Lemtrada 12 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
LEMTRADA 12 mg koncentrát pro infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje alemtuzumabum 12 mg v 1,2 ml (10 mg/ml).
Alemtuzumab je monoklonální protilátka produkovaná v suspenzní kultuře savčích buněk (ovariální buňky křečíka čínského) v živném médiu technologií rekombinantní DNA.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok (sterilní koncentrát).
Čirý, bezbarvý nebo lehce nažloutlý koncentrát s pH 7,0-7,4.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek LEMTRADA je indikován k léčbě dospělých pacientů s relabující-remitující roztroušenou sklerózou (RRRS) v aktivním stádiu onemocnění definovaném klinickými příznaky nebo nálezem daným zobrazovacími metodami (viz body 4.4 a 5.1).
4.2 Dávkování a způsob podání
Léčba přípravkem LEMTRADA má být zahájena a sledována neurologem se zkušenostmi v léčbě pacientů s RS. Musí být k dispozici odborníci a vybavení potřebné k rychlé diagnóze a zvládnutí nej častějších nežádoucích účinků, především autoimunitně podmíněných stavů a infekcí.
Je třeba mít k dispozici prostředky na zvládnutí přecitlivělosti a/nebo anafylaktických reakcí.
Pacienti léčení přípravkem LEMTRADA musí dostat Kartu pacienta a Příručku pro pacienta a musí být informováni o rizicích přípravku LEMTRADA (viz také příbalová informace).
Dávkování
Doporučená dávka přípravku LEMTRADA je 12 mg/den podávaná nitrožilní infuzí ve 2 léčebných cyklech.
• Úvodní léčebný cyklus: 12 mg/den po dobu 5 po sobě jdoucích dnů (celková dávka 60 mg)
• Druhý léčebný cyklus: 12 mg/den po dobu 3 po sobě jdoucích dnech (celková dávka 36 mg) podaných 12 měsíců po úvodním léčebném cyklu.
Zmeškané dávky nemají být podány ve stejný den jako plánovaná dávka.
Následné sledování pacientů
Léčba se doporučuje ve 2 léčebných cyklech (viz dávkování) s bezpečnostním následným sledováním pacientů od zahájení léčby až do uplynutí 48 měsíců po poslední infuzi (viz bod 4.4).
Před léčbou
Pacientům má být po dobu prvních 3 dnů každého léčebného cyklu bezprostředně před podáním přípravku LEMTRADA podána premedikace kortikosteroidy. V klinických studiích byli pacienti po dobu prvních 3 dnů každého léčebného cyklu přípravkem LEMTRADA premedikováni 1000 mg methylprednisolonu.
Před podáním přípravku LEMTRADA lze rovněž zvážit premedikaci antihistaminiky a/nebo antipyretiky.
Všem pacientům by měla být podávána perorální profylaxe herpetické infekce zahájená první den každého léčebného cyklu a trvající do uplynutí nejméně 1 měsíce od ukončení léčebného cyklu přípravkem LEMTRADA (viz také „Infekce“ v bodě 4.4). V klinických studiích byl pacientům podáván aciklovir v dávce 200 mg dvakrát denně nebo jeho ekvivalent.
Starší pacienti
Klinické studie nezahrnovaly pacienty ve věku nad 55 let a nebylo možné stanovit, zda na léčbu odpovídají jinak než mladší pacienti.
Pacienti s poruchou funkce jater nebo ledvin
Přípravek LEMTRADA nebyl hodnocen u pacientů s poruchou funkce ledvin nebo jater.
Pediatrická populace
Bezpečnost a účinnost přípravku LEMTRADA u dětí s RS ve věku od 0 do 18 let nebyla zatím stanovena. Použití alemtuzumabu k léčbě roztroušené sklerózy u dětí od narození do věku 10 let nemá žádné opodstatnění. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek LEMTRADA musí být před infuzí naředěn. Naředěný roztok se má podávat nitrožilní infuzí po dobu přibližně 4 hodin.
Návod k naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Infekce virem lidské imunodeficience (HIV).
4.4 Zvláštní upozornění a opatření pro použití
U pacientů s neaktivním onemocněním nebo stabilizovaných na stávající léčbě se přípravek LEMTRADA nedoporučuje.
Pacienti léčení přípravkem LEMTRADA musí dostat příbalovou informaci, Kartu pacienta a Příručku pro pacienta. Před léčbou musí být pacienti informováni o jejích rizicích a přínosech a o nutnosti zavázat se ke 48měsíčnímu sledování po poslední infuzi přípravku LEMTRADA.
Autoimunita
Léčba může vést k vytvoření autoprotilátek a zvýšení rizika autoimunitně podmíněných stavů, včetně imunitní trombocytopenické purpury (ITP), poruch štítné žlázy nebo ojediněle nefropatií (např. onemocnění s tvorbou protilátek proti bazální membráně glomerulů). U pacientů s jiným předchozím autoimunitním onemocněním než je RS, je třeba dbát opatrnosti, ačkoliv dostupná data naznačují, že po léčbě alemtuzumabem nedochází ke zhoršení tohoto již dříve přítomného autoimunitního onemocnění.
Imunitní trombocytopenická purpura (ITP)
U přibližně 1 % pacientů léčených v kontrolovaných klinických studiích s RS byly pozorovány závažné případy ITP. V kontrolované klinické studii pacientů s RS se u jednoho pacienta rozvinula ITP, která proběhla nepozorovaně ještě před zařazením požadavku na měsíční kontroly krve. Pacient zemřel na krvácení do mozku. Nástup ITP se obecně objevuje mezi 14. a 36. měsícem po první expozici. Příznaky ITP mohou zahrnovat (mimo jiné) snadnou tvorbu modřin, petechie, samovolné krvácení sliznic (např. epistaxe, hemoptýza), nepravidelné nebo silnější menstruační krvácení, než je obvyklé. Hemoptýza může rovněž svědčit pro anti-GBM onemocnění (viz níže) a je třeba provést odpovídající diferenciální diagnostiku. Pacientovi je třeba připomenout, aby nadále věnoval pozornost příznakům, které se mohou objevit, a aby při jakýchkoliv pochybách vyhledal okamžitou lékařskou pomoc.
Před zahájením léčby a poté v měsíčních intervalech až do uplynutí 48 měsíců od poslední infuze je vhodné odebírat kompletní krevní obraz s diferenciálem. Po tomto období má být testování prováděno na základě klinických nálezů naznačujících ITP. Kompletní krevní obraz je třeba neprodleně odebrat také tehdy, existuje-li podezření na ITP.
Pokud je nástup ITP potvrzen, je nutné ihned zahájit odpovídající lékařskou intervenci, případně odeslat pacienta neprodleně k odborníkovi. Data z klinických studií s RS ukazují, že dodržování požadavků na sledování krve a edukace ve věci rozpoznávání známek a příznaků ITP vedou k časné detekci a léčbě ITP, kdy většina případů odpovídala na léčbu první linie.
Potenciální riziko spojené s opakovanou léčbou přípravkem LEMTRADA po výskytu ITP není známo.
Nefropatie
V klinických studiích s RS byly pozorovány nefropatie, včetně onemocnění s tvorbou protilátek proti bazální membráně glomerulů (anti-GBM), u 0,3 % pacientů; obecně se objevovaly do 39 měsíců po posledním podání přípravku LEMTRADA. V průběhu klinických studií došlo ke 2 případům anti-GBM onemocnění. Oba případy byly závažné, byly zjištěny brzo díky klinickému a laboratornímu monitorování a po léčbě měly pozitivní výsledek.
Mezi klinické manifestace nefropatie může patřit zvýšení sérového kreatininu, hematurie a/nebo proteinurie. Ačkoli to nebylo v klinických studiích pozorováno, při anti-GBM onemocnění může být alveolární krvácení manifestované jako hemoptýza. Hemoptýza může rovněž svědčit pro ITP (viz výše) a je třeba provést odpovídající diferenciální diagnózu. Pacientovi je třeba připomenout, aby nadále věnoval pozornost příznakům, které se mohou objevit, a aby při jakýchkoliv pochybách vyhledal okamžitou lékařskou pomoc. Anti-GBM onemocnění může vést k selhání ledvin vyžadujícímu dialýzu a/nebo (pokud není rychle léčeno) transplantaci a ponechá-li se neléčeno, může být život ohrožující.
Před zahájením léčby a poté v měsíčních intervalech až do uplynutí 48 měsíců od poslední infuze je vhodné provádět měření hladiny sérového kreatininu. Mikroskopickou analýzu moči je vhodné provádět před zahájením a poté v měsíčních intervalech až do uplynutí 48 měsíců od poslední infuze. Pozorování klinicky významných změn od výchozí hodnoty sérového kreatininu, nevysvětlitelná hematurie a/nebo proteinurie vyžadují další vyhodnocení s ohledem na možné nefropatie, včetně případného okamžitého odeslání pacienta k odborníkovi. Časnou detekcí a léčbou nefropatií lze dosáhnout snížení rizika špatných výsledků. Po tomto období by testování mělo být provedeno na základě klinických nálezů naznačujících nemoci ledvin.
Potenciální riziko spojené s opakovanou léčbou přípravkem LEMTRADA po výskytu nefropatie není známo.
Poruchy štítné žlázy
V klinických studiích s RS (a až 48 měsíců po první expozici tomuto přípravku) byly autoimunitní poruchy štítné žlázy pozorovány odhadem u 36 % pacientů léčených přípravkem LEMTRADA 12 mg. Incidence poruch štítné žlázy byla vyšší u pacientů s anamnézou těchto poruch jak ve skupině léčené přípravkem LEMTRADA, tak ve skupině s interferonem beta 1a (IFNB-1a). U pacientů s poruchou štítné žlázy má být přípravek LEMTRADA podáván v případě, že potenciální přínos léčby převýší případná rizika. Pozorované autoimunitní poruchy štítné žlázy zahrnovaly hypertyreózu a hypotyreózu. Většina případů byla lehká až středně závažná. Před registrací došlo k vážným případům u < 1 % pacientů, přičemž pouze Basedowova choroba (také známá jako Gravesova nemoc), hypertyreóza a hypotyreóza se objevily u více než 1 pacienta. Většina případů týkající se štítné žlázy byla zvládnuta konvenční farmakoterapií, nicméně u některých pacientů byla nutná chirurgická intervence. Pacientům, u nichž se rozvinuly potíže se štítnou žlázou, nebyla v klinických studiích opakovaná léčba přípravkem LEMTRADA povolena. Ačkoli jsou zkušenosti omezené, u pacientů, kteří byli opakovaně léčeni, obecně nedocházelo ke zhoršení závažnosti poruchy štítné žlázy. Další léčbu přípravkem LEMTRADA je třeba individuálně zvažovat na základě klinického stavu daného pacienta.
Funkční testy štítné žlázy, např. hladin tyreostimulačního hormonu, by měly být provedeny před zahájením léčby a poté každé 3 měsíce až do uplynutí 48 měsíců od poslední infuze. Po tomto období by testování mělo být prováděno na základě klinických nálezů svědčících pro dysfunkci štítné žlázy.
Onemocnění štítné žlázy představuje zvláštní riziko u těhotných žen (viz bod 4.6).
V klinických studiích nebyl stav pacientových protilátek proti tyroidální peroxidáze (anti-TPO) před léčbou určující pro rozvoj nežádoucích účinků souvisejících se štítnou žlázou. U poloviny pacientů, jejichž testy na protilátky anti-TPO byly na začátku pozitivní, a u čtvrtiny pacientů, jejichž testy na protilátky anti-TPO byly na začátku negativní, se rozvinuly potíže se štítnou žlázou. Velká většina (přibližně 80 %) pacientů, u nichž byly po léčbě přítomny potíže se štítnou žlázou, měla na začátku negativní protilátky anti-TPO. Proto se, bez ohledu na stav protilátek anti-TPO před léčbou, mohou u pacientů rozvinout nežádoucí účinky na štítnou žlázu a je nezbytné všechy testy provádět pravidelně, jak bylo popsáno výše.
