Laventair 55 Mikrogramů/22 Mikrogramů
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
LAVENTAIR 55 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna inhalace poskytuje dávku (dávka, která vychází z náustku) umeclidinii bromidum 65 mikrogramů, což odpovídá umeclidinium 55 mikrogramů, a vilanterolum 22 mikrogramů (ve formě trifenatas). To odpovídá dávkované dávce umeclidinii bromidum 74,2 mikrogramů, což odpovídá umeclidinium 62,5 mikrogramů a vilanterolum 25 mikrogramů (ve formě trifenatas).
Pomocná látka se známým účinkem
Jedna dávka obsahuje přibližně 25 mg laktosy (ve formě monohydrátu).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Dávkovaný prášek k inhalaci (prášek k inhalaci).
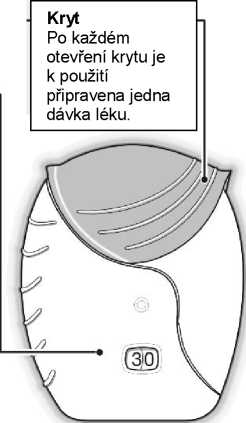
Bílý prášek ve světle šedém inhalátoru (ELLIPTA) s červeným krytem náustku a počítadlem dávek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek LAVENTAIR je indikován jako udržovací bronchodilatační léčba ke zmírnění příznaků chronické obstrukční plicní nemoci (CHOPN) u dospělých pacientů.
4.2 Dávkování a způsob podání
Dávkování
Dospělí
Doporučená dávka je jedna inhalace přípravku LAVENTAIR 55/22 mikrogramů jednou denně.
K zachování bronchodilatačního účinku je nutné přípravek LAVENTAIR podávat jednou denně, každý den ve stejnou dobu. Maximální dávka je jedna inhalace přípravku LAVENTAIR 55/22 mikrogramů jednou denně.
Zvláštní populace Starší pacienti
U pacientů starších 65 let není úprava dávkování nutná.
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin není úprava dávkování nutná.
Porucha funkce jater
U pacientů s lehkou nebo středně závažnou poruchou funkce jater není úprava dávkování nutná. Přípravek LAVENTAIR nebyl hodnocen u pacientů se závažnou poruchou funkce jater a je třeba ho u takových pacientů používat s opatrností.
Pediatrická populace
U pediatrické populace (do 18 let) neexistují žádné relevantní důvody pro používání přípravku LAVENTAIR v indikaci CHOPN.
Způsob podání
Přípravek LAVENTAIR je určen pouze k inhalačnímu podání.
Návod k použití:
Následující návod k použití pro 30dávkový inhalátor lze rovněž použít pro 7dávkový inhalátor.
Inhalátor ELLIPTA obsahuje odměřené dávky a je připravený přímo k použití.
Inhalátor je uložený v ochranné vaničce obsahující sáček s vysoušedlem, které snižuje vlhkost. Po otevření je nutné sáček s vysoušedlem vyhodit; sáček se neinhaluje ani nejí. Pacient má být poučen, aby vaničku neotevíral dříve, než bude připraven k inhalaci dávky.
Inhalátor je po prvním vyjmutí ze zatavené vaničky v „uzavřené“ pozici. Označení „Spotřebujte do“ vyjadřuje datum, které by mělo být zapsáno do štítku inhalátoru. Datum „Spotřebujte do“ je 6 týdnů od data otevření vaničky. Po tomto datu se již nemá inhalátor dále používat. Vanička může být znehodnocena po prvním otevření.
Pokud se kryt inhalátoru otevře a zavře bez toho, že by došlo k inhalaci léku, dojde ke ztrátě dávky. Ztracená dávka zůstane bezpečně uzavřená v inhalátoru, ale nebude již dostupná k inhalaci.
Při jedné inhalaci není možné náhodně použít dávku navíc ani dvojnásobnou dávku.
a) Příprava dávky
Pokud jste připraven(a) k použití dávky, otevřete kryt inhalátoru. Inhalátorem netřeste.
Stahujte víčko dolů, dokud neuslyšíte „cvaknutí“. Přípravek je nyní připraven k inhalaci.
Počítadlo dávek pro potvrzení odečetlo 1 dávku. Pokud počítadlo neodečte dávku v okamžiku, kdy uslyšíte „cvaknutí“, inhalátor neumožní inhalaci léku. Vezměte jej zpět k lékárníkovi, aby Vám poradil
b) Jak se léčivý přípravek inhaluje
Držte inhalátor dále od úst a co nejvíce vydechněte, jak je Vám pohodlné. Nevydechujte do inhalátoru.
Vložte náustek mezi rty a pevně jej svými rty stiskněte. Neblokujte vzduchové otvory prsty.
• Jednou se dlouze, rovnoměrně a zhluboka nadechněte. Zadržte dech po co nejdelší dobu (alespoň 34 sekundy).
• Vyjměte inhalátor z úst.
• Pomalu a lehce vydechněte.
Lék by neměl mít žádnou chuť ani by neměl být cítit, a to ani v případě, že se inhalátor použije správně.
c) Uzavření inhalátoru
Pokud chcete náustek očistit, otřete jej před uzavřením suchou tkaninou.
Vysuňte co nejvíce kryt zpět nahoru, až je náustek zakrytý.
4.3 Kontraindikace
Hypersenzitivita na léčivé látky nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Kombinace umeklidinium/vilanterol se nemá používat u pacientů s astmatem, protože u této populace pacientů nebyl přípravek hodnocen.
Paradoxní bronchospasmus
Stejně jako u jiné inhalační léčby, může i podání kombinace umeklidinium/vilanterol vést k paradoxnímu bronchospasmu, který může být život ohrožující. Pokud dojde k paradoxnímu bronchospasmu, je nutné léčbu kombinací umeklidinium/vilanterol okamžitě přerušit a dle potřeby zahájit alternativní léčbu.
Není určen k akutnímu užití
Kombinace umeklidinium/vilanterol není indikována k léčbě akutních epizod bronchospasmu.
Zhoršení základního onemocnění
Častější používání krátkodobě působících bronchodilatancií ke zmírnění příznaků ukazuje na zhoršení kontroly onemocnění. V případě zhoršení CHOPN v průběhu léčby kombinací umeklidinium/vilanterol je třeba přehodnotit zdravotní stav pacienta i režim léčby CHOPN.
Kardiovaskulární účinky
Při podávání antagonistů muskarinových receptorů a sympatomimetik, včetně kombinace umeklidinium/vilanterol, se mohou objevit kardiovaskulární účinky, jako srdeční arytmie, např. fibrilace síní nebo tachykardie. Pacienti s klinicky významným nekontrolovaným kardiovaskulárním onemocněním byli navíc z klinických studií vyloučeni. Proto je nutné kombinaci umeklidinium/vilanterol podávat pacientům se závažným srdečním onemocněním s opatrností.
Antimuskarinové účinky
V souladu s antimuskarinovou aktivitou je třeba kombinaci umeklidinium/vilanterol podávat pacientům s retencí moči nebo s glaukomem s úzkým úhlem s opatrností.
Hypokalemie
U některých pacientů mohou beta2-adrenergní agonisté vyvolat významnou hypokalemii, která má potenciální nežádoucí kardiovaskulární účinky. Pokles hladiny draslíku v séru je obvykle přechodný a nevyžaduje suplementaci.
V klinických studiích s umeklidinium/vilanterolem v doporučené terapeutické dávce nebyly pozorovány klinicky relevantní účinky hypokalemie. Opatrnost je zapotřebí, pokud se umeklidinium/vilanterol podává s jinými léčivými přípravky, které rovněž mohou způsobit hypokalemii (viz bod 4.5).
Hyperglykemie
U některých pacientů mohou beta2-adrenergní agonisté způsobit přechodně hyperglykemii.
Ve studiích, kde byla kombinace umeklidinium/vilanterol podávána v doporučené terapeutické dávce, nebyly pozorovány klinicky relevantní účinky na plazmatickou hladinu glukosy. Při zahájení léčby umeklidinium/vilanterolem bude pacientům s diabetem pečlivě monitorována plazmatická hladina glukosy.
Ko-existující podmínky
Umeklidinium/vilanterol se užívá s opatrností u pacientů s konvulzivními poruchami nebo tyreotoxikózou a u pacientů s neobvyklou odpovědí na beta2-adrenergní agonisty.
Pomocné látky
Tento přípravek obsahuje laktosu. Pacienti se vzácnými dědičnými problémy s intolerancí s galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Blokátory beta-adrenergních receptorů
Léčivé přípravky obsahující blokátory beta-adrenergních receptorů mohou účinky agonistů beta2-adrenergních receptorů, jako je vilanterol, oslabovat nebo antagonizovat. Současného podávání neselektivních nebo selektivních blokátorů beta-adrenergních receptorů je třeba se vyvarovat, pokud pro jejich použití není závažný důvod.
