Mepact 4 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
MEPACT 4 mg, prášek pro koncentrát pro infuzní disperzi
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje mifamurtidum 4 mg*.
Po rekonstituci obsahuje jeden ml suspenze v injekční lahvičce mifamurtidum 0,08 mg. *plně syntetický analog součásti buněčné stěny bakterie Mycobacterium sp.
Úplný seznam pomocných látek, viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro koncentrát pro infuzní disperzi.
Bílý až téměř bílý homogenní lyofylizovaný koláč nebo prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek MEPACT je indikován u dětí, dospívajících a mladých dospělých k léčbě resekovatelného osteosarkomu vysokého stupně bez metastáz po makroskopicky kompletní chirurgické resekci. Používá se v kombinaci s pooperační chemoterapií sestávající z více léčiv. Bezpečnost a účinnost byla hodnocena ve studiích u pacientů, jimž byla počáteční diagnóza stanovena mezi 2. a 30. rokem věku (viz bod 5.1).
4.2 Dávkování a způsob podání
Léčbu mifamurtidem by měli zahajovat a sledovat specializovaní lékaři zkušení v diagnostice a léčbě osteosarkomu.
Dávkování
Doporučená dávka mifamurtidu pro všechny pacienty je 2 mg/m2 tělesného povrchu. Tato dávka by měla být podávána jako adjuvantní léčba následující po resekci: dvakrát týdně s odstupem nejméně 3 dnů po dobu 12 týdnů a dále jedenkrát týdně po dobu dalších 24 týdnů, takže celkové podané množství je 48 infuzí za 36 týdnů.
Dospělí >30 let
Žádnému z pacientů, kteří byli léčeni v rámci studií týkajících se osteosarkomu, nebylo 65 či více let a do randomizované studie fáze III byli zahrnuti pouze pacienti do 30 let. Neexistuje tedy dostatek údajů, aby mohlo být používání přípravku MEPACT doporučeno u pacientů starších 30 let.
Porucha funkce ledvin nebo jater
Nebyly prokázány žádné klinicky významné účinky lehké až středně těžké poruchy funkce ledvin (clearance kreatininu (CrCL) >30 ml/min) nebo poruchy funkce jater (Child-Pugh třída A nebo B) na farmakokinetiku mifamurtidu, proto není pro tyto pacienty třeba úprava dávky.
Protože variabilita farmakokinetiky mifamurtidu je však u pacientů se středně těžkou poruchou funkce jater větší (viz bod 5.2) a údaje o bezpečnosti jsou u pacientů se středně těžkou poruchou funkce jater omezené, doporučuje se při podávání mifamurtidu pacientům se středně těžkou poruchou funkce jater opatrnost.
Protože nejsou dostupné žádné údaje týkající se farmakokinetiky mifamurtidu u pacientů s těžkou poruchou funkce ledvin nebo jater, doporučuje se při podávání mifamurtidu těmto pacientům opatrnost.
Pokud je mifamurtid používán déle než chemoterapie, doporučuje se kontinuální sledování funkce ledvin a jater, dokud není skončena veškerá léčba.
Pediatrická populace <2 roky
Bezpečnost a účinnost mifamurtidu u dětí od 0 do 2 let věku nebyla stanovena. Nejsou k dispozici žádné údaje.
Způsob podání
Přípravek MEPACT musí být před podáním rekonstituován, přefiltrován pomocí přiloženého filtru a dále naředěn. Rekonstituovaná, přefiltrovaná a naředěná infuzní suspenze je homogenní, bílá až téměř bílá neprůhledná lipozomální suspenze bez viditelných částic a bez pěny a tukových hrudek.
Po rekonstituci, přefiltrování přes přiložený filtr a dalším ředění je přípravek MEPACT podáván pomocí intravenózní infuze v průběhu 1 hodiny.
Přípravek MEPACT nesmí být podáván jako bolusová injekce.
Další instrukce týkající se rekonstituce, filtrování přes přiložený filtr a ředění před podáním viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
Současné užívání cyklosporinu nebo jiných inhibitorů kalcineurinu (viz bod 4.5).
Současné užívání vysokých dávek nesteroidních protizánětlivých léků (NSAID, inhibitory cyklooxygenázy) (viz bod 4.5).
4.4 Zvláštní upozornění a opatření pro použití
Dechová nedostatečnost
U pacientů s anamnézou astmatu nebo jiného chronického obstrukčního plicního onemocnění, by mělo být zváženo profylaktické podávání bronchodilatačních přípravků. U dvou pacientů, u kterých se již dříve projevovalo astma, se rozvinula v souvislosti s léčbou lehká až středně závažná dechová nedostatečnost (viz bod 4.8). V případě výskytu těžké dechové nedostatečnosti se má podávání mifamurtidu přerušit a zahájit odpovídající léčba.
Neutropenie
V souvislosti s podáváním mifamurtidu se běžně vyskytovala přechodná neutropenie, obvykle v případě, že byl přípravek užíván současně s chemoterapií. Epizody neutropenické horečky by měly být sledovány a adekvátně léčeny. Mifamurtid může být podáván během období neutropenie, ale následná horečka připisovaná této léčbě by měla být přísně sledována. Mělo by být vyšetřeno, zda horečka či třesavka, která přetrvává po více než 8 hodin po podání mifamurtidu, není způsobena sepsí.
Zánětlivá reakce
Spojení podávání mifamurtidu se známkami výrazné zánětlivé reakce, včetně perikarditidy a pleuritidy, bylo méně časté. Přípravek by měl být používán s opatrností u pacientů s anamnézou autoimunitního, zánětlivého nebo jiného onemocnění kolagenu. Během podávání mifamurtidu by měly být u pacientů sledovány neobvyklé známky či symptomy, jako je artritida nebo synovitida, které mohou naznačovat nekontrolované zánětlivé reakce.
Kardiovaskulární poruchy
Pacienti s anamnézou žilní trombózy, vaskulitidy nebo nestabilních kardiovaskulárních onemocnění by měli být během podávání mifamurtidu přísně sledováni. Pokud příznaky přetrvávají nebo se zhoršují, podávání přípravku by mělo být odloženo nebo přerušeno. U zvířat, jimž byly podávány velmi vysoké dávky, bylo pozorováno krvácení. Při podávání doporučené dávky se krvácení neočekává, doporučuje se však vyšetřit parametry krevní srážlivosti po první dávce a pak ještě jednou po podání několika dávek.
Alergické reakce
V souvislosti s léčbou mifamurtidem se objevily příležitostné alergické reakce, které zahrnovaly vyrážku, dušnost a hypertenzi stupně 4 (viz bod 4.8). Může být obtížné odlišit alergické reakce od výrazných zánětlivých reakcí, ale u pacientů by měl být sledován výskyt známek alergických reakcí.
Gastrointestinální toxicita
Nauzea, zvracení a ztráta chuti k jídlu jsou velmi časté nežádoucí účinky při podávání mifamurtidu (viz bod 4.8). Gastrointestinální toxicita se může zhoršovat, je-li mifamurtid používán v kombinaci s vysokými dávkami chemoterapie skládající se z více léčiv a byla-li spojena se zvýšeným používáním parenterální výživy.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Byl proveden jen omezený počet studií interakcí mifamurtidu s chemoterapií. Ačkoliv tyto studie nejsou konečné, neexistuje důkaz, že by mifamurtid ovlivňoval protinádorové účinky chemoterapie a naopak.
