Lenvima 4 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
LENVIMA 4 mg tvrdé tobolky LENVIMA 10 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
LENVIMA 4 mg tvrdé tobolky
Jedna tvrdá tobolka obsahuje lenvatinibum 4 mg (ve formě lenvatinibi mesilas). LENVIMA 10 mg tvrdé tobolky
Jedna tvrdá tobolka obsahuje lenvatinibum 10 mg (ve formě lenvatinibi mesilas). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
LENVIMA 4 mg tvrdé tobolky
Žluto-červené tělo a žluto-červené víčko, přibližná délka 14,3 mm, černým inkoustem vytištěný symbol „€“ na víčku a „LENV 4 mg“ na těle.
LENVIMA 10 mg tvrdé tobolky
Žluté tělo a žluto-červené víčko, přibližná délka 14,3 mm, černým inkoustem vytištěný symbol „€“ na víčku a „LENV 10 mg“ na těle.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek LENVIMA je indikován k léčbě dospělých pacientů s progresivním, lokálně pokročilým nebo metastatickým diferencovaným (papilárním/folikulárním karcinomem / karcinomem z Hurthleho buněk) karcinomem štítné žlázy (differentiated thyroid carcinoma - DTC), který je refrakterní na léčbu radioaktivním jódem (radioactive iodine - RAI).
4.2 Dávkování a způsob podání
Léčbu přípravkem LENVIMA má zahajovat a vést lékař se zkušenostmi v oblasti léčby zhoubných nádorů.
Dávkování
Doporučená denní dávka lenvatinibu je 24 mg (dvě 10mg tobolky a jedna 4mg tobolka) jednou denně. Denní dávka má být upravena dle potřeby podle plánu řízení dávky/toxicity.
Vynechá-li pacient dávku a nebude-li ji moci užít v průběhu 12 hodin, musí tuto dávku vynechat a další dávku užít v obvyklé době podání.
Léčba má pokračovat tak dlouho, dokud je pozorován klinický přínos nebo dokud se neobjeví neakceptovatelná toxicita.
Před jakýmkoliv přerušením léčby nebo snížením dávky lenvatinibu má být zahájena optimální léčba nauzey, zvracení a průjmu; gastrointestinální toxicitu je třeba aktivně léčit, aby se snížilo riziko rozvoje poruchy funkce ledvin nebo renálního selhání (viz bod 4.4, Porucha funkce ledvin a renální selhání).
Úprava dávkování
Zvládnutí nežádoucích účinků může vyžadovat přerušení léčby, úpravu dávkování nebo ukončení léčby lenvatinibem (viz bod 4.4). Mírné až středně závažné nežádoucí účinky (např. 1. nebo 2. stupeň) zpravidla vyžadují přerušení léčby lenvatinibem pouze v případě, že pacient léčbu netoleruje ani po její optimalizaci. Závažné (např. 3. stupeň) nebo netolerovatelné nežádoucí účinky vyžadují přerušení léčby lenvatinibem až do zlepšení reakce na 0.-1. stupeň nebo na úroveň výchozího stavu.
Při toxicitě související s užíváním lenvatinibu (viz tabulka 1) se po odeznění/zlepšení nežádoucího účinku na 0.-1. stupeň nebo na úroveň výchozího stavu má v léčbě pokračovat se sníženou dávkou lenvatinibu tak, jak je uvedeno v tabulce 2.
Léčbu je nutné ukončit v případě život ohrožujících reakcí (např. 4. stupeň) s výjimkou laboratorních abnormalit, které jsou posouzeny jako život neohrožující a v tom případě je lze zvládnout stejně jako závažnou reakci (např. 3. stupeň).
Stupně se zakládají na obecných terminologických kritériích Národního institutu pro zhoubné nádory (National Cancer Institute - NCI) definujících nežádoucí účinky (Common Terminology Criteria for Adverse Events - CTCAE).
Tabulka 1 Nežádoucí účinky vyžadující úpravu dávkování lenvatinibu
|
Nežádoucí účinek |
Závažnost |
Postup |
Úprava dávkování a pokračování v úžívání lenvatinibu |
|
Hypertenze |
3. stupeň (navzdory optimální antihypertenzní léčbě) |
Přerušte |
Do odeznění na 0., 1. nebo 2. stupeň. Viz podrobný návod v tabulce 3 v bodě 4.4. |
|
4. stupeň |
Ukončete |
Nepokračujte | |
|
Proteinurie |
> 2 g/24 hodin |
Přerušte |
Do poklesu na méně než 2 g/ 24hodin. |
|
Nefrotický syndrom |
Ukončete |
Nepokračujte | |
|
Porucha funkce ledvin nebo renální selhání |
3. stupeň |
Přerušte |
Do odeznění na 0.-1. stupeň nebo na úroveň výchozího stavu. |
|
4. stupeň* |
Ukončete |
Nepokračujte | |
|
Srdeční dysfunkce |
3. stupeň |
Přerušte |
Do odeznění na 0.-1. stupeň nebo na úroveň výchozího stavu. |
|
4. stupeň |
Ukončete |
Nepokračujte | |
|
PRES/RPLS |
Všechny stupně |
Přerušte |
Zvažte zahájení podávání snížené dávky, pokud odezní na 0.-1. stupeň |
|
Hepatotoxicita |
3. stupeň |
Přerušte |
Do odeznění na 0.-1. stupeň nebo na úroveň výchozího stavu. |
|
4. stupeň* |
Ukončete |
Nepokračujte | |
|
Arteriální tromboembolie |
Všechny stupně |
Ukončete |
Nepokračujte |
|
Krvácení |
3. stupeň |
Přerušte |
Do odeznění na 0.-1. stupeň. |
|
4. stupeň |
Ukončete |
Nepokračujte |
Tabulka 1 Nežádoucí účinky vyžadující úpravu dávkování lenvatinibu
|
Nežádoucí účinek |
Závažnost |
Postup |
Úprava dávkování a pokračování v úžívání lenvatinibu |
|
GI perforace nebo tvorba píštěle |
3. stupeň |
Přerušte |
Do odeznění na 0.-1. stupeň nebo na úroveň výchozího stavu. |
|
4. stupeň |
Ukončete |
Nepokračujte | |
|
Píštěl mimo GIT |
4. stupeň |
Ukončete |
Nepokračujte |
|
Prodloužení QT intervalu |
>500 ms |
Přerušte |
Do poklesu na <480 ms nebo na úroveň výchozího stavu. |
|
3. stupeň |
Přerušte |
Do odeznění na 0.-1. stupeň nebo na úroveň výchozího stavu. | |
|
4. stupeň (navzdory léčbě) |
Ukončete |
Nepokračujte | |
|
*Laboratorní abnorma |
lity 4. stupně, které byly posouzeny jako život neohrožující, lze léčit stejně jako | ||
závažné reakce (např. 3. stupně).
Tabulka 2_Úprava dávkování oproti doporučené denní dávce lenvatinibua
|
Úroveň dávky |
Denní dávka |
Počet tobolek |
|
Doporučená denní dávka |
24 mg perorálně jednou denně |
Dvě 10mg tobolky plus jedna 4mg tobolka |
|
První snížení dávky |
20 mg perorálně jednou denně |
Dvě 10mg tobolky |
|
Druhé snížení dávky |
14 mg perorálně jednou denně |
Jedna 10mg tobolka plus jedna 4mg tobolka |
|
Třetí snížení dávky |
10 mg perorálně jednou denněa |
Jedna 10mg tobolka |
a' Další snižování dávky je třeba zvážit dle individuálních potřeb pacienta, jelikož o dávkách do 10 mg jsou k dispozici pouze omezené údaje.
Zvláštní populace
Zdá se, že pacienti ve věku > 75 let, asijské rasy, s komorbiditami (jako například s hypertenzí a poruchou funkce jater nebo ledvin) nebo s tělesnou hmotností pod 60 kg mají sníženou snášenlivost lenvatinibu (viz bod 4.8, Další zvláštní populace). U všech ostatních pacientů, kteří netrpí těžkou poruchou funkce jater nebo ledvin (viz níže), se má léčba zahájit doporučenou dávkou 24 mg a následně má být dávka upravena v závislosti na individuální snášenlivosti.
Pacienti s hypertenzí
Před zahájením léčby lenvatinibem je nutné zajistit adekvátní kontrolu krevního tlaku a v průběhu léčby jej pravidelně kontrolovat (viz bod 4.4). Viz také bod 4.8, Další zvláštní populace.
Pacienti s poruchou funkce jater
U pacientů s mírnou (Child-Pugh skóre A) nebo středně těžkou (Child-Pugh skóre B) poruchou funkce jater není nutná žádná úprava počáteční dávky podle funkce jater. U pacientů s těžkou (Child-Pugh skóre C) poruchou funkce jater je doporučená počáteční dávka 14 mg užívaných jednou denně. Může být nezbytná další úprava dávkování v závislosti na individuální snášenlivosti. Viz také bod 4.8, Další zvláštní populace.
Pacienti s poruchou funkce ledvin
U pacientů s mírnou nebo středně těžkou poruchou funkce ledvin není nutná žádná úprava počáteční dávky podle funkce ledvin. U pacientů s těžkou renální poruchou je doporučená počáteční dávka 14 mg užívaná jednou denně. Může být nezbytná další úprava dávkování v závislosti na individuální snášenlivosti. Nebyly provedeny studie u pacientů s konečným stadiem renálního onemocnění, a proto se používání lenvatinibu u těchto pacientů nedoporučuje. Viz také bod 4.8, Další zvláštní populace.
Starší osoby
Není nutná žádná úprava počáteční dávky podle věku. O používání tohoto přípravku u pacientů ve věku > 75 let jsou dostupné pouze omezené údaje (viz také bod 4.8, Další zvláštní populace).
Pediatrická populace
Z důvodu bezpečnostních rizik identifikovaných ve studiích na zvířatech se lenvatinib nemá používat u dětí ve věku do 2 let (viz bod 5.3). Bezpečnost a účinnost lenvatinibu u dětí ve věku od 2 do < 18 let nebyla dosud stanovena (viz bod 5.1). Nejsou dostupné žádné údaje.
Rasa
Není nutná žádná úprava počáteční dávky podle rasy (viz bod 5.2). O použití u pacientů jiného než europoidního nebo asijského etnického původu jsou dostupné pouze omezené údaje (viz také bod 4.8, Další zvláštní populace).
Způsob podání
Lenvatinib je určen k perorálnímu podání. Tobolky se mají užívat s jídlem nebo bez jídla každý den přibližně ve stejnou denní dobu (viz bod 5.2). Tobolky je třeba spolknout celé a zapít vodou. Pečující osoby nesmí tobolky otevírat, aby se zabránilo jejich opakované expozici obsahu tobolky.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Hypertenze
U pacientů léčených lenvatinibem byla hlášena hypertenze, která se obvykle objevovala v počáteční fázi léčby (viz bod 4.8, Popis vybraných nežádoucích účinků). Před zahájením léčby lenvatinibem je nutné zajistit adekvátní kontrolu krevního tlaku (KT) a, pokud je známo, že pacient trpí hypertenzí, má být takový pacient léčen stabilní dávkou antihypertenziv po dobu alespoň 1 týdne před léčbou lenvatinibem. Je důležité včasné zjištění a účinná léčba hypertenze, aby byla minimalizována potřeba přerušit léčbu a snížit dávku lenvatinibu. S podáváním antihypertenziv se má začít, jakmile je potvrzen zvýšený KT. KT je třeba sledovat po 1 týdnu léčby lenvatinibem, následně každé 2 týdny po dobu prvních 2 měsíců a poté jednou za měsíc. Výběr antihypertenzní léčby má být přizpůsoben klinické situaci pacienta a má probíhat podle standardní lékařské praxe. U pacientů, kteří byli dříve normotenzní, se má při zjištění zvýšeného krevního tlaku začít s monoterapií jednou ze tříd antihypertenziv. U pacientů, kteří již užívají antihypertenzní léčbu, je možné zvýšit dávku užívaného léčiva nebo, pokud je to vhodné, lze přidat jedno léčivo nebo více léčiv z jiné třídy antihypertenziv.
