Lucentis 10 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Lucentis 10 mg/ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje ranibizumabum 10 mg*. Jedna injekční lahvička obsahuje ranibizumabum 2,3 mg v 0,23 ml roztoku.
*Ranibizumab je fragment humanizované monoklonální protilátky produkovaný buňkami Escherichia coli rekombinantní DNA technologií.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok
Čirý, bezbarvý až světle žlutý vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Lucentis je indikován u dospělých:
• k léčbě neovaskulární (vlhké) formy věkem podmíněné makulární degenerace (AMD)
• k léčbě poškození zraku způsobeného diabetickým makulárním edémem (DME)
• k léčbě poškození zraku způsobeného makulárním edémem v důsledku okluze retinální vény [uzávěr větve centrální retinální vény (BRVO) a uzávěr centrální retinální vény (CRVO)]
• k léčbě poškození zraku způsobeného choroidální neovaskularizací (CNV) sekundární k patologické myopii (PM)
4.2 Dávkování a způsob podání
Lucentis musí být aplikován kvalifikovaným oftalmologem zkušeným v podání do sklivce.
Doporučená dávka Lucentisu je 0,5 mg podávaných jako jednorázová injekce do sklivce. To odpovídá injekci o objemu 0,05 ml. Interval mezi dvěma dávkami podávanými do stejného oka má být alespoň čtyři týdny.
Léčba se zahajuje jednou injekcí za měsíc do dosažení maximální zrakové ostrosti a/nebo do vymizení příznaků aktivity onemocnění, tj. žádná změna zrakové ostrosti a ostatních příznaků a projevů onemocnění při probíhající léčbě. U pacientů s vlhkou formou AMD, DME a RVO mohou být zpočátku potřeba tři nebo více po sobě jdoucí injekce podávané jednou za měsíc.
Následně mají být lékařem určeny intervaly sledování a léčby a mají být stanoveny na základě aktivity onemocnění vyhodnocené podle zrakové ostrosti a/nebo anatomických parametrů.
Pokud zrakové a anatomické parametry ukazují na základě vyjádření lékaře, že pokračující léčba pacienta není přínosná, je třeba léčbu přípravkem Lucentis ukončit.
Sledování aktivity onemocnění může zahrnovat klinické vyšetření, funkční testy nebo zobrazovací techniky (např. optickou koherenční tomografii nebo fluorescenční angiografii).
Pokud j sou pacienti léčeni podle režimu „treat-and-extend“, pak po dosažení maximální zrakové ostrosti a/nebo vymizení příznaků aktivity onemocnění, mohou být léčebné intervaly postupně prodlouženy až do opětovného objevení se příznaků aktivity onemocnění nebo zhoršení zraku. Léčebný interval nemá být prodloužen o více než dva týdny najednou u vlhké formy AMD a může být prodloužen až o jeden měsíc najednou u DME. Při uzávěru retinální vény mohou být léčebné intervaly také postupně prodlužovány, ale nejsou dostupná dostatečná data dokládající délku těchto intervalů. Pokud se aktivita onemocnění znovu objeví, léčebný interval má být příslušně zkrácen.
V léčbě poškození zraku způsobeném CNV sekundární k PM může mnoho pacientů potřebovat pouze jednu nebo dvě injekce během prvního roku, zatímco někteří pacienti mohou potřebovat častější léčbu (viz bod 5.1).
Lucentis a laserová fotokoagulace u DME a makulárního edému v důsledku BRVO Existuje určitá zkušenost s podáním přípravku Lucentis společně s laserovou fotokoagulací (viz bod 5.1). Při podání ve stejný den by měl být Lucentis podán alespoň 30 minut po laserové fotokoagulaci. Lucentis může být podán pacientům, kteří byli léčeni předchozí laserovou fotokoagulací.
Lucentis a fotodynamická léčba Visudynem u CNV sekundární k PM Se souběžným podáváním přípravků Lucentis a Visudyne nejsou žádné zkušenosti.
Zvláštní populace
Zhoršená funkce jater
Účinky přípravku Lucentis u pacientů se zhoršenou funkcí jater nebyly hodnoceny. Pro tuto populaci však nejsou nutná žádná zvláštní opatření.
Zhoršená funkce ledvin
U pacientů se zhoršenou funkcí ledvin není nutné upravovat dávku (viz bod 5.2).
Starší jedinci
U starších jedinců není nutná úprava dávky. U pacientů s DME starších 75 let jsou omezené zkušenosti.
Pediatrická populace
Bezpečnost a účinnost Lucentisu u dětí a dospívajících pod 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Injekční lahvička na jedno použití, pouze pro podání do sklivce.
Před aplikací je nutno Lucentis vizuálně zkontrolovat, zda neobsahuje cizí částice nebo není změněna jeho barva.
Lucentis musí být aplikován za aseptických podmínek, což zahrnuje použití chirurgické desinfekce rukou, sterilních rukavic, sterilního oděvu, sterilního spekula (nebo ekvivalentní náhrady) a dostupnost sterilní paracentézy (je-li potřeba). Před aplikací injekce do sklivce je nutný pečlivý odběr anamnézy z hlediska hypersenzitivních reakcí (viz bod 4.4). Před aplikací injekce musí být podána adekvátní anestezie a použit širokospektrý lokální antimikrobiální přípravek k dezinfekci pokožky kolem očí, očního víčka a povrchu oka, v souladu s lokální praxí.
Pro informace o přípravě Lucentisu viz bod 6.6.
Injekční jehla se zasune 3,5-4,0 mm posteriomě od limbu do prostoru sklivce tak, aby směřovala do centra očního bulbu a nikoli k horizontálnímu meridiánu. Poté se aplikuje objem injekce 0,05 ml. Následující injekci je nutné aplikovat v jiném místě skléry.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Pacienti s aktivní nebo suspektní oční nebo periokulární infekcí.
Pacienti s aktivním těžkým nitroočním zánětem.
4.4 Zvláštní upozornění a opatření pro použití Reakce po podání injekce do sklivce
Podání do sklivce, včetně těch s Lucentisem, byly spojovány s endoftalmitidou, intraokulárním zánětem, rhegmatogenním odchlípením sítnice, trhlinami sítnice a iatrogenní traumatickou kataraktou (viz bod 4.8). Při aplikaci Lucentisu musí být vždy dodržena přísná pravidla asepse. V následujícím týdnu po aplikaci injekce musejí být pacienti sledováni z hlediska případného výskytu infekce, aby bylo možné zahájit včas adekvátní léčbu. Pacienty je nutno upozornit, že musejí ihned hlásit všechny příznaky možné endoftalmitidy nebo jiných výše popsaných komplikací.
Zvýšení nitroočního tlaku
Během 60 minut po injekci Lucentisu bylo pozorováno přechodné zvýšení nitroočního tlaku (IOP). Trvalá zvýšení IOP byla také zjištěna (viz bod 4.8). Je nutné monitorovat a náležitě ošetřit jak nitrooční tlak, tak i perfuzi papily očního nervu.
Léčba obou očí současně
Omezená data k léčbě obou očí přípravkem Lucentis současně (včetně podání ve stejný den) nenaznačují zvýšené riziko systémových nežádoucích účinků v porovnání s léčbou jednoho oka.
Imunogenicita
U Lucentisu existuje možnost vzniku imunogenicity. Protože existuje potenciál pro zvýšenou systémovou expozici u pacientů s DME, zvýšené riziko pro vznik hypersenzitivity u této pacientské populace nelze vyloučit. Pacienti musejí být také poučeni, aby hlásili zhoršení závažnosti nitroočního zánětu, protože se může jednat o klinický příznak charakteristický pro tvorbu nitroočních protilátek.
Současné použití jiných anti-VEGF (vaskulární endoteliální růstový faktor) léčivých přípravků Lucentis se nesmí podávat zároveň s jinými anti-VEGF léčivými přípravky (systémovými nebo očními).
Vynechání dávky Lucentisu
Dávku je nutno vynechat a v léčbě se nesmí pokračovat dříve, než je plánována další dávka v následujících případech:
• snížení nejlépe upravené ostrosti zraku (best-corrected visual acuity BCVA) o > 30 písmen ve srovnání s předchozím měřením ostrosti zraku;
• nitrooční tlak > 30 mmHg;
• poškození sítnice;
nebo je-li velikost hemoragie > 50 % celkové zákrok během uplynulých nebo následujících
• subretinální krvácení zahrnující střed fovey, plochy léze;
• provedený nebo plánovaný oční chirurgický 28 dnů.
Trhlina pigmentového epitelu sítnice
Rizikové faktory spojené s vývojem trhliny pigmentového epitelu sítnice po podání anti-VEGF léčby u vlhké formy AMD zahrnují rozsáhlé a/nebo značné odchlípení pigmentového epitelu sítnice.
U pacientů s těmito rizikovými faktory pro vznik trhlin pigmentového epitelu sítnice je třeba dbát opatrnosti při zahajování léčby Lucentisem.
Rhegmatogenní odchlípení sítnice nebo makulární díry
Léčbu je nutno přerušit u subjektů s rhegmatogenním odchlípením sítnice nebo u makulárních děr stupně 3 nebo 4.
Populace s omezenými daty
Existují pouze omezené zkušenosti s léčbou u pacientů s DME způsobeným diabetem I. typu. Lucentis nebyl studován u pacientů, kterým byla dříve podána injekce do sklivce, u pacientů s aktivními systémovými infekcemi, proliferativní diabetickou retinopatií nebo u pacientů se současnými očními onemocněními jako například odchlípení sítnice nebo makulární díra. Také nejsou zkušenosti s léčbou Lucentisem u diabetických pacientů s HbA1c nad 12 % a nekontrolovanou hypertenzí. Chybění těchto informací má být lékařem zváženo při léčbě takových pacientů.
Nejsou k dispozici dostatečné údaje k posouzení účinnosti Lucentisu u pacientů s RVO projevujícím se ireverzibilní ztrátou zraku v důsledku ischemie.
U pacientů s PM, kteří v minulosti podstoupili neúspěšnou fotodynamickou léčbu verteporfinem (vPDT) jsou k dispozici limitovaná data o účinku přípravku Lucentis. Rovněž zatímco byl pozorován odpovídající účinek u subjektů se subfoveálními a juxtafoveálními lézemi, jsou data k vyvození závěru o účinku přípravku Lucentis u subjektů s patologickou myopií s extrafoveálními lézemi nedostatečná.
Systémové účinky po podání do sklivce
Po injekční aplikaci VEGF inhibitorů do sklivce byly hlášeny systémové nežádoucí příhody včetně mimoočních krvácení a arteriálních tromboembolických příhod.
Data o bezpečnosti při léčbě pacientů s DME, makulárním edémem způsobeným RVO a CNV sekundární k patologické myopii (PM) u pacientů s mrtvicí nebo přechodnými ischemickými příhodami v anamnéze jsou omezená. U těchto pacientů je třeba dbát zvýšené opatrnosti (viz bod 4.8).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí.
Současné použití Lucentisu s fotodynamickou léčbou (PDT) verteporfinem u vlhké formy AMD a PM, viz bod 5.1.
Současné použití Lucentisu s laserovou fotokoagulací u DME a BRVO, viz body 4.2 a 5.1.
V klinických studiích zaměřených na léčbu poškození zraku způsobeného DME nebyl výsledek s ohledem na zrakovou ostrost nebo tloušťku centrální části sítnice (CSFT) u pacientů léčených přípravkem Lucentis ovlivněn současnou léčbou thiazolidindiony.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/antikoncepce u žen
Ženy ve fertilním věku musí během léčby používat účinnou antikoncepci.
Žádné údaje o podávání ranibizumabu těhotným ženám nejsou k dispozici. Studie u makaků rodu Cynomolgus nenaznačují přímé nebo nepřímé škodlivé účinky s ohledem na těhotenství nebo embryonální/fetální vývoj (viz bod 5.3). Po očním podání je nízká systémová expozice ranibizumabu, ale vzhledem k jeho mechanismu účinku je nutno ranibizumab považovat za potenciálně teratogenní a embryo-/fetotoxický. Z tohoto důvodu nesmí být ranibizumab užíván během těhotenství, aniž by očekávaný přínos převážil možné riziko pro plod. Ženám, které chtějí otěhotnět, a které byly léčeny ranibizumabem, je doporučeno vyčkat nejméně 3 měsíce po poslední dávce ranibizumabu před početím dítěte.
Kojení
Není známo, zda se Lucentis vylučuje do lidského mateřského mléka. Během léčby přípravkem Lucentis se kojení nedoporučuje.
Fertilita
Nejsou k dispozici žádné údaje vztahující se k fertilitě.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Léčba Lucentisem může vyvolat dočasné zhoršení zraku, což může ovlivnit schopnost řídit nebo obsluhovat stroje (viz bod 4.8). Pacienti, u kterých se tyto příznaky vyskytnou, nesmějí řídit nebo obsluhovat stroje, dokud tyto poruchy zraku neustoupí.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Většina nežádoucích účinků hlášených po podání Lucentisu se týká postupu podání injekce do sklivce.
Nejčastěji hlášené oční nežádoucí účinky po podání injekce Lucentisu jsou: bolest oka, oční hyperemie, zvýšený nitrooční tlak, vitritida, odloučení sklivce, hemoragie sítnice, poruchy zraku, sklivcové vločky, hemoragie spojivky, podráždění oka, pocit cizího tělesa, zvýšené slzení, zánět očního víčka, suchost oka a svědění oka.
Nejčastěji hlášené nežádoucí účinky mimo oko jsou: bolest hlavy, nazofaryngitida a artralgie.
Méně často hlášené, ale závažnější nežádoucí účinky zahrnují: endoftalmitidu, slepotu, odchlípení sítnice, trhlinu sítnice a iatrogenní traumatickou kataraktu (viz bod 4.4).
Pacienti mají být informováni o příznacích těchto potenciálních nežádoucích účinků a poučeni informovat svého lékaře, pokud se u nich objeví příznaky, jako je bolest oka nebo zvýšený oční diskomfort, zhoršující se zarudnutí oka, rozmazané nebo snížené vidění, zvýšený počet malých částic v zorném poli nebo zvýšená citlivost na světlo.
Nežádoucí účinky, které se objevily po podání Lucentisu v klinických studiích jsou uvedeny v tabulce níže.
Tabulkový seznam nežádoucích účinků#
Nežádoucí účinky jsou uvedeny podle systémově-orgánových skupin a frekvence za použití následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Infekce a infestace Velmi časté Časté |
Nazofaryngitida Infekce močových cest* |
Poruchy krve a lymfatického systému Časté Anemie
|
Poruchy imunitního systému Časté |
Hypersenzitivita |
|
Psychiatrické poruchy Časté |
Úzkost |
|
Poruchy nervového systému Velmi časté | |
|
Poruchy oka Velmi časté |
Vitritida, odloučení sklivce, hemoragie sítnice, poruchy zraku, bolest oka, sklivcové vločky, hemoragie spojivky, podráždění oka, pocit cizího tělesa, zvýšené slzení, zánět očního víčka, suchost oka, oční hyperemie, svědění oka. |
|
Časté |
Degenerace sítnice, poškození sítnice, odloučení sítnice, trhlina sítnice, odloučení pigmentového epitelu sítnice, trhlina v pigmentovém epitelu sítnice, snížení ostrosti zraku, hemoragie sklivce, poškození sklivce, uveitida, zánět duhovky, iridocyklitida, katarakta, subkapsulární katarakta, opacifikace zadního pouzdra, keratitis punctata, abraze rohovky, zarudnutí v přední části komory, rozmazané vidění, hemoragie v místě injekce, oční hemoragie, zánět spojivky, alergický zánět spojivky, výtok z oka, fotopsie, fotofobie, oční dyskomfort, otok víčka, bolestivost víčka, překrvení spojivek. |
|
Méně časté |
Slepota, endoftalmitida, hypopyon, krvácení do přední komory oka, keratopatie, adheze duhovky, korneální deposita, edém rohovky, strie rohovky, bolestivost v místě injekce, podráždění v místě injekce, abnormální pocit v oku, podráždění očního víčka. |
Respirační, hrudní a mediastinální poruchy Časté Kašel
|
Gastrointestinální poruchy Časté | |
|
Poruchy kůže a podkožní tkáně Časté |
Alergické reakce (vyrážka, kopřivka, pruritus, erytém) |
Poruchy svalové a kosterní soustavy a pojivové tkáně Velmi časté Artralgie
Vyšetření
Velmi časté Zvýšení nitroočního tlaku
# Nežádoucí účinky byly definovány jako nežádoucí příhody (u nejméně 0,5 procentních bodů pacientů), které se objevily ve vyšší míře (alespoň 2 procentní body) u pacientů léčených Lucentisem 0,5 mg než u těch, kteří byli léčeni v kontrolním rameni (simulovanou léčbou nebo fotodynamickou léčbou verteporfinem).
* pozorované pouze u populace s DME
Nežádoucí účinky související s třídou přípravku
Ve studiích fáze III s vlhkou formou AMD se mírně zvýšil celkový výskyt mimoočních krvácení u pacientů léčených ranibizumabem, což je nežádoucí účinek, který potenciálně souvisí se systémovou inhibicí VEGF (vaskulární endoteliální růstový faktor). Nicméně, jednotlivá krvácení neměla shodný charakter. Po intravitreálním podání VEGF inhibitorů existuje teoretické riziko arteriální tromboembolické příhody, včetně mrtvice a infarktu myokardu. V klinických studiích s Lucentisem byla u pacientů s AMD, DME, RVO a PM pozorována nízká incidence arteriálních tromboembolických příhod a mezi skupinami léčenými ranibizumabem nebyly ve srovnání s kontrolou významné rozdíly.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Případy předávkování byly hlášeny z klinických studií s vlhkou formou AMD a postmarketingového sledování. Nejčastěji hlášené případy nežádoucích účinků byly zvýšení nitroočního tlaku, přechodná slepota, snížená zraková ostrost, otok rohovky, bolest rohovky a bolest oka. Dojde-li k předávkování, je nutno monitorovat nitrooční tlak a v závislosti na rozhodnutí ošetřujícího lékaře případně nasadit odpovídající terapii k normalizaci nitroočního tlaku.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, látky určené k léčbě neovaskularizace, ATC kód: S01LA04
Ranibizumab je fragment humanizované monoklonální protilátky proti lidskému cévnímu endoteliálnímu růstovému faktoru A (VEGF-A). Váže se se silnou afinitou na VEGF-A isoformy (např. VEGFn0, VEGFm a VEGFJ65) a tím brání vazbě VEGF-A na receptory VEGFR-1 a VEGFR-2. Vazba VEGF-A na jeho receptory vede k proliferaci endoteliálních buněk a neovaskularizaci, jakož i k propustnosti cév. O všech těchto účincích se uvažuje jako o faktorech přispívajících k progresi neovaskulárních forem věkem podmíněné makulární degenerace, patologické myopie nebo k poškození zraku způsobeného buď diabetickým makulárním edémem, anebo makulárním edémem v důsledku RVO.
Léčba, vlhké formy AMD
Klinická bezpečnost a účinnost Lucentisu byla u vlhké formy AMD stanovena ve třech randomizovaných, dvojitě zaslepených studiích po dobu 24 měsíců se simulovanou injekcí nebo aktivním komparátorem u pacientů s neovaskulární AMD. Do dvou studií bylo zařazeno celkem 1 323 pacientů (879 na aktivní léčbě, 444 v kontrolní skupině).
Ve studii FVF2598g (MARINA) bylo 716 pacientů s minimálně klasickými nebo okultními lézemi randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali měsíčně injekce Lucentisu v dávkách 0,3 mg, injekce Lucentisu v dávkách 0,5 mg nebo simulovanou léčbu.
Ve studii FVF2587g (ANCHOR) bylo 423 pacientů s převážně klasickou CNV lézí randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali Lucentis 0,3 mg měsíčně, Lucentis 0,5 mg měsíčně nebo PDT s verteporfinem (při zahájení léčby a potom každé 3 měsíce, pokud fluoresceinová angiografie prokázala přetrvávání nebo rekurenci cévního prosakování).
Nejdůležitější výsledky měření jsou shrnuty v Tabulce 1 a na Obrázku 1.
Tabulka 1 Výsledky ve 12. a 24. měsíci studie FVF2598g (MARINA) a FVF2587g (ANCHOR)
|
FVF2598g ( |
MARINA) |
FVF2587g |
ANCHOR) | ||
|
Měřený parametr |
Měsíc |
Simulovaná léčba (n = 238) |
Lucentis 0,5 mg (n = 240) |
Verteporfin PDT (n = 143) |
Lucentis 0,5 mg (n = 140) |
|
Ztráta <15 písmen ostrosti zraku (%)a (zachování zraku, primární cíl) |
12. měsíc |
62 % |
95 % |
64 % |
96 % |
|
24. měsíc |
53 % |
90 % |
66 % |
90 % | |
|
Nárůst >15 písmen ostrosti zraku (%)a |
12. měsíc |
5 % |
34 % |
6 % |
40 % |
|
24. měsíc |
4 % |
33 % |
6 % |
41 % | |
|
Průměrná hodnota změny zrakové ostrosti (písmena) (SD)a |
12. měsíc |
-10,5 (16,6) |
+ 7,2 (14,4) |
-9,5 (16,4) |
+11,3 (14,6) |
|
24. měsíc |
-14,9 (18,7) |
+6,6 (16,5) |
-9,8 (17,6) |
+10,7 (16,5) |
a p<0.01
Obrázek 1 Průměrná hodnota změny zrakové ostrosti od výchozího stavu do 24. měsíce ve studii FVF2598g (MARINA) a ve studii FVF2587g (ANCHOR)
Studie FVF2598g (MARINA)
+6,6 'n
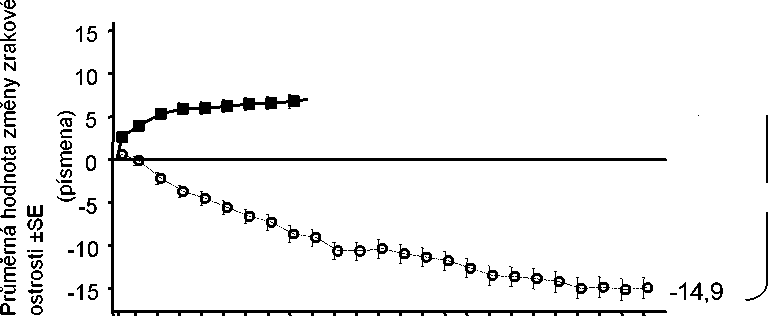
y +21,5
0 2 4 6 8 10 12 14 16 18 20 22 24
Měsíc
-CD
>
O
co
i_
N
>s
C
><D
E
N
CO
«
O
c
"O
o
-co
LU
co
+1
*2 oo
ol 2
Jr 1/5 Q_ o
15 ' 10 ■ 5
c
CD
0
V)
'cl
~-5 ■ -10 ■ -15 ■ 0
2 4 6 8
r +20,5
10 12 14 16 18 20 22 24 Měsíc
MARINA
LUCENTIS 0,5 mg (n=240) & Simulace (n=238)
ANCHOR
«- LUCENTIS 0,5 mg (n=140) • Verteporfin PDT (n=143)
Výsledky obou studií naznačují, že kontinuální léčba ranibizumabem může být také přínosem u pacientů, kteří v prvním roce léčby ztratili >15 písmen z nejlépe korigované ostrosti zraku (BCVA).
Statisticky významná zlepšení zrakových funkcí, hlášená pacienty, byla pozorována v obou studiích MARINA i ANCHOR při léčbě ranibizumabem oproti kontrolní skupině měřeno pomocí NEI VFQ-25.
Ve studii FVF192g (PIER) bylo 184 pacientů se všemi formami neovaskulární AMD randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali Lucentis 0,3 mg, Lucentis 0,5 mg nebo simulovanou léčbu v injekci jednou měsíčně po první 3 měsíce a dále dávku podávanou každý 3. měsíc. Od 14. měsíce této studie bylo pacientům se simulovanou léčbou povoleno používat ranibizumab a od 19. měsíce byla možná častější aplikace. V průměru obdrželi pacienti léčení Lucentisem ve studii PIER 10 aplikací.
Po počátečním nárůstu zrakové ostrosti (při dávkování jednou měsíčně) poklesla zraková ostrost u pacientů kteří dostávali Lucentis jednou za tři měsíce a vracela se ve 12. měsíci v průměru k počátečnímu stavu; tento účinek byl udržován ve 24. měsíci u většiny pacientů léčených ranibizumabem (82 %). Omezené údaje od osob se simulovanou léčbou, které později dostávaly ranibizumab naznačují, že předčasné zahájení léčby může být spojováno s lepším zachováním ostrosti zraku.
Data ze dvou studií (MONT BLANC, BPD952A2308 a DENALI, BPD952A2309) provedených po registraci přípravku Lucentis potvrdila jeho účinnost, ale neprokázala dodatečný účinek kombinovaného podávání verteporfinu (Visudyne PDT) a Lucentisu v porovnání s monoterapií Lucentisem.
Léčba poškození zraku způsobeného DME
Bezpečnost a účinnost Lucentisu byly hodnoceny ve třech randomizovanných kontrolovaných studiích po dobu alespoň 12 měsíců. Celkem 868 pacientů (708 aktivních a 160 kontrol) bylo zařazeno v těchto studiích.
Ve studii 2. fáze D2201 (RESOLVE) bylo léčeno 151 pacientů ranibizumabem (6 mg/ml, n = 51,
10 mg/ml, n = 51) nebo simulovanou léčbou (n = 49) intravitreální injekcí jednou měsíčně. Průměr určený z průměrných hodnot změn BCVA od 1. do 12. měsíce byl v porovnání se stavem na počátku léčby +7,8 (±7,72) písmen u shromážděných pacientů léčených ranibizumabem (n = 102), v porovnání s -0,1 (±9,77) písmen u pacientů se simulovanou léčbou a průměrná hodnota změny BCVA ve 12. měsíci od počátečního stavu byla 10,3 (±9,1) písmen v porovnání s -1,4 (±14,2) písmen, v tomto pořadí (p<0,0001 pro léčebný rozdíl).
Ve fázi III studie D2301 (RESTORE) bylo 345 pacientů randomizováno v poměru 1:1:1 do skupiny užívající ranibizumab 0,5 mg v monoterapii a simulovanou laserovou fotokoagulaci, kombinaci ranibizumabu 0,5 mg a laserové fotokoagulace nebo simulovanou injekci a laserovou fotokoagulaci. 240 pacientů, kteří předtím dokončili 12měsíční studii RESTORE, bylo zařazeno do otevřené, multicentrické 24měsíční extenze studie (extenze studie RESTORE). Pacienti byli léčeni podáním ranibizumabu 0,5 mg pro re nata (PRN) do stejného oka jako v základní studii (D2301 RESTORE).
Průměrná hodnota změny zrakové ostrosti od začátku léčby v průběhu času ve studii D2301 (RESTORE)
Obrázek 2
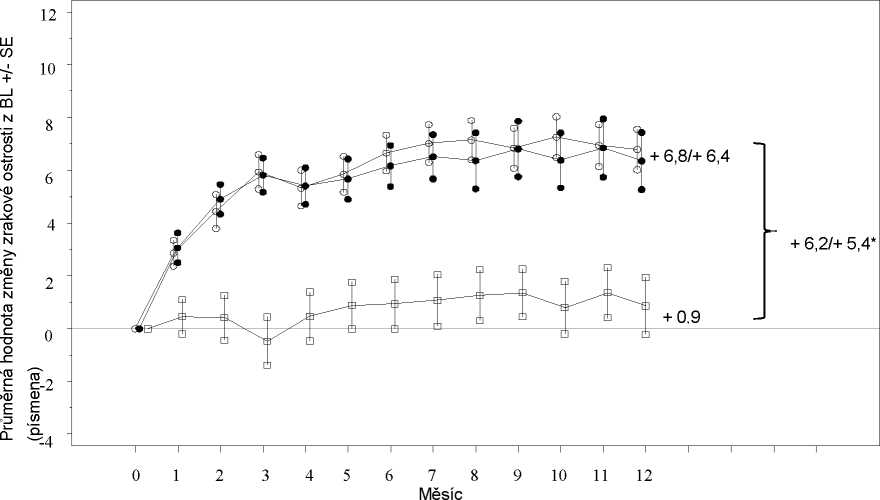
ooo Ranibizumab 0,5 mg (n=115)
Ranibizumab 0,5 mg + laser (n=118) □ □ □ Laser (n=110)
Léčebná skupina
BL=základní stav; SE=směrodatná chyba průměru
* Rozdíl v průměrech nejmenších čtverců, p<0,0001/0,0004 na základě dvoustranného stratifikovaného Cochran-Mantel-Haenszelova testu
Účinek po 12 měsících byl konzistentní ve většině podskupin. Avšak u subjektů s hodnotou BCVA >73 písmen na počátku léčby a makulárním edémem s centrální retinální tloušťkou <300 pm na počátku léčby se nezdálo, že by profitovaly z léčby ranibizumabem v porovnání s laserovou fotokoagulací.
Tabulka 2 Výsledky ve 12. měsíci ve studii D2301 (RESTORE) a ve 36. měsíci ve studii D2301-E1 (extenze studie RESTORE)
|
Výsledky měření ve 12. měsíci v porovnání se stavem na počátku léčby ve studii D2301 (RESTORE) |
Ranibizumab 0,5 mg n = 115 |
Ranibizumab 0,5 mg + laser n = 118 |
laser n = 110 |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 12. měsíce3 (+SD) |
6,1 (6,4)a |
5,9 (7,9)a |
0,8 (8,6) |
|
Průměrná hodnota změny BCVA ve 12. měsíci (+SD) |
6,8 (8,3)a |
6,4 (11,8)a |
0,9 (11,4) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 12. měsíci (%) |
22,6 |
22,9 |
8,2 |
|
Průměrný počet injekcí (0.-11. měsíc) |
7,0 |
6,8 |
7,3 (simulovaný) |
|
Výsledky měření ve 36. měsíci ve studii D2301-E1 (extenze studie RESTORE) v porovnání se stavem na počátku léčby ve studii D2301 (RESTORE) |
Před ranibizumabem 0,5 mg n = 83 |
Před ranibizumabem 0,5 mg + laser n = 83 |
Před laserem n = 74* |
|
Průměrná hodnota změny BCVA ve 24. měsíci (SD) |
7,9 (9,0) |
6,7 (7,9) |
5,4 (9,0) |
|
Průměrná hodnota změny BCVA ve 36. měsíci (SD) |
8,0 (10,1) |
6,7 (9,6) |
6,0 (9,4) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 36. měsíci (%) |
27,7 |
30,1 |
21,6 |
|
Průměrný počet injekcí (12.-35. měsíc)* |
6,8 |
6,0 |
6,5 |
ap<0,0001 pro porovnání ramen s ranibizumabem vs. rameno s laserem. n je ve studii D2301-E1 (extenze studie RESTORE) počet pacientů s hodnotou ve studii D2301 (RESTORE) na počátku léčby (měsíc 0) a při návštěvě ve 36. měsíci.
• Podíl pacientů, kteří nevyžadovali léčbu ranibizumabem během fáze prodloužení studie byl 19 % ve skupině před podáním ranibizumabu, 25 % ve skupině před podáním ranibizumabu + laseru a 20 % ve skupině před aplikací laseru.
Statisticky signifikantní přínosy pro většinu funkcí spojených se zrakem, hlášené pacienty, byly pozorovány u léčby ranibizumabem (s nebo bez laseru) oproti kontrolní skupině, měřeno pomocí NEI VFQ-25. Pro ostatní podškály tohoto dotazníku nemohly být stanoveny žádné léčebné rozdíly.
Dlouhodobý bezpečnostní profil ranibizumabu pozorovaný ve 24měsíční extenzi studie je konzistentní se známým bezpečnostním profilem přípravku Lucentis.
Ve fázi IIIb studie D2304 (RETAIN) bylo 372 pacientů randomizováno v poměru 1:1:1 k podávání:
• ranibizumabu 0,5 mg se současnou laserovou fotokoagulací v režimu „treat-and-extend“ (TE),
• ranibizumabu 0,5 mg v monoterapii v režimu TE,
• ranibizumabu 0,5 mg v monoterapii v režimu PRN.
Ve všech skupinách byl ranibizumab podáván jednou měsíčně, dokud BCVA nebyla stabilní
po alespoň tři po sobě jdoucí měsíční vyšetření. V režimu TE byl podán ranibizumab vléčebných intervalech 2-3 měsíce. Ve všech skupinách byla měsíční léčba znovu zahájena na základě snížení BCVA způsobeném progresí DME a pokračovala, dokud nebylo opět dosaženo stabilní BCVA.
Počet plánovaných léčebných návštěv po 3 počátečních injekcích byl 13 v režimu TE a 20 v režimu PRN. V obou TE režimech bylo více než 70 % pacientů schopných zachovat svou BCVA při průměrné četnosti návštěv >2 měsíce.
Nej důležitější výsledky měření jsou uvedeny v Tabulce 3.
Tabulka 3 Výsledky ve studii D2304 (RETAIN)
|
Výsledek měření porovnaný s výchozím stavem |
ranibizumab 0,5 mg + laser v režimu TE n = 117 |
ranibizumab 0,5 mg samotný v režimu TE n = 125 |
ranibizumab 0,5 mg v režimu PRN n = 117 |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 12. měsíce (SD) |
5,9 (5,5)a |
6,1 (5,7)a |
6,2 (6,0) |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 24. měsíce (SD) |
6,8 (6,0) |
6,6 (7,1) |
7,0 (6,4) |
|
Průměrná hodnota změny BCVA ve 24. měsíci (SD) |
8,3 (8,1) |
6,5 (10,9) |
8,1 (8,5) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 24. měsíci (%) |
25,6 |
28,0 |
30,8 |
|
Průměrný počet injekcí (0.-23. měsíc) |
12,4 |
12,8 |
10,7 |
ap<0,0001 pro hodnocení non-inferiority k PRN
Ve studiích zaměřených na DME bylo zlepšení BCVA doprovázeno redukcí střední CSFT v průběhu času ve všech léčebných skupinách.
Léčba poškození zraku způsobeného makulárním edémem v důsledku RVO
Klinická bezpečnost a účinnost Lucentisu u pacientů s poškozením zraku způsobeným makulárním
edémem v důsledku RVO byla hodnocena v randomizovaných dvojitě zaslepených kontrolovaných
studiích BRAVO a CRUISE, ve kterých byly zařazeny subjekty s BRVO (n = 397) a CRVO
(n = 392). V obou studiích dostávaly subjekty buď 0,3 mg nebo 0,5 mg ranibizumabu nebo injekce
simulované léčby. Pacienti v kontrolním rameni se simulovanou léčbou byli po 6 měsících přeřazeni
do ramene s ranibizumabem 0,5 mg.
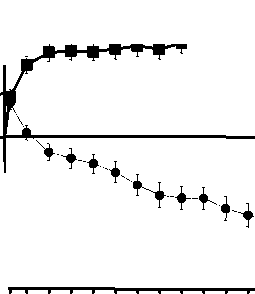
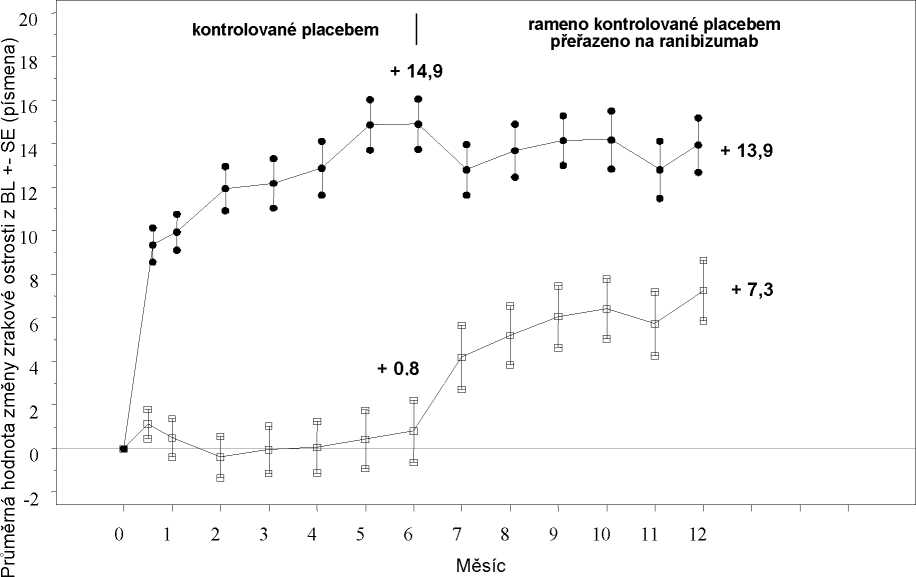
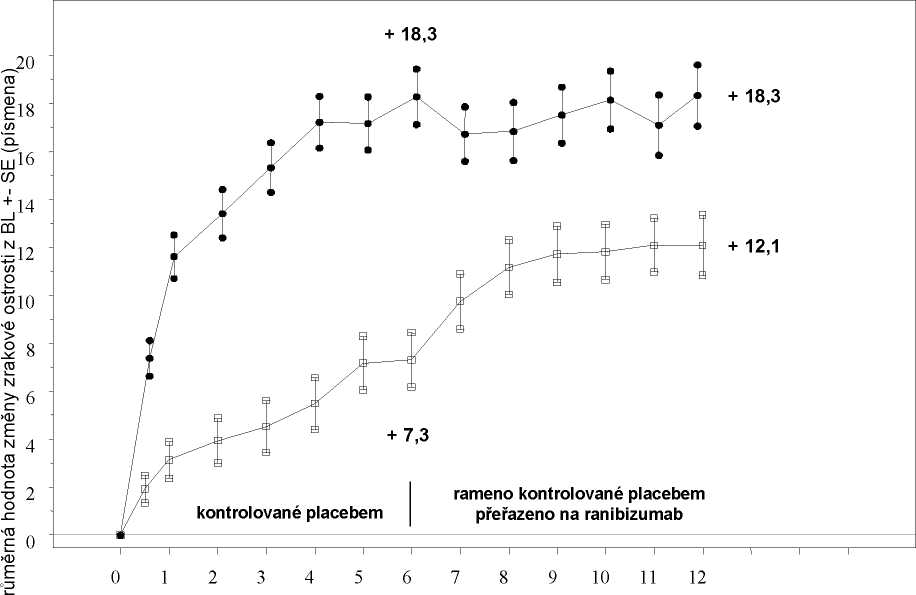
Tabulka 4 Výsledky v 6. a 12. měsíci (BRAVO a CRUISE)
|
BRAVO |
CRUISE | |||
|
Simulovaná léčba/Lucentis 0,5 mg (n = 132) |
Lucentis 0,5 mg (n = 131) |
Simulovaná léčba /Lucentis 0,5 mg (n = 130) |
Lucentis 0,5 mg (n = 130) | |
|
Průměrná hodnota změny zrakové ostrosti v 6. měsícía (písmena) (SD) (primární cíl) |
7,3 (13,0) |
18,3 (13,2) |
0,8 (16,2) |
14,9 (13,2) |
|
Průměrná hodnota změny BCVA ve 12. měsíci (písmena) (SD) |
12,1 (14,4) |
18,3 (14,6) |
7,3 (15,9) |
13,9 (14,2) |
|
Nárůst o > 15 písmen u zrakové ostrosti v 6. měsícia (%) |
28,8 |
61,1 |
16,9 |
47,7 |
|
Nárůst o > 15 písmen u zrakové ostrosti ve 12. měsíci (%) |
43,9 |
60,3 |
33,1 |
50,8 |
|
Podíl (%) pacientů léčených laserovou záchrannou terapií po 12 měsíců |
61,4 |
34,4 |
NA |
NA |
ap <0,0001 v obou studiích
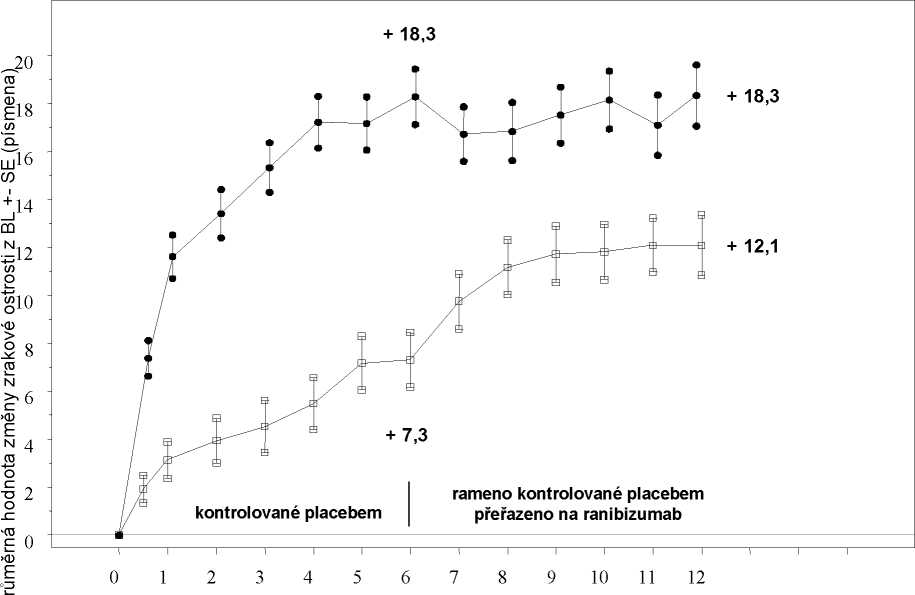
CL
Léčebná skupina
r m-m—m
Měsíc
Placebo/Ranibizumab 0,5 mg (n = 132 Ranibizumab 0,5 mg (n = 131)
BL=základní stav; SE=směrodatná chyba průměru
Léčebná skupina
Placebo/Ranibizumab 0,5 mg (n = 130) Ranibizumab 0,5 mg (n = 130)
BL=základní stav; SE=směrodatná chyba průměru
V obou studiích bylo zlepšení zraku spojeno s kontinuální a významnou redukcí makulámího edému, což bylo posuzováno podle tloušťky středu sítnice.
U pacientů s CRVO (CRUISE a extenze studie HORIZON): Subjekty léčené simulovanou léčbou v prvních 6 měsících, které následně dostávaly ranibizumab, nedosáhly srovnatelných nárůstů zrakové ostrosti (VA) do 24. měsíce (~6 písmen) v porovnání se subjekty léčenými ranibizumabem od začátku studie (~12 písmen).
Statisticky významná zlepšení v subškálách spojená s viděním na blízko a na dálku, hlášená pacienty, byla pozorována při léčbě ranibizumabem oproti kontrolní skupině, měřeno pomocí NEI VFQ-25.
Dlouhodobá (24měsíční) klinická bezpečnost a účinnost Lucentisu u pacientů s poškozením zraku způsobeného sekundárně makulárním edémem v důsledku RVO byla hodnocena ve studiích BRIGHTER (BRVO) a CRYSTAL (CRVO). V obou studiích bylo subjektům podáváno 0,5 mg ranibizumabu v režimu PRN řízeného individualizovanými kritérii stabilizace zrakové ostrosti. BRIGHTER byla 3ramenná randomizovaná aktivně kontrolovaná studie, která porovnávala 0,5 mg ranibizumabu podávaného v monoterapii nebo v kombinaci s adjuvantní laserovou fotokoagulací oproti samotné laserové fotokoagulaci. Subjektům v laserovém rameni mohl být po 6 měsících podán ranibizumab v dávce 0,5 mg. CRYSTAL byla jednoramenná studie s 0,5 mg ranibizumabu v monoterapii.
Důležité výstupy ze studií BRIGHTER a CRYSTAL jsou uvedeny v Tabulce 5. Tabulka 5 Výsledky v 6. a 24. měsíci studie (BRIGHTER a CRYSTAL)
|
BRIGHTER |
CRYSTAL | |||
|
Lucentis 0,5 mg n=180 |
Lucentis 0,5 mg + laser n=178 |
Laser* n=90 |
Lucentis 0,5 mg n=356 | |
|
Průměrná hodnota změny BCVA v 6. měsícia (písmena) (SD) |
+14,8 (10,7) |
+14,8 (11,13) |
+6,0 (14,27) |
+12,0 (13,95) |
|
Průměrná hodnota změny BCVA ve 24. měsícib (písmena) (SD) |
+15,5 (13,91) |
+17,3 (12,61) |
+11,6 (16,09) |
+12,1 (18,60) |
|
Zisk >15 písmen BCVA ve 24. měsíci (%) |
52,8 |
59,6 |
43,3 |
49,2 |
|
Průměrný počet injekcí (SD) (měsíc 0-23) |
11,4 (5,81) |
11,3 (6,02) |
NA |
13,1 (6,39) |
|
a p<0,0001 pro obě porovnání ve studii BRIGHTER v 6. měsíci: Lucentis 0,5 mg vs laser a Lucentis 0,5 mg + laser vs laser. b p<0,0001 pro nulovou hypotézu ve studii CRYSTAL, že průměrná hodnota změny ve 24. měsíci od výchozího stavu je nula. * Od 6. měsíce byla umožněna léčba 0,5 mg ranibizumabu (24 pacientů bylo léčeno pouze laserem). | ||||
Ve studii BRIGHTER byla prokázána non-inferiorita 0,5 mg ranibizumabu s podpůrnou laserovou terapií ve srovnání s ranibizumabem v monoterapii od výchozího stavu do 24. měsíce (95 % CI -2,8; 1,4).
V obou studiích byla v 1. měsíci pozorována rychlá a statisticky významná redukce tloušťky centrální části sítnice od výchozího stavu. Tento účinek přetrval až do 24. měsíce.
Účinek léčby ranibizumabem byl podobný bez ohledu na přítomnost retinální ischemie. Ve studii BRIGHTER u pacientů s ischemií (n=46) nebo bez ischemie (n=133) léčených ranibizumabem v monoterapii byla průměrná hodnota změny BCVA ve 24. měsíci od výchozího stavu +15,3; resp. +15,6 písmen. Ve studii CRYSTAL u pacientů s ischemií (n=53) nebo bez ischemie (n=300) léčených ranibizumabem v monoterapii byla průměrná hodnota změny BCVA od výchozího stavu +15,0; resp. +11,5 písmen.
V obou studiích BRIGHTER a CRYSTAL byl účinek na zlepšení zraku pozorován u všech pacientů léčených 0,5 mg ranibizumabu v monoterapii bez ohledu na délku trvání onemocnění. U pacientů
s délkou trvání onemocnění <3 měsíce byl pozorován nárůst zrakové ostrosti o 13,3; resp. 10,0 písmen v 1. měsíci a o 17,7; resp. 13,2 písmen ve 24. měsíci ve studiích BRIGHTER a CRYSTAL. Odpovídající zisk zrakové ostrosti u pacientů s délkou trvání onemocnění >12 měsíců byl v těchto studiích 8,6; resp. 8,4 písmen. Je třeba zvážit zahájení léčby v čase stanovení diagnózy.
Dlouhodobý bezpečnostní profil ranibizumabu pozorovaný ve 24měsíčních studiích je konzistentní se známým bezpečnostním profilem přípravku Lucentis.
Léčba poškození zraku způsobeného CNV sekundární k PM
Klinická bezpečnost a účinnost přípravku Lucentis u pacientů s poškozením zraku způsobeným CNV u PM byla hodnocena na základě 12měsíčních dat z dvojitě zaslepené, kontrolované pivotní studie F2301 (RADIANCE). V této studii bylo 277 pacientů randomizováno v poměru 2:2:1 do následujících ramen:
• Skupina I (ranibizumab 0,5 mg, dávkovací schéma řízené kritérii „stability“, definovanými jako žádná změna BCVA v porovnání se dvěma předchozími měsíčními vyhodnoceními).
• Skupina II (ranibizumab 0,5 mg, dávkovací schéma řízené kritérii „aktivity onemocnění“, definovanými jako poškození zraku způsobené intra- nebo subretinální tekutinou nebo aktivním prosakováním v důsledku CNV léze, jak bylo zjištěno při vyšetření OCT a/nebo FA).
• Skupina III (vPDT - pacientům bylo povoleno užívat léčbu ranibizumabem od
3. měsíce studie).
Ve skupině II, což je doporučené dávkovací schéma (viz bod 4.2), vyžadovalo 50,9 % pacientů 1 nebo 2 injekce, 34,5 % pacientů 3 až 5 injekcí a 14,7 % vyžadovalo 6 až 12 injekcí po dobu 12 měsíců trvání studie. 62,9 % pacientů ze skupiny II nevyžadovalo injekce ve druhé polovině trvání studie.
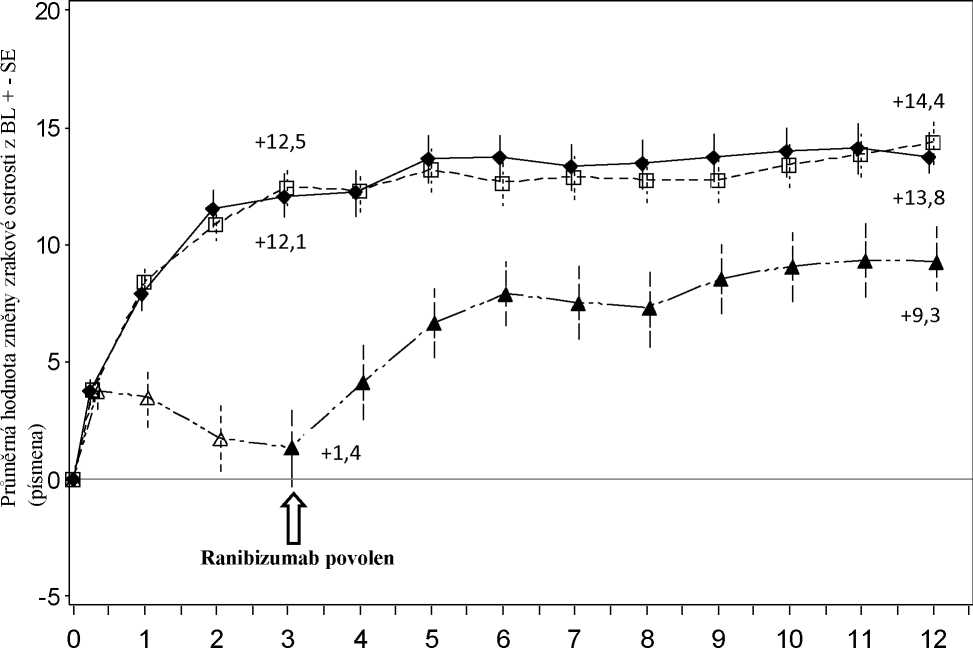
Důležité výstupy ze studie RADIANCE jsou uvedeny v Tabulce 6 a na Obrázku 5.
Tabulka 6 Výsledky ve 3. a 12. měsíci studie (RADIANCE)
|
Skupina I |
Skupina II |
Skupina | |
|
Ranibizumab |
Ranibizumab |
III | |
|
0,5 mg |
0,5 mg |
vPDTb | |
|
„stabilizace |
„aktivita | ||
|
vidění“ |
onemocnění“ | ||
|
(n = 105) |
(n = 116) |
(n = 55) | |
|
3. měsíc Průměr určený z průměrných hodnot změn BCVA od 1. do 3. měsíce studie v porovnání se základním stavema (písmena) Podíl pacientů, kteří dosáhli: >15 písmen, nebo dosáhli >84 písmen |
+10,5 |
+10,6 |
+2,2 |
|
BCVA 12. měsíc |
38,1 % |
43,1 % |
14,5 % |
|
Počet injekcí až do 12. měsíce: Průměrná hodnota |
4,6 |
3,5 |
N/A |
|
Medián |
4,0 |
2,5 |
N/A |
|
Průměr určený z průměrných hodnot změn BCVA od 1. do 12. měsíce v porovnání se základním stavem (písmena) Podíl pacientů, kteří dosáhli: |
+12,8 |
+12,5 |
N/A |
|
>15 písmen, nebo dosáhli >84 písmen BCVA |
53,3 % |
51,7 % |
N/A |
a p <0,00001 porovnání s vPDT kontrolní skupinou
b Srovnávací kontrola až do 3. měsíce studie. Pacientům randomizovaným do skupiny s vPDT bylo povoleno dostávat léčbu ranibizumabem od 3. měsíce studie (ve skupině III dostávalo ranibizumab 38 pacientů od 3. měsíce studie)
Průměrná hodnota změny BCVA od začátku léčby v průběhu času do 12. měsíce (RADIANCE)
Měsíc
___ Ranibizumab 0,5 mg Skupina I Ranibizumab 0,5 mg Skupina II
^ ^ ^ EI-EHD v i • • v v r
při aktivitě onemocněni (n = 116)
Ranibizumab 0,5 mg/PDT verteporfinem Skupina III od 3. měsíce studie dále (n = 55)
pn stabilizaci (n = 105)
a-a-a PDT verteporfinem Skupina III(n = 55)
Zlepšeni zraku bylo doprovázeno redukci tloušťky centrální části sítnice.
Prospěch hlášený pacientem byl pozorován v léčebných ramenech s ranibizumabem oproti vPDT (hodnota p <0,05) na základě zlepšení kombinovaného skóre a několika subškál (všeobecné vidění, aktivity do blízka, duševní zdraví a závislost) v NEI VFQ-25.
