Lojuxta 5 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
^ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Lojuxta 5 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje lomitapidum 5 mg ve formě lomitapidum mesilas Pomocná látka se známým účinkem
Jedna tvrdá tobolka obsahuje 70,12 mg laktózy (ve formě monohydrátu) (viz bod 4.4). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Tvrdá tobolka s oranžovou svrchní částí / oranžovou spodní částí o délce 19,4 mm, potištěná černým inkoustem s textem „5 mg“ na spodní části a „A733“ na svrchní části tobolky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Lojuxta je indikován jako podpůrný přípravek užívaný spolu s dietou s nízkým obsahem tuků a dalšími léčivými přípravky na snížení hladiny lipidů spolu s aferézou lipoproteinů o nízké hustotě (LDL) či bez ní u dospělých pacientů s homozygotní formou familiární hypercholesterolémie (HoFH).
Tam, kde je to možné, by měla být homozygotní forma familiární hypercholesterolémie potvrzena geneticky. Je nutné vyloučit jiné formy primární hyperlipoproteinémie a sekundární příčiny hypercholesterolémie (např. nefrotický syndrom, hypotyreózu).
4.2 Dávkování a způsob podání
Léčbu přípravkem Lojuxta by měl zahajovat a sledovat lékař, který má zkušenosti s léčbou poruch lipidů.
Dávkování
Doporučená zahajovací dávka přípravku je 5 mg jednou denně. Po 2 týdnech může být dávka na základě přijatelné bezpečnosti a snášenlivosti zvýšena na 10 mg a poté v minimálně čtyřtýdenních intervalech na 20 mg, 40 mg a maximální doporučenou dávku 60 mg (viz bod 4.8).
Dávka by měla být zvyšována postupně, aby se minimalizovala incidence a závažnost gastrointestinálních nežádoucích účinků a zvýšení aminotransferáz.
Podávání se stravou může u přípravku Lojuxta zvyšovat expozici. Přípravek Lojuxta by se měl užívat nalačno, minimálně dvě hodiny po večeři, protože tuk obsažený v nedávno požitém jídle může mít negativní vliv na gastrointestinální snášenlivost.
Výskyt a závažnost gastrointestinálních nežádoucích účinků souvisejících s užíváním přípravku Lojuxta se snižují při dodržování diety s nízkým obsahem tuků. Pacienti by před zahájením léčby přípravkem Lojuxta měli držet dietu, ve které je méně než 20 % energie dodáváno z tuků, a během léčby by v této dietě měli pokračovat. Mělo by být zajištěno nutriční poradenství.
Pacienti by se měli vyvarovat konzumace grapefruitového džusu (viz body 4.4 a 4.5).
U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s atorvastatinem použijte jednu z těchto variant:
• Oddělte podání obou přípravků intervalem 12 hodin NEBO
• Snižte dávku přípravku Lojuxta o polovinu.
Pacienti užívající 5 mg by měli zůstat na této dávce.
Následně lze provést opatrnou titraci podle hladiny LDL-C a bezpečnosti /snášenlivosti.
Po ukončení léčby atorvastatinem zvyšte dávku přípravku Lojuxta titrací podle hladiny LDL-C a bezpečnosti/snášenlivosti.
U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s jakýmkoliv dalším slabým inhibitorem CYP3A4 oddělte podání obou přípravků intervalem 12 hodin.
Omezení maximální dávky přípravku Lojuxta zvažte podle požadované odpovědi LDL-C.
Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta.
Na základě pozorování snížených hladin esenciálních mastných kyselin a vitamínu E v klinických studiích by pacienti po dobu léčby přípravkem Lojuxta měli také užívat denně nutriční doplňky zajišťující příjem 400 IU vitamínu E a přibližně 200 mg kyseliny linolové, 110 mg kyseliny eikosapentaenové (EPA), 210 mg kyseliny alfa-linolenové (ALA) a 80 mg kyseliny dokosahexaenové (DHA).
Starší populace
Zkušenosti s přípravkem Lojuxta jsou u pacientů ve věku 65 let a starších omezené. Proto je u těchto pacientů zapotřebí zvláštní obezřetnosti.
Protože doporučený režim dávkování zahrnuje zahájení na spodní hranici dávkovacího rozmezí a opatrné navyšování dávky podle individuální snášenlivosti pacienta, nedoporučuje se ve starším věku žádná úprava dávkovacího schématu.
Porucha jater
Přípravek Lojuxta je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater včetně pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí (viz bod 5.2).
U pacientů s mírnou poruchou funkce jater (třída A dle Child-Pugha) by neměla být překračována dávka 40 mg denně.
U pacientů v terminálním stádiu onemocnění ledvin léčených dialýzou by neměla být překračována dávka 40 mg denně (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Lojuxta u dětí mladších 18 let nebyla stanovena, a použití tohoto léčivého přípravku u dětí se proto nedoporučuje. Nejsou dostupné žádné údaje.
Způsob podání
Perorální podání.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Pacienti se středně těžkou nebo těžkou poruchou funkce jater a pacienti s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí.
• Pacienti se známým významným nebo chronickým střevním onemocněním, např. zánětlivým onemocněním střeva nebo malabsorpcí.
• Souběžné podávání více než 40 mg simvastatinu (viz bod 4.5).
• Souběžné použití přípravku Lojuxta se silnými nebo středně silnými inhibitory cytochromu P450 (CYP) 3A4 (např. antimykotiky azoly, jako např. intrakonazolem, flukonazolem, ketokonazolem, vorikonazolem a posakonazolem, makrolidovými antibiotiky, jako např.erytromycinem nebo klaritromycinem, ketolidovými antibiotiky, jako např. telitromycinem, inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem a antiarytmikem dronedaronem [viz bod 4.5]).
• Těhotenství (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Abnormality jaterních enzymů a sledování jater
Lomitapid může způsobit zvýšení hladiny alaninaminotransferázy [ALT] a aspartátaminotransferázy [AST] a jaterní steatózu. V jakém rozsahu jaterní steatóza související s lomitapidem podporuje zvýšení aminotransferáz, není známo. Ačkoli nebyly hlášeny případy jaterní dysfunkce (zvýšení aminotransferáz se zvýšením bilirubinu nebo mezinárodního normalizovaného poměru [INR] nebo jaterního selhání), existuje obava, že lomitapid může indukovat steatohepatitidu, která může za několik let progredovat do cirhózy. Není pravděpodobné, že by klinické studie na podporu bezpečnosti a účinnosti lomitapidu u HoFH byly schopné vzhledem k jejich rozsahu a době trvání tyto nežádoucí účinky detekovat.
S lomitapidem je spojeno zvýšení hladin aminotransferáz (ALT a/nebo AST) (viz bod 5.1). Nedocházelo k souběžnému nebo následnému klinicky významnému zvýšení sérového bilirubinu, INR nebo alkalické fosfatázy. Změny jaterních enzymů se nejčastěji vyskytují během navyšování dávky, ale mohou nastat kdykoli během léčby.
Sledování testů jaterních funkcí
Před zahájením léčby přípravkem Lojuxta změřte ALT, AST, alkalickou fosfatázu, celkový bilirubin, gama-glutamyltransferázu (gama-GT) a sérový albumin. Léčivý přípravek je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater a pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí. Jestliže jsou základní vyšetření jaterních testů abnormální, zvažte zahájení léčby léčivým přípravkem až poté, co hepatolog provede vhodná vyšetření a abnormality základních testů budou vysvětleny či vyřešeny.
V prvním roce měřte jatemí testy (minimálně ALT a AST) před každým zvýšením dávky nebo jednou měsíčně podle toho, co nastane dříve. Po prvním roce provádějte tyto testy nejméně jednou za 3 měsíce a před každým zvýšením dávky. Jestliže pozorujete zvýšení hladiny aminotransferáz, dávku přípravku Lojuxta snižte, a při přetrvávajícím nebo klinicky významném zvýšení ukončete léčbu (konkrétní doporučení uvádí Tabulka 1).
Modifikace dávky na základě zvýšené hladiny jaterních aminotransferáz
Tabulka 1 je shrnutím doporučení pro úpravu dávky a sledování pacientů, u kterých dojde během léčby přípravkem Lojuxta ke zvýšení hladiny aminotransferáz.
Tabulka 1: Úprava dávky a sledování pacientů se zvýšenými hladinami aminotransferáz
|
ALT nebo AST |
Doporučení týkající se léčby a sledování* |
|
>3x a <5x horní mez normálního rozsahu (ULN) |
• Zvýšení potvrďte opakovaným měřením v rámci jednoho týdne. • Jestliže se zvýšení potvrdí, snižte dávku a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). • Testy opakujte každý týden, a jestliže budou přítomny známky abnormálních jaterních funkcí (zvýšení bilirubinu či INR), jestliže aminotransferázy stoupnou nad 5x ULN nebo jestliže hladiny aminotransferáz během přibližně 4 týdnů neklesnou pod 3x ULN, přerušte léčbu. Pacienty s přetrvávajícím zvýšením hladin aminotransferáz >3x ULN odešlete k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, zvažte snížení dávky a častější sledování jaterních testů. |
|
>5x ULN |
• Přerušte léčbu a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). Jestliže během přibližně 4 týdnů hladiny aminotransferáz neklesnou pod 3x ULN, odešlete pacienta k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, snižte dávku a častěji sledujte jaterní testy. |
*Doporučení založené na ULNpřibližně 30-40 mezinárodních jednotek/l.
Jestliže je zvýšení aminotransferáz provázeno klinickými symptomy poškození jater (např. nevolností, zvracením, bolestí břicha, horečkou, žloutenkou, letargií, příznaky podobnými chřipce), vzestupem hladin bilirubinu na >2x ULN nebo aktivním onemocněním jater, přerušte léčbu přípravkem Lojuxta a odešlete pacienta k hepatologovi na další vyšetření.
Opětovné nasazení léčby lze zvážit tehdy, pokud přínosy převyšují rizika související s potenciálním onemocněním jater.
Jaterní steatóza a riziko progresivního onemocnění jater
Většina léčených pacientů vykazuje vzestup obsahu tuku v játrech, což je ve shodě s mechanismem působení lomitapidu. V otevřené studii fáze 3 došlo u 18 z 23 pacientů s HoFH k vývoji jaterní steatózy (obsah tuku v játrech > 5,56 %) dle měření nukleární magnetickou rezonanční spektroskopií (MRS) (viz bod 5.1). Medián absolutního vzestupu jaterního tuku byl dle měření pomocí MRS jak po 26 týdnech, tak po 78 týdnech léčby 6 % oproti 1 % při zahájení studie. Jaterní steatóza je rizikový faktor progresivního onemocnění jater včetně steatohepatitidy a cirhózy. Dlouhodobé následky jaterní steatózy související s léčbou přípravkem Lojuxta nejsou známy. Klinické údaje naznačují, že nahromadění tuku v játrech je po ukončení léčby přípravkem Lojuxta reverzibilní, ale není známo, zda přetrvávají histologické změny, zejména po dlouhodobém užívání.
Sledování důkazů přítomnosti progresivního onemocnění jater
Při zahájení léčby a dále každý rok by se měl provádět pravidelný screening steatohepatitidy/fibrózy s použitím následujících zobrazovacích metod a hodnocení biomarkerů:
• Zobrazení elasticity tkáně, např. fibrosken, ARFI (Acoustic Radiation Force Impulse) nebo elastografie s použitím magnetické rezonance (MR)
• Gama-GT a sérový albumin pro detekci možného poškození jater
• Minimálně jeden marker z každé z následujících kategorií:
• Vysoce senzitivní C-reaktivní protein (hs-CRP), rychlost sedimentace erytrocytů (ESR), CK-18 fragment, NashTest (zánět jater)
• Rozšířený panel pro detekci jaterní fibrózy (ELF), fibrometr, poměr AST/ALT, skóre fib-4, fibrotest (jaterní fibróza)
Provedení těchto testů a jejich interpretace by měly zahrnovat spolupráci ošetřujícího lékaře s hepatologem. U pacientů s výsledky naznačujícími přítomnost steatohepatitidy nebo fibrózy by měla být zvážena biopsie jater.
Jestliže má pacient biopticky potvrzenou steatohepatitidu nebo fibrózu, měl by být znovu zhodnocen poměr přínosů a rizik a léčba by měla být ukončena, je-li to nutné.
Souběžné užívání inhibitorů CYP3A4
Zdá se, že lomitapid je citlivým substrátem pro metabolismus cestou CYP3A4. Inhibitory CYP3A4 zvyšují expozici lomitapidu, přičemž silné inhibitory zvyšují expozici přibližně 27-krát. Souběžné užití středně silných nebo silných inhibitorů CYP3A4 a přípravku Lojuxta je kontraindikováno (viz bod 4.3). V klinických studiích s lomitapidem se u jednoho pacienta s HoFH během několika dní po zahájení užívání silného inhibitoru CYP3A4 klaritromycinu vyvinuly výrazně zvýšené hladiny aminotransferáz (ALT 24x ULN, AST 13x ULN). Jestliže se nelze vyvarovat léčby středně silnými nebo silnými inhibitory CYP3A4, mělo by být užívání přípravku Lojuxta v průběhu léčby ukončeno.
Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta má být podávána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4.
Souběžné užívání induktorů CYP3A4
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. Induktory CYP3A4 vykazují svůj účinek v závislosti na čase a dosažení maximálního účinku může trvat minimálně 2 týdny od zahájení léčby. Naopak při ukončení léčby může trvat minimálně 2 týdny, než indukce CYP3A4 odezní.
Očekává se, že souběžné podávání induktoru CYP3A4 snižuje účinek přípravku Lojuxta. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinu, pioglitazonu, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Použití třezalky tečkované současně s přípravkem Lojuxta je nutné se vyvarovat.
Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. Při vysazení induktoru CYP3A4 je nutné zvážit možnost zvýšené expozice a může být nutné snížit dávku přípravku Lojuxta.
Souběžné použití s inhibitory HMG-CoA reduktázy (..statiny")
Lomitapid zvyšuje plazmatické koncentrace statinů. U pacientů, kteří dostávají přípravek Lojuxta jako přídatnou terapii vedle statinů, by měly být sledovány nežádoucí účinky spojené s použitím vysokých dávek statinů. Statiny příležitostně mohou vyvolat myopatii. Ve vzácných případech může mít myopatie formu rhabdomyolýzy s akutním renálním selháním sekundárně způsobeným myoglobinurií nebo bez renálního selhání a může mít fatální následky. Všichni pacienti užívající přípravek Lojuxta vedle statinů by měli být informování o možném riziku myopatie a měli by být informováni, aby okamžitě hlásili nevysvětlitelnou bolest, citlivost nebo slabost svalů. S přípravkem Lojuxta by se neměly používat dávky simvastatinu > 40 mg (viz bod 4.3).
Grapefruitový džus
Ze stravy pacientů je při léčbě přípravkem Lojuxta nutno vyloučit grapefruitový džus.
Riziko supraterapeutické nebo subterapeutické antikoagulace u antikoagulačních přípravků na bázi kumarinu
Lomitapid zvyšuje plazmatické koncentrace warfarinu. Zvýšení dávky přípravku Lojuxta může vést k supraterapeutické antikoagulaci a snížení dávky může vést k subterapeutické antikoagulaci.
U jednoho z pěti pacientů užívajících souběžně warfarin přispěla obtížná kontrola INR k předčasnému ukončení studie fáze 3. Pacienti užívající warfarin by měli podstupovat pravidelné sledování INR, zejména po jakýchkoli změnách dávkování přípravku Lojuxta. Dávka warfarinu by měla být upravena dle klinické indikace.
Požívání alkoholu
Alkohol může zvyšovat hladinu tuku v játrech a může vést k indukci nebo exacerbaci poškození jater. Ve studii fáze 3 udávali 3 ze 4 pacientů se zvýšením ALT >5x ULN konzumaci alkoholu překračující limit doporučený v protokolu. Požívání alkoholu během léčby přípravkem Lojuxta se nedoporučuje.
Hepatotoxické přípravky
Opatrnosti je třeba při použití přípravku Lojuxta s dalšími léčivými přípravky se známým hepatotoxickým potenciálem, např. izotretinoinem, amiodaronem, acetaminofenem (>4 g/den po >3 dny/týden), metotrexátem, tetracyklinem a tamoxifenem. Účinek souběžného podávání přípravku Lojuxta s dalšími hepatotoxickými léčivými přípravky není znám. Může být nutné častější sledování jaterních testů.
Snížená absorpce vitamínů rozpustných v tucích a mastných kyselin v séru
Vzhledem k mechanismu působení lomitapidu v tenkém střevě může lomitapid snižovat absorpci živin rozpustných v tucích. Ve studii fáze 3 byly pacientům denně poskytovány nutriční doplňky s vitamínem E, kyselinou linolovou, ALA, EPA a DHA. V této studii se úroveň mediánu sérového vitamínu E, ALA, kyseliny linolové, EPA, DHA a kyseliny arachidonové mezi výchozím bodem studie a týdnem 26 snížila, ale zůstala nad spodním limitem referenčního rozmezí. Nežádoucí klinické důsledky tohoto snížení při léčbě lomitapidem nebyly pozorovány po dobu až 78 týdnů. Pacienti léčení přípravkem Lojuxta by měli denně užívat doplňky, které obsahují 400 mezinárodních jednotek vitamínu E a přibližně 200 mg kyseliny linolové, 210 mg ALA, 110 mg EPA a 80 mg DHA.
Antikoncepční opatření u žen ve fertilním věku
Před zahájením léčby u žen ve fertilním věku by mělo být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající
perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z důvodu průjmu a/nebo zvracení (viz bod 4.5). Perorální antikoncepční přípravky na bázi estrogenů jsou slabými inhibitory CYP3A4 (viz bod 4.2).
Pacientky by měly být informovány, že v případě otěhotnění musejí neprodleně informovat svého lékaře a ukončit užívání přípravku Lojuxta (viz bod 4.6).
Laktóza
Přípravek Lojuxta obsahuje laktózu, pacienti se vzácnými hereditárními problémy jako jsou intolerance galaktózy, vrozený deficit laktázy nebo glukózová-galaktózová malabsorpce by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
Tabulka 2: Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
Inhibitory CYP3A4 |
Při současném podávání lomitapidu v dávce 60 mg s ketokonazolem, silným inhibitorem CYP3A4, v dávce 200 mg dvakrát denně se zvýšila AUC lomitapidu přibližně 27krát a Cmax přibližně 15krát. Interakce mezi středně silnými inhibitory CYP3A4 a lomitapidem nebyly studovány. Předpokládá se, že středně silné inhibitory CYP3A4 mají významný vliv na farmakokinetiku lomitapidu. Očekává se, že souběžné použití středně silných inhibitorů CYP3A4 zvyšuje expozici lomitapidu 4-10krát podle výsledků studie se silným inhibitorem CYP3A4 podle historických údajů pro zkušební model CYP3A4 midazolam. Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při podávání lomitapidu 20 mg současně s atorvastatinem (tj. slabým inhibitorem CYP3A4) se AUC a Cmax lomitapidu zvýšily přibližně 2x. Pokud byl lomitapid podáván 12 hodin po |
Použití silných nebo středně silných inhibitorů CYP3A4 spolu s přípravkem Lojuxta je kontraindikováno. Jestliže se nelze vyvarovat léčby antimykotiky azoly (např. itrakonazolem, ketokonazolem, flukonazolem, vorikonazolem, posakonazolem), antiarytmikem dronedaronem, makrolidovými antibiotiky (např. erytromycinem, klaritromcinem), ketolidovými antibiotiky (např. telitromycinem), inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem, měla by být během této léčby léčba přípravkem Lojuxta pozastavena (viz body 4.3 a 4.4). Grapefruitový džus je středně silným inhibitorem CYP3A4 a má se za to, že podstatně zvyšuje expozici lomitapidu. Pacienti užívající přípravek Lojuxta by se měli vyvarovat konzumace grapefruitového džusu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta musí být užívána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4. Příklady slabých inhibitorů CYP3A4 zahrnují: alprazolam, amiodaron, amlodipin, atorvastatin, azitromycin, bikalutamid, cilostazol, cimetidin, cyklosporin, klotrimazol, fluoxetin, fluvoxamin, fosaprepitant, |
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
atorvastatinu, žádné klinicky významné zvýšení expozice lomitapidu nebylo pozorováno. Při podávání lomitapidu 20 mg s ethinyl estradiolem/norgestimátem (tj. slabým inhibitorem CYP3A4) žádné klinicky významné zvýšení expozice lomitapidu nebylo pozorováno, a to při užívání současném ani s intervalem 12 hodin. |
ginkgo, vodilku kanadskou, izoniazid, ivakaftor, lacidipin, lapatinib, linagliptin, nilotinib, perorální antikoncepční přípravky obsahující estrogeny, pazopanib, mátový olej, propiverin, ranitidin, ranolazin, roxithromycin, hořké pomeranče, takrolimus, tikagrelor a tolvaptan. Tento seznam není úplný a předepisující lékaři by měli zkontrolovat preskripční informace o léčivech, která mají být podávána spolu s přípravkem Lojuxta s ohledem na možné interakce zprostředkované CYP3A4. Účinek podávání více než jednoho slabého inhibitoru CYP3A4 nebyl testován, ale očekává se, že účinek na expozici lomitapidu je větší než při současném podávání jednotlivých inhibitorů s lomitapidem. Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta. | |
|
Induktory CYP3A4 |
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. To by mohlo následně snížit účinek lomitapidu. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. |
Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinnu, pioglitazonu, třezalky tečkované, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. |
|
Sekvestranty žlučových kyselin |
Interakce lomitapidu se sekvestranty žlučových kyselin (pryskyřicemi, např. kolesevelamem a cholestyraminem) nebyly testovány. |
Protože sekvestranty žlučových kyselin mohou interferovat s absorpcí perorálních léčivých přípravků, měly by se užívat minimálně 4 hodiny před přípravkem Lojuxta nebo minimálně 4 hodiny po něm. |
Účinky lomitapidu na jiné léčivé přípravky
Inhibitory HMG-CoA reduktázy („statiny“): Lomitapid zvyšuje plazmatické koncentrace statinů. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním simvastatinu 40 mg se zvýšily u kyseliny simvastatinu hodnoty AUC o 68 % a Cmax o 57 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním atorvastatinu 20 mg se zvýšily u kyseliny atorvastatinu hodnoty AUC o 52 % a Cmax o 63 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním rosuvastatinu 20 mg se zvýšila hodnota Tmax rosuvastatinu z 1 na 4 hodiny, hodnota AUC se zvýšila o 32 % a hodnota Cmax zůstala nezměněná. Riziko myopatie u simvastatinu je závislé na dávce. Použití přípravku Lojuxta je u pacientů léčených vysokými dávkami statinů (> 40 mg) kontraindikováno (viz body 4.3 a 4.4).
Kumarinové antikoagulační přípravky: Podávání lomitapidu 60 mg do dosažení ustáleného stavu a po dobu 6 dní po warfarinu 10 mg zvýšilo hodnotu INR 1,26krát. Hodnota AUC pro R(+)-warfarin se zvýšila o 25 % a pro S(-)-warfarin o 30 %. Hodnota Cmax pro R(+)-warfarin se zvýšila o 14 % a pro S(-)-warfarin o 15 %. U pacientů užívajících souběžně kumariny (např. warfarin) a přípravek Lojuxta by měla být před zahájením užívání přípravku Lojuxta stanovena hodnota INR a tato hodnota by měla být pravidelně kontrolována, přičemž dávkování kumarinů by mělo být upraveno dle klinické potřeby (viz bod 4.4).
Fenofibrát, niacin a ezetimib: Při podávání lomitapidu do dosažení ustáleného stavu před podáním mikronizovaného fenofibrátu 145 mg, niacinu s prodlouženým uvolňováním 1000 mg nebo ezetimibu 10 mg nebyly pozorovány klinicky významné účinky působení kteréhokoli z těchto léčivých přípravků. Při souběžném podávání s přípravkem Lojuxta není nutná úprava dávky.
Perorální antikoncepce: Při podávání lomitapidu 50 mg do dosažení ustáleného stavu spolu s perorálním antikoncepčním přípravkem na bázi estrogenů nebyl pozorován klinicky ani statisticky významný vliv na farmakokinetiku složek perorální antikoncepce (ethinylestradiolu a 17-deacetylnorgestimátu, metabolitu norgestimátu). Neočekává se, že by lomitapid přímo ovlivňoval účinnost perorální antikoncepce na bázi estrogenů, nicméně průjem a/nebo zvracení mohou snížit absorpci hormonů. U případů prodlouženého nebo závažného průjmu a/nebo zvracení trvajících více než 2 dny by měla být po dobu 7 dnů po ústupu symptomů použita další antikoncepční metoda.
Substráty P-gp: Lomitapid in vitro inhibuje P-gp a může zvyšovat absorpci P-gp substrátů. Souběžné podávání přípravku Lojuxta s P-pg substráty (např. aliskirenem, abrisentanem, kolchicinem, dabigatran etexilátem, dixoginem, everolimem, fexofenadinem, imatinibem, lapatinibem, maravirokem, nilotinibem, posakonazolem, ranolazinem, saxagliptinem, sirolimem, sitagliptinem, talinololem, tolvaptanem, topotekanem) může zvýšit absorpci P-gp substrátů. Při souběžném použití s přípravkem Lojuxta by mělo být zváženo snížení dávky P-gp substrátu.
In vitro hodnocení lékových interakcí: Lomitapid inhibuje CYP3A4. Lomitapide neindukuje CYP 1A2, 3A4 ani 2B6 a neinhibuje CYP 1A2, 2B6, 2C9, 2C19, 2D6 ani 2E1. Lomitapid není P-gp substrátem, ale inhibuje P-gp. Lomitapid neinhibuje protein rezistence u karcinomu prsu (BCRP).
4.6 Fertilita, těhotenství a kojení
Přípravek Lojuxta je v těhotenství kontraindikován. Neexistují spolehlivé údaje o použití přípravku u těhotných žen. Studie na zvířatech prokázaly vývojovou toxicitu (teratogenitu, embryotoxicitu, viz bod 5.3). Možné riziko pro člověka je neznámé.
Použití u žen ve fertilním věku
Před zahájením léčby u žen ve fertilním věku by mělo být vyloučeno těhotenství, mělo by být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z důvodu průjmu a/nebo zvracení. Dokud symptomy neustoupí, je nutno používat další antikoncepční metody (viz bod 4.5).
Kojení
Není známo, zda se lomitapid vylučuje do mateřského mléka. Vzhledem k potenciálu nežádoucích účinků zjištěnému na základě studií s lomitapidem na zvířatech (viz bod 5.3) je nutné rozhodnout, zda ukončit kojení nebo užívání léčivého přípravku, a to při zvážení významu léčivého přípravku pro matku.
Fertilita
U potkaních samců ani samic, kterým byl podáván lomitapid se systémovou expozicí (AUC) ve výši odhadovaného čtyř- až pětinásobku expozice u lidí při maximální doporučené dávce u člověka, nebyly pozorovány žádné nežádoucí účinky na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Lojuxta může mít mírný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejzávažnějšími nežádoucími účinky během léčby byly abnormality jaterních aminotransferáz (viz bod 4.4).
Nejčastějšími nežádoucími účinky byly gastrointestinální účinky. Nežádoucí gastrointestinální účinky byly hlášeny u 27 (93 %) z 29 pacientů klinické studie fáze 3. Průjem se objevil u 79 %, nevolnost u 65 %, dyspepsie u 38 % a zvracení u 34 % pacientů. Další účinky, které hlásilo minimálně 20 % pacientů, zahrnují bolest břicha, břišní dyskomfort, nadmutí břicha, zácpu a plynatost. Nežádoucí gastrointestinální účinky se častěji objevovaly během fáze navyšování dávky ve studii a poklesly, jakmile pacienti dosáhli maximální tolerované dávky lomitapidu.
Gastrointestinální nežádoucí účinky se závažnou intenzitou byly hlášeny u 6 (21 %) z 29 pacientů v klinické studii fáze 3, přičemž nejčastějším účinkem byl průjem (4 pacienti, 14 %), zvracení (3 pacienti, 10 %) a bolest břicha, nadmutí a/nebo dyskomfort (2 pacienti, 7 %). Gastrointestinální účinky přispěly k časnému ukončení studie u 4 (14 %) pacientů.
Nejčastěji udávanými nežádoucími účinky se závažnou intenzitou byly průjem (4 subjekty, 14 %), zvracení (3 pacienti, 10 %) a nadmutí břicha a zvýšení ALT (pokaždé 2 subjekty, 7 %).
Přehled nežádoucích účinků v tabulce
Dle frekvence jsou nežádoucí účinky definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), málo časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), s neznámou frekvencí (z dostupných údajů nelze určit).
Tabulka 3 zahrnuje veškeré nežádoucí účinky hlášené u 35 pacientů léčených ve studii fáze 2 UP1001 a ve studii fáze 3 UP1002/AEGR-733-005 nebo v rozšířené studii AEGR-733-012.
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Časté |
Gastroenteritida |
|
Poruchy metabolismu a výživy |
Velmi časté |
Snížená chuť k jídlu |
|
Poruchy nervového systému |
Časté |
Závratě Bolest hlavy Migréna |
|
Gastrointestinální poruchy |
Velmi časté |
Břišní dyskomfort Bolest horní části břicha Plynatost Distenze břicha Zácpa |
|
Časté |
Gastritida Rektální tenesmy Aerofagie Nutkání na stolici Říhání Častá stolice Dilatace žaludku Onemocnění žaludku Gastroezofageální reflux Krvácení z hemoroidů Regurgitace | |
|
Poruchy jater a žlučových cest |
Časté |
Jaterní steatóza Hepatotoxicita Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Časté |
Ekchymóza Papuly Erytematózní vyrážka Xantomy |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Únava |
|
Vyšetření |
Velmi časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Snížení tělesné hmotnosti |
|
Časté |
Zvýšený mezinárodní normalizovaný poměr Zvýšená alkalická fosfatáza v krvi Snížení draslíku v krvi Snížený karoten Abnormální mezinárodní normalizovaný poměr Abnormální testy jaterních funkcí Prodloužený protrombinový čas Zvýšené transaminázy Snížený vitamín E Snížený vitamín K |
Tabulka 4 uvádí všechny nežádoucí účinky u subjektů, které dostávaly lomitapid v monoterapii (n=291), léčených ve studiích fáze 2 u subjektů se zvýšenou hladinou LDL-C (N=462).
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Méně časté |
Gastroenteritida Gastrointestinální infekce Chřipka Nazofaryngitida Sinusitida |
|
Poruchy krve a lymfatického systému |
Méně časté | |
|
Poruchy metabolismu a výživy |
Časté |
Snížená chuť k jídlu |
|
Méně časté |
Dehydratace Zvýšená chuť k jídlu | |
|
Poruchy nervového systému |
Méně časté |
Parestézie Ospalost |
|
Poruchy oka |
Méně časté |
Otok oka |
|
Poruchy ucha a labyrintu |
Méně časté |
Závrať |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté |
Léze hltanu Syndrom kašle horních cest dýchacích |
|
Gastrointestinální poruchy |
Velmi časté |
Plynatost |
|
Časté |
Bolest horní části břicha Distenze břicha Bolest břicha Zvracení Břišní dyskomfort Říhání Bolest v podbřišku Častá stolice | |
|
Méně časté |
Sucho v ústech Tvrdá stolice Gastroezofageální reflux Citlivost břicha Dyskomfort v epigastriu Dilatace žaludku Hemateméza Krvácení v dolní části gastrointestinálního traktu Refluxní ezofagitida | |
|
Poruchy jater a žlučových cest |
Méně časté |
Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Puchýře Suchá kůže Nadměrné pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté |
Svalové spasmy |
|
Méně časté |
Bolest kloubů Bolest svalů Bolest končetin Otok kloubu Svalové záškuby | |
|
Poruchy ledvin a močových cest |
Méně časté |
Hematurie |
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Celkové poruchy a reakce v místě |
Časté |
Únava |
|
podání |
Slabost | |
|
Méně časté |
Bolest na hrudi Mrazení Rychlý pocit nasycení Poruchy chůze Malátnost Horečka | |
|
Vyšetření |
Časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Zvýšení jaterních enzymů Abnormální testy jaterních funkcí Snížený počet neutrofilů Snížený počet leukocytů |
|
Méně časté |
Snížení tělesné hmotnosti Zvýšený bilirubin v krvi Zvýšená hladina gama-glutamyltransferázy Zvýšené procento neutrofilů Protein v moči Prodloužený protrombinový čas Abnormální testy plicních funkcí Zvýšený počet leukocytů |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování neexistuje specifická léčba. U hlodavců byla dobře snášena jednorázová perorální dávka lomitapidu >600krát vyšší než maximální doporučená dávka pro člověka (1 mg/kg). Maximální dávkou podanou lidským subjektům v klinických studiích bylo 200 mg ve formě jednorázové dávky, nevyskytly se žádné nežádoucí účinky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná léčiva ovlivňující hladinu lipidů, samotná. ATC kód: C10AX12
Mechanismus účinku
Lomitapid je selektivní inhibitor mikrozomálního transferového proteinu (MTP), intracelulárního proteinu přenášejícího lipidy, který se nachází v lumen endoplazmatického retikula a zodpovídá za vazbu a převod jednotlivých lipidových molekul mezi membránami. MTP hraje klíčovou roli při sestavování lipoproteinů obsahujících apo B v játrech a střevech. Inhibice MTP snižuje sekreci lipoproteinů a cirkulující koncentrace lipidů přenášených v lipoproteinech včetně cholesterolu a triglyceridů.
Klinická účinnost a bezpečnost
Účinnost a bezpečnost lomitapidu při současném podávání diety s nízkým obsahem tuků a dalších terapií snižujících lipidy u dospělých pacientů s HoFH hodnotila otevřená studie s jedním ramenem (UP1002/AEGR-733-005). Pacienti byli instruováni, aby dodržovali dietu s nízkým obsahem tuků (< 20 % kalorického příjmu z tuků) i terapii snižující lipidy, kterou měli při vstupu do studie, včetně aferézy, byla-li tato nutná, a to po dobu 6 týdnů předcházejících výchozímu bodu studie minimálně až do týdne 26. Dávka lomitapidu byla navyšována z 5 mg na individuálně stanovenou maximální tolerovanou dávku až 60 mg. Po týdnu 26 byl pacientům lomitapid ponechán za účelem zjištění účinků dlouhodobé léčby a bylo jim umožněno změnit základní terapii snižující lipidy. Studie zahrnovala celkem 78 týdnů léčby.
Do studie bylo zařazeno 29 pacientů, z nichž 23 dokončilo týden 78. Zařazeno bylo 16 mužů (55 %) a 13 žen (45 %) s průměrným věkem 30,7 let a ve věkovém rozmezí 18 až 55 let. Průměrná dávka lomitapidu v týdnu 26 byla 45 mg a v týdnu 78 byla 40 mg. V týdnu 26 byla průměrná procentuální změna hladiny LDL-C u populace „intention-to-treat“ (ITT) oproti výchozímu bodu -40 %
(p < 0,001). Průměrná procentuální změna oproti výchozímu bodu studie v týdnu 26 s použitím metody LOCF (Last Observation Carried Forward) pro každé hodnocení je uvedena na obrázku 1.
Obrázek 1: Průměrné procentuální změny hladiny LDL-C mezi vstupní hodnotou a týdnem
26 v hlavní studii účinnosti UP1002/AEGR-733-005 (primární cíl) s použitím LOCF pro každé hodnocení (n=29)
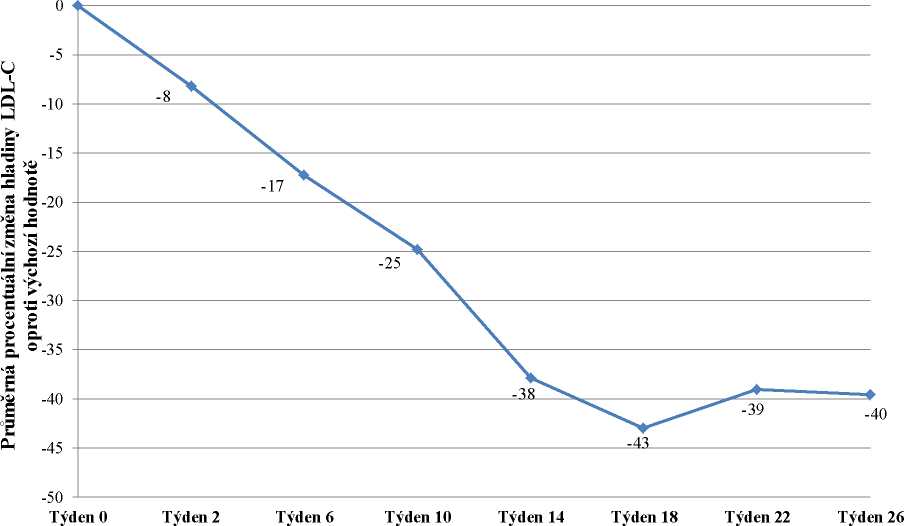
Týden studie
Změny lipidů i lipoproteinů v týdnu 26 a 78 při léčbě lomitapidem uvádí Tabulka 5.
Tabulka 5: Absolutní hodnoty a procentuální změny hladin lipidů a lipoproteinů mezi
výchozí hodnotou a týdny 26 a 78 (hlavní studie účinnosti UP1002/AEGR-733-005).
|
Parametr (jednotky) |
Výchozí bod |
Týden 26 / LOCF (n=29) |
Týden 78 (n=23) | ||||
|
Průměr (SD) |
Průměr (SD) |
% změna |
Hodnota p b |
Průměr (SD) |
% změna |
Hodnota p b | |
|
LDL-C, přímý (mg/dl) |
336 (114) |
190 (104) |
-40 |
< 0,001 |
210 (132) |
-38 |
< 0,001 |
|
Celkový cholesterol (TC) (mg/dl) |
430 (135) |
258 (118) |
-36 |
< 0,001 |
281 (149) |
-35 |
< 0,001 |
|
Apolipoprotein B (apo B) (mg/dl) |
259 (80) |
148 (74) |
-39 |
< 0,001 |
151 (89) |
-43 |
< 0,001 |
|
Triglyceridy (TG) (mg/dl)a |
92 |
57 |
-45 |
0,009 |
59 |
-42 |
0,012 |
|
Cholesterol z jiných lipoproteinů než o vysoké hustotě (non-HDL-C) (mg/dl) |
386 (132) |
217 (113) |
-40 |
< 0,001 |
239 (146) |
-39 |
< 0,001 |
|
Cholesterol z lipoproteinů o velmi nízké hustotě (VLDL-C) (mg/dl) |
21 (10) |
13 (9) |
-29 |
0,012 |
16 (15) |
-31 |
0,013 |
|
Lipoprotein (a) (Lp(a)) (nmol/l)a |
66 |
61 |
-13 |
0,094 |
72 |
-4 |
< 0,842 |
|
Cholesterol z lipoproteinů o vysoké hustotě (HDL-C) (mg/dl) |
44 (11) |
41 (13) |
-7 |
0,072 |
43 (12) |
-4,6 |
0,246 |
a Medián uvedený pro TG a Lp(a). Hodnota p je založena na průměrné procentuální změně b Hodnota p u střední procentuální změny oproti výchozímu bodu studie na základě párového t-testu
Jak v týdnu 26, tak v týdnu 78 došlo k významnému snížení hladin LDL-C, TC, apo B, TG, non-HDL-C a VLDL-C a změny u HDL-C vykazovaly v týdnu 26 trend směrem ke snížení a v týdnu 78 se vrátily k výchozím hodnotám.
Účinek přípravku Lojuxta na kardiovaskulární morbiditu a mortalitu nebyl stanoven.
Při zahájení studie užívalo 93 % pacientů statin, 76 % pacientů ezetimib, 10 % niacin, 3 % sekvestrant žlučových kyselin a 62 % pacientů bylo léčeno aferézou. U 15 z 23 (65 %) pacientů byla v týdnu 78 redukována jejich léčba na snížení lipidů, včetně plánovaných a neplánovaných snižování/přerušení. Aferéza byla přerušena u 3 ze 13 pacientů, kteří byli aferézou léčeni v týdnu 26, a u 3 pacientů byla frekvence při současném udržení nízkých hladin LDL-C i po týdnu 78 snížena. Klinický přínos omezení základní terapie snižující lipidy, včetně aferézy, není jistý.
Z 23 pacientů, kteří dokončili týden 26 studie, došlo u 19 (83 %) ke snížení LDL-C o > 25 %, přičemž 8 (35 %) z nich mělo v tomto časovém bodě LDL-C < 100 mg/dl a 1 pacient měl LDL-C < 70 mg/dl.
V této studii došlo u 10 pacientů ke zvýšení AST a/nebo ALT >3x ULN (viz Tabulka 6).
Nejvyšší výsledky testů jaterních funkcí po první dávce (hlavní studie účinnosti UP1002/AEGR-733-005)
|
Parametr/abnormalita |
N (%) |
|
ALT | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
6 (20,7) |
|
>5 až < 10 x ULN |
3 (10,3) |
|
>10 až < 20 x ULN |
1 (3,4) |
|
> 20 x ULN |
0 |
|
AST | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
5 (17,2) |
|
>5 až < 10 x ULN |
1 (3,4) |
|
>10 až < 20 x ULN |
0 |
|
> 20 x ULN |
0 |
Tabulka 6:
Zvýšení ALT a/nebo AST > 5x ULN bylo zvládnuto snížením dávky nebo dočasným pozastavením podávání lomitapidu a všichni pacienti byli schopni v léčbě studovaným léčivem pokračovat. Nebylo pozorováno klinicky významné zvýšení celkového bilirubinu ani alkalické fosfatázy. Jaterní tuk byl během klinické studie u všech dostupných pacientů prospektivně měřen s použitím MRS (Tabulka 7). Údaje od jedinců, u kterých byla po ukončení lomitapidu prováděna opakovaná měření, ukázaly, že nahromadění tuku v játrech je reverzibilní, ale není známo, zda nepřetrvávají histologické změny.
Tabulka 7: Maximální kategorické změny v % jaterního tuku (hlavní studie účinnosti
UP1002/AEGR-733-005)
|
Maximální absolutní nárůst % jaterního tuku |
Fáze účinnosti Týdny 0-26 N (%) |
Fáze bezpečnosti Týdny 26-78 N (%) |
Celá studie týdny 0-78 N (%) |
|
Počet hodnotitelných pacientů |
22 |
22 |
23 |
|
< 5% |
9 (41) |
6 (27) |
5 (22) |
|
> 5 % až < 10 % |
6 (27) |
8 (36) |
8 (35) |
|
> 10 % až < 15 % |
4 (18) |
3 (14) |
4 (17) |
|
> 15 % až < 20 % |
1 (5) |
4 (18) |
3 (13) |
|
> 20 % až < 25 % |
1 (5) |
0 |
1 (4) |
|
> 25 % |
1 (5) |
1 (5) |
2 (9) |
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lojuxta u jedné nebo více podskupin pediatrické populace s HoFH (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Absolutní perorální biologická dostupnost lomitapidu je 7 %. Absorpce není omezena prostupem léčiva přes střevní bariéru, ale je převážně ovlivněna extenzivním efektem prvního průchodu. Vrcholná plazmatická koncentrace lomitapidu byla dosažena za 4 až 8 hodin po podání perorální dávky. Farmakokinetika lomitapidu je přibližně proporcionální k dávce perorálních jednorázových dávek v terapeutickém rozmezí. U dávek vyšších než 60 mg je naznačena tendence k nelineární závislosti a tyto dávky nejsou doporučeny.
Při podání více dávek se hodnoty Cmax a AUC zvýšily přibližně proporcionálně k dávce lomitapidu. Hodnoty Cmax a AUC se zvýšily jak po vysoce tučném jídle (77 %, respektive 58 %), tak po nízkotučném jídle (70 %, respektive 28 %). Akumulace lomitapidu v plazmě byla konzistentní s hodnotou předpokládanou na základě jedné dávky po podávání jedné perorální dávky denně nad 25 mg po dobu až 4 týdnů. Interindividuální variabilita hodnoty AUC lomitapidu byla přibližně 50 %.
V ustáleném stavu byla akumulace lomitapidu 2,7 při 25 mg a 3,9 při 50 mg.
Distribuce
Po intravenózním podání byl distribuční objem lomitapidu velký (průměr = 1 200 litrů) i přes vysoký stupeň (> 99,8 %) vazby na plazmatické proteiny. Ve studiích na zvířatech se lomitapid vysoce (200-násobně) koncentroval v játrech.
Biotransformace
Lomitapid se extenzivně metabolizuje, převážně cestou CYP3A4. Izoformy CYP 2E1, 1A2, 2B6, 2C8 a 2C19 se účastní méně a izoformy 2D6 a 2C9 se metabolismu lomitapidu neúčastní.
Eliminace
Po podání dávky radioaktivně značeného perorálního roztoku zdravým subjektům bylo 93 % podané dávky vyloučeno močí a stolicí. Přibližně 33 % radioaktivity bylo vyloučeno do moči jako metabolity. Zbytek byl vyloučen stolicí, primárně jako oxidované metabolity. Eliminační poločas lomitapidu byl přibližně 29 hodin.
Zvláštní populace
Údaje ze stěžejní klinické studie byly analyzovány s ohledem na vliv možných souvisejících proměnných na expozici lomitapidu. Z vyšetřovaných parametrů (rasa, index tělesné hmotnosti (BMI), hmotnost, věk) může být jako možná související proměnná klasifikován pouze BMI.
Věk a pohlaví
Věk (18-64 let) ani pohlaví nemají na farmakokinetiku lomitapidu klinicky významný vliv.
