií s délkou léčby 24 týdnů.
Prevence anémie u předčasně narozených dětí
Roztok je podáván subkutánně v dávce 3 x 250 IU/kg tělesné hmotnosti za týden. Předčasně narozené děti u kterých již byla provedena transfuze v okamžiku zahájení léčby přípravkem NeoRecormon nebudou pravděpodobně mít takový užitek z léčby jako předčasně narozené děti, u kterých transfuze provedena nebyla. Doporučená délka léčby je 6 týdnů.
Léčba symptomatické chemoterapií indukované anémie u pacientů s nádorovým onemocněním Přípravek NeoRecormon má být podáván subkutánně pacientům s anémií (např. koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l)). Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta.
Týdenní dávka může být podána v jedné injekci jednou týdně nebo může být rozdělena na 3 až 7 jednotlivých dávek.
Doporučená úvodní dávka je 30 000 IU týdně (to odpovídá přibližně 450 IU/kg tělesné hmotnosti týdně u pacienta s průměrnou hmotností).
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Pokud dojde po 4 týdnech léčby ke zvýšení hodnot hemoglobinu o minimálně 1 g/dl (0,62 mmol/l), má se v podávání této dávky dále pokračovat. Pokud hladina hemoglobinu nestoupne o alespoň 1 g/dl (0,62 mmol/l), má být zváženo podávání dvojnásobné týdenní dávky. Pokud hladina hemoglobinu nestoupne po 8 týdnech léčby o alespoň 1 g/dl (0,62 mmol/l), je odpověď nepravděpodobná a léčba má být ukončena.
Léčba má pokračovat až 4 týdny po ukončení chemoterapie.
Maximální dávka nemá překročit 60 000 IU týdně.
Jakmile je u jednotlivého pacienta dosaženo léčebného cíle, dávka má být snížena o 25 až 50 %, aby byla udržena hladina hemoglobinu na této úrovni. Je zapotřebí vzít v úvahu titrování dávky.
Překročí-li hemoglobin hladinu 12 g/dl (7,5 mmol/l), má být dávka snížena zhruba o 25 až 50 %.
Pokud hladina hemoglobinu překročí 13 g/dl (8,1 mmol/l), má být léčba přípravkem NeoRecormon dočasně přerušena. Pokud hladina hemoglobinu klesne na 12 g/dl (7,5 mmol/l) nebo níže, má být léčba opět zahájena s dávkou přibližně o 25 % nižší, než byla předchozí dávka.
Pokud je nárůst hemoglobinu v průběhu 4 týdnů vyšší než 2 g/dl (1,3 mmol/l), dávka má být snížena o 25 až 50 %.
Pacienti mají být důkladně sledováni, přičemž je třeba ověřit, že byla použita nejnižší schválená dávka přípravku NeoRecormon postačující pro adekvátní kontrolu symptomů anémie.
Podávání pro zvýšení množství autologní krve
Přípravek je aplikován intravenózně po dobu asi 2 minut nebo subkutánně.
Přípravek NeoRecormon je podáván dvakrát týdně po dobu 4 týdnů. V případě, kdy hematokrit pacienta umožňuje odběr krve, tj. hodnota hematokritu je > 33 %, je přípravek NeoRecormon podáván v okamžiku ukončení odběru.
V průběhu celé léčby nemá být překročena hodnota hematokritu 48 %.
Dávkování musí být stanoveno chirurgickým týmem zvlášť pro každého pacienta podle požadovaného množství krve pro autologní transfuzi a endogenní rezervy červených krvinek:
1. P ožadované množství krve pro autologní transfuzi závisí na předpokládané ztrátě krve, popřípadě na použití postupů konzervování krve a na fyzické kondici pacienta.
Množství potřebné autologní krve má stačit k tomu, aby bylo možné se vyhnout transfuzi homologní krve.
Požadované množství předem odebrané krve je vyjádřeno v jednotkách, přičemž jedna jednotka v nomogramu je ekvivalentní 180 ml červených krvinek.
2. Možnost autologní transfuze krve závisí především na objemu pacientovy krve a na výchozí hodnotě hematokritu. Obě proměnné určují endogenní rezervu červených krvinek, kterou je možné vypočítat podle následujících vzorců:
Endogenní rezerva červených krvinek = objem krve [ml] x (hematokrit - 33)/100 Ženy: objem krve [ml] = 41 [ml/kg] x tělesná hmotnost [kg] + 1200 [ml]
Muži: objem krve [ml] = 44 [ml/kg] x tělesná hmotnost [kg] + 1600 [ml]
(tělesná hmotnost > 45 kg)
Indikace pro léčbu přípravkem NeoRecormon a jednotlivá dávka mají být stanoveny z požadovaného množství předem odebrané krve a endogenní rezervy červených krvinek podle následujících grafů.
Ženy
Požadované množství krve pro autologní transfuzi [jednotky]
Muži
Požadované množství krve pro autologní transfuzi [jednotky]
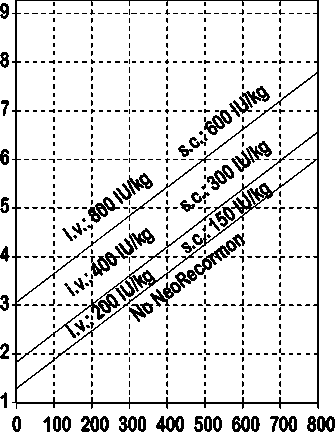
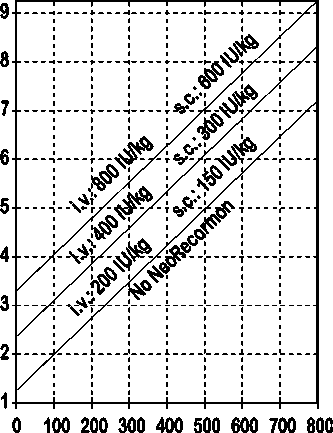
Endogenní rezerva červených krvinek [ml] Endogenní rezerva červených krvinek [ml]
Takto stanovená jednotlivá dávka je podávána dvakrát týdně po dobu 4 týdnů. Maximální dávka nemá překročit 1600 IU/kg tělesné hmotnosti a týden při intravenózním podání nebo 1200 IU/kg tělesné hmotnosti a týden při subkutánním podávání.
Způsob podání
Přípravek NeoRecormon v předplněné injekční stříkačce je připraven k použití. Aplikován může být pouze roztok, který je čirý nebo lehce opalescentní, bezbarvý a bez viditelných částic.
Přípravek NeoRecormon v předplněné injekční stříkačce je sterilní, ale neobsahuje žádné konzervační látky. Za žádných okolností proto není možno aplikovat více než jednu dávku s jednou předplněnou injekční stříkačkou; léčivý přípravek je určen pouze k jednorázovému použití.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku (uvedenou v bodě 6.1).
Špatně kontrolovatelná hypertenze.
V indikaci “podání pro zvýšení množství autologní krve”: infarkt myokardu nebo cévní mozková příhoda v průběhu jednoho měsíce před zákrokem, nestabilní angina pectoris, zvýšené riziko hluboké venózní trombózy jako např. po prodělaném tromboembolickém onemocnění.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek NeoRecormon má být užíván s opatrností u pacientů s refrakterní anémií s nadbytkem transformovaných blastů, při epilepsii, trombocytóze a chronickém selhání jater. Je nutné vyloučit nedostatek kyseliny listové a vitamínu BJ2, protože tyto stavy snižují účinek přípravku NeoRecormon.
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin, neboť vysoké kumulující se dávky epoetinu mohou mít spojitost se zvýšeným rizikem mortality, závažnými kardiovaskulárními a cerebrovaskulárními příhodami. U pacientů se slabou odpovědí hemoglobinu na epoetiny mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz body 4.2 a 5.1).
Je třeba zhodnotit u všech pacientů hladinu železa před a v průběhu léčby, aby byla zajištěna účinná erytropoéza. Může být nutná substituce železa prováděná v souladu s léčebnými doporučeními.
Závažné zatížení hliníkem, ke kterému během léčby renálního selhání může dojít, může rovněž účinek přípravku NeoRecormon snížit.
Indikace k léčbě přípravkem NeoRecormon u pacientů s nefrosklerózou, kteří ještě nejsou dialyzovaní, má být zvážena individuálně, protože u těchto pacientů nelze vyloučit urychlení postupu renálního selhání.
Čistá aplazie buněk červené krevní řady (PRCA)
PRCA způsobená neutralizujícími antierytropoetinovými protilátkami byla hlášena ve spojení s léčbou erytropoetiny, včetně přípravku NeoRecormon. Bylo prokázáno, že tyto protilátky zkříženě reagují se všemi erytropoetinovými proteiny, a proto pacienti, u kterých je podezření nebo je potvrzen výskyt neutralizujících protilátek proti erytropoetinu, nemají být převáděni na přípravek NeoRecormon (viz bod 4.8).
PRCA u pacientů s hepatitidou C
Paradoxní pokles hladiny hemoglobinu a rozvoj závažné anémie související s nízkým počtem retikulocytů má vést k okamžitému přerušení léčby epoetinem a provedení testů na protilátky proti erytropoetinu. Tyto případy byly hlášeny, pokud byly pacientům s hepatitidou C léčených interferonem a ribavirinem současně podávány epoetiny. Epoetiny nejsou schválené pro léčbu anémie související s hepatitidou C.
Monitorování krevního tlaku
Může dojít ke vzestupu krevního tlaku nebo ke zhoršení již existující hypertenze zejména v případě rychlého vzestupu hematokritu. Toto zvýšení krevního tlaku je možno upravit pomocí léků. Pokud zvýšení krevního tlaku nemůže být upraveno medikamentózně, je doporučeno krátkodobé přerušení léčby přípravkem NeoRecormon. Zvláště na začátku terapie se doporučuje pravidelné monitorování krevního tlaku, a to rovněž mezi dialýzami. Může dojít ke vzniku hypertenzní krize s příznaky napodobujícími encefalopatii, která vyžaduje okamžitou lékařskou intervenci a intenzivní péči. Zvláštní pozoronost je třeba věnovat náhlým bodavým migrenosním bolestem hlavy jako možnému varovnému příznaku.
Chronické selhání ledvin
U pacientů 5 chronickým selháním ledvin, léčených přípravkem NeoRecormon, může dojít v průběhu léčby k mírnému, na dávce závislému, nárůstu počtu krevních destiček v rozmezí normálních hodnot, zvláště pak v případě intravenózního podávání. K poklesu tohoto nárůstu dochází v dalším průběhu léčby. Je doporučeno pravidelně sledovat počet krevních destiček v průběhu prvních 8 týdnů léčby.
Koncentrace hemoglobinu
U pacientů s chronickým selháním ledvin nemá udržovací koncentrace hemoglobinu překročit horní limit cílové koncentrace hemoglobinu doporučovaný v bodě 4.2. V klinických studiích bylo pozorováno zvýšené riziko úmrtí a závažných kardiovaskulárních příhod nebo cerebrovaskulárních příhod, včetně cévní mozkové příhody, pokud byly používány přípravky stimulující erytropoézu (ESA) k dosažení cílových hodnot hemoglobinu vyšších než 12 g/dl (7,5 mmol/l).
V kontrolovaných klinických studiích se neprokázal žádný signifikantní přínos podávání epoetinů, pokud byla koncentrace hemoglobinu zvyšována nad hladinu nezbytně nutnou pro kontrolu symptomů anémie a předcházení krevní transfuze.
U předčasně narozených dětí může dojít k mírnému nárůstu počtu krevních destiček, především v období mezi 12. - 14. dnem života, proto má být počet krevních destiček sledován v pravidelných intervalech.
Účinek na růst nádorů
Epoetiny jsou růstové faktory, které primárně podporují tvorbu červených krvinek. Erytropoetinové receptory mohou být přítomny na povrchu různých nádorových buněk. Stejně jako u všech růstových faktorů je třeba mít na zřeteli, že i epoetiny mohou stimulovat rozvoj nádoru. V několika kontrolovaných klinických studiích nebylo ve spojení s epoetiny prokázáno zlepšení celkové doby přežití ani snížení rizika progrese nádorů u pacientů s anémií spojenou s nádorovým onemocněním.
V kontrolovaných klinických studiích zaměřených na použití přípravku NeoRecormon a ostatních erytropoézu stimulujících přípravků (ESA), bylo zjištěno:
- zkrácení doby do progrese nádoru u pacientů s pokročilým nádorovým onemocněním hlavy a krku, pokud byly dosahovány cílové hladiny hemoglobinu vyšší než 14 g/dl (8,7 mmol/l)
- zkrácení celkové doby přežití a zvýšení počtu úmrtí souvisejících s progresí onemocnění během 4 měsíců léčby u pacientů s metastazujícím karcinomem prsu léčených chemoterapií, pokud byly dosahovány cílové hladiny hemoglobinu 12-14 g/dl (7,5-8,7 mmol/l),
- zvýšení rizika úmrtí při dosahování cílové hladiny hemoglobinu 12 g/dl (7,5 mmol/l) u pacientů s aktivními maligními chorobami, kteří nedostávali ani chemoterapii ani ozařování. V této populaci pacientů není použití ESA indikováno.
Na základě výše uvedených skutečností má být za určitých klinických okolností při léčbě anémie u pacientů s nádorovým onemocněním upřednostněna transfuze krve. Rozhodnutí podat rekombinantní erytropoetin má být přijato na základě zvážení poměru přínosu a rizika a individuálního posouzení jednotlivého pacienta za daných specifických klinických podmínek. Další faktory, které mají být zváženy, je posouzení typu a stádia nádorového onemocnění; stupeň anémie; očekávaná délka přežití; podmínky, za kterých je pacient léčen; a vlastní volba pacienta (viz bod 5.1)
Může dojít ke vzestupu krevního tlaku, který lze farmakologicky léčit. Doporučuje se proto monitorovat krevní tlak, zejména v úvodní fázi léčby pacientů s nádorovým onemocněním.
U onkologických pacientů mají být také pravidelně sledovány počty krevních destiček a hladina hemoglobinu.
U pacientů v programu před autologní transfuzí krve může být zvýšení počtu krevních destiček, většinou v rozmezí normálních hodnot. Proto je doporučeno určovat počet krevních destiček u těchto pacientů alespoň jednou týdně. Pokud dojde ke zvýšení krevních destiček o více než 150 x 109/l nebo když nárůst přesáhne normální hodnoty, má být léčba přípravkem NeoRecormon přerušena.
Upředčasně narozených dětí nelze vyloučit možné riziko retinopatie způsobené erytropoetinem, proto je třeba zvýšené pozornosti a rozhodnutí o léčbě předčasně narozených dětí má být zvažováno na základě posouzení možného prospěchu a rizika této léčby a jiných dostupných možností.
U pacientů s chronickým selháním ledvin je v průběhu léčby přípravkem NeoRecormon při hemodialýze často nutné zvýšení dávky heparinu z důvodu zvýšeného hematokritu. Pokud není heparinizace optimální, může dojít k okluzi systému dialýzy.
U pacientů s chronickým selháním ledvin s rizikem trombózy cévního přístupu má být zvážena časná kontrola cévního přístupu a trombotická profylaxe kyselinou acetylsalicylovou.
V průběhu léčby přípravkem NeoRecormon má být pravidelně monitorována hladina sérového draslíku a fosfátů. Zvýšení kalémie bylo zaznamenáno u několika uremických pacientů léčených přípravkem NeoRecormon, i když příčinná souvislost nebyla ověřena. Pokud je pozorována zvýšená nebo stoupající hladina draslíku, pak má být zváženo přerušení podávání přípravku NeoRecormon dokud se kalémie neupraví.
Pro použití přípravku NeoRecormon v programu před autologní transfuzí krve musí být zváženy obecné zásady dárcovství krve, zvláště:
- dárci mají být pouze pacienti s hematokritem > 33 % (hemoglobin > 11g/dl [6,83 mmol/l];
- má být věnována zvláštní pozornost pacientům s hmotností nižší než 50 kg;
- jednotlivý čerpaný objem by neměl překročit cca 12 % odhadnutého objemu krve pacienta. Léčba má být vyhrazena pro pacienty, u kterých je zvlášť důležité vyhnout se transfuzi homologní krve a mají být zvážena rizika a prospěch homologní transfuze.
Zneužití
Zneužití přípravku zdravými osobami může vést k nadměrnému zvýšení hematokritu. Toto zvýšení může být spojeno s život ohrožujícími kardiovaskulárními komplikacemi.
Pomocné látky
Přípravek NeoRecormon v přeplněné injekční stříkačce obsahuje až 0,3 mg fenylalaninu v jedné injekční stříkačce jako pomocnou látku. Tato skutečnost má být brána v úvahu při léčbě pacientů trpících vážnými formami fenylketonurie.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v injekční stříkačce, tj. v podstatě je „bez sodíku“
Zpětná zjistitelnost přípravku NeoRecormon
Z důvodu snadnější zpětné zjistitelnosti látek stimulujících erytropoézu (ESAs) má být obchodní název předepsaného přípravku ESA zřetelně zaznamenán (nebo vyznačen) v pacientově dokumentaci.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Z dostupných klinických výsledků nejsou známy žádné interakce přípravku NeoRecormon s ostatními léčivými příprakvy .
Pokusy na zvířatech prokázaly, že epoetin beta nezvyšuje myelotoxicitu cytostatických léčivých látek jako jsou etoposid, cisplatina, cyklofosfamid a fluorouracil.
4.6 Fertilita, těhotenství a kojení
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3).
Nejsou k dispozici žádné klinické údaje o účincích epoetinu beta během těhotenství.
Při předepisování těhotným ženám nutno postupovat opatrně.
Kojení
Není známo, zda se epoetin beta vylučuje do lidského mateřského mléka.
Rozhodnutí, zda pokračovat v kojení/přerušit kojení nebo pokračovat v léčbě/přerušit léčbu epoetinem beta, má být zvoleno v závislosti na prospěšnosti kojení pro dítě a prospěšnosti léčby epoetinem beta u matky.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek NeoRecormon nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Na základě výsledků klinických studií s 1725 pacienty lze očekávat, že přibližně u 8 % pacientů léčených přípravkem NeoRecormon se mohou vyskytnout nežádoucí účinky.
Anemičtí pacienti s chronickým selháním ledvin
Nejčastějším nežádoucím účinkem v průběhu léčby přípravkem NeoRecormon je zvýšení krevního tlaku nebo zhoršení stávající hypertenze, zvláště pak v případech rychlého zvýšení hodnot hematokritu (viz bod 4.4). Hypertenzní krize s příznaky encefalopatie (např. bolesti hlavy a stavy zmatenosti, senzoricko-motorické poruchy jako například porucha řeči nebo zhoršení chůze až tonicko-klonické křeče) se mohou objevit také u jednotlivých pacientů s jinak normálním nebo nízkým krevním tlakem (viz bod 4.4).
Může dojít k trombóze žilního přístupu zejména u pacientů s tendencí k hypotenzi nebo s komplikacemi arteriovenózního zkratu (např. stenózy, aneuryzmata), viz bod 4.4. Ve většině případů je pozorován pokles hodnot sérového železa souběžně se vzestupem hematokritu (viz bod 4.4). Navíc byl v ojedinělých případech pozorován vzestup sérového draslíku a fosfátů (viz bod 4.4).
V ojedinělých případech byla v souvislosti s léčbou přípravkem NeoRecormon hlášena čistá aplazie červených krvinek (PRCA) způsobená neutralizujícími antitrombopoetinovými protilátkami.
V případě diagnózy čisté aplazie červených krvinek (PRCA) způsobené neutralizujícími antitrombopoetinovými protilátkami musí být léčba přípravkem NeoRecormon ukončena a pacient nemá být převáděn na léčbu jinou erytropoetickou bílkovinou (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 1.
Pacienti s rakovinou
Bolesti hlavy související s léčbou epoetinem beta a hypertenze, kterou lze medikamentózně léčit, jsou časté (viz bod 4.4).
U některých pacientů je pozorován pokles sérového železa (viz bod 4.4).
V klinických studiích byl prokázán vyšší výskyt tromboembolických událostí u pacientů s karcinomem léčených přípravkem NeoRecormon ve srovnání s neléčenými kontrolními skupinami nebo placebem. U pacientů léčených přípravkem NeoRecormon byla incidence 7 % ve srovnání s 4 % u kontrolních skupin; aniž by to bylo spojeno se zvýšenou mortalitou na tromboembolické události ve srovnání s kontrolními skupinami.
Nežádoucí účinky jsou uvedeny níže v tabulce 2.
Pacienti v programu autologního dárcovství krve
U pacientů v programu autologního dárcovství krve byla hlášena lehce zvýšená četnost tromboembolických událostí. Kauzální vztah k léčbě přípravkem NeoRecormon však nebyl prokázán.
Ve studiích kontrolovaných placebem byl více vyjádřen přechodný deficit železa u pacientů léčených přípravkem NeoRecormon než u kontrolní skupiny (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 3.
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů MedDRA a absolutní četnosti. Kategorie četnosti jsou definovány dle následující konvence:
velmi časté (> 1/10); časté (>1/100 to <1/10); méně časté (>1/1 000 to <1/100); vzácné (>1/10 000 to <1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
Tabulka 1: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů s chronickým selháním ledvin léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze Hypertenzní krize |
Časté Méně časté |
|
Poruchy nervového systému |
Časté | |
|
Poruchy krve a |
Trombóza cévního |
Vzácné |
|
lymfatického systému |
přístupu Trombocytóza |
Velmi vzácné |
Tabulka 2: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u onkologických pacientů léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze |
Časté |
|
Poruchy krve a lymfatického systému |
Tromboembolické účinky |
Časté |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Tabulka 3: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů v programu autologního dárcovství krve léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Předčasně narozené děti
Pokles hodnot sérového ferritinu je velmi častý (viz bod 4.4).
Popis vybraných nežádoucích účinků
Vzácně byly pozorovány kožní reakce jako jsou vyrážka, svědění, kopřivka nebo reakce v místě vpichu. Velmi vzácně byly popsány anafylaktoidní reakce související s léčbou epoetinem beta. V kontrolovaných klinických studiích však zvýšení výskytu hypersenzitivních reakcí nebylo popsáno.
Ve velmi vzácných případech, především na počátku léčby, byl zaznamenán výskyt příznaků podobných chřipce ("flu-like" symptomy) jako je horečka, zimnice, bolesti hlavy, bolest v končetinách, malátnost a/nebo bolest kostí. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Údaje z kontrolované klinické studie s epoetinem alfa nebo darbepoetinem alfa udávaly incidenci cévní mozkové příhody jako častou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 P ředávkování
Terapeutický rozsah přípravku NeoRecormon je velmi široký. Dokonce i při velmi vysokých hladinách v séru nebyly pozorovány žádné příznaky otravy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA01 Mechanismus účinku
Erytropoetin je glykoprotein, který stimuluje tvorbu erytrocytů z prekurzorů kmenových buněk.
Působí jako faktor stimulující mitózu kmenových buněk červené krevní řady a hormon působící na jejich diferenciaci.
Epoetin beta, léčivá látka přípravku NeoRecormon, je svým složením aminokyselin a cukrů identický s erytropoetinem, který byl izolován z moči anemických pacientů.
Biologický účinek epoetinu beta byl demonstrován po intravenózní a podkožní aplikaci u různých pokusných zvířat in vivo (normální a uremičtí potkani, polycytemické myši, psi). Po podání epoetinu beta se zvýší počet erytrocytů, hodnota Hb a retikulocytů stejně tak jako rychlost inkorporace 59Fe.
In vitro byla zjištěna zvýšená inkorporace 3H-thymidinu v erytroidních jaderných buňkách sleziny (kultura buněk sleziny myší) po inkubaci s epoetinem beta.
Pokusy na tkáňových kulturách buněk lidské kostní dřeně prokázaly, že epoetin beta stimuluje jen tvorbu červených krvinek a neovlivňuje tvorbu bílých krvinek. Nebyly zjištěny cytotoxické účinky epoetinu beta na kostní dřeň ani na lidské kožní buňky.
Po podání jednotlivé dávky epoetinu beta nebyly pozorovány žádné vlivy na chování nebo na pohybovou činnost u myší a oběhové nebo respirační funkce u psů.
Klinická účinnost a bezpečnost
V randomizované dvojitě zaslepené, placebem kontrolované studii, která hodnotila 4038 pacientů s chronickým selháním ledvin bez nutnosti dialýzy, s diabetem 2. typu a hladinami hemoglobinu < 11 g/dl, byli pacienti léčeni buď darbepoetinem alfa až do dosažení cílových hladin hemoglobinu 13 g/dl, nebo dostávali placebo (viz bod 4.4). Studie nesplnila žádný z primárních cílů, který by prokázal snížení rizika úmrtí ze všech příčin, kardiovaskulární morbidity nebo terminálního stádia onemocnění ledvin (ESRD). Analýza jednotlivých komponent složených cílových parametrů prokázala následující poměr rizik (95% CI): úmrtí 1,05 (o,92; 1,21), cévní mozková příhoda 1,92 (1,38; 2,68), městnavé srdeční selhání 0,89 (0,74; 1,08), infarkt myokardu 0,96 (0,75; 1,23), hospitalizace z důvodu ischemie myokardu 0,84 (0,55; 1,27), terminální stádium onemocnění ledvin 1,02 (0,87; 1,18).
Souhrnné post-hoc analýzy klinických studií s ESA byly provedeny u pacientů s chronickým selháním ledvin (u pacientů, kteří byli nebo nebyli na dialýze, u diabetiků a u pacientů bez diabetu). Byla pozorována tendence směrem ke zvýšení odhadovaného rizika mortality z jakýchkoli důvodů, rizika kardiovaskulárních a cerebrovaskulárních příhod spojených s vyššími kumulativními dávkami ESA, nezávisle na stavu diabetu nebo dialýze (viz body 4.2 a 4.4).
Erytropoetin je růstový faktor primárně podporující tvorbu červených krvinek. Receptory pro erytropoetin mohou být exprimovány na povrchu mnoha nádorových buněk.
Přežití a progrese nádoru byla zkoumána v pěti velkých kontrolovaných klinických studiích zahrnujících celkem 2833 pacientů; čtyři z těchto studií byly dvojitě zaslepené kontrolované placebe m, jedna studie byla otevřená. Dvou studií se účastnili pacienti léčení chemoterapií. Cílová koncentrace hemoglobinu byla ve dvou studiích > 13 g/dl; v ostatních třech studiích 12-14 g/dl.
V otevřené studii nebyl z hlediska celkového přežití zjištěn žádný rozdíl mezi pacienty léčenými rekombinantním lidským erytropoetinem a mezi kontrolní skupinou. Ve čtyřech placebem kontrolovaných studiích se poměry rizik pro celkové přežití pohybovaly mezi 1,25 a 2,47 ve prospěch kontrolních skupin. Při srovnání s kontrolními skupinami ukázaly tyto studie konzistentní nevysvětlené statisticky významné zvýšení mortality u pacientů, kteří měli anémii spojenou s různými běžnými maligními nádory a kteří dostávali rekombinantní lidský erytropoetin. Výsledek celkového přežití zjištěný v klinických studiích nebylo možné uspokojivě vysvětlit rozdílem v incidenci trombózy a přidružených komplikací mezi skupinou dostávající rekombinantní lidský erytropoetin a mezi skupinou kontrolní.
Metaanalýza založená na údajích jednotlivých pacientů zahrnovala data ze všech 12 kontrolovaných klinických studií prováděných u pacientů s anémií a maligním nádorovým onemocněním, kteří byli léčeni přípravkem NeoRecormon (n=2301), ukázala celkový odhadovaný poměr rizik pro přežití 1,13 ve prospěch kontrol (95% CI 0,87; 1,46). U pacientů, kteří měli v baseline hladinu hemoglobinu < 10 g/dl (n=899), byl odhadovaný poměr rizik pro přežití 0,98 (95% CI 0,68 až 1,40). V celkové populaci bylo pozorováno zvýšené relativní riziko tromboembolických příhod (RR 1,62, 95% CI: 1,13; 2,31).
Byla provedena analýza dat na úrovni jednotlivých nemocných u více než 13 900 pacientů se zhoubným nádorem (léčených chemoterapií, radioterapií, chemoradioterapií nebo bez terapie) zařazených do 53 kontrolovaných klinických studií zahrnujících podávání několika epoetinů. Metaanalýza údajů celkového přežití prokázala, že poměr rizik (hazard ratio) je odhadem 1,06 ve prospěch kontrolních skupin (95% CI: 1,00, 1,12; 53 studií a 13 933 pacientů) a u pacientů se zhoubným nádorem podstupujících chemoterapii byl poměr rizik (hazard ratio) celkového přežití 1,04 (95% CI: 0,97, 1,11; 38 studií a 10 441 pacientů). Meta-analýzy zároveň konzistentně poukazují na významné zvýšení relativního rizika tromboembolických příhod u pacientů se zhoubným nádorem, kteří dostávají rekombinantní lidský erytropoetin (viz bod 4.4).
Ve velmi vzácných případech se v průběhu léčby rekombinantním lidským erytropoetinem objevily neutralizující protilátky proti erytropoetinu s nebo bez čisté aplazie červené krevní řady (PRCA).
5.2 Farmakokinetické vlastnosti
Farmakokinetické sledování u zdravých dobrovolníků a uremických pacientů ukázalo, že po intravenózním podání je poločas epoetinu beta mezi 4 - 12 hodinami a že distribuční objem odpovídá jedno- až dvojnásobku plazmatického objemu. Analogické výsledky byly nalezeny v pokusech na zvířatech u normálních i uremických potkanů.
Protrahovaná absorpce epoetinu beta po subkutánním podání uremickým pacientům vede ke vzniku koncentračního plató, přičemž maximální koncentrace je dosaženo v průměru za 12 - 28 hodin. Terminální poločas je delší než po intravenózním podání s průměrem 13 - 28 hodin.
Ve srovnání s intravenózní aplikací je biologická dostupnost epoetinu beta po podkožní aplikaci mezi 23 - 42 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
Studie kancerogenity s homologním erytropoetinem u myší neodhalily žádné známky proliferativního nebo kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Močovina,
Chlorid sodný,
Polysorbát 20,
Dihydrogenfosforečnan sodný dihydrát,
Hydrogenfosforečnan sodný dodekahydrát,
Chlorid vápenatý dihydrát,
Glycin,
L-leucin,
L-isoleucin,
L-threonin,
L-kyselina glutamová,
L-fenylalanin.
Voda na injekci.
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku v původním obalu, aby byl přípravek chráněn před světlem.
Pro usnadnění ambulantního použití může pacient vyjmout přípravek z chladničky a uchovávat jej jednorázově po dobu maximálně 3 dnů při pokojové teplotě (maximálně do 25 °C).
6.5 Druh obalu a obsah balení
Předplněná injekční stříkačka (sklo typu I) s víčkem a zátkou (pryž potažená teflonem) a s injekční jehlou (27G1/2). Jedna injekční stříkačka obsahuje 0,3 ml roztoku.
Velikost balení po 1 předplněné injekční stříkačce s 1 jehlou nebo 6 předplněných injekčních stříkačkách se 6 jehlami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Nejdříve si dobře umyjte ruce!
1. Vyjměte jednu předplněnou injekční stříkačku z balení a zkontrolujte, zda roztok ve stříkačce je čirý, bezbarvý a bez viditelných částic. Odstraňte víčko z injekční stříkačky.
2. Vyjměte jednu injekční jehlu z balení, nasaďte ji na injekční stříkačku a odstraňte z jehly ochranné víčko.
3. Vytlačte vzduch z injekční stříkačky tím, že stříkačku držíte ve vzpřímené poloze a jemně tlačíte píst vzhůru. Tlačte na píst tak dlouho, dokud množství přípravku NeoRecormon v injekční stříkačce neodpovídá množství předepsanému pro aplikaci.
4. Za použití tampónu namočeného v alkoholu si očistěte kůži v místě vpichu. Vytvořte záhyb na kůži tím, že ji sevřete mezi palec a ukazováček. Stiskněte tělo injekční stříkačky v části bližší k jehle a aplikujte jehlu do záhybu kůže rychlým a pevným pohybem. Vstříkněte roztok přípravku NeoRecormon. Jehlu rychle vytáhněte a zatlačte na místo vpichu suchým a sterilním tampónem.
Tento léčivý přípravek je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/031/031 -032
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. července 1997
Datum posledního prodloužení registrace: 16. července 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
NeoRecormon 4000 IU injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s 0,3 ml injekčního roztoku obsahuje 4000 mezinárodních jednotek (IU), což odpovídá 33,2 mikrogramům epoetinu beta* (rekombinantní lidský erytropoetin). Jeden ml injekčního roztoku obsahuje 13333 IU epoetinu beta.
* Vyrobený technologií rekombinatní DNA v ovariálních buňkách čínských křečků.
Pomocné látky se známým účinkem:
Fenylalanin (až do 0,3 mg v injekční stříkačce)
Sodík (méně než 1 mmol v injekční stříkačce)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Bezbarvý, čirý až slabě opalescentní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek NeoRecormon je indikován k léčbě:
- Léčba symptomatické anémie spojené s chronickým renálním selháním u dospělých a pediatrických pacientů.
- Prevence anémie u předčasně narozených dětí s porodní váhou v rozmezí 750-1500 g, které se narodily v době před 34. týdnem těhotenství.
- Léčba symptomatické anémie u dospělých pacientů s nemyeloidními malignitami, kteří jsou léčeni chemoterapií.
- Zvýšení počtu erytrocytů před odběrem krve k autologní transfuzi krve. Použití v této indikaci musí být zváženo vzhledem k udávanému zvýšení rizika vzniku tromboembolických příhod. Použití je indikováno pouze u pacientů s mírnou anémií (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], bez nedostatku železa), pokud postupy pro konzervování krve nejsou dostupné nebo účinné nebo plánovaný zvolený velký chirurgický zákrok vyžaduje velký objem krve (4 a více jednotek krve pro ženy nebo 5 a více jednotek pro muže). Viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba přípravkem NeoRecormon může být zahájena pouze lékařem se zkušenostmi s výše uvedenými
indikacemi přípravku. Protože byly vzácně zaznamenány anafylaktoidní reakce po podání přípravku,
první dávka má být aplikována pod dohledem zdravotnického personálu.
Dávkování
Léčba symptomatické anémie u dospělých a pediatrických pacientů s chronickým selháním ledvin Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta. Přípravek NeoRecormon má být podáván buď subkutánně nebo intravenózně, tak aby byla hladina hemoglobinu zvýšena maximálně na 12 g/dl (7,5 mmol/l). U pacientů, kteří nepodstupují dialýzu, se upřednostňuje subkutánní podání, kdy není zapotřebí vpichů do periferních žil. V případě intravenózního podání má být přípravek podáván alespoň po dobu 2 minut, například přes arterio-venózní píštěl na konci dialýzy u pacientů na hemodialýze.
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu v rozsahu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Zvýšení hladiny hemoglobinu o více než 2 g/dl (1,25 mmol/l) během období čtyř týdnů není žádoucí. Pokud k němu dojde, má být dávka odpovídajícím způsobem upravena, jak je uvedeno dále. Je-li rychlost vzestupu hladiny hemoglobinu vyšší než 2 g/dl (1,25 mmol/l) za měsíc nebo pokud se hladina hemoglobinu zvýší až na hodnotu 12 g/dl (7,45 mmol/l) je třeba dávku snížit přibližně o 25 %. Pokud by se hladina hemoglobinu dále zvyšovala, má být léčba přerušena, dokud nezačne hladina opět klesat, a v tomto bodě má být znovu zahájena léčba dávkou přibližně o 25 % nižší, než byla předchozí podávaná dávka.
Pacienti mají být důkladně sledováni a je třeba ověřit, že byla použita nejnižší schválená účinná dávka přípravku NeoRecormon, která postačuje pro kontrolu symptomů anémie při zachování koncentrace hemoglobinu pod nebo na hodnotě 12 g/dl (7,45 mmol/l).
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin. U pacientů se slabou odpovědí hemoglobinu na přípravek NeoRecormon mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz bod 4.4 a 5.1).
V případě hypertenze nebo stávajících kardiovaskulárních, cerebrovaskulárních nebo periferních vaskulárních onemocnění má být týdenní zvýšení Hb a cílová hodnota Hb stanovena individuálně při zvážení klinického obrazu.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází.
1. Fáze korekční
- Subkutánní podání:
Počáteční dávka je 3 x 20 IU/kg tělesné hmotnosti týdně. Dávka může být zvýšena každé 4 týdny o 3 x 20 IU/kg za týden, jestliže není dosaženo adekvátního vzestupu Hb (< 0,25 g/dl za týden).
Týdenní dávka může být také rozdělena na denní dávky.
- Intravenózní podání:
Počáteční dávka je 3 x 40 IU/kg tělesné hmotnosti týdně. Dávka může být po 4 týdnech zvýšena na 80 IU/kg třikrát týdně a pokud je nutné další zvýšení dávky, má být o 20 IU/kg třikrát týdně v měsíčních intervalech.
Maximální dávka při obou způsobech podání nemá překročit 720 IU/kg za týden.
2. Fáze udržovací
Pro udržení Hb mezi 10 až 12 g/dl je dávka ze začátku snížena na polovinu předešlé dávky. Následně je u pacienta dávka nastavena v intervalech jednoho nebo dvou týdnů individuálně (udržovací dávka).
V případě podkožního podání může být týdenní dávka podána v jedné injekci týdně, nebo může být rozdělena do tří až sedmi dávek týdně. Pacienti, kteří jsou stabilní na režimu podávání jednou týdně, mohou být převedeni na dávkování jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Výsledky klinických studií u dětí prokázaly, že čím jsou pacienti mladší, tím v průměru vyšší dávky přípravku NeoRecormon vyžadují. Doporučený plán dávkování má však být dodržován, protože klinický účinek u jednotlivých pacientů nelze zcela předvídat.
Léčba přípravkem NeoRecormon v předplněné injekční stříkačce je obvykle dlouhodobá. Může však být, pokud je to nutné, kdykoliv přerušena. Údaje týkající se dávkovacího schématu podání jednou týdně vycházejí z klinických studií s délkou léčby 24 týdnů.
Prevence anémie u předčasně narozených dětí
Roztok je podáván subkutánně v dávce 3 x 250 IU/kg tělesné hmotnosti za týden. Předčasně narozené děti u kterých již byla provedena transfuze v okamžiku zahájení léčby přípravkem NeoRecormon nebudou pravděpodobně mít takový užitek z léčby jako předčasně narozené děti, u kterých transfuze provedena nebyla. Doporučená délka léčby je 6 týdnů.
Léčba symptomatické chemoterapií indukované anémie u pacientů s nádorovým onemocněním Přípravek NeoRecormon má být podáván subkutánně pacientům s anemémií (např.koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l)). Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta.
Týdenní dávka může být podána v jedné injekci jednou týdně nebo může být rozdělena na 3 až 7 jednotlivých dávek.
Doporučená úvodní dávka je 30 000 IU týdně (to odpovídá přibližně 450 IU/kg tělesné hmotnosti týdně u pacienta s průměrnou hmotností).
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Pokud dojde po 4 týdnech léčby ke zvýšení hodnot hemoglobinu o minimálně 1 g/dl (0,62 mmol/l), má se v podávání této dávky dále pokračovat. Pokud hladina hemoglobinu nestoupne o alespoň 1 g/dl (0,62 mmol/l), má být zváženo podávání dvojnásobné týdenní dávky. Pokud hladina hemoglobinu nestoupne po 8 týdnech léčby o alespoň 1 g/dl (0,62 mmol/l), je odpověď nepravděpodobná a léčba má být ukončena.