Pediatrická populace
Bezpečnost a účinnost ranibizumabu nebyla u pediatrických pacientů dosud studována.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lucentis u všech podskupin pediatrické populace u neovaskulární formy AMD, u poškození zraku kvůli DME, u poškození zraku kvůli makulárnímu edému v důsledku RVO a u poškození zraku v důsledku CNV sekundární k PM (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Po měsíčních aplikacích Lucentisu do sklivce jedincům s neovaskulámí AMD byly koncentrace ranibizumabu v séru obvykle nízké. Maximální koncentrace (Cmax) ranibizumabu byly obecně nižší než koncentrace ranibizumabu nutné k inhibici biologické aktivity VEGF o 50 % (11 - 27 ng/ml, stanoveno in vitro titrací buněčné proliferace). Cmax byla závislá na dávce v dávkovém rozmezí 0,05 až
1.0 mg/oko. Sérové koncentrace u limitovaného počtu pacientů s DME ukazují, že lehce vyšší systémová expozice nemůže být vyloučena ve srovnání se sérovými koncentracemi zjištěnými
u pacientů s neovaskulámí AMD. Sérové koncentrace ranibizumabu u pacientů s RVO byly podobné nebo lehce vyšší v porovnání s koncentracemi pozorovanými u pacientů s neovaskulámí AMD.
Na základě analýz populační farmakokinetiky a vymizení ranibizumabu ze séra pacientů s neovaskulámí AMD léčených dávkou 0,5 mg byl průměrný poločas eliminace ranibizumabu ze sklivce stanoven na přibližně 9 dnů. Při aplikaci Lucentisu v dávce 0,5 mg/oko jednou měsíčně je dosaženo sérové Cmax ranibizumabu, jejíž předpokládané rozmezí je mezi 0,79 a 2,90 ng/ml přibližně za 1 den po aplikaci. Cmin se předpokládá v rozmezí mezi 0,07 a 0,49 ng/l. Sérové koncentrace ranibizumabu se předpokládají přibližně 90 000násobně nižší než koncentrace ranibizumabu ve sklivci.
Pacienti se zhoršenou funkcí ledvin: Žádné cílené studie, které by hodnotily farmakokinetiku Lucentisu u pacientů se zhoršenou funkcí ledvin, nebyly provedeny. 68 % (136 ze 200) pacientů s neovaskulámí AMD mělo v populační farmakokinetické analýze zhoršenou funkci ledvin (46,5 % mírně [50 - 80 ml/min], 20 % středně [30 - 50 ml/min] a 1,5 % silně [< 30 ml/min]). 48,2 % (253 z 525) pacientů s RVO mělo zhoršenou funkci ledvin (36,4 % mírně, 9,5 % středně a 2,3 % silně). Systémová clearance byla lehce snížena, ale tento nález byl bez klinického významu.
Zhoršená funkce jater: Žádné cílené studie, které by hodnotily farmakokinetiku u pacientů se zhoršenou funkcí jater léčených Lucentisem, nebyly provedeny.
5.3 Předklinické údaje vztahující se k bezpečnosti
Oboustranná aplikace ranibizumabu do sklivce opicím cynomolgus v dávkách 0,25 mg/oko a
2.0 mg/oko jednou za dva týdny po dobu 26 týdnů vyvolala účinky na očích závislé na dávce.
Nitrooční aplikace vyvolala na dávce závislé zvýšené zarudnutí přední komory a zvýšení počtu buněk, největší za 2 dny po injekci. Závažnost zánětlivé odpovědi se obvykle zmenšila po následujících injekcích nebo během zotavení. V zadním segmentu byla pozorována infiltrace sklivce buňkami a sklivcovými vločkami, u které byla také tendence k závislosti na dávce a obvykle přetrvávala až do konce léčby. Ve 26týdenní studii se intenzita zánětu sklivce zvyšovala s počtem injekcí. Průkaz reverzibility byl však pozorován po zotavení. Povaha a načasování zánětu zadního segmentu naznačují, že se jedná o imunologicky zprostředkovanou protilátkovou odpověď, která by mohla být klinicky irelevantní. U některých zvířat byla po relativně dlouhém období intenzivního zánětu pozorována tvorba katarakty naznačující, že změny čočky byly sekundární, vyvolané silným zánětem. Bylo pozorováno přechodné zvýšení nitroočního tlaku po podání do sklivce bez ohledu na dávku.
Mikroskopické změny pozorované v oční tkáni souvisely se zánětem a nenasvědčovaly degenerativním procesům. U některých očí byly pozorovány granulomatózní zánětlivé změny na očním disku. Tyto změny zadního segmentu slábly, a v některých případech kompletně vymizely, během období zotavení.
Po podání do sklivce nebyly zjištěny žádné známky systémové toxicity. U některých zvířat byly nalezeny protilátky proti ranimizumabu v séru i sklivci.
Nejsou dostupné žádné údaje o karcinogenitě nebo mutagenitě.
U březích opic nevyvolala léčba ranibizumabem aplikovaná do sklivce , mající za následek maximální systémové expozice 0,9-7-krát nejhorší případ klinické expozice, vývojovou toxicitu ani teratogenitu, a neměla žádný vliv na hmotnost nebo strukturu placenty, i když na základě svého farmakologického účinku by měl být ranibizumab považován za potenciálně teratogenní a embryo-/fetotoxický.
Chybění účinků zprostředkovaných ranibizumabem na embryo-fetální vývoj se pravděpodobně týká hlavně neschopnosti Fab-fragmentu procházet placentou. Přesto byl popsán případ s vysokými mateřskými sérovými hladinami ranibizumabu a přítomností ranibizumabu ve fetálním séru, nasvědčující tomu, že protilátky proti ranibizumabu působily jako nosičský protein (obsahující Fc část) pro ranibizumab, a tím snižovaly jeho mateřskou sérovou clearance a umožňovaly prostup placentou. Protože embryo-fetální vývojová vyšetření byla prováděna u zdravých březích zvířat a onemocnění (jako například diabetes) mohou modifikovat prostupnost placenty pro Fab-fragment, měla by být studie interpretována s opatrností.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát a,a-trehalosy
Monohydrát histidin-hydrochloridu
Histidin
Polysorbát 20
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Před použitím může být neotevřená injekční lahvička ponechána při pokojové teplotě (25 °C) po dobu 24 hodin.
6.5 Druh obalu a obsah balení
0,23 ml sterilního injekčního roztoku v injekční lahvičce (sklo typu I) se zátkou (chlorbutylová pryž),
1 tupou jehlou s filtrem (18G x P/2", 1,2 mm x 40 mm, 5 pm), 1 injekční jehlou (30G x 'A”,
0,3 mm x 13 mm) a 1 stříkačkou (polypropylen) (1 ml). Balení obsahuje 1 injekční lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Injekční lahvička, injekční jehla, jehla s filtrem a injekční stříkačka jsou pro jednorázové použití.
Opakované použití může vést k infekci nebo jinému onemocnění/poškození. Všechny komponenty
jsou sterilní. Jakákoliv komponenta s obalem vykazující známky poškození nebo manipulace nesmí
být použita. Sterilita nemůže být zaručena, pokud nezůstane uzavření obalu komponenty neporušené.
Při přípravě přípravku Lucentis k podání do sklivce dbejte, prosím, následujících pokynů:
1. Před nasátím tekutiny do inj ekční stříkačky j e nutno desinfikovat vněj ší část pryžové zátky.
2. Jehlu s 5 pm filtrem (18G x 1%'', 1,2 mm x 40 mm, přiložena) nasaďte na 1 ml injekční stříkačku (přiložena) za použití aseptického postupu. Zasuňte jehlu s filtrem do středu zátky lahvičky, dokud se jehla nedotkne dna injekční lahvičky.
3. Nasajte veškerou tekutinu z injekční lahvičky držené ve svislé, lehce nakloněné poloze pro snadnější úplné nasátí.
4. Ujistěte se, že píst je při vyprazdňování injekční lahvičky vytažen dostatečně daleko tak, aby jehla s filtrem byla úplně vyprázdněna.
5. Jehlu s filtrem ponechte v lahvičce a stříkačku od ní odpojte. Jehla s filtrem by měla být po násátí veškerého obsahu lahvičky zlikvidována a nesmí být použita pro vlastní aplikaci do sklivce.
6. Asepticky a pevně nasaďte injekční jehlu (30G x ‘A", 0,3 mm x 13 mm, přiložena) na injekční stříkačku.
7. Opatrně odstraňte kryt z injekční jehly, aniž byste injekční jehlu odpojili od injekční stříkačky. Poznámka: Při odstraňování krytu pevně uchopte žlutou část injekční jehly.
8. Pečlivě vytlačte vzduch ze stříkačky a upravte dávku ke značce 0,05 ml uvedené na injekční stříkačce. Nyní je injekční stříkačka připravena k aplikaci.
Poznámka: Neotírejte injekční jehlu. Nevytahujte píst zpět.
Po podání injekce nenasazujte zpět kryt jehly ani jehlu neoddělujte od injekční stříkačky. Zlikvidujte použitou injekční stříkačku společně s jehlou vyhozením do nádoby na ostré předměty nebo v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/06/374/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 22. ledna 2007
Datum posledního prodloužení registrace: 24. ledna 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Lucentis 10 mg/ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje ranibizumabum 10 mg*. Jedna injekční lahvička obsahuje ranibizumabum 2,3 mg v 0,23 ml roztoku.
*Ranibizumab je fragment humanizované monoklonální protilátky produkovaný buňkami Escherichia coli rekombinantní DNA technologií.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok
Čirý, bezbarvý až světle žlutý vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Lucentis je indikován u dospělých:
• k léčbě neovaskulární (vlhké) formy věkem podmíněné makulární degenerace (AMD)
• k léčbě poškození zraku způsobeného diabetickým makulárním edémem (DME)
• k léčbě poškození zraku způsobeného makulárním edémem v důsledku okluze retinální vény [uzávěr větve centrální retinální vény (BRVO) a uzávěr centrální retinální vény (CRVO)]
• k léčbě poškození zraku způsobeného choroidální neovaskularizací (CNV) sekundární k patologické myopii (PM)
4.2 Dávkování a způsob podání
Lucentis musí být aplikován kvalifikovaným oftalmologem zkušeným v podání do sklivce.
Doporučená dávka Lucentisu je 0,5 mg podávaných jako jednorázová injekce do sklivce. To odpovídá injekci o objemu 0,05 ml. Interval mezi dvěma dávkami podávanými do stejného oka má být alespoň čtyři týdny.
Léčba se zahajuje jednou injekcí za měsíc do dosažení maximální zrakové ostrosti a/nebo do vymizení příznaků aktivity onemocnění, tj. žádná změna zrakové ostrosti a ostatních příznaků a projevů onemocnění při probíhající léčbě. U pacientů s vlhkou formou AMD, DME a RVO mohou být zpočátku potřeba tři nebo více po sobě jdoucí injekce podávané jednou za měsíc.
Následně mají být lékařem určeny intervaly sledování a léčby a mají být stanoveny na základě aktivity onemocnění vyhodnocené podle zrakové ostrosti a/nebo anatomických parametrů.
Pokud zrakové a anatomické parametry ukazují na základě vyjádření lékaře, že pokračující léčba pacienta není přínosná, je třeba léčbu přípravkem Lucentis ukončit.
Sledování aktivity onemocnění může zahrnovat klinické vyšetření, funkční testy nebo zobrazovací techniky (např. optickou koherenční tomografii nebo fluorescenční angiografii).
Pokud j sou pacienti léčeni podle režimu „treat-and-extend“, pak po dosažení maximální zrakové ostrosti a/nebo vymizení příznaků aktivity onemocnění, mohou být léčebné intervaly postupně prodlouženy až do opětovného objevení se příznaků aktivity onemocnění nebo zhoršení zraku. Léčebný interval nemá být prodloužen o více než dva týdny najednou u vlhké formy AMD a může být prodloužen až o jeden měsíc najednou u DME. Při uzávěru retinální vény mohou být léčebné intervaly také postupně prodlužovány, ale nejsou dostupná dostatečná data dokládající délku těchto intervalů. Pokud se aktivita onemocnění znovu objeví, léčebný interval má být příslušně zkrácen.
V léčbě poškození zraku způsobeném CNV sekundární k PM může mnoho pacientů potřebovat pouze jednu nebo dvě injekce během prvního roku, zatímco někteří pacienti mohou potřebovat častější léčbu (viz bod 5.1).
Lucentis a laserová fotokoagulace u DME a makulárního edému v důsledku BRVO Existuje určitá zkušenost s podáním přípravku Lucentis společně s laserovou fotokoagulací (viz bod 5.1). Při podání ve stejný den by měl být Lucentis podán alespoň 30 minut po laserové fotokoagulaci. Lucentis může být podán pacientům, kteří byli léčeni předchozí laserovou fotokoagulací.
Lucentis a fotodynamická léčba Visudynem u CNV sekundární k PM Se souběžným podáváním přípravků Lucentis a Visudyne nejsou žádné zkušenosti.
Zvláštní populace
Zhoršená funkce jater
Účinky přípravku Lucentis u pacientů se zhoršenou funkcí jater nebyly hodnoceny. Pro tuto populaci však nejsou nutná žádná zvláštní opatření.
Zhoršená funkce ledvin
U pacientů se zhoršenou funkcí ledvin není nutné upravovat dávku (viz bod 5.2).
Starší jedinci
U starších jedinců není nutná úprava dávky. U pacientů s DME starších 75 let jsou omezené zkušenosti.
Pediatrická populace
Bezpečnost a účinnost Lucentisu u dětí a dospívajících pod 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Injekční lahvička na jedno použití, pouze pro podání do sklivce.
Před aplikací je nutno Lucentis vizuálně zkontrolovat, zda neobsahuje cizí částice nebo není změněna jeho barva.
Lucentis musí být aplikován za aseptických podmínek, což zahrnuje použití chirurgické desinfekce rukou, sterilních rukavic, sterilního oděvu, sterilního spekula (nebo ekvivalentní náhrady) a dostupnost sterilní paracentézy (je-li potřeba). Před aplikací injekce do sklivce je nutný pečlivý odběr anamnézy z hlediska hypersenzitivních reakcí (viz bod 4.4). Před aplikací injekce musí být podána adekvátní anestezie a použit širokospektrý lokální antimikrobiální přípravek k dezinfekci pokožky kolem očí, očního víčka a povrchu oka, v souladu s lokální praxí.
Pro informace o přípravě Lucentisu viz bod 6.6.
Injekční jehla se zasune 3,5-4,0 mm posteriomě od limbu do prostoru sklivce tak, aby směřovala do centra očního bulbu a nikoli k horizontálnímu meridiánu. Poté se aplikuje objem injekce 0,05 ml. Následující injekci je nutné aplikovat v jiném místě skléry.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Pacienti s aktivní nebo suspektní oční nebo periokulární infekcí.
Pacienti s aktivním těžkým nitroočním zánětem.
4.4 Zvláštní upozornění a opatření pro použití Reakce po podání injekce do sklivce
Podání do sklivce, včetně těch s Lucentisem, byly spojovány s endoftalmitidou, intraokulárním zánětem, rhegmatogenním odchlípením sítnice, trhlinami sítnice a iatrogenní traumatickou kataraktou (viz bod 4.8). Při aplikaci Lucentisu musí být vždy dodržena přísná pravidla asepse. V následujícím týdnu po aplikaci injekce musejí být pacienti sledováni z hlediska případného výskytu infekce, aby bylo možné zahájit včas adekvátní léčbu. Pacienty je nutno upozornit, že musejí ihned hlásit všechny příznaky možné endoftalmitidy nebo jiných výše popsaných komplikací.
Zvýšení nitroočního tlaku
Během 60 minut po injekci Lucentisu bylo pozorováno přechodné zvýšení nitroočního tlaku (IOP). Trvalá zvýšení IOP byla také zjištěna (viz bod 4.8). Je nutné monitorovat a náležitě ošetřit jak nitrooční tlak, tak i perfuzi papily očního nervu.
Léčba obou očí současně
Omezená data k léčbě obou očí přípravkem Lucentis současně (včetně podání ve stejný den) nenaznačují zvýšené riziko systémových nežádoucích účinků v porovnání s léčbou jednoho oka.
Imunogenicita
U Lucentisu existuje možnost vzniku imunogenicity. Protože existuje potenciál pro zvýšenou systémovou expozici u pacientů s DME, zvýšené riziko pro vznik hypersenzitivity u této pacientské populace nelze vyloučit. Pacienti musejí být také poučeni, aby hlásili zhoršení závažnosti nitroočního zánětu, protože se může jednat o klinický příznak charakteristický pro tvorbu nitroočních protilátek.
Současné použití jiných anti-VEGF (vaskulární endoteliální růstový faktor) léčivých přípravků Lucentis se nesmí podávat zároveň s jinými anti-VEGF léčivými přípravky (systémovými nebo očními).
Vynechání dávky Lucentisu
Dávku je nutno vynechat a v léčbě se nesmí pokračovat dříve, než je plánována další dávka v následujících případech:
• snížení nejlépe upravené ostrosti zraku (best-corrected visual acuity BCVA) o > 30 písmen ve srovnání s předchozím měřením ostrosti zraku;
• nitrooční tlak > 30 mmHg;
• poškození sítnice;
nebo je-li velikost hemoragie > 50 % celkové zákrok během uplynulých nebo následujících
• subretinální krvácení zahrnující střed fovey, plochy léze;
• provedený nebo plánovaný oční chirurgický 28 dnů.
Trhlina pigmentového epitelu sítnice
Rizikové faktory spojené s vývojem trhliny pigmentového epitelu sítnice po podání anti-VEGF léčby u vlhké formy AMD zahrnují rozsáhlé a/nebo značné odchlípení pigmentového epitelu sítnice.
U pacientů s těmito rizikovými faktory pro vznik trhlin pigmentového epitelu sítnice je třeba dbát opatrnosti při zahajování léčby Lucentisem.
Rhegmatogenní odchlípení sítnice nebo makulární díry
Léčbu je nutno přerušit u subjektů s rhegmatogenním odchlípením sítnice nebo u makulárních děr stupně 3 nebo 4.
Populace s omezenými daty
Existují pouze omezené zkušenosti s léčbou u pacientů s DME způsobeným diabetem I. typu. Lucentis nebyl studován u pacientů, kterým byla dříve podána injekce do sklivce, u pacientů s aktivními systémovými infekcemi, proliferativní diabetickou retinopatií nebo u pacientů se současnými očními onemocněními jako například odchlípení sítnice nebo makulární díra. Také nejsou zkušenosti s léčbou Lucentisem u diabetických pacientů s HbA1c nad 12 % a nekontrolovanou hypertenzí. Chybění těchto informací má být lékařem zváženo při léčbě takových pacientů.
Nejsou k dispozici dostatečné údaje k posouzení účinnosti Lucentisu u pacientů s RVO projevujícím se ireverzibilní ztrátou zraku v důsledku ischemie.
U pacientů s PM, kteří v minulosti podstoupili neúspěšnou fotodynamickou léčbu verteporfinem (vPDT) jsou k dispozici limitovaná data o účinku přípravku Lucentis. Rovněž zatímco byl pozorován odpovídající účinek u subjektů se subfoveálními a juxtafoveálními lézemi, jsou data k vyvození závěru o účinku přípravku Lucentis u subjektů s patologickou myopií s extrafoveálními lézemi nedostatečná.
Systémové účinky po podání do sklivce
Po injekční aplikaci VEGF inhibitorů do sklivce byly hlášeny systémové nežádoucí příhody včetně mimoočních krvácení a arteriálních tromboembolických příhod.
Data o bezpečnosti při léčbě pacientů s DME, makulárním edémem způsobeným RVO a CNV sekundární k patologické myopii (PM) u pacientů s mrtvicí nebo přechodnými ischemickými příhodami v anamnéze jsou omezená. U těchto pacientů je třeba dbát zvýšené opatrnosti (viz bod 4.8).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí.
Současné použití Lucentisu s fotodynamickou léčbou (PDT) verteporfinem u vlhké formy AMD a PM, viz bod 5.1.
Současné použití Lucentisu s laserovou fotokoagulací u DME a BRVO, viz body 4.2 a 5.1.
V klinických studiích zaměřených na léčbu poškození zraku způsobeného DME nebyl výsledek s ohledem na zrakovou ostrost nebo tloušťku centrální části sítnice (CSFT) u pacientů léčených přípravkem Lucentis ovlivněn současnou léčbou thiazolidindiony.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/antikoncepce u žen
Ženy ve fertilním věku musí během léčby používat účinnou antikoncepci.
Žádné údaje o podávání ranibizumabu těhotným ženám nejsou k dispozici. Studie u makaků rodu Cynomolgus nenaznačují přímé nebo nepřímé škodlivé účinky s ohledem na těhotenství nebo embryonální/fetální vývoj (viz bod 5.3). Po očním podání je nízká systémová expozice ranibizumabu, ale vzhledem k jeho mechanismu účinku je nutno ranibizumab považovat za potenciálně teratogenní a embryo-/fetotoxický. Z tohoto důvodu nesmí být ranibizumab užíván během těhotenství, aniž by očekávaný přínos převážil možné riziko pro plod. Ženám, které chtějí otěhotnět, a které byly léčeny ranibizumabem, je doporučeno vyčkat nejméně 3 měsíce po poslední dávce ranibizumabu před početím dítěte.
Kojení
Není známo, zda se Lucentis vylučuje do lidského mateřského mléka. Během léčby přípravkem Lucentis se kojení nedoporučuje.
Fertilita
Nejsou k dispozici žádné údaje vztahující se k fertilitě.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Léčba Lucentisem může vyvolat dočasné zhoršení zraku, což může ovlivnit schopnost řídit nebo obsluhovat stroje (viz bod 4.8). Pacienti, u kterých se tyto příznaky vyskytnou, nesmějí řídit nebo obsluhovat stroje, dokud tyto poruchy zraku neustoupí.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Většina nežádoucích účinků hlášených po podání Lucentisu se týká postupu podání injekce do sklivce.
Nejčastěji hlášené oční nežádoucí účinky po podání injekce Lucentisu jsou: bolest oka, oční hyperemie, zvýšený nitrooční tlak, vitritida, odloučení sklivce, hemoragie sítnice, poruchy zraku, sklivcové vločky, hemoragie spojivky, podráždění oka, pocit cizího tělesa, zvýšené slzení, zánět očního víčka, suchost oka a svědění oka.
Nejčastěji hlášené nežádoucí účinky mimo oko jsou: bolest hlavy, nazofaryngitida a artralgie.
Méně často hlášené, ale závažnější nežádoucí účinky zahrnují: endoftalmitidu, slepotu, odchlípení sítnice, trhlinu sítnice a iatrogenní traumatickou kataraktu (viz bod 4.4).
Pacienti mají být informováni o příznacích těchto potenciálních nežádoucích účinků a poučeni informovat svého lékaře, pokud se u nich objeví příznaky, jako je bolest oka nebo zvýšený oční diskomfort, zhoršující se zarudnutí oka, rozmazané nebo snížené vidění, zvýšený počet malých částic v zorném poli nebo zvýšená citlivost na světlo.
Nežádoucí účinky, které se objevily po podání Lucentisu v klinických studiích jsou uvedeny v tabulce níže.
Tabulkový seznam nežádoucích účinků#
Nežádoucí účinky jsou uvedeny podle systémově-orgánových skupin a frekvence za použití následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Infekce a infestace
Velmi časté Nazofaryngitida
Časté Infekce močových cest*
Poruchy krve a lymfatického systému
Časté Anemie
Poruchy imunitního systému
Časté Hypersenzitivita
Psychiatrické poruchy
Časté Úzkost
Poruchy nervového systému
Velmi časté Bolest hlavy
Poruchy oka Velmi časté
Časté
Méně časté
Vitritida, odloučení sklivce, hemoragie sítnice, poruchy zraku, bolest oka, sklivcové vločky, hemoragie spojivky, podráždění oka, pocit cizího tělesa, zvýšené slzení, zánět očního víčka, suchost oka, oční hyperemie, svědění oka.
Degenerace sítnice, poškození sítnice, odloučení sítnice, trhlina sítnice, odloučení pigmentového epitelu sítnice, trhlina v pigmentovém epitelu sítnice, snížení ostrosti zraku, hemoragie sklivce, poškození sklivce, uveitida, zánět duhovky, iridocyklitida, katarakta, subkapsulární katarakta, opacifikace zadního pouzdra, keratitis punctata, abraze rohovky, zarudnutí v přední části komory, rozmazané vidění, hemoragie v místě injekce, oční hemoragie, zánět spojivky, alergický zánět spojivky, výtok z oka, fotopsie, fotofobie, oční dyskomfort, otok víčka, bolestivost víčka, překrvení spojivek.
Slepota, endoftalmitida, hypopyon, krvácení do přední komory oka, keratopatie, adheze duhovky, korneální deposita, edém rohovky, strie rohovky, bolestivost v místě injekce, podráždění v místě injekce, abnormální pocit v oku, podráždění očního víčka.
Respirační, hrudní a mediastinální poruchy Časté Kašel
Gastrointestinální poruchy
Časté Nauzea
Poruchy kůže a podkožní tkáně
Alergické reakce (vyrážka, kopřivka, pruritus, erytém)
Časté
Poruchy svalové a kosterní soustavy a pojivové tkáně Velmi časté Artralgie
Vyšetření
Velmi časté Zvýšení nitroočního tlaku
# Nežádoucí účinky byly definovány jako nežádoucí příhody (u nejméně 0,5 procentních bodů pacientů), které se objevily ve vyšší míře (alespoň 2 procentní body) u pacientů léčených Lucentisem 0,5 mg než u těch, kteří byli léčeni v kontrolním rameni (simulovanou léčbou nebo fotodynamickou léčbou verteporfinem).
* pozorované pouze u populace s DME
Nežádoucí účinky související s třídou přípravku
Ve studiích fáze III s vlhkou formou AMD se mírně zvýšil celkový výskyt mimoočních krvácení u pacientů léčených ranibizumabem, což je nežádoucí účinek, který potenciálně souvisí se systémovou inhibicí VEGF (vaskulární endoteliální růstový faktor). Nicméně, jednotlivá krvácení neměla shodný charakter. Po intravitreálním podání VEGF inhibitorů existuje teoretické riziko arteriální tromboembolické příhody, včetně mrtvice a infarktu myokardu. V klinických studiích s Lucentisem byla u pacientů s AMD, DME, RVO a PM pozorována nízká incidence arteriálních tromboembolických příhod a mezi skupinami léčenými ranibizumabem nebyly ve srovnání s kontrolou významné rozdíly.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Případy předávkování byly hlášeny z klinických studií s vlhkou formou AMD a postmarketingového sledování. Nejčastěji hlášené případy nežádoucích účinků byly zvýšení nitroočního tlaku, přechodná slepota, snížená zraková ostrost, otok rohovky, bolest rohovky a bolest oka. Dojde-li k předávkování, je nutno monitorovat nitrooční tlak a v závislosti na rozhodnutí ošetřujícího lékaře případně nasadit odpovídající terapii k normalizaci nitroočního tlaku.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, látky určené k léčbě neovaskularizace, ATC kód: S01LA04
Ranibizumab je fragment humanizované monoklonální protilátky proti lidskému cévnímu endoteliálnímu růstovému faktoru A (VEGF-A). Váže se se silnou afinitou na VEGF-A isoformy (např. VEGF110, VEGF121 a VEGF165) a tím brání vazbě VEGF-A na receptory VEGFR-1 a VEGFR-2. Vazba VEGF-A na jeho receptory vede k proliferaci endoteliálních buněk a neovaskularizaci, jakož i k propustnosti cév. O všech těchto účincích se uvažuje jako o faktorech přispívajících k progresi neovaskulárních forem věkem podmíněné makulární degenerace, patologické myopie nebo k poškození zraku způsobeného buď diabetickým makulárním edémem, anebo makulárním edémem v důsledku RVO.
Léčba vlhké formy AMD
Klinická bezpečnost a účinnost Lucentisu byla u vlhké formy AMD stanovena ve třech randomizovaných, dvojitě zaslepených studiích po dobu 24 měsíců se simulovanou injekcí nebo aktivním komparátorem u pacientů s neovaskulární AMD. Do dvou studií bylo zařazeno celkem 1 323 pacientů (879 na aktivní léčbě, 444 v kontrolní skupině).
Ve studii FVF2598g (MARINA) bylo 716 pacientů s minimálně klasickými nebo okultními lézemi randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali měsíčně injekce Lucentisu v dávkách 0,3 mg, injekce Lucentisu v dávkách 0,5 mg nebo simulovanou léčbu.
Ve studii FVF2587g (ANCHOR) bylo 423 pacientů s převážně klasickou CNV lézí randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali Lucentis 0,3 mg měsíčně, Lucentis 0,5 mg měsíčněnebo PDT s verteporfinem (při zahájení léčby a potom každé 3 měsíce, pokud fluoresceinová angiografie prokázala přetrvávání nebo rekurenci cévního prosakování).
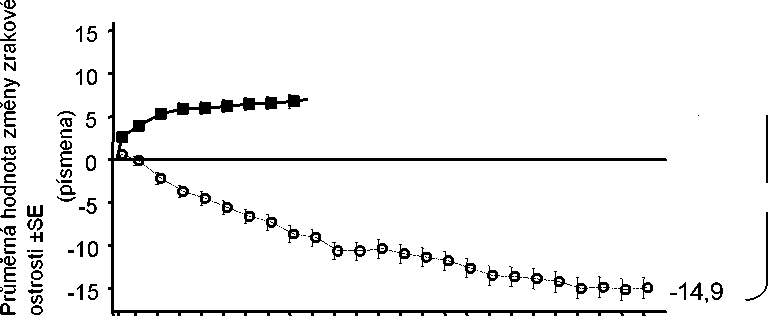
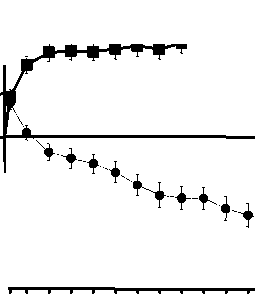
Nejdůležitější výsledky měření jsou shrnuty v Tabulce 1 a na Obrázku 1.
Tabulka 1 Výsledky ve 12. a 24. měsíci studie FVF2598g (MARINA) a FVF2587g (ANCHOR)
|
FVF2598g (MAR |
ŘINA) |
FVF2587g (ANCHOR) | |||
|
Měřený parametr |
Měsíc |
Simulovaná léčba (n = 238) |
Lucentis 0,5 mg (n = 240) |
Verteporfin PDT (n = 143) |
Lucentis 0,5 mg (n = 140) |
|
Ztráta <15 písmen ostrosti zraku (%)a (zachování zraku, primární cíl) |
12. měsíc |
62 % |
95 % |
64 % |
96 % |
|
24. měsíc |
53 % |
90 % |
66 % |
90 % | |
|
Nárůst >15 písmen ostrosti zraku (%)a |
12. měsíc |
5 % |
34 % |
6 % |
40 % |
|
24. měsíc |
4 % |
33 % |
6 % |
41 % | |
|
Průměrná hodnota změny zrakové ostrosti (písmena) (SD)a |
12. měsíc |
-10,5 (16,6) |
+ 7,2 (14,4) |
-9,5 (16,4) |
+11,3 (14,6) |
|
24. měsíc |
-14,9 (18,7) |
+6,6 (16,5) |
-9,8 (17,6) |
+10,7 (16,5) | |
p<0.01
Obrázek 1 Průměrná hodnota změny zrakové ostrosti od výchozího stavu do 24. měsíce ve studii FVF2598g (MARINA) a ve studii FVF2587g (ANCHOR)
Studie FVF2598g (MARINA)
+6,6 'n
y +21,5
0 2 4 6 8 10 12 14 16 18 20 22 24
Měsíc
-CD
>
O
co
i_
N
>s
C
><D
E
N
CO
«
O
c
"O
o
-co
LU
co
+1
*2 oo
ol 2
Jr 1/5 Q_ o
15 ' 10 ■ 5
c
CD
0
V)
'cl
~-5 ■ -10 ■ -15 ■ 0
2 4 6 8
r +20,5
10 12 14 16 18 20 22 24 Měsíc
MARINA
LUCENTIS 0,5 mg (n=240) & Simulace (n=238)
ANCHOR
«- LUCENTIS 0,5 mg (n=140) • Verteporfin PDT (n=143)
Výsledky obou studií naznačují, že kontinuální léčba ranibizumabem může být také přínosem u pacientů, kteří v prvním roce léčby ztratili >15 písmen z nejlépe korigované ostrosti zraku (BCVA).
Statisticky významná zlepšení zrakových funkcí, hlášená pacienty, byla pozorována v obou studiích MARINA i ANCHOR při léčbě ranibizumabem oproti kontrolní skupině měřeno pomocí NEI VFQ-25.
Ve studii FVF192g (PIER) bylo 184 pacientů se všemi formami neovaskulámí AMD randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali Lucentis 0,3 mg, Lucentis 0,5 mg nebo simulovanou léčbu v injekci jednou měsíčně po první 3 měsíce a dále dávku podávanou každý 3. měsíc. Od 14. měsíce této studie bylo pacientům se simulovanou léčbou povoleno používat ranibizumab a od 19. měsíce byla možná častější aplikace. V průměru obdrželi pacienti léčení Lucentisem ve studii PIER 10 aplikací.
Po počátečním nárůstu zrakové ostrosti (při dávkování jednou měsíčně) poklesla zraková ostrost u pacientů kteří dostávali Lucentis jednou za tři měsíce a vracela se ve 12. měsíci v průměru k počátečnímu stavu; tento účinek byl udržován ve 24. měsíci u většiny pacientů léčených ranibizumabem (82 %). Omezené údaje od osob se simulovanou léčbou, které později dostávaly ranibizumab naznačují, že předčasné zahájení léčby může být spojováno s lepším zachováním ostrosti zraku.
Data ze dvou studií (MONT BLANC, BPD952A2308 a DENALI, BPD952A2309) provedených po registraci přípravku Lucentis potvrdila jeho účinnost, ale neprokázala dodatečný účinek kombinovaného podávání verteporfinu (Visudyne PDT) a Lucentisu v porovnání s monoterapií Lucentisem.
Léčba poškození zraku způsobeného DME
Bezpečnost a účinnost Lucentisu byly hodnoceny ve třech randomizovanných kontrolovaných studiích po dobu alespoň 12 měsíců. Celkem 868 pacientů (708 aktivních a 160 kontrol) bylo zařazeno v těchto studiích.
Ve studii 2. fáze D2201 (RESOLVE) bylo léčeno 151 pacientů ranibizumabem (6 mg/ml, n = 51,
10 mg/ml, n = 51) nebo simulovanou léčbou (n = 49) intravitreální injekcí jednou měsíčně. Průměr určený z průměrných hodnot změn BCVA od 1. do 12. měsíce byl v porovnání se stavem na počátku léčby +7,8 (±7,72) písmen u shromážděných pacientů léčených ranibizumabem (n = 102), v porovnání s -0,1 (±9,77) písmen u pacientů se simulovanou léčbou a průměrná hodnota změny BCVA ve 12. měsíci od počátečního stavu byla 10,3 (±9,1) písmen v porovnání s -1,4 (±14,2) písmen, v tomto pořadí (p<0,0001 pro léčebný rozdíl).
Ve fázi III studie D2301 (RESTORE) bylo 345 pacientů randomizováno v poměru 1:1:1 do skupiny užívající ranibizumab 0,5 mg v monoterapii a simulovanou laserovou fotokoagulaci, kombinaci ranibizumabu 0,5 mg a laserové fotokoagulace nebo simulovanou injekci a laserovou fotokoagulaci. 240 pacientů, kteří předtím dokončili 12měsíční studii RESTORE, bylo zařazeno do otevřené, multicentrické 24měsíční extenze studie (extenze studie RESTORE). Pacienti byli léčeni podáním ranibizumabu 0,5 mg pro re nata (PRN) do stejného oka jako v základní studii (D2301 RESTORE).
Obrázek 2 Průměrná hodnota změny zrakové ostrosti od začátku léčby v průběhu času ve studii D2301 (RESTORE)
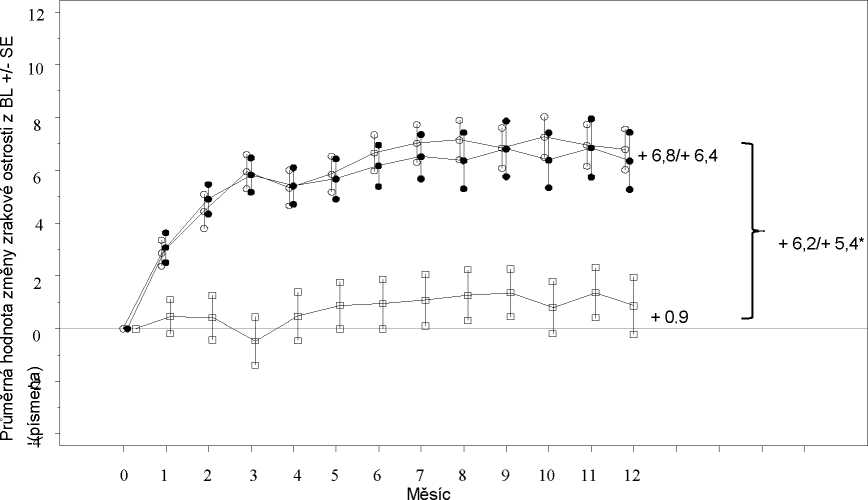
o o o Ranibizumab 0,5 mg (n=115)
Ranibizumab 0,5 mg + laser (n=118) □ □ □ Laser (n=110)
Léčebná skupina
BL=základní stav; SE=směrodatná chyba průměru
* Rozdíl v průměrech nejmenších čtverců, p<0,0001/0,0004 na základě dvoustranného stratifikovaného Cochran-Mantel-Haenszelova testu
Účinek po 12 měsících byl konzistentní ve většině podskupin. Avšak u subjektů s hodnotou BCVA >73 písmen na počátku léčby a makulárním edémem s centrální retinální tloušťkou <300 pm na počátku léčby se nezdálo, že by profitovaly z léčby ranibizumabem v porovnání s laserovou fotokoagulací.
Tabulka 2 Výsledky ve 12. měsíci ve studii D2301 (RESTORE) a ve 36. měsíci ve studii D2301-E1 (extenze studie RESTORE)
|
Výsledky měření ve 12. měsíci v porovnání se stavem na počátku léčby ve studii D2301 (RESTORE) |
Ranibizumab 0,5 mg n = 115 |
Ranibizumab 0,5 mg + laser n = 118 |
laser n = 110 |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 12. měsíce3 (+SD) |
6,1 (6,4)a |
5,9 (7,9)a |
0,8 (8,6) |
|
Průměrná hodnota změny BCVA ve 12. měsíci (+SD) |
6,8 (8,3)a |
6,4 (11,8)a |
0,9 (11,4) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 12. měsíci (%) |
22,6 |
22,9 |
8,2 |
|
Průměrný počet injekcí (0.-11. měsíc) |
7,0 |
6,8 |
7,3 (simulovaný) |
|
Výsledky měření ve 36. měsíci ve studii D2301-E1 (extenze studie RESTORE) v porovnání se stavem na počátku léčby ve studii D2301 (RESTORE) |
Před ranibizumabem 0,5 mg n = 83 |
Před ranibizumabem 0,5 mg + laser n = 83 |
Před laserem n = 74* |
|
Průměrná hodnota změny BCVA ve 24. měsíci (SD) |
7,9 (9,0) |
6,7 (7,9) |
5,4 (9,0) |
|
Průměrná hodnota změny BCVA ve 36. měsíci (SD) |
8,0 (10,1) |
6,7 (9,6) |
6,0 (9,4) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 36. měsíci (%) |
27,7 |
30,1 |
21,6 |
|
Průměrný počet injekcí (12.-35. měsíc)* |
6,8 |
6,0 |
6,5 |
ap<0,0001 pro porovnání ramen s ranibizumabem vs. rameno s laserem. n je ve studii D2301-E1 (extenze studie RESTORE) počet pacientů s hodnotou ve studii D2301 (RESTORE) na počátku léčby (měsíc 0) a při návštěvě ve 36. měsíci.
• Podíl pacientů, kteří nevyžadovali léčbu ranibizumabem během fáze prodloužení studie byl 19 % ve skupině před podáním ranibizumabu, 25 % ve skupině před podáním ranibizumabu + laseru a 20 % ve skupině před aplikací laseru.
Statisticky signifikantní přínosy pro většinu funkcí spojených se zrakem, hlášené pacienty, byly pozorovány u léčby ranibizumabem (s nebo bez laseru) oproti kontrolní skupině, měřeno pomocí NEI VFQ-25. Pro ostatní podškály tohoto dotazníku nemohly být stanoveny žádné léčebné rozdíly.
Dlouhodobý bezpečnostní profil ranibizumabu pozorovaný ve 24měsíční extenzi studie je konzistentní se známým bezpečnostním profilem přípravku Lucentis.
Ve fázi IIIb studie D2304 (RETAIN) bylo 372 pacientů randomizováno v poměru 1:1:1 k podávání:
• ranibizumabu 0,5 mg se současnou laserovou fotokoagulací v režimu „treat-and-extend“ (TE),
• ranibizumabu 0,5 mg v monoterapii v režimu TE,
• ranibizumabu 0,5 mg v monoterapii v režimu PRN.
Ve všech skupinách byl ranibizumab podáván jednou měsíčně, dokud BCVA nebyla stabilní
po alespoň tři po sobě jdoucí měsíční vyšetření. V režimu TE byl podán ranibizumab v léčebných intervalech 2-3 měsíce. Ve všech skupinách byla měsíční léčba znovu zahájena na základě snížení BCVA způsobeném progresí DME a pokračovala, dokud nebylo opět dosaženo stabilní BCVA.
Počet plánovaných léčebných návštěv po 3 počátečních injekcích byl 13 v režimu TE a 20 v režimu PRN. V obou TE režimech bylo více než 70 % pacientů schopných zachovat svou BCVA při průměrné četnosti návštěv >2 měsíce.
Nejdůležitější výsledky měření jsou uvedeny v Tabulce 3.
Tabulka 3 Výsledky ve studii D2304 (RETAIN)
|
Výsledek měření porovnaný s výchozím stavem |
ranibizumab 0,5 mg + laser v režimu TE n = 117 |
ranibizumab 0,5 mg samotný v režimu TE n = 125 |
ranibizumab 0,5 mg v režimu PRN n = 117 |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 12. měsíce (SD) |
5,9 (5,5)a |
6,1 (5,7)a |
6,2 (6,0) |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 24. měsíce (SD) |
6,8 (6,0) |
6,6 (7,1) |
7,0 (6,4) |
|
Průměrná hodnota změny BCVA ve 24. měsíci (SD) |
8,3 (8,1) |
6,5 (10,9) |
8,1 (8,5) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 24. měsíci (%) |
25,6 |
28,0 |
30,8 |
|
Průměrný počet injekcí (0.-23. měsíc) |
12,4 |
12,8 |
10,7 |
ap<0,0001 pro hodnocení non-inferiority k PRN
Ve studiích zaměřených na DME bylo zlepšení BCVA doprovázeno redukcí střední CSFT v průběhu času ve všech léčebných skupinách.
Léčba poškození zraku způsobeného makulárním edémem v důsledku RVO
Klinická bezpečnost a účinnost Lucentisu u pacientů s poškozením zraku způsobeným makulárním
edémem v důsledku RVO byla hodnocena v randomizovaných dvojitě zaslepených kontrolovaných
studiích BRAVO a CRUISE, ve kterých byly zařazeny subjekty s BRVO (n = 397) a CRVO
(n = 392). V obou studiích dostávaly subjekty buď 0,3 mg nebo 0,5 mg ranibizumabu nebo injekce
simulované léčby. Pacienti v kontrolním rameni se simulovanou léčbou byli po 6 měsících přeřazeni
do ramene s ranibizumabem 0,5 mg.
Tabulka 4 Výsledky v 6. a 12. měsíci (BRAVO a CRUISE)
|
BRAVO |
CRUISE | |||
|
Simulovaná léčba/Lucentis 0,5 mg (n = 132) |
Lucentis 0,5 mg (n = 131) |
Simulovaná léčba /Lucentis 0,5 mg (n = 130) |
