Lonquex 6 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Lonquex 6 mg injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka obsahuje 6 mg lipegfilgrastimum* v 0,6 ml roztoku.
Jeden ml injekčního roztoku obsahuje 10 mg lipegfilgrastimum.
Léčivou látkou je kovalentní konjugát filgrastimu** s methoxypolyethylenglykolem (PEG) navázaným přes karbohydrátový linker.
*Pouze na základě obsahu proteinu. Koncentrace je 20,9 mg/ml (t.j. 12,6 mg v jedné předplněné injekční stříkačce) při započtení podílu PEG a karbohydrátového linkeru.
**Filgrastim (rekombinantní methionyl humánní růstový faktor stimulující kolonie granulocytů[G-CSF]) se vyrábí pomocí technologie rekombinantní DNA v buňkách bakterií
Escherichia coli.
Sílu tohoto léčivého přípravku nelze porovnávat se silou jiného pegylovaného nebo nepegylovaného proteinu stejné terapeutické třídy. Další informace viz bod 5.1.
Pomocné látky se známým účinkem
Jedna předplněná injekční stříkačka obsahuje 30 mg sorbitolu.
Jedna předplněná injekční stříkačka obsahuje méně než 1 mmol (23 mg) sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce) Čirý bezbarvý roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Zkrácení doby trvání neutropenie a snížení výskytu febrilní neutropenie u dospělých pacientů léčených cytotoxickou chemoterapií pro maligní nádorové onemocnění (s výjimkou chronické myeloidní leukémie a myelodysplastických syndromů).
4.2 Dávkování a způsob podání
Léčba přípravkem Lonquex má být zahájena a má probíhat pod dohledem lékařů s dostatečnými zkušenostmi v onkologii či hematologii.
Dávkování
V jednom cyklu chemoterapie se doporučuje podat jednu dávku 6 mg lipegfilgrastimu (jednu předplněnou injekční stříkačku přípravku Lonquex); přípravek se podává přibližně 24 hodin po cytotoxické chemoterapii.
Zvláštní populace pacientů
Starší pacienti
V rámci klinických studií s omezeným počtem starších pacientů nebyl zjištěn žádný důležitý rozdíl
v účinnosti nebo bezpečnostních profilech lipegfilgrastimu v závislosti na věku. Proto není u starších pacientů nutná úprava dávkování.
Pacienti s poruchami funkce ledvin
V současnosti dostupné údaje jsou uvedeny v bodě 5.2, ale na jejich základě nelze učinit žádná doporučení ohledně dávkování.
Pacienti s poruchami funkce jater
V současnosti dostupné údaje jsou uvedeny v bodě 5.2, ale na jejich základě nelze učinit žádná doporučení ohledně dávkování.
Pediatrická populace
Bezpečnost a účinnost přípravku Lonquex u dětí a dospívajících ve věku do 17 roků nebyla dosud stanovena. V současnosti dostupné údaje jsou uvedeny v bodech 4.8, 5.1 a 5.2.
Způsob _ podání
Roztok se podává subkutánně (s.c.). Injekce se podávají do břicha, horní části paže nebo stehna.
Návod k zacházení s léčivým přípravkem před jeho podáním je uveden v bodě 6.6.
Přípravek Lonquex by si měli podávat sami sobě pouze pacienti, kteří jsou dobře motivovaní, dostatečně zaškolení a je pro ně dostupná rada odborníka. První aplikace přípravku Lonquex má proběhnout pod přímým dohledem lékaře.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Obecné informace
Bezpečnost a účinnost přípravku Lonquex nebyla hodnocena u pacientů léčených vysokými dávkami chemoterapie. Lonquex nesmí být používán ke zvýšení dávky cytotoxické chemoterapie nad rámec zavedených režimů dávkování.
Pro zlepšení dohledatelnosti by měly být obchodní název a číslo šarže podávaného léčivého přípravku jasně zaznamenány do dokumentace pacienta.
Alergické reakce a imunogenicita
U pacientů, kteří jsou přecitlivělí na G-CSF nebo jeho deriváty, existuje rovněž riziko reakcí přecitlivělosti na lipegfilgrastim vzhledem k možné zkřížené reaktivitě. Vzhledem k riziku zkřížené reaktivity nesmí být u těchto pacientů zahájena žádná terapie lipegfilgrastimem.
Většina biologických léčivých přípravků vyvolá určitou úroveň odpovědi v podobě protilátek. Tato protilátková odpověď může v některých případech vést k nežádoucím účinkům nebo ztrátě účinnosti. Jestliže pacient nereaguje na léčbu, měl by být dále vyšetřen.
Jestliže dojde k závažné alergické reakci, je třeba podat vhodnou léčbu a pacienta několik dní pečlivě sledovat.
Hematopoetický systém
Léčbou samotným lipegfilgrastimem nelze zabránit vzniku trombocytopenie a anémie, které se objevují jako následek myelosupresivní chemoterapie. Lipegfilgrastim může rovněž způsobit reverzibilní trombocytopenii (viz bod 4.8). Doporučuje se kontrolovat pravidelně počet krevních destiček a hematokrit. Zvláštní pozornost je třeba věnovat monoterapii nebo kombinované chemoterapii léčivými přípravky, o kterých je známo, že mohou způsobovat těžkou trombocytopenii.
Může docházet k leukocytóze (viz bod 4.8). Dosud nebyly hlášeny žádné nežádoucí účinky, které by bylo možné přímo přičíst leukocytóze. Zvýšení počtu bílých krvinek (WBC) je v souladu s farmakodynamickými účinky lipegfilgrastimu. Počet bílých krvinek má být během léčby pravidelně kontrolován vzhledem ke klinickým účinkům lipegfilgrastimu a možnosti rozvoje leukocytózy.
Jestliže počet bílých krvinek po očekávaném nadiru převyšuje 50 x 109/l, je třeba podávání lipegfilgrastimu ihned ukončit.
Zvýšení hematopoetické aktivity kostní dřeně v odpovědi na léčbu růstovým faktorem bylo spojeno s přechodnými pozitivními nálezy při zobrazovacích vyšetřeních kostí. To je třeba zvážit při interpretaci výsledků těchto vyšetření.
Pacienti s myeloidní leukémií nebo myelodysplastickým syndromem
Faktor stimulující kolonie granulocytů (G-CSF) může podporovat růst myeloidních buněk a některých nemyeloidních buněk in vitro.
Bezpečnost a účinnost podávání přípravku Lonquex pacientům s chronickou myeloidní leukémií, myelodysplastickým syndromem nebo sekundární akutní myeloidní leukémií nebyla dosud stanovena, a proto přípravek nesmí být těmto pacientům podáván. Zvláštní pozornost je třeba věnovat rozlišení diagnózy blastické transformace chronické myeloidní leukémie od akutní myeloidní leukémie.
Nežádoucí účinky na slezinu
Po podávání lipegfilgrastimu byl hlášen obecně asymptomatický výskyt splenomegalie (viz bod 4.8) a málo časté případy ruptury sleziny, z nichž některé skončily fatálně (viz bod 4.8). Proto je třeba pečlivě sledovat velikost sleziny (např. klinickým vyšetřením ultrazvukem). U pacientů, kteří si stěžují na bolesti v levém nadbřišku nebo v levém rameni, je třeba pomýšlet na diagnózu ruptury sleziny.
Nežádoucí účinky na plíce
Po podání lipegfilgrastimu byly hlášeny nežádoucí plicní účinky, zejména intersticiální pneumonie (viz bod 4.8). U pacientů s nedávným výskytem plicních infiltrátů nebo pneumonie v anamnéze může být riziko vyšší.
Příznaky onemocnění dýchacího ústrojí, jako jsou kašel, horečka a dušnost, spolu s rentgenologickým nálezem plicních infiltrátů a zhoršením plicních funkcí provázeným zvýšeným počtem neutrofilů mohou být prvními známkami syndromu akutní dechové tísně (Acute Respiratory Distress Syndrome - ARDS) (viz bod 4.8). Podávání přípravku Lonquex se musí v takovém případě na základě rozhodnutí lékaře ukončit a musí být zahájena vhodná léčba.
Cévní nežádoucí účinky
Po podání G-CSF nebo derivátů byl hlášen syndrom zvýšené permeability kapilár, který se vyznačuje hypotenzí, hypoalbuminémií, edémem a hemokoncentrací. Pacienti, u kterých se vyskytnou příznaky syndromu zvýšené permeability kapilár, mají být pečlivě sledováni a mají dostávat standardní symptomatickou léčbu, která může zahrnovat i intenzivní péči (viz bod 4.8).