Léčba má pokračovat až 4 týdny po ukončení chemoterapie.
Maximální dávka nemá překročit 60 000 IU týdně.
Jakmile je u jednotlivého pacienta dosaženo léčebného cíle, dávka má být snížena o 25 až 50 %, aby byla udržena hladina hemoglobinu na této úrovni. Je zapotřebí vzít v úvahu titrování dávky.
Překročí-li hemoglobin hladinu 12 g/dl (7,5 mmol/l), má být dávka snížena zhruba o 25 až 50 %.
Pokud hladina hemoglobinu překročí 13 g/dl (8,1 mmol/l), má být léčba přípravkem NeoRecormon dočasně přerušena. Pokud hladina hemoglobinu klesne na 12 g/dl (7,5 mmol/l) nebo níže, má být léčba opět zahájena s dávkou přibližně o 25 % nižší, než byla předchozí dávka.
Pokud je nárůst hemoglobinu v průběhu 4 týdnů vyšší než 2 g/dl (1,3 mmol/l), dávka má být snížena o 25 až 50 %.
Pacienti mají být důkladně sledováni, přičemž je třeba ověřit, že byla použita nejnižší schválená dávka přípravku NeoRecormon postačující pro adekvátní kontrolu symptomů anémie.
Podávání pro zvýšení množství autologní krve
Přípravek je aplikován intravenózně po dobu asi 2 minut nebo subkutánně.
Přípravek NeoRecormon je podáván dvakrát týdně po dobu 4 týdnů. V případě, kdy hematokrit pacienta umožňuje odběr krve, tj. hodnota hematokritu je > 33 %, je přípravek NeoRecormon podáván v okamžiku ukončení odběru.
V průběhu celé léčby nemá být překročena hodnota hematokritu 48 %.
Dávkování musí být stanoveno chirurgickým týmem zvlášť pro každého pacienta podle požadovaného množství krve pro autologní transfuzi a endogenní rezervy červených krvinek:
1. P ožadované množství krve pro autologní transfuzi závisí na předpokládané ztrátě krve, popřípadě na použití postupů konzervování krve a na fyzické kondici pacienta.
Množství potřebné autologní krve má stačit k tomu, aby bylo možné se vyhnout transfuzi homologní krve.
Požadované množství předem odebrané krve je vyjádřeno v jednotkách, přičemž jedna jednotka v nomogramu je ekvivalentní 180 ml červených krvinek.
2. Možnost autologní transfuze krve závisí především na objemu pacientovy krve a na výchozí hodnotě hematokritu. Obě proměnné určují endogenní rezervu červených krvinek, kterou je možné vypočítat podle následujících vzorců:
Endogenní rezerva červených krvinek = objem krve [ml] x (hematokrit - 33)/100 Ženy: objem krve [ml] = 41 [ml/kg] x tělesná hmotnost [kg] + 1200 [ml]
Muži: objem krve [ml] = 44 [ml/kg] x tělesná hmotnost [kg] + 1600 [ml]
(tělesná hmotnost > 45 kg)
Indikace pro léčbu přípravkem NeoRecormon a jednotlivá dávka mají být stanoveny z požadovaného množství předem odebrané krve a endogenní rezervy červených krvinek podle následujících grafů.
Ženy
Požadované množství krve pro autologní transfuzi [jednotky]
Muži
Požadované množství krve pro autologní transfuzi [jednotky]


Endogenní rezerva červených krvinek [ml] Endogenní rezerva červených krvinek [ml]
Takto stanovená jednotlivá dávka je podávána dvakrát týdně po dobu 4 týdnů. Maximální dávka nemá překročit 1600 IU/kg tělesné hmotnosti a týden při intravenózním podání nebo 1200 IU/kg tělesné hmotnosti a týden při subkutánním podávání.
Způsob podání
Přípravek NeoRecormon v předplněné injekční stříkačce je připraven k použití. Aplikován může být pouze roztok, který je čirý nebo lehce opalescentní, bezbarvý a bez viditelných částic.
Přípravek NeoRecormon v předplněné injekční stříkačce je sterilní, ale neobsahuje žádné konzervační látky. Za žádných okolností proto není možno aplikovat více než jednu dávku s jednou předplněnou injekční stříkačkou; léčivý přípravek je určen pouze k jednorázovému použití.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku (uvedenou v bodě 6.1).
Špatně kontrolovatelná hypertenze.
V indikaci “podání pro zvýšení množství autologní krve”: infarkt myokardu nebo cévní mozková příhoda v průběhu jednoho měsíce před zákrokem, nestabilní angina pectoris, zvýšené riziko hluboké venózní trombózy jako např. po prodělaném tromboembolickém onemocnění.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek NeoRecormon má být užíván s opatrností u pacientů s refrakterní anémií s nadbytkem transformovaných blastů, při epilepsii, trombocytóze a chronickém selhání jater. Je nutné vyloučit nedostatek kyseliny listové a vitamínu B12, protože tyto stavy snižují účinek přípravku NeoRecormon.
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin, neboť vysoké kumulující se dávky epoetinu mohou mít spojitost se zvýšeným rizikem mortality, závažnými kardiovaskulárními a cerebrovaskulárními příhodami. U pacientů se slabou odpovědí hemoglobinu na epoetiny mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz body 4.2 a 5.1).
Je třeba zhodnotit u všech pacientů hladinu železa před a v průběhu léčby, aby byla zajištěna účinná erytropoéza. Může být nutná substituce železa prováděná v souladu s léčebnými doporučeními.
Závažné zatížení hliníkem, ke kterému během léčby renálního selhání může dojít, může rovněž účinek přípravku NeoRecormon snížit.
Indikace k léčbě přípravkem NeoRecormon u pacientů s nefrosklerózou, kteří ještě nejsou dialyzovaní, má být zvážena individuálně, protože u těchto pacientů nelze vyloučit urychlení postupu renálního selhání.
Čistá aplazie buněk červené krevní řady (PRCA)
PRCA způsobená neutralizujícími antierytropoetinovými protilátkami byla hlášena ve spojení s léčbou erytropoetiny, včetně přípravku NeoRecormon. Bylo prokázáno, že tyto protilátky zkříženě reagují se všemi erytropoetinovými proteiny, a proto by pacienti, u kterých je podezření nebo je potvrzen výskyt neutralizujících protilátek proti erytropoetinu, nemají být převáděni na přípravek NeoRecormon (viz bod 4.8).
PRCA u pacientů s hepatitidou C
Paradoxní pokles hladiny hemoglobinu a rozvoj závažné anémie související s nízkým počtem retikulocytů má vést k okamžitému přerušení léčby epoetinem a provedení testů na protilátky proti erytropoetinu. Tyto případy byly hlášeny, pokud byly pacientům s hepatitidou C léčených interferonem a ribavirinem současně podávány epoetiny. Epoetiny nejsou schválené pro léčbu anémie související s hepatitidou C.
Monitorování krevního tlaku
Může dojít ke vzestupu krevního tlaku nebo ke zhoršení již existující hypertenze zejména v případě rychlého vzestupu hematokritu. Toto zvýšení krevního tlaku je možno upravit pomocí léků. Pokud zvýšení krevního tlaku nemůže být upraveno medikamentózně, je doporučeno krátkodobé přerušení léčby přípravkem NeoRecormon. Zvláště na začátku terapie se doporučuje pravidelné monitorování krevního tlaku, a to rovněž mezi dialýzami. Může dojít ke vzniku hypertenzní krize s příznaky napodobujícími encefalopatii, která vyžaduje okamžitou lékařskou intervenci a intenzivní péči. Zvláštní pozoronost je třeba věnovat náhlým bodavým migrenosním bolestem hlavy jako možnému varovnému příznaku.
Chronické selhání ledvin
U pacientů s chronickým selháním ledvin, léčených přípravkem NeoRecormon, může dojít v průběhu léčby k mírnému, na dávce závislému nárůstu počtu krevních destiček v rozmezí normálních hodnot, zvláště pak v případě intravenózního podávání. K poklesu tohoto nárůstu dochází v dalším průběhu léčby. Je doporučeno pravidelně sledovat počet krevních destiček v průběhu prvních 8 týdnů léčby.
Koncentrace hemoglobinu
U pacientů s chronickým selháním ledvin nemá udržovací koncentrace hemoglobinu překročit horní limit cílové koncentrace hemoglobinu doporučovaný v bodě 4.2. V klinických studiích bylo pozorováno zvýšené riziko úmrtí a závažných kardiovaskulárních příhod nebo cerebrovaskulárních příhod, včetně cévní mozkové příhody, pokud byly používány přípravky stimulující erytropoézu (ESA) k dosažení cílových hodnot hemoglobinu vyšších než 12 g/dl (7,5 mmol/l).
V kontrolovaných klinických studiích se neprokázal žádný signifikantní přínos podávání epoetinů, pokud byla koncentrace hemoglobinu zvyšována nad hladinu nezbytně nutnou pro kontrolu symptomů anémie a předcházení krevní transfuze.
U předčasně narozených dětí může dojít k mírnému nárůstu počtu krevních destiček, především v období mezi 12. - 14. dnem života, proto má být počet krevních destiček sledován v pravidelných intervalech.
Účinek na růst nádorů
Epoetiny jsou růstové faktory, které primárně podporují tvorbu červených krvinek. Erytropoetinové receptory mohou být přítomny na povrchu různých nádorových buněk. Stejně jako u všech růstových faktorů je třeba mít na zřeteli, že i epoetiny mohou stimulovat rozvoj nádoru. V několika kontrolovaných klinických studiích nebylo ve spojení s epoetiny prokázáno zlepšení celkové doby přežití ani snížení rizika progrese nádorů u pacientů s anémií spojenou s nádorovým onemocněním.
V kontrolovaných klinických studiích zaměřených na použití přípravku NeoRecormon a ostatních erytropoézu stimulujících přípravků (ESA), bylo zjištěno:
- zkrácení doby do progrese nádoru u pacientů s pokročilým nádorovým onemocněním hlavy a krku, pokud byly dosahovány cílové hladiny hemoglobinu vyšší než 14 g/dl (8,7 mmol/l)
- zkrácení celkové doby přežití a zvýšení počtu úmrtí souvisejících s progresí onemocnění během 4 měsíců léčby u pacientů s metastazujícím karcinomem prsu léčených chemoterapií, pokud byly dosahovány cílové hladiny hemoglobinu 12-14 g/dl (7,5-8,7 mmol/l),
- zvýšení rizika úmrtí při dosahování cílové hladiny hemoglobinu 12 g/dl (7,5 mmol/l) u pacientů s aktivními maligními chorobami, kteří nedostávali ani chemoterapii ani ozařování. V této populaci pacientů není použití ESA indikováno.
Na základě výše uvedených skutečností má být za určitých klinických okolností při léčbě anémie u pacientů s nádorovým onemocněním upřednostněna transfuze krve. Rozhodnutí podat rekombinantní erytropoetin má být přijato na základě zvážení poměru přínosu a rizika a individuálního posouzení jednotlivého pacienta za daných specifických klinických podmínek. Další faktory, které mají být zváženy, je posouzení typu a stádia nádorového onemocnění; stupeň anémie; očekávaná délka přežití; podmínky, za kterých je pacient léčen; a vlastní volba pacienta (viz bod 5.1)
Může dojít ke vzestupu krevního tlaku, který lze farmakologicky léčit. Doporučuje se proto monitorovat krevní tlak, zejména v úvodní fázi léčby pacientů s nádorovým onemocněním.
U onkologických pacientů mají být také pravidelně sledovány počty krevních destiček a hladina hemoglobinu.
U pacientů v programu před autologní transfuzí krve může být zvýšení počtu krevních destiček, většinou v rozmezí normálních hodnot. Proto je doporučeno určovat počet krevních destiček u těchto pacientů alespoň jednou týdně. Pokud dojde ke zvýšení krevních destiček o více než 150 x 109/l nebo když nárůst přesáhne normální hodnoty, má být léčba přípravkem NeoRecormon přerušena.
Upředčasně narozených dětí nelze vyloučit možné riziko retinopatie způsobené erytropoetinem, proto je třeba zvýšené pozornosti a rozhodnutí o léčbě předčasně narozených dětí má být zvažováno na základě posouzení možného prospěchu a rizika této léčby a jiných dostupných možností.
U pacientů s chronickým selháním ledvin je v průběhu léčby přípravkem NeoRecormon při hemodialýze často nutné zvýšení dávky heparinu z důvodu zvýšeného hematokritu. Pokud není heparinizace optimální, může dojít k okluzi systému dialýzy.
U pacientů s chronickým selháním ledvin s rizikem trombózy cévního přístupu má být zvážena časná kontrola cévního přístupu a trombotická profylaxe kyselinou acetylsalicylovou.
V průběhu léčby přípravkem NeoRecormon má být pravidelně monitorována hladina sérového draslíku a fosfátů. Zvýšení kalémie bylo zaznamenáno u několika uremických pacientů léčených přípravkem NeoRecormon, i když příčinná souvislost nebyla ověřena. Pokud je pozorována zvýšená nebo stoupající hladina draslíku, pak má být zváženo přerušení podávání přípravku NeoRecormon dokud se kalémie neupraví.
Pro použití přípravku NeoRecormon v programu před autologní transfuzí krve musí být zváženy obecné zásady dárcovství krve, zvláště:
- dárci mají být pouze pacienti s hematokritem > 33 % (hemoglobin > 11g/dl [6,83 mmol/l];
- má být věnována zvláštní pozornost pacientům s hmotností nižší než 50 kg;
- jednotlivý čerpaný objem nemá překročit cca 12 % odhadnutého objemu krve pacienta.
Léčba má být vyhrazena pro pacienty, u kterých je zvlášť důležité vyhnout se transfuzi homologní krve a má být zvážena rizika a prospěch homologní transfuze.
Zneužití
Zneužití přípravku zdravými osobami může vést k nadměrnému zvýšení hematokritu. Toto zvýšení může být spojeno s život ohrožujícími kardiovaskulárními komplikacemi.
Pomocné látky
Přípravek NeoRecormon v přeplněné injekční stříkačce obsahuje až 0,3 mg fenylalaninu v jedné injekční stříkačce jako pomocnou látku. Tato skutečnost má být brána v úvahu při léčbě pacientů trpících vážnými formami fenylketonurie.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v injekční stříkačce, tj. v podstatě je „bez sodíku“
Zpětná zjistitelnost přípravku NeoRecormon
Z důvodu snadnější zpětné zjistitelnosti látek stimulujících erytropoézu (ESAs) má být obchodní název předepsaného přípravku ESA zřetelně zaznamenán (nebo: vyznačen) v pacientově dokumentaci.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Z dostupných klinických výsledků nejsou známy žádné interakce přípravku NeoRecormon s ostatními léčivými příprakvy.
Pokusy na zvířatech prokázaly, že epoetin beta nezvyšuje myelotoxicitu cytostatických léčivých látek jako jsou etoposid, cisplatina, cyklofosfamid a fluorouracil.
4.6 Fertilita, těhotenství a kojení
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3).
Nejsou k dispozici žádné klinické údaje o účincích epoetinu beta během těhotenství.
Při předepisování těhotným ženám nutno postupovat opatrně.
Kojení
Není známo, zda se epoetin beta vylučuje do lidského mateřského mléka.
Rozhodnutí, zda pokračovat v kojení/přerušit kojení nebo pokračovat v léčbě/přerušit léčbu epoetinem beta, má být zvoleno v závislosti na prospěšnosti kojení pro dítě a prospěšnosti léčby epoetinem beta u matky.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek NeoRecormon nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Na základě výsledků klinických studií s 1725 pacienty lze očekávat, že přibližně u 8 % pacientů léčených přípravkem NeoRecormon se mohou vyskytnout nežádoucí účinky.
Anemičtí pacienti s chronickým selháním ledvin
Nejčastějším nežádoucím účinkem v průběhu léčby přípravkem NeoRecormon je zvýšení krevního tlaku nebo zhoršení stávající hypertenze, zvláště pak v případech rychlého zvýšení hodnot hematokritu (viz bod 4.4). Hypertenzní krize s příznaky encefalopatie (např. bolesti hlavy a stavy zmatenosti, senzoricko-motorické poruchy jako například porucha řeči nebo zhoršení chůze až tonicko-klonické křeče) se mohou objevit také u jednotlivých pacientů s jinak normálním nebo nízkým krevním tlakem (viz bod 4.4).
Může dojít k trombóze žilního přístupu zejména u pacientů s tendencí k hypotenzi nebo s komplikacemi arteriovenózního zkratu (např. stenózy, aneuryzmata), viz bod 4.4. Ve většině případů je pozorován pokles hodnot sérového železa souběžně se vzestupem hematokritu (viz bod 4.4). Navíc byl v ojedinělých případech pozorován vzestup sérového draslíku a hladin fosfátů (viz bod 4.4).
V ojedinělých případech byla v souvislosti s léčbou přípravkem NeoRecormon hlášena čistá aplazie červených krvinek (PRCA) způsobená neutralizujícími antitrombopoetinovými protilátkami.
V případě diagnózy čisté aplazie červených krvinek (PRCA) způsobené neutralizujícími antitrombopoetinovými protilátkami musí být léčba přípravkem NeoRecormon ukončena a pacient nemá být převáděn na léčbu jinou erytropoetickou bílkovinou (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 1.
Pacienti s rakovinou
Bolesti hlavy související s léčbou epoetinem beta a hypertenze, kterou lze medikamentózně léčit, jsou časté (viz bod 4.4).
U některých pacientů je pozorován pokles sérového železa (viz bod 4.4).
V klinických studiích byl prokázán vyšší výskyt tromboembolických událostí u pacientů s karcinomem léčených přípravkem NeoRecormon ve srovnání s neléčenými kontrolními skupinami nebo placebem. U pacientů léčených přípravkem NeoRecormon byla incidence 7 % ve srovnání s 4 % u kontrolních skupin; aniž by to bylo spojeno se zvýšenou mortalitou na tromboembolické události ve srovnání s kontrolními skupinami.
Nežádoucí účinky jsou uvedeny níže v tabulce 2.
Pacienti v programu autologního dárcovství krve
U pacientů v programu autologního dárcovství krve byla hlášena lehce zvýšená četnost tromboembolických událostí. Kauzální vztah k léčbě přípravkem NeoRecormon však nebyl prokázán.
Ve studiích kontrolovaných placebem byl více vyjádřen přechodný deficit železa u pacientů léčených přípravkem NeoRecormon než u kontrolní skupiny (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 3.
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů MedDRA a absolutní četnosti. Kategorie četnosti jsou definovány dle následující konvence:
velmi časté (> 1/10); časté (>1/100 to <1/10); méně časté (>1/1 000 to <1/100); vzácné (>1/10 000 to <1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
Tabulka 1: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů s chronickým selháním ledvin léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze Hypertenzní krize |
Časté Méně časté |
|
Poruchy nervového systému |
Časté | |
|
Poruchy krve a |
Trombóza cévního |
Vzácné |
|
lymfatického systému |
přístupu Trombocytóza |
Velmi vzácné |
Tabulka 2: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u onkologických pacientů léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze |
Časté |
|
Poruchy krve a lymfatického systému |
Tromboembolické účinky |
Časté |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Tabulka 3: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů v programu autologního dárcovství krve léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Předčasně narozené děti
Pokles hodnot sérového ferritinu je velmi častý (viz bod 4.4).
Popis vybraných nežádoucích účinků
Vzácně byly pozorovány kožní reakce jako jsou vyrážka, svědění, kopřivka nebo reakce v místě vpichu. Velmi vzácně byly popsány anafylaktoidní reakce související s léčbou epoetinem beta. V kontrolovaných klinických studiích však zvýšení výskytu hypersenzitivních reakcí nebylo popsáno.
Ve velmi vzácných případech, především na počátku léčby, byl zaznamenán výskyt příznaků podobných chřipce ("flu-like" symptomy) jako je horečka, zimnice, bolesti hlavy, bolest v končetinách, malátnost a/nebo bolest kostí. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Údaje z kontrolované klinické studie s epoetinem alfa nebo darbepoetinem alfa udávaly incidenci cévní mozkové příhody jako častou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 P ředávkování
Terapeutický rozsah přípravku NeoRecormon je velmi široký. Dokonce i při velmi vysokých hladinách v séru nebyly pozorovány žádné příznaky otravy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA01 Mechanismus účinku
Erytropoetin je glykoprotein, který stimuluje tvorbu erytrocytů z prekurzorů kmenových buněk.
Působí jako faktor stimulující mitózu kmenových buněk červené krevní řady a hormon působící na jejich diferenciaci.
Epoetin beta, léčivá látka přípravku NeoRecormon, je svým složením aminokyselin a cukrů identický s erytropoetinem, který byl izolován z moči anemických pacientů.
Biologický účinek epoetinu beta byl demonstrován po intravenózní a podkožní aplikaci u různých pokusných zvířat in vivo (normální a uremičtí potkani, polycytemické myši, psi). Po podání epoetinu beta se zvýší počet erytrocytů, hodnota Hb a retikulocytů stejně tak jako rychlost inkorporace 59Fe.
In vitro byla zjištěna zvýšená inkorporace 3H-thymidinu v erytroidních jaderných buňkách sleziny (kultura buněk sleziny myší) po inkubaci s epoetinem beta.
Pokusy na tkáňových kulturách buněk lidské kostní dřeně prokázaly, že epoetin beta stimuluje jen tvorbu červených krvinek a neovlivňuje tvorbu bílých krvinek. Nebyly zjištěny cytotoxické účinky epoetinu beta na kostní dřeň ani na lidské kožní buňky.
Po podání jednotlivé dávky epoetinu beta nebyly pozorovány žádné vlivy na chování nebo na pohybovou činnost u myší a oběhové nebo respirační funkce u psů.
Klinická účinnost a bezpečnost
V randomizované dvojitě zaslepené, placebem kontrolované studii, která hodnotila 4038 pacientů s chronickým selháním ledvin bez nutnosti dialýzy, s diabetem 2. typu a hladinami hemoglobinu < 11 g/dl, byli pacienti léčeni buď darbepoetinem alfa až do dosažení cílových hladin hemoglobinu 13 g/dl, nebo dostávali placebo (viz bod 4.4). Studie nesplnila žádný z primárních cílů, který by prokázal snížení rizika úmrtí ze všech příčin, kardiovaskulární morbidity nebo terminálního stádia onemocnění ledvin (ESRD). Analýza jednotlivých komponent složených cílových parametrů prokázala následující poměr rizik (95% CI): úmrtí 1,05 (o,92; 1,21), cévní mozková příhoda 1,92 (1,38; 2,68), městnavé srdeční selhání 0,89 (0,74; 1,08), infarkt myokardu 0,96 (0,75; 1,23), hospitalizace z důvodu ischemie myokardu 0,84 (0,55; 1,27), terminální stádium onemocnění ledvin 1,02 (0,87; 1,18).
Souhrnné post-hoc analýzy klinických studií s ESA byly provedeny u pacientů s chronickým selháním ledvin (u pacientů, kteří byli nebo nebyli na dialýze, u diabetiků a u pacientů bez diabetu). Byla pozorována tendence směrem ke zvýšení odhadovaného rizika mortality z jakýchkoli důvodů, rizika kardiovaskulárních a cerebrovaskulárních příhod spojených s vyššími kumulativními dávkami ESA, nezávisle na stavu diabetu nebo dialýze (viz body 4.2 a 4.4).
Erytropoetin je růstový faktor primárně podporující tvorbu červených krvinek. Receptory pro erytropoetin mohou být exprimovány na povrchu mnoha nádorových buněk.
Přežití a progrese nádoru byla zkoumána v pěti velkých kontrolovaných klinických studiích zahrnujících celkem 2833 pacientů; čtyři z těchto studií byly dvojitě zaslepené kontrolované placebe m, jedna studie byla otevřená. Dvou studií se účastnili pacienti léčení chemoterapií. Cílová koncentrace hemoglobinu byla ve dvou studiích >13 g/dl; v ostatních třech studiích 12-14 g/dl.
V otevřené studii nebyl z hlediska celkového přežití zjištěn žádný rozdíl mezi pacienty léčenými rekombinantním lidským erytropoetinem a mezi kontrolní skupinou. Ve čtyřech placebem kontrolovaných studiích se poměry rizik pro celkové přežití pohybovaly mezi 1,25 a 2,47 ve prospěch kontrolních skupin. Při srovnání s kontrolními skupimami ukázaly tyto studie konzistentní nevysvětlené statisticky významné zvýšení mortality u pacientů, kteří měli anémii spojenou s různými běžnými maligními nádory a kteří dostávali rekombinantní lidský erytropoetin. Výsledek celkového přežití zjištěný v klinických studiích nebylo možné uspokojivě vysvětlit rozdílem v incidenci trombózy a přidružených komplikací mezi skupinou dostávající rekombinantní lidský erytropoetin a mezi skupinou kontrolní.
Metaanalýza založená na údajích jednotlivých pacientů zahrnovalal data ze všech 12 kontrolovaných klinických studií prováděných u pacientů s anémií a maligním nádorovým onemocněním, kteří byli léčeni přípravkem NeoRecormon (n=2301), ukázala celkový odhadovaný poměr rizik pro přežití 1,13 ve prospěch kontrol (95% CI 0,87; 1,46). U pacientů, kteří měli v baseline hladinu hemoglobinu <
10 g/dl (n=899), byl odhadovaný poměr rizik pro přežití 0,98 (95% CI 0,68 až 1,40). V celkové populaci bylo pozorováno zvýšené relativní riziko tromboembolických příhod (RR 1,62, 95% CI:
1,13; 2,31).
Byla provedena analýza dat na úrovni jednotlivých nemocných u více než 13 900 pacientů se zhoubným nádorem (léčených chemoterapií, radioterapií, chemoradioterapií nebo bez terapie) zařazených do 53 kontrolovaných klinických studií zahrnujících podávání několika epoetinů. Metaanalýza údajů celkového přežití prokázala, že poměr rizik (hazard ratio) je odhadem 1,06 ve prospěch kontrolních skupin (95% CI: 1,00, 1,12; 53 studií a 13 933 pacientů) a u pacientů se zhoubným nádorem podstupujících chemoterapii byl poměr rizik (hazard ratio) celkového přežití 1,04 (95% CI: 0,97, 1,11; 38 studií a 10 441 pacientů). Meta-analýzy zároveň konzistentně poukazují na významné zvýšení relativního rizika tromboembolických příhod u pacientů se zhoubným nádorem, kteří dostávají rekombinantní lidský erytropoetin (viz bod 4.4).
Ve velmi vzácných případech se v průběhu léčby rekombinantním lidským erytropoetinem objevily neutralizující protilátky proti erytropoetinu s nebo bez čisté aplazie červené krevní řady (PRCA).
5.2 Farmakokinetické vlastnosti
Farmakokinetické sledování u zdravých dobrovolníků a uremických pacientů ukázalo, že po intravenózním podání je poločas epoetinu beta mezi 4 - 12 hodinami a že distribuční objem odpovídá jedno- až dvojnásobku plazmatického objemu. Analogické výsledky byly nalezeny v pokusech na zvířatech u normálních i uremických potkanů.
Protrahovaná absorpce epoetinu beta po subkutánním podání uremickým pacientům vede ke vzniku koncentračního plató, přičemž maximální koncentrace je dosaženo v průměru za 12 - 28 hodin. Terminální poločas je delší než po intravenózním podání s průměrem 13 - 28 hodin.
Ve srovnání s intravenózní aplikací je biologická dostupnost epoetinu beta po podkožní aplikaci mezi 23 - 42 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
Studie kancerogenity s homologním erytropoetinem u myší neodhalily žádné známky proliferativního nebo kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 S eznam pomocných látek
Močovina,
Chlorid sodný,
Polysorbát 20,
Dihydrogenfosforečnan sodný dihydrát,
Hydrogenfosforečnan sodný dodekahydrát,
Chlorid vápenatý dihydrát,
Glycin,
L-leucin,
L-isoleucin,
L-threonin,
L-kyselina glutamová,
L-fenylalanin.
Voda na injekci.
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku v původním obalu, aby byl přípravek chráněn před světlem.
Pro usnadnění ambulantního použití může pacient vyjmout přípravek z chladničky a uchovávat jej jednorázově po dobu maximálně 3 dnů při pokojové teplotě (maximálně do 25 °C).
6.5 Druh obalu a obsah balení
Předplněná injekční stříkačka (sklo typu I) s víčkem a zátkou (pryž potažená teflonem) a s injekční jehlou (27G1/2). Jedna injekční stříkačka obsahuje 0,3 ml roztoku.
Velikost balení po 1 předplněné injekční stříkačce s 1 jehlou nebo 6 předplněných injekčních stříkačkách se 6 jehlami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Nejdříve si dobře umyjte ruce!
1. Vyjměte jednu předplněnou injekční stříkačku z balení a zkontrolujte, zda roztok ve stříkačce je čirý, bezbarvý a bez viditelných částic. Odstraňte víčko z injekční stříkačky.
2. Vyjměte jednu injekční jehlu z balení, nasaďte ji na injekční stříkačku a odstraňte z jehly ochranné víčko.
3. Vytlačte vzduch z injekční stříkačky tím, že stříkačku držíte ve vzpřímené poloze a jemně tlačíte píst vzhůru. Tlačte na píst tak dlouho, dokud množství přípravku NeoRecormon v injekční stříkačce neodpovídá množství předepsanému pro aplikaci.
4. Za použití tampónu namočeného v alkoholu si očistěte kůži v místě vpichu. Vytvořte záhyb na kůži tím, že ji sevřete mezi palec a ukazováček. Stiskněte tělo injekční stříkačky v části bližší k jehle a aplikujte jehlu do záhybu kůže rychlým a pevným pohybem. Vstříkněte roztok přípravku NeoRecormon. Jehlu rychle vytáhněte a zatlačte na místo vpichu suchým a sterilním tampónem.
Tento léčivý přípravek je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/031/041 -042
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. července 1997
Datum posledního prodloužení registrace: 16. července 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
NeoRecormon 5000 IU injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s 0,3 ml injekčního roztoku obsahuje 5000 mezinárodních jednotek (IU), což odpovídá 41,5 mikrogramům epoetinu beta* (rekombinantní lidský erytropoetin). Jeden ml injekčního roztoku obsahuje 16667 IU epoetinu beta.
* Vyrobený technologií rekombinatní DNA v ovariálních buňkách čínských křečků.
Pomocné látky se známým účinkem:
Fenylalanin (až do 0,3 mg v předplněné injekční stříkačce)
Sodík (méně než 1 mmol v injekční stříkačce)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Bezbarvý, čirý až slabě opalescentní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek NeoRecormon je indikován k léčbě:
- Léčba symptomatické anémie spojené s chronickým renálním selháním u dospělých a pediatrických pacientů.
- Prevence anémie u předčasně narozených dětí s porodní váhou v rozmezí 750-1500 g, které se narodily v době před 34. týdnem těhotenství.
- Léčba symptomatické anémie u dospělých pacientů s nemyeloidními malignitami, kteří jsou léčeni chemoterapií.
- Zvýšení počtu erytrocytů před odběrem krve k autologní transfuzi krve. Použití v této indikaci musí být zváženo vzhledem k udávanému zvýšení rizika vzniku tromboembolických příhod. Použití je indikováno pouze u pacientů s mírnou anémií (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], bez nedostatku železa), pokud postupy pro konzervování krve nejsou dostupné nebo účinné nebo plánovaný zvolený velký chirurgický zákrok vyžaduje velký objem krve (4 a více jednotek krve pro ženy nebo 5 a více jednotek pro muže). Viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba přípravkem NeoRecormon může být zahájena pouze lékařem se zkušenostmi s výše uvedenými
indikacemi přípravku. Protože byly vzácně zaznamenány anafylaktoidní reakce po podání přípravku,
první dávka má být aplikována pod dohledem zdravotnického personálu.
Dávkování
Léčba symptomatické anémie u dospělých a pediatrických pacientů s chronickým selháním ledvin Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta. Přípravek NeoRecormon má být podáván buď subkutánně nebo intravenózně, tak aby byla hladina hemoglobinu zvýšena maximálně na 12 g/dl (7,5 mmol/l). U pacientů, kteří nepodstupují dialýzu, se upřednostňuje subkutánní podání, kdy není zapotřebí vpichů do periferních žil. V případě intravenózního podání má být přípravek podáván alespoň po dobu 2 minut, například přes arterio-venózní píštěl na konci dialýzy u pacientů na hemodialýze.
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu v rozsahu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Zvýšení hladiny hemoglobinu o více než 2 g/dl (1,25 mmol/l) během období čtyř týdnů není žádoucí. Pokud k němu dojde, má být dávka odpovídajícím způsobem upravena, jak je uvedeno dále. Je-li rychlost vzestupu hladiny hemoglobinu vyšší než 2 g/dl (1,25 mmol/l) za měsíc nebo pokud se hladina hemoglobinu zvýší až na hodnotu 12 g/dl (7,45 mmol/l) je třeba dávku snížit přibližně o 25 %. Pokud by se hladina hemoglobinu dále zvyšovala, má být léčba přerušena, dokud nezačne hladina opět klesat, a v tomto bodě má být znovu zahájena léčba dávkou přibližně o 25 % nižší, než byla předchozí podávaná dávka.
Pacienti mají být důkladně sledováni a je třeba ověřit, že byla použita nejnižší schválená účinná dávka přípravku NeoRecormon, která postačuje pro kontrolu symptomů anémie při zachování koncentrace hemoglobinu pod nebo na hodnotě 12 g/dl (7,45 mmol/l).
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin. U pacientů se slabou odpovědí hemoglobinu na přípravek NeoRecormon mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz bod 4.4 a 5.1).
V případě hypertenze nebo stávajících kardiovaskulárních, cerebrovaskulárních nebo periferních vaskulárních onemocnění má být týdenní zvýšení Hb a cílová hodnota Hb stanovena individuálně při zvážení klinického obrazu.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází.
1. Fáze korekční
- Subkutánní podání:
Počáteční dávka je 3 x 20 IU/kg tělesné hmotnosti týdně. Dávka může být zvýšena každé 4 týdny o 3 x 20 IU/kg za týden, jestliže není dosaženo adekvátního vzestupu Hb (< 0,25 g/dl za týden).
Týdenní dávka může být také rozdělena na denní dávky.
- Intravenózní podání :
Počáteční dávka je 3 x 40 IU/kg tělesné hmotnosti týdně. Dávka může být po 4 týdnech zvýšena na 80 IU/kg třikrát týdně a pokud je nutné další zvýšení dávky, má být o 20 IU/kg třikrát týdně v měsíčních intervalech.
Maximální dávka při obou způsobech podání nemá překročit 720 IU/kg za týden.
2. Fáze udržovací
Pro udržení Hb mezi 10 až 12 g/dl je dávka ze začátku snížena na polovinu předešlé dávky. Následně je u pacienta dávka nastavena v intervalech jednoho nebo dvou týdnů individuálně (udržovací dávka).
V případě podkožního podání může být týdenní dávka podána v jedné injekci týdně, nebo může být rozdělena do tří až sedmi dávek týdně. Pacienti, kteří jsou stabilní na režimu podávání jednou týdně, mohou být převedeni na dávkování jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Výsledky klinických studií u dětí prokázaly, že čím jsou pacienti mladší, tím v průměru vyšší dávky přípravku NeoRecormon vyžadují. Doporučený plán dávkování má však být dodržován, protože klinický účinek u jednotlivých pacientů nelze zcela předvídat.
Léčba přípravkem NeoRecormon v předplněné injekční stříkačce je obvykle dlouhodobá. Může však být, pokud je to nutné, kdykoliv přerušena. Údaje týkající se dávkovacího schématu podání jednou týdně vycházejí z klinických studií s délkou léčby 24 týdnů.
Prevence anémie u předčasně narozených dětí
Roztok je podáván subkutánně v dávce 3 x 250 IU/kg tělesné hmotnosti za týden. Předčasně narozené děti u kterých již byla provedena transfuze v okamžiku zahájení léčby přípravkem NeoRecormon nebudou pravděpodobně mít takový užitek z léčby jako předčasně narozené děti, u kterých transfuze provedena nebyla. Doporučená délka léčby je 6 týdnů.
Léčba symptomatické chemoterapií indukované anémie u pacientů s nádorovým onemocněním Přípravek NeoRecormon má být podáván subkutánně pacientům s anémií (např. koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l)). Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta.
Týdenní dávka může být podána v jedné injekci jednou týdně nebo může být rozdělena na 3 až 7 jednotlivých dávek.
Doporučená úvodní dávka je 30 000 IU týdně (to odpovídá přibližně 450 IU/kg tělesné hmotnosti týdně u pacienta s průměrnou hmotností).
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Pokud dojde po 4 týdnech léčby ke zvýšení hodnot hemoglobinu o minimálně 1 g/dl (0,62 mmol/l), má se v podávání této dávky dále pokračovat. Pokud hladina hemoglobinu nestoupne o alespoň 1 g/dl (0,62 mmol/l), má být zváženo podávání dvojnásobné týdenní dávky. Pokud hladina hemoglobinu nestoupne po 8 týdnech léčby o alespoň 1 g/dl (0,62 mmol/l), je odpověď nepravděpodobná a léčba má být ukončena.
Léčba má pokračovat až 4 týdny po ukončení chemoterapie.
Maximální dávka nemá překročit 60 000 IU týdně.
Jakmile je u jednotlivého pacienta dosaženo léčebného cíle, dávka má být snížena o 25 až 50 %, aby byla udržena hladina hemoglobinu na této úrovni. Je zapotřebí vzít v úvahu titrování dávky.
Překročí-li hemoglobin hladinu 12 g/dl (7,5 mmol/l), má být dávka snížena zhruba o 25 až 50 %.
Pokud hladina hemoglobinu překročí 13 g/dl (8,1 mmol/l), má být léčba přípravkem NeoRecormon dočasně přerušena. Pokud hladina hemoglobinu klesne na 12 g/dl (7,5 mmol/l) nebo níže, má být léčba opět zahájena s dávkou přibližně o 25 % nižší, než byla předchozí dávka.
Pokud je nárůst hemoglobinu v průběhu 4 týdnů vyšší než 2 g/dl (1,3 mmol/l), dávka má být snížena o 25 až 50 %.
Pacienti mají být důkladně sledováni, přičemž je třeba ověřit, že byla použita nejnižší schválená dávka přípravku NeoRecormon postačující pro adekvátní kontrolu symptomů anémie.
Podávání pro zvýšení množství autologní krve
Přípravek je aplikován intravenózně po dobu asi 2 minut nebo subkutánně.
Přípravek NeoRecormon je podáván dvakrát týdně po dobu 4 týdnů. V případě, kdy hematokrit pacienta umožňuje odběr krve, tj. hodnota hematokritu je > 33 %, je přípravek NeoRecormon podáván v okamžiku ukončení odběru.
V průběhu celé léčby nemá být překročena hodnota hematokritu 48 %.
Dávkování musí být stanoveno chirurgickým týmem zvlášť pro každého pacienta podle požadovaného množství krve pro autologní transfuzi a endogenní rezervy červených krvinek:
1. P ožadované množství krve pro autologní transfuzi závisí na předpokládané ztrátě krve, popřípadě na použití postupů konzervování krve a na fyzické kondici pacienta.
Množství potřebné autologní krve má stačit k tomu, aby bylo možné se vyhnout transfuzi homologní krve.
Požadované množství předem odebrané krve je vyjádřeno v jednotkách, přičemž jedna jednotka v nomogramu je ekvivalentní 180 ml červených krvinek.
