Macugen 0,3 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Macugen 0,3 mg injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka poskytuje využitelné množství pro dodání jedné dávky 90 mikrolitrů obsahující pegaptanibum natricum odpovídající 0,3 mg volného oligonukleotidu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce) Čirý bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Macugen je indikován k léčbě neovaskulární (vlhké) formy věkem podmíněné makulární degenerace (VPMD) u dospělých (viz bod 5.1).
4.2 Dávkování a způsob podání
Přípravek Macugen je určen pouze k podání očním specialistou se zkušenostmi s intravitreálním podáním.
Dávkování
Před zahájením intravitreálním aplikace je třeba u pacienta pečlivě posoudit dřívější výskyt hypersenzitivních reakcí (viz bod 4.4).
Doporučená dávka je 0,3 mg pegaptanibu ekvivalentní 90 mikrolitrům, aplikuje se injekcí intravitreálně postiženého oka jedenkrát za 6 týdnů (tzn. 9 injekcí ročně).
Po aplikaci přípravku Macugen bylo u pacientů pozorováno přechodné zvýšení nitroočního tlaku. Proto je třeba monitorovat perfuzi papily zrakového nervu a nitrooční tlak. Dále je třeba pacienty pozorně sledovat během dvou týdnů po podání injekce, kvůli případnému vzniku krvácení do sklivce a endoftalmitidy. Pacienti mají být poučeni o nutnosti okamžitého oznámení výskytu jakýchkoli příznaků, které by mohly být příznaky těchto onemocnění (viz bod 4.4).
Pokud po 2 následujících injekcích přípravku Macugen pacient při kontrole ve 12. týdnu nevykazuje léčebný přínos (úbytek méně než 15 písmen zrakové ostrosti), má být zváženo ukončení nebo přerušení léčby přípravkem Macugen.
Zvláštní skupiny pacientů
Starší pacienti:
Není třeba dodržovat žádná zvláštní opatření.
Porucha funkce jater
Účinky přípravku Macugen nebyly hodnoceny u pacientů s poruchou funkce jater. U těchto pacientů není však třeba při terapii dodržovat žádná zvláštní opatření (viz bod 5.2).
Porucha funkce ledvin
Účinky přípravku Macugen nebyly dostatečně hodnoceny u pacientů s těžkou poruchou funkce ledvin, U pacientů s lehkou až středně těžkou poruchou funkce ledvin se nedoporučuje dávkování upravovat (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Macugen nebyla u pacientů mladších 18 let dosud stanovena. Nejsou k dispozici žádné údaje.
Způsob podání
Pouze intravitreální podání (formou injekce).
Před aplikací je nutné zkontrolovat, zda přípravek Macugen neobsahuje cizí částice či není změněna jeho barva (viz bod 6.6).
Injekční podání se provádí za aseptických podmínek, včetně dezinfekce rukou, použití sterilních rukavic, sterilní roušky a sterilního spekula (nebo ekvivalentní náhrady) a dostupnosti sterilní paracentézy (je-li třeba). Před aplikací je nutné podat vhodné anestetikum a širokospektrý topický mikrobicidní přípravek.
Předplněná stříkačka je dodávána s větším obsahem přípravku než je potřebné k podání dávky. Podání celého objemu předplněné injekční stříkačky by mohlo vést k předávkování (viz body 4.8 a 4.9). Viz bod 6.6 pro instrukce k vytlačení přebytečného obsahu před podáním injekce.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Aktivní infekce nebo podezření na infekci oka a okolí.
4.4 Zvláštní upozornění a opatření pro použití
Endoftalmitida
S intravitreálním podáním je spojeno riziko vzniku endoftalmitidy, v klinických studiích s přípravkem Macugen byla incidence endoftalmitidy 0,1% na injekci (viz bod 4.2).
Zvýšení nitroočního tlaku
Po aplikaci přípravku je možné pozorovat (jako u všech podání do sklivce) přechodné zvýšení nitroočního tlaku. Následně po aplikaci má tedy být zkontrolována perfuze papily očního nervu. Zvýšení nitroočního tlaku po aplikaci injekce je nutné léčit vhodným způsobem.
Observační studie po uvedení přípravku na trh dále hlásila malé riziko pomalého trvalého nárůstu nitroočního tlaku (viz bod 4.8).
Intravitreální krvácení
Bezprostředně (v den aplikace injekce) nebo s určitým časovým odstupem po aplikaci injekce pegaptanibu se může objevit krvácení do sklivce (viz bod 4.2).
Hypersenzitivní reakce
Během postmarketingového sledování byly několik hodin po podání intravitreálního pegaptanibu pozorovány anafylaktické/anafylaktoidní reakce, včetně angioedému. V těchto případech nebyla stanovena přímá souvislost s přípravkem Macugen, ani jakýmkoli jiným léčivým přípravkem podaným v souvislosti s injekční procedurou nebo s dalšími faktory.
Systémové účinky
Systémové nežádoucí účinky včetně mimoočních krvácení a arteriálních tromboembolických příhod byly hlášeny po intravitreálním injekčním podání inhibitorů VEGF a je tedy teoretické riziko, že se mohou vztahovat k inhibici VEGF. K dispozici jsou jen omezená data ohledně bezpečnosti pacientů s mozkovou příhodou nebo tranzitomími ischemickými atakami v anamnéze. Je třeba opatrnosti při léčbě těchto pacientů (viz bod 4.8), odstavec „Skupinové nežádoucí účinky“.
Přebytečný obsah
Injekce celého obsahu předplněné stříkačky může mít za následek závažné nežádoucí účinky; proto je nutné přebytečný obsah před podáním injekce vytlačit (viz body 4.8 a 6.6).
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) na dávku, je tedy v podstatě "bez sodíku".
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie na interakce přípravku Macugen s jinými léčivými přípravky nebyly provedeny. Pegaptanib je metabolizován nukleázami, lékové interakce zprostředkované cytochromem P450 jsou tedy nepravděpodobné.
Dvě z prvních klinických studií u pacientů, kterým byl podáván přípravek Macugen samostatně či v kombinaci s fotodynamickou terapií (PDT), neodhalily žádné zjevné rozdíly v plazmatické farmakokinetice pegaptanibu.
