Lymphoseek 250 Mikrogramů
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
V Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Lymphoseek 250 mikrogramů, kit pro radiofarmakum
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Každá injekční lahvička obsahuje 250 mikrogramů tilmanoceptu. Radionuklid není součástí soupravy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Kit pro radiofarmakum.
Injekční lahvička obsahuje sterilní nepyrogenní bílý až krémově bílý lyofilizovaný prášek.
4. KLINICKÉ ÚDAJE
4.1. Terapeutické indikace
Tento přípravek je určen pouze k diagnostickým účelům.
Radioaktivně značený přípravek Lymphoseek je indikován k zobrazení a intraoperační detekci sentinelových lymfatických uzlin v drenážní oblasti primárního nádoru u dospělých pacientů s karcinomem prsu, melanomem nebo lokalizovaným spinocelulárním karcinomem dutiny ústní.
Externí zobrazení a intraoperační vyhodnocení lze provést pomocí přístroje pro detekci záření gama.
4.2. Dávkování a způsob podání
Léčivý přípravek by měli podávat pouze vyškolení zdravotničtí pracovníci se zkušenostmi s technickým provedením a interpretací postupů mapování sentinelových lymfatických uzlin.
Dávkování
Doporučená dávka je 50 mikrogramů tilmanoceptu radioaktivně značeného techneciem-99m při 18,5 MBq, pokud je chirurgický výkon proveden ve stejný den, nebo 74 MBq, pokud bude chirurgický výkon proveden následující den. Dávka 50 mikrogramů se nemá upravovat podle rozdílů v tělesné hmotnosti. Celkové množství v injekci nemá překročit 50 mikrogramů tilmanoceptu s celkovou maximální radioaktivitou 74 MBq (2,0 mCi) na dávku.
Zobrazení je doporučeno provádět po uplynutí minimálně 15 minut od podání injekce. Intraoperační lymfatické mapování je možné zahájit již 15 minut po podání injekce.
Pacientům, u nichž je chirurgický výkon plánován v den podání injekce, bude podán přípravek značený techneciem-99m 18,5 MBq. Podání by mělo proběhnout 15 hodin před plánovaným časem chirurgického výkonu a intraoperační detekce.
Pacientům, u nichž je chirurgický výkon plánován den po podání injekce, bude podán přípravek značený techneciem-99m 74 MBq. Podání by mělo proběhnout 30 hodin před plánovaným časem chirurgického výkonu a intraoperační detekce.
Pacienti s poruchou funkce jater nebo ledvin
U těchto pacientů je nutné pečlivě zvážit množství podané radioaktivity, protože u nich může dojít k větší expozici radioaktivnímu záření. Dávka radioaktivity u pacienta nepřekročí 0,69 mSv ani v případě, že nebude vyloučena žádná část dávky 74 MBq.
U běžné populace ani u zvláštních populací nebyly provedeny rozsáhlejší studie rozmezí a úprav dávky léčivého přípravku. Farmakokinetika tilmanoceptu značeného techneciem-99m nebyla u pacientů s poruchou funkce ledvin nebo jater popsána (viz bod 5.2).
Starší pacienti
V klinických studiích byli hodnoceni starší pacienti ve věku 65 a více let (32 %) a nebyla zjištěna žádná bezpečnostní rizika. Není doporučena žádná úprava dávky v závislosti na věku.
Pediatrická populace
Bezpečnost a účinnost přípravku Lymphoseek u dětí a dospívajících ve věku do 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Tento léčivý přípravek musí být před podáním pacientovi radioaktivně značen. Radioaktivně značený přípravek je čirý bezbarvý roztok bez viditelných částic.
Po radioaktivním značení lze podat v intradermální, subkutánní, intratumorózní nebo peritumorózní injekci. U melanomu je podání intradermální v jedné nebo ve více rozdělených injekcích.
U karcinomu prsu je podání intradermální, subareolární (v jedné nebo ve více rozdělených injekcích) nebo peritumorózní (ve více rozdělených injekcích).
U spinocelulárního karcinomu dutiny ústní je podání peritumorózní (ve více rozdělených injekcích).
Každá 250mikrogramová injekční lahvička obsahuje určité množství přípravku navíc. Je nicméně doporučeno připravit injekční lahvičku podle pokynů a pro jednotlivou dávku u pacienta použít alikvotní množství odpovídající dávce 50 mikrogramů.
Objem jedné injekce nemá přesáhnout 0,5 ml a nemá být menší než 0,1 ml. Celkový objem injekce nemá přesáhnout 1,0 ml a nemá být menší než 0,1 ml. Zředění přípravku v objemu nad 1,0 ml by mohlo narušit dostupnost přípravku Lymphoseek in vivo.
Návod k přípravě a kontrole radiochemické čistoty radiofarmaka je uveden v bodě 12.
Příprava pacienta viz bod 4.4.
4.3. Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na kteroukoli složku radioaktivně značeného farmaceutického přípravku.
4.4. Zvláštní upozornění a opatření pro použití
Možnost reakce z přecitlivělosti nebo anafylaktická reakce
Vždy je třeba zvážit možnost přecitlivělosti včetně závažných život ohrožujících fatálních anafylaktických/anafylaktoidních reakcí.
Pokud dojde k reakci z přecitlivělosti nebo k anafylaktické reakci, je nutné podávání léčivého přípravku okamžitě přerušit a v případě potřeby zahájit intravenózní léčbu. Aby byla v případě naléhavého stavu možná okamžitá akce, je třeba mít okamžitě k dispozici nezbytné léčivé přípravky a vybavení, např. endotracheální kanylu a ventilátor.
Individuální odůvodnění přínosů a rizik
U každého pacienta musí být působení radioaktivního záření odůvodnitelné očekávaným prospěchem. Podaná dávka radioaktivity by měla být v každém případě co nejnižší dosažitelná dávka nutná k získání požadované diagnostické informace.
Porucha funkce ledvin a jater
U těchto pacientů je nutné pečlivě zvážit poměr přínosů a rizik, protože u nich může dojít k větší expozici radioaktivnímu záření. Odhadovaná dávka radioaktivity u pacienta nepřekročí 0,69 mSv ani v případě, že nebude vyloučena žádná část dávky 74 MBq (2,0 mCi) (viz bod 4.2).
Příprava pacienta
Pacient by před zahájením vyšetření měl být dobře hydratován a během několika hodin po vyšetření by měl být podněcován k co nejčastějšímu vyprazdňování močového měchýře z důvodu snížení jeho expozice radioaktivnímu záření.
Zvláštní upozornění
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) na dávku, tedy v podstatě „neobsahuje sodík“.
Varování s ohledem na riziko pro životní prostředí viz bod 6.6.
4.5. Interakce s jinými léčivými přípravky a jiné formy interakce
Přidání velkého množství značících látek nebo jiných injekčních látek s krátkým časovým odstupem od podání přípravku Lymphoseek nebo anatomicky proximálně k místu jeho aplikace může ovlivnit dostupnost přípravku Lymphoseek in vivo. Během 30 minut od podání přípravku Lymphoseek se nemají injekčně podávat další značící látky.
Žádné další studie interakcí nebyly provedeny.
4.6. Fertilita, těhotenství a kojení
Ženy ve fertilním věku.
