Kovaltry 3000 Iu
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Kovaltry 250 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 500 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 1000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 2000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 3000 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička nominálně obsahuje 250/500/1 000/2 000/3 000 IU lidského koagulačního faktoru VIII.
• Jeden ml přípravku Kovaltry 250 IU obsahuje přibližně 100 IU (250 IU / 2,5 ml) rekombinantního lidského koagulačního faktoru VIII (INN: octocogum alfa) po rekonstituci ve vodě na injekci.
• Jeden ml přípravku Kovaltry 500 IU obsahuje přibližně 200 IU (500 IU / 2,5 ml) rekombinantního lidského koagulačního faktoru VIII (INN: octocogum alfa) po rekonstituci ve vodě na injekci.
• Jeden ml přípravku Kovaltry 1000 IU obsahuje přibližně 400 IU (1 000 IU / 2,5 ml) rekombinantního lidského koagulačního faktoru VIII (INN: octocogum alfa) po rekonstituci ve vodě na injekci.
• Jeden ml přípravku Kovaltry 2000 IU obsahuje přibližně 400 IU (2 000 IU / 5 ml) rekombinantního lidského koagulačního faktoru VIII (INN: octocogum alfa) po rekonstituci ve vodě na injekci.
• Jeden ml přípravku Kovaltry 3000 IU obsahuje přibližně 600 IU (3 000 IU / 5 ml) rekombinantního lidského koagulačního faktoru VIII (INN: octocogum alfa) po rekonstituci ve vodě na injekci.
Síla (IU) je určena chromogenní zkouškou podle Evropského lékopisu. Specifická účinnost přípravku Kovaltry je přibližně 4 000 IU/mg bílkoviny.
Oktokog alfa (rekombinantní lidský koagulační faktor VIII (rDNA) plné délky) je purifikovaný protein, který má 2 332 aminokyselin. Je vyráběn rekombinantní DNA technologií v ledvinových buňkách křeččích mláďat (BHK), do kterých byl zaveden gen pro lidský faktor VIII. Přípravek Kovaltry se vyrábí bez přidání proteinu lidského nebo zvířecího původu do procesu buněčné kultivace, purifikace nebo finální formulace.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok (zařízení Bio-Set) Prášek: pevný, bílý až nažloutlý.
Rozpouštědlo: voda na injekci, čirý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u nemocných s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek Kovaltry může být používán u všech věkových kategorií.
4.2 Dávkování a způsob podání
Léčba musí probíhat pod dozorem lékaře se zkušenostmi s léčbou hemofilie.
Dávkování
Dávka a délka substituční léčby závisí na závažnosti deficitu faktoru VIII, na lokalizaci a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek podaného faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které jsou odvozeny od současné normy WHO pro přípravky s faktorem VIII. Plazmatická aktivita faktoru VIII je vyjádřena buď jako procento (vztažené k normální lidské plazmě), nebo v mezinárodních jednotkách (odvozených z mezinárodní normy pro faktor VIII v plazmě).
Aktivita jedné mezinárodní jednotky (IU) faktoru VIII odpovídá množství faktoru VIII obsaženému v jednom ml normální lidské plazmy.
Léčba podle potřeby
Výpočet požadované dávky faktoru VIII vychází z empirické zkušenosti, že 1 mezinárodní jednotka (IU) faktoru VIII na jeden kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 1,5 % až 2,5 % normální aktivity.
Požadovaná dávka se určí pomocí následujícího vzorce:
Požadované jednotky = tělesná hmotnost (kg) x požadované zvýšení faktoru VIII (% nebo IU/dl) x reciproční hodnota pozorovaného uzdravení (tj. 0,5 pro uzdravení 2,0 %).
Podané množství a frekvence podávání mají být vždy zacíleny na klinickou účinnost požadovanou v individuálním případě.