Interakce na úrovni metabolismu a transportních systémů
Vilanterol je substrátem cytochromu P450 3A4 (CYP3A4). Společné podávání silných inhibitorů CYP3A4 (jako je např. ketokonazol, klarithromycin, itrakonazol, ritonavir, telithromycin) může inhibovat metabolismus a zvyšovat systémovou expozici vilanterolu. Společné podání s ketokonazolem (400 mg) zdravým dobrovolníkům zvýšilo průměrné AUC(0-t) vilanterolu o 65 % a Cmax vilanterolu o 22 %. Zvýšení expozice vilanterolu nebylo spojeno se zvýšením systémových účinků agonistů beta-adrenergních receptorů na srdeční frekvenci, hladiny draslíku v krvi ani QT interval (upravený s použitím metody dle Fridericia). Při společném podávání kombinace umeklidinium/vilanterol s ketokonazolem a s jinými silnými inhibitory CYP3A4 je nutná opatrnost, protože existuje riziko zvýšené expozice vilanterolu, což může vést ke zvýšení rizika výskytu nežádoucích účinků. Verapamil, středně silný inhibitor CYP3A4, neměl významný vliv na farmakokinetiku vilanterolu.
Umeklidinium je substrátem cytochromu P450 2D6 (CYP2D6). Farmakokinetika umeklidinia v rovnovážném stavu byla hodnocena u zdravých dobrovolníků s nedostatkem CYP2D6 (slabí metabolizátoři). Při podání 8násobně vyšší dávky nebyly pozorovány žádné účinky na AUC ani Cmax umeklidinia. Při podání 16násobně vyšší dávky bylo pozorováno 1,3násobné zvýšení AUC umeklidinia, bez účinku na Cmax umeklidinia. Na základě rozsahu těchto změn se při společném podávání kombinace umeklidinium/vilanterol s inhibitory CYP2D6 ani při podávání subjektům s genetickým deficitem aktivity CYP2D6 (slabí metabolizátoři) neočekávají žádné klinicky relevantní lékové interakce.
Umeklidinium i vilanterol jsou substráty transportéru pro glykoprotein P (P-gp). U zdravých dobrovolníků byl hodnocen účinek středně silného inhibitoru P-gp verapamilu (240 mg jednou denně) na farmakokinetiku umeklidinia a vilanterolu v rovnovážném stavu. Nebyly pozorovány žádné účinky verapamilu na Cmax umeklidinia ani vilanterolu. Bylo pozorováno přibližně 1,4násobné zvýšení AUC umeklidinia, bez účinku na AUC vilanterolu. Na základě rozsahu těchto změn se při podávání kombinace umeklidinium/vilanterol s inhibitory P-gp neočekávají žádné klinicky relevantní lékové interakce.
Další antimuskarinik a sympatomimetika
Společné podávání kombinace umeklidinium/vilanterol s jinými dlouhodobě účinkujícími muskarinovými antagonisty, dlouhodobě účinkujícími agonisty beta2-adrenergních receptorů, nebo s léčivými přípravky obsahujícími některé z těchto látek nebylo hodnoceno a nedoporučuje se, protože může potencovat známé nežádoucí účinky muskarinových antagonistů nebo beta2-adrenergních agonistů (viz bod 4.4 a bod 4.9).
Hypokalemie
Souběžná léčba hypokalemie deriváty methylxanthinů, steroidy nebo draslík šetřícími diuretiky může umocnit možný hypokalemický účinek beta2-adrenergních agonistů, proto musí být podány s opatrností (viz bod 4.4).
Další léčivé přípravky k léčbě CHOPN
Ačkoli nebyly provedeny formální studie lékových interakcí in vivo, byla inhalační kombinace umeklidinium/vilanterol podávána společně s jinými léčivými přípravky k léčbě CHOPN, včetně krátkodobě působících sympatomimetických bronchodilatancií a inhalačních kortikoidů, bez prokázání klinicky významných lékových interakcí.
4.6 Fertilita, těhotenství a kojení
K dispozici nejsou žádné nebo jsou pouze omezené údaje týkající se používání kombinace umeklidinium/vilanterol u těhotných žen. Studie se zvířaty prokázaly po podání vilanterolu reprodukční toxicitu (viz bod 5.3).
Kombinace umeklidinium/vilanterol se má v průběhu těhotenství používat pouze tehdy, pokud očekávaný prospěch pro matku převáží možná rizika pro plod.
Kojení
Není známo, zda se umeklidinium nebo vilanterol vylučují do mateřského mléka. Další agonisté beta2-adrenergních receptorů jsou však u člověka v mateřském mléce detekovány. Riziko pro novorozence/kojence nelze vyloučit. Při rozhodování, zda přerušit kojení nebo přerušit léčbu umeklidinium/vilanterolem, je třeba vzít v úvahu prospěch z kojení pro dítě a prospěch z léčby pro ženu.
Fertilita
K dispozici nejsou žádné údaje týkající se účinků kombinace umeklidinium/vilanterol na fertilitu u člověka. Studie na zvířatech neprokázaly žádné účinky umeklidinia ani vilanterolu na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Kombinace umeklidinium/vilanterol nemá žádný nebo má pouze zanedbatelný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nejčastěji hlášeným nežádoucím účinkem umeklidinium/vilanterolu byla nazofaryngitida (9 %).
Seznam nežádoucích účinků v tabulce
Bezpečnostní profil přípravku LAVENTAIR vychází z klinického vývojového programu na základě bezpečnostní zkušenosti s umeklidinium/vilanterolem a jednotlivými složkami zahrnující 6 855 pacientů s CHOPN a ze spontánních hlášení. Klinický vývojový program zahrnoval 2 354 pacientů, kteří obdrželi umeklidinium/vilanterol jednou denně ve fázi III klinických studií trvajících 24 týdnů nebo déle, z kterých 1 296 pacientů obdrželo doporučenou dávku 55/22 mikrogramů ve 24týdenních studiích, 832 pacientů obdrželo vyšší dávku 113/22 mikrogramů ve 24týdenních studiích a 226 pacientů obdrželo 113/22 mikrogramů ve12měsíční studii.
Četnosti přiřazené jednotlivým nežádoucím účinkům uvedeným v tabulce níže zahrnují přibližný výskyt incidence zaznamenaný v pěti 24týdenních studiích a ve 12měsíční studii.
Četnosti nežádoucích účinků jsou definovány s použitím následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < /1 000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Nežádoucí účinek |
Četnost |
|
Infekce a infestace |
infekce močových cest |
časté |
|
sinusitida |
časté | |
|
nazofaryngitida |
časté | |
|
faryngitida |
časté |
|
Třída orgánových systémů |
Nežádoucí účinek |
Četnost |
|
infekce horních cest dýchacích |
časté | |
|
Poruchy imunitního systému |
Reakce přecitlivělosti zahrnující: | |
|
vyrážku |
méně časté | |
|
anafylaxi, angioedém a kopřivku |
vzácné | |
|
Poruchy nervového systému |
časté | |
|
méně časté | ||
|
dysgeusie |
méně časté | |
|
Poruchy oka |
glaukom |
není známo |
|
Srdeční poruchy |
fibrilace síní |
méně časté |
|
supraventrikulární tachykardie |
méně časté | |
|
idioventrikulární rytmus |
méně časté | |
|
méně časté | ||
|
supraventrikulární extrasystoly |
méně časté | |
|
palpitace |
méně časté | |
|
Respirační, hrudní |
časté | |
|
a mediastinální poruchy |
orofaryngeální bolest |
časté |
|
Gastrointestinální poruchy |
zácpa |
časté |
|
sucho v ústech |
časté | |
|
Poruchy kůže a podkožní tkáně |
méně časté | |
|
Poruchy ledvin a močových |
retence moče |
vzácné |
|
cest |
vzácné | |
|
obstrukce hrdla močového měchýře |
vzácné |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Předávkování kombinací umeklidinium/vilanterol pravděpodobně povede k subjektivním a objektivním příznakům způsobeným účinky jednotlivých složek přípravku, zahrnujícím známé nežádoucí účinky inhalačních antagonistů muskarinových receptorů (např. sucho v ústech, poruchy akomodace a tachykardie), nebo příznakům předávkování agonisty beta2-adrenergních receptorů (např. arytmie, tremor, bolest hlavy, palpitace, nauzea, hyperglykemie a hypokalemie).
Jestliže dojde k předávkování, měla by být zavedena podpůrná opatření s odpovídající monitorací v případě potřeby.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčba onemocnění spojených s obstrukcí dýchacích cest, sympatomimetika v kombinaci s anticholinergiky, ATC kód: R03AL03
Mechanismus účinku
Kombinace umeklidinia a vilanterolu je kombinace inhalačního dlouhodobě působícího antagonisty muskarinových receptorů (LAMA) a dlouhodobě působícího agonisty beta2-adrenergních receptorů (LABA). Po perorální inhalaci účinkují obě složky lokálně na dýchací cesty a vedou k bronchodilataci odlišnými mechanismy.