Pacienti se srpkovitou anémií
U pacientů se srpkovitou anemií byly v souvislosti s podáváním G-CSF nebo jeho derivátů pozorovány srpkovité krize (viz bod 4.8). Lékaři by proto měli postupovat velmi opatrně, jestliže podávají Lonquex pacientům se srpkovitou anémií a měli by sledovat vhodné klinické parametry a laboratorní výsledky a věnovat pozornost možným souvislostem podávání lipegfilgrastimu se zvětšením sleziny a vazookluzivní krizí.
Hvpokalémie
Může docházet k hypokalémii (viz bod 4.8). U pacientů se zvýšeným rizikem hypokalémie v důsledku základního onemocnění nebo souběžně užívaných léků se doporučuje pečlivě sledovat hladinu draslíku v séru a v případě potřeby doplňovat příjem draslíku.
Pomocné látky se známým účinkem
Tento léčivý přípravek obsahuje sorbitol. Pacienti se vzácnou vrozenou intolerancí fruktózy by tento přípravek neměli užívat.
Tento přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné předplněné injekční stříkačce tj. v podstatě je „bez sodíku“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vzhledem k možné citlivosti rychle se dělících myeloidních buněk na cytotoxickou chemoterapii není podání přípravku Lonquex doporučováno v časovém období 24 hodin po cytotoxické chemoterapii. Současné podávání lipegfilgrastimu s jakýmkoli chemoterapeutickým léčivým přípravkem nebylo u pacientů hodnoceno. U zvířecích modelů bylo prokázáno, že souběžné podávání G-CSF a 5-fluorouracilu (5-FU) nebo jiných antimetabolitů zesiluje myelosupresi.
Bezpečnost a účinnost přípravku Lonquex nebyla hodnocena u pacientů léčených chemoterapií spojenou s opožděnou myelosupresí, jako jsou například přípravky na bázi nitrosourey.
Potenciál pro interakce s lithiem, které rovněž podporuje uvolňování neutrofilů, nebyl specificky studován. Neexistuje žádný důkaz, že by taková interakce byla škodlivá.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání lipegfilgrastimu těhotným ženám jsou velmi omezené (méně než 300 ukončených těhotenství). Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Podávání přípravku Lonquex v těhotenství se z preventivních důvodů nedoporučuje.
Kojení
Není známo, zda se lipegfilgrastim/metabolity vylučují do lidského mateřského mléka. Riziko pro kojené děti nelze vyloučit. Kojení má být během léčby přípravkem Lonquex přerušeno.
Fertilita
Nejsou dostupné žádné údaje. Studie na zvířatech s G-CSF a jeho deriváty nenaznačují škodlivé účinky na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Lonquex nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního _profilu
Nejčastěji hlášenými nežádoucími účinky jsou bolesti kostí a svalů. Bolesti kostí a svalů jsou obecně mírné nebo středně intenzivní, přechodného charakteru a u většiny pacientů je lze zvládnout běžnými analgetiky.
Syndrom zvýšené permeability kapilár, který může být v případě opožděné léčby život ohrožující, byl hlášen většinou u pacientů s nádorem podstupujících chemoterapii po podání G-CSF nebo derivátů (viz bod 4.4 a podbod „Popis vybraných nežádoucích účinků“ bodu 4.8).
Tabulkový přehled nežádoucích účinků
Bezpečnost lipegfilgrastimu byla vyhodnocena na základě výsledků klinických hodnocení u 506 pacientů a 76 zdravých dobrovolníků, kterým byl lipegfilgrastim podán nejméně jednou.
Nežádoucí účinky uvedené v tabulce 1 níže jsou rozděleny podle tříd orgánových systémů. Četnosti
jsou definovány podle následující konvence:
Velmi časté: Časté:
Méně časté: Vzácné:
Velmi vzácné:
> 1/10
> 1/100 až < 1/10
> 1/1 000 až < 1/100
> 1/10 000 až < 1/1 000 < 1/10 000
Není známo: z dostupných údajů nelze určit.
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Tabulka 1: Nežádoucí účinky | ||
|
Třída orgánových systémů |
Četnost |
Nežádoucí účinek |
|
Poruchy krve a lymfatického systému |
Časté |
Trombocytopenie*, splenomegalie* |
|
Méně časté |
Leukocytóza* | |
|
Poruchy imunitního systému |
Méně časté |
Hypersenzitivní reakce* |
|
Poruchy metabolismu a výživy |
Časté |
Hypokalémie* |
|
Poruchy nervového systému |
Časté |
Bolesti hlavy |
|
Cévní poruchy |
Není známo |
Syndrom zvýšené permeability kapilár* |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté |
Nežádoucí plicní účinky* |
|
Poruchy kůže a podkožní tkáně |
Časté |
Kožní reakce* |
|
Méně časté |
Reakce v místě vpichu* | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Velmi časté |
Bolesti kostí a svalů* |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Bolesti na hrudníku |
|
Vyšetření |
Méně časté |
Zvýšení alkalické fosfatázy v krvi*, zvýšení laktátdehydrogenázy v krvi* |
|
*Viz bod „Popis vybraných nežác |
oucích účinků“ níže | |
Popis vybraných nežádoucích účinků
Byl hlášen výskyt trombocytopenie a leukocytózy (viz bod 4.4).
Byla hlášena splenomegalie, obecně asymptomatická (viz bod 4.4)
Mohou se vyskytovat hypersenzitivní reakce, jako jsou například alergické kožní reakce, kopřivka, angioedém a závažné alergické reakce.
Byl hlášen výskyt hypokalémie (viz bod 4.4).
Byly hlášeny plicní nežádoucí účinky, zejména intersticiální pneumonie (viz bod 4.4). Mezi tyto plicní nežádoucí účinky mohou rovněž patřit plicní edém, plicní infiltráty, plicní fibróza, respirační selhání nebo ARDS (viz bod 4.4).
Mohou se vyskytovat kožní reakce jako například erytém a vyrážka.
Může docházet k reakcím v místě vpichu, jako je například zatvrdnutí a bolest v místě vpichu.
Nejčastějším nežádoucím účinkem jsou bolesti pohybového aparátu, jako například svalů a kostí. Bolesti kostí a svalů jsou obecně mírné nebo středně intenzivní, přechodného charakteru a u většiny pacientů je lze zvládnout běžnými analgetiky.
Rovněž se může vyskytovat reverzibilní, mírné až střední zvýšení alkalické fosfatázy a laktátdehydrogenázy bez doprovodných klinických projevů. Vzestup alkalické fosfatázy a laktátdehydrogenázy pochází s největší pravděpodobností ze zvýšeného počtu neutrofilů.
Některé nežádoucí účinky dosud nebyly u lipegfilgrastimu pozorovány, ale jsou obecně připisovány G-CSF a jeho derivátům:
Poruchy krve a lymfatického systému
- Ruptura sleziny, včetně několika fatálních případů (viz bod 4.4)
- Srpkovitá krize u pacientů se srpkovitou anémií (viz bod 4.4)
Cévní poruchy
- Syndrom zvýšené permeability kapilár
V postmarketingovém sledování byly hlášeny případy syndromu zvýšené permeability kapilár po podání G-CSF nebo derivátů. Obecně se vyskytly u pacientů s pokročilým stádiem zhoubného onemocnění, se sepsí, užívajících vícesložkovou chemoterapii nebo podstupujících aferézu (viz bod 4.4).
Poruchy kůže a podkožní tkáně
- Akutní febrilní neutrofilní dermatóza (Sweetův syndrom)
- Kožní vaskulitida
Pediatrická populace
Zkušenosti u dětí jsou omezené na studii fáze 1 s jednou dávkou provedenou u 21 pediatrických pacientů ve věku 2 až < 18 let (viz bod 5.1), která neukázala rozdíl v bezpečnostním profilu lipegfilgrastimu u dětí v porovnání s dospělými. Nežádoucí příhody související s léčbou byly bolest zad, bolest kostí a zvýšený počet neutrofilů (1 příhoda v každé skupině).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku.Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
S předávkováním lipegfilgrastimu neexistují žádné zkušenosti. V případě předávkování je nutno pravidelně kontrolovat počet bílých krvinek a krevních destiček a pečlivě sledovat velikost sleziny (např. pomocí klinického vyšetření a ultrazvuku).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunostimulátory, faktory stimulující kolonie, ATC kód: L03AA14
Mechanismus účinku
Lipegfilgrastim je kovalentní konjugát filgrastimu s jednou molekulou methoxypolyethylenglykolu (PEG) navázanou přes karbohydrátový linker, složený z glycinu, kyseliny N-acetylneuraminové a N-acetylgalaktosaminu. Průměrná molekulová hmotnost je přibližně 39 kDa, z toho bílkovinná frakce představuje přibližně 48 %. Humánní G-CSF je glykoprotein, který reguluje produkci a uvolňování funkčních neutrofilů z kostní dřeně. Filgrastim je neglykosylovaný rekombinantní methionyl humánní G-CSF. Lipegfilgrastim je forma filgrastimu s prodlouženou působností, které je dosaženo snížením renální clearance. Lipegfilgrastim se váže na lidský receptor pro G-CSF stejně jako filgrastim a pegfilgrastim.