Cytopenie
Vzácně byla v klinických studiích s RS hlášena podezření na autoimunitní cytopenie, jako je neutropenie, hemolytická anémie a pancytopenie. K monitorování cytopenií by měly být použity výsledky kompletního krevního obrazu (viz ITP výše). Pokud je cytopenie potvrzena, je nutné ihned zahájit odpovídající lékařskou intervenci, případně odeslat pacienta k odborníkovi.
Reakce spojené s infuzí (I AR)
V kontrolovaných klinických studiích byly reakce spojené s infuzí (IAR) definovány jako jakékoli nežádoucí příhody vyskytující se během 24 hodin od infuze přípravku LEMTRADA. K většině z nich může docházet následkem uvolnění cytokinu během infuze. Většina pacientů léčených přípravkem LEMTRADA
v kontrolovaných klinických studiích s RS prodělala lehké až středně závažné IAR během a/nebo až do 24 hodin po podání přípravku LEMTRADA 12 mg, které často zahrnovaly bolest hlavy, vyrážku, pyrexii, nauzeu, kopřivku, pruritus, insomnii, třesavku, zrudnutí, únavu, dyspnoi, dysgeuzii, hrudní diskomfort, generalizovanou vyrážku, tachykardii, bradykardii, dyspepsii, závrať a bolest. K závážným nežádoucím účinkům došlo u 3 % pacientů, a to včetně případů pyrexie, kopřivky, fibrilace síní, nauzey, hrudního diskomfortu a hypotenze. Klinická manifestace anafylaxe se může jevit stejná jako klinická manifestace reakcí souvisejících s infuzí, ale má sklon být závažnější nebo případně život ohrožující. Reakce připisované anafylaxi bývají oproti reakcím souvisejícím s infuzí hlášeny ojediněle.
Doporučuje se pacienty premedikovat, aby se zmírnil účinek reakcí na infuzi (viz bod 4.2). Většina pacientů v kontrolovaných klinických studiích dostala alespoň před jednou infuzí přípravku LEMTRADA antihistaminika a/nebo antipyretika. K IAR může u pacientů dojít navzdory premedikaci. Během infuze přípravku LEMTRADA a 2 hodiny po jejím ukončení se doporučuje sledovat, zda se u pacienta nevyskytnou reakce na infuzi. Pokud dojde k IAR, poskytněte v případě potřeby odpovídající symptomatickou léčbu. Pokud není infuze dobře tolerována, lze prodloužit její trvání. Pokud dojde k závažným reakcím na infuzi, zvažte okamžité přerušení podávání nitrožilní infuze. V klinických studiích byly anafylaxe a závažné reakce vyžadující přerušení léčby velmi vzácné.
Lékaři by měli vzít v úvahu kardiologickou anamnézu pacienta, protože reakce související s infuzí mohou zahrnovat příznaky jako je tachykardie.
Je třeba mít k dispozici prostředky na zvládnutí anafylaxe a závažných nežádoucích účinků.
Infekce
Infekce se objevily u 71 % pacientů léčených přípravkem LEMTRADA 12 mg ve srovnání s 53 % pacientů léčených subkutánním interferonem beta-1a [IFNB 1a] (44 pg 3krát týdně) v kontrolovaných klinických studiích s RS trvajících až 2 roky a byly převážně lehké až středně závažné. Infekce, které se u pacientů léčených přípravkem LEMTRADA objevovaly častěji než u pacientů na IFNB 1a, zahrnovaly nazofaryngitidu, infekci močových cest, infekci horních dýchacích cest, sinusitidu, herpes labialis, chřipku a bronchitidu. Závažné infekce se v kontrolovaných klinických studiích s RS objevily u 2,7 % pacientů léčených přípravkem LEMTRADA ve srovnání s 1 % pacientů léčených IFNB-1a. Závažné infekce ve skupině s přípravkem LEMTRADA byly následující: apendicitida, gastroenteritida, pneumonie, herpes zoster a zubní infekce. Infekce obecně trvaly obvyklou dobu a byly zvládnuty konvenční farmakoterapií.
Závažné infekce způsobené virem varicella zoster včetně primární varicelly a reaktivace viru varicella zoster se u pacientů léčených přípravkem LEMTRADA 12 mg objevovaly častěji (0,3 %) ve srovnání s léčbou IFNB-1a (0 %). U pacientek léčených přípravkem LEMTRADA 12 mg byla rovněž hlášena cervikální infekce způsobená lidskými papilomaviry (HPV) včetně cervikální dysplazie (2 %). U pacientek se doporučuje jednou ročně provádět testování HPV.
U pacientů léčených přípravkem LEMTRADA a IFNB-1a byla v kontrolovaných klinických studiích hlášena tuberkulóza. U 0,3 % pacientů léčených přípravkem LEMTRADA byla hlášena aktivní a latentní tuberkulóza (nejčastěji v endemických oblastech). V souladu s místními předpisy musí být před zahájením léčby u všech pacientů zhodnocena aktivní i neaktivní („latentní“) infekce tuberkulózou.
Listerióza/listeriózní meningitida byla hlášena u pacientů léčených přípravkem LEMTRADA zpravidla do jednoho měsíce po podání infúze s přípravekm LEMTRADA. Pro snížení tohoto rizika by se pacienti užívající přípravek LEMTRADA měli vyvarovat po dobu alespoň jednoho měsíce po podání tohoto přípravku konzumaci syrového nebo nedostatečně tepelně upraveného masa, měkkých sýrů a nepasterizovaných mléčných výrobků.
V kontrolovaných klinických studiích s RS docházelo u pacientů léčených přípravkem LEMTRADA mnohem častěji k povrchovým mykotickým infekcím, především pak k orální a vaginální kandidóze (12 %), než u pacientů léčených IFNB-1a (3 %).
Lékaři by měli zvážit oddálení zahájení podávání přípravku LEMTRADA u pacientů s aktivní infekcí až do jejího úplného zvládnutí.
První den léčby přípravkem LEMTRADA by měla být zahájena profylaxe perorálním antiherpetikem, která by měla trvat až do uplynutí nejméně 1 měsíce od ukončení každého léčebného cyklu. V klinických studiích byl pacientům podáván aciklovir v dávce 200 mg dvakrát denně nebo jeho ekvivalent.
Přípravek LEMTRADA nebyl podáván k léčbě RS souběžně s nebo v návaznosti na antineoplastickou nebo imunosupresivní léčbou. Tak jako u ostatní imunomodulační léčby je třeba při zvažování podávání přípravku LEMTRADA uvážit možné kombinované účinky na pacientův imunitní systém. Souběžné užívání přípravku LEMTRADA s jakoukoli z těchto terapií může zvýšit riziko imunosuprese.
Nejsou k dispozici žádné údaje týkající se souvislosti přípravku LEMTRADA a reaktivace viru hepatitidy B (HBV) nebo hepatitidy C (HCV), neboť pacienti s prokázanou aktivní nebo chronickou infekcí byli z klinických studií vyloučeni. Před zahájením podávání přípravku LEMTRADA je vhodné zvážit zhodnocení stavu pacientů s vysokým rizikem infekce HBV a/nebo HCV. Také je třeba dbát opatrnosti při předepisování přípravku LEMTRADA pacientům, kteří jsou nosiči HBV a/nebo HCV, neboť tito pacienti mohou být ohroženi nevratným jaterním poškozením kvůli možné reaktivaci viru v důsledku jejich předcházejícího stavu.
Malignita
Tak jako u ostatní imunomodulační léčby je třeba postupovat s opatrností při zahájení léčby přípravkem LEMTRADA u pacientů s dříve přítomnou a/nebo stávající malignitou. V současnosti není známo, zda alemtuzumab představuje větší riziko pro rozvoj malignity štítné žlázy, protože autoimunita proti štítné žláze může být sama o sobě rizikovým faktorem pro malignitu štítné žlázy.
Antikoncepce
U myší byl během březosti a po porodu pozorován placentární přenos a potenciální farmakologická aktivita. Ženy ve fertilním věku by měly během léčby a 4 měsíce po léčebném cyklu přípravkem LEMTRADA používat účinná antikoncepční opatření (viz bod 4.6).
Vakcíny
Doporučuje se, aby požadavky na lokální imunizaci byly u pacientů splněny alespoň 6 týdnů před začátkem léčby přípravkem LEMTRADA. Schopnost vyvolat imunitní odpověď na jakoukoli vakcínu po léčbě přípravkem LEMTRADA nebyla hodnocena.
Bezpečnost imunizace živými virovými vakcínami po léčebném cyklu přípravkem LEMTRADA nebyla formálně hodnocena v kontrolovaných klinických studiích s RS a tato léčba nemá být podána pacientům s RS, kteří v nedávné době podstoupili cyklus léčby přípravkem LEMTRADA.
Testování na protilátky proti viru varicella zoster / vakcinace
Tak jako u všech imunitu modulujících léčivých přípravků by před zahájením léčebného cyklu přípravkem LEMTRADA měli být pacienti, kteří dle anamnézy neprodělali plané neštovice nebo nebyli očkováni proti viru varicella zoster (VZV), otestování na protilátky proti VZV. Před zahájením léčby přípravkem LEMTRADA by měla být u pacientů s negativními protilátkami zvážena vakcinace proti VZV. Aby byl dosažen plný účinek vakcinace proti VZV, je zapotřebí odložit léčbu přípravkem LEMTRADA 6 týdnů po vakcinaci.
Doporučené laboratorní testy k monitorování pacientů
Laboratorní testy by měly být prováděny v pravidelných intervalech po dobu 48 měsíců po posledním léčebném cyklu přípravkem LEMTRADA, aby se zajistilo zjištění časných známek autoimunitního onemocnění:
• Kompletní krevní obraz s diferenciálem (před zahájením léčby a poté v měsíčních intervalech)
• Hladiny sérového kreatininu (před zahájením léčby a poté v měsíčních intervalech)
• Mikroskopická analýza moči (před zahájením léčby a poté v měsíčních intervalech)
• Test funkce štítné žlázy, jako je hladina tyreostimulačního hormonu (před zahájením léčby a poté každé 3 měsíce)
Po tomto období budou jakékoli klinické nálezy naznačující onemocnění ledvin nebo dysfunkci štítné žlázy vyžadovat další testování.
Informace získané z používání alemtuzumabu před registrací přípravku LEMTRADA mimo společností sponzorované studie
Následující nežádoucí účinky byly zjištěny před registrací přípravku LEMTRADA během používání alemtuzumabu k léčbě chronické lymfatické leukémie z B buněk (B-CLL) a také k léčbě dalších poruch, obecně při vyšších a častějších dávkách (např. 30 mg), než jaké jsou doporučeny při léčbě RS. Jelikož jsou tyto reakce hlášeny dobrovolně z populace neupřesněné velikosti, není vždy možné spolehlivě stanovit jejich četnost ani určit příčinný vztah k expozici alemtuzumabu.
Autoimunitní onemocnění
Autoimunitní příhody hlášené u pacientů léčených alemtuzumabem zahrnují neutropenii, hemolytickou anémii (včetně jednoho fatálního případu), získanou hemofilii, anti-GBM onemocnění a onemocnění štítné žlázy. U pacientů bez RS léčených alemtuzumabem byly hlášeny závažné a někdy fatální autoimunitní onemocnění, včetně autoimunitní hemolytické anémie, autoimunitní trombocytopenie, aplastické anémie, syndromu Guillain-Barré a chronické zánětlivé demyelinizační polyradikuloneuropatie. U jednoho onkologického pacienta léčeného alemtuzumabem byl hlášen pozitivní Coombsův test. U jednoho onkologického pacienta léčeného alemtuzumabem byla hlášena fatální příhoda reakce štěpu proti hostiteli související s transfuzí.