V případě následujících výskytů krvácení nemá aktivita faktoru VIII v příslušném období klesnout pod danou hladinu (v % normální hladiny). Následující tabulka může být použita jako návod pro dávkování během epizod krvácení a během operace:
abulka 1: Návod pro dávkování během krvácivých epizod a chirurgické operace
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Četnost podání dávky (hodiny)/ Délka terapie (dny) |
|
Krvácení Časné hemartrózy, krvácení do svalu nebo krvácení dutiny ústní |
20-40 |
Opakujte po 12 až 24 hodinách. Minimálně 1 den, dokud krvácení projevující se bolestí není zastaveno nebo dokud nebylo dosaženo zahojení. |
|
Intenzivnější hemartrózy, krvácení do svalu nebo tvorba hematomů |
30-60 |
Infúzi opakujte po 12 až 24 hodinách po dobu 3-4 dnů nebo déle, dokud nevymizí bolest a akutní porucha funkce. |
|
Život ohrožující hemoragie |
60-100 |
Infúzi opakujte po 8 až 24 hodinách, dokud není hrozba odvrácena. |
|
Chirurgické operace Menší operace včetně extrakce zubů |
30-60 |
Každých 24 hodin, minimálně 1 den, dokud není dosaženo zahojení. |
|
Větší operace |
80-100 (před- a pooperační) |
Infúzi opakujte po 8-24 hodinách, dokud není rána přiměřeně zahojena, poté terapie nejméně dalších 7 dnů, aby bylo udrženo 30 % až 60 % (IU/dl) aktivity faktoru VIII. |
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů se závažnou hemofilií A jsou obvykle podávány dospívajícím (>12 let věku) a dospělým pacientům dávky 20 až 40 IU přípravku Kovaltry na kg tělesné hmotnosti dva až třikrát týdně.
V některých případech, zejména u mladších pacientů, může být nutné podávání léku v kratších intervalech nebo ve vyšších dávkách.
Dříve neléčení pacienti
Bezpečnost a účinnost přípravku Kovaltry u dříve neléčených pacientů nebyla dosud stanovena. Dostupné jsou omezené údaje.
Pediatrická populace
Studie bezpečnosti a účinnosti byla provedena u dětí od 0-12 let (viz bod 5.1). U dětí do 1 roku jsou k dispozici omezené údaje.
Doporučené profylaktické dávky jsou 20-50 IU/kg dvakrát týdně, třikrát týdně nebo každé dva dny podle individuálních požadavků. U pediatrických pacientů ve věku nad 12 let jsou doporučení pro dávkování stejná jako u dospělých.
Způsob podání
Intravenózní podání.
Přípravek Kovaltry má být aplikován intravenózně po dobu 2 až 5 minut v závislosti na celkovém objemu. Rychlost aplikace má být určována podle komfortu pacienta (maximální rychlost infuze:
2 ml/min). Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6. a příbalové informaci.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Známé alergické reakce na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Při léčbě přípravkem Kovaltry může dojít ke vzniku hypersenzitivních reakcí alergického typu.
Pokud se objeví příznaky hypersenzitivity, pacientům má být doporučeno, aby okamžitě přerušili používání tohoto léčivého přípravku a kontaktovali svého lékaře.
Pacienti mají být informováni o časných příznacích hypersenzitivních reakcí, které zahrnují kopřivku, nauzeu, generalizovanou kopřivku, svíravé pocity na hrudi, sípání, hypotenzi a anafylaxi.
V případě šoku musí být provedena standardní léčba šoku.
Inhibitory
Tvorba neutralizačních protilátek (inhibitorů) proti faktoru VIII je známá komplikace v léčbě jednotlivců s hemofilií A. Tyto inhibitory jsou obvykle imunoglobuliny IgG namířené proti prokoagulační aktivitě faktoru VIII, jejichž množství je vyjádřeno v jednotkách Bethesda (BU) na ml plazmy pomocí modifikovaného testu. Riziko vývoje inhibitorů koreluje, mimo jiné, s expozicí faktoru VIII a s genetickými faktory, toto riziko je nejvyšší během prvních 20 dnů expozice. Jen vzácně se mohou inhibitory vyvinout po prvních 100 dnech expozice.
Po expozici delší než 100 dnů byly mezi dříve léčenými pacienty, kteří měli v anamnéze vývoj inhibitorů, pozorovány případy opětného výskytu inhibitorů (nízkého titru) po přechodu z faktoru VIII od jednoho výrobce na faktor VIII od jiného výrobce. Proto se po přechodu na jiný přípravek doporučuje pečlivě sledovat všechny pacienty z důvodu výskytu inhibitorů.