2. Možnost autologní transfuze krve závisí především na objemu pacientovy krve a na výchozí hodnotě hematokritu. Obě proměnné určují endogenní rezervu červených krvinek, kterou je možné vypočítat podle následujících vzorců:
Endogenní rezerva červených krvinek = objem krve [ml] x (hematokrit - 33)/100 Ženy: objem krve [ml] = 41 [ml/kg] x tělesná hmotnost [kg] + 1200 [ml]
Muži: objem krve [ml] = 44 [ml/kg] x tělesná hmotnost [kg] + 1600 [ml]
(tělesná hmotnost > 45 kg)
Indikace pro léčbu přípravkem NeoRecormon a jednotlivá dávka mají být stanoveny z požadovaného množství předem odebrané krve a endogenní rezervy červených krvinek podle následujících grafů.
Ženy
Požadované množství krve pro autologní transfuzi [jednotky]
Muži
Požadované množství krve pro autologní transfuzi [jednotky]


Endogenní rezerva červených krvinek [ml] Endogenní rezerva červených krvinek [ml]
Takto stanovená jednotlivá dávka je podávána dvakrát týdně po dobu 4 týdnů. Maximální dávka nemá překročit 1600 IU/kg tělesné hmotnosti a týden při intravenózním podání nebo 1200 IU/kg tělesné hmotnosti a týden při subkutánním podávání.
Způsob podání
Přípravek NeoRecormon v předplněné injekční stříkačce je připraven k použití. Aplikován může být pouze roztok, který je čirý nebo lehce opalescentní, bezbarvý a bez viditelných částic.
Přípravek NeoRecormon v předplněné injekční stříkačce je sterilní, ale neobsahuje žádné konzervační látky. Za žádných okolností proto není možno aplikovat více než jednu dávku s jednou předplněnou injekční stříkačkou; léčivý přípravek je určen pouze k jednorázovému použití.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku (uvedenou v bodě 6.1).
Špatně kontrolovatelná hypertenze.
V indikaci “podání pro zvýšení množství autologní krve”: infarkt myokardu nebo cévní mozková příhoda v průběhu jednoho měsíce před zákrokem, nestabilní angina pectoris, zvýšené riziko hluboké venózní trombózy jako např. po prodělaném tromboembolickém onemocnění.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek NeoRecormon má být užíván s opatrností u pacientů s refrakterní anémií s nadbytkem transformovaných blastů, při epilepsii, trombocytóze a chronickém selhání jater. Je nutné vyloučit nedostatek kyseliny listové a vitamínu BJ2, protože tyto stavy snižují účinek přípravku NeoRecormon.
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin, neboť vysoké kumulující se dávky epoetinu mohou mít spojitost se zvýšeným rizikem mortality, závažnými kardiovaskulárními a cerebrovaskulárními příhodami. U pacientů se slabou odpovědí hemoglobinu na epoetiny mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz body 4.2 a 5.1).
Je třeba zhodnotit u všech pacientů hladinu železa před a v průběhu léčby, aby byla zajištěna účinná erytropoéza. Může být nutná substituce železa prováděná v souladu s léčebnými doporučeními.
Závažné zatížení hliníkem, ke kterému během léčby renálního selhání může dojít, může rovněž účinek přípravku NeoRecormon snížit.
Indikace k léčbě přípravkem NeoRecormon u pacientů s nefrosklerózou, kteří ještě nejsou dialyzovaní, má být zvážena individuálně, protože u těchto pacientů nelze vyloučit urychlení postupu renálního selhání.
Čistá aplazie buněk červené krevní řady (PRCA)
PRCA způsobená neutralizujícími antierytropoetinovými protilátkami byla hlášena ve spojení s léčbou erytropoetiny, včetně přípravku NeoRecormon. Bylo prokázáno, že tyto protilátky zkříženě reagují se všemi erytropoetinovými proteiny, a proto pacienti, u kterých je podezření nebo je potvrzen výskyt neutralizujících protilátek proti erytropoetinu, nemají být převáděni na přípravek NeoRecormon (viz bod 4.8).
PRCA u pacientů s hepatitidou C
Paradoxní pokles hladiny hemoglobinu a rozvoj závažné anémie související s nízkým počtem retikulocytů má vést k okamžitému přerušení léčby epoetinem a provedení testů na protilátky proti erytropoetinu. Tyto případy byly hlášeny, pokud byly pacientům s hepatitidou C léčených interferonem a ribavirinem současně podávány epoetiny. Epoetiny nejsou schválené pro léčbu anémie související s hepatitidou C.
Monitorování krevního tlaku
Může dojít ke vzestupu krevního tlaku nebo ke zhoršení již existující hypertenze zejména v případě rychlého vzestupu hematokritu. Toto zvýšení krevního tlaku je možno upravit pomocí léků. Pokud zvýšení krevního tlaku nemůže být upraveno medikamentózně, je doporučeno krátkodobé přerušení léčby přípravkem NeoRecormon. Zvláště na začátku terapie se doporučuje pravidelné monitorování krevního tlaku, a to rovněž mezi dialýzami. Může dojít ke vzniku hypertenzní krize s příznaky napodobujícími encefalopatii, která vyžaduje okamžitou lékařskou intervenci a intenzivní péči. Zvláštní pozoronost je třeba věnovat náhlým bodavým migrenosním bolestem hlavy jako možnému varovnému příznaku.
Chronické selhání ledvin
U pacientů 5 chronickým selháním ledvin, léčených přípravkem NeoRecormon, může dojít v průběhu léčby k mírnému, na dávce závislému nárůstu počtu krevních destiček v rozmezí normálních hodnot, zvláště pak v případě intravenózního podávání. K poklesu tohoto nárůstu dochází v dalším průběhu léčby. Je doporučeno pravidelně sledovat počet krevních destiček v průběhu prvních 8 týdnů léčby.
Koncentrace hemoglobinu
U pacientů s chronickým selháním ledvin nemá udržovací koncentrace hemoglobinu překročit horní limit cílové koncentrace hemoglobinu doporučované v bodě 4.2. V klinických studiích bylo pozorováno zvýšené riziko úmrtí a závažných kardiovaskulárních příhod nebo cerebrovaskulárních příhod, včetně cévní mozkové příhody, pokud byly používány přípravky stimulující erytropoézu (ESA) k dosažení cílových hodnot hemoglobinu vyšších než 12 g/dl (7,5 mmol/l).
V kontrolovaných klinických studiích neprokázal žádný signifikantní přínos podávání epoetinů, pokud byla koncentrace hemoglobinu zvyšována nad hladinu nezbytně nutnou pro kontrolu symptomů anémie a předcházení krevní transfuze.
U předčasně narozených dětí může dojít k mírnému nárůstu počtu krevních destiček, především v období mezi 12. - 14. dnem života, proto má být počet krevních destiček sledován v pravidelných intervalech.
Účinek na růst nádorů
Epoetiny jsou růstové faktory, které primárně podporují tvorbu červených krvinek. Erytropoetinové receptory mohou být přítomny na povrchu různých nádorových buněk. Stejně jako u všech růstových faktorů je třeba mít na zřeteli, že i epoetiny mohou stimulovat rozvoj nádoru. V několika kontrolovaných klinických studiích nebylo ve spojení s epoetiny prokázáno zlepšení celkové doby přežití ani snížení rizika progrese nádorů u pacientů s anémií spojenou s nádorovým onemocněním.
V kontrolovaných klinických studiích zaměřených na použití přípravku NeoRecormon a ostatních erytropoézu stimulujících přípravků (ESA), bylo zjištěno:
- zkrácení doby do progrese nádoru u pacientů s pokročilým nádorovým onemocněním hlavy a krku, pokud byly dosahovány cílové hladiny hemoglobinu vyšší než 14 g/dl (8,7 mmol/l)
- zkrácení celkové doby přežití a zvýšení počtu úmrtí souvisejících s progresí onemocnění během 4 měsíců léčby u pacientů s metastazujícím karcinomem prsu léčených chemoterapií, pokud byly dosahovány cílové hladiny hemoglobinu 12-14 g/dl (7,5-8,7 mmol/l),
- zvýšení rizika úmrtí při dosahování cílové hladiny hemoglobinu 12 g/dl (7,5 mmol/l) u pacientů s aktivními maligními chorobami, kteří nedostávali ani chemoterapii ani ozařování. V této populaci pacientů není použití ESA indikováno.
Na základě výše uvedených skutečností má být za určitých klinických okolností při léčbě anémie u pacientů s nádorovým onemocněním upřednostněna transfuze krve. Rozhodnutí podat rekombinantní erytropoetin má být přijato na základě zvážení poměru přínosu a rizika a individuálního posouzení jednotlivého pacienta za daných specifických klinických podmínek. Další faktory, které mají být zváženy, je posouzení typu a stádia nádorového onemocnění; stupeň anémie; očekávaná délka přežití; podmínky, za kterých je pacient léčen; a vlastní volba pacienta (viz bod 5.1)
Může dojít ke vzestupu krevního tlaku, který lze farmakologicky léčit. Doporučuje se proto monitorovat krevní tlak, zejména v úvodní fázi léčby pacientů s nádorovým onemocněním.
U onkologických pacientů mají být také pravidelně sledovány počty krevních destiček a hladina hemoglobinu.
U pacientů v programu před autologní transfuzí krve může být zvýšení počtu krevních destiček, většinou v rozmezí normálních hodnot. Proto je doporučeno určovat počet krevních destiček u těchto pacientů alespoň jednou týdně. Pokud dojde ke zvýšení krevních destiček o více než 150 x 109/l nebo když nárůst přesáhne normální hodnoty, má být léčba přípravkem NeoRecormon přerušena.
Upředčasně narozených dětí nelze vyloučit možné riziko retinopatie způsobené erytropoetinem, proto je třeba zvýšené pozornosti a rozhodnutí o léčbě předčasně narozených dětí má být zvažováno na základě posouzení možného prospěchu a rizika této léčby a jiných dostupných možností.
U pacientů s chronickým selháním ledvin je v průběhu léčby přípravkem NeoRecormon při hemodialýze často nutné zvýšení dávky heparinu z důvodu zvýšeného hematokritu. Pokud není heparinizace optimální, může dojít k okluzi systému dialýzy.
U pacientů s chronickým selháním ledvin s rizikem trombózy cévního přístupu má být zvážena časná kontrola cévního přístupu a trombotická profylaxe kyselinou acetylsalicylovou.
V průběhu léčby přípravkem NeoRecormon má být pravidelně monitorována hladina sérového draslíku a fosfátů. Zvýšení kalémie bylo zaznamenáno u několika uremických pacientů léčených přípravkem NeoRecormon, i když příčinná souvislost nebyla ověřena. Pokud je pozorována zvýšená nebo stoupající hladina draslíku, pak má být zváženo přerušení podávání přípravku NeoRecormon dokud se kalémie neupraví.
Pro použití přípravku NeoRecormon v programu před autologní transfuzí krve musí být zváženy obecné zásady dárcovství krve, zvláště:
- dárci mají být pouze pacienti s hematokritem > 33 % (hemoglobin > 11g/dl [6,83 mmol/l];
- má být věnována zvláštní pozornost pacientům s hmotností nižší než 50 kg;
- jednotlivý čerpaný objem nemá překročit cca 12 % odhadnutého objemu krve pacienta.
Léčba má být vyhrazena pro pacienty, u kterých je zvlášť důležité vyhnout se transfuzi homologní krve a mají být zvážena rizika a prospěch homologní transfuze.
Zneužití
Zneužití přípravku zdravými osobami může vést k nadměrnému zvýšení hematokritu. Toto zvýšení může být spojeno s život ohrožujícími kardiovaskulárními komplikacemi.
Pomocné látky
Přípravek NeoRecormon v přeplněné injekční stříkačce obsahuje až 0,3 mg fenylalaninu v jedné injekční stříkačce jako pomocnou látku. Tato skutečnost má být brána v úvahu při léčbě pacientů trpících vážnými formami fenylketonurie.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v injekční stříkačce, tj. v podstatě je „bez sodíku“
Zpětná zjistitelnost přípravku NeoRecormon
Z důvodu snadnější zpětné zjistitelnosti látek stimulujících erytropoézu (ESAs) má být obchodní název předepsaného přípravku ESA zřetelně zaznamenán (nebo vyznačen) v pacientově dokumentaci.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Z dostupných klinických výsledků nejsou známy žádné interakce přípravku NeoRecormon s ostatními léčivými příprakvy.
Pokusy na zvířatech prokázaly, že epoetin beta nezvyšuje myelotoxicitu cytostatických léčivých látek k jako jsou etoposid, cisplatina, cyklofosfamid a fluorouracil.
4.6 Fertilita, těhotenství a kojení
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3).
Nejsou k dispozici žádné klinické údaje o účincích epoetinu beta během těhotenství.
Při předepisování těhotným ženám nutno postupovat opatrně.
Kojení
Není známo, zda se epoetin beta vylučuje do lidského mateřského mléka.
Rozhodnutí, zda pokračovat v kojení/přerušit kojení nebo pokračovat v léčbě/přerušit léčbu epoetinem beta, má být zvoleno v závislosti na prospěšnosti kojení pro dítě a prospěšnosti léčby epoetinem beta u matky.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek NeoRecormon nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Na základě výsledků klinických studií s 1725 pacienty lze očekávat, že přibližně u 8 % pacientů léčených přípravkem NeoRecormon se mohou vyskytnout nežádoucí účinky.
Anemičtí pacienti s chronickým selháním ledvin
Nejčastějším nežádoucím účinkem v průběhu léčby přípravkem NeoRecormon je zvýšení krevního tlaku nebo zhoršení stávající hypertenze, zvláště pak v případech rychlého zvýšení hodnot hematokritu (viz bod 4.4). Hypertenzní krize s příznaky encefalopatie (např. bolesti hlavy a stavy zmatenosti, senzoricko-motorické poruchy jako například porucha řeči nebo zhoršení chůze až tonicko-klonické křeče) se mohou objevit také u jednotlivých pacientů s jinak normálním nebo nízkým krevním tlakem (viz bod 4.4).
Může dojít k trombóze žilního přístupu zejména u pacientů s tendencí k hypotenzi nebo s komplikacemi arteriovenózního zkratu (např. stenózy, aneuryzmata), viz bod 4.4. Ve většině případů je pozorován pokles hodnot sérového železa souběžně se vzestupem hematokritu (viz bod 4.4). Navíc byl v ojedinělých případech pozorován vzestup sérového draslíku a hladin fosfátů (viz bod 4.4).
V ojedinělých případech byla v souvislosti s léčbou přípravkem NeoRecormon hlášena čistá aplazie červených krvinek (PRCA) způsobená neutralizujícími antitrombopoetinovými protilátkami.
V případě diagnózy čisté aplazie červených krvinek (PRCA) způsobené neutralizujícími antitrombopoetinovými protilátkami musí být léčba přípravkem NeoRecormon ukončena a pacient nemá být převáděn na léčbu jinou erytropoetickou bílkovinou (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 1.
Pacienti s rakovinou
Bolesti hlavy související s léčbou epoetinem beta a hypertenze, kterou lze medikamentózně léčit, jsou časté (viz bod 4.4).
U některých pacientů je pozorován pokles sérového železa (viz bod 4.4).
V klinických studiích byl prokázán vyšší výskyt tromboembolických událostí u pacientů s karcinomem léčených přípravkem NeoRecormon ve srovnání s neléčenými kontrolními skupinami nebo placebem. U pacientů léčených přípravkem NeoRecormon byla incidence 7 % ve srovnání s 4 % u kontrolních skupin; aniž by to bylo spojeno se zvýšenou mortalitou na tromboembolické události ve srovnání s kontrolními skupinami.
Nežádoucí účinky jsou uvedeny níže v tabulce 2.
Pacienti v programu autologního dárcovství krve
U pacientů v programu autologního dárcovství krve byla hlášena lehce zvýšená četnost tromboembolických událostí. Kauzální vztah k léčbě přípravkem NeoRecormon však nebyl prokázán.
Ve studiích kontrolovaných placebem byl více vyjádřen přechodný deficit železa u pacientů léčených přípravkem NeoRecormon než u kontrolní skupiny (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 3.
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů MedDRA a absolutní četnosti. Kategorie četnosti jsou definovány dle následující konvence:
velmi časté (> 1/10); časté (>1/100 to <1/10); méně časté (>1/1 000 to <1/100); vzácné (>1/10 000 to <1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
Tabulka 1: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů s chronickým selháním ledvin léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze Hypertenzní krize |
Časté Méně časté |
|
Poruchy nervového systému |
Časté | |
|
Poruchy krve a |
Trombóza cévního |
Vzácné |
|
lymfatického systému |
přístupu Trombocytóza |
Velmi vzácné |
Tabulka 2: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u onkologických pacientů léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze |
Časté |
|
Poruchy krve a lymfatického systému |
Tromboembolické účinky |
Časté |
|
Poruchynervového systému |
Bolesti hlavy |
Časté |
Tabulka 3: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů v programu autologního dárcovství krve léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Předčasně narozené děti
Pokles hodnot sérového ferritinu je velmi častý (>10 %) (viz bod 4.4).
Popis vybraných nežádoucích účinků
Vzácně byly pozorovány kožní reakce jako jsou vyrážka, svědění, kopřivka nebo reakce v místě vpichu. Velmi vzácně byly popsány anafylaktoidní reakce související s léčbou epoetinem beta. V kontrolovaných klinických studiích však zvýšení výskytu hypersenzitivních reakcí nebylo popsáno.
Ve velmi vzácných případech, především na počátku léčby, byl zaznamenán výskyt příznaků podobných chřipce ("flu-like" symptomy) jako je horečka, zimnice, bolesti hlavy, bolest v končetinách, malátnost a/nebo bolest kostí. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Údaje z kontrolované klinické studie s epoetinem alfa nebo darbepoetinem alfa udávaly incidenci cévní mozkové příhody jako častou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 P ředávkování
Terapeutický rozsah přípravku NeoRecormon je velmi široký. Dokonce i při velmi vysokých hladinách v séru nebyly pozorovány žádné příznaky otravy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA01 Mechanismus účinku
Erytropoetin je glykoprotein, který stimuluje tvorbu erytrocytů z prekurzorů kmenových buněk.
Působí jako faktor stimulující mitózu kmenových buněk červené krevní řady a hormon působící na jejich diferenciaci.
Epoetin beta, léčivá látka přípravku NeoRecormon, je svým složením aminokyselin a cukrů identický s erytropoetinem, který byl izolován z moči anemických pacientů.
Biologický účinek epoetinu beta byl demonstrován po intravenózní a podkožní aplikaci u různých pokusných zvířat in vivo (normální a uremičtí potkani, polycytemické myši, psi). Po podání epoetinu beta se zvýší počet erytrocytů, hodnota Hb a retikulocytů stejně tak jako rychlost inkorporace 59Fe.
In vitro byla zjištěna zvýšená inkorporace 3H-thymidinu v erytroidních jaderných buňkách sleziny (kultura buněk sleziny myší) po inkubaci s epoetinem beta.
Pokusy na tkáňových kulturách buněk lidské kostní dřeně prokázaly, že epoetin beta stimuluje jen tvorbu červených krvinek a neovlivňuje tvorbu bílých krvinek. Nebyly zjištěny cytotoxické účinky epoetinu beta na kostní dřeň ani na lidské kožní buňky.
Po podání jednotlivé dávky epoetinu beta nebyly pozorovány žádné vlivy na chování nebo na pohybovou činnost u myší a oběhové nebo respirační funkce u psů.
Klinická účinnost a bezpečnost
V randomizované dvojitě zaslepené, placebem kontrolované studii, která hodnotila 4038 pacientů s chronickým selháním ledvin bez nutnosti dialýzy, s diabetem 2. typu a hladinami hemoglobinu < 11 g/dl, byli pacienti léčeni buď darbepoetinem alfa až do dosažení cílových hladin hemoglobinu 13 g/dl, nebo dostávali placebo (viz bod 4.4). Studie nesplnila žádný z primárních cílů, který by prokázal snížení rizika úmrtí ze všech příčin, kardiovaskulární morbidity nebo terminálního stádia onemocnění ledvin (ESRD). Analýza jednotlivých komponent složených cílových parametrů prokázala následující poměr rizik (95% CI): úmrtí 1,05 (o,92; 1,21), cévní mozková příhoda 1,92 (1,38; 2,68), městnavé srdeční selhání 0,89 (0,74; 1,08), infarkt myokardu 0,96 (0,75; 1,23), hospitalizace z důvodu ischemie myokardu 0,84 (0,55; 1,27), terminální stádium onemocnění ledvin 1,02 (0,87; 1,18).
Souhrnné post-hoc analýzy klinických studií s ESA byly provedeny u pacientů s chronickým selháním ledvin (u pacientů, kteří byli nebo nebyli na dialýze, u diabetiků a u pacientů bez diabetu). Byla pozorována tendence směrem ke zvýšení odhadovaného rizika mortality z jakýchkoli důvodů, rizika kardiovaskulárních a cerebrovaskulárních příhod spojených s vyššími kumulativními dávkami ESA, nezávisle na stavu diabetu nebo dialýze (viz body 4.2 a 4.4).
Erytropoetin je růstový faktor primárně podporující tvorbu červených krvinek. Receptory pro erytropoetin mohou být exprimovány na povrchu mnoha nádorových buněk.
Přežití a progrese nádoru byla zkoumána v pěti velkých kontrolovaných klinických studiích zahrnujících celkem 2833 pacientů; čtyři z těchto studií byly dvojitě zaslepené kontrolované placebe m, jedna studie byla otevřená. Dvou studií se účastnili pacienti léčení chemoterapií. Cílová koncentrace hemoglobinu byla ve dvou studiích >13 g/dl; v ostatních třech studiích 12-14 g/dl.
V otevřené studii nebyl z hlediska celkového přežití zjištěn žádný rozdíl mezi pacienty léčenými rekombinantním lidským erytropoetinem a mezi kontrolní skupinou. Ve čtyřech placebem kontrolovaných studiích se poměry rizik pro celkové přežití pohybovaly mezi 1,25 a 2,47 ve prospěch kontrolních skupin. Při srovnání s kontrolními skupinami ukázaly tyto studie konzistentní nevysvětlené statisticky významné zvýšení mortality u pacientů, kteří měli anémii spojenou s různými běžnými maligními nádory a kteří dostávali rekombinantní lidský erytropoetin. Výsledek celkového přežití zjištěný v klinických studiích nebylo možné uspokojivě vysvětlit rozdílem v incidenci trombózy a přidružených komplikací mezi skupinou dostávající rekombinantní lidský erytropoetin a mezi skupinou kontrolní.
Metaanalýza založená na údajích jednotlivých pacientů zahrnovala data ze všech 12 kontrolovaných klinických studií prováděných u pacientů s anémií a maligním nádorovým onemocněním, kteří byli léčeni přípravkem NeoRecormon (n=2301), ukázala celkový odhadovaný poměr rizik pro přežití 1,13 ve prospěch kontrol (95% CI 0,87; 1,46). U pacientů, kteří měli v baseline hladinu hemoglobinu < 10 g/dl (n=899), byl odhadovaný poměr rizik pro přežití 0,98 (95% CI 0,68 až 1,40). V celkové populaci bylo pozorováno zvýšené relativní riziko tromboembolických příhod (RR 1,62, 95% CI: 1,13; 2,31).
Byla provedena analýza dat na úrovni jednotlivých nemocných u více než 13 900 pacientů se zhoubným nádorem (léčených chemoterapií, radioterapií, chemoradioterapií nebo bez terapie) zařazených do 53 kontrolovaných klinických studií zahrnujících podávání několika epoetinů. Metaanalýza údajů celkového přežití prokázala, že poměr rizik (hazard ratio) je odhadem 1,06 ve prospěch kontrolních skupin (95% CI: 1,00, 1,12; 53 studií a 13 933 pacientů) a u pacientů se zhoubným nádorem podstupujících chemoterapii byl poměr rizik (hazard ratio) celkového přežití 1,04 (95% CI: 0,97, 1,11; 38 studií a 10 441 pacientů). Meta-analýzy zároveň konzistentně poukazují na významné zvýšení relativního rizika tromboembolických příhod u pacientů se zhoubným nádorem, kteří dostávají rekombinantní lidský erytropoetin (viz bod 4.4).
Ve velmi vzácných případech se v průběhu léčby rekombinantním lidským erytropoetinem objevily neutralizující protilátky proti erytropoetinu s nebo bez čisté aplazie červené krevní řady (PRCA).
5.2 Farmakokinetické vlastnosti
Farmakokinetické sledování u zdravých dobrovolníků a uremických pacientů ukázalo, že po intravenózním podání je poločas epoetinu beta mezi 4 - 12 hodinami a že distribuční objem odpovídá jedno- až dvojnásobku plazmatického objemu. Analogické výsledky byly nalezeny v pokusech na zvířatech u normálních i uremických potkanů.
Protrahovaná absorpce epoetinu beta po subkutánním podání uremickým pacientům vede ke vzniku koncentračního plató, přičemž maximální koncentrace je dosaženo v průměru za 12 - 28 hodin. Terminální poločas je delší než po intravenózním podání s průměrem 13 - 28 hodin.
Ve srovnání s intravenózní aplikací je biologická dostupnost epoetinu beta po podkožní aplikaci mezi 23 - 42 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
Studie kancerogenity s homologním erytropoetinem u myší neodhalily žádné známky proliferativního nebo kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 S eznam pomocných látek
Močovina,
Chlorid sodný,
Polysorbát 20,
Dihydrogenfosforečnan sodný dihydrát,
Hydrogenfosforečnan sodný dodekahydrát,
Chlorid vápenatý dihydrát,
Glycin,
L-leucin,
L-isoleucin,
L-threonin,
L-kyselina glutamová,
L-fenylalanin.
Voda na injekci.
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku v původním obalu, aby byl přípravek chráněn před světlem.
Pro usnadnění ambulantního použití může pacient vyjmout přípravek z chladničky a uchovávat jej jednorázově po dobu maximálně 3 dnů při pokojové teplotě (maximálně do 25 °C).
6.5 Druh obalu a obsah balení
Předplněná injekční stříkačka (sklo typu I) s víčkem a zátkou (pryž potažená teflonem) a s injekční jehlou (27G1/2). Jedna injekční stříkačka obsahuje 0,3 ml roztoku.
Velikost balení po 1 předplněné injekční stříkačce s 1 jehlou nebo 6 předplněných injekčních stříkačkách se 6 jehlami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Nejdříve si dobře umyjte ruce!
1. Vyjměte jednu předplněnou injekční stříkačku z balení a zkontrolujte, zda roztok ve stříkačce je čirý, bezbarvý a bez viditelných částic. Odstraňte víčko z injekční stříkačky.
2. Vyjměte jednu injekční jehlu z balení, nasaďte ji na injekční stříkačku a odstraňte z jehly ochranné víčko.
3. Vytlačte vzduch z injekční stříkačky tím, že stříkačku držíte ve vzpřímené poloze a jemně tlačíte píst vzhůru. Tlačte na píst tak dlouho, dokud množství přípravku NeoRecormon v injekční stříkačce neodpovídá množství předepsanému pro aplikaci.
4. Za použití tampónu namočeného v alkoholu si očistěte kůži v místě vpichu. Vytvořte záhyb na kůži tím, že ji sevřete mezi palec a ukazováček. Stiskněte tělo injekční stříkačky v části bližší k jehle a aplikujte jehlu do záhybu kůže rychlým a pevným pohybem. Vstříkněte roztok přípravku NeoRecormon. Jehlu rychle vytáhněte a zatlačte na místo vpichu suchým a sterilním tampónem.
Tento léčivý přípravek je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/031/033 -034
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. července 1997
Datum posledního prodloužení registrace: 16. července 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
NeoRecormon 6000 IU injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s 0,3 ml injekčního roztoku obsahuje 6000 mezinárodních jednotek (IU), což odpovídá 49,8 mikrogramům epoetinu beta* (rekombinantní lidský erytropoetin). Jeden ml injekčního roztoku obsahuje 20000 IU epoetinu beta.
* Vyrobený technologií rekombinatní DNA v ovariálních buňkách čínských křečků.
Pomocné látky se známým účinkem:
Fenylalanin (až do 0,3 mg/předplněná injekční stříkačka)
Sodík (méně než 1 mmol v injekční stříkačce).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Bezbarvý, čirý až slabě opalescentní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek NeoRecormon je indikován k léčbě:
- Léčba symptomatické anémie spojené s chronickým renálním selháním u dospělých a pediatrických pacientů.
- Prevence anémie u předčasně narozených dětí s porodní váhou v rozmezí 750-1500 g, které se narodily v době před 34. týdnem těhotenství.
- Léčba symptomatické anémie u dospělých pacientů s nemyeloidními malignitami, kteří jsou léčeni chemoterapií.
- Zvýšení počtu erytrocytů před odběrem krve k autologní transfuzi krve. Použití v této indikaci musí být zváženo vzhledem k udávanému zvýšení rizika vzniku tromboembolických příhod. Použití je indikováno pouze u pacientů s mírnou anémií (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], bez nedostatku železa), pokud postupy pro konzervování krve nejsou dostupné nebo účinné nebo plánovaný zvolený velký chirurgický zákrok vyžaduje velký objem krve (4 a více jednotek krve pro ženy nebo 5 a více jednotek pro muže). Viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba přípravkem NeoRecormon může být zahájena pouze lékařem se zkušenostmi s výše uvedenými
indikacemi přípravku. Protože byly vzácně zaznamenány anafylaktoidní reakce po podání přípravku,
první dávka má být aplikována pod dohledem zdravotnického personálu.
Dávkování
Léčba symptomatické anémie u dospělých a pediatrických pacientů s chronickým selháním ledvin Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta. Přípravek NeoRecormon má být podáván buď subkutánně nebo intravenózně, tak aby byla hladina hemoglobinu zvýšena maximálně na 12 g/dl (7,5 mmol/l). U pacientů, kteří nepodstupují dialýzu, se upřednostňuje subkutánní podání, kdy není zapotřebí vpichů do periferních žil. V případě intravenózního podání má být přípravek podáván alespoň po dobu 2 minut, například přes arterio-venózní píštěl na konci dialýzy u pacientů na hemodialýze.
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu v rozsahu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Zvýšení hladiny hemoglobinu o více než 2 g/dl (1,25 mmol/l) během období čtyř týdnů není žádoucí. Pokud k němu dojde, má být dávka odpovídajícím způsobem upravena, jak je uvedeno dále. Je-li rychlost vzestupu hladiny hemoglobinu vyšší než 2 g/dl (1,25 mmol/l) za měsíc nebo pokud se hladina hemoglobinu zvýší až na hodnotu 12 g/dl (7,45 mmol/l) je třeba dávku snížit přibližně o 25 %. Pokud by se hladina hemoglobinu dále zvyšovala, má být léčba přerušena, dokud nezačne hladina opět klesat, a v tomto bodě má být znovu zahájena léčba dávkou přibližně o 25 % nižší, než byla předchozí podávaná dávka.
Pacienti mají být důkladně sledováni a je třeba ověřit, že byla použita nejnižší schválená účinná dávka přípravku NeoRecormon, která postačuje pro kontrolu symptomů anémie při zachování koncentrace hemoglobinu pod nebo na hodnotě 12 g/dl (7,45 mmol/l).
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin. U pacientů se slabou odpovědí hemoglobinu na přípravek NeoRecormon mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz bod 4.4 a 5.1).
V případě hypertenze nebo stávajících kardiovaskulárních, cerebrovaskulárních nebo periferních vaskulárních onemocnění má být týdenní zvýšení Hb a cílová hodnota Hb stanovena individuálně při zvážení klinického obrazu.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází.
1. Fáze korekční
- Subkutánní podání:
Počáteční dávka je 3 x 20 IU/kg tělesné hmotnosti týdně. Dávka může být zvýšena každé 4 týdny o 3 x 20 IU/kg za týden, jestliže není dosaženo adekvátního vzestupu Hb (< 0,25 g/dl za týden).
Týdenní dávka může být také rozdělena na denní dávky.
- Intravenózní podání :
Počáteční dávka je 3 x 40 IU/kg tělesné hmotnosti týdně. Dávka může být po 4 týdnech zvýšena na 80 IU/kg třikrát týdně a pokud je nutné další zvýšení dávky, má být o 20 IU/kg třikrát týdně v měsíčních intervalech.
Maximální dávka při obou způsobech podání nemá překročit 720 IU/kg za týden.
2. Fáze udržovací
Pro udržení Hb mezi 10 až 12 g/dl je dávka ze začátku snížena na polovinu předešlé dávky. Následně je u pacienta dávka nastavena v intervalech jednoho nebo dvou týdnů individuálně (udržovací dávka).
V případě podkožního podání může být týdenní dávka podána v jedné injekci týdně, nebo může být rozdělena do tří až sedmi dávek týdně. Pacienti, kteří jsou stabilní na režimu podávání jednou týdně, mohou být převedeni na dávkování jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Výsledky klinických studií u dětí prokázaly, že čím jsou pacienti mladší, tím v průměru vyšší dávky přípravku NeoRecormon vyžadují. Doporučený plán dávkování má však být dodržován, protože klinický účinek u jednotlivých pacientů nelze zcela předvídat.
Léčba přípravkem NeoRecormon v předplněné injekční stříkačce je obvykle dlouhodobá. Může však být, pokud je to nutné, kdykoliv přerušena. Údaje týkající se dávkovacího schématu podání jednou týdně vycházejí z klinických studií s délkou léčby 24 týdnů.
Prevence anémie u předčasně narozených dětí
Roztok je podáván subkutánně v dávce 3 x 250 IU/kg tělesné hmotnosti za týden. Předčasně narozené děti u kterých již byla provedena transfuze v okamžiku zahájení léčby přípravkem Neorecormon nebudou pravděpodobně mít takový užitek z léčby jako předčasně narozené děti, u kterých transfuze provedena nebyla. Doporučená délka léčby je 6 týdnů.
Léčba symptomatické chemoterapií indukované anémie u pacientů s nádorovým onemocněním Přípravek NeoRecormon má být podáván subkutánně pacientům s anémií (např. koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l)). Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta.
Týdenní dávka může být podána v jedné injekci jednou týdně nebo může být rozdělena na 3 až 7 jednotlivých dávek.
Doporučená úvodní dávka je 30 000 IU týdně (to odpovídá přibližně 450 IU/kg tělesné hmotnosti týdně u pacienta s průměrnou hmotností).
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže
Pokud dojde po 4 týdnech léčby ke zvýšení hodnot hemoglobinu o minimálně 1 g/dl (0,62 mmol/l), má se v podávání této dávky dále pokračovat. Pokud hladina hemoglobinu nestoupne o alespoň 1 g/dl (0,62 mmol/l), má být zváženo podávání dvojnásobné týdenní dávky. Pokud hladina hemoglobinu nestoupne po 8 týdnech léčby o alespoň 1 g/dl (0,62 mmol/l), je odpověď nepravděpodobná a léčba má být ukončena.
Léčba má pokračovat až 4 týdny po ukončení chemoterapie.
Maximální dávka nemá překročit 60 000 IU týdně.
Jakmile je u jednotlivého pacienta dosaženo léčebného cíle, dávka má být snížena o 25 až 50 %, aby byla udržena hladina hemoglobinu na této úrovni. Je zapotřebí vzít v úvahu titrování dávky.
Překročí-li hemoglobin hladinu 12 g/dl (7,5 mmol/l), má být dávka snížena zhruba o 25 až 50 %.
Pokud hladina hemoglobinu překročí 13 g/dl (8,1 mmol/l), má být léčba přípravkem NeoRecormon dočasně přerušena. Pokud hladina hemoglobinu klesne na 12 g/dl (7,5 mmol/l) nebo níže, má být léčba opět zahájena s dávkou přibližně o 25 % nižší, než byla předchozí dávka.
Pokud je nárůst hemoglobinu v průběhu 4 týdnů vyšší než 2 g/dl (1,3 mmol/l), dávka má být snížena o 25 až 50 %.
Pacienti mají být důkladně sledováni, přičemž je třeba ověřit, že byla použita nejnižší schválená dávka přípravku NeoRecormon postačující pro adekvátní kontrolu symptomů anémie.
Podávání pro zvýšení množství autologní krve
Přípravek je aplikován intravenózně po dobu asi 2 minut nebo subkutánně.
Přípravek NeoRecormon je podáván dvakrát týdně po dobu 4 týdnů. V případě, kdy hematokrit pacienta umožňuje odběr krve, tj. hodnota hematokritu je > 33 %, je přípravek NeoRecormon podáván v okamžiku ukončení odběru.
V průběhu celé léčby nemá být překročena hodnota hematokritu 48 %.
Dávkování musí být stanoveno chirurgickým týmem zvlášť pro každého pacienta podle požadovaného množství krve pro autologní transfuzi a endogenní rezervy červených krvinek:
1. P ožadované množství krve pro autologní transfuzi závisí na předpokládané ztrátě krve, popřípadě na použití postupů konzervování krve a na fyzické kondici pacienta.
Množství potřebné autologní krve má stačit k tomu, aby bylo možné se vyhnout transfuzi homologní krve.
Požadované množství předem odebrané krve je vyjádřeno v jednotkách, přičemž jedna jednotka v nomogramu je ekvivalentní 180 ml červených krvinek.
2. Možnost autologní transfuze krve závisí především na objemu pacientovy krve a na výchozí hodnotě hematokritu. Obě proměnné určují endogenní rezervu červených krvinek, kterou je možné vypočítat podle následujících vzorců:
Endogenní rezerva červených krvinek = objem krve [ml] x (hematokrit - 33)/100 Ženy: objem krve [ml] = 41 [ml/kg] x tělesná hmotnost [kg] + 1200 [ml]
Muži: objem krve [ml] = 44 [ml/kg] x tělesná hmotnost [kg] + 1600 [ml]
(tělesná hmotnost > 45 kg)
Indikace pro léčbu přípravkem NeoRecormon a jednotlivá dávka mají být stanoveny z požadovaného množství předem odebrané krve a endogenní rezervy červených krvinek podle následujících grafů.
Ženy
Požadované množství krve pro autologní transfuzi [jednotky]
Muži
Požadované množství krve pro autologní transfuzi [jednotky]


Endogenní rezerva červených krvinek [ml] Endogenní rezerva červených krvinek [ml]
Takto stanovená jednotlivá dávka je podávána dvakrát týdně po dobu 4 týdnů. Maximální dávka nemá překročit 1600 IU/kg tělesné hmotnosti a týden při intravenózním podání nebo 1200 IU/kg tělesné hmotnosti a týden při subkutánním podávání.
Způsob podání
Přípravek NeoRecormon v předplněné injekční stříkačce je připraven k použití. Aplikován může být pouze roztok, který je čirý nebo lehce opalescentní, bezbarvý a bez viditelných částic.
Přípravek NeoRecormon v předplněné injekční stříkačce je sterilní, ale neobsahuje žádné konzervační látky. Za žádných okolností proto není možno aplikovat více než jednu dávku s jednou předplněnou injekční stříkačkou; léčivý přípravek je určen pouze k jednorázovému použití.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku (uvedenou v bodě 6.1).
Špatně kontrolovatelná hypertenze.
V indikaci “podání pro zvýšení množství autologní krve”: infarkt myokardu nebo cévní mozková příhoda v průběhu jednoho měsíce před zákrokem, nestabilní angina pectoris, zvýšené riziko hluboké venózní trombózy jako např. po prodělaném tromboembolickém onemocnění.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek NeoRecormon má být užíván s opatrností u pacientů s refrakterní anémií s nadbytkem transformovaných blastů, při epilepsii, trombocytóze a chronickém selhání jater. Je nutné vyloučit nedostatek kyseliny listové a vitamínu BJ2, protože tyto stavy snižují účinek přípravku NeoRecormon.
