Lynparza 50 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Lynparza 50 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje olaparibum 50 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka
Bílá neprůhledná tvrdá tobolka o velikosti 0 s označením “OLAPARIB 50 mg” a logem AstraZeneca černým inkoustem.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Lynparza je indikován v monoterapii k udržovací léčbě dospělých pacientek s relabujícím high-grade serózním epiteliálním nádorem vaječníku, vejcovodu, nebo primárně peritoneálním s mutací BRCA (zárodečnou a/nebo somatickou) citlivým na léčbu platinou, u nichž došlo k relapsu a které odpovídají (úplně nebo částečně) na chemoterapii založenou na platině.
4.2 Dávkování a způsob podání
Léčbu přípravkem Lynparza by měl zahájit a kontrolovat lékař se zkušenostmi s protinádorovými léčivými přípravky.
Přítomnost mutace genu (buď zárodečná nebo v nádoru) náchylnosti k rakovině prsu (BRCA) musí být u pacientky potvrzena před zahájením léčby přípravkem Lynparza. Přítomnost mutace BRCA by měla být stanovena v osvědčené laboratoři za použití validovaných kontrolních metod (viz bod 5.1).
Existují pouze omezené údaje u pacientek s nádory se somatickou mutací BRCA (viz bod 5.1).
Genetické poradenství u pacientek s mutacemi BRCA se provádí podle místních doporučení.
Dávkování
Doporučená dávka přípravku Lynparza je 400 mg (osm tobolek) dvakrát denně, což odpovídá celkové denní dávce 800 mg.
Léčba pacientek přípravkem Lynparza má být zahájena nejpozději 8 trýdnů po podání poslední dávky režimu s deriváty platiny.
Doporučuje se pokračovat v léčbě do té doby, než dojde k progresi základního onemocnění. Nejsou k dispozici údaje o opětovném zahájení léčby přípravkem Lynparza po dalším relapsu onemocnění (viz bod 5.1).
Vynechaná dávka
Pokud pacientka zapomene užít dávku přípravku Lynparza, má pokračovat až následující pravidelnou dávkou.
Úprava dávkování v důsledku nežádoucích účinků
V případě výskytu nežádoucích účinků, jako je nauzea, zvracení, průjem a anémie, může být léčba přerušena a lze zvážit snížení dávkování (viz bod 4.8).
Doporučuje se snížit dávkování na 200 mg dvakrát denně (odpovídající celkové denní dávce 400 mg).
Pokud je potřeba výsledné dávkování ještě snížit, může být zváženo snížení na 100 mg dvakrát denně (odpovídající celkové denní dávce 200 mg).
Úprava dávkování s ohledem na souběžné podávání inhibitorů CYP3A
Souběžné podávání silných a středně silných inhibitorů CYP2A se nedoporučuje a má se uvažovat o alternativních látkách. Pokud musí být silné a středně silné inhibitory CYP3A podávány souběžně, doporučená snížená dávka olaparibu je 150 mg dvakrát denně (ekvivalentní celkové denní dávce 300 mg) se silným inhibitorem CYP3A nebo 200 mg dvakrát denně (ekvivalentní celkové denní dávce 400 mg) se středně silným inhibitorem CYP3A (viz body 4.4 a 4.5).
Starší pacienti
Úprava počáteční dávky u starších pacientek není potřebná. Klinické údaje o použití přípravku u pacientek ve věku 75 let a starších jsou omezené.
Pacientky s poruchou funkce ledvin
Vliv poruchy funkce ledvin na expozici přípravku Lynparza nebyl zkoumán. Přípravek Lynparza může být podáván pacientkám s mírnou poruchou funkce ledvin (clearance kreatininu >50 ml/min).
Použití přípravku Lynparza u pacientek se středně těžkou (clearance kreatininu <50 ml/min) nebo těžkou poruchou funkce ledvin (clearance kreatininu < 30 ml/min) není doporučeno vzhledem k tomu, že údaje o použití přípravku u těchto pacientek jsou omezené a bezpečnost a účinnost nebyla stanovena.
Přípravek Lynparza lze použít u pacientek se středně těžkou až těžkou poruchou funkce ledvin pouze tehdy, pokud prospěch z léčby převáží nad možnými riziky. Pacientky musí být pečlivě sledovány z pohledu funkce ledvin a výskytu nežádoucích účinků.
Pacientky s poruchou funkce jater
Vliv poruchy funkce jater na expozici přípravku Lynparza nebyl zkoumán. Použití přípravku Lynparza u pacientek s poruchou funkce jater (hladina sérového bilirubinu 1,5krát vyšší než horní hranice normálních hodnot) není doporučeno vzhledem k tomu, že bezpečnost a účinnost nebyla stanovena.
Pacientky jiné než bílé rasy
Klinické údaje o použití u pacientek jiné než bílé rasy jsou omezené. Úprava dávkování na základě etnicity však není nutná (viz bod 5.2).
Pacientky s výkonnostním stavem 2 až 4
Existují velmi omezené klinické údaje u pacientek s výkonnostním stavem 2 až 4.
Pediatrická populace
Bezpečnost a účinnost přípravku Lynparza u dětí a dospívajících nebyla dosud stanovena.
Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Lynparza je určen k perorálnímu podání.
Vzhledem k vlivu potravy na absorpci olaparibu, by pacientky měly užívat přípravek Lynparza alespoň jednu hodinu po jídle a nejíst nejlépe až dvě hodiny po užití přípravku.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Kojení v průběhu léčby a 1 měsíc po podání poslední dávky (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Hematologická toxicita
U pacientek léčených olaparibem byla hlášena hematologická toxicita zahrnující klinické diagnózy a/nebo laboratorní nálezy nejčastěji mírné nebo střední (CTCAE stupeň 1 nebo 2) anémie, neutropenie, trombocytopenie a lymfopenie. U pacientek, u kterých se projevila hematologická toxicita předchozí protinádorové léčby, nemá být terapie přípravkem Lynparza zahájena dříve, než se z tohoto stavu zotaví (hladiny hemoglobinu, trombocytů a neutrofilů mají být v normálním rozmezí či rozmezí odpovídajícím CTCAE stupni 1). Doporučuje se kompletní vyšetření krevního obrazu na počátku léčby, následně jednou měsíčně po dobu prvních 12 měsíců léčby a nadále pravidelně i po skončení tohoto období proto, aby mohly být zachyceny klinicky významné změny jeho parametrů během léčby.
Pokud se u pacientky rozvine těžká hematologická toxicita nebo potřeba krevní transfuze, léčba přípravkem Lynparza má být přerušena a mělo by být provedeno odpovídající hematologické vyšetření. Pokud krevní parametry zůstávají abnormální i 4 týdny po přerušení léčby přípravkem Lynparza, doporučuje se provést analýzu kostní dřeně a/nebo cytogenetické vyšetření krve.
Myelodysplastický syndrom/akutní myeloidní leukémie
U nízkého počtu pacientek, kterým byl podáván přípravek Lynparza samotný nebo v kombinaci s jinými protinádorovými přípravky, byl hlášen rozvoj myelodysplastického syndromu/akutní myeloidní leukémie (MDS/AML); většina těchto případů byla fatálních. Doba léčby olaparibem u pacientek, u kterých se vyvinul MDS/AML, byla < 6 měsíců až >2 roky. Jednalo se o typické případy sekundárního MDS/AML související s protinádorovou léčbou. Všechny pacientky měly potenciálně komplikující faktory pro vývoj MDS/AML; většina případů se vyskytla u nositelek zárodečné mutace BRCA a některé pacientky měly anamnézu předchozího nádorového onemocnění nebo dysplazie kostní dřeně. Všechny dříve užívaly chemoterapii založenou na derivátech platiny a mnoho z nich též jiné látky poškozující DNA nebo radioterapii. Pokud je v průběhu léčby přípravkem Lynparza potvrzen rozvoj MDS a/nebo AML, pacientka má být odpovídajícím způsobem léčena. Léčba přípravkem Lynparza má být přerušena v případě, že je doporučeno nasadit další protinádorovou léčbu. Přípravek Lynparza se nemá podávat v kombinaci s jinou protinádorovou léčbou.