4.6 Fertilita, těhotenství a kojení
Pegaptanib nebyl studován u těhotných žen. Studie se zvířaty nejsou dostatečné, ale vykázaly reprodukční toxicitu při vysokých hladinách systémové expozice (viz bod 5.3). Potenciální riziko pro člověka není známo. Lze předpokládat, že systémová expozice pegaptanibu je po jeho oční aplikaci velmi nízká. Podávání přípravku je nicméně během těhotenství doporučeno pouze v případě, kdy očekávaná prospěšnost pro matku je větší než možné riziko pro plod.
Kojení
Není známo, zda přípravek Macugen přechází do mateřského mléka. Jeho podávání během období kojení se proto nedoporučuje.
Fertilita
Data pro hodnocení účinku přípravku Macugen na fertilitu u člověka nejsou k dispozici. Při studiích na zvířatech nebyly pozorovány žádné účinky na samčí nebo samičí fertilitu u myší.Viz bod 5.3.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vzhledem k možnosti výskytu krátkodobého rozmazaného vidění po intravitreálním podání má přípravek Macugen v malé míře ovlivnit schopnost řídit a obsluhovat stroje. Pacienti mají být poučeni, že řídit nebo obsluhovat stroje je možné až po odeznění tohoto příznaku.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Většina nežádoucích účinků hlášených po podání přípravku Macugen souvisí s intravitreální injekční aplikací.
V klinických studiích jsou po podání přípravku Macugen nejčastěji hlášenými očními nežádoucími účinky: záněty v oblasti přední komory oční, bolest oka, zvýšený nitrooční tlak, keratitis punctata, sklivcové vločky a sklivcové opacity. Méně často hlášeny závažné oční nežádoucí účinky zahrnovaly endoftalmitidu, retinální krvácení, krvácení do sklivce a odchlípení sítnice.
Méně často hlášené závažné oční nežádoucí účinky zahrnovaly: endoftalmitidu, retinální krvácení, krvácení ze sklivce a odchlípení sítnice.
Tabulkový seznam nežádoucích účinků
Níže uvedené údaje o bezpečnosti přípravku shrnují všechny nežádoucí účinky potenciálně související se způsobem aplikace v rámci zmíněné skupiny 295 pacientů, kterým byla podávána dávka 0,3 mg. U každé třídy orgánových systémů je uvedeno pořadí nežádoucích účinků podle třídy orgánového systému a jejich frekvence: velmi časté (>1/10), časté (>1/100 až < 1/10), méně časté (>1/1000 až < 1/100) a není známo (z dostupných dat nelze určit).
Hlášení po uvedení přípravku na trh jsou uvedena kurzívou.
|
Třídy orgánových systémů dle MedDRA Poruchy imunitního systému Není známo |
Nežádoucí účinek anafylaktické reakce * |
|
Psychiatrické poruchy Méně časté |
noční můry, deprese |
|
Poruchy nervového systému Časté | |
|
Poruchy oka Velmi časté |
záněty v oblasti přední komory oční, bolest oka, zvýšený nitrooční tlak, keratitis punctata, sklivcové vločky a opacity |
|
Časté |
abnormální pocity v oku, katarakta, spojivkové krvácení, hyperemie spojivky, spojivkový edém, konjunktivitida, korneální dystrofie, poškození rohovkového epitelu, defekt epitelu rohovky, edém rohovky, suché oko, endoftalmitida, výtok z oka, zánět oka, podráždění oka, svědění a zčervenání oka, otok oka a očních víček, zvýšené slzení, makulární degenerace, mydriáza, oční diskomfort, oční hypertenze, periorbitální hematom, fotofobie, fotopsie, retinální krvácení, rozmazané vidění, snížení ostrosti zraku, poruchy zraku, odchlípení sklivce, poruchy sklivce |
|
Méně časté |
astenopie, blefaritida, alergická konjunktivitida, korneální depozita, oční krvácení, pruritus očních víček, keratitida, krvácení do sklivce, poruchy pupilárního reflexu, korneální abraze, retinální exsudáty, ptóza očních víček, retinální |
jizva, chalazion, eroze rohovky, snížení nitroočního tlaku, reakce v místě vpichu injekce, vesikuly v místě vpichu,
|
odchlípení sítnice, poruchy rohovky, okluze retinální arterie, trhliny v sítnici, ektropium, poruchy oční hybnosti, podráždění víčka, hyféma, pupilární poruchy, onemocnění duhovky, oční ikterus, přední uveitida, oční depozita, iritida, vyklenutí disku zrakového nervu, deformity pupily, okluze retinální žíly, prolaps sklivce | |
|
Poruchy ucha a labyrintu Méně časté |
hluchota, zhoršení Menierovy choroby, vertigo |
|
Srdeční poruchy Méně časté |
palpitace |
|
Cévní poruchy Méně časté |
hypertenze, aneuryzma aorty |
|
Respirační, hrudní a mediastinální poruchy Časté |
rinorea |
|
Méně časté |
nazofaryngitida |
|
Gastrointestinální poruchy Méně časté | |
|
Poruchy kůže a podkožní tkáně Méně časté |
kontaktní dermatitida, ekzém, změna barvy vlasů, vyrážka, svědění, noční pocení |
|
Není známo |
angioedém* |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně Méně časté | |
|
Celkové poruchy a reakce v místě aplikace Méně časté |
únava, rigor, zvýšená citlivost na dotek, bolesti na hrudi, příznaky podobné chřipce |
|
Vyšetření Méně časté |
zvýšení aktivity gama-glutamyltransferázy |
|
Poranění, otravy a procedurální komplikace Méně časté |
abraze |
* Sledování po uvedení přípravku na trh; viz „Popis vybraných nežádoucích účinků“.
Popis vybraných nežádoucích účinků
Několik hodin po podání pegaptanibu (a dalších léčivých přípravků podávaných v souvislosti s injekční procedurou) do sklivce byly u pacientů hlášeny případy anafylaktických/anafylaktoidních reakcí, včetně angioedému (viz body 4.2 a 4.4).
V případech, kdy před podáním injekce nebyl vytlačen přebytečný obsah předplněné stříkačky, byly hlášeny případy závažného zvýšení nitroočního tlaku.
V observačních studiích po uvedení přípravku na trh byl po opakovaném intravitreálním podání přípravku hlášen pomalý trvalý nárůst nitroočního tlaku (IOP. Pravděpodobnost zvýšeného IOP se s každou dodatečnou injekcí zvyšovala 1,128krát (p= 0,0003). Mezi normálními pacienty a pacienty se zvýšeným IOP nebo glaukomem nebyl nalezen statisticky významný rozdíl.