Doporučuje se časově oddělit podávání mifamurtidu a doxorubicinu či jiných lipofilních léčivých přípravků, jsou-li používány ve stejném režimu chemoterapie.
Používání mifamurtidu současně s cyklosporinem či dalšími inhibitory kalcineurinu je kontraindikováno kvůli jejich předpokládanému účinku na fagocytární funkci makrofágů a mononukleárních buněk ve slezině (viz bod 4.3).
In vitro bylo také prokázáno, že vysoké dávky NSAID (inhibitorů cyklooxygenázy) mohou blokovat aktivující účinek lipozomálního mifamurtidu na makrofágy. Užívání vysokých dávek NSAID je proto kontraindikováno (viz bod 4.3).
Protože mifamurtid působí přes stimulaci imunitního systému, mělo by být během léčby mifamurtidem vyloučeno chronické nebo pravidelné užívání kortikosteroidů.
Studie interakcí in vitro ukázaly, že lipozomální ani nelipozomální mifamurtid neinhibuje ve skupině lidských jaterních mikrozomů metabolickou aktivitu cytochromu P450. Lipozomální ani nelipozomální mifamurtid neindukuje v primárních kulturách čerstvě izolovaných lidských hepatocytů metabolickou aktivitu ani transkripci cytochromu P450. Neočekává se tedy, že by mifamurtid ovlivňoval metabolismus látek, které jsou substráty jaterního cytochromu P450.
Ve velké kontrolované randomizované studii nezvyšoval mifamurtid používaný v doporučených dávkách a podle doporučeného rozvrhu toxicitu dalších současně používaných přípravků se
známou toxicitou pro ledviny (cisplatina, ifosfamid) nebo játra (vysoké dávky methotrexátu, ifosfamid), a nebylo tudíž nutné přizpůsobovat dávku mifamurtidu.
4.6 Fertilita, těhotenství a kojení
O použití mifamurtidu u těhotných žen nejsou k dispozici žádné údaje. Pokud jde o reprodukční toxicitu, jsou údaje ze studií na zvířatech nedostačující (viz bod 5.3). Podávání mifamurtidu se v průběhu těhotenství a u žen v reprodukčním věku, které neužívají účinnou antikoncepci, nedoporučuje.
Kojení
Není známo, zda se mifamurtid vylučuje do mateřského mléka. Vylučování mifamurtidu do mléka nebylo sledováno u zvířat. Při rozhodování, zda pokračovat v kojení/přerušit kojení nebo pokračovat v léčbě/přerušit léčbu, je třeba vzít v úvahu přínos kojení pro dítě a přínos léčby mifamurtidu pro ženu.
Fertilita
S mifamurtidem nebyly provedeny žádné studie zaměřené na fertilitu (viz bod 5.3).
4.7 Účinky na schopnosti řídit a obsluhovat stroje
Nebyly provedeny žádné studie sledující vliv tohoto přípravku na schopnost řídit a obsluhovat stroje. Velmi časté a časté nežádoucí účinky léčby mifamurtidem (například točení hlavy, závratě, únava a rozmazané vidění) mohou mít dopad na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Mifamurtid byl studován jako samostatný přípravek u 248 pacientů s většinou pokročilými nádorovými onemocněními během časných jednoramenných klinických studií fáze I a II. Nejčastějšími nežádoucími účinky, které se projevily u >50 % pacientů, byly třesavka, horečka, únava, nauzea, tachykardie a bolesti hlavy. O mnoha velmi často udávaných nežádoucích účincích, jak jsou uvedeny v následující souhrnné tabulce, se předpokládá, že souvisejí s mechanismem účinku mifamurtidu (viz tabulka 1). Většina těchto příhod byla hlášena jako mírná nebo středně závažná. Tento profil je stejný, ať již bereme v úvahu všechny časné studie (n = 248) nebo pouze studie u osteosarkomu (n = 51). Je pravděpodobné, že tyto nežádoucí účinky se objevily také v uvedené velké randomizované studii, ale nebyly zaznamenány, protože u této studie byly shromažďovány pouze závažné a život ohrožující nežádoucí účinky.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky jsou řazeny podle třídy orgánových systémů a frekvence. Skupiny četností jsou stanoveny podle následující konvence: Velmi časté (>1/10), časté (> 1/100 až < 1/10). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 1. Nežádoucí účinky související s přípravkem MEPACT u > 1/100 pacientů
|
Třída oraánových systémů |
Nežádoucí účinky (preferované názvy) |
|
Infekce a infestace | |
|
Časté |
Sepse, Celulitida, Zánět nosohltanu, Infekce v místě zavedení katetru, Infekce horních dýchacích cest, Infekce močových cest, |
|
Faryngitida, Infekce virem Herpes simplex | |
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy) | |
|
Časté |
Nádorová bolest |
|
Poruchy krve a lymfatického systému | |
|
Velmi časté | |
|
Časté |
Leukopenie, Trombocytopenie, Granulocytopenie, Febrilní neutropenie |
|
Poruchy metabolismu a výživy | |
|
Velmi časté | |
|
Časté |
Dehydratace, Hypokalémie, Snížená chuť k jídlu |
|
Psychiatrické poruchy | |
|
Časté | |
|
Poruchy nervového systému | |
|
Velmi časté |
Bolest hlavy, Závrať |
|
Časté |
Parestézie, Hypestézie, Třes, Ospalost, Letargie |
|
Poruchy oka | |
|
Časté |
Rozmazané vidění |
|
Poruchy ucha a labyrintu | |
|
Časté |
Závratě, Tinitus, Ztráta sluchu |
|
Srdeční poruchy | |
|
Velmi časté | |
|
Časté |
Cyanóza, Palpitace |
|
Cévní poruchy | |
|
Velmi časté |
Hypertenze, Hypotenze |
|
Časté |
Flebitida, Návaly, Bledost |
|
Respirační, hrudní a mediastinální poruchy | |
|
Velmi časté | |
|
Časté |
Pleurální výpotek, Zhoršení dušnosti, Produktivní kašel, Hemoptýza, Sípání, Epistaxe, Námahová dušnost , kongesce sinusů, nosní kongesce, Bolest v hltanu a hrtanu |
|
Gastrointestinalní poruchy | |
|
Velmi časté |
Zvracení, Průjem, Zácpa, Bolest břicha, Nauzea |
|
Časté |
Bolest v horní části břicha, Dyspepsie, Abdominální distenze, Bolest v dolní části břicha |
|
Poruchy jater a žlučových cest | |
|
Časté |
Bolest v oblasti jater |
|
Poruchy kůže a podkožní tkáně | |
|
Velmi časté |
Hyperhidróza |
|
Časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Velmi časté |
Myalgie, Arthralgie, Bolest zad, Bolest končetin |
|
Časté |
Svalové spasmy, Bolest krku, Bolest v tříslech, Bolest kostí, Bolest ramene, Bolest v hrudní stěně, Muskuloskeletální ztuhlost |
|
Poruchy ledvin a močových cest | |
|
Časté |
Hematurie, Dysurie, Polakisurie |
|
Poruchy reprodukčního systému a prsu | |
|
Časté |
Dysmenorea |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté |
Horečka, Třesavka, Únava, Hypotermie, Bolest, Malátnost, Astenie, Bolest na hrudi |
|
Časté |
Periferní otoky, Otoky, Zánět sliznic, Erytém v místě podání infuze, Reakce v místě podání infúze, Bolest v místě zavedení katetru, Tlak na hrudi, Pocit chladu |
|
Vyšetření | |
|
Časté |
Snížení tělesné hmotnosti |
Chirurgické a léčebné postupy
Časté
Bolest po léčebném zákroku
Popis vybraných nežádoucích účinků
Poruchy krve a lymfatického systému
Anémie byla hlášena velmi často, pokud byl mifamurtid používán současně s chemoterapeutickými látkami. V randomizované kontrolované studii byla incidence myeloidních malignit (akutní myeloidní leukémie/myelodysplastický syndrom) stejná u pacientů, kteří byli léčeni přípravkem MEPACT plus chemoterapií, jako u pacientů, kteří byli léčeni pouze chemoterapií (2,1 %).