Pneumonitida
U malého počtu pacientek léčených olaparibem byl hlášen rozvoj pneumonitidy a některé z těchto případů skončily fatálně. Případy pneumonitidy neměly ucelený klinický projev a jejich rozvoj mohl souviset i s mnohými predispozičními faktory (rakovina plic a/nebo metastázy v plicích, základní plicní onemocnění, anamnéza kouření a/nebo předchozí chemoterapie a radioterapie). Pokud se u pacientek objeví nové nebo se zhorší stávající respirační příznaky, jako například dušnost, kašel a horečka, nebo se objeví abnormality na radiologických snímcích, léčba přípravkem Lynparza má být přerušena a pacientka by měla být okamžitě vyšetřena. Pokud je pneumonitida potvrzena, léčba přípravkem Lynparza má být přerušena a pacientka má být odpovídajícím způsobem léčena.
Embryofetální toxicita
Vzhledem k mechanizmu účinku (inhibice PARP), může olaparib podávaný těhotným ženám způsobit poškození plodu. V neklinických studiích prováděných na laboratorních potkanech bylo zjištěno, že olaparib má při expozicích nižších, než k jakým dochází u člověka při doporučeném dávkování 400 mg dvakrát denně, nežádoucí vliv na embryofetální přežití a vyvolává vznik fetálních malformací.
Těhotenství a antikoncepce
Přípravek Lynparza se nesmí podávat v průběhu těhotenství a ženám ve fertilním věku, které neužívají spolehlivou antikoncepci v průběhu léčby a dále ještě 1 měsíc po podání poslední dávky přípravku Lynparza (viz bod 4.6).
Interakce
Souběžné podávání olaparibu se silnými a středně silnými inhibitory CYP3A se nedoporučuje (viz bod 4.5). Pokud se silné nebo středně silné inhibitory CYP3A musí podávat souběžně, dávka olaparibu se má snížit (viz body 4.2 a 4.5).
Souběžné podávání olaparibu se silnými induktory CYP3A se nedoporučuje (viz bod 4.5). V případě, že pacientka užívající olaparib má být léčena silným induktorem CYP3A, předepisující lékař si má být vědom, že účinnost olaparibu může být zásadně snížena (viz bod 4.5).
V případě, že pacientka, která užívá olaparib, má být léčena inhibitory P-gp, je třeba pečlivě monitorovat nežádoucí účinky olaparibu a doporučuje se korigovat tyto nežádoucí účinky snížením dávky (viz bod 4.2).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Farmakodynamické interakce
Z výsledků klinických studií vyplývá, že při současném užití olaparibu s jinými protinádorovými léčivými přípravky včetně látek poškozujících DNA, dochází k potenciaci a prodloužení myelosupresivního účinku. Doporučená dávka přípravku Lynparza pro monoterapii není vhodná pro kombinaci s dalšími protinádorovými léčivými přípravky.
Kombinace olaparibu s vakcínami nebo imunosupresivy nebyla studována. Pokud se tyto léčivé přípravky podávají souběžně s olaparibem, je třeba opatrnosti a pacientky by měly být pečlivě sledovány.
Farmakokinetické interakce
Vliv jiných léčivých přípravků na olaparib
Izoenzymy převážně zodpovědné za metabolickou clearance olaparibu jsou CYP3A4/5. Klinická studie hodnotící vliv rifampicinu, známého induktoru CYP3A, prokázala, že souběžné podávání s olaparibem snížilo průměrnou hodnotu Cmax o 71 % (poměr léčeb: 0,29; 90% CI: 0,24-0,33) a průměrnou hodnotu AUC o 87 % (poměr léčeb: 0,13; 90% CI: 0,11-0,16). Z tohoto důvodu se nedoporučuje, aby se známé silné induktory tohoto isoenzymu (např. fenytoin, rifampicin, rifapentin, karbamazepin, nevirapin, fenobarbital a třezalka tečkovaná) podávaly s olaparibem, neboť je možné, že se podstatně sníží účinnost olaparibu (viz bod 4.4). Velikost účinku středně silných až silných induktorů (např. efavirenz, rifabutin) na expozici olaparibu nebyla stanovena, a proto se souběžné podávání s olaparibem nedoporučuje.
Klinická studie hodnotící vliv itrakonazolu, známého inhibitoru CYP3A, prokázala, že souběžné podávání s olaparibem zvýšilo průměrnou hodnotu Cmax olaparibu 1,42násobně (90% CI: 1,33-1,52) a průměrnou hodnotu AUC 2,70násobně (90% CI: 2,44-2,97). Z tohoto důvodu se nedoporučuje souběžně podávat známé silné inhibitory (např. itrakonazol, telithromycin, klarithromycin, proteázové inhibitory potencované ritonavirem nebo kobicistatem, boceprevirem, telaprevirem) nebo středně silné inhibitory (např. erythromycin, diltiazem, flukonazol, verapamil) tohoto isoenzymu s olaparibem (viz bod 4.4). Pokud se silné nebo středně silné inhibitory CYP3A musí podávat souběžně s olaparibem, dávka olaparibu se má snížit.
U silných inhibitorů CYP3A se doporučuje snížit dávku olaparabu na 150 mg dvakrát denně (odpovídá celkové denní dávce 300 mg), resp. na 200 mg dvakrát denně (odpovídá celkové denní dávce 400 mg) u středně silných inhibitorů CYP3A (viz body 4.2 a 4.4). V průběhu léčby olaparibem se též nedoporučuje pít grapefruitovou šťávu.
V prostředí in vitro je olaparib substrátem pro efluxní přenašeč P-gp, a proto inhibitory P-gp mohou zvyšovat expozici olaparibu (viz bod 4.4).
Vliv olaparibu na jiné léčivé přípravky
V podmínkách in vitro olaparib inhibuje CYP3A4 a předpokládá se, že je mírným inhibitorem CYP3A
v podmínkách in vivo. Proto je třeba dbát zvýšené opatrnosti v případě citlivých substrátů pro CYP3A4 nebo substrátů s úzkým terapeutickým rozmezím (např. simvastatin, cisaprid, cyklosporin, námelové alkaloidy, fentanyl, pimozid, sirolimus, takrolimus a kvetiapin), pokud jsou podávány souběžně s olaparibem. Pacienty souběžně léčené substráty pro CYP3A s úzkým terapeutickým rozmezím a olaparibem se doporučuje vhodným způsobem klinicky monitorovat.
V podmínkách in vitro byla prokázána indukce CYP1A2, 2B6 a 3A4 v pravděpodobně klinicky významném rozsahu. Potenciál olaparibu indukovat CYP2C9, CYP2C19 a P-gp nelze vyloučit. Z tohoto důvodu může olaparib při souběžném podávání se substráty těchto metabolických enzymů a transportního proteinu snížit jejich expozice. Účinnost hormonální antikoncepce může být při současném podávání s olaparibem snížena (viz též body 4.4 a 4.6).
V podmínkách in vitro olaparib inhibuje efluxní transportér P-gp (IC50 = 76 ^M), a proto nelze vyloučit klinicky relevantní lékové interakce se substráty pro P-gp (např. simvastatin, pravastatin, dabigatran, digoxin, kolchicin). U pacientů, kteří jsou souběžně léčeni těmito léčivými přípravky, se doporučuje provádět vhodné klinické monitorování.