Farmakodynamické účinky
Lipegfilgrastim a filgrastim způsobují během 24 hodin významné zvýšení počtu neutrofilů v periferní krvi spolu s lehkým zvýšením monocytů a/nebo lymfocytů. Tyto výsledky naznačují, že frakce G-CSF lipegfilgrastimu vykazuje očekávanou aktivitu tohoto růstového faktoru: stimulaci proliferace hematopoetických progenitorových buněk, diferenciaci na zralé buňky a jejich uvolnění do periferní krve. Tento účinek se netýká pouze linie neutrofilů, ale také jiných progenitorů jedné či více linií a pluripotentních hematopoetických kmenových buněk. G-CSF rovněž zvyšuje antibakteriální aktivitu neutrofilů, včetně fagocytózy.
Klinická účinnost a bezpečnost
Dávkování lipegfilgrastimu jednou za cyklus bylo zkoumáno ve dvou pivotních, randomizovaných dvojitě zaslepených klinických hodnoceních u pacientů s myelosupresivní chemoterapií.
První pivotní klinické hodnocení (fáze III) XM22-03 bylo aktivní látkou kontrolované klinické hodnocení u 202 pacientek s karcinomem prsu II. až IV. stupně, které absolvovaly až 4 cykly chemoterapie sestávající z doxorubicinu a docetaxelu. Pacientky byly randomizovány v poměru 1:1 pro podávání 6 mg lipegfilgrastimu nebo 6 mg pegfilgrastimu. Toto klinické hodnocení prokázalo, že podávání 6 mg lipegfilgrastimu nemělo horší hodnocení s ohledem na primární cílový parametr trvání závažné neutropenie (TZN) v prvním cyklu chemoterapie, než podávání 6 mg pegfilgrastimu (viz tabulka 2).
|
Tabulka 2: TZN, závažná neutropenie (ZN) a febrilní neutropenie (FN) v 1. cyklu | ||
|
studie XM22-03 (ITT) | ||
|
Pegfilgrastim 6 mg (n = 101) |
Lipegfilgrastim 6 mg (n = 101) | |
|
TZN | ||
|
Průměr ± SD (d) |
0,9 ± 0,9 |
0,7 ± 1,0 |
|
A LS průměr |
-0,186 | |
|
95 % CI |
-0,461 až 0,089 | |
|
ZN | ||
|
Incidence (%) |
51,5 |
43,6 |
|
FN | ||
|
Incidence (%) |
3,0 |
1,0 |
|
ITT = Intent-to-treat populace (všichni randomizovaní pacienti) SD = standardní odchylka d = dny CI = interval spolehlivosti A LS průměr (průměrný rozdíl lipegfilgrastim - pegfilgrastim metodou nejmenších čtverců) a interval spolehlivosti (CI) multivarianční Poissonovou regresní analýzou | ||
Druhé pivotní klinické hodnocení (fáze III) XM22-04 bylo placebem kontrolované klinické hodnocení u 375 pacientů s nemalobuněčným karcinomem plic, kteří absolvovali až 4 cykly chemoterapie sestávající z cisplatiny a etoposidu. Pacienti byli randomizováni v poměru 2:1 pro podávání buď 6 mg lipegfilgrastimu, nebo placeba. Výsledky klinického hodnocení jsou uvedeny v tabulce 3. Při dokončení hlavní studie byl výskyt úmrtí 7,2 % (placebo) a 12,5 % (lipegfilgrastim 6 mg), ačkoli po 360 denním sledování byl celkový výskyt úmrtí podobný mezi placebem a lipegfilgrastimem (44,8 % a 44,0 %; bezpečnostní populace).
|
Tabulka 3: TZN, ZN a FN v 1. cyklu studie XM22-04 (ITT) | ||
|
Placebo |
Lipegfilgrastim 6 mg | |
|
(n = 125) |
(n = 250) | |
|
FN | ||
|
Incidence (%) |
5,6 |
2,4 |
|
95% CI |
0,121 až 1,260 | |
|
p-hodnota |
0,1151 | |
|
TZN | ||
|
Průměr ± SD (d) |
2,3 ± 2,5 |
0,6 ± 1,1 |
|
A LS průměr |
-1,661 | |
|
95 % CI |
-2,089 až -1,232 | |
|
p-hodnota |
< 0,0001 | |
|
ZN | ||
|
Incidence (%) |
59,2 |
32,1 |
|
Poměr šancí |
0,325 | |
|
95% CI |
0,206 až 0,512 | |
|
p-hodnota |
<0,0001 | |
|
A LS průměr (průměrný rozdíl lipegfilgrastim - placebo), interval spolehlivosti (CI) a p-hodnota | ||
|
multivarianční Poissonovou regresní analýzou | ||
|
Poměr šancí (lipegfilgrastim/placebo), CI a p-hodnota mimo multivariační logistickou regresní | ||
|
analýzu | ||
Imunogenicita
Byla provedena analýza protilátek proti léčivé látce u 579 pacientů a zdravých dobrovolníků, kterým byl podáván lipegfilgrastim, 188 pacientů a zdravých dobrovolníků, kterým byl podáván pegfilgrastim a 121 pacientů, kterým bylo podáváno placebo. Specifické protilátky proti léčivé látce, které vznikly po začátku léčby, byly zjištěny u 0,86 % subjektů, kterým byl podáván lipegfilgrastim, u 1,06 % subjektů, kterým byl podáván pegfilgrastim a u 1,65 % subjektů, kterým bylo podáváno placebo. Nebyly pozorovány žádné neutralizující protilátky proti lipegfilgrastimu.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Lonquex u všech podskupin pediatrické populace v léčbě neutropenie navozené chemoterapií a prevenci febrilní neutropenie navozené chemoterapií (informace o použití u dětí viz bod 4.2). Ve studii fáze 1 u 21 dětí ve věku 2 až 16 let s Ewingovými nádory nebo rabdomyosarkomem byl podáván lipegfilgrastim v jedné subkutánní dávce 100 ^g/kg (maximálně 6 mg, což je fixní dávka pro dospělé) 24 hodin po ukončení poslední chemoterapie v týdnu 1 režimu léčby. Incidence FN se měnila podle věku (od 14,3 % do 71,4 %) s nejvyšší frekvencí ve skupině s nejvyšším věkem. Použití tří různých režimů léčby chemoterapií s měnícími se myelosupresivními účinky a věkovou distribucí komplikovalo srovnání účinnosti napříč věkovými skupinami. Viz bod 4.2.
5.2 Farmakokinetické vlastnosti
Obecné informace Zdraví dobrovolníci
Ve 3 studiích (XM22-01, XM22-05, XM22-06) byl u zdravých dobrovolníků po podání jedné subkutánní injekce 6 mg lipegfilgrastimu medián dosažení maximální koncentrace v krvi 30 až 36 hodin a průměrný terminální poločas byl přibližně v rozmezí 32 až 62 hodin.
Po subkutánní injekci 6 mg lipegfilgrastimu zdravým dobrovolníkům do třech různých míst (horní část paže, břicho a stehno) byla biologická dostupnost (vrcholová koncentrace a plocha pod křivkou [AUC]) po subkutánní injekci do stehna nižší než po subkutánní injekci do břicha a horní části paže. Jak biologická dostupnost lipegfilgrastimu, tak pozorované rozdíly mezi místy aplikace injekce byly v tomto limitovaném hodnocení XM22-06 větší u mužů než u žen. Farmakodynamické účinky však byly podobné a nezávisely na pohlaví a místě aplikace injekce.
Metabolismus
Lipegfilgrastim je metabolizován intracelulární nebo extracelulární degradací proteolytickými enzymy. Lipegfilgrastim je pohlcen neutrofily (nelineární proces) a poté v buňce rozložen endogenními proteolytickými enzymy. Za lineární dráhu pravděpodobně zodpovídá extracelulární degradace proteinů elastázou neutrofilů a jinými plazmatickými proteázami.
Interakce s jinými léčivými přípravky
Data získaná in vitro naznačují, že lipegfilgrastim má malý nebo nemá žádný přímý účinek nebo účinek zprostředkovaný imunitním systémem na aktivitu CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 a CYP3A4/5. Proto není pravděpodobné, že by lipegfilgrastim ovlivňoval metabolismus prostřednictvím enzymů lidského cytochromu P450.