Skupinové nežádoucí účinky
V klinické studii byla celková četnost mimoočních krvácení, jako nežádoucího účinku potenciálně souvisejícího s inhibicí systémového VEGF (vaskulární endoteliální růstový faktor), lehce zvýšená u pacientů léčených intravitreálním podáním inhibitorů VEGF. Nicméně mezi různými krváceními nebyl konzistentní vzor. Arteriální tromboembolické příhody (ATE) jsou nežádoucí účinky potenciálně související se systémovou inhibicí VEGF. Existuje teoretické riziko arteriální tromboembolické příhody včetně mozkové příhody a infarktu myokardu po intravitreálním podání VEGF inhibitorů. Několik případů arteriálních tromboembolických příhod bylo zaznamenáno v klinické studii s pegaptanibem u pacientů s VPMD a DME (diabetický makulární edém) a nebyly zaznamenány zásadní rozdíly mezi skupinou léčenou pegaptanibem a skupinou kontrolní.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V klinických studiích nebyl hlášen žádný případ předávkování přípravkem Macugen.
Předávkování zvýšeným obsahem injekce (např. když přebytečný obsah v předplněné injekční stříkačce není před podáním vytlačen) může zvýšit nitrooční tlak (viz bod 4.8). Ošetřující lékař má vždy vytlačit přebytečné množství roztoku v souladu s pokyny v bodě 6.6.
Proto má být sledován v případě předávkování nitrooční tlak, a pokud je to ošetřujícím lékařem považováno za nezbytné, má být zahájena adekvátní léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: oftalmologika, látky k terapii očních vaskulárních poruch.
ATC kód: S01LA03
Mechanizmus účinku
Pegaptanib je pegylovaný modifikovaný oligonukleotid, který se specificky (a s vysokou afinitou) váže na extracelulární vaskulární endoteliální růstový faktor (VEGF165), a inaktivuje jej. VEGF je sekreční protein, který indukuje angiogenezi, vaskulární permeabilitu a vznik zánětu. Předpokládá se, že tyto procesy přispívají k rozvoji neovaskulární (vlhké) formy VPMD.
Farmakodynamické účinky
Na patologické neovaskularizaci v oku se podílí především izoforma VEGF165. Bylo prokázáno, že selektivní inhibice navozená pegaptanibem u zvířat je stejně efektivní při supresi patologické neovaskularizace jako inhibice neselektivní, navíc díky selektivnímu účinku nedocházelo, narozdíl od neselektivní inhibice, k poškozování fyziologické vaskulatury.
U pacientů s VPMD léčených přípravkem Macugen došlo ke zpomalení růstu průměrné velikosti léze, zpomalení choroidální neovaskularizace (CNV) a snížil se únik fluoresceinu.
Klinická účinnost a bezpečnost
Účinky pegaptanibu byly ověřovány u pacientů s neovaskulární VPMD ve dvou identických kontrolovaných dvojitě slepých studiích (EOP 1003; EOP 1004). Terapii se podrobilo celkem 1190 pacientů (892 byl podáván pegaptanib, 298 simulovaná léčba), medián věku této skupiny byl 77 let. V průměru se pacienti ze všech zkušebních skupin během prvního roku studií podrobili 8,4-8,6 aplikacím (z celkem 9 možných).
Pacienti byli randomizováni do skupin, z nichž jedné byla podávána simulovaná léčba, dalším pak 0,3 mg, 1,0 mg nebo 3 mg pegaptanibu ve formě intravitreálních injekcí v šestitýdenních intervalech po dobu 48 týdnů. U pacientů s převážně klasickými lézemi byla, po zvážení hodnotiteli, povolena fotodynamická léčba verteporfinem (PDT).
Tyto dvě studie zahrnovaly pacienty se všemi podtypy lézí neovaskulární VPMD (25% predominantně klasická, 39 % okultní a 36 % minimálně klasická), s lézemi o velikosti rovnající se ploše až 12 papil, jejichž součástí bylo až u 50% subretinální krvácení a/nebo až u 25% fibrózní jizva nebo atrofie. Pacienti podstoupili dříve maximálně jednou PDT a ve studovaném oku měli výchozí ostrost vidění v rozmezí 20/40 až 20/320.
Pegaptanib 0,3 mg vykazoval v rámci obou studií po prvním roce statisticky významný léčebný prospěch v primárním cíli, tj. podílu pacientů, u nichž došlo ke snížení ostrosti zraku o méně než 15 písmen (předem specifikovaná souhrnná analýza, pegaptanib 0,3mg 70% vs. simulovaná léčba 55%, p= 0,0001; pegaptanib 0,3 mg v EOP1003 73% vs. simulovaná léčba 59%, p=0,0105; pegaptanib 0,3 mg v EOP 1004 67% vs. simulovaná léčba 52%, p=0,0031).
Vývoj průměrné ostrosti zraku v čase; Rok 1; analýza ITT (metoda LOCF)
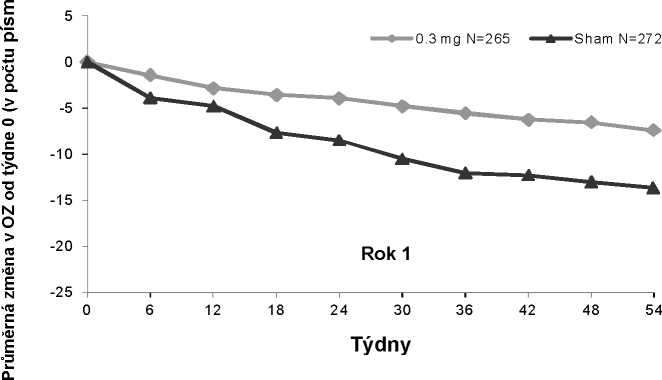
N: počet zahrnutých pacientů, OZ: ostrost zraku, Sham: simulovaná léčba
Pegaptanib 0,3 mg vykazoval léčebný přínos bez ohledu na původní typ léze, velikost léze a ostrost zraku, i bez ohledu na věk, pohlaví, pigmentaci duhovky a předchozí a/nebo současně prováděnou PDT.