Rasa
U bělochů nebo latinskoamerické populace není nutná úprava dávky. K dispozici není dostatek informací pro stanovení, zda přípravek Lojuxta vyžaduje úpravu dávky u dalších ras. Protože se ale léčivý přípravek podává způsobem postupného zvyšování dávky podle individuální bezpečnosti a snášenlivosti pacienta, úprava dávkovacího režimu na základě rasy není doporučena.
V populaci s poškozením funkce ledvin byl lomitapid studován pouze u pacientů s terminálním onemocněním ledvin (ESRD). Farmakokinetická studie u pacientů s ESRD podstupujících dialýzu ukázala 36% vzestup průměrné koncentrace lomitapidu v plazmě ve srovnání s párovanými zdravými kontrolami. Koncový poločas lomitapidu nebyl ovlivněn.
Jaterní insuficience
Pro hodnocení farmakokinetiky 60 mg lomitapidu u zdravých dobrovolníků s normálními jaterními funkcemi ve srovnání s pacienty s mírným postižením funkce jater (třída A dle Child-Pugha) a středně těžkým postižením funkce jater (třída B dle Child-Pugha) byla provedena otevřená studie s jednou dávkou. Hodnoty AUC a Cmax lomitapidu u pacientů se středně těžkým postižením funkce jater byly o 164 %, respektive 361 % vyšší než u zdravých dobrovolníků. Hodnoty AUC a Cmax lomitapidu u pacientů s mírným postižením funkce jater byly o 47 %, respektive 4 % vyšší než u zdravých dobrovolníků. U pacientů s těžkým postižením funkce jater (skóre dle Child-Pugha 10-15) nebyl přípravek Lojuxta studován.
Pediatrická populace
U dětí mladších 18 let nebyl přípravek Lojuxta studován.
Starší populace
U pacientů starších 65 let nebyl přípravek Lojuxta studován.
5.3 Předklinické údaje vztahující se k bezpečnosti
Základním zjištěním toxikologických studií léčiva s opakovanou perorální dávkou u hlodavců a psů byla akumulace lipidů v tenkém střevě a/nebo v játrech spojená s poklesem sérových hladin cholesterolu a/nebo triglyceridů. Tyto změny jsou vzhledem k mechanismu působení lomitapidu sekundární. Další změny související s játry ve studiích toxicity s opakovanou dávkou u potkanů a psů zahrnovaly zvýšené hladiny sérové aminotransferázy, subakutní zánět (pouze u potkanů) a nekrózu jednotlivých buněk. V jednoleté studii s opakovanou dávkou u psů se v játrech nevyskytovaly mikroskopické změny, ačkoli u fen byla minimálně zvýšená sérová hladina AST.
U hlodavců byla pozorována plicní histiocytóza. U psů byl pozorován pokles parametrů erytrocytů i poikilocytóza a/nebo anizocytóza. U psů byla v šestiměsíční studii při 205násobku lidské expozice (AUC) 60 mg pozorována testikulární toxicita. V roční studii u psů nebyly při 64násobku lidské expozice 60 mg pozorovány nežádoucí účinky na varlata.
Ve studii nutriční karcinogenity u myší byl lomitapid podáván po dobu až 104 týdnů v dávkách v rozmezí 0,3 až 45 mg/kg/den. Při dávkách > 1,5 mg/kg/den u samců (> 2násobek lidské expozice při 60 mg denně na základě AUC) a > 7,5 mg/kg/den u samic (> 9násobek lidské expozice při 60 mg na základě AUC) došlo ke statisticky významnému vzestupu incidence adenomu i karcinomu jater. Incidence karcinomu tenkého střeva a/nebo kombinovaného adenomu a karcinomu (u myší vzácné nádory) byla významně zvýšena při dávkách > 15 mg/kg/den u samců (> 26násobek lidské expozice při 60 mg na základě AUC) a 15 mg/kg/den u samic (22násobek lidské expozice při 60 mg na základě AUC).
Ve studii perorální kancerogenity u potkanů byl lomitapid podáván po dobu až 99 týdnů v dávkách až 7,5 mg/kg/den u samců a 2,0 mg/kg/den u samic. U samců i samic byla pozorována fokální jaterní fibróza a pouze u samců byla pozorována cystická degenerace jater. U samců s vysokou dávkou byla pozorována zvýšená incidence pankreatického acinárního adenomu při expozici 6krát vyšší než u lidí při 60 mg na základě AUC.
V panelu studií in vitro a in vivo nebyl lomitapid mutagenní ani genotoxický.
Lomitapid neměl účinek na reprodukční funkce u samic potkanů v dávkách až 1 mg/kg nebo samců potkanů v dávkách až 5 mg/kg. Systémová expozice lomitapidu v těchto dávkách byla odhadována na čtyřnásobek (samice) až pětinásobek (samci) expozice u lidí při 60 mg na základě AUC.
Lomitapid byl u potkanů teratogenní při současné absenci mateřské toxicity při expozici (AUC) odhadované na dvojnásobek lidské expozice při 60 mg. U králíků se nevyskytly známky embryofetální toxicity při trojnásobku maximální doporučené denní dávky u lidí (MRHD) 60 mg na základě plochy tělesného povrchu. Embryofetální toxicita při absenci mateřské toxicity byla pozorována u králíků při > 6,5násobku MRHD. U fretek byl lomitapid toxický pro matku i teratogenní při dávkách < 1x MRHD.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky
předbobtnalý škrob (kukuřičný) sodná sůl karboxymethylškrobu mikrokrystalická celulóza monohydrát laktózy koloidní bezvodý oxid křemičitý magnesium-stearát
Obal tobolky želatina
oxid titaničitý (E171) červený oxid železitý (E172)
Tiskařský inkoust šelak
černý oxid železitý (E172) propylenglykol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Uchovávejte v dobře uzavřené lahvičce, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Lahvička z polyetylenu o vysoké hustotě (HDPE) vybavená vložkou z polyesteru / hliníkové fólie / kartonu a s polypropylenovým šroubovacím uzávěrem.
Velikost balení:
28 tobolek
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Aegerion Pharmaceuticals Ltd Lakeside House 1 Furzeground Way Stockley Park East Uxbridge UB11 1BD Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/851/001
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. června 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
^ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Lojuxta 10 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje lomitapidum 10 mg ve formě lomitapidum mesilas Pomocná látka se známým účinkem
Jedna tvrdá tobolka obsahuje 140,23 mg laktózy (ve formě monohydrátu) (viz bod 4.4). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Tvrdá tobolka s oranžovou svrchní částí / bílou spodní částí o délce 19,4 mm, potištěná černým inkoustem s textem „10 mg“ na spodní části a „A733“ na svrchní části tobolky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Lojuxta je indikován jako podpůrný přípravek užívaný spolu s dietou s nízkým obsahem tuků a dalšími léčivými přípravky na snížení hladiny lipidů spolu s aferézou lipoproteinů o nízké hustotě (LDL) či bez ní u dospělých pacientů s homozygotní formou familiární hypercholesterolémie (HoFH).
Tam, kde je to možné, by měla být homozygotní forma familiární hypercholesterolémie potvrzena geneticky. Je nutné vyloučit jiné formy primární hyperlipoproteinémie a sekundární příčiny hypercholesterolémie (např. nefrotický syndrom, hypotyreózu).
4.2 Dávkování a způsob podání
Léčbu přípravkem Lojuxta by měl zahajovat a sledovat lékař, který má zkušenosti s léčbou poruch lipidů.
Dávkování
Doporučená zahajovací dávka přípravku je 5 mg jednou denně. Po 2 týdnech může být dávka na základě přijatelné bezpečnosti a snášenlivosti zvýšena na 10 mg a poté v minimálně čtyřtýdenních intervalech na 20 mg, 40 mg a maximální doporučenou dávku 60 mg (viz bod 4.8).
Dávka by měla být zvyšována postupně, aby se minimalizovala incidence a závažnost gastrointestinálních nežádoucích účinků a zvýšení aminotransferáz.
Podávání se stravou může u přípravku Lojuxta zvyšovat expozici. Přípravek Lojuxta by se měl užívat nalačno, minimálně dvě hodiny po večeři, protože tuk obsažený v nedávno požitém jídle může mít negativní vliv na gastrointestinální snášenlivost.
Výskyt a závažnost gastrointestinálních nežádoucích účinků souvisejících s užíváním přípravku Lojuxta se snižují při dodržování diety s nízkým obsahem tuků. Pacienti by před zahájením léčby přípravkem Lojuxta měli držet dietu, ve které je méně než 20 % energie dodáváno z tuků, a během léčby by v této dietě měli pokračovat. Mělo by být zajištěno nutriční poradenství.
Pacienti by se měli vyvarovat konzumace grapefruitového džusu (viz body 4.4 a 4.5).
U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s atorvastatinem použijte jednu z těchto variant:
• Oddělte podání obou přípravků intervalem 12 hodin NEBO
• Snižte dávku přípravku Lojuxta o polovinu.
Pacienti užívající 5 mg by měli zůstat na této dávce.
Následně lze provést opatrnou titraci podle reakce hladiny LDL-C a bezpečnosti /snášenlivosti.
Po ukončení léčby atorvastatinem zvyšte dávku přípravku Lojuxta titrací podle hladiny LDL-C a bezpečnosti/snášenlivosti.U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s jakýmkoliv dalším slabým inhibitorem CYP3A4 oddělte podání obou přípravků intervalem 12 hodin.
Omezení maximální dávky přípravku Lojuxta zvažte podle požadované odpovědi LDL-C.
Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta.
Na základě pozorování snížených hladin esenciálních mastných kyselin a vitamínu E v klinických studiích by pacienti po dobu léčby přípravkem Lojuxta měli také užívat denně nutriční doplňky zajišťující příjem 400 IU vitamínu E a přibližně 200 mg kyseliny linolové, 110 mg kyseliny eikosapentaenové (EPA), 210 mg kyseliny alfa-linolenové (ALA) a 80 mg kyseliny dokosahexaenové (DHA).
Starší populace
Zkušenosti s přípravkem Lojuxta jsou u pacientů ve věku 65 let a starších omezené. Proto je u těchto pacientů zapotřebí zvláštní obezřetnosti.
Protože doporučený režim dávkování zahrnuje zahájení na spodní hranici dávkovacího rozmezí a opatrné navyšování dávky podle individuální snášenlivosti pacienta, nedoporučuje se ve starším věku žádná úprava dávkovacího schématu.
Porucha jater
Přípravek Lojuxta je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater včetně pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí (viz bod 5.2).
U pacientů s mírnou poruchou funkce jater (třída A dle Child-Pugha) by neměla být překračována dávka 40 mg denně.
Porucha ledvin
U pacientů v terminálním stádiu onemocnění ledvin léčených dialýzou by neměla být překračována dávka 40 mg denně (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Lojuxta u dětí mladších 18 let nebyla stanovena, a použití tohoto léčivého přípravku u dětí se proto nedoporučuje. Nejsou dostupné žádné údaje.
Způsob podání
Perorální podání.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Pacienti se středně těžkou nebo těžkou poruchou funkce jater a pacienti s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí.
• Pacienti se známým významným nebo chronickým střevním onemocněním, např. zánětlivým onemocněním střeva nebo malabsorpcí.
• Souběžné podávání více než 40 mg simvastatinu (viz bod 4.5).
• Souběžné použití přípravku Lojuxta se silnými nebo středně silnými inhibitory cytochromu P450 (CYP) 3A4 (např. antimykotiky azoly, jako např. intrakonazolem, flukonazolem, ketokonazolem, vorikonazolem a posakonazolem, makrolidovými antibiotiky, jako např. erytromycinem nebo klaritromycinem, ketolidovými antibiotiky, jako např. telitromycinem, inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem a antiarytmikem dronedaronem [viz bod 4.5]).
• Těhotenství (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Abnormality jaterních enzymů a sledování jater
Lomitapid může způsobit zvýšení hladiny alaninaminotransferázy [ALT] a aspartátaminotransferázy [AST] a jaterní steatózu. V jakém rozsahu jaterní steatóza související s lomitapidem podporuje zvýšení aminotransferáz, není známo. Ačkoli nebyly hlášeny případy jaterní dysfunkce (zvýšení aminotransferáz se zvýšením bilirubinu nebo mezinárodního normalizovaného poměru [INR] nebo jaterního selhání), existuje obava, že lomitapid může indukovat steatohepatitidu, která může za několik let progredovat do cirhózy. Není pravděpodobné, že by klinické studie na podporu bezpečnosti a účinnosti lomitapidu u HoFH byly schopné vzhledem k jejich rozsahu a době trvání tyto nežádoucí účinky detekovat.
S lomitapidem je spojeno zvýšení hladin aminotransferáz (ALT a/nebo AST) (viz bod 5.1). Nedocházelo k souběžnému nebo následnému klinicky významnému zvýšení sérového bilirubinu, INR nebo alkalické fosfatázy. Změny jaterních enzymů se nejčastěji vyskytují během navyšování dávky, ale mohou nastat kdykoli během léčby.
Sledování testů jaterních funkcí
Před zahájením léčby přípravkem Lojuxta změřte ALT, AST, alkalickou fosfatázu, celkový bilirubin, gama-glutamyltransferázu (gama-GT) a sérový albumin. Léčivý přípravek je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater a pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí. Jestliže jsou základní vyšetření jaterních testů abnormální, zvažte zahájení léčby léčivým přípravkem až poté, co hepatolog provede vhodná vyšetření a abnormality základních testů budou vysvětleny či vyřešeny.
V prvním roce měřte jaterní testy (minimálně ALT a AST) před každým zvýšením dávky nebo jednou měsíčně podle toho, co nastane dříve. Po prvním roce provádějte tyto testy nejméně jednou za 3 měsíce a před každým zvýšením dávky. Jestliže pozorujete zvýšení hladiny aminotransferáz, dávku přípravku Lojuxta snižte, a při přetrvávajícím nebo klinicky významném zvýšení ukončete léčbu (konkrétní doporučení uvádí Tabulka 1).
Modifikace dávky na základě zvýšené hladiny jatemích aminotransferáz
Tabulka 1 je shrnutím doporučení pro úpravu dávky a sledování pacientů, u kterých dojde během léčby přípravkem Lojuxta ke zvýšení hladiny aminotransferáz.
Tabulka 1: Úprava dávky a sledování pacientů se zvýšenými hladinami aminotransferáz
|
ALT nebo AST |
Doporučení týkající se léčby a sledování* |
|
>3x a <5x horní mez normálního rozsahu (ULN) |
• Zvýšení potvrďte opakovaným měřením v rámci jednoho týdne. • Jestliže se zvýšení potvrdí, snižte dávku a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). • Testy opakujte každý týden, a jestliže budou přítomny známky abnormálních jaterních funkcí (zvýšení bilirubinu či INR), jestliže aminotransferázy stoupnou nad 5x ULN nebo jestliže hladiny aminotransferáz během přibližně 4 týdnů neklesnou pod 3x ULN, přerušte léčbu. Pacienty s přetrvávajícím zvýšením hladin aminotransferáz >3x ULN odešlete k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, zvažte snížení dávky a častější sledování jaterních testů. |
|
>5x ULN |
• Přerušte léčbu a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). Jestliže během přibližně 4 týdnů hladiny aminotransferáz neklesnou pod 3x ULN, odešlete pacienta k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, snižte dávku a častěji sledujte jaterní testy. |
*Doporučení založené na ULNpřibližně 30-40 mezinárodních jednotek/l.
Jestliže je zvýšení aminotransferáz provázeno klinickými symptomy poškození jater (např. nevolností, zvracením, bolestí břicha, horečkou, žloutenkou, letargií, příznaky podobnými chřipce), vzestupem hladin bilirubinu na >2x ULN nebo aktivním onemocněním jater, přerušte léčbu přípravkem Lojuxta a odešlete pacienta k hepatologovi na další vyšetření.
Opětovné nasazení léčby lze zvážit tehdy, pokud přínosy převyšují rizika související s potenciálním onemocněním jater.
Jaterní steatóza a riziko progresivního onemocnění jater
Většina léčených pacientů vykazuje vzestup obsahu tuku v játrech, což je ve shodě s mechanismem působení lomitapidu. V otevřené studii fáze 3 došlo u 18 z 23 pacientů s HoFH k vývoji jaterní steatózy (obsah tuku v játrech > 5,56 %) dle měření nukleární magnetickou rezonanční spektroskopií (MRS) (viz bod 5.1). Medián absolutního vzestupu jaterního tuku byl dle měření pomocí MRS jak po 26 týdnech, tak po 78 týdnech léčby 6 % oproti 1 % při zahájení studie. Jaterní steatóza je rizikový faktor progresivního onemocnění jater včetně steatohepatitidy a cirhózy. Dlouhodobé následky jaterní steatózy související s léčbou přípravkem Lojuxta nejsou známy. Klinické údaje naznačují, že nahromadění tuku v játrech je po ukončení léčby přípravkem Lojuxta reverzibilní, ale není známo, zda přetrvávají histologické změny, zejména po dlouhodobém užívání.
Sledování důkazů přítomnosti progresivního onemocnění jater
Při zahájení léčby a dále každý rok by se měl provádět pravidelný screening steatohepatitidy/fibrózy s použitím následujících zobrazovacích metod a hodnocení biomarkerů:
• Zobrazení elasticity tkáně, např. fibrosken, ARFI (Acoustic Radiation Force Impulse) nebo elastografie s použitím magnetické rezonance (MR)
• Gama-GT a sérový albumin pro detekci možného poškození jater
• Minimálně jeden marker z každé z následujících kategorií:
• Vysoce senzitivní C-reaktivní protein (hs-CRP), rychlost sedimentace erytrocytů (ESR), CK-18 fragment, NashTest (zánět jater)
• Rozšířený panel pro detekci jaterní fibrózy (ELF), fibrometr, poměr AST/ALT, skóre fib-4, fibrotest (jaterní fibróza)
Provedení těchto testů a jejich interpretace by měly zahrnovat spolupráci ošetřujícího lékaře s hepatologem. U pacientů s výsledky naznačujícími přítomnost steatohepatitidy nebo fibrózy by měla být zvážena biopsie jater.
Jestliže má pacient biopticky potvrzenou steatohepatitidu nebo fibrózu, měl by být znovu zhodnocen poměr přínosů a rizik a léčba by měla být ukončena, je-li to nutné.
Souběžné užívání inhibitorů CYP3A4
Zdá se, že lomitapid je citlivým substrátem pro metabolismus cestou CYP3A4. Inhibitory CYP3A4 zvyšují expozici lomitapidu, přičemž silné inhibitory zvyšují expozici přibližně 27krát. Souběžné užití středně silných nebo silných inhibitorů CYP3A4 a přípravku Lojuxta je kontraindikováno (viz bod 4.3). V klinických studiích s lomitapidem se u jednoho pacienta s HoFH během několika dní po zahájení užívání silného inhibitoru CYP3A4 klaritromycinu vyvinuly výrazně zvýšené hladiny aminotransferáz (ALT 24x ULN, AST 13x ULN). Jestliže se nelze vyvarovat léčby středně silnými nebo silnými inhibitory CYP3A4, mělo by být užívání přípravku Lojuxta v průběhu léčby ukončeno.
Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta má být podávána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4.
Souběžné užívání induktorů CYP3A4
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. Induktory CYP3A4 vykazují svůj účinek v závislosti na čase a dosažení maximálního účinku může trvat minimálně 2 týdny od zahájení léčby. Naopak při ukončení léčby může trvat minimálně 2 týdny, než indukce CYP3A4 odezní.
Očekává se, že souběžné podávání induktoru CYP3A4 snižuje účinek přípravku Lojuxta. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinu, pioglitazonu, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Použití třezalky tečkované současně s přípravkem Lojuxta je nutné se vyvarovat.
Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. Při vysazení induktoru CYP3A4 je nutné zvážit možnost zvýšené expozice a může být nutné snížit dávku přípravku Lojuxta.
Souběžné použití s inhibitory HMG-CoA reduktázv (..statinv")
Lomitapid zvyšuje plazmatické koncentrace statinů. U pacientů, kteří dostávají přípravek Lojuxta jako přídatnou terapii vedle statinů, by měly být sledovány nežádoucí účinky spojené s použitím vysokých dávek statinů. Statiny příležitostně mohou vyvolat myopatii. Ve vzácných případech může mít myopatie formu rhabdomyolýzy s akutním renálním selháním sekundárně způsobeným myoglobinurií nebo bez renálního selhání a může mít fatální následky. Všichni pacienti užívající přípravek Lojuxta vedle statinů by měli být informování o možném riziku myopatie a měli by být informováni, aby okamžitě hlásili nevysvětlitelnou bolest, citlivost nebo slabost svalů. S přípravkem Lojuxta by se neměly používat dávky simvastatinu > 40 mg (viz bod 4.3).
Grapefruitový džus
Ze stravy pacientů je při léčbě přípravkem Lojuxta nutno vyloučit grapefruitový džus.
Riziko supraterapeutické nebo subterapeutické antikoagulace u antikoagulačních přípravků na bázi kumarinu
Lomitapid zvyšuje plazmatické koncentrace warfarinu. Zvýšení dávky přípravku Lojuxta může vést k supraterapeutické antikoagulaci a snížení dávky může vést k subterapeutické antikoagulaci.
U jednoho z pěti pacientů užívajících souběžně warfarin přispěla obtížná kontrola INR k předčasnému ukončení studie fáze 3. Pacienti užívající warfarin by měli podstupovat pravidelné sledování INR, zejména po jakýchkoli změnách dávkování přípravku Lojuxta. Dávka warfarinu by měla být upravena dle klinické indikace.
Požívání alkoholu
Alkohol může zvyšovat hladinu tuku v játrech a může vést k indukci nebo exacerbaci poškození jater. Ve studii fáze 3 udávali 3 ze 4 pacientů se zvýšením ALT >5x ULN konzumaci alkoholu překračující limit doporučený v protokolu. Požívání alkoholu během léčby přípravkem Lojuxta se nedoporučuje.
Hepatotoxické přípravky
Opatrnosti je třeba při použití přípravku Lojuxta s dalšími léčivými přípravky se známým hepatotoxickým potenciálem, např. izotretinoinem, amiodaronem, acetaminofenem (>4 g/den po >3 dny/týden), metotrexátem, tetracyklinem a tamoxifenem. Účinek souběžného podávání přípravku Lojuxta s dalšími hepatotoxickými léčivými přípravky není znám. Může být nutné častější sledování jaterních testů.
Snížená absorpce vitamínů rozpustných v tucích a mastných kyselin v séru
Vzhledem k mechanismu působení lomitapidu v tenkém střevě může lomitapid snižovat absorpci živin rozpustných v tucích. Ve studii fáze 3 byly pacientům denně poskytovány nutriční doplňky s vitamínem E, kyselinou linolovou, ALA, EPA a DHA. V této studii se úroveň mediánu sérového vitamínu E, ALA, kyseliny linolové, EPA, DHA a kyseliny arachidonové mezi výchozím bodem studie a týdnem 26 snížila, ale zůstala nad spodním limitem referenčního rozmezí. Nežádoucí klinické důsledky tohoto snížení při léčbě lomitapidem nebyly pozorovány po dobu až 78 týdnů. Pacienti léčení přípravkem Lojuxta by měli denně užívat doplňky, které obsahují 400 mezinárodních jednotek vitamínu E a přibližně 200 mg kyseliny linolové, 210 mg ALA, 110 mg EPA a 80 mg DHA.
Antikoncepční opatření u žen ve fertilním věku.
Před zahájením léčby u žen ve fertilním věku by mělo být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z důvodu průjmu a/nebo zvracení (viz bod 4.5). Perorální antikoncepční přípravky na bázi estrogenů jsou slabými inhibitory CYP3A4 (viz bod 4.2).
Pacientky by měly být informovány, že v případě otěhotnění musejí neprodleně informovat svého lékaře a ukončit užívání přípravku Lojuxta (viz bod 4.6).
Laktóza
Přípravek Lojuxta obsahuje laktózu, pacienti se vzácnými hereditárními problémy jako jsou intolerance galaktózy, vrozený deficit laktázy nebo glukózová-galaktózová malabsorpce by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
Tabulka 2: Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
Inhibitory CYP3A4 |
Při současném podávání lomitapidu v dávce 60 mg s ketokonazolem, silným inhibitorem CYP3A4, v dávce 200 mg dvakrát denně se zvýšila AUC lomitapidu přibližně 27krát a Cmax přibližně 15krát. Interakce mezi středně silnými inhibitory CYP3A4 a lomitapidem nebyly studovány. Předpokládá se, že středně silné inhibitory CYP3A4 mají významný vliv na farmakokinetiku lomitapidu. Očekává se, že souběžné použití středně silných inhibitorů CYP3A4 zvyšuje expozici lomitapidu 4-10krát podle výsledků studie se silným inhibitorem CYP3A4 podle historických údajů pro zkušební model CYP3A4 midazolam. Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při podávání lomitapidu 20 mg současně s atorvastatinem (tj. |
Použití silných nebo středně silných inhibitorů CYP3A4 spolu s přípravkem Lojuxta je kontraindikováno. Jestliže se nelze vyvarovat léčby antimykotiky azoly (např. itrakonazolem, ketokonazolem, flukonazolem, vorikonazolem, posakonazolem), antiarytmikem dronedaronem, makrolidovými antibiotiky (např. erytromycinem, klaritromcinem), ketolidovými antibiotiky (např. telitromycinem), inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem, měla by být během této léčby léčba přípravkem Lojuxta pozastavena (viz body 4.3 a 4.4). Grapefruitový džus je středně silným inhibitorem CYP3A4 a má se za to, že podstatně zvyšuje expozici lomitapidu. Pacienti užívající přípravek Lojuxta by se měli vyvarovat konzumace grapefruitového džusu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta musí být užívána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4. Příklady slabých inhibitorů CYP3A4 zahrnují: alprazolam, |
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
slabým inhibitorem CYP3A4) se AUC a Cmax lomitapidu zvýšily přibližně 2x. Pokud byl lomitapid podáván 12 hodin po atorvastatinu, žádné klinicky významné zvýšení expozice lomitapidu nebylo pozorováno. Při podávání lomitapidu 20 mg s ethinyl estradiolem/norgestimátem (tj. slabým inhibitorem CYP3A4), žádné klinicky významné zvýšení expozice lomitapidu nebylo pozorováno při užívání současném ani s intervalem 12 hodin. |
amiodaron, amlodipin, atorvastatin, azitromycin, bikalutamid, cilostazol, cimetidin, cyklosporin, klotrimazol, fluoxetin, fluvoxamin, fosaprepitant, ginkgo, vodilku kanadskou, izoniazid, ivakaftor, lacidipin, lapatinib, linagliptin, nilotinib, perorální antikoncepční přípravky obsahující estrogeny, pazopanib, mátový olej, propiverin, ranitidin, ranolazin, roxithromycin, hořké pomeranče, takrolimus, tikagrelor a tolvaptan. Tento seznam není úplný a předepisující lékaři by měli zkontrolovat preskripční informace o léčivech, která mají být podávána spolu s přípravkem Lojuxta s ohledem na možné interakce zprostředkované CYP3A4. Účinek podávání více než jednoho slabého inhibitoru CYP3A4 nebyl testován, ale očekává se, že účinek na expozici lomitapidu je větší než při současném podávání jednotlivých inhibitorů s lomitapidem. Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta. | |
|
Induktory CYP3A4 |
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. To by mohlo následně snížit účinek lomitapidu. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. |
Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinnu, pioglitazonu, třezalky tečkované, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. |
|
Sekvestranty žlučových kyselin |
Interakce lomitapidu se sekvestranty žlučových kyselin (pryskyřicemi, např. kolesevelamem a cholestyraminem) nebyly testovány. |
Protože sekvestranty žlučových kyselin mohou interferovat s absorpcí perorálních léčivých přípravků, měly by se užívat minimálně 4 hodiny před přípravkem Lojuxta nebo minimálně 4 hodiny po něm. |
Účinky lomitapidu na jiné léčivé přípravky
Inhibitory HMG-CoA reduktázy („statiny“): Lomitapid zvyšuje plazmatické koncentrace statinů. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním simvastatinu 40 mg se zvýšily u kyseliny simvastatinu hodnoty AUC o 68 % a Cmax o 57 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním atorvastatinu 20 mg se zvýšily u kyseliny atorvastatinu hodnoty AUC o 52 % a Cmax o 63 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním rosuvastatinu 20 mg se zvýšila hodnota Tmax rosuvastatinu z 1 na 4 hodiny, hodnota AUC se zvýšila o 32 % a hodnota Cmax zůstala nezměněná. Riziko myopatie u simvastatinu je závislé na dávce. Použití přípravku Lojuxta je u pacientů léčených vysokými dávkami statinů (> 40 mg) kontraindikováno (viz body 4.3 a 4.4).
Kumarinové antikoagulační přípravky: Podávání lomitapidu 60 mg do dosažení ustáleného stavu a po dobu 6 dní po warfarinu 10 mg zvýšilo hodnotu INR 1,26krát. Hodnota AUC pro R(+)-warfarin se zvýšila o 25 % a pro S(-)-warfarin o 30 %. Hodnota Cmax pro R(+)-warfarin se zvýšila o 14 % a pro S(-)-warfarin o 15 %. U pacientů užívajících souběžně kumariny (např. warfarin) a přípravek Lojuxta by měla být před zahájením užívání přípravku Lojuxta stanovena hodnota INR a tato hodnota by měla být pravidelně kontrolována, přičemž dávkování kumarinů by mělo být upraveno dle klinické potřeby (viz bod 4.4).
Fenofibrát, niacin a ezetimib: Při podávání lomitapidu do dosažení ustáleného stavu před podáním mikronizovaného fenofibrátu 145 mg, niacinu s prodlouženým uvolňováním 1000 mg nebo ezetimibu 10 mg nebyly pozorovány klinicky významné účinky působení kteréhokoli z těchto léčivých přípravků. Při souběžném podávání s přípravkem Lojuxta není nutná úprava dávky.
Perorální antikoncepce: Při podávání lomitapidu 50 mg do dosažení ustáleného stavu spolu s perorálním antikoncepčním přípravkem na bázi estrogenů nebyl pozorován klinicky ani statisticky významný vliv na farmakokinetiku složek perorální antikoncepce (ethinylestradiolu a 17-deacetylnorgestimátu, metabolitu norgestimátu). Neočekává se, že by lomitapid přímo ovlivňoval účinnost perorální antikoncepce na bázi estrogenů, nicméně průjem a/nebo zvracení mohou snížit absorpci hormonů. U případů prodlouženého nebo závažného průjmu a/nebo zvracení trvajících více než 2 dny by měla být po dobu 7 dnů po ústupu symptomů použita další antikoncepční metoda.
Substráty P-gp: Lomitapid in vitro inhibuje P-gp a může zvyšovat absorpci P-gp substrátů. Souběžné podávání přípravku Lojuxta s P-pg substráty (např. aliskirenem, abrisentanem, kolchicinem, dabigatran etexilátem, dixoginem, everolimem, fexofenadinem, imatinibem, lapatinibem, maravirokem, nilotinibem, posakonazolem, ranolazinem, saxagliptinem, sirolimem, sitagliptinem, talinololem, tolvaptanem, topotekanem) může zvýšit absorpci P-gp substrátů. Při souběžném použití s přípravkem Lojuxta by mělo být zváženo snížení dávky P-gp substrátu.
In vitro hodnocení lékových interakcí: Lomitapid inhibuje CYP3A4. Lomitapide neindukuje CYP 1A2, 3A4 ani 2B6 a neinhibuje CYP 1A2, 2B6, 2C9, 2C19, 2D6 ani 2E1. Lomitapid není P-gp substrátem, ale inhibuje P-gp. Lomitapid neinhibuje protein rezistence u karcinomu prsu (BCRP).
4.6 Fertilita, těhotenství a kojení
Přípravek Lojuxta je v těhotenství kontraindikován. Neexistují spolehlivé údaje o použití přípravku u těhotných žen. Studie na zvířatech prokázaly vývojovou toxicitu (teratogenitu, embryotoxicitu, viz bod 5.3). Možné riziko pro člověka je neznámé.
Použití u žen ve fertilním věku.
Před zahájením léčby u žen ve fertilním věku by mělo být vyloučeno těhotenství, mělo by být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z důvodu průjmu a/nebo zvracení. Dokud symptomy neustoupí, je nutno používat další antikoncepční metody (viz bod 4.5).
Kojení
Není známo, zda se lomitapid vylučuje do mateřského mléka. Vzhledem k potenciálu nežádoucích účinků zjištěnému na základě studií s lomitapidem na zvířatech (viz bod 5.3) je nutné rozhodnout, zda ukončit kojení nebo užívání léčivého přípravku, a to při zvážení významu léčivého přípravku pro matku.
Fertilita
U potkaních samců ani samic, kterým byl podáván lomitapid se systémovou expozicí (AUC) ve výši odhadovaného čtyř- až pětinásobku expozice u lidí při maximální doporučené dávce u člověka, nebyly pozorovány žádné nežádoucí účinky na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Lojuxta může mít mírný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejzávažnějšími nežádoucími účinky během léčby byly abnormality jaterních aminotransferáz (viz bod 4.4).
Nejčastějšími nežádoucími účinky byly gastrointestinální účinky. Nežádoucí gastrointestinální účinky byly hlášeny u 27 (93 %) z 29 pacientů klinické studie fáze 3. Průjem se objevil u 79 %, nevolnost u 65 %, dyspepsie u 38 % a zvracení u 34 % pacientů. Další účinky, které hlásilo minimálně 20 % pacientů, zahrnují bolest břicha, břišní dyskomfort, nadmutí břicha, zácpu a plynatost.
Nežádoucí gastrointestinální účinky se častěji objevovaly během fáze navyšování dávky ve studii a poklesly, jakmile pacienti dosáhli maximální tolerované dávky lomitapidu.
Gastrointestinální nežádoucí účinky se závažnou intenzitou byly hlášeny u 6 (21 %) z 29 pacientů v klinické studii fáze 3, přičemž nejčastějším účinkem byl průjem (4 pacienti, 14 %), zvracení (3 pacienti, 10 %) a bolest břicha, nadmutí a/nebo dyskomfort (2 pacienti, 7 %). Gastrointestinální účinky přispěly k časnému ukončení studie u 4 (14 %) pacientů.
Nejčastěji udávanými nežádoucími účinky se závažnou intenzitou byly průjem (4 subjekty, 14 %), zvracení (3 pacienti, 10 %) a nadmutí břicha a zvýšení ALT (pokaždé 2 subjekty, 7 %).
Přehled nežádoucích účinků v tabulce
Dle frekvence jsou nežádoucí účinky definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), málo časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), s neznámou frekvencí (z dostupných údajů nelze určit).
Tabulka 3 zahrnuje veškeré nežádoucí účinky hlášené u 35 pacientů léčených ve studii fáze 2 UP1001 a ve studii fáze 3 UP1002/AEGR-733-005 nebo v rozšířené studii AEGR-733-012.
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Časté |
Gastroenteritida |
|
Poruchy metabolismu a výživy |
Velmi časté |
Snížená chuť k jídlu |
|
Poruchy nervového systému |
Časté |
Závratě Bolest hlavy Migréna |
|
Gastrointestinální poruchy |
Velmi časté |
Břišní dyskomfort Bolest horní části břicha Plynatost Distenze břicha Zácpa |
|
Časté |
Gastritida Rektální tenesmy Aerofagie Nutkání na stolici Říhání Častá stolice Dilatace žaludku Onemocnění žaludku Gastroezofageální reflux Krvácení z hemoroidů Regurgitace | |
|
Poruchy jater a žlučových cest |
Časté |
Jaterní steatóza Hepatotoxicita Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Časté |
Ekchymóza Papuly Erytematózní vyrážka Xantomy |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Únava |
|
Vyšetření |
Velmi časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Snížení tělesné hmotnosti |
|
Časté |
Zvýšený mezinárodní normalizovaný poměr Zvýšená alkalická fosfatáza v krvi Snížení draslíku v krvi Snížený karoten Abnormální mezinárodní normalizovaný poměr Abnormální testy jaterních funkcí Prodloužený protrombinový čas Zvýšené transaminázy Snížený vitamín E Snížený vitamín K |
Tabulka 4 uvádí všechny nežádoucí účinky u subjektů, které dostávaly lomitapid v monoterapii (n=291), léčených ve studiích fáze 2 u subjektů se zvýšenou hladinou LDL-C (N=462).
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Méně časté |
Gastroenteritida Gastrointestinální infekce Chřipka Nazofaryngitida Sinusitida |
|
Poruchy krve a lymfatického systému |
Méně časté | |
|
Poruchy metabolismu a výživy |
Časté |
Snížená chuť k jídlu |
|
Méně časté |
Dehydratace Zvýšená chuť k jídlu | |
|
Poruchy nervového systému |
Méně časté |
Parestézie Ospalost |
|
Poruchy oka |
Méně časté |
Otok oka |
|
Poruchy ucha a labyrintu |
Méně časté |
Závrať |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté |
Léze hltanu Syndrom kašle horních cest dýchacích |
|
Gastrointestinální poruchy |
Velmi časté |
Plynatost |
|
Časté |
Bolest horní části břicha Distenze břicha Bolest břicha Zvracení Břišní dyskomfort Říhání Bolest v podbřišku Častá stolice | |
|
Méně časté |
Sucho v ústech Tvrdá stolice Gastroezofageální reflux Citlivost břicha Dyskomfort v epigastriu Dilatace žaludku Hemateméza Krvácení v dolní části gastrointestinálního traktu Refluxní ezofagitida | |
|
Poruchy jater a žlučových cest |
Méně časté |
Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Puchýře Suchá kůže Nadměrné pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté |
Svalové spasmy |
|
Méně časté |
Bolest kloubů Bolest svalů Bolest končetin Otok kloubu Svalové záškuby | |
|
Poruchy ledvin a močových cest |
Méně časté |
Hematurie |
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Celkové poruchy a reakce v místě |
Časté |
Únava |
|
podání |
Slabost | |
|
Méně časté |
Bolest na hrudi Mrazení Rychlý pocit nasycení Poruchy chůze Malátnost Horečka | |
|
Vyšetření |
Časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Zvýšení jaterních enzymů Abnormální testy jaterních funkcí Snížený počet neutrofilů Snížený počet leukocytů |
|
Méně časté |
Snížení tělesné hmotnosti Zvýšený bilirubin v krvi Zvýšená hladina gama-glutamyltransferázy Zvýšené procento neutrofilů Protein v moči Prodloužený protrombinový čas Abnormální testy plicních funkcí Zvýšený počet leukocytů |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování neexistuje specifická léčba. U hlodavců byla dobře snášena jednorázová perorální dávka lomitapidu >600krát vyšší než maximální doporučená dávka pro člověka (1 mg/kg). Maximální dávkou podanou lidským subjektům v klinických studiích bylo 200 mg ve formě jednorázové dávky, nevyskytly se žádné nežádoucí účinky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná léčiva ovlivňující hladinu lipidů, samotná. ATC kód: C10AX12
Mechanismus účinku
Lomitapid je selektivní inhibitor mikrozomálního transferového proteinu (MTP), intracelulárního proteinu přenášejícího lipidy, který se nachází v lumen endoplazmatického retikula a zodpovídá za vazbu a převod jednotlivých lipidových molekul mezi membránami. MTP hraje klíčovou roli při sestavování lipoproteinů obsahujících apo B v játrech a střevech. Inhibice MTP snižuje sekreci lipoproteinů a cirkulující koncentrace lipidů přenášených v lipoproteinech včetně cholesterolu a triglyceridů.
Klinická účinnost a bezpečnost
Účinnost a bezpečnost lomitapidu při současném podávání diety s nízkým obsahem tuků a dalších terapií snižujících lipidy u dospělých pacientů s HoFH hodnotila otevřená studie s jedním ramenem (UP1002/AEGR-733-005). Pacienti byli instruováni, aby dodržovali dietu s nízkým obsahem tuků (< 20 % kalorického příjmu z tuků) i terapii snižující lipidy, kterou měli při vstupu do studie, včetně aferézy, byla-li tato nutná, a to po dobu 6 týdnů předcházejících výchozímu bodu studie minimálně až do týdne 26. Dávka lomitapidu byla navyšována z 5 mg na individuálně stanovenou maximální tolerovanou dávku až 60 mg. Po týdnu 26 byl pacientům lomitapid ponechán za účelem zjištění účinků dlouhodobé léčby a bylo jim umožněno změnit základní terapii snižující lipidy. Studie zahrnovala celkem 78 týdnů léčby.
Do studie bylo zařazeno 29 pacientů, z nichž 23 dokončilo týden 78. Zařazeno bylo 16 mužů (55 %) a 13 žen (45 %) s průměrným věkem 30,7 let a ve věkovém rozmezí 18 až 55 let. Průměrná dávka lomitapidu v týdnu 26 byla 45 mg a v týdnu 78 byla 40 mg. V týdnu 26 byla průměrná procentuální změna hladiny LDL-C u populace „intention-to-treat“ (ITT) oproti výchozímu bodu -40 %
(p < 0,001). Průměrná procentuální změna oproti výchozímu bodu studie v týdnu 26 s použitím metody LOCF (Last Observation Carried Forward) pro každé hodnocení je uvedena na obrázku 1.
Obrázek 1: Průměrné procentuální změny hladiny LDL-C mezi vstupní hodnotou a týdnem
26 v hlavní studii účinnosti UP1002/AEGR-733-005 (primární cíl) s použitím LOCF pro každé hodnocení (n=29)
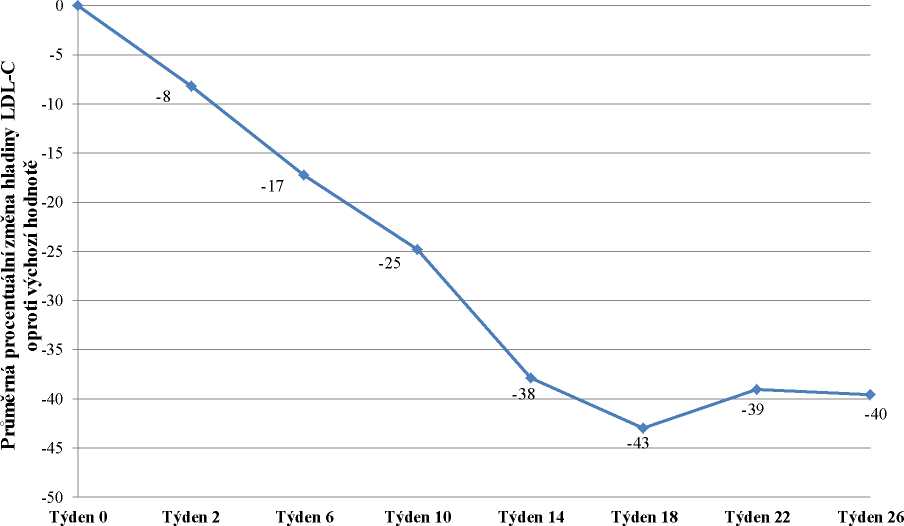
Týden studie
Změny lipidů i lipoproteinů v týdnu 26 a 78 při léčbě lomitapidem uvádí Tabulka 5.
Tabulka 5: Absolutní hodnoty a procentuální změny hladin lipidů a lipoproteinů mezi
výchozí hodnotou a týdny 26 a 78 (hlavní studie účinnosti UP1002/AEGR-733-005).
|
Parametr (jednotky) |
Výchozí bod |
Týden 26 / LOCF (n=29) |
Týden 78 (n=23) | ||||
|
Průměr (SD) |
Průměr (SD) |
% změna |
Hodnota p b |
Průměr (SD) |
% změna |
Hodnota p b | |
|
LDL-C, přímý (mg/dl) |
336 (114) |
190 (104) |
-40 |
< 0,001 |
210 (132) |
-38 |
< 0,001 |
|
Celkový cholesterol (TC) (mg/dl) |
430 (135) |
258 (118) |
-36 |
< 0,001 |
281 (149) |
-35 |
< 0,001 |
|
Apolipoprotein B (apo B) (mg/dl) |
259 (80) |
148 (74) |
-39 |
< 0,001 |
151 (89) |
-43 |
< 0,001 |
|
Triglyceridy (TG) (mg/dl)a |
92 |
57 |
-45 |
0,009 |
59 |
-42 |
0,012 |
|
Cholesterol z jiných lipoproteinů než o vysoké hustotě (non-HDL-C) (mg/dl) |
386 (132) |
217 (113) |
-40 |
< 0,001 |
239 (146) |
-39 |
< 0,001 |
|
Cholesterol z lipoproteinů o velmi nízké hustotě (VLDL-C) (mg/dl) |
21 (10) |
13 (9) |
-29 |
0,012 |
16 (15) |
-31 |
0,013 |
|
Lipoprotein (a) (Lp(a)) (nmol/l)a |
66 |
61 |
-13 |
0,094 |
72 |
-4 |
< 0,842 |
|
Cholesterol z lipoproteinů o vysoké hustotě (HDL-C) (mg/dl) |
44 (11) |
41 (13) |
-7 |
0,072 |
43 (12) |
-4,6 |
0,246 |
a Medián uvedený pro TG a Lp(a). Hodnota p je založena na průměrné procentuální změně b Hodnota p u střední procentuální změny oproti výchozímu bodu studie na základě párového t-testu
Jak v týdnu 26, tak v týdnu 78 došlo k významnému snížení hladin LDL-C, TC, apo B, TG, non-HDL-C a VLDL-C a změny u HDL-C vykazovaly v týdnu 26 trend směrem ke snížení a v týdnu 78 se vrátily k výchozím hodnotám.
Účinek přípravku Lojuxta na kardiovaskulární morbiditu a mortalitu nebyl stanoven.
