Litak 2 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
LITAK 2 mg/ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml roztoku obsahuje 2 mg cladribinum (2-CdA). Jedna ampulka obsahuje 10 mg kladribinu v 5 ml roztoku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek LITAK je indikován k léčbě trichocelulární leukémie.
4.2 Dávkování a způsob podání
Terapie přípravkem LITAK musí být zahájená kvalifikovaným lékařem se zkušeností s protinádorovou chemoterapií.
Dávkování
Doporučené dávkování k léčbě trichocelulární leukémie je jeden cyklus přípravku LITAK podaný jako subkutánní injekční bolus v denní dávce 0,14 mg / kg tělesné hmotnosti, a to 5 po sobě následujících dnů.
Odchylky od dávkování uvedeného výše se nedoporučují.
Starší pacienti
Zkušenost s pacienty staršími než 65 let je omezená. Starší pacienti by měli být léčeni podle individuálního posouzení a pečlivého monitorování krevního obrazu a renálních a jaterních funkcí. Možná rizika je nutno posoudit u každého případu zvlášť (viz bod 4.4).
Pacienti s nedostatečnou funkcí ledvin a jater
Nejsou k dispozici údaje o použití přípravku LITAK u pacientů s renálním nebo jaterním poškozením. Přípravek LITAK je kontraindikován u pacientů se středně těžkým až těžkým poškozením ledvin (clearance kreatininu < 50 ml/min) nebo se středně těžkým až těžkým poškozením jater (klasifikace Child-Pugh > 6) (viz. body 4.3, 4.4 a 5.2).
Děti a mladiství
Podávání přípravku LITAK je u pacientů mladších než 18 let kontraindikováno (viz bod 4.3).
Způsob podání
Přípravek LITAK je dodáván jako roztok připravený k injekci. Doporučená dávka se přímo natáhne stříkačkou a aplikuje se jako subkutánní bolusová injekce bez ředění. Přípravek LITAK musí být před aplikací vizuálně prohlédnut kvůli přítomnosti částic nebo zbarvení. Před aplikací nechejte přípravek LITAK zahřát na pokojovou teplotu.
Samostatné podání pacientem
Přípravek LITAK si může aplikovat pacient samostatně. Pacienti musí být poučeni a vhodně vyškoleni. Podrobné pokyny jsou uvedeny v příbalové informaci.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo kteroukoli pomocnou látku tohoto přípravku.
Těhotenství a kojení.
Pacienti mladší než 18 let.
Středně těžké až těžké poškozenímledvin (clearance kreatininu < 50 ml/min) nebo se středně těžkým až těžkým poškozením jater (Child-Pugh klasifikace > 6) — (viz. také bod 4.4).
Souběžné používání jiných myelusupresivních léčiv.
4.4 Zvláštní upozornění a zvláštní opatření pro použití
Kladribin je antineoplastická a imunosupresivní látka, která může vyvolat závažné toxické nežádoucí reakce, jako např. myelosupresi a imunosupresi, dlouhodobou lymfocytopenii a oportunní infekce. Pacienti podstupující terapii kladribinem musí být podrobně monitorováni kvůli příznakům hematologických a nehematologických toxicit.
Doporučuje se zvláštní opatrnost a zhodnocení rizik a prospěchu, pokud je aplikace kladribinu zvažována u pacientů se zvýšeným rizikem infekce, manifestním selháním nebo infiltrací kostní dřeně a s předcházející myelosupresivní léčbou, jakož i u pacientů se suspektní nebo manifestní renální nebo jaterní nedostatečností. U pacientů s aktivní infekcí musí být tato nemoc před aplikací kladribinu vyléčena. Ačkoli se antiinfekční profylaxe obecně nedoporučuje, může být vhodná pro pacienty, jejichž imunita byla před léčbou kladribinem oslabená nebo pro pacienty s již existující agranulocytózou.
Pokud se objeví závažná toxicita, lékař by měl zvážit odložení nebo přerušení terapie tímto léčivem, dokud vážné komplikace neodezní. V případě infekcí by měla být podle potřeby zahájena terapie antibiotiky.
Doporučuje se, aby byly pacientům léčeným kladribinem podány ozářené krevní buněčné komponenty/produkty, aby se při transfúzi zabránilo vzniku reakce štěpu vůči hostiteli (Ta-GVHD).
Sekundární malignity
Podobně jako terapie jinými nukleosidovými analogy je léčba kladribinem spojena s myelosupresí a silnou a dlouhodobou imunosupresí. Léčba s těmito agens je spojena s výskytem dalších malignit. Lze očekávat, že se sekundární malignity objeví u pacientů s trichocelulární leukemií. Jejich četnost se velmi liší — v rozsahu od 2 % do 21 %. Nejvyšší riziko je 2 roky po diagnóze s mediánem mezi 40 a 66 měsíci. Kumulativní frekvence sekundární malignity jsou 5 %, 10-12 % a 13-14 % následující 5, 10 a 15 let, po diagnóze trichocelulární leukémie. Po léčbě kladribinem se výskyt sekundárních malignit pohybuje v rozmezí od 0 % do 9,5 % případů s mediánem doby sledování 2,8 až 8,5 let. Četnost výskytu sekundárních malignit po léčbě přípravkem LITAK byla 3,4 % u všech 232 pacientů s trichocelulární leukemií léčených během období deseti let. Nejvyšší výskyt sekundárních malignit s přípravkem LITAK byl 6,5 % s mediánem doby sledování 8,4 let. Proto by tedy měli být pacienti léčení kladribinem pravidelně monitorováni.
Hematologická toxicita
Během prvního měsíce po léčbě je nejvýznamnější myelosuprese a může být potřebná transfuse červených krvinek nebo krevních destiček. Pacienti s příznaky deprese kostní dřeně by měli být léčeni s obezřetností, neboť lze očekávat další útlum funkce kostní dřeně. Terapeutická rizika a prospěch by měly být pečlivě zhodnoceny u pacientů s aktivními nebo suspektními infekcemi. Riziko vážné myelotoxicity a dlouhodobé imunosuprese je zvýšeno u pacientů s onemocněním související s infiltrací kostní dřeně nebo s předcházející myelosupresivní léčbou. V takových případech je vyžadováno snížení dávky a pravidelné sledování pacienta. Pancytopenie je normálně reverzibilní a intenzita aplazie kostní dřeně je závislá na dávce. Během terapie kladribinem a 6 měsíců následujících je nutno očekávat zvýšenou incidenci oportunních infekcí. Pečlivé a pravidelné sledování periferního krevního obrazu je nezbytnéé během léčby kladribinem a 2 až 4 následující měsíce, a to kvůli detekci potenciálních nežádoucích reakcí a následných komplikací (anémie, neutropenie, trombocytopenie, infekce, hemolýza nebo krvácení) a kvůli dohledu nad hematologickou regenerací. U pacientů léčených na trichocelulární leukémii se často objeví horečka neznámého původu a projevuje se převážně první 4 týdny terapie. Původ febrilních stavů by měl být vyšetřen příslušnou laboratoří a radiologickými testy. Méně než třetina febrilních stavů je spojena s průkaznou infekcí. V případě horečky mající souvislost s infekcemi nebo agranulocytózou je indikována antibiotická léčba.