Na konci prvního roku (54. týden) byli 1053 pacienti znovu randomizováni buď do skupiny, u které byla terapie přerušena, nebo do skupiny, u které se v léčbě pokračovalo do 102. týdne.
U pacientů, kteří byli randomizováni do skupiny pokračující v léčbě, obvykle léčebný přínos, včetně zachování zrakové ostrosti, přetrval do 102. týdne. Pacienti, kteří byli po prvním roce znovu randomizováni do skupiny, u které byla léčba přerušena, během druhého roku zrakovou ostrost ztratili.
Souhrn průměrných změn ostrosti zraku od výchozích hodnot do 6., 12., 54. a 102. týdne
(metoda LOCF)
|
EOP 1003 |
EOP 1004 | |||||
|
Simulace- |
Simulace- | |||||
|
0,3- |
simulace/ |
0,3- |
simulace/ | |||
|
0,3-0,3 |
ukončení |
simulace+ |
0,3-0,3 |
ukončení |
simulace+ | |
|
léčby |
ukončení |
léčby |
ukončení | |||
|
léčby |
léčby | |||||
|
N |
67 |
66 |
54 |
66 |
66 |
53 |
|
Průměrná změna |
-1,9 |
-0,0 |
-4,4 |
-1,9 |
-2,0 |
-3,4 |
|
OZ v 6. týdnu Průměrná změna | ||||||
|
OZ ve 12. týdnu Průměrná změna |
-4,3 |
-2,0 |
-4,8 |
-2,8 |
-2,2 |
-4,7 |
|
OZ v 54. týdnu Průměrná změna |
-9,6 |
-4,3 |
-11,7 |
-8,0 |
-7,6 |
-15,6 |
|
OZ ve 102. |
-10,8 |
-9,7 |
-13,1 |
-8,0 |
-12,7 |
-21,1 |
|
týdnu | ||||||
Údaje za dvouleté období naznačují, že léčba přípravkem Macugen má být zahájena co nejdříve. U onemocnění v pokročilém stádiu je třeba při zahájení a v průběhu léčby přípravkem Macugen vzít v úvahu potenciál pro alespoň částečné zachování zraku.
Terapie tímto přípravkem prováděná na obou očích současně nebyla hodnocena.
Bezpečnost a účinnost přípravku Macugen po dobu delší než 2 roky nebyla prokazována.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Macugen u všech podskupin pediatrické populace v indikaci věkem podmíněná makulární degenerace ( VPMD). Viz bod 4.2 pro informace o podání u pediatrické populace.
5.2 Farmakokinetické vlastnosti
Absorpce
U zvířat je pegaptanib po intravitreálním podání pomalu absorbován z oka do systémového oběhu. Rychlost absorpce z oka je rozhodujícím faktorem pro dostupnost pegaptanibu u zvířat, a pravděpodobně i u člověka. Průměrný zdánlivý biologický poločas pegaptanibu (± směrodatná odchylka) se po podání dávky 3 mg do jednoho oka (což je desetinásobek doporučené dávky) pohybuje okolo 10 ± 4 dnů.
Po aplikaci 3 mg do jednoho oka je u člověka maximální plazmatické koncentrace (o hodnotě cca 80 ng/ml) dosaženo v rozmezí 1 až 4 dnů. Průměrná hodnota plochy pod křivkou (AUC) se při této dávce pohybuje okolo 25 pg-h/ml. Při podávání do sklivce v šestitýdenních intervalech nedochází ke kumulaci pegaptanibu v plazmě. Při dávkách nižších než 0,5 mg/oko není pravděpodobné, že by plazmatické koncentrace pegaptanibu překročily hodnotu 10 ng/ml.
Absolutní biologická dostupnost pegaptanibu po podání do sklivce nebyla u člověka stanovena; u králíků, psů a opic dosahuje přibližně 70-100%.
Plazmatické koncentrace naměřené u zvířat, jimž byl do obou očí podáván pegaptanib v dávkách až 0,5 mg/oko, činily 0,03% až 0,15% koncentrace ve sklivci.
Distribuce.biotransformace a eliminace
U myší, potkanů, králíků, psů a opic dochází po i.v. podání pegaptanibu k jeho primární distribuci do celého objemu plazmy bez rozsáhlejšího průniku do periferních tkání. Za 24 hodin po podání radioaktivně značeného pegaptanibu do sklivce obou očí u králíků byly jako hlavní místa jeho distribuce označeny sklivec, sítnice a komorová voda. Nejvyšších koncentrací (mimo oko po aplikaci do sklivce) po intravitreální a intravenózní aplikaci u králíků dosáhl značený pegaptanib v ledvinách. Po jednorázové dávce intravitreálně i intravenózně podaného značeného pegaptanibu lze v plazmě a moči králíků detekovat nukleotid 2'-fluorouridin. Pegaptanib je metabolizován pomocí endo-a exonukleáz. U králíků je pegaptanib vylučován, nemetabolizovaný i metabolizovaný, převážně močí.
Zvláštní skupiny pacientů
Farmakokinetika pegaptanibu je podobná u ženských a mužských pacientů a dále u pacientů ve věku 50 až 90 let. Sodná sůl pegaptanibu nebyla dostatečně studována u pacientů s clearance kreatininu nižší než 20 ml/min. Při poklesu clearance kreatininu na 20 ml/min bylo pozorováno až 2,3násobné zvýšení hodnoty AUC pegaptanibu. U pacientů s clearance kreatininu vyšší než 20 ml/min, léčených doporučenou dávkou 0,3 mg sodné soli pegaptanibu, není třeba dodržovat žádná zvláštní opatření.
Farmakokinetika pegaptanibu nebyla studována u pacientů s poruchou funkce jater. Vzhledem k tomu, že i desetinásobné dávky pegaptanibu (3 mg/oko) byly dobře tolerovány, předpokládá se dobrá tolerance systémové expozice i u pacientů s poruchou funkce jater.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání a genotoxicity neodhalily žádné zvláštní riziko pro člověka. Nebyly provedeny žádné studie hodnotící karcinogenní potenciál pegaptanibu.