Obecně u pacientů, kteří jsou léčeni přípravky s koagulačním faktorem VIII, musí být pečlivě monitorován vývoj inhibitorů příslušným klinickým pozorováním a laboratorními zkouškami.
Pokud nejsou dosaženy očekávané hladiny faktoru VIII nebo pokud není krvácení zvládnuto odpovídající dávkou, mají být provedeny testy na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitorů nemusí být terapie faktorem VIII účinná a mají být zváženy jiné možnosti terapie. Léčba takových pacientů má být vedena lékaři se zkušenostmi s léčbou hemofilie a s inhibitory faktoru VIII.
Kardiovaskulární příhody
Hemofiličtí pacienti s kardiovaskulárními rizikovými faktory nebo s kardiovaskulárními chorobami mohou mít to samé riziko rozvoje kardiovaskulárních příhod jako nehemofiličtí pacienti, jestliže byla srážlivost krve normalizována léčbou faktorem VIII. Zvýšení hladiny FVIII po jeho podání, zejména u těch pacientů s přítomnými kardiovaskulárními rizikovými faktory, může pacienta vystavit stejnému riziku uzávěru cév nebo infarktu myokardu jako u nehemofilické populace. Proto mají být pacienti vyšetřeni pro přítomnost kardiovaskulárních rizikových faktorů.
Komplikace související s katetrem
Jestliže je třeba použít centrální žilní vstup (centrál venous access devices, CVAD), musí se uvážit riziko vzniku komplikací souvisejících s CVAD, včetně lokální infekce, bakteriémie a trombózy v místě vstupu katetru. Tyto komplikace nesouvisely se samotným přípravkem.
Dokumentace
Důrazně se doporučuje, aby byl vždy, když je přípravek Kovaltry podán pacientovi, zaznamenán název a číslo šarže přípravku v zájmu zachování spojení mezi pacientem a šarží přípravku.
Pediatrická populace
Uvedená upozornění a opatření platí pro dospělé i děti.
Obsah sodíku
Pro síly 250/500/1 000IU:
Tento léčivý přípravek obsahuje po rekonstituci 0,081 mmol sodíku na injekční lahvičku rekonstituovaného roztoku (což odpovídá 1,86 mg na injekční lahvičku). Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě je „bez sodíku“.
Pro síly 2 000/3 000 IU:
Tento léčivý přípravek obsahuje po rekonstituci 0,156 mmol sodíku na injekční lahvičku rekonstituovaného roztoku (což odpovídá 3,59 mg na injekční lahvičku). Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě je „bez sodíku“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly hlášeny žádné interakce přípravků obsahujících lidský koagulační faktor VIII (rDNA) s jinými léčivými přípravky.
4.6 Fertilita, těhotenství a kojení
Vzhledem k vzácnému výskytu hemofilie A u žen nejsou k dispozici žádné zkušenosti s používáním faktoru VIII během těhotenství. Reprodukční studie na zvířatech nebyly s faktorem VIII provedeny. Proto má být faktor VIII během těhotenství používán pouze, je-li to jasně indikováno.
Kojení
Není známo, zda se přípravek Kovaltry vylučuje do lidského mateřského mléka. Vylučování u zvířat nebylo hodnoceno. Proto má být faktor VIII během kojení používán pouze, je-li to jasně indikováno.
Fertilita
U přípravku Kovaltry nebyly provedeny žádné studie na zvířatech hodnotící fertilitu a jeho účinky na fertilitu u člověka nebyly stanoveny v kontrolovaných klinických studiích. Protože přípravek Kovaltry je substituční protein endogenního faktoru VIII, nejsou očekávány žádné nežádoucí účinky na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pokud pacienti zaznamenají závrať nebo jiné příznaky ovlivňující jejich schopnost soustředit se a reagovat, doporučuje se, aby neřídili ani neobsluhovali stroje, dokud tyto reakce neustoupí.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Byly pozorovány hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, zimnici, zrudnutí, generalizovanou kopřivku, bolest hlavy, vyrážku, hypotenzi, letargii, nauzeu, neklid, tachykardii, svíravý pocit na hrudi, brnění, zvracení, sípání) a v některých případech mohou vyústit v závažnou anafylaxi (včetně šoku).
V souvislosti s hypersenzitivními reakcemi se mohou vyvinout protilátky proti myšímu a křeččímu proteinu.