Umeklidinium
Umeklidinium je dlouhodobě působící antagonista muskarinových receptorů (rovněž nazývaný anticholinergikum). Je to derivát chinuklidinu s aktivitou napříč mnoha podtypy muskarinových cholinergních receptorů. Umeklidinium vykazuje svoji bronchodilatační aktivitu kompetitivní inhibicí vazby acetylcholinu s muskarinovými receptory v hladké svalovině dýchacích cest. Vykazuje pomalou reverzibilitu na M3 podtypu muskarinových receptorů u člověka in vitro a dlouhodobý účinek in vivo, pokud se v preklinických modelech podával přímo do plic.
Vilanterol
Vilanterol je selektivní dlouhodobě působící agonista beta2-adrenergních receptorů (beta2-adrenergní agonista). Farmakologické účinky agonistů beta2-adrenergních receptorů, včetně vilanterolu, jsou alespoň zčásti způsobené stimulací intracelulární adenylátcyklázy, enzymu, který katalyzuje přeměnu adenosintrifosfátu (ATP) na cyklický-3‘,5‘ adenosinmonofosfát (cAMP). Zvýšení hladin cAMP vede k relaxaci hladké svaloviny bronchiolů a inhibici uvolňování mediátorů okamžité hypersenzitivity z buněk, zejména z mastocytů.
Farmakodynamické účinky
V šestiměsíčních studiích fáze III vykazovala kombinace umeklidinium/vilanterol klinicky významné zlepšení oproti placebu při hodnocení plicních funkcí (měřeno pomocí forsírovaného expiračního objemu za jednu sekundu, FEV1) v průběhu 24 hodin po podávání jednou denně, které byly zřejmé 15 minut po podání první dávky [zlepšení proti placebu o 112 ml (p< 0,001*)]. Průměrná maximální zlepšení FEV1 v průběhu prvních 6 hodin po podání dávky oproti placebu ve 24. týdnu byla 224 ml (p< 0,001 ). V průběhu léčby nebyla v účinku přípravku LAVENTAIR zaznamenána tachyfylaxe.
Srdeční elektrofyziologie
Účinky kombinace umeklidinium/vilanterol na QT interval byly hodnoceny v placebem a aktivním komparátorem (moxifloxacin) kontrolované klinické studii hodnotící QT interval při podávání dávkovaných dávek kombinace umeklidinium/vilanterol 113/22 mikrogramů nebo 500/100 mikrogramů (dávkovaná dávka s umeklidiniem při osminásobku doporučené dávky a vilanterolem při čtyřnásobku doporučené dávky) jednou denně po dobu 10 dnů 103 zdravým dobrovolníkům. Maximální průměrný rozdíl v prodloužení QT intervalu (korigovaný pomocí metody dle Fridericia, QTcF) oproti placebu po korekci na výchozí hodnoty byl 4,3 milisekundy (90% CI = 2,2 až 6,4) zaznamenaný 10 minut po podání kombinace umeklidinium/vilanterol v dávce 113/22 mikrogramů a 8,2 milisekundy (90% CI = 6,2 až 10,2) zaznamenaný 30 minut po podání kombinace umeklidinium/vilanterol v dávce 500/100 mikrogramů. Proto nebyly při podávání kombinace umeklidinium/vilanterol v dávce 113/22 mikrogramů pozorovány žádné klinicky relevantní proarytmogenní účinky s ohledem na prodloužení QT intervalu.
Bylo rovněž pozorováno na dávce závislé zvýšení tepové frekvence. Maximální průměrný rozdíl v tepové frekvenci oproti placebu po korekci na výchozí hodnoty byl 8,4 tepu/minutu (90% CI = 7,0 až 9,8) zaznamenaný 10 minut po podání dávkované dávky kombinace umeklidinium/vilanterol 113/22 mikrogramů a 20,3 tepu/minutu (90% CI = 18,9 až 21,7) pozorovaný 10 minut po podání dávkované dávky kombinace umeklidinium/vilanterol 500/100 mikrogramů.
V průběhu 24hodinového holterovského monitorování u 53 pacientů s CHOPN léčených po dobu až
6 měsíců, kteří byli léčeni dávkou 55/22 mikrogramů umeklidinium/vilanterol jednou denně, nebo dalších 55 pacientů, kteří obdrželi dávku umeklidinium/vilanterolu 113/22 mikrogramů jednou denně v jiné šestiměsíční studii a u dalších 226 pacientů, kteří byli léčeni dávkou 113/22 mikrogramů jednou denně ve dvanáctiměsíční studii, nebyly pozorovány žádné další klinicky významné účinky na srdeční rytmus.
Klinická účinnost
Klinická účinnost umeklidinium/vilanterolu podávaného jednou denně byla hodnocena v osmi klinických studiích fáze III u 6 835 dospělých pacientů s klinicky diagnostikovanou CHOPN; 5 618 pacientů bylo v pěti 6měsíčních klinických studiích hodnotících primární účinnost [dvě placebem kontrolované a tří aktivním komparátorem (tiotropium) kontrolované], 655 pacientů bylo ve dvou 12týdenních studiích hodnotících zátěžové testy/plicní funkce a 562 pacientů bylo ve 12měsíční podpůrné studii.
Účinky na plicní funkce
Přípravek LAVENTAIR vykazoval klinicky významné zlepšení plicních funkcí (definované jako změna trough FEVi od výchozích hodnot) v několika studiích. V jedné šestiměsíční klinické studii fáze III LAVENTAIR vykazovalo statisticky významné zlepšení v trough FEV1 (primární cílový parametr) ve 24. týdnu ve srovnání s placebem a každým ramenem s monoterapií jednotlivých složek. Navíc přípravek LAVENTAIR vykazoval klinicky významné a statisticky signifikantní zlepšení v trough FEV1 ve srovnání s tiotropiem jako aktivním komparátorem ve dvou ze tří šestiměsíčních studií a vykazoval numericky větší zlepšení u tiotropia ve třetí studii s aktivním komparátorem (viz tabulka 1). V průběhu léčby nedocházelo k oslabení bronchodilatačního účinku.
Symptomatické výsledky Dušnost:
Přípravek LAVENTAIR vykazoval statisticky i klinicky významné snížení dušnosti, která byla hodnocena pomocí zvýšení indexu přechodné dušnosti TDI ve 24. týdnu (klíčový sekundární cílový parametr) ve srovnání s placebem (viz tabulka 1). Zlepšení indexu TDI ve srovnání s každou monoterapeutickou komponentou a tiotropiem nebylo statisticky signifikantní (viz tabulka 1).
Poměr pacientů, kteří reagovali alespoň minimálním klinicky významným rozdílem (MCID, minimum clinically important difference) 1 jednotky indexu TDI ve 24. týdnu, byl vyšší u přípravku LAVENTAIR (58 %) ve srovnání s placebem (41 %) a jednotlivými složkami monoterapie (53 % u umeklidinia a 51 % u vilanterolu).
Kvalita života související se zdravím:
Přípravek LAVENTAIR rovněž vykázal zlepšení kvality života související se zdravím hodnocené s použitím dotazníku SGRQ (St. George’s Respiratory Questionnaire), což bylo zaznamenáno snížením celkového skóre SGRQ ve 24. týdnu ve srovnání s placebem a jednotlivými složkami monoterapie (viz tabulka 1). Přípravek LAVENTAIR vykazoval statisticky významné snížení celkového skóre SGRQ ve srovnání s tiotropiem v jedné ze tří studií s aktivním komparátorem (viz tabulka 1).
Poměr pacientů, kteří reagovali alespoň MCID ve skóre SGQR (definovaném jako snížení o 4 jednotky od výchozích hodnot) ve 24. týdnu, byl vyšší u přípravku LAVENTAIR (49 %) ve srovnání s placebem (34 %) a jednotlivými složkami monoterapie (44 % pro umeklidinium a 48 % pro vilanterol). V jedné studii s aktivním komparátorem vyšší procentuální poměr pacientů užívajících přípravek LAVENTAIR vykazoval klinicky významné zlepšení ve skóre SGQR ve 24. týdnu (53 %) ve srovnání s tiotropiem (46 %). Ve dvou dalších studiích s aktivním komparátorem byl podobný poměr pacientů, kteří dosáhli alespoň MCID u přípravku LAVENTAIR a tiotropia; 49 % a 54 % u přípravku LAVENTAIR 55/22 mikrogramů a 52 % a 55 % u tiotropia.
Použití záchranné medikace
Přípravek LAVENTAIR ve srovnání s placebem a umeklidiniem snižoval použití záchranné medikace se salbutamolem v týdnech 1-24 (viz tabulka 1) a prokázal nárůst počtu dnů, v porovnání se stavem na počátku studie, kdy nebylo zapotřebí použít záchrannou medikaci (průměr 11,1 %) ve srovnání s placebem (průměr 0,9 %).