Poruchy metabolismu a výživy
Anorexie (21 %) byla hlášena velmi často ve studiích fáze I a II s mifamurtidem.
Poruchy nervového systému
Shodně s dalšími generalizovanými příznaky, byly velmi častými poruchami nervového systému bolest hlavy (50 %) a závratě (17 %). Jeden pacient ve studii fáze III prodělal dvě epizody epileptického záchvatu stupně 4 během léčby chemoterapií a mifamurtidem. Druhá epizoda zahrnovala opakované záchvaty typu grand mal v průběhu několika dní. V léčbě mifamurtidem bylo pokračnováno po zbývající část studie bez opětovného výskytu záchvatů.
Poruchy ucha a labyrintu
Ačkoliv ztrátu sluchu lze přisoudit ototoxické chemoterapii, např. cisplatině, není jasné, zda přípravek MEPACT podávaný současně s chemoterapií skládající se z mnoha léčiv může zvyšovat výskyt ztráty sluchu.
Vyšší četnost objektivní a subjektivní ztráty sluchu byla ve studii fáze III (popis studie (viz bod 5.1)) celkově pozorována u pacientů, kteří byli léčeni přípravkem MEPACT a chemoterapií (12 %, respektive 4 %), ve srovnání s pacienty, kteří byli léčeni pouze chemoterapií (7 % a 1 %). Všichni pacienti dostali celkovou dávku cisplatiny 480 mg/m2 jako součást režimu úvodní (neoadjuvantní) a/nebo udržovací (adjuvantní) chemoterapie.
Srdeční a cévní poruchy
V nekontrolovaných studiích s mifamurtidem byly velmi často hlášeny mírná až středně závažná tachykardie (50 %), hypertenze (26 %) a hypotenze (29 %). V časných studiích byla hlášena jedna závažná příhoda subakutní trombózy, ale ve velké randomizované kontrolované studii se
v souvislosti s mifamurtidem nevyskytly žádné závažné srdeční příhody (viz bod 4.4).
Respirační poruchy
Velmi často byly hlášeny respirační poruchy, včetně dušnosti (21 %), kašle (18 %) a tachypnoe (13 %), a ve studii fáze II se rozvinula u dvou pacientů s již dříve existujícím astmatem v souvislosti s léčbou přípravkem MEPACT mírná až středně závažná dechová nedostatečnost.
Gastrointestinální poruchy
V souvislosti s podáváním mifamurtidu se často vyskytovaly gatrointestinální poruchy, které zahrnovaly nauzeu (57 %) a zvracení (44 %) asi u poloviny pacientů, zácpu (17 %), průjem (13 %) a bolest břicha (viz bod 4.4).
Poruchy kůže a podkožní tkáně
V nekontrolovaných studiích byla u pacientů, kteří dostávali mifamurtid, velmi častá hyperhidróza (11 %).
Poruchy svalové a kosterní soustavy a pojivové tkáně
Bolest nízkého stupně byla u pacientů, kteří dostávali mifamurtid, velmi častá a zahrnovala myalgii (31 %), bolest zad (15 %), bolest končetin (12 %) a bolesti kloubů (10 %).
Celkové poruchy a reakce v místě aplikace
Většina pacientů prodělala třesavku (89 %), horečku (85 %) a únavu (53 %). Tyto příznaky byly typicky mírné až středně závažné, svou povahou přechodné a obecně dobře odpovídaly na paliativní léčbu (např. paracetamol u horečky). Ostatní celkové příznaky, které byly typicky mírné až středně závažné a velmi časté, zahrnovaly hypotermii (23 %), malátnost (13 %), bolest (15 %), astenii (13 %) a bolest na hrudi (11 %). Otoky, tlak na hrudi, lokální reakce v místě podání infuze nebo zavedeného katetru a „pocit chladu“ byly u těchto pacientů hlášeny méně často, a to zvláště u pacientů s nádorovým onemocněním pokročilého stadia.
Vyšetření
U jednoho pacienta s osteosarkomem ve studii fáze II, který měl při zařazení do studie vysokou hladinu kreatininu, se v souvislosti s užíváním mifamurtidu vyskytlo zvýšení urey a kreatininu v krvi.
Poruchy imunitního systému
V jedné studii fáze I se vyskytlo jedno hlášení závažné alergické reakce po první infuzi mifamurtidu v dávce 6 mg/m2. U pacienta se projevil třes, pocit chladu, horečka, nauzea, zvracení, nekontrolovatelný kašel, dušnost, cyanotické rty, závrať, slabost, hypotenze, tachykardie, hypertenze a hypotermie, což vedlo k přerušení studie. Vyskytlo se také jedno hlášení alergické reakce stupně 4 (hypertenze) ve studii fáze III, které si vyžádalo hospitalizaci (viz bod 4.4)
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. .
4.9 Předávkování
Během období, kdy byla schválena indikace přípravku, nebyl hlášen žádný případ předávkování. Maximální tolerovaná dávka ve studiích fáze I byla 4-6 mg/m2 s velkou variabilitou nežádoucích účinků. Známky a příznaky, které souvisely s vyššími dávkami a/nebo byly limitovány velikostí dávky, nebyly život ohrožující a zahrnovaly horečku, třesavku, únavu, nauzeu, zvracení, bolest hlavy a hypotenzi nebo hypertenzi.
Zdravý dospělý dobrovolník dostal náhodně jednu dávku 6,96 mg mifamurtidu a vyskytla se u něj reverzibilní příhoda ortostatické hypotenze související s léčbou.
V případě předávkování se doporučuje zahájit vhodnou podpůrnou léčbu. Podpůrná opatření by měla vycházet z ústavních doporučených postupů a pozorovaných klinických příznaků. Příklady zahrnují například podávání paracetamolu při horečce, třesavce a bolesti hlavy a antiemetik (jiných než steroidů) při nevolnosti a zvracení.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutické skupina: Imunostimulancia, Jiná imunostimulancia, ATC kód: L03AX15 Mechanizmus účinku.
Mifamurtid (muramyltripeptidfosfatidylethanolamin, MTP-PE) je plně syntetický derivát muramyldipeptidu (MDP), nejmenší přirozeně se vyskytující součásti buněčných stěn, která stimuluje imunitní systém a pochází z bakterie Mycobacterium sp. Má podobné imunostimulační
účinky jako přirozený MDP a navíc výhodu delšího poločasu v plazmě. MEPACT je lipozomální léková forma navržená speciálně tak, aby byla intravenózní infúzí in vivo cílena do makrofágů.