Renální a jaterní poškození
Neexistují údaje o použití přípravku LITAK u pacientů s poškozením ledvin nebo jater. Klinická zkušenost je velmi omezená a bezpečnost přípravku u těchto pacientů není dobře zhodnocena (viz body 4.3 a 5.2). Obezřetná terapie je vyžadována u pacientů se známým nebo suspektním renálním nebo jaterním poškozením. Pro všechny pacienty léčené přípravkem LITAK je doporučeno pravidelné posouzení renálních a jaterních funkcí, jak je klinicky indikováno.
Starší pacienti
Starší pacienti by měli být léčeni podle individuálního posouzení a pečlivého monitorování krevního obrazu a renálních a jaterních funkcí. Možná rizika je nutno posoudit u každého případu zvlášť (viz bod 4.2).
Prevence „tumor lysis syndrome” (rychlý rozpad nádorových buněk)
U pacientů s vysokou zátěží nádorem by měla být zahájena profylaktická terapie alopurinolem kvůli kontrole sérových hladin kyseliny močové, a to 24 hodin před začátkem chemoterapie spolu s adekvátní nebo zvýšenou hydratací. Denní perorální dávka 100 mg alopurinolu je doporučena po dobu 2 týdnů.
V případě kumulace kyseliny močové v séru nad normální rozmezí může být dávka alopurinolu zvýšena na 300 mg/den.
Fertilita
Mužům, kteří jsou léčeni kladribinem, musí být doporučeno, aby neplodili dítě do 6 měsíců po léčbě. Měli by se poradit o případném zmrazení spermií před léčbou kvůli možnosti neplodnosti v důsledku terapie kladribinem (viz body 4.6 a 5.3).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Kvůli potenciálnímu zvýšení hematologické toxicity a suprese kostní dřeně kladribin nesmí být užíván souběžně s jinými myelosupresivními léčivy. Vliv kladribinu na účinek jiných antineoplastických agens nebyl in vitro pozorován (např. doxorubicin, vinkristin, cytarabin, cyklofosfamid) ani in vivo. Avšak studie in vitro odhalila zkříženou rezistenci mezi kladribinem a chlormethinem; pro cytarabin popsal jeden autor in vivo zkříženou reakci bez ztráty aktivity.
Kvůli podobnému intracelulárnímu metabolizmu se může objevit zkřížená rezistence s jinými analogy nukleosidů, jako je např. fludarabin nebo 2'-deoxykoformycin. Proto se nedoporučuje simultánní aplikace analogů nukleosidů s kladribinem.
Ukázalo se, že kortikosteroidy, pokud jsou užívány s kladribinem, zvyšují riziko vážných infekcí a neměly by být podávány s kladribinem souběžně.
Vzhledem k tomu, že lze očekávat interakce s léčivými přípravky prodělávajícími intracelulární fosforylaci, jako jsou např. antivirová agens nebo s inhibitory příjmu adenozinu, jejich souběžné podávání s kladribinem se nedoporučuje.
4.6 Těhotenství a kojení
Kladribin způsobuje závažné vrozené vady, když je aplikován během těhotenství. Studie na zvířatech a in vitro studie lidských buněčných linií ukázaly teratogenitu a mutagenitu kladribinu. Kladribin je v těhotenství kontraindikován.
Ženy ve fertilním věku musí používat během léčby kladribinem a ještě 6 měsíců po ukončení léčby kladribinem účinnou antikoncepci. V případě otěhotnění během terapie kladribinem by žena měla být informována o potenciálním riziku pro plod.
Kojení
Není známo, zda je kladribin vylučován do mateřského mléka. Kvůli možnosti vážných nežádoucích reakcí u kojenců je kojení v průběhu léčby kladribinem a ještě 6 měsíců po poslední dávce kladribinu kontraindikováno.
Fertilita
Účinky kladribinu na fertilitu nebyly u zvířat studovány. Avšak studie toxicity provedená na opicích cynomolgus ukázala, že kladribin potlačuje zrání rychle se tvořících buněk, včetně buněk varlat. Vliv na fertilitu u lidí není znám. U antineoplastických agens, jako je např. kladribin, která interferují s DNA, RNA a syntézou proteinů, lze očekávat, že budou mít nežádoucí účinky na gametogenezi u lidí (viz bod 5.3).
Mužům, kteří jsou léčeni kladribinem, musí být doporučeno, aby neplodili dítě do 6 měsíců po léčbě. Měli by se poradit o případném zmrazení spermií před léčbou kvůli možnosti neplodnosti v důsledku terapie kladribinem (viz bod 4.4).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek LITAK má výrazný vliv na schopnost řídit a obsluhovat stroje. V připadě výskytu některých nežádoucích účinků s možným dopadem na výkon (např. závratě, velmi časté nebo ospalost, která se může objevit z důvodu anémie, která je velmi častá), musí být pacienti upozornění, že by neměli řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Velmi časté nežádoucí účinky pozorované během tří nejpodstatnějších klinických studií s kladribinem u 279 pacientů léčených kvůli různým indikacím a u 62 pacientů s trichocelulární leukémií (HCL) byly myelosuprese, obzvláště těžká neutropenie (41 % (113/279), HCL 98 % (61/62)), těžká trombocytopenie (21 % (58/279), HCL 50 % (31/62)) a těžká anémie (14 % (21/150), HCL 55 % (34/62)), jakož i těžká imunosuprese/lymfopenie (63 % (176/279), HCL 95 % (59/62)}, infekce (39 % (110/279), HCL 58 % (36/62)) a horečka (až 64 %).