U pacientů s hemofilií A se mohou vytvořit neutralizační protilátky (inhibitory) proti faktoru VIII. Pokud se objeví takovéto inhibitory, tento stav se projeví jako nedostatečná klinická odpověď.
V těchto případech se doporučuje kontaktovat specializované centrum pro léčbu hemofilie.
Tabulkový přehled nežádoucích účinků
Tabulka uvedená níže je uspořádána podle standardních tříd orgánových systémů a stanoveného vyjádření frekvence výskytu (MedDRA). Frekvence byly vyhodnoceny podle následující konvence: časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100).
V každé skupině frekvence jsou nežádoucí účinky uvedeny podle klesající závažnosti.
Tabulka 2: Frekvence nežádoucích účinků v klinických studiích
|
Podle MedDRA Třída orgánových systémů |
Nežádoucí účinky |
Frekvence |
|
Poruchy krve a lymfatického systému |
Lymfadenopatie |
Časté |
|
Srdeční poruchy |
Palpitace, sinusová tachykardie |
Časté |
|
Gastrointestinální poruchy |
Bolest břicha, abdominální dyskomfort, dyspepsie |
Časté |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie, hrudní dyskomfort, reakce v místě podání injekce* |
Časté |
|
Poruchy imunitního systému |
Hypersenzitivita |
Méně časté |
|
Poruchy nervového systému |
Bolest hlavy, závrať |
Časté |
|
Dysgeusie |
Méně časté | |
|
Psychiatrické poruchy |
Časté | |
|
Poruchy kůže a podkožní tkáně |
Pruritus, vyrážka**, alergická dermatitida |
Časté |
|
Kopřivka |
Méně časté | |
|
Cévní poruchy |
Zrudnutí |
Méně časté |
* patří sem extravazace v místě podání injekce, hematom, bolest v místě podání injekce, pruritus, otok ** vyrážka, erytematózní vyražka, pruritická vyrážka
Popis vybraných nežádoucích účinků
Imunogenicita
Imunogenicita přípravku Kovaltry byla hodnocena u dříve léčených pacientů. Během klinických studií s přípravkem Kovaltry nebyl zaznamenán žádný výskyt inhibitoru u přibližně 200 pediatrických a dospělých pacientů s diagnózou těžké hemofilie A (FVIII< 1 %) s předchozí expozicí koncentrátům faktoru VIII > 50 ED.
Pediatrická populace
V dokončených klinických studiích u 71 dříve léčených pediatrických pacientů bylo zjištěno, že frekvence, typ a závažnost nežádoucích účinků u dětí byly podobné jako u dospělých.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování humánním rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: hemostatika: krevní koagulační faktor VIII, ATC kód: B02BD02 Mechanismus účinku
Faktor VIII/von Willebrandův faktor (vWF) se skládá ze dvou molekul (faktor VIII a vWF) s různými fyziologickými funkcemi. Při aplikaci pacientovi s hemofilií se faktor VIII váže na vWF v oběhu pacienta. Aktivovaný faktor VIII působí jako kofaktor pro aktivovaný faktor IX, urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin pak přeměňuje fibrinogen na fibrin a může dojít k vytvoření sraženiny. Hemofilie A je pohlavně vázaná dědičná porucha srážlivosti krve způsobená sníženou hladinou faktoru VIII:C, následkem čehož dochází k profuznímu krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánnímu, nebo jako následek úrazu při nehodě nebo chirurgickém zákroku. Substituční léčbou se hladiny faktoru VIII v plazmě zvýší, čímž je umožněna přechodná úprava nedostatku faktoru VIII a úprava sklonů ke krvácení.
Přípravek Kovaltry neobsahuje von Willebrandův faktor.
Farmakodynamické účinky
Aktivovaný parciální tromboplastinový čas (aPTT) je prodloužený u osob s hemofilií. Stanovení aPTT je konvenční zkušební metoda in vitro pro biologickou aktivitu faktoru VIII. Léčba pomocí rFVIII normalizuje aPTT podobné těm dosaženým s faktorem VIII získaným z lidské plazmy.
Klinická účinnost a bezpečnost
Kontrola a prevence krvácení
Byly provedeny dvě multicentrické, otevřené, zkřížené, nekontrolované, randomizované studie u dříve léčených dospělých/dospívajících s těžkou hemofilií A (< 1 %) a jedna multricentrická, otevřená, nekontrolovaná studie u dříve léčených dětí ve věku < 12 let s těžkou hemofilií A.
