Lumigan 0,1 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
LUMIGAN 0,1 mg/ml oční kapky, roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml roztoku obsahuje bimatoprostum 0,1 mg.
Pomocné látky se známým účinkem:
Jeden ml roztoku obsahuje benzalkonium-chlorid 0,2 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Oční kapky, roztok. Bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Snížení zvýšeného nitroočního tlaku u chronického glaukomu s otevřeným úhlem a nitrooční hypertenze u dospělých (jako monoterapie nebo jako doplňující terapie k betablokátorům).
4.2 Dávkování a způsob podání
Dávkování
Doporučená dávka je jedna kapka do postiženého oka (očí) jedenkrát denně večer.
Dávkování jedenkrát denně by nemělo být překročeno, protože častější podávání může vést ke snížení účinku na nitrooční tlak.
Pediatrická populace :
Bezpečnost a účinnost přípravku LUMIGAN u dětí ve věku od 0 do 18 let nebyla dosud stanovena. Pacienti s poškozením funkce jater nebo ledvin:
LUMIGAN nebyl studován u pacientů s ledvinovým nebo středně závažným až závažným jaterním poškozením, a měl by proto být u těchto pacientů používán s opatrností. U pacientů s předchozím mírným jaterním onemocněním nebo abnormálními hladinami alanin aminotransferázy (ALT), aspartát aminotransferázy (AST) a/nebo bilirubinu, nemá bimatoprost 0,3 mg/ml oční kapky, roztok, nepříznivý účinek na funkci jater alespoň po dobu 24 měsíců.
Způsob podání
Pokud je používán více než jeden lokální oční přípravek, pak je mezi jejich podáním nutné zachovat časový odstup nejméně 5 minut.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodu 6.1.
LUMIGAN 0,1 mg/ml je kontraindikován u pacientů s podezřením na předchozí nežádoucí reakci na benzalkonium-chlorid, která vedla k přerušení léčby.
4.4 Zvláštní upozornění a opatření pro použití
Oční
Před zahájením léčby by měl být pacient informován o možném růstu řas, ztmavnutí kůže na víčkách a zvýšené pigmentaci duhovky, které byly pozorovány během léčby LUMIGANEM. Některé z těchto změn mohou být trvalé a mohou vést k rozdílnému vzhledu očí, pokud bylo léčeno pouze jedno z nich. Zvýšená pigmentace duhovky je pravděpodobně trvalá. Změna pigmentace je způsobena zvýšeným obsahem melaninu v melanocytech, nikoli zvýšením počtu melanocytů. Dlouhodobé účinky zvýšené pigmentace duhovky nejsou známy. Změna barvy duhovky pozorovaná při očním podání bimatoprostu nemusí být patrná po několik měsíců či let. Obvykle se hnědá pigmentace okolo zorničky rozšíří soustředně směrem k okraji duhovky a celá duhovka nebo její části více zhnědnou. Zdá se, že léčba nemá vliv na mateřská znaménkam, ani pihy na duhovce. Po 12 měsících byl zaznamenán 0,5% výskyt hyperpigmentace duhovky při používání očních kapek bimatoprost 0,1 mg/ml, roztok. Po stejné době byl výskyt tohoto účinku u očních kapek bimatoprost 0,3 mg/ml, roztok, 1,5% (viz bod 4.8, Tabulka 2) a rozsah změny se v průběhu 3leté léčby dále nezvyšoval. Podle hlášení byla u některých pacientů pigmentace periorbitální tkáně reverzibilní.
Během léčby očními kapkami obsahujícími bimatoprost 0,3 mg/ml byl méně často (>1/1000 až <1/100) zaznamenán cystoidní makulární edém. U pacientů s rizikovými faktory pro makulární edém (např. afakičtí pacienti, pseudofakičtí pacienti s trhlinou zadního pouzdra čočky) by proto měl být LUMIGAN používán s opatrností.
Existují vzácná spontánní hlášení o reaktivaci dřívějších rohovkových infiltrátů nebo očních infekcí následkem léčby očními kapkami obsahujícími bimatoprost 0,3 mg/ml. Pacienti s anamnézou předchozích závažných virových očních infekcí (jako je herpes simplex) nebo uveitidou/iritidou by měli LUMIGAN používat opatrně.
LUMIGAN nebyl studován u pacientů s očními zánětlivými stavy, glaukomem s uzavřeným úhlem neovaskulární nebo zánětlivé etiologie, kongenitálním glaukomem a glaukomem s úzkým úhlem.
Kůže
V místech, kde LUMIGAN přijde opakovaně do styku s povrchem pokožky, může docházet k růstu ochlupení. Proto je důležité LUMIGAN podávat podle pokynů a zabránit tomu, aby stékal na tvář nebo jiné oblasti kůže.
Respirační
LUMIGAN nebyl studován u pacientů s poškozenými respiračními funkcemi. Ačkoli jsou k dispozici omezené informace u pacientů s astmatem nebo CHOPN v anamnéze, bylo hlášeno zhoršení astmatu, dušnosti a CHOPN, stejně jako zprávy o astmatu v postmarketingovém období. Četnost těchto příznaků není známa. Pacienti s CHOPN, astmatem nebo sníženou respirační funkcí kvůli jiným chorobám mají být léčeni se zvýšenou opatrností.
Kardiovaskulární
LUMIGAN nebyl studován n u pacientů se srdečním blokem více než prvního stupně nebo nekontrolovaným kongestivním srdečním selháním. Existuje omezený počet spontánních hlášení o výskytu bradykardie nebo hypotenze po užití očních kapek obsahující bimatoprost 0,3 mg/ml. Pacienti s predispozicí k nízké srdeční frekvenci nebo nízkému krevnímu tlaku by měli LUMIGAN užívat opatrně.
Další informace
Ve studiích s bimatoprostem 0,3 mg/ml u pacientů s glaukomem nebo nitrooční hypertenzí bylo prokázáno, že častější expozice oka více než jedné dávce biomatorpostu denně může snížit účinnost snižování NOT (viz bod 4.5). U pacientů používajících LUMIGAN s dalšími analogy prostaglandinu mají být sledovány změny nitroočního tlaku.
LUMIGAN 0,1 mg/ml obsahuje konzervační činidlo benzalkonium-chlorid (200 ppm), který může být absorbován měkkými kontaktními čočkami. Díky jeho přítomnosti může také nastat podráždění oka a
odbarvení měkkých kontaktních čoček. Před podáním kapek by proto měly být čočky z oka vyjmuty a opět zavedeny 15 minut po podání.
Benzalkonium-chlorid, běžně používané konzervační činidlo oftalmologických přípravků, může způsobovat tečkovitou keratopatii anebo toxickou ulcerativní keratopatii. Protože LUMIGAN 0,1 mg/ml obsahuje 200 ppm benzalkonium-chloridu (což je čtyřnásobek koncentrace v očních kapkách bimatoprost 0,3 mg/ml), je třeba jej používat opatrně u pacientů, kteří trpí pocity sucha v oku nebo mají poškozenou rohovku či užívají více očních kapek obsahujících BAK (benzalkonium-chlorid). Kromě toho je třeba pacienty, kteří přípravek používají delší dobu, pečlivě sledovat.
Byla hlášena bakteriální keratitida spojená s použitím vícedávkových balení topických očních produktů. Tyto nádobky byly neúmyslně kontaminovány pacienty, kteří ve většině případů trpěli souběžným očním onemocněním. Pacientům s narušeným povrchem očního epitelu hrozí vyšší riziko bakteriální keratitidy.
Pacienty je třeba informovat, aby zabránili styku hrotu aplikační lahvičky s okem nebo okolními strukturami, aby nedošlo k poranění oka a kontaminaci roztoku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Žádné studie interakcí nebyly provedeny.
Interakce u lidí nejsou očekávány, jelikož systémová koncentrace bimatoprostu po očním podávání očních kapek obsahujících bimatoprost 0,3 mg/ml je extrémně nízká (méně než 0,2 ng/ml). Bimatoprost je přeměňován četnými enzymy a cestami, ale v preklinických studiích nebyl pozorován žádný účinek na jaterní enzymy, které metabolizují léky.
V klinických studiích byl bimatoprost 0,3 mg/ml, oční kapky, roztok, LUMIGAN používán současně s řadou různých očních betablokátorů bez známek interakcí.
Současné použití LUMIGANu s jinými antiglaukomatiky než topickými betablokátory nebylo během přídatné léčby glaukomu hodnoceno.
Účinek snižování NOT analog prostaglandinu (např. LUMIGANU) může být nižší u pacientů s glaukomem nebo oční hypertenzí, pokud zároveň používají další analoga prostaglandinu (viz bod 4.4).
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání bimatoprostu těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu při vysokých maternotoxických dávkách (viz bod 5.3).
LUMIGAN by neměl být během těhotenství podáván, pokud to není nezbytně nutné.
Kojení
Není známo, zda bimatoprost přechází do mateřského mléka. Studie na zvířatech však vylučování do mléka prokázaly. Je zapotřebí rozhodnout o ukončení kojení nebo ukončení léčby přípravkem LUMIGAN s ohledem na přínosy kojení pro dítě a přínosy léčebné terapie pro danou ženu.
Fertilita
Údaje o vlivu bimatoprostu na lidskou fertilitu nejsou k dispozici.
4.7 Účinky na schopnost řídit a obsluhovat stroje
LUMIGAN má zanedbatelný vliv na schopnost řídit a obsluhovat stroje. Jestliže, stejně jako po jiné léčbě očí, nastane po podání přechodné rozmazané vidění, měl by pacient před řízením nebo obsluhou strojů počkat, dokud není vidění ostré.
4.8 Nežádoucí účinky
Ve 12 měsíční klinické studii fáze III trpělo nežádoucími účinky asi 38% pacientů léčených očními kapkami LUMIGAN 0,1 mg/ml, roztok. Nejčastějším nežádoucím účinkem byla hyperémie spojivek (většinou stopová nebo mírná a nezánětlivé povahy), kterou trpělo 29% pacientů. Asi 4% pacientů bylo ze studie kvůli nežádoucím účinkům předčasně vyřazeno.
Během klinických studií s očními kapkami LUMIGAN 0,1 mg/ml, roztok nebo v postmarketingovém období byly zaznamenány následující nežádoucí účinky. Většina z nich byly oční, mírné nebo středně závažné, žádný z nich nebyl závažný.
Tabulka 1 ukazuje nežádoucí účinky rozdělené podle frekvence výskytu na velmi časté (>1/10), časté (>1/100 až < 1/10), méně časté >>1/1000 až <1/100), vzácné (>1/10 000 až <1/1000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit). Nežádoucí účinky jsou seřazeny podle tříd orgánových systémů a v každé skupině četnosti podle klesající závažnosti.
Tabulka 1
|
Tělesný oreán nebo skupina oreánů |
Četnost |
Nežádoucí účinek |
|
Poruchy nervového systému |
Méně časté |
Bolesti hlavy |
|
Poruchy oka |
Velmi časté |
Hyperemie spojivek |
|
Časté |
Tečkovitá keratitida, podráždění očí, svědění očí, nadměrný růst řas, bolest oka, erytém očních víček, svědění očních víček | |
|
Méně časté |
Astenopie, rozmazané vidění, poruchy spojivky, spojivkový edém, hyperpigmentace duhovky, madaróza (úplná ztráta řas), otok očních víček | |
|
Není známo |
Pigmentace víček, makulární edém, změny okolí očí a změny víčka včetně prohloubení záhybu očního víčka, syndrom suchého oka | |
|
Respirační, hrudní a mediastinální poruchy |
Není známo | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Poruchy kůže a podkožní tkáně |
Časté |
pigmentace kůže kolem očí, hypertrichóza |
|
Méně časté |
Suchá kůže, tvrdnutí okrajů víček, svědění | |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Podráždění v místě aplikace |
|
Poruchy imunitního systému |
Není známo |
Hypersenzitivní reakce včetně známek a příznaků oční alergie a alergické dermatitidy |
V klinických studiích bylo LUMIGANEM 0,3 mg/ml léčeno přes 1800 pacientů. Po sloučení údajů z fáze III monoterapie a přídatného použití LUMIGANU 0,3 mg/ml byly nejčastěji hlášenými nežádoucími účinky:
• růst řas až u 45 % pacientů v prvním roce s poklesem výskytu případů po 2 letech na 7 % a po 3
letech na 2 %
• hyperémie spojivek (většinou v náznacích nebo mírná a považována za nezánětlivou) až u
44 % pacientů v prvním roce s poklesem výskytu případů po 2 letech na 13 % a po 3 letech na 12 %
• svědění očí až u 14 % pacientů v prvním roce s poklesem výskytu případů po 2 letech na 3 % a
po 3 letech na 0 %. Méně než 9 % pacientů přerušilo léčbu kvůli jakémukoliv nežádoucímu účinku v prvním roce, ve druhém a třetím roce poklesl počet případů přerušení léčby shodně na 3 %.
Další nežádoucí účinky hlášené s LUMIGANEM 0,3 mg/ml jsou uvedeny v tabulce 2. Tabulka zahrnuje také nežádoucí účinky, které se vyskytly u obou sil přípravku, ale s různou četností výskytu. Většina z nich byly oční, mírné nebo středně závažné, žádný z nich nebyl závažný. V každé skupině četnosti jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 2
|
Tělesný oreán nebo skuDÍna orgánů (trakt) |
Četnost |
Nežádoucí účinek |
|
Poruchy nervového systému |
Časté |
Bolesti hlavy |
|
Méně časté |
Závratě | |
|
Poruchy oka |
Velmi časté |
Svědění očí, nadměrný růst řas |
|
Časté |
Eroze rohovky, pálení očí, alergická konjunktivitida, blefaritida, zhoršení zrakové ostrosti, astenopie, spojivkový edém, pocit cizího tělesa v oku, suchost očí, bolesti očí, fotofobie, slzení, výtok z očí, zhoršení zraku, rozmazané vidění, zvýšená pigmentace duhovky, ztmavnutí řas | |
|
Méně časté |
Krvácení rohovky, uveitida, cystoidní makulární edém, iritida, blefarospazmus, retrakce víček, periorbitální erytém | |
|
Cévní poruchy |
Časté |
Hypertenze |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Hirsutismus |
|
Celkové poruchy a reakce v místě aplikace |
Méně časté |
Astenie |
|
Vyšetření |
Časté |
Abnormální jaterní testy |
Nežádoucí účinky hlášené u očních kapek obsahujících fosfáty:
Velmi vzácně byly u některých pacientů s významně poškozenou rohovkou hlášeny případy korneální kalcifikace spojené s použitím očních kapek obsahujících fosfáty.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyl hlášen žádný případ předávkování a není pravděpodobné, že by po podání do oka nastal.