Při zahájení studie užívalo 93 % pacientů statin, 76 % pacientů ezetimib, 10 % niacin, 3 % sekvestrant žlučových kyselin a 62 % pacientů bylo léčeno aferézou. U 15 z 23 (65 %) pacientů byla v týdnu 78 redukována jejich léčba na snížení lipidů, včetně plánovaných a neplánovaných snižování/přerušení. Aferéza byla přerušena u 3 ze 13 pacientů, kteří byli aferézou léčeni v týdnu 26, a u 3 pacientů byla frekvence při současném udržení nízkých hladin LDL-C i po týdnu 78 snížena. Klinický přínos omezení základní terapie snižující lipidy, včetně aferézy, není jistý.
Z 23 pacientů, kteří dokončili týden 26 studie, došlo u 19 (83 %) ke snížení LDL-C o > 25 %, přičemž 8 (35 %) z nich mělo v tomto časovém bodě LDL-C < 100 mg/dl a 1 pacient měl LDL-C < 70 mg/dl.
V této studii došlo u 10 pacientů ke zvýšení AST a/nebo ALT >3x ULN (viz Tabulka 6).
Nejvyšší výsledky testů jaterních funkcí po první dávce (hlavní studie účinnosti UP1002/AEGR-733-005)
|
Parametr/abnormalita |
N (%) |
|
ALT | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
6 (20,7) |
|
>5 až < 10 x ULN |
3 (10,3) |
|
>10 až < 20 x ULN |
1 (3,4) |
|
> 20 x ULN |
0 |
|
AST | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
5 (17,2) |
|
>5 až < 10 x ULN |
1 (3,4) |
|
>10 až < 20 x ULN |
0 |
|
> 20 x ULN |
0 |
Tabulka 6:
Zvýšení ALT a/nebo AST > 5x ULN bylo zvládnuto snížením dávky nebo dočasným pozastavením podávání lomitapidu a všichni pacienti byli schopni v léčbě studovaným léčivem pokračovat. Nebylo pozorováno klinicky významné zvýšení celkového bilirubinu ani alkalické fosfatázy. Jaterní tuk byl během klinické studie u všech dostupných pacientů prospektivně měřen s použitím MRS (Tabulka 7). Údaje od jedinců, u kterých byla po ukončení lomitapidu prováděna opakovaná měření, ukázaly, že nahromadění tuku v játrech je reverzibilní, ale není známo, zda nepřetrvávají histologické změny.
Tabulka 7: Maximální kategorické změny v % jaterního tuku (hlavní studie účinnosti
UP1002/AEGR-733-005)
|
Maximální absolutní nárůst % jaterního tuku |
Fáze účinnosti Týdny 0-26 N (%) |
Fáze bezpečnosti Týdny 26-78 N (%) |
Celá studie týdny 0-78 N (%) |
|
Počet hodnotitelných pacientů |
22 |
22 |
23 |
|
< 5% |
9 (41) |
6 (27) |
5 (22) |
|
> 5 % až < 10 % |
6 (27) |
8 (36) |
8 (35) |
|
> 10 % až < 15 % |
4 (18) |
3 (14) |
4 (17) |
|
> 15 % až < 20 % |
1 (5) |
4 (18) |
3 (13) |
|
> 20 % až < 25 % |
1 (5) |
0 |
1 (4) |
|
> 25 % |
1 (5) |
1 (5) |
2 (9) |
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lojuxta u jedné nebo více podskupin pediatrické populace s HoFH (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Absolutní perorální biologická dostupnost lomitapidu je 7 %. Absorpce není omezena prostupem léčiva přes střevní bariéru, ale je převážně ovlivněna extenzivním efektem prvního průchodu. Vrcholná plazmatická koncentrace lomitapidu byla dosažena za 4 až 8 hodin po podání perorální dávky. Farmakokinetika lomitapidu je přibližně proporcionální k dávce perorálních jednorázových dávek v terapeutickém rozmezí. U dávek vyšších než 60 mg je naznačena tendence k nelineární závislosti a tyto dávky nejsou doporučeny.
Při podání více dávek se hodnoty Cmax a AUC zvýšily přibližně proporcionálně k dávce lomitapidu. Hodnoty Cmax a AUC se zvýšily jak po vysoce tučném jídle (77 %, respektive 58 %), tak po nízkotučném jídle (70 %, respektive 28 %). Akumulace lomitapidu v plazmě byla konzistentní s hodnotou předpokládanou na základě jedné dávky po podávání jedné perorální dávky denně nad 25 mg po dobu až 4 týdnů. Interindividuální variabilita hodnoty AUC lomitapidu byla přibližně 50 %.
V ustáleném stavu byla akumulace lomitapidu 2,7 při 25 mg a 3,9 při 50 mg.
Distribuce
Po intravenózním podání byl distribuční objem lomitapidu velký (průměr = 1 200 litrů) i přes vysoký stupeň (> 99,8 %) vazby na plazmatické proteiny. Ve studiích na zvířatech se lomitapid vysoce (200násobně) koncentroval v játrech.
Biotransformace
Lomitapid se extenzivně metabolizuje, převážně cestou CYP3A4. Izoformy CYP 2E1, 1A2, 2B6, 2C8 a 2C19 se účastní méně a izoformy 2D6 a 2C9 se metabolismu lomitapidu neúčastní.
Eliminace
Po podání dávky radioaktivně značeného perorálního roztoku zdravým subjektům bylo 93 % podané dávky vyloučeno močí a stolicí. Přibližně 33 % radioaktivity bylo vyloučeno do moči jako metabolity. Zbytek byl vyloučen stolicí, primárně jako oxidované metabolity. Eliminační poločas lomitapidu byl přibližně 29 hodin.
Zvláštní populace
Údaje ze stěžejní klinické studie byly analyzovány s ohledem na vliv možných souvisejících proměnných na expozici lomitapidu. Z vyšetřovaných parametrů (rasa, index tělesné hmotnosti (BMI), hmotnost, věk) může být jako možná související proměnná klasifikován pouze BMI.
Věk a pohlaví
Věk (18-64 let) ani pohlaví nemají na farmakokinetiku lomitapidu klinicky významný vliv.
Rasa
U bělochů nebo latinskoamerické populace není nutná úprava dávky. K dispozici není dostatek informací pro stanovení, zda přípravek Lojuxta vyžaduje úpravu dávky u dalších ras. Protože se ale léčivý přípravek podává způsobem postupného zvyšování dávky podle individuální bezpečnosti a snášenlivosti pacienta, úprava dávkovacího režimu na základě rasy není doporučena.
V populaci s poškozením funkce ledvin byl lomitapid studován pouze u pacientů s terminálním onemocněním ledvin (ESRD). Farmakokinetická studie u pacientů s ESRD podstupujících dialýzu ukázala 36% vzestup průměrné koncentrace lomitapidu v plazmě ve srovnání s párovanými zdravými kontrolami. Koncový poločas lomitapidu nebyl ovlivněn.
Jaterní insuficience
Pro hodnocení farmakokinetiky 60 mg lomitapidu u zdravých dobrovolníků s normálními jaterními funkcemi ve srovnání s pacienty s mírným postižením funkce jater (třída A dle Child-Pugha) a středně těžkým postižením funkce jater (třída B dle Child-Pugha) byla provedena otevřená studie s jednou dávkou. Hodnoty AUC a Cmax lomitapidu u pacientů se středně těžkým postižením funkce jater byly o 164 %, respektive 361 % vyšší než u zdravých dobrovolníků. Hodnoty AUC a Cmax lomitapidu u pacientů s mírným postižením funkce jater byly o 47 %, respektive 4 % vyšší než u zdravých dobrovolníků. U pacientů s těžkým postižením funkce jater (skóre dle Child-Pugha 10-15) nebyl přípravek Lojuxta studován.
Pediatrická populace
U dětí mladších 18 let nebyl přípravek Lojuxta studován.
Starší populace
U pacientů starších 65 let nebyl přípravek Lojuxta studován.
5.3 Předklinické údaje vztahující se k bezpečnosti
Základním zjištěním toxikologických studií léčiva s opakovanou perorální dávkou u hlodavců a psů byla akumulace lipidů v tenkém střevě a/nebo v játrech spojená s poklesem sérových hladin cholesterolu a/nebo triglyceridů. Tyto změny jsou vzhledem k mechanismu působení lomitapidu sekundární. Další změny související s játry ve studiích toxicity s opakovanou dávkou u potkanů a psů zahrnovaly zvýšené hladiny sérové aminotransferázy, subakutní zánět (pouze u potkanů) a nekrózu jednotlivých buněk. V jednoleté studii s opakovanou dávkou u psů se v játrech nevyskytovaly mikroskopické změny, ačkoli u fen byla minimálně zvýšená sérová hladina AST.
U hlodavců byla pozorována plicní histiocytóza. U psů byl pozorován pokles parametrů erytrocytů i poikilocytóza a/nebo anizocytóza. U psů byla v šestiměsíční studii při 205násobku lidské expozice (AUC) 60 mg pozorována testikulární toxicita. V roční studii u psů nebyly při 64násobku lidské expozice 60 mg pozorovány nežádoucí účinky na varlata.
Ve studii nutriční karcinogenity u myší byl lomitapid podáván po dobu až 104 týdnů v dávkách v rozmezí 0,3 až 45 mg/kg/den. Při dávkách > 1,5 mg/kg/den u samců (> 2násobek lidské expozice při 60 mg denně na základě AUC) a > 7,5 mg/kg/den u samic (> 9násobek lidské expozice při 60 mg na základě AUC) došlo ke statisticky významnému vzestupu incidence adenomu i karcinomu jater. Incidence karcinomu tenkého střeva a/nebo kombinovaného adenomu a karcinomu (u myší vzácné nádory) byla významně zvýšena při dávkách > 15 mg/kg/den u samců (> 26násobek lidské expozice při 60 mg na základě AUC) a 15 mg/kg/den u samic (22násobek lidské expozice při 60 mg na základě AUC).
Ve studii perorální kancerogenity u potkanů byl lomitapid podáván po dobu až 99 týdnů v dávkách až
7,5 mg/kg/den u samců a 2,0 mg/kg/den u samic. U samců i samic byla pozorována fokální jaterní fibróza a pouze u samců byla pozorována cystická degenerace jater. U samců s vysokou dávkou byla pozorována zvýšená incidence pankreatického acinárního adenomu při expozici 6krát vyšší než u lidí při 60 mg na základě AUC.
V panelu studií in vitro a in vivo nebyl lomitapid mutagenní ani genotoxický.
Lomitapid neměl účinek na reprodukční funkce u samic potkanů v dávkách až 1 mg/kg nebo samců potkanů v dávkách až 5 mg/kg. Systémová expozice lomitapidu v těchto dávkách byla odhadována na čtyřnásobek (samice) až pětinásobek (samci) expozice u lidí při 60 mg na základě AUC.
Lomitapid byl u potkanů teratogenní při současné absenci mateřské toxicity při expozici (AUC) odhadované na dvojnásobek lidské expozice při 60 mg. U králíků se nevyskytly známky embryofetální toxicity při trojnásobku maximální doporučené denní dávky u lidí (MRHD) 60 mg na základě plochy tělesného povrchu. Embryofetální toxicita při absenci mateřské toxicity byla pozorována u králíků při > 6,5násobku MRHD. U fretek byl lomitapid toxický pro matku i teratogenní při dávkách < 1x MRHD.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky
předbobtnalý škrob (kukuřičný) sodná sůl karboxymethylškrobu mikrokrystalická celulóza monohydrát laktózy koloidní bezvodý oxid křemičitý magnesium-stearát
Obal tobolky želatina
oxid titaničitý (E171) červený oxid železitý (E172)
Tiskařský inkoust šelak
černý oxid železitý (E172) propylenglykol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Uchovávejte v dobře uzavřené lahvičce, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Lahvička z polyetylenu o vysoké hustotě (HDPE) vybavená vložkou z polyesteru / hliníkové fólie / kartonu a s polypropylenovým šroubovacím uzávěrem.
Velikost balení:
28 tobolek
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Aegerion Pharmaceuticals Ltd Lakeside House 1 Furzeground Way Stockley Park East Uxbridge UB11 1BD Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/851/002
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. června 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
^ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Lojuxta 20 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje lomitapidum 20 mg ve formě lomitapidum mesilas. Pomocná látka se známým účinkem
Jedna tvrdá tobolka obsahuje 129,89 mg laktózy (ve formě monohydrátu) (viz bod 4.4). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Tvrdá tobolka s bílou svrchní částí / bílou spodní částí o délce 19,4 mm, potištěná černým inkoustem s textem „20 mg“ na spodní části a „A733“ na svrchní části tobolky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Lojuxta je indikován jako podpůrný přípravek užívaný spolu s dietou s nízkým obsahem tuků a dalšími léčivými přípravky na snížení hladiny lipidů spolu s aferézou lipoproteinů o nízké hustotě (LDL) či bez ní u dospělých pacientů s homozygotní formou familiární hypercholesterolémie (HoFH).
Tam, kde je to možné, by měla být homozygotní forma familiární hypercholesterolémie potvrzena geneticky. Je nutné vyloučit jiné formy primární hyperlipoproteinémie a sekundární příčiny hypercholesterolémie (např. nefrotický syndrom, hypotyreózu).
4.2 Dávkování a způsob podání
Léčbu přípravkem Lojuxta by měl zahajovat a sledovat lékař, který má zkušenosti s léčbou poruch lipidů.
Dávkování
Doporučená zahajovací dávka přípravku je 5 mg jednou denně. Po 2 týdnech může být dávka na základě přijatelné bezpečnosti a snášenlivosti zvýšena na 10 mg a poté v minimálně čtyřtýdenních intervalech na 20 mg, 40 mg a maximální doporučenou dávku 60 mg (viz bod 4.8).
Dávka by měla být zvyšována postupně, aby se minimalizovala incidence a závažnost gastrointestinálních nežádoucích účinků a zvýšení aminotransferáz.
Podávání se stravou může u přípravku Lojuxta zvyšovat expozici. Přípravek Lojuxta by se měl užívat nalačno, minimálně dvě hodiny po večeři, protože tuk obsažený v nedávno požitém jídle může mít negativní vliv na gastrointestinální snášenlivost.
Výskyt a závažnost gastrointestinálních nežádoucích účinků souvisejících s užíváním přípravku Lojuxta se snižují při dodržování diety s nízkým obsahem tuků. Pacienti by před zahájením léčby přípravkem Lojuxta měli držet dietu, ve které je méně než 20 % energie dodáváno z tuků, a během léčby by v této dietě měli pokračovat. Mělo by být zajištěno nutriční poradenství.
Pacienti by se měli vyvarovat konzumace grapefruitového džusu (viz body 4.4 a 4.5).
U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s atorvastatinem použijte jednu z těchto variant:
• Oddělte podání obou přípravků intervalem 12 hodin NEBO
• Snižte dávku přípravku Lojuxta o polovinu.
Pacienti užívající 5 mg by měli zůstat na této dávce.
Následně lze provést opatrnou titraci podle reakce hladiny LDL-C a bezpečnosti /snášenlivosti.
Po ukončení léčby atorvastatinem zvyšte dávku přípravku Lojuxta titrací podle hladiny LDL-C a bezpečnosti/snášenlivosti.
U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s jakýmkoliv dalším slabým inhibitorem CYP3A4 oddělte podání obou přípravků intervalem 12 hodin.
Omezení maximální dávky přípravku Lojuxta zvažte podle požadované odpovědi LDL-C.
Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta.
Na základě pozorování snížených hladin esenciálních mastných kyselin a vitamínu E v klinických studiích by pacienti po dobu léčby přípravkem Lojuxta měli také užívat denně nutriční doplňky zajišťující příjem 400 IU vitamínu E a přibližně 200 mg kyseliny linolové, 110 mg kyseliny eikosapentaenové (EPA), 210 mg kyseliny alfa-linolenové (ALA) a 80 mg kyseliny dokosahexaenové (DHA).
Starší populace
Zkušenosti s přípravkem Lojuxta jsou u pacientů ve věku 65 let a starších omezené. Proto je u těchto pacientů zapotřebí zvláštní obezřetnosti.
Protože doporučený režim dávkování zahrnuje zahájení na spodní hranici dávkovacího rozmezí a opatrné navyšování dávky podle individuální snášenlivosti pacienta, nedoporučuje se ve starším věku žádná úprava dávkovacího schématu.
Porucha jater
Přípravek Lojuxta je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater včetně pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí (viz bod 5.2).
U pacientů s mírnou poruchou funkce jater (třída A dle Child-Pugha) by neměla být překračována dávka 40 mg denně.
Porucha ledvin
U pacientů v terminálním stádiu onemocnění ledvin léčených dialýzou by neměla být překračována dávka 40 mg denně (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Lojuxta u dětí mladších 18 let nebyla stanovena, a použití tohoto léčivého přípravku u dětí se proto nedoporučuje. Nejsou dostupné žádné údaje.
Způsob podání
Perorální podání.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Pacienti se středně těžkou nebo těžkou poruchou funkce jater a pacienti s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí.
• Pacienti se známým významným nebo chronickým střevním onemocněním, např. zánětlivým onemocněním střeva nebo malabsorpcí.
• Souběžné podávání více než 40 mg simvastatinu (viz bod 4.5).
• Souběžné použití přípravku Lojuxta se silnými nebo středně silnými inhibitory cytochromu P450 (CYP) 3A4 (např. antimykotiky azoly, jako např. intrakonazolem, flukonazolem, ketokonazolem, vorikonazolem a posakonazolem, makrolidovými antibiotiky, jako např. erytromycinem nebo klaritromycinem, ketolidovými antibiotiky, jako např. telitromycinem, inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem a antiarytmikem dronedaronem [viz bod 4.5]).
• Těhotenství (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Abnormality jaterních enzymů a sledování jater
Lomitapid může způsobit zvýšení hladiny alaninaminotransferázy [ALT] a aspartátaminotransferázy [AST] a jaterní steatózu. V jakém rozsahu jaterní steatóza související s lomitapidem podporuje zvýšení aminotransferáz, není známo. Ačkoli nebyly hlášeny případy jaterní dysfunkce (zvýšení aminotransferáz se zvýšením bilirubinu nebo mezinárodního normalizovaného poměru [INR] nebo jaterního selhání), existuje obava, že lomitapid může indukovat steatohepatitidu, která může za několik let progredovat do cirhózy. Není pravděpodobné, že by klinické studie na podporu bezpečnosti a účinnosti lomitapidu u HoFH byly schopné vzhledem k jejich rozsahu a době trvání tyto nežádoucí účinky detekovat.
S lomitapidem je spojeno zvýšení hladin aminotransferáz (ALT a/nebo AST) (viz bod 5.1). Nedocházelo k souběžnému nebo následnému klinicky významnému zvýšení sérového bilirubinu, INR nebo alkalické fosfatázy. Změny jaterních enzymů se nejčastěji vyskytují během navyšování dávky, ale mohou nastat kdykoli během léčby.
Sledování testů jaterních funkcí
Před zahájením léčby přípravkem Lojuxta změřte ALT, AST, alkalickou fosfatázu, celkový bilirubin, gama-glutamyltransferázu (gama-GT) a sérový albumin. Léčivý přípravek je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater a pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí. Jestliže jsou základní vyšetření jaterních testů abnormální, zvažte zahájení léčby léčivým přípravkem až poté, co hepatolog provede vhodná vyšetření a abnormality základních testů budou vysvětleny či vyřešeny.
V prvním roce měřte jaterní testy (minimálně ALT a AST) před každým zvýšením dávky nebo jednou měsíčně podle toho, co nastane dříve. Po prvním roce provádějte tyto testy nejméně jednou za 3 měsíce a před každým zvýšením dávky. Jestliže pozorujete zvýšení hladiny aminotransferáz, dávku přípravku Lojuxta snižte, a při přetrvávajícím nebo klinicky významném zvýšení ukončete léčbu (konkrétní doporučení uvádí Tabulka 1).
Modifikace dávky na základě zvýšené hladiny jatemích aminotransferáz
Tabulka 1je shrnutím doporučení pro úpravu dávky a sledování pacientů, u kterých dojde během léčby přípravkem Lojuxta ke zvýšení hladiny aminotransferáz.
Tabulka 1: Úprava dávky a sledování pacientů se zvýšenými hladinami aminotransferáz
|
ALT nebo AST |
Doporučení týkající se léčby a sledování* |
|
>3x a <5x horní mez normálního rozsahu (ULN) |
• Zvýšení potvrďte opakovaným měřením v rámci jednoho týdne. • Jestliže se zvýšení potvrdí, snižte dávku a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). • Testy opakujte každý týden, a jestliže budou přítomny známky abnormálních jaterních funkcí (zvýšení bilirubinu či INR), jestliže aminotransferázy stoupnou nad 5x ULN nebo jestliže hladiny aminotransferáz během přibližně 4 týdnů neklesnou pod 3x ULN, přerušte léčbu. Pacienty s přetrvávajícím zvýšením hladin aminotransferáz >3x ULN odešlete k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, zvažte snížení dávky a častější sledování jaterních testů. |
|
>5x ULN |
• Přerušte léčbu a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). Jestliže během přibližně 4 týdnů hladiny aminotransferáz neklesnou pod 3x ULN, odešlete pacienta k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, snižte dávku a častěji sledujte jaterní testy. |
*Doporučení založené na ULN přibližně 30-40 mezinárodních jednotek/l.
Jestliže je zvýšení aminotransferáz provázeno klinickými symptomy poškození jater (např. nevolností, zvracením, bolestí břicha, horečkou, žloutenkou, letargií, příznaky podobnými chřipce), vzestupem hladin bilirubinu na >2x ULN nebo aktivním onemocněním jater, přerušte léčbu přípravkem Lojuxta a odešlete pacienta k hepatologovi na další vyšetření.
Opětovné nasazení léčby lze zvážit tehdy, pokud přínosy převyšují rizika související s potenciálním onemocněním jater.
Jaterní steatóza a riziko progresivního onemocnění jater
Většina léčených pacientů vykazuje vzestup obsahu tuku v játrech, což je ve shodě s mechanismem působení lomitapidu. V otevřené studii fáze 3 došlo u 18 z 23 pacientů s HoFH k vývoji jaterní steatózy (obsah tuku v játrech > 5,56 %) dle měření nukleární magnetickou rezonanční spektroskopií (MRS) (viz bod 5.1). Medián absolutního vzestupu jaterního tuku byl dle měření pomocí MRS jak po 26 týdnech, tak po 78 týdnech léčby 6 % oproti 1 % při zahájení studie. Jaterní steatóza je rizikový faktor progresivního onemocnění jater včetně steatohepatitidy a cirhózy. Dlouhodobé následky jaterní steatózy související s léčbou přípravkem Lojuxta nejsou známy. Klinické údaje naznačují, že nahromadění tuku v játrech je po ukončení léčby přípravkem Lojuxta reverzibilní, ale není známo, zda přetrvávají histologické změny, zejména po dlouhodobém užívání.
Sledování důkazů přítomnosti progresivního onemocnění jater
Při zahájení léčby a dále každý rok by se měl provádět pravidelný screening steatohepatitidy/fibrózy s použitím následujících zobrazovacích metod a hodnocení biomarkerů:
• Zobrazení elasticity tkáně, např. fibrosken, ARFI (Acoustic Radiation Force Impulse) nebo elastografie s použitím magnetické rezonance (MR)
• Gama-GT a sérový albumin pro detekci možného poškození jater
• Minimálně jeden marker z každé z následujících kategorií:
• Vysoce senzitivní C-reaktivní protein (hs-CRP), rychlost sedimentace erytrocytů (ESR), CK-18 fragment, NashTest (zánět jater)
• Rozšířený panel pro detekci jaterní fibrózy (ELF), fibrometr, poměr AST/ALT, skóre fib-4, fibrotest (jaterní fibróza)
Provedení těchto testů a jejich interpretace by měly zahrnovat spolupráci ošetřujícího lékaře s hepatologem. U pacientů s výsledky naznačujícími přítomnost steatohepatitidy nebo fibrózy by měla být zvážena biopsie jater.
Jestliže má pacient biopticky potvrzenou steatohepatitidu nebo fibrózu, měl by být znovu zhodnocen poměr přínosů a rizik a léčba by měla být ukončena, je-li to nutné.
Souběžné užívání inhibitorů CYP3A4
Zdá se, že lomitapid je citlivým substrátem pro metabolismus cestou CYP3A4. Inhibitory CYP3A4 zvyšují expozici lomitapidu, přičemž silné inhibitory zvyšují expozici přibližně 27krát. Souběžné užití středně silných nebo silných inhibitorů CYP3A4 a přípravku Lojuxta je kontraindikováno (viz bod 4.3). V klinických studiích s lomitapidem se u jednoho pacienta s HoFH během několika dní po zahájení užívání silného inhibitoru CYP3A4 klaritromycinu vyvinuly výrazně zvýšené hladiny aminotransferáz (ALT 24x ULN, AST 13x ULN). Jestliže se nelze vyvarovat léčby středně silnými nebo silnými inhibitory CYP3A4, mělo by být užívání přípravku Lojuxta v průběhu léčby ukončeno.
Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta má být podávána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4.
Souběžné užívání induktorů CYP3A4
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. Induktory CYP3A4 vykazují svůj účinek v závislosti na čase a dosažení maximálního účinku může trvat minimálně 2 týdny od zahájení léčby. Naopak při ukončení léčby může trvat minimálně 2 týdny, než indukce CYP3A4 odezní.
Očekává se, že souběžné podávání induktoru CYP3A4 snižuje účinek přípravku Lojuxta. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinu, pioglitazonu, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Použití třezalky tečkované současně s přípravkem Lojuxta je nutné se vyvarovat.
Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. Při vysazení induktoru CYP3A4 je nutné zvážit možnost zvýšené expozice a může být nutné snížit dávku přípravku Lojuxta.
Souběžné použití s inhibitory HMG-CoA reduktázv (..statinv")
Lomitapid zvyšuje plazmatické koncentrace statinů. U pacientů, kteří dostávají přípravek Lojuxta jako přídatnou terapii vedle statinů, by měly být sledovány nežádoucí účinky spojené s použitím vysokých dávek statinů. Statiny příležitostně mohou vyvolat myopatii. Ve vzácných případech může mít myopatie formu rhabdomyolýzy s akutním renálním selháním sekundárně způsobeným myoglobinurií nebo bez renálního selhání a může mít fatální následky. Všichni pacienti užívající přípravek Lojuxta vedle statinů by měli být informování o možném riziku myopatie a měli by být informováni, aby okamžitě hlásili nevysvětlitelnou bolest, citlivost nebo slabost svalů. S přípravkem Lojuxta by se neměly používat dávky simvastatinu > 40 mg (viz bod 4.3).
Grapefruitový džus
Ze stravy pacientů je při léčbě přípravkem Lojuxta nutno vyloučit grapefruitový džus.
Riziko supraterapeutické nebo subterapeutické antikoagulace u antikoagulačních přípravků na bázi kumarinu
Lomitapid zvyšuje plazmatické koncentrace warfarinu. Zvýšení dávky přípravku Lojuxta může vést k supraterapeutické antikoagulaci a snížení dávky může vést k subterapeutické antikoagulaci. U jednoho z pěti pacientů užívajících souběžně warfarin přispěla obtížná kontrola INR k předčasnému ukončení studie fáze 3. Pacienti užívající warfarin by měli podstupovat pravidelné sledování INR, zejména po jakýchkoli změnách dávkování přípravku Lojuxta. Dávka warfarinu by měla být upravena dle klinické indikace.
Požívání alkoholu
Alkohol může zvyšovat hladinu tuku v játrech a může vést k indukci nebo exacerbaci poškození jater. Ve studii fáze 3 udávali 3 ze 4 pacientů se zvýšením ALT >5x ULN konzumaci alkoholu překračující limit doporučený v protokolu. Požívání alkoholu během léčby přípravkem Lojuxta se nedoporučuje.
Hepatotoxické přípravky
Opatrnosti je třeba při použití přípravku Lojuxta s dalšími léčivými přípravky se známým hepatotoxickým potenciálem, např. izotretinoinem, amiodaronem, acetaminofenem (>4 g/den po >3 dny/týden), metotrexátem, tetracyklinem a tamoxifenem. Účinek souběžného podávání přípravku Lojuxta s dalšími hepatotoxickými léčivými přípravky není znám. Může být nutné častější sledování jaterních testů.
Snížená absorpce vitamínů rozpustných v tucích a mastných kyselin v séru
Vzhledem k mechanismu působení lomitapidu v tenkém střevě může lomitapid snižovat absorpci živin rozpustných v tucích. Ve studii fáze 3 byly pacientům denně poskytovány nutriční doplňky s vitamínem E, kyselinou linolovou, ALA, EPA a DHA. V této studii se úroveň mediánu sérového vitamínu E, ALA, kyseliny linolové, EPA, DHA a kyseliny arachidonové mezi výchozím bodem studie a týdnem 26 snížila, ale zůstala nad spodním limitem referenčního rozmezí. Nežádoucí klinické důsledky tohoto snížení při léčbě lomitapidem nebyly pozorovány po dobu až 78 týdnů. Pacienti léčení přípravkem Lojuxta by měli denně užívat doplňky, které obsahují 400 mezinárodních jednotek vitamínu E a přibližně 200 mg kyseliny linolové, 210 mg ALA, 110 mg EPA a 80 mg DHA.
Antikoncepční opatření u žen ve fertilním věku
Před zahájením léčby u žen ve fertilním věku by mělo být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z
důvodu průjmu a/nebo zvracení (viz bod 4.5). Perorální antikoncepční přípravky na bázi estrogenů jsou slabými inhibitory CYP3A4 (viz bod 4.2).
Pacientky by měly být informovány, že v případě otěhotnění musejí neprodleně informovat svého lékaře a ukončit užívání přípravku Lojuxta (viz bod 4.6).
Laktóza
Přípravek Lojuxta obsahuje laktózu, pacienti se vzácnými hereditárními problémy jako jsou intolerance galaktózy, vrozený deficit laktázy nebo glukózová-galaktózová malabsorpce by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
Tabulka 2: Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
Inhibitory CYP3A4 |
Při současném podávání lomitapidu v dávce 60 mg s ketokonazolem, silným inhibitorem CYP3A4, v dávce 200 mg dvakrát denně se zvýšila AUC lomitapidu přibližně 27krát a Cmax přibližně 15krát. Interakce mezi středně silnými inhibitory CYP3A4 a lomitapidem nebyly studovány. Předpokládá se, že středně silné inhibitory CYP3A4 mají významný vliv na farmakokinetiku lomitapidu. Očekává se, že souběžné použití středně silných inhibitorů CYP3A4 zvyšuje expozici lomitapidu 4-10krát podle výsledků studie se silným inhibitorem CYP3A4 podle historických údajů pro zkušební model CYP3A4 midazolam. Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při podávání lomitapidu 20 mg současně s atorvastatinem (tj. slabým inhibitorem CYP3A4) se AUC a Cmax lomitapidu zvýšily přibližně 2x. Pokud byl lomitapid podáván 12 hodin po atorvastatinu, žádné klinicky |
Použití silných nebo středně silných inhibitorů CYP3A4 spolu s přípravkem Lojuxta je kontraindikováno. Jestliže se nelze vyvarovat léčby antimykotiky azoly (např. itrakonazolem, ketokonazolem, flukonazolem, vorikonazolem, posakonazolem), antiarytmikem dronedaronem, makrolidovými antibiotiky (např. erytromycinem, klaritromcinem), ketolidovými antibiotiky (např. telitromycinem), inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem, měla by být během této léčby léčba přípravkem Lojuxta pozastavena (viz body 4.3 a 4.4). Grapefruitový džus je středně silným inhibitorem CYP3A4 a má se za to, že podstatně zvyšuje expozici lomitapidu. Pacienti užívající přípravek Lojuxta by se měli vyvarovat konzumace grapefruitového džusu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta musí být užívána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4. Příklady slabých inhibitorů CYP3A4 zahrnují: alprazolam, amiodaron, amlodipin, atorvastatin, azitromycin, bikalutamid, cilostazol, cimetidin, cyklosporin, klotrimazol, fluoxetin, fluvoxamin, fosaprepitant, ginkgo, vodilku kanadskou, izoniazid, |
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
významné zvýšení expozice lomitapidu nebylo pozorováno. Při podávání lomitapidu 20 mg s ethinyl estradiolem/norgestimátem (tj. slabým inhibitorem CYP3A4), žádné klinicky významné zvýšení expozice lomitapidu nebylo pozorováno při užívání současném ani s intervalem 12 hodin. |
ivakaftor, lacidipin, lapatinib, linagliptin, nilotinib, perorální antikoncepční přípravky obsahující estrogeny, pazopanib, mátový olej, propiverin, ranitidin, ranolazin, roxithromycin, hořké pomeranče, takrolimus, tikagrelor a tolvaptan. Tento seznam není úplný a předepisující lékaři by měli zkontrolovat preskripční informace o léčivech, která mají být podávána spolu s přípravkem Lojuxta s ohledem na možné interakce zprostředkované CYP3A4. Účinek podávání více než jednoho slabého inhibitoru CYP3A4 nebyl testován, ale očekává se, že účinek na expozici lomitapidu je větší než při současném podávání jednotlivých inhibitorů s lomitapidem. Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta. | |
|
Induktory CYP3A4 |
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. To by mohlo následně snížit účinek lomitapidu. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. |
Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinnu, pioglitazonu, třezalky tečkované, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. |
|
Sekvestranty žlučových kyselin |
Interakce lomitapidu se sekvestranty žlučových kyselin (pryskyřicemi, např. kolesevelamem a cholestyraminem) nebyly testovány. |
Protože sekvestranty žlučových kyselin mohou interferovat s absorpcí perorálních léčivých přípravků, měly by se užívat minimálně 4 hodiny před přípravkem Lojuxta nebo minimálně 4 hodiny po něm. |
Účinky lomitapidu na jiné léčivé přípravky
Inhibitory HMG-CoA reduktázy („statiny“): Lomitapid zvyšuje plazmatické koncentrace statinů. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním simvastatinu 40 mg se zvýšily u kyseliny simvastatinu hodnoty AUC o 68 % a Cmax o 57 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním atorvastatinu 20 mg se zvýšily u kyseliny atorvastatinu hodnoty AUC o 52 % a Cmax o 63 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním rosuvastatinu 20 mg se zvýšila hodnota Tmax rosuvastatinu z 1 na 4 hodiny, hodnota AUC se zvýšila o 32 % a hodnota Cmax zůstala nezměněná. Riziko myopatie u simvastatinu je závislé na dávce. Použití přípravku Lojuxta je u pacientů léčených vysokými dávkami statinů (> 40 mg) kontraindikováno (viz body 4.3 a 4.4).
Kumarinové antikoagulační přípravky: Podávání lomitapidu 60 mg do dosažení ustáleného stavu a po dobu 6 dní po warfarinu 10 mg zvýšilo hodnotu INR 1,26krát. Hodnota AUC pro R(+)-warfarin se zvýšila o 25 % a pro S(-)-warfarin o 30 %. Hodnota Cmax pro R(+)-warfarin se zvýšila o 14 % a pro S(-)-warfarin o 15 %. U pacientů užívajících souběžně kumariny (např. warfarin) a přípravek Lojuxta by měla být před zahájením užívání přípravku Lojuxta stanovena hodnota INR a tato hodnota by měla být pravidelně kontrolována, přičemž dávkování kumarinů by mělo být upraveno dle klinické potřeby (viz bod 4.4).
Fenofibrát, niacin a ezetimib: Při podávání lomitapidu do dosažení ustáleného stavu před podáním mikronizovaného fenofibrátu 145 mg, niacinu s prodlouženým uvolňováním 1000 mg nebo ezetimibu 10 mg nebyly pozorovány klinicky významné účinky působení kteréhokoli z těchto léčivých přípravků. Při souběžném podávání s přípravkem Lojuxta není nutná úprava dávky.
Perorální antikoncepce: Při podávání lomitapidu 50 mg do dosažení ustáleného stavu spolu s perorálním antikoncepčním přípravkem na bázi estrogenů nebyl pozorován klinicky ani statisticky významný vliv na farmakokinetiku složek perorální antikoncepce (ethinylestradiolu a 17-deacetylnorgestimátu, metabolitu norgestimátu). Neočekává se, že by lomitapid přímo ovlivňoval účinnost perorální antikoncepce na bázi estrogenů, nicméně průjem a/nebo zvracení mohou snížit absorpci hormonů. U případů prodlouženého nebo závažného průjmu a/nebo zvracení trvajících více než 2 dny by měla být po dobu 7 dnů po ústupu symptomů použita další antikoncepční metoda.
Substráty P-gp: Lomitapid in vitro inhibuje P-gp a může zvyšovat absorpci P-gp substrátů. Souběžné podávání přípravku Lojuxta s P-pg substráty (např. aliskirenem, abrisentanem, kolchicinem, dabigatran etexilátem, dixoginem, everolimem, fexofenadinem, imatinibem, lapatinibem, maravirokem, nilotinibem, posakonazolem, ranolazinem, saxagliptinem, sirolimem, sitagliptinem, talinololem, tolvaptanem, topotekanem) může zvýšit absorpci P-gp substrátů. Při souběžném použití s přípravkem Lojuxta by mělo být zváženo snížení dávky P-gp substrátu.
In vitro hodnocení lékových interakcí: Lomitapid inhibuje CYP3A4. Lomitapide neindukuje CYP 1A2, 3A4 ani 2B6 a neinhibuje CYP 1A2, 2B6, 2C9, 2C19, 2D6 ani 2E1. Lomitapid není P-gp substrátem, ale inhibuje P-gp. Lomitapid neinhibuje protein rezistence u karcinomu prsu (BCRP).
4.6 Fertilita, těhotenství a kojení
Přípravek Lojuxta je v těhotenství kontraindikován. Neexistují spolehlivé údaje o použití přípravku u těhotných žen. Studie na zvířatech prokázaly vývojovou toxicitu (teratogenitu, embryotoxicitu, viz bod 5.3). Možné riziko pro člověka je neznámé.
Použití u žen ve fertilním věku.
Před zahájením léčby u žen ve fertilním věku by mělo být vyloučeno těhotenství, mělo by být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z důvodu průjmu a/nebo zvracení. Dokud symptomy neustoupí, je nutno používat další antikoncepční metody (viz bod 4.5).
Kojení
Není známo, zda se lomitapid vylučuje do mateřského mléka. Vzhledem k potenciálu nežádoucích účinků zjištěnému na základě studií s lomitapidem na zvířatech (viz bod 5.3) je nutné rozhodnout, zda ukončit kojení nebo užívání léčivého přípravku, a to při zvážení významu léčivého přípravku pro matku.
Fertilita
U potkaních samců ani samic, kterým byl podáván lomitapid se systémovou expozicí (AUC) ve výši odhadovaného čtyř- až pětinásobku expozice u lidí při maximální doporučené dávce u člověka, nebyly pozorovány žádné nežádoucí účinky na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Lojuxta může mít mírný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejzávažnějšími nežádoucími účinky během léčby byly abnormality jaterních aminotransferáz (viz bod 4.4).
Nejčastějšími nežádoucími účinky byly gastrointestinální účinky. Nežádoucí gastrointestinální účinky byly hlášeny u 27 (93 %) z 29 pacientů klinické studie fáze 3. Průjem se objevil u 79 %, nevolnost u 65 %, dyspepsie u 38 % a zvracení u 34 % pacientů. Další účinky, které hlásilo minimálně 20 % pacientů, zahrnují bolest břicha, břišní dyskomfort, nadmutí břicha, zácpu a plynatost. Nežádoucí gastrointestinální účinky se častěji objevovaly během fáze navyšování dávky ve studii a poklesly, jakmile pacienti dosáhli maximální tolerované dávky lomitapidu.
Gastrointestinální nežádoucí účinky se závažnou intenzitou byly hlášeny u 6 (21 %) z 29 pacientů v klinické studii fáze 3, přičemž nejčastějším účinkem byl průjem (4 pacienti, 14 %), zvracení (3 pacienti, 10 %) a bolest břicha, nadmutí a/nebo dyskomfort (2 pacienti, 7 %). Gastrointestinální účinky přispěly k časnému ukončení studie u 4 (14 %) pacientů.
Nejčastěji udávanými nežádoucími účinky se závažnou intenzitou byly průjem (4 subjekty, 14 %), zvracení (3 pacienti, 10 %) a nadmutí břicha a zvýšení ALT (pokaždé 2 subjekty, 7 %).
Přehled nežádoucích účinků v tabulce
Dle frekvence jsou nežádoucí účinky definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), málo časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), s neznámou frekvencí (z dostupných údajů nelze určit).
Tabulka 3 zahrnuje veškeré nežádoucí účinky hlášené u 35 pacientů léčených ve studii fáze 2 UP1001 a ve studii fáze 3 UP1002/AEGR-733-005 nebo v rozšířené studii AEGR-733-012.
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Časté |
Gastroenteritida |
|
Poruchy metabolismu a výživy |
Velmi časté |
Snížená chuť k jídlu |
|
Poruchy nervového systému |
Časté |
Závratě Bolest hlavy Migréna |
|
Gastrointestinální poruchy |
Velmi časté |
Břišní dyskomfort Bolest horní části břicha Plynatost Distenze břicha Zácpa |
|
Časté |
Gastritida Rektální tenesmy Aerofagie Nutkání na stolici Říhání Častá stolice Dilatace žaludku Onemocnění žaludku Gastroezofageální reflux Krvácení z hemoroidů Regurgitace | |
|
Poruchy jater a žlučových cest |
Časté |
Jaterní steatóza Hepatotoxicita Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Časté |
Ekchymóza Papuly Erytematózní vyrážka Xantomy |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Únava |
|
Vyšetření |
Velmi časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Snížení tělesné hmotnosti |
|
Časté |
Zvýšený mezinárodní normalizovaný poměr Zvýšená alkalická fosfatáza v krvi Snížení draslíku v krvi Snížený karoten Abnormální mezinárodní normalizovaný poměr Abnormální testy jaterních funkcí Prodloužený protrombinový čas Zvýšené transaminázy Snížený vitamín E Snížený vitamín K |
Tabulka 4 uvádí všechny nežádoucí účinky u subjektů, které dostávaly lomitapid v monoterapii (n=291), léčených ve studiích fáze 2 u subjektů se zvýšenou hladinou LDL-C (N=462).
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Méně časté |
Gastroenteritida Gastrointestinální infekce Chřipka Nazofaryngitida Sinusitida |
|
Poruchy krve a lymfatického systému |
Méně časté | |
|
Poruchy metabolismu a výživy |
Časté |
Snížená chuť k jídlu |
|
Méně časté |
Dehydratace Zvýšená chuť k jídlu | |
|
Poruchy nervového systému |
Méně časté |
Parestézie Ospalost |
|
Poruchy oka |
Méně časté |
Otok oka |
|
Poruchy ucha a labyrintu |
Méně časté |
Závrať |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté |
Léze hltanu Syndrom kašle horních cest dýchacích |
|
Gastrointestinální poruchy |
Velmi časté |
Plynatost |
|
Časté |
Bolest horní části břicha Distenze břicha Bolest břicha Zvracení Břišní dyskomfort Říhání Bolest v podbřišku Častá stolice | |
|
Méně časté |
Sucho v ústech Tvrdá stolice Gastroezofageální reflux Citlivost břicha Dyskomfort v epigastriu Dilatace žaludku Hemateméza Krvácení v dolní části gastrointestinálního traktu Refluxní ezofagitida | |
|
Poruchy jater a žlučových cest |
Méně časté |
Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Puchýře Suchá kůže Nadměrné pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté |
Svalové spasmy |
|
Méně časté |
Bolest kloubů Bolest svalů Bolest končetin Otok kloubu Svalové záškuby | |
|
Poruchy ledvin a močových cest |
Méně časté |
Hematurie |
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Celkové poruchy a reakce v místě |
Časté |
Únava |
|
podání |
Slabost | |
|
Méně časté |
Bolest na hrudi Mrazení Rychlý pocit nasycení Poruchy chůze Malátnost Horečka | |
|
Vyšetření |
Časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Zvýšení jaterních enzymů Abnormální testy jaterních funkcí Snížený počet neutrofilů Snížený počet leukocytů |
|
Méně časté |
Snížení tělesné hmotnosti Zvýšený bilirubin v krvi Zvýšená hladina gama-glutamyltransferázy Zvýšené procento neutrofilů Protein v moči Prodloužený protrombinový čas Abnormální testy plicních funkcí Zvýšený počet leukocytů |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování neexistuje specifická léčba. U hlodavců byla dobře snášena jednorázová perorální dávka lomitapidu >600krát vyšší než maximální doporučená dávka pro člověka (1 mg/kg). Maximální dávkou podanou lidským subjektům v klinických studiích bylo 200 mg ve formě jednorázové dávky, nevyskytly se žádné nežádoucí účinky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná léčiva ovlivňující hladinu lipidů, samotná. ATC kód: C10AX12
Mechanismus účinku
Lomitapid je selektivní inhibitor mikrozomálního transferového proteinu (MTP), intracelulárního proteinu přenášejícího lipidy, který se nachází v lumen endoplazmatického retikula a zodpovídá za vazbu a převod jednotlivých lipidových molekul mezi membránami. MTP hraje klíčovou roli při sestavování lipoproteinů obsahujících apo B v játrech a střevech. Inhibice MTP snižuje sekreci lipoproteinů a cirkulující koncentrace lipidů přenášených v lipoproteinech včetně cholesterolu a triglyceridů.