MTP-PE se specificky váže na NOD2, receptor, který se primárně nachází na monocytech, dendritických buňkách a na makrofázích. MTP-PE je silný aktivátor monocytů a makrofágů. Aktivace lidských makrofágů mifamurtidem je spojena s produkcí cytokinů, které zahrnují tumor nekrotizující faktor (TNF-a), interleukin-1 (IL-1P), IL-6, IL-8 a IL-12, a adhezivních molekul, včetně antigenu souvisejícího s funkcí lymfocytů 1 (LFA-1) a intercelulární adhezivní molekuly 1 (ICAM-1). Lidské monocyty při léčbě in vitro ničily alogenní a autologní nádorové buňky (včetně melanomu, ovariálního karcinomu, karcinomu tlustého střeva a ledvin), ale nevykazovaly toxicitu pro normální buňky.
In vivo vedlo podávání mifamurtidu k inhibici nádorového růstu u myších a potkaních modelů pro plicní metastázy, karcinom kůže a jater a fibrosarkom. Významné zlepšení doby přežití bez přítomnosti onemocnění bylo prokázáno také při léčbě osteosarkomu a hemangiosarkomu u psů s mifamurtidem jako adjuvantní léčbou. Přesný mechanismus, jak aktivace monocytů a makrofágů mifamurtidem vede k protinádorovému účinku u zvířat a lidí, dosud není znám.
Klinická bezpečnost a účinnost
Bezpečnost lipozomálního mifamurtidu byla zhodnocena u více než 700 pacientů s různými druhy a stadii nádorového onemocnění a u 21 zdravých dospělých subjektů (viz bod 4.8).
Mifamurtid významně zvyšuje celkové přežití pacientů s nově diagnostikovaným resektabilním osteosarkomem vysokého stupně, pokud je používán současně s kombinovanou chemoterapií, ve srovnání s užíváním samotné chemoterapie. V randomizované studii fáze III u 678 pacientů (věkového rozmezí od 1,4 do 30,6 let) s nově diagnostikovaným resekovatelným osteosarkomem vysokého stupně, vedlo přidání adjuvantního mifamurtidu k chemoterapii doxorubicinem, cisplatinou a methothrexátem s ifosfamidem nebo bez něj k relativnímu snížení rizika úmrtí o 28 % (p = 0,0313, míra rizika (HR) = 0,72 [95% konfidenční interval (CI): 0,53, 0,97]).
5.2 Farmakokinetické vlastnosti
Farmakokinetika mifamurtidu byla sledována u zdravých dospělých subjektů po 4mg intravenózní infuzi a u dětských a dospělých pacientů s osteosarkomem po intravenózní infuzi 2 mg/m2.
U 21 zdravých dospělých subjektů byl mifamurtid ze séra rychle odstraňován (minuty) s poločasem 2,05 ± 0,40 hodiny, což mělo za následek velmi nízké sérové koncentrace celkového (lipozomálního a volného) mifamurtidu. Průměrná AUC byla 17,0 ± 4,86 h x nM a Cmax byla 15,7 ± 3,72 nM.
U 28 pacientů s osteosarkomem ve věku 6 až 39 let sérové koncentrace celkového mifamurtidu (lipozomálního a volného) rychle klesaly a to s průměrným poločasem 2,04 ± 0,456 hodiny. BSA normalizovaná clearance a poločas byly podobné napříč věkovým spektrem a odpovídaly hodnotám u zdravých dospělých subjektů, což podporuje doporučenou dávku 2 mg/m2.
V samostatné studii u 14 pacientů, se časové křivky průměrných koncentrací celkového a volného mifamurtidu, které byly hodnoceny po první infuzi mifamurtidu a po poslední infuzi o 11 nebo 12 týdnů později, téměř překrývaly a průměrné hodnoty AUC volného mifamurtidu po první a poslední infuzi byly obdobné. Tyto údaje naznačují, že ani celkový, ani volný mifamurtid se během období léčby neakumuluje.
6 hodin po injekci radioaktivně značených lipozomů, které obsahovaly 1 mg mifamurtidu, byla radioaktivita detekována v játrech, slezině, nosohltanu, štítné žláze a v menší míře v plicích. Lipozomy byly fagocytovány buňkami retikuloendoteliárního systému. U 2 ze 4 pacientů s plicními metastázami, byla radioaktivita asociována s plicními metastázami.
Metabolismus liposomálního MTP-PE nebyl u lidí studován.
Po injekci radioaktivně značených lipozomů obsahujících mifamurtid byl průměrný poločas radioaktivně značeného materiálu bifazický s a fází okolo 15 minut a terminální poločas přibližně 18 hodin.
Zvláštní populace
Porucha funkce ledvin
Farmakokinetika jednotlivé dávky 4 mg mifamurtidu po 1hodinové intravenózní infuzi byla hodnocena u dospělých dobrovolníků s lehkou (n=9) nebo středně těžkou (n=8) poruchou funkce ledvin a u skupiny zdravých dospělých s normálními funkcemi ledvin (n=16) vyvážené podle věku, pohlaví a tělesné hmotnosti. Nebyl zjištěn žádný účinek u lehké (50 mL/min <CLcr <80 mL/min) nebo středně těžké (30 mL/min <CLcr <50 mL/min) renální insuficience na clearance celkového MTP-PE ve srovnání s pozorováním u zdravých dospělých jedinců s normální funkcí ledvin (CLcr >80 ml/min). Dále pak systémové expozice (AUCinf) volného (nevázaného na lipozomy) MTP-PE u lehké nebo středně těžké poruchy funkce ledvin byly podobné jako u zdravých dospělých jedinců s normálními renálními funkcemi.
Porucha funkce jater
Farmakokinetika jednotlivé dávky 4 mg mifamurtidu po 1hodinové intravenózní infuzi byla hodnocena u dospělých dobrovolníků s lehkou (Child-Pugh třída A; n=9) nebo středně těžkou (Child-Pugh třída B; n=8) poruchou funkce jater a u skupiny zdravých dospělých s normálními funkcemi jater (n=19) vyvážené podle věku, pohlaví a tělesné hmotnosti. Nebyl zjištěn žádný účinek u lehké poruchy funkce jater na systémovou expozici(AUCinf) celkového MTP-PE. Středně těžká porucha funkce jater měla za následek malé zvýšení AUCinf celkového MTP-PE, s průměrným poměrem (vyjádřeno v %) pro středně těžkou poruchu funkce jater ve srovnání se skupinou s normálními jaterními funkcemi 119 % (90% CI: 94,1 % 151 %).
Farmakokinetická variabilita byla u skupiny se středně těžkou poruchou funkce jater vyšší (koeficient změn v systémové expozici (AUC inf) byl 50 % oproti <30 % v jiných skupinách hepatálních funkcí).
Průměrný biologický poločas celkového a volného MTP-PE byl u lehké poruchy funkce jater
2.02 hodiny resp. 1,99 hodiny a tyto hodnoty byly srovnatelné s hodnotami u jedinců s normálními jaterními funkcemi (2,15 hodiny resp. 2,26 hodiny). Průměrný biologický poločas celkového a volného MTP-PE u středně těžké poruchy funkce jater byl 3,21 hodiny resp. 3,15 hodiny. Dále pak byl geometrický průměr plazmatické AUCinf volného (nevázaného na liposomy) MTP-PE u mírné a středně těžké poruchy funkce jater o 47 % vyšší než odpovídající hodnoty u skupiny s normálními hepatálními funkcemi.
Tyto změny nejsou považovány za klinicky významné, protože maximální tolerovaná dávka (4-6 mg/m2) mifamurtidu je 2-3 krát vyšší než doporučená dávka (2 mg/m2).