Ve třech 6měsíčních studiích kontrolovaných aktivním komparátorem snižoval přípravek LAVENTAIR použití záchranné medikace se (salbutamolem) ve srovnání s tiotropiem, statisticky významné snížení bylo pozorováno ve dvou studiích (viz tabulka 1). Ve třech studiích přípravek LAVENTAIR rovněž prokázal nárůst počtu dnů, v porovnání se stavem na počátku studie, kdy nebylo zapotřebí použít záchrannou medikaci (průměr v rozsahu 17,6 % až 21,6 %) ve srovnání s tiotropiem (průměr v rozsahu 11,7 % až 13,4 %).
Tabulka 1. Funkce plic, příznaky a výsledky spojené s kvalitou zdraví ve 24. týdnu
|
Léčba srovnávající s přípravek LAVENTAIR 55/22 pg |
Rozdíly v léčbě1 (95% interval spolehlivosti, p-hodnota) | |||
|
FEV1 (ml) |
TDI ústřední skóre |
SGRQ ústřední skóre |
Použití záchranné medikace3 | |
|
LAVENTAIR (N = 413) proti placebu (N = 280) |
167 (128; 207) < 0,001 |
1,2 (0,7; 1,7) < 0,001 |
-5,51 (-7,88; -3,13) < 0,001* |
-0,8 (-1,3;-0,3) 0,001* |
|
LAVENTAIR (N = 413) proti umeclidiniu 55 pg (N = 418) |
52 (17; 87) 0,004 |
0,3 (-0,2; 0,7) 0,244 |
-0,82 (-2,90; 1,27) 0,441 |
-0,6 (-1,0; -0,1) 0,014* |
|
LAVENTAIR (N = 413) proti vilanterolu 22 pg (N = 421) |
95 (60; 130) < 0,001 |
0,4 (-0,1; 0,8) 0,117 |
-0,32 (-2,41; 1,78) 0,767 |
0,1 (-0,3; 0,5) 0,675 |
|
LAVENTAIR (N = 454) proti tiotropiu 18 pg (N = 451) (Studie ZEP117115) |
112 (81; 144) < 0,001 |
n/e |
-2,10 (-3,61; -0,59) 0,006 |
-0,5 (-0,7; -0,2) < 0,001 |
|
LAVENTAIR (N = 207) proti tiotropiu 18 pg (N = 203) (Studie DB2113360) |
90 (39; 141) < 0,001 |
0,12 (-0,4, 0,5) 0,817 |
0,75 (-2,12; 3,63) 0,607 |
-0,7 (-1,2; -0,1) 0,022 |
|
LAVENTAIR (N = 217) proti tiotropiu 18 pg (N = 215) (Studie DB2113374) |
60 (10, 109) 0,018* |
-0,17 (-2,85; 2,52) 0,904 |
-0,6 (-1,2; 0,0) 0,069 | |
N = počet subjektů zahrnutých v každé skupině
pg = mikrogramy
n/e = nebylo hodnoceno
1. Least squares mean
2. Společná data ze studie DB2113360 a studie DB2113374
3. Rozdíl v průměrném počtů vstřiků za den v týdnech 1 -24
Vyšší dávka umeklidinium/vilanterolu (113/22 mikrogramů) byla rovněž zkoumána ve 24. týdnu placebem kontrolované klinické studie a ve dvou ze tří aktivně kontrolovaných studiích ve 24. týdnu. Výsledky byly srovnatelné s těmi s dávkou přípravku LAVENTAIR a poskytly dodatečný podpůrný důkaz účinnosti přípravku LAVENTAIR.
Exacerbace CHOPN
Přípravek LAVENTAIR snižoval riziko exacerbace CHOPN o 50 % ve srovnání s placebem [analýza doby do první exacerbace: Hazard Ratio (HR) poměr rizik 0,5; p = 0,004*]; o 20% ve srovnáním s umeklidiniem (HR 0,8; p = 0,391); a o 30 % ve srovnání s vilanterolem (HR 0,7; p = 0,121). Ve třech studiích s aktivním komparátorem bylo riziko exacerbace CHOPN ve srovnání s tiotropinem snížené o 50 % v jedné studii (HR 0,5; p = 0,044) a ve dvou studiích bylo riziko zvýšené o 20 % a o 90 % (HR 1,2; p = 0,709 a HR 1,9; p = 0,062). Tyto studie nebyly specificky strukturovány, aby dosáhly účinné léčby exacerbace CHOPN a pacienti byli z této studie vyřazeni, pokud došlo k exacerbaci.
Zátěžové testy a plicní objem
Přípravek LAVENTAIR 55/22 mikrogramů zlepšoval dobu zátěžového vyšetřování ve srovnání s placebem, jak bylo hodnoceno pomocí testu ESWT (Endurance Shuttle Walk Test) v jedné studii, ale ne ve druhé, a zlepšoval plicní objem měřený ve srovnání s placebem v obou studiích u dospělých pacientů s CHOPN pomocí hyperinflace [funkční reziduální kapacita (FRC) > 120 %]. V první studii vykazovala léčba přípravkem LAVENTAIR 55/22 mikrogramů jednou denně statisticky významné zlepšení [na základě minimální klinicky důležitého rozdílu (MCID) mezi 45 až 85 sekundami] oproti placebu v době zátěžového vyšetřování (EET, Exercise Endurance Time) zaznamenané 3 hodiny po podání dávky ve 12. týdnu (69,4 sekundy [p = 0,003]). Zlepšení EET ve srovnání s placebem bylo pozorováno 2. den a přetrvávalo v 6. i 12. týdnu. Ve druhé studii byl léčebný rozdíl EET u přípravku LAVENTAIR 55/22 mikrogramů v porovnání s placebem 21,9 sekundy (p = 0,234) ve 12. týdnu.
Přípravek LAVENTAIR 55/22 mikrogramů vykazoval rovněž statisticky významné zlepšení ve srovnání s placebem, pokud jde o změnu plicního objemu od výchozích hodnot měřenou před podáním dávky a 3 hodiny po podání dávky ve 12. týdnu první studie (inspirační kapacita: 237 ml až 316 ml, reziduální objem: -466 ml až -643 ml a funkční reziduální kapacita: -351 ml až -522 ml, vše p < 0,001). Ve druhé studii vykazoval přípravek LAVENTAIR 55/22 mikrogramů zlepšení plicního objemu ve srovnání s placebem ve změně od výchozích hodnot před podáním a 3 hodiny po podání dávky ve 12. týdnu [inspirační kapacita:
198 ml až 238 ml, reziduální objem: -295 ml až -351 ml a funkční reziduální kapacita: -238 ml až -302 ml; vše p < 0,001*].
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s kombinací umeklidinium/vilanterol u všech podskupin pediatrické populace s CHOPN (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Pokud jsou umeklidinium a vilanterol podávané jako kombinovaná léčba inhalačně, farmakokinetika obou složek je podobná farmakokinetice pozorované u jednotlivých léčivých látek podávaných samostatně. Pro účely farmakokinetiky budou proto jednotlivé složky popsány samostatně.
Absorpce
Umeclidinium
Po inhalačním podání umeklidinia zdravým dobrovolníkům bylo Cmax dosaženo za 5 až 15 minut. Absolutní biologická dostupnost po inhalačním podání umeklidinia byla průměrně 13 % dávky, se zanedbatelným podílem z perorální absorpce. Po opakovaných inhalacích umeklidinia bylo rovnovážného stavu dosaženo během 7 až 10 dnů, s 1,5násobnou až 1,8násobnou kumulací.
Vilanterol
Po inhalačním podání vilanterolu zdravým dobrovolníkům bylo dosaženo Cmax po 5 až 15 minutách. Absolutní biologická dostupnost po inhalačním podání vilanterolu byla 27 %, se zanedbatelným podílem z perorální absorpce. Po opakovaném podávání inhalačního vilanterolu bylo rovnovážného stavu dosaženo během 6 dnů, s 2,4násobnou kumulací.
Distribuce
Umeklidinium
Po intravenózním podání zdravým subjektům byl střední distribuční objem 86 litrů. Vazba na plazmatické bílkoviny v lidské plazmě in vitro byla průměrně 89 %.
Vilanterol
Po intravenózním podání zdravým dobrovolníkům byl střední distribuční objem v rovnovážném stavu 165 litrů. Vazba na plazmatické bílkoviny v lidské plazmě in vitro byla průměrně 94 %.
Biotransformace
Umeklidinium
Studie in vitro prokázaly, že umeklidinium je metabolizováno převážně cytochromem P450 2D6 (CYP2D6) a je substrátem transportéru pro glykoprotein P (P-gp). Primárními metabolické cesty umeklidinia jsou oxidativní (hydroxylace, O-dealkylace), následované konjugací (glukuronidace, atd.) a vedou k množství metabolitů se sníženou farmakologickou aktivitou, nebo metabolitů, u kterých nebyla farmakologická aktivita stanovena. Systémová expozice těmto metabolitům je nízká.