Zvláštní populace
Pacienti s rakovinou
Ve 2 klinických hodnoceních (XM22-02 a XM22-03) u pacientek s karcinomem prsu, které užívaly chemoterapii sestávající z doxorubicinu a docetaxelu, bylo dosahováno průměrné maximální koncentrace v krvi 227 a 262 ng/ml při mediánu doby do dosažení maximální koncentrace (tmax) 44 a 48 hodin. Průměrné terminální poločasy po jedné subkutánní injekci 6 mg lipegfilgrastimu během prvního cyklu chemoterapie byly 29 a 31 hodin. Po jedné subkutánní injekci 6 mg lipegfilgrastimu během čtvrtého cyklu byly maximální koncentrace v krvi nižší než koncentrace pozorované v prvním cyklu (průměrné hodnoty byly 77 a 111 ng/ml) a medián doby do jejich dosažení tmax byl 8 hodin. Průměrné terminální poločasy ve čtvrtém cyklu byly přibližně 39 a 42 hodin.
V klinickém hodnocení (XM22-04) u pacientů s nemalobuněčným karcinomem plic, kteří užívali chemoterapii sestávající z cisplatiny a etoposidu, bylo po jedné subkutánní injekci 6 mg lipegfilgrastimu během prvního cyklu chemoterapie dosaženo průměrné maximální koncentrace v krvi 317 ng/ml po mediánu doby do jejího dosažení tmax 24 hodin, a průměrný terminální poločas byl přibližně 28 hodin. Po podání jedné subkutánní injekce 6 mg lipegfilgrastimu během čtvrtého cyklu byla průměrná maximální koncentrace v krvi 149 ng/ml, medián doby do jejího dosažení tmax byl 8 hodin a průměrný terminální poločas byl přibližně 34 hodin.
Lipegfilgrastim je zřejmě eliminován prostřednictvím clearance zprostředkované neutrofily, tento mechanismus je však při vyšších dávkách saturován. V souladu s tímto samoregulačním mechanismem clearance klesají sérové koncentrace lipegfilgrastimu pomalu během přechodného nadiru počtu neutrofilů navozeného chemoterapií a rychle při následném nástupu zotavení počtu neutrofilů (viz obrázek 1).
Obrázek 1: Profil mediánu sérové koncentrace lipegfilgrastimu a mediánu absolutního počtu neutrofilů (ANC) u pacientů léčených chemoterapií po jedné injekci 6 mg lipegfilgrastimu
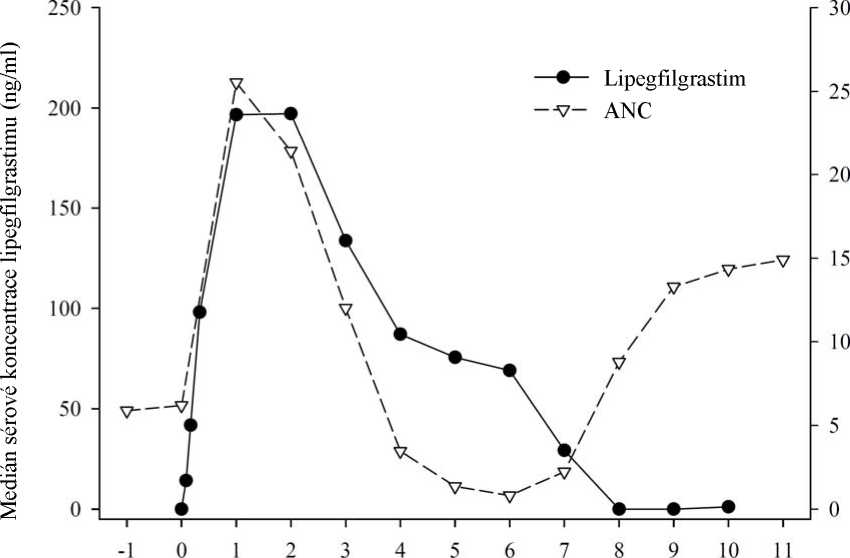
Studijní dny, injekce lipegfilgrastimu v den 0
Pacienti s poruchou funkce ledvin nebo jater
Vzhledem k mechanismu clearance, která je zprostředkována neutrofily, se neočekává, že by na farmakokinetiku lipegfilgrastimu mělo vliv poškození ledvin nebo jater.
Starší pacienti
Údaje získané na omezeném počtu pacientů nasvědčují, že farmakokinetika lipegfilgrastimu u starších pacientů (65 - 74 let) je podobná jako u mladších pacientů. Nejsou k dispozici žádné údaje o pacientech ve věku > 75 let.
Pediatrická populace
Ve studii fáze 1 (viz bod 5.1) používající pro subkutánní injekci roztok 10 mg/ml, který byl zvláště vyvinutý pro pediatrické studie, byly průměrné maximální koncentrace v krvi (Cmax) 243 ng/ml ve věkové skupině 2 až < 6 let, 255 ng/ml ve věkové skupině 6 až < 12 let a 224 ng/ml ve věkové skupině 12 až < 18 let po jedné subkutánní injekci 100 ^g/kg (maximálně 6 mg) lipegfilgrastimu při prvním cyklu chemoterapie. Maximální koncentrace v krvi bylo dosaženo po střední době (tmax) 23,9 hodin, 30,0 hodin a 95,8 hodin, v uvedeném pořadí. Viz bod 4.2.
Pacienti s nadváhou
S přírůstkem tělesné hmotnosti byl pozorován trend k nižší expozici lipegfilgrastimu. To může vést k nižším farmakodynamickým odpovědím u těžkých pacientů (> 95 kg). Následné snížení účinnosti u těchto pacientů nelze podle současných údajů vyloučit.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po jednorázovém a opakovaném podávání a lokální snášenlivosti, neodhalily žádné zvláštní riziko pro člověka.
Ve studii reprodukční a vývojové toxicity u králíků byla po vysokých dávkách lipegfilgrastimu pozorována zvýšená incidence ztrát embryí po implantaci a potratů, pravděpodobně v důsledku zvýrazněného farmakodynamického účinku specifického pro králíky. Nebyly pozorovány žádné známky teratogenních účinků lipegfilgrastimu. Tyto nálezy jsou v souladu s výsledky získanými u G-CSF a jeho derivátů. Mezi publikovanými informacemi o G-CSF a jeho derivátech nejsou žádné údaje svědčící o nežádoucích účincích na fertilitu a vývoj embrya či plodu u potkanů nebo o prenatálních či postnatálních účincích, které se zároveň nevztahují k toxicitě pro matku. Je prokázáno, že filgrastim a pegfilgrastim mohou být u potkanů v nižších dávkách transportovány přes placentu; nejsou k dispozici žádné údaje pro lipegfilgrastim. Význam těchto zjištění pro člověka není znám.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Kyselina octová, ledová Hydroxid sodný (k úpravě pH)
Sorbitol (E420)
Polysorbát 20 Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
Lonquex lze vyjmout z chladničky a uchovávat při teplotě do 25 °C nejvýše po jedno období nepřesahující délku 3 dní. Po vyjmutí z chladničky musí být léčivý přípravek spotřebován v této lhůtě nebo zlikvidován.
6.5 Druh obalu a obsah balení
0,6 ml roztoku v předplněné injekční stříkačce (sklo typu I) s pístovou zátkou [brombutylová pryž potažená kopolymerem ethylenu a tetrafluorethylenu] a fixní injekční jehlou (nerezová ocel, 29G [0,34 mm] nebo 27G [0,4 mm] x 0,5 palce [12,7 mm]).
Velikost balení: 1 předplněná injekční stříkačka s bezpečnostním systémem nebo bez něho (který zabraňuje poranění v důsledku píchnutí jehlou a opakovanému použití).
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Roztok je třeba před použitím prohlédnout. Použít lze pouze čiré bezbarvé roztoky bez částic.
Před podáním injekce nechte roztok vytemperovat (15 °C - 25 °C).
Přípravkem se nesmí silněji třepat. Nadměrné třepání může způsobit agregaci a následně biologickou inaktivaci lipegfilgrastimu.
Přípravek Lonquex neobsahuje žádné konzervační látky. Vzhledem k možnému riziku mikrobiální kontaminace jsou injekční stříkačky s přípravkem Lonquex určeny pouze k jednorázovému použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
UAB "Sicor Biotech"
Moléty pl. 5 LT-08409 Vilnius Litva
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/856/001
EU/1/13/856/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 25. červenec 2013.
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Teva Biotech GmbH DornierstraBe 10 D-89079 Ulm Německo
Název a adresa výrobců odpovědných za propouštění šarží
Teva Biotech GmbH DornierstraBe 10 D-89079 Ulm Německo
Teva Pharmaceuticals Europe B.V.