5.3 Předklinické údaje vztahující se k bezpečnosti
U citlivých druhů (králík a pes) byla nejvyšší dávka lipozomálního mifamurtidu, která nezpůsobovala nežádoucí účinky, 0,1 mg/kg, což odpovídá 1,2, respektive 2 mg/m2. Hladina, při které se u zvířat nevyskytují nežádoucí účinky mifamurtidu,přibližně odpovídá doporučené dávce 2 mg/m2 u člověka.
Údaje z šestiměsíční studie u psů, kdy byly podávány intravenózní injekce až do 0,5 mg/kg (10 mg/m2) mifamurtidu, stanovily bezpečnostní hranici kumulativní expozice pro zjevnou toxicitu na 8-19násobek zamýšlené klinické dávky u lidí. Hlavní toxické účinky související s těmito vysokými denními a kumulativními dávkami mifamurtidu byly hlavně vystupňované farmakologické účinky: pyrexie, příznaky projevující se zánětlivou reakcí, která se manifestovaly jako synovitida, bronchopneumonie, perikarditida a zánětlivá nekróza jater a kostní dřeně. Byly také pozorovány následující příhody: krvácení a prodloužení koagulačních časů, infarkty, morfologické změny ve stěně malých tepen, otoky a překrvení centrálního nervového systému, mírné srdeční účinky a lehká hyponatrémie. Mifamurtid nebyl mutagenní a u potkanů a králíků
neměl teratogenní účinky. Embryotoxické účinky byly pozorovány pouze při hladinách, které byly toxické pro matku.
Ze studií obecné toxicity nevyplynuly žádné výsledky, které by naznačovaly škodlivé účinky na mužské nebo ženské reprodukční orgány. Specifické studie týkající se reprodukčních funkcí, perinatální toxicity a karcinogenního potenciálu nebyly provedeny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
oleoylpalmitoylglycerofosfocholin (POPC) monosodná sůl dioleoylglycerofosfoserinu (OOPS)
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodu 6.6.
6.3 Doba použitelnosti
Neotevřená lahvička s práškem:
30 měsíců
Rekonstituovaná suspenze:
Chemická a fyzikální stabilita byla prokázána po dobu 6 hodin při teplotě do 25 °C.
Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání rekonstituovaného, přefiltrovaného a naředěného roztoku po otevření před použitím jsou v odpovědnosti uživatele a tato doba nesmí být delší než 6 hodin při teplotě 25 °C. Roztok chraňte před chladem a mrazem.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2-8 °C). Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání rekonstituovaného léčivého přípravku, viz bod 6.3.
6.5 Druh obalu a obsah balení
Skleněná 50ml injekční lahvička typu I s šedou pryžovou zátkou, hliníkovým uzávěrem a plastikovým víčkem, která obsahuje 4 mg mifamurtidu.
Každá krabička obsahuje jednu lahvičku a jeden jednorázový, apyrogenní, sterilní filtr na přípravek MEPACT, který je dodáván v PVC blistru.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek MEPACT musí být před podáním rekonstituován, přefiltrován pomocí přiloženého filtru a dále naředěn za použití aseptické techniky.
Každá injekční lahvička by měla být rekonstituována 50 ml 9mg/ml (0,9%) injekčního roztoku chloridu sodného. Po rekonstituci obsahuje každý ml suspenze v injekční lahvičce 0,08 mg
mifamurtidu. Objem rekonstituované suspenze odpovídající vypočtené dávce je odebrán přes dodaný filtr a dále naředěn dalšími 50 ml 9mg/ml (0,9%) injekčního roztoku chloridu sodného podle podrobného návodu, který je uveden níže.
Pokyny pro přípravu intravenózní infuze s přípravkem MEPACT
Materiál dodávaný v každém balení -
• MEPACT prášek pro koncentrát pro infuzní disperzi (injekční lahvička),
• filtr k přípravku MEPACT.
Potřebný materiál, který není dodáván -
• injekční roztok chloridu sodného 9mg/ml (0,9%), 100ml vak,
• jedna jednorázová 60- nebo 100ml injekční stříkačka s luerovou koncovkou,
• dvě sterilní injekční jehly středního průměru (18G).
Doporučuje se, aby byla rekonstituce lipozomální suspenze prováděna aseptickou technikou v digestoři s laminárním prouděním za použití sterilních rukavic.
Před rekonstitucí, filtrováním přes dodávaný filtr a ředěním se má nechat lyofylizovaný prášek dosáhnout teploty mezi asi 20-25 °C. Mělo by to trvat přibližně 30 minut.
1. Odstraňte víčko z injekční lahvičky a očistěte zátku vatovým tamponem s alkoholem.
2. Vyjměte filtr z obalu a z hrotu filtru odstraňte víčko. Pevně zasuňte hrot do septa injekční lahvičky, dokud nezapadne. V tuto chvíli neodstraňujte kryt luerové spojky filtru.
3. Připravte si 100ml vak s 9mg/ml (0,9%) injekčním roztokem chloridu sodného, jehlu a stříkačku (nejsou součástí balení).
4. Místo na vaku 9mg/ml (0,9%) injekčního roztoku chloridu sodného, kam se bude zasunovat jehla, otřete vatovým tamponem s alkoholem.
5. Pomocí jehly a stříkačky natáhněte z vaku 50 ml 9mg/ml (0,9%) injekčního roztoku chloridu sodného.
6. Po odstranění jehly ze stříkačky, připojte stříkačku k filtru tak, že otevřete uzávěr luerové spojky filtru (obrázek 1).

Obrázek 1
7. Přidejte do injekční lahvičky 9mg/ml (0,9%) injekční roztok chloridu sodného pomalým, stálým stlačováním pístu stříkačky. Filtr a stříkačka nesmí být oddělovány od injekční lahvičky.
8. Nechte injekční lahvičku v klidu stát po dobu jedné minuty, aby byla zajištěna důkladná hydratace suché látky.
9. Injekční lahvičkou poté důkladně jednu minutu třeste a udržujte filtr a stříkačku připojeny k injekční lahvičce. Během této doby se samovolně vytvoří lipozomy (obrázek 2).
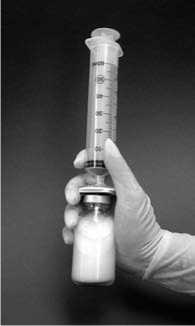
Obrázek 2
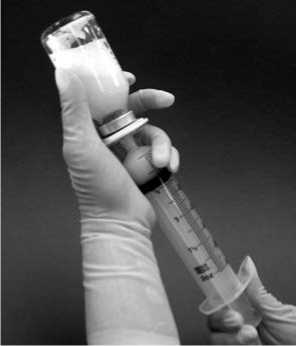
10. Požadovanou dávku lze z injekční lahvičky natáhnout obrácením injekční lahvičky a pomalým tahem za píst stříkačky (obrázek 3). Jeden ml rekonstituované suspenze obsahuje 0,08 mg mifamurtidu. Objem suspenze, která má být natažena z injekční lahvičky pro jednotlivá dávková množství, se vypočítá následovně:
Objem k natažení = [12,5 x vypočtená dávka (mg)] ml
Pro zjednodušení je uvedena následující srovnávací tabulka:
|
Dávka |
Objem |
|
1,0 mg |
12,5 ml |
|
2,0 mg |
25 ml |
|
3,0 mg |
37,5 ml |
|
4,0 mg |
50 ml |
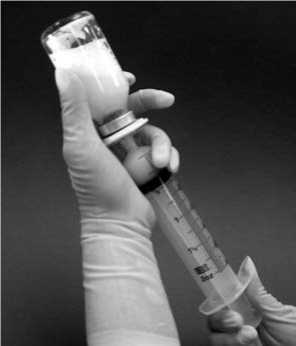
Obrázek 3
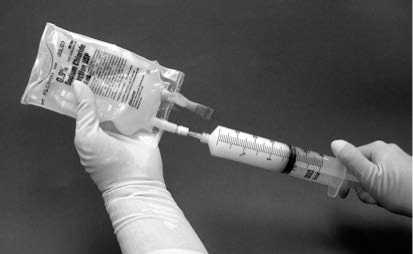
11. Stříkačku poté oddělte od filtru a na tuto stříkačku naplněnou suspenzí umístěte novou j ehlu. Otřete místo vpichu na vaku vatovým tamponem s alkoholem a vstříkněte suspenzi v stříkačce do originálního vaku, který obsahuje zbývajících 50 ml 9mg/ml (0,9%) injekčního roztoku chloridu sodného (obrázek 4).