Do výzkumného klinického programu bylo zařazeno celkem 204 pacientů, 153 pacientů bylo ve věku > 12 let a 51 pacientů bylo ve věku < 12 let. 140 pacientů bylo léčeno pod dobu minimálně 12 měsíců a 55 z těchto pacientů po střední dobu 24 měsíců.
Tabulka 3: Spotřeba a celková míra úspěšnosti (pacienti léčení pouze profylakticky)
|
Mladší děti (0 < 6 let) |
Starší děti (6 < 12let ) |
Dospívající a dospělí 12-65 let |
Celkem | |||
|
Studie 1 |
Studie 2 Dávková ní 2 x/týdně |
Study 2 Dávková ní 3 x/týdně | ||||
|
Účastníci studie |
25 |
26 |
62 |
28 |
31 |
172 |
|
Injekční dávka/profylaxe, IU/kg BW medián (min, max) |
36 IU/kg (21; 58 IU/kg) |
32 IU/kg (22; 50 IU/kg) |
31 IU/kg (21; 43 IU/kg) |
30 IU/kg (21; 34 IU/kg) |
37 IU/kg (30; 42 IU/kg) |
32 IU/kg (21; 58 IU/kg) |
|
ABR - všechna krvácení (medián, Q1,Q3) |
2,0 (0,0; 6,0) |
0,9 (0,0; 5,8) |
1,0 (0,0; 5,1) |
4,0 (0,0; 8,0) |
2,0 (0,0; 4,9) |
2,0 (0,0; 6,1) |
|
Dávka/injekce pro léčbu krvácení Medián (min; max) |
39 IU/kg (21;72 IU /kg) |
32 IU/kg (22; 50 IU/kg) |
29 IU/kg (13; 54 IU/kg) |
28 IU/kg (19; 39 IU/kg) |
31 IU/kg (21; 49IU/kg) |
31 IU/kg (13; 72 IU/kg) |
|
Četnost úspěšnosti* |
92,4 % |
86,7 % |
86,3 % |
95,0 % |
97,7 % |
91,4 % |
ABR anualizovaná četnost krvácení Q1 první kvartil; Q3 třetí kvartil BW: tělesná hmotnost
*Četnost úspěšnosti je definovaná jako % krvácení léčených úspěšně pomocí =/< 2 infuzí
5.2 Farmakokinetické vlastnosti
Farmakokinetický (FK) profil přípravku Kovaltry byl hodnocen u dříve léčených pacientů s těžkou hemofilií A po 50 IU/kg u 21 pacientů ve věku > 18 let, 5 pacientů ve věku > 12 let a < 18 let a u 19 pacientů ve věku < 12 let.
Populační FK model byl vyvinut na základě všech dostupných měření FVIII (z častých odběrů na FK vyšetření a všech obnovených vzorků) v průběhu 3 klinických studií, což umožnilo výpočet FK parametrů u pacientů v různých studiích. Tabulka 4 níže uvádí FK parametry na základě populačního FK modelu.
Tabulka 4: FK parametry (geometrický průměr (%CV)) na základě chromogenního testu.*,
|
FK parametr |
> 18 let N=109 |
12 - < 18 let N=23 |
6 - < 12 let N=27 |
0 - < 6 let N=24 |
|
Tl/2 (h) |
14,8 (34) |
13,3 (24) |
14,1 (31) |
13,3 (24) |
|
AUC (IU.h/dl)** |
1 858 (38) |
1 523 (27) |
1 242 (35) |
970 (25) |
|
CL (dl/h/kg) |
0,03 (38) |
0,03 (27) |
0,04 (35) |
0,05 (25) |
|
Vss (dl/kg) |
0,56 (14) |
0,61 (14) |
0,77 (15) |
0,92 (11) |
* Na základě populačních FK odhadů **AUC vypočtená pro dávku 50 IU/kg
Opakovaná FK měření po 6 až 12 měsících profylaktické léčby přípravkem Kovaltry neukázaly žádné významné změny FK charakteristik po dlouhodobé léčbě.