Swensweg 5 NL-2031 GA Haarlem Nizozemsko
Teva Operations Poland Sp. z o.o. ul. Mogilska 80 31-546 Kraków Polsko
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik)
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Poregistrační studie bezpečnosti k dalšímu sledování rizika progrese a mortality související s přípravkem Lonquex u pacientů s malignitou léčených cytotoxickou chemoterapií. Rizika mají být stanovena ve vztahu k zavedenému srovnávacímu přípravku a placebu, a má být provedeno objektivní hodnocení progrese onemocnění. Měl by se použít vhodně citlivý klinický model, pomocí kterého budou výše uvedená rizika vyhodnocena. Předložení konečné zprávy o klinickém hodnocení. |
31.12.2017 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Lonquex 6 mg injekční roztok v předplněné injekční stříkačce Lipegfilgrastimum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka obsahuje 6 mg lipegfilgrastimum v 0,6 ml roztoku. Jeden ml roztoku obsahuje 10 mg lipegfilgrastimum.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: Ledová kyselina octová, hydroxid sodný, sorbitol (E420), polysorbát 20 a voda na injekci.
Před použitím si přečtěte příbalovou informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 předplněná injekční stříkačka
1 předplněná injekční stříkačka s bezpečnostním systémem
5. ZPŮSOB A CESTA/CESTY PODÁNÍ_
Pouze k jednorázovému použití.
Přípravkem se nesmí silněji třepat.
Před použitím si přečtěte příbalovou informaci.
Subkutánní podání
Pouze pro předplněnou injekční stříkačku s bezpečnostním systémem:
Důležité: před manipulací s injekční stříkačkou si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UAB "Sicor Biotech" Moléty pl. 5 LT-08409 Vilnius Litva
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/856/001 1 předplněná injekční stříkačka s bezpečnostním systémem EU/1/13/856/002 1 předplněná injekční stříkačka
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku je vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Lonquex 6 mg
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU PŘEDPLNĚNÁ INJEKČNÍ STŘÍKAČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Lonquex 6 mg injekce Lipegfilgrastimum
s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,6 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele Lonquex 6 mg injekční roztok v předplněné injekční stříkačce
Lipegfilgrastimum
Tento přípravek podléhá dalšímu sledování.To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Lonquex a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Lonquex používat
3. Jak se Lonquex používá
4. Možné nežádoucí účinky
5. Jak Lonquex uchovávat
6. Obsah balení a další informace
7. Informace k injekčnímu podávání pacientem samotným
1. Co je Lonquex a k čemu se používá Co je Lonquex
Lonquex obsahuje léčivou látku lipegfilgrastim. Lipegfilgrastim je dlouhodobě působící modifikovaná bílkovina, vytvářená pomocí biotechnologie bakterií nazývanou Escherichia coli. Patří do skupiny bílkovin nazývaných cytokiny a podobá se bílkovině (faktoru stimulujícímu kolonie granulocytů [G-CSF]), která se přirozeně tvoří ve Vašem těle.
K čemu se Lonquex používá
Lékař Vám předepsal Lonquex, aby zkrátil trvání stavu zvaného neutropenie (nízký počet bílých krvinek) a snížil výskyt febrilní neutropenie (nízký počet bílých krvinek s horečkou). Tyto stavy mohou být způsobeny použitím cytotoxické chemoterapie (léků, které ničí rychle rostoucí buňky).
Jak Lonquex účinkuje
Lipegfilgrastim stimuluje kostní dřeň (tkáň, kde se tvoří nové krvinky) k vyšší produkci bílých krvinek. Bílé krvinky jsou důležité, protože pomáhají tělu bojovat s infekcí. Tyto krvinky jsou velmi citlivé na účinky chemoterapie, která může způsobit pokles jejich počtu v těle. Pokud se počet bílých krvinek příliš sníží, nemusí jich zbýt v těle dostatek k boji s bakteriemi a můžete být zvýšeně ohrožen(a) infekcí.
2. Čemu musíte věnovat pozornost, než začnete Lonquex používat Nepoužívejte Lonquex:
• jestliže jste alergický(á) na lipegfilgrastim nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Lonquex se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou
• jestliže budete mít bolesti v levém nadbřišku nebo v levém rameni. Mohlo by se jednat o následek postižení sleziny (viz bod „4. Možné nežádoucí účinky“).
• jestliže budete mít kašel, horečku a potíže s dýcháním. Mohlo by se jednat o následek postižení plic (viz bod „4. Možné nežádoucí účinky”).
• jestliže máte srpkovitou anémii, což je vrozené onemocnění, pro které je charakteristický srpkovitý tvar červených krvinek.
• jestliže jste dříve prodělal(a) alergické reakce na jiné léky, jako je tento (ze skupiny G-CSF například filgrastim, lenograstim nebo pegfilgrastim). Mohlo by být riziko stejné reakce na přípravek Lonquex.
Lékař Vám bude pravidelně vyšetřovat krev a sledovat různé složky krve a jejich hladiny.
Děti a dospívající
Nepodávejte tento přípravek dětem a dospívajícím ve věku do 18 let, protože jsou k dispozici pouze omezené zkušenosti u dětí prokazující jeho bezpečnost a účinnost u této věkové skupiny.
Další léčivé přípravky a Lonquex
Informujte svého lékaře nebo lékárníka o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
Obvykle si aplikujte pomocí injekce svou dávku přípravku Lonquex přibližně 24 hodin po poslední dávce chemoterapie na konci každého cyklu chemoterapie.
Těhotenství a kojení
Lonquex nebyl testován u těhotných žen. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, je důležité, abyste svého lékaře informovala, protože se lékař možná rozhodne, že tento přípravek nemáte užívat.
Není známo, zda se léčivá látka přítomná v tomto přípravku vylučuje do lidského mateřského mléka. Kojení má být proto během léčby přerušeno.
Řízení dopravních prostředků a obsluha strojů
Lonquex nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
Lonquex obsahuje sorbitol a sodík
Tento přípravek obsahuje sorbitol. Pokud Vám Váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
Tento přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné předplněné injekční stříkačce tj. v podstatě je „bez sodíku“.
3. Jak se Lonquex používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka přípravku je
Doporučená dávka je jedna předplněná injekční stříkačka (6 mg lipegfilgrastimu) jednou za jeden cyklus chemoterapie.
Kdy se Lonquex používá
Tento přípravek se podává přibližně 24 hodin po poslední dávce chemoterapie na konci každého cyklu chemoterapie.
Jak se aplikují injekce?
Tento přípravek se podává jako injekce z předplněné injekční stříkačky. Tato injekce se podává do tkáně těsně pod kůži (tzv. subkutánní injekce).
Lékař Vám možná navrhne, abyste se naučil(a), jak si injekce tohoto přípravku aplikovat sám/sama. Lékař nebo zdravotní sestra Vám dají pokyny, jak to provádět. Bez tohoto nácviku se nepokoušejte o samostatnou aplikaci přípravku Lonquex. Informace potřebné k použití předplněné injekční stříkačky naleznete na konci této příbalové informace (viz bod „7. Informace k injekčnímu podávání pacientem samotným“). Správná léčba Vašeho onemocnění však vyžaduje blízkou a trvalou spolupráci s lékařem.
Jestliže jste použil(a) více přípravku Lonquex, než jste měl(a)
Jestliže jste užil(a) více přípravku Lonquex, než jste měl(a), poraďte se se svým lékařem.
Jestliže jste zapomněl(a) použít Lonquex
Pokud jste zapomněl(a) na injekci, poraďte se se svým lékařem, kdy si máte aplikovat další dávku.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Nejzávažnější nežádoucí účinky
• Méně často (mohou se vyskytnout nejvýše u 1 osoby ze 100) byly hlášeny alergické reakce, jako je například kožní vyrážka, vyvýšené svědivé okrsky kůže a závažné alergické reakce se slabostí, poklesem krevního tlaku, potížemi s dýcháním a otokem obličeje. Jestliže se domníváte, že máte tento typ reakce, musíte přerušit podávání injekce přípravku Lonquex a okamžitě si zajistit lékařskou pomoc.
• U jiných léků podobných přípravku Lonquex bylo méně často (může postihnout až 1 ze
100 lidí) hlášeno zvětšení sleziny a případy ruptury (roztržení) sleziny. Některé případy ruptury sleziny byly smrtelné. Je důležité, abyste okamžitě oznámil(a) svému lékaři, jestliže pocítíte bolest v levém nadbřišku nebo v levém rameni, protože může souviset s problémem se slezinou.