Pokud je plánováno podání radiofarmak ženě ve fertilním věku, je důležité zjistit, zda není těhotná. Každou ženu, u které se nedostavila menstruace, je třeba považovat za těhotnou, pokud se neprokáže opak. Jestliže existují pochybnosti týkající se možného těhotenství (jestliže se u ženy nedostavila menstruace, nebo jestliže je menstruace velmi nepravidelná atd.), měly by být pacientce nabídnuty alternativní techniky neužívající ionizující záření (jestliže takové techniky existují).
Žádné údaje o podávání přípravku Lymphoseek těhotným ženám nejsou k dispozici. Nebyly provedeny žádné studie reprodukční toxicity u zvířat a není známo, zda může přípravek Lymphoseek při podání těhotné ženě vyvolat poškození plodu.
Při vyšetřeních s radionuklidy provedených u těhotných žen je dávce radioaktivity vystaven také plod. Proto se mají během těhotenství provádět pouze nezbytná vyšetření, jejichž pravděpodobný přínos zdaleka převyšuje riziko, které z nich pro matku a plod vyplývá.
Kojení
Není známo, zda se tilmanocept značený techneciem-99m vylučuje do mateřského mléka.
Před podáním radiofarmak kojící matce je třeba zvážit, zda nelze toto podání odložit na dobu, kdy matka přestane kojit, a jaké je nevhodnější radiofarmakum vzhledem k sekreci aktivity do mateřského mléka. Pokud je podání považováno za nezbytné, je třeba kojení přerušit na 24 hodin po podání injekce a vyloučené mléko zlikvidovat.
Fertilita
S přípravkem Lymphoseek nebyly provedeny studie fertility u zvířat.
4.7. Účinky na schopnost řídit a obsluhovat stroje
Přípravek Lymphoseek nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8. Nežádoucí účinky
V klinických studiích s 553 pacienty byly nejčastějšími nežádoucími účinky:
◦ podráždění v místě vpichu (0,7 %; 4 z 553 pacientů)
◦ bolest v místě vpichu (0,2 %; 1 z 553 pacientů)
Další nežádoucí účinky byly méně časté, mírné závažnosti a krátkého trvání.
Následující tabulka ukazuje, jak je v tomto bodě vyjádřena četnost:
Velmi časté (> 1/10)
Časté (> 1/100 až < 1/10)
Méně časté (> 1/1000 až < 1/100)
Vzácné (> 1/10 000 až < 1/1 000)
Velmi vzácné (< 1/10 000)
Není známo (z dostupných údajů nelze určit)
Klinické studie hodnotily incidenci níže uvedených nežádoucích reakcí u 553 jedinců starších 18 let, kteří dostali přípravek Lymphoseek. Tyto reakce měly časovou souvislost s podáním přípravku Lymphoseek a mohly být způsobeny jinými léčivými přípravky, které byly pacientům podány, nebo chirurgickými výkony.
|
Poruchy metabolismu a výživy |
Méně časté: hyperkalcemie |
|
Poruchy nervového systému |
Méně časté: afázie, závratě, bolest hlavy, parestezie |
|
Poruchy oka |
Méně časté: rozmazané vidění |
|
Srdeční poruchy |
Méně časté: sinusová tachykardie |
|
Cévní poruchy |
Méně časté: návaly horka |
|
Gastrointestinální poruchy |
Méně časté: nevolnost |
|
Poruchy kůže a podkožní tkáně |
Méně časté: podráždění kůže |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Méně časté: bolest v končetinách, muskuloskeletální bolest, bolest v oblasti krční páteře, bolest čelisti |
|
Poruchy ledvin a močových cest |
Méně časté: nucení na močení, polakisurie |
|
Poruchy reprodukčního systému a prsu |
Méně časté: bolest prsů |
|
Celkové poruchy a reakce v místě aplikace |
Méně časté: podráždění v místě vpichu, bolest v místě vpichu, pocit horka |
|
Poranění, otravy a procedurální komplikace |
Méně časté: bolest v místě vpichu, serom, dehiscence rány |
Expozice ionizujícímu záření je spojena s možností vzniku nádorového onemocnění a potenciálním vznikem vrozených vad. Vzhledem k tomu, že účinná dávka u dospělých (70 kg) je 0,69 mSv při podání maximální doporučené radioaktivity 74 MBq, očekává se, že k těmto nežádoucím účinkům bude docházet s nízkou pravděpodobností.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9. Předávkování
Celkový obsah injekce nemá překročit 50 mikrogramů tilmanoceptu s celkovou maximální radioaktivitou 74 MBq na dávku. Chronické nebo akutní předávkování je vzhledem k celkovému objemu injekce nepravděpodobné.
Při dávkách 3,7x vyšších, než je doporučená dávka přípravku Lymphoseek u člověka, nebo 390x vyšších, než je předpokládaná expozice tilmanoceptu u člověka, podaných zvířatům nebyly pozorovány žádné klinické následky.
V případě podání nadměrné dávky radioaktivity s přípravkem tilmanocept je třeba absorbovanou dávku u pacienta pokud možno snížit podporou vylučování radionuklidu z organismu častým močením nebo forsírovanou diurézou a častým vyprazdňováním močového měchýře.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1. Farmakodynamické vlastnosti
Farmakoterapeutická skupina: diagnostické radiofarmakum k detekci nádorů, ATC kód: V09IA09. Mechanismus účinku
Přípravek Lymphoseek je radiofarmakum cílené na receptory, které je vytvořeno tak, aby rychle procházelo lymfatickými cévami. Jeho biologickým cílem jsou primární drenážní lymfatické uzliny s klíčovým prediktivním významem (sentinelové lymfatické uzliny), ve kterých se přípravek akumuluje a zadržuje. Léčivá látka, tilmanocept, se specificky váže na proteinový receptor vázající manózu (CD206), který se nachází se na povrchu makrofágů a dendritických buněk. Makrofágy jsou ve vysoké koncentraci přítomné v lymfatických uzlinách.
Tilmanocept je makromolekula složená z více jednotek kyseliny diethylentriaminpentaoctové (DTPA) a manózy, z nichž je každá synteticky navázána na 10kDa dextranovou kostru. Manóza působí jako substrát pro receptor a DTPA slouží jako chelatační činidlo pro značení techneciem-99m. Střední průměr molekuly tilmanoceptu je 7 nm a tato malá velikost umožňuje zvýšený přestup látky do lymfatických cest, což vede k rychlé a spolehlivé clearance z místa vpichu injekce.
Po rekonstituci a značení je přípravek Lymphoseek určen k injekčnímu podání v těsné blízkosti nádoru a k lokalizaci sentinelových lymfatických uzlin v lymfatických cestách zajišťujících drenáž oblasti nádoru buď předoperačně zobrazením detekovaného záření gama pomocí stacionární gamakamery (scintigrafie), jednofotonové emisní výpočetní tomografie (SPECT) nebo SPECT/počítačové tomografie SPECT/CT a/nebo intraoperačně pomocí sondy pro detekci záření gama.
Ve studiích in vitro vykázal tilmanocept značený techneciem-99m specifickou a pevnou vazbu na lidské CD206 receptory s afinitou k primárnímu vazebnému místu Kd = 2,76 x 10-11 M. V klinických studiích fáze 1 se po 30 minutách akumulovalo v drenážních lymfatických uzlinách prostřednictvím specifické vazby přibližně 0,5 až 1,8 % dávky. Vazba tilmanoceptu značeného techneciem-99 m je nezávislá na typu a závažnosti nádoru.