Vilanterol
Studie in vitro prokázaly, že vilanterol je metabolizován převážně cestou cytochromu P450 3A4 (CYP3A4) a je substrátem transportéru pro P-gp. Primárními metabolickými cestami jsou O-dealkylace na množství metabolitů s výrazně redukovanými agonistickými účinky na beta1 a beta2-adrenergní receptory. Profily plasmatických metabolitů po perorálním podání vilanterolu u člověka ve studii s radioaktivně značeným vilanterolem byly ovlivněny vysokým metabolismem prvního průchodu. Systémová expozice těmto metabolitům je nízká.
Eliminace
Umeklidinium
Plazmatická clearance po intravenózním podání byla 151 litrů/hodinu. Po intravenózním podání je přibližně 58 % podané radioaktivně značené dávky (nebo 73 % zachycené radioaktivity) vyloučeno stolicí do 192 hodin po podání dávky. Vylučování močí se na vylučování radioaktivně značené dávky do 168 hodin podílí 22 % (27 % zachycené radioaktivity). Vylučování materiálu souvisejícího s podaným lékem stolicí po intravenózním podání dávky ukazuje na aktivní vylučování do žluče. Po perorálním podání zdravým mužům byla celková radioaktivita vyloučena primárně stolicí (92 % podané radioaktivně značené dávky nebo 99 % zachycené radioaktivity) do 168 hodin po podání dávky. Méně než 1 % perorálně podané dávky (1 % zachycené radioaktivity) bylo vyloučeno močí, což naznačuje na zanedbatelnou absorpci po perorálním podání. Plazmatický eliminační poločas umeklidinia v rovnovážném stavu po inhalačním podávání po dobu 10 dnů byl průměrně 19 hodin, se 3 % až 4 % léku vyloučeného v nezměněné formě močí.
Vilanterol
Plazmatická clearance vilanterolu po intravenózním podání byla 108 litrů/hodinu. Po perorálním podání radioaktivně značeného vilanterolu ukazovala hmotnostní bilance 70 % radioaktivně značené látky v moči a 30 % ve stolici. Primární eliminace vilanterolu byla prostřednictvím metabolismu následovaného vylučováním metabolitů močí a stolicí. Plazmatický poločas eliminace vilanterolu z plazmy po inhalačním podávání dávky po dobu 10 dnů byl průměrně 11 hodin.
Charakteristiky u zvláštních skupin subjektů nebo pacientů
Starší pacienti
Populační farmakokinetická analýza prokázala, že farmakokinetiky umeklidinia a vilanterolu byly mezi pacienty s CHOPN ve věku 65 let a staršími a pacienty s CHOPN mladšími 65 let podobné.
Porucha funkce ledvin
Pacienti se závažnou poruchou funkce ledvin nevykazovali žádné známky zvýšení systémové expozice umeklidinia ani vilanterolu (Cmax a AUC) a mezi subjekty se závažnou poruchou funkce ledvin a zdravými dobrovolníky nebyla zaznamenána porucha vazby na bílkoviny.
Porucha funkce jater
Pacienti se středně závažnou poruchou funkce jater nevykazovali žádné známky zvýšení systémové expozice umeklidinia ani vilanterolu (Cmax a AUC) a mezi subjekty se středně závažnou poruchou funkce jater a zdravými dobrovolníky nebyla zaznamenána porucha vazby na bílkoviny. Kombinace umeklidinium/vilanterol nebyla hodnocena u pacientů se závažnou poruchou funkce jater.
Další zvláštní populace
Analýza populační farmakokinetiky prokázala, že na základě věku, rasy, pohlaví, používání inhalačních kortikosteroidů ani tělesné hmotnosti není u umeklidinia ani vilanterolu nutná úprava dávkování. Studie u slabých metabolizátorů CYP2D6 neprokázala klinicky významný vliv genetického polymorfismu CYP2D6 na systémovou expozici umeklidinia.
5.3 Předklinické údaje vztahující se k bezpečnosti
V neklinických studiích s umeklidiniem a vilanterolem, samotnými i v kombinaci, byly nálezy typicky spojené s primárními farmakologickými účinky antagonistů muskarinových receptorů nebo agonistů beta2-adrenergních receptorů a/nebo lokální dráždivostí. V následujícím textu jsou uvedeny nálezy ze studií s jednotlivými složkami přípravku.
Genotoxicita a kancerogenita
Umeklidinium nebylo ve standardní baterii studií genotoxické a nebylo ani kancerogenní ve studiích celoživotní inhalace u myší ani potkanů při expozicích dosahujících > 26násobku nebo > 22násobku klinické expozice umeklidiniu při dávce 55 mikrogramů u člověka (na základě AUC).
Ve studiích genetické toxicity nebyly vilanterol (ve formě alfa-fenylcinamátu) ani kyselina trifenyloctová genotoxické, což naznačuje, že vilanterol (ve formě trifenatátu) nepředstavuje genotoxické riziko pro člověka. V souladu s nálezy u dalších agonistů beta2-adrenergních receptorů způsoboval vilanterol-trifenatát ve studiích celoživotní inhalace proliferační změny na reprodukčních orgánech u samic potkanů a myší a na hypofýze u potkanů. U potkanů ani myší nebylo prokázáno zvýšení incidence tumorů při expozicích 0,5 resp. 13násobně vyšších, než jsou dosahovány při klinické expozici vilanterolu při dávce 22 mikrogramů u člověka (na základě AUC).
Reprodukční toxicita
Umeklidinium nebylo u potkanů nebo králíků teratogenní. V pre- a postnatální studii vedlo subkutánní podávání umeklidinia potkanům k nižšímu přírůstku tělesné hmotnosti u březích samic, nižší konzumaci potravy a mírnému snížení předporodní tělesné hmotnosti mláďat při dávkách 180 mikrogramů/kg/den (přibližně 80násobek klinické expozice umeklidiniu při dávce 55 mikrogramů u člověka, na základě AUC).
Vilanterol nebyl u potkanů teratogenní. Ve studiích inhalace u králíků způsoboval vilanterol podobné účinky, jaké byly pozorovány u ostatních agonistů beta2_adrenergních receptorů (rozštěp patra, otevření očních víček, srůst jednotlivých částí hrudní kosti a flexury/malrotace končetin) při dávkách odpovídajících 6násobku klinické expozice u člověka na základě AUC. Pokud byl podáván subkutánně, nebyly tyto účinky pozorovány ani při dávkách odpovídajících 36násobku klinické expozice při dávce 22 mikrogramů vilanterolu (na základě AUC).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Monohydrát laktosy Magnesium-stearát
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
Doba použitelnosti po otevření vaničky: 6 týdnů.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C. Pokud je přípravek uchováván v chladničce, nechte inhalátor alespoň jednu hodinu před použitím ohřát na pokojovou teplotu.
Uchovávejte inhalátor v zatavené vaničce, aby byl chráněn před vlhkostí, vyjměte ho až těsně před prvním použitím.
Po prvním otevření vaničky využijte v průběhu 6 týdnů.
Na štítek inhalátoru napište datum, do kdy má být inhalátor spotřebován. Datum má být zapsáno ihned, jakmile byl inhalátor vyndán z vaničky.
6.5 Druh obalu a obsah balení
Inhalátor ELLIPTA se skládá ze světle šedého těla, červeného krytu náustku a počítadla dávek a je uložen v ochranné vaničce z laminované fólie, která obsahuje vysoušedlo. Tato vanička je zatavena odlupovacím fóliovým víčkem.
Inhalátor obsahuje dva hliníkové blistry z laminované fólie obsahující 7 nebo 30 dávek.
Inhalátor je zařízení složené z několika komponent, které jsou vyrobeny z polypropylenu, polyethylenu s vysokou hustotou, polyoxymethylenu, polybutylen-tereftalátu, akrylonitril-butadien-styrenu, polykarbonátu a nerezové oceli.
Balení obsahuje inhalátor se 7 nebo 30 dávkami.
Multipack obsahuje 3 inhalátory po 30 dávkách.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Glaxo Group Limited 980 Great West Road Brentford Middlesex
TW8 9GS Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/899/001
EU/1/14/899/002
EU/1/14/899/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 8. května 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ /VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Glaxo Operations UK Ltd. (trading as Glaxo Wellcome Operations)
Priory Street
Ware, Hertfordshire SG12 0DJ Velká Británie
Glaxo Operations UK Ltd. (trading as Glaxo Wellcome Operations),
Harmire Road,
Barnard Castle,
County Durham DL12 8DT,
Velká Británie
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku
(v rámci farmakovigilance nebo minimalizace rizik).