Lucentis 0,5 mg (n = 130) | |
|
Průměrná hodnota změny zrakové ostrosti v 6. měsícía (písmena) (SD) (primární cíl) |
7,3 (13,0) |
18,3 (13,2) |
0,8 (16,2) |
14,9 (13,2) |
|
Průměrná hodnota změny BCVA ve 12. měsíci (písmena) (SD) |
12,1 (14,4) |
18,3 (14,6) |
7,3 (15,9) |
13,9 (14,2) |
|
Nárůst o > 15 písmen u zrakové ostrosti v 6. měsícia (%) |
28,8 |
61,1 |
16,9 |
47,7 |
|
Nárůst o > 15 písmen u zrakové ostrosti ve 12. měsíci (%) |
43,9 |
60,3 |
33,1 |
50,8 |
|
Podíl (%) pacientů léčených laserovou záchrannou terapií po 12 měsíců |
61,4 |
34,4 |
NA |
NA |
ap <0,0001 v obou studiích
CL
Léčebná skupina
r m-m—m
Měsíc
Placebo/Ranibizumab 0,5 mg (n = 132 Ranibizumab 0,5 mg (n = 131)
BL=základní stav; SE=směrodatná chyba průměru
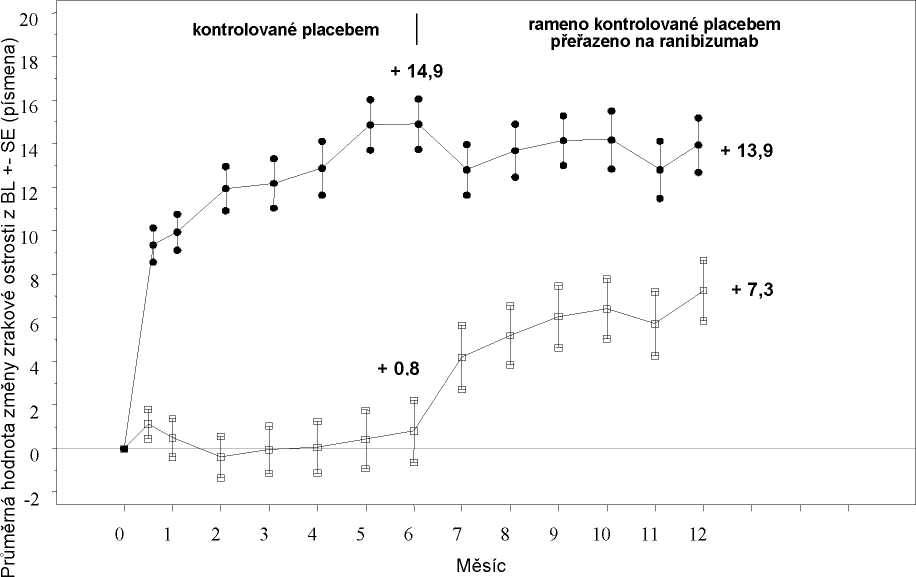
Léčebná skupina
Placebo/Ranibizumab 0,5 mg (n = 130) Ranibizumab 0,5 mg (n = 130)
BL=základní stav; SE=směrodatná chyba průměru
V obou studiích bylo zlepšení zraku spojeno s kontinuální a významnou redukcí makulámího edému, což bylo posuzováno podle tloušťky středu sítnice.
U pacientů s CRVO (CRUISE a extenze studie HORIZON): Subjekty léčené simulovanou léčbou v prvních 6 měsících, které následně dostávaly ranibizumab, nedosáhly srovnatelných nárůstů zrakové ostrosti (VA) do 24. měsíce (~6 písmen) v porovnání se subjekty léčenými ranibizumabem od začátku studie (~12 písmen).
Statisticky významná zlepšení v subškálách spojená s viděním na blízko a na dálku, hlášená pacienty, byla pozorována při léčbě ranibizumabem oproti kontrolní skupině, měřeno pomocí NEI VFQ-25.
Dlouhodobá (24měsíční) klinická bezpečnost a účinnost Lucentisu u pacientů s poškozením zraku způsobeného sekundárně makulárním edémem v důsledku RVO byla hodnocena ve studiích BRIGHTER (BRVO) a CRYSTAL (CRVO). V obou studiích bylo subjektům podáváno 0,5 mg ranibizumabu v režimu PRN řízeného individualizovanými kritérii stabilizace zrakové ostrosti. BRIGHTER byla 3ramenná randomizovaná aktivně kontrolovaná studie, která porovnávala 0,5 mg ranibizumabu podávaného v monoterapii nebo v kombinaci s adjuvantní laserovou fotokoagulací oproti samotné laserové fotokoagulaci. Subjektům v laserovém rameni mohl být po 6 měsících podán ranibizumab v dávce 0,5 mg. CRYSTAL byla jednoramenná studie s 0,5 mg ranibizumabu v monoterapii.
Důležité výstupy ze studií BRIGHTER a CRYSTAL jsou uvedeny v Tabulce 5. Tabulka 5 Výsledky v 6. a 24. měsíci studie (BRIGHTER a CRYSTAL)
|
BRIGHTER |
CRYSTAL | |||
|
Lucentis 0,5 mg n=180 |
Lucentis 0,5 mg + laser n=178 |
Laser* n=90 |
Lucentis 0,5 mg n=356 | |
|
Průměrná hodnota změny BCVA v 6. měsícia (písmena) (SD) |
+14,8 (10,7) |
+14,8 (11,13) |
+6,0 (14,27) |
+12,0 (13,95) |
|
Průměrná hodnota změny BCVA ve 24. měsícib (písmena) (SD) |
+15,5 (13,91) |
+17,3 (12,61) |
+11,6 (16,09) |
+12,1 (18,60) |
|
Zisk >15 písmen BCVA ve 24. měsíci (%) |
52,8 |
59,6 |
43,3 |
49,2 |
|
Průměrný počet injekcí (SD) (měsíc 0-23) |
11,4 (5,81) |
11,3 (6,02) |
NA |
13,1 (6,39) |
|
a p<0,0001 pro obě porovnání ve studii BRIGHTER v 6. měsíci: Lucentis 0,5 mg vs laser a Lucentis 0,5 mg + laser vs laser. b p<0,0001 pro nulovou hypotézu ve studii CRYSTAL, že průměrná hodnota změny ve 24. měsíci od výchozího stavu je nula. * Od 6. měsíce byla umožněna léčba 0,5 mg ranibizumabu (24 pacientů bylo léčeno pouze laserem). | ||||
Ve studii BRIGHTER byla prokázána non-inferiorita 0,5 mg ranibizumabu s podpůrnou laserovou terapií ve srovnání s ranibizumabem v monoterapii od výchozího stavu do 24. měsíce (95 % CI -2,8; 1,4).
V obou studiích byla v 1. měsíci pozorována rychlá a statisticky významná redukce tloušťky centrální části sítnice od výchozího stavu. Tento účinek přetrval až do 24. měsíce.
Účinek léčby ranibizumabem byl podobný bez ohledu na přítomnost retinální ischemie. Ve studii BRIGHTER u pacientů s ischemií (n=46) nebo bez ischemie (n=133) léčených ranibizumabem v monoterapii byla průměrná hodnota změny BCVA ve 24. měsíci od výchozího stavu +15,3; resp. +15,6 písmen. Ve studii CRYSTAL u pacientů s ischemií (n=53) nebo bez ischemie (n=300) léčených ranibizumabem v monoterapii byla průměrná hodnota změny BCVA od výchozího stavu +15,0; resp. +11,5 písmen.
V obou studiích BRIGHTER a CRYSTAL byl účinek na zlepšení zraku pozorován u všech pacientů léčených 0,5 mg ranibizumabu v monoterapii bez ohledu na délku trvání onemocnění. U pacientů
s délkou trvání onemocnění <3 měsíce byl pozorován nárůst zrakové ostrosti o 13,3; resp. 10,0 písmen v 1. měsíci a o 17,7; resp. 13,2 písmen ve 24. měsíci ve studiích BRIGHTER a CRYSTAL. Odpovídající zisk zrakové ostrosti u pacientů s délkou trvání onemocnění >12 měsíců byl v těchto studiích 8,6; resp. 8,4 písmen. Je třeba zvážit zahájení léčby v čase stanovení diagnózy.
Dlouhodobý bezpečnostní profil ranibizumabu pozorovaný ve 24měsíčních studiích je konzistentní se známým bezpečnostním profilem přípravku Lucentis.
Léčba poškození zraku způsobeného CNV sekundární k PM
Klinická bezpečnost a účinnost přípravku Lucentis u pacientů s poškozením zraku způsobeným CNV u PM byla hodnocena na základě 12měsíčních dat z dvojitě zaslepené, kontrolované pivotní studie F2301 (RADIANCE). V této studii bylo 277 pacientů randomizováno v poměru 2:2:1 do následujících ramen:
• Skupina I (ranibizumab 0,5 mg, dávkovací schéma řízené kritérii „stability“, definovanými jako žádná změna BCVA v porovnání se dvěma předchozími měsíčními vyhodnoceními).
• Skupina II (ranibizumab 0,5 mg, dávkovací schéma řízené kritérii „aktivity onemocnění“, definovanými jako poškození zraku způsobené intra- nebo subretinální tekutinou nebo aktivním prosakováním v důsledku CNV léze, jak bylo zjištěno při vyšetření OCT a/nebo FA).
• Skupina III (vPDT - pacientům bylo povoleno užívat léčbu ranibizumabem od 3. měsíce studie).
Ve skupině II, což je doporučené dávkovací schéma (viz bod 4.2), vyžadovalo 50,9 % pacientů 1 nebo 2 injekce, 34,5 % pacientů 3 až 5 injekcí a 14,7 % vyžadovalo 6 až 12 injekcí po dobu 12 měsíců trvání studie. 62,9 % pacientů ze skupiny II nevyžadovalo injekce ve druhé polovině trvání studie.
Důležité výstupy ze studie RADIANCE jsou uvedeny v Tabulce 6 a na Obrázku 5.
Tabulka 6 Výsledky ve 3. a 12. měsíci studie (RADIANCE)
|
Skupina I |
Skupina II |
Skupina | |
|
Ranibizumab |
Ranibizumab |
III | |
|
0,5 mg |
0,5 mg |
vPDTb | |
|
„stabilizace |
„aktivita | ||
|
vidění“ |
onemocnění“ | ||
|
(n = 105) |
(n = 116) |
(n = 55) | |
|
3. měsíc Průměr určený z průměrných hodnot změn BCVA od 1. do 3. měsíce studie v porovnání se základním stavema (písmena) Podíl pacientů, kteří dosáhli: >15 písmen, nebo dosáhli >84 písmen |
+10,5 |
+10,6 |
+2,2 |
|
BCVA 12. měsíc |
38,1 % |
43,1 % |
14,5 % |
|
Počet injekcí až do 12. měsíce: Průměrná hodnota |
4,6 |
3,5 |
N/A |
|
Medián |
4,0 |
2,5 |
N/A |
|
Průměr určený z průměrných hodnot změn BCVA od 1. do 12. měsíce v porovnání se základním stavem (písmena) Podíl pacientů, kteří dosáhli: |
+12,8 |
+12,5 |
N/A |
|
>15 písmen, nebo dosáhli >84 písmen BCVA |
53,3 % |
51,7 % |
N/A |
a p <0,00001 porovnání s vPDT kontrolní skupinou
b Srovnávací kontrola až do 3. měsíce studie. Pacientům randomizovaným do skupiny s vPDT bylo povoleno dostávat léčbu ranibizumabem od 3. měsíce studie (ve skupině III dostávalo ranibizumab 38 pacientů od 3. měsíce studie)
Průměrná hodnota změny BCVA od začátku léčby v průběhu času do 12. měsíce (RADIANCE)
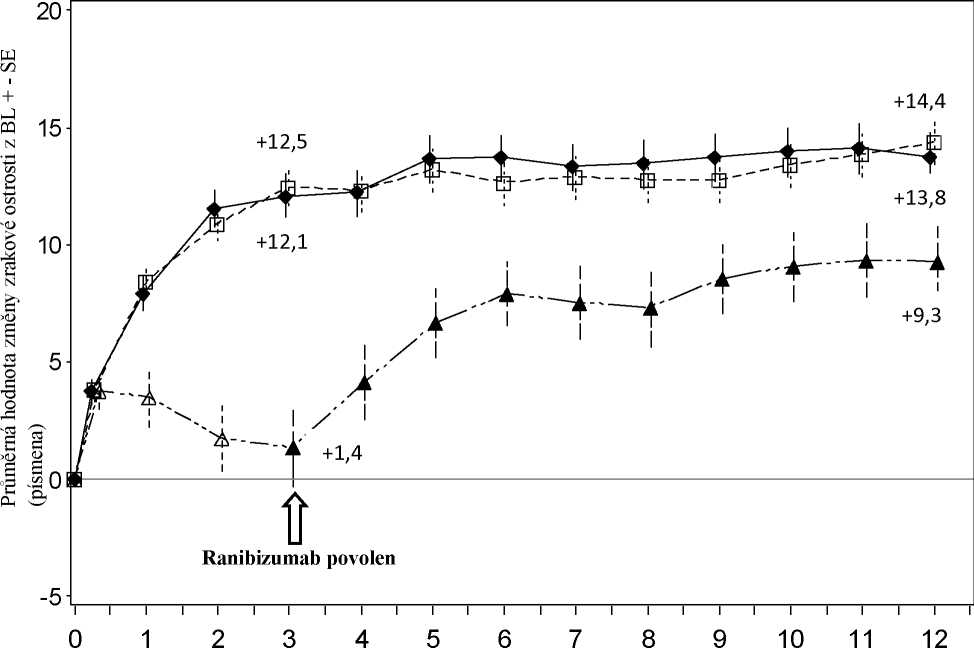
Měsíc
___ Ranibizumab 0,5 mg Skupina I Ranibizumab 0,5 mg Skupina II
^ ^ ^ EI-EHD v i • • v v r
při aktivitě onemocněni (n = 116)
Ranibizumab 0,5 mg/PDT verteporfinem Skupina III od 3. měsíce studie dále (n = 55)
pn stabilizaci (n = 105)
a-a-a PDT verteporfinem Skupina III(n = 55)
Zlepšeni zraku bylo doprovázeno redukci tloušťky centrální části sítnice.
Prospěch hlášený pacientem byl pozorován v léčebných ramenech s ranibizumabem oproti vPDT (hodnota p <0,05) na základě zlepšení kombinovaného skóre a několika subškál (všeobecné vidění, aktivity do blízka, duševní zdraví a závislost) v NEI VFQ-25.
Pediatrická populace
Bezpečnost a účinnost ranibizumabu nebyla u pediatrických pacientů dosud studována.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lucentis u všech podskupin pediatrické populace u neovaskulární formy AMD, u poškození zraku kvůli DME, u poškození zraku kvůli makulárnímu edému v důsledku RVO a u poškození zraku v důsledku CNV sekundární k PM (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Po měsíčních aplikacích Lucentisu do sklivce jedincům s neovaskulámí AMD byly koncentrace ranibizumabu v séru obvykle nízké. Maximální koncentrace (Cmax) ranibizumabu byly obecně nižší než koncentrace ranibizumabu nutné k inhibici biologické aktivity VEGF o 50 % (11 - 27 ng/ml, stanoveno in vitro titrací buněčné proliferace). Cmax byla závislá na dávce v dávkovém rozmezí 0,05 až
1.0 mg/oko. Sérové koncentrace u limitovaného počtu pacientů s DME ukazují, že lehce vyšší systémová expozice nemůže být vyloučena ve srovnání se sérovými koncentracemi zjištěnými
u pacientů s neovaskulámí AMD. Sérové koncentrace ranibizumabu u pacientů s RVO byly podobné nebo lehce vyšší v porovnání s koncentracemi pozorovanými u pacientů s neovaskulámí AMD.
Na základě analýz populační farmakokinetiky a vymizení ranibizumabu ze séra pacientů s neovaskulámí AMD léčených dávkou 0,5 mg byl průměrný poločas eliminace ranibizumabu ze sklivce stanoven na přibližně 9 dnů. Při aplikaci Lucentisu v dávce 0,5 mg/oko jednou měsíčně je dosaženo sérové Cmax ranibizumabu, jejíž předpokládané rozmezí je mezi 0,79 a 2,90 ng/ml přibližně za 1 den po aplikaci. Cmin se předpokládá v rozmezí mezi 0,07 a 0,49 ng/l. Sérové koncentrace ranibizumabu se předpokládají přibližně 90 000násobně nižší než koncentrace ranibizumabu ve sklivci.
Pacienti se zhoršenou funkcí ledvin: Žádné cílené studie, které by hodnotily farmakokinetiku Lucentisu u pacientů se zhoršenou funkcí ledvin, nebyly provedeny. 68 % (136 ze 200) pacientů s neovaskulámí AMD mělo v populační farmakokinetické analýze zhoršenou funkci ledvin (46,5 % mírně [50 - 80 ml/min], 20 % středně [30 - 50 ml/min] a 1,5 % silně [< 30 ml/min]). 48,2 % (253 z 525) pacientů s RVO mělo zhoršenou funkci ledvin (36,4 % mírně, 9,5 % středně a 2,3 % silně). Systémová clearance byla lehce snížena, ale tento nález byl bez klinického významu.
Zhoršená funkce jater: Žádné cílené studie, které by hodnotily farmakokinetiku u pacientů se zhoršenou funkcí jater léčených Lucentisem, nebyly provedeny.
5.3 Předklinické údaje vztahující se k bezpečnosti
Oboustranná aplikace ranibizumabu do sklivce opicím cynomolgus v dávkách 0,25 mg/oko a
2.0 mg/oko jednou za dva týdny po dobu 26 týdnů vyvolala účinky na očích závislé na dávce.
Nitrooční aplikace vyvolala na dávce závislé zvýšené zarudnutí přední komory a zvýšení počtu buněk, největší za 2 dny po injekci. Závažnost zánětlivé odpovědi se obvykle zmenšila po následujících injekcích nebo během zotavení. V zadním segmentu byla pozorována infiltrace sklivce buňkami a sklivcovými vločkami, u které byla také tendence k závislosti na dávce a obvykle přetrvávala až do konce léčby. Ve 26týdenní studii se intenzita zánětu sklivce zvyšovala s počtem injekcí. Průkaz reverzibility byl však pozorován po zotavení. Povaha a načasování zánětu zadního segmentu naznačují, že se jedná o imunologicky zprostředkovanou protilátkovou odpověď, která by mohla být klinicky irelevantní. U některých zvířat byla po relativně dlouhém období intenzivního zánětu pozorována tvorba katarakty naznačující, že změny čočky byly sekundární, vyvolané silným zánětem. Bylo pozorováno přechodné zvýšení nitroočního tlaku po podání do sklivce bez ohledu na dávku.
Mikroskopické změny pozorované v oční tkáni souvisely se zánětem a nenasvědčovaly degenerativním procesům. U některých očí byly pozorovány granulomatózní zánětlivé změny na očním disku. Tyto změny zadního segmentu slábly, a v některých případech kompletně vymizely, během období zotavení.
Po podání do sklivce nebyly zjištěny žádné známky systémové toxicity. U některých zvířat byly nalezeny protilátky proti ranimizumabu v séru i sklivci.
Nejsou dostupné žádné údaje o karcinogenitě nebo mutagenitě.
U březích opic nevyvolala léčba ranibizumabem aplikovaná do sklivce , mající za následek maximální systémové expozice 0,9-7-krát nejhorší případ klinické expozice, vývojovou toxicitu ani teratogenitu, a neměla žádný vliv na hmotnost nebo strukturu placenty, i když na základě svého farmakologického účinku by měl být ranibizumab považován za potenciálně teratogenní a embryo-/fetotoxický.
Chybění účinků zprostředkovaných ranibizumabem na embryo-fetální vývoj se pravděpodobně týká hlavně neschopnosti Fab-fragmentu procházet placentou. Přesto byl popsán případ s vysokými mateřskými sérovými hladinami ranibizumabu a přítomností ranibizumabu ve fetálním séru, nasvědčující tomu, že protilátky proti ranibizumabu působily jako nosičský protein (obsahující Fc část) pro ranibizumab, a tím snižovaly jeho mateřskou sérovou clearance a umožňovaly prostup placentou. Protože embryo-fetální vývojová vyšetření byla prováděna u zdravých březích zvířat a onemocnění (jako například diabetes) mohou modifikovat prostupnost placenty pro Fab-fragment, měla by být studie interpretována s opatrností.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát a,a-trehalosy
Monohydrát histidin-hydrochloridu
Histidin
Polysorbát 20
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Před použitím může být neotevřená injekční lahvička ponechána při pokojové teplotě (25 °C) po dobu 24 hodin.
6.5 Druh obalu a obsah balení
0,23 ml sterilního injekčního roztoku v injekční lahvičce (sklo typu I) se zátkou (chlorbutylová pryž). Balení obsahuje 1 injekční lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Injekční lahvička je pro jednorázové použití. Po podání injekce musí být veškerý nepoužitý léčivý přípravek zlikvidován. Jakákoliv injekční lahvička vykazující známky poškození nebo manipulace nesmí být použita. Sterilita nemůže být zaručena, pokud nezůstane uzávěr obalu neporušený.
Pro přípravu a podání injekce do sklivce jsou potřebné následující zdravotnické pomůcky pro jednorázové použití:
- jehla s 5 pm filtrem (18G)
- 1 ml sterilní injekční stříkačka
- injekční jehla (30G x 14").
Tyto zdravotnické pomůcky nejsou obsaženy v balení přípravku Lucentis.
Při přípravě přípravku Lucentis k podání do sklivce dbejte, prosím, následujících pokynů:
1. Před nasátím tekutiny do injekční stříkačky je nutno desinfikovat vněj ší část pryžové zátky.
2. Jehlu s 5 pm filtrem (18G) nasaďte na 1 ml injekční stříkačku za použití aseptického postupu. Zasuňte jehlu s filtrem do středu zátky lahvičky, dokud se jehla nedotkne dna injekční lahvičky.
3. Nasajte veškerou tekutinu z injekční lahvičky držené ve svislé, lehce nakloněné poloze pro snadnější úplné nasátí.
4. Ujistěte se, že píst je při vyprazdňování injekční lahvičky vytažen dostatečně daleko tak, aby jehla s filtrem byla úplně vyprázdněna.
5. Jehlu s filtrem ponechte v lahvičce a stříkačku od ní odpojte. Jehla s filtrem by měla být po násátí veškerého obsahu lahvičky zlikvidována a nesmí být použita pro vlastní aplikaci do sklivce.
6. Asepticky a pevně nasaďte injekční jehlu (30G x ‘A”) na injekční stříkačku.
7. Opatrně odstraňte kryt z injekční jehly, aniž byste injekční jehlu odpojili od injekční stříkačky. Poznámka: Při odstraňování krytu pevně uchopte část injekční jehly.
8. Pečlivě vytlačte vzduch ze stříkačky a upravte dávku ke značce 0,05 ml uvedené na injekční stříkačce. Nyní je injekční stříkačka připravena k aplikaci.
Poznámka: Neotírejte injekční jehlu. Nevytahujte píst zpět.
Po podání injekce nenasazujte zpět kryt jehly ani jehlu neoddělujte od injekční stříkačky. Zlikvidujte použitou injekční stříkačku společně s jehlou vyhozením do nádoby na ostré předměty nebo v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/06/374/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 22. ledna 2007
Datum posledního prodloužení registrace: 24. ledna 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Lucentis 10 mg/ml injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje ranibizumabum 10 mg*. Jedna předplněná injekční stříkačka obsahuje 0,165 ml, odpovídajících ranibizumabum 1,65 mg. Extrahovatelný objem jedné předplněné injekční stříkačky je 0,1 ml. To zajišťuje použitelné množství k podání jednorázové dávky 0,05 ml, obsahující ranibizumabum 0,5 mg.
*Ranibizumab je fragment humanizované monoklonální protilátky produkovaný buňkami Escherichia coli rekombinantní DNA technologií.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok
Čirý, bezbarvý až světle žlutý vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Lucentis je indikován u dospělých:
• k léčbě neovaskulární (vlhké) formy věkem podmíněné makulární degenerace (AMD)
• k léčbě poškození zraku způsobeného diabetickým makulárním edémem (DME)
• k léčbě poškození zraku způsobeného makulárním edémem v důsledku okluze retinální vény [uzávěr větve centrální retinální vény (BRVO) a uzávěr centrální retinální vény (CRVO)]
• k léčbě poškození zraku způsobeného choroidální neovaskularizací (CNV) sekundární k patologické myopii (PM)
4.2 Dávkování a způsob podání
Lucentis musí být aplikován kvalifikovaným oftalmologem zkušeným v podání do sklivce.
Doporučená dávka Lucentisu je 0,5 mg podávaných jako jednorázová injekce do sklivce. To odpovídá injekci o objemu 0,05 ml. Interval mezi dvěma dávkami podávanými do stejného oka má být alespoň čtyři týdny.
Léčba se zahajuje jednou injekcí za měsíc do dosažení maximální zrakové ostrosti a/nebo do vymizení příznaků aktivity onemocnění, tj. žádná změna zrakové ostrosti a ostatních příznaků a projevů onemocnění při probíhající léčbě. U pacientů s vlhkou formou AMD, DME a RVO mohou být zpočátku potřeba tři nebo více po sobě jdoucí injekce podávané jednou za měsíc.
Následně mají být lékařem určeny intervaly sledování a léčby a mají být stanoveny na základě aktivity onemocnění vyhodnocené podle zrakové ostrosti a/nebo anatomických parametrů.
Pokud zrakové a anatomické parametry ukazují na základě vyjádření lékaře, že pokračující léčba pacienta není přínosná, je třeba léčbu přípravkem Lucentis ukončit.
Sledování aktivity onemocnění může zahrnovat klinické vyšetření, funkční testy nebo zobrazovací techniky (např. optickou koherenční tomografii nebo fluorescenční angiografii).
Pokud j sou pacienti léčeni podle režimu „treat-and-extend“, pak po dosažení maximální zrakové ostrosti a/nebo vymizení příznaků aktivity onemocnění, mohou být léčebné intervaly postupně prodlouženy až do opětovného objevení se příznaků aktivity onemocnění nebo zhoršení zraku. Léčebný interval nemá být prodloužen o více než dva týdny najednou u vlhké formy AMD a může být prodloužen až o jeden měsíc najednou u DME. Při uzávěru retinální vény mohou být léčebné intervaly také postupně prodlužovány, ale nejsou dostupná dostatečná data dokládající délku těchto intervalů. Pokud se aktivita onemocnění znovu objeví, léčebný interval má být příslušně zkrácen.
V léčbě poškození zraku způsobeném CNV sekundární k PM může mnoho pacientů potřebovat pouze jednu nebo dvě injekce během prvního roku, zatímco někteří pacienti mohou potřebovat častější léčbu (viz bod 5.1).
Lucentis a laserová fotokoagulace u DME a makulárního edému v důsledku BRVO Existuje určitá zkušenost s podáním přípravku Lucentis společně s laserovou fotokoagulací (viz bod 5.1). Při podání ve stejný den by měl být Lucentis podán alespoň 30 minut po laserové fotokoagulaci. Lucentis může být podán pacientům, kteří byli léčeni předchozí laserovou fotokoagulací.
Lucentis a fotodynamická léčba Visudynem u CNV sekundární k PM Se souběžným podáváním přípravků Lucentis a Visudyne nejsou žádné zkušenosti.
Zvláštní populace
Zhoršená funkce jater
Účinky přípravku Lucentis u pacientů se zhoršenou funkcí jater nebyly hodnoceny. Pro tuto populaci však nejsou nutná žádná zvláštní opatření.
Zhoršená funkce ledvin
U pacientů se zhoršenou funkcí ledvin není nutné upravovat dávku (viz bod 5.2).
Starší jedinci
U starších jedinců není nutná úprava dávky. U pacientů s DME starších 75 let jsou omezené zkušenosti.
Pediatrická populace
Bezpečnost a účinnost Lucentisu u dětí a dospívajících pod 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Předplněná injekční stříkačka na jedno použití, pouze pro podání do sklivce. Předplněná injekční stříkačka obsahuje více než doporučenou dávku 0,5 mg. Extrahovatelný objem předplněné injekční stříkačky (0,1 ml) není určen k celkovému podání. Nadbytečný objem je třeba před podáním injekce vytlačit. Při podání celého objemu předplněné injekční stříkačky by mohlo dojít k předávkování.
K vytlačení vzduchové bubliny spolu s nadbytečným léčivým přípravkem pomalu tlačte píst, dokud okraj pod vyklenutím pryžové zarážky není vyrovnaný s černou dávkovací linkou na injekční stříkačce (ekvivalentní 0,05 ml, tj. 0,5 mg ranibizumabu).
Před aplikací je nutno Lucentis vizuálně zkontrolovat, zda neobsahuje cizí částice nebo není změněna jeho barva.
Lucentis musí být aplikován za aseptických podmínek, což zahrnuje použití chirurgické desinfekce rukou, sterilních rukavic, sterilního oděvu, sterilního spekula (nebo ekvivalentní náhrady) a dostupnost sterilní paracentézy (je-li potřeba). Před aplikací injekce do sklivce je nutný pečlivý odběr anamnézy z hlediska hypersenzitivních reakcí (viz bod 4.4). Před aplikací injekce musí být podána adekvátní anestezie a použit širokospektrý lokální antimikrobiální přípravek k dezinfekci pokožky kolem očí, očního víčka a povrchu oka, v souladu s lokální praxí.
Pro informace o přípravě Lucentisu viz bod 6.6.
Injekční jehla se zasune 3,5-4,0 mm posteriorně od limbu do prostoru sklivce tak, aby směřovala do centra očního bulbu a nikoli k horizontálnímu meridiánu. Poté se aplikuje objem injekce 0,05 ml. Následující injekci je nutné aplikovat v jiném místě skléry. Každou předplněnou injekční stříkačku je třeba použít k léčbě pouze jednoho oka.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Pacienti s aktivní nebo suspektní oční nebo periokulární infekcí.
Pacienti s aktivním těžkým nitroočním zánětem.
4.4 Zvláštní upozornění a opatření pro použití Reakce po podání injekce do sklivce
Podání do sklivce, včetně těch s Lucentisem, byly spojovány s endoftalmitidou, intraokulárním zánětem, rhegmatogenním odchlípením sítnice, trhlinami sítnice a iatrogenní traumatickou kataraktou (viz bod 4.8). Při aplikaci Lucentisu musí být vždy dodržena přísná pravidla asepse. V následujícím týdnu po aplikaci injekce musejí být pacienti sledováni z hlediska případného výskytu infekce, aby bylo možné zahájit včas adekvátní léčbu. Pacienty je nutno upozornit, že musejí ihned hlásit všechny příznaky možné endoftalmitidy nebo jiných výše popsaných komplikací.
Zvýšení nitroočního tlaku
Během 60 minut po injekci Lucentisu bylo pozorováno přechodné zvýšení nitroočního tlaku (IOP). Trvalá zvýšení IOP byla také zjištěna (viz bod 4.8). Je nutné monitorovat a náležitě ošetřit jak nitrooční tlak, tak i perfuzi papily očního nervu.
Léčba obou očí současně
Omezená data k léčbě obou očí přípravkem Lucentis současně (včetně podání ve stejný den) nenaznačují zvýšené riziko systémových nežádoucích účinků v porovnání s léčbou jednoho oka.
Imunogenicita
U Lucentisu existuje možnost vzniku imunogenicity. Protože existuje potenciál pro zvýšenou systémovou expozici u pacientů s DME, zvýšené riziko pro vznik hypersenzitivity u této pacientské populace nelze vyloučit. Pacienti musejí být také poučeni, aby hlásili zhoršení závažnosti nitroočního zánětu, protože se může jednat o klinický příznak charakteristický pro tvorbu nitroočních protilátek.
Současné použití jiných anti-VEGF (vaskulární endoteliální růstový faktor) léčivých přípravků Lucentis se nesmí podávat zároveň s jinými anti-VEGF léčivými přípravky (systémovými nebo očními).
Vynechání dávky Lucentisu
Dávku je nutno vynechat a v léčbě se nesmí pokračovat dříve, než je plánována další dávka v následujících případech:
• snížení nejlépe upravené ostrosti zraku (best-corrected visual acuity BCVA) o > 30 písmen ve srovnání s předchozím měřením ostrosti zraku;
• nitrooční tlak > 30 mmHg;
• poškození sítnice;
• subretinální krvácení zahrnující střed fovey, nebo je-li velikost hemoragie > 50 % celkové plochy léze;
• provedený nebo plánovaný oční chirurgický zákrok během uplynulých nebo následujících 28 dnů.
Trhlina pigmentového epitelu sítnice
Rizikové faktory spojené s vývojem trhliny pigmentového epitelu sítnice po podání anti-VEGF léčby u vlhké formy AMD zahrnují rozsáhlé a/nebo značné odchlípení pigmentového epitelu sítnice.
U pacientů s těmito rizikovými faktory pro vznik trhlin pigmentového epitelu sítnice je třeba dbát opatrnosti při zahajování léčby Lucentisem.
Rhegmatogenní odchlípení sítnice nebo makulární díry
Léčbu je nutno přerušit u subjektů s rhegmatogenním odchlípením sítnice nebo u makulárních děr stupně 3 nebo 4.
Populace s omezenými daty
Existují pouze omezené zkušenosti s léčbou u pacientů s DME způsobeným diabetem I. typu. Lucentis nebyl studován u pacientů, kterým byla dříve podána injekce do sklivce, u pacientů s aktivními systémovými infekcemi, proliferativní diabetickou retinopatií nebo u pacientů se současnými očními onemocněními jako například odchlípení sítnice nebo makulární díra. Také nejsou zkušenosti s léčbou Lucentisem u diabetických pacientů s HbA1c nad 12 % a nekontrolovanou hypertenzí. Chybění těchto informací má být lékařem zváženo při léčbě takových pacientů.
Nejsou k dispozici dostatečné údaje k posouzení účinnosti Lucentisu u pacientů s RVO projevujícím se ireverzibilní ztrátou zraku v důsledku ischemie.
U pacientů s PM, kteří v minulosti podstoupili neúspěšnou fotodynamickou léčbu verteporfinem (vPDT) jsou k dispozici limitovaná data o účinku přípravku Lucentis. Rovněž zatímco byl pozorován odpovídající účinek u subjektů se subfoveálními a juxtafoveálními lézemi, jsou data k vyvození závěru o účinku přípravku Lucentis u subjektů s patologickou myopií s extrafoveálními lézemi nedostatečná.
Systémové účinky po podání do sklivce
Po injekční aplikaci VEGF inhibitorů do sklivce byly hlášeny systémové nežádoucí příhody včetně mimoočních krvácení a arteriálních tromboembolických příhod.
Data o bezpečnosti při léčbě pacientů s DME, makulárním edémem způsobeným RVO a CNV sekundární k patologické myopii (PM) u pacientů s mrtvicí nebo přechodnými ischemickými příhodami v anamnéze jsou omezená. U těchto pacientů je třeba dbát zvýšené opatrnosti (viz bod 4.8).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí.
Současné použití Lucentisu s fotodynamickou léčbou (PDT) verteporfinem u vlhké formy AMD a PM, viz bod 5.1.
Současné použití Lucentisu s laserovou fotokoagulací u DME a BRVO, viz body 4.2 a 5.1.
V klinických studiích zaměřených na léčbu poškození zraku způsobeného DME nebyl výsledek s ohledem na zrakovou ostrost nebo tloušťku centrální části sítnice (CSFT) u pacientů léčených přípravkem Lucentis ovlivněn současnou léčbou thiazolidindiony.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/antikoncepce u žen
Ženy ve fertilním věku musí během léčby používat účinnou antikoncepci.
Žádné údaje o podávání ranibizumabu těhotným ženám nejsou k dispozici. Studie u makaků rodu Cynomolgus nenaznačují přímé nebo nepřímé škodlivé účinky s ohledem na těhotenství nebo embryonální/fetální vývoj (viz bod 5.3). Po očním podání je nízká systémová expozice ranibizumabu, ale vzhledem k jeho mechanismu účinku je nutno ranibizumab považovat za potenciálně teratogenní a embryo-/fetotoxický. Z tohoto důvodu nesmí být ranibizumab užíván během těhotenství, aniž by očekávaný přínos převážil možné riziko pro plod. Ženám, které chtějí otěhotnět, a které byly léčeny ranibizumabem, je doporučeno vyčkat nejméně 3 měsíce po poslední dávce ranibizumabu před početím dítěte.
Kojení
Není známo, zda se Lucentis vylučuje do lidského mateřského mléka. Během léčby přípravkem Lucentis se kojení nedoporučuje.
Fertilita
Nejsou k dispozici žádné údaje vztahující se k fertilitě.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Léčba Lucentisem může vyvolat dočasné zhoršení zraku, což může ovlivnit schopnost řídit nebo obsluhovat stroje (viz bod 4.8). Pacienti, u kterých se tyto příznaky vyskytnou, nesmějí řídit nebo obsluhovat stroje, dokud tyto poruchy zraku neustoupí.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Většina nežádoucích účinků hlášených po podání Lucentisu se týká postupu podání injekce do sklivce.
Nejčastěji hlášené oční nežádoucí účinky po podání injekce Lucentisu jsou: bolest oka, oční hyperemie, zvýšený nitrooční tlak, vitritida, odloučení sklivce, hemoragie sítnice, poruchy zraku, sklivcové vločky, hemoragie spojivky, podráždění oka, pocit cizího tělesa, zvýšené slzení, zánět očního víčka, suchost oka a svědění oka.
Nejčastěji hlášené nežádoucí účinky mimo oko jsou: bolest hlavy, nazofaryngitida a artralgie.
Méně často hlášené, ale závažnější nežádoucí účinky zahrnují: endoftalmitidu, slepotu, odchlípení sítnice, trhlinu sítnice a iatrogenní traumatickou kataraktu (viz bod 4.4).
Pacienti mají být informováni o příznacích těchto potenciálních nežádoucích účinků a poučeni informovat svého lékaře, pokud se u nich objeví příznaky, jako je bolest oka nebo zvýšený oční diskomfort, zhoršující se zarudnutí oka, rozmazané nebo snížené vidění, zvýšený počet malých částic v zorném poli nebo zvýšená citlivost na světlo.
Nežádoucí účinky, které se objevily po podání Lucentisu v klinických studiích jsou uvedeny v tabulce níže.
Tabulkový seznam nežádoucích účinků#
Nežádoucí účinky jsou uvedeny podle systémově-orgánových skupin a frekvence za použití následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Infekce a infestace
Velmi časté Časté
Nazofaryngitida Infekce močových cest*
Poruchy krve a lymfatického systému
Časté Anemie
Poruchy imunitního systému
Časté Hypersenzitivita
Psychiatrické poruchy
Časté Úzkost
Poruchy nervového systému
Velmi časté Bolest hlavy
Poruchy oka
Velmi časté
Časté
Méně časté
Vitritida, odloučení sklivce, hemoragie sítnice, poruchy zraku, bolest oka, sklivcové vločky, hemoragie spojivky, podráždění oka, pocit cizího tělesa, zvýšené slzení, zánět očního víčka, suchost oka, oční hyperemie, svědění oka.
Degenerace sítnice, poškození sítnice, odloučení sítnice, trhlina sítnice, odloučení pigmentového epitelu sítnice, trhlina v pigmentovém epitelu sítnice, snížení ostrosti zraku, hemoragie sklivce, poškození sklivce, uveitida, zánět duhovky, iridocyklitida, katarakta, subkapsulární katarakta, opacifikace zadního pouzdra, keratitis punctata, abraze rohovky, zarudnutí v přední části komory, rozmazané vidění, hemoragie v místě injekce, oční hemoragie, zánět spojivky, alergický zánět spojivky, výtok z oka, fotopsie, fotofobie, oční dyskomfort, otok víčka, bolestivost víčka, překrvení spojivek.
Slepota, endoftalmitida, hypopyon, krvácení do přední komory oka, keratopatie, adheze duhovky, korneální deposita, edém rohovky, strie rohovky, bolestivost v místě injekce, podráždění v místě injekce, abnormální pocit v oku, podráždění očního víčka.
Respirační, hrudní a mediastinální poruchy Časté Kašel
Gastrointestinální poruchy
Časté Nauzea
Poruchy kůže a podkožní tkáně
Časté Alergické reakce (vyrážka, kopřivka, pruritus, erytém)
Poruchy svalové a kosterní soustavy a pojivové tkáně Velmi časté Artralgie
Vyšetření
Velmi časté Zvýšení nitroočního tlaku
# Nežádoucí účinky byly definovány jako nežádoucí příhody (u nejméně 0,5 procentních bodů pacientů), které se objevily ve vyšší míře (alespoň 2 procentní body) u pacientů léčených Lucentisem 0,5 mg než u těch, kteří byli léčeni v kontrolním rameni (simulovanou léčbou nebo fotodynamickou léčbou verteporfinem).
* pozorované pouze u populace s DME
Nežádoucí účinky související s třídou přípravku
Ve studiích fáze III s vlhkou formou AMD se mírně zvýšil celkový výskyt mimoočních krvácení u pacientů léčených ranibizumabem, což je nežádoucí účinek, který potenciálně souvisí se systémovou inhibicí VEGF (vaskulární endoteliální růstový faktor). Nicméně, jednotlivá krvácení neměla shodný charakter. Po intravitreálním podání VEGF inhibitorů existuje teoretické riziko arteriální tromboembolické příhody, včetně mrtvice a infarktu myokardu. V klinických studiích s Lucentisem byla u pacientů s AMD, DME, RVO a PM pozorována nízká incidence arteriálních tromboembolických příhod a mezi skupinami léčenými ranibizumabem nebyly ve srovnání s kontrolou významné rozdíly.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Případy předávkování byly hlášeny z klinických studií s vlhkou formou AMD a postmarketingového sledování. Nejčastěji hlášené případy nežádoucích účinků byly zvýšení nitroočního tlaku, přechodná slepota, snížená zraková ostrost, otok rohovky, bolest rohovky a bolest oka. Dojde-li k předávkování, je nutno monitorovat nitrooční tlak a v závislosti na rozhodnutí ošetřujícího lékaře případně nasadit odpovídající terapii k normalizaci nitroočního tlaku.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, látky určené k léčbě neovaskularizace, ATC kód: S01LA04
Ranibizumab je fragment humanizované monoklonální protilátky proti lidskému cévnímu endoteliálnímu růstovému faktoru A (VEGF-A). Váže se se silnou afinitou na VEGF-A isoformy (např. VEGF110, VEGF121 a VEGF165) a tím brání vazbě VEGF-A na receptory VEGFR-1 a VEGFR-2. Vazba VEGF-A na jeho receptory vede k proliferaci endoteliálních buněk a neovaskularizaci, jakož i k propustnosti cév. O všech těchto účincích se uvažuje jako o faktorech přispívajících k progresi neovaskulárních forem věkem podmíněné makulární degenerace, patologické myopie nebo k poškození zraku způsobeného buď diabetickým makulárním edémem, anebo makulárním edémem v důsledku RVO.
Léčba vlhké formy AMD
Klinická bezpečnost a účinnost Lucentisu byla u vlhké formy AMD stanovena ve třech randomizovaných, dvojitě zaslepených studiích po dobu 24 měsíců se simulovanou injekcí nebo aktivním komparátorem u pacientů s neovaskulární AMD. Do dvou studií bylo zařazeno celkem 1 323 pacientů (879 na aktivní léčbě, 444 v kontrolní skupině).
Ve studii FVF2598g (MARINA) bylo 716 pacientů s minimálně klasickými nebo okultními lézemi randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali měsíčně injekce Lucentisu v dávkách 0,3 mg, injekce Lucentisu v dávkách 0,5 mg nebo simulovanou léčbu.
Ve studii FVF2587g (ANCHOR) bylo 423 pacientů s převážně klasickou CNV lézí randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali Lucentis 0,3 mg měsíčně, Lucentis 0,5 mg měsíčně nebo PDT s verteporfinem (při zahájení léčby a potom každé 3 měsíce, pokud fluoresceinová angiografie prokázala přetrvávání nebo rekurenci cévního prosakování).
Nejdůležitější výsledky měření jsou shrnuty v Tabulce 1 a na Obrázku 1.
Tabulka 1 Výsledky ve 12. a 24. měsíci studie FVF2598g (MARINA) a FVF2587g (ANCHOR)
|
FVF2598g (MAR |
ŘINA) |
FVF2587g (ANCHOR) | |||
|
Měřený parametr |
Měsíc |
Simulovaná léčba (n = 238) |
Lucentis 0,5 mg (n = 240) |
Verteporfin PDT (n = 143) |
Lucentis 0,5 mg (n = 140) |
|
Ztráta <15 písmen ostrosti zraku (%)a (zachování zraku, primární cíl) |
12. měsíc |
62 % |
95 % |
64 % |
96 % |
|
24. měsíc |
53 % |
90 % |
66 % |
90 % | |
|
Nárůst >15 písmen ostrosti zraku (%)a |
12. měsíc |
5 % |
34 % |
6 % |
40 % |
|
24. měsíc |
4 % |
33 % |
6 % |
41 % | |
|
Průměrná hodnota změny zrakové ostrosti (písmena) (SD)a |
12. měsíc |
-10,5 (16,6) |
+ 7,2 (14,4) |
-9,5 (16,4) |
+11,3 (14,6) |
|
24. měsíc |
-14,9 (18,7) |
+6,6 (16,5) |
-9,8 (17,6) |
+10,7 (16,5) | |
p<0.01
Obrázek 1 Průměrná hodnota změny zrakové ostrosti od výchozího stavu do 24. měsíce ve studii FVF2598g (MARINA) a ve studii FVF2587g (ANCHOR)
-CD
>
O
co
i_
N
>s
C
><D
E
N
CO
«
O
15 ' 10 ■ 1o 5 '
c
CD
0
_ JS)
■o o.
Žiu^-5
I - -io
>CD w
E 2
°3 -1 ■
S_ (/) -l5
CL O