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin, neboť vysoké kumulující se dávky epoetinu mohou mít spojitost se zvýšeným rizikem mortality, závažnými kardiovaskulárními a cerebrovaskulárními příhodami. U pacientů se slabou odpovědí hemoglobinu na epoetiny mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz body 4.2 a 5.1).
Je třeba zhodnotit u všech pacientů hladinu železa před a v průběhu léčby, aby byla zajištěna účinná erytropoéza. Může být nutná substituce železa prováděná v souladu s léčebnými doporučeními.
Závažné zatížení hliníkem, ke kterému během léčby renálního selhání může dojít, může rovněž účinek přípravku NeoRecormon snížit.
Indikace k léčbě přípravkem NeoRecormon u pacientů s nefrosklerózou, kteří ještě nejsou dialyzovaní, má být zvážena individuálně, protože u těchto pacientů nelze vyloučit urychlení postupu renálního selhání.
Čistá aplazie buněk červené krevní řady (PRCA)
PRCA způsobená neutralizujícími antierytropoetinovými protilátkami byla hlášena ve spojení s léčbou erytropoetiny, včetně přípravku NeoRecormon. Bylo prokázáno, že tyto protilátky zkříženě reagují se všemi erytropoetinovými proteiny, a proto pacienti, u kterých je podezření nebo je potvrzen výskyt neutralizujících protilátek proti erytropoetinu, nemají být převáděni na přípravek NeoRecormon (viz bod 4.8).
PRCA u pacientů s hepatitidou C
Paradoxní pokles hladiny hemoglobinu a rozvoj závažné anémie související s nízkým počtem retikulocytů má vést k okamžitému přerušení léčby epoetinem a provedení testů na protilátky proti erytropoetinu. Tyto případy byly hlášeny, pokud byly pacientům s hepatitidou C léčených interferonem a ribavirinem současně podávány epoetiny. Epoetiny nejsou schválené pro léčbu anémie související s hepatitidou C.
Monitorování krevního tlaku
Může dojít ke vzestupu krevního tlaku nebo ke zhoršení již existující hypertenze zejména v případě rychlého vzestupu hematokritu. Toto zvýšení krevního tlaku je možno upravit pomocí léků. Pokud zvýšení krevního tlaku nemůže být upraveno medikamentózně, je doporučeno krátkodobé přerušení léčby přípravkem NeoRecormon. Zvláště na začátku terapie se doporučuje pravidelné monitorování krevního tlaku, a to rovněž mezi dialýzami. Může dojít ke vzniku hypertenzní krize s příznaky napodobujícími encefalopatii, která vyžaduje okamžitou lékařskou intervenci a intenzivní péči. Zvláštní pozoronost je třeba věnovat náhlým bodavým migrenosním bolestem hlavy jako možnému varovnému příznaku.
Chronické selhání ledvin
U pacientů 5 chronickým selháním ledvin, léčených přípravkem NeoRecormon, může dojít v průběhu léčby k mírnému, na dávce závislému, nárůstu počtu krevních destiček v rozmezí normálních hodnot, zvláště pak v případě intravenózního podávání. K poklesu tohoto nárůstu dochází v dalším průběhu léčby. Je doporučeno pravidelně sledovat počet krevních destiček v průběhu prvních 8 týdnů léčby.
Koncentrace hemoglobinu
U pacientů s chronickým selháním ledvin nemá udržovací koncentrace hemoglobinu překročit horní limit cílové koncentrace hemoglobinu doporučovaný v bodě 4.2. V klinických studiích bylo pozorováno zvýšené riziko úmrtí a závažných kardiovaskulárních příhod nebo cerebrovaskulárních příhod, včetně cévní mozkové příhody, pokud byly používány přípravky stimulující erytropoézu (ESA) k dosažení cílových hodnot hemoglobinu vyšších než 12 g/dl (7,5 mmol/l).
V kontrolovaných klinických studiích se neprokázal žádný signifikantní přínos podávání epoetinů, pokud byla koncentrace hemoglobinu zvyšována nad hladinu nezbytně nutnou pro kontrolu symptomů anémie a předcházení krevní transfuze.
U předčasně narozených dětí může dojít k mírnému nárůstu počtu krevních destiček, především v období mezi 12. - 14. dnem života, proto má být počet krevních destiček sledován v pravidelných intervalech.
Účinek na růst nádorů
Epoetiny jsou růstové faktory, které primárně podporují tvorbu červených krvinek. Erytropoetinové receptory mohou být přítomny na povrchu různých nádorových buněk. Stejně jako u všech růstových faktorů je třeba mít na zřeteli, že i epoetiny mohou stimulovat rozvoj nádoru. V několika kontrolovaných klinických studiích nebylo ve spojení s epoetiny prokázáno zlepšení celkové doby přežití ani snížení rizika progrese nádorů u pacientů s anémií spojenou s nádorovým onemocněním.
V kontrolovaných klinických studiích zaměřených na použití přípravku NeoRecormon a ostatních erytropoézu stimulujících přípravků (ESA), bylo zjištěno:
- zkrácení doby do progrese nádoru u pacientů s pokročilým nádorovým onemocněním hlavy a krku, pokud byly dosahovány cílové hladiny hemoglobinu vyšší než 14 g/dl (8,7 mmol/l)
- zkrácení celkové doby přežití a zvýšení počtu úmrtí souvisejících s progresí onemocnění během 4 měsíců léčby u pacientů s metastazujícím karcinomem prsu léčených chemoterapií, pokud byly dosahovány cílové hladiny hemoglobinu 12-14 g/dl (7,5-8,7 mmol/l),
- zvýšení rizika úmrtí při dosahování cílové hladiny hemoglobinu 12 g/dl (7,5 mmol/l) u pacientů s aktivními maligními chorobami, kteří nedostávali ani chemoterapii ani ozařování. V této populaci pacientů není použití ESA indikováno.
Na základě výše uvedených skutečností má být za určitých klinických okolností při léčbě anémie u pacientů s nádorovým onemocněním upřednostněna transfuze krve. Rozhodnutí podat rekombinantní erytropoetin má být přijato na základě zvážení poměru přínosu a rizika a individuálního posouzení jednotlivého pacienta za daných specifických klinických podmínek. Další faktory, které mají být zváženy, je posouzení typu a stádia nádorového onemocnění; stupeň anémie; očekávaná délka přežití; podmínky, za kterých je pacient léčen; a vlastní volba pacienta (viz bod 5.1)
Může dojít ke vzestupu krevního tlaku, který lze farmakologicky léčit. Doporučuje se proto monitorovat krevní tlak, zejména v úvodní fázi léčby pacientů s nádorovým onemocněním.
U onkologických pacientů mají být také pravidelně sledovány počty krevních destiček a hladina hemoglobinu.
U pacientů v programu před autologní transfuzí krve může být zvýšení počtu krevních destiček, většinou v rozmezí normálních hodnot. Proto je doporučeno určovat počet krevních destiček u těchto pacientů alespoň jednou týdně. Pokud dojde ke zvýšení krevních destiček o více než 150 x 109/l nebo když nárůst přesáhne normální hodnoty, má být léčba přípravkem NeoRecormon přerušena.
U předčasně narozených dětí nelze vyloučit možné riziko retinopatie způsobené erytropoetinem, proto je třeba zvýšené pozornosti a rozhodnutí o léčbě předčasně narozených dětí má být zvažováno na základě posouzení možného prospěchu a rizika této léčby a jiných dostupných možností.
U pacientů 5 chronickým selháním ledvin je v průběhu léčby přípravkem NeoRecormon při hemodialýze často nutné zvýšení dávky heparinu z důvodu zvýšeného hematokritu. Pokud není heparinizace optimální, může dojít k okluzi systému dialýzy.
U pacientů s chronickým selháním ledvin s rizikem trombózy cévního přístupu má být zvážena časná kontrola cévního přístupu a trombotická profylaxe kyselinou acetylsalicylovou.
V průběhu léčby přípravkem NeoRecormon má být pravidelně monitorována hladina sérového draslíku a fosfátů. Zvýšení kalémie bylo zaznamenáno u několika uremických pacientů léčených přípravkem NeoRecormon, i když příčinná souvislost nebyla ověřena. Pokud je pozorována zvýšená nebo stoupající hladina draslíku, pak má být zváženo přerušení podávání přípravku NeoRecormon dokud se kalémie neupraví.
Pro použití přípravku NeoRecormon v programu před autologní transfuzí krve musí být zváženy obecné zásady dárcovství krve, zvláště:
- dárci mají být pouze pacienti s hematokritem > 33 % (hemoglobin > 11g/dl [6,83 mmol/l];
- má být věnována zvláštní pozornost pacientům s hmotností nižší než 50 kg;
- jednotlivý čerpaný objem nemá překročit cca 12 % odhadnutého objemu krve pacienta.
Léčba má být vyhrazena pro pacienty, u kterých je zvlášť důležité vyhnout se transfuzi homologní krve a mají být zvážena rizika a prospěch homologní transfuze.
Zneužití
Zneužití přípravku zdravými osobami může vést k nadměrnému zvýšení hematokritu. Toto zvýšení může být spojeno s život ohrožujícími kardiovaskulárními komplikacemi.
Pomocné látky
Přípravek NeoRecormon v přeplněné injekční stříkačce obsahuje až 0,3 mg fenylalaninu v jedné injekční stříkačce jako pomocnou látku. Tato skutečnost má být brána v úvahu při léčbě pacientů trpících vážnými formami fenylketonurie.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v injekční stříkačce, tj. v podstatě je „bez sodíku“
Zpětná zjistitelnost přípravku NeoRecormon
Z důvodu snadnější zpětné zjistitelnosti látek stimulujících erytropoézu (ESAs) má být obchodní název předepsaného přípravku ESA zřetelně zaznamenán (nebo vyznačen) v pacientově dokumentaci.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Z dostupných klinických výsledků nejsou známy žádné interakce přípravku NeoRecormon s ostatními léčivými příprakvy.
Pokusy na zvířatech prokázaly, že epoetin beta nezvyšuje myelotoxicitu cytostatických léčivých látek jako jsou etoposid, cisplatina, cyklofosfamid a fluorouracil.
4.6 Fertilita, těhotenství a kojení
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3).
Nejsou k dispozici žádné klinické údaje o účincích epoetinu beta během těhotenství.
Při předepisování těhotným ženám nutno postupovat opatrně.
Kojení
Není známo, zda se epoetin beta vylučuje do lidského mateřského mléka.
Rozhodnutí, zda pokračovat v kojení/přerušit kojení nebo pokračovat v léčbě/přerušit léčbu epoetinem beta, má být zvoleno v závislosti na prospěšnosti kojení pro dítě a prospěšnosti léčby epoetinem beta u matky.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek NeoRecormon nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Na základě výsledků klinických studií s 1725 pacienty lze očekávat, že přibližně u 8 % pacientů léčených přípravkem NeoRecormon se mohou vyskytnout nežádoucí účinky.
Anemičtí pacienti s chronickým selháním ledvin
Nejčastějším nežádoucím účinkem v průběhu léčby přípravkem NeoRecormon je zvýšení krevního tlaku nebo zhoršení stávající hypertenze, zvláště pak v případech rychlého zvýšení hodnot hematokritu (viz bod 4.4). Hypertenzní krize s příznaky encefalopatie (např. bolesti hlavy a stavy zmatenosti, senzoricko-motorické poruchy jako například porucha řeči nebo zhoršení chůze až tonicko-klonické křeče) se mohou objevit také u jednotlivých pacientů s jinak normálním nebo nízkým krevním tlakem (viz bod 4.4).
Může dojít k trombóze žilního přístupu zejména u pacientů s tendencí k hypotenzi nebo s komplikacemi arteriovenózního zkratu (např. stenózy, aneuryzmata), viz bod 4.4. Ve většině případů je pozorován pokles hodnot sérového železa souběžně se vzestupem hematokritu (viz bod 4.4). Navíc byl v ojedinělých případech pozorován vzestup sérového draslíku a hladin fosfátů (viz bod 4.4).
V ojedinělých případech byla v souvislosti s léčbou přípravkem NeoRecormon hlášena čistá aplazie červených krvinek (PRCA) způsobená neutralizujícími antitrombopoetinovými protilátkami.
V případě diagnózy čisté aplazie červených krvinek (PRCA) způsobené neutralizujícími antitrombopoetinovými protilátkami musí být léčba přípravkem NeoRecormon ukončena a pacient nemá být převáděn na léčbu jinou erytropoetickou bílkovinou (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 1.
Pacienti s rakovinou
Bolesti hlavy související s léčbou epoetinem beta a hypertenze, kterou lze medikamentózně léčit, jsou časté (viz bod 4.4).
U některých pacientů je pozorován pokles sérového železa (viz bod 4.4).
V klinických studiích byl prokázán vyšší výskyt tromboembolických událostí u pacientů s karcinomem léčených přípravkem NeoRecormon ve srovnání s neléčenými kontrolními skupinami nebo placebem. U pacientů léčených přípravkem NeoRecormon byla incidence 7 % ve srovnání s 4 % u kontrolních skupin; aniž by to bylo spojeno se zvýšenou mortalitou na tromboembolické události ve srovnání s kontrolními skupinami.
Nežádoucí účinky jsou uvedeny níže v tabulce 2.
Pacienti v programu autologního dárcovství krve
U pacientů v programu autologního dárcovství krve byla hlášena lehce zvýšená četnost tromboembolických událostí. Kauzální vztah k léčbě přípravkem NeoRecormon však nebyl prokázán.
Ve studiích kontrolovaných placebem byl více vyjádřen přechodný deficit železa u pacientů léčených přípravkem NeoRecormon než u kontrolní skupiny (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 3.
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů MedDRA a absolutní četnosti. Kategorie četnosti jsou definovány dle následující konvence:
velmi časté (> 1/10); časté (>1/100 to <1/10); méně časté (>1/1 000 to <1/100); vzácné (>1/10 000 to <1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
Tabulka 1: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů s chronickým selháním ledvin léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze Hypertenzní krize |
Časté Méně časté |
|
Poruchy nervového systému |
Časté | |
|
Poruchy krve a |
Trombóza cévního |
Vzácné |
|
lymfatického systému |
přístupu Trombocytóza |
Velmi vzácné |
Tabulka 2: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u onkologických pacientů léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze |
Časté |
|
Poruchy krve a lymfatického systému |
Tromboembolické události |
Časté |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Tabulka 3: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů v programu autologního dárcovství krve léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Předčasně narozené děti
Pokles hodnot sérového ferritinu je velmi častý (viz bod 4.4).
Popis vybraných nežádoucích účinků
Vzácně byly pozorovány kožní reakce jako jsou vyrážka, svědění, kopřivka nebo reakce v místě vpichu. Velmi vzácně byly popsány anafylaktoidní reakce související s léčbou epoetinem beta. V kontrolovaných klinických studiích však zvýšení výskytu hypersenzitivních reakcí nebylo popsáno.
Ve velmi vzácných případech, především na počátku léčby, byl zaznamenán výskyt příznaků podobných chřipce ("flu-like" symptomy) jako je horečka, zimnice, bolesti hlavy, bolest v končetinách, malátnost a/nebo bolest kostí. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Údaje z kontrolované klinické studie s epoetinem alfa nebo darbepoetinem alfa udávaly incidenci cévní mozkové příhody jako častou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 P ředávkování
Terapeutický rozsah přípravku NeoRecormon je velmi široký. Dokonce i při velmi vysokých hladinách v séru nebyly pozorovány žádné příznaky otravy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA01 Mechanismu účinku
Erytropoetin je glykoprotein, který stimuluje tvorbu erytrocytů z prekurzorů kmenových buněk.
Působí jako faktor stimulující mitózu kmenových buněk červené krevní řady a hormon působící na jejich diferenciaci.
Epoetin beta, léčivá látka přípravku NeoRecormon, je svým složením aminokyselin a cukrů identický s erytropoetinem, který byl izolován z moči anemických pacientů.
Biologický účinek epoetinu beta byl demonstrován po intravenózní a podkožní aplikaci u různých pokusných zvířat in vivo (normální a uremičtí potkani, polycytemické myši, psi). Po podání epoetinu beta se zvýší počet erytrocytů, hodnota Hb a retikulocytů stejně tak jako rychlost inkorporace 59Fe.
In vitro byla zjištěna zvýšená inkorporace 3H-thymidinu v erytroidních jaderných buňkách sleziny (kultura buněk sleziny myší) po inkubaci s epoetinem beta.
Pokusy na tkáňových kulturách buněk lidské kostní dřeně prokázaly, že epoetin beta stimuluje jen tvorbu červených krvinek a neovlivňuje tvorbu bílých krvinek. Nebyly zjištěny cytotoxické účinky epoetinu beta na kostní dřeň ani na lidské kožní buňky.
Po podání jednotlivé dávky epoetinu beta nebyly pozorovány žádné vlivy na chování nebo na pohybovou činnost u myší a oběhové nebo respirační funkce u psů.
Klinická účinnost a bezpečnost
V randomizované dvojitě zaslepené, placebem kontrolované studii, která hodnotila 4038 pacientů s chronickým selháním ledvin bez nutnosti dialýzy, s diabetem 2. typu a hladinami hemoglobinu < 11 g/dl, byli pacienti léčeni buď darbepoetinem alfa až do dosažení cílových hladin hemoglobinu 13 g/dl, nebo dostávali placebo (viz bod 4.4). Studie nesplnila žádný z primárních cílů, který by prokázal snížení rizika úmrtí ze všech příčin, kardiovaskulární morbidity nebo terminálního stádia onemocnění ledvin (ESRD). Analýza jednotlivých komponent složených cílových parametrů prokázala následující poměr rizik (95% CI): úmrtí 1,05 (o,92; 1,21), cévní mozková příhoda 1,92 (1,38; 2,68), městnavé srdeční selhání 0,89 (0,74; 1,08), infarkt myokardu 0,96 (0,75; 1,23), hospitalizace z důvodu ischemie myokardu 0,84 (0,55; 1,27), terminální stádium onemocnění ledvin 1,02 (0,87; 1,18).
Souhrnné post-hoc analýzy klinických studií s ESA byly provedeny u pacientů s chronickým selháním ledvin (u pacientů, kteří byli nebo nebyli na dialýze, u diabetiků a u pacientů bez diabetu). Byla pozorována tendence směrem ke zvýšení odhadovaného rizika mortality z jakýchkoli důvodů, rizika kardiovaskulárních a cerebrovaskulárních příhod spojených s vyššími kumulativními dávkami ESA, nezávisle na stavu diabetu nebo dialýze (viz body 4.2 a 4.4).
Erytropoetin je růstový faktor primárně podporující tvorbu červených krvinek. Receptory pro erytropoetin mohou být exprimovány na povrchu mnoha nádorových buněk.
Přežití a progrese nádoru byla zkoumána v pěti velkých kontrolovaných klinických studiích zahrnujících celkem 2833 pacientů; čtyři z těchto studií byly dvojitě zaslepené kontrolované placebe m, jedna studie byla otevřená. Dvou studií se účastnili pacienti léčení chemoterapií. Cílová koncentrace hemoglobinu byla ve dvou studiích >13 g/dl; v ostatních třech studiích 12-14 g/dl.
V otevřené studii nebyl z hlediska celkového přežití zjištěn žádný rozdíl mezi pacienty léčenými rekombinantním lidským erytropoetinem a mezi kontrolní skupinou. Ve čtyřech placebem kontrolovaných studiích se poměry rizik pro celkové přežití pohybovaly mezi 1,25 a 2,47 ve prospěch kontrolních skupin. Při srovnání s kontrolními skupinami ukázaly tyto studie konzistentní nevysvětlené statisticky významné zvýšení mortality u pacientů, kteří měli anémii spojenou s různými běžnými maligními nádory a kteří dostávali rekombinantní lidský erytropoetin. Výsledek celkového přežití zjištěný v klinických studiích nebylo možné uspokojivě vysvětlit rozdílem v incidenci trombózy a přidružených komplikací mezi skupinou dostávající rekombinantní lidský erytropoetin a mezi skupinou kontrolní.
Metaanalýza založená na údajích jednotlivých pacientů zahrnovala data ze všech 12 kontrolovaných klinických studií prováděných u pacientů s anémií a maligním nádorovým onemocněním, kteří byli léčeni přípravkem NeoRecormon (n=2301), ukázala celkový odhadovaný poměr rizik pro přežití 1,13 ve prospěch kontrol (95% CI 0,87; 1,46). U pacientů, kteří měli v baseline hladinu hemoglobinu <
10 g/dl (n=899), byl odhadovaný poměr rizik pro přežití 0,98 (95% CI 0,68 až 1,40). V celkové populaci bylo pozorováno zvýšené relativní riziko tromboembolických příhod (RR 1,62, 95% CI:
1,13; 2,31).
Byla provedena analýza dat na úrovni jednotlivých nemocných u více než 13 900 pacientů se zhoubným nádorem (léčených chemoterapií, radioterapií, chemoradioterapií nebo bez terapie) zařazených do 53 kontrolovaných klinických studií zahrnujících podávání několika epoetinů. Metaanalýza údajů celkového přežití prokázala, že poměr rizik (hazard ratio) je odhadem 1,06 ve prospěch kontrolních skupin (95% CI: 1,00, 1,12; 53 studií a 13 933 pacientů) a u pacientů se zhoubným nádorem podstupujících chemoterapii byl poměr rizik (hazard ratio) celkového přežití 1,04 (95% CI: 0,97, 1,11; 38 studií a 10 441 pacientů). Meta-analýzy zároveň konzistentně poukazují na významné zvýšení relativního rizika tromboembolických příhod u pacientů se zhoubným nádorem, kteří dostávají rekombinantní lidský erytropoetin (viz bod 4.4).
Ve velmi vzácných případech se v průběhu léčby rekombinantním lidským erytropoetinem objevily neutralizující protilátky proti erytropoetinu s nebo bez čisté aplazie červené krevní řady (PRCA).
5.2 Farmakokinetické vlastnosti
Farmakokinetické sledování u zdravých dobrovolníků a uremických pacientů ukázalo, že po intravenózním podání je poločas epoetinu beta mezi 4 - 12 hodinami a že distribuční objem odpovídá jedno- až dvojnásobku plazmatického objemu. Analogické výsledky byly nalezeny v pokusech na zvířatech u normálních i uremických potkanů.
Protrahovaná absorpce epoetinu beta po subkutánním podání uremickým pacientům vede ke vzniku koncentračního plató, přičemž maximální koncentrace je dosaženo v průměru za 12 - 28 hodin. Terminální poločas je delší než po intravenózním podání s průměrem 13 - 28 hodin.
Ve srovnání s intravenózní aplikací je biologická dostupnost epoetinu beta po podkožní aplikaci mezi 23 - 42 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
Studie kancerogenity s homologním erytropoetinem u myší neodhalily žádné známky proliferativního nebo kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 S eznam pomocných látek
Močovina,
Chlorid sodný,
Polysorbát 20,
Dihydrogenfosforečnan sodný dihydrát,
Hydrogenfosforečnan sodný dodekahydrát,
Chlorid vápenatý dihydrát,
Glycin,
L-leucin,
L-isoleucin,
L-threonin,
L-kyselina glutamová,
L-fenylalanin.
Voda na injekci.
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku v původním obalu, aby byl přípravek chráněn před světlem.
Pro usnadnění ambulantního použití může pacient vyjmout přípravek z chladničky a uchovávat jej jednorázově po dobu maximálně 3 dnů při pokojové teplotě (maximálně do 25 °C).
6.5 Druh obalu a obsah balení
Předplněná injekční stříkačka (sklo typu I) s víčkem a zátkou (pryž potažená teflonem) a s injekční jehlou (27G1/2). Jedna injekční stříkačka obsahuje 0,3 ml roztoku.
Velikost balení po 1 předplněné injekční stříkačce s 1 jehlou nebo 6 předplněných injekčních stříkačkách se 6 jehlami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Nejdříve si dobře umyjte ruce!
1. Vyjměte jednu předplněnou injekční stříkačku z balení a zkontrolujte, zda roztok ve stříkačce je čirý, bezbarvý a bez viditelných částic. Odstraňte víčko z injekční stříkačky.
2. Vyjměte jednu injekční jehlu z balení, nasaďte ji na injekční stříkačku a odstraňte z jehly ochranné víčko.
3. Vytlačte vzduch z injekční stříkačky tím, že stříkačku držíte ve vzpřímené poloze a jemně tlačíte píst vzhůru. Tlačte na píst tak dlouho, dokud množství přípravku NeoRecormon v injekční stříkačce neodpovídá množství předepsanému pro aplikaci.
4. Za použití tampónu namočeného v alkoholu si očistěte kůži v místě vpichu. Vytvořte záhyb na kůži tím, že ji sevřete mezi palec a ukazováček. Stiskněte tělo injekční stříkačky v části bližší k jehle a aplikujte jehlu do záhybu kůže rychlým a pevným pohybem. Vstříkněte roztok přípravku NeoRecormon. Jehlu rychle vytáhněte a zatlačte na místo vpichu suchým a sterilním tampónem.
Tento léčivý přípravek je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/031/043 -044
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. července 1997
Datum posledního prodloužení registrace: 16. července 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
NeoRecormon 10 000 IU injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s 0,6 ml injekčního roztoku obsahuje 10 000 mezinárodních jednotek (IU), což odpovídá 83 mikrogramům epoetinu beta* (rekombinantní lidský erytropoetin). Jeden ml injekčního roztoku obsahuje 16 667 IU epoetinu beta.
* Vyrobený technologií rekombinatní DNA v ovariálních buňkách čínských křečků.
Pomocné látky se známým účinkem:
Fenylalanin (až do 0,3 mg/předplněná injekční stříkačka)
Sodík (méně než 1 mmol v injekční stříkačce)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Bezbarvý, čirý až slabě opalescentní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek NeoRecormon je indikován k léčbě:
- Léčba symptomatické anémie spojené s chronickým renálním selháním u dospělých a pediatrických pacientů.
- Prevence anémie u předčasně narozených dětí s porodní váhou v rozmezí 750-1500 g, které se narodily v době před 34. týdnem těhotenství.
- Léčba symptomatické anémie u dospělých pacientů s nemyeloidními malignitami, kteří jsou léčeni chemoterapií.
- Zvýšení počtu erytrocytů před odběrem krve k autologní transfuzi krve. Použití v této indikaci musí být zváženo vzhledem k udávanému zvýšení rizika vzniku tromboembolických příhod. Použití je indikováno pouze u pacientů s mírnou anémií (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], bez nedostatku železa), pokud postupy pro konzervování krve nejsou dostupné nebo účinné nebo plánovaný zvolený velký chirurgický zákrok vyžaduje velký objem krve (4 a více jednotek krve pro ženy nebo 5 a více jednotek pro muže). Viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba přípravkem NeoRecormon může být zahájena pouze lékařem se zkušenostmi s výše uvedenými
indikacemi přípravku. Protože byly vzácně zaznamenány anafylaktoidní reakce po podání přípravku,
první dávka má být aplikována pod dohledem zdravotnického personálu.
Dávkování
Léčba symptomatické anémie u dospělých a pediatrických pacientů s chronickým selháním ledvin Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta. Přípravek NeoRecormon má být podáván buď subkutánně nebo intravenózně, tak aby byla hladina hemoglobinu zvýšena maximálně na 12 g/dl (7,5 mmol/l). U pacientů, kteří nepodstupují dialýzu, se upřednostňuje subkutánní podání, kdy není zapotřebí vpichů do periferních žil. V případě intravenózního podání má být přípravek podáván alespoň po dobu 2 minut, například přes arterio-venózní píštěl na konci dialýzy u pacientů na hemodialýze.
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu v rozsahu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Zvýšení hladiny hemoglobinu o více než 2 g/dl (1,25 mmol/l) během období čtyř týdnů není žádoucí. Pokud k němu dojde, má být dávka odpovídajícím způsobem upravena, jak je uvedeno dále. Je-li rychlost vzestupu hladiny hemoglobinu vyšší než 2 g/dl (1,25 mmol/l) za měsíc nebo pokud se hladina hemoglobinu zvýší až na hodnotu 12 g/dl (7,45 mmol/l) je třeba dávku snížit přibližně o 25 %. Pokud by se hladina hemoglobinu dále zvyšovala, má být léčba přerušena, dokud nezačne hladina opět klesat, a v tomto bodě má být znovu zahájena léčba dávkou přibližně o 25 % nižší, než byla předchozí podávaná dávka.
Pacienti mají být důkladně sledováni a je třeba ověřit, že byla použita nejnižší schválená účinná dávka přípravku NeoRecormon, která postačuje pro kontrolu symptomů anémie při zachování koncentrace hemoglobinu pod nebo na hodnotě 12 g/dl (7,45 mmol/l).
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin. U pacientů se slabou odpovědí hemoglobinu na přípravek NeoRecormon mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz bod 4.4 a 5.1).
V případě hypertenze nebo stávajících kardiovaskulárních, cerebrovaskulárních nebo periferních vaskulárních onemocnění má být týdenní zvýšení Hb a cílová hodnota Hb stanovena individuálně při zvážení klinického obrazu.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází.
1. Fáze korekční
- Subkutánní podání:
Počáteční dávka je 3 x 20 IU/kg tělesné hmotnosti týdně. Dávka může být zvýšena každé 4 týdny o 3 x 20 IU/kg za týden, jestliže není dosaženo adekvátního vzestupu Hb (< 0,25 g/dl za týden).
Týdenní dávka může být také rozdělena na denní dávky.
- Intravenózní podání :
Počáteční dávka je 3 x 40 IU/kg tělesné hmotnosti týdně. Dávka může být po 4 týdnech zvýšena na 80 IU/kg třikrát týdně a pokud je nutné další zvýšení dávky, má být o 20 IU/kg třikrát týdně v měsíčních intervalech.
Maximální dávka při obou způsobech podání nemá překročit 720 IU/kg za týden.
2. Fáze udržovací
Pro udržení Hb mezi 10až12 g/dl je dávka ze začátku snížena na polovinu předešlé dávky. Následně je u pacienta dávka nastavena v intervalech jednoho nebo dvou týdnů individuálně (udržovací dávka).
V případě podkožního podání může být týdenní dávka podána v jedné injekci týdně, nebo může být rozdělena do tří až sedmi dávek týdně. Pacienti, kteří jsou stabilní na režimu podávání jednou týdně, mohou být převedeni na dávkování jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Výsledky klinických studií u dětí prokázaly, že čím jsou pacienti mladší, tím v průměru vyšší dávky přípravku NeoRecormon vyžadují. Doporučený plán dávkování má však být dodržován, protože klinický účinek u jednotlivých pacientů nelze zcela předvídat.
Léčba přípravkem NeoRecormon v předplněné injekční stříkačce je obvykle dlouhodobá. Může však být, pokud je to nutné, kdykoliv přerušena. Údaje týkající se dávkovacího schématu podání jednou týdně vycházejí z klinických studií s délkou léčby 24 týdnů.
Prevence anémie u předčasně narozených dětí
Roztok je podáván subkutánně v dávce 3 x 250 IU/kg tělesné hmotnosti za týden. Předčasně narozené děti u kterých již byla provedena transfuze v okamžiku zahájení léčby přípravkem NeoRecormon nebudou pravděpodobně mít takový užitek z léčby jako předčasně narozené děti, u kterých transfuze provedena nebyla. Doporučená délka léčby je 6 týdnů.
Léčba symptomatické chemoterapií indukované anémie u pacientů s nádorovým onemocněním Přípravek NeoRecormon má být podáván subkutánně pacientům s anémií (např. koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l)). Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta.
Týdenní dávka může být podána v jedné injekci jednou týdně nebo může být rozdělena na 3 až 7 jednotlivých dávek.
Doporučená úvodní dávka je 30 000 IU týdně (to odpovídá přibližně 450 IU/kg tělesné hmotnosti týdně u pacienta s průměrnou hmotností).
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Pokud dojde po 4 týdnech léčby ke zvýšení hodnot hemoglobinu o minimálně 1 g/dl (0,62 mmol/l), má se v podávání této dávky dále pokračovat. Pokud hladina hemoglobinu nestoupne o alespoň 1 g/dl (0,62 mmol/l), má být zváženo podávání dvojnásobné týdenní dávky. Pokud hladina hemoglobinu nestoupne po 8 týdnech léčby o alespoň 1 g/dl (0,62 mmol/l), je odpověď nepravděpodobná a léčba má být ukončena.
Léčba má pokračovat až 4 týdny po ukončení chemoterapie.
Maximální dávka nemá překročit 60 000 IU týdně.
Jakmile je u jednotlivého pacienta dosaženo léčebného cíle, dávka má být snížena o 25 až 50 %, aby byla udržena hladina hemoglobinu na této úrovni. Je zapotřebí vzít v úvahu titrování dávky.
Překročí-li hemoglobin hladinu 12 g/dl (7,5 mmol/l), má být dávka snížena zhruba o 25 až 50 %.
Pokud hladina hemoglobinu překročí 13 g/dl (8,1 mmol/l), má být léčba přípravkem NeoRecormon dočasně přerušena. Pokud hladina hemoglobinu klesne na 12 g/dl (7,5 mmol/l) nebo níže, má být léčba opět zahájena s dávkou přibližně o 25 % nižší, než byla předchozí dávka.
Pokud je nárůst hemoglobinu v průběhu 4 týdnů vyšší než 2 g/dl (1,3 mmol/l), dávka má být snížena o 25 až 50 %.
Pacienti mají být důkladně sledováni, přičemž je třeba ověřit, že byla použita nejnižší schválená dávka přípravku NeoRecormon postačující pro adekvátní kontrolu symptomů anémie.
Podávání pro zvýšení množství autologní krve
Přípravek je aplikován intravenózně po dobu asi 2 minut nebo subkutánně.
Přípravek NeoRecormon je podáván dvakrát týdně po dobu 4 týdnů. V případě, kdy hematokrit pacienta umožňuje odběr krve, tj. hodnota hematokritu je > 33 %, je přípravek NeoRecormon podáván v okamžiku ukončení odběru.
V průběhu celé léčby nemá být překročena hodnota hematokritu 48 %.
Dávkování musí být stanoveno chirurgickým týmem zvlášť pro každého pacienta podle požadovaného množství krve pro autologní transfuzi a endogenní rezervy červených krvinek:
1. P ožadované množství krve pro autologní transfuzi závisí na předpokládané ztrátě krve, popřípadě na použití postupů konzervování krve a na fyzické kondici pacienta.
Množství potřebné autologní krve má stačit k tomu, aby bylo možné se vyhnout transfuzi homologní krve.
Požadované množství předem odebrané krve je vyjádřeno v jednotkách, přičemž jedna jednotka v nomogramu je ekvivalentní 180 ml červených krvinek.
2. Možnost autologní transfuze krve závisí především na objemu pacientovy krve a na výchozí hodnotě hematokritu. Obě proměnné určují endogenní rezervu červených krvinek, kterou je možné vypočítat podle následujících vzorců:
Endogenní rezerva červených krvinek = objem krve [ml] x (hematokrit - 33)/100 Ženy: objem krve [ml] = 41 [ml/kg] x tělesná hmotnost [kg] + 1200 [ml]
Muži: objem krve [ml] = 44 [ml/kg] x tělesná hmotnost [kg] + 1600 [ml]
(tělesná hmotnost > 45 kg)
Indikace pro léčbu přípravkem NeoRecormon a jednotlivá dávka mají být stanoveny z požadovaného množství předem odebrané krve a endogenní rezervy červených krvinek podle následujících grafů.
Ženy
Požadované množství krve pro autologní transfuzi [jednotky]
Muži
Požadované množství krve pro autologní transfuzi [jednotky]

Endogenní rezerva červených krvinek [ml]

Endogenní rezerva červených krvinek [ml]
Takto stanovená jednotlivá dávka je podávána dvakrát týdně po dobu 4 týdnů. Maximální dávka nemá překročit 1600 IU/kg tělesné hmotnosti a týden při intravenózním podání nebo 1200 IU/kg tělesné hmotnosti a týden při subkutánním podávání.
Způsob podání
Přípravek NeoRecormon v předplněné injekční stříkačce je připraven k použití. Aplikován může být pouze roztok, který je čirý nebo lehce opalescentní, bezbarvý a bez viditelných částic.
Přípravek NeoRecormon v předplněné injekční stříkačce je sterilní, ale neobsahuje žádné konzervační látky. Za žádných okolností proto není možno aplikovat více než jednu dávku s jednou předplněnou injekční stříkačkou; léčivý přípravek je určen pouze k jednorázovému použití.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku (uvedenou v bodě 6.1).
Špatně kontrolovatelná hypertenze.
V indikaci “podání pro zvýšení množství autologní krve”: infarkt myokardu nebo cévní mozková příhoda v průběhu jednoho měsíce před zákrokem, nestabilní angina pectoris, zvýšené riziko hluboké venózní trombózy jako např. po prodělaném tromboembolickém onemocnění.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek NeoRecormon má být užíván s opatrností u pacientů s refrakterní anémií s nadbytkem transformovaných blastů, při epilepsii, trombocytóze a chronickém selhání jater. Je nutné vyloučit nedostatek kyseliny listové a vitamínu B12, protože tyto stavy snižují účinek přípravku NeoRecormon.
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin, neboť vysoké kumulující se dávky epoetinu mohou mít spojitost se zvýšeným rizikem mortality, závažnými kardiovaskulárními a cerebrovaskulárními příhodami. U pacientů se slabou odpovědí hemoglobinu na epoetiny mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz body 4.2 a 5.1).
Je třeba zhodnotit u všech pacientů hladinu železa před a v průběhu léčby, aby byla zajištěna účinná erytropoéza. Může být nutná substituce železa prováděná v souladu s léčebnými doporučeními.
Závažné zatížení hliníkem, ke kterému během léčby renálního selhání může dojít, může rovněž účinek přípravku NeoRecormon snížit.
Indikace k léčbě přípravkem NeoRecormon u pacientů s nefrosklerózou, kteří ještě nejsou dialyzovaní, má být zvážena individuálně, protože u těchto pacientů nelze vyloučit urychlení postupu renálního selhání.
Čistá aplazie buněk červené krevní řady (PRCA)
PRCA způsobená neutralizujícími antierytropoetinovými protilátkami byla hlášena ve spojení s léčbou erytropoetiny, včetně přípravku NeoRecormon. Bylo prokázáno, že tyto protilátky zkříženě reagují se všemi erytropoetinovými proteiny, a proto pacienti, u kterých je podezření nebo je potvrzen výskyt neutralizujících protilátek proti erytropoetinu, nemají být převáděni na přípravek NeoRecormon (viz bod 4.8).
PRCA u pacientů s hepatitidou C
Paradoxní pokles hladiny hemoglobinu a rozvoj závažné anémie související s nízkým počtem retikulocytů má vést k okamžitému přerušení léčby epoetinem a provedení testů na protilátky proti erytropoetinu. Tyto případy byly hlášeny, pokud byly pacientům s hepatitidou C léčených interferonem a ribavirinem současně podávány epoetiny. Epoetiny nejsou schválené pro léčbu anémie související s hepatitidou C.