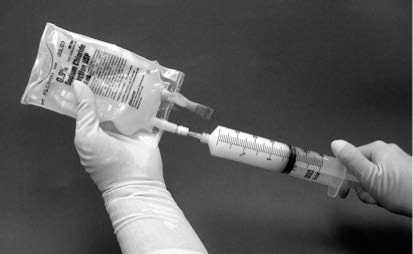
Obrázek 4
12. Jemně vakem otáčejte, aby se roztok smísil.
13. Vak obsahující rekonstituovanou, přefiltrovanou a naředěnou lipozomální suspenzi označte identifikací pacienta, časem a datem.
14. Chemická a fyzikální stabilita po otevření před použitím byla prokázána po dobu 6 hodin při pokojové teplotě (mezi přibližně 20-25 °C).
15. Z mikrobiologického hlediska má být roztok použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání po otevření před použitím jsou v odpovědnosti uživatele a tato doba nesmí být delší než 6 hodin při pokojové teplotě.
16. Lipozomální suspenze se podává intravenózní infuzí v průběhu jedné hodiny.
Likvidace
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Takeda France SAS Immeuble Pacific 11-13 cours Valmy 92800 - Puteaux Francie
8. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/502/001
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace: 6. března 2009 Datum posledního prodloužení registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravkyEMA http://www.ema.europa.eu.
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců odpovědných za propouštění šarží
Takeda Austria GmbH St. Peter-StraPe 25 A-4020 Linz Rakousko
Takeda Italia S.p.A Via Crosa, 86 28065 Cerano (NO)
Itálie
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU VNĚJŠÍ OBAL
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
MEPACT 4 mg, prášek pro koncentrát pro infuzní disperzi Mifamurtidum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje mifamurtidum 4 mg. Po rekonstituci obsahuje jeden ml suspenze v injekční lahvičce mifamurtidum 0,08 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: oleoylpalmitoylglycerofosfocholin (POPC), monosodná sůl dioleoylglycerofosfoserinu (OOPS)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro koncentrát pro infuzní disperzi
Balení 1 injekční lahvička s práškem, 1 sterilní filtr k přípravku MEPACT
5. ZPŮSOB A CESTA / CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Intravenózní podání po rekonstituci, přefiltrování přes dodaný filtr a po dalším naředění.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Takeda France SAS Immeuble Pacific 11-13 cours Valmy 92800 - Puteaux Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/502/001
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU OZNAČENÍ INJEKČNÍ LAHVIČKY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
MEPACT 4 mg, prášek pro koncentrát pro infuzní disperzi Mifamurtidum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje mifamurtidum 4 mg. Po rekonstituci obsahuje jeden ml suspenze v injekční lahvičce mifamurtidu 0,08 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: oleoylpalmitoylglycerofosfocholin (POPC), monosodná sůl dioleoylglycerofosfoserinu (OOPS)
4. LÉKOVÁ FORMA A VELIKOST BALENÍ
Prášek pro koncentrát pro infuzní disperzi 4 mg mifamurtidu
5. ZPŮSOB A CESTA / CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Intravenózní podání po rekonstituci, přefiltrování přes dodaný filtr a po dalším naředění.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Takeda France SAS Immeuble Pacific 11-13 cours Valmy 92800 - Puteaux Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/502/001
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
MEPACT 4 mg, prášek pro koncentrát pro infuzní disperzi
Mifamurtidum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek MEPACT a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek MEPACT užívat
3. Jak se přípravek MEPACT užívá
4. Možné nežádoucí účinky
5. Jak přípravek MEPACT uchovávat
6. Obsah balení a další informace
1. Co je přípravek MEPACT a k čemu se používá
MEPACT obsahuje léčivou látku mifamurtid, která je podobná součásti buněčné stěny určité bakterie. Stimuluje Váš imunitní systém, aby Vašemu tělu pomohl zničit nádorové buňky.
Přípravek MEPACT se používá k léčbě osteosarkomu (nádorové onemocnění kostí) u dětí, dospívajících a mladých dospělých (mezi 2 a 30 roky). Používá se poté, co u Vás byl proveden chirurgický zákrok k odstranění nádoru, současně s chemoterapií, aby zničil zbývající nádorové buňky, a tím snížil riziko znovuobjevení se nádoru.
2. Čemu musíte věnovat pozornost, než začnete přípravek MEPACT používat
Nepoužívejte přípravek MEPACT
- jestliže jste alergický(á) na mifamurtid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže užíváte léčivé přípravky, které obsahují cyklosporin nebo takrolimus nebo vysoké dávky nesteroidních antirevmatik (NSAID) (viz Vzájemné působení s dalšími léčivými přípravky níže).
Upozornění a opatření
Před použitím přípravku MEPACT se poraďte se svým lékařem:
- jestliže máte nebo jste měl(a) problémy se srdcem či cévami, například krevní sraženiny (trombózu), krvácení (hemoragii) nebo zánět žil (vaskulitidu). Při používání přípravku MEPACT byste měl(a) být přísněji sledován(a). Máte-li dlouhodobé nebo zhoršující se příznaky, oznamte to svému lékaři, protože léčbu přípravkem MEPACT možná bude potřeba odložit nebo přerušit.
- jestliže máte anamnézu astmatu nebo jiných poruch dýchání. Před začátkem používání přípravku MEPACT, konzultujte se svým lékařem, zda byste zároveň s používáním přípravku MEPACT měl(a) brát také léky na astma.
- j estliže j ste prodělala zánětlivé nebo autoimunitní onemocnění nebo pokud j ste byl(a) léčen(a) kortikosteroidy nebo jinými léčivými přípravky, které mohou ovlivňovat Váš imunitní systém.jestliže máte alergickou reakci na nějaký lék, jako je vyrážka, potíže s dýcháním nebo
máte vysoký krevní tlak. Jestliže se Vaše příznaky zhoršují, kontaktujte svého lékaře, neboť mohou být způsobeny přípravkem MEPACT.
- jesliže máte zažívací potíže jako je pocit na zvracení, zvracení, nedostatečná chuť k jídlu. Jestliže se Vaše potíže zhoršují, kontaktujte svého lékaře, neboť mohou být způsobeny přípravkem MEPACT, pokud je používán současně s chemoterapií.