Kultivačně negativní horečka po léčbě kladribinem se objevila u 10-40 % pacientů s trichocelulární leukémií a je zřídka pozorována u pacientů s jinými neoplastickými chorobami. Kožní vyrážky (2-31 %) jsou popsány hlavně u pacientů s jinými souběžně podávanými léčivými přípravky známými jako příčina vyrážky (antibiotika a/nebo alopurinol). Gastrointestinální nežádoucí účinky jako nausea (5-28 %), zvracení (1-13 %) a průjem (3-12 %), jakož i únava (2-48 %), bolest hlavy (1-23 %) a snížená chuť k jídlu (1-22 %) byly hlášeny během léčby kladribinem. Není pravděpodobné, že by kladribin způsoboval alopecii; mírná a přechodná alopecie trvající několik dnů byla pozorována u 4/523 pacientů během terapie, ale nemohla být jednoznačně dána do souvislosti s kladribinem.
Nežádoucí reakce, které byly hlášeny, jsou uvedeny v tabulce dole podle kategorií četnosti a tříd orgánových systémů. Četnosti jsou definovány následovně: Velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10 000 až <1/1000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit). Jejich závažnost je uvedena v textu pod tabulkou.
|
Infekce a infestace |
Velmi časté: infekce* (např. pneumonie*, septikémie*) |
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy) |
Časté: další malignity* Vzácné: syndrom z rozpadu nádoru* |
|
Poruchy krve a lymfatického systému |
Velmi časté: pancytopenie/myelosuprese*, neutropenie, trombocytopenie, anémie, lymfopenie Méně časté: hemolytická anémie* Vzácné: hypereosinofilie Velmi vzácné: amyloidóza |
|
Poruchy imunitního systému |
Velmi časté: imunosuprese* Vzácné: reakce štěpu vůči hostiteli*, |
|
Poruchy metabolizmu a výživy |
Velmi časté: snížená chuť k jídlu Méně časté: kachexie |
|
Poruchy nervového systému |
Velmi časté: bolest hlavy, závrať Časté: nespavost, úzkost Méně časté: ospalost, parestézie, letargie, polyneuropatie, zmatenost, ataxie Vzácné: apoplexe, neurologické poruchy řeči a polykání Velmi vzácné: deprese, epileptický záchvat |
|
Poruchy oka |
Méně časté: konjunktivitida Velmi vzácné: blefaritida |
|
Srdeční poruchy |
Časté: tachykardie, srdeční šelest, hypotenze, epistaxe, ischémie myokardu* Vzácné: srdeční selhání, atriální fibrilace, srdeční dekompenzace |
|
Cévní poruchy |
Velmi časté: purpura Časté: petechie, hemoragie* Méně časté: flebitida |
|
Respirační, hrudní a mediastinální poruchy |
Velmi časté: abnormální dýchací šelesty, abnormální hrudní šelesty, kašel Časté: dechová nedostatečnost, pulmonární intersticiální infiltrace většinou infekčního původu, mukozitida Méně časté: faryngitida Velmi vzácné: plicní embolie |
|
Gastrointestinální poruchy |
Velmi časté: nausea, zvracení, zácpa, průjem Časté: gastrointestinální bolest, flatulence Vzácné: žloutenka |
|
Poruchy jater a žlučových cest |
Časté: reverzibilní, většinou mírné zvýšení hodnot bilirubinu a transamináz Vzácné: jaterní selhání Velmi vzácné: cholecystitida |
|
Poruchy kůže a podkožní tkáně |
Velmi časté: vyrážka, lokalizovaný exantém, diaforéza Časté: pruritus, bolest kůže, erytém, kopřivka Vzácné: Stevens-Johnsonův syndrom/Lyellův syndrom |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté: myalgia, arthralgia, arthritis, bolest kostí |
|
Poruchy ledvin a močových cest |
Vzácné: renální selhání |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté: reakce v místě vpichu, horečka, únava, zimnice, asthenia Časté: edém, nevolnost, bolest |
* viz. popis níže.
Nehematologické nežádoucí účinky
Nehematologické nežádoucí účinky jsou obecně mírné až středně těžké. Léčba nevolnosti antiemetiky není obvykle nutná. Nežádoucí účinky související s kůží a podkožní tkání jsou většinou mírné nebo středně těžké a jsou přechodné, obvykle odeznívají ve 30 denním intervalu.
Krevní obraz
Vzhledem k tomu, že pacienti s aktivní trichocelulární leukemií mají většinou snížené parametry krevního obrazu, zejména nízký počet neutrofilů, více než 90 % případů má přechodnou těžkou neutropenii (< 1,0 x 109/l). Použití hematopoetických růstových faktorů ani nezlepší regeneraci počtu neutrofilů ani nesníží výskyt horečky. Těžké trombocytopenie (< 50 x 109/l) jsou pozorovány u asi 20 % až 30 % všech pacientů. Lze očekávat lymfocytopeni trvající několik měsíců a imunosupresi se zvýšeným rizikem infekcí. Regenerace cytotoxických T-lymfocytů a NK buněk se objeví do 3 až 12 měsíců. Úplná regenerace T-helperů a B-lymfocytů je zpožděna až do 2 let. Kladribin vyvolává vážné a dlouhodobé snížení počtu CD4+ a CD8+ T-lymfocytů. V současné době neexistují zkušenosti s možnými dlouhodobými důsledky této imunosuprese.
Infekce
Vzácně byly hlášeny závažné dlouhotrvající lymfocytopenie, které však nemohly být dány do souvislosti s pozdními infekčními komplikacemi. Velmi časté závažné komplikace, v některých případech s fatálními následky, jsou oportunní infekce (např. Pneumocystis carinii, Toxoplasma gondii, listeria, kandida, herpesviry, cytomegalovirus a atypické mykobaktérie). Čtyřicet procent pacientů, kteří byli léčení přípravkem LITAK v dávce 0,7 mg / kg tělesné hmotnosti / cyklus trpělo infekcemi. Tyto byly v průměru závažnější než infekce manifestující se u 27 % všech pacientů, kteří dostávali sníženou dávku 0,5 mg / kg tělesné hmotnosti na cyklus. Čtyřicet tři pacientů s trichocelulární leukemií mělo infekční komplikace při standardním režimu dávkování. Třetina těchto infekcí musí být považována za závažné (např. septikémie, pneumonie). Bylo hlášeno při nejmenším 10 případů s akutní autoimunní hemolytickou anémií. Všichni pacienti byli úspěšně léčeni kortikosteroidy.