Monitorování krevního tlaku
Může dojít ke vzestupu krevního tlaku nebo ke zhoršení již existující hypertenze zejména v případě rychlého vzestupu hematokritu. Toto zvýšení krevního tlaku je možno upravit pomocí léků. Pokud zvýšení krevního tlaku nemůže být upraveno medikamentózně, je doporučeno krátkodobé přerušení léčby přípravkem NeoRecormon. Zvláště na začátku terapie se doporučuje pravidelné monitorování krevního tlaku, a to rovněž mezi dialýzami. Může dojít ke vzniku hypertenzní krize s příznaky napodobujícími encefalopatii, která vyžaduje okamžitou lékařskou intervenci a intenzivní péči. Zvláštní pozoronost je třeba věnovat náhlým bodavým migrenosním bolestem hlavy jako možnému varovnému příznaku.
Chronické selhání ledvin
U pacientů 5 chronickým selháním ledvin, léčených přípravkem NeoRecormon, může dojít v průběhu léčby k mírnému, na dávce závislému nárůstu počtu krevních destiček v rozmezí normálních hodnot, zvláště pak v případě intravenózního podávání. K poklesu tohoto nárůstu dochází v dalším průběhu léčby. Je doporučeno pravidelně sledovat počet krevních destiček v průběhu prvních 8 týdnů léčby.
Koncentrace hemoglobinu
U pacientů s chronickým selháním ledvin nemá udržovací koncentrace hemoglobinu překročit horní limit cílové koncentrace hemoglobinu doporučované v bodě 4.2. V klinických studiích bylo pozorováno zvýšené riziko úmrtí a závažných kardiovaskulárních příhod nebo cerebrovaskulárních příhod, včetně cévní mozkové příhody, pokud byly používány přípravky stimulující erytropoézu (ESA) k dosažení cílových hodnot hemoglobinu vyšších než 12 g/dl (7,5 mmol/l).
V kontrolovaných klinických studiích se neprokázal žádný signifikantní přínos podávání epoetinů, pokud byla koncentrace hemoglobinu zvyšována nad hladinu nezbytně nutnou pro kontrolu symptomů anémie a předcházení krevní transfuze.
U předčasně narozených dětí může dojít k mírnému nárůstu počtu krevních destiček, především v období mezi 12. - 14. dnem života, proto má být počet krevních destiček sledován v pravidelných intervalech.
Účinek na růst nádorů
Epoetiny jsou růstové faktory, které primárně podporují tvorbu červených krvinek. Erytropoetinové receptory mohou být přítomny na povrchu různých nádorových buněk. Stejně jako u všech růstových faktorů je třeba mít na zřeteli, že i epoetiny mohou stimulovat rozvoj nádoru. V několika kontrolovaných klinických studiích nebylo ve spojení s epoetiny prokázáno zlepšení celkové doby přežití ani snížení rizika progrese nádorů u pacientů s anémií spojenou s nádorovým onemocněním.
V kontrolovaných klinických studiích zaměřených na použití přípravku NeoRecormon a ostatních erytropoézu stimulujících přípravků (ESA), bylo zjištěno:
- zkrácení doby do progrese nádoru u pacientů s pokročilým nádorovým onemocněním hlavy a krku, pokud byly dosahovány cílové hladiny hemoglobinu vyšší než 14 g/dl (8,7 mmol/l)
- zkrácení celkové doby přežití a zvýšení počtu úmrtí souvisejících s progresí onemocnění během 4 měsíců léčby u pacientů s metastazujícím karcinomem prsu léčených chemoterapií, pokud byly dosahovány cílové hladiny hemoglobinu 12-14 g/dl (7,5-8,7 mmol/l),
- zvýšení rizika úmrtí při dosahování cílové hladiny hemoglobinu 12 g/dl (7,5 mmol/l) u pacientů s aktivními maligními chorobami, kteří nedostávali ani chemoterapii ani ozařování. V této populaci pacientů není použití ESA indikováno.
Na základě výše uvedených skutečností má být za určitých klinických okolností při léčbě anémie u pacientů s nádorovým onemocněním upřednostněna transfuze krve. Rozhodnutí podat rekombinantní erytropoetin má být přijato na základě zvážení poměru přínosu a rizika a individuálního posouzení jednotlivého pacienta za daných specifických klinických podmínek. Další faktory, které mají být zváženy, je posouzení typu a stádia nádorového onemocnění; stupeň anémie; očekávaná délka přežití; podmínky, za kterých je pacient léčen; a vlastní volba pacienta (viz bod 5.1)
Může dojít ke vzestupu krevního tlaku, který lze farmakologicky léčit. Doporučuje se proto monitorovat krevní tlak, zejména v úvodní fázi léčby pacientů s nádorovým onemocněním.
U onkologických pacientů mají být také pravidelně sledovány počty krevních destiček a hladina hemoglobinu.
U pacientů v programu před autologní transfuzí krve může být zvýšení počtu krevních destiček, většinou v rozmezí normálních hodnot. Proto je doporučeno určovat počet krevních destiček u těchto pacientů alespoň jednou týdně. Pokud dojde ke zvýšení krevních destiček o více než 150 x 109/l nebo když nárůst přesáhne normální hodnoty, má být léčba přípravkem NeoRecormon přerušena.
U předčasně narozených dětí nelze vyloučit možné riziko retinopatie způsobené erytropoetinem, proto je třeba zvýšené pozornosti a rozhodnutí o léčbě předčasně narozených dětí má být zvažováno na základě posouzení možného prospěchu a rizika této léčby a jiných dostupných možností.
U pacientů 5 chronickým selháním ledvin je v průběhu léčby přípravkem NeoRecormon při hemodialýze často nutné zvýšení dávky heparinu z důvodu zvýšeného hematokritu. Pokud není heparinizace optimální, může dojít k okluzi systému dialýzy.
U pacientů s chronickým selháním ledvin s rizikem trombózy cévního přístupu má být zvážena časná kontrola cévního přístupu a trombotická profylaxe kyselinou acetylsalicylovou.
V průběhu léčby přípravkem NeoRecormon má být pravidelně monitorována hladina sérového draslíku a fosfátů. Zvýšení kalémie bylo zaznamenáno u několika uremických pacientů léčených přípravkem NeoRecormon, i když příčinná souvislost nebyla ověřena. Pokud je pozorována zvýšená nebo stoupající hladina draslíku, pak má být zváženo přerušení podávání přípravku NeoRecormon dokud se kalémie neupraví.
Pro použití přípravku NeoRecormon v programu před autologní transfuzí krve musí být zváženy obecné zásady dárcovství krve, zvláště:
- dárci mají být pouze pacienti s hematokritem > 33 % (hemoglobin > 11g/dl [6,83 mmol/l];
- má být věnována zvláštní pozornost pacientům s hmotností nižší než 50 kg;
- jednotlivý čerpaný objem nemá překročit cca 12 % odhadnutého objemu krve pacienta.
Léčba má být vyhrazena pro pacienty, u kterých je zvlášť důležité vyhnout se transfuzi homologní krve a mají být zvážena rizika a prospěch homologní transfuze.
Zneužití
Zneužití přípravku zdravými osobami může vést k nadměrnému zvýšení hematokritu. Toto zvýšení může být spojeno s život ohrožujícími kardiovaskulárními komplikacemi.
Pomocné látky
Přípravek NeoRecormon v přeplněné injekční stříkačce obsahuje až 0,3 mg fenylalaninu v jedné injekční stříkačce jako pomocnou látku. Tato skutečnost má být brána v úvahu při léčbě pacientů trpících vážnými formami fenylketonurie.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v injekční stříkačce, tj. v podstatě je „bez sodíku“
Zpětná zjistitelnost přípravku NeoRecormon
Z důvodu snadnější zpětné zjistitelnosti látek stimulujících erytropoézu (ESAs) má být obchodní název předepsaného přípravku ESA zřetelně zaznamenán (nebo vyznačen) v pacientově dokumentaci.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Z dostupných klinických výsledků nejsou známy žádné interakce přípravku NeoRecormon s ostatními léčivými příprakvy.
Pokusy na zvířatech prokázaly, že epoetin beta nezvyšuje myelotoxicitu cytostatických léčivých látek cisplatina, cyklofosfamid a fluorouracil.
4.6 Fertilita, těhotenství a kojení
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3).
Nejsou k dispozici žádné klinické údaje o účincích epoetinu beta během těhotenství.
Při předepisování těhotným ženám nutno postupovat opatrně.
Kojení
Není známo, zda se epoetin beta vylučuje do lidského mateřského mléka.
Rozhodnutí, zda pokračovat v kojení/přerušit kojení nebo pokračovat v léčbě/přerušit léčbu epoetinem beta, má být zvoleno v závislosti na prospěšnosti kojení pro dítě a prospěšnosti léčby epoetinem beta u matky.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek NeoRecormon nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Na základě výsledků klinických studií s 1725 pacienty lze očekávat, že přibližně u 8 % pacientů léčených přípravkem NeoRecormon se mohou vyskytnout nežádoucí účinky.
Anemičtí pacienti s chronickým selháním ledvin
Nejčastějším nežádoucím účinkem v průběhu léčby přípravkem NeoRecormon je zvýšení krevního tlaku nebo zhoršení stávající hypertenze, zvláště pak v případech rychlého zvýšení hodnot hematokritu (viz bod 4.4). Hypertenzní krize s příznaky encefalopatie (např. bolesti hlavy a stavy zmatenosti, senzoricko-motorické poruchy jako například porucha řeči nebo zhoršení chůze až tonicko-klonické křeče) se mohou objevit také u jednotlivých pacientů s jinak normálním nebo nízkým krevním tlakem (viz bod 4.4).
Může dojít k trombóze žilního přístupu zejména u pacientů s tendencí k hypotenzi nebo s komplikacemi arteriovenózního zkratu (např. stenózy, aneuryzmata), viz bod 4.4. Ve většině případů je pozorován pokles hodnot sérového železa souběžně se vzestupem hematokritu (viz bod 4.4). Navíc byl v ojedinělých případech pozorován vzestup sérového draslíku a hladin fosfátů (viz bod 4.4).
V ojedinělých případech byla v souvislosti s léčbou přípravkem NeoRecormon hlášena čistá aplazie červených krvinek (PRCA) způsobená neutralizujícími antitrombopoetinovými protilátkami.
V případě diagnózy čisté aplazie červených krvinek (PRCA) způsobené neutralizujícími antitrombopoetinovými protilátkami musí být léčba přípravkem NeoRecormon ukončena a pacient by neměl být převáděn na léčbu jinou erytropoetickou bílkovinou (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 1.
Pacienti s rakovinou
Bolesti hlavy související s léčbou epoetinem beta a hypertenze, kterou lze medikamentózně léčit, jsou časté (viz bod 4.4).
U některých pacientů je pozorován pokles sérového železa (viz bod 4.4).
V klinických studiích byl prokázán vyšší výskyt tromboembolických událostí u pacientů s karcinomem léčených přípravkem NeoRecormon ve srovnání s neléčenými kontrolními skupinami nebo placebem. U pacientů léčených přípravkem NeoRecormon byla incidence 7 % ve srovnání s 4 % u kontrolních skupin; aniž by to bylo spojeno se zvýšenou mortalitou na tromboembolické události ve srovnání s kontrolními skupinami.
Nežádoucí účinky jsou uvedeny níže v tabulce 2.
Pacienti v programu autologního dárcovství krve
U pacientů v programu autologního dárcovství krve byla hlášena lehce zvýšená četnost tromboembolických událostí. Kauzální vztah k léčbě přípravkem NeoRecormon však nebyl prokázán.
Ve studiích kontrolovaných placebem byl více vyjádřen přechodný deficit železa u pacientů léčených přípravkem NeoRecormon než u kontrolní skupiny (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 3.
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů MedDRA a absolutní četnosti. Kategorie četnosti jsou definovány dle následující konvence:
velmi časté (> 1/10); časté (>1/100 to <1/10); méně časté (>1/1 000 to <1/100); vzácné (>1/10 000 to <1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
Tabulka 1: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů s chronickým selháním ledvin léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze Hypertenzní krize |
Časté Méně časté |
|
Poruchy nervového systému |
Časté | |
|
Poruchy krve a |
Trombóza cévního |
Vzácné |
|
lymfatického systému |
přístupu Trombocytóza |
Velmi vzácné |
Tabulka 2: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u onkologických pacientů léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze |
Časté |
|
Poruchy krve a lymfatického systému |
Tromboembolické účinky |
Časté |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Tabulka 3: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů v programu autologního dárcovství krve léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Předčasně narozené děti
Pokles hodnot sérového ferritinu je velmi častý (viz bod 4.4).
Popis vybraných nežádoucích účinků
Vzácně byly pozorovány kožní reakce jako jsou vyrážka, svědění, kopřivka nebo reakce v místě vpichu. Velmi vzácně byly popsány anafylaktoidní reakce související s léčbou epoetinem beta. V kontrolovaných klinických studiích však zvýšení výskytu hypersenzitivních reakcí nebylo popsáno.
Ve velmi vzácných případech, především na počátku léčby, byl zaznamenán výskyt příznaků podobných chřipce ("flu-like" symptomy) jako je horečka, zimnice, bolesti hlavy, bolest v končetinách, malátnost a/nebo bolest kostí. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Údaje z kontrolované klinické studie s epoetinem alfa nebo darbepoetinem alfa udávaly incidenci cévní mozkové příhody jako častou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 P ředávkování
Terapeutický rozsah přípravku NeoRecormon je velmi široký. Dokonce i při velmi vysokých hladinách v séru nebyly pozorovány žádné příznaky otravy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA01 Mechanismus účinku
Erytropoetin je glykoprotein, který stimuluje tvorbu erytrocytů z prekurzorů kmenových buněk.
Působí jako faktor stimulující mitózu kmenových buněk červené krevní řady a hormon působící na jejich diferenciaci.
Epoetin beta, léčivá látka přípravku NeoRecormon, je svým složením aminokyselin a cukrů identický s erytropoetinem, který byl izolován z moči anemických pacientů.
Biologický účinek epoetinu beta byl demonstrován po intravenózní a podkožní aplikaci u různých pokusných zvířat in vivo (normální a uremičtí potkani, polycytemické myši, psi). Po podání epoetinu beta se zvýší počet erytrocytů, hodnota Hb a retikulocytů stejně tak jako rychlost inkorporace 59Fe.
In vitro byla zjištěna zvýšená inkorporace 3H-thymidinu v erytroidních jaderných buňkách sleziny (kultura buněk sleziny myší) po inkubaci s epoetinem beta.
Pokusy na tkáňových kulturách buněk lidské kostní dřeně prokázaly, že epoetin beta stimuluje jen tvorbu červených krvinek a neovlivňuje tvorbu bílých krvinek. Nebyly zjištěny cytotoxické účinky epoetinu beta na kostní dřeň ani na lidské kožní buňky.
Po podání jednotlivé dávky epoetinu beta nebyly pozorovány žádné vlivy na chování nebo na pohybovou činnost u myší a oběhové nebo respirační funkce u psů.
Klinická účinnost a bezpečnost
V randomizované dvojitě zaslepené, placebem kontrolované studii, která hodnotila 4038 pacientů s chronickým selháním ledvin bez nutnosti dialýzy, s diabetem 2. typu a hladinami hemoglobinu < 11 g/dl, byli pacienti léčeni buď darbepoetinem alfa až do dosažení cílových hladin hemoglobinu 13 g/dl, nebo dostávali placebo (viz bod 4.4). Studie nesplnila žádný z primárních cílů, který by prokázal snížení rizika úmrtí ze všech příčin, kardiovaskulární morbidity nebo terminálního stádia onemocnění ledvin (ESRD). Analýza jednotlivých komponent složených cílových parametrů prokázala následující poměr rizik (95% CI): úmrtí 1,05 (o,92; 1,21), cévní mozková příhoda 1,92 (1,38; 2,68), městnavé srdeční selhání 0,89 (0,74; 1,08), infarkt myokardu 0,96 (0,75; 1,23), hospitalizace z důvodu ischemie myokardu 0,84 (0,55; 1,27), terminální stádium onemocnění ledvin 1,02 (0,87; 1,18).
Souhrnné post-hoc analýzy klinických studií s ESA byly provedeny u pacientů s chronickým selháním ledvin (u pacientů, kteří byli nebo nebyli na dialýze, u diabetiků a u pacientů bez diabetu). Byla pozorována tendence směrem ke zvýšení odhadovaného rizika mortality z jakýchkoli důvodů, rizika kardiovaskulárních a cerebrovaskulárních příhod spojených s vyššími kumulativními dávkami ESA, nezávisle na stavu diabetu nebo dialýze (viz body 4.2 a 4.4).
Erytropoetin je růstový faktor primárně podporující tvorbu červených krvinek. Receptory pro erytropoetin mohou být exprimovány na povrchu mnoha nádorových buněk.
Přežití a progrese nádoru byla zkoumána v pěti velkých kontrolovaných klinických studiích zahrnujících celkem 2833 pacientů; čtyři z těchto studií byly dvojitě zaslepené kontrolované placebe m, jedna studie byla otevřená. Dvou studií se účastnili pacienti léčení chemoterapií. Cílová koncentrace hemoglobinu byla ve dvou studiích >13 g/dl; v ostatních třech studiích 12-14 g/dl.
V otevřené studii nebyl z hlediska celkového přežití zjištěn žádný rozdíl mezi pacienty léčenými rekombinantním lidským erytropoetinem a mezi kontrolní skupinou. Ve čtyřech placebem kontrolovaných studiích se poměry rizik pro celkové přežití pohybovaly mezi 1,25 a 2,47 ve prospěch kontrolních skupin. Při srovnání s kontrolními skupinami ukázaly tyto studie konzistentní nevysvětlené statisticky významné zvýšení mortality u pacientů, kteří měli anémii spojenou s různými běžnými maligními nádory a kteří dostávali rekombinantní lidský erytropoetin. Výsledek celkového přežití zjištěný v klinických studiích nebylo možné uspokojivě vysvětlit rozdílem v incidenci trombózy a přidružených komplikací mezi skupinou dostávající rekombinantní lidský erytropoetin a mezi skupinou kontrolní.
Metaanalýza založená na údajích jednotlivých pacientů zahrnovala data ze všech 12 kontrolovaných klinických studií prováděných u pacientů s anémií a maligním nádorovým onemocněním, kteří byli léčeni přípravkem NeoRecormon (n=2301), ukázala celkový odhadovaný poměr rizik pro přežití 1,13 ve prospěch kontrol (95% CI 0,87; 1,46). U pacientů, kteří měli v baseline hladinu hemoglobinu < 10 g/dl (n=899), byl odhadovaný poměr rizik pro přežití 0,98 (95% CI 0,68 až 1,40). V celkové populaci bylo pozorováno zvýšené relativní riziko tromboembolických příhod (RR 1,62, 95% CI: 1,13; 2,31).
Byla provedena analýza dat na úrovni jednotlivých nemocných u více než 13 900 pacientů se zhoubným nádorem (léčených chemoterapií, radioterapií, chemoradioterapií nebo bez terapie) zařazených do 53 kontrolovaných klinických studií zahrnujících podávání několika epoetinů. Metaanalýza údajů celkového přežití prokázala, že poměr rizik (hazard ratio) je odhadem 1,06 ve prospěch kontrolních skupin (95% CI: 1,00, 1,12; 53 studií a 13 933 pacientů) a u pacientů se zhoubným nádorem podstupujících chemoterapii byl poměr rizik (hazard ratio) celkového přežití 1,04 (95% CI: 0,97, 1,11; 38 studií a 10 441 pacientů). Meta-analýzy zároveň konzistentně poukazují na významné zvýšení relativního rizika tromboembolických příhod u pacientů se zhoubným nádorem, kteří dostávají rekombinantní lidský erytropoetin (viz bod 4.4).
Ve velmi vzácných případech se v průběhu léčby rekombinantním lidským erytropoetinem objevily neutralizující protilátky proti erytropoetinu s nebo bez čisté aplazie červené krevní řady (PRCA).
5.2 Farmakokinetické vlastnosti
Farmakokinetické sledování u zdravých dobrovolníků a uremických pacientů ukázalo, že po intravenózním podání je poločas epoetinu beta mezi 4 - 12 hodinami a že distribuční objem odpovídá jedno- až dvojnásobku plazmatického objemu. Analogické výsledky byly nalezeny v pokusech na zvířatech u normálních i uremických potkanů.
Protrahovaná absorpce epoetinu beta po subkutánním podání uremickým pacientům vede ke vzniku koncentračního plató, přičemž maximální koncentrace je dosaženo v průměru za 12 - 28 hodin. Terminální poločas je delší než po intravenózním podání s průměrem 13 - 28 hodin.
Ve srovnání s intravenózní aplikací je biologická dostupnost epoetinu beta po podkožní aplikaci mezi 23 - 42 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
Studie kancerogenity s homologním erytropoetinem u myší neodhalily žádné známky proliferativního nebo kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 S eznam pomocných látek
Močovina,
Chlorid sodný,
Polysorbát 20,
Dihydrogenfosforečnan sodný dihydrát,
Hydrogenfosforečnan sodný dodekahydrát,
Chlorid vápenatý dihydrát,
Glycin,
L-leucin,
L-isoleucin,
L-threonin,
L-kyselina glutamová,
L-fenylalanin.
Voda na injekci.
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku v původním obalu, aby byl přípravek chráněn před světlem.
Pro usnadnění ambulantního použití může pacient vyjmout přípravek z chladničky a uchovávat jej jednorázově po dobu maximálně 3 dnů při pokojové teplotě (maximálně do 25 °C).
6.5 Druh obalu a obsah balení
Předplněná injekční stříkačka (sklo typu I) s víčkem a zátkou (pryž potažená teflonem) a s injekční jehlou (27G1/2). Jedna injekční stříkačka obsahuje 0,6 ml roztoku.
Velikost balení po 1 předplněné injekční stříkačce s 1 jehlou nebo 6 předplněných injekčních stříkačkách se 6 jehlami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Nejdříve si dobře umyjte ruce!
1. Vyjměte jednu předplněnou injekční stříkačku z balení a zkontrolujte, zda roztok ve stříkačce je čirý, bezbarvý a bez viditelných částic. Odstraňte víčko z injekční stříkačky.
2. Vyjměte jednu injekční jehlu z balení, nasaďte ji na injekční stříkačku a odstraňte z jehly ochranné víčko.
3. Vytlačte vzduch z injekční stříkačky tím, že stříkačku držíte ve vzpřímené poloze a jemně tlačíte píst vzhůru. Tlačte na píst tak dlouho, dokud množství přípravku NeoRecormon v injekční stříkačce neodpovídá množství předepsanému pro aplikaci.
4. Za použití tampónu namočeného v alkoholu si očistěte kůži v místě vpichu. Vytvořte záhyb na kůži tím, že ji sevřete mezi palec a ukazováček. Stiskněte tělo injekční stříkačky v části bližší k jehle a aplikujte jehlu do záhybu kůže rychlým a pevným pohybem. Vstříkněte roztok přípravku NeoRecormon. Jehlu rychle vytáhněte a zatlačte na místo vpichu suchým a sterilním tampónem.
Tento léčivý přípravek je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/031/035 -036
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. července 1997
Datum posledního prodloužení registrace: 16. července 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
NeoRecormon 20 000 IU injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s 0,6 ml injekčního roztoku obsahuje 20 000 mezinárodních jednotek (IU), což odpovídá 166 mikrogramům epoetinu beta* (rekombinantní lidský erytropoetin). Jeden ml injekčního roztoku obsahuje 33 333 IU epoetinu beta.
* Vyrobený technologií rekombinatní DNA v ovariálních buňkách čínských křečků.
Pomocné látky se známým účinkem:
Fenylalanin (až do 0,3 mg/předplněná injekční stříkačka)
Sodík (méně než 1 mmol v injekční stříkačce)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Bezbarvý, čirý až slabě opalescentní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek NeoRecormon je indikován k léčbě:
- Léčba symptomatické anémie spojené s chronickým renálním selháním u dospělých a pediatrických pacientů.
- Prevence anémie u předčasně narozených dětí s porodní váhou v rozmezí 750-1500 g, které se narodily v době před 34. týdnem těhotenství.
- Léčba symptomatické anémie u dospělých pacientů s nemyeloidními malignitami, kteří jsou léčeni chemoterapií.
- Zvýšení počtu erytrocytů před odběrem krve k autologní transfuzi krve. Použití v této indikaci musí být zváženo vzhledem k udávanému zvýšení rizika vzniku tromboembolických příhod. Použití je indikováno pouze u pacientů s mírnou anémií (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], bez nedostatku železa), pokud postupy pro konzervování krve nejsou dostupné nebo účinné nebo plánovaný zvolený velký chirurgický zákrok vyžaduje velký objem krve (4 a více jednotek krve pro ženy nebo 5 a více jednotek pro muže). Viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba přípravkem NeoRecormon může být zahájena pouze lékařem se zkušenostmi s výše uvedenými
indikacemi přípravku. Protože byly vzácně zaznamenány anafylaktoidní reakce po podání přípravku,
první dávka má být aplikována pod dohledem zdravotnického personálu.
Dávkování
Léčba symptomatické anémie u dospělých a pediatrických pacientů s chronickým selháním ledvin Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta. Přípravek NeoRecormon má být podáván buď subkutánně nebo intravenózně, tak aby byla hladina hemoglobinu zvýšena maximálně na 12 g/dl (7,5 mmol/l). U pacientů, kteří nepodstupují dialýzu, se upřednostňuje subkutánní podání, kdy není zapotřebí vpichů do periferních žil. V případě intravenózního podání má být přípravek podáván alespoň po dobu 2 minut, například přes arterio-venózní píštěl na konci dialýzy u pacientů na hemodialýze.
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu v rozsahu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Zvýšení hladiny hemoglobinu o více než 2 g/dl (1,25 mmol/l) během období čtyř týdnů není žádoucí. Pokud k němu dojde, má být dávka odpovídajícím způsobem upravena, jak je uvedeno dále. Je-li rychlost vzestupu hladiny hemoglobinu vyšší než 2 g/dl (1,25 mmol/l) za měsíc nebo pokud se hladina hemoglobinu zvýší až na hodnotu 12 g/dl (7,45 mmol/l) je třeba dávku snížit přibližně o 25 %. Pokud by se hladina hemoglobinu dále zvyšovala, má být léčba přerušena, dokud nezačne hladina opět klesat, a v tomto bodě má být znovu zahájena léčba dávkou přibližně o 25 % nižší, než byla předchozí podávaná dávka.
Pacienti mají být důkladně sledováni a je třeba ověřit, že byla použita nejnižší schválená účinná dávka přípravku NeoRecormon, která postačuje pro kontrolu symptomů anémie při zachování koncentrace hemoglobinu pod nebo na hodnotě 12 g/dl (7,45 mmol/l).
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin. U pacientů se slabou odpovědí hemoglobinu na přípravek NeoRecormon mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz bod 4.4 a 5.1).
V případě hypertenze nebo stávajících kardiovaskulárních, cerebrovaskulárních nebo periferních vaskulárních onemocnění má být týdenní zvýšení Hb a cílová hodnota Hb stanovena individuálně při zvážení klinického obrazu.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází.
1. Fáze korekční
- Subkutánní podání:
Počáteční dávka je 3 x 20 IU/kg tělesné hmotnosti týdně. Dávka může být zvýšena každé 4 týdny o 3 x 20 IU/kg za týden, jestliže není dosaženo adekvátního vzestupu Hb (< 0,25 g/dl za týden).
Týdenní dávka může být také rozdělena na denní dávky.
- Intravenózní podání:
Počáteční dávka je 3 x 40 IU/kg tělesné hmotnosti týdně. Dávka může být po 4 týdnech zvýšena na 80 IU/kg třikrát týdně a pokud je nutné další zvýšení dávky, má být o 20 IU/kg třikrát týdně v měsíčních intervalech.
Maximální dávka při obou způsobech podání nemá překročit 720 IU/kg za týden.
2. Fáze udržovací
Pro udržení Hb mezi 10 až 12 g/dl je dávka ze začátku snížena na polovinu předešlé dávky. Následně je u pacienta dávka nastavena v intervalech jednoho nebo dvou týdnů individuálně (udržovací dávka).
V případě podkožního podání může být týdenní dávka podána v jedné injekci týdně, nebo může být rozdělena do tří až sedmi dávek týdně. Pacienti, kteří jsou stabilní na režimu podávání jednou týdně, mohou být převedeni na dávkování jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Výsledky klinických studií u dětí prokázaly, že čím jsou pacienti mladší, tím v průměru vyšší dávky přípravku NeoRecormon vyžadují. Doporučený plán dávkování má však být dodržován, protože klinický účinek u jednotlivých pacientů nelze zcela předvídat.
Léčba přípravkem NeoRecormon v předplněné injekční stříkačce je obvykle dlouhodobá. Může však být, pokud je to nutné, kdykoliv přerušena. Údaje týkající se dávkovacího schématu podání jednou týdně vycházejí z klinických studií s délkou léčby 24 týdnů.
Prevence anémie u předčasně narozených dětí
Roztok je podáván subkutánně v dávce 3 x 250 IU/kg tělesné hmotnosti za týden. Předčasně narozené děti u kterých již byla provedena transfuze v okamžiku zahájení léčby přípravkem NeoRecormon nebudou pravděpodobně mít takový užitek z léčby jako předčasně narozené děti, u kterých transfuze provedena nebyla. Doporučená délka léčby je 6 týdnů.
Léčba symptomatické chemoterapií indukované anémie u pacientů s nádorovým onemocněním Přípravek NeoRecormon má být podáván subkutánně pacientům s anémií (např. koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l)). Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta.
Týdenní dávka může být podána v jedné injekci jednou týdně nebo může být rozdělena na 3 až 7 jednotlivých dávek.
Doporučená úvodní dávka je 30 000 IU týdně (to odpovídá přibližně 450 IU/kg tělesné hmotnosti týdně u pacienta s průměrnou hmotností).
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Pokud dojde po 4 týdnech léčby ke zvýšení hodnot hemoglobinu o minimálně 1 g/dl (0,62 mmol/l), má se v podávání této dávky dále pokračovat. Pokud hladina hemoglobinu nestoupne o alespoň 1 g/dl (0,62 mmol/l), má být zváženo podávání dvojnásobné týdenní dávky. Pokud hladina hemoglobinu nestoupne po 8 týdnech léčby o alespoň 1 g/dl (0,62 mmol/l), je odpověď nepravděpodobná a léčba má být ukončena.
Léčba má pokračovat až 4 týdny po ukončení chemoterapie.
Maximální dávka nemá překročit 60 000 IU týdně.
Jakmile je u jednotlivého pacienta dosaženo léčebného cíle, dávka má být snížena o 25 až 50 %, aby byla udržena hladina hemoglobinu na této úrovni. Je zapotřebí vzít v úvahu titrování dávky.
Překročí-li hemoglobin hladinu 12 g/dl (7,5 mmol/l), má být dávka snížena zhruba o 25 až 50 %.
Pokud hladina hemoglobinu překročí 13 g/dl (8,1 mmol/l), má být léčba přípravkem NeoRecormon dočasně přerušena. Pokud hladina hemoglobinu klesne na 12 g/dl (7,5 mmol/l) nebo níže, má být léčba opět zahájena s dávkou přibližně o 25 % nižší, než byla předchozí dávka.
Pokud je nárůst hemoglobinu v průběhu 4 týdnů vyšší než 2 g/dl (1,3 mmol/l), dávka má být snížena o 25 až 50 %.
Pacienti mají být důkladně sledováni, přičemž je třeba ověřit, že byla použita nejnižší schválená dávka přípravku NeoRecormon postačující pro adekvátní kontrolu symptomů anémie.
Podávání pro zvýšení množství autologní krve
Přípravek je aplikován intravenózně po dobu asi 2 minut nebo subkutánně.
Přípravek NeoRecormon je podáván dvakrát týdně po dobu 4 týdnů. V případě, kdy hematokrit pacienta umožňuje odběr krve, tj. hodnota hematokritu je > 33 %, je přípravek NeoRecormon podáván v okamžiku ukončení odběru.
V průběhu celé léčby nemá být překročena hodnota hematokritu 48 %.
Dávkování musí být stanoveno chirurgickým týmem zvlášť pro každého pacienta podle požadovaného množství krve pro autologní transfuzi a endogenní rezervy červených krvinek:
1. P ožadované množství krve pro autologní transfuzi závisí na předpokládané ztrátě krve, popřípadě na použití postupů konzervování krve a na fyzické kondici pacienta.
Množství potřebné autologní krve má stačit k tomu, aby bylo možné se vyhnout transfuzi homologní krve.
Požadované množství předem odebrané krve je vyjádřeno v jednotkách, přičemž jedna jednotka v nomogramu je ekvivalentní 180 ml červených krvinek.
2. Možnost autologní transfuze krve závisí především na objemu pacientovy krve a na výchozí hodnotě hematokritu. Obě proměnné určují endogenní rezervu červených krvinek, kterou je možné vypočítat podle následujících vzorců:
Endogenní rezerva červených krvinek = objem krve [ml] x (hematokrit - 33)/100 Ženy: objem krve [ml] = 41 [ml/kg] x tělesná hmotnost [kg] + 1200 [ml]
Muži: objem krve [ml] = 44 [ml/kg] x tělesná hmotnost [kg] + 1600 [ml]
(tělesná hmotnost > 45 kg)
Indikace pro léčbu přípravkem NeoRecormon a jednotlivá dávka mají být stanoveny z požadovaného množství předem odebrané krve a endogenní rezervy červených krvinek podle následujících grafů.
Ženy
Požadované množství krve pro autologní transfuzi [jednotky]
Muži
Požadované množství krve pro autologní transfuzi [jednotky]


Endogenní rezerva červených krvinek [ml] Endogenní rezerva červených krvinek [ml]
Takto stanovená jednotlivá dávka je podávána dvakrát týdně po dobu 4 týdnů. Maximální dávka nemá překročit 1600 IU/kg tělesné hmotnosti a týden při intravenózním podání nebo 1200 IU/kg tělesné hmotnosti a týden při subkutánním podávání.
Způsob podání
Přípravek NeoRecormon v předplněné injekční stříkačce je připraven k použití. Aplikován může být pouze roztok, který je čirý nebo lehce opalescentní, bezbarvý a bez viditelných částic.
Přípravek NeoRecormon v předplněné injekční stříkačce je sterilní, ale neobsahuje žádné konzervační látky. Za žádných okolností proto není možno aplikovat více než jednu dávku s jednou předplněnou injekční stříkačkou; léčivý přípravek je určen pouze k jednorázovému použití.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku (uvedenou v bodě 6.1).
Špatně kontrolovatelná hypertenze.
V indikaci “podání pro zvýšení množství autologní krve”: infarkt myokardu nebo cévní mozková příhoda v průběhu jednoho měsíce před zákrokem, nestabilní angina pectoris, zvýšené riziko hluboké venózní trombózy jako např. po prodělaném tromboembolickém onemocnění.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek NeoRecormon má být užíván s opatrností u pacientů s refrakterní anémií s nadbytkem transformovaných blastů, při epilepsii, trombocytóze a chronickém selhání jater. Je nutné vyloučit nedostatek kyseliny listové a vitamínu BJ2, protože tyto stavy snižují účinek přípravku NeoRecormon.
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin, neboť vysoké kumulující se dávky epoetinu mohou mít spojitost se zvýšeným rizikem mortality, závažnými kardiovaskulárními a cerebrovaskulárními příhodami. U pacientů se slabou odpovědí hemoglobinu na epoetiny mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz body 4.2 a 5.1).
Je třeba zhodnotit u všech pacientů hladinu železa před a v průběhu léčby, aby byla zajištěna účinná erytropoéza. Může být nutná substituce železa prováděná v souladu s léčebnými doporučeními.
Závažné zatížení hliníkem, ke kterému během léčby renálního selhání může dojít, může rovněž účinek přípravku NeoRecormon snížit.
Indikace k léčbě přípravkem NeoRecormon u pacientů s nefrosklerózou, kteří ještě nejsou dialyzovaní, má být zvážena individuálně, protože u těchto pacientů nelze vyloučit urychlení postupu renálního selhání.
Čistá aplazie buněk červené krevní řady (PRCA)
PRCA způsobená neutralizujícími antierytropoetinovými protilátkami byla hlášena ve spojení s léčbou erytropoetiny, včetně přípravku NeoRecormon. Bylo prokázáno, že tyto protilátky zkříženě reagují se všemi erytropoetinovými proteiny, a proto pacienti, u kterých je podezření nebo je potvrzen výskyt neutralizujících protilátek proti erytropoetinu, nemají být převáděni na přípravek NeoRecormon (viz bod 4.8).
PRCA u pacientů s hepatitidou C
Paradoxní pokles hladiny hemoglobinu a rozvoj závažné anémie související s nízkým počtem retikulocytů má vést k okamžitému přerušení léčby epoetinem a provedení testů na protilátky proti erytropoetinu. Tyto případy byly hlášeny, pokud byly pacientům s hepatitidou C léčených interferonem a ribavirinem současně podávány epoetiny. Epoetiny nejsou schválené pro léčbu anémie související s hepatitidou C.
Monitorování krevního tlaku
Může dojít ke vzestupu krevního tlaku nebo ke zhoršení již existující hypertenze zejména v případě rychlého vzestupu hematokritu. Toto zvýšení krevního tlaku je možno upravit pomocí léků. Pokud zvýšení krevního tlaku nemůže být upraveno medikamentózně, je doporučeno krátkodobé přerušení léčby přípravkem NeoRecormon. Zvláště na začátku terapie se doporučuje pravidelné monitorování krevního tlaku, a to rovněž mezi dialýzami. Může dojít ke vzniku hypertenzní krize s příznaky napodobujícími encefalopatii, která vyžaduje okamžitou lékařskou intervenci a intenzivní péči. Zvláštní pozoronost je třeba věnovat náhlým bodavým migrenosním bolestem hlavy jako možnému varovnému příznaku.
Chronické selhání ledvin
U pacientů 5 chronickým selháním ledvin, léčených přípravkem NeoRecormon, může dojít v průběhu léčby k mírnému, na dávce závislému, nárůstu počtu krevních destiček v rozmezí normálních hodnot, zvláště pak v případě intravenózního podávání. K poklesu tohoto nárůstu dochází v dalším průběhu léčby. Je doporučeno pravidelně sledovat počet krevních destiček v průběhu prvních 8 týdnů léčby.
Koncentrace hemoglobinu
U pacientů s chronickým selháním ledvin nemá udržovací koncentrace hemoglobinu překročit horní limit cílové koncentrace hemoglobinu doporučované v bodě 4.2. V klinických studiích bylo pozorováno zvýšené riziko úmrtí a závažných kardiovaskulárních příhod nebo cerebrovaskulárních příhod, včetně cévní mozkové příhody, pokud byly používány přípravky stimulující erytropoézu (ESA) k dosažení cílových hodnot hemoglobinu vyšších než 12 g/dl (7,5 mmol/l).
V kontrolovaných klinických studiích se neprokázal žádný signifikantní přínos podávání epoetinů, pokud byla koncentrace hemoglobinu zvyšována nad hladinu nezbytně nutnou pro kontrolu symptomů anémie a předcházení krevní transfuze.
U předčasně narozených dětí může dojít k mírnému nárůstu počtu krevních destiček, především v období mezi 12. - 14. dnem života, proto má být počet krevních destiček sledován v pravidelných intervalech.
Účinek na růst nádorů
Epoetiny jsou růstové faktory, které primárně podporují tvorbu červených krvinek. Erytropoetinové receptory mohou být přítomny na povrchu různých nádorových buněk. Stejně jako u všech růstových faktorů je třeba mít na zřeteli, že i epoetiny mohou stimulovat rozvoj nádoru. V několika kontrolovaných klinických studiích nebylo ve spojení s epoetiny prokázáno zlepšení celkové doby přežití ani snížení rizika progrese nádorů u pacientů s anémií spojenou s nádorovým onemocněním.