Reakce spojené s infuzí
U pacientů bez RS léčených alemtuzumabem byly při vyšších a častějších dávkách, než se používají u RS, pozorovány závažné a někdy fatální IAR, včetně bronchospasmu, hypoxie, synkopy, plicních infiltrátů, syndromu akutní respirační tísně, respirační zástavy, infarktu myokardu, arytmií, akutní srdeční nedostatečnosti a srdeční zástavy. Rovněž byla hlášena těžká anafylaxe a další hypersenzitivní reakce včetně anafylaktického šoku a angioedému.
Infekce a infestace
U pacientů bez RS léčených alemtuzumabem vyššími a častějšími dávkami, než se používají u RS, byly hlášeny závažné a někdy fatální virové, bakteriální, protozoální a mykotické infekce, včetně těch, které jsou důsledkem reaktivace latentních infekcí. U pacientů s B-CLL s léčbou i bez léčby alemtuzumabem byla hlášena progresivní multifokální leukoencefalopatie (PML). Četnost PML u pacientů s B-CLL léčených alemtuzumabem není vyšší než četnost z normálního prostředí.
Poruchy krve a lymfatického systému U pacientů bez RS byly hlášeny vážné krvácivé reakce.
Srdeční poruchy
U pacientů bez RS léčených alemtuzumabem, kteří byli dříve léčeni potenciálně kardiotoxickými látkami, bylo hlášeno městnavé srdeční selhání, kardiomyopatie a snížená ejekční frakce.
Lymfoproliferativní poruchy spojované s virem Epsteina-Barrové
Mimo společností sponzorované studie byly pozorovány lymfoproliferativní poruchy spojované s virem Epsteina-Barrové.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
U pacientů s RS nebyly vedeny žádné formální studie lékových interakcí s přípravkem LEMTRADA za použití doporučené dávky. V kontrolované klinické studii u pacientů s RS nedávno léčených beta interferonem a glatiramer acetátem bylo nutné léčbu přerušit 28 dnů před zahájením léčby přípravkem LEMTRADA.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Sérové koncentrace byly nízké nebo nedetekovatelné během cca 30 dnů po každé léčebné kúře. Proto by ženy ve fertilním věku měly během léčebného cyklu přípravkem LEMTRADA a dále po dobu 4 měsíců po jejím ukončení používat účinná antikoncepční opatření.
Údaje o podávání přípravku LEMTRADA těhotným ženám jsou omezené. Přípravek LEMTRADA lze podávat během těhotenství pouze v případě, že potenciální přínos odůvodní možné riziko pro plod.
Je známo, že lidský IgG prostupuje placentární bariérou; alemtuzumab může placentární bariérou prostupovat také, a tím představovat potenciální riziko pro plod. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Není známo, zda alemtuzumab může způsobit poškození plodu, je-li podáván těhotným ženám, nebo zda může ovlivnit schopnost reprodukce.
Onemocnění štítné žlázy (viz bod 4.4 Poruchy štítné žlázy) představuje u těhotných žen zvláštní riziko. Bez léčby hypotyreózy existuje v těhotenství zvýšené riziko spontánního potratu a účinků na plod ve smyslu mentální retardace a nanismu. U matek s Gravesovou nemocí se protilátky proti receptorům tyreostimulačního hormonu matky mohou přenášet do vyvíjejícího se plodu a způsobit přechodnou novorozeneckou Gravesovu nemoc.
Kojení
Alemtuzumab byl detekován v mléce a u mláďat kojících samic myší.
Není známo, zda je alemtuzumab vylučován do lidského mateřského mléka. Riziko pro kojené dítě nelze vyloučit. Proto by během každého léčebného cyklu přípravkem LEMTRADA a také po dobu 4 měsíců po podání poslední infuze každého léčebného cyklu mělo být kojení přerušeno. Přínos imunity získané z mateřského mléka však může u kojeného dítěte převážit riziko z potenciální expozice alemtuzumabu.
Fertilita
Nejsou k dispozici žádné adekvátní klinické bezpečnostní údaje o vlivu přípravku LEMTRADA na fertilitu. V podstudii u 13 pacientů (mužů) léčených alemtuzumabem (léčených buď 12 mg, nebo 24 mg) nebyly zjištěny žádné známky aspermie, azoospermie, konzistentně sníženého počtu spermií, poruch motility ani zvýšení počtu morfologických abnormalit spermií.
Je známo, že CD52 je přítomný v reprodukčních tkáních lidí a hlodavců. Údaje získané ve studiích na zvířatech ukazovaly účinky na fertilitu humanizovaných myší (viz bod 5.3), nicméně potenciální vliv na lidskou fertilitu během období expozice není na základě dostupných údajů znám.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyla provedena žádná studie účinků přípravku LEMTRADA na schopnost řídit a obsluhovat stroje.
Většina pacientů prodělá IAR, které se objeví do 24 hodin od léčby přípravkem LEMTRADA. Některé z IAR (např. závrať) mohou dočasně ovlivnit schopnost pacienta řídit a obsluhovat stroje a je třeba dbát opatrnosti až do jejích odeznění.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Celkem 1188 pacientů s relabující-remitující RS (RRRS) léčených přípravkem LEMTRADA (12 mg nebo 24 mg) představovalo bezpečnostní populaci souhrnné analýzy kontrolovaných klinických studií s výsledkem 2363 pacientoroků bezpečnostního následného sledování a s mediánem následného sledování 24 měsíců.
Nejdůležitější nežádoucí účinky jsou autoimunitní (ITP, poruchy štítné žlázy, nefropatie, cytopenie), IAR a infekce. Jsou popsány v bodě 4.4.
Nejčastějšími nežádoucími účinky přípravku LEMTRADA (u > 20 % pacientů) jsou vyrážka, bolest hlavy, pyrexie a infekce dýchacích cest.
Seznam nežádoucích účinků v tabulce
Níže uvedená tabulka je založena na souhrnných bezpečnostních údajích až za 24 měsíců od pacientů s RRRS léčených přípravkem LEMTRADA 12 mg/den po dobu 5 po sobě jdoucích dnů při vstupu do studie a po dobu 3 po sobě jdoucích dnů ve 12. měsíci studie. Nežádoucí účinky, které se vyskytly u > 0,5 % pacientů, jsou uvedeny dle tříd orgánových systémů (SOC) a preferované terminologie (PT) MedDRA. Četnosti jsou definovány dle následujících konvencí: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 1: Nežádoucí účinky ve studii 1, 2 a 3 pozorované u > 0,5 % pacientů léčených přípravkem LEMTRADA 12 mg
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
|
Infekce a infestace |
Infekce horních cest dýchacích, infekce močových cest |
Infekce dolních cest dýchacích, herpes zoster, gastroenteritida, herpes labialis, orální kandidóza, vulvovaginální kandidóza, chřipka, ušní infekce |
Zubní infekce, herpes genitalis, onychomykóza |
|
Poruchy krve a lymfatického systému |
Lymfopenie, leukopenie |
Lymfadenopatie |
Imunitní trombocytopenická purpura, trombocytopenie, snížený hemoglobin, snížený hematokrit |
|
Poruchy imunitního systému |
Syndrom uvolňování cytokinů | ||
|
Endokrinní poruchy |
Basedowova choroba, hypertyreóza, autoimunitní tyroiditida, hypotyreóza, struma, pozitivní protilátky proti štítné žláze | ||
|
Psychiatrické poruchy |
Insomnie*, úzkost | ||
|
Poruchy nervového systému |
Bolest hlavy* |
Relaps RS, závrať*, hypestezie, parestezie, tremor, dysgeuzie* |
Smyslové poruchy, hyperestezie |
|
Poruchy oka |
Rozmazané vidění |
Konjunktivitida | |
|
Poruchy ucha a labyrintu | |||
|
Srdeční poruchy |
Tachykardie*, bradykardie*, palpitace | ||
|
Cévní poruchy |
Zrudnutí* |
Hypotenze*, hypertenze | |
|
Respirační, hrudní a mediastinální poruchy |
Dyspnoe*, kašel, epistaxe, orofaryngeální bolest |
Pocit sevření hrdla, škytavka, podráždění v krku, | |
|
Gastrointestinální poruchy |
Nauzea* |
Bolest břicha, zvracení, průjem, dyspepsie*, stomatitida |
Zácpa, gastroesofageální reflux, krvácení z dásní, dysfagie |
|
Poruchy jater a žlučových cest |
zvýšená aspartátaminotransferáza | ||
|
Poruchy kůže a podkožní tkáně |
Kopřivka*, vyrážka*, pruritus* |
Vyrážka na kůži celého těla*, erytém, ekchymóza, alopecie, hyperhidróza, akné |
Puchýř, noční poty |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Myalgie, svalová slabost, artralgie, bolest zad, bolest v končetině, svalové spazmy, bolest krku | ||
|
Poruchy ledvin a močových cest |
Proteinurie, hematurie | ||
|
Poruchy |
Menoragie, nepravidelná |
Cervikální dysplazie, |
|
reprodukčního systému a prsu |
menstruace |
amenorea | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie*, únava* |
Hrudní diskomfort*, třesavka*, bolest*, periferní edém, astenie, příznaky připomínající chřipku, malátnost, bolest v místě infuze | |
|
Vyšetření |
Pokles tělesné hmotnosti | ||
|
Poranění, otravy a procedurální komplikace |
Kontuze |
Popis vybraných nežádoucích účinků
Pojmy označené hvězdičkou (*) v tabulce 1 označují nežádoucí účinky uváděné jako reakce spojené s infuzí. Mezi IAR rovněž patří fibrilace síní a anafylaxe, které se objevovaly v četnosti pod hranicí 0,5 % souvisejících událostí (viz bod 4.4.).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Dva pacienti s RS v kontrolovaných klinických studiích nedopatřením dostali až 60 mg přípravku LEMTRADA (tj. celkovou dávku pro úvodní léčebný cyklus) v jedné infuzi a prodělali závažné reakce (bolest hlavy, vyrážka a buď hypotenze, nebo sinusová tachykardie). Dávky přípravku LEMTRADA vyšší než testované v klinických studiích mohou zvyšovat intenzitu a/nebo trvání nežádoucích účinků souvisejících s infuzí nebo zvyšovat jejich účinek na imunitu.
Antidotum při předávkování alemtuzumabem není známo. Léčba zahrnuje přerušení podávání léčivého přípravku a podpůrnou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Selektivní imunosupresiva, ATC kód: L04AA34. Mechanismus účinku
Alemtuzumab je humanizovaná monoklonální protilátka odvozená z rekombinantní DNA zaměřená proti povrchovému glykoproteinu CD52 o hmotnosti 21-28 kD. Alemtuzumab je protilátka typu IgG1 kappa s lidskou variabilní částí a konstantními oblastmi. Oblasti určující komplementaritu pocházejí z myší (potkaní) monoklonální protilátky. Protilátka má molekulární hmotnost přibližně 150 kD.
Alemtuzumab se váže na CD52, povrchový antigen přítomný ve vysokých hladinách na T (CD3+) a B (CD19+) lymfocytech a v nižších hladinách na NK buňkách, monocytech a makrofázích. Na neutrofilech, plazmatických buňkách nebo kmenových buňkách kostní dřeně je CD52 detekován jen velmi málo nebo vůbec. Alemtuzumab účinkuje prostřednictvím buněčné cytolýzy závislé na protilátkách a komplementem zprostředkované lýzy po navázání na buněčný povrch T a B lymfocytů.
Mechanismus, jakým přípravek LEMTRADA uplatňuje svůj účinek u RS, není zcela vysvětlen. Výzkumy však ukazují na imunomodulační účinky prostřednictvím deplece a repopulace lymfocytů, včetně:
- Alterací v počtu, proporcích a vlastnostech některých podskupin lymfocytů po léčbě.
- Zvýšeného zastoupení podskupin regulačních T buněk.
- Zvýšeného zastoupení paměťových T a B lymfocytů.
- Přechodných účinků na složky vrozené imunity (tj. neutrofily, makrofágy, NK buňky).
Snížení hladiny cirkulujících B a T buněk prostřednictvím přípravku LEMTRADA a následná repopulace mohou snížit riziko relapsu, což v konečném důsledku zpozdí progresi onemocnění.