Závažné nežádoucí účinky
Závažné nežádoucí účinky jako žloutenka, závažné jaterní selhání, renální selhání, srdeční selhání, atriální fibrilace, srdeční dekompenzace, apoplexe, neurologické poruchy řeči a polykání, „tumor lysis syndrome” (náhlá smrt buněk) s akutním renálním selháním, transfúzní reakce štěpu na příjemce, Stevens-Johnsonův syndrom/Lyellův syndrom (toxická epidermální nekrolýza), hemolytická anémie, hypereosinofilie (s erytematózní kožní vyrážkou, svěděním a faciálním edémem) jsou vzácné.
Fatální konec
Většina úmrtí souvisejících s léčivem nastává kvůli infekčním komplikacím. Další vzácné případy s fatálním koncem popsané v souvislosti s chemoterapií přípravkem LITAK byly sekundární malignity, mozkové a kardiovaskulární infarkty, reakce štěpu proti příjemci způsobená mnohočetnými transfúzemi neozářené krve, jakož i „tumour lysis syndromem” s hyperurikémií, metabolickou acidózou a akutním selháním ledvin.
4.9 Předávkování
Nejčastěji pozorované příznaky předávkování jsou nausea, zvracení, průjem, těžká deprese kostní dřeně, (včetně anémie, trombocytopenie, leukopenie a agranulocytózy), akutní renální insuficience, jakož i ireverzibilní neurologická toxicita (paraparéza/kvadriparéza), Guillain-Barrého syndrom a Brown-Séquardův syndrom. Akutní ireverzibilní neurotoxicita a nefrotoxicita byly popsány u jednotlivých pacientů léčených dávkou, která byla > 4krát vyšší než doporučený režim pro trichocelulární leukemii.
Neexistuje žádné speciální antidotum. Okamžité přerušení léčby, pečlivé pozorování a zahájení příslušných podpůrných opatření (krevní transfúze, dialýza, hemofiltrace, antiinfekční terapie) je indikovaná terapie při předávkování kladribinem. Pacienti, kteří byly předávkováni kladribinem, by měli být hematologicky monitorováni při nejmenším ještě čtyři týdny.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Analogy purinů, ATC kód: L01BB04
Kladribin je analog purinového nukleosidu, který působí jako antimetabolit. Jednoduchá substituce vodíku chlorem na pozici 2 odlišuje kladribin od jeho přirozeného protějšku 2'-deoxyadenozinu a činí molekulu rezistentní k deaminaci adenozindeaminázou.
Mechanizmus účinku
Kladribin je „prodrug” (proléčivo), které je po parenterálním podání rychle přijímáno buňkami a je intracelulárně fosforylováno na aktivní nukleotid 2-chlorodeoxyadenozin-5'-trifosfát (CdATP) pomocí deoxycytidinkinázy (dCK). Kumulace aktivního CdATP je pozorována především u buněk s vysokou aktivitou dCK a nízkou aktivitou deoxynukleotidázy, zejména v lymfocytech a v jiných hemopoetických buňkách. Cytotoxicita kladribinu je závislá na dávce. Zdá se, že nehematologické tkáně nejsou postiženy, což vysvětluje nízký výskyt nehematopoetické toxicity kladribinu.
Na rozdíl od jiných analogů nukleosidů je kladribin toxický v rychle proliferujících buňkách stejně jako v buňkách v klidovém stavu. Žádné cytotoxické účinky kladribinu není možno pozorovat v buněčných liniích solidních nádorů. Mechanizmus účinku kladribinu se přisuzuje inkorporaci CdATP do řetězců DNA: syntéza nové DNA v dělících se buňkách je blokována a reparační mechanizmus DNA je inhibovaný, což má za následek kumulaci zlomů řetězců DNA a snížení NAD (nikotinamidadenindinukleotid) a koncentrace ATP dokonce i v buňkách v klidovém stavu. Navíc CdATP inhibuje ribonukleotidreduktázu, enzym zodpovědný za přeměnu ribonukleotidů na deoxyribonukleotidy. Buněčná smrt nastává v důsledku deplece energie a apoptózy.
Klinická účinnost
V klinické zkoušce se subkutánně užívaným přípravkem LITAK bylo léčeno 63 pacientů s trichocelulární leukemií (33 nově diagnostifikovaných pacientů a 30 pacientů s relapsujícím nebo progresivním onemocněním). Celková četnost odpovědi byla 97 % s dlouhodobou remisí, přičemž 73 % pacientů zůstalo v celkové remisi po dobu následujících čtyř let.
5.2 Farmakokinetické vlastnosti
Absorbce
Kladribin vykazuje úplnou biologickou dostupnost po parenterálním podání; průměrná plocha pod křivkou koncentrace v plazmě a času (AUC) je srovnatelná po kontinuální nebo intermitentní dvouhodinové intravenózní infúzi a po subkutánní injekci.
Distribuce
Po subkutánní bolusové injekci dávky 0,14 mg/kg kladribinu je maximální plazmatické koncentrace Cmax 91 ng/ml dosaženo v průměru už po 20 minutách. V jiné studii, kde byla použita dávka 0,10 mg / kg tělesné hmotnosti / den, byla maximální plazmatická koncentrace Cmax po kontinuální intravenózní infuzi 5,1 ng/ml (tmax: 12 hodin) ve srovnání s 51 ng/ml po subkutánní bolusové injekci (tmax:
25 minut).
Intracelulární koncentrace kladribinu přesahuje jeho plazmatickou koncentraci 128 až 375krát.
Průměrný distribuční objem kladribinu je 9,2 l/kg. Vazba kladribinu na plazmatické proteiny je v průměru 25 % s širokou interindividuální variabilitou (5-50 %).
Metabolizmus
Prodrug kladribin je metabolizován intracelulárně přednostně pomocí deoxycytidinkinázy na 2-chlorodeoxyadenosin-5'-monofosfát, který je dále fosforylován na difosfát pomocí nukleosidmonofosátkinázy a na aktivní metabolit 2-chlorodeoxyadenosin.5'-trifosfát (CdATP) pomocí nukleosiddifosfátkinázy.
Eliminace
Farmakokinetické studie u lidí ukázaly, že křivka plazmatické koncentrace kladribinu vyhovuje 2-nebo 3-kompártmentovému modelu s a- a P-poločasy v průměru 35 minut respektive 6,7 hodin. Biexponenciální pokles sérové koncentrace kladribinu po subkutánní bolusové injekci je srovnatelný s eliminačními parametry po dvouhodinové intravenózní infuzi s počátečním a terminálním poločasem přibližně 2 hodiny a 11 hodin. Čas intracelulární retence kladribinových nukleotidů in vivo je zřetelně prodloužen v porovnání s časem retence v plazmě: poločasy t1/2 nejprve 15 hodin a následně více než 30 hodin byly naměřeny v leukemických buňkách.