V mezinárodní studii provedené ve 41 klinických laboratořích byla hodnocena účinnost přípravku Kovaltry v testech FVIII:C a porovnána s přípravkem obsahujícím rFVIII plné délky. Byly stanoveny konzistentní výsledky pro oba přípravky. FVIII:C u přípravku Kovalty může být změřen v plazmě pomocí jednofázového koagulačního testu a rovněž pomocí chromogenního testu při použití běžných laboratorních metod.
Analýza všech uváděných inkrementálních uzdravení u dříve léčených pacientů ukázala medián zvýšení o > 2 % (> 2 IU/dl) na IU/kg tělesné váhy pro přípravek Kovaltry. Tento výsledek je podobný hodnotám zaznamenaným pro faktor VIII získaný z lidské plazmy. Během 6-12měsíční léčebné fáze nedošlo k žádné významné změně.
Tabulka 5: Výsledky inkrementálních uzdravení z fáze III
|
Účastníci studie |
N=115 |
|
Výsledky chromogenního testu Medián; (Q1; Q3) (IU/dl / IU/kg) |
2,3 (1,8; 2,6) |
|
Výsledky jednofázového testu Medián; (Q1; Q3) (IU/dl / IU/kg) |
2,2 (1,8; 2,4) |
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě bezpečnostních farmakologických in vitro genotoxických studií a krátkodobých studií toxicity po opakovaném podávání neodhalily žádné zvláštní riziko pro člověka. Studie opakované toxicity delší než 5 dnů, studie reprodukční toxicity a karcinogenity nebyly provedeny. Takové studie nejsou považovány za významné v důsledku tvorby protilátek proti heterogennímu humánnímu proteinu u zvířat. FVIII je také vnitřní protein a není známo, že vyvolává jakékoli reprodukční nebo karcinogenní účinky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Sacharosa
Histidin
Glycin
Chlorid sodný Chlorid vápenatý Polysorbát 80
Rozpouštědlo Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
K rekonstituci a k aplikaci mohou být použity pouze dodávané infuzní soupravy, protože může dojít k selhání léčby v důsledku adsorpce lidského rekombinantního koagulačního faktoru VIII na vnitřní povrchy některého infuzního zařízení.
6.3 Doba použitelnosti
30 měsíců
Chemická a fyzikální stabilita po otevření před použitím po rekonstituci byla prokázána po dobu 3 hodin při pokojové teplotě.
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
Po zředění chraňte před chladem.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C-8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku a předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
V rámci své celkové doby použitelnosti v délce 30 měsíců může být přípravek uchováván ve vnějším obalu při pokojové teplotě (do 25 °C) po omezenou dobu 12 měsíců. V takovém případě skončí doba použitelnosti na konci tohoto 12měsíčního období, nebo po uplynutí data použitelnosti uvedeného na injekční lahvičce, podle toho, co nastane dříve. Nová doba použitelnosti musí být uvedena na krabičce.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení a zvláštní vybavení pro použití, podání nebo implantaci
Jedno balení přípravku Kovaltry obsahuje:
• jednu injekční lahvičku s víčkem na rekonstituci (zařízení Bio-Set) obsahující prášek (injekční lahvička typu 1 z čirého skla o objemu 10 ml se zátkou z šedé halogenobutylové pryžové směsi a víčkem pro rekonstituci)
• jednu předplněnou injekční stříkačku s 2,5 ml (pro 250 IU, 500 IU a 1 000 IU) nebo 5 ml (pro
2 000 IU a 3 000 IU) rozpouštědla (lahvička cylindrického typu 1 z čirého skla se zátkou z šedé brombutylové pryžové směsi)
• píst pro injekční stříkačku
• jednu venepunkční sadu
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Podrobné pokyny pro přípravu a podání jsou obsaženy v příbalové informaci k přípravku Kovaltry. Rekonstituovaný léčivý přípravek je čirý a bezbarvý roztok.
Prášek přípravku Kovaltry musí být rekonstituován pouze s dodaným rozpouštědlem (2,5 ml nebo 5 ml vody na injekci) v předplněné injekční stříkačce a integrovaným těsněním a součástí pro přenos tekutiny. Pro infuzi musí být přípravek připravován v aseptických podmínkách. Pokud je kterákoli součást balení otevřená nebo poškozená, nepoužívejte ji.