Farmakodynamické účinky
Přípravek LEMTRADA ničí cirkulující T a B lymfocyty po každém léčebném cyklu; nejnižší hodnoty byly pozorovány 1 měsíc po léčebném cyklu (nejčasnější časový bod po léčbě ve studiích fáze 3). U lymfocytů dochází v průběhu času k repopulaci s obnovou B buněk dokončenou obvykle do 6 měsíců. Počty CD3+ a CD4+ lymfocytů narůstají směrem k normálu pomaleji, ale obecně se do 12 měsíců po léčbě nevrátí k výchozí hodnotě. Přibližně 40 % pacientů mělo celkový počet lymfocytů na spodním limitu normálu (LLN) za 6 měsíců po každém léčebném cyklu a přibližně 80 % pacientů mělo celkový počet lymfocytů na LLN za 12 měsíců po každém cyklu léčby.
Neutrofily, monocyty, eozinofily, bazofily a NK buňky jsou přípravkem LEMTRADA ovlivněny pouze přechodně.
Klinická účinnost a bezpečnost
Bezpečnost a účinnost přípravku LEMTRADA byly hodnoceny u pacientů s RRRS ve 3 randomizovaných, pro posuzovatele zaslepených klinických studiích s aktivním komparátorem.
Design studie, demografické údaje a výsledky pro studii 1 a 2 jsou uvedeny v tabulce 2 a 3
|
Tabulka 2: Design studií 1 a 2 a výchozí charakteristiky | ||
|
Studie 1 |
Studie 2 | |
|
Název studie |
CAMMS323 (CARE-MS I) |
CAMMS32400507 (CARE-MS II) |
|
Design studie | ||
|
Anamnéza onemocnění |
Pacienti s aktivní RS, definovanou alespoň 2 relapsy během předchozích 2 let. | |
|
Sledování |
2 roky | |
|
Studijní populace |
Dosud neléčení pacienti |
Pacienti s neadekvátní odpovědí na předchozí léčbu* |
|
Výchozí charakteristiky | ||
|
Průměrný věk (roky) |
33 |
35 |
|
Průměrné trvání/medián trvání onemocnění |
2,0/1,6 roku |
4,5/3,8 roku |
|
Průměrné trvání předchozí léčby RS (> 1 použité léčivo) |
Žádné |
36 měsíců |
|
% dostávajících > 2 předchozí léčby RS |
Neuplatňuje se. |
28 % |
|
Průměrné skóre EDSS na počátku |
2,0 |
2,7 |
* Definováno jako pacienti, kteří prodělali alespoň 1 relaps během léčby beta interferonem nebo glatiramer acetátem poté, co byli na léčbě tímto léčivým přípravkem nejméně 6 měsíců.
Tabulka 3: Klíčové klinické a MRI cílové parametry ze studií 1 a 2
|
Studie 1 |
Studie 2 | |||
|
Název studie |
CAMMS323 (CARE-MS I) |
CAMMS32400507 (CARE-MS II) | ||
|
Klinické cílové parametry |
LEMTRADA 12 mg (N = 376) |
SC IFNB-1a (N = 187) |
LEMTRADA 12 mg (N = 426) |
SC IFNB-1a (N = 202) |
|
Výskyt relapsu1 Roční výskyt relapsů (ARR) (95% CI) |
0,18 (0,13; 0,23) |
0,39 (0,29; 0,53) |
0,26 (0,21; 0,33) |
0,52 (0,41; 0,66) |
|
Poměr frekvence (95% CI) Snížení rizika |
0,45 (0,32; 0,63) 54,9 (p < 0,0001) |
0,51 (0,39; 0,65) 49,4 (p < 0,0001) | ||
|
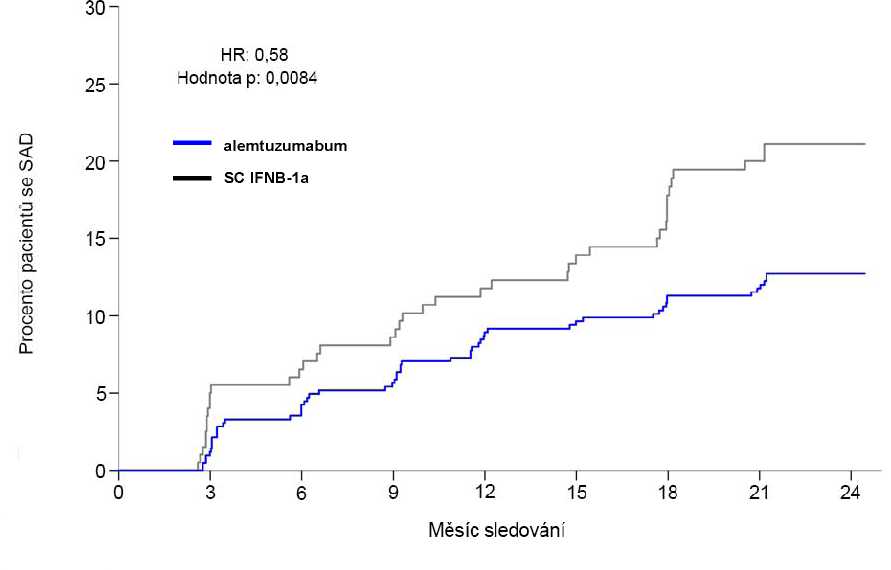
Postižení2 (Trvalá akumulace postižení [SAD] > 6 měsíců1) Pacienti s 6měsíčním SAD (95% CI) |
8,0 % (5,7; 11,2) |
11,1 % (7,3; 16,7) |
12,7 % (9,9; 16,3) |
21,1 % (15,9; 27,7) |
|
Poměr rizik (95% CI) |
0,70 (0,40; 1,23) (p = 0,22) |
0,58 (0,38; 0,87) (p = 0,0084) | ||
|
Pacienti, kteří jsou ve 2. roce bez relapsu (95% CI) |
77,6 % (72,9; 81,6) (p < 0,0001) |
58,7 % (51,1; 65,5) |
65,4 % (60,6; 69,7) (p < 0,0001) |
46,7 % (39,5; 53,5) |
|
Změna od základní hodnoty v EDSS ve 2. roce Odhad (95% CI) |
-0,14 (-0,25; -0,02) (p = 0,42) |
-0,14 (-0,29; 0,01) |
-0,17 (-0,29; -0,05) (p < 0,0001) |
0,24 (0,07; 0,41) |
|
MRI cílové parametry (0-2 roky) | ||||
|
Medián % změny v objemu léze T2 dle MRI |
-9,3 (-19,6; -0,2) (p = 0,31) |
-6,5 (-20,7; 2,5) |
-1,3 (p = 0,14) |
-1,2 |
|
Pacienti s novými nebo zvětšujícími se lézemi T2 během 2. roku |
48,5 % (p = 0,035) |
57,6 % |
46,2 % (p < 0,0001) |
67,9 % |
|
Pacienti s lézemi zvýrazněnými gadoliniem během 2. roku |
15,4 % (p = 0,001) |
27,0 % |
18,5 % (p < 0,0001) |
34,2 % |
|
Pacienti s novými T1 hypointenzivními lézemi během 2. roku |
24,0 % (p = 0,055) |
31,4 % |
19,9 % (p < 0,0001) |
38,0 % |
|
Medián % změny frakce mozkového parenchymu |
-0,867 (p < 0,0001) |
-1,488 |
-0,615 (p = 0,012) |
-0,810 |
|
1 Koprimární cílové parametry: ARR a SAD. Studie byla prohlášena za úspěšnou, pokud byl splněn alespoň jeden ze dvou koprimárních cílových parametrů. 2 Doba do propuknutí SAD byla definována jako zvýšení alespoň 1 bodu na škále EDSS z jejího základního skóre > 1,0 (zvýšení 1,5 bodu u pacientů se základní EDSS 0), které se udrželo po dobu 6 měsíců. | ||||
Obrázek 1: Doba do 6měsíční trvalé akumulace postižení ve studii 2
Závažnost relapsu
V souladu s účinkem na četnost relapsů podpůrné analýzy ze studie 1 (CAMMS323) ukázaly, že přípravek LEMTRADA 12 mg/den vedl u jím léčených pacientů k významně nižšímu počtu závažných relapsů (61% snížení, p = 0,0056) a významně nižšímu počtu relapsů, jež následně vedly k léčbě steroidy (58% snížení, p < 0,0001), v porovnání s IFNB-1a.
Podpůrné analýzy ze studie 2 (CAMMS32400507) ukázaly, že přípravek LEMTRADA 12 mg/den vedl u jím léčených pacientů k významně nižšímu počtu závažných relapsů (48% snížení, p = 0,0121) a významně nižšímu počtu relapsů, jež následně vedly k léčbě steroidy (56% snížení, p < 0,0001) nebo hospitalizaci (55% snížení, p = 0,0045), v porovnání s IFNB-1a.
Trvalá redukce postižení (SRD)
Doba do nástupu SRD byla definována jako snížení alespoň jednoho bodu na škále EDSS z jejího základního skóre > 2, které se udrželo alespoň po dobu 6 měsíců. SRD je měřítkem pro trvalé zlepšení postižení. Ve studii 2 dosáhlo SRD 29 % pacientů léčených přípravkem LEMTRADA, zatímco u pacientů léčených subkutánně IFNB-1a dosáhlo tohoto cílového parametru pouze 13 %. Rozdíl byl statisticky významný
(p = 0,0002).
Studie 3 (studie fáze 2 CAMMS223) hodnotila bezpečnost a účinnost přípravku LEMTRADA u pacientů s RRRS v průběhu 5 let. Pacienti měli při vstupu do studie EDSS od 0-3,0, nejméně 2 klinické epizody RS v předchozích 2 letech a > 1 gadoliniem zvýrazněnou lézi. Pacienti předtím nedostávali léčbu na RS. Pacienti byli léčeni přípravkem LEMTRADA 12 mg/den (N = 108) nebo 24 mg/den (N = 108) podávaným jednou denně po dobu 5 po sobě jdoucích dnů v 0. měsíci a po dobu 3 dnů v 12. měsíci nebo subkutánně IFNB-1a 44 pg (N = 107) podávaným 3krát týdně po 3 roky. Čtyřicetšest pacientů podstoupilo třetí cyklus léčby přípravkem LEMTRADA 12 mg/den nebo 24/mg den po dobu 3 dnů ve 24. měsíci.
Po 3 letech přípravek LEMTRADA snížil riziko 6měsíčního SAD o 76 % (poměr rizik 0,24 [95% CI: 0,110; 0,545], p < 0,0006) a dále snížil ARR o 67 % (poměr výskytu 0,33 [95% CI: 0,196; 0,552], p < 0,0001) v porovnání se subkutánním IFNB-1a. Alemtuzumab 12 mg/den vedl k významně nižším skóre EDSS (zlepšení ve srovnání se základní hodnotou) v průběhu 2 let sledování ve srovnání s IFNB-1a (p < 0,0001).
Po 5 letech přípravek LEMTRADA snížil riziko SAD o 69 % (poměr rizik 0,31 [95% CI: 0,161; 0,598], p = 0,0005) a dále snížil ARR o 66 % (poměr frekvence 0,34 [95% CI: 0,202; 0,569], p < 0,0001) v porovnání se subkutánním IFNB-1a.
V klinických studiích s přípravkem LEMTRADA s otevřeným sledováním dostávali někteří pacienti „dle potřeby“ další léčbu přípravkem LEMTRADA dle zdokumentované souhrnné aktivity onemocnění RS. Další cykly léčby přípravkem LEMTRADA byly podávány takto: 12 mg/den po dobu 3 po sobě jdoucích dnů (celková dávka činila 36 mg) alespoň 12 měsíců po předchozím léčebném cyklu. Přínosy a rizika > 2 léčebných cyklů nebyly zcela prokázány, výsledky však naznačují, že bezpečnostní profil se s dalšími cykly léčby zřejmě nemění. Pokud mají být podány další léčebné cykly, musí být podány alespoň 12 měsíců po cyklu předchozím.