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Předloží závěrečné zprávy klinické studie o poregistrační bezpečnostní observační skupinové studii (PAS) k vyčíslení výskytu srovnatelné bezpečnosti vybraných kardiovaskulárních a cerebrovaskulárních příhod u pacientů s CHOPN přípravkem Laventair v porovnání s tiotropiem (studie 201038) podle protokolu schváleného PRAC. |
do 3Q 2024 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
KRABIČKA (POUZE JEDNOTLIVÁ BALENÍ A MULTIPACK)
55 mikrogramů/22 mikrogramů_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
LAVENTAIR 55 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci umeclidinium/vilanterolum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna podaná dávka obsahuje umeclidinium 55 mikrogramů (což odpovídá umeclidinii bromidum 65 mikrogramů) a vilanterolum 22 mikrogramů (ve formě trifenatas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje rovněž laktosu a magnesium-stearát. Další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Dávkovaný prášek k inhalaci, ELLIPTA 1 inhalátor po 7 dávkách 1 inhalátor po 30 dávkách
Multipack: 90 dávek (3 balení po 30 dávkách) 3x 30 dávek
5 ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Inhalační podání, pouze jednou denně.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Doba použitelnosti po prvním otevření: 6 týdnů.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH
PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11 NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, Velká Británie Logo Glaxo Group Ltd 12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/899/001 1 inhalátor po 7 dávkách EU/1/14/899/002 1 inhalátor po 30 dávkách EU/1/14/899/003 Multipack: 90 dávek (3 balení po 30 dávkách)
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ 16 INFORMACE V BRAILLOVĚ PÍSMU laventair ellipta
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VNITŘNÍ KRABIČKA (POUZE MULTIPACK BEZ BLUE-BOXU) 55 mikrogramů/22 mikrogramů_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
LAVENTAIR 55 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci umeclidinium/vilanterolum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna podaná dávka obsahuje umeclidinium 55 mikrogramů (což odpovídá umeclidinii bromidum 65 mikrogramů) a vilanterolum 22 mikrogramů (ve formě trifenatas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje rovněž laktosu a magnesium-stearát. Další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
1 inhalátor po 30 dávkách ELLIPTA
Součást multipacku, samostatně neprodejné.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Inhalační podání, pouze jednou denně.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Doba použitelnosti po prvním otevření: 6 týdnů.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10 ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, Velká Británie Logo Glaxo Group Ltd
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/899/003
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ 16. INFORMACE V BRAILLOVE PÍSMU laventair ellipta
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH VÍČKO VLOŽKY Z LAMINOVANÉ FÓLIE
55 mikrogramů/22 mikrogramů_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
LAVENTAIR 55/22 pg prášek k inhalaci umeclidinium/vilanterolum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Logo Glaxo Group Ltd
3. POUŽITELNOST
EXP
4 ČÍSLO ŠARŽE
Lot
5. JINÉ
Neotevírejte, dokud nejste připraven(a) k inhalaci. Doba použitelnosti po prvním otevření: 6 týdnů.
7 dávek 30 dávek ELLIPTA
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK INHALÁTORU
55 mikrogramů/22 mikrogramů_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
LAVENTAIR 55/22 pg prášek k inhalaci umeclidinium/vilanterolum
Inhalační podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4 ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
7 dávek 30 dávek
6. JINÉ
Doba použitelnosti po prvním otevření: 6 týdnů. Spotřebujte do:
ELLIPTA
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
LAVENTAIR 55 mikrogramů/22 mikrogramů dávkovaný prášek k inhalaci
umeclidinium/vilanterolum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek LAVENTAIR a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek LAVENTAIR používat
3. Jak se přípravek LAVENTAIR používá
4. Možné nežádoucí účinky
5. Jak přípravek LAVENTAIR uchovávat
6. Obsah balení a další informace Podrobný návod k použití
1. Co je přípravek LAVENTAIR a k čemu se používá Co je přípravek LAVENTAIR
Přípravek LAVENTAIR obsahuje dvě léčivé látky: umeklidinium a vilanterol, které patří do skupiny léků zvaných bronchodilatancia.
K čemu se přípravek LAVENTAIR používá
Přípravek LAVENTAIR se používá k léčbě chronické plicní obstrukční nemoci (CHOPN) u dospělých. CHOPN je dlouhodobé onemocnění, při kterém dochází k potížím s dýcháním, které se pomalu zhoršují.
Při CHOPN se svaly dýchacích cest stahují. Tento lék brání stahování této svaloviny v plicích a tím pomáhá udržet otevřené dýchací cesty, což usnadňuje proudění vzduchu do plic i z plic. Pokud se používá pravidelně, pomáhá ke kontrole dechových obtíží a snižuje vliv CHOPN na Váš každodenní život.
Přípravek LAVENTAIR se nesmí používat k úlevě při náhlém záchvatu dusnosti nebo sípotu.
Pokud se u Vás objeví takový druh záchvatu, musíte použít inhalátor s rychle účinkujícím přípravkem (jako např. salbutamol).
2. Čemu musíte věnovat pozornost, než začnete přípravek LAVENTAIR používat Nepoužívejte přípravek LAVENTAIR
- jestliže j ste alergický(á) na umeklidinium, vilanterol nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
Pokud si myslíte, že se Vás to týká, nepoužívejte tento léčivý přípravek, dokud se neporadíte se svým lékařem.
Upozornění a opatření
Před použitím tohoto přípravku se poraďte se svým lékařem:
- jestliže máte astma (nepoužívejte přípravek LAVENTAIR k léčbě astmatu);
- jestliže máte problémy se srdcem nebo vysoký krevní tlak;
- jestliže máte problém s očima nazývaný glaukom (zelený zákal) s úzkým úhlem;
- jestliže máte zbytnělou prostatu, obtíže s močením nebo překážku v močovém měchýři;
- jestliže trpíte epilepsii;
- jestliže máte problémy se štítnou žlázou;
- jestliže máte cukrovku;
- jestliže trpíte závažným onemocněním jater.
Pokud si myslíte, že se Vás něco z tohoto může týkat, poraďte se se svým lékařem.
Okamžité dýchací potíže
Pokud se bezprostředně po použití přípravku LAVENTAIR objeví pocit tlaku na hrudi, kašel, sípání nebo dušnost:
Přestaňte tento přípravek používat a neprodleně vyhledejte lékařskou pomoc, protože můžete mít závažnou zdravotní komplikaci, která se nazývá paradoxní bronchospasmus.
Oční problémy v průběhu léčby přípravkem LAVENTAIR
Pokud se u Vás objeví bolest oka nebo nepříjemný pocit v oku, dočasně rozmazané vidění, zrakové haló nebo barevné obrazy v souvislosti se zarudnutím očí v průběhu léčby přípravkem LAVENTAIR:
Přestaňte tento lék používat a okamžitě vyhledejte lékařskou pomoc, protože to mohou být příznaky akutního záchvatu glaukomu (zeleného zákalu) s úzkým úhlem.
Děti a dospívající
Tento přípravek se nemá podávat dětem a dospívajícím mladším 18 let.
Další léčivé přípravky a přípravek LAVENTAIR
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Některé léky mohou ovlivnit způsob, jakým přípravek LAVENTAIR účinkuje, nebo mohou zvýšit pravděpodobnost, že se objeví nežádoucí účinky. Mezi tyto léky patří:
- léky nazývané beta-blokátory (jako je propranolol), používané k léčbě vysokého krevního tlaku nebo onemocnění srdce;
- ketokonazol nebo itrakonazol, používané k léčbě plísňových infekcí;
- klarithromycin nebo telithromycin, používané k léčbě bakteriálních infekcí;
- ritonavir, používaný k léčbě HIV infekce;
- přípravky, které sníží množství draslíku ve Vaší krvi, jako některá diuretika (přípravky na močení);
- jiné léky s dlouhodobým účinkem podobné tomuto přípravku, které se používají k léčbě dýchacích obtíží, jako je např. tiotropium, indakaterol. Nepoužívejte přípravek LAVENTAIR, pokud již tyto léky užíváte.
Pokud některý z těchto léků užíváte, sdělte to svému lékaři nebo lékárníkovi.
Těhotenství, kojení a plodnost
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek používat. Jste-li těhotná, nepoužívejte tento přípravek, dokud Vám to lékař nedovolí.
Není známo, zda složky přípravku LAVENTAIR mohou procházet do mateřského mléka. Pokud kojíte, musíte se před použitím přípravku LAVENTAIR poradit se svým lékařem. Jestliže kojíte, nepoužívejte tento přípravek, dokud Vám to lékař nedovolí.
Řízení dopravních prostředků a obsluha strojů
Není pravděpodobné, že by přípravek LAVENTAIR ovlivnil schopnost řídit nebo obsluhovat stroje. Přípravek LAVENTAIR obsahuje laktózu
Pokud Vám někdy lékař řekl, že trpíte nesnášenlivostí některých cukrů nebo mléčných bílkovin, sdělte to svému lékaři dříve, než začnete používat tento lék.
3. Jak se přípravek LAVENTAIR používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka přípravku je jedna inhalace každý den ve stejnou dobu. Tento přípravek je třeba inhalovat jednou denně, protože jeho účinky přetrvávají po dobu 24 hodin.