V případě potřeby zahajte léčbu hypertenze postupem doporučeným v tabulce 3.
Tabulka 3
Doporučený postup léčby hy
pertenze
|
Hodnota krevního tlaku (KT) |
Doporučený postup |
|
Systolický KT >140 mmHg až < 160 mmHg nebo diastolický KT > 90 mmHg až <100 mmHg |
Pokračujte v podávání lenvatinibu a zahajte anihypertenzní léčbu, nebyla-li již zahájena. NEBO Pokračujte v podávání lenvatinibu a zvyšte dávku stávající antihypertenzní léčby nebo zahajte léčbu dalšími antihypertenzivy. |
|
Systolický KT > 160 mmHg nebo diastolický KT >100 mmHg navzdory optimální léčbě antihypertenzivy |
1. Přerušte léčbu lenvatinibem. 2. Bude-li systolický KT <150 mmHg a diastolický KT < 95 mmHg a pacient bude užívat stabilní dávku antihypertenzní léčby po dobu minimálně 48 hodin, obnovte léčbu lenvatinibem ve snížené dávce (viz bod 4.2). |
|
Život ohrožující stavy (maligní hypertenze, neurologický deficit nebo hypertenzní krize) |
Je indikována okamžitá intervence. Přerušte podávání lenvatinibu a zahajte odpovídající léčbu. |
Ženy ve fertilním věku
Ženy ve fertilním věku musí během léčby lenvatinibem a ještě jeden měsíc po jejím ukončení používat vysoce účinnou antikoncepci (viz bod 4.6). V současné době není známo, zda lenvatinib v kombinaci s perorální antikoncepcí zvyšuje riziko tromboembolických příhod.
Proteinurie
U pacientů léčených lenvatinibem byla zaznamenána proteinurie, která se obvykle objevovala v počáteční fázi léčby (viz bod 4.8, Popis vybraných nežádoucích účinků). Je třeba pravidelně sledovat množství bílkovin v moči. Bude-li testovací proužek indikovat proteinurii > 2+, může být nutné přerušit léčbu, upravit dávkování nebo léčbu zcela ukončit (viz bod 4.2). Léčba lenvatinibem se má ukončit v případě nefrotického syndromu.
Porucha funkce ledvin a renální selhání
U pacientů léčených lenvatinibem byla zaznamenána porucha funkce ledvin a renální selhání (viz bod 4.8, Popis vybraných nežádoucích účinků). Primárním identifikovaným rizikovým faktorem byla dehydratace a/nebo hypovolemie následkem gastrointestinální toxicity. Gastrointestinální toxicitu je třeba aktivně léčit, aby se snížilo riziko rozvoje renální poruchy nebo renálního selhání. Může být nutné přerušit léčbu, upravit dávkování nebo léčbu zcela ukončit (viz bod 4.2).
U pacientů s těžkou renální poruchou je nutné počáteční dávku lenvatinibu upravit (viz body 4.2 a 5.2).
Srdeční dysfunkce
U pacientů léčených lenvatinibem bylo zaznamenáno srdeční selhání (< 1 %) a snížená ejekční frakce levé srdeční komory (viz bod 4.8, Popis vybraných nežádoucích účinků). U pacientů je nutné sledovat klinické příznaky nebo známky srdeční dekompenzace, neboť může být nutné přerušit léčbu, upravit dávkování nebo léčbu zcela ukončit (viz bod 4.2).
Syndrom posteriomí reverzibilní encefalopatie (posterior reversible encephalopathy syndrome -PRES) / Syndrom reverzibilní posteriomí leukoencefalopatie (reversible _posterior leucoencephalopathy syndrome -RPLS)
U pacientů léčených lenvatinibem byl zaznamenán PRES, rovněž známý jako RPLS (< 1 %; viz bod 4.8, Popis vybraných nežádoucích účinků). PRES je neurologická porucha, která se může projevovat bolestmi hlavy, záchvaty, letargií, zmateností, změněnou mentální funkcí, slepotou a jinými poruchami vidění nebo neurologickými poruchami. Může být přítomna mírná až těžká hypertenze. Pro potvrzení diagnózy PRES je nutné zobrazení pomocí magnetické rezonance. Je nutné zajistit adekvátní kontrolu krevního tlaku (viz bod 4.4, Hypertenze). U pacientů se známkami nebo příznaky PRES může být nutné přerušit léčbu, upravit dávkování nebo léčbu zcela ukončit (viz bod 4.2).
Hepatotoxicita
Nežádoucí účinky související s játry, které byly nejčastěji hlášeny u pacientů léčených lenvatinibem, zahrnovaly zvýšení alaninaminotransferázy, zvýšení aspartátaminotransferázy a zvýšení bilirubinu v krvi. U pacientů léčených lenvatinibem bylo zaznamenáno selhání jater a akutní hepatitida (< 1 %; viz bod 4.8, Popis vybraných nežádoucích účinků). Případy selhání jater byly zpravidla zaznamenány u pacientů s progresivními jaterními metastázami. Funkční jaterní testy je nutné sledovat před zahájením léčby, následně každé 2 týdny po dobu prvních 2 měsíců a poté v průběhu léčby jednou za měsíc. V případě hepatotoxicity může být nutné přerušit léčbu, upravit dávkování nebo léčbu zcela ukončit (viz bod 4.2).
U pacientů s těžkou poruchou funkce jater je nutné počáteční dávku lenvatinibu upravit (viz body 4.2 a 5.2).
Arteriální tromboembolie
U pacientů léčených lenvatinibem byla zaznamenána arteriální tromboembolie (cévní mozková příhoda, tranzitorní ischemická ataka a infarkt myokardu) (viz bod 4.8, Popis vybraných nežádoucích účinků). Lenvatinib nebyl hodnocen u pacientů, u kterých se vyskytla arteriální tromboembolie v předcházejících 6 měsících, a proto je třeba jej u takových pacientů používat s opatrností. Rozhodnutí o léčbě má vycházet z posouzení přínosů a rizik u každého pacienta. Léčba lenvatinibem se má ukončit po arteriální trombotické příhodě.
Krvácení
Závažné krvácení související s nádorem, včetně fatálních hemoragických příhod, se objevilo v klinických studiích a bylo zaznamenáno po uvedení přípravku na trh (viz bod 4.8, Popis vybraných nežádoucích účinků). Během sledování po uvedení přípravku na trh byly pozorovány případy závažného a fatálního krvácení z karotid u pacientů s anaplastickým karcinomem štítné žlázy (anaplastic thyroid carcinoma - ATC) častěji než u pacientů s DTC nebo jiným typem nádoru. Stupeň invaze/infiltrace nádoru do velkých cév (např. karotid) se má zvážit kvůli potenciálnímu riziku závažného krvácení spojeného se zmenšením/nekrózou nádoru po terapii lenvatinibem. Některé případy krvácení se objevily sekundárně po zmenšení nádoru a vytvoření píštěle, např. tracheoezofageální píštěle._U některých pacientů s mozkovými metastázami nebo bez mozkových metastáz byly zaznamenány případy fatálního intrakraniálního krvácení. Také byly zaznamenány případy krvácení v jiných částech těla než mozku (např. v trachei, v břišní dutině, v plicích).
V případě krvácení může být nutné přerušit léčbu, upravit dávkování nebo léčbu ukončit (viz bod 4.2, tabulka 2).
Gastrointestinální perforace a tvorba píštělí
U pacientů léčených lenvatinibem byla zaznamenána gastrointestinální perforace a tvorba píštělí (viz bod 4.8). Ve většině případů se gastrointestinální perforace a tvorba píštělí objevily u pacientů s rizikovými faktory, jako jsou předchozí operace nebo radioterapie. V případě gastrointestinální perforace nebo píštělí může být nutné přerušit léčbu, upravit dávkování nebo léčbu zcela ukončit (viz bod 4.2).
Píštěl mimo gastrointestinální trakt
U pacientů léčených lenvatinibem může být zvýšené riziko vzniku píštělí. Během klinických studií a po uvedení přípravku na trh byly pozorovány případy tvorby píštělí nebo jejich rozšíření, které zahrnovaly další části těla než žaludek nebo střeva (např. píštělí tracheálních, tracheoezofageálních, ezofageálních, kožních, postihujících ženské pohlavní ústrojí). Další přispívající rizikové faktory mohou být předchozí operace a radioterapie. U pacientů s píštělí se nemá zahájit podávání lenvatinibu, aby nedošlo ke zhoršení stavu, a podávání lenvatinibu se má trvale ukončit u pacientů s ezofageálním nebo tracheobronchiálním postižením a jakoukoliv píštělí 4. stupně (viz bod 4.2); ohledně přerušní léčby a úpravy dávkování při řešení jiných příhod jsou dostupné omezené informace, ale bylo pozorováno zhoršení některých případů a je třeba dbát opatrnosti. Lenvatinib může nepříznivě ovlivnit proces hojení ran jako další terapeutika ze stejné skupiny.
Prodloužení QT intervalu
Prodloužení QT/QTc intervalu bylo u pacientů léčených lenvatinibem hlášeno s vyšší incidencí než u pacientů léčeným placebem (viz bod 4.8, Popis vybraných nežádoucích účinků). U všech pacientů je třeba sledovat elektrokardiogramy, přičemž zvláštní pozornost se má věnovat pacientům s vrozeným syndromem dlouhého QT, s městnavým srdečním selháním, bradyarytmiemi a u pacientů užívajících léčivé přípravky, o nichž je známo, že prodlužují QT interval, včetně antiarytmik třídy Ia a III. Podávání lenvatinibu se má přerušit v případě rozvoje prodloužení QT intervalu na více než 500 ms.
V léčbě lenvatinibem se má pokračovat se sníženou dávkou, když se zmenší prodloužení QTc na < 480 ms nebo na úroveň výchozího stavu.
Elektrolytové poruchy, jako například hypokalémie, hypokalcémie nebo hypomagnezémie, zvyšují riziko prodloužení QT intervalu, a proto je u všech pacientů před zahájením léčby a poté pravidelně v průběhu léčby nutné monitorovat a korigovat abnormální hladiny elektrolytů. V průběhu léčby se má se zvážit pravidelné sledování EKG a elektrolytů (hořčíku, draslíku a vápníku). Hladina vápníku v krvi se má sledovat nejméně jednou měsíčně a vápník se má během léčby lenvatinibem v případě potřeby doplňovat. Podávání lenvatinibu se má přerušit nebo se má upravit dávkování v závislosti na závažnosti, přítomnosti změn na EKG a přetrvávající hypokalcémii.