Při i.v. aplikaci pegaptanibu myším v dávkách od 1 do 40 mg/kg/den nebyla zjištěna toxicita pro matku, teratogenita ani zvýšení úmrtnosti myších plodů. Byl pozorován pokles tělesné hmotnosti (5 %) a minimální zpoždění osifikace článků předních končetin; a to pouze při takové expozici, která odpovídá AUC více než 300x převyšující hodnotu AUC očekávanou u člověka. Předpokládá se proto, že tyto nálezy mají omezený klinický význam. U skupiny používající dávku 40 mg/kg/den činila koncentrace pegaptanibu v amniotické tekutině 0,05% koncentrace pegaptanibu v plazmě matky. Nebyly prováděny studie reprodukční toxicity u králíků.
Neexistují údaje pro zhodnocení vlivu na páření nebo fertilitu ani u samců, ani u samic.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný
Monohydrát dihydrogenfosforečnanu sodného Heptahydrát hydrogenfosforečnanu sodného Hydroxid sodný (k úpravě pH)
Kyselina chlorovodíková (k úpravě pH)
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Před podáním musí být roztok temperován na pokojovou teplotu (25 °C).
Tento léčivý přípravek musí být zlikvidován, pokud byl uchováván při pokojové teplotě po dobu delší než dva týdny. Aby se zabránilo kontaminaci, injekční stříkačka nesmí být vyjmuta ze sáčku dříve, než je pacient připraven k aplikaci přípravku.
6.5 Druh obalu a obsah balení
Jedno balení obsahuje sáček v krabičce obsahující 1 ml předplněnou injekční stříkačku ze skla třídy I, utěsněnou elastomerní (brombutylová pryž) zarážkou pístu a připevněný píst, zajištěný plastovou svorkou. Injekční stříkačka má připevněný plastový adaptér luer lock z polykarbonátu, jehož špička je utěsněná elastomerním (brombutyl/syntetický isopren) víčkem.
Jedna předplněná injekční stříkačka obsahuje přibližně 0,25-0,27 ml roztoku.
Jedna krabička obsahuje jednu předplněnou injekční stříkačku v sáčku (jednodávkové balení).
Jehla není součástí balení.
6.6 Zvláštní opatření pro likvidaci přípravku a zacházení s ním
Přípravek Macugen je určen k jednorázovému použití. Dávku přípravku Macugen nepodávejte, je-li zakalený, obsahuje-li viditelné částice nebo je-li stříkačka poškozena, chybí-li plastová svorka nebo není-li připevněna ke stříkačce.
Před podáním se stříkačka vyjme z plastové svorky a sejme se víčko. Na luer lock adaptér se nasadí jehla o velikosti 27 nebo 30 G x !4 palce, aby bylo možno léčivý přípravek podat (viz obr. 1 níže).
UPOZORNĚNÍ: Protože předplněná injekční stříkačka obsahuje více léčivého přípravku (250-270 mikrolitrů), než je doporučená dávka (90 mikrolitrů), musí být část obsahu v injekční stříkačce před podáním zlikvidována. Následujte níže uvedené instrukce k vytlačení přebytečného obsahu před podáním injekce.
Obr 1. Před vytlačením vzduchových bublin a přebytečného obsahu

ryska značící dávku 3. kroužek zarážky pístu (horní okraj)
(tvorba bublinek se liší případ od případu)
Injekční stříkačku s jehlou směřující vzhůru je nutno zkontrolovat na přítomnost bublinek. Pokud se objeví bublinky, je nutno na injekční stříkačku lehce klepat prstem, dokud bublinky nevystoupí ke špičce injekční stříkačky
POMALU stlačte píst, aby se odstranily všechny bublinky a vytlačil se přebytečný obsah přípravku; horní okraj 3. kroužku zarážky pístu se musí krýt s předtištěnou černou ryskou značící dávku (viz obr. 2 níže). Zarážka pístu se nezatahuje zpět.
Obr 2. Po vytlačení vzduchových bublin a přebytečného obsahu

ryska značící dávku zároveň s horním okrajem 3. kroužku zarážky pístu
V tuto chvíli je možné podat zbývající obsah stříkačky.
Veškerý nepoužitý léčivý přípravek a odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00 Praha 7 Česká republika
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/05/325/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 31. ledna.2006
Datum posledního prodloužení: 19. listopadu 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Pfizer Manufacturing Belgium NV Rijksweg 12 B-2870 Puurs Belgie
B PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které
mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je třeba je předložit současně.
• Další opatření k minimalizaci rizik
Před zahájením v každém členském státě musí držitel rozhodnutí o registraci (MAH) předložit finální verzi vzdělávacích materiálů k odsouhlasení příslušné národní autoritě.
MAH musí - po diskuzi a odsouhlasení příslušnou národní autoritou v každém členském státě, kde se přípravek Macugen uvádí na trh - zajistit, aby byl při zahájení a po zahájení všem oftalmologickým klinikám, kde se předpokládá použití přípravku Macugen, poskytnut aktuální informační balíček pro lékaře, který obsahuje následující složky:
• souhrn údajů o přípravku,
• bezpečnostní příručka pro lékaře,
• videozáznam postupu intravitreální injekce,
• piktogram postupu intravitreální injekce,
• informace pro pacienta.
Bezpečnostní příručka pro lékaře má obsahovat následující klíčové údaje:
a) postup intravitreální injekce, jak byl prováděn v hlavních klinických studiích, spolu s jakýmikoli technickými vylepšeními,
b) použití jodovaného povidonu,
c) provádění očištění víčka,
d) podání anestetika k zajištění pohodlí pacienta,
e) sterilní techniky k minimalizaci rizika infekce,
f) podání antibiotik,
g) techniky intravitreální injekce,
h) klíčové známky a příznaky nežádoucích účinků intravitreální injekce, včetně endoftalmitidy, zvýšeného nitroočního tlaku, poranění sítnice, nitroočního krvácení, traumatické katarakty, hypersenzitivity a injekce nadměrného objemu,
i) regulace nitroočního tlaku,
j) léčba endoftalmitidy,
k) porozumění rizikovým faktorům vývoje endoftalmitidy,
l) hlášení závažných nežádoucích účinků (upozorňovací pomoc).
Informace pro pacienta mají obsahovat následující klíčové údaje:
m) klíčové známky a příznaky nežádoucích účinků spojených s postupem intravitreální injekce, včetně endoftalmitidy, zvýšeného nitroočního tlaku, poranění sítnice, nitroočního krvácení, traumatické katarakty, hypersenzitivity a injekce nadměrného objemu,
n) kdy je třeba naléhavě vyhledat lékařské ošetření.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU Krabička
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Macugen 0,3 mg injekční roztok pegaptanibum
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jedna předplněná injekční stříkačka poskytuje využitelné množství pro dodání jedné dávky obsahující pegaptanibum natricum 90 mikrolitrů, což odpovídá 0,3 mg volného oligonukleotidu.