• Kašel, horečka a potíže s dýcháním nebo bolest při dýchání mohou být známkou méně častých (mohou se vyskytnout nejvýše u 1 osoby ze 100) závažných plicních nežádoucích účinků, jako je například pneumonie nebo syndrom akutní dechové tísně, které mohou být smrtelné. Jestliže máte horečku nebo kterýkoli z těchto příznaků, je důležité, abyste to okamžitě oznámil(a) svému lékaři.
• Je důležité, abyste ihned kontaktoval(a) svého lékaře, pokud se u Vás vyskytnou následující příznaky: otok nebo opuchlina, které mohou být spojeny s méně častým močením, dušnost, otok břicha a pocit plnosti a celkový pocit únavy. Tyto příznaky mají obvykle rychlý nástup.
Mohou to být příznaky onemocnění hlášeného u jiných léků podobných přípravku Lonquex a nazývaného „syndrom zvýšené permeability kapilár“, který způsobuje prosakování krve
z malých cév do Vašeho těla a vyžaduje okamžitou lékařskou pomoc.
Další nežádoucí účinky
Velmi časté (mohou se vyskytnout u více než 1 osoby z 10)
• Muskuloskeletální bolesti, jako jsou bolesti kostí a kloubů, svalů, končetin, hrudníku, krku nebo zad. Lékař Vám řekne, co byste mohl(a) udělat pro zmírnění těchto bolestí.
Časté (mohou se vyskytnout nejvýše u 1 osoby z 10)
• Snížení počtu krevních destiček, které zvyšuje riziko krvácení nebo vzniku modřin.
• Bolesti hlavy.
• Kožní reakce, jako je například zarudnutí nebo vyrážka.
• Nízké hladiny draslíku v krvi, které mohou způsobovat svalovou slabost, svalové záškuby nebo abnormální srdeční rytmus.
• Bolesti na hrudníku.
Méně časté (mohou se vyskytnout nejvýše u 1 osoby ze 100)
• Zvýšení počtu bílých krvinek.
• Místní reakce v místě vpichu, jako je například bolest nebo zatvrdnutí.
• Může dojít k některým změnám v krvi, ty však budou zjištěny běžnými krevními testy.
Nežádoucí účinky, které byly pozorovány u podobných léků, u přípravku Lonquex však zatím nikoli
• Srpkovité krize u pacientů se srpkovitou anémií.
• Švestkově zbarvené, vyvýšené, bolestivé afekce na kůži končetin a někdy i na obličeji a krku s horečkou (Sweetův syndrom).
• Zánět krevních cév v kůži.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Lonquex uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku na předplněné injekční stříkačce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
Lonquex lze vyjmout z chladničky a uchovávat při teplotě do 25 °C nejvýše po jedno období nepřesahující délku 3 dní. Po vyjmutí z chladničky musí být lék v této lhůtě spotřebován nebo zlikvidován.
Nepoužívejte tento přípravek, pokud si všimnete, že je zakalený nebo v něm jsou přítomny částice. Zlikvidujte tento přípravek v souladu s pokyny svého lékaře, lékárníka nebo zdravotní sestry.
6. Obsah balení a další informace Co Lonquex obsahuje
• Léčivou látkou je lipegfilgrastim. Jedna předplněná injekční stříkačka obsahuje 6 mg lipegfilgrastimu. Jeden ml roztoku obsahuje 10 mg lipegfilgrastimu.
• Dalšími složkami (pomocnými látkami) jsou ledová kyselina octová, hydroxid sodný, sorbitol (E420), polysorbát 20 a voda na injekci.
Jak Lonquex vypadá a co obsahuje toto balení
Lonquex je injekční roztok (injekce) v předplněné injekční stříkačce s fixní injekční jehlou v blistru. Lonquex je čirý bezbarvý roztok Jedna předplněná injekční stříkačka obsahuje 0,6 ml roztoku.
Jedno balení obsahuje: 1 předplněná injekční stříkačka s bezpečnostním systémem nebo bez něho.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
UAB "Sicor Biotech"
Moléty pl. 5 LT-08409 Vilnius Litva
Výrobce
Teva Biotech GmbH DornierstraBe 10 89079 Ulm Německo
Teva Pharmaceuticals Europe B.V. Swensweg 5 2031 GA Haarlem Nizozemsko
Teva Operations Poland Sp. z o.o. ul. Mogilska 80 31-546 Kraków Polsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Teva Pharma Belgium N.V./S.A./AG Tél/Tel: +32 3 820 73 73
Btnrapun
TeBa OapMacroTHKMC Etnrapnn EOOfl Ten: +359 2 489 95 82
Česká republika
Teva Pharmaceuticals CR, s.r.o. Tel: +420 251 007 111
Danmark
Teva Denmark A/S Tlf: +45 44 98 55 11
Deutschland
TEVA GmbH Tel: +49 731 402 08
Eesti
UAB "Sicor Biotech" Eesti filiaal Tel: +372 661 0801
Lietuva
UAB "Sicor Biotech"
Tel: +370 5 266 0203
Luxembourg/Luxemburg
Teva Pharma Belgium N.V./S.A./AG,
Belgique/Belgien
Tél/Tel: +32 3 820 73 73
Magyarország
Teva Gyógyszergyár Zrt.
Tel.: +36 1 288 64 00
Malta
Teva Pharmaceuticals Ireland, L-Irlanda Tel: +353 51 321740
Nederland
Teva Nederland B.V.
Tel: +31 800 0228 400
Norge
Teva Norway AS Tlf: +47 66 77 55 90
|
EXXáSa Teva EAAág A.E. Tn^: +30 210 72 79 099 |
Osterreich ratiopharm Arzneimittel Vertriebs-GmbH Tel: +43 1 97 007 |
|
Espaňa Teva Pharma, S.L.U. Tel: +34 91 387 32 80 |
Polska Teva Pharmaceuticals Polska Sp. z o.o. Tel.: +48 22 345 93 00 |
|
France Teva Santé Tél: +33 1 55 91 78 00 |
Portugal Teva Pharma - Produtos Farmaceuticos, Lda Tel: +351 21 476 75 50 |
|
Hrvatska Pliva Hrvatska d.o.o. Tel: +385 1 37 20 000 |
Románia Teva Pharmaceuticals S.R.L Tel: +40 21 230 65 24 |
|
Ireland Teva Pharmaceuticals Ireland Tel: +353 51 321740 |
Slovenija Pliva Ljubljana d.o.o. Tel: +386 1 58 90 390 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika TEVA Pharmaceuticals Slovakia s.r.o. Tel: +421 2 57 26 79 11 |
|
Italia Teva Italia S.r.l. Tel: +39 02 89 17 98 1 |
Suomi/Finland Teva Finland Puh/Tel: +358 20 180 5900 |
|
Kúnpoq Teva EÍlág A.E., EMáSa Tn^: +30 210 72 79 099 |
Sverige Teva Sweden AB Tel: +46 42 12 11 00 |
|
Latvija UAB "Sicor Biotech" filiale Latvija Tel: +371 673 23 666 |
United Kingdom Teva UK Limited Tel: +44 1977 628500 |
Tato příbalová informace byla naposledy revidována {měsíc RRRR}.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tato část obsahuje informace k podkožnímu podávání injekcí přípravku Lonquex pacientem sobě samému. Je důležité, abyste se nepokoušel(a) aplikovat si injekci sám/sama, dokud nebudete dostatečně zaškolen(a) svým lékařem nebo zdravotní sestrou. Jestliže si nejste jistý(á), jak si aplikovat injekci nebo máte jakékoliv otázky, požádejte o pomoc svého lékaře nebo zdravotní sestru.
Jak se Lonquex používá
Injekci si musíte podat do tkáně těsně pod kůží. Tomuto způsobu aplikace se říká podkožní (subkutánní) injekce.
Potřebné vybavení
K podání injekce sobě samému do tkáně pod kůží budete potřebovat toto vybavení:
• předplněná injekční stříkačka s přípravkem Lonquex,
• čtvereček navlhčený alkoholem,
• kus gázového obvazu nebo sterilní gázový tampon,
• schránka na ostré předměty (zhotovená z plastu a poskytnutá nemocnicí nebo lékárnou) k bezpečné likvidaci použitých injekčních stříkaček.
Jak postupovat před injekcí
1. Vyjměte lék z chladničky.
2. Otevřete blistr a vyjměte předplněnou injekční stříkačku z blistru. Předplněnou injekční stříkačku neberte za píst ani za chránič jehly.
3. Zkontrolujte datum použitelnosti na štítku předplněné injekční stříkačky (EXP). Přípravek nepoužívejte po uplynutí posledního dne uvedeného měsíce.
4. Zkontrolujte vzhled přípravku Lonquex. Roztok musí být čirý a bezbarvý. Pokud obsahuje částice nebo je zakalený, nesmíte jej použít.