V podmínkách in vitro bylo prokázáno, že olaparib inhibuje OATP1B1, OCT1, OCT2, OAT3, MATE1 a MATE2K. Nemůže být vyloučeno, že olaparib může zvyšovat expozici substrátů pro OATP1B1 (např. bosentan, glibenklamid, repaglinid, statiny a valsartan), OCT1 (např. metformin), OCT2 (např. sérový kreatinin), OAP3 (např. furosemid a methotrexát), MATE1 (např. metformin) a MATE2K (např. metformin). Zvýšené opatrnosti je třeba dbát zejména v situaci, kdy je olaparib podáván souběžně s některým ze statinů.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/antikoncepce u žen
Ženy ve fertilním věku nesmí otěhotnět v průběhu léčby přípravkem Lynparza a nesmí být těhotné na počátku léčby. U všech premenopauzálních žen musí být před zahájením léčby proveden těhotenský test. Ženy ve fertilním věku musí v průběhu léčby a ještě 1 měsíc po podání poslední dávky přípravku Lynparza užívat spolehlivou antikoncepci. Vzhledem ktomu, že nelze vyloučit snížení expozice substrátů pro CYP3A olaparibem cestou enzymové indukce, může být účinnost hormonální antikoncepce snížena při souběžném užívání olaparibu. Proto je třeba zvážit dodatečnou nehormonální metodu antikoncepce a pravidelné provádění těhotenských testů v průběhu léčby (viz bod 4.5).
Studie na zvířatech prokázaly reprodukční toxicitu včetně závažných teratogenních účinků a účinků na embryofetální přežívání u laboratorních potkanů při systémových expozicích nižších než při použití terapeutických dávek u člověka (viz bod 5.3). Neexistují údaje o použití olaparibu u těhotných žen, avšak s ohledem na mechanismus účinku olaparibu, by přípravek Lynparza neměl být užíván v průběhu těhotenství a ženami ve fertilním věku, které neužívají spolehlivou metodu antikoncepce v průběhu léčby a ještě 1 měsíc po podání poslední dávky přípravku Lynparza (viz předchozí odstavec: „Ženy ve fertilním věku/antikoncepce u žen“ ohledně dalších informací o kontrole početí a těhotenských testech).
Kojení
Neexistují studie na zvířatech studující vylučování olaparibu do mateřského mléka. Není známo, zda jsou olaparib nebo jeho metabolity vylučovány do mateřského mléka u člověka. Přípravek Lynparza je kontraindikován v průběhu kojení a ještě 1 měsíc po podání poslední dávky s ohledem na farmakologické vlastnosti přípravku (viz bod 4.3).
Fertilita
Neexistují žádné klinické údaje o vlivu na fertilitu. Studie fertility u samic laboratorních potkanů však prokázala negativní vlivy na embryofetální přežití (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Během léčby přípravkem Lynparza byly hlášeny případy tělesné slabosti, únavy a závratí a pacientky, u nichž se tyto stavy rozvinou, by měly být při řízení nebo obsluze strojů obezřetné.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Monoterapie olaparibem byla provázena nežádoucími účinky nejčastěji mírné nebo střední závažnosti (CTCAE 1 nebo 2), které ve většině případů nevedly k nutnosti přerušit léčbu. Nejčastěji pozorovanými nežádoucími účinky v klinických studiích u pacientek užívajících monoterapii olaparibem (> 10 %) byly nauzea, zvracení, průjem, dyspepsie, únava, bolesti hlavy, poruchy chuti, snížená chuť k jídlu, závratě, anémie, neutropenie, lymfopenie, zvýšení středního objemu erytrocytů a zvýšený kreatinin.
Tabulkové seznamy nežádoucích účinků
V průběhu klinických studií se u pacientek užívajících monoterapii přípravkem Lynparza vyskytly níže uvedené nežádoucí účinky. Jejich četnost je uvedena podle klasifikace četnosti CIOMS III a dále jsou seřazeny podle MedDRA SOC (třídy orgánových systémů) a preferenčních termínů. Četnosti výskytu nežádoucích účinků jsou definovány jako: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100); vzácné (> 1/10000 až < 1/1000); velmi vzácné (< 1/10000). Tato část obsahuje pouze údaje získané z dokončených studií se známou expozicí pacientek.
Představuje četnost laboratorních nálezů, nikoliv četnost hlášených nežádoucích účinků.
Tabulka 1 Tabulkový přehled nežádoucích účinků
|
Nežádoucí účinek | ||
|
MedDRA třídy orgánových systémů |
Frekvence všech stupňů CTCAE |
Četnost CTCAE stupeň 3 a vyšší |
|
Poruchy metabolismu a výživy |
Velmi časté Snížení chuti k jídlu |
Méně časté Snížení chuti k jídlu |
|
Poruchy nervového systému |
Velmi časté Bolest hlavy, závrať, poruchy chuti |
Méně časté Závrať, bolest hlavy |
|
Gastrointestinální poruchy |
Velmi časté Nauzea, zvracení, průjem, dyspepsie Časté Bolest horní části břicha, stomatitida |
Časté Nauzea, zvracení, průjem Méně časté Bolest horní části břicha, stomatitida |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté Únava (včetně slabosti) |
Časté Únava (včetně slabosti) |
|
Vyšetření |
Velmi časté Anémie (snížené množství hemoglobinu)3, b Neutropenie (snížení absolutního počtu neutrofilů)a, b Lymfopenie (snížený počet lymfocytů)a, b Zvýšení hladiny kreatininu v krvia, d Zvýšení středního objemu erytrocytů1, c Časté Trombocytopenie (snížený počet krevních destiček) a, b |
Velmi časté Anémie (snížené množství hemoglobinu)a, b Lymfopenie (snížený počet lymfocytů)a, b Časté Neutropenie (snížení absolutního počtu neutrofilů)a, b Trombocytopenie (snížený počet krevních destiček) a, b Méně časté Zvýšení hladiny kreatininu v krvi^ d |
a
b
Snížení množství hemoglobinu, absolutního počtu neutrofilů, trombocytů a lymfocytů odpovídající CTCAE stupni 2 nebo vyšší.
c
d
Zvýšení středního objemu erytrocytů z původní hodnoty nad hodnotu ULN (horní hranice normálních hodnot). Hodnoty se po přerušení léčby vrátily do normálního rozmezí a klinické důsledky nebyly zaznamenány.
Údaje získané z dvojitě zaslepené placebem kontrolované studie ukázaly medián zvýšení až 23 % (procentuální změna ve srovnání s výchozí hodnotou), které bylo po dobu studie stálé a k původním hodnotám se navrátilo po přerušení léčby, aniž by byly pozorovány klinické důsledky. Původní hodnoty odpovídaly u 90 % pacientek CTCAE stupni 0 a u 10 % CtCAE stupni 1.
Popis vybraných nežádoucích účinků
Gastrointestinální toxicita je při terapii olaparibem hlášena často, většinou je nízkého stupně (CTCAE stupeň 1 nebo 2), je intermitentní a ustupuje po přerušení užívání přípravku, snížení dávky a/nebo souběžné léčbě (např. antiemetická terapie). Antiemetická profylaxe není nutná.
Anémie a další hematologické toxicity jsou většinou nízkého stupně (CTCAE stupeň 1 nebo 2), avšak jsou hlášeny i případy CTCAE stupně 3 a vyšších. Doporučuje se kompletní vyšetření krevního obrazu na počátku léčby, následně jednou měsíčně po dobu prvních 12 měsíců léčby a dále pravidelně i po skončení tohoto období proto, aby mohly být během léčby zachyceny klinicky významné změny některého z parametrů, které by vyžadovaly přerušení léčby, snížení dávky a/nebo další léčbu.
Pediatrická _ populace U dětí nebyly provedeny žádné studie.