Po rekonstituci je roztok čirý. Léčivé přípravky pro parenterální podání je nutno před podáním vizuálně zkontrolovat, zda se v nich nenalézají částice nebo zda nezměnily barvu. Nepoužívejte přípravek Kovaltry, pokud jsou v roztoku viditelné částečky látky nebo zákal.
Po rekonstituci je roztok natažen zpět do injekční stříkačky. Přípravek Kovaltry je nutno rekonstituovat a podat s použitím pomůcek (předplněná injekční stříkačka, venepunkční sada), které jsou dodány v jednotlivých balíčcích.
Rekonstituovaný přípravek je nutno před podáním přefiltrovat, aby byly z roztoku odstraněny případné přítomné částice. Filtrace se provádí podle podrobných pokynů pro přípravu a/nebo podání, které jsou obsaženy v příbalové informaci, která je dodávaná s přípravkem Kovaltry. K podání je důležité použít venepunkční sadu dodanou spolu s přípravkem, protože její součástí je in-line filtr.
V situacích, kdy nelze použít dodanou venepunkční sadu (např. při infuzi do periferního nebo centrálního katetru), je třeba použít samostatný filtr, kompatibilní s přípravkem Kovaltry. Tyto kompatibilní filtry mají polyakrylový kryt s adaptérem typu luer s integrovaným filtračním prvkem, který obsahuje polyamidovou síťku s velikostí ok 5-20 mikrometrů.
Dodaná venepunkční sada nesmí být použita k odběru krve, protože obsahuje in-line filtr. Jestliže je třeba před infuzí provést odběr krve, použijte aplikační sadu bez filtru a pak podejte infuzi přípravku Kovaltry přes injekční filtr.
Máte-li jakékoli dotazy, které se týkají přípravku Kovaltry a kompatibilních samostatných filtrů, obraťte se na společnost Bayer Pharma AG.
Pouze pro jednorázové použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlín Německo
8. REGISTRAČNÍ ČÍSLA
|
EU/1/15/1076/001 |
- Kovaltry 250 IU |
|
EU/1/15/1076/003 |
- Kovaltry 500 IU |
|
EU/1/15/1076/005 EU/1/15/1076/007 EU/1/15/1076/009 |
- Kovaltry 1000 IU - Kovaltry 2000 IU - Kovaltry 3000 IU |
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Kovaltry 250 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 500 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 1000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 2000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 3000 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička nominálně obsahuje 250/500/1 000/2 000/3 000 IU lidského koagulačního faktoru VIII.
• Jeden ml přípravku Kovaltry 250 IU obsahuje přibližně 100 IU (250 IU / 2,5 ml) rekombinantního lidského koagulačního faktoru VIII (INN: octocogum alfa) po rekonstituci ve vodě na injekci.
• Jeden ml přípravku Kovaltry 500 IU obsahuje přibližně 200 IU (500 IU / 2,5 ml) rekombinantního lidského koagulačního faktoru VIII (INN: octocogum alfa) po rekonstituci ve vodě na injekci.
• Jeden ml přípravku Kovaltry 1000 IU obsahuje přibližně 400 IU (1 000 IU / 2,5 ml) rekombinantního lidského koagulačního faktoru VIII (INN: octocogum alfa) po rekonstituci ve vodě na injekci.
• Jeden ml přípravku Kovaltry 2000 IU obsahuje přibližně 400 IU (2 000 IU / 5 ml) rekombinantního lidského koagulačního faktoru VIII (INN: octocogum alfa) po rekonstituci ve vodě na injekci.
• Jeden ml přípravku Kovaltry 3000 IU obsahuje přibližně 600 IU (3 000 IU / 5 ml) rekombinantního lidského koagulačního faktoru VIII (INN: octocogum alfa) po rekonstituci ve vodě na injekci.
Síla (IU) je určena chromogenní zkouškou podle Evropského lékopisu. Specifická účinnost přípravku Kovaltry je přibližně 4 000 IU/mg bílkoviny.
Oktokog alfa (rekombinantní lidský koagulační faktor VIII (rDNA) plné délky) je purifikovaný protein, který má 2 332 aminokyselin. Je vyráběn rekombinantní DNA technologií v ledvinových buňkách křeččích mláďat (BHK), do kterých byl zaveden gen pro lidský faktor VIII. Přípravek Kovaltry se vyrábí bez přidání proteinu lidského nebo zvířecího původu do procesu buněčné kultivace, purifikace nebo finální formulace.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok. (adaptér na injekční lahvičku) Prášek: pevný, bílý až nažloutlý.