5. Přípravkem Lonquex se nesmí silněji třepat, protože to může mít vliv na jeho účinnost.
6. Aby byla aplikace injekce příjemnější, nechejte předplněnou injekční stříkačku odstát po dobu 30 minut, aby dosáhla pokojové teploty (nejvýše 25 °C) nebo předplněnou injekční stříkačku po dobu několika minut opatrně podržte v ruce. Nezahřívejte přípravek Lonquex žádným jiným způsobem (například jej nezahřívejte v mikrovlnné troubě nebo horké vodě).
7. Chránič jehly nesnímejte z injekční stříkačky, dokud nejste připraven(a) k aplikaci injekce.
8. Najděte si pohodlné dobře osvětlené místo. Uložte všechny pomůcky tak, abyste na ně pohodlně dosáhl(a) (předplněnou injekční stříkačku s přípravkem Lonquex, čtvereček navlhčený alkoholem, kus gázového obvazu nebo sterilní gázový tampon a schránku na ostré předměty).
9. Důkladně si umyjte ruce.
Jak se připravit k podání injekce
Než si aplikujete injekci přípravku Lonquex, musíte provést následující kroky:
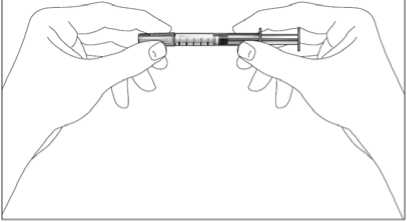
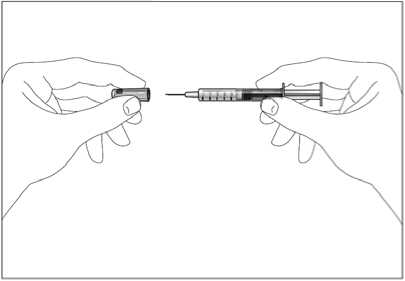
1. Uchopte injekční stříkačku a bez kroucení opatrně sejměte chránič z jehly. Táhněte rovně, jak ukazují obrázky 1 a 2. Nedotýkejte se jehly a netlačte na píst.
2. Můžete si všimnout malých bublinek vzduchu v předplněné injekční stříkačce. Pokud jsou přítomny vzduchové bublinky, jemně poklepejte prsty na injekční stříkačku, aby vystoupaly k její horní části. Ze stříkačky směřující vzhůru vytlačte vzduch pomalým stlačením pístu.
3. Nyní můžete předplněnou injekční stříkačku použít.
1
2
Kam si podat injekci
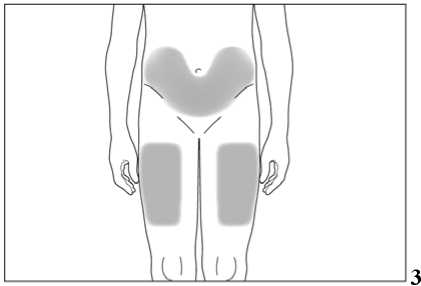
Nejvhodnějšími místy pro podání injekce sobě samému jsou:
• horní část stehen,
• břicho (viz šedé plochy na obrázku 3) s výjimkou kůže v bezprostředním okolí pupku.
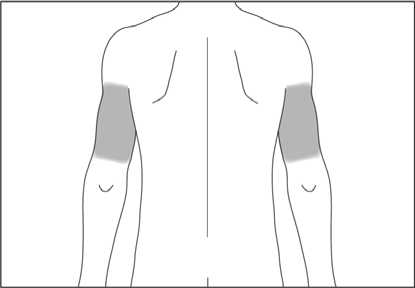
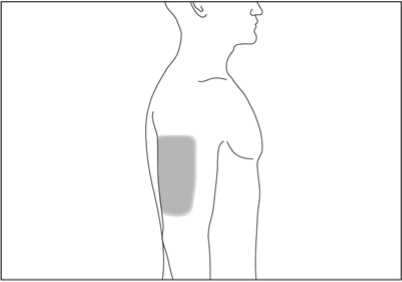
Pokud Vám podává injekci druhá osoba, může rovněž injekci aplikovat do horní části Vaší paže zezadu nebo ze strany (viz šedé plochy na obrázku 4 a 5).
4
5
Jak podat injekci sám/sama sobě
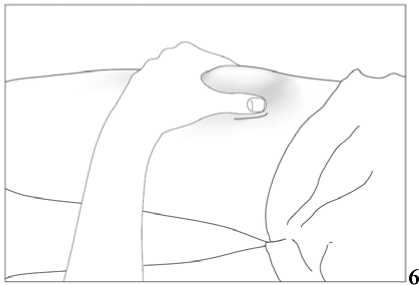
1. Místo vpichu pod kůži vydezinfikujte pomocí čtverečku navlhčeného alkoholem a volně, bez mačkání, uchopte mezi palec a ukazováček kožní řasu (viz obrázek 6).
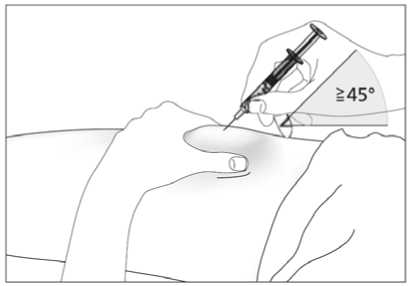
2. Zaveďte celou jehlu do kůže, jak Vám ukázal lékař nebo zdravotní sestra. Úhel mezi injekční stříkačkou a kůží nesmí být příliš úzký (nejméně 45°, viz obrázek 7).
3. Opatrně zatáhněte za píst, abyste se ujistil(a), že nebyla nabodnuta céva. Spatříte-li ve stříkačce krev, jehlu vytáhněte a zaveďte do jiného místa.
4. Pomalu a rovnoměrně vstříkněte kapalinu do tkáně. Nadále držte kožní řasu.
5. Po vstříknutí kapaliny vytáhněte jehlu a pusťte kůži.
6. Na několik sekund přitlačte na místo vpichu kus gázového obvazu nebo sterilní gázový tampon.
7. Každou injekční stříkačku použijte pouze na jednu injekci. Případný zbytek přípravku Lonquex v injekční stříkačce nepoužívejte.
Pamatujte
Máte-li jakýkoli problém, požádejte o radu či pomoc svého lékaře nebo zdravotní sestru.
Likvidace použitých injekčních stříkaček
• Nenasazujte chránič zpět na použité jehly.
• Použité injekční stříkačky vložte do schránky na ostré předměty a tuto schránku uchovávejte mimo dohled a dosah dětí.
• Až se schránka naplní, zlikvidujte ji podle pokynů svého lékaře, lékárníka nebo zdravotní sestry.
• Nikdy nevyhazujte použité injekční stříkačky do odpadkového koše na běžný domácí odpad.
Tato část obsahuje informace k podkožnímu podávání injekcí přípravku Lonquex pacientem sobě samému. Je důležité, abyste se nepokoušel(a) aplikovat si injekci sám/sama, dokud nebudete dostatečně zaškolen(a) svým lékařem nebo zdravotní sestrou. Jestliže si nejste jistý(á), jak si aplikovat injekci nebo máte jakékoliv otázky, požádejte o pomoc svého lékaře nebo zdravotní sestru.
Jak se Lonquex používá
Injekci si musíte podat do tkáně těsně pod kůží. Tomuto způsobu aplikace se říká podkožní (subkutánní) injekce.
Potřebné vybavení
K podání injekce sobě samému do tkáně pod kůží budete potřebovat toto vybavení:
• předplněná injekční stříkačka s přípravkem Lonquex,
• čtvereček navlhčený alkoholem,
• kus gázového obvazu nebo sterilní gázový tampon.
Jak postupovat před injekcí
1. Vyjměte lék z chladničky.
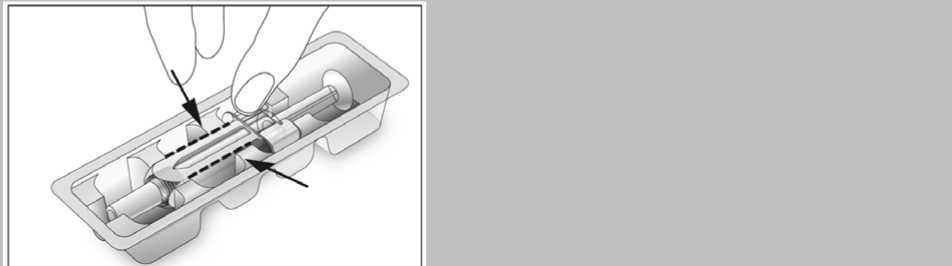
2. Otevřete blistr a vyjměte předplněnou injekční stříkačku z blistru (viz obrázek 1). Předplněnou injekční stříkačku neberte za píst ani za chránič jehly. Mohl by se poškodit bezpečnostní systém.