Jiné zvláštní _populace
U starších pacientek (věk > 75 let) a u pacientek jiné než bílé rasy jsou k dispozici pouze omezené údaje o bezpečnosti.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Neexistuje specifická léčba při předávkování přípravkem Lynparza a příznaky předávkování nejsou stanoveny. Lékaři by v případě předávkování měli dodržovat obecná podpůrná opatření a poskytovat symptomatickou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: jiné antineoplastické látky, ATC kód: L01XX46 Mechanismus účinku a farmakodynamické účinky
Přípravek Lynparza je silným inhibitorem skupiny lidských enzymů poly (ADP-ribózo) polymeráza (PARP-1, PARP-2 a PARP-3) a byl prokázán jeho inhibiční účinek na růst určitých nádorových buněčných linií v prostředí invitro a na růst nádoru v prostředí invivo jak v monoterapii, tak v kombinaci se zavedenými metodami chemoterapie.
Enzymy PARP jsou potřebné k účinné reparaci jednovláknových zlomů DNA a pro uskutečnění opravy indukované PARP je důležité, aby se PARP po modifikaci chromatinu sám pozměnil, odloučil se tak od DNA a usnadnil tím přístup enzymům zprostředkovávajícím opravy DNA prostřednictvím excize bází (base excision repair, BER). Pokud je přípravek Lynparza navázán na aktivní místo komplexu PARP-DNA, brání odloučení PARP a udržuje tak vazbu na DNA, což znemožňuje reparaci. V množících se buňkách to vede k dvouvláknovým zlomům DNA (double strand breaks, DSB) ve chvíli, kdy se replikační vidlice setkává s komplexem PARP-DNA. V normálních buňkách jsou tyto dvouvláknové zlomy DNA účinně reparovány pomocí homologní rekombinační opravy (homologous recombination repair, HRR), pro kterou jsou však potřebné funkční geny BRCA1 a 2. Pokud nejsou funkční geny BRCA1 nebo 2 přítomny, dvouvláknové zlomy DNA nemohou být pomocí HRR opraveny. Namísto toho se aktivují alternativní cesty náchylné ke vzniku chyb jako například cesta nehomologního připojení konců (non-homologous end joining, NHEJ), které vedou ke zvýšení genomové nestability. Po několika cyklech replikace může genomová nestabilita dosáhnout neúnosné úrovně a vyústit tak ve smrt nádorové buňky, neboť nádorové buňky obsahují větší množství poškození DNA oproti buňkám normálním.
V invivo modelech s chybějící funkcí BRCA vedl olaparib podávaný v návaznosti na léčbu platinou k oddálení nádorové progrese a k prodloužení celkové doby přežití ve srovnání se samotnou léčbou platinou.
Detekce mutace BRCA
Léčba přípravkem Lynparza je vhodná pro pacientky s potvrzenou patogenní nebo suspektní patogenní mutací BRCA (tj. mutací, která narušuje normální funkci genu) v zárodečné buněčné linii nebo nádorové buněčné linii (diagnostikovanou pomocí schváleného testu).
Klinická účinnost
Bezpečnost a účinnost olaparibu v udržovací terapii pacientek léčených pro relabující high-grade serózní ovariální karcinom, včetně vejcovodu nebo primárně peritoneální senzitivní k platině (platinum-sensitive relapsed, PSR) byla hodnocena v randomizované dvojitě zaslepené placebem kontrolované studii fáze II (studie 19), která následovala po dvou nebo více léčebných cyklech chemoterapie obsahujících platinu. Studie srovnávala účinnost udržovací léčby olaparibem užívaným do progrese ve srovnání s placebem u 265 pacientek (136 pacientek na olaparibu a 129 pacientek na placebu) sPSR serózním ovariálním karcinomen buď kompletně odpovídajících (CR [kompletní odpověď]) nebo částečně odpovídajících (PR [částečná odpověď]) potvrzené podle RECIST a/nebo CA-125 kritérií definovaných mezinárodní skupinou pro gynekologické nádory (Gynecologic Cancer InterGroup) (GCIG) (nejméně 50% snížení hladiny CA-125 ve srovnání s posledním vzorkem před léčbou, potvrzené o 28 dnů později) na dva nebo více ukončené cykly chemoterapie obsahující platinu. Primárním cílovým parametrem byla doba přežití bez progrese (PFS) hodnocená řešitelem podle RECIST 1.0. Mezi sekundární cílové ukazatele účinnosti patřily celkové přežití (OS), míra kontroly onemocnění (DCR) definovaná jako potvrzené CR/PR + SD (stabilizovaná nemoc), kvalita života související se zdravím (HRQoL) a příznaky související s onemocněním. Dále byly provedeny exploratorní analýzy doby do první následné terapie nebo úmrtí (TFST) a doba do druhé následné terapie nebo úmrtí (TSST - odhad PFS2).
Do studie byly zařazeny pouze PSR pacientky s částečnou odpovědí na platinu (interval bez platiny od 6 do 12 měsíců) a pacientky s úplnou odpovědí na platinu (interval bez platiny > 12 měsíců), které odpovídaly na platinu při ukončení poslední chemoterapie založené na platině. Pacientky nesměly být v předchozím období léčeny olaparibem ani jiným inhibitorem PARP. Pacientky mohly být léčeny bevacizumabem s výjimkou podání v režimu, který bezprostředně předcházel randomizaci. Opětovná léčba olaparibem nebyla povolena, pokud pacientka progredovala na olaparibu.
Pacientky byly randomizovány do studie s mediánem 40 dnů od ukončení předchozí chemoterapie platinou. Byly léčeny v průměru 3 předchozími režimy (rozmezí 2-11), resp. 2,6 režimy (rozmezí 2-8) chemoterapie založené na platině.
Pacientky ve skupině užívající olaparib byly léčeny déle, než pacientky ve skupině užívající placebo. Celkem bylo 54 pacientek (39,7 %) ze skupiny užívající olaparib léčeno > 12 měsíců ve srovnání se 14 pacientkami (10,9 %) ze skupiny užívající placebo.
Studie splnila svůj primární cíl, když v celkové studijní populaci prokázala významné prodloužení PFS při udržovací léčbě olaparibem ve srovnání s placebem (HR0,35; 95% CI 0,25-0,49; p< 0,00001), navíc pak předem naplánovaná analýza podskupin podle přítomnosti mutace BRCA identifikovala pacientky s ovariálním karcinomem a přítomnou mutací BRCA (n=136, 51,3%) jako podskupinu, která měla z udržovací monoterapie olaparibem největší klinický přínos.
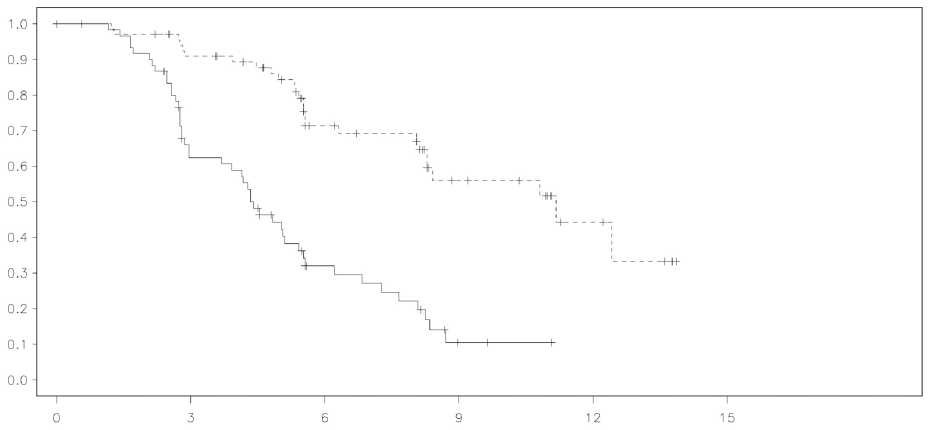
U pacientek s přítomnou mutací BRCA (n = 136) došlo ke statisticky významnému prodloužení PFS, TFST a TSST. Medián prodloužení PFS byl 6,9 měsíce oproti placebu ve prospěch pacientek léčených olaparibem (HR 0,18; 95% CI 0,10-0,31; p < 0,00001; medián 11,2 měsíce vs 4,3 měsíce). Hodnocení PFS řešiteli bylo v souladu s nezávislým zaslepeným centrálním radiologickým hodnocením PFS. Období od randomizace do začátku první následující léčby nebo úmrtí (TFST) bylo o 9,4 měsíce delší u pacientek užívajících olaparib (HR0,33; 95% CI 0,22-0,50; p< 0,00001; medián 15,6 měsíce vs 6,2 měsíce). Období od randomizace do začátku druhé následující léčby nebo úmrtí (TSST) bylo o 8,6 měsíce delší u pacientek užívajících olaparib (HR0,44; 95% CI 0,29-0,67; p = 0,00013; medián 23,8 měsíce vs 15,2 měsíce). Nebyl popsán statisticky významný rozdíl v OS (HR0,73; 95% CI 0,45-1,17; p = 0,19; medián 34,9 měsíce vs 31,9 měsíce). Míra kontroly onemocnění byla v rámci populace s přítomnou mutací BRCA ve 24. týdnu 57 % u pacientek užívajících olaparib, resp. 24 % u pacientek užívajících placebo.