Studie FVF2598g (MARINA)
+6,6

> +21,5
0 2 4 6
8 10 12 14 16 18 20 22 24
Měsíc

E U -10 I 2 -15 ■
- 1/5 Q_ o
-9'8
r +20,5
0 2 4 6 8
10 12 14 16 18 20 22 24 Měsíc
MARINA
■*■ LUCENTIS 0,5 mg (n=240) & Simulace (n=238)
ANCHOR
«- LUCENTIS 0,5 mg (n=140) * Verteporfin PDT (n=143)
Výsledky obou studií naznačují, že kontinuální léčba ranibizumabem může být také přínosem u pacientů, kteří v prvním roce léčby ztratili >15 písmen z nejlépe korigované ostrosti zraku (BCVA).
Statisticky významná zlepšení zrakových funkcí, hlášená pacienty, byla pozorována v obou studiích MARINA i ANCHOR při léčbě ranibizumabem oproti kontrolní skupině měřeno pomocí NEI VFQ-25.
Ve studii FVF192g (PIER) bylo 184 pacientů se všemi formami neovaskulámí AMD randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali Lucentis 0,3 mg, Lucentis 0,5 mg nebo simulovanou léčbu v injekci jednou měsíčně po první 3 měsíce a dále dávku podávanou každý 3. měsíc. Od 14. měsíce této studie bylo pacientům se simulovanou léčbou povoleno používat ranibizumab a od 19. měsíce byla možná častější aplikace. V průměru obdrželi pacienti léčení Lucentisem ve studii PIER 10 aplikací.
Po počátečním nárůstu zrakové ostrosti (při dávkování jednou měsíčně) poklesla zraková ostrost u pacientů kteří dostávali Lucentis jednou za tři měsíce a vracela se ve 12. měsíci v průměru k počátečnímu stavu; tento účinek byl udržován ve 24. měsíci u většiny pacientů léčených ranibizumabem (82 %). Omezené údaje od osob se simulovanou léčbou, které později dostávaly ranibizumab naznačují, že předčasné zahájení léčby může být spojováno s lepším zachováním ostrosti zraku.
Data ze dvou studií (MONT BLANC, BPD952A2308 a DENALI, BPD952A2309) provedených po registraci přípravku Lucentis potvrdila jeho účinnost, ale neprokázala dodatečný účinek kombinovaného podávání verteporfinu (Visudyne PDT) a Lucentisu v porovnání s monoterapií Lucentisem.
Léčba poškození zraku způsobeného DME
Bezpečnost a účinnost Lucentisu byly hodnoceny ve třech randomizovanných kontrolovaných studiích po dobu alespoň 12 měsíců. Celkem 868 pacientů (708 aktivních a 160 kontrol) bylo zařazeno v těchto studiích.
Ve studii 2. fáze D2201 (RESOLVE) bylo léčeno 151 pacientů ranibizumabem (6 mg/ml, n = 51,
10 mg/ml, n = 51) nebo simulovanou léčbou (n = 49) intravitreální injekcí jednou měsíčně. Průměr určený z průměrných hodnot změn BCVA od 1. do 12. měsíce byl v porovnání se stavem na počátku léčby +7,8 (±7,72) písmen u shromážděných pacientů léčených ranibizumabem (n = 102), v porovnání s -0,1 (±9,77) písmen u pacientů se simulovanou léčbou a průměrná hodnota změny BCVA ve 12. měsíci od počátečního stavu byla 10,3 (±9,1) písmen v porovnání s -1,4 (±14,2) písmen, v tomto pořadí (p<0,0001 pro léčebný rozdíl).
Ve fázi III studie D2301 (RESTORE) bylo 345 pacientů randomizováno v poměru 1:1:1 do skupiny užívající ranibizumab 0,5 mg v monoterapii a simulovanou laserovou fotokoagulaci, kombinaci ranibizumabu 0,5 mg a laserové fotokoagulace nebo simulovanou injekci a laserovou fotokoagulaci. 240 pacientů, kteří předtím dokončili 12měsíční studii RESTORE, bylo zařazeno do otevřené, multicentrické 24měsíční extenze studie (extenze studie RESTORE). Pacienti byli léčeni podáním ranibizumabu 0,5 mg pro re nata (PRN) do stejného oka jako v základní studii (D2301 RESTORE).
Nejdůležitější výsledky měření jsou uvedeny v Tabulce 2 (RESTORE a extenze studie) a na Obrázku 2 (RESTORE).
Obrázek 2 Průměrná hodnota změny zrakové ostrosti od začátku léčby v průběhu času ve studii D2301 (RESTORE)

Ranibizumab 0,5 mg (n=115) Ranibizumab 0,5 mg + laser (n=118) Laser (n=110)
Léčebná skupina
□ □ □
BL=základní stav; SE=směrodatná chyba průměru
* Rozdíl v průměrech nejmenších čtverců, p<0,0001/0,0004 na základě dvoustranného stratifikovaného Cochran-Mantel-Haenszelova testu
Účinek po 12 měsících byl konzistentní ve většině podskupin. Avšak u subjektů s hodnotou BCVA >73 písmen na počátku léčby a makulámím edémem s centrální retinální tloušťkou <300 pm na počátku léčby se nezdálo, že by profitovaly z léčby ranibizumabem v porovnání s laserovou fotokoagulací.
Tabulka 2 Výsledky ve 12. měsíci ve studii D2301 (RESTORE) a ve 36. měsíci ve studii D2301-E1 (extenze studie RESTORE)
|
Výsledky měření ve 12. měsíci v porovnání se stavem na počátku léčby ve studii D2301 (RESTORE) |
Ranibizumab 0,5 mg n = 115 |
Ranibizumab 0,5 mg + laser n = 118 |
laser n = 110 |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 12. měsíce3 (+SD) |
6,1 (6,4)a |
5,9 (7,9)a |
0,8 (8,6) |
|
Průměrná hodnota změny BCVA ve 12. měsíci (+SD) |
6,8 (8,3)a |
6,4 (11,8)a |
0,9 (11,4) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 12. měsíci (%) |
22,6 |
22,9 |
8,2 |
|
Průměrný počet injekcí (0.-11. měsíc) |
7,0 |
6,8 |
7,3 (simulovaný) |
|
Výsledky měření ve 36. měsíci ve studii D2301-E1 (extenze studie RESTORE) v porovnání se stavem na počátku léčby ve studii D2301 (RESTORE) |
Před ranibizumabem 0,5 mg n = 83 |
Před ranibizumabem 0,5 mg + laser n = 83 |
Před laserem n = 74* |
|
Průměrná hodnota změny BCVA ve 24. měsíci (SD) |
7,9 (9,0) |
6,7 (7,9) |
5,4 (9,0) |
|
Průměrná hodnota změny BCVA ve 36. měsíci (SD) |
8,0 (10,1) |
6,7 (9,6) |
6,0 (9,4) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 36. měsíci (%) |
27,7 |
30,1 |
21,6 |
|
Průměrný počet injekcí (12.-35. měsíc)* |
6,8 |
6,0 |
6,5 |
ap<0,0001 pro porovnání ramen s ranibizumabem vs. rameno s laserem. n je ve studii D2301-E1 (extenze studie RESTORE) počet pacientů s hodnotou ve studii D2301 (RESTORE) na počátku léčby (měsíc 0) a při návštěvě ve 36. měsíci.
• Podíl pacientů, kteří nevyžadovali léčbu ranibizumabem během fáze prodloužení studie byl 19 % ve skupině před podáním ranibizumabu, 25 % ve skupině před podáním ranibizumabu + laseru a 20 % ve skupině před aplikací laseru.
Statisticky signifikantní přínosy pro většinu funkcí spojených se zrakem, hlášené pacienty, byly pozorovány u léčby ranibizumabem (s nebo bez laseru) oproti kontrolní skupině, měřeno pomocí NEI VFQ-25. Pro ostatní podškály tohoto dotazníku nemohly být stanoveny žádné léčebné rozdíly.
Dlouhodobý bezpečnostní profil ranibizumabu pozorovaný ve 24měsíční extenzi studie je konzistentní se známým bezpečnostním profilem přípravku Lucentis.
Ve fázi IlIb studie D2304 (RETAIN) bylo 372 pacientů randomizováno v poměru 1:1:1 k podávání:
• ranibizumabu 0,5 mg se současnou laserovou fotokoagulací v režimu „treat-and-extend“ (TE) ,
• ranibizumabu 0,5 mg v monoterapii v režimu TE,
• ranibizumabu 0,5 mg v monoterapii v režimu PRN.
Ve všech skupinách byl ranibizumab podáván jednou měsíčně, dokud BCVA nebyla stabilní po alespoň tři po sobě jdoucí měsíční vyšetření. V režimu TE byl podán ranibizumab v léčebných intervalech 2-3 měsíce. Ve všech skupinách byla měsíční léčba znovu zahájena na základě snížení BCVA způsobeném progresí DME a pokračovala, dokud nebylo opět dosaženo stabilní BCVA.
Počet plánovaných léčebných návštěv po 3 počátečních injekcích byl 13 v režimu TE a 20 v režimu PRN. V obou TE režimech bylo více než 70 % pacientů schopných zachovat svou BCVA při průměrné četnosti návštěv >2 měsíce.
Nejdůležitější výsledky měření jsou uvedeny v Tabulce 3.
Tabulka 3 Výsledky ve studii D2304 (RETAIN)
|
Výsledek měření porovnaný s výchozím stavem |
ranibizumab 0,5 mg + laser v režimu TE n = 117 |
ranibizumab 0,5 mg samotný v režimu TE n = 125 |
ranibizumab 0,5 mg v režimu PRN n = 117 |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 12. měsíce (SD) |
5,9 (5,5)a |
6,1 (5,7)a |
6,2 (6,0) |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 24. měsíce (SD) |
6,8 (6,0) |
6,6 (7,1) |
7,0 (6,4) |
|
Průměrná hodnota změny BCVA ve 24. měsíci (SD) |
8,3 (8,1) |
6,5 (10,9) |
8,1 (8,5) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 24. měsíci (%) |
25,6 |
28,0 |
30,8 |
|
Průměrný počet injekcí (0.-23. měsíc) |
12,4 |
12,8 |
10,7 |
ap<0,0001 pro hodnocení non-inferiority k PRN
Ve studiích zaměřených na DME bylo zlepšení BCVA doprovázeno redukcí střední CSFT v průběhu času ve všech léčebných skupinách.
Léčba poškození zraku způsobeného makulámím edémem v důsledku RVO
Klinická bezpečnost a účinnost Lucentisu u pacientů s poškozením zraku způsobeným makulárním
edémem v důsledku RVO byla hodnocena v randomizovaných dvojitě zaslepených kontrolovaných
studiích BRAVO a CRUISE, ve kterých byly zařazeny subjekty s BRVO (n = 397) a CRVO
(n = 392). V obou studiích dostávaly subjekty buď 0,3 mg nebo 0,5 mg ranibizumabu nebo injekce
simulované léčby. Pacienti v kontrolním rameni se simulovanou léčbou byli po 6 měsících přeřazeni
do ramene s ranibizumabem 0,5 mg.
Nejdůležitější výsledky měření ze studií BRAVO a CRUISE jsou shrnuty v Tabulce 4 a na Obrázcích 3 a 4.
Tabulka 4 Výsledky v 6. a 12. měsíci (BRAVO a CRUISE)
|
BRAVO |
CRUISE | |||
|
Simulovaná léčba/Lucentis 0,5 mg (n = 132) |
Lucentis 0,5 mg (n = 131) |
Simulovaná léčba /Lucentis 0,5 mg (n = 130) |
Lucentis 0,5 mg (n = 130) | |
|
Průměrná hodnota změny zrakové ostrosti v 6. měsícía (písmena) (SD) (primární cíl) |
7,3 (13,0) |
18,3 (13,2) |
0,8 (16,2) |
14,9 (13,2) |
|
Průměrná hodnota změny BCVA ve 12. měsíci (písmena) (SD) |
12,1 (14,4) |
18,3 (14,6) |
7,3 (15,9) |
13,9 (14,2) |
|
Nárůst o > 15 písmen u zrakové ostrosti v 6. měsícia (%) |
28,8 |
61,1 |
16,9 |
47,7 |
|
Nárůst o > 15 písmen u zrakové ostrosti ve 12. měsíci (%) |
43,9 |
60,3 |
33,1 |
50,8 |
|
Podíl (%) pacientů léčených laserovou záchrannou terapií po 12 měsíců |
61,4 |
34,4 |
NA |
NA |
ap <0,0001 v obou studiích