Kladribin je vylučován hlavně ledvinami. Renální exkrece nemetabolizovaného kladribinu nastává do 24 hodin a činí 15 % a 18 % dávky po dvouhodinovém intravenózním a subkutánním podání. Osud zbytku není znám. Průměrná plazmatická clearance dosahuje 794 ml/min po intravenózní infuzi a 814 ml/min po subkutánní bolusové injekci při dávce 0,10 mg / kg tělesné hmotnosti / den.
Speciální populace
Renální a jaterní poškození
Nejsou k dispozici žádné studie s použitím kladribinu u pacientů s poškozením ledvin nebo jater (viz. také body 4.2 a 4.4, Zvláštní varování a zvláštní opatření pro užití). Klinické zkušenosti jsou velmi omezené a bezpečnost přípravku LITAK není u těchto pacientů dobře určena. Přípravek LITAK je kontraindikován u pacientů se středně těžkým až těžkým poškozením ledvin nebo se středně těžkým až těžkým poškozením jater (viz bod 4.3).
Děti
Použití přípravku LITAK u dětí nebylo zkoumáno (see section 4.2).
Starší pacienti
Zkušenosti s pacienty staršími než 65 let jsou velmi omezené. Pacienti staršího věku by měli být léčeni podle individuálního posouzení a pečlivého sledování krevního obrazu a renálních a jaterních funkcí.
5.3 Předklinické údaje vztahující se k bezpečnosti
Kladribin je středně akutně toxický pro myši s LD50 1 50 mg/kg po intraperitoneální aplikaci.
Ve studiích s opicemi cynomolgus, které dostávaly 7denní až 14denní kontinuální intravenózní infuze, byly cílovými orgány imunitní systém (> 0,3 mg/kg/den), kostní dřeň, kůže, sliznice, nervový systém a varlata (> 0,6 mg/kg/den) a ledviny (> 1 mg/kg/den). Až na fatální případy výsledky ukazovaly, že většina těchto účinků by byla po ukončení expozice pomalu reverzibilní.
Kladribin je teratogenní u myší (v dávkách 1,5-3,0 mg/kg/den podaných v 6. až 15. dni gestace).
Vlivy na sternální osifikaci byly pozorovány při dávkách 1,5 a 3,0 mg/kg/den. Zvýšená resorpce, snížení počtu živých zvířat ve vrhu, snížená hmotnost plodů a zvýšení fetálních malformací hlavy, trupu a končetin byly pozorovány při dávce 3,0 mg/kg/den. U králíků je kladribin teratogenní v dávkách 3,0 mg/kg/den (podaných 7.-19. den gestace). Při této dávce byly pozorovány vážné abnormality končetin, jakož i signifikantní snížení průměrné hmotnosti plodů. Snížená osifikace byla pozorována při dávce 1,0 mg/kg/den.
Kancerogenita/mutagenita
Dlouhodobé studie u zvířat prováděné kvůli zhodnocení kancerogenního potenciálu kladribinu nebyly provedeny. Na základě údajů, které jsou k dispozici, nemůže být provedeno zhodnocení kancerogenního rizika kladribinu u lidí.
Kladribin je cytotoxické léčivo, které je mutagenní pro kultivované savčí buňky. Kladribin je inkorporován do řetězců DNA a inhibuje syntézu DNA a reparace. Vystavení vlivu kladribinu indukuje fragmentaci DNA a buněčnou smrt u různých normálních a leukemických buněk a buněčných linií při koncentracích 5 nM až 20 pM.
Fertilita
Účinky kladribinu na fertilitu nebyly u zvířat studovány. Avšak studie toxicity provedená na opicích cynomolgus ukázala, že kladribin potlačuje zrání rychle se tvořících buněk, včetně buněk varlat. Vliv na fertilitu u lidí není znám. U antineoplastických agens, jako je např. kladribin, která interferují s DNA, RNA a syntézou proteinů, lze očekávat, že budou mít nežádoucí účinky na gametogenezi u lidí (viz. body 4.4 a 4.6).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný
Hydroxid sodný (k úpravě pH)
Kyselina chlorovodíková (k úpravě pH)
Voda na injekci
6.2 Inkompatibility
Přípravek LITAK nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti 4 roky.
Z mikrobiologického hlediska, pokud otevření nevyloučí riziko mikrobiologické kontaminace, přípravek má být použit okamžitě. Pokud není použit okamžitě, doba a podmínky uchovávání přípravku před použitím jsou v odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C-8 °C).
Chraňte před mrazem.
6.5 Druh obalu a velikost balení
10 ml injekční lahvička ze skla typu I s pryžovou zátkou (bromobutyl) a snímatelným hliníkovým víčkem.
Balení obsahuje 1 nebo 5 injekčních lahviček, jedna lahvička obsahuje 5 ml roztoku. Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Musí být použity postupy pro příslušný způsob zacházení s antineoplastickými léčivy a pro jejich likvidaci. S cytotoxickými léčivy by se mělo zacházet s opatrností. Nesmí s nimi přijít do styku těhotné ženy. Při zacházení a aplikaci přípravku LITAK se doporučuje použití rukavic na jednorázové použití a ochranný oděv. Pokud dojde ke kontaktu přípravku LITAK s kůží nebo sliznicemi, ihned důkladně opláchněte postižené místo vodou.
Parenterální léčiva musí být před aplikací vizuálně prohlédnuta kvůli přítomnosti částic nebo zbarvení.
Injekční lahvičky jsou pouze k jednorázovému použití. Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Lipomed GmbH Hegenheimer Strasse 2 D-79576 Weil/Rhein Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/275/001
EU/1/04/275/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 14/04/2004 Datum posledního prodloužení: 19/04/2009
10. DATUM REVIZE TEXTU
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY REGISTRACE
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Lipomed GmbH Hegenheimer Strasse 2 D-79576 Weil/Rhein Německo
B. PODMÍNKY REGISTRACE
• PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ, KLADENÉ NA DRŽITELE ROZHODNUTÍ O REGISTRACI
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
• PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
Neuplatňuje se.