Klinická účinnost a bezpečnost
Účinnost a bezpečnost lomitapidu při současném podávání diety s nízkým obsahem tuků a dalších terapií snižujících lipidy u dospělých pacientů s HoFH hodnotila otevřená studie s jedním ramenem (UP1002/AEGR-733-005). Pacienti byli instruováni, aby dodržovali dietu s nízkým obsahem tuků (< 20 % kalorického příjmu z tuků) i terapii snižující lipidy, kterou měli při vstupu do studie, včetně aferézy, byla-li tato nutná, a to po dobu 6 týdnů předcházejících výchozímu bodu studie minimálně až do týdne 26. Dávka lomitapidu byla navyšována z 5 mg na individuálně stanovenou maximální tolerovanou dávku až 60 mg. Po týdnu 26 byl pacientům lomitapid ponechán za účelem zjištění účinků dlouhodobé léčby a bylo jim umožněno změnit základní terapii snižující lipidy. Studie zahrnovala celkem 78 týdnů léčby.
Do studie bylo zařazeno 29 pacientů, z nichž 23 dokončilo týden 78. Zařazeno bylo 16 mužů (55 %) a 13 žen (45 %) s průměrným věkem 30,7 let a ve věkovém rozmezí 18 až 55 let. Průměrná dávka lomitapidu v týdnu 26 byla 45 mg a v týdnu 78 byla 40 mg. V týdnu 26 byla průměrná procentuální změna hladiny LDL-C u populace „intention-to-treat“ (ITT) oproti výchozímu bodu -40 %
(p < 0,001). Průměrná procentuální změna oproti výchozímu bodu studie v týdnu 26 s použitím metody LOCF (Last Observation Carried Forward) pro každé hodnocení je uvedena na obrázku 1.
Obrázek 1: Průměrné procentuální změny hladiny LDL-C mezi vstupní hodnotou a týdnem
26 v hlavní studii účinnosti UP1002/AEGR-733-005 (primární cíl) s použitím LOCF pro každé hodnocení (n=29)
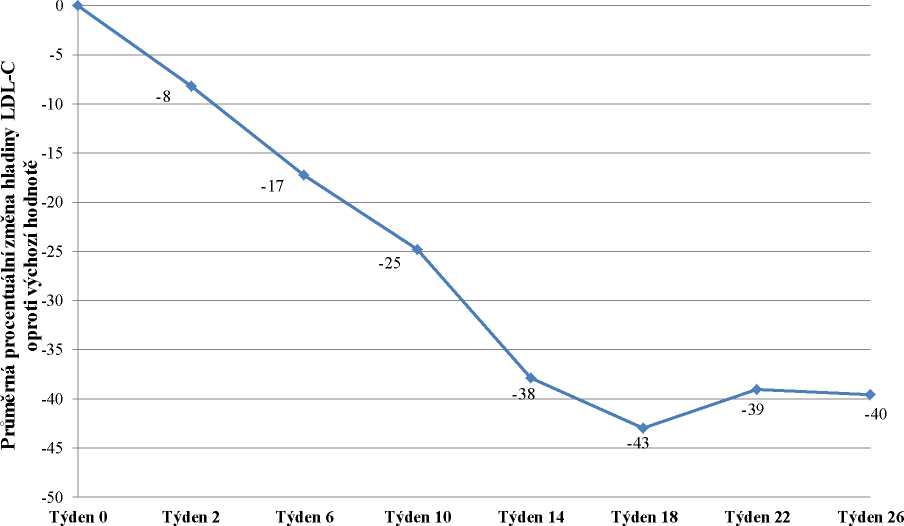
Týden studie
Změny lipidů i lipoproteinů v týdnu 26 a 78 při léčbě lomitapidem uvádí Tabulka 5.
Tabulka 5: Absolutní hodnoty a procentuální změny hladin lipidů a lipoproteinů mezi
výchozí hodnotou a týdny 26 a 78 (hlavní studie účinnosti UP1002/AEGR-733-005).
|
Parametr (jednotky) |
Výchozí bod |
Týden 26 / LOCF (n=29) |
Týden 78 (n=23) | ||||
|
Průměr (SD) |
Průměr (SD) |
% změna |
Hodnota p b |
Průměr (SD) |
% změna |
Hodnota p b | |
|
LDL-C, přímý (mg/dl) |
336 (114) |
190 (104) |
-40 |
< 0,001 |
210 (132) |
-38 |
< 0,001 |
|
Celkový cholesterol (TC) (mg/dl) |
430 (135) |
258 (118) |
-36 |
< 0,001 |
281 (149) |
-35 |
< 0,001 |
|
Apolipoprotein B (apo B) (mg/dl) |
259 (80) |
148 (74) |
-39 |
< 0,001 |
151 (89) |
-43 |
< 0,001 |
|
Triglyceridy (TG) (mg/dl)a |
92 |
57 |
-45 |
0,009 |
59 |
-42 |
0,012 |
|
Cholesterol z jiných lipoproteinů než o vysoké hustotě (non-HDL-C) (mg/dl) |
386 (132) |
217 (113) |
-40 |
< 0,001 |
239 (146) |
-39 |
< 0,001 |
|
Cholesterol z lipoproteinů o velmi nízké hustotě (VLDL-C) (mg/dl) |
21 (10) |
13 (9) |
-29 |
0,012 |
16 (15) |
-31 |
0,013 |
|
Lipoprotein (a) (Lp(a)) (nmol/l)a |
66 |
61 |
-13 |
0,094 |
72 |
-4 |
< 0,842 |
|
Cholesterol z lipoproteinů o vysoké hustotě (HDL-C) (mg/dl) |
44 (11) |
41 (13) |
-7 |
0,072 |
43 (12) |
-4,6 |
0,246 |
a Medián uvedený pro TG a Lp(a). Hodnota p je založena na průměrné procentuální změně b Hodnota p u střední procentuální změny oproti výchozímu bodu studie na základě párového t-testu
Jak v týdnu 26, tak v týdnu 78 došlo k významnému snížení hladin LDL-C, TC, apo B, TG, non-HDL-C a VLDL-C a změny u HDL-C vykazovaly v týdnu 26 trend směrem ke snížení a v týdnu 78 se vrátily k výchozím hodnotám.
Účinek přípravku Lojuxta na kardiovaskulární morbiditu a mortalitu nebyl stanoven.
Při zahájení studie užívalo 93 % pacientů statin, 76 % pacientů ezetimib, 10 % niacin, 3 % sekvestrant žlučových kyselin a 62 % pacientů bylo léčeno aferézou. U 15 z 23 (65 %) pacientů byla v týdnu 78 redukována jejich léčba na snížení lipidů, včetně plánovaných a neplánovaných snižování/přerušení. Aferéza byla přerušena u 3 ze 13 pacientů, kteří byli aferézou léčeni v týdnu 26, a u 3 pacientů byla frekvence při současném udržení nízkých hladin LDL-C i po týdnu 78 snížena. Klinický přínos omezení základní terapie snižující lipidy, včetně aferézy, není jistý.
Z 23 pacientů, kteří dokončili týden 26 studie, došlo u 19 (83 %) ke snížení LDL-C o > 25 %, přičemž 8 (35 %) z nich mělo v tomto časovém bodě LDL-C < 100 mg/dl a 1 pacient měl LDL-C < 70 mg/dl.
V této studii došlo u 10 pacientů ke zvýšení AST a/nebo ALT >3x ULN (viz Tabulka 6).
Nejvyšší výsledky testů jaterních funkcí po první dávce (hlavní studie účinnosti UP1002/AEGR-733-005)
|
Parametr/abnormalita |
N (%) |
|
ALT | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
6 (20,7) |
|
>5 až < 10 x ULN |
3 (10,3) |
|
>10 až < 20 x ULN |
1 (3,4) |
|
> 20 x ULN |
0 |
|
AST | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
5 (17,2) |
|
>5 až < 10 x ULN |
1 (3,4) |
|
>10 až < 20 x ULN |
0 |
|
> 20 x ULN |
0 |
Tabulka 6:
Zvýšení ALT a/nebo AST > 5x ULN bylo zvládnuto snížením dávky nebo dočasným pozastavením podávání lomitapidu a všichni pacienti byli schopni v léčbě studovaným léčivem pokračovat. Nebylo pozorováno klinicky významné zvýšení celkového bilirubinu ani alkalické fosfatázy. Jaterní tuk byl během klinické studie u všech dostupných pacientů prospektivně měřen s použitím MRS (Tabulka 7). Údaje od jedinců, u kterých byla po ukončení lomitapidu prováděna opakovaná měření, ukázaly, že nahromadění tuku v játrech je reverzibilní, ale není známo, zda nepřetrvávají histologické změny.
Tabulka 7: Maximální kategorické změny v % jaterního tuku (hlavní studie účinnosti UP1002/AEGR-733-005)
|
Maximální absolutní nárůst % jaterního tuku |
Fáze účinnosti Týdny 0-26 N (%) |
Fáze bezpečnosti Týdny 26-78 N (%) |
Celá studie týdny 0-78 N (%) |
|
Počet hodnotitelných pacientů |
22 |
22 |
23 |
|
< 5% |
9 (41) |
6 (27) |
5 (22) |
|
> 5 % až < 10 % |
6 (27) |
8 (36) |
8 (35) |
|
> 10 % až < 15 % |
4 (18) |
3 (14) |
4 (17) |
|
> 15 % až < 20 % |
1 (5) |
4 (18) |
3 (13) |
|
> 20 % až < 25 % |
1 (5) |
0 |
1 (4) |
|
> 25 % |
1 (5) |
1 (5) |
2 (9) |
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lojuxta u jedné nebo více podskupin pediatrické populace s HoFH (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Absolutní perorální biologická dostupnost lomitapidu je 7 %. Absorpce není omezena prostupem léčiva přes střevní bariéru, ale je převážně ovlivněna extenzivním efektem prvního průchodu. Vrcholná plazmatická koncentrace lomitapidu byla dosažena za 4 až 8 hodin po podání perorální dávky. Farmakokinetika lomitapidu je přibližně proporcionální k dávce perorálních jednorázových dávek v terapeutickém rozmezí. U dávek vyšších než 60 mg je naznačena tendence k nelineární závislosti a tyto dávky nejsou doporučeny.
Při podání více dávek se hodnoty Cmax a AUC zvýšily přibližně proporcionálně k dávce lomitapidu. Hodnoty Cmax a AUC se zvýšily jak po vysoce tučném jídle (77 %, respektive 58 %), tak po nízkotučném jídle (70 %, respektive 28 %). Akumulace lomitapidu v plazmě byla konzistentní s hodnotou předpokládanou na základě jedné dávky po podávání jedné perorální dávky denně nad 25 mg po dobu až 4 týdnů. Interindividuální variabilita hodnoty AUC lomitapidu byla přibližně 50 %.
V ustáleném stavu byla akumulace lomitapidu 2,7 při 25 mg a 3,9 při 50 mg.
Distribuce
Po intravenózním podání byl distribuční objem lomitapidu velký (průměr = 1 200 litrů) i přes vysoký stupeň (> 99,8 %) vazby na plazmatické proteiny. Ve studiích na zvířatech se lomitapid vysoce (200násobně) koncentroval v játrech.
Biotransformace
Lomitapid se extenzivně metabolizuje, převážně cestou CYP3A4. Izoformy CYP 2E1, 1A2, 2B6, 2C8 a 2C19 se účastní méně a izoformy 2D6 a 2C9 se metabolismu lomitapidu neúčastní.
Eliminace
Po podání dávky radioaktivně značeného perorálního roztoku zdravým subjektům bylo 93 % podané dávky vyloučeno močí a stolicí. Přibližně 33 % radioaktivity bylo vyloučeno do moči jako metabolity. Zbytek byl vyloučen stolicí, primárně jako oxidované metabolity. Eliminační poločas lomitapidu byl přibližně 29 hodin.
Zvláštní populace
Údaje ze stěžejní klinické studie byly analyzovány s ohledem na vliv možných souvisejících proměnných na expozici lomitapidu. Z vyšetřovaných parametrů (rasa, index tělesné hmotnosti (BMI), hmotnost, věk) může být jako možná související proměnná klasifikován pouze BMI.
Věk a pohlaví
Věk (18-64 let) ani pohlaví nemají na farmakokinetiku lomitapidu klinicky významný vliv.
Rasa
U bělochů nebo latinskoamerické populace není nutná úprava dávky. K dispozici není dostatek informací pro stanovení, zda přípravek Lojuxta vyžaduje úpravu dávky u dalších ras. Protože se ale léčivý přípravek podává způsobem postupného zvyšování dávky podle individuální bezpečnosti a snášenlivosti pacienta, úprava dávkovacího režimu na základě rasy není doporučena.
V populaci s poškozením funkce ledvin byl lomitapid studován pouze u pacientů s terminálním onemocněním ledvin (ESRD). Farmakokinetická studie u pacientů s ESRD podstupujících dialýzu ukázala 36% vzestup průměrné koncentrace lomitapidu v plazmě ve srovnání s párovanými zdravými kontrolami. Koncový poločas lomitapidu nebyl ovlivněn.
Jaterní insuficience
Pro hodnocení farmakokinetiky 60 mg lomitapidu u zdravých dobrovolníků s normálními jaterními funkcemi ve srovnání s pacienty s mírným postižením funkce jater (třída A dle Child-Pugha) a středně těžkým postižením funkce jater (třída B dle Child-Pugha) byla provedena otevřená studie s jednou dávkou. Hodnoty AUC a Cmax lomitapidu u pacientů se středně těžkým postižením funkce jater byly o 164 %, respektive 361 % vyšší než u zdravých dobrovolníků. Hodnoty AUC a Cmax lomitapidu u pacientů s mírným postižením funkce jater byly o 47 %, respektive 4 % vyšší než u zdravých dobrovolníků. U pacientů s těžkým postižením funkce jater (skóre dle Child-Pugha 10-15) nebyl přípravek Lojuxta studován.
Pediatrická populace
U dětí mladších 18 let nebyl přípravek Lojuxta studován.
Starší populace
U pacientů starších 65 let nebyl přípravek Lojuxta studován.
5.3 Předklinické údaje vztahující se k bezpečnosti
Základním zjištěním toxikologických studií léčiva s opakovanou perorální dávkou u hlodavců a psů byla akumulace lipidů v tenkém střevě a/nebo v játrech spojená s poklesem sérových hladin cholesterolu a/nebo triglyceridů. Tyto změny jsou vzhledem k mechanismu působení lomitapidu sekundární. Další změny související s játry ve studiích toxicity s opakovanou dávkou u potkanů a psů zahrnovaly zvýšené hladiny sérové aminotransferázy, subakutní zánět (pouze u potkanů) a nekrózu jednotlivých buněk. V jednoleté studii s opakovanou dávkou u psů se v játrech nevyskytovaly mikroskopické změny, ačkoli u fen byla minimálně zvýšená sérová hladina AST.
U hlodavců byla pozorována plicní histiocytóza. U psů byl pozorován pokles parametrů erytrocytů i poikilocytóza a/nebo anizocytóza. U psů byla v šestiměsíční studii při 205násobku lidské expozice (AUC) 60 mg pozorována testikulární toxicita. V roční studii u psů nebyly při 64násobku lidské expozice 60 mg pozorovány nežádoucí účinky na varlata.
Ve studii nutriční karcinogenity u myší byl lomitapid podáván po dobu až 104 týdnů v dávkách v rozmezí 0,3 až 45 mg/kg/den. Při dávkách > 1,5 mg/kg/den u samců (> 2násobek lidské expozice při 60 mg denně na základě AUC) a > 7,5 mg/kg/den u samic (> 9násobek lidské expozice při 60 mg na základě AUC) došlo ke statisticky významnému vzestupu incidence adenomu i karcinomu jater. Incidence karcinomu tenkého střeva a/nebo kombinovaného adenomu a karcinomu (u myší vzácné nádory) byla významně zvýšena při dávkách > 15 mg/kg/den u samců (> 26násobek lidské expozice při 60 mg na základě AUC) a 15 mg/kg/den u samic (22násobek lidské expozice při 60 mg na základě AUC).
Ve studii perorální kancerogenity u potkanů byl lomitapid podáván po dobu až 99 týdnů v dávkách až
7,5 mg/kg/den u samců a 2,0 mg/kg/den u samic. U samců i samic byla pozorována fokální jaterní fibróza a pouze u samců byla pozorována cystická degenerace jater. U samců s vysokou dávkou byla pozorována zvýšená incidence pankreatického acinárního adenomu při expozici 6krát vyšší než u lidí při 60 mg na základě AUC.
V panelu studií in vitro a in vivo nebyl lomitapid mutagenní ani genotoxický.
Lomitapid neměl účinek na reprodukční funkce u samic potkanů v dávkách až 1 mg/kg nebo samců potkanů v dávkách až 5 mg/kg. Systémová expozice lomitapidu v těchto dávkách byla odhadována na čtyřnásobek (samice) až pětinásobek (samci) expozice u lidí při 60 mg na základě AUC.
Lomitapid byl u potkanů teratogenní při současné absenci mateřské toxicity při expozici (AUC) odhadované na dvojnásobek lidské expozice při 60 mg. U králíků se nevyskytly známky embryofetální toxicity při trojnásobku maximální doporučené denní dávky u lidí (MRHD) 60 mg na základě plochy tělesného povrchu. Embryofetální toxicita při absenci mateřské toxicity byla pozorována u králíků při > 6,5násobku MRHD. U fretek byl lomitapid toxický pro matku i teratogenní při dávkách < 1x MRHD.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky
předbobtnalý škrob (kukuřičný) sodná sůl karboxymethylškrobu mikrokrystalická celulóza monohydrát laktózy koloidní bezvodý oxid křemičitý magnesium-stearát
Obal tobolky želatina
oxid titaničitý (E171)
Tiskařský inkoust šelak
černý oxid železitý (E172) propylenglykol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Uchovávejte v dobře uzavřené lahvičce, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Lahvička z polyetylenu o vysoké hustotě (HDPE) vybavená vložkou z polyesteru / hliníkové fólie / kartonu a s polypropylenovým šroubovacím uzávěrem.
Velikost balení:
28 tobolek
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Aegerion Pharmaceuticals Ltd Lakeside House 1 Furzeground Way Stockley Park East Uxbridge UB11 1BD Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/851/003
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. června 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
^ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Lojuxta 30 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje lomitapidum 30 mg ve formě lomitapidum mesilas Pomocná látka se známým účinkem:
Jedna tvrdá tobolka obsahuje 194,84 mg laktózy (ve formě monohydrátu) (viz bod 4.4). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Tvrdá tobolka s oranžovou svrchní částí / žlutou spodní částí o délce 21,6 mm, potištěná černým inkoustem s textem „30 mg“ na spodní části a „A733“ na svrchní části tobolky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Lojuxta je indikován jako podpůrný přípravek užívaný spolu s dietou s nízkým obsahem tuků a dalšími léčivými přípravky na snížení hladiny lipidů spolu s aferézou lipoproteinů o nízké hustotě (LDL) či bez ní u dospělých pacientů s homozygotní formou familiární hypercholesterolémie (HoFH).
Tam, kde je to možné, by měla být homozygotní forma familiární hypercholesterolémie potvrzena geneticky. Je nutné vyloučit jiné formy primární hyperlipoproteinémie a sekundární příčiny hypercholesterolémie (např. nefrotický syndrom, hypotyreózu).
4.2 Dávkování a způsob podání
Léčbu přípravkem Lojuxta by měl zahajovat a sledovat lékař, který má zkušenosti s léčbou poruch lipidů.
Dávkování
Doporučená zahajovací dávka přípravku je 5 mg jednou denně. Po 2 týdnech může být dávka na základě přijatelné bezpečnosti a snášenlivosti zvýšena na 10 mg a poté v minimálně čtyřtýdenních intervalech na 20 mg, 40 mg a maximální doporučenou dávku 60 mg (viz bod 4.8).
Dávka by měla být zvyšována postupně, aby se minimalizovala incidence a závažnost gastrointestinálních nežádoucích účinků a zvýšení aminotransferáz.
Podávání se stravou může u přípravku Lojuxta zvyšovat expozici. Přípravek Lojuxta by se měl užívat nalačno, minimálně dvě hodiny po večeři, protože tuk obsažený v nedávno požitém jídle může mít negativní vliv na gastrointestinální snášenlivost.
Výskyt a závažnost gastrointestinálních nežádoucích účinků souvisejících s užíváním přípravku Lojuxta se snižují při dodržování diety s nízkým obsahem tuků. Pacienti by před zahájením léčby přípravkem Lojuxta měli držet dietu, ve které je méně než 20 % energie dodáváno z tuků, a během léčby by v této dietě měli pokračovat. Mělo by být zajištěno nutriční poradenství.
Pacienti by se měli vyvarovat konzumace grapefruitového džusu (viz body 4.4 a 4.5).
U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s atorvastatinem použijte jednu z těchto variant:
• Oddělte podání obou přípravků intervalem 12 hodin NEBO
• Snižte dávku přípravku Lojuxta o polovinu.
Pacienti užívající 5 mg by měli zůstat na této dávce.
Následně lze provést opatrnou titraci podle reakce hladiny LDL-C a bezpečnosti /snášenlivosti.
Po ukončení léčby vysazení atorvastatinem zvyšte dávku přípravku Lojuxta titrací podle hladiny LDL-C a bezpečnosti/snášenlivosti.
U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s jakýmkoliv dalším slabým inhibitorem CYP3A4 oddělte podání obou přípravků intervalem 12 hodin.
Omezení maximální dávky přípravku Lojuxta zvažte podle požadované odpovědi LDL-C.
Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta.
Na základě pozorování snížených hladin esenciálních mastných kyselin a vitamínu E v klinických studiích by pacienti po dobu léčby přípravkem Lojuxta měli také užívat denně nutriční doplňky zajišťující příjem 400 IU vitamínu E a přibližně 200 mg kyseliny linolové, 110 mg kyseliny eikosapentaenové (EPA), 210 mg kyseliny alfa-linolenové (ALA) a 80 mg kyseliny dokosahexaenové (DHA).
Starší populace
Zkušenosti s přípravkem Lojuxta jsou u pacientů ve věku 65 let a starších omezené. Proto je u těchto pacientů zapotřebí zvláštní obezřetnosti.
Protože doporučený režim dávkování zahrnuje zahájení na spodní hranici dávkovacího rozmezí a opatrné navyšování dávky podle individuální snášenlivosti pacienta, nedoporučuje se ve starším věku žádná úprava dávkovacího schématu.
Porucha jater
Přípravek Lojuxta je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater včetně pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí (viz bod 5.2).
U pacientů s mírnou poruchou funkce jater (třída A dle Child-Pugha) by neměla být překračována dávka 40 mg denně.
Porucha ledvin
U pacientů v terminálním stádiu onemocnění ledvin léčených dialýzou by neměla být překračována dávka 40 mg denně (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Lojuxta u dětí mladších 18 let nebyla stanovena, a použití tohoto léčivého přípravku u dětí se proto nedoporučuje. Nejsou dostupné žádné údaje.
Způsob podání
Perorální podání.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Pacienti se středně těžkou nebo těžkou poruchou funkce jater a pacienti s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí.
• Pacienti se známým významným nebo chronickým střevním onemocněním, např. zánětlivým onemocněním střeva nebo malabsorpcí.
• Souběžné podávání více než 40 mg simvastatinu (viz bod 4.5).
• Souběžné použití přípravku Lojuxta se silnými nebo středně silnými inhibitory cytochromu P450 (CYP) 3A4 (např. antimykotiky azoly, jako např. intrakonazolem, flukonazolem, ketokonazolem, vorikonazolem a posakonazolem, makrolidovými antibiotiky, jako např. erytromycinem nebo klaritromycinem, ketolidovými antibiotiky, jako např. telitromycinem, inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem a antiarytmikem dronedaronem [viz bod 4.5]).
• Těhotenství (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Abnormality jaterních enzymů a sledování jater
Lomitapid může způsobit zvýšení hladiny alaninaminotransferázy [ALT] a aspartátaminotransferázy [AST] a jaterní steatózu. V jakém rozsahu jaterní steatóza související s lomitapidem podporuje zvýšení aminotransferáz, není známo. Ačkoli nebyly hlášeny případy jaterní dysfunkce (zvýšení aminotransferáz se zvýšením bilirubinu nebo mezinárodního normalizovaného poměru [INR] nebo jaterního selhání), existuje obava, že lomitapid může indukovat steatohepatitidu, která může za několik let progredovat do cirhózy. Není pravděpodobné, že by klinické studie na podporu bezpečnosti a účinnosti lomitapidu u HoFH byly schopné vzhledem k jejich rozsahu a době trvání tyto nežádoucí účinky detekovat.
S lomitapidem je spojeno zvýšení hladin aminotransferáz (ALT a/nebo AST) (viz bod 5.1). Nedocházelo k souběžnému nebo následnému klinicky významnému zvýšení sérového bilirubinu, INR nebo alkalické fosfatázy. Změny jaterních enzymů se nejčastěji vyskytují během navyšování dávky, ale mohou nastat kdykoli během léčby.
Sledování testů jaterních funkcí
Před zahájením léčby přípravkem Lojuxta změřte ALT, AST, alkalickou fosfatázu, celkový bilirubin, gama-glutamyltransferázu (gama-GT) a sérový albumin. Léčivý přípravek je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater a pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí. Jestliže jsou základní vyšetření jaterních testů abnormální, zvažte zahájení léčby léčivým přípravkem až poté, co hepatolog provede vhodná vyšetření a abnormality základních testů budou vysvětleny či vyřešeny.
V prvním roce měřte jaterní testy (minimálně ALT a AST) před každým zvýšením dávky nebo jednou měsíčně podle toho, co nastane dříve. Po prvním roce provádějte tyto testy nejméně jednou za 3 měsíce a před každým zvýšením dávky. Jestliže pozorujete zvýšení hladiny aminotransferáz, dávku přípravku Lojuxta snižte, a při přetrvávajícím nebo klinicky významném zvýšení ukončete léčbu (konkrétní doporučení uvádí Tabulka 1).
Modifikace dávky na základě zvýšené hladiny jatemích aminotransferáz
Tabulka 1 je shrnutím doporučení pro úpravu dávky a sledování pacientů, u kterých dojde během léčby přípravkem Lojuxta ke zvýšení hladiny aminotransferáz.
Tabulka 1: Úprava dávky a sledování pacientů se zvýšenými hladinami aminotransferáz
|
ALT nebo AST |
Doporučení týkající se léčby a sledování* |
|
>3x a <5x horní mez normálního rozsahu (ULN) |
• Zvýšení potvrďte opakovaným měřením v rámci jednoho týdne. • Jestliže se zvýšení potvrdí, snižte dávku a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). • Testy opakujte každý týden, a jestliže budou přítomny známky abnormálních jaterních funkcí (zvýšení bilirubinu či INR), jestliže aminotransferázy stoupnou nad 5x ULN nebo jestliže hladiny aminotransferáz během přibližně 4 týdnů neklesnou pod 3x ULN, přerušte léčbu. Pacienty s přetrvávajícím zvýšením hladin aminotransferáz >3x ULN odešlete k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, zvažte snížení dávky a častější sledování jaterních testů. |
|
>5x ULN |
• Přerušte léčbu a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). Jestliže během přibližně 4 týdnů hladiny aminotransferáz neklesnou pod 3x ULN, odešlete pacienta k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, snižte dávku a častěji sledujte jaterní testy. |
*Doporučení založené na ULNpřibližně 30-40 mezinárodních jednotek/l.
Jestliže je zvýšení aminotransferáz provázeno klinickými symptomy poškození jater (např. nevolností, zvracením, bolestí břicha, horečkou, žloutenkou, letargií, příznaky podobnými chřipce), vzestupem hladin bilirubinu na >2x ULN nebo aktivním onemocněním jater, přerušte léčbu přípravkem Lojuxta a odešlete pacienta k hepatologovi na další vyšetření.
Opětovné nasazení léčby lze zvážit tehdy, pokud přínosy převyšují rizika související s potenciálním onemocněním jater.
Jaterní steatóza a riziko progresivního onemocnění jater
Většina léčených pacientů vykazuje vzestup obsahu tuku v játrech, což je ve shodě s mechanismem působení lomitapidu. V otevřené studii fáze 3 došlo u 18 z 23 pacientů s HoFH k vývoji jaterní steatózy (obsah tuku v játrech > 5,56 %) dle měření nukleární magnetickou rezonanční spektroskopií (MRS) (viz bod 5.1). Medián absolutního vzestupu jaterního tuku byl dle měření pomocí MRS jak po 26 týdnech, tak po 78 týdnech léčby 6 % oproti 1 % při zahájení studie. Jaterní steatóza je rizikový faktor progresivního onemocnění jater včetně steatohepatitidy a cirhózy. Dlouhodobé následky jaterní steatózy související s léčbou přípravkem Lojuxta nejsou známy. Klinické údaje naznačují, že nahromadění tuku v játrech je po ukončení léčby přípravkem Lojuxta reverzibilní, ale není známo, zda přetrvávají histologické změny, zejména po dlouhodobém užívání.
Sledování důkazů přítomnosti progresivního onemocnění jater
Při zahájení léčby a dále každý rok by se měl provádět pravidelný screening steatohepatitidy/fibrózy s použitím následujících zobrazovacích metod a hodnocení biomarkerů:
• Zobrazení elasticity tkáně, např. fibrosken, ARFI (Acoustic Radiation Force Impulse) nebo elastografie s použitím magnetické rezonance (MR)
• Gama-GT a sérový albumin pro detekci možného poškození jater
• Minimálně jeden marker z každé z následujících kategorií:
• Vysoce senzitivní C-reaktivní protein (hs-CRP), rychlost sedimentace erytrocytů (ESR), CK-18 fragment, NashTest (zánět jater)
• Rozšířený panel pro detekci jaterní fibrózy (ELF), fibrometr, poměr AST/ALT, skóre fib-4, fibrotest (jaterní fibróza)
Provedení těchto testů a jejich interpretace by měly zahrnovat spolupráci ošetřujícího lékaře s hepatologem. U pacientů s výsledky naznačujícími přítomnost steatohepatitidy nebo fibrózy by měla být zvážena biopsie jater.
Jestliže má pacient biopticky potvrzenou steatohepatitidu nebo fibrózu, měl by být znovu zhodnocen poměr přínosů a rizik a léčba by měla být ukončena, je-li to nutné.
Souběžné užívání inhibitorů CYP3A4
Zdá se, že lomitapid je citlivým substrátem pro metabolismus cestou CYP3A4. Inhibitory CYP3A4 zvyšují expozici lomitapidu, přičemž silné inhibitory zvyšují expozici přibližně 27krát. Souběžné užití středně silných nebo silných inhibitorů CYP3A4 a přípravku Lojuxta je kontraindikováno (viz bod 4.3). V klinických studiích s lomitapidem se u jednoho pacienta s HoFH během několika dní po zahájení užívání silného inhibitoru CYP3A4 klaritromycinu vyvinuly výrazně zvýšené hladiny aminotransferáz (ALT 24x ULN, AST 13x ULN). Jestliže se nelze vyvarovat léčby středně silnými nebo silnými inhibitory CYP3A4, mělo by být užívání přípravku Lojuxta v průběhu léčby ukončeno.
Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta má být podávána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4.
Souběžné užívání induktorů CYP3A4
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. Induktory CYP3A4 vykazují svůj účinek v závislosti na čase a dosažení maximálního účinku může trvat minimálně 2 týdny od zahájení léčby. Naopak při ukončení léčby může trvat minimálně 2 týdny, než indukce CYP3A4 odezní.
Očekává se, že souběžné podávání induktoru CYP3A4 snižuje účinek přípravku Lojuxta. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinu, pioglitazonu, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Použití třezalky tečkované současně s přípravkem Lojuxta je nutné se vyvarovat.
Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. Při vysazení induktoru CYP3A4 je nutné zvážit možnost zvýšené expozice a může být nutné snížit dávku přípravku Lojuxta.
Souběžné použití s inhibitory HMG-CoA reduktázv (..statinv")
Lomitapid zvyšuje plazmatické koncentrace statinů. U pacientů, kteří dostávají přípravek Lojuxta jako přídatnou terapii vedle statinů, by měly být sledovány nežádoucí účinky spojené s použitím vysokých dávek statinů. Statiny příležitostně mohou vyvolat myopatii. Ve vzácných případech může mít myopatie formu rhabdomyolýzy s akutním renálním selháním sekundárně způsobeným myoglobinurií nebo bez renálního selhání a může mít fatální následky. Všichni pacienti užívající přípravek Lojuxta vedle statinů by měli být informování o možném riziku myopatie a měli by být informováni, aby okamžitě hlásili nevysvětlitelnou bolest, citlivost nebo slabost svalů. S přípravkem Lojuxta by se neměly používat dávky simvastatinu > 40 mg (viz bod 4.3).
Grapefruitový džus
Ze stravy pacientů je při léčbě přípravkem Lojuxta nutno vyloučit grapefruitový džus.
Riziko supraterapeutické nebo subterapeutické antikoagulace u antikoagulačních přípravků na bázi kumarinu
Lomitapid zvyšuje plazmatické koncentrace warfarinu. Zvýšení dávky přípravku Lojuxta může vést k supraterapeutické antikoagulaci a snížení dávky může vést k subterapeutické antikoagulaci.
U jednoho z pěti pacientů užívajících souběžně warfarin přispěla obtížná kontrola INR k předčasnému ukončení studie fáze 3. Pacienti užívající warfarin by měli podstupovat pravidelné sledování INR, zejména po jakýchkoli změnách dávkování přípravku Lojuxta. Dávka warfarinu by měla být upravena dle klinické indikace.
Požívání alkoholu
Alkohol může zvyšovat hladinu tuku v játrech a může vést k indukci nebo exacerbaci poškození jater. Ve studii fáze 3 udávali 3 ze 4 pacientů se zvýšením ALT >5x ULN konzumaci alkoholu překračující limit doporučený v protokolu. Požívání alkoholu během léčby přípravkem Lojuxta se nedoporučuje.
Hepatotoxické přípravky
Opatrnosti je třeba při použití přípravku Lojuxta s dalšími léčivými přípravky se známým hepatotoxickým potenciálem, např. izotretinoinem, amiodaronem, acetaminofenem (>4 g/den po >3 dny/týden), metotrexátem, tetracyklinem a tamoxifenem. Účinek souběžného podávání přípravku Lojuxta s dalšími hepatotoxickými léčivými přípravky není znám. Může být nutné častější sledování jaterních testů.
Snížená absorpce vitamínů rozpustných v tucích a mastných kyselin v séru
Vzhledem k mechanismu působení lomitapidu v tenkém střevě může lomitapid snižovat absorpci živin rozpustných v tucích. Ve studii fáze 3 byly pacientům denně poskytovány nutriční doplňky s vitamínem E, kyselinou linolovou, ALA, EPA a DHA. V této studii se úroveň mediánu sérového vitamínu E, ALA, kyseliny linolové, EPA, DHA a kyseliny arachidonové mezi výchozím bodem studie a týdnem 26 snížila, ale zůstala nad spodním limitem referenčního rozmezí. Nežádoucí klinické důsledky tohoto snížení při léčbě lomitapidem nebyly pozorovány po dobu až 78 týdnů. Pacienti léčení přípravkem Lojuxta by měli denně užívat doplňky, které obsahují 400 mezinárodních jednotek vitamínu E a přibližně 200 mg kyseliny linolové, 210 mg ALA, 110 mg EPA a 80 mg DHA.
Antikoncepční opatření u žen ve fertilním věku.
Před zahájením léčby u žen ve fertilním věku by mělo být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z důvodu průjmu a/nebo zvracení (viz bod 4.5). Perorální antikoncepční přípravky na bázi estrogenů jsou slabými inhibitory CYP3A4 (viz bod 4.2).
Pacientky by měly být informovány, že v případě otěhotnění musejí neprodleně informovat svého lékaře a ukončit užívání přípravku Lojuxta (viz bod 4.6).
Laktóza
Přípravek Lojuxta obsahuje laktózu, pacienti se vzácnými hereditárními problémy jako jsou intolerance galaktózy, vrozený deficit laktázy nebo glukózová-galaktózová malabsorpce by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
Tabulka 2: Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
Inhibitory CYP3A4 |
Při současném podávání lomitapidu v dávce 60 mg s ketokonazolem, silným inhibitorem CYP3A4, v dávce 200 mg dvakrát denně se zvýšila AUC lomitapidu přibližně 27krát a Cmax přibližně 15krát. Interakce mezi středně silnými inhibitory CYP3A4 a lomitapidem nebyly studovány. Předpokládá se, že středně silné inhibitory CYP3A4 mají významný vliv na farmakokinetiku lomitapidu. Očekává se, že souběžné použití středně silných inhibitorů CYP3A4 zvyšuje expozici lomitapidu 4-10krát podle výsledků studie se silným inhibitorem CYP3A4 podle historických údajů pro zkušební model CYP3A4 midazolam. Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při podávání lomitapidu 20 mg současně s atorvastatinem (tj. |
Použití silných nebo středně silných inhibitorů CYP3A4 spolu s přípravkem Lojuxta je kontraindikováno. Jestliže se nelze vyvarovat léčby antimykotiky azoly (např. itrakonazolem, ketokonazolem, flukonazolem, vorikonazolem, posakonazolem), antiarytmikem dronedaronem, makrolidovými antibiotiky (např. erytromycinem, klaritromcinem), ketolidovými antibiotiky (např. telitromycinem), inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem, měla by být během této léčby léčba přípravkem Lojuxta pozastavena (viz body 4.3 a 4.4). Grapefruitový džus je středně silným inhibitorem CYP3A4 a má se za to, že podstatně zvyšuje expozici lomitapidu. Pacienti užívající přípravek Lojuxta by se měli vyvarovat konzumace grapefruitového džusu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta musí být užívána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4. Příklady slabých inhibitorů CYP3A4 zahrnují: alprazolam, |
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
slabým inhibitorem CYP3A4) se AUC a Cmax lomitapidu zvýšily přibližně 2x. Pokud byl lomitapid podáván 12 hodin po atorvastatinu, žádné klinicky významné zvýšení expozice lomitapidu nebylo pozorováno. Při podávání lomitapidu 20 mg s ethinyl estradiolem/norgestimátem (tj. slabým inhibitorem CYP3A4), žádné klinicky významné zvýšení expozice lomitapidu nebylo pozorováno při užívání současném ani s intervalem 12 hodin. |
amiodaron, amlodipin, atorvastatin, azitromycin, bikalutamid, cilostazol, cimetidin, cyklosporin, klotrimazol, fluoxetin, fluvoxamin, fosaprepitant, ginkgo, vodilku kanadskou, izoniazid, ivakaftor, lacidipin, lapatinib, linagliptin, nilotinib, perorální antikoncepční přípravky obsahující estrogeny, pazopanib, mátový olej, propiverin, ranitidin, ranolazin, roxithromycin, hořké pomeranče, takrolimus, tikagrelor a tolvaptan. Tento seznam není úplný a předepisující lékaři by měli zkontrolovat preskripční informace o léčivech, která mají být podávána spolu s přípravkem Lojuxta s ohledem na možné interakce zprostředkované CYP3A4. Účinek podávání více než jednoho slabého inhibitoru CYP3A4 nebyl testován, ale očekává se, že účinek na expozici lomitapidu je větší než při současném podávání jednotlivých inhibitorů s lomitapidem. Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta. | |
|
Induktory CYP3A4 |
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. To by mohlo následně snížit účinek lomitapidu. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. |
Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinnu, pioglitazonu, třezalky tečkované, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. |
|
Sekvestranty žlučových kyselin |
Interakce lomitapidu se sekvestranty žlučových kyselin (pryskyřicemi, např. kolesevelamem a cholestyraminem) nebyly testovány. |
Protože sekvestranty žlučových kyselin mohou interferovat s absorpcí perorálních léčivých přípravků, měly by se užívat minimálně 4 hodiny před přípravkem Lojuxta nebo minimálně 4 hodiny po něm. |
Účinky lomitapidu na jiné léčivé přípravky
Inhibitory HMG-CoA reduktázy („statiny“): Lomitapid zvyšuje plazmatické koncentrace statinů. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním simvastatinu 40 mg se zvýšily u kyseliny simvastatinu hodnoty AUC o 68 % a Cmax o 57 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním atorvastatinu 20 mg se zvýšily u kyseliny atorvastatinu hodnoty AUC o 52 % a Cmax o 63 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním rosuvastatinu 20 mg se zvýšila hodnota Tmax rosuvastatinu z 1 na 4 hodiny, hodnota AUC se zvýšila o 32 % a hodnota Cmax zůstala nezměněná. Riziko myopatie u simvastatinu je závislé na dávce. Použití přípravku Lojuxta je u pacientů léčených vysokými dávkami statinů (> 40 mg) kontraindikováno (viz body 4.3 a 4.4).
Kumarinové antikoagulační přípravky: Podávání lomitapidu 60 mg do dosažení ustáleného stavu a po dobu 6 dní po warfarinu 10 mg zvýšilo hodnotu INR 1,26krát. Hodnota AUC pro R(+)-warfarin se zvýšila o 25 % a pro S(-)-warfarin o 30 %. Hodnota Cmax pro R(+)-warfarin se zvýšila o 14 % a pro S(-)-warfarin o 15 %. U pacientů užívajících souběžně kumariny (např. warfarin) a přípravek Lojuxta by měla být před zahájením užívání přípravku Lojuxta stanovena hodnota INR a tato hodnota by měla být pravidelně kontrolována, přičemž dávkování kumarinů by mělo být upraveno dle klinické potřeby (viz bod 4.4).
Fenofibrát, niacin a ezetimib: Při podávání lomitapidu do dosažení ustáleného stavu před podáním mikronizovaného fenofibrátu 145 mg, niacinu s prodlouženým uvolňováním 1000 mg nebo ezetimibu 10 mg nebyly pozorovány klinicky významné účinky působení kteréhokoli z těchto léčivých přípravků. Při souběžném podávání s přípravkem Lojuxta není nutná úprava dávky.
Perorální antikoncepce: Při podávání lomitapidu 50 mg do dosažení ustáleného stavu spolu s perorálním antikoncepčním přípravkem na bázi estrogenů nebyl pozorován klinicky ani statisticky významný vliv na farmakokinetiku složek perorální antikoncepce (ethinylestradiolu a 17-deacetylnorgestimátu, metabolitu norgestimátu). Neočekává se, že by lomitapid přímo ovlivňoval účinnost perorální antikoncepce na bázi estrogenů, nicméně průjem a/nebo zvracení mohou snížit absorpci hormonů. U případů prodlouženého nebo závažného průjmu a/nebo zvracení trvajících více než 2 dny by měla být po dobu 7 dnů po ústupu symptomů použita další antikoncepční metoda.
Substráty P-gp: Lomitapid in vitro inhibuje P-gp a může zvyšovat absorpci P-gp substrátů. Souběžné podávání přípravku Lojuxta s P-pg substráty (např. aliskirenem, abrisentanem, kolchicinem, dabigatran etexilátem, dixoginem, everolimem, fexofenadinem, imatinibem, lapatinibem, maravirokem, nilotinibem, posakonazolem, ranolazinem, saxagliptinem, sirolimem, sitagliptinem, talinololem, tolvaptanem, topotekanem) může zvýšit absorpci P-gp substrátů. Při souběžném použití s přípravkem Lojuxta by mělo být zváženo snížení dávky P-gp substrátu.
In vitro hodnocení lékových interakcí: Lomitapid inhibuje CYP3A4. Lomitapide neindukuje CYP 1A2, 3A4 ani 2B6 a neinhibuje CYP 1A2, 2B6, 2C9, 2C19, 2D6 ani 2E1. Lomitapid není P-gp substrátem, ale inhibuje P-gp. Lomitapid neinhibuje protein rezistence u karcinomu prsu (BCRP).
4.6 Fertilita, těhotenství a kojení
Přípravek Lojuxta je v těhotenství kontraindikován. Neexistují spolehlivé údaje o použití přípravku u těhotných žen. Studie na zvířatech prokázaly vývojovou toxicitu (teratogenitu, embryotoxicitu, viz bod 5.3). Možné riziko pro člověka je neznámé.
Použití u žen ve fertilním věku.
Před zahájením léčby u žen ve fertilním věku by mělo být vyloučeno těhotenství, mělo by být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z důvodu průjmu a/nebo zvracení. Dokud symptomy neustoupí, je nutno používat další antikoncepční metody (viz bod 4.5).
Kojení
Není známo, zda se lomitapid vylučuje do mateřského mléka. Vzhledem k potenciálu nežádoucích účinků zjištěnému na základě studií s lomitapidem na zvířatech (viz bod 5.3) je nutné rozhodnout, zda ukončit kojení nebo užívání léčivého přípravku, a to při zvážení významu léčivého přípravku pro matku.
Fertilita
U potkaních samců ani samic, kterým byl podáván lomitapid se systémovou expozicí (AUC) ve výši odhadovaného čtyř- až pětinásobku expozice u lidí při maximální doporučené dávce u člověka, nebyly pozorovány žádné nežádoucí účinky na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Lojuxta může mít mírný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejzávažnějšími nežádoucími účinky během léčby byly abnormality jaterních aminotransferáz (viz bod 4.4).
Nejčastějšími nežádoucími účinky byly gastrointestinální účinky. Nežádoucí gastrointestinální účinky byly hlášeny u 27 (93 %) z 29 pacientů klinické studie fáze 3. Průjem se objevil u 79 %, nevolnost u 65 %, dyspepsie u 38 % a zvracení u 34 % pacientů. Další účinky, které hlásilo minimálně 20 % pacientů, zahrnují bolest břicha, břišní dyskomfort, nadmutí břicha, zácpu a plynatost.
Nežádoucí gastrointestinální účinky se častěji objevovaly během fáze navyšování dávky ve studii a poklesly, jakmile pacienti dosáhli maximální tolerované dávky lomitapidu.