V kontrolovaných klinických studiích zaměřených na použití přípravku NeoRecormon a ostatních erytropoézu stimulujících přípravků (ESA), bylo zjištěno:
- zkrácení doby do progrese nádoru u pacientů s pokročilým nádorovým onemocněním hlavy a krku, pokud byly dosahovány cílové hladiny hemoglobinu vyšší než 14 g/dl (8,7 mmol/l)
- zkrácení celkové doby přežití a zvýšení počtu úmrtí souvisejících s progresí onemocnění během 4 měsíců léčby u pacientů s metastazujícím karcinomem prsu léčených chemoterapií, pokud byly dosahovány cílové hladiny hemoglobinu 12-14 g/dl (7,5-8,7 mmol/l),
- zvýšení rizika úmrtí při dosahování cílové hladiny hemoglobinu 12 g/dl (7,5 mmol/l) u pacientů s aktivními maligními chorobami, kteří nedostávali ani chemoterapii ani ozařování. V této populaci pacientů není použití ESA indikováno.
Na základě výše uvedených skutečností má být za určitých klinických okolností při léčbě anémie u pacientů s nádorovým onemocněním upřednostněna transfuze krve. Rozhodnutí podat rekombinantní erytropoetin má být přijato na základě zvážení poměru přínosu a rizika a individuálního posouzení jednotlivého pacienta za daných specifických klinických podmínek. Další faktory, které mají být zváženy, je posouzení typu a stádia nádorového onemocnění; stupeň anémie; očekávaná délka přežití; podmínky, za kterých je pacient léčen; a vlastní volba pacienta (viz bod 5.1)
Může dojít ke vzestupu krevního tlaku, který lze farmakologicky léčit. Doporučuje se proto monitorovat krevní tlak, zejména v úvodní fázi léčby pacientů s nádorovým onemocněním.
U onkologických pacientů mají být také pravidelně sledovány počty krevních destiček a hladina hemoglobinu.
U pacientů v programu před autologní transfuzí krve může být zvýšení počtu krevních destiček, většinou v rozmezí normálních hodnot. Proto je doporučeno určovat počet krevních destiček u těchto pacientů alespoň jednou týdně. Pokud dojde ke zvýšení krevních destiček o více než 150 x 109/l nebo když nárůst přesáhne normální hodnoty, má být léčba přípravkem NeoRecormon přerušena.
U předčasně narozených dětí nelze vyloučit možné riziko retinopatie způsobené erytropoetinem, proto je třeba zvýšené pozornosti a rozhodnutí o léčbě předčasně narozených dětí má být zvažováno na základě posouzení možného prospěchu a rizika této léčby a jiných dostupných možností.
U pacientů 5 chronickým selháním ledvin je v průběhu léčby přípravkem NeoRecormon při hemodialýze často nutné zvýšení dávky heparinu z důvodu zvýšeného hematokritu. Pokud není heparinizace optimální, může dojít k okluzi systému dialýzy.
U pacientů s chronickým selháním ledvin s rizikem trombózy cévního přístupu má být zvážena časná kontrola cévního přístupu a trombotická profylaxe kyselinou acetylsalicylovou.
V průběhu léčby přípravkem NeoRecormon má být pravidelně monitorována hladina sérového draslíku a fosfátů. Zvýšení kalémie bylo zaznamenáno u několika uremických pacientů léčených přípravkem NeoRecormon, i když příčinná souvislost nebyla ověřena. Pokud je pozorována zvýšená nebo stoupající hladina draslíku, pak má být zváženo přerušení podávání přípravku NeoRecormon dokud se kalémie neupraví.
Pro použití přípravku NeoRecormon v programu před autologní transfuzí krve musí být zváženy obecné zásady dárcovství krve, zvláště:
- dárci mají být pouze pacienti s hematokritem > 33 % (hemoglobin > 11g/dl [6,83 mmol/l];
- má být věnována zvláštní pozornost pacientům s hmotností nižší než 50 kg;
- jednotlivý čerpaný objem by neměl překročit cca 12 % odhadnutého objemu krve pacienta. Léčba má být vyhrazena pro pacienty, u kterých je zvlášť důležité vyhnout se transfuzi homologní krve a mají být zvážena rizika a prospěch homologní transfuze.
Zneužití
Zneužití přípravku zdravými osobami může vést k nadměrnému zvýšení hematokritu. Toto zvýšení může být spojeno s život ohrožujícími kardiovaskulárními komplikacemi.
Pomocné látky
Přípravek NeoRecormon v přeplněné injekční stříkačce obsahuje až 0,3 mg fenylalaninu v jedné injekční stříkačce jako pomocnou látku. Tato skutečnost má být brána v úvahu při léčbě pacientů trpících vážnými formami fenylketonurie.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v injekční stříkačce, tj. v podstatě je „bez sodíku“
Zpětná zjistitelnost přípravku NeoRecormon
Z důvodu snadnější zpětné zjistitelnosti látek stimulujících erytropoézu (ESAs) má být obchodní název předepsaného přípravku ESA zřetelně zaznamenán (nebo vyznačen) v pacientově dokumentaci.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Z dostupných klinických výsledků nejsou známy žádné interakce přípravku NeoRecormon s ostatními léčivými příprakvy.
Pokusy na zvířatech prokázaly, že epoetin beta nezvyšuje myelotoxicitu cytostatických léčivých látek cisplatina, cyklofosfamid a fluorouracil.
4.6 Fertilita, těhotenství a kojení
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3).
Nejsou k dispozici žádné klinické údaje o účincích epoetinu beta během těhotenství.
Při předepisování těhotným ženám nutno postupovat opatrně.
Kojení
Není známo, zda se epoetin beta vylučuje do lidského mateřského mléka.
Rozhodnutí, zda pokračovat v kojení/přerušit kojení nebo pokračovat v léčbě/přerušit léčbu epoetinem beta, má být zvoleno v závislosti na prospěšnosti kojení pro dítě a prospěšnosti léčby epoetinem beta u matky.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek NeoRecormon nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Na základě výsledků klinických studií s 1725 pacienty lze očekávat, že přibližně u 8 % pacientů léčených přípravkem NeoRecormon se mohou vyskytnout nežádoucí účinky.
Anemičtí pacienti s chronickým selháním ledvin
Nejčastějším nežádoucím účinkem v průběhu léčby přípravkem NeoRecormon je zvýšení krevního tlaku nebo zhoršení stávající hypertenze, zvláště pak v případech rychlého zvýšení hodnot hematokritu (viz bod 4.4). Hypertenzní krize s příznaky encefalopatie (např. bolesti hlavy a stavy zmatenosti, senzoricko-motorické poruchy jako například porucha řeči nebo zhoršení chůze až tonicko-klonické křeče) se mohou objevit také u jednotlivých pacientů s jinak normálním nebo nízkým krevním tlakem (viz bod 4.4).
Může dojít k trombóze žilního přístupu zejména u pacientů s tendencí k hypotenzi nebo s komplikacemi arteriovenózního zkratu (např. stenózy, aneuryzmata), viz bod 4.4. Ve většině případů je pozorován pokles hodnot sérového železa souběžně se vzestupem hematokritu (viz bod 4.4). Navíc byl v ojedinělých případech pozorován vzestup sérového draslíku a hladin fosfátů (viz bod 4.4).
V ojedinělých případech byla v souvislosti s léčbou přípravkem NeoRecormon hlášena čistá aplazie červených krvinek (PRCA) způsobená neutralizujícími antitrombopoetinovými protilátkami.
V případě diagnózy čisté aplazie červených krvinek (PRCA) způsobené neutralizujícími antitrombopoetinovými protilátkami musí být léčba přípravkem NeoRecormon ukončena a pacient nemá být převáděn na léčbu jinou erytropoetickou bílkovinou (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 1.
Pacienti s rakovinou
Bolesti hlavy související s léčbou epoetinem beta a hypertenze, kterou lze medikamentózně léčit, jsou časté (viz bod 4.4).
U některých pacientů je pozorován pokles sérového železa (viz bod 4.4).
V klinických studiích byl prokázán vyšší výskyt tromboembolických událostí u pacientů s karcinomem léčených přípravkem NeoRecormon ve srovnání s neléčenými kontrolními skupinami nebo placebem. U pacientů léčených přípravkem NeoRecormon byla incidence 7 % ve srovnání s 4 % u kontrolních skupin; aniž by to bylo spojeno se zvýšenou mortalitou na tromboembolické události ve srovnání s kontrolními skupinami.
Nežádoucí účinky jsou uvedeny níže v tabulce 2.
Pacienti v programu autologního dárcovství krve
U pacientů v programu autologního dárcovství krve byla hlášena lehce zvýšená četnost tromboembolických událostí. Kauzální vztah k léčbě přípravkem NeoRecormon však nebyl prokázán.
Ve studiích kontrolovaných placebem byl více vyjádřen přechodný deficit železa u pacientů léčených přípravkem NeoRecormon než u kontrolní skupiny (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 3.
Shrnutí nežádoucích účinků
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů MedDRA a absolutní četnosti. Kategorie četnosti jsou definovány dle následující konvence:
velmi časté (> 1/10); časté (>1/100 to <1/10); méně časté (>1/1 000 to <1/100); vzácné (>1/10 000 to <1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
Tabulka 1: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů s chronickým selháním ledvin léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze Hypertenzní krize |
Časté Méně časté |
|
Poruchy nervového systému |
Časté | |
|
Poruchy krve a |
Trombóza cévního |
Vzácné |
|
lymfatického systému |
přístupu Trombocytóza |
Velmi vzácné |
Tabulka 2: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u onkologických pacientů léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy í |
Hypertenze |
Časté |
|
Poruchy krve a lymfatického systému |
Tromboembolické účinky |
Časté |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Tabulka 3: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů v programu autologního dárcovství krve léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Poruchy í nervového systému |
Bolesti hlavy |
Časté |
Předčasně narozené děti
Pokles hodnot sérového ferritinu je velmi častý (viz bod 4.4).
Popis vybraných nežádoucích účinků
Vzácně byly pozorovány kožní reakce jako jsou vyrážka, svědění, kopřivka nebo reakce v místě vpichu. Velmi vzácně byly popsány anafylaktoidní reakce související s léčbou epoetinem beta. V kontrolovaných klinických studiích však zvýšení výskytu hypersenzitivních reakcí nebylo popsáno.
Ve velmi vzácných případech, především na počátku léčby, byl zaznamenán výskyt příznaků podobných chřipce ("flu-like" symptomy) jako je horečka, zimnice, bolesti hlavy, bolest v končetinách, malátnost a/nebo bolest kostí. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Údaje z kontrolované klinické studie s epoetinem alfa nebo darbepoetinem alfa udávaly incidenci cévní mozkové příhody jako častou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 P ředávkování
Terapeutický rozsah přípravku NeoRecormon je velmi široký. Dokonce i při velmi vysokých hladinách v séru nebyly pozorovány žádné příznaky otravy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA01 Mechanismus účinku
Erytropoetin je glykoprotein, který stimuluje tvorbu erytrocytů z prekurzorů kmenových buněk.
Působí jako faktor stimulující mitózu kmenových buněk červené krevní řady a hormon působící na jejich diferenciaci.
Epoetin beta, léčivá látka přípravku NeoRecormon, je svým složením aminokyselin a cukrů identický s erytropoetinem, který byl izolován z moči anemických pacientů.
Biologický účinek epoetinu beta byl demonstrován po intravenózní a podkožní aplikaci u různých pokusných zvířat in vivo (normální a uremičtí potkani, polycytemické myši, psi). Po podání epoetinu beta se zvýší počet erytrocytů, hodnota Hb a retikulocytů stejně tak jako rychlost inkorporace 59Fe.
In vitro byla zjištěna zvýšená inkorporace 3H-thymidinu v erytroidních jaderných buňkách sleziny (kultura buněk sleziny myší) po inkubaci s epoetinem beta.
Pokusy na tkáňových kulturách buněk lidské kostní dřeně prokázaly, že epoetin beta stimuluje jen tvorbu červených krvinek a neovlivňuje tvorbu bílých krvinek. Nebyly zjištěny cytotoxické účinky epoetinu beta na kostní dřeň ani na lidské kožní buňky.
Po podání jednotlivé dávky epoetinu beta nebyly pozorovány žádné vlivy na chování nebo na pohybovou činnost u myší a oběhové nebo respirační funkce u psů.
Klinická účinnost a bezpečnost
V randomizované dvojitě zaslepené, placebem kontrolované studii, která hodnotila 4038 pacientů s chronickým selháním ledvin bez nutnosti dialýzy, s diabetem 2. typu a hladinami hemoglobinu < 11 g/dl, byli pacienti léčeni buď darbepoetinem alfa až do dosažení cílových hladin hemoglobinu 13 g/dl, nebo dostávali placebo (viz bod 4.4). Studie nesplnila žádný z primárních cílů, který by prokázal snížení rizika úmrtí ze všech příčin, kardiovaskulární morbidity nebo terminálního stádia onemocnění ledvin (ESRD). Analýza jednotlivých komponent složených cílových parametrů prokázala následující poměr rizik (95% CI): úmrtí 1,05 (o,92; 1,21), cévní mozková příhoda 1,92 (1,38; 2,68), městnavé srdeční selhání 0,89 (0,74; 1,08), infarkt myokardu 0,96 (0,75; 1,23), hospitalizace z důvodu ischemie myokardu 0,84 (0,55; 1,27), terminální stádium onemocnění ledvin 1,02 (0,87; 1,18).
Souhrnné post-hoc analýzy klinických studií s ESA byly provedeny u pacientů s chronickým selháním ledvin (u pacientů, kteří byli nebo nebyli na dialýze, u diabetiků a u pacientů bez diabetu). Byla pozorována tendence směrem ke zvýšení odhadovaného rizika mortality z jakýchkoli důvodů, rizika kardiovaskulárních a cerebrovaskulárních příhod spojených s vyššími kumulativními dávkami ESA, nezávisle na stavu diabetu nebo dialýze (viz body 4.2 a 4.4).
Erytropoetin je růstový faktor primárně podporující tvorbu červených krvinek. Receptory pro erytropoetin mohou být exprimovány na povrchu mnoha nádorových buněk.
Přežití a progrese nádoru byla zkoumána v pěti velkých kontrolovaných klinických studiích zahrnujících celkem 2833 pacientů; čtyři z těchto studií byly dvojitě zaslepené kontrolované placebe m, jedna studie byla otevřená. Dvou studií se účastnili pacienti léčení chemoterapií. Cílová koncentrace hemoglobinu byla ve dvou studiích > 13 g/dl; v ostatních třech studiích 12-14 g/dl.
V otevřené studii nebyl z hlediska celkového přežití zjištěn žádný rozdíl mezi pacienty léčenými rekombinantním lidským erytropoetinem a mezi kontrolní skupinou. Ve čtyřech placebem kontrolovaných studiích se poměry rizik pro celkové přežití pohybovaly mezi 1,25 a 2,47 ve prospěch kontrolních skupin. Při srovnání s kontrolními skupinami ukázaly tyto studie konzistentní nevysvětlené statisticky významné zvýšení mortality u pacientů, kteří měli anémii spojenou s různými běžnými maligními nádory a kteří dostávali rekombinantní lidský erytropoetin. Výsledek celkového přežití zjištěný v klinických studiích nebylo možné uspokojivě vysvětlit rozdílem v incidenci trombózy a přidružených komplikací mezi skupinou dostávající rekombinantní lidský erytropoetin a mezi skupinou kontrolní.
Metaanalýza založená na údajích jednotlivých pacientů zahrnovala data ze všech 12 kontrolovaných klinických studií prováděných u pacientů s anémií a maligním nádorovým onemocněním, kteří byli léčeni přípravkem NeoRecormon (n=2301), ukázala celkový odhadovaný poměr rizik pro přežití 1,13 ve prospěch kontrol (95% CI 0,87; 1,46). U pacientů, kteří měli v baseline hladinu hemoglobinu <
10 g/dl (n=899), byl odhadovaný poměr rizik pro přežití 0,98 (95% CI 0,68 až 1,40). V celkové populaci bylo pozorováno zvýšené relativní riziko tromboembolických příhod (RR 1,62, 95% CI:
1,13; 2,31).
Byla provedena analýza dat na úrovni jednotlivých nemocných u více než 13 900 pacientů se zhoubným nádorem (léčených chemoterapií, radioterapií, chemoradioterapií nebo bez terapie) zařazených do 53 kontrolovaných klinických studií zahrnujících podávání několika epoetinů. Metaanalýza údajů celkového přežití prokázala, že poměr rizik (hazard ratio) je odhadem 1,06 ve prospěch kontrolních skupin (95% CI: 1,00, 1,12; 53 studií a 13 933 pacientů) a u pacientů se zhoubným nádorem podstupujících chemoterapii byl poměr rizik (hazard ratio) celkového přežití 1,04 (95% CI: 0,97, 1,11; 38 studií a 10 441 pacientů). Meta-analýzy zároveň konzistentně poukazují na významné zvýšení relativního rizika tromboembolických příhod u pacientů se zhoubným nádorem, kteří dostávají rekombinantní lidský erytropoetin (viz bod 4.4).
Ve velmi vzácných případech se v průběhu léčby rekombinantním lidským erytropoetinem objevily neutralizující protilátky proti erytropoetinu s nebo bez čisté aplazie červené krevní řady (PRCA).
5.2 Farmakokinetické vlastnosti
Farmakokinetické sledování u zdravých dobrovolníků a uremických pacientů ukázalo, že po intravenózním podání je poločas epoetinu beta mezi 4 - 12 hodinami a že distribuční objem odpovídá jedno- až dvojnásobku plazmatického objemu. Analogické výsledky byly nalezeny v pokusech na zvířatech u normálních i uremických potkanů.
Protrahovaná absorpce epoetinu beta po subkutánním podání uremickým pacientům vede ke vzniku koncentračního plató, přičemž maximální koncentrace je dosaženo v průměru za 12 - 28 hodin. Terminální poločas je delší než po intravenózním podání s průměrem 13 - 28 hodin.
Ve srovnání s intravenózní aplikací je biologická dostupnost epoetinu beta po podkožní aplikaci mezi 23 - 42 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
Studie kancerogenity s homologním erytropoetinem u myší neodhalily žádné známky proliferativního nebo kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 S eznam pomocných látek
Močovina,
Chlorid sodný,
Polysorbát 20,
Dihydrogenfosforečnan sodný dihydrát,
Hydrogenfosforečnan sodný dodekahydrát,
Chlorid vápenatý dihydrát,
Glycin,
L-leucin,
L-isoleucin,
L-threonin,
L-kyselina glutamová,
L-fenylalanin.
Voda na injekci.
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku v původním obalu, aby byl přípravek chráněn před světlem.
Pro usnadnění ambulantního použití může pacient vyjmout přípravek z chladničky a uchovávat jej jednorázově po dobu maximálně 3 dnů při pokojové teplotě (maximálně do 25 °C).
6.5 Druh obalu a obsah balení
Předplněná injekční stříkačka (sklo typu I) s víčkem a zátkou (pryž potažená teflonem) a s injekční jehlou (27G1/2). Jedna injekční stříkačka obsahuje 0,6 ml roztoku.
Velikost balení po 1 předplněné injekční stříkačce s 1 jehlou nebo 6 předplněných injekčních stříkačkách se 6 jehlami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Nejdříve si dobře umyjte ruce!
1. Vyjměte jednu předplněnou injekční stříkačku z balení a zkontrolujte, zda roztok ve stříkačce je čirý, bezbarvý a bez viditelných částic. Odstraňte víčko z injekční stříkačky.
2. Vyjměte jednu injekční jehlu z balení, nasaďte ji na injekční stříkačku a odstraňte z jehly ochranné víčko.
3. Vytlačte vzduch z injekční stříkačky tím, že stříkačku držíte ve vzpřímené poloze a jemně tlačíte píst vzhůru. Tlačte na píst tak dlouho, dokud množství přípravku NeoRecormon v injekční stříkačce neodpovídá množství předepsanému pro aplikaci.
4. Za použití tampónu namočeného v alkoholu si očistěte kůži v místě vpichu. Vytvořte záhyb na kůži tím, že ji sevřete mezi palec a ukazováček. Stiskněte tělo injekční stříkačky v části bližší k jehle a aplikujte jehlu do záhybu kůže rychlým a pevným pohybem. Vstříkněte roztok přípravku NeoRecormon. Jehlu rychle vytáhněte a zatlačte na místo vpichu suchým a sterilním tampónem.
Tento léčivý přípravek je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/031/037 -038
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. července 1997
Datum posledního prodloužení registrace: 16. července 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
NeoRecormon 30 000 IU injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s 0,6 ml injekčního roztoku obsahuje 30 000 mezinárodních jednotek (IU), což odpovídá 250 mikrogramům epoetinu beta* (rekombinantní lidský erytropoetin). Jeden ml injekčního roztoku obsahuje 50 000 IU epoetinu beta.
* Vyrobený technologií rekombinatní DNA v ovariálních buňkách čínských křečků.
Pomocné látky se známým účinkem:
Fenylalanin (až do 0,3 mg/předplněná injekční stříkačka)
Sodík (méně než 1 mmol v injekční lahvičce)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Bezbarvý, čirý až slabě opalescentní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek NeoRecormon je indikován k léčbě:
- Léčba symptomatické anémie spojené s chronickým renálním selháním u dospělých a pediatrických pacientů.
- Prevence anémie u předčasně narozených dětí s porodní váhou v rozmezí 750-1500 g, které se narodily v době před 34. týdnem těhotenství.
- Léčba symptomatické anémie u dospělých pacientů s nemyeloidními malignitami, kteří jsou léčeni chemoterapií.
- Zvýšení počtu erytrocytů před odběrem krve k autologní transfuzi krve. Použití v této indikaci musí být zváženo vzhledem k udávanému zvýšení rizika vzniku tromboembolických příhod. Použití je indikováno pouze u pacientů s mírnou anémií (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], bez nedostatku železa), pokud postupy pro konzervování krve nejsou dostupné nebo účinné nebo plánovaný zvolený velký chirurgický zákrok vyžaduje velký objem krve (4 a více jednotek krve pro ženy nebo 5 a více jednotek pro muže). Viz bod 5.1.
4.2 Dávkování a způsob podání
Léčba přípravkem NeoRecormon může být zahájena pouze lékařem se zkušenostmi s výše uvedenými
indikacemi přípravku. Protože byly vzácně zaznamenány anafylaktoidní reakce po podání přípravku,
první dávka má být aplikována pod dohledem zdravotnického personálu.
Dávkování
Léčba symptomatické anémie u dospělých a pediatrických pacientů s chronickým selháním ledvin Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta. Přípravek NeoRecormon má být podáván buď subkutánně nebo intravenózně, tak aby byla hladina hemoglobinu zvýšena maximálně na 12 g/dl (7,5 mmol/l). U pacientů, kteří nepodstupují dialýzu, se upřednostňuje subkutánní podání, kdy není zapotřebí vpichů do periferních žil. V případě intravenózního podání má být přípravek podáván alespoň po dobu 2 minut, například přes arterio-venózní píštěl na konci dialýzy u pacientů na hemodialýze.
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu v rozsahu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Zvýšení hladiny hemoglobinu o více než 2 g/dl (1,25 mmol/l) během období čtyř týdnů není žádoucí. Pokud k němu dojde, má být dávka odpovídajícím způsobem upravena, jak je uvedeno dále. Je-li rychlost vzestupu hladiny hemoglobinu vyšší než 2 g/dl (1,25 mmol/l) za měsíc nebo pokud se hladina hemoglobinu zvýší až na hodnotu 12 g/dl (7,45 mmol/l) je třeba dávku snížit přibližně o 25 %. Pokud by se hladina hemoglobinu dále zvyšovala, má být léčba přerušena, dokud nezačne hladina opět klesat, a v tomto bodě má být znovu zahájena léčba dávkou přibližně o 25 % nižší, než byla předchozí podávaná dávka.
Pacienti mají být důkladně sledováni a je třeba ověřit, že byla použita nejnižší schválená účinná dávka přípravku NeoRecormon, která postačuje pro kontrolu symptomů anémie při zachování koncentrace hemoglobinu pod nebo na hodnotě 12 g/dl (7,45 mmol/l).
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin. U pacientů se slabou odpovědí hemoglobinu na přípravek NeoRecormon mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz bod 4.4 a 5.1).
V případě hypertenze nebo stávajících kardiovaskulárních, cerebrovaskulárních nebo periferních vaskulárních onemocnění má být týdenní zvýšení Hb a cílová hodnota Hb stanovena individuálně při zvážení klinického obrazu.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází.
1. Fáze korekční
- Subkutánní podání:
Počáteční dávka je 3 x 20 IU/kg tělesné hmotnosti týdně. Dávka může být zvýšena každé 4 týdny o 3 x 20 IU/kg za týden, jestliže není dosaženo adekvátního vzestupu Hb (< 0,25 g/dl za týden).
Týdenní dávka může být také rozdělena na denní dávky.
- Intravenózní podání :
Počáteční dávka je 3 x 40 IU/kg tělesné hmotnosti týdně. Dávka může být po 4 týdnech zvýšena na 80 IU/kg třikrát týdně a pokud je nutné další zvýšení dávky, má být o 20 IU/kg třikrát týdně v měsíčních intervalech.
Maximální dávka při obou způsobech podání nemá překročit 720 IU/kg za týden.
2. Fáze udržovací
Pro udržení Hb mezi 10 až 12 g/dl je dávka ze začátku snížena na polovinu předešlé dávky. Následně je u pacienta dávka nastavena v intervalech jednoho nebo dvou týdnů individuálně (udržovací dávka).
V případě podkožního podání může být týdenní dávka podána v jedné injekci týdně, nebo může být rozdělena do tří až sedmi dávek týdně. Pacienti, kteří jsou stabilní na režimu podávání jednou týdně, mohou být převedeni na dávkování jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Výsledky klinických studií u dětí prokázaly, že čím jsou pacienti mladší, tím v průměru vyšší dávky přípravku NeoRecormon vyžadují. Doporučený plán dávkování má však být dodržován, protože klinický účinek u jednotlivých pacientů nelze zcela předvídat.
Léčba přípravkem NeoRecormon v předplněné injekční stříkačce je obvykle dlouhodobá. Může však být, pokud je to nutné, kdykoliv přerušena. Údaje týkající se dávkovacího schématu podání jednou týdně vycházejí z klinických studií s délkou léčby 24 týdnů.
Prevence anémie u předčasně narozených dětí
Roztok je podáván subkutánně v dávce 3 x 250 IU/kg tělesné hmotnosti za týden. Předčasně narozené děti u kterých již byla provedena transfuze v okamžiku zahájení léčby přípravkem NeoRecormon nebudou pravděpodobně mít takový užitek z léčby jako předčasně narozené děti, u kterých transfuze provedena nebyla. Doporučená délka léčby je 6 týdnů.
Léčba symptomatické chemoterapií indukované anémie u pacientů s nádorovým onemocněním Přípravek NeoRecormon má být podáván subkutánně pacientům s anémií (např. koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l)). Příznaky a následky anémie se mohou lišit v závislosti na věku, pohlaví a celkovém zatížení chorobou; je nutné, aby lékař individuálně zhodnotil klinický průběh a stav pacienta.
Týdenní dávka může být podána v jedné injekci jednou týdně nebo může být rozdělena na 3 až 7 jednotlivých dávek.
Doporučená úvodní dávka je 30 000 IU týdně (to odpovídá přibližně 450 IU/kg tělesné hmotnosti týdně u pacienta s průměrnou hmotností).
Kvůli variabilitě mezi pacienty mohou být někdy pozorovány individuální hladiny hemoglobinu nad nebo pod požadovanou hodnotou. Variabilita hemoglobinu má být usměrněna úpravou dávkování s ohledem na cílovou hladinu hemoglobinu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Nemá dojít k trvalému zvýšení hladiny hemoglobinu nad 12 g/dl (7,5 mmol/l); doporučení pro vhodnou úpravu dávkování při hodnotách hemoglobinu překračujících 12 g/dl (7,5 mmol/l) jsou popsána níže.
Pokud dojde po 4 týdnech léčby ke zvýšení hodnot hemoglobinu o minimálně 1 g/dl (0,62 mmol/l), má se v podávání této dávky dále pokračovat. Pokud hladina hemoglobinu nestoupne o alespoň 1 g/dl (0,62 mmol/l), má být zváženo podávání dvojnásobné týdenní dávky. Pokud hladina hemoglobinu nestoupne po 8 týdnech léčby o alespoň 1 g/dl (0,62 mmol/l), je odpověď nepravděpodobná a léčba má být ukončena.
Léčba má pokračovat až 4 týdny po ukončení chemoterapie.
Maximální dávka nemá překročit 60 000 IU týdně.
Jakmile je u jednotlivého pacienta dosaženo léčebného cíle, dávka má být snížena o 25 až 50 %, aby byla udržena hladina hemoglobinu na této úrovni. Je zapotřebí vzít v úvahu titrování dávky.
Překročí-li hemoglobin hladinu 12 g/dl (7,5 mmol/l), má být dávka snížena zhruba o 25 až 50 %.
Pokud hladina hemoglobinu překročí 13 g/dl (8,1 mmol/l), má být léčba přípravkem NeoRecormon dočasně přerušena. Pokud hladina hemoglobinu klesne na 12 g/dl (7,5 mmol/l) nebo níže, má být léčba opět zahájena s dávkou přibližně o 25 % nižší, než byla předchozí dávka.
Pokud je nárůst hemoglobinu v průběhu 4 týdnů vyšší než 2 g/dl (1,3 mmol/l), dávka má být snížena o 25 až 50 %.
Pacienti mají být důkladně sledováni, přičemž je třeba ověřit, že byla použita nejnižší schválená dávka přípravku NeoRecormon postačující pro adekvátní kontrolu symptomů anémie.
Podávání pro zvýšení množství autologní krve
Přípravek je aplikován intravenózně po dobu asi 2 minut nebo subkutánně.
Přípravek NeoRecormon je podáván dvakrát týdně po dobu 4 týdnů. V případě, kdy hematokrit pacienta umožňuje odběr krve, tj. hodnota hematokritu je > 33 %, je přípravek NeoRecormon podáván v okamžiku ukončení odběru.
V průběhu celé léčby nemá být překročena hodnota hematokritu 48 %.
Dávkování musí být stanoveno chirurgickým týmem zvlášť pro každého pacienta podle požadovaného množství krve pro autologní transfuzi a endogenní rezervy červených krvinek:
1. P ožadované množství krve pro autologní transfuzi závisí na předpokládané ztrátě krve, popřípadě na použití postupů konzervování krve a na fyzické kondici pacienta.
Množství potřebné autologní krve má stačit k tomu, aby bylo možné se vyhnout transfuzi homologní krve.
Požadované množství předem odebrané krve je vyjádřeno v jednotkách, přičemž jedna jednotka v nomogramu je ekvivalentní 180 ml červených krvinek.
2. Možnost autologní transfuze krve závisí především na objemu pacientovy krve a na výchozí hodnotě hematokritu. Obě proměnné určují endogenní rezervu červených krvinek, kterou je možné vypočítat podle následujících vzorců:
Endogenní rezerva červených krvinek = objem krve [ml] x (hematokrit - 33)/100 Ženy: objem krve [ml] = 41 [ml/kg] x tělesná hmotnost [kg] + 1200 [ml]
Muži: objem krve [ml] = 44 [ml/kg] x tělesná hmotnost [kg] + 1600 [ml]
(tělesná hmotnost > 45 kg)
Indikace pro léčbu přípravkem NeoRecormon a jednotlivá dávka mají být stanoveny z požadovaného množství předem odebrané krve a endogenní rezervy červených krvinek podle následujících grafů.
Ženy
Požadované množství krve pro autologní transfuzi [jednotky]
Muži
Požadované množství krve pro autologní transfuzi [jednotky]


Endogenní rezerva červených krvinek [ml] Endogenní rezerva červených krvinek [ml]
Takto stanovená jednotlivá dávka je podávána dvakrát týdně po dobu 4 týdnů. Maximální dávka nemá překročit 1600 IU/kg tělesné hmotnosti a týden při intravenózním podání nebo 1200 IU/kg tělesné hmotnosti a týden při subkutánním podávání.
Způsob podání
Přípravek NeoRecormon v předplněné injekční stříkačce je připraven k použití. Aplikován může být pouze roztok, který je čirý nebo lehce opalescentní, bezbarvý a bez viditelných částic.
Přípravek NeoRecormon v předplněné injekční stříkačce je sterilní, ale neobsahuje žádné konzervační látky. Za žádných okolností proto není možno aplikovat více než jednu dávku s jednou předplněnou injekční stříkačkou; léčivý přípravek je určen pouze k jednorázovému použití.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku (uvedenou v bodě 6.1).
Špatně kontrolovatelná hypertenze.
V indikaci “podání pro zvýšení množství autologní krve”: infarkt myokardu nebo cévní mozková příhoda v průběhu jednoho měsíce před zákrokem, nestabilní angina pectoris, zvýšené riziko hluboké venózní trombózy jako např. po prodělaném tromboembolickém onemocnění.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek NeoRecormon má být užíván s opatrností u pacientů s refrakterní anémií s nadbytkem transformovaných blastů, při epilepsii, trombocytóze a chronickém selhání jater. Je nutné vyloučit nedostatek kyseliny listové a vitamínu BJ2, protože tyto stavy snižují účinek přípravku NeoRecormon.
Opatrnosti je třeba při zvyšování dávek přípravku NeoRecormon u pacientů s chronickým selháním ledvin, neboť vysoké kumulující se dávky epoetinu mohou mít spojitost se zvýšeným rizikem mortality, závažnými kardiovaskulárními a cerebrovaskulárními příhodami. U pacientů se slabou odpovědí hemoglobinu na epoetiny mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz body 4.2 a 5.1).
Je třeba zhodnotit u všech pacientů hladinu železa před a v průběhu léčby, aby byla zajištěna účinná erytropoéza. Může být nutná substituce železa prováděná v souladu s léčebnými doporučeními.
Závažné zatížení hliníkem, ke kterému během léčby renálního selhání může dojít, může rovněž účinek přípravku NeoRecormon snížit.
Indikace k léčbě přípravkem NeoRecormon u pacientů s nefrosklerózou, kteří ještě nejsou dialyzovaní, má být zvážena individuálně, protože u těchto pacientů nelze vyloučit urychlení postupu renálního selhání.
Čistá aplazie buněk červené krevní řady (PRCA)
PRCA způsobená neutralizujícími antierytropoetinovými protilátkami byla hlášena ve spojení s léčbou erytropoetiny, včetně přípravku NeoRecormon. Bylo prokázáno, že tyto protilátky zkříženě reagují se všemi erytropoetinovými proteiny, a proto pacienti, u kterých je podezření nebo je potvrzen výskyt neutralizujících protilátek proti erytropoetinu, nemají být převáděni na přípravek NeoRecormon (viz bod 4.8).
PRCA u pacientů s hepatitidou C
Paradoxní pokles hladiny hemoglobinu a rozvoj závažné anémie související s nízkým počtem retikulocytů má vést k okamžitému přerušení léčby epoetinem a provedení testů na protilátky proti erytropoetinu. Tyto případy byly hlášeny, pokud byly pacientům s hepatitidou C léčených interferonem a ribavirinem současně podávány epoetiny. Epoetiny nejsou schválené pro léčbu anémie související s hepatitidou C.
Monitorování krevního tlaku
Může dojít ke vzestupu krevního tlaku nebo ke zhoršení již existující hypertenze zejména v případě rychlého vzestupu hematokritu. Toto zvýšení krevního tlaku je možno upravit pomocí léků. Pokud zvýšení krevního tlaku nemůže být upraveno medikamentózně, je doporučeno krátkodobé přerušení léčby přípravkem NeoRecormon. Zvláště na začátku terapie se doporučuje pravidelné monitorování krevního tlaku, a to rovněž mezi dialýzami. Může dojít ke vzniku hypertenzní krize s příznaky napodobujícími encefalopatii, která vyžaduje okamžitou lékařskou intervenci a intenzivní péči. Zvláštní pozoronost je třeba věnovat náhlým bodavým migrenosním bolestem hlavy jako možnému varovnému příznaku.
Chronické selhání ledvin
U pacientů s chronickým selháním ledvin, léčených přípravkem NeoRecormon, může dojít v průběhu léčby k mírnému, na dávce závislému nárůstu počtu krevních destiček v rozmezí normálních hodnot, zvláště pak v případě intravenózního podávání. K poklesu tohoto nárůstu dochází v dalším průběhu léčby. Je doporučeno pravidelně sledovat počet krevních destiček v průběhu prvních 8 týdnů léčby.
Koncentrace hemoglobinu
U pacientů s chronickým selháním ledvin nemá udržovací koncentrace hemoglobinu překročit horní limit cílové koncentrace hemoglobinu doporučované v bodě 4.2. V klinických studiích bylo pozorováno zvýšené riziko úmrtí a závažných kardiovaskulárních příhod nebo cerebrovaskulárních příhod, včetně cévní mozkové příhody, pokud byly používány přípravky stimulující erytropoézu (ESA) k dosažení cílových hodnot hemoglobinu vyšších než 12 g/dl (7,5 mmol/l).
V kontrolovaných klinických studiích se neprokázal žádný signifikantní přínos podávání epoetinů, pokud byla koncentrace hemoglobinu zvyšována nad hladinu nezbytně nutnou pro kontrolu symptomů anémie a předcházení krevní transfuze.
U předčasně narozených dětí může dojít k mírnému nárůstu počtu krevních destiček, především v období mezi 12. - 14. dnem života, proto má být počet krevních destiček sledován v pravidelných intervalech.
Účinek na růst nádorů
Epoetiny jsou růstové faktory, které primárně podporují tvorbu červených krvinek. Erytropoetinové receptory mohou být přítomny na povrchu různých nádorových buněk. Stejně jako u všech růstových faktorů je třeba mít na zřeteli, že i epoetiny mohou stimulovat rozvoj nádoru. V několika kontrolovaných klinických studiích nebylo ve spojení s epoetiny prokázáno zlepšení celkové doby přežití ani snížení rizika progrese nádorů u pacientů s anémií spojenou s nádorovým onemocněním.
V kontrolovaných klinických studiích zaměřených na použití přípravku NeoRecormon a ostatních erytropoézu stimulujících přípravků (ESA), bylo zjištěno:
- zkrácení doby do progrese nádoru u pacientů s pokročilým nádorovým onemocněním hlavy a krku, pokud byly dosahovány cílové hladiny hemoglobinu vyšší než 14 g/dl (8,7 mmol/l)
- zkrácení celkové doby přežití a zvýšení počtu úmrtí souvisejících s progresí onemocnění během 4 měsíců léčby u pacientů s metastazujícím karcinomem prsu léčených chemoterapií, pokud byly dosahovány cílové hladiny hemoglobinu 12-14 g/dl (7,5-8,7 mmol/l),
- zvýšení rizika úmrtí při dosahování cílové hladiny hemoglobinu 12 g/dl (7,5 mmol/l) u pacientů s aktivními maligními chorobami, kteří nedostávali ani chemoterapii ani ozařování. V této populaci pacientů není použití ESA indikováno.