Klinická, účinnost
V klinických studiích fáze 3 byl tilmanocept značený techneciem-99m detekovatelný v sentinelových lymfatických uzlinách do 10 minut. Při analýze externího zobrazení záření gama se ukázalo, že navázaný tilmanocept značený techneciem-99m je zadržen ve stejných drenážních lymfatických uzlinách až 30 hodin. Předoperační lymfoscintigrafie byla provedena u 100 % pacientů s melanomem, u 100 % pacientů se spinocelulárním karcinomem hlavy a krku a u 82 % pacientů s karcinomem prsu. Celkový podíl shody mezi lokalizací lymfatických uzlin (určenou detekcí radioaktivity) při předoperační scintigrafii a intraoperačním hledáním lymfatických uzlin činil pro všechny pacienty 97,8 %.
V klinických studiích fáze 3 u pacientů s karcinomem prsu, u nichž bylo mapování provedeno jak pomocí tilmanoceptu značeného techneciem-99m, tak pomocí vitální modři, byl podle metaanalýz s modelem fixních efektů tilmanocept značený techneciem-99m vychytáván u 99,91 % pacientů, a to průměrně ve 2,08 sentinelových lymfatických uzlinách na pacienta. Tento podíl byl statisticky významně větší (p < 0,0001) než podíly vychytávání dle metaanalýz s modelem náhodných efektů publikované v literatuře, dosažené
u koloidních látek pro lymfatické mapování, které se používají v klinické praxi v Evropě. V metaanalýze s modelem fixních efektů dvou studií fáze 3 se tilmanocept značený techneciem-99m vychytával v 99,99 % excidovaných lymfatických uzlin zbarvených modře pomocí vitální modři (shoda). Vitální modř byla naopak nalezena v 66,96 % excidovaných lymfatických uzlin detekovaných pomocí tilmanoceptu značeného techneciem-99m (reverzní shoda).
V klinických studiích fáze 3 u pacientů s melanomem, u nichž bylo mapování provedeno jak pomocí tilmanoceptu značeného techneciem-99m, tak pomocí vitální modři, byl tilmanocept značený techneciem-99m podle metaanalýz s modelem fixních efektů vychytáván u 99,89 % pacientů, a to průměrně ve 2,30 lokalizovaných sentinelových lymfatických uzlinách na pacienta. Tento podíl byl statisticky významně větší (p < 0,0001) než podíly vychytávání dle metaanalýz s modelem náhodných efektů publikované v literatuře, dosažené u koloidních látek pro lymfatické mapování, které se používají v klinické praxi v Evropě.
V metaanalýze s modelem pevných efektů dvou studií fáze 3 se tilmanocept značený techneciem-99m vychytával v 99,99 % excidovaných lymfatických uzlin zbarvených modře pomocí vitální modři (shoda). Vitální modř byla naopak nalezena v 63,50 % excidovaných lymfatických uzlin detekovaných pomocí tilmanoceptu značeného techneciem-99m (reverzní shoda).
V jedné klinické studii fáze 3 u pacientů s intraorálním nebo kožním spinocelulárním karcinomem se tilmanocept značený techneciem-99m vychytával v sentinelových lymfatických uzlinách u 97,59 % pacientů, kteří podstoupili vyšetření lymfatických uzlin. Pokud jde o patologický status lymfatických uzlin odstraněných při jejich úplné disekci, tilmanocept značený techneciem-99m se správně vychytával
v sentinelových lymfatických uzlinách s predikovanou metastázou nádoru u 38 z 39 pacientů, což představuje podíl falešně negativních výsledků 2,56 %. Celková přesnost identifikace skutečně pozitivních a skutečně negativních pacientů pomocí tilmanoceptu značeného techneciem-99m vzhledem k patologickému nálezu v lymfatických uzlinách, ve kterých se vychytával, byla 98,80 %.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lymphoseek u jedné nebo více podskupin pediatrické populace týkající se vizualizace lymfatické drenáže solidních maligních nádorů za diagnostickým účelem (informace o použití u dětí viz bod 4.2).
5.2. Farmakokinetické vlastnosti
Byly provedeny dvě klinické studie fáze 1 u pacientů s karcinomem prsu a jedna studie fáze 1 u pacientů s melanomem. Účelem těchto studií bylo zhodnocení radiofarmakokinetiky přípravku Lymphoseek.
Distribuce
V jedné studii fáze 1 u pacientů s karcinomem prsu vykázal přípravek Lymphoseek ve všech třech testovaných dávkách (4, 20 a 100 mikrogramů) rychlou clearance z místa podání injekce (rychlostní konstanty eliminace v rozmezí 0,222/hod až 0,278/hod). Vychytávání tilmanoceptu značeného techneciem-99m v primární sentinelové uzlině se zvyšovalo v závislosti na dávce (p = 0,009): Po injekci přípravku Lymphoseek v dávce 4, resp. 20, resp. 100 mikrogramů bylo dosaženo hladiny tilmanoceptu značeného techneciem-99m v primární sentinelové uzlině (L SN) 0,09 ± 0,20 pmol, resp. 6,53 ± 2,52 pmol, resp. 10,58 ± 8,43 pmol. Procentuální podíl injekčně podané dávky, která dosáhla primární sentinelové uzliny (%ID SN) ve skupinách s dávkou přípravku Lymphoseek 4, resp. 20, resp. 100 mikrogramů činil 0,05 % ± 0,10 %, resp. 0,52 % ± 0,38 %, resp. 0,21 % ± 0,17 %. Plazmatická hodnota %ID na gram u dvou dávek dosáhla vrcholu za 4 hodiny, přičemž průměrné hodnoty u dávek 4, resp. 100 mikrogramů byly 0,0090 %/g ± 0,0048 %/g, resp. 0,0039 %/g ± 0,0046 %/g. U dávky 20 mikrogramů bylo dosaženo vrcholu za 2,5 hodiny s průměrnou hodnotou %ID 0,0023 %/g ± 0,0005 %/g.
Ve druhé studii fáze 1 u pacientů s karcinomem prsu, ve které bylo pacientům injekčně podáno 20 mikrogramů přípravku Lymphoseek, činila průměrná rychlostní konstanta eliminace tilmanoceptu značeného techneciem-99m 0,299/hod a poločas léčiva v místě vpichu byl 2,6 hodiny. Hodnota %IDSN byla ve skupině s podáním přípravku Lymphoseek 3 hodiny před chirurgickým výkonem 1,68 % ± 1,22 % a ve skupině s podáním 16 hodin před chirurgickým výkonem 1,81 % ± 2,19 %.