Imunogenicita
Tak jako u všech léčebných proteinů existuje i zde možnost imunogenicity. Údaje odráží procento pacientů, jejichž výsledky testů pomocí enzymové imunoanalýzy na pevné fázi (ELISA) byly považovány za pozitivní na protilátky proti alemtuzumabu a byly potvrzeny analýzou s využitím kompetitivní vazby. Pozitivní vzorky byly dále hodnoceny na známky inhibice in vitro pomocí analýzy průtokovou cytometrií. Pacienti v kontrolovaných klinických studiích s RS měli vzorky séra odebrané 1, 3 a 12 měsíců po každém léčebném cyklu, aby bylo možné stanovit protilátky proti alemtuzumabu. Přibližně 85 % pacientů dostávajících přípravek LEMTRADA bylo během studie v testech pozitivní na protilátky proti alemtuzumabu, 92 % těchto pacientů bylo v testech rovněž pozitivní na protilátky, které inhibovaly navázání přípravku LEMTRADA in vitro. U pacientů, u nichž se vytvořily protilátky proti alemtuzumabu, se tak stalo za 15 měsíců od úvodní expozice. Neexistovala souvislost přítomnosti protilátek proti alemtuzumabu nebo inhibičních protilátek proti alemtuzumabu a snížení účinnosti, změny ve farmakodynamice ani výskytu nežádoucích účinků, včetně reakcí spojených s infuzí.
Incidence protilátek je významně závislá na citlivosti a specificitě analýzy. Dále může být pozorovaná incidence pozitivity protilátek (včetně inhibičních protilátek) v analýze ovlivněna několika faktory, včetně metodologie analýzy, manipulace se vzorkem, načasováním odběru vzorku, souběžně podávanými léky a základním onemocněním. Z těchto důvodů může být srovnání incidence protilátek proti přípravku LEMTRADA s incidencí protilátek proti jiným produktům zavádějící.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s alemtuzumabem u dětí od narození do věku 10 let k léčbě roztroušené sklerózy (informace o použití u dětí viz bod 4.2).
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem LEMTRADA u jedné nebo více podskupin pediatrické populace při léčbě RRRS (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti přípravku LEMTRADA byly vyhodnoceny celkem u 216 pacientů s RRRS, kteří dostávali intravenózní infuze buď s 12 mg/den, nebo s 24 mg/den po dobu 5 po sobě jdoucích dnů a následně po dobu 3 po sobě jdoucích dnů 12 měsíců po úvodním cyklu léčby. Sérové koncentrace narůstají s každou následující dávkou v průběhu léčebného cyklu, přičemž nejvyšší pozorované koncentrace se vyskytují po poslední infuzi každého cyklu léčby. Podávání dávky 12 mg/den vedlo k průměrné Cmax 3014 ng/ml 5. den úvodního léčebného cyklu a 2276 ng/ml 3. den druhého léčebného cyklu. Alfa poločas se blížil 4-5 dnům a byl srovnatelný mezi jednotlivými cykly vedoucími k nízkým nebo nedetekovatelným sérovým koncentracím během cca 30 dnů po každém léčebném cyklu.
Alemtuzumab je protein, jehož očekávanou metabolickou cestou je degradace na malé peptidy a jednotlivé aminokyseliny prostřednictvím široce rozšířených proteolytických enzymů. Klasické biotransformační studie nebyly provedeny.
Z dostupných údajů nelze učinit závěry týkající se vlivu rasy a pohlaví na farmakokinetiku přípravku LEMTRADA. Farmakokinetika přípravku LEMTRADA nebyla u pacientů ve věku 55 let a starších zjišťována.
5.3 Předklinické údaje vztahující se k bezpečnosti
Kancerogeneze a mutageneze
Nebyly provedeny žádné studie ke zhodnocení kancerogenních nebo mutagenních účinků alemtuzumabu. Fertilita a reprodukce
Intravenózní léčba alemtuzumabem při dávkách až 10 mg/kg/den podávaná po dobu 5 po sobě jdoucích dnů (AUC 7,1násobně převyšující expozici při doporučené denní dávce u člověka) neměla žádný vliv na fertilitu ani reprodukční schopnost u huCD52 transgenních samců myší. Počet normálních spermií byl významně snížen (< 10 %) vzhledem ke kontrolám a procento abnormálních spermií (oddělené hlavičky nebo bez hlaviček) bylo výrazně zvýšené (až 3 %). Tyto změny však neovlivnily fertilitu a nebyly proto považovány za nepříznivé.
U samic myší, jimž byly podávány dávky alemtuzumabu až 10 mg/kg/den intravenózně (AUC 4,7násobně převyšující expozici při doporučené denní dávce u člověka) po dobu 5 po sobě jdoucích dnů před kohabitací s „wild-type“ samcem, byl průměrný počet žlutých tělísek a míst implantace na jednu myš výrazně snížen ve srovnání se zvířaty léčenými vehikulem. U březích myší s dávkami 10 mg/kg/den byl v průběhu březosti pozorován snížený přírůstek tělesné hmotnosti vzhledem ke kontrolám s vehikulem.
Studie reprodukční toxicity u březích myší vystavených intravenózním dávkám alemtuzumabu až 10 mg/kg/den (AUC 2,4násobně převyšující expozici u člověka při doporučené dávce 12 mg/den) po dobu 5 po sobě jdoucích dnů během období březosti vedly k výrazně zvýšenému počtu samic, jejichž všechny zárodky zemřely nebo zanikly, spolu se souběžným snížením počtu samic se životaschopnými plody. Při dávkách až 10 mg/kg/den nebyly pozorovány žádné vnější malformace, malformace měkkých tkání ani kosterní malformace či odchylky.
U myší byl během období březosti a po porodu pozorován placentární přenos a potenciální farmakologická aktivita alemtuzumabu. Ve studiích na myších byly pozorovány změny v počtu lymfocytů u mláďat vystavených alemtuzumabu během období březosti při dávkách 3 mg/kg/den podávaných po dobu 5 po sobě jdoucích dnů (AUC je 0,6násobně převyšující expozici u člověka při doporučené denní dávce 12 mg/kg). Kognitivní, fyzický a sexuální vývoj mláďat vystavených alemtuzumabu během kojení nebyl ovlivněn při dávkách alemtuzumabu až do 10 mg/kg/den.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát hydrogenfosforečnanu sodného (E339)
Dihydrát dinatrium-edetátu Chlorid draselný (E508)
Dihydrogen fosforečnan draselný (E340)
Polysorbát 80 (E433)
Chlorid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Koncentrát 3 roky
Naředěný roztok
Chemická a fyzikální stabilita po otevření před použitím byla prokázána po dobu 8 hodin při teplotě 2 °C -8 °C.
Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 8 hodin při teplotě 2 °C - 8 °C a přípravek musí být ochráněn před světlem.
6.4 Zvláštní opatření pro uchovávání
Koncentrát
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Přípravek LEMTRADA je dodáván v čiré 2ml skleněné injekční lahvičce s butylovou gumovou zátkou a hliníkovým těsněním s plastovým flip-off odtrhávacím víčkem.
Velikosti balení: krabička s 1 injekční lahvičkou.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Obsah injekční lahvičky je třeba před podáním zkontrolovat, zda se v něm nevytvořily částice nebo nedošlo ke změně barvy. Pokud jsou přítomny částice nebo pokud má koncentrát změněnou barvu, výrobek nepoužívejte.
Před použitím injekční lahvičku neprotřepávejte.
K intravenóznímu podání: za použití aseptické techniky natáhněte z injekční lahvičky 1,2 ml přípravku LEMTRADA do stříkačky. Vstříkněte do 100 ml infuzního roztoku chloridu sodného 9 mg/ml (0,9 %) nebo glukózy (5 %). Tento léčivý přípravek nesmí být ředěn jinými rozpouštědly. Roztok promíchejte opatrným otočením vaku.
Přípravek LEMTRADA neobsahuje žádné antimikrobiální konzervační látky, a proto je třeba dbát, aby byla zachována sterilita připraveného roztoku. Doporučuje se naředěný výrobek podat okamžitě. Všechny injekční lahvičky jsou určeny pouze k jednorázovému použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Genzyme Therapeutics Ltd 4620 Kingsgate Cascade Way
Oxford Business Park South Oxford OX4 2SU Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/869/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 12. září 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA
PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce/výrobců biologické léčivé látky
Boehringer Ingelheim Pharma GmbH & Co. KG Birkendorfer Strafle 65 88397 Biberach an der Riss NĚMECKO
Název a adresa výrobců odpovědných za propouštění šarží
Genzyme Limited 37 Hollands Road Haverhill Suffolk CB9 8PU Velká Británie
Genzyme Ireland Limited IDA Industrial Park Old Kilmeaden Road Waterford Irsko
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Před uvedením na trh v jednotlivých členských státech se držitel rozhodnutí o registraci dohodne s příslušným státním úřadem na vzdělávacím programu pro zdravotnické pracovníky a pacienty.
Držitel rozhodnutí o registraci zajistí dle smlouvy s příslušnými státními úřady v jednotlivých členských státech, kde bude přípravek LEMTRADA uveden na trh, aby při uvedení na trh a po něm všichni lékaři, kteří chtějí přípravek LEMTRADA předepisovat, dostali aktualizovaný edukační balíček pro lékaře obsahující následující prvky:
• Souhrn údajů o přípravku
• Příručka pro zdravotnické pracovníky
• Kontrolní seznam pro předepisující lékaře
• Příručka pro pacienta
• Karta pacienta
Příručka pro zdravotnické pracovníky bude obsahovat následující klíčové informace:
1. Popis rizik spojených s používáním přípravku LEMTRADA, jmenovitě:
• imunitní trombocytopenická purpura (ITP),
• nefropatie, včetně onemocnění s tvorbou protilátek proti bazální membráně glomerulů (anti-GBM),
• poruchy štítné žlázy.
2. Doporučení, jak tato rizika omezit prostřednictvím správného poradenství, monitorování a vedení pacienta.
3. Část Často kladené otázky.
Kontrolní seznam pro předepisující lékaře bude obsahovat následující klíčové body:
1. seznam testů, které je třeba provést při úvodním screeningu pacienta,
2. plán očkování, který je třeba dokončit 6 týdnů před zahájením léčby,
3. kontrola premedikace, celkového zdravotního stavu, těhotenství a antikoncepce těsně před léčbou,
4. monitorování činností v průběhu léčby a 4 roky po jejím ukončení,
5. odkaz na skutečnost, že byl pacient informován o riziku závažných autoimunitních onemocnění, infekcí a malignit, plně těmto rizikům rozumí a že byl také poučen, jak je minimalizovat.
Příručka pro pacienta bude obsahovat následující klíčové informace:
1. Popis rizik spojených s používáním přípravku LEMTRADA, jmenovitě:
• imunitní trombocytopenická purpura (ITP),
• nefropatie, včetně onemocnění s tvorbou protilátek proti bazální membráně glomerulů (anti-GBM),
• poruchy štítné žlázy,
• závažné infekce.
2. Popis známek a příznaků autoimunitních rizik
3. Popis optimálního postupu, pokud se zámky a příznaky těchto rizik objeví (např. jak se spojit s lékařem)
4. Doporučení pro plánování rozvrhu sledování
Karta pacienta bude obsahovat následující klíčové informace:
1. Varovné oznámení určené pro zdravotnického pracovníka, který bude pacienta ošetřovat (a to za jakýchkoli okolností, včetně akutních stavů), že pacient podstoupil léčbu přípravkem LEMTRADA.
2. Léčba přípravkem Lemtrada může zvyšovat riziko rozvoje následujících onemocnění:
• imunitní trombocytopenická purpura (ITP),
• nefropatie, včetně onemocnění s tvorbou protilátek proti bazální membráně glomerulů (anti-GBM),
• poruchy štítné žlázy,
• závažné infekce.