Na základě výše uvedených skutečností má být za určitých klinických okolností při léčbě anémie u pacientů s nádorovým onemocněním upřednostněna transfuze krve. Rozhodnutí podat rekombinantní erytropoetin má být přijato na základě zvážení poměru přínosu a rizika a individuálního posouzení jednotlivého pacienta za daných specifických klinických podmínek. Další faktory, které mají být zváženy, je posouzení typu a stádia nádorového onemocnění; stupeň anémie; očekávaná délka přežití; podmínky, za kterých je pacient léčen; a vlastní volba pacienta (viz bod 5.1)
Může dojít ke vzestupu krevního tlaku, který lze farmakologicky léčit. Doporučuje se proto monitorovat krevní tlak, zejména v úvodní fázi léčby pacientů s nádorovým onemocněním.
U onkologických pacientů mají být také pravidelně sledovány počty krevních destiček a hladina hemoglobinu.
U pacientů v programu před autologní transfuzí krve může být zvýšení počtu krevních destiček, většinou v rozmezí normálních hodnot. Proto je doporučeno určovat počet krevních destiček u těchto pacientů alespoň jednou týdně. Pokud dojde ke zvýšení krevních destiček o více než 150 x 109/l nebo když nárůst přesáhne normální hodnoty, má být léčba přípravkem NeoRecormon přerušena.
U předčasně narozených dětí nelze vyloučit možné riziko retinopatie způsobené erytropoetinem, proto je třeba zvýšené pozornosti a rozhodnutí o léčbě předčasně narozených dětí má být zvažováno na základě posouzení možného prospěchu a rizika této léčby a jiných dostupných možností.
U pacientů 5 chronickým selháním ledvin je v průběhu léčby přípravkem NeoRecormon při hemodialýze často nutné zvýšení dávky heparinu z důvodu zvýšeného hematokritu. Pokud není heparinizace optimální, může dojít k okluzi systému dialýzy.
U pacientů s chronickým selháním ledvin s rizikem trombózy cévního přístupu má být zvážena časná kontrola cévního přístupu a trombotická profylaxe kyselinou acetylsalicylovou.
V průběhu léčby přípravkem NeoRecormon má být pravidelně monitorována hladina sérového draslíku a fosfátů. Zvýšení kalémie bylo zaznamenáno u několika uremických pacientů léčených přípravkem NeoRecormon, i když příčinná souvislost nebyla ověřena. Pokud je pozorována zvýšená nebo stoupající hladina draslíku, pak má být zváženo přerušení podávání přípravku NeoRecormon dokud se kalémie neupraví.
Pro použití přípravku NeoRecormon v programu před autologní transfuzí krve musí být zváženy obecné zásady dárcovství krve, zvláště:
- dárci mají být pouze pacienti s hematokritem > 33 % (hemoglobin > 11g/dl [6,83 mmol/l];
- má být věnována zvláštní pozornost pacientům s hmotností nižší než 50 kg;
- jednotlivý čerpaný objem nemá překročit cca 12 % odhadnutého objemu krve pacienta.
Léčba má být vyhrazena pro pacienty, u kterých je zvlášť důležité vyhnout se transfuzi homologní krve a mají být zvážena rizika a prospěch homologní transfuze.
Zneužití
Zneužití přípravku zdravými osobami může vést k nadměrnému zvýšení hematokritu. Toto zvýšení může být spojeno s život ohrožujícími kardiovaskulárními komplikacemi.
Pomocné látky
Přípravek NeoRecormon v přeplněné injekční stříkačce obsahuje až 0,3 mg fenylalaninu v jedné injekční stříkačce jako pomocnou látku. Tato skutečnost má být brána v úvahu při léčbě pacientů trpících vážnými formami fenylketonurie.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v injekční stříkačce, tj. v podstatě je „bez sodíku“
Zpětná zjistitelnost přípravku NeoRecormon
Z důvodu snadnější zpětné zjistitelnosti látek stimulujících erytropoézu (ESAs) má být obchodní název předepsaného přípravku ESA zřetelně zaznamenán (nebo vyznačen) v pacientově dokumentaci.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Z dostupných klinických výsledků nejsou známy žádné interakce přípravku NeoRecormon s ostatními léčivými příprakvy.
Pokusy na zvířatech prokázaly, že epoetin beta nezvyšuje myelotoxicitu cytostatických léčivých látek cisplatina, cyklofosfamid a fluorouracil.
4.6 Fertilita, těhotenství a kojení
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3).
Nejsou k dispozici žádné klinické údaje o účincích epoetinu beta během těhotenství.
Při předepisování těhotným ženám nutno postupovat opatrně.
Kojení
Není známo, zda se epoetin beta vylučuje do lidského mateřského mléka.
Rozhodnutí, zda pokračovat v kojení/přerušit kojení nebo pokračovat v léčbě/přerušit léčbu epoetinem beta, má být zvoleno v závislosti na prospěšnosti kojení pro dítě a prospěšnosti léčby epoetinem beta u matky.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek NeoRecormon nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Na základě výsledků klinických studií s 1725 pacienty lze očekávat, že přibližně u 8 % pacientů léčených přípravkem NeoRecormon se mohou vyskytnout nežádoucí účinky.
Anemičtí pacienti s chronickým selháním ledvin
Nejčastějším nežádoucím účinkem v průběhu léčby přípravkem NeoRecormon je zvýšení krevního tlaku nebo zhoršení stávající hypertenze, zvláště pak v případech rychlého zvýšení hodnot hematokritu (viz bod 4.4). Hypertenzní krize s příznaky encefalopatie (např. bolesti hlavy a stavy zmatenosti, senzoricko-motorické poruchy jako například porucha řeči nebo zhoršení chůze až tonicko-klonické křeče) se mohou objevit také u jednotlivých pacientů s jinak normálním nebo nízkým krevním tlakem (viz bod 4.4).
Může dojít k trombóze žilního přístupu zejména u pacientů s tendencí k hypotenzi nebo s komplikacemi arteriovenózního zkratu (např. stenózy, aneuryzmata), viz bod 4.4. Ve většině případů je pozorován pokles hodnot sérového železa souběžně se vzestupem hematokritu (viz bod 4.4). Navíc byl v ojedinělých případech pozorován vzestup sérového draslíku a hladin fosfátů (viz bod 4.4).
V ojedinělých případech byla v souvislosti s léčbou přípravkem NeoRecormon hlášena čistá aplazie červených krvinek (PRCA) způsobená neutralizujícími antitrombopoetinovými protilátkami.
V případě diagnózy čisté aplazie červených krvinek (PRCA) způsobené neutralizujícími antitrombopoetinovými protilátkami musí být léčba přípravkem NeoRecormon ukončena a pacient nemá být převáděn na léčbu jinou erytropoetickou bílkovinou (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 1.
Pacienti s rakovinou
Bolesti hlavy související s léčbou epoetinem beta a hypertenze, kterou lze medikamentózně léčit, jsou časté (viz bod 4.4).
U některých pacientů je pozorován pokles sérového železa (viz bod 4.4).
V klinických studiích byl prokázán vyšší výskyt tromboembolických událostí u pacientů s karcinomem léčených přípravkem NeoRecormon ve srovnání s neléčenými kontrolními skupinami nebo placebem. U pacientů léčených přípravkem NeoRecormon byla incidence 7 % ve srovnání s 4 % u kontrolních skupin; aniž by to bylo spojeno se zvýšenou mortalitou na tromboembolické události ve srovnání s kontrolními skupinami.
Nežádoucí účinky jsou uvedeny níže v tabulce 2.
Pacienti v programu autologního dárcovství krve
U pacientů v programu autologního dárcovství krve byla hlášena lehce zvýšená četnost tromboembolických událostí. Kauzální vztah k léčbě přípravkem NeoRecormon však nebyl prokázán.
Ve studiích kontrolovaných placebem byl více vyjádřen přechodný deficit železa u pacientů léčených přípravkem NeoRecormon než u kontrolní skupiny (viz bod 4.4).
Nežádoucí účinky jsou uvedeny níže v tabulce 3.
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů MedDRA a absolutní četnosti. Kategorie četnosti jsou definovány dle následující konvence:
velmi časté (> 1/10); časté (>1/100 to <1/10); méně časté (>1/1 000 to <1/100); vzácné (>1/10 000 to <1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
Tabulka 1: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů s chronickým selháním ledvin léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze Hypertenzní krize |
Časté Méně časté |
|
Poruchy nervového systému |
Časté | |
|
Poruchy krve a |
Trombóza cévního |
Vzácné |
|
lymfatického systému |
přístupu Trombocytóza |
Velmi vzácné |
Tabulka 2: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u onkologických pacientů léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Cévní poruchy |
Hypertenze |
Časté |
|
Poruchy krve a lymfatického systému |
Tromboembolické účinky |
Časté |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
Tabulka 3: Nežádoucí účinky vyskytující se v kontrolovaných klinických studií u pacientů v programu autologního dárcovství krve léčených přípravkem NeoRecormon_
|
Třídy orgánových systémů |
Nežádoucí účinek |
Frekvence výskytu |
|
Onemocnění nervového systému |
Bolesti hlavy |
Časté |
Předčasně narozené děti
Pokles hodnot sérového ferritinu je velmi častý (viz bod 4.4).
Popis vybraných nežádoucích účinků
Vzácně byly pozorovány kožní reakce jako jsou vyrážka, svědění, kopřivka nebo reakce v místě vpichu. Velmi vzácně byly popsány anafylaktoidní reakce související s léčbou epoetinem beta. V kontrolovaných klinických studiích však zvýšení výskytu hypersenzitivních reakcí nebylo popsáno.
Ve velmi vzácných přípradech, především na počátku léčby, byl zaznamenán výskyt příznaků podobných chřipce ("flu-like" symptomy) jako je horečka, zimnice, bolesti hlavy, bolest v končetinách, malátnost a/nebo bolest kostí. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Údaje z kontrolované klinické studie s epoetinem alfa nebo darbepoetinem alfa udávaly incidenci cévní mozkové příhody jako častou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky,
aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 P ředávkování
Terapeutický rozsah přípravku NeoRecormon je velmi široký. Dokonce i při velmi vysokých hladinách v séru nebyly pozorovány žádné příznaky otravy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antianemikum, ATC kód: B03XA01 Mechanismus účinku
Erytropoetin je glykoprotein, který stimuluje tvorbu erytrocytů z prekurzorů kmenových buněk.
Působí jako faktor stimulující mitózu kmenových buněk červené krevní řady a hormon působící na jejich diferenciaci.
Epoetin beta, léčivá látka přípravku NeoRecormon, je svým složením aminokyselin a cukrů identický s erytropoetinem, který byl izolován z moči anemických pacientů.
Biologický účinek epoetinu beta byl demonstrován po intravenózní a podkožní aplikaci u různých pokusných zvířat in vivo (normální a uremičtí potkani, polycytemické myši, psi). Po podání epoetinu beta se zvýší počet erytrocytů, hodnota Hb a retikulocytů stejně tak jako rychlost inkorporace 59Fe.
In vitro byla zjištěna zvýšená inkorporace 3H-thymidinu v erytroidních jaderných buňkách sleziny (kultura buněk sleziny myší) po inkubaci s epoetinem beta.
Pokusy na tkáňových kulturách buněk lidské kostní dřeně prokázaly, že epoetin beta stimuluje jen tvorbu červených krvinek a neovlivňuje tvorbu bílých krvinek. Nebyly zjištěny cytotoxické účinky epoetinu beta na kostní dřeň ani na lidské kožní buňky.
Po podání jednotlivé dávky epoetinu beta nebyly pozorovány žádné vlivy na chování nebo na pohybovou činnost u myší a oběhové nebo respirační funkce u psů.
Klinická účinnost a bezpečnost
V randomizované dvojitě zaslepené, placebem kontrolované studii, která hodnotila 4038 pacientů s chronickým selháním ledvin bez nutnosti dialýzy, s diabetem 2. typu a hladinami hemoglobinu < 11 g/dl, byli pacienti léčeni buď darbepoetinem alfa až do dosažení cílových hladin hemoglobinu 13 g/dl, nebo dostávali placebo (viz bod 4.4). Studie nesplnila žádný z primárních cílů, který by prokázal snížení rizika úmrtí ze všech příčin, kardiovaskulární morbidity nebo terminálního stádia onemocnění ledvin (ESRD). Analýza jednotlivých komponent složených cílových parametrů prokázala následující poměr rizik (95% CI): úmrtí 1,05 (o,92; 1,21), cévní mozková příhoda 1,92 (1,38; 2,68), městnavé srdeční selhání 0,89 (0,74; 1,08), infarkt myokardu 0,96 (0,75; 1,23), hospitalizace z důvodu ischemie myokardu 0,84 (0,55; 1,27), terminální stádium onemocnění ledvin 1,02 (0,87; 1,18).
Souhrnné post-hoc analýzy klinických studií s ESA byly provedeny u pacientů s chronickým selháním ledvin (u pacientů, kteří byli nebo nebyli na dialýze, u diabetiků a u pacientů bez diabetu). Byla pozorována tendence směrem ke zvýšení odhadovaného rizika mortality z jakýchkoli důvodů, rizika kardiovaskulárních a cerebrovaskulárních příhod spojených s vyššími kumulativními dávkami ESA, nezávisle na stavu diabetu nebo dialýze (viz body 4.2 a 4.4).
Erytropoetin je růstový faktor primárně podporující tvorbu červených krvinek. Receptory pro erytropoetin mohou být exprimovány na povrchu mnoha nádorových buněk.
Přežití a progrese nádoru byla zkoumána v pěti velkých kontrolovaných klinických studiích zahrnujících celkem 2833 pacientů; čtyři z těchto studií byly dvojitě zaslepené kontrolované placebe m, jedna studie byla otevřená. Dvou studií se účastnili pacienti léčení chemoterapií. Cílová koncentrace hemoglobinu byla ve dvou studiích > 13 g/dl; v ostatních třech studiích 12-14 g/dl.
V otevřené studii nebyl z hlediska celkového přežití zjištěn žádný rozdíl mezi pacienty léčenými rekombinantním lidským erytropoetinem a mezi kontrolní skupinou. Ve čtyřech placebem kontrolovaných studiích se poměry rizik pro celkové přežití pohybovaly mezi 1,25 a 2,47 ve prospěch kontrolních skupin. Při srovnání s kontrolními skupinami ukázaly tyto studie konzistentní nevysvětlené statisticky významné zvýšení mortality u pacientů, kteří měli anémii spojenou s různými běžnými maligními nádory a kteří dostávali rekombinantní lidský erytropoetin. Výsledek celkového přežití zjištěný v klinických studiích nebylo možné uspokojivě vysvětlit rozdílem v incidenci trombózy a přidružených komplikací mezi skupinou dostávající rekombinantní lidský erytropoetin a mezi skupinou kontrolní.
Metaanalýza založená na údajích jednotlivých pacientů zahrnovala data ze všech 12 kontrolovaných klinických studií prováděných u pacientů s anémií a maligním nádorovým onemocněním, kteří byli léčeni přípravkem NeoRecormon (n=2301), ukázala celkový odhadovaný poměr rizik pro přežití 1,13 ve prospěch kontrol (95% CI 0,87; 1,46). U pacientů, kteří měli v baseline hladinu hemoglobinu <
10 g/dl (n=899), byl odhadovaný poměr rizik pro přežití 0,98 (95% CI 0,68 až 1,40). V celkové populaci bylo pozorováno zvýšené relativní riziko tromboembolických příhod (RR 1,62, 95% CI:
1,13; 2,31).
Byla provedena analýza dat na úrovni jednotlivých nemocných u více než 13 900 pacientů se zhoubným nádorem (léčených chemoterapií, radioterapií, chemoradioterapií nebo bez terapie) zařazených do 53 kontrolovaných klinických studií zahrnujících podávání několika epoetinů. Metaanalýza údajů celkového přežití prokázala, že poměr rizik (hazard ratio) je odhadem 1,06 ve prospěch kontrolních skupin (95% CI: 1,00, 1,12; 53 studií a 13 933 pacientů) a u pacientů se zhoubným nádorem podstupujících chemoterapii byl poměr rizik (hazard ratio) celkového přežití 1,04 (95% CI: 0,97, 1,11; 38 studií a 10 441 pacientů). Meta-analýzy zároveň konzistentně poukazují na významné zvýšení relativního rizika tromboembolických příhod u pacientů se zhoubným nádorem, kteří dostávají rekombinantní lidský erytropoetin (viz bod 4.4).
Ve velmi vzácných případech se v průběhu léčby rekombinantním lidským erytropoetinem objevily neutralizující protilátky proti erytropoetinu s nebo bez čisté aplazie červené krevní řady (PRCA).
5.2 Farmakokinetické vlastnosti
Farmakokinetické sledování u zdravých dobrovolníků a uremických pacientů ukázalo, že po intravenózním podání je poločas epoetinu beta mezi 4 - 12 hodinami a že distribuční objem odpovídá jedno- až dvojnásobku plazmatického objemu. Analogické výsledky byly nalezeny v pokusech na zvířatech u normálních i uremických potkanů.
Protrahovaná absorpce epoetinu beta po subkutánním podání uremickým pacientům vede ke vzniku koncentračního plató, přičemž maximální koncentrace je dosaženo v průměru za 12 - 28 hodin. Terminální poločas je delší než po intravenózním podání s průměrem 13 - 28 hodin.
Ve srovnání s intravenózní aplikací je biologická dostupnost epoetinu beta po podkožní aplikaci mezi 23 - 42 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
Studie kancerogenity s homologním erytropoetinem u myší neodhalily žádné známky proliferativního nebo kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 S eznam pomocných látek
Močovina,
Chlorid sodný,
Polysorbát 20,
Dihydrogenfosforečnan sodný dihydrát,
Hydrogenfosforečnan sodný dodekahydrát,
Chlorid vápenatý dihydrát,
Glycin,
L-leucin,
L-isoleucin,
L-threonin,
L-kyselina glutamová,
L-fenylalanin.
Voda na injekci.
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku v původním obalu, aby byl přípravek chráněn před světlem.
Pro usnadnění ambulantního použití může pacient vyjmout přípravek z chladničky a uchovávat jej jednorázově po dobu maximálně 3 dnů při pokojové teplotě (maximálně do 25 °C).
6.5 Druh obalu a obsah balení
Předplněná injekční stříkačka (sklo typu I) s víčkem a zátkou (pryž potažená teflonem) a s injekční jehlou (27G1/2). Jedna injekční stříkačka obsahuje 0,6 ml roztoku.
Velikost balení po 1 předplněné injekční stříkačce s 1 jehlou nebo 4 předplněných injekčních stříkačkách se 4 jehlami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Nejdříve si dobře umyjte ruce!
1. Vyjměte jednu předplněnou injekční stříkačku z balení a zkontrolujte, zda roztok ve stříkačce je čirý, bezbarvý a bez viditelných částic. Odstraňte víčko z injekční stříkačky.
2. Vyjměte jednu injekční jehlu z balení, nasaďte ji na injekční stříkačku a odstraňte z jehly ochranné víčko.
3. Vytlačte vzduch z injekční stříkačky tím, že stříkačku držíte ve vzpřímené poloze a jemně tlačíte píst vzhůru. Tlačte na píst tak dlouho, dokud množství přípravku NeoRecormon v injekční stříkačce neodpovídá množství předepsanému pro aplikaci.
4. Za použití tampónu namočeného v alkoholu si očistěte kůži v místě vpichu. Vytvořte záhyb na kůži tím, že ji sevřete mezi palec a ukazováček. Stiskněte tělo injekční stříkačky v části bližší k jehle a aplikujte jehlu do záhybu kůže rychlým a pevným pohybem. Vstříkněte roztok přípravku NeoRecormon. Jehlu rychle vytáhněte a zatlačte na místo vpichu suchým a sterilním tampónem.
Tento léčivý přípravek je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/031/045 -046
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. července 1997
Datum posledního prodloužení registrace: 16. července 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY/BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY/BIOLOGICKÝCH
LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa vvrobce/vvrobců biologické léčivé látky/biologických léčivých látek
Roche Diagnostics GmbH Nonnenwald 2 D-82377 Penzberg Německo
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
NeoRecormon vícedávkový 50 000 IU lyofilizát a rozpouštědlo pro přípravu injekčního roztoku Epoetinum beta
1 injekční lahvička obsahuje 50 000 IU epoetinu beta.
1 injekční lahvička obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný, hydrogenfosforečnan sodný, chlorid vápenatý, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou a L-fenylalanin.
1 ampulka obsahuje rozpouštědlo (benzylalkohol a benzalkonium chlorid ve vodě na injekce)
Léčivý přípravek obsahuje fenylalanin a sodík a rozpouštědlo obsahuje benzylalkohol, pro další informace viz příbalová informace
Lyofilizát a rozpouštědlo pro přípravu injekčního roztoku
1 injekční lahvička s lyofilizátem (50 000 IU) pro přípravu injekčního roztoku, 1 ampulka s konzervačním rozpouštědlem (10 ml), 1 rekonstituční zařízení pro manipulaci při rozpouštění a nabírání roztoku, 1 jednorázová injekční stříkačka (10 ml), 1 jehla (21G2)
Subkutánní nebo intravenózní podání po rekonstituci dle návodu. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
Roztok po rekonstituci je použitelný po dobu 1 měsíce při uchovávání při teplotě 2 °C - 8 °C (v chladničce)
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/019
13. ČÍSLO ŠARŽE č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ (na vnitřní straně obalu)
Návod k přípravě a použití přípravku viz příbalová informace
Označení na stojánku NeoRecormon vícedávkový 50 000 IU 5000 IU/ml
Uchovávejte při +2°C až +8°C
Vždy dodržujte zásady aseptické aplikace
Pro každou dávku použijte sterilní stříkačku a jehlu
|
5000 IU/ml | |||||||||
|
500 IU |
1000IU |
1500IU |
2000 IU |
2500IU |
3000IU |
3500IU |
4000IU |
4500 IU |
5000IU |
|
0.1 ml |
0.2 ml |
0.3 ml |
0.4 ml |
0.5 ml |
0.6 ml |
0.7 ml |
0.8 ml |
0.9 ml |
1.0 ml |
neorecormon 50 000 IU
2D čárový kód s jedinečným identifikátorem.
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
NeoRecormon vícedávkový 50 000 IU lyofilizát na injekci
1. v./s.c.podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
EXP
Datum rekonstituce lili
EXP J_i_i_i_i_i_L
Po otevření smí být rekonstituovaný roztok uchován po dobu nejvýše 1 měsíce při teplotě 2 °C - 8 °C
|
4. |
ČÍSLO ŠARŽE |
|
Lot | |
|
5. |
OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET |
|
5000 IU/1 ml | |
|
6. |
JINÉ |
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
10 ml rozpouštědla pro NeoRecormon vícedávkový 50 000 IU (voda na injekce, benzylalkohol, benzalkonium-chlorid)
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
NeoRecormon 500 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 500 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
1 předplněná injekční stříkačka (0,3 ml) a 1 jehla (30G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/025
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 500 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
NeoRecormon 500 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 500 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
6 předplněných injekčních stříkaček (0,3 ml) a 6 jehel (30G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA)
EU/1/97/031/026
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMATION IN BRAILLE
neorecormon 500 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
NeoRecormon 500 IU injekční roztok v předplněné injekční stříkačce Epoetin beta
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU Nálepk a
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
NeoRecormon 500 IU injekce
1. v./s.c. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,3 ml
6. JINÉ
NeoRecormon 2000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 2000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
1 předplněná injekční stříkačka (0,3 ml) a 1 jehla (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/029
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 2000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
NeoRecormon 2000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 2000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
6 předplněných injekčních stříkaček (0,3 ml) a 6 jehel (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/030
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 2000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
NeoRecormon 2000 IU injekční roztok v předplněné injekční stříkačce Epoetin beta
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU Nálepk a
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
NeoRecormon 2000 IU injekce
1. v./s.c. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,3 ml
6. JINÉ
NeoRecormon 3000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 3000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
1 předplněná injekční stříkačka (0,3 ml) a 1 jehla (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/031
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 3000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
NeoRecormon 3000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 3000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
6 předplněných injekčních stříkaček (0,3 ml) a 6 jehel (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/032
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 3000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
NeoRecormon 3000 IU injekční roztok v předplněné injekční stříkačce Epoetin beta
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU Nálepk a
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
NeoRecormon 3000 IU injekce
1. v./s.c. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,3 ml
6. JINÉ
NeoRecormon 4000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 4000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
1 předplněná injekční stříkačka (0,3 ml) a 1 jehla (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/041
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 4000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
NeoRecormon 4000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 4000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
6 předplněných injekčních stříkaček (0,3 ml) a 6 jehel (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/042
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 4000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
NeoRecormon 4000 IU injekční roztok v předplněné injekční stříkačce Epoetin beta
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU Nálepk a
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
NeoRecormon 4000 IU injekce
1. v./s.c. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,3 ml
6. JINÉ
NeoRecormon 5000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 5000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
1 předplněná injekční stříkačka (0,3 ml) a 1 jehla (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/033
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 5000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
NeoRecormon 5000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 5000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
6 předplněných injekčních stříkaček (0,3 ml) a 6 jehel (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/034
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 5000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
NeoRecormon 5000 IU injekční roztok v předplněné injekční stříkačce Epoetin beta
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU Nálepk a
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
NeoRecormon 5000 IU injekce
1. v./s.c. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,3 ml
6. JINÉ
NeoRecormon 6000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 6000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
1 předplněná injekční stříkačka (0,3 ml) a 1 jehla (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/043
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 6000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
NeoRecormon 6000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 6000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
6 předplněných injekčních stříkaček (0,3 ml) a 6 jehel (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/044
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 6000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
NeoRecormon 6000 IU injekční roztok v předplněné injekční stříkačce Epoetin beta
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU Nálepk a
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
NeoRecormon 6000 IU injekce
1. v./s.c. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,3 ml
6. JINÉ
NeoRecormon 10 000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 10 000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
1 předplněná injekční stříkačka (0,6 ml) a 1 jehla (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/035
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 10 000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
NeoRecormon 10 000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 10 000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
6 předplněných injekčních stříkaček (0,6 ml) a 6 jehel (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/036
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 10 000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
NeoRecormon 10 000 IU injekční roztok v předplněné injekční stříkačce Epoetin beta
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU Nálepk a
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
NeoRecormon 10 000 IU injekce
1. v./s.c. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,6 ml
6. JINÉ
NeoRecormon 20 000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 20 000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
1 předplněná injekční stříkačka (0,6 ml) a 1 jehla (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/037
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 20 000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
NeoRecormon 20 000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 20 000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
6 předplněných injekčních stříkaček (0,6 ml) a 6 jehel (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/038
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 20 000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
NeoRecormon 20 000 IU injekční roztok v předplněné injekční stříkačce Epoetin beta
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU Nálepk a
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
NeoRecormon 20 000 IU injekce
1. v./s.c. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,6 ml
6. JINÉ
NeoRecormon 30 000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 30 000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
1 předplněná injekční stříkačka (0,6 ml) a 1 jehla (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/045
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 30 000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
NeoRecormon 30 000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
1 předplněná injekční stříkačka obsahuje 30 000 IU epoetinu beta.
1 injekční stříkačka obsahuje: Močovinu, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselinu glutamovou, L-fenylalanin a vodu na injekce.
Léčivý přípravek obsahuje fenylalanin a sodík, pro další informace viz příbalová informace.
Injekční roztok
4 předplněné injekční stříkačky (0,6 ml) a 4 jehly (27G1/2)
Subkutánní a intravenózní podání. Před použitím si přečtěte příbalovou informaci
Uchovávejte mimo dohled a dosah dětí
Použitelné do:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/97/031/046
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
neorecormon 30 000 IU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
NeoRecormon 30 000 IU injekční roztok v předplněné injekční stříkačce Epoetin beta
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU Nálepk a
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
NeoRecormon 30 000 IU injekce
1. v./s.c. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,6 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
NeoRecormon vícedávkový 50 000 IU lyofilizát a rozpouštědlo pro přípravu injekčního roztoku
Epoetinum beta
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoliv další otázky, zeptejte se svého lékaře nebo lékárníka.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek NeoRecormon a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek NeoRecormon používat
3. Jak se přípravek NeoRecormon používá
4. Možné nežádoucí účinky
5. Jak přípravek NeoRecormon uchovávat
6. Obsah balení a další informace
1. Co je přípravek NeoRecormon a k čemu se používá
Tato léková forma přípravku NeoRecormon obsahuje bílý lyofilizát a rozpouštědlo. Po rozpuštění je přípravek NeoRecormon aplikován prostřednictvím podkožní injekce (subkutánně) nebo nitrožilně
(intravenózně).
Přípravek obsahuje hormon nazývaný epoetin beta, který stimuluje tvorbu červených krvinek. Epoetin beta je vyráběn technikami genetického inženýrství a působí naprosto stejným způsobem jako přirozený hormon erytropoetin.
Informujte svého lékaře, pokud se nebudete cítit lépe nebo pokud se budete cítit hůře.
Přípravek NeoRecormon je indikován k:
• Léčbě symptomatické anémie spojené s chronickým selháním ledvin (renální anémie) u pacientů podstupujících dialýzu nebo u pacientů, kteří ještě nepodstupují dialýzu.
• Léčbě anémie a přidružených symptomů u dospělých pacientů se zhoubnými nádory, kteří jsou léčeni chemoterapií.
• Léčbě pacientů darujících vlastní krev před chirurgickým zákrokem. Injekce epoetinu beta umožní zvýšení objemu krve, která Vám může být odebrána před plánovaným chirurgickým zákrokem a kterou v případě potřeby dostanete zpět ve formě transfuze v průběhu operace nebo po jejím skončení (toto je nazýváno autologní transfuze).
2. Čemu musíte věnovat pozornost, než začnete přípravek NeoRecormon používat Nepoužívejte přípravek NeoRecormon
• jestliže jste alergický(á) na epoetin beta nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
• jestliže trpíte špatně kontrolovatelným vysokým tlakem
• pokud darujete vlastní krev před operací a:
• prodělal(a) jste v průběhu 1 měsíce před léčbou infarkt myokardu nebo cévní mozkovou příhodu
• trpíte nestabilní anginou pectoris - náhle vzniklá nebo zvyšující se bolest na hrudi
• je u Vás zvýšené riziko vzniku krevní sraženiny v cévách (hluboká venózní trombóza) například pokud jste prodělal(a) tromboembolické onemocnění žil.
• u kojenců nebo dětí do 3 let věku, neboť rozpouštědlo pro přípravek NeoRecormon vícedávkový obsahuje benzylalkohol jako konzervační látku.
Jestliže se Vás týká cokoliv z výše uvedeného, informujte svého lékaře.
Upozornění a opatření
Před užitím přípravku NeoRecormon informujte svého lékaře
• jestliže trpíte anémií, která se léčbou epoetinem beta nezlepší
• jestliže máte nedostatek některých B vitamínů (kyselina listová nebo vitamín B12)
• jestliže máte zvýšenou koncentraci hliníku v krvi
• jestliže máte vysoký počet krevních destiček
• jestliže trpíte chronickým onemocněním jater
• jestliže trpíte epilepsií
• jestliže u Vás došlo k rozvoji antierytropoetinových protilátek a čisté aplazie červené krevní řady (snížení nebo zastavení tvorby červených krevních buněk) v průběhu předchozí léčby přípravky obsahujícími erytropoetin.V tomto případě byste neměl(a) být léčen(a) přípravkem NeoRecormon.
Dbejte zvláštní opatrnosti při použití jiných přípravků, které stimulují tvorbu červených krvinek:
Přípravek NeoRecormon je jedním ze skupiny přípravků, které stimulují tvorbu červených krvinek podobně jako lidský protein erytropoetin. Váš lékař vždy přesně zaznamená přípravek, který je Vám podáván.
Zvláštní upozornění
Během léčby přípravkem NeoRecormon
Pokud jste pacient s chronickým onemocněním ledvin, a zejména pokud odpovídajícím způsobem nereagujete na léčbu přípravkem NeoRecormon, Váš lékař zkontroluje Vaši dávku přípravku NeoRecormon, neboť opakované zvyšování této dávky v případě, že neodpovídáte na léčbu, může zvýšit riziko problémů se srdcem nebo s krevními cévami a může také zvýšit riziko srdečního infarktu, cévní mozkové příhody a úmrtí.
Pokud máte nádorové onemocnění, měl(a) byste vědět, že přípravek NeoRecormon jako růstový faktor může, za některých okolností, toto Vaše onemocnění negativně ovlivnit. V závislosti na Vaší individuální situaci by mohla být upřednostněna léčba transfuzí krve. Prosím, poraďte se o tom se svým lékařem.
Pokud trpíte nefrosklerózou a ještě nepodstupujete dialýzu, rozhodne Váš lékař, zda je tento typ léčby pro Vás vhodný. Důvodem je možné riziko urychlení postupu selhání ledvin, které nelze s jistotou vyloučit.
Lékař Vám bude provádět pravidelná vyšetření krve, aby bylo možno zkontrolovat:
• koncentrace draslíku. Pokud máte vysoké nebo zvyšující se koncentrace draslíku, může lékař zvážit další způsob léčby.
• počet krevních destiček. V průběhu léčby epoetinem beta může dojít k mírnému až střednímu nárůstu počtu krevních destiček, který může ovlivnit srážlivost krve.
Pokud podstupujete dialýzu, může Váš lékař rozhodnout o úpravě dávky heparinu. Tím zabrání možnému zablokování systému dialýzy.
Jestliže trpíte onemocněním ledvin a podstupujete dialýzu, je u Vás riziko trombózy cévního přístupu, kdy může dojít k nahromadění krevních sraženin (trombů) v cévním přístupu (céva užívaná k
propoje ní se systémem dialýzy). Lékař Vám proto může předepsat kyselinu acetylsalicylovou nebo rozhodnout o úpravě cévního přístupu.
Pokud darujete vlastní krev před operací, bude třeba, aby Váš lékař:
• posoudil, zda můžete krev darovat, zvláště vážíte-li méně než 50 kg
• posoudil, zda máte dostatečný počet červených krevních buněk (hodnota hemoglobinu minimálně 11 g/dl)
• zajistil, že množství jednorázově darované krve nepřekročí 12 % celkového objemu Vaší krve Nezneužívejte přípravek NeoRecormon
Zneužití přípravku NeoRecormon zdravými osobami může vést k nadměrnému zvýšení počtu krevních buněk a následně ke zhoustnutí krve. Tento stav může být následně spojen s život ohrožujícími srdečními nebo cévními komplikacemi.
Další léčivé přípravky a přípravek NeoRecormon
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, a to i o lécích, které jsou dostupné bez lékařského předpisu.
Těhotenství, kojení a plodnost
Není mnoho zkušeností s podáváním přípravku NeoRecormon ženám v průběhu těhotenství nebo ženám, které kojí. Požádejte svého lékaře nebo lékárníka o radu předtím, než začnete užívat jakýkoliv lék.
Nebyl prokázán dopad přípravku NeoRecormon na plodnost u zvířat. Možné riziko u lidí není známo. Řízení dopravních prostředků a obsluha strojů
Nebyl zaznamenán žádný vliv na schopnost řídit motorová vozidla a obsluhovat stroje.
Přípravek NeoRecormon obsahuje fenylalanin, benzylalkohol a sodík.
Tento lék obsahuje fenylalanin. Pro osoby trpící fenylketonurií může být škodlivý. Trpíte-li fenylketonurií, informujte svého lékaře v případě, že zvažuje léčbu přípravkem NeoRecormon.
Přípravek NeoRecormon obsahuje až 40 mg benzylalkoholu jako konzervační látky v ampulce s rozpouštědlem, a proto nesmí být podán kojencům a dětem do 3 let.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v dávce, tj. v podstatě je „bez sodíku“
3. Jak se přípravek NeoRecormon používá
Léčbu přípravkem NeoRecormon zahájí lékař, který dobře zná Váš zdravotní stav. První dávka Vám bude podána pod lékařským dohledem z důvodu možného vzniku alergické reakce.
Injekci přípravku NeoRecormon může podat zdravotní sestra nebo lékař. (Viz Návod k použití na konci této příbalové informace)
Dávkování přípravku NeoRecormon
Dávky přípravku NeoRecormon závisí na stavu Vašeho onemocnění, způsobu podání injekce (podkožně nebo nitrožilně) a Vaší tělesné hmotnosti. Váš lékař stanoví Vaši správnou dávku.
Váš lékař použije nejnižší účinnou dávku ke kontrole příznaků anémie.
Pokud odpovídajícím způsobem nereagujete na léčbu přípravkem NeoRecormon, lékař bude kontrolovat Vaši dávku přípravku NeoRecormon a bude Vás informovat v případě, že bude třeba změna dávky přípravku NeoRecormon.
• Symptomatická anémie způsobená chronickým selháním ledvin
Injekce jsou podávány podkožně nebo nitrožilně. V případě nitrožilního podání má být přípravek podáván alespoň po dobu 2 minut, např. přes arterio-venózní píštěl na konci dialýzy u pacientů na hemodialýze.
Nedialyzovaným pacientům jsou injekce obvykle podávány podkožně.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází:
a) Úprava anémie
Úvodní dávka u podkožního podání je 20 IU v jedné injekci na každý 1 kg Vaší tělesné hmotnosti, podávaná třikrát týdně.
Po 4 týdnech lékař provede testy a pokud odpověď na léčbu není uspokojivá, dávka může být zvýšena až na 40 IU/kg v jedné injekci, podávané třikrát týdně. Pokud je to nezbytné, může lékař v měsíčních intervalech pokračovat ve zvyšování dávky.
Týdenní dávka může být také rozdělena na denní dávky.
Úvodní dávka u nitrožilního podání je 40 IU v jedné injekci na každý 1 kg Vaší tělesné hmotnosti, podávaná třikrát týdně.
Po 4 týdnech lékař provede testy a pokud odpověď na léčbu není uspokojivá, dávka může být zvýšena až na 80 IU/kg v jedné injekci, podávané třikrát týdně. Pokud je to nezbytné, může lékař v měsíčních intervalech pokračovat ve zvyšování dávky.
Týdenní dávka může být také rozdělena na denní dávky.
Pro oba způsoby podání nemá maximální dávka překročit 720 IU na každý 1 kg Vaší tělesné hmotnosti týdně.
b) Udržení uspokojivých hladin buněk červené krevní řady
Udržovací dávka: Jakmile bylo dosaženo uspokojivé hladiny buněk červené krevní řady, je dávka snížena na polovinu původní dávky nutné k úpravě anémie. Týdenní dávka může být podána jednou týdně, nebo rozdělena do tří nebo sedmi dávek týdně. Pokud je dosaženo stabilní hladiny buněk červené krevní řady při dávkovacím režimu jednou týdně, je možno přistoupit k podání dávky jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Jednou týdně nebo jednou za dva týdny může lékař přistoupit k úpravě dávky, aby určil Vaši osobní udržovací dávku.
Děti mohou být léčeny dle výše uvedeného dávkování. V klinických studiích bylo prokázáno, že děti obvykle potřebovaly vyšší dávky přípravku NeoRecormon (čím mladší dítě, tím vyšší dávka).
Léčba přípravkem NeoRecormon je obvykle dlouhodobá. Nicméně, pokud je třeba, může být kdykoliv přerušena.
• Dospělí pacienti se symptomatickou anémií a se zhoubnými nádory Injekce jsou podávány podkožně.
Váš lékař může zahájit léčbu přípravkem NeoRecormon, jestliže Vaše hladina hemoglobinu je 10 g/dl nebo nižší. Po zahájení léčby bude Váš lékař udržovat hladinu Vašeho hemoglobinu mezi 10 až 12 g/dl.