Ve studii fáze 1 u pacientů s melanomem vykázal přípravek Lymphoseek ve všech třech testovaných dávkách (20, 100 a 200 mikrogramů) clearance z místa podání injekce s rychlostními konstantami eliminace v rozmezí 0,227/hod až 0,396/hod, což znamená poločas léčiva v místě vpichu 1,75 až 3,05 hod. Vychytávání tilmanoceptu značeného techneciem-99m v primární sentinelové uzlině se zvyšovalo v závislosti na dávce: Po injekci přípravku Lymphoseek v dávce 20, resp. 100, resp. 200 mikrogramů bylo dosaženo hodnoty LSN tilmanoceptu značeného techneciem-99m 5,01 ± 8,02 pmol, resp. 17,5 ± 13,7 pmol, resp. 58,2 ± 41,2 pmol. Hodnota %IDSN pro vychytání v primární lymfatické uzlině činila 0,50 % u dávky 20 mikrogramů, 0,35 % u dávky 100 mikrogramů a 0,58 % u dávky 200 mikrogramů přípravku Lymphoseek. Plazmatická hodnota %ID na gram u dvou dávek dosáhla vrcholu za 15 minut, přičemž průměrné hodnoty u dávek 20, resp. 200 mikrogramů byly 0,0104 %/g ± 0,0135 %/g, resp. 0,0065 %/g ± 0,0082 %/g. U dávky 100 mikrogramů bylo dosaženo vrcholu za 1 a za 2 hodiny s průměrnou hodnotou %ID 0,0018 %/g ± 0,001 %/g v obou časových okamžicích.
Eliminace
Tilmanocept značený techneciem-99m se vylučuje primárně ledvinami. Metabolismus tilmanoceptu značeného techneciem-99m nebyl experimentálně zkoumán. Tilmanocept může být metabolizován v játrech na jednotlivé molekuly, ze kterých je složen, jmenovitě dextran (který se vylučuje ledvinami a/nebo je dále metabolizován na glukózu), manózu (endogenní cukr) a kyselinu diethylentriaminpentaoctovou (která se vylučuje ledvinami). Stejně jako u všech metabolitů obecně, zvláště u těch, kde v eliminaci mají měřitelnou úlohu játra, je vylučování určité části žlučí pravděpodobné také u tilmanoceptu značeného techneciem-99m.
Hodnota %ID v játrech, ledvinách a močovém měchýři vypočítaná z celotělového zobrazení u pacientů s karcinomem prsu za 1, 2,5 a 12 hodin po podání byla vždy nižší než 2,6 % (u všech dávek). Hodnota %ID v játrech, ledvinách a močovém měchýři vypočítaná z celotělového zobrazení u pacientů s melanomem za 1 a za 12 hodin po podání se pohybovala v rozmezí 1,1 % až 3,1 % za 1 hodinu a za 12 hodin vždy klesla na méně než 1 %.
5.3. Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po akutním a opakovaném podávání a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1. Seznam pomocných látek
Dihydrát trehalosy Glycin (E640)
Natrium-askorbát (E301)
Dihydrát chloridu cínatého(E512)
Hydroxid sodný (E524)
Kyselina chlorovodíková 10% (E507)
6.2. Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6 a 12.
6.3. Doba použitelnosti
2 roky
Po radioaktivním značení
6 hodin. Uchovávejte při teplotě do 25 °C. Uchovávejte za použití vhodného stínění radioaktivity.
Z mikrobiologického hlediska je třeba přípravek použít okamžitě. Pokud není použit okamžitě, za dobu a podmínky uchovávání před použitím zodpovídá uživatel.
6.4. Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho radioaktivním značení jsou uvedeny v bodě 6.3. Uchovávání radiofarmak musí být v souladu s národními předpisy pro radioaktivní látky.
6.5. Druh obalu a obsah balení
5ml injekční lahvička ze skla typu I s butyl pryžovou zátkou a odtrhovacím uzávěrem. Každá injekční lahvička obsahuje 250 mikrogramů tilmanoceptu.
Balení obsahuje 5 injekčních lahviček.
6.6. Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Obecné upozornění
Radiofarmaka smějí přejímat, používat a podávat pouze oprávněné osoby v určených zdravotnických zařízeních. Jejich příjem, skladování, použití, přeprava a likvidace podléhají předpisům a příslušným povolením kompetentních oficiálních organizací.
Radioaktivní léčivé přípravky by měly být připraveny způsobem, který vyhovuje požadavkům jak na radiační bezpečnost, tak na farmaceutickou kvalitu. Měla by být přijata vhodná aseptická opatření.
Obsah injekční lahvičky je určen pouze k přípravě a radioaktivnímu značení přípravku Lymphoseek a není určen k podání přímo pacientovi bez předchozího provedení přípravného postupu. Každá 250mikrogramová injekční lahvička obsahuje určité množství přípravku navíc. Je nicméně doporučeno připravit injekční lahvičku podle pokynů a pro jednotlivou dávku u pacienta použít alikvotní množství odpovídající dávce 50 mikrogramů, přičemž veškerý zbývající materiál je třeba po rekonstituci a použití zlikvidovat.
Návod k rekonstituci a radioaktivnímu značení léčivého přípravku před jeho podáním je uveden v bodě 12. Radioaktivně značený přípravek je čirý bezbarvý roztok bez viditelných částic.
Jestliže je kdykoli při přípravě tohoto přípravku narušena integrita injekční lahvičky, neměl by se přípravek používat.
Postup při podání přípravku by měl být proveden takovým způsobem, aby minimalizoval kontaminaci léčivého přípravku a ozáření obsluhy. Je povinné odpovídající stínění.
Obsah kitu není před bezprostřední přípravou radioaktivní. Po přidání technecistanu-(99m Tc) sodného dle Evropského lékopisu je nicméně nutné provádět závěrečnou přípravu při odpovídajícím stínění.
Podání radiofarmak vede u dalších osob k riziku vnějšího ozáření nebo kontaminace z rozlité moči, zvratků apod. Musí být proto podniknuta opatření v rámci radiační ochrany v souladu s národními nařízeními.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Navidea Biopharmaceuticals Limited 30 Upper High Street Thame, OX9 3EZ Spojené království
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/955/001
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
10. DATUM REVIZE TEXTU
11. DOZIMETRIE
Technecium-99m se vyrábí prostřednictvím generátoru (99Mo/99mTc) a rozpadá se za emise záření gama s průměrnou energií 140 keV s poločasem 6,02 hodiny na technecium-99, které lze z důvodu jeho dlouhého poločasu 2,13 x 105 roku považovat za kvazistabilní.
Odhad dávky radioaktivního záření pro řadu orgánů vychází z referenčních hodnot MIRD (Medical Internal Radiation Dose) u člověka a hodnot MIRD S a byl vypočítán z biologických údajů o vychytávání v orgánech a clearance z krve.
Dávky radioaktivity pro orgány a tkáně u průměrného pacienta (70 kg) na MBq radioaktivně značeného přípravku Lymphoseek uvádí tabulka 1 a tabulka 2.