3. Kontaktní údaje na lékaře předepisujícího přípravek LEMTRADA.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
LEMTRADA 12 mg koncentrát pro infuzní roztok alemtuzumabum
Jedna injekční lahvička obsahuje alemtuzumabum 12 mg v 1,2 ml (10 mg/ml).
E339, dihydrát dinatrium-edetátu, E508, E340, E433, chlorid sodný, voda na injekci
Koncentrát pro infuzní roztok 1 injekční lahvička 12 mg/1,2 ml
Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
Podejte do 8 hodin po naředění.
Uchovávejte mimo dohled a dosah dětí.
EXP
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem. Uchovávejte v chladničce.
Chraňte před mrazem a neprotřepávejte.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Genzyme Therapeutics Ltd 4620 Kingsgate Cascade Way
Oxford Business Park South Oxford OX4 2SU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/869/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
<Nevyžaduje se - odůvodnění přijato>
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
LEMTRADA 12 mg sterilní koncentrát
alemtuzumabum
i.v.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1,2 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
LEMTRADA 12 mg koncentrát pro infuzní roztok
alemtuzumabum
^FtciUo přípravek podléhá dalšímu sledování.To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte
v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek LEMTRADA a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude přípravek LEMTRADA podán
3. Jak se přípravek LEMTRADA podává
4. Možné nežádoucí účinky
5. Jak přípravek LEMTRADA uchovávat
6. Obsah balení a další informace
1. Co je přípravek LEMTRADA a k čemu se používá
Přípravek LEMTRADA obsahuje léčivou látku alemtuzumab, která se používá k léčbě jedné z forem roztroušené sklerózy (RS) u dospělých pacientů zvané relabující-remitující roztroušená skleróza (RRRS). Přípravek LEMTRADA onemocnění RS nevyléčí, ale může snížit počet relapsů. Tento přípravek může rovněž pomoci zpomalit nebo zvrátit některé známky a příznaky RS. V klinických studiích měli pacienti léčení přípravkem LEMTRADA méně relapsů s nižší pravděpodobností výskytu zhoršení postižení v porovnání s pacienty léčenými interferonem beta aplikovaným injekčně několikrát týdně.
Co je roztroušená skleróza?
RS je autoimunitní onemocnění, které postihuje centrální nervový systém (tzn. mozek a míchu). U RS Váš imunitní systém chybně útočí na ochrannou vrstvu (myelin) obalující nervová vlákna a způsobuje zánět. Jakmile začne tento zánět způsobovat příznaky, jsou tyto často označovány jako „ataka“ nebo „relaps“.
U pacientů s RRRS je relaps následován obdobím zotavení.
Příznaky, které zažíváte, se mohou lišit v závislosti na tom, která část centrálního nervového systému je zasažena. Poškození nervů způsobené tímto zánětem bývá reverzibilní (zvratné), ale v průběhu onemocnění se s přibývajícími relapsy může stát trvalým.
Jak přípravek LEMTRADA účinkuje
Přípravek LEMTRADA upravuje funkci imunitního systému a brání mu napadat nervový systém.
2. Čemu musíte věnovat pozornost, než Vám bude přípravek LEMTRADA podán
NEPOUŽÍVEJTE přípravek LEMTRADA:
- jestliže jste alergický(á) na alemtuzumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
- jestliže trpíte infekcí virem lidské imunodeficience (HIV).
Upozornění a opatření
Před podáním přípravku LEMTRADA se poraďte se svým lékařem. Po absolvování cyklu léčby přípravkem LEMTRADA u Vás může existovat vyšší riziko rozvoje dalších autoimunitních onemocnění nebo závažných infekcí. Je důležité těmto rizikům rozumět a rozpoznat jejich případný výskyt. Dostanete Kartu pacienta a pokyny pro pacienta s dalšími informacemi. Je důležité, abyste si Kartu pacienta ponechal(a) po celou dobu léčby a dále také po dobu 4 let od poslední infuze přípravku LEMTRADA, neboť nežádoucí účinky se mohou objevit i po mnoha letech od ukončení léčby. Pokud budete podstupovat jakoukoli jinou léčbu (i jiného onemocnění než RS), ukažte svému lékaři Kartu pacienta.
Před zahájením léčby přípravkem LEMTRADA Vám lékař provede krevní testy. Pomocí nich se zjistí, zda jste pro jeho podávání vhodnými kandidáty. Před zahájením léčby přípravkem LEMTRADA se Váš lékař rovněž bude muset ujistit, že netrpíte některými jinými chorobami.
• Autoimunitní choroby
Léčba přípravkem LEMTRADA může zvyšovat riziko rozvoje autoimunitních onemocnění. Jde o onemocnění, při kterých Váš imunitní systém chybně útočí na Vaše vlastní tělo. Informace o některých konkrétních chorobách, které byly pozorovány u pacientů s RS léčených přípravkem LEMTRADA, jsou uvedeny níže.
Autoimunitní choroby se mohou objevit i po uplynutí mnoha let od ukončení léčby přípravkem LEMTRADA. Z tohoto důvodu je nutné po dobu 4 let od poslední infuze přípravku pravidelně testovat vzorky krve a moči. Toto testování je nezbytné i v případě, že se cítíte dobře a příznaky RS jsou pod kontrolou. Dále existují určité známky a příznaky, které musíte sledovat sám(a). Podrobnosti o těchto známkách a příznacích, o testování a o činnostech, které musíte provést, jsou popsány v bodě 4 -autoimunitní choroby.
Více užitečných informací o těchto autoimunitních onemocněních (a o jejich testování) naleznete v Příručce pro pacienty užívající přípravek LEMTRADA.
o Imunitní trombocytopenická purpura (ITP)
Méně často se u pacientů objevila krvácivá porucha způsobená nízkou hladinou krevních destiček zvaná imunitní trombocytopenická purpura (ITP). Tuto poruchu je nutné odhalit a léčit včas, neboť by jinak mohlo dojít k závažným nebo i smrtelným následkům. Známky a příznaky ITP jsou popsány v bodě 4.
o Poruchy ledvin (jako např. onemocnění s anti-GBM protilátkami)
Vzácně se u pacientů objevily autoimunitní problémy související s ledvinami, jako např. onemocnění ledvin s protilátkami proti bazální membráně glomerulů (onemocnění s anti-GBM protilátkami). Známky a příznaky onemocnění ledvin jsou popsány v bodě 4. Pokud není léčeno, může způsobit selhání ledvin vyžadující dialýzu nebo transplantaci a může vést k úmrtí.
o Poruchy štítné žlázy
Velmi často se u pacientů objevilo autoimunitní onemocnění štítné žlázy ovlivňující schopnost žlázy tvořit nebo řídit tvorbu hormonů, které jsou důležité pro metabolismus.
Přípravek LEMTRADA může způsobovat různé typy poruch štítné žlázy, včetně následujících:
• Nadměrně aktivní štítná žláza (hypertyreóza), kdy štítná žláza produkuje příliš mnoho hormonu
• Málo aktivní štítná žláza (hypotyreóza), kdy štítná žláza neprodukuje dostatečné množství hormonu.
Známky a příznaky onemocnění štítné žlázy jsou popsány v bodě 4.
Pokud se u Vás rozvine porucha štítné žlázy, je vysoce pravděpodobné, že budete muset být po zbytek života léčen(a) pomocí přípravků upravujících funkci štítné žlázy; v některých případech může být nezbytné štítnou žlázu odstranit.
Správná léčba poruchy štítné žlázy je velmi důležitá, a to zejména v případě, že po podávání přípravku LEMTRADA otěhotníte. Neléčená porucha štítné žlázy může poškodit ještě nenarozené dítě a může ublížit i dítěti po porodu.
o Jiné autoimunitní choroby
Vzácně se u pacientů objevily autoimunitní choroby spojené se stavem červených nebo bílých krvinek. Tato onemocnění je možné diagnostikovat pomocí krevních testů, které budete pravidelně absolvovat po léčbě přípravkem LEMTRADA. Pokud se u Vás některá z těchto poruch objeví, lékař Vás o tom bude informovat a provede příslušná léčebná opatření.
• Reakce na infuzi
U většiny pacientů léčených přípravkem LEMTRADA se objeví nežádoucí účinky v průběhu infuze nebo během 24 hodin po jejím ukončení. Váš lékař Vám podá další léčivé přípravky, které slouží k minimalizaci těchto reakcí (viz bod 4 - reakce na infuzi).
• Infekce
U pacientů léčených přípravkem LEMTRADA existuje vyšší riziko výskytu závažných infekcí (viz bod 4 -infekce). Obecně lze tyto infekce léčit standardními prostředky.
Váš lékař ověří, zda jiné užívané přípravky nemohou v tomto ohledu ovlivňovat Váš imunitní systém. Proto je důležité, abyste svému lékaři řekl(a) o všech přípravcích, které užíváte.
Pokud máte před zahájením léčby přípravkem LEMTRADA infekci, může Váš lékař zvážit odložení začátku léčby do doby, než bude tato infekce pod kontrolou nebo zcela vyléčená.
Pacienti léčení přípravkem LEMTRADA mají větší riziko rozvoje herpetické infekce (např. oparu rtu). Obecně platí, že jakmile má pacient herpetickou infekci, je u něj zvýšené riziko rozvoje další infekce. Je také možné, že k rozvoji herpetické infekce dojde poprvé. Doporučuje se, aby Vám lékař předepsal léky ke snížení pravděpodobnosti rozvoje herpetické infekce, které je třeba užívat po dobu léčby přípravkem LEMTRADA a jeden měsíc po léčbě.
Dále je možný výskyt infekcí, které mohou mít za následek abnormality děložního čípku. Proto se doporučuje, aby všechny ženy absolvovaly pravidelné roční screeningové vyšetření, jakým je např. stěr z čípku. Váš lékař Vám sdělí, které testy budete muset absolvovat.
Pacienti kteří jsou léčeni přípravkem LEMTRADA jsou vystaveni vyššímu riziku vzniku listeriální infekce/listeriozního zánětu mozkových blan . Pro snížení tohoto rizika by se pacienti měli vyhnout po dobu alespoň jednoho měsíce po podání přípravku LEMTRADA konzumaci syrového nebo nedostatečně tepelně upraveného masa, měkkých sýrů a nepasterizovaných mléčných výrobků.
Pokud žijete v oblasti, kde se často vyskytují tuberkulózní infekce, může u Vás existovat vyšší riziko výskytu této infekce. Váš lékař Vám naplánuje screeningové vyšetření na tuberkulózu.
Pokud jste přenašeč(ka) žloutenky typu B nebo C (nemoci postihující játra), bude před zahájením léčby přípravkem LEMTRADA nutné dbát zvláštní opatrnosti, neboť není známo, zda léčba nemůže toto onemocnění aktivovat a žloutenka pak může následně poškodit játra.
• Dříve diagnostikované nádorové onemocnění
Pokud Vám v minulosti bylo diagnostikováno nádorové onemocnění, informujte o tom svého lékaře.
• Očkování
Není známo, zda přípravek LEMTRADA může ovlivnit Vaši odpověď na očkování. Pokud nemáte dokončené standardní očkovací schéma dle kalendáře, Váš lékař rozhodne, zda je máte podstoupit ještě před zahájením léčby přípravkem LEMTRADA. Lékař rovněž zváží, zda bude třeba očkovat Vás proti planým neštovicím, pokud jste je dosud neprodělal(a). Veškerá očkování je nutné provést minimálně 6 týdnů před zahájením cyklu léčby přípravkem LEMTRADA.
Pokud Vám byl v nedávné době podán přípravek LEMTRADA, NESMÍTE podstoupit určité typy očkování (živé virové vakcíny).
Děti a dospívající
Přípravek LEMTRADA není určen k použití u dětí ani dospívajících mladších 18 let, neboť jeho účinek nebyl u pacientů s RS mladších 18 let studován.
Další léčivé přípravky a přípravek LEMTRADA
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které plánujete užívat (včetně jakéhokoli očkování nebo rostlinných přípravků).