Pokud dojde k předávkování, léčba by měla být symptomatická a podpůrná. Jestliže je LUMIGAN náhodně požit, mohou být užitečné následující informace: během dvoutýdenní studie u potkanů a myší při perorálních dávkách až do 100 mg/kg/den nevznikla žádná toxicita. Tato dávka vyjádřená v mg/m2 je nejméně 210krát vyšší než množství přípravku v jedné lahvičce očních kapek LUMIGAN 0,1 mg/ml pro 10 kg dítě.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, analoga prostaglandinu, ATC kód: S01EE03 Mechanismus účinku
Mechanismem účinku, kterým bimatoprost redukuje nitrooční tlak u lidí, je zvýšený odtok nitrooční tekutiny trámčinou komorového úhlu a zvýšený odtok uveosklerální cestou. Snižování nitroočního tlaku začíná přibližně 4 hodiny po prvním podání a maximálního účinku je dosaženo přibližně během 8 až 12 hodin. Snížení nitroočního tlaku přetrvává nejméně 24 hodin.
Bimatoprost je silné oční hypotenzivum. Je to syntetický prostamid, strukturálně blízký prostaglandinu F2a (PGF2a), který nepracuje cestou známých prostaglandinových receptorů. Bimatoprost selektivně napodobuje účinek nově objevených biosyntetizovaných substancí nazývaných prostamidy. Nicméně prostamidové receptory nebyly ještě dosud strukturálně identifikovány.
Během 12 měsíční pivotní studie očních kapek LUMIGAN 0,1 mg/ml u dospělých se průměrné denní hodnoty IOP měřené během všech návštěv během 12 měsíčního období studie nelišily o víc než 1,1 mmHg během dne a nikdy nepřekročily 17,7 mmHg.
LUMIGAN 0,1 mg/ml oční kapky obsahuje BAK v koncentraci 200 ppm.
S použitím LUMIGANU u pacientů s glaukomem s otevřeným úhlem, s pseudoexfoliativním a pigmentovým glaukomem a u pacientů s chronickým glaukomem s uzavřeným úhlem s provedenou iridotomií jsou omezené zkušenosti.
Během klinických studií nebyl pozorován žádný klinicky relevantní účinek na srdeční frekvenci a krevní tlak.
Pediatrická populace
Bezpečnost a účinnost přípravku LUMIGAN u dětí od 0 do méně než 18 let nebyla dosud stanovena.
5.2 Farmakokinetické vlastnosti
Vstřebávání
Bimatoprost in vitro velmi dobře penetruje lidskou rohovkou a sklérou. Po očním podání dospělým pacientům je systémová expozice bimatoprostu velmi nízká bez akumulace během doby podávání. Při podávání jedenkrát denně po jedné kapce LUMIGANU 0,3 mg/ml bimatoprostu do obou očí po dobu dvou týdnů je dosaženo vrcholové koncentrace v krvi během 10 minut po podání a následné snížení na nejnižší detekovatelnou hodnotu (0,025 ng/ml) během 1,5 hodiny po aplikaci. Střední Cmax a AUC 0-24hod. hodnoty byly 7. a 14. den podobné, přibližně 0,08 ng/ml respektive 0,09 ng^hod/ml, což ukazuje, že rovnovážného stavu koncentrace bimatoprostu bylo dosaženo během prvního týdne očního podávání.
Distribuce
Bimatoprost je mírně distribuován do tělesných tkání a systémová hladina je ustálena na 0,67 l/kg. V lidské krvi je bimatoprost především v plazmě. Vazba bimatoprostu na plazmatické bílkoviny je přibližně 88 %.
Biotransformace
Jakmile je po očním podání dosaženo systémové cirkulace, je bimatoprost hlavní cirkulující částí v krvi. Bimatoprost podléhá oxidaci, N-deetylaci a glukuronidaci a tvoří se řada různých metabolitů.
Eliminace
Bimatoprost je primárně eliminován ledvinami, více než 67 % z intravenózní dávky zdravým dobrovolníkům bylo vyloučeno močí, 25 % bylo vyloučeno stolicí. Poločas eliminace určený po intravenózním podání byl přibližně 45 minut. Celková clearance krve byla,5 l/hod/kg.
Charakteristika u starších pacientů
U starších pacientů (65 let a více) byla při dávkování bimatroprostu 0,3 mg/ml dvakrát denně byla střední hodnota AUC 0-24hod bimatoprostu 0,0634 ng^hod/ml, což je signifikantně více než 0,0218 ng^hod/ml u mladých zdravých dospělých osob. Nicméně toto zjištění není klinicky relevantní, protože systémová expozice starších i mladších osob byla při očním podávání velmi nízká. Během užívání nedocházelo ke kumulaci bimatoprostu v krvi a bezpečnostní profil pro starší i mladé pacienty je podobný.
5.3 Předklinické údaje vztahující se k bezpečnosti
Účinky v neklinických studiích byly pozorovány pouze po expozicích dostatečně převyšujících maximální expozici u člověka, což svědčí o malém významu při klinickém použití.
Oční podávání bimatoprostu opicím v koncentraci >D0,3mg/ml denně po dobu jednoho roku způsobilo zvýšení pigmentace duhovky a reverzibilní na dávce závislý periokulární efekt charakterizovaný prominující horní a/nebo dolní rýhou a rozšířením palpebrální štěrbiny. Zdá se, že zvýšení pigmentace duhovky je způsobeno zvýšenou stimulací produkce melaninu v melanocytech, a ne zvýšením počtu melanocytů. Žádné funkční ani mikroskopické změny ve vztahu k periokulárnímu efektu nebyly pozorovány, mechanizmus vlivu na periokulární změny není znám.
Bimatoprost nebyl v sérii in vitro a in vivo studií mutagenní nebo karcinogenní.
Bimatoprost nenarušoval u potkanů až do dávky 0,6 mg/kg/den (nejméně 103násobná předpokládaná humánní expozice) fertilitu. V embryo/fetální vývojové studii abortů nebyl ale pozorován vývojový účinek u myší ani potkanů při dávkách, které byly nejméně 860krát respektive 1700krát vyšší než humánní. Tyto dávky byly výsledně při systémovém podávání nejméně 33 respektive 97krát vyšší než množství určené pro člověka. V peri/postnatálních studiích u potkanů způsobila mateřská toxicita redukci gestačního času, fetální smrt a snížení tělesné hmotnosti mláďat o > 0,3 mg/kg/den (nejméně 41krát vyšší než předpokládaná humánní expozice). Neurobehaviorální funkce potomků nebyly postiženy.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Benzalkonium-chlorid Chlorid sodný
Heptahydrát hydrogenfosforečnanu sodného
Monohydrát kyseliny citrónové
Roztok hydroxidu sodného 1 mol/l nebo roztok kyseliny chlorovodíkové 1 mol/l (k udržení pH) Čištěná voda
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
4 týdny po prvním otevření.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a velikost balení
Bílá, neprůhledná lahvička z polyetylenu s polystyrenovým uzávěrem se závitem. Jedna lahvička je naplněna 3 ml.
Dostupné jsou následující velikosti balení: krabička obsahující 1 nebo 3 lahvičky po 3 ml roztoku. Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a zacházení s ním
Žádné zvláštní požadavky na likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co. Mayo
Irsko
8. REGISTRAČNÍ ČÍSLA
EU/1/02/205/003-004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
7. ledna 2010
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agenturypro léčivé přípravky: http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
LUMIGAN 0,3 mg/ml oční kapky, roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml roztoku obsahuje bimatoprostum 0,3 mg.
Pomocné látky se známým účinkem:
Jeden ml roztoku obsahuje benzalkonium-chlorid 0,05 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Oční kapky, roztok.
Bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Snížení zvýšeného nitroočního tlaku u chronického glaukomu s otevřeným úhlem a nitrooční hypertenze u dospělých (jako monoterapie nebo jako doplňující terapie k betablokátorům).
4.2 Dávkování a způsob podání
Dávkování
Doporučená dávka je jedna kapka do postiženého oka (očí) jedenkrát denně večer.
Dávkování jedenkrát denně by nemělo být překročeno, protože častější podávání může vést ke snížení účinku na nitrooční tlak.
Pediatrická populace :
Bezpečnost a účinnost přípravku LUMIGAN u dětí od 0 do 18 let nebyla dosud stanovena.
Pacienti s poškozením funkce jater nebo ledvin:
LUMIGAN nebyl studován u pacientů s ledvinovým nebo středně závažným až závažným jaterním poškozením a měl by proto být u těchto pacientů používán s opatrností. U pacientů s předchozím mírným jaterním onemocněním nebo abnormálními hladinami alanin aminotransferázy (ALT), aspartát aminotransferázy (AST) a/nebo bilirubinu, nemá bimatoprost 0,3 mg/ml oční kapky, roztok nepříznivý účinek na funkci jater alespoň po dobu 24 měsíců.
Způsob podání
Pokud je používán více než jeden lokální oční přípravek, pak je mezi jejich podáním nutné zachovat časový odstup nejméně 5 minut.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodu 6.1.
LUMIGAN 0,3 mg/ml je kontraindikován u pacientů s předchozím podezřením na nežádoucí reakci na benzalkonium chlorid, které vedlo k ukončení jeho podávání.
4.4 Zvláštní upozornění a opatření pro použití
Oční
Před zahájením léčby by měl být pacient informován o možném růstu řas, ztmavnutí kůže na víčkách a zvýšené pigmentaci duhovky, které byly pozorovány během léčby LUMIGANEM. Některé z těchto změn mohou být trvalé a mohou vést k rozdílnému vzhledu očí, pokud bylo léčeno pouze jedno z nich. Zvýšená pigmentace duhovky je pravděpodobně trvalá. Změna pigmentace je způsobena zvýšeným obsahem melaninu v melanocytech, nikoli zvýšením počtu melanocytů. Dlouhodobé účinky zvýšené pigmentace duhovky nejsou známy. Změna barvy duhovky pozorovaná při očním podání bimatoprostu nemusí být patrná po několik měsíců či let. Obvykle se hnědá pigmentace okolo zorničky rozšíří soustředně směrem k okraji duhovky a celá duhovka nebo její části více zhnědnou. Zdá se, že léčba nemá vliv na mateřská znaménka ani pihy na duhovce. Po 12 měsících byl při používání bimatoprostu 0,3 mg/ml výskyt pigmentace duhovky 1,5 % (viz část 4.8) a v průběhu 3leté léčby se dále nezvyšoval. Podle hlášení byla u některých pacientů pigmentace periorbitální tkáně reverzibilní.
Během léčby očními kapkami obsahujícími bimatoprost 0,3 mg/ml byl méně často (>1/1000 až <1/100) zaznamenám cystoidní makulární edém. U pacientů s rizikovými faktory pro makulární edém (např. afakičtí pacienti, pseudofakičtí pacienti s trhlinou zadního pouzdra čočky) by proto měl být LUMIGAN používán s opatrností.
Existují vzácná spontánní hlášení o reaktivaci dřívějších rohovkových infiltrátů nebo očních infekcí následkem léčby očními kapkami obsahujícími bimatoprost 0,3 mg/ml. Pacienti s anamnézou předchozích závažných virových očních infekcí (jako je herpes simplex) nebo uveitidou/iritidou by měli LUMIGAN užívat opatrně.
LUMIGAN nebyl studován u pacientů s očními zánětlivými stavy, glaukomem s uzavřeným úhlem neovaskulární nebo zánětlivé etiologie, kongenitálním glaukomem a glaukomem s úzkým úhlem.
Kůže
V místech, kde LUMIGAN přijde opakovaně do styku s povrchem pokožky, může docházet k růstu ochlupení. Proto je důležité LUMIGAN podávat podle pokynů a zabránit tomu, aby stékal na tvář nebo jiné oblasti kůže.
Respirační
LUMIGAN nebyl studován u pacientů s poškozenými respiračními funkcemi, a měl by být proto u nich používán s opatrností. Ačkoli jsou k dispozici omezené informace u pacientů s astmatem nebo CHOPN v anamnéze, bylo hlášeno zhoršení astmatu, dušnosti a CHOPN, stejně jako zprávy o astmatu v postmarketingovém období. Četnost těchto příznaků není známa. Pacienti s CHOPN, astmatem nebo sníženou respirační funkcí kvůli jiným chorobám mají být léčeni se zvýšenou opatrností.
Kardiovaskulární
LUMIGAN nebyl studován n u pacientů se srdečním blokem více než prvního stupně nebo nekontrolovaným kongestivním srdečním selháním. Existuje omezený počet spontánních hlášení o výskytu bradykardie nebo hypotenze po užití očních kapek obsahující bimatoprost 0,3 mg/ml. Pacienti s predispozicí k nízké srdeční frekvenci nebo nízkému krevnímu tlaku by měli LUMIGAN užívat opatrně.
Další informace
Ve studiích s bimatoprostem 0,3 mg/ml u pacientů s glaukomem nebo nitrooční hypertenzí bylo prokázáno, že častější expozice oka více než jedné dávce biomatorpostu denně může snížit účinnost snižování NOT (viz část 4.5). U pacientů užívajících LUMIGAN s dalšími analogy prostaglandinu by měly být sledovány změny nitroočního tlaku.
Oční kapky bimatoprost 0,3 mg/ml obsahují konzervační činidlo benzalkonium-chlorid, který může být absorbován měkkými kontaktními čočkami. Díky jeho přítomnosti může také nastat podráždění oka a odbarvení měkkých kontaktních čoček. Před podáním kapek by proto měly být čočky z oka vyjmuty a opět zavedeny 15 minut po podání.
Benzalkonium-chlorid, který je běžně používaným konzervačním činidlem oftalmologických přípravků. V souvislosti s benzalkonium-chloridem byl hlášen výskyt tečkovité keratopatie a/nebo toxické ulcerativní keratopatie. Protože LUMIGAN benzalkonium chlorid obsahuje, je při jeho častém nebo dlouhodobém užívání třeba pečlivě sledovat pacienty, kteří trpí pocity suchého oka nebo mají poškozenou rohovku.
Byla hlášena bakteriální keratitida spojená s použitím vícedávkových balení topických očních produktů. Tyto nádobky byly neúmyslně kontaminovány pacienty, kteří měli ve většině případů souběžné oční onemocnění. Pacientům s narušeným povrchem očního epitelu hrozí vyšší riziko bakteriální keratitidy.