CL
Léčebná skupina
r m-m—m
Měsíc
Placebo/Ranibizumab 0,5 mg (n = 132 Ranibizumab 0,5 mg (n = 131)
BL=základní stav; SE=směrodatná chyba průměru

Léčebná skupina
Placebo/Ranibizumab 0,5 mg (n = 130) Ranibizumab 0,5 mg (n = 130)
BL=základní stav; SE=směrodatná chyba průměru
V obou studiích bylo zlepšení zraku spojeno s kontinuální a významnou redukcí makulámího edému, což bylo posuzováno podle tloušťky středu sítnice.
U pacientů s CRVO (CRUISE a extenze studie HORIZON): Subjekty léčené simulovanou léčbou v prvních 6 měsících, které následně dostávaly ranibizumab, nedosáhly srovnatelných nárůstů zrakové ostrosti (VA) do 24. měsíce (~6 písmen) v porovnání se subjekty léčenými ranibizumabem od začátku studie (~12 písmen).
Statisticky významná zlepšení v subškálách spojená s viděním na blízko a na dálku, hlášená pacienty, byla pozorována při léčbě ranibizumabem oproti kontrolní skupině, měřeno pomocí NEI VFQ-25.
Dlouhodobá (24měsíční) klinická bezpečnost a účinnost Lucentisu u pacientů s poškozením zraku způsobeného sekundárně makulárním edémem v důsledku RVO byla hodnocena ve studiích BRIGHTER (BRVO) a CRYSTAL (CRVO). V obou studiích bylo subjektům podáváno 0,5 mg ranibizumabu v režimu PRN řízeného individualizovanými kritérii stabilizace zrakové ostrosti. BRIGHTER byla 3ramenná randomizovaná aktivně kontrolovaná studie, která porovnávala 0,5 mg ranibizumabu podávaného v monoterapii nebo v kombinaci s adjuvantní laserovou fotokoagulací oproti samotné laserové fotokoagulaci. Subjektům v laserovém rameni mohl být po 6 měsících podán ranibizumab v dávce 0,5 mg. CRYSTAL byla jednoramenná studie s 0,5 mg ranibizumabu v monoterapii.
Důležité výstupy ze studií BRIGHTER a CRYSTAL jsou uvedeny v Tabulce 5. Tabulka 5 Výsledky v 6. a 24. měsíci studie (BRIGHTER a CRYSTAL)
|
BRIGHTER |
CRYSTAL | |||
|
Lucentis 0,5 mg n=180 |
Lucentis 0,5 mg + laser n=178 |
Laser* n=90 |
Lucentis 0,5 mg n=356 | |
|
Průměrná hodnota změny BCVA v 6. měsícia (písmena) (SD) |
+14,8 (10,7) |
+14,8 (11,13) |
+6,0 (14,27) |
+12,0 (13,95) |
|
Průměrná hodnota změny BCVA ve 24. měsícib (písmena) (SD) |
+15,5 (13,91) |
+17,3 (12,61) |
+11,6 (16,09) |
+12,1 (18,60) |
|
Zisk >15 písmen BCVA ve 24. měsíci (%) |
52,8 |
59,6 |
43,3 |
49,2 |
|
Průměrný počet injekcí (SD) (měsíc 0-23) |
11,4 (5,81) |
11,3 (6,02) |
NA |
13,1 (6,39) |
|
a p<0,0001 pro obě porovnání ve studii BRIGHTER v 6. měsíci: Lucentis 0,5 mg vs laser a Lucentis 0,5 mg + laser vs laser. b p<0,0001 pro nulovou hypotézu ve studii CRYSTAL, že průměrná hodnota změny ve 24. měsíci od výchozího stavu je nula. * Od 6. měsíce byla umožněna léčba 0,5 mg ranibizumabu (24 pacientů bylo léčeno pouze laserem). | ||||
Ve studii BRIGHTER byla prokázána non-inferiorita 0,5 mg ranibizumabu s podpůrnou laserovou terapií ve srovnání s ranibizumabem v monoterapii od výchozího stavu do 24. měsíce (95 % CI -2,8; 1,4).
V obou studiích byla v 1. měsíci pozorována rychlá a statisticky významná redukce tloušťky centrální části sítnice od výchozího stavu. Tento účinek přetrval až do 24. měsíce.
Účinek léčby ranibizumabem byl podobný bez ohledu na přítomnost retinální ischemie. Ve studii BRIGHTER u pacientů s ischemií (n=46) nebo bez ischemie (n=133) léčených ranibizumabem v monoterapii byla průměrná hodnota změny BCVA ve 24. měsíci od výchozího stavu +15,3; resp. +15,6 písmen. Ve studii CRYSTAL u pacientů s ischemií (n=53) nebo bez ischemie (n=300) léčených ranibizumabem v monoterapii byla průměrná hodnota změny BCVA od výchozího stavu +15,0; resp. +11,5 písmen.
V obou studiích BRIGHTER a CRYSTAL byl účinek na zlepšení zraku pozorován u všech pacientů léčených 0,5 mg ranibizumabu v monoterapii bez ohledu na délku trvání onemocnění. U pacientů
s délkou trvání onemocnění <3 měsíce byl pozorován nárůst zrakové ostrosti o 13,3; resp. 10,0 písmen v 1. měsíci a o 17,7; resp. 13,2 písmen ve 24. měsíci ve studiích BRIGHTER a CRYSTAL. Odpovídající zisk zrakové ostrosti u pacientů s délkou trvání onemocnění >12 měsíců byl v těchto studiích 8,6; resp. 8,4 písmen. Je třeba zvážit zahájení léčby v čase stanovení diagnózy.
Dlouhodobý bezpečnostní profil ranibizumabu pozorovaný ve 24měsíčních studiích je konzistentní se známým bezpečnostním profilem přípravku Lucentis.
Léčba poškození zraku způsobeného CNV sekundární k PM
Klinická bezpečnost a účinnost přípravku Lucentis u pacientů s poškozením zraku způsobeným CNV u PM byla hodnocena na základě 12měsíčních dat z dvojitě zaslepené, kontrolované pivotní studie F2301 (RADIANCE). V této studii bylo 277 pacientů randomizováno v poměru 2:2:1 do následujících ramen:
• Skupina I (ranibizumab 0,5 mg, dávkovací schéma řízené kritérii „stability“, definovanými jako žádná změna BCVA v porovnání se dvěma předchozími měsíčními vyhodnoceními).
• Skupina II (ranibizumab 0,5 mg, dávkovací schéma řízené kritérii „aktivity onemocnění“, definovanými jako poškození zraku způsobené intra- nebo subretinální tekutinou nebo aktivním prosakováním v důsledku CNV léze, jak bylo zjištěno při vyšetření OCT a/nebo FA).
• Skupina III (vPDT - pacientům bylo povoleno užívat léčbu ranibizumabem od 3. měsíce studie).
Ve skupině II, což je doporučené dávkovací schéma (viz bod 4.2), vyžadovalo 50,9 % pacientů 1 nebo 2 injekce, 34,5 % pacientů 3 až 5 injekcí a 14,7 % vyžadovalo 6 až 12 injekcí po dobu 12 měsíců trvání studie. 62,9 % pacientů ze skupiny II nevyžadovalo injekce ve druhé polovině trvání studie.
Důležité výstupy ze studie RADIANCE jsou uvedeny v Tabulce 6 a na Obrázku 5.
Tabulka 6 Výsledky ve 3. a 12. měsíci studie (RADIANCE)
|
Skupina I |
Skupina II |
Skupina | |
|
Ranibizumab |
Ranibizumab |
III | |
|
0,5 mg |
0,5 mg |
vPDTb | |
|
„stabilizace |
„aktivita | ||
|
vidění“ |
onemocnění“ | ||
|
(n = 105) |
(n = 116) |
(n = 55) | |
|
3. měsíc Průměr určený z průměrných hodnot změn BCVA od 1. do 3. měsíce studie v porovnání se základním stavema (písmena) Podíl pacientů, kteří dosáhli: >15 písmen, nebo dosáhli >84 písmen |
+10,5 |
+10,6 |
+2,2 |
|
BCVA 12. měsíc |
38,1 % |
43,1 % |
14,5 % |
|
Počet injekcí až do 12. měsíce: Průměrná hodnota |
4,6 |
3,5 |
N/A |
|
Medián |
4,0 |
2,5 |
N/A |
|
Průměr určený z průměrných hodnot změn BCVA od 1. do 12. měsíce v porovnání se základním stavem (písmena) Podíl pacientů, kteří dosáhli: >15 písmen, nebo dosáhli >84 písmen |
+12,8 |
+12,5 |
N/A |
|
BCVA |
53,3 % |
51,7 % |
N/A |
a p <0,00001 porovnání s vPDT kontrolní skupinou
b Srovnávací kontrola až do 3. měsíce studie. Pacientům randomizovaným do skupiny s vPDT bylo povoleno dostávat léčbu ranibizumabem od 3. měsíce studie (ve skupině III dostávalo ranibizumab 38 pacientů od 3. měsíce studie)
Průměrná hodnota změny BCVA od začátku léčby v průběhu času do 12. měsíce (RADIANCE)

|
Měsíc | ||
|
___ Ranibizumab 0,5 mg Skupina I ^ ^ ^ v , . . pn stabilizaci (n = 105) |
EI-EHD |
Ranibizumab 0,5 mg Skupina II při aktivitě onemocnění (n = 116) |
|
a-a-a PDT verteporfinem Skupina III(n = 55) |
A-A-A |
Ranibizumab 0,5 mg/PDT verteporfinem Skupina III od 3. měsíce studie dále (n = 55) |
Zlepšení zraku bylo doprovázeno redukcí tloušťky centrální části sítnice.
Prospěch hlášený pacientem byl pozorován v léčebných ramenech s ranibizumabem oproti vPDT (hodnota p <0,05) na základě zlepšení kombinovaného skóre a několika subškál (všeobecné vidění, aktivity do blízka, duševní zdraví a závislost) v NEI VFQ-25.
Pediatrická populace
Bezpečnost a účinnost ranibizumabu nebyla u pediatrických pacientů dosud studována.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lucentis u všech podskupin pediatrické populace u neovaskulární formy AMD, u poškození zraku kvůli DME, u poškození zraku kvůli makulárnímu edému v důsledku RVO a u poškození zraku v důsledku CNV sekundární k PM (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Po měsíčních aplikacích Lucentisu do sklivce jedincům s neovaskulámí AMD byly koncentrace ranibizumabu v séru obvykle nízké. Maximální koncentrace (Cmax) ranibizumabu byly obecně nižší než koncentrace ranibizumabu nutné k inhibici biologické aktivity VEGF o 50 % (11 - 27 ng/ml, stanoveno in vitro titrací buněčné proliferace). Cmax byla závislá na dávce v dávkovém rozmezí 0,05 až
1.0 mg/oko. Sérové koncentrace u limitovaného počtu pacientů s DME ukazují, že lehce vyšší systémová expozice nemůže být vyloučena ve srovnání se sérovými koncentracemi zjištěnými
u pacientů s neovaskulámí AMD. Sérové koncentrace ranibizumabu u pacientů s RVO byly podobné nebo lehce vyšší v porovnání s koncentracemi pozorovanými u pacientů s neovaskulámí AMD.
Na základě analýz populační farmakokinetiky a vymizení ranibizumabu ze séra pacientů s neovaskulámí AMD léčených dávkou 0,5 mg byl průměrný poločas eliminace ranibizumabu ze sklivce stanoven na přibližně 9 dnů. Při aplikaci Lucentisu v dávce 0,5 mg/oko jednou měsíčně je dosaženo sérové Cmax ranibizumabu, jejíž předpokládané rozmezí je mezi 0,79 a 2,90 ng/ml přibližně za 1 den po aplikaci. Cmin se předpokládá v rozmezí mezi 0,07 a 0,49 ng/l. Sérové koncentrace ranibizumabu se předpokládají přibližně 90 000násobně nižší než koncentrace ranibizumabu ve sklivci.
Pacienti se zhoršenou funkcí ledvin: Žádné cílené studie, které by hodnotily farmakokinetiku Lucentisu u pacientů se zhoršenou funkcí ledvin, nebyly provedeny. 68 % (136 ze 200) pacientů s neovaskulámí AMD mělo v populační farmakokinetické analýze zhoršenou funkci ledvin (46,5 % mírně [50 - 80 ml/min], 20 % středně [30 - 50 ml/min] a 1,5 % silně [< 30 ml/min]). 48,2 % (253 z 525) pacientů s RVO mělo zhoršenou funkci ledvin (36,4 % mírně, 9,5 % středně a 2,3 % silně). Systémová clearance byla lehce snížena, ale tento nález byl bez klinického významu.
Zhoršená funkce jater: Žádné cílené studie, které by hodnotily farmakokinetiku u pacientů se zhoršenou funkcí jater léčených Lucentisem, nebyly provedeny.
5.3 Předklinické údaje vztahující se k bezpečnosti
Oboustranná aplikace ranibizumabu do sklivce opicím cynomolgus v dávkách 0,25 mg/oko a
2.0 mg/oko jednou za dva týdny po dobu 26 týdnů vyvolala účinky na očích závislé na dávce.
Nitrooční aplikace vyvolala na dávce závislé zvýšené zarudnutí přední komory a zvýšení počtu buněk, největší za 2 dny po injekci. Závažnost zánětlivé odpovědi se obvykle zmenšila po následujících injekcích nebo během zotavení. V zadním segmentu byla pozorována infiltrace sklivce buňkami a sklivcovými vločkami, u které byla také tendence k závislosti na dávce a obvykle přetrvávala až do konce léčby. Ve 26týdenní studii se intenzita zánětu sklivce zvyšovala s počtem injekcí. Průkaz reverzibility byl však pozorován po zotavení. Povaha a načasování zánětu zadního segmentu naznačují, že se jedná o imunologicky zprostředkovanou protilátkovou odpověď, která by mohla být klinicky irelevantní. U některých zvířat byla po relativně dlouhém období intenzivního zánětu pozorována tvorba katarakty naznačující, že změny čočky byly sekundární, vyvolané silným zánětem. Bylo pozorováno přechodné zvýšení nitroočního tlaku po podání do sklivce bez ohledu na dávku.
Mikroskopické změny pozorované v oční tkáni souvisely se zánětem a nenasvědčovaly degenerativním procesům. U některých očí byly pozorovány granulomatózní zánětlivé změny na očním disku. Tyto změny zadního segmentu slábly, a v některých případech kompletně vymizely, během období zotavení.
Po podání do sklivce nebyly zjištěny žádné známky systémové toxicity. U některých zvířat byly nalezeny protilátky proti ranimizumabu v séru i sklivci.
Nejsou dostupné žádné údaje o karcinogenitě nebo mutagenitě.
U březích opic nevyvolala léčba ranibizumabem aplikovaná do sklivce , mající za následek maximální systémové expozice 0,9-7-krát nejhorší případ klinické expozice, vývojovou toxicitu ani teratogenitu, a neměla žádný vliv na hmotnost nebo strukturu placenty, i když na základě svého farmakologického účinku by měl být ranibizumab považován za potenciálně teratogenní a embryo-/fetotoxický.
Chybění účinků zprostředkovaných ranibizumabem na embryo-fetální vývoj se pravděpodobně týká hlavně neschopnosti Fab-fragmentu procházet placentou. Přesto byl popsán případ s vysokými mateřskými sérovými hladinami ranibizumabu a přítomností ranibizumabu ve fetálním séru, nasvědčující tomu, že protilátky proti ranibizumabu působily jako nosičský protein (obsahující Fc část) pro ranibizumab, a tím snižovaly jeho mateřskou sérovou clearance a umožňovaly prostup placentou. Protože embryo-fetální vývojová vyšetření byla prováděna u zdravých březích zvířat a onemocnění (jako například diabetes) mohou modifikovat prostupnost placenty pro Fab-fragment, měla by být studie interpretována s opatrností.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát a,a-trehalosy
Monohydrát histidin-hydrochloridu
Histidin
Polysorbát 20
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte předplněnou injekční stříkačku v jejím zataveném obalu v krabičce, aby byl přípravek chráněn před světlem.
Před použitím může být neotevřený obal ponechán při pokojové teplotě (25 °C) po dobu 24 hodin.
6.5 Druh obalu a obsah balení
Sterilní roztok o objemu 0,165 ml v předplněné injekční stříkačce (sklo třídy I) s bromobutylovou pryžovou pístovou zarážkou a víčkem, skládajícím se z bílého, pevného a proti nedovolené manipulaci odolného těsného uzávěru s šedou bromobutylovou pryžovou čepičkou včetně adaptéru typu Luer lock. Předplněná injekční stříkačka má pístové táhlo a držadlo, a je zabalena v zataveném obalu.
Balení o velikosti jedné předplněné injekční stříkačky.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Předplněná injekční stříkačka je pouze pro jednorázové použití. Předplněná injekční stříkačka je sterilní. Přípravek nepoužívejte, pokud je balení poškozeno. Sterilita předplněné injekční stříkačky nemůže být zaručena, pokud nezůstane obal zatavený. Předplněnou injekční stříkačku nepoužívejte, pokud má roztok změněnou barvu, je zakalený nebo obsahuje částice.
Předplněná injekční stříkačka obsahuje více než doporučenou dávku 0,5 mg. Extrahovatelný objem předplněné injekční stříkačky (0,1 ml) není určen k celkovému podání. Nadbytečný objem je třeba před podáním injekce vytlačit. Při podání celého objemu předplněné injekční stříkačky by mohlo dojít k předávkování. K vytlačení vzduchové bubliny spolu s nadbytečným léčivým přípravkem pomalu tlačte píst, dokud okraj pod vyklenutím pryžové zarážky není vyrovnaný s černou dávkovací linkou na injekční stříkačce (ekvivalentní 0,05 ml, tj. 0,5 mg ranibizumabu).
Pro podání do sklivce je třeba použít sterilní injekční jehlu 30G x 'A".
Při přípravě přípravku Lucentis k podání do sklivce dbejte, prosím, pokynů k použití:
Před použitím předplněné injekční stříkačky si pečlivě přečtěte všechny pokyny. Předplněná injekční stříkačka je pouze pro jednorázové použití. Předplněná injekční stříkačka je sterilní. Přípravek nepoužívejte, pokud je balení poškozeno. Otevření zataveného obalu a všechny následující kroky musí být provedeny za aseptických podmínek.
Poznámka: dávka musí být nastavena na 0,05 ml._
Čepička
Označení dávky
Držadlo

Adaptér Luerlock
Pryžová zarážka
Pístové táhlo
Obrázek 1
|
Příprava |
1. Ujistěte se, že balení obsahuje: • sterilní předplněnou injekční stříkačku v zataveném obalu. 2. Sloupněte víčko zataveného obalu injekční stříkačky a za použití aseptické techniky opatrně vyjměte injekční stříkačku. | |
|
Zkontrolujte injekční stříkačku |
3. Zkontrolujte, že: • čepička injekční stříkačky není oddělena od adaptéru Luer lock. • injekční stříkačka není poškozena. • injekční roztok je čirý, bezbarvý až světle žlutý a neobsahuje žádné částice. 4. Pokud některý údaj uvedený výše nesouhlasí, znehodnoťte předplněnou injekční stříkačku a použijte novou. | |
5. Ulomte (neotáčejte ani nekraťte) čepičku injekční stříkačky (viz Obrázek 2).
6. Zlikvidujte čepičku injekční stříkačky (viz Obrázek 3).

Obrázek 2

Obrázek 3
7. Pevně nasaďte sterilní injekční jehlu 30G x Í4'' na injekční stříkačku těsným zašroubováním do adaptéru Luer lock (viz Obrázek 4).
8. Opatrně odstraňte kryt j ehly j eho přímým vytažením (viz Obrázek 5).
Poznámka: Jehlu nikdy neotírejte.

0

Držte injekční stříkačku svisle.
Pokud jsou přítomny vzduchové bubliny, jemně poklepejte injekční stříkačku prstem, dokud bubliny stoupají nahoru (viz Obrázek 6).
Obrázek 4 Obrázek 5

Obrázek 6
Nastavte dávku
11. Držte injekční stříkačku v úrovni očí a opatrně tlačte píst, dokud není okraj pod vyklenutím pryžové zarážky vyrovnaný s označením dávky (viz Obrázek 7). Tak se vytlačí vzduch a nadbytek injekčního roztoku a nastaví se dávka na 0,05 ml.
Poznámka: Pístové táhlo není připojeno k pryžové zarážce - to aby se předešlo natažení vzduchu zpět do injekční stříkačky.

Obrázek 7
Podejte injekci
Postup podání injekce má být proveden za aseptických podmínek.
12. Injekční jehla se zasune 3,5-4,0 mm posteriorně k limbu do prostoru sklivce tak, aby směřovala do centra očního bulbu a nikoli k horizontálnímu meridiánu.
13. Injekci podávejte pomalu, dokud pryžová zarážka nedosáhne dna injekční stříkačky, aby byl podán objem 0,05 ml.
14. Následující injekce aplikujte do odlišných míst skléry.
15. Po podání inj ekce nenasazujte zpět kryt j ehly ani j ehlu neoddělujte od injekční stříkačky. Zlikvidujte použitou injekční stříkačku společně
s jehlou vyhozením do nádoby na ostré předměty nebo v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/06/374/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 22. ledna 2007
Datum posledního prodloužení registrace: 24. ledna 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Lucentis 10 mg/ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje ranibizumabum 10 mg*. Jedna injekční lahvička obsahuje ranibizumabum 2,3 mg v 0,23 ml roztoku.
*Ranibizumab je fragment humanizované monoklonální protilátky produkovaný buňkami Escherichia coli rekombinantní DNA technologií.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok
Čirý, bezbarvý až světle žlutý vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Lucentis je indikován u dospělých:
• k léčbě neovaskulární (vlhké) formy věkem podmíněné makulární degenerace (AMD)
• k léčbě poškození zraku způsobeného diabetickým makulárním edémem (DME)
• k léčbě poškození zraku způsobeného makulárním edémem v důsledku okluze retinální vény [uzávěr větve centrální retinální vény (BRVO) a uzávěr centrální retinální vény (CRVO)]
• k léčbě poškození zraku způsobeného choroidální neovaskularizací (CNV) sekundární k patologické myopii (PM)
4.2 Dávkování a způsob podání
Lucentis musí být aplikován kvalifikovaným oftalmologem zkušeným v podání do sklivce.
Doporučená dávka Lucentisu je 0,5 mg podávaných jako jednorázová injekce do sklivce. To odpovídá injekci o objemu 0,05 ml. Interval mezi dvěma dávkami podávanými do stejného oka má být alespoň čtyři týdny.
Léčba se zahajuje jednou injekcí za měsíc do dosažení maximální zrakové ostrosti a/nebo do vymizení příznaků aktivity onemocnění, tj. žádná změna zrakové ostrosti a ostatních příznaků a projevů onemocnění při probíhající léčbě. U pacientů s vlhkou formou AMD, DME a RVO mohou být zpočátku potřeba tři nebo více po sobě jdoucí injekce podávané jednou za měsíc.
Následně mají být lékařem určeny intervaly sledování a léčby a mají být stanoveny na základě aktivity onemocnění vyhodnocené podle zrakové ostrosti a/nebo anatomických parametrů.
Pokud zrakové a anatomické parametry ukazují na základě vyjádření lékaře, že pokračující léčba pacienta není přínosná, je třeba léčbu přípravkem Lucentis ukončit.
Sledování aktivity onemocnění může zahrnovat klinické vyšetření, funkční testy nebo zobrazovací techniky (např. optickou koherenční tomografii nebo fluorescenční angiografii).
Pokud j sou pacienti léčeni podle režimu „treat-and-extend“, pak po dosažení maximální zrakové ostrosti a/nebo vymizení příznaků aktivity onemocnění, mohou být léčebné intervaly postupně prodlouženy až do opětovného objevení se příznaků aktivity onemocnění nebo zhoršení zraku. Léčebný interval nemá být prodloužen o více než dva týdny najednou u vlhké formy AMD a může být prodloužen až o jeden měsíc najednou u DME. Při uzávěru retinální vény mohou být léčebné intervaly také postupně prodlužovány, ale nejsou dostupná dostatečná data dokládající délku těchto intervalů. Pokud se aktivita onemocnění znovu objeví, léčebný interval má být příslušně zkrácen.
V léčbě poškození zraku způsobeném CNV sekundární k PM může mnoho pacientů potřebovat pouze jednu nebo dvě injekce během prvního roku, zatímco někteří pacienti mohou potřebovat častější léčbu (viz bod 5.1).
Lucentis a laserová fotokoagulace u DME a makulárního edému v důsledku BRVO Existuje určitá zkušenost s podáním přípravku Lucentis společně s laserovou fotokoagulací (viz bod 5.1). Při podání ve stejný den by měl být Lucentis podán alespoň 30 minut po laserové fotokoagulaci. Lucentis může být podán pacientům, kteří byli léčeni předchozí laserovou fotokoagulací.
Lucentis a fotodynamická léčba Visudynem u CNV sekundární k PM Se souběžným podáváním přípravků Lucentis a Visudyne nejsou žádné zkušenosti.
Zvláštní populace
Zhoršená funkce jater
Účinky přípravku Lucentis u pacientů se zhoršenou funkcí jater nebyly hodnoceny. Pro tuto populaci však nejsou nutná žádná zvláštní opatření.
Zhoršená funkce ledvin
U pacientů se zhoršenou funkcí ledvin není nutné upravovat dávku (viz bod 5.2).
Starší jedinci
U starších jedinců není nutná úprava dávky. U pacientů s DME starších 75 let jsou omezené zkušenosti.
Pediatrická populace
Bezpečnost a účinnost Lucentisu u dětí a dospívajících pod 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Injekční lahvička na jedno použití, pouze pro podání do sklivce.
Před aplikací je nutno Lucentis vizuálně zkontrolovat, zda neobsahuje cizí částice nebo není změněna jeho barva.
Lucentis musí být aplikován za aseptických podmínek, což zahrnuje použití chirurgické desinfekce rukou, sterilních rukavic, sterilního oděvu, sterilního spekula (nebo ekvivalentní náhrady) a dostupnost sterilní paracentézy (je-li potřeba). Před aplikací injekce do sklivce je nutný pečlivý odběr anamnézy z hlediska hypersenzitivních reakcí (viz bod 4.4). Před aplikací injekce musí být podána adekvátní anestezie a použit širokospektrý lokální antimikrobiální přípravek k dezinfekci pokožky kolem očí, očního víčka a povrchu oka, v souladu s lokální praxí.
Pro informace o přípravě Lucentisu viz bod 6.6.
Injekční jehla se zasune 3,5-4,0 mm posteriomě od limbu do prostoru sklivce tak, aby směřovala do centra očního bulbu a nikoli k horizontálnímu meridiánu. Poté se aplikuje objem injekce 0,05 ml. Následující injekci je nutné aplikovat v jiném místě skléry.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Pacienti s aktivní nebo suspektní oční nebo periokulární infekcí.
Pacienti s aktivním těžkým nitroočním zánětem.
4.4 Zvláštní upozornění a opatření pro použití Reakce po podání injekce do sklivce
Podání do sklivce, včetně těch s Lucentisem, byly spojovány s endoftalmitidou, intraokulárním zánětem, rhegmatogenním odchlípením sítnice, trhlinami sítnice a iatrogenní traumatickou kataraktou (viz bod 4.8). Při aplikaci Lucentisu musí být vždy dodržena přísná pravidla asepse. V následujícím týdnu po aplikaci injekce musejí být pacienti sledováni z hlediska případného výskytu infekce, aby bylo možné zahájit včas adekvátní léčbu. Pacienty je nutno upozornit, že musejí ihned hlásit všechny příznaky možné endoftalmitidy nebo jiných výše popsaných komplikací.
Zvýšení nitroočního tlaku
Během 60 minut po injekci Lucentisu bylo pozorováno přechodné zvýšení nitroočního tlaku (IOP). Trvalá zvýšení IOP byla také zjištěna (viz bod 4.8). Je nutné monitorovat a náležitě ošetřit jak nitrooční tlak, tak i perfuzi papily očního nervu.
Léčba obou očí současně
Omezená data k léčbě obou očí přípravkem Lucentis současně (včetně podání ve stejný den) nenaznačují zvýšené riziko systémových nežádoucích účinků v porovnání s léčbou jednoho oka.
Imunogenicita
U Lucentisu existuje možnost vzniku imunogenicity. Protože existuje potenciál pro zvýšenou systémovou expozici u pacientů s DME, zvýšené riziko pro vznik hypersenzitivity u této pacientské populace nelze vyloučit. Pacienti musejí být také poučeni, aby hlásili zhoršení závažnosti nitroočního zánětu, protože se může jednat o klinický příznak charakteristický pro tvorbu nitroočních protilátek.
Současné použití jiných anti-VEGF (vaskulární endoteliální růstový faktor) léčivých přípravků Lucentis se nesmí podávat zároveň s jinými anti-VEGF léčivými přípravky (systémovými nebo očními).
Vynechání dávky Lucentisu
Dávku je nutno vynechat a v léčbě se nesmí pokračovat dříve, než je plánována další dávka v následujících případech:
• snížení nejlépe upravené ostrosti zraku (best-corrected visual acuity BCVA) o > 30 písmen ve srovnání s předchozím měřením ostrosti zraku;
• nitrooční tlak > 30 mmHg;
• poškození sítnice;
nebo je-li velikost hemoragie > 50 % celkové zákrok během uplynulých nebo následujících
• subretinální krvácení zahrnující střed fovey, plochy léze;
• provedený nebo plánovaný oční chirurgický 28 dnů.
Trhlina pigmentového epitelu sítnice
Rizikové faktory spojené s vývojem trhliny pigmentového epitelu sítnice po podání anti-VEGF léčby u vlhké formy AMD zahrnují rozsáhlé a/nebo značné odchlípení pigmentového epitelu sítnice.
U pacientů s těmito rizikovými faktory pro vznik trhlin pigmentového epitelu sítnice je třeba dbát opatrnosti při zahajování léčby Lucentisem.
Rhegmatogenní odchlípení sítnice nebo makulární díry
Léčbu je nutno přerušit u subjektů s rhegmatogenním odchlípením sítnice nebo u makulárních děr stupně 3 nebo 4.
Populace s omezenými daty
Existují pouze omezené zkušenosti s léčbou u pacientů s DME způsobeným diabetem I. typu. Lucentis nebyl studován u pacientů, kterým byla dříve podána injekce do sklivce, u pacientů s aktivními systémovými infekcemi, proliferativní diabetickou retinopatií nebo u pacientů se současnými očními onemocněními jako například odchlípení sítnice nebo makulární díra. Také nejsou zkušenosti s léčbou Lucentisem u diabetických pacientů s HbA1c nad 12 % a nekontrolovanou hypertenzí. Chybění těchto informací má být lékařem zváženo při léčbě takových pacientů.
Nejsou k dispozici dostatečné údaje k posouzení účinnosti Lucentisu u pacientů s RVO projevujícím se ireverzibilní ztrátou zraku v důsledku ischemie.
U pacientů s PM, kteří v minulosti podstoupili neúspěšnou fotodynamickou léčbu verteporfinem (vPDT) jsou k dispozici limitovaná data o účinku přípravku Lucentis. Rovněž zatímco byl pozorován odpovídající účinek u subjektů se subfoveálními a juxtafoveálními lézemi, jsou data k vyvození závěru o účinku přípravku Lucentis u subjektů s patologickou myopií s extrafoveálními lézemi nedostatečná.
Systémové účinky po podání do sklivce
Po injekční aplikaci VEGF inhibitorů do sklivce byly hlášeny systémové nežádoucí příhody včetně mimoočních krvácení a arteriálních tromboembolických příhod.
Data o bezpečnosti při léčbě pacientů s DME, makulárním edémem způsobeným RVO a CNV sekundární k patologické myopii (PM) u pacientů s mrtvicí nebo přechodnými ischemickými příhodami v anamnéze jsou omezená. U těchto pacientů je třeba dbát zvýšené opatrnosti (viz bod 4.8).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí.
Současné použití Lucentisu s fotodynamickou léčbou (PDT) verteporfinem u vlhké formy AMD a PM, viz bod 5.1.
Současné použití Lucentisu s laserovou fotokoagulací u DME a BRVO, viz body 4.2 a 5.1.
V klinických studiích zaměřených na léčbu poškození zraku způsobeného DME nebyl výsledek s ohledem na zrakovou ostrost nebo tloušťku centrální části sítnice (CSFT) u pacientů léčených přípravkem Lucentis ovlivněn současnou léčbou thiazolidindiony.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/antikoncepce u žen
Ženy ve fertilním věku musí během léčby používat účinnou antikoncepci.
Žádné údaje o podávání ranibizumabu těhotným ženám nejsou k dispozici. Studie u makaků rodu Cynomolgus nenaznačují přímé nebo nepřímé škodlivé účinky s ohledem na těhotenství nebo embryonální/fetální vývoj (viz bod 5.3). Po očním podání je nízká systémová expozice ranibizumabu, ale vzhledem k jeho mechanismu účinku je nutno ranibizumab považovat za potenciálně teratogenní a embryo-/fetotoxický. Z tohoto důvodu nesmí být ranibizumab užíván během těhotenství, aniž by očekávaný přínos převážil možné riziko pro plod. Ženám, které chtějí otěhotnět, a které byly léčeny ranibizumabem, je doporučeno vyčkat nejméně 3 měsíce po poslední dávce ranibizumabu před početím dítěte.
Kojení
Není známo, zda se Lucentis vylučuje do lidského mateřského mléka. Během léčby přípravkem Lucentis se kojení nedoporučuje.
Fertilita
Nejsou k dispozici žádné údaje vztahující se k fertilitě.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Léčba Lucentisem může vyvolat dočasné zhoršení zraku, což může ovlivnit schopnost řídit nebo obsluhovat stroje (viz bod 4.8). Pacienti, u kterých se tyto příznaky vyskytnou, nesmějí řídit nebo obsluhovat stroje, dokud tyto poruchy zraku neustoupí.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Většina nežádoucích účinků hlášených po podání Lucentisu se týká postupu podání injekce do sklivce.
Nejčastěji hlášené oční nežádoucí účinky po podání injekce Lucentisu jsou: bolest oka, oční hyperemie, zvýšený nitrooční tlak, vitritida, odloučení sklivce, hemoragie sítnice, poruchy zraku, sklivcové vločky, hemoragie spojivky, podráždění oka, pocit cizího tělesa, zvýšené slzení, zánět očního víčka, suchost oka a svědění oka.
Nejčastěji hlášené nežádoucí účinky mimo oko jsou: bolest hlavy, nazofaryngitida a artralgie.
Méně často hlášené, ale závažnější nežádoucí účinky zahrnují: endoftalmitidu, slepotu, odchlípení sítnice, trhlinu sítnice a iatrogenní traumatickou kataraktu (viz bod 4.4).
Pacienti mají být informováni o příznacích těchto potenciálních nežádoucích účinků a poučeni informovat svého lékaře, pokud se u nich objeví příznaky, jako je bolest oka nebo zvýšený oční diskomfort, zhoršující se zarudnutí oka, rozmazané nebo snížené vidění, zvýšený počet malých částic v zorném poli nebo zvýšená citlivost na světlo.
Nežádoucí účinky, které se objevily po podání Lucentisu v klinických studiích jsou uvedeny v tabulce níže.
Tabulkový seznam nežádoucích účinků#
Nežádoucí účinky jsou uvedeny podle systémově-orgánových skupin a frekvence za použití následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Infekce a infestace
Velmi časté Nazofaryngitida
Časté Infekce močových cest*
Poruchy krve a lymfatického systému Časté Anemie
Poruchy imunitního systému
Časté Hypersenzitivita
Psychiatrické poruchy
Časté Úzkost
Poruchy nervového systému
Velmi časté Bolest hlavy
Poruchy oka Velmi časté
Časté
Méně časté
Vitritida, odloučení sklivce, hemoragie sítnice, poruchy zraku, bolest oka, sklivcové vločky, hemoragie spojivky, podráždění oka, pocit cizího tělesa, zvýšené slzení, zánět očního víčka, suchost oka, oční hyperemie, svědění oka.
Degenerace sítnice, poškození sítnice, odloučení sítnice, trhlina sítnice, odloučení pigmentového epitelu sítnice, trhlina v pigmentovém epitelu sítnice, snížení ostrosti zraku, hemoragie sklivce, poškození sklivce, uveitida, zánět duhovky, iridocyklitida, katarakta, subkapsulární katarakta, opacifikace zadního pouzdra, keratitis punctata, abraze rohovky, zarudnutí v přední části komory, rozmazané vidění, hemoragie v místě injekce, oční hemoragie, zánět spojivky, alergický zánět spojivky, výtok z oka, fotopsie, fotofobie, oční dyskomfort, otok víčka, bolestivost víčka, překrvení spojivek.
Slepota, endoftalmitida, hypopyon, krvácení do přední komory oka, keratopatie, adheze duhovky, korneální deposita, edém rohovky, strie rohovky, bolestivost v místě injekce, podráždění v místě injekce, abnormální pocit v oku, podráždění očního víčka.
Respirační, hrudní a mediastinální poruchy Časté Kašel
Gastrointestinální poruchy
Časté Nauzea
Poruchy kůže a podkožní tkáně
Alergické reakce (vyrážka, kopřivka, pruritus, erytém)
Časté
Poruchy svalové a kosterní soustavy a pojivové tkáně Velmi časté Artralgie
Vyšetření
Velmi časté Zvýšení nitroočního tlaku
# Nežádoucí účinky byly definovány jako nežádoucí příhody (u nejméně 0,5 procentních bodů pacientů), které se objevily ve vyšší míře (alespoň 2 procentní body) u pacientů léčených Lucentisem 0,5 mg než u těch, kteří byli léčeni v kontrolním rameni (simulovanou léčbou nebo fotodynamickou léčbou verteporfinem).
* pozorované pouze u populace s DME
Nežádoucí účinky související s třídou přípravku
Ve studiích fáze III s vlhkou formou AMD se mírně zvýšil celkový výskyt mimoočních krvácení u pacientů léčených ranibizumabem, což je nežádoucí účinek, který potenciálně souvisí se systémovou inhibicí VEGF (vaskulární endoteliální růstový faktor). Nicméně, jednotlivá krvácení neměla shodný charakter. Po intravitreálním podání VEGF inhibitorů existuje teoretické riziko arteriální tromboembolické příhody, včetně mrtvice a infarktu myokardu. V klinických studiích s Lucentisem byla u pacientů s AMD, DME, RVO a PM pozorována nízká incidence arteriálních tromboembolických příhod a mezi skupinami léčenými ranibizumabem nebyly ve srovnání s kontrolou významné rozdíly.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Případy předávkování byly hlášeny z klinických studií s vlhkou formou AMD a postmarketingového sledování. Nejčastěji hlášené případy nežádoucích účinků byly zvýšení nitroočního tlaku, přechodná slepota, snížená zraková ostrost, otok rohovky, bolest rohovky a bolest oka. Dojde-li k předávkování, je nutno monitorovat nitrooční tlak a v závislosti na rozhodnutí ošetřujícího lékaře případně nasadit odpovídající terapii k normalizaci nitroočního tlaku.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, látky určené k léčbě neovaskularizace, ATC kód: S01LA04
Ranibizumab je fragment humanizované monoklonální protilátky proti lidskému cévnímu endoteliálnímu růstovému faktoru A (VEGF-A). Váže se se silnou afinitou na VEGF-A isoformy (např. VEGFn0, VEGFm a VEGFJ65) a tím brání vazbě VEGF-A na receptory VEGFR-1 a VEGFR-2. Vazba VEGF-A na jeho receptory vede k proliferaci endoteliálních buněk a neovaskularizaci, jakož i k propustnosti cév. O všech těchto účincích se uvažuje jako o faktorech přispívajících k progresi neovaskulárních forem věkem podmíněné makulární degenerace, patologické myopie nebo k poškození zraku způsobeného buď diabetickým makulárním edémem, anebo makulárním edémem v důsledku RVO.
Léčba, vlhké formy AMD
Klinická bezpečnost a účinnost Lucentisu byla u vlhké formy AMD stanovena ve třech randomizovaných, dvojitě zaslepených studiích po dobu 24 měsíců se simulovanou injekcí nebo aktivním komparátorem u pacientů s neovaskulární AMD. Do dvou studií bylo zařazeno celkem 1 323 pacientů (879 na aktivní léčbě, 444 v kontrolní skupině).
Ve studii FVF2598g (MARINA) bylo 716 pacientů s minimálně klasickými nebo okultními lézemi randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali měsíčně injekce Lucentisu v dávkách 0,3 mg, injekce Lucentisu v dávkách 0,5 mg nebo simulovanou léčbu.
Ve studii FVF2587g (ANCHOR) bylo 423 pacientů s převážně klasickou CNV lézí randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali Lucentis 0,3 mg měsíčně, Lucentis 0,5 mg měsíčně nebo PDT s verteporfinem (při zahájení léčby a potom každé 3 měsíce, pokud fluoresceinová angiografie prokázala přetrvávání nebo rekurenci cévního prosakování).
Nejdůležitější výsledky měření jsou shrnuty v Tabulce 1 a na Obrázku 1.
Tabulka 1 Výsledky ve 12. a 24. měsíci studie FVF2598g (MARINA) a FVF2587g (ANCHOR)
|
FVF2598g (MA] |
ŘINA) |
FVF2587g (ANCHOR) | |||
|
Měřený parametr |
Měsíc |
Simulovaná léčba (n = 238) |
Lucentis 0,5 mg (n = 240) |
Verteporfin PDT (n = 143) |
Lucentis 0,5 mg (n = 140) |
|
Ztráta <15 písmen ostrosti zraku (%)a (zachování zraku, primární cíl) |
12. měsíc |
62 % |
95 % |
64 % |
96 % |
|
24. měsíc |
53 % |
90 % |
66 % |
90 % | |
|
Nárůst >15 písmen ostrosti zraku (%)a |
12. měsíc |
5 % |
34 % |
6 % |
40 % |
|
24. měsíc |
4 % |
33 % |
6 % |
41 % | |
|
Průměrná hodnota změny zrakové ostrosti (písmena) (SD)a |
12. měsíc |
-10,5 (16,6) |
+ 7,2 (14,4) |
-9,5 (16,4) |
+11,3 (14,6) |
|
24. měsíc |
-14,9 (18,7) |
+6,6 (16,5) |
-9,8 (17,6) |
+10,7 (16,5) | |
p<0.01
Obrázek 1 Průměrná hodnota změny zrakové ostrosti od výchozího stavu do 24. měsíce ve studii FVF2598g (MARINA) a ve studii FVF2587g (ANCHOR)
Studie FVF2598g (MARINA)
+6,6 'n