3. SEZNAM POMOCNÝCH LÁTEK
Chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, hydroxid sodný a kyselina chlorovodíková (k úpravě pH), voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
Jedna dávka 0,3 mg v 90 mikrolitrech.
Balení s jednou předplněnou injekční stříkačkou, zarážkou pístu a připevněným pístem. Jehla není součástí balení.
5. ZPŮSOB A CESTA PODÁNÍ
Pouze k jednorázovému použití.
Před použitím si přečtěte příbalovou informaci. Intravitreální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
UPOZORNĚNÍ: Před podáním vytlačte přebytečný obsah stříkačky.
Horní okraj 3. kroužku zarážky pístu se musí krýt s předtištěnou černou ryskou značící dávku.
8. POUŽITELNOST
EXP:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem.
10 ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11 NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00 Praha 7 Česká republika
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/05/325/002
13. ČÍSLO ŠARŽE
Č.šarže:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16 INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato.
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU Předplněná injekční stříkačka_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Macugen 0,3 mg injekce pegaptanibum
2. CESTA PODÁNÍ
3. POUŽITELNOST
4. ČÍSLO ŠARŽE
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
Jedna dávka: 0,3 mg/90 pl
6. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU
Sáček s předplněnou injekční stříkačkou, zarážkou pístu a připevněným pístem
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Macugen 0,3 mg injekční roztok
pegaptanibum
Intravitreální podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
Jedna dávka: 0,3 mg/90 ^l
6. JINÉ
Sáček nesmí být otevřen dříve, než je pacient připraven k aplikaci léku.
UPOZORNĚNÍ: Před podáním vytlačte přebytečný obsah stříkačky.
Horní okraj 3. kroužku zarážky pístu se musí krýt s předtištěnou černou ryskou značící dávku.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Macugen 0,3 mg injekční roztok
pegaptanibum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka nebo nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Macugen a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Macugen podán
3. Jak Vám bude přípravek Macugen podáván
4. Možné nežádoucí účinky
5 Jak přípravek Macugen uchovávat
6. Obsah balení a další informace
1. Co je přípravek Macugen a k čemu se používá
Macugen je roztok, který se podává v injekci do oka. Pegaptanib, léčivá látka tohoto přípravku, blokuje aktivitu faktoru, známého jako vaskulární endoteliální růstový faktor165 (VEGF165), který se podílí na abnormální tvorbě nových krevních cév v oku.
Přípravek Macugen je určen k léčbě vlhké formy věkem podmíněné makulární degenerace (VPMD). U pacientů postižených tímto onemocněním dochází ke ztrátě zraku v důsledku poškození střední části sítnice (nazývané makula), v zadní části oka. Makula umožňuje oku rozlišovat drobné detaily, což je zapotřebí např. při řízení motorových vozidel, četbě tištěného textu a podobných činnostech.
Vlhká forma VPMD se projevuje abnormálním růstem krevních cév v sítnici a v oblasti makuly. Tyto nové cévy často krvácí a propouští tekutinu, v důsledku čehož může dojít až k vyklenutí či nadzvednutí makuly. To má za následek zkreslení až poškození zraku, které může být velmi závažné a může mít rychlý nástup. Přípravek Macugen zastavuje růst těchto abnormálních cév a zároveň brání krvácení a prosakování tekutiny. Přípravek se používá u dospělých pacientů s VPMD k léčbě růstu abnormálních cév všech typů.
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Macugen podán Přípravek Macugen Vám nesmí být podán:
Jestliže jste alergický(á) na léčivou látku nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6).
Jestliže máte aktivní infekci v oku nebo jeho okolí nebo podezření na ni.
Upozornění a opatření
Než je Vám přípravek Macugen podán, poraďte se svým lékařem.
Někdy se po injekci přípravku Macugen (během 2 následujících týdnů) vyskytne infekce nebo krvácení v oku. Je důležité tyto stavy co nejdříve rozpoznat a léčit. Oznamte ihned svému lékaři, pokud se u Vás vyskytne jakýkoliv z těchto příznaků: nepříjemné pocity nebo bolest v oku, zhoršující se zarudnutí oka, rozmazané nebo zhoršené vidění, zvýšená citlivost na světlo, zvýšené množství „mušek“, malých částic v zorném poli Vašeho oka. Jestliže Váš ošetřující lékař není z jakéhokoliv důvodu k zastižení, okamžitě kontaktujte jiného lékaře.
U některých pacientů dochází bezprostředně po aplikaci injekce na krátkou dobu ke zvýšení nitroočního tlaku. Lékař může Váš nitrooční tlak po každé injekci kontrolovat.
Po podání injekce se mohou vyskytnout závažné alergické reakce. Příznaky, které byste mohl(a) pocítit a instrukce, co dělat v takových případech, jsou popsány v bodě 4 této příbalové informace.
Děti a dospívající
Přípravek Macugen není určen k podání dětem a dospívajícím do 18 let.
Další léčivé přípravky a přípravek Macugen
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Jestliže jste těhotná nebo kojíte, myslíte si, že byste mohla být těhotná nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než bude zahájena léčba přípravkem Macugen.
• S použitím přípravku Macugen u těhotných žen nejsou žádné zkušenosti. Přípravek Macugen nesmí být používán během těhotenství, pokud potenciální přínos nepřeváží možné riziko pro nenarozené dítě. Pokud jste těhotná, poraďte se se svým lékařem před léčbou přípravkem Macugen.
• Podávání přípravku Macugen se během kojení nedoporučuje, protože není známo, zda přechází do mateřského mléka. Poraďte se se svým lékařem nebo lékárníkem dříve, než bude léčba přípravkem Macugen zahájena.
Řízení dopravních prostředků a obsluha strojů
Po aplikaci přípravku se u Vás může dočasně vyskytnout rozmazané vidění. Až do odeznění tohoto příznaku neřiďte nebo neobsluhujte stroje.
Důležité informace o některých složkách přípravku Macugen
Tento přípravek obsahuje v j edné dávce o 90 mikrolitrech méně než 1 mmol sodíku (23 mg), tj. v podstatě je bez sodíku (viz bod 6).