Pacienty je třeba informovat, aby zabránili styku hrotu aplikační lahvičky s okem nebo okolními strukturami, aby nedošlo k poranění oka a kontaminaci roztoku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Žádné studie interakcí nebyly provedeny.
Interakce u lidí nejsou očekávány, jelikož systémová koncentrace bimatoprostu po očním podávání očních kapek obsahujících bimatoprost 0,3 mg/ml je extrémně nízká (méně než 0,2 ng/ml). Bimatoprost je přeměňován četnými enzymy a cestami, ale v preklinických studiích nebyl pozorován žádný účinek na jaterní enzymy, které metabolizují léky.
V klinických studiích byl LUMIGAN používán současně s řadou různých očních betablokátorů bez známek interakcí.
Současné použití LUMIGANu s jinými antiglaukomatiky než topickými betablokátory nebylo během přídatné léčby glaukomu hodnoceno.
Účinek snižování NOT analogů prostaglandinu (např. LUMIGANU) může být nižší u pacientů s glaukomem nebo oční hypertenzí, pokud zároveň užívají další analogy prostaglandinu (viz část 4.4).
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání bimatoprostu těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu při vysokých maternotoxických dávkách (viz bod 5.3).
LUMIGAN by neměl být během těhotenství podáván, pokud to není nezbytně nutné.
Kojení
Není známo, zda bimatoprost přechází do mateřského mléka. Studie na zvířatech však vylučování do mléka prokázaly. Je zapotřebí rozhodnout o ukončení kojení nebo ukončení léčby přípravkem LUMIGAN s ohledem na přínosy kojení pro dítě a přínosy léčebné terapie pro danou ženu.
Fertilita
Údaje o vlivů bimatoprostu na lidskou fertilitu nejsou k dispozici.
4.7 Účinky na schopnost řídit a obsluhovat stroje
LUMIGAN má zanedbatelný vliv na schopnost řídit a obsluhovat stroje. Jestliže, stejně jako po jiné léčbě očí, nastane po podání přechodné rozmazané vidění, měl by pacient před řízením nebo obsluhou strojů počkat, dokud není vidění ostré.
4.8 Nežádoucí účinky
V klinických studiích bylo očními kapkami LUMIGAN 0,3 mg/ml léčeno přes 1800 pacientů. Po sloučení údajů z fáze III monoterapie a přídatného použití očních kapek LUMIGAN 0,3 mg/ml byly nejčastěji hlášenými nežádoucími účinky: růst řas až u 45 % pacientů v prvním roce s poklesem výskytu případů po 2 letech na 7 % a po 3 letech na 2 %, hyperémie spojivek (většinou v náznacích nebo mírná a považována za nezánětlivou) až u 44 % pacientů v prvním roce s poklesem výskytu případů po 2 letech na 13 % a po 3 letech na 12 % a svědění očí až u 14 % pacientů v prvním roce s poklesem výskytu případů po 2 letech na 3 % a po 3 letech na 0 %. Méně než 9 % pacientů přerušilo léčbu kvůli jakémukoliv nežádoucímu účinku v prvním roce, ve druhém a třetím roce poklesl počet případů přerušení léčby shodně na 3 %.
Během klinických studií s očními kapkami LUMIGAN 0,3 mg/ml, roztok nebo v postmarketingovém období byly zaznamenány následující nežádoucí účinky. Většina z nich byly oční, mírné nebo středně závažné, žádný z nich nebyl závažný:
Tabulka 1 ukazuje nežádoucí účinky rozdělené podle frekvence výskytu na velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10 000 až <1/1000), velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit). Nežádoucí účinky jsou seřazeny podle tříd orgánových systémů a v každé skupině četnosti podle klesající závažnosti.
|
Oreánovv systém |
F rekvence |
Nežádoucí účinek |
|
Poruchy nervového systému |
Časté |
Bolesti hlavy |
|
Méně časté |
Závratě | |
|
Poruchy oka |
Velmi časté |
Hyperemie spojivek, svědění očí, nadměrný růst řas |
|
Časté |
Povrchová tečkovitá keratitida, eroze rohovky, pálení očí, podráždění očí, alergická konjunktivitida, blefaritida, zhoršení zrakové ostrosti, astenopie, spojivkový edém, pocit cizího tělesa v oku, suchost očí, bolesti očí, fotofobie, slzení, výtok z očí, zhoršení zraku/rozmazané vidění, zvýšená pigmentace duhovky, ztmavnutí řas, erytém očních víček, svědění očních víček | |
|
Méně časté |
Krvácení rohovky, uveitida, cystoidní makulární edém, iritida, blefarospazmus, retrakce víček, periorbitální erytém, edém víček | |
|
Není známo |
Změny okolí očí a změny víčka včetně prohloubení záhybu očního víčka | |
|
Cévní poruchy |
Časté |
Hypertenze |
|
Respirační, hrudní a mediastinální poruchy |
Není známo | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Poruchy kůže a podkožní tkáně |
Časté |
Pigmentace kůže kolem očí |
|
Méně časté |
Hirsutismus | |
|
Celkové poruchy a reakce v místě aplikace |
Méně časté |
Astenie |
|
Vyšetření |
Časté |
Abnormální jaterní testy |
|
Poruchy imunitního systému |
Není známo |
Hypersenzitivní reakce včetně známek a příznaků oční alergie a alergické dermatitidy |
Nežádoucí účinky hlášené u očních kapek obsahujících fosfáty:
Velmi vzácně byly u některých pacientů s významně poškozenou rohovkou hlášeny případy komeální kalcifikace spojené s použitím očních kapek obsahujících fosfáty.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyl hlášen žádný případ předávkování a není pravděpodobné, že by po podání do oka nastal.
Pokud dojde k předávkování, léčba by měla být symptomatická a podpůrná. Jestliže je LUMIGAN náhodně požit, mohou být užitečné následující informace: během dvoutýdenní studie u potkanů a myší při perorálních dávkách až do 100 mg/kg/den nevznikla žádná toxicita. Tato dávka vyjádřená v mg/m2 je nejméně 70krát vyšší než množství přípravku v jedné lahvičce očních kapek LUMIGAN 0,3 mg/ml pro 10 kg dítě.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, analoga prostaglandinu, ATC kód: S01EE03 Mechanismus účinku
Mechanismem účinku, kterým bimatoprost redukuje nitrooční tlak u lidí, je zvýšený odtok nitrooční tekutiny trámčinou komorového úhlu a zvýšený odtok uveosklerální cestou. Snižování nitroočního tlaku začíná přibližně 4 hodiny po prvním podání a maximálního účinku je dosaženo přibližně během 8 až 12 hodin. Snížení nitroočního tlaku přetrvává nejméně 24 hodin.
Bimatoprost je silné oční hypotenzivum. Je to syntetický prostamid, strukturálně blízký prostaglandinu F2a (PGF2a), který nepracuje cestou známých prostaglandinových receptorů. Bimatoprost selektivně napodobuje účinek nově objevených biosyntetizovaných substancí nazývaných prostamidy. Nicméně prostamidové receptory nebyly ještě dosud strukturálně identifikovány.
Během 12 měsíční monoterapie LUMIGANEM 0,3 mg/ml u dospělých je proti timololu hlavní změna v ranní základní hodnotě (08:00) nitroočního tlaku v rozmezí od -7,9 do -8,8 mm Hg. Průměrné denní hodnoty IOP, měřené při každé návštěvě po celou dobu 12 měsíční studie, se nelišily o více než 1,3 mm Hg během dne a nikdy nebyly vyšší než 18,0 mm Hg.
V 6 měsíční klinické studii LUMIGANU 0,3 mg/ml bylo, oproti latanoprostu, bylo statisticky největší snížení ranních hodnot IOP (hodnoty od -7,6 do -8,2 mm Hg u bimatoprostu oproti -6,0 do -
7.2 mm Hg u latanoprostu) pozorováno v průběhu všech kontrol během studie. Hyperémie spojivek, růst řas a svědění očí byly statisticky signifikantně častější u bimatoprostu než u latanoprostu, nicméně případy přerušení léčby z důvodu nežádoucích účinků byly ojedinělé a bez statisticky signifikantního rozdílu.
Ve srovnání s léčbou samotnými betablokátory, snížila kombinovaná terapie betablokátor a LUMIGAN 0,3 mg/ml ranní (08:00) průměrný nitrooční tlak o -6,5 až -8,1 mm Hg.
S použitím u pacientů s glaukomem s otevřeným úhlem, s pseudoexfoliativním a pigmentovým glaukomem a u pacientů s chronickým glaukomem s uzavřeným úhlem s provedenou iridotomií jsou omezené zkušenosti.
Během klinických studií nebyl pozorován žádný klinicky relevantní účinek na srdeční frekvenci a krevní tlak.
Pediatrická populace
Bezpečnost a účinnost přípravku LUMIGAN u dětí od 0 do méně než 18 let nebyla dosud stanovena.
5.2 Farmakokinetické vlastnosti Vstřebávání
Bimatoprost in vitro velmi dobře penetruje lidskou rohovkou a sklérou. Po očním podání dospělým pacientům je systémová expozice bimatoprostu velmi nízká bez akumulace během doby podávání. Při podávání jedenkrát denně po jedné kapce LUMIGANU 0,3 mg/ml do obou očí po dobu dvou týdnů je dosaženo vrcholové koncentrace v krvi během 10 minut po podání a následné snížení na nejnižší detekovatelnou hodnotu (0,025 ng/ml) během 1,5 hodiny po aplikaci. Střední Cmax a AUC 0-24hod. hodnoty byly 7. a 14. den podobné, přibližně 0,08 ng/ml respektive 0,09 ng^hod/ml, což ukazuje, že rovnovážného stavu koncentrace bimatoprostu bylo dosaženo během prvního týdne očního podávání.
Distribuce
Bimatoprost je mírně distribuován do tělesných tkání a systémová hladina je ustálena na 0,67 l/kg.
V lidské krvi je bimatoprost především v plazmě. Vazba bimatoprostu na plazmatické bílkoviny je přibližně 88 %.
Biotransformace
Jakmile je po očním podání dosaženo systémové cirkulace, je bimatoprost hlavní cirkulující částí v krvi. Bimatoprost podléhá oxidaci, N-deetylaci a glukuronidaci a tvoří se řada různých metabolitů.
Eliminace
Bimatoprost je primárně eliminován ledvinami, více než 67 % z intravenózní dávky zdravým dospělým dobrovolníkům bylo vyloučeno močí, 25 % bylo vyloučeno stolicí. Poločas eliminace určený po intravenózním podání byl přibližně 45 minut. Celková clearance krve byla 1,5 l/hod/kg.
Charakteristika u starších pacientů
U starších pacientů (65 let a více) byla při dávkování LUMIGANU 0,3 mg/ml dvakrát denně střední hodnota AUC 0-24hod bimatoprostu 0,0634 ng^hod/ml, což je signifikantně více než 0,0218 ng^hod/ml u mladých zdravých dospělých osob. Nicméně toto zjištění není klinicky relevantní, protože systémová expozice starších i mladších osob byla při očním podávání velmi nízká. Během užívání nedocházelo ke kumulaci bimatoprostu v krvi a bezpečnostní profil pro starší i mladé pacienty je podobný.
5.3 Předklinické údaje vztahující se k bezpečnosti
Účinky v neklinických studiích byly pozorovány pouze po expozicích dostatečně převyšujících maximální expozici u člověka, což svědčí o malém významu při klinickém použití.
Oční podávání bimatoprostu opicím v koncentraci >0,3 mg/ml denně po dobu jednoho roku způsobilo zvýšení pigmentace duhovky a reverzibilní na dávce závislý periokulární efekt charakterizovaný prominující horní a/nebo dolní rýhou a rozšířením palpebrální štěrbiny. Zdá se, že zvýšení pigmentace duhovky je způsobeno zvýšenou stimulací produkce melaninu v melanocytech, a ne zvýšením počtu melanocytů. Žádné funkční ani mikroskopické změny ve vztahu k periokulárnímu efektu nebyly pozorovány, mechanizmus vlivu na periokulární změny není znám.
Bimatoprost nebyl v sérii in vitro a in vivo studií mutagenní nebo karcinogenní.
Bimatoprost nenarušoval u potkanů až do dávky 0,6 mg/kg/den (nejméně 103násobná předpokládaná humánní expozice) fertilitu. V embryo/fetální vývojové studii abortů nebyl ale pozorován vývojový účinek u myší ani potkanů při dávkách, které byly nejméně 860krát respektive 1700krát vyšší než humánní. Tyto dávky byly výsledně při systémovém podávání nejméně 33 respektive 97krát vyšší než množství určené pro člověka. V peri/postnatálních studiích u potkanů způsobila mateřská toxicita redukci gestačního času, fetální smrt a snížení tělesné hmotnosti mláďat o > 0,3 mg/kg/den (nejméně 41krát vyšší než předpokládaná humánní expozice). Neurobehaviorální funkce potomků nebyly postiženy.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Benzalkonium-chlorid Chlorid sodný
Heptahydrát hydrogenfosforečnanu sodného Monohydrát kyseliny citrónové
Roztok hydroxidu sodného 1 mol/l nebo roztok kyseliny chlorovodíkové 1 mol/l (k udržení pH) Čištěná voda
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
4 týdny po prvním otevření.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a velikost balení
Bílá, neprůhledná lahvička z polyetylenu s polystyrenovým uzávěrem se závitem. Jedna lahvička je naplněna 3 ml.
Dostupné jsou následující velikosti balení: krabička obsahující 1 nebo 3 lahvičky po 3 ml roztoku. Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a zacházení s ním
Žádné zvláštní požadavky na likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co. Mayo
Irsko
8. REGISTRAČNÍ ČÍSLA
EU/1/02/205/001 -002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
8. březen 2002/ 20.únor 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agenturypro léčivé přípravky: http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
LUMIGAN 0,3 mg/ml oční kapky, roztok, v jednodávkovém obalu
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml roztoku obsahuje bimatoprostum 0,3 mg (bimatoprost). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Oční kapky, roztok, v jednodávkovém obalu Bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Snížení zvýšeného nitroočního tlaku u chronického glaukomu s otevřeným úhlem a nitrooční hypertenze u dospělých (jako monoterapie nebo jako doplňující terapie k betablokátorům).
4.2 Dávkování a způsob podání
Dávkování
Doporučená dávka je jedna kapka do postiženého oka (očí) jedenkrát denně večer.
Dávkování jedenkrát denně by nemělo být překročeno, protože častější podávání může vést ke snížení účinku na nitrooční tlak.