Porucha suprese tyreostimulačního hormonu / Porucha funkce štítné žlázy Hypotyreóza byla zaznamenána u pacientů léčených lenvatinibem (viz bod 4.8, Popis vybraných nežádoucích účinků). Funkce štítné žlázy se má monitorovat před zahájením léčby lenvatinibem a také pravidelně během této léčby. Hypotyreóza se léčí podle standardní lékařské praxe, aby se zachoval eutyreoidní stav.
Lenvatinib narušuje exogenní supresi štítné žlázy (viz bod 4.8, Popis vybraných nežádoucích účinků). Je třeba pravidelně kontrolovat hladiny tyreostimulačního hormonu (thyroid stimulating hormone -TSH) a upravit podávání tyreoidálních hormonů, aby se dosáhlo přiměřené hladiny TSH podle léčebného cíle pacienta.
U pacientů léčených lenvatinibem byl často hlášen průjem, který se obvykle objevoval v počáteční fázi léčby (viz bod 4.8, Popis vybraných nežádoucích účinků). Aby se předešlo dehydrataci, je třeba okamžitě zahájit léčbu průjmu. Podávání lenvatinibu se musí přerušit v případě přetrvávání průjmu 4. stupně navzdory poskytnutí léčby.
Zvláštní populace
U pacientů jiného než europoidního či asijského etnického původu a u pacientů ve věku >75 let jsou dostupné omezené údaje. U těchto pacientů se má lenvatinib používat s opatrností, vzhledem ke
snížené snášenlivosti lenvatinibu u pacientů asijského etnika a u starších pacientů (viz bod 4.8, Další zvláštní populace).
Neexistují žádné údaje o použití lenvatinibu ihned po sorafenibu nebo jiné protinádorové léčbě a může existovat riziko aditivních toxicit, pokud mezi léčbami není dodrženo přiměřené vymývací období. Minimální vymývací období v klinických studiích bylo 4 týdny.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Účinky jiných léčivých přípravků na lenvatinib Chemoterapeutika
Současné podání lenvatinibu, karboplatiny a paklitaxelu nemělo žádný významný vliv na farmakokinetiku jakékoliv z těchto 3 látek.
Vliv lenvatinibu na jiné léčivé přípravky
Nejsou dostupné žádné údaje, které by bylo možné použít k vyloučení rizika možné indukce CYP3A4 nebo P-gp lenvatinibem v gastrointestinálním traktu. To může vést ke snížené expozici perorálně podávaných substrátů CYP3A4/P-gp. To je nutné vzít v úvahu při souběžném perorálním podávání substrátů CYP3A4/P-gp, u nichž je zachování účinnosti velmi důležité. Substráty CYP3A4, o kterých je známo, že mají úzký terapeutický index (např. astemizol, terfenadin, cisaprid, pimozid, chinidin, bepridil nebo námelové alkaloidy [ergotamin, dihydroergotamin]), je proto u pacientů užívajících lenvatinib nutné podávat s opatrností.
Perorální antikoncepce
V současné době není známo, zda může lenvatinib snížit účinnost hormonální antikoncepce, a proto mají ženy užívající perorální hormonální antikoncepci používat ještě bariérovou ochranu (viz bod 4.6).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku.
Ženy ve fertilním věku se musí během léčby lenvatinibem a ještě minimálně jeden měsíc po ukončení terapie vyvarovat otěhotnění a musí používat vysoce účinnou antikoncepci. V současné době není známo, zda může lenvatinib snížit účinnost hormonální antikoncepce, a proto mají ženy užívající perorální hormonální antikoncepci používat ještě bariérovou ochranu.
Údaje o podávání lenvatinibu těhotným ženám nejsou k dispozici. Lenvatinib byl embryotoxický a teratogenní při podání potkanům a králíkům (viz bod 5.3).
Lenvatinib lze v těhotenství použít pouze tehdy, když je to naprosto nezbytné a až po pečlivém zvážení potřeb matky a rizika pro plod.
Kojení
Není známo, zda se lenvatinib vylučuje do lidského mateřského mléka. Lenvatinib a jeho metabolity se vylučují do mateřského mléka potkanů (viz bod 5.3). Riziko pro novorozence nebo kojence nelze vyloučit, a proto je podávání lenvatinibu během kojení kontraindikováno (viz bod 4.3).
Fertilita
Účinky u člověka nejsou známy. U potkanů, psů a opic však byla pozorována testikulární a ovariální toxicita (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Lenvatinib má kvůli nežádoucím účinkům, jako jsou únava a závratě, malý vliv na schopnost řídit nebo obsluhovat stroje. Pacienti, u kterých se vyskytnou tyto příznaky, by měli při řízení nebo obsluze strojů dbát zvýšené opatrnosti.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nejčastěji hlášenými nežádoucími účinky (vyskytujícími se u > 30 % pacientů) jsou hypertenze (68,6 %), průjem (62,8 %), snížená chuť k jídlu (51,5 %), snížená hmotnost (49,1 %), únava (45,8 %), nauzea (44,5 %), proteinurie (36,9 %), stomatitida (35,8 %), zvracení (34,5 %), dysfonie (34,1 %), bolest hlavy (34,1 %) a syndrom palmoplantární erytrodysestézie (PPE) (32,7 %). Hypertenze a proteinurie se obvykle vyskytují v časných fázích léčby lenvatinibem (viz body 4.4 a 4.8, Popis vybraných nežádoucích účinků). Většina nežádoucích účinků 3. nebo 4. stupně se objevila během prvních 6 měsíců léčby s výjimkou průjmu, který se objevoval v průběhu celé léčby, a váhového úbytku, který měl tendenci v průběhu času narůstat.
Nejdůležitějšími závažnými nežádoucími účinky byly renální porucha a selhání (2,4 %), arteriální tromboembolie (3,9 %), srdeční selhání (0,7 %), krvácení z intrakraniálního nádoru (0,7 %) PRES/RPLS (0,2 %), selhání jater (0,2 %) a arteriální tromboembolie (cévní mozková příhoda [1,1 %], tranzitorní ischemická ataka [0,7 %] a infarkt myokardu [0,9 %]).
U 452 pacientů s DTC refrakterním na léčbu RAI bylo kvůli nežádoucímu účinku u 63,1 % pacientů přistoupeno ke snížení dávky a u 19,5 % pacientů k ukončení léčby. Nežádoucími účinky, které nejčastěji vedly ke snížení dávky (u > 5 % pacientů), byly hypertenze, proteinurie, průjem, únava, PPE, snížená tělesná hmotnost a snížená chuť k jídlu. Nežádoucími účinky, které nejčastěji vedly k ukončení léčby lenvatinibem, byly proteinurie, astenie, hypertenze, cévní mozková příhoda, průjem aplicní embolie.
Souhrn nežádoucích účinků v tabulce
Tabulka 4 uvádí kategorie frekvencí nežádoucích účinků pozorovaných v klinických studiích.
Frekvence jsou definovány takto:
• Velmi časté (> 1/10)
• Časté (> 1/100 až < 1/10)
• Méně časté (> 1/1000 až < 1/100)
• Není známo (z dostupných údajů nelze určit)
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Tabulka 4 N |
ežádoucí účinky hlášené u pacientů v průběhu klinických stut |
ií | ||
|
Třída orgánového systému (Terminologie MedDRA*) |
Velmi časté |
Časté |
Méně časté |
Není známo |
|
Infekce a infestace |
Infekce močových cest |
Perineální absces | ||
|
Poruchy krve a lymfatického systému |
Trombocytopeniea |
Lymfopeniea |
Infarkt sleziny | |
|
Třída orgánového systému (Terminologie MedDRA*) |
Velmi časté |
Časté |
Méně časté |
Není známo |
|
Endokrinní poruchy |
Hypotyreóza Zvýšená hladina tyreostimulačního hormonu v krvi* | |||
|
Poruchy metabolismu a výživy |
Hypokalcémie* Hypokalémie Snížená tělesná hmotnost Snížená chuť k jídlu |
Dehydratace Hypomagnezémieb Hypercholesterolé mie | ||
|
Psychiatrické poruchy |
Insomnie | |||
|
Poruchy nervového systému |
Závratě Bolest hlavy Dysgeuzie |
Cévní mozková příhoda |
Syndrom posteriorní reverzibilní encefalopatie Monoparéza Tranzitorní ischemická ataka | |
|
Srdeční poruchy |
Infarkt myokardu0’* Srdeční selhání Prodloužení QT intervalu na elektrokardiogram u Snížená ejekční frakce | |||
|
Cévní poruchy |
Krvácení4 *’* Hypertenze^* Hypotenze | |||
|
Respirační, hrudní a mediastinální poruchy |
Dysfonie |
Plicní embolie*’ | ||
|
Gastrointestinální poruchy |
Gastrointestinální a břišní bolestf Zvracení Nauzea Zánět v dutině ústníg Bolest v dutině ústníh Zácpa Dyspepsie Sucho v ústech |
Anální píštěl Flatulence |
|
Třída orgánového systému (Terminologie MedDRA*) |
Velmi časté |
Časté |
Méně časté |
Není známo |
|
Poruchy jater a žlučových cest |
Zvýšená aspartátaminotrans feráza* Hypoalbuminemie* Zvýšená alaninaminotransfe ráza* Zvýšená alkalická fosfatáza v krvi Abnormální jaterní funkce Zvýšená gamaglutamyltrans feráza Zvýšený bilirubin v krvi* |
Hepatocelulární poškození a hepatitidai | ||
|
Poruchy kůže a podkožní tkáně |
Syndrom palmoplantární erytrodysestézie Alopecie |
Hyperkeratóza | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolest zad Artralgie Myalgie Bolest v končetině Muskuloskeletální bolest | |||
|
Poruchy ledvin a močových cest |
Proteinurie* |
Případy renálního selháníj ^ Porucha funkce ledvin Zvýšení kreatininu v krvi Zvýšená močovina v krvi | ||
|
Celkové poruchy a reakce v místě aplikace |
Únava Astenie Periferní edém |
Píštěl mimo gastro-intestinální trakú | ||
|
*: Slovník lékařské terminologie pro regu |
ační činnosti (MedDRA) verze 16.1. Preferované termíny | |||
byly přiřazeny do třídy orgánového systému (SOC) nejrelevantnější pro cílový orgán.
^ Zahrnuje případy s fatálními následky.
{: Podrobnější charakteristika viz bod 4.8 Popis vybraných nežádoucích účinků.