Kromě přípravku LEMTRADA existují i další formy léčby (včetně léčby RS nebo jiných onemocnění), které mohou ovlivňovat imunitní systém a mít vliv na schopnost bojovat s infekcemi. Pokud užíváte takový lék, lékař Vás může před zahájením léčby přípravkem LEMTRADA požádat o jeho vysazení.
Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než Vám bude tento přípravek podán.
Ženy, které mohou otěhotnět, musí v průběhu každého cyklu léčby přípravkem LEMTRADA a po dobu 4 měsíců po každém cyklu používat účinnou metodu antikoncepce.
Pokud po léčbě přípravkem LEMTRADA otěhotníte a během těhotenství se u Vás rozvine porucha štítné žlázy, je nutná zvláštní pozornost. Poruchy štítné žlázy mohou být škodlivé pro dítě (viz bod 2 Upozornění a opatření - autoimunitní choroby).
Kojení
Není známo, zda přípravek LEMTRADA přechází do mateřského mléka, ale existuje možnost, že tomu tak je. Proto se nedoporučuje v průběhu každého cyklu léčby přípravkem LEMTRADA ani po dobu 4 měsíců po dokončení každého cyklu kojit. Kojení však může být přínosné (mateřské mléko pomáhá chránit dítě před infekcemi), proto pokud plánujete své dítě kojit, obraťte se na svého lékaře. Váš lékař Vám poradí, co je pro Vás a Vaše dítě nejlepší.
Plodnost
Přípravek LEMTRADA může v průběhu cyklu léčby a také po dobu následujících 4 měsíců přetrvávat ve Vašem těle. Není známo, zda přípravek LEMTRADA má během této doby vliv na plodnost. Pokud v této době plánujete otěhotnět, promluvte si se svým lékařem.
Řízení dopravních prostředků a obsluha strojů
Mnoho pacientů může mít nežádoucí účinky v průběhu infuze nebo do 24 hodin od podání přípravku LEMTRADA a při některých z nich (např. závratích) může být nebezpečné řídit dopravní prostředky či obsluhovat stroje. Pokud se u Vás takové účinky objeví, těmto činnostem se vyhněte, dokud se nebudete cítit lépe.
Přípravek LEMTRADA obsahuje draslík a sodík
Tento léčivý přípravek obsahuje méně než 1 mmol draslíku (39 mg) v jedné infuzi, tj. je v podstatě „bez obsahu draslíku“.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné infuzi, tj. je v podstatě „bez obsahu sodíku“.
3. Jak se přípravek LEMTRADA podává
Lékař Vám vysvětlí, jak Vám bude přípravek LEMTRADA podáván. Máte-li jakékoli otázky, zeptejte se svého lékaře.
Během prvního cyklu léčby budete dostávat jednu infuzi denně po dobu 5 dní (cyklus 1).
O jeden rok později budete dostávat jednu infuzi denně po dobu 3 dní (cyklus 2).
Mezi těmito dvěma cykly nebudete přípravek LEMTRADA dostávat.
Maximální denní dávku představuje jedna infuze.
Přípravek LEMTRADA bude podáván formou infuze do žíly. Každá infuze bude trvat přibližně 4 hodiny. U většiny pacientů sníží 2 cykly této léčby aktivitu RS na 2 roky. Ve sledování nežádoucích účinků a pravidelném testování je však třeba pokračovat po dobu 4 let po poslední infuzi.
Schéma uvedené níže srozumitelně ilustruje dobu trvání účinků léčby a dobu nutného sledování.
Začněte s testováním krve a moči
před léčbou-- a pokračujte po dobu 4 let po poslední infuzi
Léčebná kúra 1 Léčebná kúra 2
um ni
Sdenní léčba 3denní léčba
Rok 1 Rok 2 Rok 3 Rok 4 Rok 5
POZNÁMKA U většiny pacientů mají 2 cykly vliv 2 roky nebo déle.
Sledování po ukončení léčby přípravkem LEMTRADA
Po podání přípravku LEMTRADA budete muset podstoupit pravidelné testování, které slouží k zajištění včasné diagnózy a léčby možných nežádoucích účinků. Tato testování je nutné pravidelně provádět po dobu 4 let po podání poslední infuze. Blíže jsou popsána v bodě 4 - nejdůležitější nežádoucí účinky.
Jestliže jste dostal(a) více přípravku LEMTRADA, než jste měl(a)
U pacientů, kterým bylo omylem podáno více přípravku LEMTRADA v jedné infuzi, se objevily závažné nežádoucí účinky jako např. bolesti hlavy, vyrážka, nízký krevní tlak nebo zvýšená srdeční frekvence. Podání vyšší než doporučené dávky může mít za následek závažnější nebo déle trvající reakce na infuzi (viz bod 4) nebo silnější účinek na imunitní systém. V těchto případech je nutné přerušit podávání přípravku LEMTRADA a příznaky léčit.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i přípravek LEMTRADA nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nejdůležitější nežádoucí účinky jsou autoimunitní poruchy popsané v bodě 2, mezi které patří:
• ITP (krvácivá porucha), (méně častá - může se objevit až u 1 pacienta ze 100): může se projevovat jako malé rozptýlené tečky na kůži červené, růžové nebo fialové barvy; snadno se tvořícími modřinami; obtížně zastavitelným krvácením z řezných ran; silnějším, delším nebo častějším menstruačním krvácením, než je obvyklé, krvácením mimo menstruační cyklus; krvácením z dásní nebo nosu, které je u pacienta nové nebo jej lze hůře zastavit; nebo vykašláváním krve.
• poruchy ledvin (vzácné - mohou se objevit až u 1 pacienta z 1000): mohou se projevovat jako krev v moči (moč má červené nebo „čajové“ zbarvení), otoky končetin nebo chodidel. Může také způsobit poškození plic vedoucí k vykašlávání krve.
Pokud se u Vás objeví známky nebo příznaky krvácivých poruch nebo poruch ledvin, ihned tyto příznaky nahlaste svému lékaři. Pokud Váš lékař není dostupný, ihned vyhledejte lékařskou pomoc.
• poruchy štítné žlázy (velmi časté - mohou se objevit u více než 1 pacienta z 10): mohou se projevovat jako nadměrné pocení; nevysvětlitelný pokles nebo nárůst tělesné hmotnosti; otoky očí; nervozita; rychlý srdeční tep; pocit chladu; zhoršující se únava; nebo zácpa (u pacienta nová).
• poruchy červené a bílé krevní řady (vzácné - mohou se objevit až u 1 pacienta z 1000) diagnostikované z krevních testů.
Souhrn testů na autoimunitní poruchy:
|
Test |
Kdy? |
Jak dlouho? |
|
Krevní test (pro diagnostiku všech výše uvedených důležitých závažných nežádoucích účinků) |
Před zahájením léčby a každý měsíc po jejím ukončení |
Po dobu 4 let po poslední infuzi přípravku LEMTRADA |
|
Testy moči (dodatečný test pro diagnostiku poruch ledvin) |
Před zahájením léčby a každý měsíc po jejím ukončení |
Po dobu 4 let po poslední infuzi přípravku LEMTRADA |
Tyto závažné nežádoucí účinky se mohou objevit i mnoho let po ukončení léčby přípravkem LEMTRADA. Pokud se u Vás některé známky nebo příznaky objeví, ihned o nich informujte svého lékaře. Budete rovněž podstupovat pravidelné testy vzorků krve a moči, aby mohly být tyto poruchy včas zjištěny a rychle léčeny._
Pokud se u Vás po tomto období projeví příznaky ITP, poruchy ledvin nebo štítné žlázy, Váš lékař provede další testy. Je zapotřebí, abyste nadále po dobu čtyř let sledoval(a) známky a příznaky nežádoucích účinků, jak je podrobně uvedeno v pokynech pro pacienta, a měl(a) byste s sebou nadále nosit Kartu pacienta.
Dalším důležitým nežádoucím účinkem je zvýšené riziko vzniku infekcí (informace o četnosti výskytu infekcí u pacientů viz níže). Ve většině případů jsou tyto infekce mírné, ale mohou se vyskytnout i závažné infekce.
Pokud zpozorujete některou z následujících známek infekce, ihned se obraťte na svého lékaře: • horečka a/nebo třesavka
_• zduřelé lymfatické uzliny_
Za účelem snížení rizika vzniku některých infekcí může Váš lékař zvážit očkování proti planým neštovicím a/nebo další očkování, která bude považovat za nezbytná (viz bod 2: Čemu musíte věnovat pozornost, než Vám bude přípravek LEMTRADA podán - Očkování). Lékař Vám může rovněž předepsat léky na opar (viz bod 2: Čemu musíte věnovat pozornost, než Vám bude přípravek LEMTRADA podán - Infekce).
Nejčastější nežádoucí účinky jsou reakce na infuzi (informace o četnosti těchto nežádoucích účinků viz níže), které se objevují během podávání infuze nebo do 24 hodin po jejím podání. Ve většině případů jsou tyto reakce mírné, ale mohou se vyskytnout i závažné reakce. Ojediněle se mohou objevit i alergické reakce.
Pro omezení výskytu reakcí na infuzi Vám Váš lékař podá před každou z prvních 3 infuzí v rámci cyklu léčby přípravkem LEMTRADA další léčivé přípravky (kortikosteroidy). Před podáním infuze nebo při výskytu příznaků mohou být použity další formy léčby k omezení těchto reakcí. V průběhu infuze a po dobu 2 hodin po jejím dokončení budete pod kontrolou. V případě závažných reakcí může být infuze zpomalena nebo přerušena.
Více informací o těchto případech naleznete v Pokynech pro pacienta užívajícího přípravek LEMTRADA.
Nežádoucí účinky, které se u Vás mohou objevit, jsou:
Velmi časté nežádoucí účinky (mohou se objevit u více než 1 pacienta z 10):
• Reakce na infuzi, které se mohou vyskytnout během podávání infuze nebo do 24 hodin po jejím dokončení: bolesti hlavy, vyrážka, horečka, nevolnost, kopřivka, svědění, zarudnutí obličeje a krku, pocit únavy;
• Infekce: infekce dýchacích cest jako např. nachlazení, záněty dutin, cystitida;
• Snížení počtu bílých krvinek (lymfocytů).
Časté nežádoucí účinky (mohou se objevit až u 1 pacienta z 10)
• Reakce na infuzi, které se mohou vyskytnout během podávání infuze nebo do 24 hodin po jejím dokončení: změny v srdeční frekvenci, porucha trávení, třesavka, nepříjemný pocit na hrudi, bolest, závratě, změny chuti, problémy se spaním, ztížené dýchání nebo dušnost, vyrážka po celém těle, nízký krevní tlak;
• Infekce: kašel, infekce ucha, příznaky připomínající chřipku, bronchitida, pneumonie, moučnivka
v ústech nebo v pochvě, pásový opar, plané neštovice, opar na rtu, zduřelé nebo zvětšené lymfatické uzliny;
• bolest v místě infuze, bolesti zad, krku nebo paží či nohou, bolesti svalů, křeče svalů, bolesti kloubů, bolesti úst nebo krku;
• zánět úst/dásní/jazyka;
• celkový pocit nemoci, slabost, zvracení, průjem, bolesti břicha, střevní chřipka;
• pálení žáhy;
• abnormality zjištěné při vyšetřeních: krev nebo bílkovina v moči, snížení srdeční frekvence, nepravidelná srdeční frekvence nebo abnormální srdeční tep, vysoký krevní tlak;
• relaps RS;
• chvění, ztráta citlivosti, pocit pálení nebo svědění;
• nadměrně nebo málo aktivní štítná žláza nebo struma (otok štítné žlázy v krku);
• otok paží a/nebo nohou;
• problémy se zrakem;
• pocit úzkosti;
• abnormálně silná, prodloužená nebo nepravidelná menstruace;
• akné, erytém kůže, nadměrné pocení;
• krvácení z nosu, tvorba modřin;
• vypadávání vlasů.