y +21,5
0 2 4 6 8 10 12 14 16 18 20 22 24
Měsíc
-CD
>
O
co
i_
N
>s
C
><D
E
N
CO
«
O
c
"O
o
-co
LU
co
+1
*2 oo
ol 2
Jr 1/5 Q_ o
15 ' 10 ■ 5
c
CD
0
V)
'cl
~-5 ■ -10 ■ -15 ■ 0

2 4 6 8
r +20,5
10 12 14 16 18 20 22 24 Měsíc
MARINA
LUCENTIS 0,5 mg (n=240) & Simulace (n=238)
ANCHOR
«- LUCENTIS 0,5 mg (n=140) • Verteporfin PDT (n=143)
Výsledky obou studií naznačují, že kontinuální léčba ranibizumabem může být také přínosem u pacientů, kteří v prvním roce léčby ztratili >15 písmen z nejlépe korigované ostrosti zraku (BCVA).
Statisticky významná zlepšení zrakových funkcí, hlášená pacienty, byla pozorována v obou studiích MARINA i ANCHOR při léčbě ranibizumabem oproti kontrolní skupině měřeno pomocí NEI VFQ-25.
Ve studii FVF192g (PIER) bylo 184 pacientů se všemi formami neovaskulámí AMD randomizováno v poměru 1:1:1 do skupin, ve kterých pacienti dostávali Lucentis 0,3 mg, Lucentis 0,5 mg nebo simulovanou léčbu v injekci jednou měsíčně po první 3 měsíce a dále dávku podávanou každý 3. měsíc. Od 14. měsíce této studie bylo pacientům se simulovanou léčbou povoleno používat ranibizumab a od 19. měsíce byla možná častější aplikace. V průměru obdrželi pacienti léčení Lucentisem ve studii PIER 10 aplikací.
Po počátečním nárůstu zrakové ostrosti (při dávkování jednou měsíčně) poklesla zraková ostrost u pacientů kteří dostávali Lucentis jednou za tři měsíce a vracela se ve 12. měsíci v průměru k počátečnímu stavu; tento účinek byl udržován ve 24. měsíci u většiny pacientů léčených ranibizumabem (82 %). Omezené údaje od osob se simulovanou léčbou, které později dostávaly ranibizumab naznačují, že předčasné zahájení léčby může být spojováno s lepším zachováním ostrosti zraku.
Data ze dvou studií (MONT BLANC, BPD952A2308 a DENALI, BPD952A2309) provedených po registraci přípravku Lucentis potvrdila jeho účinnost, ale neprokázala dodatečný účinek kombinovaného podávání verteporfinu (Visudyne PDT) a Lucentisu v porovnání s monoterapií Lucentisem.
Léčba poškození zraku způsobeného DME
Bezpečnost a účinnost Lucentisu byly hodnoceny ve třech randomizovanných kontrolovaných studiích po dobu alespoň 12 měsíců. Celkem 868 pacientů (708 aktivních a 160 kontrol) bylo zařazeno v těchto studiích.
Ve studii 2. fáze D2201 (RESOLVE) bylo léčeno 151 pacientů ranibizumabem (6 mg/ml, n = 51,
10 mg/ml, n = 51) nebo simulovanou léčbou (n = 49) intravitreální injekcí jednou měsíčně. Průměr určený z průměrných hodnot změn BCVA od 1. do 12. měsíce byl v porovnání se stavem na počátku léčby +7,8 (±7,72) písmen u shromážděných pacientů léčených ranibizumabem (n = 102), v porovnání s -0,1 (±9,77) písmen u pacientů se simulovanou léčbou a průměrná hodnota změny BCVA ve 12. měsíci od počátečního stavu byla 10,3 (±9,1) písmen v porovnání s -1,4 (±14,2) písmen, v tomto pořadí (p<0,0001 pro léčebný rozdíl).
Ve fázi III studie D2301 (RESTORE) bylo 345 pacientů randomizováno v poměru 1:1:1 do skupiny užívající ranibizumab 0,5 mg v monoterapii a simulovanou laserovou fotokoagulaci, kombinaci ranibizumabu 0,5 mg a laserové fotokoagulace nebo simulovanou injekci a laserovou fotokoagulaci. 240 pacientů, kteří předtím dokončili 12měsíční studii RESTORE, bylo zařazeno do otevřené, multicentrické 24měsíční extenze studie (extenze studie RESTORE). Pacienti byli léčeni podáním ranibizumabu 0,5 mg pro re nata (PRN) do stejného oka jako v základní studii (D2301 RESTORE).
Obrázek 2 Průměrná hodnota změny zrakové ostrosti od začátku léčby v průběhu času ve studii D2301 (RESTORE)

Ranibizumab 0,5 mg (n=115) Ranibizumab 0,5 mg + laser (n=118) Laser (n=110)
Léčebná skupina
□ □ □
BL=základní stav; SE=směrodatná chyba průměru
* Rozdíl v průměrech nejmenších čtverců, p<0,0001/0,0004 na základě dvoustranného stratifikovaného Cochran-Mantel-Haenszelova testu
Účinek po 12 měsících byl konzistentní ve většině podskupin. Avšak u subjektů s hodnotou BCVA >73 písmen na počátku léčby a makulámím edémem s centrální retinální tloušťkou <300 pm na počátku léčby se nezdálo, že by profitovaly z léčby ranibizumabem v porovnání s laserovou fotokoagulací.
Tabulka 2 Výsledky ve 12. měsíci ve studii D2301 (RESTORE) a ve 36. měsíci ve studii D2301-E1 (extenze studie RESTORE)
|
Výsledky měření ve 12. měsíci v porovnání se stavem na počátku léčby ve studii D2301 (RESTORE) |
Ranibizumab 0,5 mg n = 115 |
Ranibizumab 0,5 mg + laser n = 118 |
laser n = 110 |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 12. měsíce3 (+SD) |
6,1 (6,4)a |
5,9 (7,9)a |
0,8 (8,6) |
|
Průměrná hodnota změny BCVA ve 12. měsíci (+SD) |
6,8 (8,3)a |
6,4 (11,8)a |
0,9 (11,4) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 12. měsíci (%) |
22,6 |
22,9 |
8,2 |
|
Průměrný počet injekcí (0.-11. měsíc) |
7,0 |
6,8 |
7,3 (simulovaný) |
|
Výsledky měření ve 36. měsíci ve studii D2301-E1 (extenze studie RESTORE) v porovnání se stavem na počátku léčby ve studii D2301 (RESTORE) |
Před ranibizumabem 0,5 mg n = 83 |
Před ranibizumabem 0,5 mg + laser n = 83 |
Před laserem n = 74* |
|
Průměrná hodnota změny BCVA ve 24. měsíci (SD) |
7,9 (9,0) |
6,7 (7,9) |
5,4 (9,0) |
|
Průměrná hodnota změny BCVA ve 36. měsíci (SD) |
8,0 (10,1) |
6,7 (9,6) |
6,0 (9,4) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 36. měsíci (%) |
27,7 |
30,1 |
21,6 |
|
Průměrný počet injekcí (12.-35. měsíc)* |
6,8 |
6,0 |
6,5 |
ap<0,0001 pro porovnání ramen s ranibizumabem vs. rameno s laserem. n je ve studii D2301-E1 (extenze studie RESTORE) počet pacientů s hodnotou ve studii D2301 (RESTORE) na počátku léčby (měsíc 0) a při návštěvě ve 36. měsíci.
• Podíl pacientů, kteří nevyžadovali léčbu ranibizumabem během fáze prodloužení studie byl 19 % ve skupině před podáním ranibizumabu, 25 % ve skupině před podáním ranibizumabu + laseru a 20 % ve skupině před aplikací laseru.
Statisticky signifikantní přínosy pro většinu funkcí spojených se zrakem, hlášené pacienty, byly pozorovány u léčby ranibizumabem (s nebo bez laseru) oproti kontrolní skupině, měřeno pomocí NEI VFQ-25. Pro ostatní podškály tohoto dotazníku nemohly být stanoveny žádné léčebné rozdíly.
Dlouhodobý bezpečnostní profil ranibizumabu pozorovaný ve 24měsíční extenzi studie je konzistentní se známým bezpečnostním profilem přípravku Lucentis.
Ve fázi IlIb studie D2304 (RETAIN) bylo 372 pacientů randomizováno v poměru 1:1:1 k podávání:
• ranibizumabu 0,5 mg se současnou laserovou fotokoagulací v režimu „treat-and-extend“ (TE),
• ranibizumabu 0,5 mg v monoterapii v režimu TE,
• ranibizumabu 0,5 mg v monoterapii v režimu PRN.
Ve všech skupinách byl ranibizumab podáván jednou měsíčně, dokud BCVA nebyla stabilní po alespoň tři po sobě jdoucí měsíční vyšetření. V režimu TE byl podán ranibizumab v léčebných intervalech 2-3 měsíce. Ve všech skupinách byla měsíční léčba znovu zahájena na základě snížení BCVA způsobeném progresí DME a pokračovala, dokud nebylo opět dosaženo stabilní BCVA.
Počet plánovaných léčebných návštěv po 3 počátečních injekcích byl 13 v režimu TE a 20 v režimu PRN. V obou TE režimech bylo více než 70 % pacientů schopných zachovat svou BCVA při průměrné četnosti návštěv >2 měsíce.
Nejdůležitější výsledky měření jsou uvedeny v Tabulce 3.
Tabulka 3 Výsledky ve studii D2304 (RETAIN)
|
Výsledek měření porovnaný s výchozím stavem |
ranibizumab 0,5 mg + laser v režimu TE n = 117 |
ranibizumab 0,5 mg samotný v režimu TE n = 125 |
ranibizumab 0,5 mg v režimu PRN n = 117 |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 12. měsíce (SD) |
5,9 (5,5)a |
6,1 (5,7)a |
6,2 (6,0) |
|
Průměr určený z průměrných hodnot změn BCVA od 1. měsíce do 24. měsíce (SD) |
6,8 (6,0) |
6,6 (7,1) |
7,0 (6,4) |
|
Průměrná hodnota změny BCVA ve 24. měsíci (SD) |
8,3 (8,1) |
6,5 (10,9) |
8,1 (8,5) |
|
Nárůst o >15 písmen nebo BCVA >84 písmen ve 24. měsíci (%) |
25,6 |
28,0 |
30,8 |
|
Průměrný počet injekcí (0.-23. měsíc) |
12,4 |
12,8 |
10,7 |
ap<0,0001 pro hodnocení non-inferiority k PRN
Ve studiích zaměřených na DME bylo zlepšení BCVA doprovázeno redukcí střední CSFT v průběhu času ve všech léčebných skupinách.
Léčba poškození zraku způsobeného makulárním edémem v důsledku RVO
Klinická bezpečnost a účinnost Lucentisu u pacientů s poškozením zraku způsobeným makulárním
edémem v důsledku RVO byla hodnocena v randomizovaných dvojitě zaslepených kontrolovaných
studiích BRAVO a CRUISE, ve kterých byly zařazeny subjekty s BRVO (n = 397) a CRVO
(n = 392). V obou studiích dostávaly subjekty buď 0,3 mg nebo 0,5 mg ranibizumabu nebo injekce
simulované léčby. Pacienti v kontrolním rameni se simulovanou léčbou byli po 6 měsících přeřazeni
do ramene s ranibizumabem 0,5 mg.
Tabulka 4 Výsledky v 6. a 12. měsíci (BRAVO a CRUISE)
|
BRAVO |
CRUISE | |||
|
Simulovaná léčba/Lucentis 0,5 mg (n = 132) |
Lucentis 0,5 mg (n = 131) |
Simulovaná léčba /Lucentis 0,5 mg (n = 130) |
Lucentis 0,5 mg (n = 130) | |
|
Průměrná hodnota změny zrakové ostrosti v 6. měsícía (písmena) (SD) (primární cíl) |
7,3 (13,0) |
18,3 (13,2) |
0,8 (16,2) |
14,9 (13,2) |
|
Průměrná hodnota změny BCVA ve 12. měsíci (písmena) (SD) |
12,1 (14,4) |
18,3 (14,6) |
7,3 (15,9) |
13,9 (14,2) |
|
Nárůst o > 15 písmen u zrakové ostrosti v 6. měsícia (%) |
28,8 |
61,1 |
16,9 |
47,7 |
|
Nárůst o > 15 písmen u zrakové ostrosti ve 12. měsíci (%) |
43,9 |
60,3 |
33,1 |
50,8 |
|
Podíl (%) pacientů léčených laserovou záchrannou terapií po 12 měsíců |
61,4 |
34,4 |
NA |
NA |
ap <0,0001 v obou studiích

CL
Léčebná skupina
r m-m—m
Měsíc
Placebo/Ranibizumab 0,5 mg (n = 132 Ranibizumab 0,5 mg (n = 131)
BL=základní stav; SE=směrodatná chyba průměru

Léčebná skupina
Placebo/Ranibizumab 0,5 mg (n = 130) Ranibizumab 0,5 mg (n = 130)
BL=základní stav; SE=směrodatná chyba průměru
V obou studiích bylo zlepšení zraku spojeno s kontinuální a významnou redukcí makulámího edému, což bylo posuzováno podle tloušťky středu sítnice.
U pacientů s CRVO (CRUISE a extenze studie HORIZON): Subjekty léčené simulovanou léčbou v prvních 6 měsících, které následně dostávaly ranibizumab, nedosáhly srovnatelných nárůstů zrakové ostrosti (VA) do 24. měsíce (~6 písmen) v porovnání se subjekty léčenými ranibizumabem od začátku studie (~12 písmen).
Statisticky významná zlepšení v subškálách spojená s viděním na blízko a na dálku, hlášená pacienty, byla pozorována při léčbě ranibizumabem oproti kontrolní skupině, měřeno pomocí NEI VFQ-25.
Dlouhodobá (24měsíční) klinická bezpečnost a účinnost Lucentisu u pacientů s poškozením zraku způsobeného sekundárně makulárním edémem v důsledku RVO byla hodnocena ve studiích BRIGHTER (BRVO) a CRYSTAL (CRVO). V obou studiích bylo subjektům podáváno 0,5 mg ranibizumabu v režimu PRN řízeného individualizovanými kritérii stabilizace zrakové ostrosti. BRIGHTER byla 3ramenná randomizovaná aktivně kontrolovaná studie, která porovnávala 0,5 mg ranibizumabu podávaného v monoterapii nebo v kombinaci s adjuvantní laserovou fotokoagulací oproti samotné laserové fotokoagulaci. Subjektům v laserovém rameni mohl být po 6 měsících podán ranibizumab v dávce 0,5 mg. CRYSTAL byla jednoramenná studie s 0,5 mg ranibizumabu v monoterapii.
Důležité výstupy ze studií BRIGHTER a CRYSTAL jsou uvedeny v Tabulce 5. Tabulka 5 Výsledky v 6. a 24. měsíci studie (BRIGHTER a CRYSTAL)
|
BRIGHTER |
CRYSTAL | |||
|
Lucentis 0,5 mg n=180 |
Lucentis 0,5 mg + laser n=178 |
Laser* n=90 |
Lucentis 0,5 mg n=356 | |
|
Průměrná hodnota změny BCVA v 6. měsícia (písmena) (SD) |
+14,8 (10,7) |
+14,8 (11,13) |
+6,0 (14,27) |
+12,0 (13,95) |
|
Průměrná hodnota změny BCVA ve 24. měsícib (písmena) (SD) |
+15,5 (13,91) |
+17,3 (12,61) |
+11,6 (16,09) |
+12,1 (18,60) |
|
Zisk >15 písmen BCVA ve 24. měsíci (%) |
52,8 |
59,6 |
43,3 |
49,2 |
|
Průměrný počet injekcí (SD) (měsíc 0-23) |
11,4 (5,81) |
11,3 (6,02) |
NA |
13,1 (6,39) |
|
a p<0,0001 pro obě porovnání ve studii BRIGHTER v 6. měsíci: Lucentis 0,5 mg vs laser a Lucentis 0,5 mg + laser vs laser. b p<0,0001 pro nulovou hypotézu ve studii CRYSTAL, že průměrná hodnota změny ve 24. měsíci od výchozího stavu je nula. * Od 6. měsíce byla umožněna léčba 0,5 mg ranibizumabu (24 pacientů bylo léčeno pouze laserem). | ||||
Ve studii BRIGHTER byla prokázána non-inferiorita 0,5 mg ranibizumabu s podpůrnou laserovou terapií ve srovnání s ranibizumabem v monoterapii od výchozího stavu do 24. měsíce (95 % CI -2,8; 1,4).
V obou studiích byla v 1. měsíci pozorována rychlá a statisticky významná redukce tloušťky centrální části sítnice od výchozího stavu. Tento účinek přetrval až do 24. měsíce.
Účinek léčby ranibizumabem byl podobný bez ohledu na přítomnost retinální ischemie. Ve studii BRIGHTER u pacientů s ischemií (n=46) nebo bez ischemie (n=133) léčených ranibizumabem v monoterapii byla průměrná hodnota změny BCVA ve 24. měsíci od výchozího stavu +15,3; resp. +15,6 písmen. Ve studii CRYSTAL u pacientů s ischemií (n=53) nebo bez ischemie (n=300) léčených ranibizumabem v monoterapii byla průměrná hodnota změny BCVA od výchozího stavu +15,0; resp. +11,5 písmen.
V obou studiích BRIGHTER a CRYSTAL byl účinek na zlepšení zraku pozorován u všech pacientů léčených 0,5 mg ranibizumabu v monoterapii bez ohledu na délku trvání onemocnění. U pacientů
s délkou trvání onemocnění <3 měsíce byl pozorován nárůst zrakové ostrosti o 13,3; resp. 10,0 písmen v 1. měsíci a o 17,7; resp. 13,2 písmen ve 24. měsíci ve studiích BRIGHTER a CRYSTAL. Odpovídající zisk zrakové ostrosti u pacientů s délkou trvání onemocnění >12 měsíců byl v těchto studiích 8,6; resp. 8,4 písmen. Je třeba zvážit zahájení léčby v čase stanovení diagnózy.
Dlouhodobý bezpečnostní profil ranibizumabu pozorovaný ve 24měsíčních studiích je konzistentní se známým bezpečnostním profilem přípravku Lucentis.
Léčba poškození zraku způsobeného CNV sekundární k PM
Klinická bezpečnost a účinnost přípravku Lucentis u pacientů s poškozením zraku způsobeným CNV u PM byla hodnocena na základě 12měsíčních dat z dvojitě zaslepené, kontrolované pivotní studie F2301 (RADIANCE). V této studii bylo 277 pacientů randomizováno v poměru 2:2:1 do následujících ramen:
• Skupina I (ranibizumab 0,5 mg, dávkovací schéma řízené kritérii „stability“, definovanými jako žádná změna BCVA v porovnání se dvěma předchozími měsíčními vyhodnoceními).
• Skupina II (ranibizumab 0,5 mg, dávkovací schéma řízené kritérii „aktivity onemocnění“, definovanými jako poškození zraku způsobené intra- nebo subretinální tekutinou nebo aktivním prosakováním v důsledku CNV léze, jak bylo zjištěno při vyšetření OCT a/nebo FA).
• Skupina III (vPDT - pacientům bylo povoleno užívat léčbu ranibizumabem od 3. měsíce studie).
Ve skupině II, což je doporučené dávkovací schéma (viz bod 4.2), vyžadovalo 50,9 % pacientů 1 nebo 2 injekce, 34,5 % pacientů 3 až 5 injekcí a 14,7 % vyžadovalo 6 až 12 injekcí po dobu 12 měsíců trvání studie. 62,9 % pacientů ze skupiny II nevyžadovalo injekce ve druhé polovině trvání studie.
Důležité výstupy ze studie RADIANCE jsou uvedeny v Tabulce 6 a na Obrázku 5.
Tabulka 6 Výsledky ve 3. a 12. měsíci studie (RADIANCE)
|
Skupina I |
Skupina II |
Skupina | |
|
Ranibizumab |
Ranibizumab |
III | |
|
0,5 mg |
0,5 mg |
vPDTb | |
|
„stabilizace |
„aktivita | ||
|
vidění“ |
onemocnění“ | ||
|
(n = 105) |
(n = 116) |
(n = 55) | |
|
3. měsíc Průměr určený z průměrných hodnot změn BCVA od 1. do 3. měsíce studie v porovnání se základním stavema (písmena) Podíl pacientů, kteří dosáhli: >15 písmen, nebo dosáhli >84 písmen |
+10,5 |
+10,6 |
+2,2 |
|
BCVA 12. měsíc |
38,1 % |
43,1 % |
14,5 % |
|
Počet injekcí až do 12. měsíce: Průměrná hodnota |
4,6 |
3,5 |
N/A |
|
Medián |
4 |
2,0 |
N/A |
|
Průměr určený z průměrných hodnot změn v BCVA od 1. do 12. měsíce v porovnání se základním stavem (písmena) Podíl pacientů, kteří dosáhli: |
+12,8 |
+12,5 |
N/A |
|
>15 písmen, nebo dosáhli >84 písmen BCVA |
53,3 % |
51,7 % |
N/A |
a p <0,00001 porovnání s vPDT kontrolní skupinou
b Srovnávací kontrola až do 3. měsíce studie. Pacientům randomizovaným do skupiny s vPDT bylo povoleno dostávat léčbu ranibizumabem od 3. měsíce studie (ve skupině III dostávalo ranibizumab 38 pacientů od 3. měsíce studie)
Průměrná hodnota změny BCVA od začátku léčby v průběhu času do 12. měsíce (RADIANCE)