Úvodní týdenní dávka je 30 000 IU. Tato dávka může být podána jednou injekcí jednou týdně nebo v rozdělených dávkách ve 3 až 7 injekcích za týden. Lékař Vám bude provádět pravidelné kontroly krevních vzorků. Na základě výsledků těchto testů může přistoupit ke zvýšení nebo snížení dávky, nebo přerušit léčbu. Hladiny hemoglobinu by neměly překročit hodnotu 12 g/dl.
Léčba má pokračovat po dobu následujících 4 týdnů od ukončení chemoterapie.
Maximální dávka nemá přesáhnout 60 000 IU týdně.
• Pacienti darující vlastní krev před chirurgickým zákrokem
Injekce jsou aplikovány nitrožilně po dobu minimálně 2 minut, nebo podkožně.
Dávka přípravku NeoRecormon je závislá na Vašem zdravotním stavu, množství buněk červené krevní řady a množství krve, které Vám bude odebráno před operací.
Dávka přípravku určená lékařem Vám bude podávána dvakrát týdně po dobu 4 týdnů. Přípravek NeoRecormon Vám bude podán v okamžiku ukončení odběru krve.
Maximální dávka nemá přesáhnout
• u nitrožilního podání: 1600 IU na 1 kg Vaší tělesné hmotnosti týdně
• u podkožního podání: 1200 IU na 1 kg Vaší tělesné hmotnosti týdně.
Jestliže jste použil(a) více přípravku NeoRecormon, než jste měl(a)
Nezvyšujte dávku určenou lékařem. Jestliže se domníváte, že jste si aplikoval(a) více přípravku NeoRecormon, než jste měl(a), obraťte se na svého lékaře. Je nepravděpodobné, že by Váš stav byl závažný. Dokonce i při velmi vysokých hladinách v krvi nebyly pozorovány žádné příznaky otravy.
Jestliže jste zapomněl(a) použít přípravek NeoRecormon
Jestliže jste si zapomněl(a) aplikovat injekci nebo jste si aplikoval(a) nižší dávku, poraďte se se svým lékařem.
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Nežádoucí účinky, které se mohou vyskytnout u kteréhokoliv pacienta
• Většina pacientů (velmi časté nežádoucí účinky mohou postihnout více než 1 pacienta z 10) má snížené koncentrace železa v krvi. Téměř všichni pacienti dostávali v průběhu léčby NeoRecormonem doplňky obsahující železo.
• Vzácně (mohou postihnout až 1 pacienta z 1 000) se vyskytovaly alergické nebo kožní reakce, jako je vyrážka nebo kopřivka, nebo reakce v místě vpichu injekce.
• Velmi vzácně (mohou postihnout až 1 pacienta z 10 000) došlo k výskytu závažných alergických reakcí, především bezprostředně po podání injekce. V tomto případě je třeba okamžitá léčba. Pokud u Vás dojde k neobvyklému sípavému dýchání nebo se Vám těžce dýchá, máte oteklý jazyk, tvář nebo krk, nebo otok v místě vpichu; pociťujete lehkou bolest hlavy nebo máte pocit slabosti nebo v případě, že omdlíváte, je nezbytné ihned zavolat lékaře.
• Velmi vzácně (mohou postihnout až 1 pacienta z 10 000) někteří pacienti pociťovali příznaky podobné chřipce, především na počátku léčby. Tyto příznaky většinou zahrnovaly horečku, zimnici, bolesti hlavy, bolest v končetinách, bolest kostí a/nebo pocit celkové slabosti. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Další n ežádoucí účinky u pacientů s chronickým onemocněním ledvin (renální anémií)
• Zvýšení krevního tlaku, zhoršení již existujícího zvýšeného krevního tlaku a bolest hlavy
patřily mezi nejčastěji se vyskytující nežádoucí účinky (velmi časté nežádoucí účinky mohou p ostihnout více než 1 pacienta z 10). Lékař bude pravidelně kontrolovat Váš krevní tlak, především na počátku léčby. Váš lékař bude zvýšený krevní tlak léčit odpovídajícími léky nebo může dočasně přerušit léčbu přípravkem NeoRecormon.
• Ihned kontaktujte svého lékaře, jakmile pocítíte bolest hlavy, zvláště pokud je bolest náhlá, bodavá a připomíná migrénu, pokud máte pocit zmatenosti, poruchu řeči, nejistou chůzi, návaly nebo křeče. Toto mohou být příznaky závažného zvýšení krevního tlaku (hypertenzní krize), a to i přesto, že máte krevní tlak normální nebo nízký. V tomto případě je nezbytné zahájit okamžitou léčbu.
• Pokud máte nízký krevní tlak nebo problémy s cévním přístupem, může u Vás být zvýšené riziko trombózy cévního přístupu (vznik krevních sraženin a ucpání cévy, která slouží k napojení na systém dialýzy).
• Velmi vzácně (mohou postihnout až 1 pacienta z 10 000) byly u některých pacientů zaznamenány zvýšené koncentrace draslíku nebo fosfátů v krvi. Váš lékař Vás v tomto případě bude odpovídajícím způsobem léčit.
• V průběhu léčby erytropoetinem byla zaznamenána čistá aplazie buněk červené krevní řady (PRCA) způsobená neutralizujícími protilátkami, včetně hlášení ojedinělých případů při léčbě přípravkem NeoRecormon. PRCA znamená, že se ve Vašem těle zastavila nebo snížila tvorba červených krvinek. Toto způsobí závažnou anémii, doprovázenou příznaky zahrnujícími neobvyklou únavu a nedostatek energie. Jestliže jsou ve Vašem těle vytvářeny neutralizující protilátky, ošetřující lékař přeruší léčbu přípravkem NeoRecormon a určí nejvhodnější postup léčby Vaší anémie.
Další nežádoucí účinky u dospělých pacientů podstupujících chemoterapeutickou léčbu zhoubných nádorů
• V některých případech může dojít ke zvýšení krevního tlaku a bolesti hlavy. Zvýšení krevního tlaku bude upraveno léky předepsanými Vaším lékařem.
• Byl zaznamenán nárůst výskytu krevních sraženin.
Další nežádoucí účinky u dospělých pacientů darujících vlastní krev před chirurgickým zákrokem
• Byl zaznamenán mírný nárůst výskytu krevních sraženin.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek NeoRecormon uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Přípravek NeoRecormon nepoužívejte po uplynutí doby použitelnosti uvedené na krabičce a nálepce injekční lahvičky.
• Uchovávejte v chladničce (2 °C - 8 °C).
• Injekční lahvička může být vyjmuta z chladničky a uchována jednorázově po dobu maximálně 5 dnů při pokojové teplotě (do 25 °C).
• Připravený roztok může být uložen v chladničce při 2 °C - 8 °C po dobu jednoho měsíce.
• Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
• Léčivé přípravky se nesmí vyhazovat do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak máte likvidovat přípravky, které již nepotřebujete. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek NeoRecormon obsahuje
• Léčivou látkou je epoetinum beta. Jedna injekční lahvička obsahuje 50 000 IU (mezinárodních jednotek) epoetinu beta.
• Dalšími složkami jsou:
V lyofilizátu: močovina, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný, hydrogenfosforečnan sodný, chlorid vápenatý, glycin, L-leucin, L-isoleucin, L-threonin, L-kyselina glutamová a L-fenylalanin.
• V rozpouštědle: benzylalkohol a benzalkonium-chlorid jako konzervační látky a vodu na injekci.
Jak přípravek NeoRecormon vypadá a co obsahuje toto balení
Přípravek NeoRecormon vícedávkový je lyofilizát a ampulka s rozpouštědlem pro přípravu injekčního roztoku. Lyofilizát je bílý a rozpouštědlo je čiré a bezbarvé.
Balení obsahuje 1 injekční lahvičku s 50 000 IU epoetinu beta, 1 ampulku obsahující 10 ml rozpouštědla, 1 rekonstituční zařízení pro manipulaci při rozpouštění a nabírání roztoku, 1 jehlu (21G2) a 1 jednorázovou injekční stříkačku.
Držitel rozhodnutí o registraci
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
Výrobce
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
N.V. Roche S.A.
Tél/Tel: +32 (0) 2 525 82 11
Eunrapnu
Pom Etnrapua EOOfl Ten: +359 2 818 44 44
Česká republika
Roche s. r. o.
Tel: +420 -2 20382111
Danmark
Roche a/s
Tlf: +45 - 36 39 99 99
Lietuva
UAB “Roche Lietuva”
Tel: +370 5 2546799
Luxembourg/Luxemburg
(Voir/siehe Belgique/Belgien)
Magyarország
Roche (Magyarország) Kft. Tel: +36 - 23 446 800
Malta
(See United Kingdom)
|
Deutschland Roche Pharma AG Tel: +49 (0) 7624 140 |
Nederland Roche Nederland B.V. Tel: +31 (0) 348 438050 |
|
Eesti Roche Eesti OU Tel: + 372 -6 177 380 |
Norge Roche Norge AS Tlf: +47 - 22 78 90 00 |
|
EXXáSa Roche (Hellas) A.E. Tn^: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
Espaňa Roche Farma S.A. Tel: +34 - 91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 -22 345 18 88 |
|
France Roche Tél: +33 (0)1 47 61 40 00 |
Portugal Roche Farmaceutica Química, Lda Tel: +351 -21 425 70 00 |
|
Hrvatska Roche d.o.o Tel: +385 1 4722 333 |
Románia Roche Románia S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska družba d.o.o. Tel: +386 - 1 360 26 00 |
|
Ísland Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 -2 52638201 |
|
Italia Roche S.p.A. Tel: +39 -039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
|
Kúrcpog r.A.Zxa^áxn? & £ra AxS. Tn^: +357 - 22 76 62 76 |
Sverige Roche AB Tel: +46 (0) 8 726 1200 |
|
Latvija Roche Latvija SIA Tel: +371 -6 7039831 |
United Kingdom Roche Products Ltd. Tel: +44 (0) 1707 366000 |
Tato příbalová informace byla naposledy revidována {měsíc RRRR}.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace je určena pouze pro zdravotnické pracovníky
NeoRecormon vícedávkový je přípravek vtzv. mnohodávkové formě, tzn. že během 1 měsíce po rekonstituci lze z 1 lahvičky odebrat několik jednotlivých dávek pro několik pacientů. Aby bylo zabráněno riziku přenosu infekce, měl(a) byste vždy dodržovat aseptické podmínky a pro každé použití používat vždy sterilní jednorázové injekční stříkačky a jehly. Prosím, ujistěte se, že je vždy připravena pro použití (tj. rekonstituována) pouze jedna injekční lahvička léčivého přípravku NeoRecormon vícedávkový.
Nemíchejte NeoRecomon s jinými injekčními nebo infuzními roztoky.
Pro injekci používejte pouze umělohmotné materiály.
Návod k použití
Nejprve si umyjte ruce!
Příprava roztoku přípravku NeoRecormon vícedávkový
(1) Vyjměte lahvičku s lyofilizátem z obalu. Označte štítek datem rekonstituce a datem ukončení doby použitelnosti (expirace), což je přesně 1 měsíc po rekonstituci.
(2) Sejměte víčko z umělé hmoty z injekční lahvičky.
(3) Dezinfikujte gumovou zátku alkoholem.
(4) Vyjměte z obalu speciální rekonstituční zařízení pro rozpouštění a odběr (umožňuje sterilní výměnu vzduchu) a odstraňte ochrannou čepičku ze vpichovacího trnu.
(5) Nasaďte tento uzávěr na lahvičku a protlačte ho gumovou zátkou. Uzávěr musí zaskočit.
(6) Nasaďte zelenou jehlu na přibalenou stříkačku a odstraňte kryt jehly.
(7) Podržte OPC (One-Point-Cut) ampulku modrou tečkou dopředu. Protřepejte ampulkou nebo na ni poklepejte, aby vytekla všechna kapalina ze zúženého místa dolů. Ulomte ampulku v místě jejího zúžení směrem dozadu. Nasajte všechnu kapalinu do stříkačky. Dezinfikujte gumovou zátku zařízení alkoholem.
(8) Propíchněte jehlu zátkou asi do hloubky 1 cm a vstříkněte pomalu všechno rozpouštědlo do injekční lahvičky. Poté odstraňte injekční stříkačku s jehlou ze zařízení.
(9) Lehce otáčejte lahvičkou,až se lyofilizát rozpustí. Netřepejte. Ujistěte se, že je roztok čirý, bezbarvý a bez jakýchkoli částic. Pokud ne, injekci neaplikujte. Nasaďte ochrannou čepičku nahoru na rekonstituční zařízení.
(10) Před a po rekonstituci musí být lahvička s přípravkem NeoRecormon vícedávkový uchovávána v chladničce při 2°C až 8° C.
Příprava jednotlivé dávky
(1) Dezinfikujte gumovou zátku uzávěru rekonstitučního zařízení alkoholem před každým odběrem.
(2) Nasaďte jehlu 26G pevně na dodanou jednorázovou stříkačku (obsah 1 ml).
(3) Odstraňte kryt z jehly a propíchněte jehlu pryžovou zátkou rekonstitučního zařízení. Nasajte roztok přípravku NeoRecormon do stříkačky a vypusťte vzduch ze stříkačky do injekční lahvičky a upravte množství roztoku přípravku NeoRecormon ve stříkačce na předepsanou dávku. Odstraňte injekční stříkačku (s jehlou) z rekonstitučního zařízení.
(4) Jehlu nahraďte novou jehlou (nová jehla má mít stejnou velikost jako jehla, kterou běžně používáte pro aplikaci injekce).
(5) Sejměte ochranný kryt z jehly a pečlivě odstraňte z jehly vzduch tím, že podržíte stříkačku svisle vzhůru a budete lehce tlačit píst nahoru, až vystoupí kapka kapaliny ze špičky jehly.
Před podkožní injekcí očistěte kůži v místě vpichu tampónem s alkoholem. Pomocí palce a ukazováku utvořte kožní řasu. Uchopte stříkačku blízko jehly a rychle vpíchněte jehlu do kůže. Injikujte roztok s přípravkem NeoRecormon. Rychle vytáhněte jehlu a přitlačte na místo vpichu suchý sterilní tampón.
Příbalová informace: informace pro uživatele NeoRecormon 500 IU NeoRecormon 2 000 IU NeoRecormon 3 000 IU NeoRecormon 4 000 IU NeoRecormon 5 000 IU NeoRecormon 6 000 IU NeoRecormon 10 000 IU NeoRecormon 20 000 IU NeoRecormon 30 000 IU injekční roztok v předplněné injekční stříkačce Epoetinum beta
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat,
protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li případně jakékoliv další otázky, zeptejte se svého lékaře nebo lékárníka.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek NeoRecormon a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek NeoRecormon používat
3. Jak se přípravek NeoRecormon používá
4. Možné nežádoucí účinky
5. Jak se přípravek NeoRecormon uchovává
6. Obsah balení a další informace 1. Co je přípravek NeoRecormon a k čemu se používá
Přípravek NeoRecormon je čirý, bezbarvý roztok pro podkožní (subkutánní) nebo nitrožilní
(intravenózní) injekci. Přípravek obsahuje léčivou látku epoetin beta, hormon stimulující tvorbu
červených krevních buněk. Epoetin beta je vyráběn technikami genetického inženýrství a působí
naprosto stejným způsobem jako přirozený hormon erytropoetin.
Informujte svého lékaře, pokud se nebudete cítit lépe nebo pokud se budete cítit hůře.
Přípravek NeoRecormon je indikován k:
• Léčbě symptomatické anémie spojené s chronickým selháním ledvin (renální anémie) u pacientů podstupujících dialýzu nebo u pacientů, kteří ještě nepodstupují dialýzu.
• Prevenci anémie u předčasně narozených dětí (vážících 750 až 1500 g a narozených před 34. týdnem těhotenství).
• Léčbě anémie a přidružených symptomů u dospělých pacientů se zhoubnými nádory, kteří jsou léčeni chemoterapií.
• Léčbě pacientů darujících vlastní krev před chirurgickým zákrokem. Injekce epoetinu beta umožní zvýšení objemu krve, která Vám může být odebrána před plánovaným chirurgickým zákrokem a kterou v případě potřeby dostanete zpět ve formě transfuze v průběhu operace nebo po jejím skončení (toto je nazýváno autologní transfuze).
2. Čemu musíte věnovat pozornost, než začnete přípravek NeoRecormon používat Nepouž ívejte přípravek NeoRecormon
• jestliže jste alergický(á) na epoetin beta nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
• jestliže trpíte špatně kontrolovatelným vysokým tlakem
• pokud darujete vlastní krev před operací a:
• prodělal(a) jste v průběhu 1 měsíce před léčbou infarkt myokardu nebo cévní mozkovou příhodu
• trpíte nestabilní anginou pectoris - náhle vzniklá nebo zvyšující se bolest na hrudi
• je u Vás zvýšené riziko vzniku krevní sraženiny v cévách (hluboká venózní trombóza) například pokud jste prodělal(a) tromboembolické onemocnění žil.
Jestliže se Vás týká nebo může týkat cokoliv z výše uvedeného, informujte ihned svého lékaře.
Upozornění a opatření
Před užitím přípravku NeoRecomon informujte svého lékaře
• jestliže Vaše předčasně narozené dítě potřebuje léčbu přípravkem NeoRecormon, bude pečlivě monitorováno pro případ možných poruch oka
• jestliže trpíte anémií, která se léčbou epoetinem beta nezlepší
• jestliže máte nedostatek některých B vitamínů (kyselina listová nebo vitamín B12)
• jestliže máte zvýšenou koncentraci hliníku v krvi
• jestliže máte vysoký počet krevních destiček
• jestliže trpíte chronickým onemocněním jater
• jestliže trpíte epilepsií
• jestliže u Vás došlo k rozvoji antierytropoetinových protilátek a čisté aplazie červené krevní řady (snížení nebo zastavení tvorby červených krevních buněk) v průběhu předchozí léčby přípravky obsahujícími erytropoetin. V tomto případě byste neměl(a) být léčen(a) přípravkem NeoRecormon.
Dbejte zvláštní opatrnosti při použití jiných přípravků, které stimulují tvorbu červených krvinek:
Přípravek NeoRecormon je jedním ze skupiny přípravků, které stimulují tvorbu červených krvinek podobně jako lidský protein erytropoetin. Váš lékař vždy přesně zaznamená přípravek, který je Vám podáván.
Zvláštní upozornění
Během léčby přípravkem NeoRecormon
Pokud jste pacient s chronickým onemocněním ledvin, a zejména pokud odpovídajícím způsobem nereagujete na léčbu přípravkem NeoRecormon, Váš lékař zkontroluje Vaši dávku přípravku NeoRecormon, neboť opakované zvyšování této dávky v případě, že neodpovídáte na léčbu, může zvýšit riziko problémů se srdcem nebo s krevními cévami a může také zvýšit riziko srdečního infarktu, cévní mozkové příhody a úmrtí.
Pokud máte nádorové onemocnění, měl(a) byste vědět, že přípravek NeoRecormon jako růstový faktor může, za některých okolností, toto Vaše onemocnění negativně ovlivnit. V závislosti na Vaší individuální situaci by mohla být upřednostněna léčba transfuzí krve. Prosím, poraďte se o tom se svým lékařem.
Pokud trpíte nefrosklerózou a ještě nepodstupujete dialýzu, rozhodne Váš lékař, zda je tento typ léčby pro Vás vhodný. Důvodem je možné riziko urychlení postupu selhání ledvin, které nelze s jistotou vyloučit.
Lékař Vám bude provádět pravidelná vyšetření krve, aby bylo možno zkontrolovat:
• koncentrace draslíku. Pokud máte vysoké nebo zvyšující se koncentrace draslíku, může lékař zvážit další způsob léčby.
• počet krevních destiček. V průběhu léčby epoetinem beta může dojít k mírnému až střednímu nárůstu počtu krevních destiček, který může ovlivnit srážlivost krve.
Pokud podstupujete dialýzu, může Váš lékař rozhodnout o úpravě dávky heparinu. Tím zabrání možnému zablokování systému dialýzy.
Jestliže trpíte onemocněním ledvin a podstupujete dialýzu, je u Vás riziko trombózy cévního přístupu, kdy může dojít k nahromadění krevních sraženin (trombů) v cévním přístupu (céva užívaná k propojení se systémem dialýzy). Lékař Vám proto může předepsat kyselinu acetylsalicylovou nebo rozhodnout o úpravě cévního přístupu.
Pokud darujete vlastní krev před operací, bude třeba, aby Váš lékař:
• posoudil, zda můžete krev darovat, zvláště vážíte-li méně než 50 kg
• posoudil, zda máte dostatečný počet červených krevních buněk (hodnota hemoglobinu minimálně 11 g/dl)
• zajistil, že množství jednorázově darované krve nepřekročí 12 % celkového objemu Vaší krve Nezneužívejte přípravek NeoRecormon
Zneužití přípravku NeoRecormon zdravými osobami může vést k nadměrnému zvýšení počtu krevních buněk a následně ke zhoustnutí krve. Tento stav může být následně spojen s život ohrožujícími srdečními nebo cévními komplikacemi.
Další léčivé přípravky a přípravek NeoRecormon
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, a to i o lécích, které jsou dostupné bez lékařského předpisu.
Těhotenství, kojení a plodnost
Není mnoho zkušeností s podáváním přípravku NeoRecormon ženám v průběhu těhotenství nebo ženám, které kojí. Požádejte svého lékaře nebo lékárníka o radu předtím, než začnete užívat jakýkoliv lék.
Nebyl prokázán dopad přípravku NeoRecormon na plodnost u zvířat. Možné riziko u lidí není známo. Řízení dopravních prostředků a obsluha strojů
Nebyl zaznamenán žádný vliv na schopnost řídit motorová vozidla a obsluhovat stroje.
Přípravek NeoRecormon obsahuje fenylalanin a sodík.
Tento lék obsahuje fenylalanin. Pro osoby trpící fenylketonurií může být škodlivý. Trpíte-li fenylketonurií, informujte svého lékaře v případě, že zvažuje léčbu přípravkem NeoRecormon.
Tento léčivý přípravek obsahuje v jedné dávce méně než 1 mmol (23 mg) sodíku, tj. v podstatě je „bez sodíku“.
3. Jak se přípravek NeoRecormon používá
Léčbu přípravkem NeoRecormon zahájí lékař, který dobře zná Váš zdravotní stav. První dávka Vám bude podána pod lékařským dohledem z důvodu možného vzniku alergické reakce.
Injekci přípravku NeoRecormon může podat zdravotní sestra nebo lékař. Poté, co absolvujete instruktáž, můžete si injekci aplikovat sám(a).
Přípravek NeoRecormon v předplněné injekční stříkačce je připraven k použití. Jedna předplněná injekční stříkačka by měla být použita pouze jednorázově. Nemíchejte přípravek NeoRecormon s jinými injekčními nebo infuzními roztoky.
Návod k použití
Nejdříve si umyjte ruce!
1. Vyjměte jednu předplněnou injekční stříkačku z balení.
Zkontrolujte tekutinu v předplněné injekční stříkačce:
• je čirá?
• je bezbarvá?
• je bez viditelných částic?
Jestliže odpověď na některou z otázek je NE, neaplikujte injekci.
Injekční stříkačku zlikvidujte předepsaným způsobem a začněte znovu s jinou předplněnou injekční stříkačkou.
Jestliže odpověď na všechny tři otázky zní ano, sejměte víčko z předplněné injekční stříkačky a pokračujte krokem 2.
2. Vyjměte jehlu z balení, nasaďte ji bezpečně na předplněnou injekční stříkačku a sejměte ochranné víčko z jehly.
3. Vytlačte vzduch z předplněné injekční stříkačky a jehly. Toto proveďte lehkým poklepáváním na horní část předplněné injekční stříkačky. Všechny vzduchové bubliny vystoupají do horní části. Poté držte předplněnou injekční stříkačku ve svislé poloze jehlou vzhůru a jemně stlačte píst směrem vzhůru. Držte píst stlačený do té doby, než množství přípravku NeoRecormon v předplněné injekční stříkačce nedosáhne předepsané dávky.
4. Otřete kůži v místě vpichu tampónem s alkoholem. Vytvořte záhyb na kůži tím, že ji sevřete mezi palec a ukazováček.
5. Uchopte předplněnou injekční stříkačku v části bližší k jehle, vpíchněte jehlu do záhybu kůže rychlým a pevným pohybem.Vstříkněte roztok přípravku NeoRecormon. Jehlu rychle vytáhněte a zatlačte na místo vpichu suchým a sterilním tampónem.
Dávkování přípravku NeoRecormon
Dávky přípravku NeoRecormon závisí na stavu Vašeho onemocnění, způsobu podání injekce (pod kůži nebo do žíly) a Vaší tělesné hmotnosti. Váš lékař stanoví Vaši správnou dávku.
Váš lékař použije nejnižší účinnou dávku ke kontrole příznaků anémie.
Pokud odpovídajícím způsobem nereagujete na léčbu přípravkem NeoRecormon, lékař bude kontrolovat Vaši dávku přípravku NeoRecormon a bude Vás informovat v případě, že bude třeba změna dávky přípravku NeoRecormon.
• Symptomatická anémie způsobená chronickým selháním ledvin
Injekce jsou podávány podkožně nebo nitrožilně. Pokud je injekce aplikována do žíly, má být podávána po dobu minimálně 2 minut, např. u pacientů na dialýze, kteří budou dostávat injekci přes arterio-venózní píštěl na konci dialýzy.
Pacientům, kteří nepodstupují dialýzu, budou injekce podávány podkožně.
Léčba přípravkem NeoRecormon je rozdělena do dvou fází: a) Úprava anémie
Úvodní dávka pro podkožní injekci je 20 IU v jedné injekci na každý 1 kg Vaší tělesné hmotnosti, podávaná třikrát týdně.
Po 4 týdnech lékař provede testy a pokud odpověď na léčbu není uspokojivá, dávka může být zvýšena až na 40 IU/kg v jedné injekci, podávané třikrát týdně. Pokud je to nezbytné, může lékař v měsíčních intervalech pokračovat ve zvyšování dávky.
Týdenní dávka může být také rozdělena na denní dávky.
Úvodní dávka pro nitrožilní injekci je 40 IU v jedné injekci na 1 kg Vaší tělesné hmotnosti, podávaná třikrát týdně.
Po 4 týdnech lékař provede testy a pokud odpověď na léčbu není uspokojivá, dávka může být zvýšena až na 80 IU/kg v jedné injekci, podávané třikrát týdně. Pokud je to nezbytné, může lékař v měsíčních intervalech pokračovat ve zvyšování dávky.
Pro oba způsoby podání injekce by maximální dávka neměla překročit 720 IU na 1 kg Vaší tělesné hmotnosti za týden.
b) Udržení uspokojivých hladin buněk červené krevní řady
Udržovací dávka: Jakmile bylo dosaženo uspokojivé hladiny buněk červené krevní řady, je dávka snížena na polovinu původní dávky nutné k úpravě anémie. Týdenní dávka může být podána jednou týdně, nebo rozdělena do tří nebo sedmi dávek týdně. Pokud je dosaženo stabilní hladiny buněk červené krevní řady při dávkovacím režimu jednou týdně, je možno přistoupit k podání dávky jednou za dva týdny. V tomto případě může být nezbytné zvýšení dávky.
Jednou týdně nebo jednou za dva týdny může lékař přistoupit k úpravě dávky, aby určil Vaši osobní udržovací dávku.
Děti mohou být léčeny dle výše uvedeného dávkování. V klinických studiích bylo prokázáno, že děti obvykle potřebovaly vyšší dávky přípravku NeoRecormon (čím mladší dítě, tím vyšší dávka).
Léčba přípravkem NeoRecormon je obvykle dlouhodobá. Nicméně, pokud je třeba, může být kdykoliv přerušena.
• Anémie u předčasně narozených dětí Injekce jsou podávány podkožně.
Úvodní dávka je 250 IU na 1 kg tělesné hmotnosti dítěte, podávaná třikrát týdně.
U předčasně narozených dětí, u nichž byla provedena transfuze před zahájením léčby přípravkem NeoRecormon, není pravděpodobný takový prospěch léčby jako u dětí, u nichž nebyla transfuze provedena.
Doporučená délka léčby je 6 týdnů.
• Dospělí pacienti se symptomatickou anémií a se zhoubnými nádory Injekce jsou podávány podkožně.
Váš lékař může zahájit léčbu přípravkem NeoRecormon, jestliže Vaše hladina hemoglobinu je 10 g/dl nebo nižší. Po zahájení léčby bude Váš lékař udržovat hladinu Vašeho hemoglobinu mezi 10 až 12 g/dl.
Úvodní týdenní dávka je 30 000 IU. Tato dávka může být podána jednou injekcí jednou týdně nebo v rozdělených dávkách ve 3 až 7 injekcích za týden. Lékař Vám bude provádět pravidelné kontroly krevních vzorků. Na základě výsledků těchto testů může přistoupit ke zvýšení nebo snížení dávky, nebo přerušit léčbu. Hladiny hemoglobinu nemají překročit hodnotu 12 g/dl.
Léčba má pokračovat po dobu 4 týdnů po skončení chemoterapie.
Maximální dávka nemá přesáhnout 60 000 IU týdně.
• Pacienti darující vlastní krev před chirurgickým zákrokem Injekce jsou aplikovány nitrožilně po dobu 2 minut nebo podkožně.
Dávka přípravku NeoRecormon je závislá na Vašem zdravotním stavu, množství buněk červené krevní řady a množství krve, které Vám bude odebráno před operací.
Dávka přípravku NeoRecormon určená lékařem Vám bude podávána dvakrát týdně po dobu 4 týdnů. Dávka Vám bude podána v okamžiku ukončení odběru krve.
Maxim ální dávka nemá přesáhnout
• při nitrožilním podání: 1600 IU na 1 kg Vaší tělesné hmotnosti týdně
• při podkožním podání: 1200 IU na 1 kg Vaší tělesné hmotnosti týdně.
Jestliže jste použil(a) více přípravku NeoRecormon, než jste měl(a)
Nezvyšujte dávku určenou lékařem. Jestliže se domníváte, že jste si aplikoval(a) více přípravku NeoRecormon, než jste měl(a), obraťte se na svého lékaře. Je nepravděpodobné, že by Váš stav byl závažný. Dokonce i při velmi vysokých hladinách v krvi nebyly pozorovány žádné příznaky otravy.
Jestliže jste zapomněl(a) použít přípravek NeoRecormon
Jestliže jste si zapomněl(a) aplikovat injekci nebo jste si aplikoval(a) nižší dávku, poraďte se se svým lékařem.
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Nežádoucí účinky, které se mohou vyskytnout u kteréhokoliv pacienta
• Většina pacientů (velmi časté nežádoucí účinky mohou postihnout více než 1 pacienta z 10) má snížené koncentrace železa v krvi. Téměř všichni pacienti dostávali v průběhu léčby přípravkem NeoRecormon doplňky obsahující železo.
• Vzácně (mohou postihnout až 1 pacienta z 1 000) se vyskytovaly alergické nebo kožní reakce, jako je vyrážka nebo kopřivka, svědění nebo reakce v místě vpichu injekce.
• Velmi vzácně (mohou postihnout až 1 pacienta z 10 000) došlo k výskytu závažných alergických reakcí, především bezprostředně po podání injekce. V tomto případě je třeba okamžitá léčba. Pokud u Vás dojde k neobvyklému sípavému dýchání nebo se Vám těžce dýchá, máte oteklý jazyk, tvář nebo krk, nebo otok v místě vpichu; pociťujete lehkou bolest hlavy nebo máte pocit slabosti nebo v případě, že omdlíváte, je nezbytné ihned zavolat lékaře.
• Velmi vzácně (mohou postihnout až 1 pacienta z 10 000) někteří pacienti pociťovali příznaky podobné chřipce, především na počátku léčby. Tyto příznaky většinou zahrnovaly horečku, zimnici, bolesti hlavy, bolest v končetinách, bolest kostí a/nebo pocit celkové slabosti. Tyto příznaky byly mírné až střední intenzity a odezněly v průběhu několika hodin nebo dnů.
Další nežádoucí účinky u pacientů s chronickým onemocněním ledvin (renální anémií)
• Zvýšení krevního tlaku, zhoršení již existujícího zvýšeného krevního tlaku a bolest hlavy
patřily mezi nejčastěji se vyskytující nežádoucí účinky (velmi časté nežádoucí účinky mohou postihnout více než 1 pacienta z 10). Lékař bude pravidelně kontrolovat Váš krevní tlak, především na počátku léčby. Váš lékař bude zvýšený krevní tlak léčit odpovídajícími léky nebo může dočasně přerušit léčbu přípravkem NeoRecormon.
• Ihned kontaktujte svého lékaře, jakmile pocítíte bolest hlavy, zvláště pokud je bolest náhlá, bodavá a připomíná migrénu, pokud máte pocit zmatenosti, poruchu řeči, nejistou chůzi, návaly nebo křeče. Toto mohou být příznaky závažného zvýšení krevního tlaku (hypertenzní krize), a to i přesto, že máte krevní tlak normální nebo nízký. V tomto případě je nezbytné zahájit okamžitou léčbu.
• Pokud máte nízký krevní tlak nebo problémy s cévním přístupem, může u Vás být zvýšené riziko trombózy cévního přístupu (vznik krevních sraženin a ucpání cévy, která slouží k napojení na systém dialýzy).
• Velmi vzácně (mohou postihnout až 1 pacienta z 10 000) byly u některých pacientů zaznamenány zvýšené koncentrace draslíku nebo fosfátů v krvi. Váš lékař Vás v tomto případě bude odpovídajícím způsobem léčit.
• V průběhu léčby erytropoetinem byla zaznamenána čistá aplazie buněk červené krevní řady (PRCA) způsobená neutralizujícími protilátkami, včetně hlášení ojedinělých případů při léčbě přípravkem NeoRecormon. PRCA znamená, že se ve Vašem těle zastavila nebo snížila tvorba červených krvinek. Toto způsobí závažnou anémii, doprovázenou příznaky zahrnujícími neobvyklou únavu a nedostatek energie. Jestliže jsou ve Vašem těle vytvářeny neutralizující protilátky, ošetřující lékař přeruší léčbu přípravkem NeoRecormon a určí nejvhodnější postup léčby Vaší anémie.
Další nežádoucí účinky u dospělých pacientů podstupujících chemoterapeutickou léčbu zhoubných nádorů
• V některých případech může dojít ke zvýšení krevního tlaku a bolesti hlavy.
Zvýšení krevního tlaku bude upraveno léky předepsanými Vaším lékařem.
• Byl zaznamenán nárůst výskytu krevních sraženin.
Další nežádoucí účinky u dospělých pacientů darujících vlastní krev před chirurgickým zákrokem
• Byl zaznamenán mírný nárůst výskytu krevních sraženin.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak se přípravek NeoRecormon uchovává
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Přípravek NeoRecormon nepoužívejte po uplynutí doby použitelnosti uvedené na krabičce a nálepce injekční stříkačky.
• Uchovávejte v chladničce (2 °C - 8 °C).
• Injekční stříkačka může být vyjmuta z chladničky a uchována jednorázově po dobu maximálně 3 dnů při pokojové teplotě (do 25 °C).
• Předplněné injekční stříkačky uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
• Léky by neměly být likvidovány prostřednictvím odpadní vody ani v domácím odpadu. Zeptejte se svého lékárníka, jak likvidovat léky, které už dále nepotřebujete. Toto opatření pomáhá chránit životní prostředí.
6. Obsah balení a další informace Co přípravek NeoRecormon obsahuje
• Léčivou látkou je epoetinum beta. Jedna předplněná injekční stříkačka obsahuje 500, 2 000,
3 000, 4 000, 5 000, 6 000, 10 000, 20 000 nebo 30 000 IU (mezinárodních jednotek) epoetinu beta v 0,3 ml nebo 0,6 ml roztoku.
• Dalšími složkami jsou močovina, chlorid sodný, polysorbát 20, dihydrogenfosforečnan sodný
dihydrát, hydrogenfosforečnan sodný dodekahydrát, chlorid vápenatý dihydrát, glycin, L-leucin, L -isoleucin, L-threonin, L-kyselina glutamová a L-fenylalanin a voda na injekce.
Jak přípravek NeoRecormon vypadá a co obsahuje toto balení
Přípravek NeoRecormon je injekční roztok v předplněné injekční stříkačce.
Roztok je bezbarvý, čirý až lehce opalescentní.
Přípravek NeoRecormon 500 IU, 2000 IU, 3000 IU, 4000 IU, 5000 IU a 6000 IU: Jedna předplněná injekční stříkačka obsahuje 0,3 ml roztoku.
Přípravek NeoRecormon 10 000 IU, 20 000 IU a 30 000 IU: Jedna předplněná injekční stříkačka obsahuje 0,6 ml roztoku.
Přípravek NeoRecormon je dodáván v následujících velikostech balení:
NeoRecormon 500 IU
1 předplněná injekční stříkačka s 1 jehlou (30G1/2) nebo 6 předplněných injekčních stříkaček se 6 jehlami (30G1/2).
NeoRecormon 2000 IU, 3000 IU, 4000 IU, 5000 IU, 6000 IU, 10 000 IU a 20 000 IU 1 předplněná injekční stříkačka s 1 jehlou (27G1/2) nebo 6 předplněných injekčních stříkaček se 6 jehlami (27G1/2).
NeoRecormon 30 000 IU
1 předplněná injekční stříkačka s 1 jehlou (27G1/2) nebo 4 předplněné injekční stříkačky se 4 jehlami (27G1/2).
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
Výrobce
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
N.V. Roche S.A.
Tél/Tel: +32 (0) 2 525 82 11
Bt^rapuu
Pom EtnrapHH EOOfl Ten: +359 2 818 44 44
Česká republika
Roche s. r. o.
Tel: +420 -2 20382111
Lietuva
UAB “Roche Lietuva”
Tel: +370 5 2546799
Luxembourg/Luxemburg
(Voir/siehe Belgique/Belgien)
Magyarország
Roche (Magyarország) Kft. Tel: +36 - 23 446 800
|
Danmark Roche a/s Tlf: +45 - 36 39 99 99 |
Malta (See United Kingdom) |
|
Deutschland Roche Pharma AG Tel: +49 (0) 7624 140 |
Nederland Roche Nederland B.V. Tel: +31 (0) 348 438050 |
|
Eesti Roche Eesti OU Tel: + 372 -6 177 380 |
Norge Roche Norge AS Tlf: +47 - 22 78 90 00 |
|
EXXáSa Roche (Hellas) A.E. Tn^: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
Espaňa Roche Farma S.A. Tel: +34 - 91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 -22 345 18 88 |
|
France Roche Tél: +33 (0)1 47 61 40 00 |
Portugal Roche Farmaceutica Química, Lda Tel: +351 -21 425 70 00 |
|
Hrvatska Roche d.o.o Tel: +385 1 4722 333 |
Románia Roche Románia S.R.L. Tel: +40 21 206 47 01 |
Ireland Slovenija
Roche Products (Ireland) Ltd. Roche farmacevtska družba d.o.o
Tel: +353 (0) 1 469 0700 Tel: +386 - 1 360 26 00
|
Ísland Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 -2 52638201 |
|
Italia Roche S.p.A. Tel: +39 -039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
|
Kúrcpog r.A.Zxa^áxn? & £ra AxS. Tn^: +357 - 22 76 62 76 |
Sverige Roche AB Tel: +46 (0) 8 726 1200 |
|
Latvija Roche Latvija SIA Tel: +371 -6 7039831 |
United Kingdom Roche Products Ltd. Tel: +44 (0) 1707 366000 |
Tato příbalová informace byla naposledy revidována {měsíc RRRR}.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http ://www.ema.europa.eu
214