V oblasti symptomů popisovaných pacientem nebo HRQoL, měřených na škále zlepšení a zhoršení příznaků v rámci FACT/NCCN indexu ovariálních symptomů (FOSI), indexu výsledků studie (TOI) a funkčního hodnocení terapie nádorů-celkového ovariálního skóre (FACT-O celkově) nebyly mezi skupinou užívající olaparib a skupinou užívající placebo pozorovány statisticky významné rozdíly.
Klíčové údaje týkající se účinnosti zjištěné ve studii 19 u pacientek s přítomnou mutací BRCA jsou uvedeny v tabulce 2 a grafech 1 a 2.
HR = poměr rizik. Hodnota < 1 upřednostňuje olaparib. Analýza byla provedena pomocí modelu proporcionálních rizik COX a zahrnovala údaje o léčbě, období do začátku progrese onemocnění po předchozím předposledním cyklu terapie platinou, objektivní odpověď na předchozí poslední cyklus terapie platinou a židovský původ.
Tabulka 2 Souhrn klíčových údajů týkajících se účinnosti zjištěných ve studii 19 u pacientek s přítomnou mutací BRCA a PSR ovariálním karcinomem
|
PFS |
N (příhody/pacientky) (%) |
Medián PFS (měsíce) |
HRa |
95% CI |
hodnota p |
|
Olaparib 400 mg dvakrát denně |
26/74 (35 %) |
11,2 |
0,18 |
0,10-0,31 |
<0,00001 |
|
Placebo |
46/62 (74 %) |
4,3 | |||
|
TSST-odhad PFS2 |
N |
Medián TSST (měsíce) |
HRa |
95% CI |
hodnota p |
|
Olaparib 400 mg dvakrát denně |
42/74 (57 %) |
23,8 |
0,44 |
0,29-0,67 |
0,00013 |
|
Placebo |
49/62 (79 %) |
15,2 | |||
|
Průběžná hodnota OS (52% úplnost) |
N |
Medián OS (měsíce) |
HRa |
95% CI |
hodnota p |
|
Olaparib 400 mg dvakrát denně |
37/74 (50 %) |
34,9 |
0,73 |
0,45-1,17 |
0,19 |
|
Placebob |
34/62 (55 %) |
31,9 |
a
b
N
Přibližně čtvrtina pacientek z podskupiny s přítomnou mutací BRCA užívajících placebo (14/62;
22,6 %) dostala následně inhibitor pArP.
Počet příhod/počet randomizovaných pacientek, OS = celková doba přežití, PFS = doba přežití bez progrese , CI = interval spolehlivosti, TSST = doba od randomizace do začátku druhé následné terapie nebo úmrtí.
Studie 19: Graf Kaplan-Meier pro PFS u pacientek s mutací BRCA (hodnocení řešitelů při 53% úplnosti údajů)
Obrázek 1
|
měsíce |
0 |
3 |
6 |
9 |
12 |
15 |
|
n-olaparib |
74 |
59 |
34 |
15 |
5 |
0 |
|
n-placebo |
62 |
35 |
13 |
2 |
0 |
0 |
-----olaparib 400 mg dvakrát denně,_placebo, osa x = čas od randomizace v měsících, osa y = PFS (doba
bez progrese onemocnění), n-olaparib = počet rizikových pacientek-olaparib, n-placebo = počet rizikových pacientek-placebo
Obrázek 2 Studie 19: Graf Kaplan-Meier pro OS u pacientek s mutací BRCA (52% úplnost údajů)

1 .0 0.9 0.8 0.7 0.6 0.5 0.4 0.5 0.2 0.1 0.0
0 3 6 9 12 15 18 21 24 27 30 33 36 39 42 45 48 51
|
měsíce |
0 |
3 |
6 |
9 |
12 |
15 |
18 |
21 |
24 |
27 |
30 |
33 |
36 |
39 |
42 |
45 |
48 |
51 |
|
n-olaparib |
74 |
71 |
69 |
67 |
65 |
62 |
56 |
53 |
50 |
48 |
39 |
36 |
26 |
12 |
7 |
0 |
0 |
0 |
|
n-placebo |
62 |
62 |
58 |
52 |
50 |
46 |
39 |
36 |
33 |
29 |
29 |
27 |
21 |
10 |
4 |
0 |
0 |
0 |
-----olaparib 400 mg dvakrát denně,_placebo, osa x = čas od randomizace v měsících, osa y = OS
(celková doba přežití), n-olaparib = počet rizikových pacientek-olaparib, n-placebo = počet rizikových pacientek-placebo
Ve studii 19 mělo 18 pacientek somatickou mutaci BRCA (mutace v nádoru, ale divoký typ v zárodečných buňkách). Omezené údaje u těchto pacientek se somatickou mutací BRCA (sBRCA) ukazují, že ve skupině
pacientek s olaparibem ve srovnání s placebem bylo zaznamenáno méně progresí onemocnění nebo úmrtí (Tabulka 3).
Tabulka 3 Souhrn doby přežití bez progrese a celkového přežití: populace s mutací sBRCA ve studii 19
|
N příhody/pacientky (%) | |
|
PFS | |
|
Olaparib 400 mg dvakrát denně |
3/8 (38 %) |
|
Placebo |
6/10 (60 %) |
|
OS | |
|
Olaparib 400 mg dvakrát denně |
4/8 (50 %) |
|
Placebo |
6/10 (60 %) |
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lynparza u všech podskupin pediatrické populace v indikaci ovariálního karcinomu (s výjimkou rabdomyosarkomu a nádorů ze zárodečných buněk). (Informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika olaparibu při dávkování 400 mg dvakrát denně ve formě tobolek je charakterizována zdánlivou plazmatickou clearance přibližně 8,6 l/h, zdánlivým distribučním objemem přibližně 167 l a terminálním plazmatickým poločasem 11,9 hodiny.
Absorpce
Po perorálním podání olaparibu ve formě tobolek dochází k rychlé absorpci a maximální plazmatické koncentrace je dosaženo za 1 až 3 hodiny po podání. Po opakovaném podání nebyla zaznamenána kumulace a ustáleného stavu je dosaženo během asi 3 až 4 dnů.
Současné užití s potravou zpomalovalo rychlost vstřebávání (tmax opožděno o 2 hodiny) a hraničně zvyšovalo rozsah absorpce olaparibu (zvětšení AUC o přibližně 20 %). Z tohoto důvodu se doporučuje, aby pacienti užívali přípravek Lynparza alespoň jednu hodinu po jídle a nejedli nejlépe až dvě hodiny po užití přípravku (viz bod 4.2).