3. Jak Vám bude přípravek MACUGEN podáván
Všechny injekce aplikuje lékař.
Přípravek Macugen se podává jednorázovou injekcí (0,3 mg) do oka každých 6 týdnů (tzn. 9x ročně). Injekce se podává do očního sklivce, což je gelovitá hmota uvnitř oční bulvy. Váš lékař bude sledovat Váš zdravotní stav a rozhodne, jak dlouho máte být přípravkem Macugen léčen(a).
Před zahájením léčby Vás lékař může požádat, abyste použil(a) oční kapky s obsahem antibiotika, nebo pečlivě vyčistil(a) oči. Váš lékař Vám také podá lokální anestetikum (přípravek pro znecitlivění). Tím se sníží nebo se zamezí případné bolesti během aplikace injekce.
Pokud jste alergický(á) na kteroukoliv složku přípravku, prosím nezapomeňte sdělit tuto skutečnost svému lékaři.
Po aplikaci každé injekce Vám lékař může předepsat oční kapky s obsahem antibiotika (nebo jinou formu antibiotické léčby), aby se zabránilo vzniku oční infekce.
Jestliže Vám bylo podáno více přípravku Macugen, než mělo být
V případě podání nadbytečného množství přípravku Macugen může dojít k výraznému nárůstu nitroočního tlaku. Vždy, když se u Vás vyskytnou poruchy vidění, nepříjemný pocit v oku / bolest, zarudnutí oka nebo pocit na zvracení a zvracení, ihned informujte svého lékaře a sdělte mu Vaše příznaky.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Po podání injekce byly hlášeny závažné alergické reakce včetně anafylaktické reakce a angioedému, jejichž příznaky jsou popsány níže. Vyhledejte okamžitou lékařskou pomoc, pokud se u Vás bezprostředně po podání injekce vyskytne kterákoli z následujících obtíží: náhlá dušnost nebo sípot, otok úst, obličeje, rukou nebo nohou, svědění kůže, mdloby, zrychlený tep, žaludeční křeče, pocit na zvracení, zvracení nebo průjem. Četnost výskytu těchto nežádoucích účinků nelze odhadnout z dostupných údajů.
Méně často se v následujících 2 týdnech po podání přípravku Macugen může vyskytnout infekce ve vnitřní části oka. Příznaky, které můžete zaznamenat, jsou popsány v bodu 2 této příbalové informace (Upozornění a opatření). Přečtěte si bod 2. Dozvíte se, co máte udělat, zaznamenáte-li některý z těchto příznaků.
Další možné nežádoucí účinky jsou následující:
Velmi časté (mohou postihnout více než 1 z 10 pacientů):
Tyto nežádoucí účinky s největší pravděpodobností souvisejí se způsobem aplikace, spíše než s negativním působením přípravku:
• zánět oka,
• bolest oka,
• zvýšení nitroočního tlaku,
• drobné tečky na rohovce (keratitis punctata),
• malé částečky nebo tečky ve Vašem zorném poli (tzv. „mušky“).
Časté (mohou postihnout až 1 z 10 pacientů):
Další časté oční nežádoucí účinky, které mohou být způsobeny přípravkem nebo způsobem aplikace přípravku jsou:
• rozmazané vidění,
• poruchy zraku,
• nepříjemný pocit v oku,
• zhoršené vidění,
• zvýšená citlivost na světlo, mžitky před očima,
• mžitky před očima,
• krvácení v okolí oka (periorbitální krvácení),
• překrvení oka (krvácení do spojivek),
• poškození gelovité hmoty uvnitř oka (porucha sklivce), jako je posunutí nebo odtržení
(odchlípení sklivce),
• šedý zákal čočky (katarakta),
• poruchy vrstvy na povrchu oka (rohovky),
• otok nebo zánět očního víčka, otok vnitřní strany víček nebo okraje oka (spojivka),
• zánět oka, slzení, zánět spojivky (konjunktivitida), suché oko, výtok z oka, podráždění oka, svědění oka, zčervenání oka nebo rozšíření zornice.
Jiné časté nežádoucí účinky (mimo nežádoucí účinky oční), které mohou být způsobeny přípravkem nebo způsobem aplikace přípravku, jsou:
• bolesti hlavy,
• výtok z nosu.
Méně časté (mohou postihnout až 1 ze 100 pacientů)
Méně časté oční nežádoucí účinky, které mohou být způsobeny léčivou látkou nebo způsobem aplikace přípravku, zahrnují:
• zánět oka nebo jeho povrchu,
• krvácení do oka nebo do jeho vnitřní části (sklivce),
• napětí v oku,
• zánět rohovky (keratitida),
• usazeniny na oku nebo na jeho povrchu (rohovka), usazeniny na očním pozadí,
• svědění očních víček,
• poruchy reakce oka na světlo (poruchy zornicového reflexu),
• drobné narušení střední části povrchu oka (rohovky),
• pokleslá víčka,
• jizvička uvnitř oka (jizva sítnice),
• malá bulka zánětlivého původu na víčku (ječné zrno),
• snížení nitroočního tlaku,
• reakce v místě vpichu injekce, puchýřky v místě vpichu,
• odchlípení nebo odtržení vrstvy na očním pozadí (sítnice),
• poruchy zornice nebo zabarvené části oka (duhovka),
• uzávěr retinální tepny,
• převrácení očního víčka, poruchy oční hybnosti, podráždění víčka,
• krev v oku, změny v zabarvení oka, usazeniny v oku,
• zánět oka (duhovky),
• vyklenutí disku zrakového nervu,
• deformita pupily,
• uzávěr žíly v zadní části oka,
• výtok sklivce (vnitřní gelovité hmoty oka).
Méně časté nežádoucí účinky, které nesouvisí se zrakem a které mohou být způsobeny léčivou látkou nebo způsobem aplikace přípravku, zahrnují:
• noční můry, deprese, hluchota, závratě,
• bušení srdce, zvýšení krevního tlaku, rozšíření aorty (srdečnice),
• zánět horních cest dýchacích, zvracení, poruchy trávení,
• podráždění a záněty kůže, změna barvy vlasů, vyrážka, svědění,
• noční pocení, bolesti zad, únava, třesavka, zvýšená citlivost na dotek, bolesti na hrudi, náhlá horečka, příznaky podobné chřipce (celková zchvácenost a bolesti),
• zvýšené hodnoty jaterních enzymů, oděrka.