Následující pojmy byly zkombinovány:
a: Trombocytopenie zahrnuje trombocytopenii a snížený počet trombocytů. Lymfopenie zahrnuje lymfopenii a snížený počet lymfocytů.
b: Hypomagnezémie zahrnuje hypomagnezémii a snížený hořčík v krvi. Hypercholesterolémie zahrnuje hypercholesterolémii a zvýšený cholesterol v krvi.
c: Infarkt myokardu zahrnuje infarkt myokardu a akutní infarkt myokardu.
d: Krvácení zahrnuje epistaxi, hemoptýzu, hematurii, kontuzi, hematochezii, krvácení z dásně, petechii, plicní krvácení, rektální krvácení, přítomnou krev v moči, hematom, vaginální krvácení, krvácení spojivky, hemoroidální krvácení, intrakraniální nádorové krvácení, krvácení hrtanu, ekchymózu, zvýšenou náchylnost k tvoření modřin, krvácení po výkonu, purpuru, krvácení kůže,
rupturu aneurysmatu, arteriální krvácení, oční krvácení, žaludeční krvácení, hemoragickou gastroduodenitidu, gastrointestinální krvácení, hematemézu, hemoragii, hemoragickou mozkovou příhodu, melenu, metroragii, krvácení nehtového lůžka, pleurální krvácení, postmenopauzální krvácení, hemoragickou proktitidu, renální hematom, krvácení ze sleziny, třískovitou hemoragii, subarachnoidální krvácení, krvácení z trachey a nádorové krvácení. e: Hypertenze zahrnuje hypertenzi, hypertenzní krizi, zvýšený diastolický krevní tlak a zvýšený krevní tlak.
f: Gastrointestinální a břišní bolest zahrnuje břišní diskomfort, bolest břicha, bolest dolní poloviny břicha, bolest horní poloviny břicha, břišní citlivost, epigastrický diskomfort a gastrointestinální bolest.
g: Zánět v dutině ústní zahrnuje aftózní stomatitidu, stomatitidu, glositidu, vřed úst a zánět sliznice. h: Bolest v dutině ústní zahrnuje bolest v dutině ústní, glosodynii a orofaryngeální bolest. i: Hepatocelulární poškození a hepatitida zahrnuje polékové poškození jater, jaterní steatózu a cholestatické poranění jater.
j: Případy renálního selhání zahrnují akutní prerenální selhání, renální selhání, akutní renální selhání a renální tubulární nekrózu.
k: Píštěle mimo gastrointestinální trakt zahrnují případy píštělí, které se vyskytly mimo žaludek
a střevo, jako píštěle tracheální, tracheoezofageální, ezofageální, postihující ženské pohlavní ústrojí a kožní píštěl.
Popis vybraných nežádoucích účinků
Hypertenze (viz bod 4.4)
V pivotní studii SELECT fáze III (viz bod 5.1) byla hypertenze (včetně hypertenze, hypertenzní krize, zvýšeného diastolického krevního tlaku a zvýšeného krevního tlaku) zaznamenána u 72,8 % pacientů léčených lenvatinibem a 16,0 % pacientů ve skupině léčené placebem. Medián doby do nástupu
u pacientů léčených lenvatinibem byl 16 dnů. Reakce 3. nebo vyššího stupně (včetně 1 reakce 4. stupně) se objevily u 44,4 % pacientů léčených lenvatinibem ve srovnání s 3,8 % pacientů léčených placebem. Ve většině případů reakce ustoupily nebo byly vyřešeny po přerušení léčby nebo snížení dávky, ke kterému bylo přistoupeno u 13,0 %, resp. 13,4 % pacientů. U 1,1 % pacientů vedla hypertenze k trvalému ukončení léčby.
Proteinurie (viz bod 4.4)
V pivotní studii SELECT fáze III (viz bod 5.1) byla proteinurie zaznamenána u 33,7 % pacientů léčených lenvatinibem a u 3,1 % pacientů ve skupině léčené placebem. Medián doby do nástupu byl 6,7 týdnů. Reakce 3. stupně se objevily u 10,7 % pacientů léčených lenvatinibem; u pacientů léčených placebem se neobjevily žádné. Ve většině případů reakce ustoupily nebo byly vyřešeny po přerušení léčby nebo snížení dávky, ke kterému bylo přistoupeno u 16,9 %, resp. 10,7 % pacientů. U 0,8 % pacientů vedla proteinurie k trvalému ukončení léčby.
Renální selhání a porucha funkce ledvin (viz bod 4.4)
V pivotní studii SELECT fáze III (viz bod 5.1) se u 5,0 % pacientů rozvinulo renální selhání au 1,9 % se rozvinula porucha funkce ledvin (u 3,1 % pacientů se objevily příhody renálního selhání nebo poruchy funkce ledvin > 3. stupně). Ve skupině užívající placebo se u 0,8 % pacientů rozvinulo renální selhání nebo porucha funkce ledvin (0,8 % bylo > 3. stupně).
Srdeční dysfunkce (viz bod 4.4)
V pivotní studii SELECT fáze III (viz bod 5.1) byl zaznamenán pokles ejekční frakce / srdeční selhání u 6,5 % pacientů (1,5 % bylo > 3. stupně) ve skupině léčené lenvatinibem a 2,3 % ve skupině užívající placbo (žádné nebyly > 3. stupně).
Syndrom posteriorní reverzibilní encefalopatie (PRES) / Syndrom reverzibilní posteriorní leukoencefalopatie (RPLS)
V pivotní studii SELECT fáze III (viz bod 5.1) se ve skupině léčené lenvatinibem objevil 1 případ PRES (2. stupně) a žádný případ nebyl hlášen ve skupině užívající placebo.
Mezi 1 166 pacienty léčenými lenvatinibem byly 4 případy (0,3 %) PRES (0,3 % byly 3. nebo
4. stupně), přičemž po léčbě a/nebo vynechání dávky nebo trvalém ukončení podávání všechny odezněly.
Hepatotoxicita (viz bod 4.4)
V pivotní studii SELECT fáze III (viz bod 5.1) byly nejčastěji hlášenými nežádoucími účinky souvisejícími s játry hypoalbuminémie (9,6 % lenvatinib vs. 1,5 % placebo) a zvýšené jaterní enzymy, včetně zvýšené alaninaminotransferázy (7,7 % lenvatinib vs. 0 placebo), zvýšené aspartátaminotransferázy (6,9 % lenvatinib vs. 1,5 % placebo) a zvýšeného bilirubinu v krvi (1,9 % lenvatinib vs. 0 placebo). Medián doby do nástupu jaterních reakcí u pacientů léčených lenvatinibem byl 12,1 týdne. Reakce související s játry 3. nebo vyššího stupně (včetně 1 případu selhání jater
5. stupně) se objevily u 5,4 % pacientů léčených lenvatinibem ve srovnání s 0,8 % pacientů léčených placebem. Reakce související s játry vedly k přerušení léčby u 4,6 % pacientů, k úpravě dávkování
u 2,7 % pacientů a k trvalému ukončení léčby u 0,4 % pacientů.
Mezi 1 166 pacienty léčenými lenvatinibem byly 3 případy (0,3 %) selhání jater, všechny s fatálními následky. K jednomu případu došlo u pacienta, který neměl jaterní metastázy. Rovněž se objevil případ akutní hepatitidy u pacienta bez jaterních metastáz.
Arteriální tromboembolie (viz bod 4.4)
V pivotní studii SELECT fáze III (viz bod 5.1) byly u 5,4 % pacientů ve skupině léčené lenvatinibem a 2,3 % pacientů ve skupině užívající placebo zaznamenány případy arteriální tromboembolie.
Mezi 1 166 pacienty léčenými lenvatinibem bylo 5 případů (0,4 %) arteriální tromboembolie (3 případy infarktu myokardu a 2 případy cévní mozkové příhody) s fatálními následky.
Krvácení (viz bod 4.4)
V pivotní studii SELECT fáze III (viz bod 5.1) bylo krvácení zaznamenáno u 34,9 % pacientů léčených lenvatinibem (1,9 % bylo > 3. stupně) a 18,3 % pacientů léčených placebem (3,1 % bylo > 3. stupně). Mezi reakce, které se objevily s incidencí > 0,75 % v porovnání s placebem patřily: epistaxe (11,9 %), hematurie (6,5 %), kontuze (4,6 %), krvácení z dásně (2,3 %), hematochezie (2,3 %), rektální krvácení (1,5 %), hematom (1,1 %), hemoroidální krvácení (1,1 %), laryngeální krvácení (1,1 %), petechie (1,1 %) a intrakraniální nádorové krvácení (0,8 %). Během této studie se mezi 16 pacienty, kterým byl podáván lenvatinib a na začátku studie měli metastázy v CNS, vyskytl 1 případ fatálního intrakraniálního krvácení.
Medián doby do prvního nástupu u pacientů léčených lenvatinibem byl 10,1 týdne. Mezi pacienty léčenými lenvatinibem a pacienty léčenými placebem nebyly pozorovány žádné rozdíly v incidenci závažných reakcí (3,4 % vs. 3,8 %), reakcí vedoucích k předčasnému ukončení léčby (1,1 % vs. 1,5 %) nebo reakcí vedoucích k přerušení léčby (3,4 % vs. 3,8 %) nebo ke snížení dávky (0,4 % vs. 0).
Mezi 1 166 pacienty léčenými lenvatinibem bylo u 2 % pacientů zaznamenáno krvácení 3. nebo vyššího stupně, u 3 pacientů (0,3 %) se objevilo krvácení 4. stupně a u 5 pacientů (0,4 %) reakce 5. stupně, včetně arteriálního krvácení, hemoragické cévní mozkové příhody, intrakraniálního nádorového krvácení, hemoptýzy a nádorového krvácení.
Hypokalcémie (viz bod 4.4, Prodloužení QT intervalu)
V pivotní studii SELECT fáze III (viz bod 5.1) byla hypokalcémie zaznamenána u 12,6 % pacientů léčených lenvatinibem; u pacientů ve skupině léčené placebem nebyly zaznamenány žádné případy. Medián doby do prvního nástupu u pacientů léčených lenvatinibem byl 11,1 týdne. Reakce 3. nebo 4. stupně závažnosti se objevily u 5,0 %pacientů léčených lenvatinibem; u pacientů léčených placebem se neobjevily žádné. Většina reakcí vymizela po podpůrné léčbě, aniž by muselo dojít
k přerušení léčby nebo snížení dávky; ke kterému bylo přikročeno u 1,5 %, resp. 1,1 % pacientů; u 1 pacienta s hypokalcémií 4. stupně byla léčba trvale ukončena.
Gastrointestinální perforace a tvorba píštělí (viz bod 4.4)
V pivotní studii SELECT fáze III (viz bod 5.1) byly zaznamenány případy gastrointestinální perforace nebo tvorby píštělí u 1,9 % pacientů ve skupině léčené lenvatinibem a u 0,8 % pacientů ve skupině užívající placebo.
Píštěle mimo gastrointestinální trakt (viz bod 4.4)
Používání lenvatinibu bylo spojeno s případy tvorby píštělí včetně reakcí končících úmrtím. Případy píštělí v jiných částech těla než žaludku a střevě byly pozorovány u různých indikací. Reakce byly zaznamenány v různých fázích během léčby v rozmezí od dvou týdnů do více než 1 roku od zahájení podávání lenvatinibu. Medián latence je přibližně 3 měsíce.
Prodloužení QT intervalu (viz bod 4.4)
V pivotní studii SELECT fáze III (viz bod 5.1) bylo zaznamenáno prodloužení QT/QTc u 8,8 % pacientů ve skupině léčené lenvatinibem a u 1,5 % pacientů ve skupině užívající placebo. Incidence prodloužení QT intervalu na více než 500 ms byla ve skupině léčené lenvatinibem 2 % ve srovnání s žádným případem ve skupině užívající placebo.
Zvýšený tyreostimulační hormon v krvi (viz bod 4.4, Porucha suprese tyreostimulačního hormonu / Porucha funkce štítné žlázy)
V pivotní studii SELECT fáze III (viz bod 5.1) mělo 88 % všech pacientů výchozí hladinu TSH nižší nebo rovnu 0,5 mU/l. U pacientů s normální výchozí hladinou TSH bylo po zahájení léčby pozorováno zvýšení hladiny TSH nad 0,5 mU/l u 57 % pacientů léčených lenvatinibem ve srovnání se 14 % pacientů léčených placebem.