Nepoužívejte více přípravku, než Vám řekl Váš lékař.
Používejte přípravek LAVENTAIR pravidelně
Je velmi důležité, abyste přípravek LAVENTAIR používal(a) každý den tak, jak Vám doporučil Váš lékař. To Vám pomáhá zajistit odstranění příznaků onemocnění v průběhu dne i noci.
Přípravek LAVENTAIR se nesmí používat k úlevě od náhlého záchvatu dusnosti nebo sípotu. Pokud se u Vás objeví tento druh záchvatu, musíte použít inhalátor s rychle účinkujícím přípravkem (jako je např. salbutamol).
Jak se inhalátor používá
Úplné informace naleznete v části „Návod krok za krokem" této příbalové informace.
Přípravek LAVENTAIR se vdechuje ústy do plic pomocí inhalátoru ELLIPTA.
Pokud se příznaky nezlepšují
Pokud se příznaky CHOPN (dušnost, sípot, kašel) nezlepšují, nebo pokud se zhoršují, nebo pokud musíte používat přípravek s rychle účinkujícím přípravkem častěji: kontaktujte co nejdříve svého lékaře.
Jestliže jste použil(a) více přípravku LAVENTAIR, než jste měl(a)
Pokud omylem použijete více tohoto přípravku, než Vám doporučil lékař, ihned se poraďte se svým lékařem nebo lékárníkem, jelikož můžete potřebovat lékařskou pomoc. Je-li to možné, ukažte mu inhalátor, balení přípravku nebo tuto příbalovou informaci. Můžete zaznamenat rychlejší tlukot srdce než obvykle, třes, poruchy zraku, sucho v ústech nebo bolest hlavy.
Jestliže jste zapomněl(a) použít přípravek LAVENTAIR
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Vezměte si pouze následující dávku v obvyklý čas.
Pokud se objeví dušnost nebo sípot, použijte inhalátor s rychle účinkujícím přípravkem (např. salbutamol) a poté se poraďte s lékařem.
Jestliže jste přestal(a) používat přípravek LAVENTAIR
Používejte tento přípravek tak dlouho, jak Vám doporučil Váš lékař. Tento přípravek bude účinný pouze tak dlouho, jak dlouho jej budete používat. Nepřestávejte používat tento přípravek dříve, než Vám to doporučí lékař, a to ani v případě, že se budete cítit lépe, jelikož Vaše příznaky se mohou zhoršit.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Alergické reakce na přípravek ANORO jsou méně časté (mohou postihnout méně než 1 osobu ze 100).
Pokud se u Vás při používání přípravku ANORO objeví následující příznaky, přestaňte tento lék používat a ihned vyhledejte svého lékaře.
• kožní vyrážka (kopřivka) nebo zarudnutí;
• otok, někdy obličeje nebo úst (angioedém);
• zhoršující se sípání, kašel nebo potíže s dýcháním;
• náhlý pocit slabosti nebo závratě (což může vést ke kolapsu nebo ztrátě vědomí).
Okamžité dýchací potíže
Pokud se ihned po použití tohoto přípravku objeví pocit tlaku na hrudi, kašel, sípot nebo dušnost:
přestaňte tento přípravek používat a ihned vyhledejte lékařskou pomoc, protože se u Vás může objevit závažná komplikace nazývaná paradoxní bronchospasmus.
Časté nežádoucí účinky
Mohou postihnout až 1 osobu z 10
• bolestivé a časté močení (může být známkou infekce močových cest);
• kombinace bolesti v krku a rýmy;
• bolest v krku;
• pocit tlaku nebo bolesti ve tvářích a na čele (mohou být známkami zánětu vedlejších dutin, kterému se říká sinusitida);
• bolest hlavy;
• kašel;
• bolest a podráždění zadní části úst a hrdla;
• zácpa;
• sucho v ústech;
• infekce horních cest dýchacích.
Méně časté nežádoucí účinky
Mohou postihnout až 1 osobu ze 100
• nepravidelný tlukot srdce;
• zrychlený tlukot srdce;
• uvědomování si bušení srdce (palpitace);
• vyrážka;
• třes;
• porucha chuti.
Vzácné nežádoucí účinky
Mohou postihnout až 1 osobu z 1 000
• obtíže a bolest při močení - mohou to být příznaky neprůchodnosti hrdla močového měchýře nebo zadržování moči.
Další nežádoucí účinky
Další nežádoucí účinky se mohou vyskytnou u velmi malého počtu pacientů, ale jejich četnost výskytu není známa.
• Snížení zraku nebo bolest očí v důsledku vysokého tlaku (možný příznak glaukomu).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek LAVENTAIR uchovávat Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za „EXP“:
Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte inhalátor v zatavené ochranné vaničce, aby byl chráněn před vlhkostí. Neotevírejte fóliové víčko ochranné vaničky, dokud nejste připraven(a) k prvnímu použití. Jakmile je vanička otevřena, inhalátor může být použit po dobu 6 týdnů od data otevření. Napište na štítek inhalátoru datum, do kdy má být inhalátor spotřebován. Datum zapište ihned, jakmile vyjmete inhalátor z vaničky.
Uchovávejte při teplotě do 30 °C.
Pokud inhalátor uchováváte v chladničce, nechejte jej alespoň hodinu před použitím ohřát na pokojovou teplotu.
Nevyhazujte žádné léčivé přípravky do domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek LAVENTAIR obsahuje
Léčivými látkami jsou umeclidinii bromidum a vilanterolum.
Jedna inhalace poskytne dávku (dávka, která vyjde z náustku) umeclidinium 55 mikrogramů (což odpovídá umeclidinii bromidum 65 mikrogramů) a vilanterolum 22 mikrogramů (ve formě trifenatas)
Dalšími složkami jsou monohydrát laktosy a magnesium-stearát.
Jak přípravek LAVENTAIR vypadá a co obsahuje toto balení
Samotný přístroj se skládá z šedého plastového těla, červeného krytu náustku a počítadla dávek. Je zabalený ve vložce z laminované fólie s odlupovacím fóliovým víčkem. Vložka obsahuje vysoušedlo, které snižuje vlhkost uvnitř balení.
Léčivá látka je přítomna ve formě bílého prášku v oddělených blistrem uvnitř inhalátoru. Inhalátor obsahuje 7 nebo 30 dávek. K dispozici je rovněž multipack obsahující 90 dávek (3 inhalátory po 30 dávkách).
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci:
Glaxo Group Limited 980 Great West Road Brentford
Middlesex TW8 9GS Velká Británie
Výrobce:
Glaxo Operations UK Limited (trading as Glaxo Wellcome Operations)
Priory Street
Ware
Hertfordshire, SG12 0DJ Velká Británie
Glaxo Operations UK Limited (trading as Glaxo Wellcome Operations)
Harmire Road
Barnard Castle
County Durham
DL12 8DT
Velká Británie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Česká republika
GlaxoSmithKline s.r.o. Tel: + 420 222 001 111 cz.info@gsk.com
Lietuva
GlaxoSmithKline Lietuva UAB Tel: + 370 5 264 90 00 info.lt@gsk.com
Luxembourg/Luxemburg
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Belgique/Belgien
Tél/Tel: + 32 (0) 10 85 52 00
Magyarország
GlaxoSmithKline Kft.
Tel.: + 36 1 225 5300
Danmark
GlaxoSmithKline Pharma A/S Tlf: + 45 36 35 91 00 dk-info@gsk.com
Deutschland
GlaxoSmithKline GmbH & Co. KG Tel.: + 49 (0)89 36044 8701 produkt.info@gsk.com
Eesti
GlaxoSmithKline Eesti OU Tel: + 372 6676 900 estonia@gsk.com
EXXába
GlaxoSmithKline A.E.B.E.
Tr|U + 30 210 68 82 100
Espaňa
GlaxoSmithKline, S.A. Tel: + 34 902 202 700 es-ci@gsk.com
Nederland
GlaxoSmithKline BV Tel: + 31 (0)30 6938100 nlinfo@gsk.com
Norge
GlaxoSmithKline AS Tlf: + 47 22 70 20 00 firmapost@gsk.no
Osterreich
GlaxoSmithKline Pharma GmbH Tel: + 43 (0)1 97075 0 at.info@gsk.com
Polska
GSK Services Sp. z o.o.