Méně časté nežádoucí účinky (mohou se objevit až u 1 pacienta ze 100):
• Infekce: genitální opar, infekce oka, infekce zubu;
• problémy s krevní srážlivostí, chudokrevnost;
• atletická noha;
• abnormální vaginální výtěr;
• deprese;
• zvýšená citlivost;
• obtížné polykání;
• škytavka;
• snížení tělesné hmotnosti;
• zácpa;
• krvácení z dásní;
• abnormální jaterní testy;
• puchýře.
Kartu pacienta a tuto příbalovou informaci ukažte každému lékaři, který se podílí na Vaší léčbě, nikoli jen svému neurologovi.
Tyto informace naleznete rovněž na Kartě pacienta a v Pokynech pro pacienta, které Vám předal Váš lékař. Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek LEMTRADA uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku lahvičky a na krabičce za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Z důvodu možného rizika mikrobiální kontaminace je doporučeno přípravek použít ihned po naředění. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 8 hodin při 2 °C až 8 °C, za předpokladu, že je přípravek chráněn před světlem.
Nepoužívejte tento přípravek, pokud si všimnete přítomnosti částic v roztoku nebo změny barvy roztoku v injekční lahvičce.
Nevyhazujte žádné léčivé přípravky do odpadních vod. Pomůžete tak chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek LEMTRADA obsahuje Léčivou látkou je alemtuzumab.
Jedna lahvička obsahuje 12 mg alemtuzumabum v 1,2 ml.
Dalšími složkami jsou:
• dihydrát hydrogenfosforečnanu sodného (E339)
• dihydrát dinatrium-edetátu
• chlorid draselný (E508)
• dihydrogenfosforečnan draselný (E340)
• polysorbát 80 (E433)
• chlorid sodný
• voda na injekci
Jak přípravek LEMTRADA vypadá a co obsahuje toto balení
Přípravek LEMTRADA je čirý, bezbarvý až světle žlutý koncentrát pro infuzní roztok (sterilní koncentrát) dodávaný ve skleněné lahvičce se zátkou.
Každé balení obsahuje 1 injekční lahvičku.
Držitel rozhodnutí o registraci
Genzyme Therapeutics Ltd, 4620 Kingsgate, Cascade Way, Oxford Business Park South, Oxford, OX4 2SU, Velká Británie
Výrobce
Genzyme Ltd., 37 Hollands Road, Haverhill, Suffolk CB9 8PU, Velká Británie.
Genzyme Ireland Limited, IDA Industrial Park, Old Kilmeaden Road, Waterford, Irsko.
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien/ Luxemburg/Luxembourg Sanofi Belgium Tél/Tel: + 32 2 710 54 00 |
Lietuva UAB „SANOFI-AVENTIS LIETUVA Tel. +370 5 275 5224 |
|
Btarapna Sanofi-Aventis Bulgaria EOOD Ten: +359 2 9705300 |
Magyarország sanofi-aventis Zrt Tel: +36 1 505 0050 |
|
Česká republika sanofi-aventis, s.r.o. Tel: +420 233086 111 |
Malta Sanofi-Aventis Malta Ltd Tel: +356 21493022 |
|
Danmark sanofi-aventis Denmark A/S Tlf: +45 45 16 70 00 |
Nederland Genzyme Europe B.V. Tel: +31 35 699 1200 |
|
Deutschland Genzyme Therapeutics Ltd. Tel: +49 (0) 6102 3674 451 |
Norge sanofi-aventis Norge AS Tlf: + 47 67 10 71 00 |
|
Eesti sanofi-aventis Estonia OU Tel. +372 6 273 488 |
Ósterreich sanofi-aventis GmbH Tel: + 43 1 80 185 - 0 |
|
Ekkáda/Kúnpo^ sanofi-aventis AEBE (EkkáSa) T pk: +30 210 900 1600 |
Polska sanofi-aventis Sp. z o.o. Tel.: +48 22 280 00 00 |
|
Espaňa Genzyme, S.L.U. Tel: +34 93 485 94 00 sanofi-aventis, S.A. Tel: +34 93 485 94 00 |
Portugal Sanofi - Produtos Farmaceuticos, Lda. Tel: +351 21 422 0100 |
|
France Genzyme S.A.S. Tél : +33 (0) 825 825 863 |
Romania sanofi-aventis Romania S.R.L. Tel: +40 (0) 21 317 31 36 |
|
Hrvatska sanofi-aventis Croatia d.o.o. Tel: +385 1 6003 400 |
Slovenija sanofi-aventis d.o.o. Tel: +386 1 560 4800 |
Ísland
Slovenská republika
sanofi-aventis Pharma Slovakia s.r.o. Tel.: +421 2 33 100 100
Suomi/Finland
sanofi-aventis Oy Puh/Tel: + 358 201 200 300
Sverige
sanofi-aventis AB Tel: +46 (0)8 634 50 00
United Kingdom
Genzyme Therapeutics Ltd. (United Kingdom)
Tel: +44 (0) 1865 405200
Vistorhf.
Sími: +354 535 7000 Ireland
GenzymeTherapeutics Ltd. (United Kingdom) Tel: +44 (0) 1865 405200
Italia
Genzyme Srl Tel: +39 059 349 811
Latvija
sanofi-aventis Latvia SIA Tel: +371 67 33 24 51
Tato příbalová informace byla naposledy revidována Další zdroje informací
Pro edukaci pacientů ohledně možných nežádoucích účinků a pokynů, co dělat v případě výskytu určitých nežádoucích účinků, jsou k dispozici následující materiály pro minimalizaci možných rizik:
1 Karta pacienta: Pacient tuto kartu předloží dalším zdravotnickým pracovníkům a informuje je tak, že
mu byl podán přípravek LEMTRADA
2 Pokyny pro pacienta: Obsahují podrobné informace o autoimunitních reakcích, infekcích a další
informace.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese: http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Informace o minimalizaci možných rizik - autoimunitní stavy
• Je mimořádně důležité, aby pacient pochopil nutnost provádění pravidelného testování (po dobu 4 let po poslední infuzi přípravku), a to i v případě, že je pacient asymptomatický a RS je dobře kontrolovaná.
• Ve spolupráci s pacientem naplánujte pravidelné sledování a dodržujte je.
• U nespolupracujících pacientů může být nutný další pohovor, kde zdůrazníte rizika vynechání naplánovaných testů.
• Sledujte výsledky testů a hledejte příznaky nežádoucích účinků.
• Projděte si Pokyny pro pacienta a příbalovou informaci přípravku LEMTRADA spolu s pacientem. Připomeňte pacientovi, aby nezapomínal sledovat příznaky související s autoimunitními stavy a aby v případě jakýchkoli obav vyhledal lékařskou pomoc.
K dispozici jsou rovněž výukové materiály pro zdravotnické pracovníky:
• Pokyny k přípravku LEMTRADA pro zdravotnické pracovníky
• Školicí modul k přípravku LEMTRADA
• Kontrolní seznam pro předepisování přípravku LEMTRADA
Další informace si přečtěte v souhrnu údajů o přípravku (k dispozici na výše uvedené webové stránce agentury EMA).
Informace o přípravě přípravku LEMTRADA k podání a o sledování pacientů
• Těsně před podáním infuze přípravku LEMTRADA je nutné pacienty první 3 dny cyklu léčby premedikovat kortikosteroidy. Rovněž je možné před podáním přípravku LEMTRADA zvážit premedikaci podáním antihistaminik a/nebo antipyretik.
• Všem pacientům je nutné v průběhu cyklu léčby a po dobu 1 měsíce po cyklu podávat perorální antivirotika proti viru herpes simplex. V klinických studiích bylo pacientům podáváno 200 mg acikloviru dvakrát denně (nebo ekvivalentní dávka).
• Proveďte vstupní testy a screening dle popisu v Souhrnu údajů o přípravku, bod 4.
• Obsah injekční lahvičky je nutné před podáním zkontrolovat - ujistěte se, že neobsahuje částice nebo nedošlo ke změně zbarvení. Pokud koncentrát obsahuje částice nebo pokud vykazuje změnu zbarvení, přípravek nepoužívejte.
PŘED POUŽITÍM INJEKČNÍ LAHVIČKY NEPROTŘEPÁVEJTE.
• Asepticky odeberte 1,2 ml přípravku LEMTRADA z injekční lahvičky a vstříkněte jej do 100 ml infuzního roztoku chloridu sodného 9 mg/ml (0,9% roztok) nebo do infuzního roztoku glukózy (5% roztok). Roztok promíchejte opatrným převrácením infuzního vaku. Je nutné dávat pozor na zajištění sterility připraveného roztoku, neboť tento roztok neobsahuje žádné konzervační látky.
• Infuzní roztok přípravku LEMTRADA aplikujte intravenózní infuzí po dobu přibližně 4 hodin.
• Do infuzního roztoku přípravku LEMTRADA se nesmí přidávat žádné další léčivé přípravky, ani se tyto přípravky nesmí podávat přes stejnou intravenózní linku.
• Doporučuje se přípravek použít ihned po naředění z důvodu možného rizika mikrobiální kontaminace. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 8 hodin při 2°C až 8°C, za předpokladu, že je přípravek chráněn před světlem.
• Dodržujte postupy pro správnou manipulaci a likvidaci. Veškerý rozlitý nebo odpadní materiál je nutné zlikvidovat v souladu s místními požadavky.
• Po každé infuzi je u pacienta nutné po dobu 2 hodin sledovat výskyt reakcí souvisejících s infuzí.
V případě potřeby je možné zahájit symptomatickou léčbu - viz Souhrn údajů o přípravku. Pacienta testujte každý měsíc na výskyt autoimunitních poruch, a to až do uplynutí 4 let od poslední infuze přípravku. Další informace naleznete v Pokynech k přípravku LEMTRADA pro zdravotnické pracovníky nebo si přečtěte Souhrn údajů o přípravku, který je k dispozici na výše uvedené webové stránce agentury EMA.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY V REGISTRACI
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizovaných zpráv o bezpečnosti (PSUR) alemtuzumabu dospěl výbor CHMP k těmto vědeckým závěrům:
Listerióza / Listeriózní meningitida
Léky, které mají modulační účinek na imunitní systém jako přípravek Lemtrada, mohou být spojeny se zvýšeným rizikem oportunních infekcí. Bylo identifikováno celkem 5 hlášení z EU. U jednoho pacienta léčeného alemtuzumabem pro MS , zahrnutého do klinické studie CAMMS223, se rozvinula listeriózní meningitida a u čtyř spontánně hlášených post-marketingových případů se objevila buď systémová listerióza, nebo meningitida způsobená Listeria monocytogenes.
Bradykardie jako nežádoucí účinek související s infuzí
Sedmdesát jedna případů (u 55 pacientů) bradykardie (z nichž dva případy byly hodnoceny jako závažné, zbytek jako nezávažné) bylo hlášeno v klinických studiích. V těchto studiích bylo celkem 1505 pacientů vystaveno alemtuzumabu. Navíc třicet devět případů bradykardie (osm z nich bylo vyhodnoceno jako závažné, zbytek jako nezávažné) bylo hlášeno po uvedení přípravku na trh ke dni 1. května 2015. Každý z deseti závažných případů zahrnujících bradykardii se vyskytl v kontextu infuzní reakce
Proto s ohledem na předložené údaje v hodnoceném PSUR má PRAC za to, že změny v informacích o přípravku léčivých přípravků obsahujících alemtuzumab jsou odůvodněné. Bod 4.4 Souhrnu údajů o přípravku a odpovídající body Příbalové informace jsou aktualizovány.
Výbor CHMP souhlasí s vědeckými závěry - výboru PRAC.
Zdůvodnění doporučující změnu podmínek rozhodnutí o registraci
Na základě vědeckých závěrů týkajících se alemtuzumabu výbor CHMP zastává stanovisko, že poměr přínosů a rizik léčivého přípravku obsahujícího alemtuzumab zůstává nezměněný, a to pod podmínkou, že v údajích o přípravku budou provedeny navrhované změny.
Výbor CHMP doporučuje změnu v registraci.
41