Průjem (viz bod 4.4)
V pivotní studii SELECT fáze III (viz bod 5.1) byl zaznamenán průjem u 67,4 % pacientů ve skupině léčené lenvatinibem (9,2 % bylo > 3. stupně) au 16,8 % pacientů ve skupině užívající palcebo (žádný nebyl > 3. stupně).
Pediatrická populace
U této populace nejsou dosud dostupné žádné klinické údaje (viz bod 4.2).
Další zvláštní populace
Starší pacienti
U pacientů ve věku >75 let byla větší pravděpodobnost výskytu hypertenze 3. nebo 4. stupně, proteinurie, snížené chutě k jídlu a dehydratace.
Pohlaví
U žen se více vyskytovala hypertenze (včetně hypertenze 3. nebo 4. stupně), proteinurie a PPE, zatímco u mužů se častěji vyskytovala snížená ejekční frakce, gastrointestinální perforace a tvorba píštělí.
Etnický původ
Incidence periferního edému, hypertenze, únavy, PPE, proteinurie, trombocytopenie a zvýšeného tyreostimulačního hormonu v krvi byla vyšší u pacientů asijského etnického původu ve srovnání s pacienty europoidního etnika.
Hypertenze ve výchozím stavu
U pacientů s hypertenzí ve výchozím stavu byla incidence hypertenze, proteinurie, průjmu a dehydratace 3. nebo 4. stupně vyšší a tito pacienti zaznamenali více závažných případů dehydratace, hypotenze, plicní embolie, maligní pleurální efuze, atriální fibrilace a gastrointestinálních příznaků (bolest břicha, průjem a zvracení).
Porucha funkce jater
U pacientů s poruchou funkce jater ve výchozím stavu byla vyšší incidence hypertenze a PPE a vyšší incidence hypertenze, astenie, únavy a hypokalcémie 3. nebo 4. stupně ve srovnání s pacienty s normální funkcí jater.
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin ve výchozím stavu byla vyšší incidence hypertenze, proteinurie, únavy, stomatitidy, periferního edému, trombocytopenie, dehydratace, prodlouženého QT intervalu na elektrokardiogramu, hypotyreózy, hyponatremie, zvýšeného tyreostimulačního hormonu v krvi a pneumonie 3. nebo 4. stupně ve srovnání s pacienty s normální funkcí ledvin. U těchto pacientů byla také zaznamenána vyšší incidence renálních reakcí a tendence k vyšší incidenci jaterních reakcí.
Pacienti s tělesnou hmotností < 60 kg
U pacientů s nízkou tělesnou hmotností (< 60 kg) byla zaznamenána vyšší incidence PPE, proteinurie, hypokalcémie a hyponatremie 3. nebo 4. stupně a tendence k vyšší incidenci snížené chuti k jídlu 3. nebo 4. stupně.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nejvyšší dávky lenvatinibu podávaného v klinických studiích byly 32 mg a 40 mg denně.
V klinických studiích se také objevily náhodné chyby v medikaci vedoucí k jednorázovým dávkám 40 až 48 mg. Nejčastěji pozorovanými nežádoucími účinky při těchto dávkách byly hypertenze, nauzea, průjem, únava, stomatitida, proteinurie, bolest hlavy a zhoršení PPE. Byly rovněž hlášeny případy předávkování lenvatinibem zahrnující jednorázová podání 6- až 10násobku doporučené denní dávky. Tyto případy byly spojeny s výskytem nežádoucích reakcí ve shodě se známým bezpečnostním profilem lenvatinibu (tj. renální a srdeční selhání) nebo byly bez nežádoucích reakcí.
Příznaky a léčba
Na předávkování lenvatinibem neexistuje žádné specifické antidotum. V případě podezření na předávkování se má přerušit podávání lenvatinibu a poskytnout vhodná podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatika, inhibitory proteinkinázy, ATC kód: L01XE29
Lenvatinib je multikinázový inhibitor, u kterého byly in vitro a in vivo prokázány zejména antiangiogenní vlastnosti a na modelech in vitro byla také pozorována přímá inhibice růstu nádoru.
Mechanismus účinku
Lenvatinib je inhibitor receptorové tyrosinkinázy (RTK), který selektivně inhibuje kinázovou aktivitu receptorů pro vaskulární endoteliální růstový faktor VEGFR1 (FLT1), VEGFR2 (KDR) a VEGFR3 (FLT4), kromě jiných RTK souvisejících s proangiogenní a onkogenní dráhou, včetně receptorů pro růstový faktor pro fibroblasty FGFR1, 2, 3 a 4, receptoru pro růstový faktor pro krevní destičky PDGFRa, KIT a RET.
Přestože mechanismus účinku pro hypertenzi nebyl studován přímo s lenvatinibem, předpokládá se, že je zprostředkován inhibicí VEGFR2 v endoteliálních buňkách cév. Podobně přestože nebyl přímo studován, se předpokládá, že mechanismus účinku pro proteinurii je zprostředkován regulací VEGFR1 a VEGFR2 v podocytech glomerulu směrem dolů.
Mechanismus účinku pro hypotyreózu není zcela objasněn.
Klinická, účinnost
Diferencovaný karcinom štítné žlázy refrakterní na léčbu radioaktivním jódem Studie SELECT byla multicentrická, randomizovaná, dvojitě zaslepená, placebem kontrolovaná klinická studie, která byla provedena na 392 pacientech s diferencovaným karcinomem štítné žlázy refrakterním na léčbu radioaktivním jódem s nezávisle provedeným, centrálně hodnoceným radiografickým průkazem progrese onemocnění do 12 měsíců (+1 měsíc) před zařazením do studie. Refrakterita na léčbu radioaktivním jódem byla definována jako jedna nebo více měřitelných lézí buď s nedostatečným vychytáváním jódu, nebo s progresí i přes léčbu radioaktivním jódem (RAI), nebo s kumulativní aktivitou RAI > 600 mCi nebo 22 GBq u poslední dávky minimálně 6 měsíců před vstupem do studie. Randomizace byla stratifikována dle geografických oblastí (Evropa, Severní Amerika a ostatní), předchozí cílené léčby VEGF/VEGFR (pacienti nepodstoupili žádnou nebo podstoupili 1 předchozí cílenou léčbu VEGF/VEGFR) a věku (< 65 let nebo > 65 let). Hlavním ukazatelem účinnosti bylo přežití bez progrese onemocnění (PFS) dle hodnocení pomocí zaslepeného nezávislého radiologického posudku s použitím kritérií hodnocení odpovědi u solidních nádorů RECIST (Response Evaluation Criteria in Solid Tumours) 1.1. Sekundárním ukazatelem účinnosti byla celková četnost odpovědí a celkové přežití. Pacienti ve skupině léčené placebem si mohli vybrat léčbu lenvatinibem v době potvrzení progrese onemocnění.
Vybraní pacienti s měřitelným onemocněním dle kritérií RECIST 1.1 byli randomizováni v poměru 2: 1 do skupiny užívající 24 mg lenvatinibu jednou denně (n = 261) nebo do skupiny užívající placebo (n = 131). Vstupní demografické údaje a charakteristiky onemocnění byly vyvážené v obou léčených skupinách. Z 392 randomizovaných pacientů bylo 76,3 % neléčených předchozí cílenou léčbou VEGF/VEGFR, 49,0 % představovalo ženy, 49,7 % byli Evropané a medián věku činil 63 let.
Z histologického hlediska mělo 66,1 % potvrzenou diagnózu papilárního karcinomu štítné žlázy a 33,9 % mělo folikulární karcinom štítné žlázy, který byl ve 14,8 % z Hůrthleho buněk a ve 3,8 % ze světlých buněk. Metastázy byly přítomny u 99 % pacientů: v plicích u 89,3 %, lymfatických uzlinách u 51,5 %, kostech u 38,8 %, játrech u 18,1 %, pohrudnici u 16,3 % a v mozku u 4,1 %. U většiny pacientů odpovídalo skóre ECOG hodnotě 0; u 42,1 % bylo dosaženo hodnoty 1 a u 3,9 % hodnoty větší než 1. Medián kumulativní aktivity RAI podávaného před vstupem do studie byl 350 mCi (12,95 GBq).
Statisticky významné prodloužení PFS bylo prokázáno u pacientů léčených lenvatinibem v porovnání s pacienty užívajícími placebo (p < 0,0001) (viz obrázek 1). Pozitivní účinek na PFS byl pozorován ve všech podskupinách dle věku (nad nebo pod 65 let), pohlaví, rasy, histologického subtypu, geografické oblasti a u pacientů, kteří nepodstoupili žádnou nebo podstoupili 1 předchozí cílenou léčbu VEGF/VEGFR. Po nezávislém potvrzení progrese onemocnění přešlo v době primární analýzy účinnosti 109 (83,2 %) pacientů randomizovaných do skupiny užívající placebo do skupiny užívající lenvatinib v otevřeném uspořádání.
Četnost objektivních odpovědí (úplná odpověď [CR] plus částečná odpověď [PR]) dle nezávislého radiologického hodnocení byla významně (p < 0,0001) vyšší u skupiny léčené lenvatinibem (64,8 %) v porovnání se skupinou léčenou placebem (1,5 %). Čtyři (1,5 %) pacienti léčení lenvatinibem dosáhli CR a 165 pacientů (63,2 %) dosáhlo PR, zatímco žádní pacienti léčení placebem nedosáhli CR a 2 (1,5 %) pacienti dosáhli PR.
Medián doby do prvního snížení dávky činil 2,8 měsíců. Medián doby do objektivní odpovědi činil 2,0 (95% CI: 1,9; 3,5) měsíce; avšak z pacientů, kteří dosáhli úplné nebo částečné odpovědi na lenvatinib, bylo u 70,4 % pozorováno dosažení odpovědi 30. dne nebo do 30 dnů po zahájení 24mg dávky.
Analýza celkového přežití byla negována skutečností, že pacienti léčení placebem s potvrzenou progresí onemocnění mohli přejít do skupiny užívající lenvatinib v otevřeném uspořádání. V celkovém přežití nebyl mezi léčenými skupinami v době primární analýzy účinnosti žádný statisticky významný rozdíl (HR= 0,73; 95% CI: 0,50; 1,07; p = 0,1032). Medián celkového přežití nebyl dosažen skupinou užívající lenvatinib, ani skupinou přecházející z placeba.