• DALŠÍ PODMÍNKY
Držitel rozhodnutí o registraci je povinen informovat Evropskou komisi o plánu uvádění léčivého přípravku registrovaného tímto rozhodnutím na trh.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
LITAK 2 mg/ml injekční roztok cladribinum
Jeden ml roztoku obsahuje 2 mg kladribinu 10 mg/5 ml
Obsahuje chlorid sodný, hydroxid sodný, kyselinu chlorovodíkovou a vodu na injekci
1 injekční lahvička obsahující 5 ml injekčního roztoku
Subkutánní podání
Před použitím si přečtěte příbalový leták.
Uchovávejte mimo dosah a dohled dětí.
Cytotoxické. Zvláštní opatření pro zacházení s přípravkem (viz příbalový leták) Pouze k jednorázovému použití
EXP
Uchovávejte v chladničce
Chraňte před mrazem
10. zvláštní opatřeni pro likvidaci nepoužitých lecivych přípravku
NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Lipomed GmbH Hegenheimer Strasse 2 D-79576 Weil/Rhein Německo
12. REGISTRAČNÍ ČÍSLO
EU/1/04/275/001 13 ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
LITAK 2 mg/ml injekční roztok cladribinum
Jeden ml roztoku obsahuje 2 mg kladribinu 10 mg/5 ml
Obsahuje chlorid sodný, hydroxid sodný, kyselinu chlorovodíkovou a vodu pro injekce
5 injekčních lahviček, jedna injekční lahvička obsahuje 5 ml injekčního roztoku
Subkutánní podání
Před použitím si přečtěte příbalový leták.
Uchovávejte mimo dosah a dohled dětí.
Cytotoxické. Zvláštní opatření pro zacházení s přípravkem (viz příbalový leták) Pouze pro jednorázové použití
EXP
Uchovávejte v chladničce Chraňte před mrazem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Lipomed GmbH Hegenheimer Strasse 2 D-79576 Weil/Rhein Německo
EU/1/04/275/002
č.š.:
Výdej léčivého přípravku vázán na lékařský předpis
Nevyžaduje se - odůvodnění přijato
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK NA LAHVIČCE
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
LITAK 2 mg/ml injekční roztok
cladribinum
Subkutánní podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalový leták
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
č.s.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
10 mg/5 ml
6. JINÉ
Cytotoxické
LITAK 2 mg/ml injekční roztok
cladribinum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Pokud se kterýkoli z nežádoucích účinků vyskytne v závažné míře, nebo pokud si všimnete jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci, prosím, sdělte to svému lékaři nebo lékárníkovi.
V příbalové informaci naleznete:
1. Co je přípravek LITAK a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek LITAK užívat
3. Jak se přípravek LITAK užívá
4. Možné nežádoucí účinky
5. Jak přípravek LITAK uchovávat
6. Další informace
1. CO JE PŘÍPRAVEK LITAK A K ČEMU SE POUŽÍVÁ
LITAK obsahuje léčivou látku kladribin. Kladribin je cytostatická látka. Ovlivňuje růst maligních (rakovinných) bílých krvinek, které hrají roli při vzniku trichocelulární leukémie (HCL). LITAK se užívá k léčbě tohoto onemocnění.
2. ČEMU MUSÍTE VĚNOVAT POZORNOST, NEŽ ZAČNETE PŘÍPRAVEK LITAK
UŽÍVAT
Neužívejte přípravek LITAK:
- jestliže jste alergický/á (přecitlivělý/á) na kladribin nebo na kteroukoli další složku přípravku LITAK,
- jestliže jste těhotná nebo kojíte,
- jestliže jste mladší než 18 let,
- jestliže máte středně těžkou až těžkou poruchu ledvin nebo jater,
- jestliže užíváte jiné léčivé přípravky, které ovlivňují tvorbu krevních buněk v kostní dřeni (myelosuprese).
Zvláštní opatrnosti při použití přípravku LITAK je zapotřebí
Informujte svého lékaře, jestliže jste měl/a nebo máte:
- problémy s játry nebo ledvinami
- infekci
■ jestliže trpíte infekcí, bude tato léčena ještě předtím, než začnete užívat přípravek LITAK.
■ jestliže zaznamenáte během nebo po léčbě přípravkem LITAK jakýkoli příznak horečky nebo infekce (jako např. příznaky podobné chřipce), ihned informujte svého lékaře.
- horečku.
Před a během podávání přípravku LITAK podstoupíte pravidelné krevní testy, aby se zjistilo, zda je pro vás bezpečné pokračovat s touto léčbou. Lékař může rozhodnout, že musíte obdržet krevní transfuze, aby se zlepšil váš krevní obraz. Navíc bude přezkoušena správná funkce jater a ledvin.
Pokud chcete zplodit dítě, sdělte to prosím svému lékaři před započetím léčby přípravkem LITAK. Dítě nesmí být během léčby a do šesti měsíců po léčbě přípravkem LITAK zplozeno. Lékař může navrhnout uchování hluboce zmrazeného spermatu (kryokonzervace).
Vzájemné působení s dalšími léčivými přípravky
Prosím, informujte svého lékaře o všech lécích, které užíváte nebo jste užíval(a) v nedávné době, a to i o lécích, které jsou dostupné bez lékařského předpisu. Svého lékaře informujte zejména, jestliže užíváte léčivé přípravky obsahující:
- kortikosteroidy, obvykle používané k léčbě zánětů
- antivirová agens, používaná k léčbě virových infekcí
Přípravek LITAK nesmíte užívat současně s dalšími léčivými přípravky, které ovlivňují tvrobu krevních buněk v kostní dřeni (myelosuprese).
Těhotenství a kojení
Přípravek LITAK nesmíte užívat, pokud jste těhotná. Musíte používat adekvátní antikoncepční opatření během terapie a nejméně šest měsíců po poslední dávce přípravku LITAK. Pokud otěhotníte během léčby, okamžitě o tom informujte svého lékaře.
Během léčby a nejméně šest měsíců po poslední dávce přípravku LITAK nesmíte kojit.
Řízení dopravních prostředků a obsluha strojů
Přípravek LITAK má velký vliv na schopnost řídit a obsluhovat stroje., Pokud cítíte ospalost, což může být zapříčiněno nízkým počtem červených krvinek způsobeným léčbou přípravkem LITAK, nebo pokud pociťujete závrať neřiďte ani neobsluhujte stroje.
3. JAK SE PŘÍPRAVEK LITAK UŽÍVÁ
Vždy užívejte přípravek LITAK přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem. Lékař vypočte potřebnou dávku podle vaší tělesné hmotnosti a vysvětlí podrobně plán léčby. Doporučená denní dávka je 0,14 mg na kg tělesné hmotnosti po dobu pěti po sobě následujících dnů (jeden léčebný cyklus).