Tabulka 1. Odhadovaná dávka absorbovaná z přípravku Lymphoseek u pacientů s karcinomem prsua
|
Odhadovaná dávka absorbované radioaktivity u karcinomu prsu, mGy/MBq | |
|
Cílový orgán |
Dospělí |
|
mozek |
0,0002 |
|
prs (místo vpichu injekce) |
0,0897 |
|
stěna žlučníku |
0,0019 |
|
stěna dolní části tlustého střeva |
0,0007 |
|
tenké střevo |
0,0005 |
|
žaludek |
0,0010 |
|
stěna horní části tlustého střeva |
0,0007 |
|
ledviny |
0,0101 |
|
játra |
0,0018 |
|
plíce |
0,0020 |
|
sval |
0,0005 |
|
vaječníky |
0,0101 |
|
červená kostní dřeň |
0,0007 |
|
kost |
0,0010 |
|
slezina |
0,0015 |
|
varlata |
0,0027 |
|
brzlík |
0,0063 |
|
štítná žláza |
0,0048 |
|
močový měchýř |
0,0032 |
|
celý organismus (krev)b |
0,0011 |
|
Účinná dávka (E) (muži, mSv/MBq) |
0,01600 |
|
Účinná dávka (E) (ženy, mSv/MBq) |
0,01785 |
a Vypočteno dle údajů od 18 pacientů s karcinomem prsu, kteří dostali čtyři peritumorózní injekce přípravku Lymphoseek v dávkách 4, 20 a 100 mikrogramů.
b Krev představuje expozici celého organismu bez ohledu na nezávislá měření dalších orgánů a tkání. Tab. 2. Odhadovaná dávka absorbovaná z přípravku Lymphoseek u pacientů s melanomema
|
Odhadovaná dávka absorbované radioaktivity u melanomu, mGy/MBq | |
|
Cílový orgán |
Dospělí s melanomem |
|
mozek |
0,0050 |
|
prs (místo vpichu injekce) |
0,0427 |
|
stěna žlučníku |
0,0038 |
|
stěna dolní části tlustého střeva |
0,0031 |
|
tenké střevo |
0,0032 |
|
žaludek |
0,0030 |
|
stěna horní části tlustého střeva |
0,0031 |
|
ledviny |
0,0150 |
|
játra |
0,0050 |
|
plíce |
0,0032 |
|
sval |
0,0024 |
|
vaječníky |
0,0162 |
|
červená kostní dřeň |
0,0027 |
|
kost |
0,0047 |
|
slezina |
0,0032 |
|
varlata |
0,0056 |
|
brzlík |
0,0031 |
|
štítná žláza |
0,0025 |
|
močový měchýř |
0,0076 |
|
celý organismus (krev)b |
0,0030 |
|
Účinná dávka (E) (muži, mSv/MBq) |
0,01094 |
|
Účinná dávka (E) (ženy, mSv/MBq) |
0,01357 |
a Vypočteno dle údajů od 18 pacientů s melanomem, kteří dostali čtyři intradermální injekce přípravku Lymphoseek v dávkách 20, 100 a 200 mikrogramů.
b Krev představuje expozici celého organismu bez ohledu na nezávislá měření dalších orgánů a tkání.
12. NÁVOD PRO PŘÍPRAVU RADIOFARMAK
Bezpečnost z hlediska radioaktivity - manipulace s přípravkem
Při manipulaci s přípravkem Lymphoseek používejte voděodolné rukavice, účinné stínění radioaktivity a vhodná bezpečnostní opatření, abyste předešli zbytečné expozici pacienta, zaměstnanců, klinického personálu a dalších osob radioaktivitě.
Radiofarmaka by měli používat nebo na jejich použití dohlížet kvalifikovaní zdravotničtí pracovníci se speciálním školením a zkušenostmi s bezpečným používáním radionuklidů a manipulací s nimi, jejichž zkušenosti a školení schválila příslušná vládní agentura oprávněná poskytovat licenci k používání radionuklidů.
Pokyny k radioaktivnímu značení 250 mikrogramů práškového tilmanoceptu v injekční lahvičce techneciem-99m
Obecná ustanovení
Složky kitu v injekční lahvičce jsou sterilní, nepyrogenní a jsou určeny výhradně k použití při přípravě přípravku Lymphoseek. Nepodávejte nepřipravené složky kitu v injekční lahvičce přímo pacientovi.
Během přípravy a podání dodržujte aseptické postupy.
Během přípravy a podání dodržujte vhodná bezpečnostní opatření z hlediska radioaktivity. U radioaktivně značeného přípravku Lymphoseek používejte stínění radioaktivity z důvodu prevence expozice radioaktivnímu záření.
Používejte pouze eluát z generátoru technecia-99m, který byl předtím eluován po dobu 8 hodin. Nejvyšší radiochemické čistoty dosáhnete při rekonstituci s čerstvě eluovaným eluátem z generátoru technecia-99m.
Reakce značení techneciem-99m závisí na udržení cínatého iontu v redukovaném stavu. K rekonstituci tohoto kitu se nemají používat injekce technecistanu-(99m Tc) sodného obsahující oxidační činidla. Injekční lahvičky jsou uzavírány v dusíkové atmosféře; vzduch nebo kyslík obsah injekční lahvičky poškozuje, a proto by se do injekční lahvičky neměl dostat vzduch.
Radioaktivně označený injekční roztok přípravku Lymphoseek má být podán do 6 hodin od rekonstituce. Dávka musí v době podání obsahovat minimální požadovanou hladinu radioaktivity technecia-99m pro případ provedení chirurgického výkonu ve stejný den (18,5 MBq) nebo pro případ provedení chirurgického výkonu následující den (74 MBq).
Stanovení objemu injekce
Přípravek Lymphoseek může být pacientovi podán v jedné injekci nebo ve více injekcích. Před přípravou stanovte plánovanou techniku podání injekce a počet injekcí, které u daného pacienta použijete. Pro každou injekci připravte samostatnou injekční stříkačku. Na základě plánovaného počtu injekčních stříkaček a plánovaného celkového objemu injekce na pacienta určete (podle níže uvedené tabulky 3) objem radioaktivně značeného přípravku Lymphoseek v injekční lahvičce po rekonstituci.
Každá injekční lahvička s přípravkem Lymphoseek by po rekonstituci a radioaktivním značení provedeném podle pokynů měla obsahovat 250 mikrogramů přípravku a měla by být podána podle tabulky 3. Celkový obsah injekční lahvičky by neměl být podán jedinému pacientovi. Radioaktivně značený přípravek se má použít do 6 hodin od přípravy. Nespotřebovaný přípravek zlikvidujte.
Tabulka 3: Injekce přípravku Lymphoseek podle objemu injekce
|
Požadovaný počet injekcí |
Celkový objem určený k injekčnímu podání |
Celkový objem přípravku Lymphoseek v injekční lahvičce po rekonstituci |
|
injekce 1 x 0,1 ml |
0,1 ml |
0,5 ml |
|
injekce 5 x 0,1 ml nebo injekce 2 x 0,25 ml nebo injekce 1 x 0,5 ml |
0,5 ml |
2,5 ml |
|
injekce 5 x 0,2 ml nebo injekce 4 x 0,25 ml nebo injekce 2 x 0,5 ml |
1,0 ml |
5,0 ml |
Způsob přípravy
Příprava radioaktivně značeného injekčního roztoku přípravku Lymphoseek z kitu se provádí podle
následujícího aseptického postupu:
a. Před radioaktivním označením prohlédněte injekční lahvičku s práškovým tilmanoceptem, jestli není nijak poškozená. Pokud se vám zdá, že je injekční lahvička poškozená, nepoužívejte ji.
b. K radioaktivnímu značení použijte roztok technecistanu-(Tc 99m) sodného z generátoru technecia-99m do 8 hodin po jeho eluci.
c. Před radioaktivním značením ani v jeho průběhu by se do injekční lahvičky s práškovým tilmanoceptem
neměl dostat vzduch.