Gastrointestinální nežádoucí účinky se závažnou intenzitou byly hlášeny u 6 (21 %) z 29 pacientů v klinické studii fáze 3, přičemž nejčastějším účinkem byl průjem (4 pacienti, 14 %), zvracení (3 pacienti, 10 %) a bolest břicha, nadmutí a/nebo dyskomfort (2 pacienti, 7 %). Gastrointestinální účinky přispěly k časnému ukončení studie u 4 (14 %) pacientů.
Nejčastěji udávanými nežádoucími účinky se závažnou intenzitou byly průjem (4 subjekty, 14 %), zvracení (3 pacienti, 10 %) a nadmutí břicha a zvýšení ALT (pokaždé 2 subjekty, 7 %).
Přehled nežádoucích účinků v tabulce
Dle frekvence jsou nežádoucí účinky definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), málo časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), s neznámou frekvencí (z dostupných údajů nelze určit).
Tabulka 3 zahrnuje veškeré nežádoucí účinky hlášené u 35 pacientů léčených ve studii fáze 2 UP1001 a ve studii fáze 3 UP1002/AEGR-733-005 nebo v rozšířené studii AEGR-733-012.
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Časté |
Gastroenteritida |
|
Poruchy metabolismu a výživy |
Velmi časté |
Snížená chuť k jídlu |
|
Poruchy nervového systému |
Časté |
Závratě Bolest hlavy Migréna |
|
Gastrointestinální poruchy |
Velmi časté |
Břišní dyskomfort Bolest horní části břicha Plynatost Distenze břicha Zácpa |
|
Časté |
Gastritida Rektální tenesmy Aerofagie Nutkání na stolici Říhání Častá stolice Dilatace žaludku Onemocnění žaludku Gastroezofageální reflux Krvácení z hemoroidů Regurgitace | |
|
Poruchy jater a žlučových cest |
Časté |
Jaterní steatóza Hepatotoxicita Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Časté |
Ekchymóza Papuly Erytematózní vyrážka Xantomy |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Únava |
|
Vyšetření |
Velmi časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Snížení tělesné hmotnosti |
|
Časté |
Zvýšený mezinárodní normalizovaný poměr Zvýšená alkalická fosfatáza v krvi Snížení draslíku v krvi Snížený karoten Abnormální mezinárodní normalizovaný poměr Abnormální testy jaterních funkcí Prodloužený protrombinový čas Zvýšené transaminázy Snížený vitamín E Snížený vitamín K |
Tabulka 4 uvádí všechny nežádoucí účinky u subjektů, které dostávaly lomitapid v monoterapii (n=291), léčených ve studiích fáze 2 u subjektů se zvýšenou hladinou LDL-C (N=462).
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Méně časté |
Gastroenteritida Gastrointestinální infekce Chřipka Nazofaryngitida Sinusitida |
|
Poruchy krve a lymfatického systému |
Méně časté | |
|
Poruchy metabolismu a výživy |
Časté |
Snížená chuť k jídlu |
|
Méně časté |
Dehydratace Zvýšená chuť k jídlu | |
|
Poruchy nervového systému |
Méně časté |
Parestézie Ospalost |
|
Poruchy oka |
Méně časté |
Otok oka |
|
Poruchy ucha a labyrintu |
Méně časté |
Závrať |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté |
Léze hltanu Syndrom kašle horních cest dýchacích |
|
Gastrointestinální poruchy |
Velmi časté |
Plynatost |
|
Časté |
Bolest horní části břicha Distenze břicha Bolest břicha Zvracení Břišní dyskomfort Říhání Bolest v podbřišku Častá stolice | |
|
Méně časté |
Sucho v ústech Tvrdá stolice Gastroezofageální reflux Citlivost břicha Dyskomfort v epigastriu Dilatace žaludku Hemateméza Krvácení v dolní části gastrointestinálního traktu Refluxní ezofagitida | |
|
Poruchy jater a žlučových cest |
Méně časté |
Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Puchýře Suchá kůže Nadměrné pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté |
Svalové spasmy |
|
Méně časté |
Bolest kloubů Bolest svalů Bolest končetin Otok kloubu Svalové záškuby | |
|
Poruchy ledvin a močových cest |
Méně časté |
Hematurie |
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Celkové poruchy a reakce v místě |
Časté |
Únava |
|
podání |
Slabost | |
|
Méně časté |
Bolest na hrudi Mrazení Rychlý pocit nasycení Poruchy chůze Malátnost Horečka | |
|
Vyšetření |
Časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Zvýšení jaterních enzymů Abnormální testy jaterních funkcí Snížený počet neutrofilů Snížený počet leukocytů |
|
Méně časté |
Snížení tělesné hmotnosti Zvýšený bilirubin v krvi Zvýšená hladina gama-glutamyltransferázy Zvýšené procento neutrofilů Protein v moči Prodloužený protrombinový čas Abnormální testy plicních funkcí Zvýšený počet leukocytů |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování neexistuje specifická léčba. U hlodavců byla dobře snášena jednorázová perorální dávka lomitapidu >600krát vyšší než maximální doporučená dávka pro člověka (1 mg/kg). Maximální dávkou podanou lidským subjektům v klinických studiích bylo 200 mg ve formě jednorázové dávky, nevyskytly se žádné nežádoucí účinky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná léčiva ovlivňující hladinu lipidů, samotná. ATC kód: C10AX12
Mechanismus účinku
Lomitapid je selektivní inhibitor mikrozomálního transferového proteinu (MTP), intracelulárního proteinu přenášejícího lipidy, který se nachází v lumen endoplazmatického retikula a zodpovídá za vazbu a převod jednotlivých lipidových molekul mezi membránami. MTP hraje klíčovou roli při sestavování lipoproteinů obsahujících apo B v játrech a střevech. Inhibice MTP snižuje sekreci lipoproteinů a cirkulující koncentrace lipidů přenášených v lipoproteinech včetně cholesterolu a triglyceridů.
Klinická účinnost a bezpečnost
Účinnost a bezpečnost lomitapidu při současném podávání diety s nízkým obsahem tuků a dalších terapií snižujících lipidy u dospělých pacientů s HoFH hodnotila otevřená studie s jedním ramenem (UP1002/AEGR-733-005). Pacienti byli instruováni, aby dodržovali dietu s nízkým obsahem tuků (< 20 % kalorického příjmu z tuků) i terapii snižující lipidy, kterou měli při vstupu do studie, včetně aferézy, byla-li tato nutná, a to po dobu 6 týdnů předcházejících výchozímu bodu studie minimálně až do týdne 26. Dávka lomitapidu byla navyšována z 5 mg na individuálně stanovenou maximální tolerovanou dávku až 60 mg. Po týdnu 26 byl pacientům lomitapid ponechán za účelem zjištění účinků dlouhodobé léčby a bylo jim umožněno změnit základní terapii snižující lipidy. Studie zahrnovala celkem 78 týdnů léčby.
Do studie bylo zařazeno 29 pacientů, z nichž 23 dokončilo týden 78. Zařazeno bylo 16 mužů (55 %) a 13 žen (45 %) s průměrným věkem 30,7 let a ve věkovém rozmezí 18 až 55 let. Průměrná dávka lomitapidu v týdnu 26 byla 45 mg a v týdnu 78 byla 40 mg. V týdnu 26 byla průměrná procentuální změna hladiny LDL-C u populace „intention-to-treat“ (ITT) oproti výchozímu bodu -40 %
(p < 0,001). Průměrná procentuální změna oproti výchozímu bodu studie v týdnu 26 s použitím metody LOCF (Last Observation Carried Forward) pro každé hodnocení je uvedena na obrázku 1.
Obrázek 1: Průměrné procentuální změny hladiny LDL-C mezi vstupní hodnotou a týdnem
26 v hlavní studii účinnosti UP1002/AEGR-733-005 (primární cíl) s použitím LOCF pro každé hodnocení (n=29)

Týden studie
Změny lipidů i lipoproteinů v týdnu 26 a 78 při léčbě lomitapidem uvádí Tabulka 5.
Tabulka 5: Absolutní hodnoty a procentuální změny hladin lipidů a lipoproteinů mezi
výchozí hodnotou a týdny 26 a 78 (hlavní studie účinnosti UP1002/AEGR-733-005).
|
Parametr (jednotky) |
Výchozí bod |
Týden 26 / LOCF (n=29) |
Týden 78 (n=23) | ||||
|
Průměr (SD) |
Průměr (SD) |
% změna |
Hodnota p b |
Průměr (SD) |
% změna |
Hodnota p b | |
|
LDL-C, přímý (mg/dl) |
336 (114) |
190 (104) |
-40 |
< 0,001 |
210 (132) |
-38 |
< 0,001 |
|
Celkový cholesterol (TC) (mg/dl) |
430 (135) |
258 (118) |
-36 |
< 0,001 |
281 (149) |
-35 |
< 0,001 |
|
Apolipoprotein B (apo B) (mg/dl) |
259 (80) |
148 (74) |
-39 |
< 0,001 |
151 (89) |
-43 |
< 0,001 |
|
Triglyceridy (TG) (mg/dl)a |
92 |
57 |
-45 |
0,009 |
59 |
-42 |
0,012 |
|
Cholesterol z jiných lipoproteinů než o vysoké hustotě (non-HDL-C) (mg/dl) |
386 (132) |
217 (113) |
-40 |
< 0,001 |
239 (146) |
-39 |
< 0,001 |
|
Cholesterol z lipoproteinů o velmi nízké hustotě (VLDL-C) (mg/dl) |
21 (10) |
13 (9) |
-29 |
0,012 |
16 (15) |
-31 |
0,013 |
|
Lipoprotein (a) (Lp(a)) (nmol/l)a |
66 |
61 |
-13 |
0,094 |
72 |
-4 |
< 0,842 |
|
Cholesterol z lipoproteinů o vysoké hustotě (HDL-C) (mg/dl) |
44 (11) |
41 (13) |
-7 |
0,072 |
43 (12) |
-4,6 |
0,246 |
a Medián uvedený pro TG a Lp(a). Hodnota p je založena na průměrné procentuální změně b Hodnota p u střední procentuální změny oproti výchozímu bodu studie na základě párového t-testu
Jak v týdnu 26, tak v týdnu 78 došlo k významnému snížení hladin LDL-C, TC, apo B, TG, non-HDL-C a VLDL-C a změny u HDL-C vykazovaly v týdnu 26 trend směrem ke snížení a v týdnu 78 se vrátily k výchozím hodnotám.
Účinek přípravku Lojuxta na kardiovaskulární morbiditu a mortalitu nebyl stanoven.
Při zahájení studie užívalo 93 % pacientů statin, 76 % pacientů ezetimib, 10 % niacin, 3 % sekvestrant žlučových kyselin a 62 % pacientů bylo léčeno aferézou. U 15 z 23 (65 %) pacientů byla v týdnu 78 redukována jejich léčba na snížení lipidů, včetně plánovaných a neplánovaných snižování/přerušení. Aferéza byla přerušena u 3 ze 13 pacientů, kteří byli aferézou léčeni v týdnu 26, a u 3 pacientů byla frekvence při současném udržení nízkých hladin LDL-C i po týdnu 78 snížena. Klinický přínos omezení základní terapie snižující lipidy, včetně aferézy, není jistý.
Z 23 pacientů, kteří dokončili týden 26 studie, došlo u 19 (83 %) ke snížení LDL-C o > 25 %, přičemž 8 (35 %) z nich mělo v tomto časovém bodě LDL-C < 100 mg/dl a 1 pacient měl LDL-C < 70 mg/dl.
V této studii došlo u 10 pacientů ke zvýšení AST a/nebo ALT >3x ULN (viz Tabulka 6).
Nejvyšší výsledky testů jaterních funkcí po první dávce (hlavní studie účinnosti UP1002/AEGR-733-005)
|
Parametr/abnormalita |
N (%) |
|
ALT | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
6 (20,7) |
|
>5 až < 10 x ULN |
3 (10,3) |
|
>10 až < 20 x ULN |
1 (3,4) |
|
> 20 x ULN |
0 |
|
AST | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
5 (17,2) |
|
>5 až < 10 x ULN |
1 (3,4) |
|
>10 až < 20 x ULN |
0 |
|
> 20 x ULN |
0 |
Tabulka 6:
Zvýšení ALT a/nebo AST > 5x ULN bylo zvládnuto snížením dávky nebo dočasným pozastavením podávání lomitapidu a všichni pacienti byli schopni v léčbě studovaným léčivem pokračovat. Nebylo pozorováno klinicky významné zvýšení celkového bilirubinu ani alkalické fosfatázy. Jaterní tuk byl během klinické studie u všech dostupných pacientů prospektivně měřen s použitím MRS (Tabulka 7). Údaje od jedinců, u kterých byla po ukončení lomitapidu prováděna opakovaná měření, ukázaly, že nahromadění tuku v játrech je reverzibilní, ale není známo, zda nepřetrvávají histologické změny.
Tabulka 7: Maximální kategorické změny v % jaterního tuku (hlavní studie účinnosti
UP1002/AEGR-733-005)
|
Maximální absolutní nárůst % jaterního tuku |
Fáze účinnosti Týdny 0-26 N (%) |
Fáze bezpečnosti Týdny 26-78 N (%) |
Celá studie týdny 0-78 N (%) |
|
Počet hodnotitelných pacientů |
22 |
22 |
23 |
|
< 5% |
9 (41) |
6 (27) |
5 (22) |
|
> 5 % až < 10 % |
6 (27) |
8 (36) |
8 (35) |
|
> 10 % až < 15 % |
4 (18) |
3 (14) |
4 (17) |
|
> 15 % až < 20 % |
1 (5) |
4 (18) |
3 (13) |
|
> 20 % až < 25 % |
1 (5) |
0 |
1 (4) |
|
> 25 % |
1 (5) |
1 (5) |
2 (9) |
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lojuxta u jedné nebo více podskupin pediatrické populace s HoFH (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Absolutní perorální biologická dostupnost lomitapidu je 7 %. Absorpce není omezena prostupem léčiva přes střevní bariéru, ale je převážně ovlivněna extenzivním efektem prvního průchodu. Vrcholná plazmatická koncentrace lomitapidu byla dosažena za 4 až 8 hodin po podání perorální dávky. Farmakokinetika lomitapidu je přibližně proporcionální k dávce perorálních jednorázových dávek v terapeutickém rozmezí. U dávek vyšších než 60 mg je naznačena tendence k nelineární závislosti a tyto dávky nejsou doporučeny.
Při podání více dávek se hodnoty Cmax a AUC zvýšily přibližně proporcionálně k dávce lomitapidu. Hodnoty Cmax a AUC se zvýšily jak po vysoce tučném jídle (77 %, respektive 58 %), tak po nízkotučném jídle (70 %, respektive 28 %). Akumulace lomitapidu v plazmě byla konzistentní s hodnotou předpokládanou na základě jedné dávky po podávání jedné perorální dávky denně nad 25 mg po dobu až 4 týdnů. Interindividuální variabilita hodnoty AUC lomitapidu byla přibližně 50 %.
V ustáleném stavu byla akumulace lomitapidu 2,7 při 25 mg a 3,9 při 50 mg.
Distribuce
Po intravenózním podání byl distribuční objem lomitapidu velký (průměr = 1 200 litrů) i přes vysoký stupeň (> 99,8 %) vazby na plazmatické proteiny. Ve studiích na zvířatech se lomitapid vysoce (200násobně) koncentroval v játrech.
Biotransformace
Lomitapid se extenzivně metabolizuje, převážně cestou CYP3A4. Izoformy CYP 2E1, 1A2, 2B6, 2C8 a 2C19 se účastní méně a izoformy 2D6 a 2C9 se metabolismu lomitapidu neúčastní.
Eliminace
Po podání dávky radioaktivně značeného perorálního roztoku zdravým subjektům bylo 93 % podané dávky vyloučeno močí a stolicí. Přibližně 33 % radioaktivity bylo vyloučeno do moči jako metabolity. Zbytek byl vyloučen stolicí, primárně jako oxidované metabolity. Eliminační poločas lomitapidu byl přibližně 29 hodin.
Zvláštní populace
Údaje ze stěžejní klinické studie byly analyzovány s ohledem na vliv možných souvisejících proměnných na expozici lomitapidu. Z vyšetřovaných parametrů (rasa, index tělesné hmotnosti (BMI), hmotnost, věk) může být jako možná související proměnná klasifikován pouze BMI.
Věk a pohlaví
Věk (18-64 let) ani pohlaví nemají na farmakokinetiku lomitapidu klinicky významný vliv.
Rasa
U bělochů nebo latinskoamerické populace není nutná úprava dávky. K dispozici není dostatek informací pro stanovení, zda přípravek Lojuxta vyžaduje úpravu dávky u dalších ras. Protože se ale léčivý přípravek podává způsobem postupného zvyšování dávky podle individuální bezpečnosti a snášenlivosti pacienta, úprava dávkovacího režimu na základě rasy není doporučena.
V populaci s poškozením funkce ledvin byl lomitapid studován pouze u pacientů s terminálním onemocněním ledvin (ESRD). Farmakokinetická studie u pacientů s ESRD podstupujících dialýzu ukázala 36% vzestup průměrné koncentrace lomitapidu v plazmě ve srovnání s párovanými zdravými kontrolami. Koncový poločas lomitapidu nebyl ovlivněn.
Jaterní insuficience
Pro hodnocení farmakokinetiky 60 mg lomitapidu u zdravých dobrovolníků s normálními jaterními funkcemi ve srovnání s pacienty s mírným postižením funkce jater (třída A dle Child-Pugha) a středně těžkým postižením funkce jater (třída B dle Child-Pugha) byla provedena otevřená studie s jednou dávkou. Hodnoty AUC a Cmax lomitapidu u pacientů se středně těžkým postižením funkce jater byly o 164 %, respektive 361 % vyšší než u zdravých dobrovolníků. Hodnoty AUC a Cmax lomitapidu u pacientů s mírným postižením funkce jater byly o 47 %, respektive 4 % vyšší než u zdravých dobrovolníků. U pacientů s těžkým postižením funkce jater (skóre dle Child-Pugha 10-15) nebyl přípravek Lojuxta studován.
Pediatrická populace
U dětí mladších 18 let nebyl přípravek Lojuxta studován.
Starší populace
U pacientů starších 65 let nebyl přípravek Lojuxta studován.
5.3 Předklinické údaje vztahující se k bezpečnosti
Základním zjištěním toxikologických studií léčiva s opakovanou perorální dávkou u hlodavců a psů byla akumulace lipidů v tenkém střevě a/nebo v játrech spojená s poklesem sérových hladin cholesterolu a/nebo triglyceridů. Tyto změny jsou vzhledem k mechanismu působení lomitapidu sekundární. Další změny související s játry ve studiích toxicity s opakovanou dávkou u potkanů a psů zahrnovaly zvýšené hladiny sérové aminotransferázy, subakutní zánět (pouze u potkanů) a nekrózu jednotlivých buněk. V jednoleté studii s opakovanou dávkou u psů se v játrech nevyskytovaly mikroskopické změny, ačkoli u fen byla minimálně zvýšená sérová hladina AST.
U hlodavců byla pozorována plicní histiocytóza. U psů byl pozorován pokles parametrů erytrocytů i poikilocytóza a/nebo anizocytóza. U psů byla v šestiměsíční studii při 205násobku lidské expozice (AUC) 60 mg pozorována testikulární toxicita. V roční studii u psů nebyly při 64násobku lidské expozice 60 mg pozorovány nežádoucí účinky na varlata.
Ve studii nutriční karcinogenity u myší byl lomitapid podáván po dobu až 104 týdnů v dávkách v rozmezí 0,3 až 45 mg/kg/den. Při dávkách > 1,5 mg/kg/den u samců (> 2násobek lidské expozice při 60 mg denně na základě AUC) a > 7,5 mg/kg/den u samic (> 9násobek lidské expozice při 60 mg na základě AUC) došlo ke statisticky významnému vzestupu incidence adenomu i karcinomu jater. Incidence karcinomu tenkého střeva a/nebo kombinovaného adenomu a karcinomu (u myší vzácné nádory) byla významně zvýšena při dávkách > 15 mg/kg/den u samců (> 26násobek lidské expozice při 60 mg na základě AUC) a 15 mg/kg/den u samic (22násobek lidské expozice při 60 mg na základě AUC).
Ve studii perorální kancerogenity u potkanů byl lomitapid podáván po dobu až 99 týdnů v dávkách až
7,5 mg/kg/den u samců a 2,0 mg/kg/den u samic. U samců i samic byla pozorována fokální jaterní fibróza a pouze u samců byla pozorována cystická degenerace jater. U samců s vysokou dávkou byla pozorována zvýšená incidence pankreatického acinárního adenomu při expozici 6krát vyšší než u lidí při 60 mg na základě AUC.
V panelu studií in vitro a in vivo nebyl lomitapid mutagenní ani genotoxický.
Lomitapid neměl účinek na reprodukční funkce u samic potkanů v dávkách až 1 mg/kg nebo samců potkanů v dávkách až 5 mg/kg. Systémová expozice lomitapidu v těchto dávkách byla odhadována na čtyřnásobek (samice) až pětinásobek (samci) expozice u lidí při 60 mg na základě AUC.
Lomitapid byl u potkanů teratogenní při současné absenci mateřské toxicity při expozici (AUC) odhadované na dvojnásobek lidské expozice při 60 mg. U králíků se nevyskytly známky embryofetální toxicity při trojnásobku maximální doporučené denní dávky u lidí (MRHD) 60 mg na základě plochy tělesného povrchu. Embryofetální toxicita při absenci mateřské toxicity byla pozorována u králíků při > 6,5násobku MRHD. U fretek byl lomitapid toxický pro matku i teratogenní při dávkách < 1x MRHD.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky
předbobtnalý škrob (kukuřičný) sodná sůl karboxymethylškrobu mikrokrystalická celulóza monohydrát laktózy koloidní bezvodý oxid křemičitý magnesium-stearát
Obal tobolky želatina
oxid titaničitý (E171) červený oxid železitý (E172) žlutý oxid železitý (E172)
Tiskařský inkoust šelak
černý oxid železitý (E172) propylenglykol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Uchovávejte v dobře uzavřené lahvičce, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Lahvička z polyetylenu o vysoké hustotě (HDPE) vybavená vložkou z polyesteru / hliníkové fólie / kartonu a s polypropylenovým šroubovacím uzávěrem.
Velikost balení:
28 tobolek
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Aegerion Pharmaceuticals Ltd Lakeside House 1 Furzeground Way Stockley Park East Uxbridge UB11 1BD Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/851/004
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
^ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Lojuxta 40 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje lomitapidum 40 mg ve formě lomitapidum mesilas Pomocná látka se známým účinkem:
Jedna tvrdá tobolka obsahuje 259,79 mg laktózy (ve formě monohydrátu) (viz bod 4.4). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Tvrdá tobolka se žlutou svrchní částí / bílou spodní částí o délce 23,4 mm, potištěná černým inkoustem s textem „40 mg“ na spodní části a „A733“ na svrchní části tobolky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Lojuxta je indikován jako podpůrný přípravek užívaný spolu s dietou s nízkým obsahem tuků a dalšími léčivými přípravky na snížení hladiny lipidů spolu s aferézou lipoproteinů o nízké hustotě (LDL) či bez ní u dospělých pacientů s homozygotní formou familiární hypercholesterolémie (HoFH).
Tam, kde je to možné, by měla být homozygotní forma familiární hypercholesterolémie potvrzena geneticky. Je nutné vyloučit jiné formy primární hyperlipoproteinémie a sekundární příčiny hypercholesterolémie (např. nefrotický syndrom, hypotyreózu).
4.2 Dávkování a způsob podání
Léčbu přípravkem Lojuxta by měl zahajovat a sledovat lékař, který má zkušenosti s léčbou poruch lipidů.
Dávkování
Doporučená zahajovací dávka přípravku je 5 mg jednou denně. Po 2 týdnech může být dávka na základě přijatelné bezpečnosti a snášenlivosti zvýšena na 10 mg a poté v minimálně čtyřtýdenních intervalech na 20 mg, 40 mg a maximální doporučenou dávku 60 mg (viz bod 4.8).
Dávka by měla být zvyšována postupně, aby se minimalizovala incidence a závažnost gastrointestinálních nežádoucích účinků a zvýšení aminotransferáz.
Podávání se stravou může u přípravku Lojuxta zvyšovat expozici. Přípravek Lojuxta by se měl užívat nalačno, minimálně dvě hodiny po večeři, protože tuk obsažený v nedávno požitém jídle může mít negativní vliv na gastrointestinální snášenlivost.
Výskyt a závažnost gastrointestinálních nežádoucích účinků souvisejících s užíváním přípravku Lojuxta se snižují při dodržování diety s nízkým obsahem tuků. Pacienti by před zahájením léčby přípravkem Lojuxta měli držet dietu, ve které je méně než 20 % energie dodáváno z tuků, a během léčby by v této dietě měli pokračovat. Mělo by být zajištěno nutriční poradenství.
Pacienti by se měli vyvarovat konzumace grapefruitového džusu (viz body 4.4 a 4.5).
U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s atorvastatinem použijte jednu z těchto variant:
• Oddělte podání obou přípravků intervalem 12 hodin NEBO
• Snižte dávku přípravku Lojuxta o polovinu.
Pacienti užívající 5 mg by měli zůstat na této dávce.
Následně lze provést opatrnou titraci podle reakce hladiny LDL-C a bezpečnosti /snášenlivosti.
Po ukončení léčby atorvastatinem zvyšte dávku přípravku Lojuxta titrací podle hladiny LDL-C a bezpečnosti/snášenlivosti.
U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s jakýmkoliv dalším slabým inhibitorem CYP3A4 oddělte podání obou přípravků intervalem 12 hodin Omezení maximální dávky přípravku Lojuxta zvažte podle požadované odpovědi LDL-C.
Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta.
Na základě pozorování snížených hladin esenciálních mastných kyselin a vitamínu E v klinických studiích by pacienti po dobu léčby přípravkem Lojuxta měli také užívat denně nutriční doplňky zajišťující příjem 400 IU vitamínu E a přibližně 200 mg kyseliny linolové, 110 mg kyseliny eikosapentaenové (EPA), 210 mg kyseliny alfa-linolenové (ALA) a 80 mg kyseliny dokosahexaenové (DHA).
Starší populace
Zkušenosti s přípravkem Lojuxta jsou u pacientů ve věku 65 let a starších omezené. Proto je u těchto pacientů zapotřebí zvláštní obezřetnosti.
Protože doporučený režim dávkování zahrnuje zahájení na spodní hranici dávkovacího rozmezí a opatrné navyšování dávky podle individuální snášenlivosti pacienta, nedoporučuje se ve starším věku žádná úprava dávkovacího schématu.
Porucha jater
Přípravek Lojuxta je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater včetně pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí (viz bod 5.2).
U pacientů s mírnou poruchou funkce jater (třída A dle Child-Pugha) by neměla být překračována dávka 40 mg denně.
Porucha ledvin
U pacientů v terminálním stádiu onemocnění ledvin léčených dialýzou by neměla být překračována dávka 40 mg denně (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Lojuxta u dětí mladších 18 let nebyla stanovena, a použití tohoto léčivého přípravku u dětí se proto nedoporučuje. Nejsou dostupné žádné údaje.
Způsob podání
Perorální podání.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Pacienti se středně těžkou nebo těžkou poruchou funkce jater a pacienti s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí.
• Pacienti se známým významným nebo chronickým střevním onemocněním, např. zánětlivým onemocněním střeva nebo malabsorpcí.
• Souběžné podávání více než 40 mg simvastatinu (viz bod 4.5).
• Souběžné použití přípravku Lojuxta se silnými nebo středně silnými inhibitory cytochromu P450 (CYP) 3A4 (např. antimykotiky azoly, jako např. intrakonazolem, flukonazolem, ketokonazolem, vorikonazolem a posakonazolem, makrolidovými antibiotiky, jako např. erytromycinem nebo klaritromycinem, ketolidovými antibiotiky, jako např. telitromycinem, inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem a antiarytmikem dronedaronem [viz bod 4.5]).
• Těhotenství (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Abnormality jaterních enzymů a sledování jater
Lomitapid může způsobit zvýšení hladiny alaninaminotransferázy [ALT] a aspartátaminotransferázy [AST] a jaterní steatózu. V jakém rozsahu jaterní steatóza související s lomitapidem podporuje zvýšení aminotransferáz, není známo. Ačkoli nebyly hlášeny případy jaterní dysfunkce (zvýšení aminotransferáz se zvýšením bilirubinu nebo mezinárodního normalizovaného poměru [INR] nebo jaterního selhání), existuje obava, že lomitapid může indukovat steatohepatitidu, která může za několik let progredovat do cirhózy. Není pravděpodobné, že by klinické studie na podporu bezpečnosti a účinnosti lomitapidu u HoFH byly schopné vzhledem k jejich rozsahu a době trvání tyto nežádoucí účinky detekovat.
S lomitapidem je spojeno zvýšení hladin aminotransferáz (ALT a/nebo AST) (viz bod 5.1). Nedocházelo k souběžnému nebo následnému klinicky významnému zvýšení sérového bilirubinu, INR nebo alkalické fosfatázy. Změny jaterních enzymů se nejčastěji vyskytují během navyšování dávky, ale mohou nastat kdykoli během léčby.
Sledování testů jaterních funkcí
Před zahájením léčby přípravkem Lojuxta změřte ALT, AST, alkalickou fosfatázu, celkový bilirubin, gama-glutamyltransferázu (gama-GT) a sérový albumin. Léčivý přípravek je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater a pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí. Jestliže jsou základní vyšetření jaterních testů abnormální, zvažte zahájení léčby léčivým přípravkem až poté, co hepatolog provede vhodná vyšetření a abnormality základních testů budou vysvětleny či vyřešeny.
V prvním roce měřte jaterní testy (minimálně ALT a AST) před každým zvýšením dávky nebo jednou měsíčně podle toho, co nastane dříve. Po prvním roce provádějte tyto testy nejméně jednou za 3 měsíce a před každým zvýšením dávky. Jestliže pozorujete zvýšení hladiny aminotransferáz, dávku přípravku Lojuxta snižte, a při přetrvávajícím nebo klinicky významném zvýšení ukončete léčbu (konkrétní doporučení uvádí Tabulka 1).
Modifikace dávky na základě zvýšené hladiny jatemích aminotransferáz
Tabulka 1 je shrnutím doporučení pro úpravu dávky a sledování pacientů, u kterých dojde během léčby přípravkem Lojuxta ke zvýšení hladiny aminotransferáz.
Tabulka 1: Úprava dávky a sledování pacientů se zvýšenými hladinami aminotransferáz
|
ALT nebo AST |
Doporučení týkající se léčby a sledování* |
|
>3x a <5x horní mez normálního rozsahu (ULN) |
• Zvýšení potvrďte opakovaným měřením v rámci jednoho týdne. • Jestliže se zvýšení potvrdí, snižte dávku a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). • Testy opakujte každý týden, a jestliže budou přítomny známky abnormálních jaterních funkcí (zvýšení bilirubinu či INR), jestliže aminotransferázy stoupnou nad 5x ULN nebo jestliže hladiny aminotransferáz během přibližně 4 týdnů neklesnou pod 3x ULN, přerušte léčbu. Pacienty s přetrvávajícím zvýšením hladin aminotransferáz >3x ULN odešlete k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, zvažte snížení dávky a častější sledování jaterních testů. |
|
>5x ULN |
• Přerušte léčbu a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). Jestliže během přibližně 4 týdnů hladiny aminotransferáz neklesnou pod 3x ULN, odešlete pacienta k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, snižte dávku a častěji sledujte jaterní testy. |
*Doporučení založené na ULNpřibližně 30-40 mezinárodních jednotek/l.
Jestliže je zvýšení aminotransferáz provázeno klinickými symptomy poškození jater (např. nevolností, zvracením, bolestí břicha, horečkou, žloutenkou, letargií, příznaky podobnými chřipce), vzestupem hladin bilirubinu na >2x ULN nebo aktivním onemocněním jater, přerušte léčbu přípravkem Lojuxta a odešlete pacienta k hepatologovi na další vyšetření.
Opětovné nasazení léčby lze zvážit tehdy, pokud přínosy převyšují rizika související s potenciálním onemocněním jater.
Jaterní steatóza a riziko progresivního onemocnění jater
Většina léčených pacientů vykazuje vzestup obsahu tuku v játrech, což je ve shodě s mechanismem působení lomitapidu. V otevřené studii fáze 3 došlo u 18 z 23 pacientů s HoFH k vývoji jaterní steatózy (obsah tuku v játrech > 5,56 %) dle měření nukleární magnetickou rezonanční spektroskopií (MRS) (viz bod 5.1). Medián absolutního vzestupu jaterního tuku byl dle měření pomocí MRS jak po 26 týdnech, tak po 78 týdnech léčby 6 % oproti 1 % při zahájení studie. Jaterní steatóza je rizikový faktor progresivního onemocnění jater včetně steatohepatitidy a cirhózy. Dlouhodobé následky jaterní steatózy související s léčbou přípravkem Lojuxta nejsou známy. Klinické údaje naznačují, že nahromadění tuku v játrech je po ukončení léčby přípravkem Lojuxta reverzibilní, ale není známo, zda přetrvávají histologické změny, zejména po dlouhodobém užívání.
Sledování důkazů přítomnosti progresivního onemocnění jater
Při zahájení léčby a dále každý rok by se měl provádět pravidelný screening steatohepatitidy/fibrózy s použitím následujících zobrazovacích metod a hodnocení biomarkerů:
• Zobrazení elasticity tkáně, např. fibrosken, ARFI (Acoustic Radiation Force Impulse) nebo elastografie s použitím magnetické rezonance (MR)
• Gama-GT a sérový albumin pro detekci možného poškození jater
• Minimálně jeden marker z každé z následujících kategorií:
• Vysoce senzitivní C-reaktivní protein (hs-CRP), rychlost sedimentace erytrocytů (ESR), CK-18 fragment, NashTest (zánět jater)
• Rozšířený panel pro detekci jaterní fibrózy (ELF), fibrometr, poměr AST/ALT, skóre fib-4, fibrotest (jaterní fibróza)
Provedení těchto testů a jejich interpretace by měly zahrnovat spolupráci ošetřujícího lékaře s hepatologem. U pacientů s výsledky naznačujícími přítomnost steatohepatitidy nebo fibrózy by měla být zvážena biopsie jater.
Jestliže má pacient biopticky potvrzenou steatohepatitidu nebo fibrózu, měl by být znovu zhodnocen poměr přínosů a rizik a léčba by měla být ukončena, je-li to nutné.
Souběžné užívání inhibitorů CYP3A4
Zdá se, že lomitapid je citlivým substrátem pro metabolismus cestou CYP3A4. Inhibitory CYP3A4 zvyšují expozici lomitapidu, přičemž silné inhibitory zvyšují expozici přibližně 27krát. Souběžné užití středně silných nebo silných inhibitorů CYP3A4 a přípravku Lojuxta je kontraindikováno (viz bod 4.3). V klinických studiích s lomitapidem se u jednoho pacienta s HoFH během několika dní po zahájení užívání silného inhibitoru CYP3A4 klaritromycinu vyvinuly výrazně zvýšené hladiny aminotransferáz (ALT 24x ULN, AST 13x ULN). Jestliže se nelze vyvarovat léčby středně silnými nebo silnými inhibitory CYP3A4, mělo by být užívání přípravku Lojuxta v průběhu léčby ukončeno.
Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta má být podávána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4.
Souběžné užívání induktorů CYP3A4
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. Induktory CYP3A4 vykazují svůj účinek v závislosti na čase a dosažení maximálního účinku může trvat minimálně 2 týdny od zahájení léčby. Naopak při ukončení léčby může trvat minimálně 2 týdny, než indukce CYP3A4 odezní.
Očekává se, že souběžné podávání induktoru CYP3A4 snižuje účinek přípravku Lojuxta. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinu, pioglitazonu, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Použití třezalky tečkované současně s přípravkem Lojuxta je nutné se vyvarovat.
Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. Při vysazení induktoru CYP3A4 je nutné zvážit možnost zvýšené expozice a může být nutné snížit dávku přípravku Lojuxta.
Souběžné použití s inhibitory HMG-CoA reduktázv (..statinv")
Lomitapid zvyšuje plazmatické koncentrace statinů. U pacientů, kteří dostávají přípravek Lojuxta jako přídatnou terapii vedle statinů, by měly být sledovány nežádoucí účinky spojené s použitím vysokých dávek statinů. Statiny příležitostně mohou vyvolat myopatii. Ve vzácných případech může mít myopatie formu rhabdomyolýzy s akutním renálním selháním sekundárně způsobeným myoglobinurií nebo bez renálního selhání a může mít fatální následky. Všichni pacienti užívající přípravek Lojuxta vedle statinů by měli být informování o možném riziku myopatie a měli by být informováni, aby okamžitě hlásili nevysvětlitelnou bolest, citlivost nebo slabost svalů. S přípravkem Lojuxta by se neměly používat dávky simvastatinu > 40 mg (viz bod 4.3).
Grapefruitový džus
Ze stravy pacientů je při léčbě přípravkem Lojuxta nutno vyloučit grapefruitový džus.
Riziko supraterapeutické nebo subterapeutické antikoagulace u antikoagulačních přípravků na bázi kumarinu
Lomitapid zvyšuje plazmatické koncentrace warfarinu. Zvýšení dávky přípravku Lojuxta může vést k supraterapeutické antikoagulaci a snížení dávky může vést k subterapeutické antikoagulaci.
U jednoho z pěti pacientů užívajících souběžně warfarin přispěla obtížná kontrola INR k předčasnému ukončení studie fáze 3. Pacienti užívající warfarin by měli podstupovat pravidelné sledování INR, zejména po jakýchkoli změnách dávkování přípravku Lojuxta. Dávka warfarinu by měla být upravena dle klinické indikace.
Požívání alkoholu
Alkohol může zvyšovat hladinu tuku v játrech a může vést k indukci nebo exacerbaci poškození jater. Ve studii fáze 3 udávali 3 ze 4 pacientů se zvýšením ALT >5x ULN konzumaci alkoholu překračující limit doporučený v protokolu. Požívání alkoholu během léčby přípravkem Lojuxta se nedoporučuje.
Hepatotoxické přípravky
Opatrnosti je třeba při použití přípravku Lojuxta s dalšími léčivými přípravky se známým hepatotoxickým potenciálem, např. izotretinoinem, amiodaronem, acetaminofenem (>4 g/den po >3 dny/týden), metotrexátem, tetracyklinem a tamoxifenem. Účinek souběžného podávání přípravku Lojuxta s dalšími hepatotoxickými léčivými přípravky není znám. Může být nutné častější sledování jaterních testů.
Snížená absorpce vitamínů rozpustných v tucích a mastných kyselin v séru
Vzhledem k mechanismu působení lomitapidu v tenkém střevě může lomitapid snižovat absorpci živin rozpustných v tucích. Ve studii fáze 3 byly pacientům denně poskytovány nutriční doplňky s vitamínem E, kyselinou linolovou, ALA, EPA a DHA. V této studii se úroveň mediánu sérového vitamínu E, ALA, kyseliny linolové, EPA, DHA a kyseliny arachidonové mezi výchozím bodem studie a týdnem 26 snížila, ale zůstala nad spodním limitem referenčního rozmezí. Nežádoucí klinické důsledky tohoto snížení při léčbě lomitapidem nebyly pozorovány po dobu až 78 týdnů. Pacienti léčení přípravkem Lojuxta by měli denně užívat doplňky, které obsahují 400 mezinárodních jednotek vitamínu E a přibližně 200 mg kyseliny linolové, 210 mg ALA, 110 mg EPA a 80 mg DHA.
Antikoncepční opatření u žen ve fertilním věku.
Před zahájením léčby u žen ve fertilním věku by mělo být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z důvodu průjmu a/nebo zvracení (viz bod 4.5). Perorální antikoncepční přípravky na bázi estrogenů jsou slabými inhibitory CYP3A4 (viz bod 4.2).
Pacientky by měly být informovány, že v případě otěhotnění musejí neprodleně informovat svého lékaře a ukončit užívání přípravku Lojuxta (viz bod 4.6).
Laktóza
Přípravek Lojuxta obsahuje laktózu, pacienti se vzácnými hereditárními problémy jako jsou intolerance galaktózy, vrozený deficit laktázy nebo glukózová-galaktózová malabsorpce by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
Tabulka 2: Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
Inhibitory CYP3A4 |
Při současném podávání lomitapidu v dávce 60 mg s ketokonazolem, silným inhibitorem CYP3A4, v dávce 200 mg dvakrát denně se zvýšila AUC lomitapidu přibližně 27krát a Cmax přibližně 15krát. Interakce mezi středně silnými inhibitory CYP3A4 a lomitapidem nebyly studovány. Předpokládá se, že středně silné inhibitory CYP3A4 mají významný vliv na farmakokinetiku lomitapidu. Očekává se, že souběžné použití středně silných inhibitorů CYP3A4 zvyšuje expozici lomitapidu 4-10krát podle výsledků studie se silným inhibitorem CYP3A4 podle historických údajů pro zkušební model CYP3A4 midazolam. Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při podávání lomitapidu 20 mg současně s atorvastatinem (tj. |
Použití silných nebo středně silných inhibitorů CYP3A4 spolu s přípravkem Lojuxta je kontraindikováno. Jestliže se nelze vyvarovat léčby antimykotiky azoly (např. itrakonazolem, ketokonazolem, flukonazolem, vorikonazolem, posakonazolem), antiarytmikem dronedaronem, makrolidovými antibiotiky (např. erytromycinem, klaritromcinem), ketolidovými antibiotiky (např. telitromycinem), inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem, měla by být během této léčby léčba přípravkem Lojuxta pozastavena (viz body 4.3 a 4.4). Grapefruitový džus je středně silným inhibitorem CYP3A4 a má se za to, že podstatně zvyšuje expozici lomitapidu. Pacienti užívající přípravek Lojuxta by se měli vyvarovat konzumace grapefruitového džusu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta musí být užívána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4. Příklady slabých inhibitorů CYP3A4 zahrnují: alprazolam, |
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
slabým inhibitorem CYP3A4) se AUC a Cmax lomitapidu zvýšily přibližně 2x. Pokud byl lomitapid podáván 12 hodin po atorvastatinu, žádné klinicky významné zvýšení expozice lomitapidu nebylo pozorováno. Při podávání lomitapidu 20 mg s ethinyl estradiolem/norgestimátem (tj. slabým inhibitorem CYP3A4), žádné klinicky významné zvýšení expozice lomitapidu nebylo pozorováno při užívání současném ani s intervalem 12 hodin. |
amiodaron, amlodipin, atorvastatin, azitromycin, bikalutamid, cilostazol, cimetidin, cyklosporin, klotrimazol, fluoxetin, fluvoxamin, fosaprepitant, ginkgo, vodilku kanadskou, izoniazid, ivakaftor, lacidipin, lapatinib, linagliptin, nilotinib, perorální antikoncepční přípravky obsahující estrogeny, pazopanib, mátový olej, propiverin, ranitidin, ranolazin, roxithromycin, hořké pomeranče, takrolimus, tikagrelor a tolvaptan. Tento seznam není úplný a předepisující lékaři by měli zkontrolovat preskripční informace o léčivech, která mají být podávána spolu s přípravkem Lojuxta s ohledem na možné interakce zprostředkované CYP3A4. Účinek podávání více než jednoho slabého inhibitoru CYP3A4 nebyl testován, ale očekává se, že účinek na expozici lomitapidu je větší než při současném podávání jednotlivých inhibitorů s lomitapidem. Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta. | |
|
Induktory CYP3A4 |
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. To by mohlo následně snížit účinek lomitapidu. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. |
Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinnu, pioglitazonu, třezalky tečkované, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. |
|
Sekvestranty žlučových kyselin |
Interakce lomitapidu se sekvestranty žlučových kyselin (pryskyřicemi, např. kolesevelamem a cholestyraminem) nebyly testovány. |
Protože sekvestranty žlučových kyselin mohou interferovat s absorpcí perorálních léčivých přípravků, měly by se užívat minimálně 4 hodiny před přípravkem Lojuxta nebo minimálně 4 hodiny po něm. |
Účinky lomitapidu na jiné léčivé přípravky
Inhibitory HMG-CoA reduktázy („statiny“): Lomitapid zvyšuje plazmatické koncentrace statinů. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním simvastatinu 40 mg se zvýšily u kyseliny simvastatinu hodnoty AUC o 68 % a Cmax o 57 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním atorvastatinu 20 mg se zvýšily u kyseliny atorvastatinu hodnoty AUC o 52 % a Cmax o 63 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním rosuvastatinu 20 mg se zvýšila hodnota Tmax rosuvastatinu z 1 na 4 hodiny, hodnota AUC se zvýšila o 32 % a hodnota Cmax zůstala nezměněná. Riziko myopatie u simvastatinu je závislé na dávce. Použití přípravku Lojuxta je u pacientů léčených vysokými dávkami statinů (> 40 mg) kontraindikováno (viz body 4.3 a 4.4).
Kumarinové antikoagulační přípravky: Podávání lomitapidu 60 mg do dosažení ustáleného stavu a po dobu 6 dní po warfarinu 10 mg zvýšilo hodnotu INR 1,26krát. Hodnota AUC pro R(+)-warfarin se zvýšila o 25 % a pro S(-)-warfarin o 30 %. Hodnota Cmax pro R(+)-warfarin se zvýšila o 14 % a pro S(-)-warfarin o 15 %. U pacientů užívajících souběžně kumariny (např. warfarin) a přípravek Lojuxta by měla být před zahájením užívání přípravku Lojuxta stanovena hodnota INR a tato hodnota by měla být pravidelně kontrolována, přičemž dávkování kumarinů by mělo být upraveno dle klinické potřeby (viz bod 4.4).