Měsíc
___ Ranibizumab 0,5 mg Skupina I Ranibizumab 0,5 mg Skupina II
^ ^ ^ EI-EHD v i • • v v r
při aktivitě onemocněni (n = 116)
Ranibizumab 0,5 mg/PDT verteporfinem Skupina III od 3. měsíce studie dále (n = 55)
pn stabilizaci (n = 105)
a-a-a PDT verteporfinem Skupina III(n = 55)
Zlepšeni zraku bylo doprovázeno redukci tloušťky centrální části sítnice.
Prospěch hlášený pacientem byl pozorován v léčebných ramenech s ranibizumabem oproti vPDT (hodnota p <0,05) na základě zlepšení kombinovaného skóre a několika subškál (všeobecné vidění, aktivity do blízka, duševní zdraví a závislost) v NEI VFQ-25.
Pediatrická populace
Bezpečnost a účinnost ranibizumabu nebyla u pediatrických pacientů dosud studována.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lucentis u všech podskupin pediatrické populace u neovaskulární formy AMD, u poškození zraku kvůli DME, u poškození zraku kvůli makulárnímu edému v důsledku RVO a u poškození zraku v důsledku CNV sekundární k PM (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Po měsíčních aplikacích Lucentisu do sklivce jedincům s neovaskulámí AMD byly koncentrace ranibizumabu v séru obvykle nízké. Maximální koncentrace (Cmax) ranibizumabu byly obecně nižší než koncentrace ranibizumabu nutné k inhibici biologické aktivity VEGF o 50 % (11 - 27 ng/ml, stanoveno in vitro titrací buněčné proliferace). Cmax byla závislá na dávce v dávkovém rozmezí 0,05 až
1.0 mg/oko. Sérové koncentrace u limitovaného počtu pacientů s DME ukazují, že lehce vyšší systémová expozice nemůže být vyloučena ve srovnání se sérovými koncentracemi zjištěnými
u pacientů s neovaskulámí AMD. Sérové koncentrace ranibizumabu u pacientů s RVO byly podobné nebo lehce vyšší v porovnání s koncentracemi pozorovanými u pacientů s neovaskulámí AMD.
Na základě analýz populační farmakokinetiky a vymizení ranibizumabu ze séra pacientů s neovaskulámí AMD léčených dávkou 0,5 mg byl průměrný poločas eliminace ranibizumabu ze sklivce stanoven na přibližně 9 dnů. Při aplikaci Lucentisu v dávce 0,5 mg/oko jednou měsíčně je dosaženo sérové Cmax ranibizumabu, jejíž předpokládané rozmezí je mezi 0,79 a 2,90 ng/ml přibližně za 1 den po aplikaci. Cmin se předpokládá v rozmezí mezi 0,07 a 0,49 ng/l. Sérové koncentrace ranibizumabu se předpokládají přibližně 90 000násobně nižší než koncentrace ranibizumabu ve sklivci.
Pacienti se zhoršenou funkcí ledvin: Žádné cílené studie, které by hodnotily farmakokinetiku Lucentisu u pacientů se zhoršenou funkcí ledvin, nebyly provedeny. 68 % (136 ze 200) pacientů s neovaskulámí AMD mělo v populační farmakokinetické analýze zhoršenou funkci ledvin (46,5 % mírně [50 - 80 ml/min], 20 % středně [30 - 50 ml/min] a 1,5 % silně [< 30 ml/min]). 48,2 % (253 z 525) pacientů s RVO mělo zhoršenou funkci ledvin (36,4 % mírně, 9,5 % středně a 2,3 % silně). Systémová clearance byla lehce snížena, ale tento nález byl bez klinického významu.
Zhoršená funkce jater: Žádné cílené studie, které by hodnotily farmakokinetiku u pacientů se zhoršenou funkcí jater léčených Lucentisem, nebyly provedeny.
5.3 Předklinické údaje vztahující se k bezpečnosti
Oboustranná aplikace ranibizumabu do sklivce opicím cynomolgus v dávkách 0,25 mg/oko a
2.0 mg/oko jednou za dva týdny po dobu 26 týdnů vyvolala účinky na očích závislé na dávce.
Nitrooční aplikace vyvolala na dávce závislé zvýšené zarudnutí přední komory a zvýšení počtu buněk, největší za 2 dny po injekci. Závažnost zánětlivé odpovědi se obvykle zmenšila po následujících injekcích nebo během zotavení. V zadním segmentu byla pozorována infiltrace sklivce buňkami a sklivcovými vločkami, u které byla také tendence k závislosti na dávce a obvykle přetrvávala až do konce léčby. Ve 26týdenní studii se intenzita zánětu sklivce zvyšovala s počtem injekcí. Průkaz reverzibility byl však pozorován po zotavení. Povaha a načasování zánětu zadního segmentu naznačují, že se jedná o imunologicky zprostředkovanou protilátkovou odpověď, která by mohla být klinicky irelevantní. U některých zvířat byla po relativně dlouhém období intenzivního zánětu pozorována tvorba katarakty naznačující, že změny čočky byly sekundární, vyvolané silným zánětem. Bylo pozorováno přechodné zvýšení nitroočního tlaku po podání do sklivce bez ohledu na dávku.
Mikroskopické změny pozorované v oční tkáni souvisely se zánětem a nenasvědčovaly degenerativním procesům. U některých očí byly pozorovány granulomatózní zánětlivé změny na očním disku. Tyto změny zadního segmentu slábly, a v některých případech kompletně vymizely, během období zotavení.
Po podání do sklivce nebyly zjištěny žádné známky systémové toxicity. U některých zvířat byly nalezeny protilátky proti ranimizumabu v séru i sklivci.
Nejsou dostupné žádné údaje o karcinogenitě nebo mutagenitě.
U březích opic nevyvolala léčba ranibizumabem aplikovaná do sklivce , mající za následek maximální systémové expozice 0,9-7-krát nejhorší případ klinické expozice, vývojovou toxicitu ani teratogenitu, a neměla žádný vliv na hmotnost nebo strukturu placenty, i když na základě svého farmakologického účinku by měl být ranibizumab považován za potenciálně teratogenní a embryo-/fetotoxický.
Chybění účinků zprostředkovaných ranibizumabem na embryo-fetální vývoj se pravděpodobně týká hlavně neschopnosti Fab-fragmentu procházet placentou. Přesto byl popsán případ s vysokými mateřskými sérovými hladinami ranibizumabu a přítomností ranibizumabu ve fetálním séru, nasvědčující tomu, že protilátky proti ranibizumabu působily jako nosičský protein (obsahující Fc část) pro ranibizumab, a tím snižovaly jeho mateřskou sérovou clearance a umožňovaly prostup placentou. Protože embryo-fetální vývojová vyšetření byla prováděna u zdravých březích zvířat a onemocnění (jako například diabetes) mohou modifikovat prostupnost placenty pro Fab-fragment, měla by být studie interpretována s opatrností.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát a,a-trehalosy
Monohydrát histidin-hydrochloridu
Histidin
Polysorbát 20
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Před použitím může být neotevřená injekční lahvička ponechána při pokojové teplotě (25 °C) po dobu 24 hodin.
6.5 Druh obalu a obsah balení
0,23 ml sterilního injekčního roztoku v injekční lahvičce (sklo typu I) se zátkou (chlorbutylová pryž) a 1 tupou jehlou s filtrem (18G x P/2", 1,2 mm x 40 mm, 5 pm). Balení obsahuje 1 injekční lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Injekční lahvička a jehla s filtrem jsou pro jednorázové použití. Opakované použití může vést k infekci nebo jinému onemocnění/poškození. Všechny komponenty jsou sterilní. Jakákoliv komponenta s obalem vykazující známky poškození nebo manipulace nesmí být použita. Sterilita nemůže být zaručena, pokud nezůstane uzavření obalu komponenty neporušené.
Pro přípravu a injekci do sklivce jsou potřeba následující zdravotnické prostředky pro jednorázové použití:
- 5 pm jehla s filtrem (18G x 1%'', 1,2 mm x 40 mm, přiložena)
- 1 ml sterilní injekční stříkačka (není zahrnuta v balení přípravku Lucentis)
- injekční jehla (30G x /"; není zahrnuta v balení přípravku Lucentis)
Při přípravě přípravku Lucentis k podání do sklivce dbejte, prosím, následujících pokynů:
1. Před nasátím tekutiny do injekční stříkačky je nutno desinfikovat vnější část pryžové zátky.
2. Jehlu s 5 pm filtrem (18G x 1/", 1,2 mm x 40 mm, přiložena) nasaďte na 1 ml injekční stříkačku za použití aseptického postupu. Zasuňte jehlu s filtrem do středu zátky lahvičky, dokud se jehla nedotkne dna injekční lahvičky.
3. Nasajte veškerou tekutinu z injekční lahvičky držené ve svislé, lehce nakloněné poloze pro snadnější úplné nasátí.
4. Ujistěte se, že píst je při vyprazdňování injekční lahvičky vytažen dostatečně daleko tak, aby jehla s filtrem byla úplně vyprázdněna.
5. Jehlu s filtrem ponechte v lahvičce a stříkačku od ní odpojte. Jehla s filtrem by měla být po násátí veškerého obsahu lahvičky zlikvidována a nesmí být použita pro vlastní aplikaci do sklivce.
6. Asepticky a pevně nasaďte injekční jehlu (30G x /") na injekční stříkačku.
7. Opatrně odstraňte kryt z injekční jehly, aniž byste injekční jehlu odpojili od injekční stříkačky. Poznámka: Při odstraňování krytu pevně uchopte část injekční jehly.
8. Pečlivě vytlačte vzduch ze stříkačky a upravte dávku ke značce 0,05 ml uvedené na injekční stříkačce. Nyní je injekční stříkačka připravena k aplikaci.
Poznámka: Neotírejte injekční jehlu. Nevytahujte píst zpět.
Po podání injekce nenasazujte zpět kryt jehly ani jehlu neoddělujte od injekční stříkačky. Zlikvidujte použitou injekční stříkačku společně s jehlou vyhozením do nádoby na ostré předměty nebo v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/06/374/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 22. ledna 2007
Datum posledního prodloužení registrace: 24. ledna 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců biologické léčivé látky
Genentech, Inc.
1 DNA Way
South San Francisco, CA 94080-4990 USA
Roche Singapore Technical Operations Pte. Ltd.
10 Tuas Bay Link Singapore 637394 Singapur
Název a adresa výrobce odpovědného za propouštění šarží
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2. registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci (MAH) je povinen odsouhlasit konečný edukační materiál s národní kompetentní autoritou před uvedením přípravku na trh v každém členském státě.
MAH má zajistit, aby následně po diskusích a schváleních národními kompetentními autoritami v každém členském státě, kde je Lucentis na trhu, byl všem oftalmologickým klinikám, kde se předpokládá použití Lucentisu, při uvedení na trh a po uvedení na trh, poskytnut aktualizovaný soubor informací pro lékaře obsahující následující složky:
• Informace pro lékaře
• Videozáznam metodiky podání do sklivce
• Piktogram procedury podání do sklivce
• Soubory informací pro pacienta
Informace pro lékaře mají obsahovat následující nejdůležitější složky:
• Souhrn údajů o přípravku
• Sterilní techniky, včetně periokulární a okulární dezinfekce, aby se minimalizovalo riziko infekce
• Použití jodovaného povidonu nebo rovnocenné náhrady
• Požadavek k vytlačení nadbytečného objemu z předplněné injekční stříkačky před podáním přípravku Lucentis, aby se předešlo předávkování
• Techniky podání do sklivce
• Monitorování pacienta po podání intravitreální injekce
• Hlavní známky a příznaky nežádoucích účinků souvisejících s podáním do sklivce, zahrnující zvýšený nitrooční tlak, traumatickou kataraktu a endoftalmitidu
• Zvládnutí podání do sklivce související s nežádoucími účinky
Soubor informací pro pacienta má být zajištěn v obou formách, jako informační brožury pro pacienta a jako audio-CD, a obsahuje následující nejdůležitější složky:
• Příbalová informace pro pacienta
• Jak se připravit na léčbu Lucentisem
• Jaké jsou kroky, které následují po podání Lucentisu
• Hlavní známky a příznaky závažných nežádoucích účinků léčby, zahrnující zvýšený nitrooční tlak, traumatickou kataraktu a endoftalmitidu
• Kdy vyhledat okamžitou pomoc u poskytovatele zdravotní péče
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
INJEKČNÍ LAHVIČKA + JEHLA S FILTREM + INJEKČNÍ JEHLA + INJEKČNÍ STŘÍKAČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Lucentis 10 mg/ml injekční roztok Ranibizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje ranibizumabum 10 mg. Injekční lahvička obsahuje ranibizumabum 2,3 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Dále obsahuje: Dihydrát a,a-trehalosy, monohydrát histidin-hydrochloridu, histidin, polysorbát 20, vodu na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
1 injekční lahvička s 0,23 ml injekčního roztoku, 1 injekční jehla (30G x 'A'', 0,3 mm x 13 mm), 1 jehla s filtrem (18G x PA", 1,2 mm x 40 mm, 5 ^m), 1 injekční stříkačka (1 ml).
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Podání do sklivce.
Injekční lahvička, jehly a injekční stříkačka pouze pro jednorázové použití. Před použitím si přečtěte příbalovou informaci.
Jehla s filtrem není určena k aplikaci injekce.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Použité jehly odložte do nádoby na ostré předměty.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/06/374/001
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK
INJEKČNÍ LAHVIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Lucentis 10 mg/ml injekční roztok
Ranibizumabum
Podání do sklivce
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 injekční lahvička = ranibizumabum 2,3 mg.
6. JINÉ
KRABIČKA
INJEKČNÍ LAHVIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Lucentis 10 mg/ml injekční roztok Ranibizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje ranibizumabum 10 mg. Injekční lahvička obsahuje ranibizumabum 2,3 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Dále obsahuje: Dihydrát a,a-trehalosy, monohydrát histidin-hydrochloridu, histidin, polysorbát 20, vodu na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
1 injekční lahvička s 0,23 ml injekčního roztoku.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Podání do sklivce.
Injekční lahvička pouze pro jednorázové použití. Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/06/374/002
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK
INJEKČNÍ LAHVIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Lucentis 10 mg/ml injekční roztok
Ranibizumabum
Podání do sklivce
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 injekční lahvička = ranibizumabum 2,3 mg.
6. JINÉ
PŘEDPLNĚNÁ INJEKČNÍ STŘÍKAČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Lucentis 10 mg/ml injekční roztok v předplněné injekční stříkačce Ranibizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka o objemu 0,165 ml injekčního roztoku obsahuje ranibizumabum 1,65 mg (10 mg/ml).
3. SEZNAM POMOCNÝCH LÁTEK
Dále obsahuje: Dihydrát a,a-trehalosy, monohydrát histidin-hydrochloridu, histidin, polysorbát 20, vodu na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 předplněná injekční stříkačka o objemu 0,165 ml. Jednotlivá dávka 0,5 mg/0,05 ml.
Nadbytečný objem je třeba před podáním injekce vytlačit.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze pro jednorázové použití. Otevření zataveného obalu provádějte za aseptických podmínek. Nastavte dávku na označení dávky 0,05 ml.
Před použitím si přečtěte příbalovou informaci.
Podání do sklivce.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte předplněnou injekční stříkačku v jejím zataveném obalu v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/06/374/003
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU BLISTR
PŘEDPLNĚNÁ INJEKČNÍ STŘÍKAČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Lucentis 10 mg/ml injekční roztok v předplněné injekční stříkačce
Ranibizumabum
Podání do sklivce
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
0,165 ml
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK
PŘEDPLNĚNÁ INJEKČNÍ STŘÍKAČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Lucentis 10 mg/ml injekční roztok
Ranibizumabum
Podání do sklivce
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,165 ml
6. JINÉ
INJEKČNÍ LAHVIČKA + JEHLA S FILTREM
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Lucentis 10 mg/ml injekční roztok Ranibizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje ranibizumabum 10 mg. Injekční lahvička obsahuje ranibizumabum 2,3 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Dále obsahuje: Dihydrát a,a-trehalosy, monohydrát histidin-hydrochloridu, histidin, polysorbát 20, vodu na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
1 injekční lahvička s 0,23 ml injekčního roztoku, 1 jehla s filtrem (18G x PA”, 1,2 mm x 40 mm, 5 ^m).
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Podání do sklivce.
Injekční lahvička a jehla s filtrem pouze pro jednorázové použití. Před použitím si přečtěte příbalovou informaci.
Jehla s filtrem není určena k aplikaci injekce.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Použitou jehlu odložte do nádoby na ostré předměty.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/06/374/004
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK
INJEKČNÍ LAHVIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Lucentis 10 mg/ml injekční roztok
Ranibizumabum
Podání do sklivce
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 injekční lahvička = ranibizumabum 2,3 mg.
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Lucentis 10 mg/ml injekční roztok
Ranibizumabum
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Co naleznete v této příbalové informaci
1. Co je Lucentis a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude Lucentis podán
3. Jak se Lucentis podává
4. Možné nežádoucí účinky
5. Jak Lucentis uchovávat
6. Obsah balení a další informace
1. Co je Lucentis a k čemu se používá Co je Lucentis
Lucentis je roztok, který se aplikuje injekcí do oka. Lucentis patří do skupiny léků, nazvané antineovaskularizační látky. Obsahuje léčivou látku zvanou ranibizumab.
K čemu se Lucentis používá
Lucentis se používá u dospělých k léčbě několika očních onemocnění, způsobujících poškození zraku.
Tato onemocnění jsou výsledkem poškození sítnice (na světlo citlivé vrstvy v zadní části oka), způsobeným:
- růstem propustných, abnormálních krevních cév (choroidální neovaskularizace, CNV). Toto se pozoruje u onemocnění, jako například věkem podmíněná makulární degenerace (AMD) nebo patologická myopie (PM).
- makulárním edémem (otokem centrální části sítnice). Tento otok může být způsoben cukrovkou (onemocnění zvané diabetický makulární edém (DME)) nebo blokádou sítnicových žil (onemocnění zvané okluze retinální vény, RVO)).
Jak Lucentis působí
Lucentis specificky rozpoznává a váže se na bílkovinu zvanou lidský růstový faktor cévního endotelu A (VEGF-A), přítomnou v oku. VEGF-A způsobuje navíc abnormální růst krevních cév a otok v oku, který může vést k poškození zraku u onemocnění, jako jsou AMD, PM, DME nebo RVO. Lucentis může vazbou na VEGF-A blokovat jeho funkce a předcházet tomuto abnormálnímu růstu a otoku.
U těchto onemocnění může Lucentis pomoci stabilizovat a v mnoha případech zlepšit Váš zrak.
Lucentis Vám nesmí být podán
- jestliže jste alergický(á) na ranibizumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže máte infekci v oku nebo kolem očí.
- jestliže Vás bolí oči nebo je máte zarudlé (těžký nitrooční zánět).
Upozornění a opatření
Před podáním přípravku Lucentis se poraďte se svým lékařem.
- Lucentis je podáván jako injekce do oka. Po léčbě Lucentisem se může někdy vyvinout infekce vnitřní části oka, bolest oka nebo zarudnutí (zánět), odchlípení nebo natržení jedné z vrstev
v zadní části oka (odchlípení nebo natržení sítnice a odchlípení nebo natržení pigmentového epitelu sítnice), zákal čočky (katarakta). Je důležité rozeznat a léčit tuto infekci nebo odchlípení sítnice co možná nejdříve. Řekněte, prosím, svému lékaři ihned, pokud se u Vás vyskytnou příznaky jako bolest v oku, nepříjemný pocit v oku, zhoršení zarudnutí, rozmazané nebo snížené vidění. Informujte také neprodleně svého lékaře pokud máte pocit, že před okem vidíte zvýšený počet malých částic (teček) nebo pokud se u Vás vyskytne zvýšená citlivost na světlo.
- U některých pacientů může dojít vzápětí po injekci ke krátkodobému zvýšení nitroočního tlaku. Zvýšení nitroočního tlaku se u Vás nemusí projevit žádnými příznaky, ošetřující lékař však může nitrooční tlak po každé injekci kontrolovat.
- Informujte svého lékaře, jestliže jste někdy v minulosti měl(a) onemocnění oka nebo podstoupil(a) léčbu oka, nebo prodělal(a) mrtvici nebo přechodné příznaky mrtvice (slabost nebo ochrnutí končetin nebo obličeje, potíže při mluvení nebo porozumění). Tato informace bude brána v úvahu při zhodnocení, zda je Lucentis pro Vás vhodnou léčbou.
Děti a dospívající (pod 18 let věku)
Použití Lucentisu u dětí a dospívajících nebylo studováno, a proto se nedoporučuje.
Další léčivé přípravky a Lucentis
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které
možná budete užívat.
Těhotenství a kojení
- Ženám, které mohou otěhotnět se doporučuje používat během léčby účinnou antikoncepci.
- S použitím přípravku Lucentis u těhotných žen nej sou zkušenosti, proto případné riziko není známo. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, prodiskutujte to se svým lékařem před léčbou přípravkem Lucentis.
- Lucentis se nedoporučuje během kojení, protože není známo, zda Lucentis přestupuje do mateřského mléka. Poraďte se se svým lékařem nebo lékárníkem před léčbou přípravkem Lucentis.
Řízení dopravních prostředků a obsluha strojů
Po aplikaci přípravku Lucentis můžete mít dočasné problémy s viděním. Pokud se Vám toto stane,
neřiďte nebo neobsluhujte stroje, dokud poruchy zraku nevymizí.
Lucentis je podáván v místním znecitlivění jako injekce do oka Vaším očním lékařem. Obvyklá dávka v jedné injekci je 0,05 ml (které obsahují 0,5 mg léčivé látky). Interval mezi dvěma dávkami podanými do stejného oka má být alespoň čtyři týdny. Všechny injekce Vám vždy bude aplikovat oční lékař.
Váš lékař Vám před injekcí pečlivě vypláchne oko, aby zabránil infekci. Dostanete také odpovídající místní umrtvení, aby se zmenšila případná bolest oka při injekci nebo se jí předešlo.
Léčba se zahajuje podáním jedné injekce Lucentisu za měsíc. Lékař bude sledovat stav Vašeho oka a podle odpovědi na léčbu rozhodne, zda a kdy potřebujete dostat další léčbu.
Podrobné pokyny pro používání jsou uvedeny na konci příbalové informace pod „Jak připravit a jak aplikovat Lucentis“.
Starší pacienti (věk 65 let a více)
Lucentis může být použit u osob ve věku 65 let a starších bez úpravy dávkování.
Jestliže jste zmeškal(a) dávku přípravku Lucentis
Kontaktujte co nejdříve ošetřujícího lékaře nebo nemocnici, abyste si domluvil(a) další návštěvu.
Před ukončením léčby přípravkem Lucentis
Jestliže uvažujete o ukončení léčby přípravkem Lucentis, jděte na další návštěvu a poraďte se se svým lékařem. Lékař Vám poradí a rozhodne, jak dlouho budete přípravkem Lucentis léčen(a).
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky spojené s podáním Lucentisu jsou způsobené buď vlastním přípravkem nebo podáním injekce a většinou postihují oko.
Nejzávažnější nežádoucí účinky jsou popsány níže:
Časté závažné nežádoucí účinky (mohou postihovat až 1 z 10 osob): Odchlípení nebo trhlina vrstvy v zadní části oka (odchlípení sítnice nebo trhlina), vedoucí k zábleskům světla se sklivcovými vločkami a postupující do přechodné ztráty zraku nebo zakalení čočky (katarakta).
Méně časté závažné nežádoucí účinky (mohou postihovat až 1 ze 100 osob): Slepota, infekce oční bulvy (endoftalmitida) se zánětem vnitřní strany oka.
Příznaky, které se u Vás mohou objevit jsou popsány v bodu 2 této příbalové informace (prosím čtěte bod 2 „Co potřebujete vědět, než Vám bude Lucentis podán“). Sdělte, prosím, ihned svému lékaři, pokud se u Vás objeví kterýkoli z těchto nežádoucích účinků.
Nejčastěji hlášené nežádoucí účinky jsou uvedeny níže:
Velmi časté nežádoucí účinky (mohou postihovat více než 1 z 10 osob)
Oční nežádoucí účinky zahrnují: zánět oka, krvácení v zadní části oka (krvácení do sítnice), poruchy vidění, bolestivost oka, vidění malých částic nebo bodů (vloček), krvavé body v oku, dráždění oka a pocit cizího tělesa v oku, zvýšené slzení, zánět nebo infekce okrajů očního víčka, suchost oka, zarudnutí nebo svědění oka a zvýšení nitroočního tlaku.
Nežádoucí účinky mimo oko zahrnují: bolest v krku, zduření nosní sliznice, rýma, bolest hlavy a bolest kloubů.
Ostatní nežádoucí účinky, které se mohou vyskytnout po léčbě Lucentisem jsou popsány níže:
Časté nežádoucí účinky
Oční nežádoucí účinky zahrnují: Snížení ostrosti zraku, otoky některých částí oka (cévnatky, rohovky), zánět rohovky (přední část oka), malé tečky na povrchu oka, neostré vidění, krvácení v místě podání injekce, krvácení do oka, výtok z oka se svěděním, zarudnutí a otok (zánět spojivek), světloplachost, nepříjemný pocit v oku, otok očního víčka, bolestivost očního víčka.
Nežádoucí účinky mimo oko zahrnují: infekce močových cest, nízký počet červených krvinek (s příznaky jako je únava, ztížené dýchání, závrať, bledá kůže), pocit úzkosti, kašel, nevolnost, alergické rakce jako vyrážka, kopřivka, svědění a zčervenání kůže.
Méně časté nežádoucí účinky
Oční nežádoucí účinky zahrnují: Zánět a krvácení v přední části oka, hnisavý váček na oku, změny ve střední části povrchu oka, bolest nebo podráždění v místě injekce, abnormální citlivost oka, dráždění očního víčka.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Lucentis uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a na injekční lahvičce za Použitelné do:/EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
- Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
- Před použitím může být neotevřená injekční lahvička ponechána při pokojové teplotě (25 °C) po dobu 24 hodin.
- Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
- Nepoužívejte žádné balení, které je poškozeno.
Co Lucentis obsahuje
- Léčivou látkou je ranibizumabum (10 mg/ml). Jeden ml obsahuje 10 mg ranibizumabu.
- Dalšími složkami jsou dihydrát a,a-trehalosy, monohydrát histidin-hydrochloridu, histidin, polysorbát 20, voda na injekci.
Jak Lucentis vypadá a co obsahuje toto balení
Lucentis je injekční roztok dodávaný v injekční lahvičce (0,23 ml). Roztok je čirý, bezbarvý až světle žlutý a vodný.
Lucentis je dodáván v balení, které obsahuje: jednu skleněnou injekční lahvičku ranibizumabu s chlorbutylovou pryžovou zátkou, jednu tupou jehlu s filtrem (18G x 1/", 1,2 mm x 40 mm,
5 mikrometrů) pro nasátí obsahu injekční lahvičky, jednu injekční jehlu (30G x /", 0,3 mm x 13 mm) a jednu stříkačku (1 ml) pro nasátí obsahu injekční lahvičky a pro podání do sklivce. Všechny komponenty jsou pouze pro jednorázové použití.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11 Tel: +370 5 269 16 50
Etarapnu
Novartis Pharma Services Inc. Tea.: +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
EXXáSa
Novartis (Hellas) A.E.B.E. Tr(k: +30 210 281 17 12
Osterreich
Novartis Pharma GmbH Tel: +43 1 86 6570
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A. Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Island Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kúnpoq Novartis Pharma Services Inc Tn^: +357 22 690 690 |
Sverige . Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc Tel: +371 67 887 070 |
United Kingdom . Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
Viz také bod 3. „Jak se Lucentis podává“.
Jak připravit a jak aplikovat Lucentis
Jednorázové podání do sklivce
Lucentis musí být aplikován kvalifikovaným oftalmologem zkušeným v aplikaci do sklivce.
U vlhké formy AMD a u poškození zraku kvůli DME, pro makulární edém v důsledku RVO nebo pro CNV sekundární k PM je doporučená dávka Lucentisu 0,5 mg, podávaná jako jednorázová injekce do sklivce. To odpovídá injekci o objemu 0,05 ml. Interval mezi dvěma dávkami podávanými do stejného oka má být alespoň čtyři týdny.
Léčba se zahajuje jednou injekcí za měsíc do dosažení maximální zrakové ostrosti a/nebo do vymizení příznaků aktivity onemocnění, tj. žádná změna zrakové ostrosti a ostatních příznaků a projevů onemocnění při probíhající léčbě. U pacientů s vlhkou formou AMD, DME a RVO mohou být zpočátku potřeba tři nebo více po sobě jdoucí injekce podávané jednou za měsíc.
Následně mají být lékařem určeny intervaly sledování a léčby a mají být stanoveny na základě aktivity onemocnění, vyhodnocené podle zrakové ostrosti a/nebo anatomických parametrů.
Pokud zrakové a anatomické parametry ukazují na základě vyjádření lékaře, že pokračující léčba pacienta není přínosná, je třeba léčbu přípravkem Lucentis ukončit.
Sledování aktivity onemocnění může zahrnovat klinické vyšetření, funkční testy nebo zobrazovací techniky (např. optickou koherenční tomografii nebo fluorescenční angiografii).
Pokud j sou pacienti léčeni podle režimu „treat-and-extend“, pak po dosažení maximální zrakové ostrosti a/nebo vymizení příznaků aktivity onemocnění, mohou být léčebné intervaly postupně prodlouženy až do opětovného objevení se příznaků aktivity onemocnění nebo zhoršení zraku. Léčebný interval nemá být prodloužen o více než dva týdny najednou u vlhké formy AMD a může být prodloužen až o jeden měsíc najednou u DME. Při uzávěru retinální vény mohou být léčebné intervaly také postupně prodlužovány, ale nejsou dostupná dostatečná data dokládající délku těchto intervalů. Pokud se aktivita onemocnění znovu objeví, léčebný interval má být příslušně zkrácen.
V léčbě poškození zraku způsobeném CNV sekundární k PM může mnoho pacientů potřebovat pouze jednu nebo dvě injekce během prvního roku, zatímco někteří pacienti mohou potřebovat častější léčbu.
Lucentis a laserová fotokoagulace u DME a makulárního edému v důsledku BRVO Existuje určitá zkušenost s podáním přípravku Lucentis společně s laserovou fotokoagulací. Při podání ve stejný den má být Lucentis podán alespoň 30 minut po laserové fotokoagulaci. Lucentis může být podán pacientům, kteří byli léčeni předchozí laserovou fotokoagulací.
Lucentis a fotodynamická léčba Visudynem u CNV sekundární k PM Se souběžným podáváním přípravků Lucentis a Visudyne nejsou žádné zkušenosti.
Lucentis je nutno před aplikací vizuálně zkontrolovat, zda neobsahuje cizí částice nebo není změněna jeho barva.
Lucentis musí být aplikován za aseptických podmínek, což zahrnuje použití chirurgické desinfekce rukou, sterilních rukavic, sterilního oděvu, sterilního spekula (nebo ekvivalentní náhrady) a dostupnost sterilní paracentézy (je-li potřeba). Před aplikací injekce do sklivce je nutný pečlivý odběr anamnézy z hlediska hypersenzitivních reakcí. Před aplikací injekce musí být podána adekvátní anestezie a použit širokospektrý lokální antimikrobiální přípravek k dezinfekci pokožky v okolí oka, očního víčka a povrchu oka, v souladu s lokální praxí.
Při přípravě přípravku Lucentis k podání do sklivce dbejte, prosím, následujících pokynů:
Všechny komponenty jsou sterilní a pouze pro jednorázové použití. Jakákoliv komponenta s obalem vykazující známky poškození nebo manipulace nesmí být použita. Sterilita nemůže být zaručena, pokud nezůstane uzavření obalu komponenty neporušené. Opakované použití může vést k infekci nebo jinému onemocnění/poškození.


1. Před nasátím tekutiny do injekční stříkačky je nutno desinfikovat vnější část pryžové zátky.
2. Jehlu s 5 pm filtrem (18G x 1%'', 1,2 mm x 40 mm, 5 pm, přiložena) nasaďte na 1 ml injekční stříkačku (přiložena) za použití aseptického postupu. Zasuňte jehlu s filtrem do středu zátky lahvičky, dokud se jehla nedotkne dna injekční lahvičky.
3. Nasajte veškerou tekutinu z injekční lahvičky držené ve svislé, lehce nakloněné poloze pro snadnější úplné nasátí.
4. Ujistěte se, že píst je při vyprazdňování injekční lahvičky vytažen dostatečně daleko tak, aby jehla s filtrem byla úplně vyprázdněna.
5. Jehlu s filtrem ponechte v lahvičce a injekční stříkačku od ní odpojte. Jehla s filtrem by měla být po nasátí veškerého obsahu lahvičky zlikvidována a nesmí být použita pro vlastní aplikaci do sklivce.
6. Asepticky a pevně nasaďte injekční jehlu (30G x IV',
0,3 mm x 13 mm, přiložena) na stříkačku.
7. Opatrně odstraňte kryt z injekční jehly, aniž byste injekční jehlu odpojili od injekční stříkačky.
Poznámka: Při odstraňování krytu pevně uchopte žlutou část injekční jehly.
8. Důkladně vytlačte vzduch ze stříkačky a upravte dávku ke značce 0,05 ml uvedené na injekční stříkačce. Nyní je injekční stříkačka připravena k aplikaci.

Poznámka: Neotírejte injekční jehlu. Nevytahujte píst zpět.
Injekční jehla se zasune 3,5-4,0 mm posteriorně od limbu do prostoru sklivce tak, aby směřovala do centra očního bulbu a nikoli k horizontálnímu meridiánu. Poté se aplikuje objem injekce 0,05 ml. Následující injekci je nutné aplikovat v jiném místě skléry.
Po podání injekce nenasazujte zpět kryt jehly ani jehlu neoddělujte od injekční stříkačky. Zlikvidujte použitou injekční stříkačku společně s jehlou vyhozením do nádoby na ostré předměty nebo v souladu s místními požadavky.
Lucentis 10 mg/ml injekční roztok
Ranibizumabum
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Co naleznete v této příbalové informaci
1. Co je Lucentis a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude Lucentis podán
3. Jak se Lucentis podává
4. Možné nežádoucí účinky
5. Jak Lucentis uchovávat
6. Obsah balení a další informace
1. Co je Lucentis a k čemu se používá Co je Lucentis
Lucentis je roztok, který se aplikuje injekcí do oka. Lucentis patří do skupiny léků, nazvané antineovaskularizační látky. Obsahuje léčivou látku zvanou ranibizumab.
K čemu se Lucentis používá
Lucentis se používá u dospělých k léčbě několika očních onemocnění, způsobujících poškození zraku.
Tato onemocnění jsou výsledkem poškození sítnice (na světlo citlivé vrstvy v zadní části oka), způsobeným:
- růstem propustných, abnormálních krevních cév (choroidální neovaskularizace, CNV). Toto se pozoruje u onemocnění, jako například věkem podmíněná makulární degenerace (AMD) nebo patologická myopie (PM).
- makulárním edémem (otokem centrální části sítnice). Tento otok může být způsoben cukrovkou (onemocnění zvané diabetický makulární edém (DME)) nebo blokádou sítnicových žil (onemocnění zvané okluze retinální vény, RVO)).
Jak Lucentis působí
Lucentis specificky rozpoznává a váže se na bílkovinu zvanou lidský růstový faktor cévního endotelu A (VEGF-A), přítomnou v oku. VEGF-A způsobuje navíc abnormální růst krevních cév a otok v oku, který může vést k poškození zraku u onemocnění, jako jsou AMD, PM, DME nebo RVO. Lucentis může vazbou na VEGF-A blokovat jeho funkce a předcházet tomuto abnormálnímu růstu a otoku.
U těchto onemocnění může Lucentis pomoci stabilizovat a v mnoha případech zlepšit Váš zrak.
Lucentis Vám nesmí být podán
- jestliže jste alergický(á) na ranibizumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže máte infekci v oku nebo kolem očí.
- jestliže Vás bolí oči nebo je máte zarudlé (těžký nitrooční zánět).
Upozornění a opatření
Před podáním přípravku Lucentis se poraďte se svým lékařem.
- Lucentis je podáván jako injekce do oka. Po léčbě Lucentisem se může někdy vyvinout infekce vnitřní části oka, bolest oka nebo zarudnutí (zánět), odchlípení nebo natržení jedné z vrstev
v zadní části oka (odchlípení nebo natržení sítnice a odchlípení nebo natržení pigmentového epitelu sítnice), zákal čočky (katarakta). Je důležité rozeznat a léčit tuto infekci nebo odchlípení sítnice co možná nejdříve. Řekněte, prosím, svému lékaři ihned, pokud se u Vás vyskytnou příznaky jako bolest v oku, nepříjemný pocit v oku, zhoršení zarudnutí, rozmazané nebo snížené vidění. Informujte také neprodleně svého lékaře pokud máte pocit, že před okem vidíte zvýšený počet malých částic (teček) nebo pokud se u Vás vyskytne zvýšená citlivost na světlo.
- U některých pacientů může dojít vzápětí po injekci ke krátkodobému zvýšení nitroočního tlaku. Zvýšení nitroočního tlaku se u Vás nemusí projevit žádnými příznaky, ošetřující lékař však může nitrooční tlak po každé injekci kontrolovat.
- Informujte svého lékaře, jestliže jste někdy v minulosti měl(a) onemocnění oka nebo podstoupil(a) léčbu oka, nebo prodělal(a) mrtvici nebo přechodné příznaky mrtvice (slabost nebo ochrnutí končetin nebo obličeje, potíže při mluvení nebo porozumění). Tato informace bude brána v úvahu při zhodnocení, zda je Lucentis pro Vás vhodnou léčbou.
Děti a dospívající (pod 18 let věku)
Použití Lucentisu u dětí a dospívajících nebylo studováno, a proto se nedoporučuje.
Další léčivé přípravky a Lucentis
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které
možná budete užívat.
Těhotenství a kojení
- Ženám, které mohou otěhotnět se doporučuje používat během léčby účinnou antikoncepci.
- S použitím přípravku Lucentis u těhotných žen nej sou zkušenosti, proto případné riziko není známo. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, prodiskutujte to se svým lékařem před léčbou přípravkem Lucentis.
- Lucentis se nedoporučuje během kojení, protože není známo, zda Lucentis přestupuje do mateřského mléka. Poraďte se se svým lékařem nebo lékárníkem před léčbou přípravkem Lucentis.
Řízení dopravních prostředků a obsluha strojů
Po aplikaci přípravku Lucentis můžete mít dočasné problémy s viděním. Pokud se Vám toto stane,
neřiďte nebo neobsluhujte stroje, dokud poruchy zraku nevymizí.
Lucentis je podáván v místním znecitlivění jako injekce do oka Vaším očním lékařem. Obvyklá dávka v jedné injekci je 0,05 ml (které obsahují 0,5 mg léčivé látky). Interval mezi dvěma dávkami podanými do stejného oka má být alespoň čtyři týdny. Všechny injekce Vám vždy bude aplikot oční lékař.
Váš lékař Vám před injekcí pečlivě vypláchneoko, aby zabránil infekci. Dostanete také odpovídající místní umrtvení, aby se zmenšila případná bolest oka při injekci nebo se jí předešlo.
Léčba se zahajuje podáním jedné injekce Lucentisu za měsíc. Lékař bude sledovat stav Vašeho oka a podle odpovědi na léčbu rozhodne, zda a kdy potřebujete dostat další léčbu.
Podrobné pokyny pro používání jsou uvedeny na konci příbalové informace pod „Jak připravit a jak aplikovat Lucentis“.
Starší pacienti (věk 65 let a více)
Lucentis může být použit u osob ve věku 65 let a starších bez úpravy dávkování.
Jestliže jste zmeškal(a) dávku přípravku Lucentis
Kontaktujte co nejdříve ošetřujícího lékaře nebo nemocnici, abyste si domluvil(a) další návštěvu.
Před ukončením léčby přípravkem Lucentis
Jestliže uvažujete o ukončení léčby přípravkem Lucentis, jděte na další návštěvu a poraďte se se svým lékařem. Lékař Vám poradí a rozhodne, jak dlouho budete přípravkem Lucentis léčen(a).
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky spojené s podáním Lucentisu jsou způsobené buď vlastním přípravkem nebo podáním injekce a většinou postihují oko.
Nejzávažnější nežádoucí účinky jsou popsány níže:
Časté závažné nežádoucí účinky (mohou postihovat až 1 z 10 osob): Odchlípení nebo trhlina vrstvy v zadní části oka (odchlípení sítnice nebo trhlina), vedoucí k zábleskům světla se sklivcovými vločkami a postupující do přechodné ztráty zraku nebo zakalení čočky (katarakta).
Méně časté závažné nežádoucí účinky (mohou postihovat až 1 ze 100 osob): Slepota, infekce oční bulvy (endoftalmitida) se zánětem vnitřní strany oka.
Příznaky, které se u Vás mohou objevit jsou popsány v bodu 2 této příbalové informace (prosím čtěte bod 2 „Co potřebujete vědět, než Vám bude Lucentis podán“). Sdělte, prosím, ihned svému lékaři, pokud se u Vás objeví kterýkoli z těchto nežádoucích účinků.
Nejčastěji hlášené nežádoucí účinky jsou uvedeny níže:
Velmi časté nežádoucí účinky (mohou postihovat více než 1 z 10 osob)
Oční nežádoucí účinky zahrnují: zánět oka, krvácení v zadní části oka (krvácení do sítnice), poruchy vidění, bolestivost oka, vidění malých částic nebo bodů (vloček), krvavé body v oku, dráždění oka a pocit cizího tělesa v oku, zvýšené slzení, zánět nebo infekce okrajů očního víčka, suchost oka, zarudnutí nebo svědění oka a zvýšení nitroočního tlaku.
Nežádoucí účinky mimo oko zahrnují: bolest v krku, zduření nosní sliznice, rýma, bolest hlavy a bolest kloubů.
Ostatní nežádoucí účinky, které se mohou vyskytnout po léčbě Lucentisem jsou popsány níže:
Časté nežádoucí účinky
Oční nežádoucí účinky zahrnují: Snížení ostrosti zraku, otoky některých částí oka (cévnatky, rohovky), zánět rohovky (přední část oka), malé tečky na povrchu oka, neostré vidění, krvácení v místě podání injekce, krvácení do oka, výtok z oka se svěděním, zarudnutí a otok (zánět spojivek), světloplachost, nepříjemný pocit v oku, otok očního víčka, bolestivost očního víčka.
Nežádoucí účinky mimo oko zahrnují: infekce močových cest, nízký počet červených krvinek (s příznaky jako je únava, ztížené dýchání, závrať, bledá kůže), pocit úzkosti, kašel, nevolnost, alergické rakce jako vyrážka, kopřivka, svědění a zčervenání kůže.
Méně časté nežádoucí účinky
Oční nežádoucí účinky zahrnují: Zánět a krvácení v přední části oka, hnisavý váček na oku, změny ve střední části povrchu oka, bolest nebo podráždění v místě injekce, abnormální citlivost oka, dráždění očního víčka.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Lucentis uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a na injekční lahvičce za Použitelné do:/EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
- Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
- Před použitím může být neotevřená injekční lahvička ponechána při pokojové teplotě (25 °C) po dobu 24 hodin.
- Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
- Nepoužívejte žádné balení, které je poškozeno.
6. Obsah balení a další informace
Co Lucentis obsahuje
- Léčivou látkou je ranibizumabum (10 mg/ml). Jeden ml obsahuje 10 mg ranibizumabu.
- Dalšími složkami j sou dihydrát a,a-trehalosy, monohydrát histidin-hydrochloridu, histidin, polysorbát 20, voda na injekci.
Jak Lucentis vypadá a co obsahuje toto balení
Lucentis je injekční roztok dodávaný v injekční lahvičce (0,23 ml). Roztok je čirý, bezbarvý až světle žlutý a vodný.
Lucentis je dodáván v balení, které obsahuje: jednu skleněnou injekční lahvičku ranibizumabu s chlorbutylovou pryžovou zátkou. Injekční lahvička je pouze pro jednorázové použití.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11 Tel: +370 5 269 16 50
Etarapnu
Novartis Pharma Services Inc. Tea.: +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A. Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kónpoq Novartis Pharma Services Inc Tn^: +357 22 690 690 |
Sverige . Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc Tel: +371 67 887 070 |
United Kingdom . Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
EXXáSa
Novartis (Hellas) A.E.B.E. Tr(k: +30 210 281 17 12
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Viz také bod 3. „Jak se Lucentis podává“.
Jak připravit a jak aplikovat Lucentis
Jednorázové podání do sklivce
Lucentis musí být aplikován kvalifikovaným oftalmologem zkušeným v aplikaci do sklivce.
U vlhké formy AMD a u poškození zraku kvůli DME, pro makulární edém v důsledku RVO nebo pro CNV sekundární k PM je doporučená dávka Lucentisu 0,5 mg, podávaná jako jednorázová injekce do sklivce. To odpovídá injekci o objemu 0,05 ml. Interval mezi dvěma dávkami podávanými do stejného oka má být alespoň čtyři týdny.
Léčba se zahajuje jednou injekcí za měsíc do dosažení maximální zrakové ostrosti a/nebo do vymizení příznaků aktivity onemocnění, tj. žádná změna zrakové ostrosti a ostatních příznaků a projevů onemocnění při probíhající léčbě. U pacientů s vlhkou formou AMD, DME a RVO mohou být zpočátku potřeba tři nebo více po sobě jdoucí injekce podávané jednou za měsíc.
Následně mají být lékařem určeny intervaly sledování a léčby a mají být stanoveny na základě aktivity onemocnění, vyhodnocené podle zrakové ostrosti a/nebo anatomických parametrů.
Pokud zrakové a anatomické parametry ukazují na základě vyjádření lékaře, že pokračující léčba pacienta není přínosná, je třeba léčbu přípravkem Lucentis ukončit.
Sledování aktivity onemocnění může zahrnovat klinické vyšetření, funkční testy nebo zobrazovací techniky (např. optickou koherenční tomografii nebo fluorescenční angiografii).
Pokud j sou pacienti léčeni podle režimu „treat-and-extend“, pak po dosažení maximální zrakové ostrosti a/nebo vymizení příznaků aktivity onemocnění, mohou být léčebné intervaly postupně prodlouženy až do opětovného objevení se příznaků aktivity onemocnění nebo zhoršení zraku. Léčebný interval nemá být prodloužen o více než dva týdny najednou u vlhké formy AMD a může být prodloužen až o jeden měsíc najednou u DME. Při uzávěru retinální vény mohou být léčebné intervaly také postupně prodlužovány, ale nejsou dostupná dostatečná data dokládající délku těchto intervalů. Pokud se aktivita onemocnění znovu objeví, léčebný interval má být příslušně zkrácen.
V léčbě poškození zraku způsobeném CNV sekundární k PM může mnoho pacientů potřebovat pouze jednu nebo dvě injekce během prvního roku, zatímco někteří pacienti mohou potřebovat častější léčbu.
Lucentis a laserová fotokoagulace u DME a makulárního edému v důsledku BRVO Existuje určitá zkušenost s podáním přípravku Lucentis společně s laserovou fotokoagulací. Při podání ve stejný den má být Lucentis podán alespoň 30 minut po laserové fotokoagulaci. Lucentis může být podán pacientům, kteří byli léčeni předchozí laserovou fotokoagulací.
Lucentis a fotodynamická léčba Visudynem u CNV sekundární k PM Se souběžným podáváním přípravků Lucentis a Visudyne nejsou žádné zkušenosti.
Lucentis je nutno před aplikací vizuálně zkontrolovat, zda neobsahuje cizí částice nebo není změněna jeho barva.
Lucentis musí být aplikován za aseptických podmínek, což zahrnuje použití chirurgické desinfekce rukou, sterilních rukavic, sterilního oděvu, sterilního spekula (nebo ekvivalentní náhrady) a dostupnost sterilní paracentézy (je-li potřeba). Před aplikací injekce do sklivce je nutný pečlivý odběr anamnézy z hlediska hypersenzitivních reakcí. Před aplikací injekce musí být podána adekvátní anestezie a použit širokospektrý lokální antimikrobiální přípravek k dezinfekci pokožky v okolí oka, očního víčka a povrchu oka, v souladu s lokální praxí.
Při přípravě přípravku Lucentis k podání do sklivce dbejte, prosím, následujících pokynů:
Injekční lahvička je pouze pro jednorázové použití. Po podání injekce musí být veškerý nepoužitý léčivý přípravek zlikvidován. Jakákoliv injekční lahvička vykazující známky poškození nebo manipulace nesmí být použita. Sterilita nemůže být zaručena, pokud nezůstane uzávěr obalu neporušený.
Pro přípravu a podání injekce do sklivce jsou potřebné následující zdravotnické pomůcky pro jednorázové použití:
- jehla s 5 pm filtrem (18G)
- 1 ml sterilní injekční stříkačka
- injekční jehla (30G x 14").
Tyto zdravotnické pomůcky nejsou obsaženy v balení přípravku Lucentis.


1. Před nasátím tekutiny do injekční stříkačky je nutno desinfikovat vnější část pryžové zátky.
2. Jehlu s 5 pm filtrem (18G) nasaďte na 1 ml injekční stříkačku za použití aseptického postupu. Zasuňte jehlu s filtrem do středu zátky lahvičky, dokud se jehla nedotkne dna injekční lahvičky.
3. Nasajte veškerou tekutinu z injekční lahvičky držené ve svislé, lehce nakloněné poloze pro snadnější úplné nasátí.
4. Ujistěte se, že píst je při vyprazdňování injekční lahvičky vytažen dostatečně daleko tak, aby jehla s filtrem byla úplně vyprázdněna.
5. Jehlu s filtrem ponechte v lahvičce a injekční stříkačku od ní odpojte. Jehla s filtrem by měla být po nasátí veškerého obsahu lahvičky zlikvidována a nesmí být použita pro vlastní aplikaci do sklivce.
6. Asepticky a pevně nasaďte injekční jehlu (30G x 4") na stříkačku.
7. Opatrně odstraňte kryt z injekční jehly, aniž byste injekční jehlu odpojili od injekční stříkačky.
Poznámka: Při odstraňování krytu pevně uchopte část injekční jehly.
8. Důkladně vytlačte vzduch ze stříkačky a upravte dávku ke značce 0,05 ml uvedené na injekční stříkačce. Nyní je injekční stříkačka připravena k aplikaci.