d. Pomocí sterilní injekční stříkačky asepticky odeberte přibližně 92,5 MBq nebo 370 MBq roztoku technecistanu-(Tc 99m) sodného buď do objemu asi 0,35 ml (pro objem injekční lahvičky po rekonstituci 0,5 ml), nebo do objemu asi 0,70 ml (pro objem injekční lahvičky po rekonstituci 2,5 ml nebo 5 ml). Stanovte radioaktivitu technecia-99m v injekční stříkačce pomocí kalibrátoru dávky.
e. Před radioaktivním značením zapište do určeného místa na štítku injekční lahvičky s radioaktivním přípravkem množství radioaktivity, objem injekční lahvičky po rekonstituci, datum a čas, dobu použitelnosti a číslo šarže a štítek nalepte na injekční lahvičku s práškovým tilmanoceptem. Injekční lahvičku vložte za radiační štít a septum vydezinfikujte tamponem namočeným v alkoholu.
f. Do injekční lahvičky s práškovým tilmanoceptem asepticky přidejte roztok technecistanu-(Tc 99m) sodného (z kroku d uvedeného výše). Aniž byste vytáhli jehlu, odeberte z horního prostoru injekční lahvičky stejný objem plynu. Nevpouštějte vzduch.
g. Vytáhněte jehlu, jemným zatřepáním promíchejte obsah injekční lahvičky a poté injekční lahvičku ponechte nejméně 15 minut stát při pokojové teplotě.
h. Před odebráním dávky pro daného pacienta do injekční stříkačky/injekčních stříkaček přidejte
k radioaktivně značenému přípravku v injekční lahvičce s práškovým tilmanoceptem aseptickým postupem sterilní injekční roztok chloridu sodného 9 mg/ml (0,9%) tak, abyste dosáhli objemu v injekční lahvičce po rekonstituci 0,5 ml, 2,5 ml nebo 5 ml. Kvůli vyrovnání tlaku odeberte z horní části injekční lahvičky stejné množství plynu.
i. Stanovte celkovou radioaktivitu radioaktivně značené injekční lahvičky pomocí kalibrátoru dávky. Na štítek stínicího štítu obsažený v kitu zapište koncentraci radioaktivity, celkový objem, datum a čas stanovení, dobu použitelnosti a číslo šarže technecia-99m. Štítek nalepte na stínicí štít.
j. Stanovte radiochemickou čistotu radioaktivně značeného přípravku, jak je popsáno níže.
k. Do požadovaného počtu injekčních stříkaček odeberte požadovaný objem radioaktivně značeného přípravku. Stanovte radioaktivitu injekční stříkačky/stříkaček v kalibrátoru dávky. Na štítek injekční stříkačky zapište množství radioaktivity, datum a čas stanovení, objem a dobu použitelnosti (nemá přesáhnout 6 hodin od doby přípravy) a štítek nalepte na injekční stříkačku/stříkačky.
l. Radioaktivně značený přípravek uchovávejte za stínicím štítem. Uchovávejte při teplotě do 25 °C.
Použijte před uplynutím doby použitelnosti uvedené na štítku.
Stanovení radiochemické čistoty radioaktivně značeného přípravku Lymphoseek.
Radiochemickou čistotu radioaktivně značeného přípravku Lymphoseek stanovte instantní chromatografií na tenké vrstvě (ITCL) s využitím papíru Whatman stupně 1, 3 mm, 31ET Chr nebo Biodex 150-001 Red Strips (celulózového chromatografického papíru) pomocí následujícího postupu:
a. Tužkou označte na chromatografickém proužku start, střední linii a čelní linii rozpouštědla, jak je znázorněno níže:
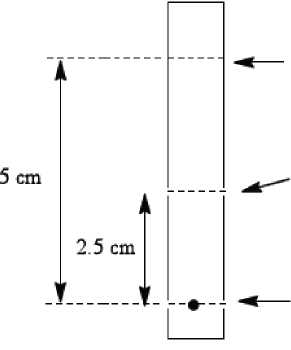
Střední linie
Chromatografický proužek Whatman stupeň 1, 3 mm,
Čelní linie rozpouštědla
jí
Startovní linie 1 cm od dolního konce
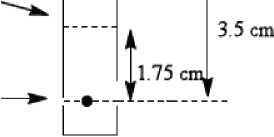
Biodex 150-001 Red Strips
31ET
b. Do středu startovní linie na chromatografickém proužku kápněte malou kapku (3-10 mikrolitrů) radioaktivně značeného přípravku.
c. Proužek vložte do chromatografické komory s obsahem 1 ml acetonu jako vyvíjecího rozpouštědla. Rozpouštědlo nechejte dosáhnout jeho čelní linie (5 cm od dolního konce u proužku Whatman a 3,5 cm u proužku Biodex). Vyjměte proužek z komory, nechte ho uschnout a přestřihněte ho v polovině. U obou polovin proužku stanovte radioaktivitu pomocí vhodného přístroje pro počítání radioaktivních impulzů (kalibrátoru dávky nebo vícekanálového analyzátoru).
d. Procento radiochemické čistoty (% RCP) vypočítejte následujícím způsobem: počet impulzů (aktivita) v dolní
% RCP
x 100
polovině_
počet impulzů (aktivita) v dolní polovině + počet impulzů (aktivita) v horní polovině
e. Pokud je radiochemická čistota nižší než 90 %, radioaktivně značený přípravek Lymphoseek nepoužívejte.
Získání zobrazení / mapování sentinelových lymfatických uzlin
Při použití u karcinomu prsu, melanomu a spinocelulárního karcinomu dutiny ústní u dospělých:
• V klinických studiích dostávali pacienti přípravek Lymphoseek až 30 hodin před chirurgickým
výkonem. K intraoperační identifikaci sentinelových lymfatických uzlin s vychytáváním technecia-99m byl použit ruční čítač záření gama (např. jakákoli ruční sonda detekující záření gama). V klinických studiích s přípravkem Lymphoseek použili zkoušející lékaři pravidlo prahu pozitivity vychytávání technecia-99m odhadnutého podle množství radioaktivity na pozadí plus tři směrodatné odchylky od průměrné hladiny radioaktivity pozadí (tj. pravidlo tří sigma, což znamená > 99,7% pravděpodobnost rozdílu od pozadí) [viz tabulka 4]. Radioaktivita pozadí byla v typickém případě stanovena v tkáni ve vzdálenosti nejméně 20 cm distálně od místa vpichu injekce.
Tabulka 4: Příklad pravidla prahu tří sigma
|
Radioaktivita pozadí3 |
Hodnota prahu tří sigma |
|
5 |
11,71 |
|
10 |
19,49 |
|
15 |
26,62 |
|
20 |
33,42 |
|
25 |
40,00 |
a Průměr ze tří počtů za 2 sekundy nebo jednoho počtu za 10 sekund
• Všechny přípravky pro mapování lymfatických cest využívají k distribuci prvky lymfatického systému. Zobrazení a detekce sentinelových lymfatických uzlin při použití přípravku Lymphoseek závisí na specifickém molekulárním zacílení přípravku a jeho vazbě na retikuloendoteliální buňky v lymfatických uzlinách. Poškození anatomické struktury a funkce lymfatického systému dané oblasti předchozím rozsáhlým chirurgickým výkonem, ozařováním nebo metastázou může vést ke sníženému vychytávání přípravku Lymphoseek v lymfatických uzlinách. Podle klinických studií nezávisí míra vychytávání (procento všech pacientů s nejméně jednou horkou uzlinou) a stupeň vychytávání (průměrný počet horkých uzlin na pacienta) přípravku Lymphoseek na technice injekce radiofarmaka. Použití přípravku Lymphoseek je určeno k doplnění palpačního a vizuálního vyšetření a dalších postupů důležitých pro lokalizaci lymfatických uzlin. Intraoperační lymfatické mapování pomocí detekce záření gama lze zahájit již za 15 minut po injekci a do 30 hodin (při chirurgickém výkonu následující den) po podání přípravku Lymphoseek.