- jestliže začnete mít zimnici, třesavku nebo pocit tepla. Měl(a) byste si změřit tělesnou teplotu, zda nemáte horečku. Horečka a snížený počet bílých krvinek (neutropenie) může být známkou závážné infekce.
Podrobné informace o upozorněních a varováních týkajících se nežádoucích účinků, které se mohou objevit během používání tohoto přípravku, jsou uvedeny v bodě 4.
Děti a dospívající
Tento přípravek se nedoporučuje podávat dětem mladším 2 let, protože pro tuto věkovou skupinu nejsou dostupné informace o bezpečnosti a a účinnosti tohoto přípravku.
Další léčivé přípravky a MEPACT
Prosím, informujte svého lékaře o všech lécích, které užíváte nebo jste užíval(a) v nedávné době, a to i o lécích, které jsou dostupné bez lékařského předpisu. Je obzvláště důležité informovat lékaře, že užíváte léky, které obsahují některou z následujících léčivých látek:
- cyklosporin, takrolimus, používané po transplantaci k zabránění rejekce (odmítnutí) transplantovaných orgánů, nebo jiná imunosupresiva používaná např. k léčbě lupénky (kožní onemocnění),
- nesteroidní protizánětlivé léky (NSAID), jako je kyselina acetylsalicylová, ibuprofen nebo diklofenak, používané k léčbě bolestí hlavy a jiných bolestí nebo k léčbě horečky; přípravek MEPACT nesmí být používán současně s vysokými dávkami NSAID,
- kortikosteroidy, používané k léčbě zánětů, alergií nebo astmatu. Při podávání přípravku MEPACT je třeba vyloučit pravidelné užívání kortikosteroidů, neboť to může ovlivnit účinek léku.
Doporučuje se časově oddělit podávání přípravku MEPACT a doxorubicinu či jiných léčivých přípravků, jsou-li používány ve stejném režimu chemoterapie.
Těhotenství ,kojení a plodnost
MEPACT nebyl testován u těhotných žen. Přípravek MEPACT by proto neměl být používán v průběhu těhotenství a u žen, které neužívají účinnou antikoncepci. Pokud jste léčena přípravkem MEPACT, měla byste užívat účinnou antikoncepci. Pokud jste těhotná, myslíte si, že můžete být těhotná nebo plánujete otěhotnět, je důležité oznámit to svému lékaři.
Není známo, zda přípravek MEPACT přechází do lidského mléka. Pokud kojíte, konzultujte tuto skutečnost se svým lékařem.
Řízení dopravních prostředků a obsluha strojů
Některé velmi časté a časté nežádoucí účinky léčby přípravkem MEPACT (jako točení hlavy, závratě, únava a rozmazané vidění) mohou mít dopad na schopnost řídit a obsluhovat stroje.
3. Jak se přípravek MEPACT používá Dávkování a trvání léčby
MEPACT Vám bude podáván pouze pod dohledem odborného specialisty. Vždy používejte tento lék přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), zeptejte se svého lékaře.
Dávka přípravku MEPACT je 2 mg mifamurtidu/m2 tělesného povrchu. Bude Vám podáván dvakrát týdně (s nejméně třídenním odstupem) v prvních 12 týdnech a poté jedenkrát týdně po dobu dalších 24 týdnů.
Rozpis podávání Vaší léčby přípravkem MEPACT může být přizpůsoben tak, aby vyhovoval Vašemu rozpisu podávání chemoterapie. Pokud je odloženo podávání chemoterapie, není nutné přerušovat podávání přípravku MEPACT; měl(a) byste dokončit 36 týdnů (9 měsíců) léčby přípravkem MEPACT bez přerušení.
Jak se přípravek MEPACT podává
Mrazem vysušený (lyofilizovaný) prášek se musí před použitím rekonstituovat na tekutou suspenzi, přefiltrovat přes přiložený filtr a dále naředit. Přípravek MEPACT Vám pak bude podán infuzí do žíly (intravenózně) v průběhu jedné hodiny. Tento úkon provádí Váš lékař nebo sestra, který/á Vás během této doby bude také sledovat. Kvůli podávání přípravku MEPACT nemusíte být hospitalizován(a). Přípravek může být podáván také ambulantně.
Jestliže jste použil(a) více přípravku MEPACT, než jste měl(a)
Můžete pociťovat závažnější nežádoucí účinky, které mohou zahrnovat horečku, třesavku, únavu, pocit na zvracení, zvracení, bolest hlavy a nízký nebo vysoký krevní tlak. V případě předávkování kontaktujte svého lékaře nebo nejbližší nemocnici.
Jestliže jste přestal(a) používat přípravek MEPACT
Neukončujte léčbu přípravkem MEPACT bez ukončení léčebného cyklu a předchozí porady se svým lékařem.Máte-li jakékoliv další otázky, týkající se používání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i přípravek MEPACT nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Většina pacientů pociťuje třesavku, horečku a únavu, zvláště pak při prvním podání přípravku MEPACT. Typicky jsou mírné až středně závažné a přechodné a obvykle mohou být léčeny Vaším lékařem, např. podáním paracetamolu při horečce.
Léčba přípravkem MEPACT, pokud je používán společně s chemoterapií, může často způsobit žaludeční potíže jako je pocit na zvracení, zvracení a ztráta chuti k jídlu.
Ihned se spojte se svým lékařem,:
- pokud máte přetrvávající horečku nebo třesavku déle než 8 hodin poté, co jste dostal(a) dávku přípravku MEPACT, protože může jít o známku infekce nebo
- pokud se u Vás objevila vyrážka nebo jakékoli dechové potíže (sípání) nebo
- pokud máte žaludeční problémy
Velmi časté nežádoucí účinky (mohou se vyskytnout u více než 1 pacienta z 10):
- horečka, třesavka/zimnice, slabost, únava nebo celkový pocit nemoci
- pocit na zvracení a/nebo zvracení, průjem nebo zácpa,
- bolest hlavy nebo závratě,
- rychlé bušení srdce,
- vysoký krevní tlak nebo nízký krevní tlak,
- ztráta chuti k jídlu,
- pocení,
- bolest, včetně celkové bolesti, bolesti ve svalech a/nebo kloubech a bolest v zádech, na hrudi, v břiše, v pažích či nohou,
- kašel, potíže s dýcháním nebo zrychlené dýchání
- nízká tělesná teplota,
- nízký počet červených krvinek.
Časté nežádoucí účinky (mohou se vyskytnout až u 1 pacienta z 10):
- modravé zbarvení tkání jako je kůže nebo dásně způsobené nízkým obsahem kyslíku,
- citelný vzestup frekvence nebo síly srdečního tepu,
- otoky paží nebo nohou nebo jiné otoky,
- nepříjemný pocit na hrudi,
- podrážděný žaludek, snížení chuti k jídlu nebo pokles tělesné hmotnosti,
- zčervenání místa injekce nebo místa zavedení katetru, otok, infekce nebo jiná lokální reakce,vyrážka nebo zčervenání, zánět kůže, svědění, suchá kůže, bledost nebo přechodné zčervenání,
- zánět kůže, šlach, svalů nebo podobných podpůrných tkání,
- zánět žil,
- bolest horní části břicha nebo hrudní stěny, pocit nadmutí nebo bolest břicha,
- jiná bolest, včetně bolesti krku, ramen nebo hrdla,
- svalové křeče nebo napětí,
- pocit chladu,
- pocit únavy, otupělosti nebo ospalosti,
- pocit pálení, píchání/brnění nebo snížené citlivosti
- mimovolní pohyby (třes),
- dehydratace,
- zánět sliznic,
- překrvení nebo zánět nosu, hrdla nebo vedlejších nosních dutin,
- infekce horních dýchacích cest (jako je nachlazení) nebo močových cest (jako je zánět močového měchýře),
- generalizovaná infekce,
- infekce virem Herpes simplex,
- produktivní kašel, sípání nebo námahová dušnost nebo zhoršení dušnosti,
- plivání krve nebo krvácení z nosu,
- tekutina v dutině plicní,
- krev v moči, obtížné nebo bolestivé močení nebo časté močení,
- spánkové obtíže, deprese, úzkost nebo zmatenost,
- závrať,
- zvonění v uších,
- rozmazané vidění,
- vypadávání vlasů,
- bolestivá menstruace,
- ztráta sluchu,
- nízký počet bílých krvinek s horečkou nebo bez ní, nízký počet krevních destiček.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek MEPACT uchovávat
Uchovávejte mimo dohled a dosah dětí.