Distribuce
V podmínkách in vitro při koncentracích odpovídajích plazmatickým hladinám dosaženým při dávkování 400 mg dvakrát denně je vazba olaparibu na proteiny přibližně 82 %.
Olaparib je středně vázán na HSA (lidský sérový albumin) nesaturovatelným způsobem (přibližně z 55 %) a slabě vázán na AAG (kyselý alfa-1 glykoprotein) (přibližně z 35 %).
Biotransformace
Bylo prokázáno, že za metabolizmus olaparibu je v prostředí in vitro primárně zodpovědný CYP3A4 (viz bod 4.5).
Po perorálním podávání 14C-olaparibu pacientkám tvořil nezměněný olaparib většinu cirkulující radioaktivity v plazmě (70 %) a byl hlavní složkou nalezenou jak v moči, tak ve stolici (15 %, resp. 6 % dávky). Olaparib podléhá intenzivnímu metabolismu. Hlavní část metabolismu probíhá jako oxidační reakce s tvorbou množství látek, které následně podléhají konjugaci s kyselinou glukuronovou nebo kyselinou sírovou.
V plazmě, moči a stolici bylo detekováno až 20, resp. 37, resp. 20 metabolitů, přičemž většina z nich představovala méně než 1 % podané dávky. Hydroxycyklopropylový derivát s otevřeným kruhem a dva monooxygenované metabolity (každý přibližně 10 %) tvořily většinu cirkulujících látek, přičemž jeden z těchto monooxygenovaných metabolitů byl rovněž hlavním metabolitem obsaženým v exkretech (tvořil 6 % radioaktivity moči, resp. 5 % radioaktivity stolice).
V podmínkách in vitro působil olaparib malou/nebo žádnou inhibici cytochromu CYP 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 nebo 2E1 a nepředpokládá se, že by měl klinicky významný inhibiční vliv na enzymy P450 v závislosti na čase. Údaje získané v podmínkách in vitro rovněž prokázaly, že olaparib není substrátem pro OATP1B1, OATP1B3, OCT1, BCRP nebo MRP2 ani inhibitorem OATP1B3, OAT1 nebo MRP2.
Eliminace
Po aplikaci jednotlivé dávky 14C-olaparibu se během 7denního období vyloučilo přibližně 86 % podané dávky radioaktivity a to přibližně 44 % močí a přibližně 42 % stolicí. Většina látky se vyloučila ve formě metabolitů.
Zvláštní _ populace Porucha funkce ledvin
Vliv poruchy funkce ledvin na expozici olaparibu nebyl testován. Přípravek Lynparza může být podáván pacientkám s mírnou poruchou funkce ledvin (clearance kreatininu > 50 ml/min), ale o použití u pacientek se středně těžkou (clearance kreatininu <50 ml/min) nebo těžkou poruchou funkce (clearance kreatininu <30 ml/min) jsou omezené údaje (viz bod 4.2).
Porucha funkce jater
Vliv poruchy funkce jater na expozici olaparibu nebyl testován. Použití olaparibu u pacientek s poruchou funkce jater (hladina sérového bilirubinu > 1,5krát vyšší než horní hranice normálních hodnot) není doporučeno.
Starší pacienti
Údaje o použití přípravku u pacientek ve věku 75 let a starších jsou omezené. Populační analýza dostupných údajů neprokázala vztah mezi plazmatickými koncentracemi olaparibu a věkem pacientky.
Hmotnost
Údaje o použití přípravku u pacientek obézních (BMI >30 kg/m2) a podvyživených (BMI <18 kg/m2) nejsou dostupné. Populační analýza dostupných dat neodhalila žádné údaje, že by hmotnost pacientky ovlivňovala plazmatické koncentrace olaparibu.
Rasa
Pro hodnocení možného vlivu rasy na farmakokinetiku olaparibu nejsou dostatečné údaje, neboť klinická zkušenost vychází převážně z použití u pacientek bílé rasy (94 % pacientek zahrnutých v populační analýze bylo bílé rasy). V tomto omezeném množství údajů nebyl zaznamenán výrazný rozdíl ve farmakokinetice olaparibu mezi Japonkami a pacientkami bílé rasy.
Pediatrická _ populace
Studie zaměřené na farmakokinetiku olaparibu nebyly u dětí provedeny.
5.3 Předklinické údaje vztahující se k bezpečnosti
Genotoxicita
Olaparib nevykazoval mutagenní potenciál, ale v podmínkách in vitro měl klastogenní účinky na savčí buňky. Po perorálním podávání laboratorním potkanům indukoval olaparib tvorbu mikrojader v kostní dřeni. Tato klastogenicita odpovídá známému farmakologickému účinku olaparibu a svědčí pro možný genotoxický potenciál i u člověka.
Toxicita po opakovaném podání
Ve studiích toxicity po opakovaném podání laboratorním potkanům a psům po dobu až 6 měsíců byly denní dávky olaparibu dobře snášeny. Hlavním primárním cílovým orgánem postiženým toxicitou byla u obou těchto živočišných druhů kostní dřeň s odpovídajícími změnami hematologických parametrů periferní krve. Tyto nálezy se vyskytly při expozicích nižších, než jsou expozice klinické a z velké části byly reverzibilní během 4 týdnů po ukončení podávání. Studie v podmínkách ex vivo za použití lidské kostní dřeně rovněž potvrdily, že olaparib má cytotoxický účinek na buňky lidské kostní dřeně.
Reprodukční toxicita
Ve studii fertility u samic laboratorních potkanů při podávání až do okamžiku implantace, nebyla ovlivněna schopnost rozmnožování a četnost gravidit, třebaže u některých zvířat byla pozorována prodloužená doba říje. Embryofetální přežití však bylo mírně sníženo.
Ve studiích embryofetálního vývoje u laboratorních potkanů při dávkách, které nevyvolaly významnou toxicitu u samic, způsoboval olaparib snížení embryofetálního přežití, včetně velkých očních malformací (např. anoftalmie, mikroftalmie), malformací páteře/žeber a viscerální a skeletální abnormality.
Kancerogenita
Studie kancerogenity olaparibu nebyly provedeny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky Glyceromakrogol-laurát
Obal tobolky Hypromelosa Oxid titaničitý (E171)
Gelanová klovatina (E418)
Kalium-acetát
Inkoust k potisku Šelak
Černý oxid železitý (E172)
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
6.5 Druh obalu a obsah balení
Plastová HDPE lahvička s dětským bezpečnostním uzávěrem obsahující 112 tvrdých tobolek.
Balení obsahuje 448 tobolek (4 lahvičky po 112 tobolkách).
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
AstraZeneca AB SE-151 85 Sodertálje Švédsko
8. REGISTRAČNÍ ČÍSLO
EU/1/14/959/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. prosince 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
AstraZeneca UK Limited Silk Road Business Park Macclesfield, Cheshire SK10 2NA Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
PAES: Držitel rozhodnutí o registraci předloží závěrečnou analýzu celkového přežití (OS) ve studii D0810C00019, randomizovaná dvojitě zaslepená multicentrická studie fáze II za účelem další charakterizace dlouhodobé účinnosti olaparibu u pacientek s relabujícím BRCA mutovaným high grade serózním ovariálním karcinomem citlivým na léčbu platinou. Závěrečná zpráva ze studie bude předložena do: |
červen 2017 |
|
PAES: Držitel rozhodnutí o registraci předloží výsledky studie D0816C00002, randomizovaná dvojitě zaslepená placebem kontrolovaná multicentrická studie fáze III za účelem potvrzení účinnosti olaparibu u pacientek s relabujícím BRCA mutovaným high grade serózním ovariálním karcinomem citlivým na léčbu platinou. Závěrečná zpráva ze studie bude předložena do: |
prosinec 2018 |
|
PAES: Držitel rozhodnutí o registraci provede studii fáze IV a předloží výsledky otevřené nerandomizované multicentrické studie s jedním ramenem u pacientek s relabujícím high grade serózním karcinomem vaječníků se somatickou mutací BRCA citlivým na léčbu platinou za účelem další charakterizace účinnosti olaparibu u pacientek, které úplně nebo částečně reagovaly na léčbu platinou a které nemají funkční zárodečnou nebo somatickou mutaci BRCA. Závěrečná zpráva ze studie bude předložena do: |
září 2018 |
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Lynparza 50 mg tvrdé tobolky olaparibum
2 OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje olaparibum 50 mg. 3 SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdá tobolka
448 tobolek (4 lahvičky po 112 tobolkách)
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Cytotoxická látka 8 POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 30 °C.