Tabulka 5 Výsledky hodnocení účinnosti
|
Lenvatinib N = 261 |
Placebo N = 131 | |
|
Přežití bez progrese onemocnění (PFS)a | ||
|
Počet případů progrese nebo úmrtí (%) |
107 (41,0) |
113 (86,3) |
|
Medián PFS v měsících (95% CI) |
18,3 (15,1; NE) |
3,6 (2,2; 3,7) |
|
Poměr rizik (99% CI)b,c |
0,21 (0,14; 0,31) | |
|
Hodnota pb |
<0,0001 | |
|
Pacienti, kteří nepodstoupili předchozí cílenou léčbu VEGF/VEGFR (%) |
195 (74,7) |
104 (79,4) |
|
Počet případů progrese nebo úmrtí |
76 |
88 |
|
Medián PFS v měsících (95% CI) |
18,7 (16,4; NE) |
3,6 (2,1; 5,3) |
|
Poměr rizik (95% CI)b,c |
0,20 (0,14; 0,27) | |
|
Pacienti, kteří podstoupili 1 předchozí cílenou léčbu VEGF/VEGFR (%) |
66 (25,3) |
27 (20,6) |
|
Počet případů progrese nebo úmrtí |
31 |
25 |
|
Medián PFS v měsících (95% CI) |
15,1 (8,8; NE) |
3,6 (1,9; 3,7) |
|
Poměr rizik (95% CI)b,c |
0,22 (0,12; 0,41) | |
|
Četnost objektivních odpovědi | ||
|
Počet objektivních pacientů s odpovědí na léčbu (%) |
169 (64,8) |
2 (1,5) |
|
(95% CI) |
(59,0; 70,5) |
(0,0; 3,6) |
|
Hodnota pb |
<0,0001 | |
|
Počet úplných odpovědí |
4 |
0 |
|
Počet částečných odpovědí |
165 |
2 |
|
Medián doby do objektivní odpovědi,d měsíce (95% CI) |
2,0 (1,9; 3,5) |
5,6 (1,8; 9,4) |
|
Medián doby trvání odpovědi,d měsíce (95% CI) |
NE (16,8; NE) |
NE (NE; NE) |
|
Celkové přežití | ||
|
Počet případů úmrtí (%) |
71 (27,2) |
47 (35,9) |
|
Medián celkového přežití, v měsících (95% CI) |
NE (22,0; NE) |
NE (20,3; NE) |
|
Poměr rizik (95% CI)b,e |
0,73 (0,50; 1,07) | |
|
Hodnota pb’e |
0,1032 | |
CI, interval spolehlivosti; NE, neodhadnutelné; OS, celkové přežití, PFS, přežití bez progrese onemocnění; RPSFT, strukturální model doby selhání se zachováním stupně; VEGF/VEGFR, vaskulární endoteliální růstový faktor / receptor pro vaskulární endoteliální růstový faktor. a: Nezávislý radiologický posudek.
b: Stratifikováno dle regionů (Evropa vs. Severní Amerika vs. ostatní), věkových skupin (< 65 let
vs. > 65 let) a předchozí cílené léčby VEGF/VEGFR (0 vs. 1) c: Vypočteno pomocí Coxova modelu proporcionálních rizik.
d: Vypočteno pomocí Kaplan-Meierovy metody; 95% CI bylo sestaveno pomocí zobecněné
Brookmeyerovy a Crowleyho metody u pacientů s nejlepším celkovým počtem úplných odpovědí nebo částečných odpovědí. e: Bez úpravy vzhledem k vlivu změny skupiny.
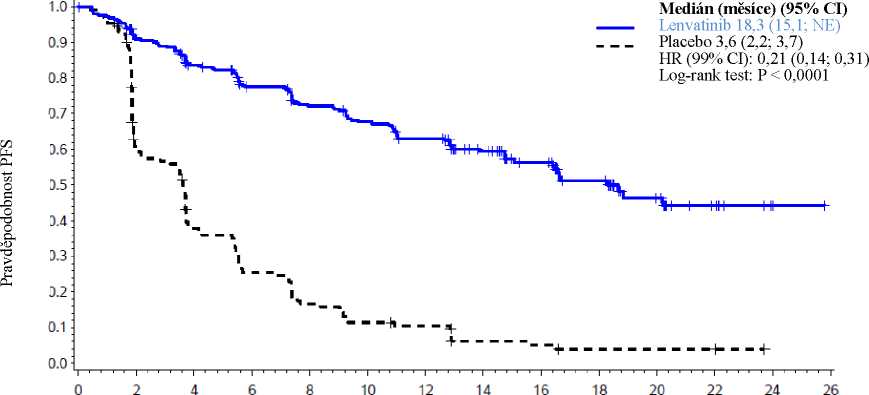
Počet pacientů v riziku: Čas (měsíce)
Lerwatinib 261 225 198 176 159 148 136 92 66 44 24 11 3 0
Placebo 131 71 43 29 19 13 11 5 4 2 2 2 0 0
CI, interval spolehlivosti; NE, neodhadnutelné.
Prodloužení QT intervalu
Dle výsledků z podrobné studie intervalu QT u zdravých dobrovolníků jedna 32mg dávka lenvatinibu neprodloužila QT/QTc interval; nicméně u pacientů léčených lenvatinibem byla zaznamenána vyšší incidence prodloužení QT/QTc intervalu než u pacientů léčených placebem (viz body 4.4 a 4.8).
Pediatrická populace
Evropská agentura pro léčivé přípravky (EMA) udělila odklad povinnosti předložit výsledky studií s lenvatinibem u jedné nebo více podskupin pediatrické populace v léčbě diferencovaného karcinomu štítné žlázy refrakterního na léčbu radioaktivním jódem.
5.2 Farmakokinetické vlastnosti
Farmakokinetické parametry lenvatinibu byly hodnoceny u zdravých dospělých subjektů, dospělých subjektů s poruchou jater, renální poruchou a solidními nádory.
Absorpce
Lenvatinib je rychle absorbován po perorálním podání s tmax zpravidla pozorovaným od 1 do 4 hodin po podání dávky. Jídlo nemá vliv na rozsah absorpce, zpomaluje ale rychlost absorpce. Maximální plazmatická koncentrace při podání zdravým dobrovolníkům spolu s jídlem je opožděna o 2 hodiny. Absolutní biologická dostupnost nebyla u člověka stanovena; údaje ze studie hmotnostní bilance však naznačují, že je v řádu 85 %. Lenvatinib vykazoval dobrou biologickou dostupnost po perorálním podání u psů (70,4 %) a opic (78,4 %).
Distribuce
Vazba lenvatinibu in vitro na lidské plazmatické proteiny je vysoká a pohybuje se od 98 % do 99 % (0,3-30 pg/ml, mesylát). Tato vazba byla hlavně na albumin s menší vazbou na a1-kyselý glykoprotein a y-globulin.
In vitro se poměr koncentrací lenvatinibu v plazmě a v krvi pohyboval od 0,598 do 0,608 (0,110 pg/ml, mesylát).
Lenvatinib je substrátem P-glykoproteinu a BCRP. Lenvatinib není substrátem OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2 nebo BSEP.
U pacientů se medián hodnoty zdánlivého distribučního objemu (Vz/F) první dávky pohyboval v rozmezí 50,5 l až 92l a byl obecně konzistentní ve všech skupinách s dávkami od 3,2 mg do 32 mg. Analogický zdánlivý distribuční objem v ustáleném stavu (Vz/Fss) byl rovněž obecně konzistentní a pohyboval se od 43,2l do 1211.
Biotransformace
Bylo prokázáno, že cytochrom P450 3A4 je in vitro převládající (> 80 %) izoformou podílející se na metabolismu lenvatinibu zprostředkovaném P450. Údaje in vivo však naznačují, že významný podíl celkové metabolizace lenvatinibu je zprostředkován jinou cestou než P-450. Z toho vyplývá, že induktory a inhibitory CYP3A4 in vivo měly minimální vliv na expozici lenvatinibu (viz bod 4.5).
V lidských jaterních mikrosomech byla demethylovaná forma lenvatinibu (M2) identifikována jako hlavní metabolit. M2’ a M3’, hlavní metabolity v lidské stolici, vznikly za přispění aldehyd-oxidázy z M2, resp. z lenvatinibu.
Ve vzorcích plazmy odebraných do 24 hodin po podání tvořil lenvatinib 97 % radioaktivity v radiochromatogramech plazmy, zatímco metabolit M2 tvořil dalších 2,5 %. Podle AUC(0 -inf) tvořil lenvatinib 60 % celkové radioaktivity v plazmě a 64 % v krvi.
Údaje ze studie hmotnostní bilance / studie vylučování naznačují, že se lenvatinib u člověka v rozsáhlé míře metabolizuje. Hlavní metabolické cesty u člověka byly identifikovány jako oxidace aldehyd-oxidázou, demethylace cestou CYP3A4, glutathionový konjugát s eliminací O -arylové skupiny (chlorbenzylová složka) a kombinace těchto cest následované dalšími biotransformacemi (např. glukuronidace, hydroláza glutathionové složky, degradace cysteinové složky a intramolekulární přeskupení cysteinylglycinových a cysteinových konjugátů s následnou dimerizací). Tyto in vivo metabolické cesty jsou v souladu s údaji získanými v in vitro studiích s využitím lidských biomateriálů.
In vitro studie transportérů
U následujících transportérů byla vyloučena klinicky relevantní inhibice na základě vyřazení hodnot
IC50 > 50 X Cmax,unbound.
Lenvatinib prokázal minimální nebo neprokázal žádnou inhibiční aktivitu vůči transportní činnosti zprostředkované P-gp a BCRP. Podobně nebyla pozorována žádná indukce exprese P-gp mRNA.
Lenvatinib prokázal minimální nebo neprokázal žádný inhibiční účinek na OATP1B3. U lidského jaterního cytosolu lenvatinib neinhiboval aktivitu aldehyd-oxidázy.
Eliminace
Koncentrace v plazmě klesají biexponenciálně po dosažení Cmax. Střední terminální exponenciální poločas lenvatinibu činí přibližně 28 hodin.
Po podání radioaktivně značeného lenvatinibu 6 pacientům se solidními nádory byly přibližně dvě třetiny radioaktivně značeného lenvatinibu eliminovány ve stolici a jedna čtvrtina v moči. Převládajícím analytem v exkrementech byl metabolit M3 (~ 17 % dávky), následovaný metabolitem M2’ (~ 11 % dávky) a metabolitem M2 (~ 4,4 % dávky).
Linearita/nelinearita
Závislost na dávce a akumulace
U pacientů se solidními nádory, kterým byla podána jednorázová dávka a opakované dávky lenvatinibu jednou denně, se expozice lenvatinibu (Cmax a AUC) zvýšila přímo úměrně k podané dávce v rozmezí 3,2 až 32 mg jednou denně.
Lenvatinib vykazuje minimální akumulaci v ustáleném stavu. V tomto rozmezí se medián indexu kumulace (Rac) pohyboval od 0,96 (20 mg) do 1,54 (6,4 mg).
Zvláštní populace
Porucha funkce jater
Farmakokinetika lenvatinibu po jednorázové 10mg dávce byla hodnocena u 6 subjektů s mírnou až středně těžkou poruchou funkce jater (Child-Pugh skóre A, resp. Child-Pugh skóre B). 5mg dávka byla hodnocena u 6 subjektů s těžkou poruchou jater (Child-Pugh skóre C). Osm zdravých subjektů se shodnými demografickými parametry sloužilo jako kontrolní skupina a užívaly 10mg dávku. Medián poločasu byl srovnatelný u subjektů s mírnou, středně těžkou a těžkou poruchou jater, jakož i u pacientů s normální funkcí jater a pohyboval se v rozmezí od 26 hodin do 31 hodin. Procento dávky lenvatinibu vyloučeného močí bylo nízké ve všech kohortách (< 2,16 % ve všech léčených kohortách).
Expozice lenvatinibu, podle hodnot AUC0-t a AUC0-inf upravených dle dávky, byla 119 % normálu u pacientů s mírnou poruchou jater, 107 % normálu u pacientů se středně těžkou poruchou a 180 % normálu u pacientů s těžkou poruchou funkce jater. Není známo, zda u subjektů s poruchou funkce jater dochází ke změně ve vazbě na plazmatické bílkoviny. Doporučené dávkování viz bod 4.2
Porucha funkce ledvin
Farmakokinetika lenvatinibu po jednorázové 24mg dávce byla hodnocena u 6 subjektů s mírnou, středně těžkou a těžkou renální poruchou v porovnání s 8 zdravými subjekty se shodnými demografickými parametry. Subjekty s konečným stadiem renálního onemocnění nebyly hodnoceny.