Fenofibrát, niacin a ezetimib: Při podávání lomitapidu do dosažení ustáleného stavu před podáním mikronizovaného fenofibrátu 145 mg, niacinu s prodlouženým uvolňováním 1000 mg nebo ezetimibu 10 mg nebyly pozorovány klinicky významné účinky působení kteréhokoli z těchto léčivých přípravků. Při souběžném podávání s přípravkem Lojuxta není nutná úprava dávky.
Perorální antikoncepce: Při podávání lomitapidu 50 mg do dosažení ustáleného stavu spolu s perorálním antikoncepčním přípravkem na bázi estrogenů nebyl pozorován klinicky ani statisticky významný vliv na farmakokinetiku složek perorální antikoncepce (ethinylestradiolu a 17-deacetylnorgestimátu, metabolitu norgestimátu). Neočekává se, že by lomitapid přímo ovlivňoval účinnost perorální antikoncepce na bázi estrogenů, nicméně průjem a/nebo zvracení mohou snížit absorpci hormonů. U případů prodlouženého nebo závažného průjmu a/nebo zvracení trvajících více než 2 dny by měla být po dobu 7 dnů po ústupu symptomů použita další antikoncepční metoda.
Substráty P-gp: Lomitapid in vitro inhibuje P-gp a může zvyšovat absorpci P-gp substrátů. Souběžné podávání přípravku Lojuxta s P-pg substráty (např. aliskirenem, abrisentanem, kolchicinem, dabigatran etexilátem, dixoginem, everolimem, fexofenadinem, imatinibem, lapatinibem, maravirokem, nilotinibem, posakonazolem, ranolazinem, saxagliptinem, sirolimem, sitagliptinem, talinololem, tolvaptanem, topotekanem) může zvýšit absorpci P-gp substrátů. Při souběžném použití s přípravkem Lojuxta by mělo být zváženo snížení dávky P-gp substrátu.
In vitro hodnocení lékových interakcí: Lomitapid inhibuje CYP3A4. Lomitapide neindukuje CYP 1A2, 3A4 ani 2B6 a neinhibuje CYP 1A2, 2B6, 2C9, 2C19, 2D6 ani 2E1. Lomitapid není P-gp substrátem, ale inhibuje P-gp. Lomitapid neinhibuje protein rezistence u karcinomu prsu (BCRP).
4.6 Fertilita, těhotenství a kojení
Přípravek Lojuxta je v těhotenství kontraindikován. Neexistují spolehlivé údaje o použití přípravku u těhotných žen. Studie na zvířatech prokázaly vývojovou toxicitu (teratogenitu, embryotoxicitu, viz bod 5.3). Možné riziko pro člověka je neznámé.
Použití u žen ve fertilním věku.
Před zahájením léčby u žen ve fertilním věku by mělo být vyloučeno těhotenství, mělo by být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z důvodu průjmu a/nebo zvracení. Dokud symptomy neustoupí, je nutno používat další antikoncepční metody (viz bod 4.5).
Kojení
Není známo, zda se lomitapid vylučuje do mateřského mléka. Vzhledem k potenciálu nežádoucích účinků zjištěnému na základě studií s lomitapidem na zvířatech (viz bod 5.3) je nutné rozhodnout, zda ukončit kojení nebo užívání léčivého přípravku, a to při zvážení významu léčivého přípravku pro matku.
Fertilita
U potkaních samců ani samic, kterým byl podáván lomitapid se systémovou expozicí (AUC) ve výši odhadovaného čtyř- až pětinásobku expozice u lidí při maximální doporučené dávce u člověka, nebyly pozorovány žádné nežádoucí účinky na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Lojuxta může mít mírný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejzávažnějšími nežádoucími účinky během léčby byly abnormality jaterních aminotransferáz (viz bod 4.4).
Nejčastějšími nežádoucími účinky byly gastrointestinální účinky. Nežádoucí gastrointestinální účinky byly hlášeny u 27 (93 %) z 29 pacientů klinické studie fáze 3. Průjem se objevil u 79 %, nevolnost u 65 %, dyspepsie u 38 % a zvracení u 34 % pacientů. Další účinky, které hlásilo minimálně 20 % pacientů, zahrnují bolest břicha, břišní dyskomfort, nadmutí břicha, zácpu a plynatost.
Nežádoucí gastrointestinální účinky se častěji objevovaly během fáze navyšování dávky ve studii a poklesly, jakmile pacienti dosáhli maximální tolerované dávky lomitapidu.
Gastrointestinální nežádoucí účinky se závažnou intenzitou byly hlášeny u 6 (21 %) z 29 pacientů v klinické studii fáze 3, přičemž nejčastějším účinkem byl průjem (4 pacienti, 14 %), zvracení (3 pacienti, 10 %) a bolest břicha, nadmutí a/nebo dyskomfort (2 pacienti, 7 %). Gastrointestinální účinky přispěly k časnému ukončení studie u 4 (14 %) pacientů.
Nejčastěji udávanými nežádoucími účinky se závažnou intenzitou byly průjem (4 subjekty, 14 %), zvracení (3 pacienti, 10 %) a nadmutí břicha a zvýšení ALT (pokaždé 2 subjekty, 7 %).
Přehled nežádoucích účinků v tabulce
Dle frekvence jsou nežádoucí účinky definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), málo časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), s neznámou frekvencí (z dostupných údajů nelze určit).
Tabulka 3 zahrnuje veškeré nežádoucí účinky hlášené u 35 pacientů léčených ve studii fáze 2 UP1001 a ve studii fáze 3 UP1002/AEGR-733-005 nebo v rozšířené studii AEGR-733-012.
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Časté |
Gastroenteritida |
|
Poruchy metabolismu a výživy |
Velmi časté |
Snížená chuť k jídlu |
|
Poruchy nervového systému |
Časté |
Závratě Bolest hlavy Migréna |
|
Gastrointestinální poruchy |
Velmi časté |
Břišní dyskomfort Bolest horní části břicha Plynatost Distenze břicha Zácpa |
|
Časté |
Gastritida Rektální tenesmy Aerofagie Nutkání na stolici Říhání Častá stolice Dilatace žaludku Onemocnění žaludku Gastroezofageální reflux Krvácení z hemoroidů Regurgitace | |
|
Poruchy jater a žlučových cest |
Časté |
Jaterní steatóza Hepatotoxicita Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Časté |
Ekchymóza Papuly Erytematózní vyrážka Xantomy |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Únava |
|
Vyšetření |
Velmi časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Snížení tělesné hmotnosti |
|
Časté |
Zvýšený mezinárodní normalizovaný poměr Zvýšená alkalická fosfatáza v krvi Snížení draslíku v krvi Snížený karoten Abnormální mezinárodní normalizovaný poměr Abnormální testy jaterních funkcí Prodloužený protrombinový čas Zvýšené transaminázy Snížený vitamín E Snížený vitamín K |
Tabulka 4 uvádí všechny nežádoucí účinky u subjektů, které dostávaly lomitapid v monoterapii (n=291), léčených ve studiích fáze 2 u subjektů se zvýšenou hladinou LDL-C (N=462).
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Méně časté |
Gastroenteritida Gastrointestinální infekce Chřipka Nazofaryngitida Sinusitida |
|
Poruchy krve a lymfatického systému |
Méně časté | |
|
Poruchy metabolismu a výživy |
Časté |
Snížená chuť k jídlu |
|
Méně časté |
Dehydratace Zvýšená chuť k jídlu | |
|
Poruchy nervového systému |
Méně časté |
Parestézie Ospalost |
|
Poruchy oka |
Méně časté |
Otok oka |
|
Poruchy ucha a labyrintu |
Méně časté |
Závrať |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté |
Léze hltanu Syndrom kašle horních cest dýchacích |
|
Gastrointestinální poruchy |
Velmi časté |
Plynatost |
|
Časté |
Bolest horní části břicha Distenze břicha Bolest břicha Zvracení Břišní dyskomfort Říhání Bolest v podbřišku Častá stolice | |
|
Méně časté |
Sucho v ústech Tvrdá stolice Gastroezofageální reflux Citlivost břicha Dyskomfort v epigastriu Dilatace žaludku Hemateméza Krvácení v dolní části gastrointestinálního traktu Refluxní ezofagitida | |
|
Poruchy jater a žlučových cest |
Méně časté |
Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Puchýře Suchá kůže Nadměrné pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté |
Svalové spasmy |
|
Méně časté |
Bolest kloubů Bolest svalů Bolest končetin Otok kloubu Svalové záškuby | |
|
Poruchy ledvin a močových cest |
Méně časté |
Hematurie |
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Celkové poruchy a reakce v místě |
Časté |
Únava |
|
podání |
Slabost | |
|
Méně časté |
Bolest na hrudi Mrazení Rychlý pocit nasycení Poruchy chůze Malátnost Horečka | |
|
Vyšetření |
Časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Zvýšení jaterních enzymů Abnormální testy jaterních funkcí Snížený počet neutrofilů Snížený počet leukocytů |
|
Méně časté |
Snížení tělesné hmotnosti Zvýšený bilirubin v krvi Zvýšená hladina gama-glutamyltransferázy Zvýšené procento neutrofilů Protein v moči Prodloužený protrombinový čas Abnormální testy plicních funkcí Zvýšený počet leukocytů |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování neexistuje specifická léčba. U hlodavců byla dobře snášena jednorázová perorální dávka lomitapidu >600krát vyšší než maximální doporučená dávka pro člověka (1 mg/kg). Maximální dávkou podanou lidským subjektům v klinických studiích bylo 200 mg ve formě jednorázové dávky, nevyskytly se žádné nežádoucí účinky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná léčiva ovlivňující hladinu lipidů, samotná. ATC kód: C10AX12
Mechanismus účinku
Lomitapid je selektivní inhibitor mikrozomálního transferového proteinu (MTP), intracelulárního proteinu přenášejícího lipidy, který se nachází v lumen endoplazmatického retikula a zodpovídá za vazbu a převod jednotlivých lipidových molekul mezi membránami. MTP hraje klíčovou roli při sestavování lipoproteinů obsahujících apo B v játrech a střevech. Inhibice MTP snižuje sekreci lipoproteinů a cirkulující koncentrace lipidů přenášených v lipoproteinech včetně cholesterolu a triglyceridů.
Klinická účinnost a bezpečnost
Účinnost a bezpečnost lomitapidu při současném podávání diety s nízkým obsahem tuků a dalších terapií snižujících lipidy u dospělých pacientů s HoFH hodnotila otevřená studie s jedním ramenem (UP1002/AEGR-733-005). Pacienti byli instruováni, aby dodržovali dietu s nízkým obsahem tuků (< 20 % kalorického příjmu z tuků) i terapii snižující lipidy, kterou měli při vstupu do studie, včetně aferézy, byla-li tato nutná, a to po dobu 6 týdnů předcházejících výchozímu bodu studie minimálně až do týdne 26. Dávka lomitapidu byla navyšována z 5 mg na individuálně stanovenou maximální tolerovanou dávku až 60 mg. Po týdnu 26 byl pacientům lomitapid ponechán za účelem zjištění účinků dlouhodobé léčby a bylo jim umožněno změnit základní terapii snižující lipidy. Studie zahrnovala celkem 78 týdnů léčby.
Do studie bylo zařazeno 29 pacientů, z nichž 23 dokončilo týden 78. Zařazeno bylo 16 mužů (55 %) a 13 žen (45 %) s průměrným věkem 30,7 let a ve věkovém rozmezí 18 až 55 let. Průměrná dávka lomitapidu v týdnu 26 byla 45 mg a v týdnu 78 byla 40 mg. V týdnu 26 byla průměrná procentuální změna hladiny LDL-C u populace „intention-to-treat“ (ITT) oproti výchozímu bodu -40 %
(p < 0,001). Průměrná procentuální změna oproti výchozímu bodu studie v týdnu 26 s použitím metody LOCF (Last Observation Carried Forward) pro každé hodnocení je uvedena na obrázku 1.
Obrázek 1: Průměrné procentuální změny hladiny LDL-C mezi vstupní hodnotou a týdnem
26 v hlavní studii účinnosti UP1002/AEGR-733-005 (primární cíl) s použitím LOCF pro každé hodnocení (n=29)

Týden studie
Změny lipidů i lipoproteinů v týdnu 26 a 78 při léčbě lomitapidem uvádí Tabulka 5.
Tabulka 5: Absolutní hodnoty a procentuální změny hladin lipidů a lipoproteinů mezi
výchozí hodnotou a týdny 26 a 78 (hlavní studie účinnosti UP1002/AEGR-733-005).
|
Parametr (jednotky) |
Výchozí bod |
Týden 26 / LOCF (n=29) |
Týden 78 (n=23) | ||||
|
Průměr (SD) |
Průměr (SD) |
% změna |
Hodnota p b |
Průměr (SD) |
% změna |
Hodnota p b | |
|
LDL-C, přímý (mg/dl) |
336 (114) |
190 (104) |
-40 |
< 0,001 |
210 (132) |
-38 |
< 0,001 |
|
Celkový cholesterol (TC) (mg/dl) |
430 (135) |
258 (118) |
-36 |
< 0,001 |
281 (149) |
-35 |
< 0,001 |
|
Apolipoprotein B (apo B) (mg/dl) |
259 (80) |
148 (74) |
-39 |
< 0,001 |
151 (89) |
-43 |
< 0,001 |
|
Triglyceridy (TG) (mg/dl)a |
92 |
57 |
-45 |
0,009 |
59 |
-42 |
0,012 |
|
Cholesterol z jiných lipoproteinů než o vysoké hustotě (non-HDL-C) (mg/dl) |
386 (132) |
217 (113) |
-40 |
< 0,001 |
239 (146) |
-39 |
< 0,001 |
|
Cholesterol z lipoproteinů o velmi nízké hustotě (VLDL-C) (mg/dl) |
21 (10) |
13 (9) |
-29 |
0,012 |
16 (15) |
-31 |
0,013 |
|
Lipoprotein (a) (Lp(a)) (nmol/l)a |
66 |
61 |
-13 |
0,094 |
72 |
-4 |
< 0,842 |
|
Cholesterol z lipoproteinů o vysoké hustotě (HDL-C) (mg/dl) |
44 (11) |
41 (13) |
-7 |
0,072 |
43 (12) |
-4,6 |
0,246 |
a Medián uvedený pro TG a Lp(a). Hodnota p je založena na průměrné procentuální změně b Hodnota p u střední procentuální změny oproti výchozímu bodu studie na základě párového t-testu
Jak v týdnu 26, tak v týdnu 78 došlo k významnému snížení hladin LDL-C, TC, apo B, TG, non-HDL-C a VLDL-C a změny u HDL-C vykazovaly v týdnu 26 trend směrem ke snížení a v týdnu 78 se vrátily k výchozím hodnotám.
Účinek přípravku Lojuxta na kardiovaskulární morbiditu a mortalitu nebyl stanoven.
Při zahájení studie užívalo 93 % pacientů statin, 76 % pacientů ezetimib, 10 % niacin, 3 % sekvestrant žlučových kyselin a 62 % pacientů bylo léčeno aferézou. U 15 z 23 (65 %) pacientů byla v týdnu 78 redukována jejich léčba na snížení lipidů, včetně plánovaných a neplánovaných snižování/přerušení. Aferéza byla přerušena u 3 ze 13 pacientů, kteří byli aferézou léčeni v týdnu 26, a u 3 pacientů byla frekvence při současném udržení nízkých hladin LDL-C i po týdnu 78 snížena. Klinický přínos omezení základní terapie snižující lipidy, včetně aferézy, není jistý.
Z 23 pacientů, kteří dokončili týden 26 studie, došlo u 19 (83 %) ke snížení LDL-C o > 25 %, přičemž 8 (35 %) z nich mělo v tomto časovém bodě LDL-C < 100 mg/dl a 1 pacient měl LDL-C < 70 mg/dl.
V této studii došlo u 10 pacientů ke zvýšení AST a/nebo ALT >3x ULN (viz Tabulka 6).
Nejvyšší výsledky testů jaterních funkcí po první dávce (hlavní studie účinnosti UP1002/AEGR-733-005)
|
Parametr/abnormalita |
N (%) |
|
ALT | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
6 (20,7) |
|
>5 až < 10 x ULN |
3 (10,3) |
|
>10 až < 20 x ULN |
1 (3,4) |
|
> 20 x ULN |
0 |
|
AST | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
5 (17,2) |
|
>5 až < 10 x ULN |
1 (3,4) |
|
>10 až < 20 x ULN |
0 |
|
> 20 x ULN |
0 |
Tabulka 6:
Zvýšení ALT a/nebo AST > 5x ULN bylo zvládnuto snížením dávky nebo dočasným pozastavením podávání lomitapidu a všichni pacienti byli schopni v léčbě studovaným léčivem pokračovat. Nebylo pozorováno klinicky významné zvýšení celkového bilirubinu ani alkalické fosfatázy. Jaterní tuk byl během klinické studie u všech dostupných pacientů prospektivně měřen s použitím MRS (Tabulka 7). Údaje od jedinců, u kterých byla po ukončení lomitapidu prováděna opakovaná měření, ukázaly, že nahromadění tuku v játrech je reverzibilní, ale není známo, zda nepřetrvávají histologické změny.
Tabulka 7: Maximální kategorické změny v % jaterního tuku (hlavní studie účinnosti
UP1002/AEGR-733-005)
|
Maximální absolutní nárůst % jaterního tuku |
Fáze účinnosti Týdny 0-26 N (%) |
Fáze bezpečnosti Týdny 26-78 N (%) |
Celá studie týdny 0-78 N (%) |
|
Počet hodnotitelných pacientů |
22 |
22 |
23 |
|
< 5% |
9 (41) |
6 (27) |
5 (22) |
|
> 5 % až < 10 % |
6 (27) |
8 (36) |
8 (35) |
|
> 10 % až < 15 % |
4 (18) |
3 (14) |
4 (17) |
|
> 15 % až < 20 % |
1 (5) |
4 (18) |
3 (13) |
|
> 20 % až < 25 % |
1 (5) |
0 |
1 (4) |
|
> 25 % |
1 (5) |
1 (5) |
2 (9) |
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lojuxta u jedné nebo více podskupin pediatrické populace s HoFH (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Absolutní perorální biologická dostupnost lomitapidu je 7 %. Absorpce není omezena prostupem léčiva přes střevní bariéru, ale je převážně ovlivněna extenzivním efektem prvního průchodu. Vrcholná plazmatická koncentrace lomitapidu byla dosažena za 4 až 8 hodin po podání perorální dávky. Farmakokinetika lomitapidu je přibližně proporcionální k dávce perorálních jednorázových dávek v terapeutickém rozmezí. U dávek vyšších než 60 mg je naznačena tendence k nelineární závislosti a tyto dávky nejsou doporučeny.
Při podání více dávek se hodnoty Cmax a AUC zvýšily přibližně proporcionálně k dávce lomitapidu. Hodnoty Cmax a AUC se zvýšily jak po vysoce tučném jídle (77 %, respektive 58 %), tak po nízkotučném jídle (70 %, respektive 28 %). Akumulace lomitapidu v plazmě byla konzistentní s hodnotou předpokládanou na základě jedné dávky po podávání jedné perorální dávky denně nad 25 mg po dobu až 4 týdnů. Interindividuální variabilita hodnoty AUC lomitapidu byla přibližně 50 %.
V ustáleném stavu byla akumulace lomitapidu 2,7 při 25 mg a 3,9 při 50 mg.
Distribuce
Po intravenózním podání byl distribuční objem lomitapidu velký (průměr = 1 200 litrů) i přes vysoký stupeň (> 99,8 %) vazby na plazmatické proteiny. Ve studiích na zvířatech se lomitapid vysoce (200násobně) koncentroval v játrech.
Biotransformace
Lomitapid se extenzivně metabolizuje, převážně cestou CYP3A4. Izoformy CYP 2E1, 1A2, 2B6, 2C8 a 2C19 se účastní méně a izoformy 2D6 a 2C9 se metabolismu lomitapidu neúčastní.
Eliminace
Po podání dávky radioaktivně značeného perorálního roztoku zdravým subjektům bylo 93 % podané dávky vyloučeno močí a stolicí. Přibližně 33 % radioaktivity bylo vyloučeno do moči jako metabolity. Zbytek byl vyloučen stolicí, primárně jako oxidované metabolity. Eliminační poločas lomitapidu byl přibližně 29 hodin.
Zvláštní populace
Údaje ze stěžejní klinické studie byly analyzovány s ohledem na vliv možných souvisejících proměnných na expozici lomitapidu. Z vyšetřovaných parametrů (rasa, index tělesné hmotnosti (BMI), hmotnost, věk) může být jako možná související proměnná klasifikován pouze BMI.
Věk a pohlaví
Věk (18-64 let) ani pohlaví nemají na farmakokinetiku lomitapidu klinicky významný vliv.
Rasa
U bělochů nebo latinskoamerické populace není nutná úprava dávky. K dispozici není dostatek informací pro stanovení, zda přípravek Lojuxta vyžaduje úpravu dávky u dalších ras. Protože se ale léčivý přípravek podává způsobem postupného zvyšování dávky podle individuální bezpečnosti a snášenlivosti pacienta, úprava dávkovacího režimu na základě rasy není doporučena.
V populaci s poškozením funkce ledvin byl lomitapid studován pouze u pacientů s terminálním onemocněním ledvin (ESRD). Farmakokinetická studie u pacientů s ESRD podstupujících dialýzu ukázala 36% vzestup průměrné koncentrace lomitapidu v plazmě ve srovnání s párovanými zdravými kontrolami. Koncový poločas lomitapidu nebyl ovlivněn.
Jaterní insuficience
Pro hodnocení farmakokinetiky 60 mg lomitapidu u zdravých dobrovolníků s normálními jaterními funkcemi ve srovnání s pacienty s mírným postižením funkce jater (třída A dle Child-Pugha) a středně těžkým postižením funkce jater (třída B dle Child-Pugha) byla provedena otevřená studie s jednou dávkou. Hodnoty AUC a Cmax lomitapidu u pacientů se středně těžkým postižením funkce jater byly o 164 %, respektive 361 % vyšší než u zdravých dobrovolníků. Hodnoty AUC a Cmax lomitapidu u pacientů s mírným postižením funkce jater byly o 47 %, respektive 4 % vyšší než u zdravých dobrovolníků. U pacientů s těžkým postižením funkce jater (skóre dle Child-Pugha 10-15) nebyl přípravek Lojuxta studován.
Pediatrická populace
U dětí mladších 18 let nebyl přípravek Lojuxta studován.
Starší populace
U pacientů starších 65 let nebyl přípravek Lojuxta studován.
5.3 Předklinické údaje vztahující se k bezpečnosti
Základním zjištěním toxikologických studií léčiva s opakovanou perorální dávkou u hlodavců a psů byla akumulace lipidů v tenkém střevě a/nebo v játrech spojená s poklesem sérových hladin cholesterolu a/nebo triglyceridů. Tyto změny jsou vzhledem k mechanismu působení lomitapidu sekundární. Další změny související s játry ve studiích toxicity s opakovanou dávkou u potkanů a psů zahrnovaly zvýšené hladiny sérové aminotransferázy, subakutní zánět (pouze u potkanů) a nekrózu jednotlivých buněk. V jednoleté studii s opakovanou dávkou u psů se v játrech nevyskytovaly mikroskopické změny, ačkoli u fen byla minimálně zvýšená sérová hladina AST.
U hlodavců byla pozorována plicní histiocytóza. U psů byl pozorován pokles parametrů erytrocytů i poikilocytóza a/nebo anizocytóza. U psů byla v šestiměsíční studii při 205násobku lidské expozice (AUC) 60 mg pozorována testikulární toxicita. V roční studii u psů nebyly při 64násobku lidské expozice 60 mg pozorovány nežádoucí účinky na varlata.
Ve studii nutriční karcinogenity u myší byl lomitapid podáván po dobu až 104 týdnů v dávkách v rozmezí 0,3 až 45 mg/kg/den. Při dávkách > 1,5 mg/kg/den u samců (> 2násobek lidské expozice při 60 mg denně na základě AUC) a > 7,5 mg/kg/den u samic (> 9násobek lidské expozice při 60 mg na základě AUC) došlo ke statisticky významnému vzestupu incidence adenomu i karcinomu jater. Incidence karcinomu tenkého střeva a/nebo kombinovaného adenomu a karcinomu (u myší vzácné nádory) byla významně zvýšena při dávkách > 15 mg/kg/den u samců (> 26násobek lidské expozice při 60 mg na základě AUC) a 15 mg/kg/den u samic (22násobek lidské expozice při 60 mg na základě AUC).
Ve studii perorální kancerogenity u potkanů byl lomitapid podáván po dobu až 99 týdnů v dávkách až
7,5 mg/kg/den u samců a 2,0 mg/kg/den u samic. U samců i samic byla pozorována fokální jaterní fibróza a pouze u samců byla pozorována cystická degenerace jater. U samců s vysokou dávkou byla pozorována zvýšená incidence pankreatického acinárního adenomu při expozici 6krát vyšší než u lidí při 60 mg na základě AUC.
V panelu studií in vitro a in vivo nebyl lomitapid mutagenní ani genotoxický.
Lomitapid neměl účinek na reprodukční funkce u samic potkanů v dávkách až 1 mg/kg nebo samců potkanů v dávkách až 5 mg/kg. Systémová expozice lomitapidu v těchto dávkách byla odhadována na čtyřnásobek (samice) až pětinásobek (samci) expozice u lidí při 60 mg na základě AUC.
Lomitapid byl u potkanů teratogenní při současné absenci mateřské toxicity při expozici (AUC) odhadované na dvojnásobek lidské expozice při 60 mg. U králíků se nevyskytly známky embryofetální toxicity při trojnásobku maximální doporučené denní dávky u lidí (MRHD) 60 mg na základě plochy tělesného povrchu. Embryofetální toxicita při absenci mateřské toxicity byla pozorována u králíků při > 6,5násobku MRHD. U fretek byl lomitapid toxický pro matku i teratogenní při dávkách < 1x MRHD.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky
předbobtnalý škrob (kukuřičný) sodná sůl karboxymethylškrobu mikrokrystalická celulóza monohydrát laktózy koloidní bezvodý oxid křemičitý magnesium-stearát
Obal tobolky želatina
oxid titaničitý (E171) žlutý oxid železitý (E172)
Tiskařský inkoust šelak
černý oxid železitý (E172) propylenglykol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Uchovávejte v dobře uzavřené lahvičce, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Lahvička z polyetylenu o vysoké hustotě (HDPE) vybavená vložkou z polyesteru / hliníkové fólie / kartonu a s polypropylenovým šroubovacím uzávěrem.
Velikost balení:
28 tobolek
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Aegerion Pharmaceuticals Ltd Lakeside House 1 Furzeground Way Stockley Park East Uxbridge UB11 1BD Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/851/005
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
^ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Lojuxta 60 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje lomitapidum 60 mg ve formě lomitapidum mesilas Pomocná látka se známým účinkem:
Jedna tvrdá tobolka obsahuje 389,68 mg laktózy (ve formě monohydrátu) (viz bod 4.4). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Tvrdá tobolka se žlutou svrchní částí / žlutou spodní částí o délce 23,4 mm, potištěná černým inkoustem s textem „60 mg“ na spodní části a „A733“ na svrchní části tobolky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Lojuxta je indikován jako podpůrný přípravek užívaný spolu s dietou s nízkým obsahem tuků a dalšími léčivými přípravky na snížení hladiny lipidů spolu s aferézou lipoproteinů o nízké hustotě (LDL) či bez ní u dospělých pacientů s homozygotní formou familiární hypercholesterolémie (HoFH).
Tam, kde je to možné, by měla být homozygotní forma familiární hypercholesterolémie potvrzena geneticky. Je nutné vyloučit jiné formy primární hyperlipoproteinémie a sekundární příčiny hypercholesterolémie (např. nefrotický syndrom, hypotyreózu).
4.2 Dávkování a způsob podání
Léčbu přípravkem Lojuxta by měl zahajovat a sledovat lékař, který má zkušenosti s léčbou poruch lipidů.
Dávkování
Doporučená zahajovací dávka přípravku je 5 mg jednou denně. Po 2 týdnech může být dávka na základě přijatelné bezpečnosti a snášenlivosti zvýšena na 10 mg a poté v minimálně čtyřtýdenních intervalech na 20 mg, 40 mg a maximální doporučenou dávku 60 mg (viz bod 4.8).
Dávka by měla být zvyšována postupně, aby se minimalizovala incidence a závažnost gastrointestinálních nežádoucích účinků a zvýšení aminotransferáz.
Podávání se stravou může u přípravku Lojuxta zvyšovat expozici. Přípravek Lojuxta by se měl užívat nalačno, minimálně dvě hodiny po večeři, protože tuk obsažený v nedávno požitém jídle může mít negativní vliv na gastrointestinální snášenlivost.
Výskyt a závažnost gastrointestinálních nežádoucích účinků souvisejících s užíváním přípravku Lojuxta se snižují při dodržování diety s nízkým obsahem tuků. Pacienti by před zahájením léčby přípravkem Lojuxta měli držet dietu, ve které je méně než 20 % energie dodáváno z tuků, a během léčby by v této dietě měli pokračovat. Mělo by být zajištěno nutriční poradenství.
Pacienti by se měli vyvarovat konzumace grapefruitového džusu (viz body 4.4 a 4.5).
U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s atorvastatinem použijte jednu z těchto variant:
• Oddělte podání obou přípravků intervalem 12 hodin NEBO
• Snižte dávku přípravku Lojuxta o polovinu.
Pacienti užívající 5 mg by měli zůstat na této dávce.
Následně lze provést opatrnou titraci podle reakce hladiny LDL-C a bezpečnosti /snášenlivosti.
Po ukončení léčby atorvastatinem zvyšte dávku přípravku Lojuxta titrací podle hladiny LDL-C a bezpečnosti/snášenlivosti.
U pacientů užívajících stabilní udržovací dávku přípravku Lojuxta v kombinaci s jakýmkoliv dalším slabým inhibitorem CYP3A4 oddělte podání obou přípravků intervalem 12 hodin.
Omezení maximální dávky přípravku Lojuxta zvažte podle požadované odpovědi LDL-C.
Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta.
Na základě pozorování snížených hladin esenciálních mastných kyselin a vitamínu E v klinických studiích by pacienti po dobu léčby přípravkem Lojuxta měli také užívat denně nutriční doplňky zajišťující příjem 400 IU vitamínu E a přibližně 200 mg kyseliny linolové, 110 mg kyseliny eikosapentaenové (EPA), 210 mg kyseliny alfa-linolenové (ALA) a 80 mg kyseliny dokosahexaenové (DHA).
Starší populace
Zkušenosti s přípravkem Lojuxta jsou u pacientů ve věku 65 let a starších omezené. Proto je u těchto pacientů zapotřebí zvláštní obezřetnosti.
Protože doporučený režim dávkování zahrnuje zahájení na spodní hranici dávkovacího rozmezí a opatrné navyšování dávky podle individuální snášenlivosti pacienta, nedoporučuje se ve starším věku žádná úprava dávkovacího schématu.
Porucha jater
Přípravek Lojuxta je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater včetně pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí (viz bod 5.2).
U pacientů s mírnou poruchou funkce jater (třída A dle Child-Pugha) by neměla být překračována dávka 40 mg denně.
Porucha ledvin
U pacientů v terminálním stádiu onemocnění ledvin léčených dialýzou by neměla být překračována dávka 40 mg denně (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Lojuxta u dětí mladších 18 let nebyla stanovena, a použití tohoto léčivého přípravku u dětí se proto nedoporučuje. Nejsou dostupné žádné údaje.
Způsob podání
Perorální podání.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Pacienti se středně těžkou nebo těžkou poruchou funkce jater a pacienti s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí.
• Pacienti se známým významným nebo chronickým střevním onemocněním, např. zánětlivým onemocněním střeva nebo malabsorpcí.
• Souběžné podávání více než 40 mg simvastatinu (viz bod 4.5).
• Souběžné použití přípravku Lojuxta se silnými nebo středně silnými inhibitory cytochromu P450 (CYP) 3A4 (např. antimykotiky azoly, jako např. intrakonazolem, flukonazolem, ketokonazolem, vorikonazolem a posakonazolem, makrolidovými antibiotiky, jako např. erytromycinem nebo klaritromycinem, ketolidovými antibiotiky, jako např. telitromycinem, inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem a antiarytmikem dronedaronem [viz bod 4.5]).
• Těhotenství (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Abnormality jaterních enzymů a sledování jater
Lomitapid může způsobit zvýšení hladiny alaninaminotransferázy [ALT] a aspartátaminotransferázy [AST] a jaterní steatózu. V jakém rozsahu jaterní steatóza související s lomitapidem podporuje zvýšení aminotransferáz, není známo. Ačkoli nebyly hlášeny případy jaterní dysfunkce (zvýšení aminotransferáz se zvýšením bilirubinu nebo mezinárodního normalizovaného poměru [INR] nebo jaterního selhání), existuje obava, že lomitapid může indukovat steatohepatitidu, která může za několik let progredovat do cirhózy. Není pravděpodobné, že by klinické studie na podporu bezpečnosti a účinnosti lomitapidu u HoFH byly schopné vzhledem k jejich rozsahu a době trvání tyto nežádoucí účinky detekovat.
S lomitapidem je spojeno zvýšení hladin aminotransferáz (ALT a/nebo AST) (viz bod 5.1). Nedocházelo k souběžnému nebo následnému klinicky významnému zvýšení sérového bilirubinu, INR nebo alkalické fosfatázy. Změny jaterních enzymů se nejčastěji vyskytují během navyšování dávky, ale mohou nastat kdykoli během léčby.
Sledování testů jaterních funkcí
Před zahájením léčby přípravkem Lojuxta změřte ALT, AST, alkalickou fosfatázu, celkový bilirubin, gama-glutamyltransferázu (gama-GT) a sérový albumin. Léčivý přípravek je kontraindikován u pacientů se středně těžkou nebo těžkou poruchou funkce jater a pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí. Jestliže jsou základní vyšetření jaterních testů abnormální, zvažte zahájení léčby léčivým přípravkem až poté, co hepatolog provede vhodná vyšetření a abnormality základních testů budou vysvětleny či vyřešeny.
V prvním roce měřte jaterní testy (minimálně ALT a AST) před každým zvýšením dávky nebo jednou měsíčně podle toho, co nastane dříve. Po prvním roce provádějte tyto testy nejméně jednou za 3 měsíce a před každým zvýšením dávky. Jestliže pozorujete zvýšení hladiny aminotransferáz, dávku přípravku Lojuxta snižte, a při přetrvávajícím nebo klinicky významném zvýšení ukončete léčbu (konkrétní doporučení uvádí Tabulka 1).
Modifikace dávky na základě zvýšené hladiny jatemích aminotransferáz
Tabulka 1 je shrnutím doporučení pro úpravu dávky a sledování pacientů, u kterých dojde během léčby přípravkem Lojuxta ke zvýšení hladiny aminotransferáz.
Tabulka 1: Úprava dávky a sledování pacientů se zvýšenými hladinami aminotransferáz
|
ALT nebo AST |
Doporučení týkající se léčby a sledování* |
|
>3x a <5x horní mez normálního rozsahu (ULN) |
• Zvýšení potvrďte opakovaným měřením v rámci jednoho týdne. • Jestliže se zvýšení potvrdí, snižte dávku a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). • Testy opakujte každý týden, a jestliže budou přítomny známky abnormálních jaterních funkcí (zvýšení bilirubinu či INR), jestliže aminotransferázy stoupnou nad 5x ULN nebo jestliže hladiny aminotransferáz během přibližně 4 týdnů neklesnou pod 3x ULN, přerušte léčbu. Pacienty s přetrvávajícím zvýšením hladin aminotransferáz >3x ULN odešlete k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, zvažte snížení dávky a častější sledování jaterních testů. |
|
>5x ULN |
• Přerušte léčbu a proveďte další jaterní testy, pokud již tyto nebyly provedeny (např. na alkalickou fosfatázu, celkový bilirubin a INR). Jestliže během přibližně 4 týdnů hladiny aminotransferáz neklesnou pod 3x ULN, odešlete pacienta k hepatologovi na další vyšetření. • Pokud bude léčba přípravkem Lojuxta po poklesu hladin aminotransferáz pod <3x ULN znovu zahájena, snižte dávku a častěji sledujte jaterní testy. |
*Doporučení založené na ULNpřibližně 30-40 mezinárodních jednotek/l.
Jestliže je zvýšení aminotransferáz provázeno klinickými symptomy poškození jater (např. nevolností, zvracením, bolestí břicha, horečkou, žloutenkou, letargií, příznaky podobnými chřipce), vzestupem hladin bilirubinu na >2x ULN nebo aktivním onemocněním jater, přerušte léčbu přípravkem Lojuxta a odešlete pacienta k hepatologovi na další vyšetření.
Opětovné nasazení léčby lze zvážit tehdy, pokud přínosy převyšují rizika související s potenciálním onemocněním jater.
Jaterní steatóza a riziko progresivního onemocnění jater
Většina léčených pacientů vykazuje vzestup obsahu tuku v játrech, což je ve shodě s mechanismem působení lomitapidu. V otevřené studii fáze 3 došlo u 18 z 23 pacientů s HoFH k vývoji jaterní steatózy (obsah tuku v játrech > 5,56 %) dle měření nukleární magnetickou rezonanční spektroskopií (MRS) (viz bod 5.1). Medián absolutního vzestupu jaterního tuku byl dle měření pomocí MRS jak po 26 týdnech, tak po 78 týdnech léčby 6 % oproti 1 % při zahájení studie. Jaterní steatóza je rizikový faktor progresivního onemocnění jater včetně steatohepatitidy a cirhózy. Dlouhodobé následky jaterní steatózy související s léčbou přípravkem Lojuxta nejsou známy. Klinické údaje naznačují, že nahromadění tuku v játrech je po ukončení léčby přípravkem Lojuxta reverzibilní, ale není známo, zda přetrvávají histologické změny, zejména po dlouhodobém užívání.
Sledování důkazů přítomnosti progresivního onemocnění jater
Při zahájení léčby a dále každý rok by se měl provádět pravidelný screening steatohepatitidy/fibrózy s použitím následujících zobrazovacích metod a hodnocení biomarkerů:
• Zobrazení elasticity tkáně, např. fibrosken, ARFI (Acoustic Radiation Force Impulse) nebo elastografie s použitím magnetické rezonance (MR)
• Gama-GT a sérový albumin pro detekci možného poškození jater
• Minimálně jeden marker z každé z následujících kategorií:
• Vysoce senzitivní C-reaktivní protein (hs-CRP), rychlost sedimentace erytrocytů (ESR), CK-18 fragment, NashTest (zánět jater)
• Rozšířený panel pro detekci jaterní fibrózy (ELF), fibrometr, poměr AST/ALT, skóre fib-4, fibrotest (jaterní fibróza)
Provedení těchto testů a jejich interpretace by měly zahrnovat spolupráci ošetřujícího lékaře s hepatologem. U pacientů s výsledky naznačujícími přítomnost steatohepatitidy nebo fibrózy by měla být zvážena biopsie jater.
Jestliže má pacient biopticky potvrzenou steatohepatitidu nebo fibrózu, měl by být znovu zhodnocen poměr přínosů a rizik a léčba by měla být ukončena, je-li to nutné.
Souběžné užívání inhibitorů CYP3A4
Zdá se, že lomitapid je citlivým substrátem pro metabolismus cestou CYP3A4. Inhibitory CYP3A4 zvyšují expozici lomitapidu, přičemž silné inhibitory zvyšují expozici přibližně 27krát. Souběžné užití středně silných nebo silných inhibitorů CYP3A4 a přípravku Lojuxta je kontraindikováno (viz bod 4.3). V klinických studiích s lomitapidem se u jednoho pacienta s HoFH během několika dní po zahájení užívání silného inhibitoru CYP3A4 klaritromycinu vyvinuly výrazně zvýšené hladiny aminotransferáz (ALT 24x ULN, AST 13x ULN). Jestliže se nelze vyvarovat léčby středně silnými nebo silnými inhibitory CYP3A4, mělo by být užívání přípravku Lojuxta v průběhu léčby ukončeno.
Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta má být podávána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4.
Souběžné užívání induktorů CYP3A4
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. Induktory CYP3A4 vykazují svůj účinek v závislosti na čase a dosažení maximálního účinku může trvat minimálně 2 týdny od zahájení léčby. Naopak při ukončení léčby může trvat minimálně 2 týdny, než indukce CYP3A4 odezní.
Očekává se, že souběžné podávání induktoru CYP3A4 snižuje účinek přípravku Lojuxta. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinu, pioglitazonu, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Použití třezalky tečkované současně s přípravkem Lojuxta je nutné se vyvarovat.
Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. Při vysazení induktoru CYP3A4 je nutné zvážit možnost zvýšené expozice a může být nutné snížit dávku přípravku Lojuxta.
Souběžné použití s inhibitory HMG-CoA reduktázv (..statinv")
Lomitapid zvyšuje plazmatické koncentrace statinů. U pacientů, kteří dostávají přípravek Lojuxta jako přídatnou terapii vedle statinů, by měly být sledovány nežádoucí účinky spojené s použitím vysokých dávek statinů. Statiny příležitostně mohou vyvolat myopatii. Ve vzácných případech může mít myopatie formu rhabdomyolýzy s akutním renálním selháním sekundárně způsobeným myoglobinurií nebo bez renálního selhání a může mít fatální následky. Všichni pacienti užívající přípravek Lojuxta vedle statinů by měli být informování o možném riziku myopatie a měli by být informováni, aby okamžitě hlásili nevysvětlitelnou bolest, citlivost nebo slabost svalů. S přípravkem Lojuxta by se neměly používat dávky simvastatinu > 40 mg (viz bod 4.3).
Grapefruitový džus
Ze stravy pacientů je při léčbě přípravkem Lojuxta nutno vyloučit grapefruitový džus.
Riziko supraterapeutické nebo subterapeutické antikoagulace u antikoagulačních přípravků na bázi kumarinu
Lomitapid zvyšuje plazmatické koncentrace warfarinu. Zvýšení dávky přípravku Lojuxta může vést k supraterapeutické antikoagulaci a snížení dávky může vést k subterapeutické antikoagulaci.
U jednoho z pěti pacientů užívajících souběžně warfarin přispěla obtížná kontrola INR k předčasnému ukončení studie fáze 3. Pacienti užívající warfarin by měli podstupovat pravidelné sledování INR, zejména po jakýchkoli změnách dávkování přípravku Lojuxta. Dávka warfarinu by měla být upravena dle klinické indikace.
Požívání alkoholu
Alkohol může zvyšovat hladinu tuku v játrech a může vést k indukci nebo exacerbaci poškození jater. Ve studii fáze 3 udávali 3 ze 4 pacientů se zvýšením ALT >5x ULN konzumaci alkoholu překračující limit doporučený v protokolu. Požívání alkoholu během léčby přípravkem Lojuxta se nedoporučuje.
Hepatotoxické přípravky
Opatrnosti je třeba při použití přípravku Lojuxta s dalšími léčivými přípravky se známým hepatotoxickým potenciálem, např. izotretinoinem, amiodaronem, acetaminofenem (>4 g/den po >3 dny/týden), metotrexátem, tetracyklinem a tamoxifenem. Účinek souběžného podávání přípravku Lojuxta s dalšími hepatotoxickými léčivými přípravky není znám. Může být nutné častější sledování jaterních testů.
Snížená absorpce vitamínů rozpustných v tucích a mastných kyselin v séru
Vzhledem k mechanismu působení lomitapidu v tenkém střevě může lomitapid snižovat absorpci živin rozpustných v tucích. Ve studii fáze 3 byly pacientům denně poskytovány nutriční doplňky s vitamínem E, kyselinou linolovou, ALA, EPA a DHA. V této studii se úroveň mediánu sérového vitamínu E, ALA, kyseliny linolové, EPA, DHA a kyseliny arachidonové mezi výchozím bodem studie a týdnem 26 snížila, ale zůstala nad spodním limitem referenčního rozmezí. Nežádoucí klinické důsledky tohoto snížení při léčbě lomitapidem nebyly pozorovány po dobu až 78 týdnů. Pacienti léčení přípravkem Lojuxta by měli denně užívat doplňky, které obsahují 400 mezinárodních jednotek vitamínu E a přibližně 200 mg kyseliny linolové, 210 mg ALA, 110 mg EPA a 80 mg DHA.
Antikoncepční opatření u žen ve fertilním věku.
Před zahájením léčby u žen ve fertilním věku by mělo být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z důvodu průjmu a/nebo zvracení (viz bod 4.5). Perorální antikoncepční přípravky na bázi estrogenů jsou slabými inhibitory CYP3A4 (viz bod 4.2).
Pacientky by měly být informovány, že v případě otěhotnění musejí neprodleně informovat svého lékaře a ukončit užívání přípravku Lojuxta (viz bod 4.6).