Přípravek LITAK se musí aplikovat injekcí pod kůži (subkutánní podání), v přibližně stejnou dobu každý den. Pokud si vstřikujete přípravek LITAk samostatně, musí Vás lékař nebo zdravotní sestra řádně zaškolit. podrobné pokyny k samostatnému podání naleznete na konci této příbalové informace
Můžete také obdržet další léčivý přípravek obsahující léčivou látku alopurinol, aby se snížila hladina kyseliny močové v séru.
Jestliže jste užil(a) více přípravku LITAK než jste měl(a)
Pokud si aplikujete nesprávnou dávku, ihned informujte svého lékaře.
Jestliže jste zapomněl(a) užít přípravek LITAK
Nevstřikujte si dvojitou dávku, abyste nahradili zapomenutou dávku. V případě, že vynecháte dávku, ihned informujte svého lékaře.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. MOŽNÉ NEŽÁDOUCÍ ÚČINKY
Podobně jako všechny léky, může mít i přípravek LITAK nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Informujte ihned svého lékaře, pokud se během léčby nebo po léčbě přípravkem LITAK objeví:
- jakýkoli příznak infekce (například symptomy podobné chřipce)
- horečka
Opakovaný výskyt maligní (zhoubné) nemoci nelze vyloučit. Tím je míněno, že riziko rozvoje maligního onemocnění v budoucnosti je u Vás o trochu vyšší, než u zdravých lidí. Toto lehce vyšší riziko může být z důvodu trichocelulární leukémie nebo z důvodu terapií použitých k léčbě tohoto onemocnění, včetně přípravku LITAK.
Nežádoucí účinky se vyskytují v určité četnosti, která je definována následujícím způsobem:
- velmi časté: týkají se více než 1 osoby z 10
- časté: týkají se 1 až 10 osob ze 100
- méně časté: týkají se 1 až 10 osob z 1 000
- vzácné: týkají se 1 až 10 osob z 10 000
- velmi vzácné: týkají se méně než 1 osoby z 10 000
- není známo: z dostupných údajů nelze určit.
Velmi časté nežádoucí účinky
■ infekce,
■ horečka,
■ nízky počet bílých krvinek (neutrofilů a lymfocytů) a krevních destiček v krevních testech,
■ nízký počet červených krvinek, který může mít za následek anémii, s příznaky jako je únava a ospalost,
■ snížená funkce imunitního systému těla,
■ bolest hlavy, závratě,
■ abnormální dýchací šelesty, abnormální hrudní šelesty, kašel,
■ pocit nevolnosti, zvracení, zácpa, průjem,
■ kožní vyrážka, otoky, zarudnutí a také bolestivost v místě vpichu injekce, pocení; kožní reakce jsou většinou mírné až středně závažné a obvykle se ztratí během několika dnů,
■ únava, zimnice, snížená chuť k jídlu.
■ slabost.
Časté nežádoucí účinky
■ opakovaný výskyt maligního (zhoubného) onemocnění,
■ nízký počet krevních destiček, který může být příčinou neobvyklého krvácení (například z nosu nebo kůže),
■ nespavost, pocit úzkosti,
■ zrychlený srdeční tep, abnormální srdeční šelest, nízký krevní tlak, snížený přísun krve do srdečního svalu,
■ dušnost, otoky plicní tkáně z důvodu infekce, zánět úst a jazyka,
■ bolest břicha a zvýšená plynatost v žaludku nebo střevech, většinou mírně zvýšené hodnoty jaterních testů (bilirubin, transaminázy), které se vrátí na původní hodnoty, jakmile je léčba ukončena,
■ svědění, svědivá kožní vyrážka (kopřivka), zarudnutí a bolestivost pokožky,
■ otoky tkání (edém), pocit nemoci, bolest (bolest svalů, kloubů a kostí).
Méně časté nežádoucí účinky
■ anémie způsobená rozpadem červených krvinek,
■ ospalost, snížená necitlivost kůže nebo mravenčení, ochablost, netečnost, poruchy periferních nervů, zmatenost, poruchy koordinace pohybů,
■ oční zánět,
■ bolest v krku,.
■ zánět žil,
■ velký úbytek hmnotnosti.
Vzácné nežádoucí účinky
■ snížená funkce jater,
■ snížená funkce ledvin,
■ komplikace způsobené léčbou rakoviny z důvodu rozpadu rakovinových buněk,
■ odmítavá reakce na transfuzi krve,
■ zvýšený počet určitých bílých krvinek (eozinofilů),
■ mrtvice.
■ poruchy řeči a polykání,
■ srdeční selhání,
■ abnormální srdeční rytmus,
■ neschopnost srdce udržovat potřebný krevní oběh,
■ neprůchodnost střev,
■ závažné alergické kožní reakce (Stevens-Johnsonův syndrom nebo Lyellův syndrom).
Velmi vzácné nežádoucí účinky
■ deprese, epileptický záchvat,
■ otok očního víčka,
■ krevní sraženina v plicích,
■ zánět zlučníku,
■ snížená funkce orgánů kvůli vysokému objemu specifické substance produkované tělem (glykoprotein).
Pokud se kterýkoli z nežádoucích účinků vyskytne v závažné míře, nebo pokud si všimnete jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci, prosím, sdělte to svému lékaři nebo lékárníkovi.
5. JAK PŘÍPRAVEK LITAK UCHOVÁVAT
Uchovávejte mimo dosah a dohled dětí.
Uchovávejte v chladničce (2 °C-8 °C). Chraňte před mrazem.
Přípravek LITAK nepoužívejte po uplynutí doby použitelnosti, uvedené na štítku injekční lahvičky a krabičky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Z mikrobiologického hlediska, pokud otevření nevyloučí riziko mikrobiologické kontaminace, přípravek má být použit okamžitě. Pokud není použit okamžitě, doba a podmínky uchovávání přípravku před použitím jsou v odpovědnosti uživatele.
Nepoužívejte přípravek LITAK, pokud si všimnete, že je injekční lahvička poškozená, nebo že roztok není čirý nebo obsahuje částice.
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
6. DALŠÍ INFORMACE Co přípravek LITAK obsahuje
- Léčivou látkou je kladribin. Jeden ml roztoku obsahuje 2 mg kladribinu. Jedna injekční lahvička obsahuje 10 mg kladribinu v 5 ml roztoku.