Poznámka: Neotírejte injekční jehlu. Nevytahujte píst zpět.
Injekční jehla se zasune 3,5-4,0 mm posteriorně od limbu do prostoru sklivce tak, aby směřovala do centra očního bulbu a nikoli k horizontálnímu meridiánu. Poté se aplikuje objem injekce 0,05 ml. Následující injekci je nutné aplikovat v jiném místě skléry.
Po podání injekce nenasazujte zpět kryt jehly ani jehlu neoddělujte od injekční stříkačky. Zlikvidujte použitou injekční stříkačku společně s jehlou vyhozením do nádoby na ostré předměty nebo v souladu s místními požadavky.
Příbalová informace: informace pro pacienta
Lucentis 10 mg/ml injekční roztok v předplněné injekční stříkačce
Ranibizumabum
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Co naleznete v této příbalové informaci
1. Co je Lucentis a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude Lucentis podán
3. Jak se Lucentis podává
4. Možné nežádoucí účinky
5. Jak Lucentis uchovávat
6. Obsah balení a další informace
1. Co je Lucentis a k čemu se používá Co je Lucentis
Lucentis je roztok, který se aplikuje injekcí do oka. Lucentis patří do skupiny léků, nazvané antineovaskularizační látky. Obsahuje léčivou látku zvanou ranibizumab.
K čemu se Lucentis používá
Lucentis se používá u dospělých k léčbě několika očních onemocnění, způsobujících poškození zraku.
Tato onemocnění jsou výsledkem poškození sítnice (na světlo citlivé vrstvy v zadní části oka), způsobeným:
- růstem propustných, abnormálních krevních cév (choroidální neovaskularizace, CNV). Toto se pozoruje u onemocnění, jako například věkem podmíněná makulární degenerace (AMD) nebo patologická myopie (PM).
- makulárním edémem (otokem centrální části sítnice). Tento otok může být způsoben cukrovkou (onemocnění zvané diabetický makulární edém (DME)) nebo blokádou sítnicových žil (onemocnění zvané okluze retinální vény, RVO)).
Jak Lucentis působí
Lucentis specificky rozpoznává a váže se na bílkovinu zvanou lidský růstový faktor cévního endotelu A (VEGF-A), přítomnou v oku. VEGF-A způsobuje navíc abnormální růst krevních cév a otok v oku, který může vést k poškození zraku u onemocnění, jako jsou AMD, PM, DME nebo RVO. Lucentis může vazbou na VEGF-A blokovat jeho funkce a předcházet tomuto abnormálnímu růstu a otoku.
U těchto onemocnění může Lucentis pomoci stabilizovat a v mnoha případech zlepšit Váš zrak.
Lucentis Vám nesmí být podán
- jestliže jste alergický(á) na ranibizumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže máte infekci v oku nebo kolem očí.
- jestliže Vás bolí oči nebo je máte zarudlé (těžký nitrooční zánět).
Upozornění a opatření
Před podáním přípravku Lucentis se poraďte se svým lékařem.
- Lucentis je podáván jako injekce do oka. Po léčbě Lucentisem se může někdy vyvinout infekce vnitřní části oka, bolest oka nebo zarudnutí (zánět), odchlípení nebo natržení jedné z vrstev
v zadní části oka (odchlípení nebo natržení sítnice a odchlípení nebo natržení pigmentového epitelu sítnice), zákal čočky (katarakta). Je důležité rozeznat a léčit tuto infekci nebo odchlípení sítnice co možná nejdříve. Řekněte, prosím, svému lékaři ihned, pokud se u Vás vyskytnou příznaky jako bolest v oku, nepříjemný pocit v oku, zhoršení zarudnutí, rozmazané nebo snížené vidění. Informujte také neprodleně svého lékaře pokud máte pocit, že před okem vidíte zvýšený počet malých částic (teček) nebo pokud se u Vás vyskytne zvýšená citlivost na světlo.
- U některých pacientů může dojít vzápětí po injekci ke krátkodobému zvýšení nitroočního tlaku. Zvýšení nitroočního tlaku se u Vás nemusí projevit žádnými příznaky, ošetřující lékař však může nitrooční tlak po každé injekci kontrolovat.
- Informujte svého lékaře, jestliže jste někdy v minulosti měl(a) onemocnění oka nebo podstoupil(a) léčbu oka, nebo prodělal(a) mrtvici nebo přechodné příznaky mrtvice (slabost nebo ochrnutí končetin nebo obličeje, potíže při mluvení nebo porozumění). Tato informace bude brána v úvahu při zhodnocení, zda je Lucentis pro Vás vhodnou léčbou.
Děti a dospívající (pod 18 let věku)
Použití Lucentisu u dětí a dospívajících nebylo studováno, a proto se nedoporučuje.
Další léčivé přípravky a Lucentis
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které
možná budete užívat.
Těhotenství a kojení
- Ženám, které mohou otěhotnět se doporučuje používat během léčby účinnou antikoncepci.
- S použitím přípravku Lucentis u těhotných žen nej sou zkušenosti, proto případné riziko není známo. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, prodiskutujte to se svým lékařem před léčbou přípravkem Lucentis.
- Lucentis se nedoporučuje během kojení, protože není známo, zda Lucentis přestupuje do mateřského mléka. Poraďte se se svým lékařem nebo lékárníkem před léčbou přípravkem Lucentis.
Řízení dopravních prostředků a obsluha strojů
Po aplikaci přípravku Lucentis můžete mít dočasné problémy s viděním. Pokud se Vám toto stane,
neřiďte nebo neobsluhujte stroje, dokud poruchy zraku nevymizí.
Lucentis je podáván v místním znecitlivění jako injekce do oka Vaším očním lékařem. Obvyklá dávka v jedné injekci je 0,05 ml (které obsahují 0,5 mg léčivé látky). Předplněná injekční stříkačka obsahuje více než doporučenou dávku 0,5 mg. Extrahovatelný objem není určen k celkovému podání. Nadbytečný objem je třeba před podáním injekce vytlačit. Při podání celého objemu předplněné injekční stříkačky by mohlo dojít k předávkování.
Interval mezi dvěma dávkami podanými do stejného oka má být alespoň čtyři týdny. Všechny injekce Vám vždy bude aplikovat oční lékař.
Váš lékař Vám před injekcí pečlivě vypláchneoko, aby zabránil infekci. Dostanete také odpovídající místní umrtvení, aby se zmenšila případná bolest oka při injekci nebo se jí předešlo.
Léčba se zahajuje podáním jedné injekce Lucentisu za měsíc. Lékař bude sledovat stav Vašeho oka a podle odpovědi na léčbu rozhodne, zda a kdy potřebujete dostat další léčbu.
Podrobné pokyny pro používání jsou uvedeny na konci příbalové informace pod „Jak připravit a jak aplikovat Lucentis“.
Starší pacienti (věk 65 let a více)
Lucentis může být použit u osob ve věku 65 let a starších bez úpravy dávkování.
Jestliže jste zmeškal(a) dávku přípravku Lucentis
Kontaktujte co nejdříve ošetřujícího lékaře nebo nemocnici, abyste si domluvil(a) další návštěvu.
Před ukončením léčby přípravkem Lucentis
Jestliže uvažujete o ukončení léčby přípravkem Lucentis, jděte na další návštěvu a poraďte se se svým lékařem. Lékař Vám poradí a rozhodne, jak dlouho budete přípravkem Lucentis léčen(a).
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky spoj ené s podáním Lucentisu jsou způsobené buď vlastním přípravkem nebo podáním injekce a většinou postihují oko.
Nejzávažnější nežádoucí účinky jsou popsány níže:
Časté závažné nežádoucí účinky (mohou postihovat až 1 z 10 osob): Odchlípení nebo trhlina vrstvy v zadní části oka (odchlípení sítnice nebo trhlina), vedoucí k zábleskům světla se sklivcovými vločkami a postupující do přechodné ztráty zraku nebo zakalení čočky (katarakta).
Méně časté závažné nežádoucí účinky (mohou postihovat až 1 ze 100 osob): Slepota, infekce oční bulvy (endoftalmitida) se zánětem vnitřní strany oka.
Příznaky, které se u Vás mohou objevit jsou popsány v bodu 2 této příbalové informace (prosím čtěte bod 2 „Co potřebujete vědět, než Vám bude Lucentis podán“). Sdělte, prosím, ihned svému lékaři, pokud se u Vás objeví kterýkoli z těchto nežádoucích účinků.
Nejčastěji hlášené nežádoucí účinky jsou uvedeny níže:
Velmi časté nežádoucí účinky (mohou postihovat více než 1 z 10 osob)
Oční nežádoucí účinky zahrnují: zánět oka, krvácení v zadní části oka (krvácení do sítnice), poruchy vidění, bolestivost oka, vidění malých částic nebo bodů (vloček), krvavé body v oku, dráždění oka a pocit cizího tělesa v oku, zvýšené slzení, zánět nebo infekce okrajů očního víčka, suchost oka, zarudnutí nebo svědění oka a zvýšení nitroočního tlaku.
Nežádoucí účinky mimo oko zahrnují: bolest v krku, zduření nosní sliznice, rýma, bolest hlavy a bolest kloubů.
Ostatní nežádoucí účinky, které se mohou vyskytnout po léčbě Lucentisem jsou popsány níže:
Časté nežádoucí účinky
Oční nežádoucí účinky zahrnují: Snížení ostrosti zraku, otoky některých částí oka (cévnatky, rohovky), zánět rohovky (přední část oka), malé tečky na povrchu oka, neostré vidění, krvácení v místě podání injekce, krvácení do oka, výtok z oka se svěděním, zarudnutí a otok (zánět spojivek), světloplachost, nepříjemný pocit v oku, otok očního víčka, bolestivost očního víčka.
Nežádoucí účinky mimo oko zahrnují: infekce močových cest, nízký počet červených krvinek (s příznaky jako je únava, ztížené dýchání, závrať, bledá kůže), pocit úzkosti, kašel, nevolnost, alergické rakce jako vyrážka, kopřivka, svědění a zčervenání kůže.
Méně časté nežádoucí účinky
Oční nežádoucí účinky zahrnují: Zánět a krvácení v přední části oka, hnisavý váček na oku, změny ve střední části povrchu oka, bolest nebo podráždění v místě injekce, abnormální citlivost oka, dráždění očního víčka.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Lucentis uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a na štítku předplněné injekční stříkačky za Použitelné do:/EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
- Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
- Před použitím může být zatavený obal ponechán při pokojové teplotě (25 °C) po dobu 24 hodin.
- Uchovávejte předplněnou injekční stříkačku v jejím neotevřeném obalu v krabičce, aby byl přípravek chráněn před světlem.
- Nepoužívejte žádné balení, které je poškozeno.
6. Obsah balení a další informace
Co Lucentis obsahuje
- Léčivou látkou je ranibizumabum (10 mg/ml). Jeden ml obsahuje 10 mg ranibizumabu.
- Dalšími složkami jsou dihydrát a,a-trehalosy, monohydrát histidin-hydrochloridu, histidin, polysorbát 20, voda na injekci.
Jak Lucentis vypadá a co obsahuje toto balení
Lucentis je injekční roztok dodávaný v předplněné injekční stříkačce. Předplněná injekční stříkačka obsahuje 0,165 ml sterilního, čirého, bezbarvého až světle žlutého vodného roztoku. Předplněná injekční stříkačka obsahuje více než doporučenou dávku 0,5 mg. Extrahovatelný objem není určen k celkovému podání. Nadbytečný objem je třeba před podáním injekce vytlačit. Při podání celého objemu předplněné injekční stříkačky by mohlo dojít k předávkování.
Balení o velikosti jedné předplněné injekční stříkačky, baleno v zataveném obalu. Předplněná injekční stříkačka je pouze pro jednorázové použití.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11 Tel: +370 5 269 16 50
Etarapnu
Novartis Pharma Services Inc. Tea.: +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A. Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Island Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kúnpoq Novartis Pharma Services Inc Tn^: +357 22 690 690 |
Sverige . Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc Tel: +371 67 887 070 |
United Kingdom . Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
EXXáSa
Novartis (Hellas) A.E.B.E. Tr(k: +30 210 281 17 12
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
Viz také bod 3. „Jak se Lucentis podává“.
Jak připravit a jak aplikovat Lucentis
Předplněná injekční stříkačka pouze pro jednorázové podání do sklivce
Lucentis musí být aplikován kvalifikovaným oftalmologem zkušeným v aplikaci do sklivce.
U vlhké formy AMD a u poškození zraku kvůli DME, pro makulární edém v důsledku RVO nebo pro CNV sekundární k PM je doporučená dávka Lucentisu 0,5 mg, podávaná jako jednorázová injekce do sklivce. To odpovídá injekci o objemu 0,05 ml. Interval mezi dvěma dávkami podávanými do stejného oka má být alespoň čtyři týdny.
Léčba se zahajuje jednou injekcí za měsíc do dosažení maximální zrakové ostrosti a/nebo do vymizení příznaků aktivity onemocnění, tj. žádná změna zrakové ostrosti a ostatních příznaků a projevů onemocnění při probíhající léčbě. U pacientů s vlhkou formou AMD, DME a RVO mohou být zpočátku potřeba tři nebo více po sobě jdoucí injekce podávané jednou za měsíc.
Následně mají být lékařem určeny intervaly sledování a léčby a mají být stanoveny na základě aktivity onemocnění, vyhodnocené podle zrakové ostrosti a/nebo anatomických parametrů.
Pokud zrakové a anatomické parametry ukazují na základě vyjádření lékaře, že pokračující léčba pacienta není přínosná, je třeba léčbu přípravkem Lucentis ukončit.
Sledování aktivity onemocnění může zahrnovat klinické vyšetření, funkční testy nebo zobrazovací techniky (např. optickou koherenční tomografii nebo fluorescenční angiografii).
Pokud j sou pacienti léčeni podle režimu „treat-and-extend“, pak po dosažení maximální zrakové ostrosti a/nebo vymizení příznaků aktivity onemocnění, mohou být léčebné intervaly postupně prodlouženy až do opětovného objevení se příznaků aktivity onemocnění nebo zhoršení zraku. Léčebný interval nemá být prodloužen o více než dva týdny najednou u vlhké formy AMD a může být prodloužen až o jeden měsíc najednou u DME. Při uzávěru retinální vény mohou být léčebné intervaly také postupně prodlužovány, ale nejsou dostupná dostatečná data dokládající délku těchto intervalů. Pokud se aktivita onemocnění znovu objeví, léčebný interval má být příslušně zkrácen.
V léčbě poškození zraku způsobeném CNV sekundární k PM může mnoho pacientů potřebovat pouze jednu nebo dvě injekce během prvního roku, zatímco někteří pacienti mohou potřebovat častější léčbu.
Lucentis a laserová fotokoagulace u DME a makulárního edému v důsledku BRVO Existuje určitá zkušenost s podáním přípravku Lucentis společně s laserovou fotokoagulací. Při podání ve stejný den má být Lucentis podán alespoň 30 minut po laserové fotokoagulaci. Lucentis může být podán pacientům, kteří byli léčeni předchozí laserovou fotokoagulací.
Lucentis a fotodynamická léčba Visudynem u CNV sekundární k PM Se souběžným podáváním přípravků Lucentis a Visudyne nejsou žádné zkušenosti.
Lucentis je nutno před aplikací vizuálně zkontrolovat, zda neobsahuje cizí částice nebo není změněna jeho barva.
Lucentis musí být aplikován za aseptických podmínek, což zahrnuje použití chirurgické desinfekce rukou, sterilních rukavic, sterilního oděvu, sterilního spekula (nebo ekvivalentní náhrady) a dostupnost sterilní paracentézy (je-li potřeba). Před aplikací injekce do sklivce je nutný pečlivý odběr anamnézy z hlediska hypersenzitivních reakcí. Před aplikací injekce musí být podána adekvátní anestezie a použit širokospektrý lokální antimikrobiální přípravek k dezinfekci pokožky v okolí oka, očního víčka a povrchu oka, v souladu s lokální praxí.
Předplněná injekční stříkačka je pouze pro jednorázové použití. Předplněná injekční stříkačka je sterilní. Přípravek nepoužívejte, pokud je balení poškozeno. Sterilita předplněné injekční stříkačky nemůže být zaručena, pokud nezůstane obal zatavený. Předplněnou injekční stříkačku nepoužívejte, pokud má roztok změněnou barvu, je zakalený nebo obsahuje částice.
Předplněná injekční stříkačka obsahuje více než doporučenou dávku 0,5 mg. Extrahovatelný objem předplněné injekční stříkačky (0,1 ml) není určen k celkovému podání. Nadbytečný objem je třeba před podáním injekce vytlačit. Při podání celého objemu předplněné injekční stříkačky by mohlo dojít k předávkování. K vytlačení vzduchové bubliny spolu s nadbytečným léčivým přípravkem pomalu tlačte píst, dokud okraj pod vyklenutím pryžové zarážky není vyrovnaný s černou dávkovací linkou na injekční stříkačce (ekvivalentní 0,05 ml, tj. 0,5 mg ranibizumabu).
Pro podání do sklivce je třeba použít sterilní injekční jehlu 30G x ^".
Při přípravě přípravku Lucentis k podání do sklivce dbejte, prosím, pokynů k použití:
Před použitím předplněné injekční stříkačky si pečlivě přečtěte všechny pokyny. Předplněná injekční stříkačka je pouze pro jednorázové použití. Předplněná injekční stříkačka je sterilní. Přípravek nepoužívejte, pokud je balení poškozeno. Otevření zataveného obalu a všechny následující kroky musí být provedeny za aseptických podmínek.
Poznámka: dávka musí být nastavena na 0,05 ml._
Čepička Označení dávky Držadlo
0,05 ml

Adaptér Pryžová zarážka Pístové táhlo Luer lock
2.
Ujistěte se, že balení obsahuje:
sterilní předplněnou injekční stříkačku v zataveném obalu.
Sloupněte víčko zataveného obalu injekční stříkačky a za použití aseptické techniky opatrně vyjměte injekční stříkačku.
|
Zkontrolujte |
3. |
|
injekční |
• |
|
stříkačku |
• |
|
• | |
|
4. | |
|
Odstraňte |
5. |
|
čepičku |
6. |
Zkontrolujte, že:
čepička injekční stříkačky není oddělena od adaptéru Luer lock. injekční stříkačka není poškozena. injekční roztok je čirý, bezbarvý až světle žlutý a neobsahuje žádné částice.
Pokud některý údaj uvedený výše nesouhlasí, znehodnoťte předplněnou injekční stříkačku a použijte novou._
Ulomte (neotáčejte ani nekruťte) čepičku injekční stříkačky (viz Obrázek 2). Zlikvidujte čepičku injekční stříkačky (viz Obrázek 3).

Obrázek 2

Obrázek 3
Nasaďte jehlu
7. Pevně nasaďte sterilní injekční jehlu 30G x Í4'' na injekční stříkačku těsným zašroubováním do adaptéru Luer lock (viz Obrázek 4).
8. Opatrně odstraňte kryt j ehly j eho přímým vytažením (viz Obrázek 5).
Poznámka: Jehlu nikdy neotírejte.

Obrázek 4 Obrázek 5
9. Držte injekční stříkačku svisle.
10. Pokud jsou přítomny vzduchové bubliny, jemně poklepejte injekční stříkačku prstem, dokud bubliny stoupají nahoru (viz Obrázek 6).

Obrázek 6
11. Držte injekční stříkačku v úrovni očí a opatrně tlačte píst, dokud není okraj pod vyklenutím pryžové zarážky vyrovnaný s označením dávky (viz Obrázek 7). Tak se vytlačí vzduch a nadbytek injekčního roztoku a nastaví se dávka na 0,05 ml.
Poznámka: Pístové táhlo není připojeno k pryžové zarážce - to aby se předešlo natažení vzduchu zpět do injekční stříkačky.

Obrázek 7
Postup podání injekce má být proveden za aseptických podmínek.
12. Injekční jehla se zasune 3,5-4,0 mm posteriorně k limbu do prostoru sklivce tak, aby směřovala do centra očního bulbu a nikoli k horizontálnímu meridiánu.
13. Injekci podávejte pomalu, dokud pryžová zarážka nedosáhne dna injekční stříkačky, aby byl podán objem 0,05 ml.
14. Následující injekce aplikujte do odlišných míst skléry.
15. Po podání injekce nenasazujte zpět kryt jehly ani jehlu neoddělujte od injekční stříkačky. Zlikvidujte použitou injekční stříkačku společně
s jehlou vyhozením do nádoby na ostré předměty nebo v souladu s místními požadavky._
Lucentis 10 mg/ml injekční roztok
Ranibizumabum
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Co naleznete v této příbalové informaci
1. Co je Lucentis a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude Lucentis podán
3. Jak se Lucentis podává
4. Možné nežádoucí účinky
5. Jak Lucentis uchovávat
6. Obsah balení a další informace
1. Co je Lucentis a k čemu se používá Co je Lucentis
Lucentis je roztok, který se aplikuje injekcí do oka. Lucentis patří do skupiny léků, nazvané antineovaskularizační látky. Obsahuje léčivou látku zvanou ranibizumab.
K čemu se Lucentis používá
Lucentis se používá u dospělých k léčbě několika očních onemocnění, způsobujících poškození zraku.
Tato onemocnění jsou výsledkem poškození sítnice (na světlo citlivé vrstvy v zadní části oka), způsobeným:
- růstem propustných, abnormálních krevních cév (choroidální neovaskularizace, CNV). Toto se pozoruje u onemocnění, jako například věkem podmíněná makulární degenerace (AMD) nebo patologická myopie (PM).
- makulárním edémem (otokem centrální části sítnice). Tento otok může být způsoben cukrovkou (onemocnění zvané diabetický makulární edém (DME)) nebo blokádou sítnicových žil (onemocnění zvané okluze retinální vény, RVO)).
Jak Lucentis působí
Lucentis specificky rozpoznává a váže se na bílkovinu zvanou lidský růstový faktor cévního endotelu A (VEGF-A), přítomnou v oku. VEGF-A způsobuje navíc abnormální růst krevních cév a otok v oku, který může vést k poškození zraku u onemocnění, jako jsou AMD, PM, DME nebo RVO. Lucentis může vazbou na VEGF-A blokovat jeho funkce a předcházet tomuto abnormálnímu růstu a otoku.
U těchto onemocnění může Lucentis pomoci stabilizovat a v mnoha případech zlepšit Váš zrak.
Lucentis Vám nesmí být podán
- jestliže jste alergický(á) na ranibizumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže máte infekci v oku nebo kolem očí.
- jestliže Vás bolí oči nebo je máte zarudlé (těžký nitrooční zánět).
Upozornění a opatření
Před podáním přípravku Lucentis se poraďte se svým lékařem.
- Lucentis je podáván jako injekce do oka. Po léčbě Lucentisem se může někdy vyvinout infekce vnitřní části oka, bolest oka nebo zarudnutí (zánět), odchlípení nebo natržení jedné z vrstev
v zadní části oka (odchlípení nebo natržení sítnice a odchlípení nebo natržení pigmentového epitelu sítnice), zákal čočky (katarakta). Je důležité rozeznat a léčit tuto infekci nebo odchlípení sítnice co možná nejdříve. Řekněte, prosím, svému lékaři ihned, pokud se u Vás vyskytnou příznaky jako bolest v oku, nepříjemný pocit v oku, zhoršení zarudnutí, rozmazané nebo snížené vidění. Informujte také neprodleně svého lékaře pokud máte pocit, že před okem vidíte zvýšený počet malých částic (teček) nebo pokud se u Vás vyskytne zvýšená citlivost na světlo.
- U některých pacientů může dojít vzápětí po injekci ke krátkodobému zvýšení nitroočního tlaku. Zvýšení nitroočního tlaku se u Vás nemusí projevit žádnými příznaky, ošetřující lékař však může nitrooční tlak po každé injekci kontrolovat.
- Informujte svého lékaře, jestliže jste někdy v minulosti měl(a) onemocnění oka nebo podstoupil(a) léčbu oka, nebo prodělal(a) mrtvici nebo přechodné příznaky mrtvice (slabost nebo ochrnutí končetin nebo obličeje, potíže při mluvení nebo porozumění). Tato informace bude brána v úvahu při zhodnocení, zda je Lucentis pro Vás vhodnou léčbou.
Děti a dospívající (pod 18 let věku)
Použití Lucentisu u dětí a dospívajících nebylo studováno, a proto se nedoporučuje.
Další léčivé přípravky a Lucentis
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které
možná budete užívat.
Těhotenství a kojení
- Ženám, které mohou otěhotnět se doporučuje používat během léčby účinnou antikoncepci.
- S použitím přípravku Lucentis u těhotných žen nej sou zkušenosti, proto případné riziko není známo. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, prodiskutujte to se svým lékařem před léčbou přípravkem Lucentis.
- Lucentis se nedoporučuje během kojení, protože není známo, zda Lucentis přestupuje do mateřského mléka. Poraďte se se svým lékařem nebo lékárníkem před léčbou přípravkem Lucentis.
Řízení dopravních prostředků a obsluha strojů
Po aplikaci přípravku Lucentis můžete mít dočasné problémy s viděním. Pokud se Vám toto stane,
neřiďte nebo neobsluhujte stroje, dokud poruchy zraku nevymizí.
Lucentis je podáván v místním znecitlivění jako injekce do oka Vaším očním lékařem. Obvyklá dávka v jedné injekci je 0,05 ml (které obsahují 0,5 mg léčivé látky). Interval mezi dvěma dávkami podanými do stejného oka má být alespoň čtyři týdny. Všechny injekce Vám vždy bude aplikovat oční lékař.
Váš lékař Vám před injekcí pečlivě vypláchne oko, aby zabránil infekci. Dostanete také odpovídající místní umrtvení, aby se zmenšila případná bolest oka při injekci nebo se jí předešlo.
Léčba se zahajuje podáním jedné injekce Lucentisu za měsíc. Lékař bude sledovat stav Vašeho oka a podle odpovědi na léčbu rozhodne, zda a kdy potřebujete dostat další léčbu.
Podrobné pokyny pro používání jsou uvedeny na konci příbalové informace pod „Jak připravit a jak aplikovat Lucentis“.
Starší pacienti (věk 65 let a více)
Lucentis může být použit u osob ve věku 65 let a starších bez úpravy dávkování.
Jestliže jste zmeškal(a) dávku přípravku Lucentis
Kontaktujte co nejdříve ošetřujícího lékaře nebo nemocnici, abyste si domluvil(a) další návštěvu.
Před ukončením léčby přípravkem Lucentis
Jestliže uvažujete o ukončení léčby přípravkem Lucentis, jděte na další návštěvu a poraďte se se svým lékařem. Lékař Vám poradí a rozhodne, jak dlouho budete přípravkem Lucentis léčen(a).
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky spojené s podáním Lucentisu jsou způsobené buď vlastním přípravkem nebo podáním injekce a většinou postihují oko.
Nejzávažnější nežádoucí účinky jsou popsány níže:
Časté závažné nežádoucí účinky (mohou postihovat až 1 z 10 osob): Odchlípení nebo trhlina vrstvy v zadní části oka (odchlípení sítnice nebo trhlina), vedoucí k zábleskům světla se sklivcovými vločkami a postupující do přechodné ztráty zraku nebo zakalení čočky (katarakta).
Méně časté závažné nežádoucí účinky (mohou postihovat až 1 ze 100 osob): Slepota, infekce oční bulvy (endoftalmitida) se zánětem vnitřní strany oka.
Příznaky, které se u Vás mohou objevit jsou popsány v bodu 2 této příbalové informace (prosím čtěte bod 2 „Co potřebujete vědět, než Vám bude Lucentis podán“). Sdělte, prosím, ihned svému lékaři, pokud se u Vás objeví kterýkoli z těchto nežádoucích účinků.
Nejčastěji hlášené nežádoucí účinky jsou uvedeny níže:
Velmi časté nežádoucí účinky (mohou postihovat více než 1 z 10 osob)
Oční nežádoucí účinky zahrnují: zánět oka, krvácení v zadní části oka (krvácení do sítnice), poruchy vidění, bolestivost oka, vidění malých částic nebo bodů (vloček), krvavé body v oku, dráždění oka a pocit cizího tělesa v oku, zvýšené slzení, zánět nebo infekce okrajů očního víčka, suchost oka, zarudnutí nebo svědění oka a zvýšení nitroočního tlaku.
Nežádoucí účinky mimo oko zahrnují: bolest v krku, zduření nosní sliznice, rýma, bolest hlavy a bolest kloubů.
Ostatní nežádoucí účinky, které se mohou vyskytnout po léčbě Lucentisem jsou popsány níže:
Časté nežádoucí účinky
Oční nežádoucí účinky zahrnují: Snížení ostrosti zraku, otoky některých částí oka (cévnatky, rohovky), zánět rohovky (přední část oka), malé tečky na povrchu oka, neostré vidění, krvácení v místě podání injekce, krvácení do oka, výtok z oka se svěděním, zarudnutí a otok (zánět spojivek), světloplachost, nepříjemný pocit v oku, otok očního víčka, bolestivost očního víčka.
Nežádoucí účinky mimo oko zahrnují: infekce močových cest, nízký počet červených krvinek (s příznaky jako je únava, ztížené dýchání, závrať, bledá kůže), pocit úzkosti, kašel, nevolnost, alergické rakce jako vyrážka, kopřivka, svědění a zčervenání kůže.
Méně časté nežádoucí účinky
Oční nežádoucí účinky zahrnují: Zánět a krvácení v přední části oka, hnisavý váček na oku, změny ve střední části povrchu oka, bolest nebo podráždění v místě injekce, abnormální citlivost oka, dráždění očního víčka.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Lucentis uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a na injekční lahvičce za Použitelné do:/EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
- Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
- Před použitím může být neotevřená injekční lahvička ponechána při pokojové teplotě (25 °C) po dobu 24 hodin.
- Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
- Nepoužívejte žádné balení, které je poškozeno.
6. Obsah balení a další informace
Co Lucentis obsahuje
- Léčivou látkou je ranibizumabum (10 mg/ml). Jeden ml obsahuje 10 mg ranibizumabu.
- Dalšími složkami jsou dihydrát a,a-trehalosy, monohydrát histidin-hydrochloridu, histidin, polysorbát 20, voda na injekci.
Jak Lucentis vypadá a co obsahuje toto balení
Lucentis je injekční roztok dodávaný v injekční lahvičce (0,23 ml). Roztok je čirý, bezbarvý až světle
žlutý a vodný.
Lucentis je dodáván v balení, které obsahuje: jednu skleněnou injekční lahvičku ranibizumabu s chlorbutylovou pryžovou zátkou a jednu tupou jehlu s filtrem (18G x 1/", 1,2 mm x 40 mm,
5 mikrometrů) pro nasátí obsahu injekční lahvičky. Všechny komponenty jsou pouze pro jednorázové použití.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Btarapnu
Novartis Pharma Services Inc. Ten.: +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
EXXáda
Novartis (Hellas) A.E.B.E. Tpp +30 210 281 17 12
Espaňa
Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00
Lietuva
Novartis Pharma Services Inc. Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Osterreich
Novartis Pharma GmbH Tel: +43 1 86 6570
Polska
Novartis Poland Sp. z o.o.
Tel.: +48 22 375 4888
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A. Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Island Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kúnpoq Novartis Pharma Services Inc TpA,: +357 22 690 690 |
Sverige . Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc Tel: +371 67 887 070 |
United Kingdom . Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Viz také bod 3. „Jak se Lucentis podává“.
Jak připravit a jak aplikovat Lucentis Jednorázové podání do sklivce
Lucentis musí být aplikován kvalifikovaným oftalmologem zkušeným v aplikaci do sklivce.
U vlhké formy AMD a u poškození zraku kvůli DME, pro makulární edém v důsledku RVO nebo pro CNV sekundární k PM je doporučená dávka Lucentisu 0,5 mg, podávaná jako jednorázová injekce do sklivce. To odpovídá injekci o objemu 0,05 ml. Interval mezi dvěma dávkami podávanými do stejného oka má být alespoň čtyři týdny.
Léčba se zahajuje jednou injekcí za měsíc do dosažení maximální zrakové ostrosti a/nebo do vymizení příznaků aktivity onemocnění, tj. žádná změna zrakové ostrosti a ostatních příznaků a projevů onemocnění při probíhající léčbě. U pacientů s vlhkou formou AMD, DME a RVO mohou být zpočátku potřeba tři nebo více po sobě jdoucí injekce podávané jednou za měsíc.
Následně mají být lékařem určeny intervaly sledování a léčby a mají být stanoveny na základě aktivity onemocnění, vyhodnocené podle zrakové ostrosti a/nebo anatomických parametrů.
Pokud zrakové a anatomické parametry ukazují na základě vyjádření lékaře, že pokračující léčba pacienta není přínosná, je třeba léčbu přípravkem Lucentis ukončit.
Sledování aktivity onemocnění může zahrnovat klinické vyšetření, funkční testy nebo zobrazovací techniky (např. optickou koherenční tomografii nebo fluorescenční angiografii).
Pokud jsou pacienti léčeni podle režimu „treat-and-extend“, pak po dosažení maximální zrakové ostrosti a/nebo vymizení příznaků aktivity onemocnění, mohou být léčebné intervaly postupně prodlouženy až do opětovného objevení se příznaků aktivity onemocnění nebo zhoršení zraku. Léčebný interval nemá být prodloužen o více než dva týdny najednou u vlhké formy AMD a může být prodloužen až o jeden měsíc najednou u DME. Při uzávěru retinální vény mohou být léčebné intervaly také postupně prodlužovány, ale nejsou dostupná dostatečná data dokládající délku těchto intervalů. Pokud se aktivita onemocnění znovu objeví, léčebný interval má být příslušně zkrácen.
V léčbě poškození zraku způsobeném CNV sekundární k PM může mnoho pacientů potřebovat pouze jednu nebo dvě injekce během prvního roku, zatímco někteří pacienti mohou potřebovat častější léčbu.
Lucentis a laserová fotokoagulace u DME a makulárního edému v důsledku BRVO Existuje určitá zkušenost s podáním přípravku Lucentis společně s laserovou fotokoagulací. Při podání ve stejný den má být Lucentis podán alespoň 30 minut po laserové fotokoagulaci. Lucentis může být podán pacientům, kteří byli léčeni předchozí laserovou fotokoagulací.
Lucentis a fotodynamická léčba Visudynem u CNV sekundární k PM Se souběžným podáváním přípravků Lucentis a Visudyne nejsou žádné zkušenosti.
Lucentis je nutno před aplikací vizuálně zkontrolovat, zda neobsahuje cizí částice nebo není změněna jeho barva.
Lucentis musí být aplikován za aseptických podmínek, což zahrnuje použití chirurgické desinfekce rukou, sterilních rukavic, sterilního oděvu, sterilního spekula (nebo ekvivalentní náhrady) a dostupnost sterilní paracentézy (je-li potřeba). Před aplikací injekce do sklivce je nutný pečlivý odběr anamnézy z hlediska hypersenzitivních reakcí. Před aplikací injekce musí být podána adekvátní anestezie a použit širokospektrý lokální antimikrobiální přípravek k dezinfekci pokožky v okolí oka, očního víčka a povrchu oka, v souladu s lokální praxí.
Při přípravě přípravku Lucentis k podání do sklivce dbejte, prosím, následujících pokynů:
Všechny komponenty jsou sterilní a pouze pro jednorázové použití. Jakákoliv komponenta s obalem vykazující známky poškození nebo manipulace nesmí být použita. Sterilita nemůže být zaručena, pokud nezůstane uzavření obalu komponenty neporušené. Opakované použití může vést k infekci nebo jinému onemocnění/poškození.
Pro přípravu a injekci do sklivce jsou potřeba následující zdravotnické prostředky pro jednorázové použití:
- 5 pm jehla s filtrem (18G x 1%'', 1,2 mm x 40 mm, přiložena)
- 1 ml sterilní injekční stříkačka (není zahrnuta v balení přípravku Lucentis)
- injekční jehla (30G x /"; není zahrnuta v balení přípravku Lucentis)


1. Před nasátím tekutiny do injekční stříkačky je nutno desinfikovat vnější část pryžové zátky.
2. Jehlu s 5 pm filtrem (18G x 1/", 1,2 mm x 40 mm, 5 pm, přiložena) nasaďte na 1 ml injekční stříkačku za použití aseptického postupu. Zasuňte jehlu s filtrem do středu zátky lahvičky, dokud se jehla nedotkne dna injekční lahvičky.
3. Nasajte veškerou tekutinu z injekční lahvičky držené ve svislé, lehce nakloněné poloze pro snadnější úplné nasátí.
4. Ujistěte se, že píst je při vyprazdňování injekční lahvičky vytažen dostatečně daleko tak, aby jehla s filtrem byla úplně vyprázdněna.
5. Jehlu s filtrem ponechte v lahvičce a injekční stříkačku od ní odpojte. Jehla s filtrem by měla být po nasátí veškerého obsahu lahvičky zlikvidována a nesmí být použita pro vlastní aplikaci do sklivce.
6. Asepticky a pevně nasaďte injekční jehlu (30G x /") na stříkačku.
7. Opatrně odstraňte kryt z injekční jehly, aniž byste injekční jehlu odpojili od injekční stříkačky.
Poznámka: Při odstraňování krytu pevně uchopte část injekční jehly.
8. Důkladně vytlačte vzduch ze stříkačky a upravte dávku ke značce 0,05 ml uvedené na injekční stříkačce. Nyní je injekční stříkačka připravena k aplikaci.

Poznámka: Neotírejte injekční jehlu. Nevytahujte píst zpět.
Injekční jehla se zasune 3,5-4,0 mm posteriorně od limbu do prostoru sklivce tak, aby směřovala do centra očního bulbu a nikoli k horizontálnímu meridiánu. Poté se aplikuje objem injekce 0,05 ml. Následující injekci je nutné aplikovat v jiném místě skléry.
Po podání injekce nenasazujte zpět kryt jehly ani jehlu neoddělujte od injekční stříkačky. Zlikvidujte použitou injekční stříkačku společně s jehlou vyhozením do nádoby na ostré předměty nebo v souladu s místními požadavky.
150