Po opakovaných injekcích do oka existuje malé riziko trvalého mírného zvýšení tlaku uvnitř oka. Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Macugen uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a na krabičce za zkratkou EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Přípravek musí být zlikvidován, pokud je uchováván při pokojové teplotě po dobu delší než 2 týdny.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Macugen obsahuje
- Léčivou látkou je pegaptanibum. Jedna jednodávková předplněná stříkačka obsahuje dávku pegaptanibum 0,3 mg v 90 mikrolitrech.
- Dalšími složkami jsou chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, hydroxid sodný a kyselina chlorovodíková (k úpravě pH) a voda na injekci. Další informace týkající se obsahu sodíku v přípravku Macugen, viz bod 2.
Jak přípravek Macugen vypadá a co obsahuje toto balení
Přípravek Macugen injekční roztok je dodáván v balení pro jednu dávku.
Jedno balení obsahuje sáček v krabičce, obsahující předplněnou injekční stříkačku ze skla třídy I naplněnou 0,25-0,27 ml roztoku, utěsněnou elastomerní zarážkou pístu a připevněný píst, zajištěný plastovou svorkou. Injekční stříkačka má připevněný plastový adaptér luer lock z polykarbonátu, jehož špička je utěsněná elastomerním víčkem.
Jehla není součástí balení.
Držitel rozhodnutí o registraci
PharmaSwiss Česká republika s.r.o. Jankovcova 1569/2c 170 00 Praha 7 Česká republika
Výrobce
Pfizer Manufacturing Belgium NV Rijksweg 12 B-2870 Puurs Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
Belgie/Belgique/Belgien
Bausch & Lomb Pharma nv/sa, Belgium Tél/Tel: + 32 (0)3 280 82 84
Bt^rapnn
PharmaSwiss EOOD Ten.: + 359 2 89 52 110
Lietuva
PharmaSwiss UAB Tel. + 370 5 279 0762
Luxembourg/Luxemburg
Bausch & Lomb Pharma nv/sa, Belgium Tél/Tel: + 32 (0)3 280 82 84
|
Česká republika PharmaSwiss Česká republika s.r.o. Tel: + 420 234 719 600 |
Magyarország Valeant Pharma Magyarország Kft. Tel. +36 1 345 5900 |
|
Danmark Bausch & Lomb Nordic AB Tlf: 80 88 82 68 Tlf (fra udlandet): +46 8 616 95 85 |
Malta Laboratoire Chauvin, France Tél: + 33 (0)4 67 12 30 30 |
|
Deutschland Bausch & Lomb GmbH Tel: + 49 (0)30 33093 0 |
Nederland Bausch & Lomb Pharma nv/sa, Belgium Tel: + 32 (0)3 280 82 84 |
|
Eesti PharmaSwiss Eesti OU Tel: +372 6 827 400 |
Norge Bausch & Lomb Nordic AB Tlf: 800 19 841 Fra utlandet Tlf.: +46 8 616 95 85 |
|
EXlába Pharmaswiss Hellas A.E. T^U +30 210 8108 460 |
Osterreich Bausch & Lomb GmbH Tel: + 49 (0)30 33093 0 |
|
Espaňa Bausch & Lomb, S.A. Tel: + 34 91 657 63 00 |
Polska Valeant sp. z o.o. sp. j. Tel.: +48 17 865 51 00 |
|
France Laboratoire Chauvin SAS Tél: + 33 (0)4 67 12 30 30 |
Portugal Bausch & Lomb, S.A. (Sucursal Portugal) Tel: + 351 21 424 15 10 |
|
Hrvatska PharmaSwiss d.o.o. Tel: +385 1 6311 833 |
Románia Valeant Pharma S.R.L. Tel: +40 374 102 600 |
|
Ireland Bausch & Lomb UK Ltd. Tel: +44 (0) 1748 828864 |
Slovenija PharmaSwiss d.o.o. Tel: + 386 1 2364 700 |
|
Ísland Bausch & Lomb UK Ltd. Sími frá útlondum: +44 (0) 1748 828864 |
Slovenská republika Valeant Slovakia s.r.o. Tel: +421 2 3233 4900 |
|
Italia Bausch & Lomb-IOM S.p.A. Tel: + 39 (0)2 27407300 |
Suomi/Finland Bausch & Lomb Nordic AB Puh./Tel: 0800 773 851 Ulkomailta/Frán utomlands: +46 8 616 95 85 |
|
Kúrcpog Kypropharm Ltd. TnU + 357 22 43 46 99 |
Sverige Bausch & Lomb Nordic AB Tel: 020 088 3496 Frán utomlands: +46 8 616 95 85 |
|
Latvija SIA PharmaSwiss Latvia Tel: + 371 67502185 |
United Kingdom Bausch & Lomb UK Ltd. Tel: +44 (0) 1748 828864 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese: http://www.ema.europa.eu/
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
UPOZORNĚNÍ: Protože předplněná injekční stříkačka obsahuje více léčivého přípravku (250-270 mikrolitrů), než je doporučená dávka (90 mikrolitrů), musí být část obsahu v injekční stříkačce před podáním zlikvidována. Následujte níže uvedené instrukce k vytlačení přebytečného obsahu před podáním injekce.
Obr 1. Před vytlačením vzduchových bublinek a přebytečného obsahu

ryska značící dávku
3. kroužek zarážky pístu (horní okraj)
(tvorba bublinek se liší případ od případu)
Injekční stříkačku s jehlou směřující vzhůru je nutno zkontrolovat na přítomnost bublinek. Pokud se objeví bublinky, je nutno na injekční stříkačku jemně klepat prstem, dokud bublinky nevystoupí ke špičce injekční stříkačky.
POMALU stlačte píst, aby se odstranily všechny bublinky a vytlačil se přebytečný obsah přípravku; horní okraj 3. kroužku zarážky pístu se musí krýt zároveň s předtištěnou černou ryskou značící dávku (viz obr. 2 níže). Zarážka pístu se nesmí zatáhnout zpět.
Obr 2. Po vytlačení vzduchových bublin a přebytečného obsahu

ryska značící dávku zároveň s horním okrajem 3. kroužku zarážky pístu
V tuto chvíli je možné podat zbývající obsah stříkačky.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
29