Tel.: + 48 (0)22 576 9000
|
France Laboratoire GlaxoSmithKline Tél.: + 33 (0)1 39 17 84 44 diam@gsk.com |
Portugal GlaxoSmithKline - Produtos Farmaceuticos, Lda. Tel: + 351 21 412 95 00 |
|
Hrvatska GlaxoSmithKline d.o.o. Tel: + 385 1 6051 999 |
Románia GlaxoSmithKline (GSK) S.R.L. Tel: + 4021 3028 208 |
|
Ireland GlaxoSmithKline (Ireland) Limited Tel: + 353 (0)1 4955000 |
Slovenija GlaxoSmithKline d.o.o. Tel: + 386 (0)1 280 25 00 medical.x.si@gsk.com |
|
Ísland Vistor hf. Sími: + 354 535 7000 |
Slovenská republika GlaxoSmithKline Slovakia s. r. o. Tel: + 421 (0)2 48 26 11 11 recepcia.sk@gsk.com |
|
Italia A.Menarini Industrie Farmaceutiche Riunite s.r.l. Tel: +39 (0)55 56801 |
Suomi/Finland GlaxoSmithKline Oy Puh/Tel: + 358 (0)10 30 30 30 Finland.tuoteinfo@gsk.com |
|
Kónpoq GlaxoSmithKline (Cyprus) Ltd Tr^: + 357 22 39 70 00 gskcyprus@gsk.com |
Sverige GlaxoSmithKline AB Tel: + 46 (0)8 638 93 00 info.produkt@gsk.com |
|
Latvija GlaxoSmithKline Latvia SIA Tel: + 371 67312687 lv-epasts@gsk.com |
United Kingdom GlaxoSmithKline UK Tel: + 44 (0)800 221441 customercontactuk@gsk.com |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Návod krok za krokem
Co je inhalátor?
Před prvním použitím přípravku LAVENTAIR nemusíte, zda inhalátor funguje správně. Inhalátor obsahuje dávkované dávky a je připravený přímo k použití.
Inhalátor je zabalen v ochranné vaničce obsahující sáček s vysoušedlem, které snižuje vlhkost. Tento sáček vyhoďte - nejezte ho ani neinhalujte.
Když vyndáte inhalátor ze zatavené vaničky, je v „uzavřené“ pozici. Neotevírejte jej, dokud nejste připraven(a) k inhalaci léku. Jakmile je vanička otevřena, napište na štítek inhalátoru datum „Spotřebujte do“, do kdy má být inhalátor spotřebován. Datum „Spotřebujte do“ je 6 týdnů od data otevření vaničky. Po tomto datu se již nemá inhalátor dále používat. Vaničku lze po prvním otevření vyhodit.
Návod k použití inhalátoru uvedený níže lze použít pro inhalátor s 30 dávkami i se 7 dávkami.
Před použitím si přečtěte následující informace
Pokud kryt inhalátoru otevřete a zavřete bez toho, že byste inhaloval(a) lék, dojde ke ztrátě dávky.
Ztracená dávka zůstane bezpečně uzavřená v inhalátoru, ale nebude již dostupná k inhalaci.
Při jedné inhalaci není možné náhodně použít dávku navíc ani dvojnásobnou dávku.
Počítadlo dávek
Počítadlo ukazuje, kolik dávek léku v inhalátoru ještě zbývá.
Před prvním použitím inhalátoru ukazuje počítadlo přesně 30 dávek.
Při každém otevření krytu inhalátoru se 1 dávka odečte.
Pokud v inhalátoru zbývá méně než 10 dávek, polovina počítadla ukazuje červeně.
Po použití poslední dávky ukazuje polovina počítadla červeně a zobrazuje 0. Inhalátor je nyní prázdný. Pokud poté otevřete kryt inhalátoru, počítadlo změní barvu z původní z poloviny červené na zcela červenou.
1) Příprava dávky
Počkejte s otevřením krytu, dokud nejste připraven(a) k použití své dávky. Inhalátorem netřeste.
• Stahujte víčko dolů, dokud neuslyšíte „cvaknutí“.
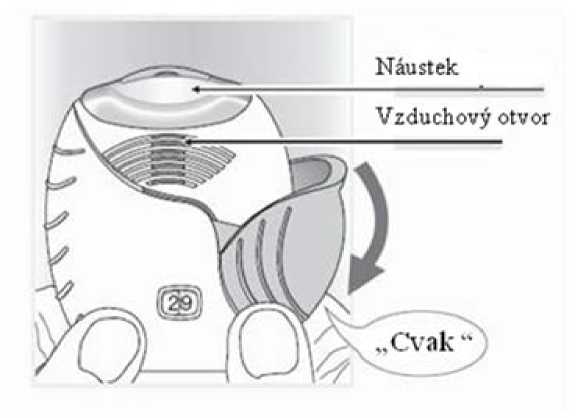
Lék je nyní připraven k inhalaci.
Počítadlo dávky pro potvrzení odečetlo 1 dávku.
• Pokud počítadlo neodečte dávku v okamžiku, kdy uslyšíte „cvaknutí,“ inhalátor neumožní inhalaci léku.
Vezměte jej zpět k lékárníkovi, aby Vám poradil.
2) Inhalace léku
• Držte inhalátor dále od úst a co nejvíce vydechněte (jak je Vám pohodlné).
Nevydechujte do inhalátoru.
• Vložte náustek mezi rty a pevně jej svými rty stiskněte.
Neblokujte vzduchový otvor prsty.
Při inhalaci dávky držte své rty přitisknuté na tvarovaném okraji náustku. Nezakrývejte prsty vzduchový otvor.
• Jednou se dlouze, rovnoměrně a zhluboka nadechněte. Zadržte dech po co nejdelší dobu (alespoň 3 -4 sekundy).
• Vyjměte inhalátor z úst.
• Pomalu a lehce vydechněte.
Lék by neměl mít žádnou chuť, ani byste jej neměl(a) cítit, a to ani v případě, že jste inhalátor použil(a) správně.
3) Uzavření inhalátoru
Pokud chcete náustek očistit, otřete jej před uzavřením suchou tkaninou.
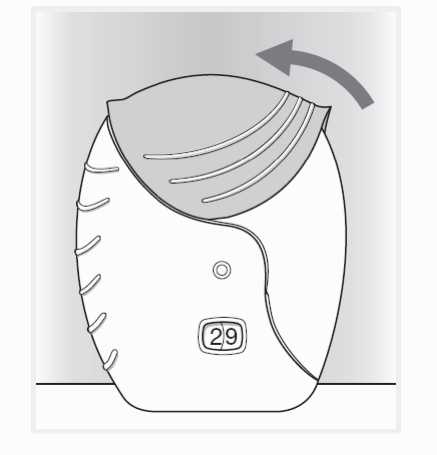
Vysuňte kryt zpět nahoru co nejvíce, až je náustek zakrytý.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY V REGISTRACI
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizovaných zpráv o bezpečnosti (PSUR) pro umedidinii bromidum/vilanterolum, dospěl výbor CHMP k těmto vědeckým závěrům:
V souvislosti s užíváním přípravků obsahujících umeclidinium bromidum/vilanterolum (UMEC/VI) bylo zaznamenáno celkem 30 spontánních hlášení popisujících 31 nežádoucích účinků v souladu s předem definovanými preferovanými termíny (PT) pro nežádoucí účinky u močových cest. .
Z nich celkem 12 spontánních hlášení obsahovalo dostatečně podrobné informace pro jejich úplné posouzení. V těchto 12 hlášeních bylo popsáno 13 nežádoucích účinků týkajících se močových cest:
8 závažných případů retence moči, 2 závažné případy obstrukce močových cest, 2 nezávazné případy snížení průtoku moči a 1 případ snížení frekvence močení. Krátká doba mezi zahájením léčby UMEC/VI a nástupem účinku (< 2 týdny) byla hlášena u 7 z 12 hodnocených případů. V 8 případech bylo prokázáno vymizení projevů po vysazení UMEC/VI (pozitivní dechallenge).
„Retence moči“ je známý skupinový účinek (class effect) skupiny anticholinergik (Halpina, 2015 Stephenson, 2011; Afonso, 2011) a v souhrnu údajů o přípravku je již obsaženo upozornění doporučující použití UMEC/VI s opatrností u pacientů s retencí moči.
Na základě výsledků tohoto souhrnného hodnocení a s ohledem na pravděpodobný farmakologický anticholinergní účinek na funkce močových cest je dostatečně podložené, že je nutné vyžadovat aktualizaci informací o přípravku s uvedením „retenci moči“, obstrukci hrdla močového měchýře a dysurii“ jako nežádoucích účinků spojených s užíváním UMEC/VI.
Proto se s ohledem na předložené údaje v hodnoceném PSUR výbor PRAC domnívá, že změny v informacích o léčivých přípravcích obsahujících umeclidini bromidum/vilanterolum jsou oprávněné. Skupina CHMP souhlasí s vědeckými závěry výboru PRAC.
Zdůvodnění změny v registraci
Na základě vědeckých závěrů týkajících se umeclidinii bromidum/vilanterolum zastává výbor CHMP stanovisko, že poměr přínosů a rizik léčivých přípravků obsahujících léčivé látky umeclidinii bromidum/vilanterolum je příznivý za předpokladu, že v údajích o přípravku budou provedeny navržené změny.
Výbor CHMP doporučuje změnit podmínky rozhodnutí o registraci.
38