Přípravek MEPACT nepoužívejte po uplynutí doby použitelnosti, uvedené na štítku injekční lahvičky a na krabičce.
Neotevřená injekční lahvička
Uchovávejte v chladničce (2-8 °C). Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem. Rekonstituovaná suspenze
Po rekonstituování v 9mg/ml (0,9 %) roztoku chloridu sodného uchovávejte při pokojové teplotě (přibližně 20-25 °C) a spotřebujte do 6 hodin.
6. Obsah balení a další informace Co přípravek MEPACT obsahuje
- Léčivá látka je mifamurtidum. Jedna injekční lahvička obsahuje mifamurtidum 4 mg. Po rekonstituci obsahuje jeden ml suspenze 0,08 mg mifamurtidu.
- Pomocnými látkami j sou oleoylpalmitoylglycerofosfocholin (POPC) a monosodná sůl dioleoylglycerofosfoserinu (OOPS).
Jak přípravek MEPACT vypadá a co obsahuje toto balení
Přípravek MEPACT je bílý až téměř bílý homogenní koláč nebo prášek pro koncentrát pro infuzní disperzi.
Přípravek MEPACT se dodává v krabičce, která obsahuje
• Jednu 50 ml lahvičku se šedou butylovou zátkou, hliníkovým uzávěrem a plastovým víčkem.
• Jeden sterilní filtr bez obsahu latexu k přípravku MEPACT dodávaný v blistru.
Držitelem rozhodnutí o registraci je:
Takeda France SAS Immeuble Pacific 11-13 cours Valmy 92800 - Puteaux Francie
Výrobcem přípravku je:
Takeda Austria GmbH St. Peter-StraPe 25 A-4020 Linz Rakousko
Takeda Italia S.p.A Via Crosa, 86 28065 Cerano (NO)
Itálie
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky EMA: http://www.ema.europa.eu/
Následující informace je určena pouze pro zdravotnické pracovníky:
Pokyny pro přípravu intravenózní infuze přípravku MEPACT
Materiál dodávaný v každém balení -
• 1 injekční lahvička přípravku MEPACT (mifamurtid),
• 1 filtr k přípravku MEPACT.
Potřebný materiál, který není dodáván -
• injekční roztok chloridu sodného 9 mg/ml (0,9%), 100ml vak,
• jedna jednorázová 60- nebo 100ml injekční stříkačka s luerovou koncovkou,
• dvě sterilní injekční jehly středního průměru (18G).
Doporučuje se, aby byla rekonstituce lipozomální suspenze prováděna aseptickou technikou v digestoři s laminárním prouděním za použití sterilních rukavic.
Před rekonstitucí, filtrováním přes dodávaný filtr a ředěním má lyofilizovaný prášek dosáhnout teploty mezi asi 20-25 °C. Mělo by to trvat přibližně 30 minut.
1. Odstraňte víčko z injekční lahvičky a očistěte zátku vatovým tamponem s alkoholem.
2. Vyjměte filtr z obalu a z hrotu filtru odstraňte víčko. Pevně zasuňte hrot do septa injekční lahvičky, dokud nezapadne. V tuto chvíli neodstraňujte kryt luerové spojky filtru.
3. Připravte si 100ml vak s 9mg/ml (0,9%) injekčním roztokem chloridu sodného, jehlu a stříkačku (nejsou součástí balení).
4. Místo na vaku 9mg/ml (0,9%) injekčního roztoku chloridu sodného, kam se bude zasunovat jehla, otřete vatovým tamponem s alkoholem.
5. Pomocí jehly a stříkačky natáhněte z vaku 50 ml 9mg/ml (0,9%) injekčního roztoku chloridu sodného.
6. Po odstranění jehly ze stříkačky, připojte stříkačku k filtru tak, že otevřete uzávěr luerové spojky filtru (obrázek 1).

Obrázek 1
7. Přidejte do injekční lahvičky 9mg/ml (0,9%) injekční roztok chloridu sodného pomalým, stálým stlačováním pístu stříkačky. Filtr a stříkačka nesmí být oddělovány od injekční lahvičky.
8. Nechte injekční lahvičku v klidu stát po dobu jedné minuty, aby byla zajištěna důkladná hydratace suché látky.
9. Injekční lahvičkou poté důkladně jednu minutu třeste a udržujte filtr a stříkačku připojeny k injekční lahvičce. Během této doby se samovolně vytvoří lipozomy (obrázek 2).
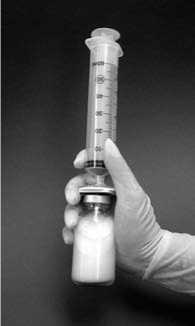
Obrázek 2
10. Požadovanou dávku lze z injekční lahvičky natáhnout obrácením injekční lahvičky a pomalým tahem za píst stříkačky (obrázek 3) Každý ml rekonstituované suspenze obsahuje 0,08 mg mifamurtidu. Objem suspenze, která má být natažena z injekční lahvičky pro jednotlivá dávková množství, se vypočítá následovně:
Objem k natažení = [12,5 x vypočtená dávka (mg)] ml
Pro zjednodušení je uvedena následující srovnávací tabulka:
|
Dávka |
Objem |
|
1,0 mg |
12,5 ml |
|
2,0 mg |
25 ml |
|
3,0 mg |
37,5 ml |
|
4,0 mg |
50 ml |
Obrázek 3
11. Stříkačku poté oddělte od filtru a na tuto stříkačku naplněnou suspenzí umístěte novou j ehlu. Otřete místo vpichu na vaku vatovým tamponem s alkoholem a vstříkněte suspenzi v stříkačce do originálního vaku, který obsahuje zbývajících 50 ml 9mg/ml (0,9%) injekčního roztoku chloridu sodného (obrázek 4).
Obrázek 4
12. Jemně vakem otáčejte, aby se roztok smísil.
13. Vak obsahující rekonstituovanou, přefiltrovanou a naředěnou suspenzi označte identifikací pacienta, časem a datem.
14. Chemická a fyzikální stabilita po otevření před použitím byla prokázána po dobu 6 hodin při pokojové teplotě (mezi přibližně 20-25 °C).
15. Z mikrobiologického hlediska má být roztok použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání po otevření před použitím jsou v odpovědnosti uživatele a tato doba nesmí být delší než 6 hodin při pokojové teplotě.
Likvidace
Žádné zvláštní požadavky.
32