Laktóza
Přípravek Lojuxta obsahuje laktózu, pacienti se vzácnými hereditárními problémy jako jsou intolerance galaktózy, vrozený deficit laktázy nebo glukózová-galaktózová malabsorpce by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
Tabulka 2: Interakce přípravku Lojuxta s jinými léčivými přípravky a jiné formy interakce
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
Inhibitory CYP3A4 |
Při současném podávání lomitapidu v dávce 60 mg s ketokonazolem, silným inhibitorem CYP3A4, v dávce 200 mg dvakrát denně se zvýšila AUC lomitapidu přibližně 27krát a Cmax přibližně 15krát. Interakce mezi středně silnými inhibitory CYP3A4 a lomitapidem nebyly studovány. Předpokládá se, že středně silné inhibitory CYP3A4 mají významný vliv na farmakokinetiku lomitapidu. Očekává se, že souběžné použití středně silných inhibitorů CYP3A4 zvyšuje expozici lomitapidu 4-10krát podle výsledků studie se silným inhibitorem CYP3A4 podle historických údajů pro zkušební model CYP3A4 midazolam. Slabé inhibitory CYP3A4 při současném podávání budou patrně zvyšovat expozici lomitapidu. Při podávání lomitapidu 20 mg současně s atorvastatinem (tj. |
Použití silných nebo středně silných inhibitorů CYP3A4 spolu s přípravkem Lojuxta je kontraindikováno. Jestliže se nelze vyvarovat léčby antimykotiky azoly (např. itrakonazolem, ketokonazolem, flukonazolem, vorikonazolem, posakonazolem), antiarytmikem dronedaronem, makrolidovými antibiotiky (např. erytromycinem, klaritromcinem), ketolidovými antibiotiky (např. telitromycinem), inhibitory HIV proteáz, blokátory vápníkových kanálů diltiazemem a verapamilem, měla by být během této léčby léčba přípravkem Lojuxta pozastavena (viz body 4.3 a 4.4). Grapefruitový džus je středně silným inhibitorem CYP3A4 a má se za to, že podstatně zvyšuje expozici lomitapidu. Pacienti užívající přípravek Lojuxta by se měli vyvarovat konzumace grapefruitového džusu. Při současném podávání s atorvastatinem má být interval mezi oběma přípravky 12 hodin nebo se má snížit dávka přípravku Lojuxta o polovinu (viz bod 4.2). Lojuxta musí být užívána 12 hodin po jakémkoliv dalším slabém inhibitoru CYP3A4. Příklady slabých inhibitorů CYP3A4 zahrnují: alprazolam, |
|
Léčivé přípravky |
Účinek na hladinu lomitapidu |
Doporučení týkající se souběžného podávání s přípravkem Lojuxta |
|
slabým inhibitorem CYP3A4) se AUC a Cmax lomitapidu zvýšily přibližně 2x. Pokud byl lomitapid podáván 12 hodin po atorvastatinu, žádné klinicky významné zvýšení expozice lomitapidu nebylo pozorováno. Při podávání lomitapidu 20 mg s ethinyl estradiolem/norgestimátem (tj. slabým inhibitorem CYP3A4), žádné klinicky významné zvýšení expozice lomitapidu nebylo pozorováno při užívání současném ani s intervalem 12 hodin. |
amiodaron, amlodipin, atorvastatin, azitromycin, bikalutamid, cilostazol, cimetidin, cyklosporin, klotrimazol, fluoxetin, fluvoxamin, fosaprepitant, ginkgo, vodilku kanadskou, izoniazid, ivakaftor, lacidipin, lapatinib, linagliptin, nilotinib, perorální antikoncepční přípravky obsahující estrogeny, pazopanib, mátový olej, propiverin, ranitidin, ranolazin, roxithromycin, hořké pomeranče, takrolimus, tikagrelor a tolvaptan. Tento seznam není úplný a předepisující lékaři by měli zkontrolovat preskripční informace o léčivech, která mají být podávána spolu s přípravkem Lojuxta s ohledem na možné interakce zprostředkované CYP3A4. Účinek podávání více než jednoho slabého inhibitoru CYP3A4 nebyl testován, ale očekává se, že účinek na expozici lomitapidu je větší než při současném podávání jednotlivých inhibitorů s lomitapidem. Opatrnost je dále nutná při podávání více než jednoho slabého inhibitoru CYP3A4 s přípravkem Lojuxta. | |
|
Induktory CYP3A4 |
Dá se očekávat, že léčivé přípravky indukující CYP3A4 mohou zvyšovat rychlost a rozsah metabolismu lomitapidu. To by mohlo následně snížit účinek lomitapidu. Jakýkoli vliv na účinnost je pravděpodobně proměnlivý. |
Při souběžném podávání induktorů CYP3A4 (tj. aminoglutetimidu, nafcilinu, non-nukleosidových inhibitorů reverzní transkriptázy, fenobarbitalu, rifampicinu, karbamazepinnu, pioglitazonu, třezalky tečkované, glukokortikoidů, modafinilu a fenytoinu) a přípravku Lojuxta je nutné zvážit možnost vzájemné interakce mezi léčivy s vlivem na účinnost. Jestliže je induktor CYP3A4 určen k chronickému použití, doporučuje se zvýšit frekvenci hodnocení LDL-C během souběžného použití a zvážit zvýšení dávky přípravku Lojuxta, aby bylo zajištěno udržení požadované úrovně účinnosti. |
|
Sekvestranty žlučových kyselin |
Interakce lomitapidu se sekvestranty žlučových kyselin (pryskyřicemi, např. kolesevelamem a cholestyraminem) nebyly testovány. |
Protože sekvestranty žlučových kyselin mohou interferovat s absorpcí perorálních léčivých přípravků, měly by se užívat minimálně 4 hodiny před přípravkem Lojuxta nebo minimálně 4 hodiny po něm. |
Účinky lomitapidu na jiné léčivé přípravky
Inhibitory HMG-CoA reduktázy („statiny“): Lomitapid zvyšuje plazmatické koncentrace statinů. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním simvastatinu 40 mg se zvýšily u kyseliny simvastatinu hodnoty AUC o 68 % a Cmax o 57 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním atorvastatinu 20 mg se zvýšily u kyseliny atorvastatinu hodnoty AUC o 52 % a Cmax o 63 %. Při podávání lomitapidu 60 mg do dosažení ustáleného stavu před podáním rosuvastatinu 20 mg se zvýšila hodnota Tmax rosuvastatinu z 1 na 4 hodiny, hodnota AUC se zvýšila o 32 % a hodnota Cmax zůstala nezměněná. Riziko myopatie u simvastatinu je závislé na dávce. Použití přípravku Lojuxta je u pacientů léčených vysokými dávkami statinů (> 40 mg) kontraindikováno (viz body 4.3 a 4.4).
Kumarinové antikoagulační přípravky: Podávání lomitapidu 60 mg do dosažení ustáleného stavu a po dobu 6 dní po warfarinu 10 mg zvýšilo hodnotu INR 1,26krát. Hodnota AUC pro R(+)-warfarin se zvýšila o 25 % a pro S(-)-warfarin o 30 %. Hodnota Cmax pro R(+)-warfarin se zvýšila o 14 % a pro S(-)-warfarin o 15 %. U pacientů užívajících souběžně kumariny (např. warfarin) a přípravek Lojuxta by měla být před zahájením užívání přípravku Lojuxta stanovena hodnota INR a tato hodnota by měla být pravidelně kontrolována, přičemž dávkování kumarinů by mělo být upraveno dle klinické potřeby (viz bod 4.4).
Fenofibrát, niacin a ezetimib: Při podávání lomitapidu do dosažení ustáleného stavu před podáním mikronizovaného fenofibrátu 145 mg, niacinu s prodlouženým uvolňováním 1000 mg nebo ezetimibu 10 mg nebyly pozorovány klinicky významné účinky působení kteréhokoli z těchto léčivých přípravků. Při souběžném podávání s přípravkem Lojuxta není nutná úprava dávky.
Perorální antikoncepce: Při podávání lomitapidu 50 mg do dosažení ustáleného stavu spolu s perorálním antikoncepčním přípravkem na bázi estrogenů nebyl pozorován klinicky ani statisticky významný vliv na farmakokinetiku složek perorální antikoncepce (ethinylestradiolu a 17-deacetylnorgestimátu, metabolitu norgestimátu). Neočekává se, že by lomitapid přímo ovlivňoval účinnost perorální antikoncepce na bázi estrogenů, nicméně průjem a/nebo zvracení mohou snížit absorpci hormonů. U případů prodlouženého nebo závažného průjmu a/nebo zvracení trvajících více než 2 dny by měla být po dobu 7 dnů po ústupu symptomů použita další antikoncepční metoda.
Substráty P-gp: Lomitapid in vitro inhibuje P-gp a může zvyšovat absorpci P-gp substrátů. Souběžné podávání přípravku Lojuxta s P-pg substráty (např. aliskirenem, abrisentanem, kolchicinem, dabigatran etexilátem, dixoginem, everolimem, fexofenadinem, imatinibem, lapatinibem, maravirokem, nilotinibem, posakonazolem, ranolazinem, saxagliptinem, sirolimem, sitagliptinem, talinololem, tolvaptanem, topotekanem) může zvýšit absorpci P-gp substrátů. Při souběžném použití s přípravkem Lojuxta by mělo být zváženo snížení dávky P-gp substrátu.
In vitro hodnocení lékových interakcí: Lomitapid inhibuje CYP3A4. Lomitapide neindukuje CYP 1A2, 3A4 ani 2B6 a neinhibuje CYP 1A2, 2B6, 2C9, 2C19, 2D6 ani 2E1. Lomitapid není P-gp substrátem, ale inhibuje P-gp. Lomitapid neinhibuje protein rezistence u karcinomu prsu (BCRP).
4.6 Fertilita, těhotenství a kojení
Přípravek Lojuxta je v těhotenství kontraindikován. Neexistují spolehlivé údaje o použití přípravku u těhotných žen. Studie na zvířatech prokázaly vývojovou toxicitu (teratogenitu, embryotoxicitu, viz bod 5.3). Možné riziko pro člověka je neznámé.
Použití u žen ve fertilním věku.
Před zahájením léčby u žen ve fertilním věku by mělo být vyloučeno těhotenství, mělo by být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být zahájeno podávání účinné antikoncepce. Pacientky užívající perorální antikoncepci na bázi estrogenů by měly být informovány o případné ztrátě účinnosti z důvodu průjmu a/nebo zvracení. Dokud symptomy neustoupí, je nutno používat další antikoncepční metody (viz bod 4.5).
Kojení
Není známo, zda se lomitapid vylučuje do mateřského mléka. Vzhledem k potenciálu nežádoucích účinků zjištěnému na základě studií s lomitapidem na zvířatech (viz bod 5.3) je nutné rozhodnout, zda ukončit kojení nebo užívání léčivého přípravku, a to při zvážení významu léčivého přípravku pro matku.
Fertilita
U potkaních samců ani samic, kterým byl podáván lomitapid se systémovou expozicí (AUC) ve výši odhadovaného čtyř- až pětinásobku expozice u lidí při maximální doporučené dávce u člověka, nebyly pozorovány žádné nežádoucí účinky na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Lojuxta může mít mírný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejzávažnějšími nežádoucími účinky během léčby byly abnormality jaterních aminotransferáz (viz bod 4.4).
Nejčastějšími nežádoucími účinky byly gastrointestinální účinky. Nežádoucí gastrointestinální účinky byly hlášeny u 27 (93 %) z 29 pacientů klinické studie fáze 3. Průjem se objevil u 79 %, nevolnost u 65 %, dyspepsie u 38 % a zvracení u 34 % pacientů. Další účinky, které hlásilo minimálně 20 % pacientů, zahrnují bolest břicha, břišní dyskomfort, nadmutí břicha, zácpu a plynatost.
Nežádoucí gastrointestinální účinky se častěji objevovaly během fáze navyšování dávky ve studii a poklesly, jakmile pacienti dosáhli maximální tolerované dávky lomitapidu.
Gastrointestinální nežádoucí účinky se závažnou intenzitou byly hlášeny u 6 (21 %) z 29 pacientů v klinické studii fáze 3, přičemž nejčastějším účinkem byl průjem (4 pacienti, 14 %), zvracení (3 pacienti, 10 %) a bolest břicha, nadmutí a/nebo dyskomfort (2 pacienti, 7 %). Gastrointestinální účinky přispěly k časnému ukončení studie u 4 (14 %) pacientů.
Nejčastěji udávanými nežádoucími účinky se závažnou intenzitou byly průjem (4 subjekty, 14 %), zvracení (3 pacienti, 10 %) a nadmutí břicha a zvýšení ALT (pokaždé 2 subjekty, 7 %).
Přehled nežádoucích účinků v tabulce
Dle frekvence jsou nežádoucí účinky definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), málo časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), s neznámou frekvencí (z dostupných údajů nelze určit).
Tabulka 3 zahrnuje veškeré nežádoucí účinky hlášené u 35 pacientů léčených ve studii fáze 2 UP1001 a ve studii fáze 3 UP1002/AEGR-733-005 nebo v rozšířené studii AEGR-733-012.
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Časté |
Gastroenteritida |
|
Poruchy metabolismu a výživy |
Velmi časté |
Snížená chuť k jídlu |
|
Poruchy nervového systému |
Časté |
Závratě Bolest hlavy Migréna |
|
Gastrointestinální poruchy |
Velmi časté |
Břišní dyskomfort Bolest horní části břicha Plynatost Distenze břicha Zácpa |
|
Časté |
Gastritida Rektální tenesmy Aerofagie Nutkání na stolici Říhání Častá stolice Dilatace žaludku Onemocnění žaludku Gastroezofageální reflux Krvácení z hemoroidů Regurgitace | |
|
Poruchy jater a žlučových cest |
Časté |
Jaterní steatóza Hepatotoxicita Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Časté |
Ekchymóza Papuly Erytematózní vyrážka Xantomy |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Únava |
|
Vyšetření |
Velmi časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Snížení tělesné hmotnosti |
|
Časté |
Zvýšený mezinárodní normalizovaný poměr Zvýšená alkalická fosfatáza v krvi Snížení draslíku v krvi Snížený karoten Abnormální mezinárodní normalizovaný poměr Abnormální testy jaterních funkcí Prodloužený protrombinový čas Zvýšené transaminázy Snížený vitamín E Snížený vitamín K |
Tabulka 4 uvádí všechny nežádoucí účinky u subjektů, které dostávaly lomitapid v monoterapii (n=291), léčených ve studiích fáze 2 u subjektů se zvýšenou hladinou LDL-C (N=462).
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Infekce a infestace |
Méně časté |
Gastroenteritida Gastrointestinální infekce Chřipka Nazofaryngitida Sinusitida |
|
Poruchy krve a lymfatického systému |
Méně časté | |
|
Poruchy metabolismu a výživy |
Časté |
Snížená chuť k jídlu |
|
Méně časté |
Dehydratace Zvýšená chuť k jídlu | |
|
Poruchy nervového systému |
Méně časté |
Parestézie Ospalost |
|
Poruchy oka |
Méně časté |
Otok oka |
|
Poruchy ucha a labyrintu |
Méně časté |
Závrať |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté |
Léze hltanu Syndrom kašle horních cest dýchacích |
|
Gastrointestinální poruchy |
Velmi časté |
Plynatost |
|
Časté |
Bolest horní části břicha Distenze břicha Bolest břicha Zvracení Břišní dyskomfort Říhání Bolest v podbřišku Častá stolice | |
|
Méně časté |
Sucho v ústech Tvrdá stolice Gastroezofageální reflux Citlivost břicha Dyskomfort v epigastriu Dilatace žaludku Hemateméza Krvácení v dolní části gastrointestinálního traktu Refluxní ezofagitida | |
|
Poruchy jater a žlučových cest |
Méně časté |
Hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Puchýře Suchá kůže Nadměrné pocení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté |
Svalové spasmy |
|
Méně časté |
Bolest kloubů Bolest svalů Bolest končetin Otok kloubu Svalové záškuby | |
|
Poruchy ledvin a močových cest |
Méně časté |
Hematurie |
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinky |
|
Celkové poruchy a reakce v místě |
Časté |
Únava |
|
podání |
Slabost | |
|
Méně časté |
Bolest na hrudi Mrazení Rychlý pocit nasycení Poruchy chůze Malátnost Horečka | |
|
Vyšetření |
Časté |
Zvýšená alaninaminotransferáza Zvýšená aspartátaminotransferáza Zvýšení jaterních enzymů Abnormální testy jaterních funkcí Snížený počet neutrofilů Snížený počet leukocytů |
|
Méně časté |
Snížení tělesné hmotnosti Zvýšený bilirubin v krvi Zvýšená hladina gama-glutamyltransferázy Zvýšené procento neutrofilů Protein v moči Prodloužený protrombinový čas Abnormální testy plicních funkcí Zvýšený počet leukocytů |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování neexistuje specifická léčba. U hlodavců byla dobře snášena jednorázová perorální dávka lomitapidu >600krát vyšší než maximální doporučená dávka pro člověka (1 mg/kg). Maximální dávkou podanou lidským subjektům v klinických studiích bylo 200 mg ve formě jednorázové dávky, nevyskytly se žádné nežádoucí účinky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná léčiva ovlivňující hladinu lipidů, samotná. ATC kód: C10AX12
Mechanismus účinku
Lomitapid je selektivní inhibitor mikrozomálního transferového proteinu (MTP), intracelulárního proteinu přenášejícího lipidy, který se nachází v lumen endoplazmatického retikula a zodpovídá za vazbu a převod jednotlivých lipidových molekul mezi membránami. MTP hraje klíčovou roli při sestavování lipoproteinů obsahujících apo B v játrech a střevech. Inhibice MTP snižuje sekreci lipoproteinů a cirkulující koncentrace lipidů přenášených v lipoproteinech včetně cholesterolu a triglyceridů.
Klinická účinnost a bezpečnost
Účinnost a bezpečnost lomitapidu při současném podávání diety s nízkým obsahem tuků a dalších terapií snižujících lipidy u dospělých pacientů s HoFH hodnotila otevřená studie s jedním ramenem (UP1002/AEGR-733-005). Pacienti byli instruováni, aby dodržovali dietu s nízkým obsahem tuků (< 20 % kalorického příjmu z tuků) i terapii snižující lipidy, kterou měli při vstupu do studie, včetně aferézy, byla-li tato nutná, a to po dobu 6 týdnů předcházejících výchozímu bodu studie minimálně až do týdne 26. Dávka lomitapidu byla navyšována z 5 mg na individuálně stanovenou maximální tolerovanou dávku až 60 mg. Po týdnu 26 byl pacientům lomitapid ponechán za účelem zjištění účinků dlouhodobé léčby a bylo jim umožněno změnit základní terapii snižující lipidy. Studie zahrnovala celkem 78 týdnů léčby.
Do studie bylo zařazeno 29 pacientů, z nichž 23 dokončilo týden 78. Zařazeno bylo 16 mužů (55 %) a 13 žen (45 %) s průměrným věkem 30,7 let a ve věkovém rozmezí 18 až 55 let. Průměrná dávka lomitapidu v týdnu 26 byla 45 mg a v týdnu 78 byla 40 mg. V týdnu 26 byla průměrná procentuální změna hladiny LDL-C u populace „intention-to-treat“ (ITT) oproti výchozímu bodu -40 %
(p < 0,001). Průměrná procentuální změna oproti výchozímu bodu studie v týdnu 26 s použitím metody LOCF (Last Observation Carried Forward) pro každé hodnocení je uvedena na obrázku 1.
Obrázek 1: Průměrné procentuální změny hladiny LDL-C mezi vstupní hodnotou a týdnem
26 v hlavní studii účinnosti UP1002/AEGR-733-005 (primární cíl) s použitím LOCF pro každé hodnocení (n=29)

Týden studie
Změny lipidů i lipoproteinů v týdnu 26 a 78 při léčbě lomitapidem uvádí Tabulka 5.
Tabulka 5: Absolutní hodnoty a procentuální změny hladin lipidů a lipoproteinů mezi
výchozí hodnotou a týdny 26 a 78 (hlavní studie účinnosti UP1002/AEGR-733-005).
|
Parametr (jednotky) |
Výchozí bod |
Týden 26 / LOCF (n=29) |
Týden 78 (n=23) | ||||
|
Průměr (SD) |
Průměr (SD) |
% změna |
Hodnota p b |
Průměr (SD) |
% změna |
Hodnota p b | |
|
LDL-C, přímý (mg/dl) |
336 (114) |
190 (104) |
-40 |
< 0,001 |
210 (132) |
-38 |
< 0,001 |
|
Celkový cholesterol (TC) (mg/dl) |
430 (135) |
258 (118) |
-36 |
< 0,001 |
281 (149) |
-35 |
< 0,001 |
|
Apolipoprotein B (apo B) (mg/dl) |
259 (80) |
148 (74) |
-39 |
< 0,001 |
151 (89) |
-43 |
< 0,001 |
|
Triglyceridy (TG) (mg/dl)a |
92 |
57 |
-45 |
0,009 |
59 |
-42 |
0,012 |
|
Cholesterol z jiných lipoproteinů než o vysoké hustotě (non-HDL-C) (mg/dl) |
386 (132) |
217 (113) |
-40 |
< 0,001 |
239 (146) |
-39 |
< 0,001 |
|
Cholesterol z lipoproteinů o velmi nízké hustotě (VLDL-C) (mg/dl) |
21 (10) |
13 (9) |
-29 |
0,012 |
16 (15) |
-31 |
0,013 |
|
Lipoprotein (a) (Lp(a)) (nmol/l)a |
66 |
61 |
-13 |
0,094 |
72 |
-4 |
< 0,842 |
|
Cholesterol z lipoproteinů o vysoké hustotě (HDL-C) (mg/dl) |
44 (11) |
41 (13) |
-7 |
0,072 |
43 (12) |
-4,6 |
0,246 |
a Medián uvedený pro TG a Lp(a). Hodnota p je založena na průměrné procentuální změně b Hodnota p u střední procentuální změny oproti výchozímu bodu studie na základě párového t-testu
Jak v týdnu 26, tak v týdnu 78 došlo k významnému snížení hladin LDL-C, TC, apo B, TG, non-HDL-C a VLDL-C a změny u HDL-C vykazovaly v týdnu 26 trend směrem ke snížení a v týdnu 78 se vrátily k výchozím hodnotám.
Účinek přípravku Lojuxta na kardiovaskulární morbiditu a mortalitu nebyl stanoven.
Při zahájení studie užívalo 93 % pacientů statin, 76 % pacientů ezetimib, 10 % niacin, 3 % sekvestrant žlučových kyselin a 62 % pacientů bylo léčeno aferézou. U 15 z 23 (65 %) pacientů byla v týdnu 78 redukována jejich léčba na snížení lipidů, včetně plánovaných a neplánovaných snižování/přerušení. Aferéza byla přerušena u 3 ze 13 pacientů, kteří byli aferézou léčeni v týdnu 26, a u 3 pacientů byla frekvence při současném udržení nízkých hladin LDL-C i po týdnu 78 snížena. Klinický přínos omezení základní terapie snižující lipidy, včetně aferézy, není jistý.
Z 23 pacientů, kteří dokončili týden 26 studie, došlo u 19 (83 %) ke snížení LDL-C o > 25 %, přičemž 8 (35 %) z nich mělo v tomto časovém bodě LDL-C < 100 mg/dl a 1 pacient měl LDL-C < 70 mg/dl.
V této studii došlo u 10 pacientů ke zvýšení AST a/nebo ALT >3x ULN (viz Tabulka 6).
Nejvyšší výsledky testů jaterních funkcí po první dávce (hlavní studie účinnosti UP1002/AEGR-733-005)
|
Parametr/abnormalita |
N (%) |
|
ALT | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
6 (20,7) |
|
>5 až < 10 x ULN |
3 (10,3) |
|
>10 až < 20 x ULN |
1 (3,4) |
|
> 20 x ULN |
0 |
|
AST | |
|
Počet vyšetřených pacientů |
29 |
|
>3 až < 5 x ULN |
5 (17,2) |
|
>5 až < 10 x ULN |
1 (3,4) |
|
>10 až < 20 x ULN |
0 |
|
> 20 x ULN |
0 |
Tabulka 6:
Zvýšení ALT a/nebo AST > 5x ULN bylo zvládnuto snížením dávky nebo dočasným pozastavením podávání lomitapidu a všichni pacienti byli schopni v léčbě studovaným léčivem pokračovat. Nebylo pozorováno klinicky významné zvýšení celkového bilirubinu ani alkalické fosfatázy. Jaterní tuk byl během klinické studie u všech dostupných pacientů prospektivně měřen s použitím MRS (Tabulka 7). Údaje od jedinců, u kterých byla po ukončení lomitapidu prováděna opakovaná měření, ukázaly, že nahromadění tuku v játrech je reverzibilní, ale není známo, zda nepřetrvávají histologické změny.
Tabulka 7: Maximální kategorické změny v % jaterního tuku (hlavní studie účinnosti
UP1002/AEGR-733-005)
|
Maximální absolutní nárůst % jaterního tuku |
Fáze účinnosti Týdny 0-26 N (%) |
Fáze bezpečnosti Týdny 26-78 N (%) |
Celá studie týdny 0-78 N (%) |
|
Počet hodnotitelných pacientů |
22 |
22 |
23 |
|
< 5% |
9 (41) |
6 (27) |
5 (22) |
|
> 5 % až < 10 % |
6 (27) |
8 (36) |
8 (35) |
|
> 10 % až < 15 % |
4 (18) |
3 (14) |
4 (17) |
|
> 15 % až < 20 % |
1 (5) |
4 (18) |
3 (13) |
|
> 20 % až < 25 % |
1 (5) |
0 |
1 (4) |
|
> 25 % |
1 (5) |
1 (5) |
2 (9) |
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lojuxta u jedné nebo více podskupin pediatrické populace s HoFH (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Absolutní perorální biologická dostupnost lomitapidu je 7 %. Absorpce není omezena prostupem léčiva přes střevní bariéru, ale je převážně ovlivněna extenzivním efektem prvního průchodu. Vrcholná plazmatická koncentrace lomitapidu byla dosažena za 4 až 8 hodin po podání perorální dávky. Farmakokinetika lomitapidu je přibližně proporcionální k dávce perorálních jednorázových dávek v terapeutickém rozmezí. U dávek vyšších než 60 mg je naznačena tendence k nelineární závislosti a tyto dávky nejsou doporučeny.
Při podání více dávek se hodnoty Cmax a AUC zvýšily přibližně proporcionálně k dávce lomitapidu. Hodnoty Cmax a AUC se zvýšily jak po vysoce tučném jídle (77 %, respektive 58 %), tak po nízkotučném jídle (70 %, respektive 28 %). Akumulace lomitapidu v plazmě byla konzistentní s hodnotou předpokládanou na základě jedné dávky po podávání jedné perorální dávky denně nad 25 mg po dobu až 4 týdnů. Interindividuální variabilita hodnoty AUC lomitapidu byla přibližně 50 %.
V ustáleném stavu byla akumulace lomitapidu 2,7 při 25 mg a 3,9 při 50 mg.
Distribuce
Po intravenózním podání byl distribuční objem lomitapidu velký (průměr = 1 200 litrů) i přes vysoký stupeň (> 99,8 %) vazby na plazmatické proteiny. Ve studiích na zvířatech se lomitapid vysoce (200násobně) koncentroval v játrech.
Biotransformace
Lomitapid se extenzivně metabolizuje, převážně cestou CYP3A4. Izoformy CYP 2E1, 1A2, 2B6, 2C8 a 2C19 se účastní méně a izoformy 2D6 a 2C9 se metabolismu lomitapidu neúčastní.
Eliminace
Po podání dávky radioaktivně značeného perorálního roztoku zdravým subjektům bylo 93 % podané dávky vyloučeno močí a stolicí. Přibližně 33 % radioaktivity bylo vyloučeno do moči jako metabolity. Zbytek byl vyloučen stolicí, primárně jako oxidované metabolity. Eliminační poločas lomitapidu byl přibližně 29 hodin.
Zvláštní populace
Údaje ze stěžejní klinické studie byly analyzovány s ohledem na vliv možných souvisejících proměnných na expozici lomitapidu. Z vyšetřovaných parametrů (rasa, index tělesné hmotnosti (BMI), hmotnost, věk) může být jako možná související proměnná klasifikován pouze BMI.
Věk a pohlaví
Věk (18-64 let) ani pohlaví nemají na farmakokinetiku lomitapidu klinicky významný vliv.
Rasa
U bělochů nebo latinskoamerické populace není nutná úprava dávky. K dispozici není dostatek informací pro stanovení, zda přípravek Lojuxta vyžaduje úpravu dávky u dalších ras. Protože se ale léčivý přípravek podává způsobem postupného zvyšování dávky podle individuální bezpečnosti a snášenlivosti pacienta, úprava dávkovacího režimu na základě rasy není doporučena.
V populaci s poškozením funkce ledvin byl lomitapid studován pouze u pacientů s terminálním onemocněním ledvin (ESRD). Farmakokinetická studie u pacientů s ESRD podstupujících dialýzu ukázala 36% vzestup průměrné koncentrace lomitapidu v plazmě ve srovnání s párovanými zdravými kontrolami. Koncový poločas lomitapidu nebyl ovlivněn.
Jaterní insuficience
Pro hodnocení farmakokinetiky 60 mg lomitapidu u zdravých dobrovolníků s normálními jaterními funkcemi ve srovnání s pacienty s mírným postižením funkce jater (třída A dle Child-Pugha) a středně těžkým postižením funkce jater (třída B dle Child-Pugha) byla provedena otevřená studie s jednou dávkou. Hodnoty AUC a Cmax lomitapidu u pacientů se středně těžkým postižením funkce jater byly o 164 %, respektive 361 % vyšší než u zdravých dobrovolníků. Hodnoty AUC a Cmax lomitapidu u pacientů s mírným postižením funkce jater byly o 47 %, respektive 4 % vyšší než u zdravých dobrovolníků. U pacientů s těžkým postižením funkce jater (skóre dle Child-Pugha 10-15) nebyl přípravek Lojuxta studován.
Pediatrická populace
U dětí mladších 18 let nebyl přípravek Lojuxta studován.
Starší populace
U pacientů starších 65 let nebyl přípravek Lojuxta studován.
5.3 Předklinické údaje vztahující se k bezpečnosti
Základním zjištěním toxikologických studií léčiva s opakovanou perorální dávkou u hlodavců a psů byla akumulace lipidů v tenkém střevě a/nebo v játrech spojená s poklesem sérových hladin cholesterolu a/nebo triglyceridů. Tyto změny jsou vzhledem k mechanismu působení lomitapidu sekundární. Další změny související s játry ve studiích toxicity s opakovanou dávkou u potkanů a psů zahrnovaly zvýšené hladiny sérové aminotransferázy, subakutní zánět (pouze u potkanů) a nekrózu jednotlivých buněk. V jednoleté studii s opakovanou dávkou u psů se v játrech nevyskytovaly mikroskopické změny, ačkoli u fen byla minimálně zvýšená sérová hladina AST.
U hlodavců byla pozorována plicní histiocytóza. U psů byl pozorován pokles parametrů erytrocytů i poikilocytóza a/nebo anizocytóza. U psů byla v šestiměsíční studii při 205násobku lidské expozice (AUC) 60 mg pozorována testikulární toxicita. V roční studii u psů nebyly při 64násobku lidské expozice 60 mg pozorovány nežádoucí účinky na varlata.
Ve studii nutriční karcinogenity u myší byl lomitapid podáván po dobu až 104 týdnů v dávkách v rozmezí 0,3 až 45 mg/kg/den. Při dávkách > 1,5 mg/kg/den u samců (> 2násobek lidské expozice při 60 mg denně na základě AUC) a > 7,5 mg/kg/den u samic (> 9násobek lidské expozice při 60 mg na základě AUC) došlo ke statisticky významnému vzestupu incidence adenomu i karcinomu jater. Incidence karcinomu tenkého střeva a/nebo kombinovaného adenomu a karcinomu (u myší vzácné nádory) byla významně zvýšena při dávkách > 15 mg/kg/den u samců (> 26násobek lidské expozice při 60 mg na základě AUC) a 15 mg/kg/den u samic (22násobek lidské expozice při 60 mg na základě AUC).
Ve studii perorální kancerogenity u potkanů byl lomitapid podáván po dobu až 99 týdnů v dávkách až
7,5 mg/kg/den u samců a 2,0 mg/kg/den u samic. U samců i samic byla pozorována fokální jaterní fibróza a pouze u samců byla pozorována cystická degenerace jater. U samců s vysokou dávkou byla pozorována zvýšená incidence pankreatického acinárního adenomu při expozici 6krát vyšší než u lidí při 60 mg na základě AUC.
V panelu studií in vitro a in vivo nebyl lomitapid mutagenní ani genotoxický.
Lomitapid neměl účinek na reprodukční funkce u samic potkanů v dávkách až 1 mg/kg nebo samců potkanů v dávkách až 5 mg/kg. Systémová expozice lomitapidu v těchto dávkách byla odhadována na čtyřnásobek (samice) až pětinásobek (samci) expozice u lidí při 60 mg na základě AUC.
Lomitapid byl u potkanů teratogenní při současné absenci mateřské toxicity při expozici (AUC) odhadované na dvojnásobek lidské expozice při 60 mg. U králíků se nevyskytly známky embryofetální toxicity při trojnásobku maximální doporučené denní dávky u lidí (MRHD) 60 mg na základě plochy tělesného povrchu. Embryofetální toxicita při absenci mateřské toxicity byla pozorována u králíků při > 6,5násobku MRHD. U fretek byl lomitapid toxický pro matku i teratogenní při dávkách < 1x MRHD.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky
předbobtnalý škrob (kukuřičný) sodná sůl karboxymethylškrobu mikrokrystalická celulóza monohydrát laktózy koloidní bezvodý oxid křemičitý magnesium-stearát
Obal tobolky želatina
oxid titaničitý (E171) žlutý oxid železitý (E172)
Tiskařský inkoust šelak
černý oxid železitý (E172) propylenglykol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Uchovávejte v dobře uzavřené lahvičce, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Lahvička z polyetylenu o vysoké hustotě (HDPE) vybavená vložkou z polyesteru / hliníkové fólie / kartonu a s polypropylenovým šroubovacím uzávěrem.
Velikost balení:
28 tobolek
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Aegerion Pharmaceuticals Ltd Lakeside House 1 Furzeground Way Stockley Park East Uxbridge UB11 1BD Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/851/006
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ
POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO
REGISTRACI PŘÍPRAVKU ZA VÝJIMEČNÝCH OKOLNOSTÍ
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Catalent UK Packaging Limited
Lancaster Way
Wingates Industrial Estate
Westhoughton
Bolton
Lancashire
BL5 3XX
Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s „omezením“ (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Před uvedením přípravku na trh by měl držitel rozhodnutí o registraci poskytnout edukační balíček všem lékařům, u kterých se očekává, že budou lomitapid předepisovat/používat.
Edukační balíček pro lékaře by měl obsahovat:
• souhrn údajů o přípravku,
• pokyny pro předepisujícího lékaře,
• bezpečnostní karty pacienta,
• brožury pro pacienty.
Držitel rozhodnutí o registraci se musí před distribucí tohoto přípravku v daném teritoriu dohodnout na obsahu a formátu edukačních materiálů a komunikačního plánu s příslušným vnitrostátním orgánem v každém členském státě.
Pokyny pro předepisující lékaře by měly zahrnovat následující klíčové prvky:
Vhodný výběr pacientů
• Přípravek Lojuxta je indikován k použití pouze u dospělých pacientů s HoFH;
• bezpečnost a účinnost přípravku Lojuxta u dětí mladších 18 let nebyla stanovena;
• léčbu přípravkem Lojuxta by měl zahajovat a sledovat lékař, který má zkušenosti s léčbou poruch lipidů;
• v neklinických studiích byl přípravek Lojuxta teratogenní a ženy ve fertilním věku nesmějí být před zahájením léčby těhotné a musejí užívat účinnou antikoncepci.
Gastrointestinální (GIT) účinky
• Informace o nežádoucích účincích, včetně průjmu, nevolnosti, nadýmání, bolesti břicha nebo břišního dyskomfortu, nadmutí břicha, zvracení, dyspepsie, říhání a snížené chuti k jídlu;
• kontraindikace použití u pacientů se známým významným nebo chronickým onemocněním střeva, např. zánětlivým onemocněním střeva nebo malabsorpcí;
• doporučení navyšovat dávku přípravku Lojuxta postupně pro dosažení lepší snášenlivosti léčivého přípravku;
• doporučení pacientům týkající se:
- nutnosti dodržovat dietu s nízkým obsahem tuků (tj. pacienti by měli dodržovat dietu, ve které
pochází méně než 20 % energie z tuků);
- načasování podávání léčivého přípravku (přípravek Lojuxta by se měl užívat nalačno, minimálně dvě hodiny po večeři);
- potřeby užívat denně nutriční doplňky (tj. 400 IU vitamínu E, přibližně 200 mg kyseliny linolové, 110 mg kyseliny eikosapentaenové (EPA), 210 mg alfa-linolenové kyseliny (ALA) a 80 mg dokosahexaenové kyseliny (DHA) denně).
Jaterní příhody související se zvýšením aminotransferáz a progresivní onemocnění jater
• Informace o kontraindikaci u pacientů se středně těžkým nebo těžkým preexistujícím postižením funkce jater/onemocněním jater včetně pacientů s neobjasněnými přetrvávajícími abnormálními testy jaterních funkcí;
• informace o klinických nálezech (tj. zvýšení jaterních enzymů a steatóze) u subjektů léčených přípravkem Lojuxta během fáze vývoje;
• doporučení nabádající k opatrnosti při používání přípravku Lojuxta s dalšími hepatotoxickými léčivy a doporučení zvážit častější sledování jaterních testů;
• doporučení pro pacienty týkající se rizika souběžného požívání alkoholu;
• doporučení týkající se sledování jaterních funkcí (měření jaterních enzymů a celkového bilirubinu) před léčbou přípravkem Lojuxta a během ní a doporučení týkající se rutinního screeningu pro detekci přítomnosti steatohepatitidy a jaterní fibrózy včetně specifických detailů screeningových testů při zahájení léčby a dále každoročně takto:
- zobrazení elasticity tkáně, např. fibrosken, ARFI (Acoustic Radiation Force Impulse) nebo elastografie s použitím magnetické rezonance (MR);
- měření biomarkerů a/nebo skórovací metody. Tyto metody zahrnují minimálně jeden marker z každé z následujících kategorií:
> gama-GT, sérový albumin (poškození jater);
> vysoce senzitivní C-reaktivní protein (hs-CRP), rychlost sedimentace erytrocytů (ESR), CK-18 fragment, NashTest (zánět jater);
> rozšířený panel pro detekci jaterní fibrózy (ELF), fibrometr, poměr AST/ALT, skóre fib-4, fibroteset (jaterní fibróza).
Ženy v reprodukčním věku
• Lomitapid byl v neklinických studiích teratogenní a je kontraindikován u žen, které jsou těhotné nebo mohou otěhotnět. Ženám, které otěhotní, by mělo být poskytnuto poradenství a měly by být odeslány ke specialistovi na teratologii;
• před zahájením léčby žen ve fertilním věku:
- mělo by být potvrzeno, že žena není těhotná;
- mělo by být poskytnuto vhodné poradenství o účinných metodách antikoncepce a mělo by být
zahájeno podávání účinné antikoncepce;
• upozornění na možnou ztrátu účinnosti perorálních antikoncepčních přípravků v důsledku průjmu či zvracení a na nutnost používat další antikoncepci po dobu 7 dní po ústupu symptomů;
• ženy by měly okamžitě informovat svého lékaře, pokud se domnívají, že by mohly být těhotné.
Lékové interakce
• Informace o interakcích s inhibitory a induktory CYP3A4, kumarinovými antikoagulanty, statiny, P-gp substráty, perorálními antikoncepčními přípravky, sekvestranty žlučových kyselin a grapefruitovým džusem;
• význam suplementace mastných kyselin a vitamínů rozpustných v tucích;
• dodržování režimu suplementace by mělo být ověřováno na pravidelných kontrolách dle rozvrhu a význam suplementace by měl být zdůrazněn.
Edukační materiály pro pacienty
Informace, že edukační materiály pro pacienty, které jsou součástí balíčku pro předepisujícího lékaře, mohou být použity pro konzultaci s pacientem.
Všem pacientům by v čase zahájení léčby přípravkem Lojuxta měla být poskytnuta kopie brožury pro pacienta a bezpečnostní karta pacienta.
Pacienti by měli být informováni, že bezpečnostní kartu pacienta je nutné nosit při sobě a ukázat ji všem lékařům, kteří je léčí.
Registr LOWER (Lomitapide Observational Worldwide Evaluation Registry)
Informace o existenci a významu celosvětového observačního registru určeného k systematickému sběru informací o výsledcích z hlediska bezpečnosti a účinnosti u pacientů léčených lomitapidem. Předepisující lékaři jsou vyzýváni, aby všechny pacienty léčené přípravkem Lojuxta zařadili do globálního registru.
Brožura pro pacienty
Brožura pro pacienty by měla zahrnovat následující klíčové prvky:
• neužívat přípravek Lojuxta, jestliže má pacient problémy s játry nebo neobjasněné abnormální jaterní testy;
• informace, že přípravek Lojuxta může způsobit problémy s játry;
• nutnost informovat lékaře, pokud měl pacient v minulosti problémy s játry;
• nutnost informovat lékaře o veškerých léčivých přípravcích, které pacient užívá, jelikož je nutné věnovat zvláštní pozornost situaci, kdy jsou zároveň užívána jiná léčiva, která mohou způsobit problémy s játry;
• symptomy onemocnění jater, u kterých by měl pacient vyhledat lékaře;
• vysvětlení všech typů prováděných testů (zobrazovacích metod i krevních testů) pro kontrolu jaterních funkcí a význam jejich pravidelného provádění;
• informace, že přípravek Lojuxta byl v neklinických studiích teratogenní a neměl by se užívat během těhotenství nebo u pacientek, které se snaží otěhotnět;
• ženy v reprodukčním věku by měly užívat adekvátní antikoncepci, a pokud mají podezření, že by mohly být těhotné, musejí neprodleně informovat svého lékaře;
• přípravek Lojuxta může způsobit průjem a zvracení a pokud tato situace nastane, musejí pacientky užívající perorální antikoncepci používat další antikoncepční metody po dobu 7 dní po ústupu
symptomů;
• informace o interakcích s inhibitory a induktory CYP3A4, kumarinovými antikoagulanty, statiny, P-gp substráty, perorálními antikoncepčními přípravky, sekvestranty žlučových kyselin;
• nutnost vyvarovat se alkoholu;
• nutnost vyvarovat se grapefruitového džusu;
• význam suplementace mastných kyselin a vitamínů rozpustných v tucích (vitamínu E);
• informace o významu dodržování diety s nízkým obsahem tuků (diety dodávající méně než 20 % energie z tuku);
• informace o užívání přípravku Lojuxta ve večerních hodinách spolu s vodou minimálně 2 hodiny po večeři a bez jídla;
• informace o existenci a významu celosvětového observačního registru pro hodnocení lomitapidu (Lomitapide Observational Worldwide Evaluation Registry) určeného k systematickému sběru informací o výsledcích z hlediska bezpečnosti a účinnosti u pacientů léčených lomitapidem.
Bezpečnostní karta pacienta
Účelem bezpečnostní karty pacienta je informovat zdravotnické profesionály před předepsáním dalšího léčiva o možných vzájemných interakcích mezi léčivy. Pacientům bude doporučeno, aby tuto kartu nosili při sobě a všem ošetřujícím lékařům ji ukázali.
Tato karta bude poskytovat informace o interakcích s: o inhibitory CYP3A4;
o induktory CYP3A4;
o kumarinovými antikoagulancii; o statiny; o substráty P-gp;
o perorálními antikoncepčními přípravky obsahujícími estrogeny.
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedené opatření:
|
Popis |
Termín splnění |
|
Na základě protokolu schváleného výborem CHMP má žadatel provést klinickou studii s adekvátními nepřímými cílovými parametry týkajícími se hodnocení cév s použitím zobrazovacích technik pro sledování cévních funkcí, stabilizace onemocnění a/nebo jeho regrese. |
Závěrečná zpráva studie by měla být předložena do 31. prosince 2021. |
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO REGISTRACI PŘÍPRAVKU ZA VÝJIMEČNÝCH OKOLNOSTÍ
Tato registrace byla schválena za výjimečných okolností, a proto podle článku 14(8) nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
Žadatel spustí dlouhodobou prospektivní observační studii za účelem systematického sběru informací o výsledcích z hlediska bezpečnosti a účinnosti u pacientů léčených lomitapidem. Studie má následující cíle: • hodnotit u pacientů léčených lomitapidem výskyt následujících situací: o jaterních příhod; o gastrointestinálních příhod; o nádorů tenkého střeva, jater, kolorekta a slinivky; o příhod souvisejících s koagulopatií; o závažných nežádoucích srdečních příhod (MACE); o úmrtí včetně příčiny úmrtí; • hodnotit výskyt a výsledky těhotenství u žen v reprodukčním věku léčených lomitapidem, které se rozhodnou po informacích od teratologa v těhotenství pokračovat; • hodnotit dlouhodobou účinnost lomitapidu v rámci udržování kontroly hladiny sérových lipidů v klinické praxi; • hodnotit, zda lékaři předepisující lomitapid dodržují screeningová a monitorovací doporučení specifikovaná v informacích o přípravku a v edukačních materiálech. |
V čase každoročního přehodnocení budou předloženy výroční zprávy. |
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU A VNITŘNÍM OBALU KRABIČKA A LAHVIČKA (5 mg, 10 mg, 20 mg, 30 mg, 40 mg a 60 mg)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Lojuxta 5 mg tvrdé tobolky Lojuxta 10 mg tvrdé tobolky Lojuxta 20 mg tvrdé tobolky Lojuxta 30 mg tvrdé tobolky Lojuxta 40 mg tvrdé tobolky Lojuxta 60 mg tvrdé tobolky lomitapidum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje lomitapidum mesilas ekvivalentní lomitapidum 5 mg. Jedna tvrdá tobolka obsahuje lomitapidum mesilas ekvivalentní lomitapidum 10 mg. Jedna tvrdá tobolka obsahuje lomitapidum mesilas ekvivalentní lomitapidum 20 mg. Jedna tvrdá tobolka obsahuje lomitapidum mesilas ekvivalentní lomitapidum 30 mg. Jedna tvrdá tobolka obsahuje lomitapidum mesilas ekvivalentní lomitapidum 40 mg. Jedna tvrdá tobolka obsahuje lomitapidum mesilas ekvivalentní lomitapidum 60 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
Podrobnější informace jsou uvedeny v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 tvrdých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 30 °C.