Pouze k jednorázovému použití, obsah jedné nádobky je dostačující k ošetření obou očí. Veškerý nespotřebovaný roztok je zapotřebí bezprostředně po použití zlikvidovat.
Pediatrická populace :
Bezpečnost a účinnost přípravku LUMIGAN u dětí od 0 do 18 let nebyla dosud stanovena.
Pacienti s poškozením funkce jater nebo ledvin:
LUMIGAN nebyl studován u pacientů s ledvinovým nebo středně závažným až závažným jaterním poškozením a měl by proto být u těchto pacientů používán s opatrností. U pacientů s předchozím mírným jaterním onemocněním nebo abnormálními hladinami alanin aminotransferázy (ALT), aspartát aminotransferázy (AST) a/nebo bilirubinu, nemá bimatoprost 0,3 mg/ml oční kapky (vícedávkové balení), roztok nepříznivý účinek na funkci jater alespoň po dobu 24 měsíců.
Způsob podání
Pokud je používán více než jeden lokální oční přípravek, pak je mezi jejich podáním nutné zachovat časový odstup nejméně 5 minut.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodu 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Oční
Před zahájením léčby by měl být pacient informován o možném růstu řas, ztmavnutí kůže na víčkách a zvýšené pigmentaci duhovky, které byly pozorovány během léčby LUMIGANEM. Některé z těchto změn mohou být trvalé a mohou vést k rozdílnému vzhledu očí, pokud bylo léčeno pouze jedno z nich. Zvýšená pigmentace duhovky je pravděpodobně trvalá. Změna pigmentace je způsobena zvýšeným obsahem melaninu v melanocytech, nikoli zvýšením počtu melanocytů. Dlouhodobé účinky zvýšené pigmentace duhovky nejsou známy. Změna barvy duhovky pozorovaná při očním podání bimatoprostu nemusí být patrná po několik měsíců či let. Obvykle se hnědá pigmentace okolo zorničky rozšíří soustředně směrem k okraji duhovky a celá duhovka nebo její části více zhnědnou. Zdá se, že léčba nemá vliv na mateřská znaménka ani pihy na duhovce. Po třech měsících byl při používání bimatoprostu 0,3 mg/ml v jednodávkovém obalu výskyt pigmentace duhovky 0,3 %. Po 12 měsících byl při používání bimatoprostu 0,3 mg/ml (vícedávkové balení) výskyt pigmentace duhovky
1,5 % (viz část 4.8) a v průběhu 3leté léčby se dále nezvyšoval. Podle hlášení byla u některých pacientů pigmentace periorbitální tkáně reverzibilní.
Během léčby očními kapkami obsahujícími bimatoprost 0,3 mg/ml (vícedávkové balení) byl méně často (>1/1 000 až <1/100) zaznamenám cystoidní makulární edém. U pacientů s rizikovými faktory pro makulární edém (např. afakičtí pacienti, pseudofakičtí pacienti s trhlinou zadního pouzdra čočky) by proto měl být LUMIGAN používán s opatrností.
Existují vzácná spontánní hlášení o reaktivaci dřívějších rohovkových infiltrátů nebo očních infekcí následkem léčby očními kapkami obsahujícími bimatoprost 0,3 mg/ml (vícedávkové balení). Pacienti s anamnézou předchozích závažných virových očních infekcí (jako je herpes simplex) nebo uveitidou/iritidou by měli LUMIGAN užívat opatrně.
LUMIGAN nebyl studován u pacientů s očními zánětlivými stavy, glaukomem s uzavřeným úhlem neovaskulární nebo zánětlivé etiologie, kongenitálním glaukomem a glaukomem s úzkým úhlem.
Kůže
V místech, kde LUMIGAN přijde opakovaně do styku s povrchem pokožky, může docházet k růstu ochlupení. Proto je důležité LUMIGAN podávat podle pokynů a zabránit tomu, aby stékal na tvář nebo jiné oblasti kůže.
Respirační
LUMIGAN nebyl studován u pacientů s poškozenými respiračními funkcemi, a měl by být proto u nich používán s opatrností. Ačkoli jsou k dispozici omezené informace u pacientů s astmatem nebo CHOPN v anamnéze, bylo hlášeno zhoršení astmatu, dušnosti a CHOPN, stejně jako zprávy o astmatu v postmarketingovém období. Četnost těchto příznaků není známa. Pacienti s CHOPN, astmatem nebo sníženou respirační funkcí kvůli jiným chorobám mají být léčeni se zvýšenou opatrností.
Kardiovaskulární
LUMIGAN nebyl studován n u pacientů se srdečním blokem více než prvního stupně nebo nekontrolovaným kongestivním srdečním selháním. Existuje omezený počet spontánních hlášení o výskytu bradykardie nebo hypotenze po užití očních kapek obsahující bimatoprost 0,3 mg/ml (vícedávkové balení). Pacienti s predispozicí k nízké srdeční frekvenci nebo nízkému krevnímu tlaku by měli LUMIGAN užívat opatrně.
Další informace
Ve studiích s bimatoprostem 0,3 mg/ml u pacientů s glaukomem nebo nitrooční hypertenzí bylo prokázáno, že častější expozice oka více než jedné dávce biomatorpostu denně může snížit účinnost snižování NOT. U pacientů užívajících LUMIGAN s dalšími analogy prostaglandinu by měly být sledovány změny nitroočního tlaku.
Použití přípravku LUMIGAN 0,3 mg/ml v jednodávkovém obalu nebylo u pacientů používajících kontaktní čočky studováno.
Před podáním kapek by proto měly být čočky z oka vyjmuty a opět zavedeny 15 minut po podání.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Žádné studie interakcí nebyly provedeny.
Interakce u lidí nejsou očekávány, jelikož systémová koncentrace bimatoprostu po očním podávání očních kapek obsahujících bimatoprost 0,3 mg/ml (vícedávkové balení) je extrémně nízká (méně než 0,2 ng/ml). Bimatoprost je přeměňován četnými enzymy a cestami, ale v preklinických studiích nebyl pozorován žádný účinek na jaterní enzymy, které metabolizují léky.
V klinických studiích byl LUMIGAN 0,3 mg/ml (vícedávkové balení) používán současně s řadou různých očních betablokátorů bez známek interakcí.
Současné použití LUMIGANu s jinými antiglaukomatiky než topickými betablokátory nebylo během přídatné léčby glaukomu hodnoceno.
Účinek snižování NOT analogů prostaglandinu (např. LUMIGANU) může být nižší u pacientů s glaukomem nebo oční hypertenzí, pokud zároveň užívají další analogy prostaglandinu (viz část 4.4).
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání bimatoprostu těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu při vysokých maternotoxických dávkách (viz bod 5.3).
LUMIGAN by neměl být během těhotenství podáván, pokud to není nezbytně nutné.
Kojení
Není známo, zda bimatoprost přechází do mateřského mléka. Studie na zvířatech však vylučování do mléka prokázaly. Je zapotřebí rozhodnout o ukončení kojení nebo přerušení léčby přípravkem LUMIGAN s ohledem na přínosy kojení pro dítě a přínosy léčebné terapie pro danou ženu.
Fertilita
Údaje o vlivů bimatoprostu na lidskou fertilitu nejsou k dispozici.
4.7 Účinky na schopnost řídit a obsluhovat stroje
LUMIGAN má zanedbatelný vliv na schopnost řídit a obsluhovat stroje. Jestliže, stejně jako po jiné léčbě očí, nastane po podání přechodné rozmazané vidění, měl by pacient před řízením nebo obsluhou strojů počkat, dokud není vidění ostré.
4.8 Nežádoucí účinky
Ve tříměsíční klinické studii došlo k rozvoji nežádoucích účinků přibližně u 29 % pacientů léčených přípravkem LUMIGAN 0,3 mg/ml v jednodávkovém obalu. Nejčastěji udávanými nežádoucími reakcemi byla hyperemie spojivky (většinou v náznacích nebo mírná a nezánětlivé povahy), ke které došlo u 24 % pacientů, a svědění očí, které se vyskytlo u 4 % pacientů. Přibližně 0,7 % pacientů ve skupině používající LUMIGAN 0,3 mg/ml v jednodávkovém obalu ukončilo účast ve tříměsíční studii kvůli nežádoucí příhodě.
Během klinických studií s očními kapkami LUMIGAN 0,3 mg/ml v jednodávkovém obalu nebo v postmarketingovém období byly zaznamenány následující nežádoucí účinky. Většina z nich byly oční, mírné a žádný z nich nebyl závažný:
Tabulka 1 ukazuje nežádoucí účinky rozdělené podle frekvence výskytu na velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10 000 až <1/1000), velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit). Nežádoucí účinky jsou seřazeny podle tříd orgánových systémů a v každé skupině četnosti podle klesající závažnosti.
Tabulka 1
|
Oreánovv systém |
F rekvence |
Nežádoucí účinek |
|
Poruchy nervového systému |
Méně časté |
Bolesti hlavy |
|
Poruchy oka |
Velmi časté |
Hyperemie spojivek |
|
Časté |
Povrchová tečkovitá keratitida, podráždění očí, pocit cizího tělesa v oku, suchost očí, bolesti očí, svědění očí, růst řas, erytém očního víčka | |
|
Méně časté |
Astenopie, otok spojivek, fotofobie, zvýšené slzení, hyperpigmentace duhovky, rozmazané vidění, svědění očního víčka, otok očního víčka | |
|
Respirační, hrudní a mediastinální poruchy |
Není známo | |
|
Poruchy kůže a podkožní tkáně |
Časté |
Hyperpigmentace kůže (okolo očí) |
|
Méně časté |
Abnormální růst chloupků | |
|
Poruchy imunitního systému |
Není známo |
Přecitlivělé reakce včetně projevů a příznaků oční alergie a alergické dermatitidy |
V klinických studiích bylo očními kapkami LUMIGAN 0,3 mg/ml (vícedávkové balení) léčeno přes 1 800 pacientů. Po sloučení údajů z fáze III monoterapie a přídatného použití očních kapek LUMIGAN 0,3 mg/ml (vícedávkové balení) byly nejčastěji hlášenými nežádoucími reakcemi:
• růst řas až u 45 % pacientů v prvním roce s poklesem výskytu případů po 2 letech na 7 % a po 3 letech na 2 %
• hyperemie spojivek (většinou v náznacích nebo mírná a považována za nezánětlivou) až u
44 % pacientů v prvním roce s poklesem výskytu případů po 2 letech na 13 % a po 3 letech na 12 %
• svědění očí až u 14 % pacientů v prvním roce s poklesem výskytu případů po 2 letech na 3 % a po 3 letech na 0 %.
Méně než 9 % pacientů přerušilo léčbu kvůli nežádoucímu účinku v prvním roce, ve druhém a třetím roce poklesl počet případů přerušení léčby shodně na 3 %.
Tabulka 2 uvádí nežádoucí účinky pozorované ve dvanáctiměsíční klinické studii přípravku LUMIGAN 0,3 mg/ml (vícedávkové balení), které byly udávány častěji než u přípravku LUMIGAN 0,3 mg/ml (v jednodávkovém obalu). Většinou se jednalo o účinky oční, mírné až středně závažné a žádný nebyl závažný.
Tabulka 2
|
Oreánovv systém |
F rekvence |
Nežádoucí účinek |
|
Poruchy nervového systému |
Časté |
Bolesti hlavy |
|
Poruchy oka |
Velmi časté |
Svědění očí, růst řas |
|
Časté |
Astenopie, otok spojivek, fotofobie, slzení, zvýšená pigmentace duhovky; rozmazané vidění | |
|
Poruchy kůže a podkožní tkáně |
Časté |
Svědění očního víčka |
Navíc k nežádoucím účinkům pozorovaným u přípravku LUMIGAN 0,3 mg/ml v jednodávkovém obalu uvádí tabulka 3 další nežádoucí účinky pozorované u přípravku LUMIGAN 0,3 mg/ml (vícedávkové balení). Většinou se jednalo o účinky oční, mírné až středně závažné a žádný nebyl závažný.
Tabulka 3
|
Oreánovv systém |
F rekvence |
Nežádoucí účinek |
|
Poruchy nervového systému |
Méně časté |
Závratě |
|
Poruchy oka |
Časté |
Eroze rohovky, pálení očí, alergická konjunktivitida, blefaritida, zhoršení zrakové ostrosti, výtok z oka, poruchy vidění, ztmavnutí očních řas |
|
Méně časté |
Krvácení do sítnice, uveitida, cystoidní makulární edém, iritida, blefarospasmus, retrakce očního víčka | |
|
Není známo |
Změny okolí očí a změny víčka včetně zhoršení poklesu očního víčka | |
|
Cévní poruchy |
Časté |
Hypertenze |
|
Gastrointestinální poruchy |
Méně časté | |
|
Poruchy kůže a podkožní tkáně |
Není známo |
Periorbitální erytém |
|
Celkové poruchy a reakce v místě aplikace |
Méně časté |
Astenie |
|
Vyšetření |
Časté |
Abnormality jaterních testů |
Nežádoucí účinky hlášené u očních kapek obsahujících fosfáty:
Velmi vzácně byly u některých pacientů s významně poškozenou rohovkou hlášeny případy korneální kalcifikace spojené s použitím očních kapek obsahujících fosfáty.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
K dispozici nejsou žádné informace o předávkování u lidí. Předávkování není při podání do oka pravděpodobné.
Pokud dojde k předávkování, léčba by měla být symptomatická a podpůrná. Jestliže je LUMIGAN 0,3 mg/ml v jednodávkovém obalu náhodně požit, mohou být užitečné následující informace:
V krátkodobých perorálních studiích u myší a potkanů dávky bimatoprostu až 100 mg/kg/den (podávané sondou) nevedly k žádné toxicitě. Tato dávka je alespoň 22 krát vyšší než dávka celého obsahu balení přípravku LUMIGAN 0,3 mg/ml v jednodávkovém obalu (30 x 0,4ml jednorázová nádobka; 12 ml) náhodně požitá dítětem o tělesné hmotnosti 10 kg.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, analoga prostaglandinu, ATC kód: S01EE03 Mechanismus účinku
Mechanismem účinku, kterým bimatoprost redukuje nitrooční tlak u lidí, je zvýšený odtok nitrooční tekutiny trámčinou komorového úhlu a zvýšený odtok uveosklerální cestou. Snižování nitroočního tlaku začíná přibližně 4 hodiny po prvním podání a maximálního účinku je dosaženo přibližně během 8 až 12 hodin. Snížení nitroočního tlaku přetrvává nejméně 24 hodin.