• Po injekční aplikaci přípravku Lymphoseek lze provést zevní detekci a zobrazení záření gama. Doporučená doba provedení předoperačních zobrazovacích postupů je 15 minut po injekci, ale postupy lze zahájit již po 10 minutách. Mezi účinné předoperační postupy zobrazení patří planární scintigrafie gamakamerou, SPECT a SPECT/CT. Vzhledem k tomu, že se jedná o doplněk k intraoperačnímu vyšetření gamasondou, nelze takto získaná zobrazení považovat za náhradu profesionálně a pečlivě provedeného intraoperačního vyšetření ruční sondou pro detekci záření gama.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ / VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ / VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného / výrobců odpovědných za propouštění šarží
Penn Pharmaceutical Services Ltd.
23-24 Tafarnaubach Industrial Estate Tredegar, Gwent NP22 3AA South Wales Spojené království
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU Vnější papírový obal
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Lymphoseek 250 mikrogramů, kit pro radiofarmakum Tilmanocept
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Každá injekční lahvička obsahuje 250 mikrogramů tilmanoceptu.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky Dihydrát trehalosy Glycin (E640)
Natrium-askorbát (E301)
Dihydrát chloridu cínatého (E512) Hydroxid sodný (E524)
Kyselina chlorovodíková 10% (E507)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Kit pro radiofarmakum 5 injekčních lahviček
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím čtěte příbalovou informaci.
Součástí tohoto balení jsou pokyny pro rekonstituci a radioaktivní značení přípravku.
Pro injekční aplikaci po radioaktivním značení.
Intradermální, subkutánní, intratumorózní nebo peritumorózní podání po značení technecistanem-(99mTc) sodným.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP:
Radioaktivně značený roztok lze použít po dobu 6 hodin, pokud je uchováván při teplotě do 25 °C.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 30 °C.
Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Navidea Biopharmaceuticals Limited 30 Upper High Street
Thame, Oxon, OX9 3EZ, Spojené království
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/955/001
13. ČÍSLO ŠARŽE
Šarže:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Lymphoseek 250 mikrogramů, kit pro radiofarmakum Tilmanocept
2. ZPŮSOB PODÁNÍ
Pro injekční aplikaci po radioaktivním značení technecistanem-(99mTc) sodným. 3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Šarže:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Před použitím si přečtěte příbalovou informaci. Navidea Biopharmaceuticals Ltd
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Lymphoseek 250 mikrogramů, injekční roztok tilmanocept značený techneciem-99m
Intradermální, subkutánní, intratumorózní nebo peritumorózní podání.
2. ZPŮSOB PODÁNÍ_
Pro injekce
3. POUŽITELNOST_
Podejte do 6 hodin po radioaktivním značení.
EXP:_čas/datum
4. ČÍSLO ŠARŽE_
Šarže:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
Celková aktivita:_MBq
Celkový objem:_ml
Čas kalibrace:_čas/datum
6. JINÉ_
Uchovávejte při teplotě do 25°C.

POZOR
RADIOAKTIVNÍ
MATERIÁL
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: Informace pro pacienta
Lymphoseek 250 mikrogramů, kit pro radiofarmakum
Tilmanocept
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než je vám tento léčivý přípravek podán, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého specialisty na nukleární medicínu, který bude na výkon dohlížet.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému specialistovi na nukleární medicínu. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Lymphoseek a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Lymphoseek používat
3. Jak se přípravek Lymphoseek používá
4. Možné nežádoucí účinky
5. Jak přípravek Lymphoseek uchovávat
6. Obsah balení a další informace
1. Co je přípravek Lymphoseek a k čemu se používá
Tento léčivý přípravek je určen pouze k diagnostickým účelům. To znamená, že se používá u karcinomu prsu, melanomu nebo nádorových onemocnění ústní dutiny k vyšetření onemocnění. Nejedná se o léčbu vašeho onemocnění.
Před použitím se prášek v injekční lahvičce, který obsahuje tilmanocept, smíchá s radioaktivním přípravkem zvaným technecistan sodný (obsahujícím technecium-99m) a vznikne látka zvaná tilmanocept značený techneciem-99m.
Vzhledem k tomu, že tilmanocept značený techneciem-99m obsahuje malé množství radioaktivity, může během vyšetření zviditelnit určitá místa v organismu pro lékaře a pomoci jim zjistit, zda se nádorové onemocnění rozšířilo do míst zvaných „lymfatické uzliny“ nacházejících se v blízkosti nádoru. Lymfatické uzliny nejblíže nádoru se nazývají „sentinelové“ lymfatické uzliny. Tyto lymfatické uzliny jsou místa, kam se nádorové buňky s největší pravděpodobností šíří. Když jsou pomocí přípravku Lymphoseek vyhledány sentinelové lymfatické uzliny, je možné je odstranit a provést vyšetření, které ukáže, zda se v nich nacházejí nádorové buňky. Přípravek Lymphoseek vyhledá lymfatické uzliny, které lze zachytit pomocí speciální kamery nebo detektoru.
Při podání přípravku Lymphoseek jste vystaven(a) malému množství radioaktivity. Váš lékař a specialista na nukleární medicínu dospěli k názoru, že klinický přínos, který pro vás bude mít výkon s radiofarmakem, převyšuje riziko způsobené ozářením.
2. Čemu musíte věnovat pozornost, než začnete přípravek Lymphoseek používat Přípravek Lymphoseek se nesmí použít
jestliže j ste alergický/á na tilmanocept nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6) nebo jakoukoliv ze složek radioaktivně značeného přípravku.
Upozornění a opatření
Zvláštní opatrnosti při použití přípravku Lymphoseek je zapotřebí:
• jestliže j ste těhotná nebo se domníváte, že byste těhotná mohla být;
• jestliže kojíte.
Před podáním přípravku Lymphoseek:
Řiďte se pokyny lékaře nebo specialisty na nukleární medicínu.
Děti a dospívající
Tento léčivý přípravek není určen k použití u dětí a dospívajících, kteří nedosáhli 18 let věku, protože nebyl u této věkové skupiny hodnocen.
Další léčivé přípravky a přípravek Lymphoseek
Informujte specialistu na nukleární medicínu o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Týká se to i léčivých přípravků, které jsou dostupné bez lékařského předpisu, a rostlinných léčivých přípravků.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se specialistou na nukleární medicínu dříve, než vám bude tento léčivý přípravek podán.
Pokud je možné, že jste těhotná, pokud se u vás opozdila menstruace či pokud kojíte, musíte o tom před podáním přípravku Lymphoseek informovat svého specialistu na nukleární medicínu.