- Pomocnými látkami jsou chlorid sodný, hydroxid sodný (k úpravě pH), kyselina chlorovodíková (k úpravě pH) a voda na injekci.
Jak přípravek LITAK vypadá a co obsahuje toto balení
LITAK se dodává v injekční lahvičce ze skla s obsahem 5 ml čirého, bezbarvého injekčního roztoku. Balení obsahuje 1 nebo 5 injekčních lahviček. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce Lipomed GmbH Hegenheimer Strasse 2 D-79576 Weil/Rhein
Německo
Další informace o tomto přípravku získáte u držitele rozhodnutí o registraci. Tato příbalová informace byla naposledy schválena
POKYNY K PODÁNÍ INJEKCE
Tato část obsahuje informace o aplikaci injekce přípravku LITAK. Injekci neaplikujte sami, pokud jste nebyli poučeni lékařem nebo zdravotní sestrou. Lékař vám řekne, jaké množství přípravku LITAK potřebujete a jak často a kdy musíte injekci aplikovat. LITAk se aplikuje do tkáně přímo pod kůží (subkutánní injekce). Pokud máte jakýkoli dotaz týkající se aplikace injekce, požádejte, prosím, o pomoc svého lékaře nebo zdravotní sestru.
Přípravek LITAK je cytotoxický a mělo by se s ním proto zacházet opatrně. Pokud není přípravek LITAK aplikován samostatně, doporučuje se použit rukavice na jednorázové použití a ochranný oděv. Pokud dojde ke kontaktu přípravku LITAK s kůží nebo sliznicemi, ihned důkladně opláchněte postižené místo vodou. S přípravkem LITAK nesmí přijít do kontaktu těhotné ženy.
Co je potřeba k aplikaci injekce?
Abyste si mohli injekci aplikovat sami, budete potřebovat následující položky:
- jednu injekční lahvičku přípravku LITAK (nebo dvě injekční lahvičky, pokud potřebujete aplikovat více než 5 ml),
Nepoužívejte injekční lahvičky, které jsou poškozené, nebo pokud roztok není čirý nebo obsahuje částice.
- jednu sterilní injekční stříkačku (např. stříkačku 10 ml LUER),
- jednu sterilní injekční jehlu (např. 0,5 x 19 mm, 25 G x %"),
- tampony navlhčené v alkoholu,
- nádobu odolnou proti propíchnutí k bezpečnému odstranění použitých stříkaček.
Co bych měl(a) udělat před aplikací subkutánní injekce přípravku LITAK?
1. Před injekcí nechejte přípravek LITAK zahřát na pokojovou teplotu.
2. Důkladně si umyjte ruce.
3. Najděte si pohodlné, dobře osvětlené místo a položte vše, co potřebujete, na dosah ruky.
Jak připravím injekci?
Před aplikací přípravku LITAK musíte provést následující:
1. Odstraňte červené ochranné víčko z injekční lahvičky s přípravkem LITAK. Pryžovou zátku lahvičky neodstraňujte. Očistěte pryžový vršek lahvičky tamponem navlhčeným v alkoholu. Vyjměte stříkačku z obalu, aniž byste se dotkli hrotu stříkačky. Vyjměte injekční jehlu z obalu a upevněte ji na hrot stříkačky. Odstraňte kryt jehly, aniž byste se jehly dotkli.
2. Protlačte jehlu přes pryžovou zátku lahvičky a otočte ampulku a stříkačku dnem nahoru. Ujistěte se, že hrot jehly je ponořený v roztoku.
3. Vytažením pístu natáhněte do stříkačky správný objem přípravku LITAK (požadované množství přípravku LITAK v mililitrech vám sdělí lékař).
4. Vytáhněte jehlu z lahvičky.
5. Přesvědčte se, že ve stříkačce není žádný vzduch: obraťte jehlu nahoru a vzduch vytlačte.
6. Zkontrolujte, že máte správný objem.
7. Injekci ihned aplikujte.
Kam si mám injekci aplikovat?
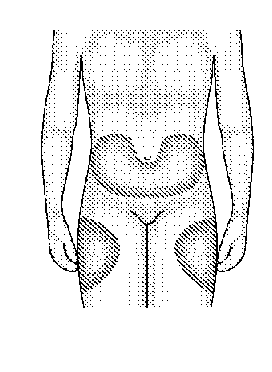
Jak si mám injekci aplikovat?
Nejvhodnější místa pro aplikaci injekce sami sobě jsou znázorněna zde:horní oblast vašich stehen a břicho, s výjimkou okolí pupku. Pokud vám injekci aplikuje někdo jiný, může také využít vnější povrch nadloktí nebo hýždí.
1. Desinfikujte svoji kůži pomocí tamponu namočeného v alkoholu, vyčkejte až oblast uschne a uchopte kůži mezi palec
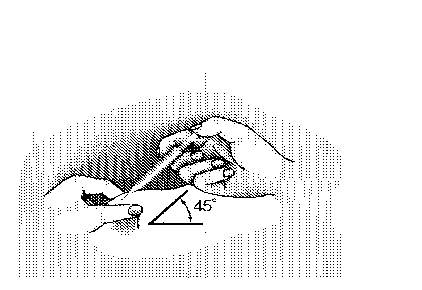
a ukazováček, ale nestiskujte ji.
2. Jehlu zcela vpíchněte do kůže pod úhlem asi 45°, jak je znázorněno na obrázku.
3. Jemně povytáhněte píst, abyste zjistili, zda jste nenapíchnuli žádnou krevní cévu. Pokud vidíte ve stříkačce krev, vytáhněte jehlu a vpich proveďte znovu na jiném místě.
4.
Pomalu a rovnoměrně aplikujte roztok po dobu přibližně jedné minuty, přičemž kůži neustále přidržujte.
5. Po aplikaci roztoku jehlu vytáhněte.
6. Vložte použitou stříkačku do nádoby odolné proti propíchnutí. Ke každé aplikaci použijte novou
stříkačku a injekční jehlu. Injekční lahvičky jsou pouze na jednorázové použití. Vraťte jakékoli zbývající množství roztoku lékaři nebo lékárníkovi, aby mohl být řádně zlikvidován.
Odstranění použitých stříkaček
Vložte použité stříkačky do nádoby odolné proti propíchnutí a uchovávejte ji mimo dosah a dohled dětí.
Nádobu odolnou proti propíchnutí zlikvidujte podle pokynů lékaře, zdravotní sestry nebo lékárníka. Nedávejte použité stříkačky do koše na normální odpad.
28