Bimatoprost je silné oční hypotenzivum. Je to syntetický prostamid, strukturálně blízký prostaglandinu F2a (PGF2a), který nepracuje cestou známých prostaglandinových receptorů. Bimatoprost selektivně napodobuje účinek nově objevených biosyntetizovaných substancí nazývaných prostamidy. Nicméně prostamidové receptory nebyly ještě dosud strukturálně identifikovány.
Klinická, účinnost.
12 týdnů trvající klinická studie (dvojitě zaslepená, randomizovaná, s paralelními skupinami) srovnávala účinnost a bezpečnost přípravku LUMIGAN 0,3 mg/ml v jednodávkovém obalu a přípravku LUMIGAN 0,3 mg/ml ve vícedávkovém balení. LUMIGAN 0,3 mg/ml v jednodávkovém obalu nevykazoval horší účinnost při snižování NOT než LUMIGAN 0,3 mg/ml (vícedávkové balení) u změny NOT v horším oku oproti vstupnímu stavu u pacientů s glaukomem nebo oční hypertenzí. LUMIGAN 0,3 mg/ml v jednodávkovém obalu dosahoval stejné účinnosti při snižování NOT jako LUMIGAN 0,3 mg/ml (vícedávkové balení) v průměrném NOT při všech dalších časových bodech po 2, 6 a 12 týdnech.
Během 12 měsíční monoterapie LUMIGANEM 0,3 mg/ml (vícedávkové balení) u dospělých je proti timololu hlavní změna v ranní základní hodnotě (08:00) nitroočního tlaku v rozmezí od -7,9 do -
8,8 mm Hg. Průměrné denní hodnoty IOP, měřené při každé návštěvě po celou dobu 12 měsíční studie, se nelišily o více než 1,3 mm Hg během dne a nikdy nebyly vyšší než 18,0 mm Hg.
V 6 měsíční klinické studii LUMIGANU 0,3 mg/ml (vícedávkové balení) bylo, oproti latanoprostu, bylo statisticky největší snížení ranních hodnot IOP (hodnoty od -7,6 do -8,2 mm Hg u bimatoprostu oproti -6,0 do -7,2 mmHg u latanoprostu) pozorováno v průběhu všech kontrol během studie. Hyperémie spojivek, růst řas a svědění očí byly statisticky signifikantně častější u bimatoprostu než u latanoprostu, nicméně případy přerušení léčby z důvodu nežádoucích účinků byly ojedinělé a bez statisticky signifikantního rozdílu.
Ve srovnání s léčbou samotnými betablokátory, snížila kombinovaná terapie betablokátor a LUMIGAN 0,3 mg/ml (vícedávkové balení) ranní (08:00) průměrný nitrooční tlak o 6,5 až 8,1 mmHg.
S použitím u pacientů s glaukomem s otevřeným úhlem, s pseudoexfoliativním a pigmentovým glaukomem a u pacientů s chronickým glaukomem s uzavřeným úhlem s provedenou iridotomií jsou omezené zkušenosti.
Během klinických studií nebyl pozorován žádný klinicky relevantní účinek na srdeční frekvenci a krevní tlak.
Pediatrická populace
Bezpečnost a účinnost přípravku LUMIGAN u dětí od 0 do 18 let nebyla dosud stanovena.
5.2 Farmakokinetické vlastnosti
Vstřebávání
Bimatoprost in vitro velmi dobře penetruje lidskou rohovkou a sklérou. Po očním podání dospělým pacientům je systémová expozice bimatoprostu velmi nízká bez akumulace během doby podávání. Při podávání jedenkrát denně po jedné kapce LUMIGANU 0,3 mg/ml do obou očí po dobu dvou týdnů je dosaženo vrcholové koncentrace v krvi během 10 minut po podání a následné snížení na nejnižší detekovatelnou hodnotu (0,025 ng/ml) během 1,5 hodiny po aplikaci. Střední Cmax a AUC 0-24hod. hodnoty byly 7. a 14. den podobné, přibližně 0,08 ng/ml respektive 0,09 ng^hod/ml, což ukazuje, že rovnovážného stavu koncentrace bimatoprostu bylo dosaženo během prvního týdne očního podávání.
Distribuce
Bimatoprost je mírně distribuován do tělesných tkání a systémová hladina je ustálena na 0,67 l/kg.
V lidské krvi je bimatoprost především v plazmě. Vazba bimatoprostu na plazmatické bílkoviny je přibližně 88 %.
Biotransformace
Jakmile je po očním podání dosaženo systémové cirkulace, je bimatoprost hlavní cirkulující částí v krvi. Bimatoprost podléhá oxidaci, N-deetylaci a glukuronidaci a tvoří se řada různých metabolitů.
Eliminace
Bimatoprost je primárně eliminován ledvinami, více než 67 % z intravenózní dávky zdravým dospělým dobrovolníkům bylo vyloučeno močí, 25 % bylo vyloučeno stolicí. Poločas eliminace určený po intravenózním podání byl přibližně 45 minut. Celková clearance krve byla 1,5 l/hod/kg.
Charakteristika u starších pacientů
U starších pacientů (65 let a více) byla při dávkování LUMIGANU 0,3 mg/ml dvakrát denně střední hodnota AUC 0-24hod bimatoprostu 0,0634 ng^hod/ml, což je signifikantně více než 0,0218 ng^hod/ml u mladých zdravých dospělých osob. Nicméně toto zjištění není klinicky relevantní, protože systémová expozice starších i mladších osob byla při očním podávání velmi nízká. Během užívání nedocházelo ke kumulaci bimatoprostu v krvi a bezpečnostní profil pro starší i mladé pacienty je podobný.
5.3 Předklinické údaje vztahující se k bezpečnosti
Účinky v neklinických studiích byly pozorovány pouze po expozicích dostatečně převyšujících maximální expozici u člověka, což svědčí o malém významu při klinickém použití.
Oční podávání bimatoprostu opicím v koncentraci >D0,3mg/ml denně po dobu jednoho roku způsobilo zvýšení pigmentace duhovky a reverzibilní na dávce závislý periokulární efekt charakterizovaný prominující horní a/nebo dolní rýhou a rozšířením palpebrální štěrbiny. Zdá se, že zvýšení pigmentace duhovky je způsobeno zvýšenou stimulací produkce melaninu v melanocytech, a ne zvýšením počtu melanocytů. Žádné funkční ani mikroskopické změny ve vztahu k periokulárnímu efektu nebyly pozorovány, mechanizmus vlivu na periokulární změny není znám.
Bimatoprost nebyl v sérii in vitro a in vivo studií mutagenní nebo karcinogenní.
Bimatoprost nenarušoval u potkanů až do dávky 0,6 mg/kg/den (nejméně 103násobná předpokládaná humánní expozice) fertilitu. V embryo/fetální vývojové studii abortů nebyl ale pozorován vývojový účinek u myší ani potkanů při dávkách, které byly nejméně 860krát respektive 1700krát vyšší než humánní. Tyto dávky byly výsledně při systémovém podávání nejméně 33 respektive 97krát vyšší než množství určené pro člověka. V peri/postnatálních studiích u potkanů způsobila mateřská toxicita redukci gestačního času, fetální smrt a snížení tělesné hmotnosti mláďat o > 0,3 mg/kg/den (nejméně 41krát vyšší než předpokládaná humánní expozice). Neurobehaviorální funkce potomků nebyly postiženy.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný
Heptahydrát hydrogenfosforečnanu sodného Monohydrát kyseliny citronové
Roztok hydroxidu sodného 1 mol/l nebo roztok kyseliny chlorovodíkové 1 mol/l (k udržení pH) Čištěná voda
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
balení s 5 jednodávkovými nádobkami - 12 měsíců balení s 30 jednodávkovými nádobkami - 18 měsíců balení s 90 jednodávkovými nádobkami - 18 měsíců Po otevření plata spotřebujte jednodávkové nádobky do 30 dnů
Otevřenou jednodávkovou nádobku zlikvidujte bezprostředně po použití.
6.4 Zvláštní opatření pro uchovávání
balení s 5 jednodávkovými nádobkami - Neuchovávejte při teplotě nad 25 °C
balení s 30 jednodávkovými nádobkami -Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
balení s 90 jednodávkovými nádobkami - Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a velikost balení
Průhledné, jednodávkové LDPE nádobky s víčkem k ukroucení.
Jedna jednodávková nádobka obsahuje 0,4 ml roztoku.
Dostupné jsou následující velikosti balení: krabičky obsahující 5 jednodávkových nádobek krabička obsahující 30 jednodávkových nádobek v platu krabička obsahující 90 jednodávkových nádobek ve třech platech.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a zacházení s ním
Žádné zvláštní požadavky na likvidaci.
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co. Mayo
Irsko
8. REGISTRAČNÍ ČÍSLA
EU/1/02/205/005-007
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
8. března 2002/ 20. února 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
County Mayo
Irsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v Modulu 1.8.2. registrace a ve veškerých schválených následných aktualizací RMP.
Aktualizovaný RMP je třeba předložit:
• Na žádost Evropské agentury pro léčivé přípravky
• Při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
LUMIGAN 0,1 mg/ml, oční kapky, roztok Bimatoprostum
Jeden ml roztoku obsahuje bimatoprostum 0,1 mg
Benzalkonium-chlorid, heptahydrát hydrogenfosforečnanu sodného, monohydrát kyseliny citrónové, chlorid sodný, kyselina chlorovodíková nebo hydroxid sodný (k udržení pH) a čištěná voda
Oční kapky, roztok 1 x 3 ml
Oční podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
Před použitím vyjměte kontaktní čočky.
Použitelné do:
Zlikvidujte za čtyři týdny po prvním otevření. Otevřeno:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co.Mayo
Irsko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/02/205/003
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
LUMIGAN 0,1 mg/ml
LUMIGAN 0,1 mg/ml, oční kapky, roztok Bimatoprostum
Jeden ml roztoku obsahuje 0,1 mg bimatoprostum
Benzalkonium-chlorid, heptahydrát hydrogenfosforečnanu sodného, monohydrát kyseliny citrónové, chlorid sodný, kyselina chlorovodíková nebo hydroxid sodný (k udržení pH) a čištěná voda
Oční kapky, roztok 3 x 3 ml
Oční podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
Před použitím vyjměte kontaktní čočky.
EXP
Zlikvidujte za čtyři týdny po prvním otevření. Otevřeno (1):
Otevřeno (2):
Otevřeno (3):
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co.Mayo
Irsko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/02/205/004
13. ČÍSLO ŠARŽE
Lot:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
LUMIGAN 0,1 mg/ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
LUMIGAN 0,1 mg/ml, oční kapky, roztok
Bimatoprostum
Oční podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
Exp:
Zlikvidujte za 4 týdny po prvním otevření.
4. ČÍSLO ŠARŽE
Lot:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
3 ml
6. JINÉ
LUMIGAN 0,3 mg/ml, oční kapky, roztok Bimatoprostum
Jeden ml roztoku obsahuje 0,3 mg bimatoprostum
Benzalkonium-chlorid, heptahydrát hydrogenfosforečnanu sodného, monohydrát kyseliny citrónové, chlorid sodný, kyselina chlorovodíková nebo hydroxid sodný (k udržení pH) a čištěná voda
Oční kapky, roztok 1 x 3 ml
Oční podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
Před použitím vyjměte kontaktní čočky.
EXP
Zlikvidujte za čtyři týdny po prvním otevření. Otevřeno:
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co.Mayo
Irsko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/02/205/001
13. ČÍSLO ŠARŽE
Lot:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
LUMIGAN 0,3 mg/ml
LUMIGAN 0,3 mg/ml, oční kapky, roztok Bimatoprostum
Jeden ml roztoku obsahuje 0,3 mg bimatoprostum
Benzalkonium-chlorid, heptahydrát hydrogenfosforečnanu sodného, monohydrát kyseliny citronové, chlorid sodný, kyselina chlorovodíková nebo hydroxid sodný (k udržení pH) a čištěná voda
Oční kapky, roztok 3 x 3 ml
Oční podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
Před použitím vyjměte kontaktní čočky.
EXP
Zlikvidujte za čtyři týdny po prvním otevření. Otevřeno (1):
Otevřeno (2):
Otevřeno (3):
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co.Mayo
Irsko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/02/205/002
13. ČÍSLO ŠARŽE
Lot:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
LUMIGAN 0,3 mg/ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
LUMIGAN 0,3 mg/ml, oční kapky, roztok
Bimatoprostum
Oční podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
Zlikvidujte za 4 týdny po prvním otevření. Exp:
4. ČÍSLO ŠARŽE
Lot:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
3 ml
6. JINÉ
LUMIGAN 0,3 mg/ml, oční kapky, roztok, v jednodávkovém obalu Bimatoprostum
Jeden ml roztoku obsahuje 0,3 mg bimatoprostum.
Heptahydrát hydrogenfosforečnanu sodného, monohydrát kyseliny citrónové, chlorid sodný, kyselina chlorovodíková nebo hydroxid sodný (k úpravě pH) a čištěná voda.
Oční kapky, roztok 5 x 0,4 ml
Před použitím si přečtěte příbalovou informaci. Oční podání.
Uchovávejte mimo dohled a dosah dětí.
EXP
Neuchovávejte při teplotě nad 25 °C.
Otevřenou jednodávkovou nádobku zlikvidujte neprodleně po použití.
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co.Mayo
Irsko
EU/1/02/205/005
Lot:
Výdej léčivého přípravku vázán na lékařský předpis
Pouze k jednorázovému použití
LUMIGAN 0,3 mg/ml jednodávkový
LUMIGAN 0,3 mg/ml, oční kapky, roztok, v jednodávkovém obalu Bimatoprostum
Jeden ml roztoku obsahuje 0,3 mg bimatoprostum.
Heptahydrát hydrogenfosforečnanu sodného, monohydrát kyseliny citrónové, chlorid sodný, kyselina chlorovodíková nebo hydroxid sodný (k úpravě pH) a čištěná voda.
Oční kapky, roztok 30 x 0,4 ml
Před použitím si přečtěte příbalovou informaci. Oční podání.
Uchovávejte mimo dohled a dosah dětí.
EXP
Po otevření plata spotřebujte jednodávkové nádobky do 30 dnů.
Otevřenou jednodávkovou nádobku zlikvidujte neprodleně po použití.