Pokud máte pochybnosti, je důležité je probrat s vaším specialistou na nukleární medicínu, který bude dohlížet na výkon.
Pokud jste těhotná, podá vám specialista na nukleární medicínu tento přípravek během těhotenství, pouze pokud bude předpokládat, že jeho přínos převýší rizika.
Pokud kojíte, je třeba zlikvidovat mateřské mléko vyloučené po dobu 24 hodin po podání přípravku Lymphoseek.
Prosím zeptejte se svého specialisty na nukleární medicínu, kdy můžete kojení obnovit.
Řízení dopravních prostředků a obsluha strojů
Je považováno za nepravděpodobné, že přípravek Lymphoseek naruší vaši schopnost řídit nebo obsluhovat stroje. Váš lékař a specialista na nukleární medicínu vám sdělí, kdy bude po chirurgickém výkonu bezpečné řídit.
Přípravek Lymphoseek obsahuje sodík:
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) na dávku. To znamená, že v podstatě „neobsahuje sodík“.
3. Jak se přípravek Lymphoseek používá
Existují přísné zákony týkající se manipulace s radiofarmaky a jejich použití a likvidace. Přípravek Lymphoseek se bude používat pouze ve zvláštních kontrolovaných prostorech. S tímto přípravkem budou manipulovat a budou vám ho podávat pouze osoby, které jsou školeny na jeho bezpečné použití. Tyto osoby se zvláště starají o bezpečné použití tohoto přípravku a budou vás informovat o své činnosti.
Specialista na nukleární medicínu dohlížející na výkon rozhodne, jaké množství přípravku Lymphoseek bude ve vašem případě použito. Bude to nejmenší množství potřebné k tomu, aby byla získána požadovaná informace.
Obvykle doporučované množství pro podání u dospělých se pohybuje od 18,5 do 74 MBq (megabequerelů,
což je jednotka používaná k vyjádření radioaktivity).
Dávka může být rozdělena do menších množství. To znamená, že vám může lékař do oblasti v okolí nádoru podat více než jednu injekci.
Podání přípravku Lymphoseek a provedení výkonu
Přípravek Lymphoseek se injekčně podává pod kůži, pod bradavku nebo do okolí nádoru. Místo závisí na typu nádoru.
Přípravek Lymphoseek se podává buď den před výkonem, nebo v den výkonu.
Trvání výkonu
Specialista na nukleární medicínu vám sdělí, jak dlouho bude výkon přibližně trvat.
Specialista na nukleární medicínu používá k vyhledání přípravku Lymphoseek v těle speciální kameru. Chirurg použije zhotovené snímky ke zjištění, kde se nacházejí sentinelové lymfatické uzliny. Chirurg použije také přístroj, který vyhledá technecium-99m, které je součástí léčivého přípravku. Technecium-99m ukáže chirurgovi, kde se nacházejí sentinelové lymfatické uzliny.
Když je sentinelová uzlina nalezena, chirurg ji odstraní. Pokud je sentinelových uzlin více, chirurg je odstraní všechny. Sentinelové lymfatické uzliny jsou poté vyšetřeny, aby se zjistilo, zda se do nich nádorové buňky rozšířily.
Co byste měl(a) dělat po podání přípravku Lymphoseek?
Specialista na nukleární medicínu vás bude informovat, zda u vás budou po podání tohoto léčivého přípravku nutná zvláštní opatření. Jestliže máte jakékoli dotazy, obraťte se na svého lékaře.
Jestliže jste dostal(a) více přípravku Lymphoseek, než jste měl(a)
Předávkování je nepravděpodobné, protože dostanete speciálně odměřené množství přípravku Lymphoseek, které pečlivě zkontroluje lékař, který na výkon dohlíží. V případě předávkování nicméně dostanete odpovídající léčbu.
Pokud máte jakékoli další otázky týkající se použití přípravku Lymphoseek, prosím zeptejte se specialisty na nukleární medicínu, který na výkon dohlíží.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. U tohoto léčivého přípravku se mohou objevit následující nežádoucí účinky:
Méně časté (mohou postihnout až 1 ze 100 osob):
• podráždění nebo bolest v místě podání injekce (včetně prsu a kůže);
• bolest v ráně, otevření rány a hromadění tekutiny v místě chirurgického výkonu;
• nevolnost (nauzea) nebo závrať;
• rozmazané vidění;
• obtížné mluvení;
• bolest hlavy;
• zvýšená tepová frekvence;
• časté nebo naléhavé nucení na močení;
• pocit tepla, pocit píchání nebo mravenčení nebo bolest v končetině, v rameni, v oblasti krční páteře nebo v čelisti;
• návaly horka;
• vysoká hladina vápníku v krvi.
Toto radiofarmakum bude vydávat malé množství ionizujícího záření, které je spojené s minimálním rizikem nádorového onemocnění nebo vrozených vad.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému specialistovi na nukleární medicínu. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře.
Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Lymphoseek uchovávat
Tento léčivý přípravek nemusíte uchovávat. Za uchovávání tohoto léčivého přípravku odpovídá specialista v příslušném zařízení. Uchovávání radiofarmak musí být v souladu s národními předpisy pro radioaktivní látky.
Následující informace jsou určeny pouze pro odborníky.
Nepoužívejte přípravek Lymphoseek po uplynutí doby použitelnosti uvedené na krabičce a štítku za označením „Exp“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 30 °C. Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Radioaktivně značený roztok je stabilní po dobu 6 hodin při 25 °C.
Radioaktivně značený přípravek je čirý bezbarvý roztok bez viditelných částic. Nepoužívejte, pokud pozorujete pevné částice a/nebo změnu zabarvení.
6. Obsah balení a další informace Co přípravek Lymphoseek obsahuje
• Léčivou látkou je tilmanocept. Každá injekční lahvička obsahuje 250 mikrogramů tilmanoceptu.
• Dalšími složkami jsou dihydrát trehalosy, glycin (E640), natrium-askorbát (E301), dihydrát chloridu cínatého, hydroxid sodný (E524) a kyselina chlorovodíková (E507).
Jak přípravek Lymphoseek vypadá a co obsahuje toto balení
Před použitím se prášek v injekční lahvičce, který obsahuje tilmanocept, smíchá s jiným léčivým přípravkem zvaných technecistan sodný a vznikne látka zvaná tilmanocept značený techneciem-99m.
Velikosti balení
Skleněné injekční lahvičky jsou dodávány v krabičce obsahující 5 injekčních lahviček.
Držitel rozhodnutí o registraci
Navidea Biopharmaceuticals Limited 30 Upper High Street Thame, OX9 3EZ Spojené království
Tel. +1 614 822 2415 Fax: +1-614-822-2409
Výrobce
Penn Pharmaceutical Services Ltd 23-24 Tafarnaubach Ind. Est. Tredegar, Wales NP22 3AA. Spojené království
Tato příbalová informace byla naposledy revidována
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Úplný souhrn údajů o přípravku Lymphoseek naleznete v odtrhávací části na konci vytištěné příbalové informace v balení přípravku. Jeho cílem je poskytnout zdravotnickým pracovníkům další dodatečné odborné a praktické informace o podání a používání tohoto radiofarmaka.
Viz souhrn údajů o přípravku [souhrn údajů o přípravku musí být přiložen v krabičce].
30