Expozice lenvatinibu podle hodnot AUC0_inf byla u subjektů s mírnou poruchou ledvin 101 %, u pacientů se středně těžkou poruchou ledvin 90 % au pacientů s těžkou poruchou funkce ledvin 122 %. Není známo, zda u subjektů s renální poruchou dochází ke změně ve vazbě na plazmatické bílkoviny. Doporučené dávkování viz bod 4.2.
Věk, pohlaví, hmotnost, rasa
Podle farmakokinetické populační analýzy dat pacientů užívajících 24 mg lenvatinibu jednou denně neměl věk, pohlaví, hmotnost a rasa (japonská vs. ostatní, europoidní vs. ostatní) žádný významný vliv na clearance (viz bod 4.2).
Pediatrická populace
U pediatrických pacientů nebyly provedeny studie.
5.3 Předklinické údaje vztahující se k bezpečnosti
Ve studiích toxicity po opakovaných dávkách (až do 39 týdnů) způsobil lenvatinib toxikologické změny v různých orgánech a tkáních ve vztahu k očekávanému farmakologickému účinku lenvatinibu včetně glomerulopatie, testikulární hypocelularity, atrezie ovariálních folikulů, gastrointestinálních změn, změn kostí, změn nadledvin (potkani a psi) a arteriálních (arteriální fibrinoidní nekróza, mediální degenerace nebo krvácení) lézí u potkanů, psů a opic rodu cynomolgus. U potkanů, psů a opic byly rovněž pozorovány zvýšené hladiny transaminázy spojené se známkami hepatotoxicity.
U všech druhů zvířat byla na konci 4týdenního období rekonvalescence pozorována reverzibilita toxikologických změn.
Genotoxicita
Lenvatinib nebyl genotoxický.
U lenvatinibu nebyly provedeny studie kancerogenity.
Reprodukční a vývojová toxicita
Nebyly provedeny žádné specifické studie na zvířatech pro zjištění vlivu lenvatinibu na fertilitu. Ve studiích toxicity po opakovaných dávkách u zvířat byly ale pozorovány testikulární (hypocelularita semenotvorného epitelu) a ovariální změny (atrezie folikulů) při expozicích 11 až 15násobku (potkan) nebo 0,6- až 7násobku (opice) očekávané klinické expozice (podle AUC) při maximální tolerované dávce pro člověka. Tyto nálezy byly reverzibilní na konci 4týdenního období rekonvalescence.
Podání lenvatinibu v období organogeneze vedlo k embryoletalitě a teratogenitě u potkanů (externí a skeletální anomálie plodu) při expozicích pod klinickou expozicí (podle AUC) při maximální tolerované dávce pro člověka a u králíků (externí, viscerální nebo skeletální anomálie plodu) podle plochy povrchu těla; mg/m2 při maximální tolerované dávce pro člověka. Tyto nálezy naznačují, že lenvatinib má teratogenní potenciál, pravděpodobně související s farmakologickou antiangiogenní aktivitou lenvatinibu.
Lenvatinib a jeho metabolity se vylučují do mateřského mléka potkanů.
Studie toxicity na nedospělých zvířatech
Příčinou mortality byla dávku limitující toxicita u nedospělých potkanů, u nichž bylo dávkování zahájeno postnatálním dnem (PND) 7 nebo PND 21. Byla pozorována při expozicích, které byly 125-, resp. 12krát nižší v porovnání s expozicí, při níž byla pozorována mortalita u dospělých potkanů, což naznačuje zvýšenou citlivost k toxicitě s klesajícím věkem. Proto lze mortalitu přisoudit komplikacím spojeným s primárními duodenálními lézemi s možným přispěním aditivních toxicit u nezralých cílových orgánů.
Toxicita lenvatinibu byla výraznější u mladších potkanů (dávkování zahájeno v PND 7) v porovnání s potkany, u nichž bylo dávkování zahájeno v PND 21 a mortalita a některé toxicity byly pozorovány dříve u nedospělých potkanů při dávce 10 mg/kg v porovnání s dospělými potkany, kterým byla podávána stejná úroveň dávky. U nedospělých potkanů byly rovněž pozorovány retardace růstu, sekundární opoždění fyzického vývoje a léze, které lze přisoudit farmakologickým účinkům (řezáky, femur [epifyzární růstová ploténka], ledviny, nadledviny aduodenum).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky Uhličitan vápenatý Mannitol
Mikrokrystalická celulosa Hyprolosa
Částečně substituovaná hyprolosa Mastek
Obal tobolky Hypromelosa Oxid titaničitý (E171)
Žlutý oxid železitý (E172)
Červený oxid železitý (E172)
Barva na potisk Šelak
Černý oxid železitý (E172)
Hydroxid draselný Propylenglykol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 25 °C. Uchovávejte v původním blistru, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Polyamid/Al/PVC/Al blistry obsahující 10 tobolek. Jedna krabička obsahuje 30 tobolek.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Pečující osoby nesmí tobolky otevírat, aby se zabránilo jejich opakované expozici obsahu tobolky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Eisai Europe Ltd.
European Knowledge Centre
Mosquito Way
Hatfield
Herts AL10 9SN Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1002/001
EU/1/15/1002/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 28. května 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží Eisai Manufacturing Ltd
European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace ave veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
LENVIMA 4 mg tvrdé tobolky lenvatinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje lenvatinibum 4 mg (ve formě lenvatinibi mesilas).
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
30 tvrdých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 25 °C. Uchovávejte v původním blistru, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Eisai Europe Ltd. Mosquito Way Hatfield
Herts AL10 9SN Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1002/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
LENVIMA 4 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
LENVIMA 4 mg tvrdé tobolky lenvatinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Eisai Europe Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
LENVIMA 10 mg tvrdé tobolky lenvatinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje lenvatinibum 10 mg (ve formě lenvatinibi mesilas).
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
30 tvrdých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 25 °C. Uchovávejte v původním blistru, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Eisai Europe Ltd. Mosquito Way Hatfield
Herts AL10 9SN Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1002/002
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
LENVIMA 10 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
LENVIMA 10 mg tvrdé tobolky lenvatinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Eisai Europe Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
LENVIMA 4 mg tvrdé tobolky LENVIMA 10 mg tvrdé tobolky
lenvatinibum
'V Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek LENVIMA a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek LENVIMA užívat
3. Jak se přípravek LENVIMA užívá
4. Možné nežádoucí účinky
5. Jak přípravek LENVIMA uchovávat
6. Obsah balení a další informace
1. Co je přípravek LENVIMA a k čemu se používá Co je přípravek LENVIMA
LENVIMA je léčivý přípravek obsahující léčivou látku lenvatinib. Používá se k léčbě progresivního nebo pokročilého nádorového onemocnění štítné žlázy u dospělých, u nichž se nepodařilo onemocnění zastavit léčbou radioaktivním jódem.
Jak přípravek LENVIMA působí
Přípravek LENVIMA blokuje působení bílkovin, které se označují jako receptorové tyrozinkinázy (RTK). Ty se účastní vývoje nových krevních cév, které dodávají kyslík a živiny do buněk a pomáhají jim růst. Tyto bílkoviny mohou být přítomné ve velkém množství v nádorových buňkách a zablokováním jejich působení může přípravek LENVIMA zpomalit dělení nádorových buněk a rychlost růstu nádoru a může pomoci zastavit zásobování nádorových buněk krví, kterou tyto buňky potřebují.
2. Čemu musíte věnovat pozornost, než začnete přípravek LENVIMA užívat Neužívejte přípravek LENVIMA:
• jestliže jste alergický(á) na lenvatinib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6);
• jestliže kojíte (viz níže uvedený bod Antikoncepce, těhotenství a kojení).
Upozornění a opatření
Před užitím přípravku LENVIMA se poraďte se svým lékařem, jestliže:
• máte vysoký krevní tlak;
• jste žena v plodném věku (viz níže uvedený bod Antikoncepce, těhotenství a kojení);
• měl(a) jste v minulosti srdeční potíže nebo cévní mozkovou příhodu;
• máte problémy s játry nebo ledvinami;
• jste v nedávné době prodělal(a) operaci nebo léčbu ozařováním;
• je vám více než 75 let;
• patříte do jiné než bílé nebo asijské etnické skupiny;
• vážíte méně než 60 kg;
• máte v anamnéze abnormální kanálky (známé jako píštěle) spojující různé orgány v těle nebo orgán a kůži.
Před užitím přípravku LENVIMA Vám lékař může provést vyšetření krve, například zkontroluje krevní tlak a funkci jater a ledvin, zjistí, zda nemáte nízkou hladinu soli a vysokou hladinu hormonu stimulujícího štítnou žlázu. Váš lékař s Vámi probere výsledky těchto vyšetření a rozhodne, zda Vám může být přípravek LENVIMA podáván. Můžete potřebovat další léčbu jinými léky, užívat nižší dávku přípravku LENVIMA nebo dostávat speciální péči v důsledku zvýšeného rizika nežádoucích účinků.
Pokud si nejste jistý(á), informujte před užitím přípravku LENVIMA svého lékaře.
Děti a dospívající
Přípravek LENVIMA se nedoporučuje podávat dětem a dospívajícím. Účinky přípravku LENVIMA u osob mladších 18 let nejsou známy.
Další léčivé přípravky a přípravek LENVIMA
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. To se vztahuje i na rostlinné přípravky a volně prodejné léčivé přípravky.
Antikoncepce, těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
• Jestliže můžete otěhotnět, používejte po dobu léčby tímto přípravkem a nejméně jeden měsíc po jejím ukončení vysoce účinnou antikoncepci. Není známo, zda může přípravek LENVIMA snížit účinek antikoncepčních pilulek podávaných ústy. Pokud je toto Vaše běžná metoda antikoncepce a v průběhu léčby přípravkem LEVIMA jste sexuálně aktivní, je nutné přidat také bariérovou metodu, jako je pesar nebo kondom.
• Neužívejte přípravek LENVIMA, pokud v době léčby plánujete otěhotnět. Mohlo by dojít k závažnému ohrožení Vašeho dítěte.
• Jestliže otěhotníte v průběhu léčby přípravkem LENVIMA, ihned to sdělte svému lékaři. Lékař Vám pomůže rozhodnout, zda máte v léčbě pokračovat.
• Během užívání přípravku LENVIMA nekojte. Mohlo by dojít k závažnému ohrožení Vašeho kojeného dítěte, protože se přípravek vylučuje do mateřského mléka.
Řízení dopravních prostředků a obsluha strojů
Přípravek LENVIMA může způsobovat nežádoucí účinky, které mohou ovlivnit Vaši schopnost řídit nebo obsluhovat stroje. Neřiďte ani neobsluhujte stroje, pokud pociťujete závratě nebo únavu.
3. Jak se přípravek LENVIMA užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Kolik přípravku se užívá
• Doporučená dávka přípravku LENVIMA je obvykle 24 mg jednou denně (2 tobolky 10 mg a 1 tobolka 4 mg).
• Máte-li závažné potíže s játry nebo ledvinami, je doporučená dávka 14 mg jednou denně
(1 tobolka 10 mg a 1 tobolka 4 mg).
V případě problémů s nežádoucími účinky Vám může lékař dávku snížit.