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co.Mayo
Irsko
EU/1/02/205/006
Lot:
Výdej léčivého přípravku vázán na lékařský předpis
Pouze k jednorázovému použití
LUMIGAN 0,3 mg/ml jednodávkový
LUMIGAN 0,3 mg/ml, oční kapky, roztok, v jednodávkovém obalu Bimatoprostum
Jeden ml roztoku obsahuje 0,3 mg bimatoprostum
Heptahydrát hydrogenfosforečnanu sodného, monohydrát kyseliny citronové, chlorid sodný, kyselina chlorovodíková nebo hydroxid sodný (k úpravě pH) a čištěná voda
Oční kapky, roztok 90 x 0,4 ml
Před použitím si přečtěte příbalovou informaci. Oční podání.
Uchovávejte mimo dohled a dosah dětí.
EXP
Po otevření plata spotřebujte jednodávkové nádobky do 30 dnů
Otevřenou jednodávkovou nádobku zlikvidujte neprodleně po použití.
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co. Mayo
Irsko
EU/1/02/205/007
Lot:
Výdej léčivého přípravku vázán na lékařský předpis
Pouze k jednorázovému použití.
LUMIGAN 0,3 mg/ml jednodávkový
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU BALENÍ S 30 A 90 JEDNODÁVKOVÝMI NÁDOBKAMI
VÍKO PLATA OBSAHUJÍCÍHO 30 JEDNODÁVKOVÝCH NÁDOBEK
LUMIGAN 0,3 mg/ml, oční kapky, roztok, v jednodávkovém obalu Bimatoprostum
Jeden ml roztoku obsahuje 0,3 mg bimatoprostum.
Heptahydrát hydrogenfosforečnanu sodného, monohydrát kyseliny citronové, chlorid sodný, kyselina chlorovodíková nebo hydroxid sodný (k úpravě pH) a čištěná voda
Oční kapky, roztok 30 x 0,4 ml
Před použitím si přečtěte příbalovou informaci. Oční podání.
Uchovávejte mimo dohled a dosah dětí.
EXP
Po otevření plata spotřebujte jednodávkové nádobky do 30 dnů
Otevřenou jednodávkovou nádobku zlikvidujte neprodleně po použití.
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co. Mayo
Irsko
Lot:
Výdej léčivého přípravku vázán na lékařský předpis
Pouze k jednorázovému použití.
Nevyžaduje se - odůvodnění přijato
ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU JEDNODÁVKOVÁ NÁDOBKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
LUMIGAN 0,3 mg/ml Bimatoprost
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta LUMIGAN 0,1 mg/ml, oční kapky, roztok
Bimatoprostum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li případně další otázky, zeptejte se, prosím, svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte i v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je LUMIGAN 0,1 mg/ml a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete LUMIGAN 0,1 mg/ml používat
3. Jak se LUMIGAN 0,1 mg/ml používá
4. Možné nežádoucí účinky
5. Jak LUMIGAN 0,1 mg/ml uchovávat
6. Obsah balení a další informace
1. Co je LUMIGAN 0,1 mg/ml a k čemu se používá
LUMIGAN je přípravek k léčbě glaukomu. Patří do skupiny přípravků nazývané prostamidy.
Oční kapky LUMIGAN se používají ke snížení zvýšeného tlaku v oku. Tento přípravek se může používat samostatně nebo s jinými očními kapkami nazývanými betablokátory, které také snižují tlak.
Vaše oko obsahuje průzračnou vodnatou tekutinu, která vyživuje vnitřní části oka. Tato tekutina je stále odváděna z oka a je nahrazována novou. Jestliže tekutina nemůže dostatečně rychle odtékat, zvyšuje se tlak uvnitř oka. Tento přípravek zvyšuje množství odváděné tekutiny. Tak se snižuje tlak uvnitř oka. Pokud není tlak snižován, může se rozvinout onemocnění zvané glaukom a nakonec může dojít k poškození zraku.
2. Čemu musíte věnovat pozornost, než začnete LUMIGAN 0,1 mg/ml používat Nepoužívejte LUMIGAN 0,1 mg/ml
- jestliže jste alergický(á) (přecitlivělý(á)) na bimatoprost nebo na kteroukoliv další složku LUMIGANU.
- Pokud jste museli v minulosti přestat používat oční kapky kvůli nežádoucím účinkům konzervačního činidla benzalkonium-chlorid.
Upozornění a opatření
Před použitím přípravku LUMIGAN 0,1 mg/ml se poraďte se svým lékařem nebo lékárníkem Informujte Vašeho lékaře, jestliže:
- máte potíže s dýcháním.
- máte potíže s játry nebo ledvinami.
- jste v minulosti podstoupil(a) operaci katarakty
- máte sucho v očích
- máte nebo jste měl(a) problémy s rohovkou (přední průhledná část oka)
- používáte kontaktní čočky (viz „Důležité informace o některých složkách LUMIGANU 0,1 mg/ml“)
- Pokud trpíte nízkou srdeční frekvencí nebo nízkým krevním tlakem
- Pokud jste měl(a) virovou infekci nebo zánět oka
LUMIGAN může způsobit ztmavnutí řas, jejich růst, a také ztmavnutí kůže v oblasti víček. Vaše duhovka může také časem ztmavnout. Tyto změny mohou být trvalé a výraznější, pokud je léčeno pouze jedno oko.
Děti a dospívající
LUMIGAN nebyl testován u dětí do 18 let a proto jej osoby mladší 18 let nemají používat.
Další léčivé přípravky a LUMIGAN::
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete užívat jakýkoliv přípravek.
LUMIGAN může přecházet do mateřského mléka. Jestliže používáte LUMIGAN, neměla byste kojit.
Řízení dopravních prostředků a obsluha strojů:
Krátkou dobu po použití LUMIGANu může být Váš zrak rozmazaný. Dokud tyto příznaky nevymizí, neměli byste řídit motorová vozidla a obsluhovat stroje.
Důležité informace o některých složkách LUMIGANU 0,1 mg/ml
Nepoužívejte kapky, pokud máte nasazeny kontaktní čočky. Po použití kapek počkejte 15 minut a teprve potom si nasaďte kontaktní čočky zpět. Konzervační látka benzalkonium-chlorid obsažená v LUMIGANU může způsobit podráždění oka nebo změnu barvy měkké kontaktní čočky.
3. Jak se LUMIGAN 0,1 mg/ml používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
LUMIGAN je určen pouze k použití do očí. Doporučená dávka LUMIGANU je vkápnutí jedné kapky denně do léčeného oka, vždy každý den večer.
Pokud užíváte přípravek LUMIGAN s jiným očním lékem, počkejte mezi použitím přípravku LUMIGAN a dalšího očního léku alespoň 5 minut.
Nepoužívejte častěji než jednou denně, protože může dojít ke snížení účinnosti léčby.
Návod k použití:
Lahvičku nesmíte použít, pokud je před prvním otevřením porušena bezpečnostní pečeť.
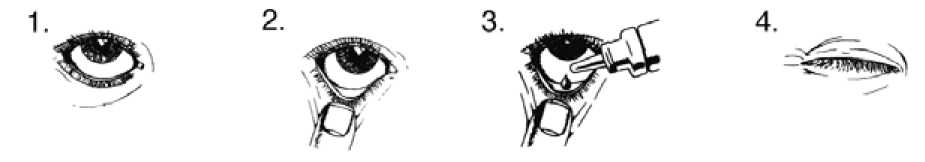
1. Umyjte si ruce. Zakloňte hlavu a podívejte se na strop.
2. Jemně stáhněte dolní víčko a vytvořte tak malou kapsu.
3. Otočte lahvičku dnem vzhůru a zmáčkněte, aby se uvolnila jedna kapka do každého léčeného oka.
4. Uvolněte dolní víčko a zavřete na 30 vteřin oči.
Setřete veškeré zbytky stékající po tváři.
Jestliže kapka Vaše oko minula, opakujte postup znovu.
Abyste zabránili infekci a poranění oka, neměl by se hrot lahvičky při kapání dotknout oka nebo něčeho jiného. Ihned po použití zavřete lahvičku víčkem.
Jestliže jste použil(a) více LUMIGANU 0,1 mg/ml než jste měl(a)
Jestliže jste použil(a) více LUMIGANU než jste měl(a), je nepravděpodobné, že si vážněji ublížíte. Další dávku si vkápněte v obvyklém čase. Jestliže máte obavy, poraďte se s Vaším lékařem nebo lékárníkem.
Jestliže jste zapomněl(a) LUMIGAN 0,1 mg/ml použít
Jestliže jste zapomněl(a) LUMIGAN použít, vkápněte si jednu kapku hned, jakmile si vzpomenete, a pak se vraťte ke svému pravidelnému dávkování. Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat LUMIGAN 0,1 mg/ml
Pro správnou funkci se LUMIGAN musí používat denně. Pokud léčbu přerušíte, může se zvýšit váš nitrooční tlak, proto se před přerušením léčby poraďte se svým lékařem.
Pokud máte jakékoliv další dotazy k užívání tohoto přípravku, obraťte se na svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Velmi časté nežádoucí účinky
Mohou postihnout 1 nebo více osob z 10 osob používajících přípravek Účinky na. oko
• lehké zčervenání (až u 29 % osob)
Časté nežádoucí účinky
Mohou postihnout 1 - 9 osob ze 100 uživatelů Účinky na. oko
• malé oděrky na povrchu oka se zánětem nebo bez něj
• podráždění
• svědění očí
• prodloužení řas
• podráždění po nakapání do oka
• bolest oka
Účinky na. kůži
• zčervenání a svědění očních víček
• ztmavnutí kůže kolem oka
• růst chloupků kolem oka
Méně časté nežádoucí účinky
Mohou postihnout 1 - 9 osob z každých 1 000 osob používajících přípravek Postižení oka
• ztmavnutí duhovky
• unavené oči
• otok povrchu oka
• rozmazané vidění
• ztráta řas
Postižení kůže
• suchá kůže
• tvorba krust na okraji víček
• otoky očních víček
• svědění
Postižení těla
• bolení hlavy
• pocit nevolnosti
Nežádoucí účinky, jejichž četnost výskytu není známa
Postižení oka
• makulární edém (otok sítnice v zadní části oka, který může vést k zhoršení zraku)
• tmavší barva očního víčka
• oči vypadají zapadle
• pocit suchého oka
Postižení těla
• astma
• zhoršení astmatu
• zhoršení plicního onemocnění zvaného chronická obstrukční plicní nemoc (CHOPN)
• dušnost
• projevy alergické reakce (otok, zarudnutí oka a vyrážka na kůži)
Kromě nežádoucích účinků LUMIGANU 0,1 mg/ml byly u vyšší síly přípravku (obsahujícího 0,3 mg/ml bimatoprostu) pozorovány následující nežádoucí účinky:
• závratě
• pálení očí;
• alergická reakce oka;
• zánět očních víček
• rozmazané vidění
• zhoršení zraku
• otok průhledné vrstvy kryjící povrch oka
• pocit cizího tělesa v oku;
• citlivost na světlo
• slzení
• lepkavé oči;
• ztmavnutí řas
• krvácení do sítnice.
zánět v oku;
cystoidní makulámí edém (otok sítnice v oku vedoucí ke zhoršení zraku); záškuby očního víčka
stažení očního víčka, posun očního víčka od povrchu oka
zarudnutí kůže okolo oka zvýšený krevní tlak; slabost
zvýšené hodnoty jaterních testů
Další nežádoucí účinky hlášené u očních kapek obsahujících fosfáty
Ve velmi vzácných případech došlo u některých pacientů se závažným poškozením průhledné vrsty přední části oka (rohovky) ke vzniku zakalených vápenatých skvrn na rohovce, které jsou způsobené léčbou.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak LUMIGAN 0,1 mg/ml uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na štítku lahvičky a na krabičce za označením Použitelné do: Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Nejpozději po 4 týdnech od prvního otevření lahvičku zlikvidujte, i přesto, že v ní nějaké kapky ještě zůstaly. Tím zabráníte infekci. Pro lepší zapamatování si zapište datum otevření na označené místo na krabičce.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co LUMIGAN 0,1 mg/ml obsahuje
- Léčivou látkou je bimatoprostum. Jeden ml roztoku obsahuje bimatoprostum 0,1 mg.
- Pomocné látky jsou benzalkonium-chlorid (konzervační činidlo), chlorid sodný, heptahydrát hydrogenfosforečnanu sodného, monohydrát kyseliny citronové a čištěná voda. Pro udržení kyselosti (hladiny pH) může být přidáno malé množství kyseliny chlorovodíkové nebo hydroxidu sodného.
Jak LUMIGAN 0,1 mg/ml vypadá a co obsahuje toto balení
LUMIGAN je bezbarvý čirý roztok očních kapek v balení obsahujícím 1 nebo 3 plastové lahvičky se šroubovacím uzávěrem. Každá lahvička je naplněna přibližně z poloviny a obsahuje 3 mililitry roztoku. To je dostačující množství pro používání na dobu 4 týdnů. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co. Mayo
Irsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Luxembourg/Luxemburg/Nederland Allergan n.v. Tél/Tel: +32 (0)2 351 24 24 |
Island Actavis ehf. Sími: +354 550 3300 |
|
Btarapnu AaepraH Etarapna EOOfl Tea.: +359 (0) 800 20 280 |
Italia Allergan S.p.A Tel: +39 06 509 561 |
|
Česká republika Allergan CZ s.r.o. Tel: +420 800 188 818 |
Latvija Allergan Baltics UAB Tel: +371 27331152 |
|
Danmark/Norge/Suomi/Finland/Sverige Allergan Norden AB Tlf/Puh/Tel: +46 (0)8 594 100 00 |
Lietuva Allergan Baltics UAB Tel: +370 62660247 |
|
Deutschland Pharm-Allergan GmbH Tel: +49 69 92038 10 |
Magyarország Allergan Hungary Kft. Tel: +36 80 100 101 |
|
Eesti Allergan Baltics UAB Tel: + 372 56955144 |
Osterreich Pharm-Allergan GmbH Tel: +43 1 99460 6355 |
|
EXláSa/Kónpoq Allergan Hellas Pharmaceuticals S.A. TpA.: +30 210 74 73 300 |
Polska Allergan Sp. z o.o. Tel: +48 22 256 37 00 |
|
Espaňa Allergan S.A. Tel: +34 91 807 6130 |
Portugal Profarin Lda. Tel: +351 21 425 3242 |
|
France Allergan France SAS Tel: +33 (0)1 49 07 83 00 |
Románia Allergan S.R.L. Tel.: +40 21 301 53 02 |
|
Hrvatska Ewopharma d.o.o. Tel: +385 1 6646 563 |
Slovenija Ewopharma d.o.o. Tel: +386 (0) 590 848 40 |
|
Ireland/Malta/United Kingdom Allergan Ltd Tel: +44 (0)1628 494026 |
Slovenská republika Allergan SK s.r.o. Tel: +421 800 221 223 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu/
Příbalová informace: informace pro uživatele
LUMIGAN 0,3 mg/ml, oční kapky, roztok
Bimatoprostum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li případně další otázky, zeptejte se, prosím, svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je LUMIGAN 0,3 mg/ml a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete LUMIGAN 0,3 mg/ml používat
3. Jak se LUMIGAN 0,3 mg/ml používá
4. Možné nežádoucí účinky
5. Jak LUMIGAN 0,3 mg/ml uchovávat
6. Obsah balení a další informace
1. Co je LUMIGAN 0,3 mg/ml a k čemu se používá
LUMIGAN je přípravek k léčbě glaukomu. Patří do skupiny nazývané prostamidy.