10 ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. 1L NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
AstraZeneca AB SE-151 85 Sodertálje Švédsko
EU/1/14/959/001 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis. 15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
lynparza 50 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Lynparza 50 mg tvrdé tobolky olaparibum
2 OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje olaparibum 50 mg. 3 SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdá tobolka 112 tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Cytotoxická látka 8 POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 30 °C.
10 ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. 1L NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
AstraZeneca AB SE-151 85 Sodertalje Švédsko
EU/1/14/959/001 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis. 15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
B. PŘÍBALOVÁ INFORMACE
Lynparza 50 mg tvrdé tobolky
olaparibum
VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, nebo lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Lynparza a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Lynparza užívat
3. Jak se přípravek Lynparza užívá
4. Možné nežádoucí účinky
5 Jak přípravek Lynparza uchovávat
6. Obsah balení a další informace
1. Co je přípravek Lynparza a k čemu se používá Co je přípravek Lynparza a jak účinkuje
Přípravek Lynparza tvrdé tobolky obsahuje léčivou látku nazývanou olaparib. Olaparib je druhem protinádorové léčivé látky označované jako inhibitor PARP (poly [adenosin ribózo-difosfát] polymeráza).
U pacientek s mutacemi (změnami) některých genů označovaných jako BRCA (gen nádoru prsu), u kterých existuje riziko vývoje některých forem nádorů, mohou inhibitory PARP vyvolat smrt nádorových buněk tím, že zablokují enzym, který pomáhá opravovat DNA.
K čemu se přípravek Lynparza používá
Lynparza se používá k léčbě určitého typu nádoru vaječníků označovaného jako „BRCA-mutovaný karcinom vaječníků“. Používá se v případě, že nádor reagoval na předchozí standardní léčbu chemoterapií založenou na platině. K určení BRCA-mutovaného nádoru se používá vyšetřovací test.
2. Čemu musíte věnovat pozornost, než začnete přípravek Lynparza užívat Neužívejte přípravek Lynparza:
- jestliže jste alergická na olaparib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Neužívejte přípravek Lynparza, pokud se kterýkoli z údajů výše vztahuje také na Vás. V případě nejistoty se poraďte s lékařem, lékárníkem nebo zdravotní sestrou předtím, než začnete užívat přípravek Lynparza.
Upozornění a opatření
Před užitím přípravku nebo v průběhu léčby přípravkem Lynparza se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou:
• Jestliže bylo při vyšetření krve zjištěno, že máte málo krevních buněk. Může se jednat o malý počet červených krvinek (anémie), malý počet bílých krvinek (neutropenie) nebo malý počet krevních destiček (trombocytopenie). Více informací o těchto nežádoucích účincích najdete v bodě 4. Těchto příznaků a projevů je třeba si všímat (horečka nebo infekce, tvorba modřin nebo krvácení). Může se vzácně jednat o projevy závažnějších problémů s kostní dření jako je „myelodysplastický syndrom“ (MDS) nebo „akutní myeloidní leukémie“ (AML). Váš lékař může provádět vyšetření kostní dřeně, aby mohl kontrolovat tyto problémy.
• Jestliže zaznamenáte nové příznaky nebo zhoršení příznaků dušnosti, kašle nebo sípání. U malého počtu pacientek léčených přípravkem Lynparza byl hlášen zánět plic (pneumonitida). Pneumonitida je závažný stav, u kterého je často vyžadována nemocniční léčba.
Pokud se některé údaje uvedené výše vztahují také na Vás (nebo pokud si nejste jistá), poraďte se s lékařem, nebo lékárníkem nebo zdravotní sestrou.
Vyšetření a kontroly
Váš lékař bude provádět vyšetření krve před léčbou a v průběhu léčby přípravkem Lynparza.
Krevní testy budou provedeny:
• před léčbou
• každý měsíc první rok léčby
• v pravidelných intrvalech podle rozhodnutí lékaře po prvním roce léčby.
Pokud dojde ke snížení počtů krevních buněk na nízkou úroveň, může být nutná transfuze krve (kdy dostanete novou krev nebo část krve od dárce).
Další léčivé přípravky a přípravek Lynparza
Informujte svého lékaře, nebo lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užívala nebo které možná budete užívat, včetně léků volně prodejných a rostlinných přípravků. Důvodem je fakt, že přípravek Lynparza může ovlivňovat účinek jiných léků. Též jiné léky mohou ovlivňovat účinek přípravku Lynparza.
Neužívejte přípravek Lynparza, pokud užíváte jiné protinádorové přípravky. Informujte lékaře, lékárníka nebo zdravotní sestru, jestliže plánujete očkování očkovací látkou nebo budete užívat přípravek, který potlačuje imunitu, neboť může být potřebné pečlivé sledování.
Informujte lékaře nebo lékárníka, pokud užíváte kterýkoli z následujích léčivých přípravků:
• itrakonazol, flukonazol - k léčbě plísňových onemocnění
• telithromycin, klarithromycin, erythromycin - k léčbě bakteriálních infekcí
• inhibitory proteáz potencované ritonavirem nebo kobicistatem, boceprevirem, telaprevirem, nevirapinem, efavirenz - k léčbě virových infekcí, včetně HIV
• rifampicin, rifapentin, rifabutin - k léčbě bakteriálních infekcí, včetně tuberkulózy (TBC)
• fenytoin, karbamazepin, fenobarbital - jako sedativa (uklidňující léky) nebo k léčbě křečí a epilepsie
• třezalka tečkovaná (Hypericum perforatum) - rostlinný přípravek používaný zejména k léčbě deprese
• digoxin, diltiazem, furosemid, verapamil, valsartan - k léčbě poruch srdce nebo vysokého krevního tlaku
• bosentan - k léčbě plicní hypertenze
• statiny, např. simvastatin, pravastatin - k léčběvysokých hladin cholesterolu v krvi
• dabigatran - k snížení srážlivosti krve
• glibenklamid, metformin, repaglinid - k léčbě cukrovky
• námelové alkaloidy - k léčbě migrény a bolesti hlavy
• fentanyl - k léčbě nádorové bolesti
• pimozid - k léčbě schizofrenie
• kvetiapin - k léčbě schizofrenie a bipolární poruchy
• cisaprid - k léčbě žaludečních poruch
• kolchicin - k léčbě dny
• methotrexát - k léčbě nádorů, revmatoidní artritidy (revmatický zánět kloubů) a lupénky
Lynparza s pitím
Po celou dobu léčby přípravkem Lynparza nepijte grapefruitovou šťávu. Mohlo by to mít vliv na účinek
léčivého přípravku.
Těhotenství a kojení
• Neužívejte přípravek Lynparza, pokud jste těhotná nebo můžete být těhotná, neboť by mohlo dojít k poškození ještě nenarozeného dítěte.
• V průběhu léčby tímto léčivým přípravkem nesmíte otěhotnět. V průběhu léčby tímto přípravkem a ještě 1 měsíc po ukončení léčby přípravkem Lynparza musíte používat účinné metody zabraňující početí (antikoncepce). Není známo, zda přípravek Lynparza může ovlivnit účinnost některých perorálních antikoncepčních přípravků. Informujte lékaře, pokud užíváte pororální antikoncepci (podávanou ústy), neboť lékař Vám může doporučit další nehormonální antikoncepční metodu.