Užívání tohoto přípravku
• Tobolky lze užívat s jídlem nebo bez jídla.
• Tobolky polykejte celé a zapijte vodou.
• Užívejte tobolky každý den přibližně ve stejnou denní dobu.
• Pečující osoby nesmí otvírat tobolky, aby sevyhnulyexpozici obsahu tobolky.
Jak dlouho se přípravek LENVIMA užívá
Obvykle budete tento přípravek užívat do té doby, dokud pro Vás bude léčba přínosná.
Jestliže jste užil(a) více přípravku LENVIMA, než jste měl(a)
Jestliže jste užil(a) více přípravku LENVIMA, než jste měl(a), sdělte to ihned svému lékaři nebo lékárníkovi. Vezměte si s sebou balení tohoto léku.
Jestliže jste zapomněl(a) užít přípravek LENVIMA
Nezdvojnásobujte následující dávku (neužívejte dvě dávky najednou), abyste nahradil(a) vynechanou dávku.
Co máte dělat, jestliže jste zapomněl(a) užít svou dávku, závisí na tom, za jak dlouho máte užít další dávku.
• Jestliže máte další dávku užít za 12 nebo více hodin, užijte vynechanou dávku ihned, jak si vzpomenete. Pak pokračujte další dávkou v obvyklou dobu podání.
• Jestliže máte další dávku užít za méně než 12 hodin, vynechanou dávku neužívejte. Pak pokračujte další dávkou v obvyklou dobu.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Při užívání tohoto přípravku se mohou vyskytnout níže uvedené nežádoucí účinky.
Ihned informujte svého lékaře, pokud se u Vás vyskytne kterýkoli z následujících nežádoucích účinků - můžete potřebovat naléhavou lékařskou péči:
• pocit snížené citlivosti nebo slabosti na jedné straně těla, těžká bolest hlavy, záchvat, zmatenost, potíže s mluvením, změny vidění nebo pocit závratí - to mohou být známky cévní mozkové příhody, krvácení do mozku nebo vlivu závažného zvýšení krevního tlaku na mozek;
• bolest nebo tlak na hrudi, bolest paží, zad, krku nebo čelisti, dušnost, rychlá nebo nepravidelná srdeční frekvence, kašlání, modré zbarvení rtů nebo prstů na rukou, pocit silné únavy - to mohou být známky srdečních potíží nebo krevní sraženiny v plicích;
• silná bolest v břiše - důvodem může být proděravění střevní stěny nebo píštěl (otvor ve střevech, který je trubicovým vývodem propojen s další částí Vašeho těla nebo kůží);
• černá, dehtovitá nebo krvavá stolice nebo vykašlávání krve - to mohou být známky vnitřního krvácení v těle;
• průjem, pocit nevolnosti nebo zvracení - to jsou velmi časté nežádoucí účinky, které mohou být závažné, pokud v jejich důsledku dojde k dehydrataci (nedostatku vody v organismu)
a následnému selhání ledvin. Lékař Vám může poskytnout léky, které tyto nežádoucí účinky omezují.
Ihned informujte svého lékaře, pokud se u Vás vyskytne kterýkoli z výše uvedených nežádoucích účinků.
Mezi další nežádoucí účinky patří:
Velmi časté (mohou postihnout více než 1 osobu z 10)
• vysoký nebo nízký krevní tlak
• ztráta chuti k jídlu nebo váhový úbytek
• pocit nevolnosti nebo zvracení, zácpa, průjem, bolest břicha, porucha trávení
• pocit silné únavy nebo slabosti
• chraptivý hlas
• otok nohou
• vyrážka
• suchá, bolestivá nebo zanícená ústa, pocit zvláštní chuti v ústech
• bolest kloubů nebo svalů
• pocit závratí
• vypadávání vlasů
• krvácení (nejčastěji krvácení z nosu, ale také další typy krvácení, např. krev v moči, podlitiny, krvácení z dásní nebo ze stěny střeva)
• obtíže se spánkem
• změny hladin bílkovin (vysoké) v testech z moči a močové infekce (zvýšená četnost močení a bolestivé močení)
• bolest hlavy a zad
• zarudnutí, bolest a otok kůže na rukou a nohou (syndrom ruka-noha)
• změny výsledků krevních testů draslíku (nízké) a hladiny vápníků (nízké)
• nízké hladiny krevních destiček v krvi, což může vést k tvorbě modřin a obtížnému hojení ran
Časté (mohou postihnout až 1 osobu z 10)
• ztráta tělesných tekutin (dehydratace)
• bušení srdce
• suchá kůže, ztluštění a svědění kůže
• pocit nadýmání nebo přítomnost plynu ve střevě (plynatost)
• nedostatečná činnost štítné žlázy (únava, zvýšení tělesné hmotnosti, zácpa, pocit chladu, suchá kůže)
• srdeční potíže nebo krevní sraženiny v plicích (obtížné dýchání, bolest na hrudi) nebo v jiných orgánech
• pocit nemoci
• cévní mozková příhoda
• řitní píštěl (kanálek, který se vytvoří mezi řití a okolní kůží)
• změny výsledků krevních testů jater, ledvin, bílých krvinek (nízké), sodíku a hořčíku v krvi (nízké), cholesterolu (vysoké) a hormonu stimulujícího štítnou žlázu (vysoké)
• změny výsledků krevních testů funkce ledvin a selhání ledvin
Méně časté (mohou postihnout až 1 osobu ze 100)
• bolestivá infekce nebo podráždění v okolí konečníku
• malá cévní mozková příhoda
• poškození jater
• závažná bolest v levé horní části břicha, která může být spojená s horečkou, zimnicí, pocitem na zvracení a zvracením
Není známo (následující nežádoucí účinky byly zaznamenány po uvedení přípravku LENVIMA na trh, ale frekvence jejich výskytu není známa)
• další typy píštělí (abnormální spojení mezi různými orgány v těle nebo mezi orgánem a kůží nebo strukturou ležící pod ní, např. jícnem nebo průdušnicí). Příznaky závisí na místě, kde se píštěl nachází. Informujte svého lékaře, pokud pocítíte jakékoliv nové nebo nezvyklé příznaky, např. kašel při polykání.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek LENVIMA uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a každém blistru za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Neuchovávejte při teplotě nad 25 °C. Uchovávejte v původním blistru, aby byl přípravek chráněn před vlhkostí.
• Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek LENVIMA obsahuje
• Léčivou látkou je lenvatinibum.
- LENVIMA 4 mg tvrdá tobolka: Jedna tvrdá tobolka obsahuje lenvatinibum 4 mg (ve formě lenvatinibi mesilas).
- LENVIMA 10 mg tvrdá tobolka: Jedna tvrdá tobolka obsahuje lenvatinibum 10 mg (ve formě lenvatinibi mesilas).
• Dalšími složkami jsou uhličitan vápenatý, mannitol, mikrokrystalická celulosa, hyprolosa, částečně substituovaná hyprolosa, mastek. Obal tobolky obsahuje hypromelosu, oxid titaničitý (E171), žlutý oxid železitý (E172), červený oxid železitý (E172). Inkoust na potisk obsahuje šelak, černý oxid železitý (E172), hydroxid draselný, propylenglykol.
Jak přípravek LENVIMA vypadá a co obsahuje toto balení
• 4mg tobolka má žluto-červené tělo a žluto-červené víčko, přibližná délka je 14,3 mm, černým inkoustem je vytištěný symbol „€“ na víčku a „LENV 4 mg“ na těle.
• 10mg tobolka má žluté tělo a žluto-červené víčko, přibližná délka je 14,3 mm, černým inkoustem je vytištěný symbol „€“ na víčku a „LENV 10 mg“ na těle.
• Tobolky jsou baleny v polyamid/Al/PVC blistrech s protlačovací hliníkovou fólií v krabičkách obsahujících 30 tobolek.
Držitel rozhodnutí o registraci
Eisai Europe Limited, European Knowledge Centre, Mosquito Way, Hatfield, Herts AL10 9SN,
Velká Británie
E-mail: EUmedinfo@eisai.net
Výrobce
Eisai Manufacturing Ltd, Mosquito Way, Hatfield, Herts AL10 9SN, Velká Británie.
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Eisai SA/NV Eisai Europe Ltd.
Tél/Tel: + 32 (0) 2 502 58 04 Tel. + 44 (0) 208 600 1400
(Jungtiné Karalysté)
|
Btarapna Eisai Europe Ltd. Ten.: +44 (0)20 8600 1400 (OóegmeHO Kpancrao) |
Luxembourg/Luxemburg Eisai SA/NV Tél/Tel: + 32 (0) 2 502 58 04 (Belgique/Belgien) |
|
Česká republika Eisai GesmbH organizační složka Tel.: + 420 242 485 839 |
Magyarország Eisai Europe Ltd. Tel.: + 44 (0) 20 8600 1400 (Egyesult Királyság) |
|
Danmark Eisai AB Tlf: + 46 (0) 8 501 01 600 (Sverige) |
Malta Eisai Europe Ltd. Tel: +44 (0) 20 8600 1400 (United Kingdom) |
|
Deutschland Eisai GmbH Tel: + 49 (0) 69 66 58 50 |
Nederland Eisai B.V. Tél/Tel: + 31 (0) 900 575 3340 |
|
Eesti Eisai Europe Ltd. Tel: + 44 (0) 208 600 1400 (Ůhendkuningriik) |
Norge Eisai AB Tlf: + 46 (0) 8 501 01 600 (Sverige) |
|
EXláSa Eisai Europe Ltd. Tel: + 44 (0) 208 600 1400 (Hvm^évo BaoíXsto) |
Osterreich Eisai GesmbH Tel: + 43 (0) 1 535 1980-0 |
|
Espaňa Eisai Farmacéutica, S.A. Tel: + (34) 91 455 94 55 |
Polska Eisai Europe Ltd. Tel.: + 44 (0) 208 600 1400 (Wielka Brytania) |
|
France Eisai SAS Tél: + (33) 1 47 67 00 05 |
Portugal Eisai Farmacéutica, Unipessoal Lda Tel: + 351 214 875 540 |
|
Hrvatska Eisai Europe Ltd. Tel: + 44 (0) 20 8600 1400 (Velika Britanija) |
Románia Eisai Europe Ltd. Tel: +44 (0) 20 8600 1400 (Marea Britanie) |
|
Ireland Eisai Europe Ltd. Tel: + 44 (0) 208 600 1400 (United Kingdom) |
Slovenija Eisai Europe Ltd. Tel: +44 (0) 20 8600 1400 (Velika Britanija) |
|
Ísland Eisai AB Sími: + 46 (0) 8 501 01 600 (Svífróó) |
Slovenská republika Eisai GesmbH organizační složka Tel.: +420 242 485 839 (Česká republika) |
Italia
Eisai S.r.l.
Tel: + 39 02 5181401
Kúrcpog
Eisai Europe Ltd.
Tqk +44 (0) 20 8600 1400 (Hvro^évo BaoíXsto)
Latvija
Eisai Europe Ltd.
Tel: +44 (0) 20 8600 1400 (Anglija)
Suomi/Finland
Eisai AB
Puh/Tel: + 46 (0) 8 501 01 600 (Ruotsi/Sverige)
Sverige
Eisai AB
Tel: + 46 (0) 8 501 01 600
United Kingdom
Eisai Europe Ltd.
Tel: + 44 (0) 208 600 1400
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
41