LUMIGAN se používá ke snížení zvýšeného tlaku v oku. Tento přípravek se může používat samostatně nebo s jinými očními kapkami nazývanými betablokátory, které také snižují tlak.
Vaše oko obsahuje průzračnou vodovitou tekutinu, která vyživuje vnitřní části oka. Tato tekutina je stále odváděna z oka a je nahrazována novou. Jestliže tekutina nemůže dostatečně rychle odtékat, zvyšuje se tlak uvnitř oka. Tak se snižuje tlak uvnitř oka. Pokud není tlak snižován, může se rozvinout onemocnění zvané glaukom a nakonec může dojít k poškození zraku.
2. Čemu musíte věnovat pozornost, než začnete LUMIGAN 0,3 mg/ml používat Nepoužívejte LUMIGAN 0,3 mg/ml
- jestliže jste alergický(á) (přecitlivělý(á)) na bimatoprost nebo na kteroukoliv další složku LUMIGANU
- jestliže jste v minulosti musel(a) přestat používat oční kapky kvůli nežádoucím účinkům konzervačního prostředku benzalkonium chloridu.
Upozornění a opatření:
Před použitím přípravku LUMIGAN 0,3 mg/ml se poraďte se svým lékařem nebo lékárníkem:
- Informujte Vašeho lékaře, jestliže:
- máte potíže s dýcháním.
- máte potíže s játry nebo ledvinami.
- jste v minulosti podstoupil(a) operaci katarakty.
- máte sucho v očích
- Máte nebo j ste měl(a) problémy s rohovkou (přední průhledná část oka)
- používáte kontaktní čočky (viz „Důležité informace o některých přísadách LUMIGANU 0,3 mg/ml“)
- Pokud trpíte nízkou srdeční frekvencí nebo nízkým krevním tlakem
- Pokud j ste měli virovou infekci nebo zánět oka
LUMIGAN může způsobit ztmavnutí řas, jejich růst, a také ztmavnutí kůže v oblasti víček. Vaše duhovka může také časem ztmavnout. Tyto změny mohou být trvalé a výraznější, pokud je léčeno pouze jedno oko.
Děti a dospívající
LUMIGAN nebyl testován u dětí do 18 let a proto by osoby mladší 18 let neměly LUMIGAN používat.
Další léčivé přípravky a LUMIGAN:
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Pokud používáte LUMIGAN s dalšími očními kapkami, mezi vkápnutím jednotlivých přípravků dodržte odstup nejméně pěti minut.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete užívat jakýkoliv přípravek.
LUMIGAN může přecházet do mateřského mléka. Jestliže používáte LUMIGAN, neměla byste kojit.
Řízení dopravních prostředků a obsluha strojů:
Krátkou dobu po použití LUMIGANu může být Váš zrak rozmazaný. Dokud tyto příznaky nevymizí, neměli byste řídit motorová vozidla a obsluhovat stroje.
Důležité informace o některých složkách LUMIGANU 0,3 mg/ml
Nepoužívejte kapky, pokud nosíte kontaktní čočky. Po použití kapek počkejte 15 minut a teprve potom si nasaďte kontaktní čočky zpět. Konzervační látka benzalkonium-chlorid obsažená v LUMIGANU může způsobit podráždění oka nebo změnu barvy měkké kontaktní čočky.
3. Jak se LUMIGAN 0,3 mg/ml používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
LUMIGAN může být používán pouze do očí. Doporučená dávka LUMIGANU je vkápnutí jedné kapky denně do léčeného oka, vždy každý den večer.
Pokud užíváte přípravek LUMIGAN s jiným očním lékem, počkejte mezi použitím přípravku LUMIGAN a dalšího očního léku alespoň 5 minut.
Nepoužívejte častěji než jednou denně, protože může dojít ke snížení účinnosti léčby.
Návod k použití:
Lahvičku nesmíte použít, pokud je před prvním otevřením porušena bezpečnostní pečeť.
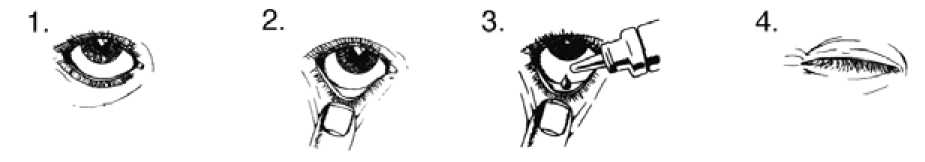
1. Umyjte si ruce. Zakloňte hlavu a podívejte se na strop.
2. Jemně stáhněte dolní víčko a vytvořte tak malou kapsu.
3. Otočte lahvičku dnem vzhůru a zmáčkněte, aby se uvolnila jedna kapka do každého léčeného oka.
4. Uvolněte dolní víčko a zavřete na 30 vteřin oči.
Setřete veškeré zbytky stékající po tváři.
Jestliže kapka Vaše oko minula, opakujte postup znovu.
Abyste zabránili infekci a poranění oka, neměl by se hrot lahvičky při kapání dotknout oka nebo něčeho jiného. Ihned po použití zavřete lahvičku víčkem.
Jestliže jste použil(a) více LUMIGANU 0,3 mg/ml než jste měl(a)
Jestliže jste použil(a) více LUMIGANU než jste měl(a), je nepravděpodobné, že si vážněji ublížíte. Další dávku si vkápněte v obvyklém čase. Jestliže máte obavy, poraďte se s Vaším lékařem nebo lékárníkem.
Jestliže jste zapomněl(a) LUMIGAN 0,3 mg/ml použít
Jestliže jste zapomněl(a) LUMIGAN použít, vkápněte si jednu kapku hned, jakmile si vzpomenete, a pak se vraťte ke svému pravidelnému dávkování. Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat LUMIGAN 0,3 mg/ml
Pro správnou funkci se LUMIGAN musí používat denně. Pokud léčbu přerušíte, může se zvýšit váš nitrooční tlak, proto se před přerušením léčby poraďte se svým lékařem.
Pokud máte jakékoliv další dotazy k užívání tohoto přípravku, obraťte se na svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Velmi časté nežádoucí účinky
Mohou postihnout 1 nebo více osob z každých 10 uživatelů Účinky na. oko
delší řasy (až 45 % lidí) lehké zčervenání (až 44 % lidí) svědění (až 14 % lidí)
Časté nežádoucí účinky
Mohou postihnout 1 - 9 osob ze 100 uživatelů Účinky na. oko
• alergická reakce na oku
• unavené oči
• citlivost na světlo
• tmavší zbarvení kůže v okolí oka
• tmavší řasy
• bolest
• pocit cizího tělesa v oku
• zalepené oči
• tmavší barva duhovky
• potíže s ostrým viděním
• podráždění
• pálení
• zanícená, červená a svědící oční víčka
• slzení
• pocit sucha
• zhoršení zraku
• rozmazané vidění
• otok průhledné vrstvy kryjící povrch oka
• malé oděrky na povrchu oka s nebo bez zánětu
Celkové účinky
• bolest hlavy
• zvýšení hodnot krevních testů, které ukazují, jak pracují Vaše játra
• zvýšení krevního tlaku
Méně časté nežádoucí účinky
Mohou postihnout 1 - 9 osob z každých 1000 osob používajících přípravek Postižení oka
• Cystoidní makulární edém (otok sítnice oka vedoucí ke zhoršenému vidění)
• Zánět některých vnitřních částí oka
• Retinální hemoragie(krvácení do sítnice)
• Oteklá oční víčka
• Záškuby očního víčka
• Stažení očního víčka, posun očního víčka od povrchu oka
• Zarudnutí kůže okolo oka
Celkové účinky
• Nevolnost
• Závratě
• Slabost
• Růst chloupků kolem oka
Nežádoucí účinky, jejichž četnost výskytu není známa
Účinky na. oko
• Oči vypadají zapadlé
Postižení těla
• Astma
• Zhoršení astmatu
• Zhoršení plicního onemocnění zvaného chronická obstrukční plicní nemoc (CHOPN)
• Dušnost
• projevy alergické reakce (otok, zarudnutí oka a vyrážka na kůži)
Další nežádoucí účinky hlášené u očních kapek obsahujících fosfáty
Ve velmi vzácných případech došlo u některých pacientů se závažným poškozením průhledné vrsty přední části oka (rohovky) ke vzniku zakalených vápenatých skvrn na rohovce, které jsou způsobené léčbou.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak LUMIGAN 0,3 mg/ml uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na štítku lahvičky a na krabičce pod označením EXP: Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Nejpozději po 4 týdnech od prvního otevření lahvičku zlikvidujte, i přesto, že v ní nějaké kapky ještě zůstaly. Tím zabráníte infekci. Pro lepší zapamatování si zapište datum otevření na označené místo na krabičce.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co LUMIGAN 0,3 mg/ml obsahuje
- Léčivou látkou je Bimatoprostum. Jeden ml roztoku obsahuje 0,3 mg bimatoprostu.
- Pomocné látky jsou benzalkonium-chlorid (konzervační látka), chlorid sodný, heptahydrát hydrogenfosforečnanu sodného, monohydrát kyseliny citronové a čištěná voda. Pro udržení kyselosti (hladiny pH) může být přidáno malé množství kyseliny chlorovodíkové nebo hydroxidu sodného.
Jak LUMIGAN 0,3 mg/ml vypadá a co obsahuje toto balení
LUMIGAN je bezbarvý čirý roztok očních kapek v balení obsahujícím 1 nebo 3 plastové lahvičky se šroubovacím uzávěrem. Každá lahvička je naplněna přibližně z poloviny a obsahuje 3 mililitry roztoku. To je dostačující množství pro používání na dobu 4 týdnů. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Allergan Pharmaceuticals Ireland
Castlebar Road
Westport
Co. Mayo
Irsko
|
Další informace o tomto přípravku získáte |
u místního zástupce držitele rozhodnutí o registraci. |
|
Belgie/Belgique/Belgien Luxembourg/Luxemburg/Nederland Allergan n.v. Tél/Tel: +32 (0)2 351 24 24 |
Island Actavis ehf. Sími: +354 550 3300 |
|
Etarapna AaepraH Etarapua EOOfl Tea.: +359 (0) 800 20 280 |
Italia Allergan S.p.A Tel: +39 06 509 561 |
|
Česká republika Allergan CZ s.r.o. Tel: +420 800 188 818 |
Latvija Allergan Baltics UAB Tel: +371 27331152 |
|
Danmark/Norge/Suomi/Finland/Sverige Allergan Norden AB Tlf/Puh/Tel: +46 (0)8 594 100 00 |
Lietuva Allergan Baltics UAB Tel: +370 62660247 |
|
Deutschland Pharm-Allergan GmbH Tel: +49 69 92038 10 |
Magyarország Allergan Hungary Kft. Tel: +36 80 100 101 |
|
Eesti Allergan Baltics UAB Tel: + 372 56955144 |
Osterreich Pharm-Allergan GmbH Tel: +43 1 99460 6355 |
|
EXlába/Kónpoq Allergan Hellas Pharmaceuticals S.A. TpA.: +30 210 74 73 300 |
Polska Allergan Sp. z o.o. Tel: +48 22 256 37 00 |
|
Espaňa Allergan S.A. Tel: +34 91 807 6130 |
Portugal Profarin Lda. Tel: +351 21 425 3242 |
|
France Allergan France SAS Tel: +33 (0)1 49 07 83 00 |
Románia Allergan S.R.L. Tel.: +40 21 301 53 02 |
|
Hrvatska Ewopharma d.o.o. Tel: +385 1 6646 563 |
Slovenija Ewopharma d.o.o. Tel: +386 (0) 590 848 40 |
|
Ireland/Malta/United Kingdom Allergan Ltd Tel: +44 (0)1628 494026 |
Slovenská republika Allergan SK s.r.o. Tel: +421 800 221 223 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu/
Příbalová informace: informace pro uživatele
LUMIGAN 0,3 mg/ml, oční kapky, roztok, v jednodávkovém obalu
Bimatoprostum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li případně další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je LUMIGAN 0,3 mg/ml v j ednodávkovém obalu a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete LUMIGAN 0,3 mg/ml v jednodávkovém obalu používat
3. Jak se LUMIGAN 0,3 mg/ml v jednodávkovém obalu používá
4. Možné nežádoucí účinky
5. Jak LUMIGAN 0,3 mg/ml v jednodávkovém obalu uchovávat
6. Obsah balení a další informace
1. Co je LUMIGAN 0,3 mg/ml v jednodávkovém obalu a k čemu se používá
LUMIGAN 0,3 mg/ml v jednodávkovém obalu je přípravek k léčbě glaukomu. Patří do skupiny nazývané prostamidy.
LUMIGAN 0,3 mg/ml v jednodávkovém obalu se používá ke snížení zvýšeného tlaku v oku. Tento přípravek se může používat samostatně nebo s jinými očními kapkami nazývanými betablokátory, které také snižují tlak.
Vaše oko obsahuje průzračnou vodovitou tekutinu, která vyživuje vnitřní části oka. Tato tekutina je stále odváděna z oka a je nahrazována novou. Jestliže tekutina nemůže dostatečně rychle odtékat, zvyšuje se tlak uvnitř oka. Tak se snižuje tlak uvnitř oka. Pokud není tlak snižován, může se rozvinout onemocnění zvané glaukom a nakonec může dojít k poškození zraku.
Neobsahuje konzervační látky.