• Před zahájením léčby přípravkem Lynparza a v pravidelných intervalech v průběhu léčby a 1 měsíc po ukončení léčby musíte provádět testy na kontrolu těhotenství. Pokud byste otěhotněla v průběhu této doby, musíte se ihned poradit s lékařem.
• Není známo, zda se přípravek Lynparza vylučuje do mateřského mléka. V průběhu léčby přípravkem Lynparza a 1 měsíc po podání poslední dávky přípravku Lynparza nekojte. Pokud plánujete kojit, poraďte se nejdříve s lékařem.
Řízení dopravních prostředků a obsluha strojů
Přípravek Lynparza může ovlivňovat Vaši schopnost řídit a obsluhovat stroje. Pokud pociťujete závrať,
slabost nebo jste unavená v průběhu léčby přípravkem Lynparza, neřiďte ani nepoužívejte nástroje nebo
stroje.
3. Jak se přípravek Lynparza užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře, lékárníka nebo zdravotní sestry. Pokud si nejste jistá, poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Jakou dávku přípravku užívat
• Doporučená dávka přípravku je 8 tobolek (400 mg) podávaných ústy dvakrát denně (celkem
16 tobolek za den). Je důležité, abyste užívala celou doporučenou dávku a pokračovala tak podle rady lékaře, lékárníka nebo zdravotní sestry.
Jak přípravek užívat
• Užívejte jednu dávku (8 tobolek) přípravku Lynparza ústy a zapijte vodou, jednu dávku ráno a jednu dávku večer.
• Užívejte přípravek Lynparza nejméně jednu hodinu po jídle. Nejezte nejlépe až dvě hodiny po užití přípravku Lynparza.
Jestliže si všimnete nežádoucích účinků, lékař Vám může předepsat přípravek Lynparza v nižší dávce. Jestliže jste užila více přípravku Lynparza, než jste měla
Jestliže jste užila více přípravku Lynparza než je Vaše obvyklá dávka, kontaktujte svého lékaře nebo nejbližší nemocnici.
Jestliže jste zapomněla užít přípravek Lynparza
Jestliže jste zapomněla užít přípravek Lynparza, užijte Vaši obvyklou dávku v pravidelnou dobu. Nezdvojnásobujte následující dávku, abyste nahradila vynechanou dávku.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Je důležité, abyste věděla, jaké nežádoucí účinky to mohou být.
Lékař Vám může též předepsat další léky, které pomohou kontrolovat tyto nežádoucí účinky.
Pokud si všimnete následujících nežádoucích účinků, informujte o tom svého lékaře - můžete potřebovat rychlou lékařskou pomoc:
Velmi časté (mohou se vyskytnout u více než 1 z 10 lidí):
• horečka nebo infekce - může jít o známky nízkého počtu bílých krvinek (neutropenie nebo lymfopenie).
• dušnost, pocit velké únavy, zblednutí kůže, nebo zrychlená činnost srdce - může jít o známky nízkého počtu červených krvinek (anémie).
Časté (mohou se vyskytnout až u 1 z 10 lidí):
• tvorba modřin nebo krvácení, které při poranění trvá déle než obvykle - jde o známky nízkého počtu krevních destiček (trombocytopenie).
Informujte ihned svého lékaře, pokud zaznamenáte kterýkoli z těchto nežádoucích účinků.
Další nežádoucí účinky zahrnují:
Velmi časté
• bolest hlavy
• pocit závratě
• ztráta chuti k jídlu
• pocit únavy nebo slabost
• nevolnost (pocit na zvracení)
• zvracení
• změny chuti
• potíže s trávením nebo pálení žáhy (dyspepsie)
• průjem. Pokud se stav zhorší, informujte ihned lékaře
• zvýšení hladin kreatininu v krvi pozorované při laboratorním vyšetření ukazuje, jak dobře pracují Vaše ledviny
• vyšetření krve prokazující zvětšení velikosti červených krvinek
Časté
• bolest v ústech (stomatitida)
• bolest v oblasti žaludku v podžebří
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi nebo zdravotní sestře. To zahrnuje i jakýkoli nežádoucí účinek, který není uveden v této příbalové informaci. Lékař Vám může předepsat léky k léčbě příznaků jako je pocit nucení na zvracení, zvracení, průjem a dyspepsie.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Lynparza uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a lahvičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 30 °C.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Lynparza obsahuje
Léčivou látkou je olaparib. Jedna tvrdá tobolka obsahuje olaparibum 50 mg.
Dalšími složkami/pomocnými látkami jsou:
• Obsah tobolek: glyceromakrogol-laurát.
• Obal tobolky: hypromelosa, oxid titaničitý (E171), gelanová klovatina (E418), kalium-acetát.
• Inkoust k potisku: šelak, černý oxid železitý (E172).
Jak přípravek Lynparza vypadá a co obsahuje toto balení
Přípravek Lynparza 50 mg je bílá neprůhledné tvrdá tobolka označená černým inkoustem „OLAPARIB 50 mg“ a logem AstraZeneca.
Přípravek Lynparza se dodává v plastových HDPE lahvičkách obsahujících 112 tvrdých tobolek. Balení obsahuje 448 tvrdých tobolek (4 lahvičky po 112 tvrdých tobolkách).
Držitel rozhodnutí o registraci a výrobce
AstraZeneca AB SE-151 85 Sodertálje Švédsko
Výrobce
AstraZeneca UK Limited Silk Road Business Park Macclesfield, Cheshire, SK10 2NA Velká Británie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Ten.: +359 2 44 55 000
Česká republika
AstraZeneca Czech Republic s.r.o.
Tel: +420 222 807 111
Danmark
AstraZeneca A/S
Malta
Associated Drug Co. Ltd
|
Tlf: +45 43 66 64 62 |
Tel: +356 2277 8000 |
|
Deutschland AstraZeneca GmbH Tel: +49 41 03 7080 |
Nederland AstraZeneca BV Tel: +31 79 363 2222 |
|
Eesti AstraZeneca Tel: +372 6549 600 |
Norge AstraZeneca AS Tlf: +47 21 00 64 00 |
|
EXláSa AstraZeneca A.E. Tn^: +30 2 106871500 |
Osterreich AstraZeneca Osterreich GmbH Tel: +43 1 711 31 0 |
|
Espaňa AstraZeneca Farmacéutica Spain, S.A. Tel: +34 91 301 91 00 |
Polska AstraZeneca Pharma Poland Sp. z o.o. Tel.: +48 22 874 35 00 |
|
France AstraZeneca Tél: +33 1 41 29 40 00 |
Portugal AstraZeneca Produtos Farmaceuticos, Lda. Tel: +351 21 434 61 00 |
|
Hrvatska AstraZeneca d.o.o. Tel: +385 1 4628 000 |
Románia AstraZeneca Pharma SRL Tel: +40 21 317 60 41 |
|
Ireland AstraZeneca Pharmaceuticals (Ireland) Ltd Tel: +353 1609 7100 |
Slovenija AstraZeneca UK Limited Tel: +386 1 51 35 600 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika AstraZeneca AB, o.z. Tel: +421 2 5737 7777 |
|
Italia AstraZeneca S.p.A. Tel: +39 02 9801 1 |
Suomi/Finland AstraZeneca Oy Puh/Tel: +358 10 23 010 |
|
Kúnpoq A^skt^p Oap^aKeuxiKq ArS Tn^: +357 22490305 |
Sverige AstraZeneca AB Tel: +46 8 553 26 000 |
|
Latvija SIA AstraZeneca Latvija Tel: +371 67377100 |
United Kingdom AstraZeneca UK Ltd Tel: +44 1582 836 836 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
32