Uchovávejte v dobře uzavřené lahvičce, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Aegerion Pharmaceuticals Ltd Lakeside House 1 Furzeground Way Stockley Park East Uxbridge UB11 1BD Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/851/001
EU/1/13/851/002
EU/1/13/851/003
EU/1/13/851/004
EU/1/13/851/005
EU/1/13/851/006
13. ČÍSLO ŠARŽE
č.S.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
lojuxta 5 mg lojuxta 10 mg lojuxta 20 mg lojuxta 30 mg lojuxta 40 mg lojuxta 60 mg
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Lojuxta 5 mg tvrdé tobolky Lojuxta 10 mg tvrdé tobolky Lojuxta 20 mg tvrdé tobolky
lomitapidum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou.
Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Lojuxta a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Lojuxta užívat
3. Jak se přípravek Lojuxta užívá
4. Možné nežádoucí účinky
5. Jak přípravek Lojuxta uchovávat
6. Obsah balení a další informace
1. Co je přípravek Lojuxta a k čemu se používá
Přípravek Lojuxta obsahuje léčivou látku zvanou lomitapid. Lomitapid je „látka modifikující hladinu tuků“, která funguje tak, že blokuje působení „mikrozomálního triglyceridového transportního proteinu“. Tento protein se nachází v jaterních buňkách a buňkách zažívacího traktu, kde se účastní sestavování mastných látek do větších částic, které jsou poté uvolňovány do krevního řečiště. Blokádou tohoto proteinu tento léčivý přípravek snižuje hladinu tuků a cholesterolu (lipidů) v krvi.
Přípravek Lojuxta se používá k léčbě dospělých pacientů s velmi vysokým cholesterolem z důvodu onemocnění přenášeného v rodině (homozygotní formy familiární hypercholesterolémie neboli HoFH). Onemocnění je v typickém případě zděděno jak od otce, tak od matky, kteří také zdědili vysokou hladinu cholesterolu od svých rodičů. Hladiny „špatného“ cholesterolu u pacientů jsou od velmi raného věku velmi vysoké. „Špatný“ cholesterol může vést již v časném věku k srdečnímu infarktu, mozkové mrtvici nebo dalším příhodám. Přípravek Lojuxta se používá společně s dietou s nízkým obsahem tuků a další léčbou snižující koncentraci tuků v krvi ke snížení hladiny cholesterolu.
Přípravek Lojuxta může snížit krevní hladiny:
• cholesterolu z lipoproteinů o nízké hustotě (LDL) („špatného“ cholesterolu),
• celkového cholesterolu,
• apolipoproteinu B, proteinu, který přenáší „špatný cholesterol“ v krvi,
• triglyceridů (tuku přenášeného v krvi).
2. Čemu musíte věnovat pozornost, než začnete přípravek Lojuxta užívat Neužívejte přípravek Lojuxta:
• jestliže jste alergický(á) na lomitapid nebo kteroukoli další složku tobolek přípravku Lojuxta (uvedenou v bodě 6),
• jestliže máte problémy s játry nebo neobjasněné abnormální jaterní testy,
• jestliže máte střevní problémy nebo nemůžete vstřebávat stravu ze střeva,
• jestliže užíváte denně více než 40 mg simvastatinu (dalšího léčivého přípravku používaného ke snižování cholesterolu),
• jestliže užíváte některý z těchto léčivých přípravků, které ovlivňují způsob odbourávání lomitapidu v těle:
o itrakonazol, ketokonazol, flukonazol, vorikonazol, posakonazol (na plísňové infekce), o telitromycin, klaritromycin, erytromycin (na bakteriální infekce), o indinavir, nelfinavir, sakvinavir (na infekci HIV),
o diltiazem, verapamil (na vysoký krevní tlak nebo anginu pectoris) a dronedaron (na regulaci srdečního rytmu),
• jestliže jste těhotná, snažíte se otěhotnět nebo se domníváte, že byste mohla být těhotná (viz bod: „Těhotenství a kojení“).
Upozornění a opatření
Před užitím přípravku Lojuxta se poraďte se svým lékařem nebo lékárníkem, jestliže:
• jste měl/a problémy s játry, včetně problémů s játry při užívání jiných léčivých přípravků.
Tyto tobolky mohou způsobit nežádoucí účinky, které mohou být také příznaky problémů s játry. Tyto nežádoucí účinky jsou uvedeny v bodě 4, a pokud se u vás jakýkoli z těchto příznaků a známek objeví, musíte neprodleně informovat svého lékaře, protože mohou být způsobeny poškozením jater. Před zahájením léčby, při zvyšování dávky a pravidelně během léčby vám bude lékař provádět krevní testy za účelem kontroly jater. Tyto krevní testy pomáhají vašemu lékaři upravit dávku. Jestliže vaše testy ukazují na problémy s játry, může se lékař rozhodnout dávku snížit nebo léčbu ukončit.
Děti a dospívající
U dětí a dospívajících mladších 18 let nebyly provedeny žádné studie. Použití tohoto léčivého přípravku u dětí a dospívajících se proto nedoporučuje.
Další léčivé přípravky a přípravek Lojuxta
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Další léčivé přípravky mohou ovlivnit způsob fungování přípravku Lojuxta. Společně s přípravkem Lojuxta neužívejte následující léčivé přípravky:
• některé léčivé přípravky na bakteriální, plísňové infekce nebo infekci HIV (viz bod: „Neužívejte přípravek Lojuxta“)
• některé léčivé přípravky na vysoký krevní tlak, anginu pectoris nebo nepravidelný srdeční rytmus (viz bod: „Neužívejte přípravek Lojuxta“).
Musíte také informovat svého lékaře nebo lékárníka, pokud užíváte některý z následujících léčivých přípravků, protože může být nutné změnit vaši dávku přípravku Lojuxta:
• léčivé přípravky, které snižují cholesterol (např. atorvastatin),
• kombinované perorální antikoncepční přípravky (např. ethinylestradiol, norgestimát),
• glukokortikoidy (např. beklometazon, prednizolon), steroidní léčivé přípravky používané k léčbě zánětu u onemocnění, jako je závažné astma, artritida,
• léčivé přípravky určené k léčbě nádorových onemocnění (např. bikalutamid, lapatinib, metotrexát, nilotinib, pazopanib, tamoxifen) nebo nevolnosti/zvracení při léčbě nádorových onemocnění (např. fosaprepitant),
• léčivé přípravky určené ke snížení aktivity imunitního systému (např. cyklosporin, takrolimus),
• léčivé přípravky určené k léčbě bakteriálních či plísňových infekcí (např. nafcilin, azitromycin, roxitromycin, klotrimazol),
• léčivé přípravky určené k léčbě a prevenci krevních sraženin (např. cilostazol, tikagrelor),
• léčivé přípravky určené k léčbě anginy pectoris - bolesti na hrudi srdečního původu (např. ranolazin),
• léčivé přípravky na snížení krevního tlaku (např. amlodipin, lacidipin),
• léčivé přípravky na regulaci srdečního rytmu (např. amiodaron),
• léčivé přípravky určené k léčbě epilepsie (např. fenobarbital, karbamazepin, fenytoin),
• léčivé přípravky určené k léčbě diabetu (např. pioglitazon, linagliptin),
• léčivé přípravky určené k léčbě tuberkulózy (např. izoniazid, rifampicin),
• tetracyklinová antibiotika určené k léčbě infekcí, jako jsou infekce močového traktu,
• léčivé přípravky určené k léčbě úzkostných poruch a deprese (např. alprazolam, fluoxetin, fluvoxamin),
• antacida (např. ranitidin, cimetidin),
• aminoglutetimid - léčivý přípravek používaný k léčbě Cushingova syndromu,
• léčivé přípravky určené k léčbě závažného akné (např. izotretinoin),
• paracetamol - určený k léčbě bolesti,
• léčivé přípravky určené k léčbě cystické fibrózy (např. ivakaflor),
• léčivé přípravky určené k léčbě močové inkontinence (např. propiverin),
• léčivé přípravky používané ke snížení hladin sodíku v krvi (např. tolvaptan),
• léčivé přípravky určené k léčbě nadměrné denní spavosti (např. modafinil),
• některé rostlinné přípravky:
o třezalka tečkovaná (na depresi), o ginkgo (na zlepšení paměti), o vodilka kanadská (na záněty a infekce).
Přípravek Lojuxta může ovlivnit způsob fungování dalších léčivých přípravků. Informujte svého lékaře nebo lékárníka, pokud užíváte některý z následujících léčivých přípravků:
• perorální antikoncepční přípravky (viz bod 2: „Těhotenství a kojení“),
• další léčivé přípravky používané na snížení cholesterolu, j ako j sou:
o statiny, např. simvastatin. Riziko poškození jater se zvyšuje, když je tento léčivý přípravek užíván zároveň se statiny. Může dojít k nárůstu bolesti a pobolívání svalů (myalgie) nebo se může vyskytnout svalová slabost (myopatie). Neprodleně kontaktujte svého lékaře, pokud se u vás objeví nevysvětlené pobolívání a bolest svalů, citlivost nebo svalová slabost. Při užívání přípravku Lojuxta byste neměli užívat více než 40 mg simvastatinu (viz bod „Neužívejte přípravek Lojuxta“),
• kumarinová antikoagulancia na ředění krve (např. warfarin),
• léčivé přípravky určené k léčbě nádorových onemocnění (např. everolimus, imatinib, lapatinib, nilotinib, topotekan),
• léčivé přípravky na snížení aktivity imunitního systému (např. sirolimus),
• léčivé přípravky určené k léčbě HIV (napr. maravirok),
• léčivé přípravky určené k léčbě a prevenci krevních sraženin (např. dabigatran etexilát),
• léčivé přípravky určené k léčbě anginy pectoris - bolesti na hrudi srdečního původu (např. ranolazin),
• léčivé přípravky na snížení krevního tlaku (např. talinolol, aliskiren, ambrisentan),
• léčivé přípravky na regulaci srdečního rytmu (např. digoxin),
• léčivé přípravky určené k léčbě diabetu (např. saxagliptin, sitagliptin),
• léčivé přípravky určené k léčbě dny (např. kolchicin),
• léčivé přípravky na snížení hladin sodíku v krvi (např. tolvaptan),
• antihistaminika, přípravky určené k léčbě senné rýmy (např. fexofenadin).
Přípravek Lojuxta s jídlem, pitím a alkoholem
• Nepijte grapefruitový džus.
• Požívání alkoholu během léčby přípravkem Lojuxta se nedoporučuje.
• Pokud užíváte mátový olej nebo hořké pomeranče, může být u vás nutná úprava dávky přípravku Lojuxta.
• Pro snížení pravděpodobnosti břišních potíží musíte během užívání tohoto léčivého přípravku dodržovat dietu s nízkým obsahem tuků. Abyste zjistili, co můžete během užívání přípravku Lojuxta jíst, spolupracujte s nutričním poradcem.
Těhotenství a kojení
Neužívejte tento léčivý přípravek, jestliže jste těhotná, snažíte se otěhotnět nebo se domníváte, že byste mohla být těhotná, protože existuje možnost, že by tento přípravek mohl poškodit nenarozené dítě. Jestliže při používání tohoto léčivého přípravku otěhotníte, okamžitě zavolejte svému lékaři a přestaňte tobolky užívat.
• Před zahájením léčby byste se měla ujistit, že nejste těhotná a že užíváte účinnou antikoncepci dle doporučení svého lékaře. Jestliže užíváte antikoncepční pilulky a prodělala jste epizodu průjmu či zvracení, která trvala více než 2 dny, musíte užívat alternativní metodu antikoncepce (např. kondomy, pesar) po dobu 7 dní po ústupu příznaků.
• Jestliže se po zahájení léčby přípravkem Lojuxta rozhodnete otěhotnět, informujte prosím svého lékaře, protože bude nutné změnit vaši léčbu.
Kojení
• Není známo, zda přípravek Lojuxta přechází do mateřského mléka. Prosím informujte svého lékaře, pokud kojíte nebo plánujete kojit. Váš lékař vám může doporučit, abyste přestala užívat přípravek Lojuxta nebo abyste přestala kojit.
Řízení dopravních prostředků a obsluha strojů
Vaše léčba může ovlivnit vaši schopnost řídit dopravní prostředky nebo obsluhovat stroje. Pokud během léčby pocítíte závrať, neřiďte dopravní prostředky ani neobsluhujte stroje, dokud se nebudete cítit lépe.
Přípravek Lojuxta obsahuje laktózu.
Pokud vám lékař sdělil, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
3. Jak se přípravek Lojuxta užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem. Tyto tobolky by vám měl podávat lékař se zkušenostmi s léčbou poruch lipidů, který vás také bude pravidelně sledovat.
Doporučená počáteční dávka je tobolka 5 mg jednou denně. Váš lékař může postupem času pomalu dávku zvyšovat až na maximální dávku 60 mg jednou denně. Váš lékař vás bude informovat:
• jakou dávku užívat a po jak dlouhou dobu,
• kdy zvýšit či snížit vaši dávku.
Dávku si samovolně neměňte. 1
Lékař může změnit čas užívání léků, aby nedošlo k vzájemnému ovlivňování jejich účinků, případně může snížit užívanou dávku přípravku Lojuxta. Informujte lékaře o jakýchkoli změnách v užívaných lécích.
Při užívání tohoto léčivého přípravku budete také muset denně užívat doplňky s vitamínem E a esenciálními mastnými kyselinami (omega-3 a omega-6). Obvyklá dávka, kterou budete muset užívat, je uvedena dále. Zeptejte se svého lékaře nebo nutričního poradce, kde tyto doplňky získat. Viz bod 2: Přípravek Lojuxta s jídlem, pitím a alkoholem.
|
Denní množství | |
|
Vitamín E |
400 IU1 |
|
Omega-3 |
Přibližně |
|
EPA |
110 mg1 |
|
DHA |
80 mg |
|
ALA |
210 mg |
|
Omega-6 | |
|
Kyselina linolová |
200 mg |
* IU - mezinárodní jednotky, mg - miligramy
Jestliže jste užil(a) více přípravku Lojuxta, než jste měl(a)
Okamžitě kontaktujte svého lékaře nebo lékárníka.
Jestliže jste zapomněl(a) užít přípravek Lojuxta
Vezměte si normální dávku přípravku v obvyklém čase další den. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Lojuxta
Jestliže přestanete užívat tento léčivý přípravek, váš cholesterol se může opět zvýšit. Než přestanete tento léčivý přípravek užívat, měli byste kontaktovat svého lékaře.
Máte-li jakékoli další otázky týkající se užívání tohoto léčivého přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Závažné nežádoucí účinky:
• Často byly udávány abnormální krevní testy jaterních funkcí (mohou se vyskytnout až u 1 z 10 osob). Tyto příznaky problémů s játry zahrnují:
o nevolnost (pocit na zvracení), o zvracení, o bolest břicha, o bolest a pobolívání svalů, o horečku,
o změnu zbarvení kůže nebo očního bělma do žluta, o větší únavu než obvykle, o pocit, jako by měl člověk chřipku.
Informujte svého lékaře okamžitě, pokud máte kterékoli z těchto příznaků, protože lékař může rozhodnout o ukončení léčby.
Vyskytly se také následující nežádoucí účinky:
Velmi časté (mohou postihnout více než 1 z 10 osob):
• průjem,
• nevolnost a zvracení (pocit na zvracení nebo zvracení),
• bolest břicha, nevolnost nebo nadýmání,
• snížená chuť k jídlu,
• poruchy zažívání,
• plynatost (větry),
• zácpa,
• úbytek hmotnosti.
Časté (mohou postihnout až 1 z 10 osob):
• zánět žaludku a střev, který způsobuje průjem a zvracení,
• regurgitace (návrat nestrávené potravy zpět do úst),
• říhání,
• pocit nedostatečného vyprázdnění (stolice), urgentní potřeba vyprázdnění stolice,
• krvácení z rekta (konečníku) nebo krev ve stolici,
• závrať, bolest hlavy, migréna,
• únava, nedostatek energie nebo celková slabost,
• zvětšení, poškození nebo ztukovatění jater,
• změna zbarvení kůže do fialova, tuhé boule na kůži, vyrážka, žluté boule na kůži,
• změny testů krevní srážlivosti,
• změny krevního obrazu,
• snížená hladina draslíku, karotenu, vitamínu E, vitamínu K v krvi,
• svalové spasmy.
Méně časté (mohou postihnout až 1 ze 100 osob):
• chřipka či nachlazení, horečka, zánět vedlejších dutin nosních, kašel,
• nízký počet červených krvinek (anémie),
• dehydratace, sucho v ústech,
• zvýšená chuť k jídlu,
• pálení nebo svědění kůže,
• otok oka,
• vřed nebo bolestivé místo v hrdle,
• zvracení krve,
• suchá kůže,
• puchýře,
• nadměrné pocení,
• bolest nebo otok kloubů, bolest rukou nebo nohou,
• bolest svalů,
• krev nebo bílkovina v moči,
• bolest na hrudi,
• změny chůze,
• abnormální test plicních funkcí.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Jak přípravek Lojuxta uchovávat
5.
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti (EXP) uvedené na štítku nebo na krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 30 °C.
Uchovávejte v dobře uzavřené lahvičce, aby byl přípravek chráněn před vlhkostí.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Lojuxta obsahuje
• Léčivou látkou je lomitapid.
Lojuxta 5 mg: Jedna tvrdá tobolka obsahuje lomitapid mesylát ekvivalentní 5 mg lomitapidu. Lojuxta 10 mg: Jedna tvrdá tobolka obsahuje lomitapid mesylát ekvivalentní 10 mg lomitapidu. Lojuxta 20 mg: Jedna tvrdá tobolka obsahuje lomitapid mesylát ekvivalentní 20 mg lomitapidu.
• Pomocnými látkami j sou: předbobtnalý škrob, sodná sůl karboxymethylškrobu, mikrokrystalická celulóza, monohydrát laktózy, koloidní bezvodý oxid křemičitý a magnesium-stearát (informace o monohydrátu laktózy viz bod 2: Přípravek Lojuxta obsahuje laktózu).
Obal tobolky:
• Obal tobolek 5 mg a 10 mg obsahuje želatinu, oxid titaničitý (E171) a červený oxid železitý
(E172).
• Obal tobolek 20 mg obsahuje želatinu a oxid titaničitý (E171).
• Všechny tobolky jsou potištěny jedlým černým inkoustem.
Jak přípravek Lojuxta vypadá a co obsahuje toto balení
• Přípravek Lojuxta 5 mg jsou tvrdé tobolky s oranžovou svrchní i spodní částí potištěné černým
inkoustem s textem „5 mg“ na spodní části a „A733“ na svrchní části tobolky.
• Přípravek Lojuxta 10 mg jsou tvrdé tobolky s oranžovou svrchní a bílou spodní částí potištěné
černým inkoustem s textem „10 mg“ na spodní části a „A733“ na svrchní části tobolky.
• Přípravek Lojuxta 20 mg jsou tvrdé tobolky s bílou svrchní i spodní částí potištěné černým
inkoustem s textem „20 mg“ na spodní části a „A733“ na svrchní části tobolky.
Velikosti balení:
- 28 tobolek přípravku Lojuxta 5 mg,
- 28 tobolek přípravku Lojuxta 10 mg,
- 28 tobolek přípravku Lojuxta 20 mg.
Držitel rozhodnutí o registraci
Aegerion Pharmaceuticals Ltd Lakeside House 1 Furzeground Way Stockley Park East Uxbridge UB11 1BD Velká Británie
Výrobce
Catalent UK Packaging Limited
Lancaster Way, Wingates Industrial Estate, Westhoughton, Bolton, Lancashire, BL5 3XX Velká Británie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Aegerion Pharmaceuticals Ltd Tél/Tel: 80023437466 |
Lietuva AOP Orphan Pharmaceuticals AG Tel: 80023437466 |
|
Btarapna Aegerion Pharmaceuticals Ltd Ten.: 80023437466 |
Luxembourg/Luxemburg Aegerion Pharmaceuticals Ltd Tél/Tel: 80023437466 |
|
Česká republika AOP Orphan Pharmaceuticals AG Tel: 80023437466 |
Magyarország AOP Orphan Pharmaceuticals AG Tel.: 80023437466 |
|
Danmark Aegerion Pharmaceuticals Ltd Tlf: 80023437466 |
Malta Aegerion Pharmaceuticals Ltd Tel: 80023437466 |
|
Deutschland Aegerion Pharmaceuticals GmbH Tel: 80023437466 |
Nederland Aegerion Pharmaceuticals GmbH Tel: 80023437466 |
|
Eesti Aegerion Pharmaceuticals Ltd Tel: 80023437466 |
Norge Aegerion Pharmaceuticals A/S Tlf: + 4721034389 |
|
EXláSa Aegerion Pharmaceuticals Ltd T^:+ 302111983992 |
Osterreich AOP Orphan Pharmaceuticals AG Tel: 80023437466 |
|
Espaňa Praxis Pharmaceutical S.A. Tel: +34 916565657 |
Polska Aegerion Pharmaceuticals Ltd Tel.: 80023437466 |
|
France Aegerion Pharmaceuticals SAS Tél: 80023437466 |
Portugal PRX Pharma, Lda Tel: +351 214414777 |
|
Hrvatska Aegerion Pharmaceuticals Ltd Tel: + 38517776525 |
Románia Aegerion Pharmaceuticals Ltd Tel: + 40316300768 |
|
Ireland Aegerion Pharmaceuticals Ltd Tel: 80023437466 |
Slovenija Aegerion Pharmaceuticals Ltd Tel: 80023437466 |
|
Ísland Aegerion Pharmaceuticals Ltd Sími: 80023437466 |
Slovenská republika AOP Orphan Pharmaceuticals AG Tel: 80023437466 |
Italia
Aegerion Pharmaceuticals Srl Tel: 80023437466
Kúnpoq
Aegerion Pharmaceuticals Ltd T^: 80023437466
Latvija
AOP Orphan Pharmaceuticals AG Tel: 80023437466
Suomi/Finland
Aegerion Pharmaceuticals Ltd Puh/Tel: 80023437466
Sverige
Aegerion Pharmaceuticals Ltd Tel: 80023437466
United Kingdom
Aegerion Pharmaceuticals Ltd Tel: 80023437466
Tato příbalová informace byla naposledy revidována <{MM.RRRR}> <{měsíc RRRR}>.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
Příbalová informace: informace pro uživatele
Lojuxta 30 mg tvrdé tobolky Lojuxta 40 mg tvrdé tobolky Lojuxta 60 mg tvrdé tobolky
lomitapidum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou.
Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Lojuxta a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Lojuxta užívat
3. Jak se přípravek Lojuxta užívá
4. Možné nežádoucí účinky
5. Jak přípravek Lojuxta uchovávat
6. Obsah balení a další informace
1. Co je přípravek Lojuxta a k čemu se používá
Přípravek Lojuxta obsahuje léčivou látku zvanou lomitapid. Lomitapid je „látka modifikující hladinu tuků“, která funguje tak, že blokuje působení „mikrozomálního triglyceridového transportního proteinu“. Tento protein se nachází v jaterních buňkách a buňkách zažívacího traktu, kde se účastní sestavování mastných látek do větších částic, které jsou poté uvolňovány do krevního řečiště. Blokádou tohoto proteinu tento léčivý přípravek snižuje hladinu tuků a cholesterolu (lipidů) v krvi.
Přípravek Lojuxta se používá k léčbě dospělých pacientů s velmi vysokým cholesterolem z důvodu onemocnění přenášeného v rodině (homozygotní formy familiární hypercholesterolémie neboli HoFH). Onemocnění je v typickém případě zděděno jak od otce, tak od matky, kteří také zdědili vysokou hladinu cholesterolu od svých rodičů. Hladiny „špatného“ cholesterolu u pacientů jsou od velmi raného věku velmi vysoké. „Špatný“ cholesterol může vést již v časném věku k srdečnímu infarktu, mozkové mrtvici nebo dalším příhodám. Přípravek Lojuxta se používá společně s dietou s nízkým obsahem tuků a další léčbou snižující koncentraci tuků v krvi ke snížení hladiny cholesterolu.
Přípravek Lojuxta může snížit krevní hladiny:
• cholesterolu z lipoproteinů o nízké hustotě (LDL) („špatného“ cholesterolu),
• celkového cholesterolu,
• apolipoproteinu B, proteinu, který přenáší „špatný cholesterol“ v krvi,
• triglyceridů (tuku přenášeného v krvi)
2. Čemu musíte věnovat pozornost, než začnete přípravek Lojuxta užívat Neužívejte přípravek Lojuxta:
• jestliže jste alergický(á) na lomitapid nebo kteroukoli další složku tobolek přípravku Lojuxta (uvedenou v bodě 6),
• jestliže máte problémy s játry nebo neobjasněné abnormální jaterní testy,
• jestliže máte střevní problémy nebo nemůžete vstřebávat stravu ze střeva,
• jestliže užíváte denně více než 40 mg simvastatinu (dalšího léčivého přípravku používaného ke snižování cholesterolu),
• jestliže užíváte některý z těchto léčivých přípravků, které ovlivňují způsob odbourávání lomitapidu v těle:
o itrakonazol, ketokonazol, flukonazol, vorikonazol, posakonazol (na plísňové infekce), o telitromycin, klaritromycin, erytromycin (na bakteriální infekce), o indinavir, nelfinavir, sakvinavir (na infekci HIV),
o diltiazem, verapamil (na vysoký krevní tlak nebo anginu pectoris) a dronedaron (na regulaci srdečního rytmu),
• jestliže jste těhotná, snažíte se otěhotnět nebo se domníváte, že byste mohla být těhotná (viz bod: „Těhotenství a kojení“).
Upozornění a opatření
Před užitím přípravku Lojuxta se poraďte se svým lékařem nebo lékárníkem, jestliže:
• j ste měl/a problémy s j átry, včetně problémů s j átry při užívání j iných léčivých přípravků.
Tyto tobolky mohou způsobit nežádoucí účinky, které mohou být také příznaky problémů s játry. Tyto nežádoucí účinky jsou uvedeny v bodě 4, a pokud se u vás jakýkoli z těchto příznaků a známek objeví, musíte neprodleně informovat svého lékaře, protože mohou být způsobeny poškozením jater. Před zahájením léčby, při zvyšování dávky a pravidelně během léčby vám bude lékař provádět krevní testy za účelem kontroly jater. Tyto krevní testy pomáhají vašemu lékaři upravit dávku. Jestliže vaše testy ukazují na problémy s játry, může se lékař rozhodnout dávku snížit nebo léčbu ukončit.
Děti a dospívající
U dětí a dospívajících mladších 18 let nebyly provedeny žádné studie. Použití tohoto léčivého přípravku u dětí a dospívajících se proto nedoporučuje.
Další léčivé přípravky a přípravek Lojuxta
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Další léčivé přípravky mohou ovlivnit způsob fungování přípravku Lojuxta. Společně s přípravkem Lojuxta neužívejte následující léčivé přípravky:
• některé léčivé přípravky na bakteriální, plísňové infekce nebo infekci HIV (viz bod: „Neužívejte přípravek Lojuxta“)
• některé léčivé přípravky na vysoký krevní tlak, anginu pectoris nebo nepravidelný srdeční rytmus (viz bod: „Neužívejte přípravek Lojuxta“)
Musíte také informovat svého lékaře nebo lékárníka, pokud užíváte některý z následujících léčivých přípravků, protože může být nutné změnit vaši dávku přípravku Lojuxta:
• léčivé přípravky, které snižují cholesterol (např. atorvastatin),
• kombinované perorální antikoncepční přípravky (např. ethinylestradiol, norgestimát),
• glukokortikoidy (např. beklometazon, prednizolon), steroidní léčivé přípravky používané k léčbě zánětu u onemocnění, jako je závažné astma, artritida,
• léčivé přípravky určené k léčbě nádorových onemocnění (např. bikalutamid, lapatinib, metotrexát, nilotinib, pazopanib, tamoxifen) nebo nevolnosti/zvracení při léčbě nádorových onemocnění (např. fosaprepitant),
• léčivé přípravky určené ke snížení aktivity imunitního systému (např. cyklosporin, takrolimus),
• léčivé přípravky určené k léčbě bakteriálních či plísňových infekcí (např. nafcilin, azitromycin, roxitromycin, klotrimazol),
• léčivé přípravky určené k léčbě a prevenci krevních sraženin (např. cilostazol, tikagrelor),
• léčivé přípravky určené k léčbě anginy pectoris - bolesti na hrudi srdečního původu (např. ranolazin),
• léčivé přípravky na snížení krevního tlaku (např. amlodipin, lacidipin),
• léčivé přípravky na regulaci srdečního rytmu (např. amiodaron),
• léčivé přípravky určené k léčbě epilepsie (např. fenobarbital, karbamazepin, fenytoin),
• léčivé přípravky určené k léčbě diabetu (např. pioglitazon, linagliptin),
• léčivé přípravky určené k léčbě tuberkulózy (např. izoniazid, rifampicin),
• tetracyklinová antibiotika určené k léčbě infekcí, jako jsou infekce močového traktu,
• léčivé přípravky určené k léčbě úzkostných poruch a deprese (např. alprazolam, fluoxetin, fluvoxamin),
• antacida (např. ranitidin, cimetidin),
• aminoglutetimid - léčivý přípravek používaný k léčbě Cushingova syndromu,
• léčivé přípravky určené k léčbě závažného akné (např. izotretinoin),
• paracetamol - určený k léčbě bolesti,
• léčivé přípravky určené k léčbě cystické fibrózy (např. ivakaflor),
• léčivé přípravky určené k léčbě močové inkontinence (např. propiverin),
• léčivé přípravky používané ke snížení hladin sodíku v krvi (např. tolvaptan),
• léčivé přípravky určené k léčbě nadměrné denní spavosti (např. modafinil),
• některé rostlinné přípravky:
o třezalka tečkovaná (na depresi), o ginkgo (na zlepšení paměti), o vodilka kanadská (na záněty a infekce)
Přípravek Lojuxta může ovlivnit způsob fungování dalších léčivých přípravků. Informujte svého lékaře nebo lékárníka, pokud užíváte některý z následujících léčivých přípravků:
• perorální antikoncepční přípravky (viz bod 2: „Těhotenství a kojení“),
• další léčivé přípravky používané na snížení cholesterolu, j ako j sou:
o statiny, např. simvastatin. Riziko poškození jater se zvyšuje, když je tento léčivý přípravek užíván zároveň se statiny. Může dojít k nárůstu bolesti a pobolívání svalů (myalgie) nebo se může vyskytnout svalová slabost (myopatie). Neprodleně kontaktujte svého lékaře, pokud se u vás objeví nevysvětlené pobolívání a bolest svalů, citlivost nebo svalová slabost. Při užívání přípravku Lojuxta byste neměli užívat více než 40 mg simvastatinu (viz bod „Neužívejte přípravek Lojuxta“),
• kumarinová antikoagulancia na ředění krve (např. warfarin),
• léčivé přípravky určené k léčbě nádorových onemocnění (např. everolimus, imatinib, lapatinib, nilotinib, topotekan),
• léčivé přípravky na snížení aktivity imunitního systému (např. sirolimus),
• léčivé přípravky určené k léčbě HIV (napr. maravirok),
• léčivé přípravky určené k léčbě a prevenci krevních sraženin (např. dabigatran etexilát),
• léčivé přípravky určené k léčbě anginy pectoris - bolesti na hrudi srdečního původu (např. ranolazin),
• léčivé přípravky na snížení krevního tlaku (např. talinolol, aliskiren, ambrisentan),
• léčivé přípravky na regulaci srdečního rytmu (např. digoxin),
• léčivé přípravky určené k léčbě diabetu (např. saxagliptin, sitagliptin),
• léčivé přípravky určené k léčbě dny (např. kolchicin),
• léčivé přípravky na snížení hladin sodíku v krvi (např. tolvaptan),
• antihistaminika, přípravky určené k léčbě senné rýmy (např. fexofenadin).
Přípravek Lojuxta s jídlem, pitím a alkoholem
• Nepijte grapefruitový džus.
• Požívání alkoholu během léčby přípravkem Lojuxta se nedoporučuje.
• Pokud užíváte mátový olej nebo hořké pomeranče, může být u vás nutná úprava dávky přípravku Lojuxta.
• Pro snížení pravděpodobnosti břišních potíží musíte během užívání tohoto léčivého přípravku dodržovat dietu s nízkým obsahem tuků. Abyste zjistili, co můžete během užívání přípravku Lojuxta jíst, spolupracujte s nutričním poradcem.
Těhotenství a kojení
Neužívejte tento léčivý přípravek, jestliže jste těhotná, snažíte se otěhotnět nebo se domníváte, že byste mohla být těhotná, protože existuje možnost, že by tento přípravek mohl poškodit nenarozené dítě. Jestliže při používání tohoto léčivého přípravku otěhotníte, okamžitě zavolejte svému lékaři a přestaňte tobolky užívat.
• Před zahájením léčby byste se měla ujistit, že nejste těhotná a že užíváte účinnou antikoncepci dle doporučení svého lékaře. Jestliže užíváte antikoncepční pilulky a prodělala jste epizodu průjmu či zvracení, která trvala více než 2 dny, musíte užívat alternativní metodu antikoncepce (např. kondomy, pesar) po dobu 7 dní po ústupu příznaků.
• Jestliže se po zahájení léčby přípravkem Lojuxta rozhodnete otěhotnět, informujte prosím svého lékaře, protože bude nutné změnit vaši léčbu.
Kojení
• Není známo, zda přípravek Lojuxta přechází do mateřského mléka. Prosím informujte svého lékaře, pokud kojíte nebo plánujete kojit. Váš lékař vám může doporučit, abyste přestala užívat přípravek Lojuxta nebo abyste přestala kojit.
Řízení dopravních prostředků a obsluha strojů
Vaše léčba může ovlivnit vaši schopnost řídit dopravní prostředky nebo obsluhovat stroje. Pokud během léčby pocítíte závrať, neřiďte dopravní prostředky ani neobsluhujte stroje, dokud se nebudete cítit lépe.
Přípravek Lojuxta obsahuje laktózu.
Pokud vám lékař sdělil, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
3. Jak se přípravek Lojuxta užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem. Tyto tobolky by vám měl podávat lékař se zkušenostmi s léčbou poruch lipidů, který vás také bude pravidelně sledovat.
Doporučená počáteční dávka je tobolka 5 mg jednou denně. Váš lékař může postupem času pomalu dávku zvyšovat až na maximální dávku 60 mg jednou denně. Váš lékař vás bude informovat:
• jakou dávku užívat a po jak dlouhou dobu,
• kdy zvýšit či snížit vaši dávku.
Dávku si samovolně neměňte. 2
Lékař může změnit čas užívání léků, aby nedošlo k vzájemnému ovlivňování jejich účinků, případně může snížit užívanou dávku přípravku Lojuxta. Informujte lékaře o jakýchkoli změnách v užívaných lécích.
Při užívání tohoto léčivého přípravku budete také muset denně užívat doplňky s vitamínem E a esenciálními mastnými kyselinami (omega-3 a omega-6). Obvyklá dávka, kterou budete muset užívat, je uvedena dále. Zeptejte se svého lékaře nebo nutričního poradce, kde tyto doplňky získat. Viz bod 2: Přípravek Lojuxta s jídlem, pitím a alkoholem.
|
Denní množství | |
|
Vitamín E |
400 IU2 |
|
Omega-3 |
Přibližně |
|
EPA |
110 mg2 |
|
DHA |
80 mg |
|
ALA |
210 mg |
|
Omega-6 | |
|
Kyselina linolová |
200 mg |
* IU - mezinárodní jednotky, mg - miligramy
Jestliže jste užil(a) více přípravku Lojuxta, než jste měl(a)
Okamžitě kontaktujte svého lékaře nebo lékárníka.
Jestliže jste zapomněl(a) užít přípravek Lojuxta
Vezměte si normální dávku přípravku v obvyklém čase další den. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Lojuxta
Jestliže přestanete užívat tento léčivý přípravek, váš cholesterol se může opět zvýšit. Než přestanete tento léčivý přípravek užívat, měli byste kontaktovat svého lékaře.
Máte-li jakékoli další otázky týkající se užívání tohoto léčivého přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Závažné nežádoucí účinky:
• Často byly udávány abnormální krevní testy jaterních funkcí (mohou se vyskytnout až u 1 z 10 osob). Tyto příznaky problémů s játry zahrnují:
o nevolnost (pocit na zvracení), o zvracení, o bolest břicha, o bolest a pobolívání svalů, o horečku,
o změnu zbarvení kůže nebo očního bělma do žluta, o větší únavu než obvykle, o pocit, jako by měl člověk chřipku.
Informujte svého lékaře okamžitě, pokud máte kterékoli z těchto příznaků, protože lékař může rozhodnout o ukončení léčby.
Vyskytly se také následující nežádoucí účinky:
Velmi časté (mohou postihnout více než 1 z 10 osob):
• průjem,
• nevolnost a zvracení (pocit na zvracení nebo zvracení),
• bolest břicha, nevolnost nebo nadýmání,
• snížená chuť k jídlu,
• poruchy zažívání,
• plynatost (větry),
• zácpa,
• úbytek hmotnosti.
Časté (mohou postihnout až 1 z 10 osob):
• zánět žaludku a střev, který způsobuje průjem a zvracení,
• regurgitace (návrat nestrávené potravy zpět do úst),
• říhání,
• pocit nedostatečného vyprázdnění (stolice), urgentní potřeba vyprázdnění stolice,
• krvácení z rekta (konečníku) nebo krev ve stolici,
• závrať, bolest hlavy, migréna,
• únava, nedostatek energie nebo celková slabost,
• zvětšení, poškození nebo ztukovatění jater,
• změna zbarvení kůže do fialova, tuhé boule na kůži, vyrážka, žluté boule na kůži,
• změny testů krevní srážlivosti,
• změny krevního obrazu,
• snížená hladina draslíku, karotenu, vitamínu E, vitamínu K v krvi,
• svalové spasmy.
Méně časté (mohou postihnout až 1 ze 100 osob):
• chřipka či nachlazení, horečka, zánět vedlejších dutin nosních, kašel,
• nízký počet červených krvinek (anémie),
• dehydratace, sucho v ústech,
• zvýšená chuť k jídlu,
• pálení nebo svědění kůže,
• otok oka,
• vřed nebo bolestivé místo v hrdle,
• zvracení krve,
• suchá kůže,
• puchýře,
• nadměrné pocení,
• bolest nebo otok kloubů, bolest rukou nebo nohou,
• bolest svalů,
• krev nebo bílkovina v moči,
• bolest na hrudi,
• změny chůze,
• abnormální test plicních funkcí.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Jak přípravek Lojuxta uchovávat
5.
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti (EXP) uvedené na štítku nebo na krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 30 °C.
Uchovávejte v dobře uzavřené lahvičce, aby byl přípravek chráněn před vlhkostí.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Lojuxta obsahuje
• Léčivou látkou je lomitapid.
Lojuxta 30 mg: Jedna tvrdá tobolka obsahuje lomitapid mesylát ekvivalentní 30 mg lomitapidu. Lojuxta 40 mg: Jedna tvrdá tobolka obsahuje lomitapid mesylát ekvivalentní 40 mg lomitapidu. Lojuxta 60 mg: Jedna tvrdá tobolka obsahuje lomitapid mesylát ekvivalentní 60 mg lomitapidu.
• Pomocnými látkami j sou: předbobtnalý škrob, sodná sůl karboxymethylškrobu, mikrokrystalická celulóza, monohydrát laktózy, koloidní bezvodý oxid křemičitý a magnesium-stearát (informace o monohydrátu laktózy viz bod 2: Přípravek Lojuxta obsahuje laktózu).
Obal tobolky:
a červený oxid železitý (E172) a
a žlutý oxid železitý (E172). a žlutý oxid železitý (E172).
• Obal tobolek 30 mg obsahuje želatinu, oxid titaničitý (E171)
žlutý oxid železitý (E172).
• Obal tobolek 40 mg obsahuje želatinu, oxid titaničitý (E171)
• Obal tobolek 60 mg obsahuje želatinu, oxid titaničitý (E171)
• Všechny tobolky j sou potištěny jedlým černým inkoustem.
Jak přípravek Lojuxta vypadá a co obsahuje toto balení
• Přípravek Lojuxta 30 mg jsou tvrdé tobolky s oranžovou svrchní a žlutou spodní částí potištěné
černým inkoustem s textem „30 mg“ na spodní části a „A733“ na svrchní části tobolky.
• Přípravek Lojuxta 40 mg jsou tvrdé tobolky se žlutou svrchní a bílou spodní částí potištěné
černým inkoustem s textem „40 mg“ na spodní části a „A733“ na svrchní části tobolky.
• Přípravek Lojuxta 60 mg jsou tvrdé tobolky se žlutou svrchní i spodní částí potištěné černým
inkoustem s textem „60 mg“ na spodní části a „A733“ na svrchní části tobolky.
Velikosti balení:
- 28 tobolek přípravku Lojuxta 30 mg,
- 28 tobolek přípravku Lojuxta 40 mg,
- 28 tobolek přípravku Lojuxta 60 mg.
Držitel rozhodnutí o registraci
Aegerion Pharmaceuticals Ltd Lakeside House 1 Furzeground Way Stockley Park East Uxbridge UB11 1BD Velká Británie
Výrobce
Catalent UK Packaging Limited
Lancaster Way, Wingates Industrial Estate, Westhoughton, Bolton, Lancashire, BL5 3XX Velká Británie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Aegerion Pharmaceuticals Ltd Tél/Tel: 80023437466 |
Lietuva AOP Orphan Pharmaceuticals AG Tel: 80023437466 |
|
Btarapna Aegerion Pharmaceuticals Ltd Ten.: 80023437466 |
Luxembourg/Luxemburg Aegerion Pharmaceuticals Ltd Tél/Tel: 80023437466 |
|
Česká republika AOP Orphan Pharmaceuticals AG Tel: 80023437466 |
Magyarország AOP Orphan Pharmaceuticals AG Tel.: 80023437466 |
|
Danmark Aegerion Pharmaceuticals Ltd Tlf: 80023437466 |
Malta Aegerion Pharmaceuticals Ltd Tel: 80023437466 |
|
Deutschland Aegerion Pharmaceuticals GmbH Tel: 80023437466 |
Nederland Aegerion Pharmaceuticals GmbH Tel: 80023437466 |
|
Eesti Aegerion Pharmaceuticals Ltd Tel: 80023437466 |
Norge Aegerion Pharmaceuticals A/S Tlf: + 4721034389 |
|
EXláSa Aegerion Pharmaceuticals Ltd T^:+ 302111983992 |
Osterreich AOP Orphan Pharmaceuticals AG Tel: 80023437466 |
|
Espaňa Praxis Pharmaceutical S.A. Tel: +34 916565657 |
Polska Aegerion Pharmaceuticals Ltd Tel.: 80023437466 |
|
France Aegerion Pharmaceuticals SAS Tél: 80023437466 |
Portugal PRX Pharma, Lda Tel: +351 214414777 |
|
Hrvatska Aegerion Pharmaceuticals Ltd Tel: + 38517776525 |
Románia Aegerion Pharmaceuticals Ltd Tel: + 40316300768 |
|
Ireland Aegerion Pharmaceuticals Ltd Tel: 80023437466 |
Slovenija Aegerion Pharmaceuticals Ltd Tel: 80023437466 |
|
Ísland Aegerion Pharmaceuticals Ltd Sími: 80023437466 |
Slovenská republika AOP Orphan Pharmaceuticals AG Tel: 80023437466 |
Italia
Aegerion Pharmaceuticals Srl Tel: 80023437466
Kúnpoq
Aegerion Pharmaceuticals Ltd T^: 80023437466
Latvija
AOP Orphan Pharmaceuticals AG Tel: 80023437466
Suomi/Finland
Aegerion Pharmaceuticals Ltd Puh/Tel: 80023437466
Sverige
Aegerion Pharmaceuticals Ltd Tel: 80023437466
United Kingdom
Aegerion Pharmaceuticals Ltd Tel: 80023437466
Tato příbalová informace byla naposledy revidována <{MM.RRRR}> <{měsíc RRRR}>.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
150
Tento léčivý přípravek užívejte jednou denně před spaním spolu s vodou minimálně 2 hodiny po večeři (viz bod 2: „Přípravek Lojuxta s jídlem, pitím a alkoholem“).
• Tento léčivý přípravek neužívejte s jídlem, protože užívání těchto tobolek s jídlem může způsobit žaludeční problémy.
• Jestliže užíváte další léčivý přípravek snižující cholesterol vazbou na žlučové kyseliny, jako je kolesevelam nebo cholestyramin, užívejte léčivý přípravek vázající žlučové kyseliny minimálně 4 hodiny před užíváním přípravku Lojuxta nebo 4 hodiny po něm.
Tento léčivý přípravek užívejte jednou denně před spaním spolu s vodou minimálně 2 hodiny po večeři (viz bod 2: „Přípravek Lojuxta s jídlem, pitím a alkoholem“).
• Tento léčivý přípravek neužívejte s jídlem, protože užívání těchto tobolek s jídlem může způsobit žaludeční problémy.
• Jestliže užíváte další léčivý přípravek snižující cholesterol vazbou na žlučové kyseliny, jako je kolesevelam nebo cholestyramin, užívejte léčivý přípravek vázající žlučové kyseliny minimálně 4 hodiny před užíváním přípravku Lojuxta nebo 4 hodiny po něm.