2. Čemu musíte věnovat pozornost, než začnete LUMIGAN 0,3 mg/ml v jednodávkovém obalu používat
Tento přípravek nepoužívejte:
- Jestliže jste alergický(á) na léčivou látku bimatoprost nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření:
Před použitím přípravku LUMIGAN 0,3 mg/ml v jednodávkovém obalu se poraďte se svým lékařem nebo lékárníkem:
Informujte Vašeho lékaře nebo lékárníka, jestliže:
- máte potíže s dýcháním.
- máte potíže s játry nebo ledvinami.
- jste v minulosti podstoupil(a) operaci katarakty (šedého zákalu).
- Pokud trpíte nízkou srdeční frekvencí nebo nízkým krevním tlakem
- Pokud jste měli virovou infekci nebo zánět oka
LUMIGAN 0,3 mg/ml vjednodávkovém obalu může způsobit ztmavnutí řas, jejich růst, a také ztmavnutí kůže v oblasti víček. Vaše duhovka může také časem ztmavnout. Tyto změny mohou být trvalé a výraznější, pokud je léčeno pouze jedno oko.
Děti a dospívající
LUMIGAN 0,3 mg/ml v jednodávkovém obalu nebyl testován u dětí do 18 let a proto by jej osoby mladší 18 let neměly používat.
Další léčivé přípravky a LUMIGAN 0,3 mg/ml v jednodávkovém obalu:
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Pokud používáte LUMIGAN s dalšími očními kapkami, mezi vkápnutím jednotlivých přípravků dodržte odstup nejméně pěti minut.
Těhotenství a kojení a fertilita
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
LUMIGAN 0,3 mg/ml v jednodávkovém obalu může přecházet do mateřského mléka. Jestliže používáte tento léčivý přípravek, neměla byste kojit.
Řízení dopravních prostředků a obsluha strojů:
Krátkou dobu po použití LUMIGANu 0,3 mg/ml v jednodávkovém obalu může být Váš zrak rozmazaný. Dokud tyto příznaky nevymizí, neměli byste řídit motorová vozidla a obsluhovat stroje.
3. Jak se LUMIGAN 0,3 mg/ml v jednodávkovém obalu používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka je vkápnutí jedné kapky jednou denně do léčeného oka, vždy každý den večer. LUMIGAN 0,3 mg/ml v jednodávkovém obalu může být používán pouze do očí.
Pokud užíváte přípravek LUMIGAN 0,3 mg/ml v jednodávkovém obalu s jiným očním lékem, počkejte mezi použitím přípravku LUMIGAN 0,3 mg/ml v jednodávkovém obalu a dalšího očního léku alespoň 5 minut.
Nepoužívejte častěji než jednou denně, protože může dojít ke snížení účinnosti léčby.
Součástí balení je kalendář, očíslovaný 1-31 na každý den v měsíci. Umístěte kalendář na plastovou vaničku a zatlačte, aby zapadl na své místo. Oddělte od sebe 30 jednodávkových nádobek z balení a jednotlivě je zasuňte do očíslovaných otvorů. Začněte u otvoru s číslem dnešního data. Pokud vám nádobky zbudou, pokračujte od čísla 1 - tuto dávku použijete první den příštího měsíce. Každý den odeberte nádobku z čísla odpovídajícího aktuálnímu datu - tak zajistíte, že dávku nevynecháte.
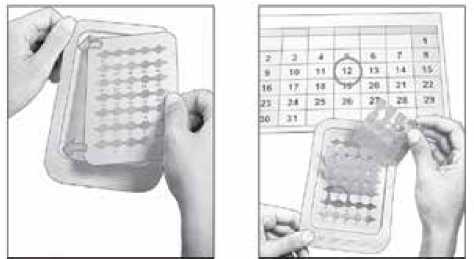
Před použitím si umyjte ruce. Před použitím se ujistěte, že je jednodávková nádobka neporušená. Roztok je zapotřebí použít bezprostředně po otevření. Aby nedošlo ke kontaminaci, zabraňte tomu, aby se otevřená část jednodávkové nádobky dotkla oka nebo čehokoli jiného.
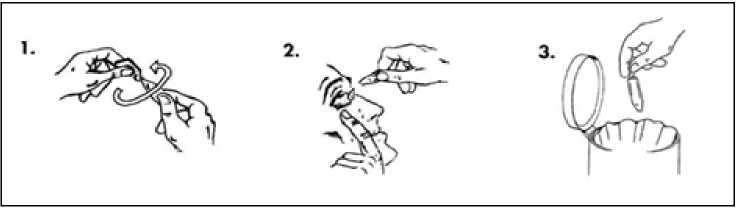
1. Vezměte jednu jednodávkovou nádobku z otvoru s číslem shodujícím se s daným dnem
v kalendáři a podržte ji tak, aby její kryt směřoval vzhůru, a kroucením odtrhněte kryt.
2. Jemně zatáhněte za spodní víčko a vytvořte tak malou kapsu. Otočte jednodávkovou nádobku dnem vzhůru a stlačte ji, aby do postiženého oka (očí) kápla 1 kapka. Několikrát mrkněte.
3. Jednodávkovou nádobku po použití zlikvidujte, i když se v ní nachází zbytek přípravku.
Setřete veškeré zbytky stékající po tváři.
Pokud používáte kontaktní čočky, před použitím tohoto přípravku je vyjměte. Počkejte 15 minut po aplikaci přípravku a poté si čočky znovu nasaďte.
Jestliže jste použil(a) více LUMIGANU 0,3 mg/ml v jednodávkovém obalu, než jste měl(a)
Jestliže jste použil(a) více tohoto přípravku, než jste měl(a), je nepravděpodobné, že si vážněji ublížíte. Další dávku si vkápněte v obvyklém čase. Jestliže máte obavy, poraďte se s Vaším lékařem nebo lékárníkem.
Jestliže jste zapomněl(a) LUMIGAN 0,3 mg/ml v jednodávkovém obalu použít
Jestliže jste zapomněl(a) LUMIGAN 0,3 mg/ml v jednodávkovém obalu použít, vkápněte si jednu kapku hned, jakmile si vzpomenete, a pak se vraťte ke svému pravidelnému dávkování. Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat LUMIGAN 0,3 mg/ml v jednodávkovém obalu
Pro správnou funkci se LUMIGAN 0,3 mg/ml v jednodávkovém obalu musí používat denně. Pokud léčbu přerušíte, může se zvýšit váš nitrooční tlak, proto se před přerušením léčby poraďte se svým lékařem.
Pokud máte jakékoliv další dotazy k používání tohoto přípravku, obraťte se na svého lékaře nebo lékárníka.
Možné nežádoucí účinky
4.
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Velmi časté nežádoucí účinky
Mohou postihnout 1 nebo více osob z každých 10 použivatelů Účinky na oko
• lehké zčervenání (až 24 % lidí)
Časté nežádoucí účinky
Mohou postihnout 1 - 9 osob ze 100 použivatelů Účinky na. oko
• malé oděrky na povrchu oka, se zánětem či bez zánětu
• podráždění očí
• svědící oči
• bolest
• suchost
• pocit cizího tělesa v oku
• delší řasy
• tmavší kůže okolo očí
• červené oči
Méně časté nežádoucí účinky
Mohou postihnout 1 až 9 z 1 000 použivatelů Účinky na. oko:
• unavené oči
• citlivost na světlo
• tmavší barva duhovky
• svědící a oteklá oční víčka
• slzení
• otok průhledné vrstvy kryjící povrch oka
• rozmazané vidění
Celkové účinky
• bolest hlavy
• růst chloupků okolo očí
Nežádoucí účinky, jejichž četnost výskytu není známa
Postižení těla
• astma
• zhoršení astmatu
• zhoršení plicního onemocnění zvaného chronická obstrukční plicní nemoc (CHOPN)
• dušnost
• projevy alergické reakce (otok, zarudnutí oka a vyrážka na kůži)
Kromě těchto nežádoucích účinků spojených s přípravkem LUMIGAN 0,3 mg/ml v jednodávkovém obalu byly u konzervovaného přípravku LUMIGAN 0,3 mg/ml ve vícedávkovém balení pozorovány následující nežádoucí účinky, které se mohou vyskytnout u pacientů používajících přípravek LUMIGAN 0,3 mg/ml v jednodávkovém obalu:
• závratě
• pocit pálení v oku
• alergické reakce oka
• zánět očních víček
• rozmazané vidění
• slepení očních víček
• poruchy zraku
• tmavší řasy
• retinální krvácení (krvácení do sítnice)
• zánět v oku
• cystoidní makulární edém (otok sítnice v oku vedoucí ke zhoršení vidění)
• zánět duhovky
• záškuby očního víčka
• stažení očního víčka, posun očního víčka od povrchu oka
• oči vypadají zapadlé
• zvýšený krevní tlak
• nevolnost
• zarudnutí kůže okolo očí
• slabost
• zvýšení výsledků krevních testů ukazujících funkci jater Další nežádoucí účinky hlášené u očních kapek obsahujících fosfáty
Ve velmi vzácných případech došlo u některých pacientů se závažným poškozením průhledné vrsty přední části oka (rohovky) ke vzniku zakalených vápenatých skvrn na rohovce, které jsou způsobené léčbou.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak LUMIGAN 0,3 mg/ml vjednodávkovém obalu uchovávat Uchovávejte tento přípravek mimo dohled a dosah dětí.
Tento přípravek je určen k jednorázovému použití a neobsahuje konzervační látky. Neuchovávejte žádný nevyužitý roztok.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na jednodávkové nádobce a na platu za EXP: Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání. Po otevření však má být balení spotřebováno do 30 dnů.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co LUMIGAN 0,3 mg/ml v jednodávkovém obalu obsahuje
- Léčivou látkou je bimatoprostum. Jeden ml roztoku obsahuje 0,3 mg bimatoprostu.
- Pomocné látky jsou chlorid sodný, heptahydrát hydrogenfosforečnanu sodného, monohydrát kyseliny citronové a čištěná voda. Pro udržení kyselosti (hladiny pH) může být přidáno malé množství kyseliny chlorovodíkové nebo hydroxidu sodného.
Jak LUMIGAN 0,3 mg/ml v jednodávkovém obalu vypadá a co obsahuje toto balení
LUMIGAN 0,3 mg/ml v jednodávkovém obalu je čirý bezbarvý roztok dodávaný v plastových jednodávkových nádobkách, z nichž každá obsahuje 0,4 ml roztoku.
Jedno balení obsahuje 5, 30 nebo 90 jednodávkových nádobek. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Allergan Pharmaceuticals Ireland Castlebar Road Westport Co. Mayo Irsko
u místního zástupce držitele rozhodnutí o registraci.
Island
Actavis ehf.
Sími: +354 550 3300
Další informace o tomto přípravku získáte
Belgie/Belgique/Belgien Luxembourg/Luxemburg/Nederland
Allergan n.v.
Tél/Tel: +32 (0)2 351 24 24
|
Bt^rapnn AnepraH Etnrapua EOOfl Ten.: +359 (0) 800 20 280 |
Italia Allergan S.p.A Tel: +39 06 509 561 |
|
Česká republika Allergan CZ s.r.o. Tel: +420 800 188 818 |
Latvija Allergan Baltics UAB Tel: +371 27331152 |
|
Danmark/Norge/Suomi/Finland/Sverige Allergan Norden AB Tlf/Puh/Tel: +46 (0)8 594 100 00 |
Lietuva Allergan Baltics UAB Tel: +370 62660247 |
|
Deutschland Pharm-Allergan GmbH Tel: +49 69 92038 10 |
Magyarország Allergan Hungary Kft. Tel: +36 80 100 101 |
|
Eesti Allergan Baltics UAB Tel: + 372 56955144 |
Osterreich Pharm-Allergan GmbH Tel: +43 1 99460 6355 |
|
EXlába/Kónpoq Allergan Hellas Pharmaceuticals S.A. TqA.: +30 210 74 73 300 |
Polska Allergan Sp. z o.o. Tel: +48 22 256 37 00 |
Espaňa
Allergan S.A.
Tel: +34 91 807 6130
Portugal
Profarin Lda.
Tel: +351 21 425 3242
France
Allergan France SAS Tel: +33 (0)1 49 07 83 00
Románia
Allergan S.R.L.
Tel.: +40 21 301 53 02
Hrvatska Slovenija
Ewopharma d.o.o. Ewopharma d.o.o.
Tel: +385 1 6646 563 Tel: +386 (0) 590 848 40
Ireland/Malta/United Kingdom Slovenská republika
Allergan Ltd Allergan SK s.r.o.
Tel: +44 (0)1628 494026 Tel: +421 800 221 223
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu/
Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY PODMÍNEK ROZHODNUTÍ O
REGISTRACI
Vědecké závěry
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizovaných zpráv o bezpečnosti (PSUR) k bimatoprostu dospěl výbor CHMP k těmto vědeckým závěrům:
Během ohlašovacího období této zprávy PSUR byl hlášen určitý počet případů z rutinních farmakovigilančních aktivit a byl stanoven příčinný vztah s použitím léčivého přípravku obsahujícího bimatoprost. Na základě dostupných údajů a analýzy předložené v této zprávě PSUR došel výbor PRAC k závěru, že je zapotřebí upravit souhrn údajů o přípravku a příbalovou informaci léčivých přípravků obsahujících bimatoprost (všechny formulace) tak, aby odrážely následující nežádoucí účinky léčivých přípravků: hypersenzitivní reakce včetně známek a příznaků oční alergie a alergické dermatitidy, astmatu, exacerbace astmatu, exacerbace CHOPN a dyspnoe, s četností není známo. Souhrn údajů o přípravku a příbalová informace u přípravků obsahujících bimatoprost (formulace s koncentrací 0,01 %) mají být upraveny tak, aby odrážely následující nežádoucí účinky léčivých přípravků: hyperpigmentace duhovky s méně častou četností a makulární edém, pigmentace očních víček, změny okolí očí a očního víčka zahrnující prohloubení záhybu očního víčka a syndrom suchého oka s četností není známo.
Proto, s ohledem na data prezentovaná v revidované zprávě PSUR / revidovaných zprávách PSUR má výbor PRAC za to, že je nutné provést tyto změny v údajích o léčivých přípravcích obsahujících bimatoprost.
CHMP souhlasí s vědeckými závěry výboru PRAC.
Zdůvodnění změny podmínek rozhodnutí o registraci
Na základě vědeckých závěrů týkajících se bimatoprostu zastává výbor CHMP stanovisko, že poměr přínosů a rizik léčivých přípravků obsahujících bimatoprost je příznivý pod podmínkou, že v údajích o přípravku budou provedeny navržené změny.
Výbor CHMP doporučuje změnit podmínky rozhodnutí o registraci.
72