3. Zkontrolujte datum použitelnosti na štítku předplněné injekční stříkačky (EXP). Přípravek nepoužívejte po uplynutí posledního dne uvedeného měsíce.
4. Zkontrolujte vzhled přípravku Lonquex. Roztok musí být čirý a bezbarvý. Pokud obsahuje částice nebo je zakalený, nesmíte jej použít.
5. Přípravkem Lonquex se nesmí silněji třepat, protože to může mít vliv na jeho účinnost.
6. Aby byla aplikace injekce příjemnější, nechejte předplněnou injekční stříkačku odstát po dobu 30 minut, aby dosáhla pokojové teploty (nejvýše 25 °C) nebo předplněnou injekční stříkačku po dobu několika minut opatrně podržte v ruce. Nezahřívejte přípravek Lonquex žádným jiným způsobem (například jej nezahřívejte v mikrovlnné troubě nebo horké vodě).
7. Chránič jehly nesnímejte z injekční stříkačky, dokud nejste připraven(a) k aplikaci injekce.
8. Najděte si pohodlné dobře osvětlené místo. Uložte všechny pomůcky tak, abyste na ně pohodlně dosáhl(a) (předplněnou injekční stříkačku s přípravkem Lonquex, čtvereček navlhčený alkoholem a kus gázového obvazu nebo sterilní gázový tampon).
9. Důkladně si umyjte ruce.
1
Jak se připravit k podání injekce
Než si aplikujete injekci přípravku Lonquex, musíte provést následující kroky:
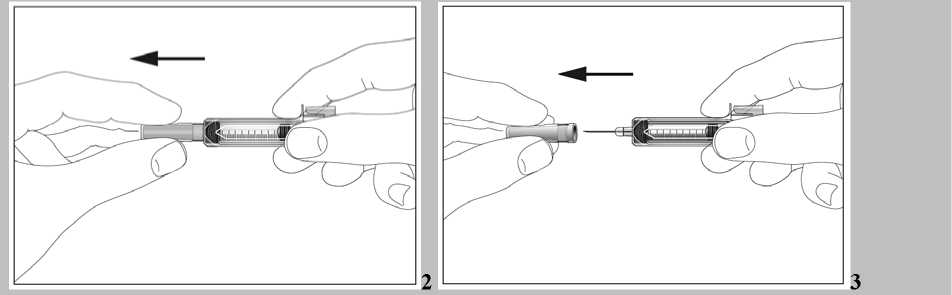
1. Uchopte injekční stříkačku a bez kroucení opatrně sejměte chránič z jehly. Táhněte rovně, jak ukazují obrázky 2 a 3. Nedotýkejte se jehly a netlačte na píst.
2. Můžete si všimnout malých bublinek vzduchu v předplněné injekční stříkačce. Pokud jsou přítomny vzduchové bublinky, jemně poklepejte prsty na injekční stříkačku, aby vystoupaly k její horní části. Ze stříkačky směřující vzhůru vytlačte vzduch pomalým stlačením pístu.
3. Nyní můžete předplněnou injekční stříkačku použít.
Kam si podat injekci
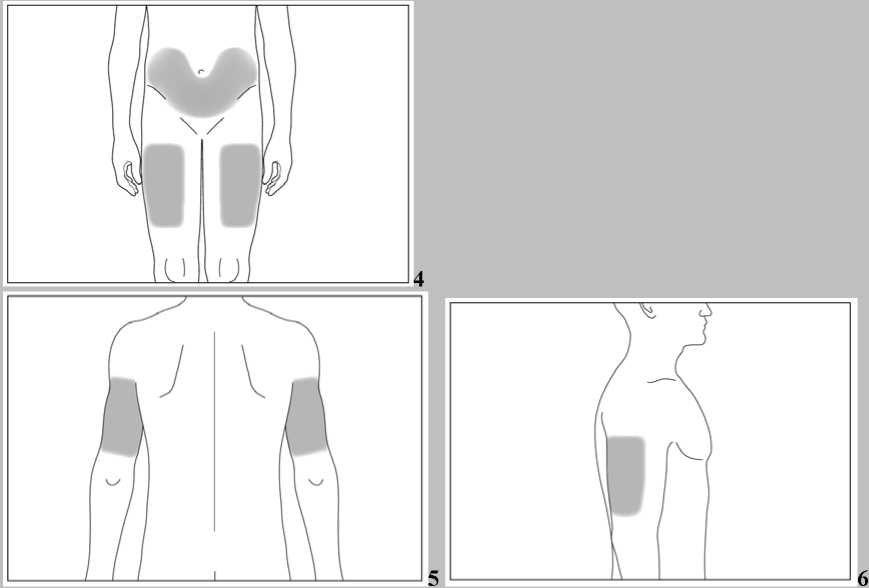
Nejvhodnějšími místy pro podání injekce sobě samému jsou:
• horní část stehen,
• břicho (viz šedé plochy na obrázku 4) s výjimkou kůže v bezprostředním okolí pupku.
Pokud Vám podává injekci druhá osoba, může rovněž injekci aplikovat do horní části paže zezadu nebo ze strany (viz šedé plochy na obrázku 5 a 6).
Jak podat injekci sám/sama sobě
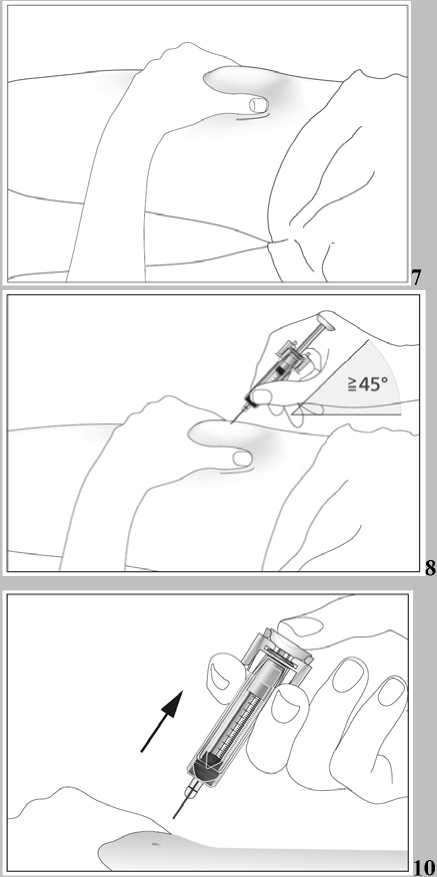
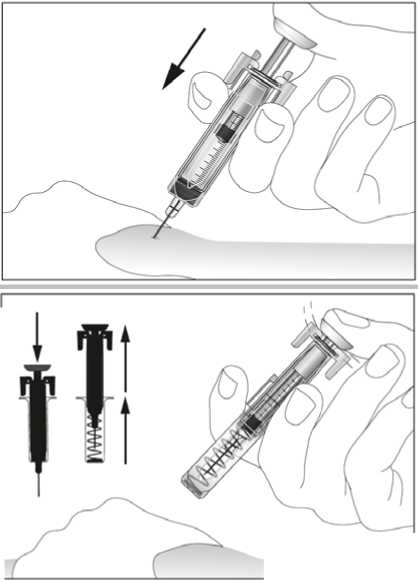
1. Místo vpichu pod kůži vydezinfikujte pomocí čtverečku navlhčeného alkoholem a volně, bez mačkání, uchopte mezi palec a ukazováček kožní řasu (viz obrázek 7).
2. Zaveďte celou jehlu do kůže, jak Vám ukázal lékař nebo zdravotní sestra. Úhel mezi injekční stříkačkou a kůží nesmí být příliš úzký (nejméně 45°, viz obrázek 8).
3. Opatrně zatáhněte za píst, abyste se ujistil(a), že nebyla nabodnuta céva. Spatříte-li ve stříkačce krev, jehlu vytáhněte a zaveďte do jiného místa.
4. Pomalu a rovnoměrně vstříkněte kapalinu do tkáně. Nadále držte kožní řasu (viz obrázek 9).
5. Zatlačte píst co nejdále, aby se vstříkla všechna kapalina. S pístem stále stlačeným do krajní polohy vytáhněte jehlu z kůže (viz obrázek 10). Poté uvolněte píst. Bezpečnostní systém se ihned aktivuje. Celá jehla a stříkačka budou okamžitě automaticky vtaženy zpět do chrániče, takže se nemůžete píchnout (viz obrázek 11).
6. Na několik sekund přitlačte na místo vpichu kus gázového obvazu nebo sterilní gázový tampon.
7. Každou přeplněnou injekční stříkačku použijte pouze na jednu injekci.
35