Rozpouštědlo: voda na injekci, čirý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u nemocných s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek Kovaltry může být používán u všech věkových kategorií.
4.2 Dávkování a způsob podání
Léčba musí probíhat pod dozorem lékaře se zkušenostmi s léčbou hemofilie.
Dávkování
Dávka a délka substituční léčby závisí na závažnosti deficitu faktoru VIII, na lokalizaci a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek podaného faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které jsou odvozeny od současné normy WHO pro přípravky s faktorem VIII. Plazmatická aktivita faktoru VIII je vyjádřena buď jako procento (vztažené k normální lidské plazmě), nebo v mezinárodních jednotkách (odvozených z mezinárodní normy pro faktor VIII v plazmě).
Aktivita jedné mezinárodní jednotky (IU) faktoru VIII odpovídá množství faktoru VIII obsaženému v jednom ml normální lidské plazmy.
Léčba podle potřeby
Výpočet požadované dávky faktoru VIII vychází z empirické zkušenosti, že 1 mezinárodní jednotka (IU) faktoru VIII na jeden kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 1,5 % až 2,5 % normální aktivity.
Požadovaná dávka se určí pomocí následujícího vzorce:
Požadované jednotky = tělesná hmotnost (kg) x požadované zvýšení faktoru VIII (% nebo IU/dl) x reciproční hodnota pozorovaného uzdravení (tj. 0,5 pro uzdravení 2,0 %).
Podané množství a frekvence podávání mají být vždy zacíleny na klinickou účinnost požadovanou v individuálním případě.
V případě následujících výskytů krvácení nemá aktivita faktoru VIII v příslušném období klesnout pod danou hladinu (v % normální hladiny). Následující tabulka může být použita jako návod pro dávkování během epizod krvácení a během operace:
abulka 1: Návod pro dávkování během krvácivých epizod a chirurgické operace
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Četnost podání dávky (hodiny)/ Délka terapie (dny) |
|
Krvácení Časné hemartrózy, krvácení do svalu nebo krvácení dutiny ústní |
20-40 |
Opakujte po 12 až 24 hodinách. Minimálně 1 den, dokud krvácení projevující se bolestí není zastaveno nebo dokud nebylo dosaženo zahojení. |
|
Intenzivnější hemartrózy, krvácení do svalu nebo tvorba hematomů |
30-60 |
Infúzi opakujte po 12 až 24 hodinách po dobu 3-4 dnů nebo déle, dokud nevymizí bolest a akutní porucha funkce. |
|
Život ohrožující hemoragie |
60-100 |
Infúzi opakujte po 8 až 24 hodinách, dokud není hrozba odvrácena. |
|
Chirurgické operace Menší operace včetně extrakce zubů |
30-60 |
Každých 24 hodin, minimálně 1 den, dokud není dosaženo zahojení. |
|
Větší operace |
80-100 (před- a pooperační) |
Infuzi opakujte po 8-24 hodinách, dokud není rána přiměřeně zahojena, poté terapie nejméně dalších 7 dnů, aby bylo udrženo 30 % až 60 % (IU/dl) aktivity faktoru VIII. |
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů se závažnou hemofilií A jsou obvykle podávány dospívajícím (>12 let věku) a dospělým pacientům dávky 20 až 40 IU přípravku Kovaltry na kg tělesné hmotnosti dva až třikrát týdně.
V některých případech, zejména u mladších pacientů, může být nutné podávání léku v kratších intervalech nebo ve vyšších dávkách.
Dříve neléčení pacienti
Bezpečnost a účinnost přípravku Kovaltry u dříve neléčených pacientů nebyla dosud stanovena. Dostupné jsou omezené údaje.
Pediatrická populace
Studie bezpečnosti a účinnosti byla provedena u dětí od 0-12 let (viz bod 5.1). U dětí do 1 roku jsou k dispozici omezené údaje.
Doporučené profylaktické dávky jsou 20-50 IU/kg dvakrát týdně, třikrát týdně nebo každé dva dny podle individuálních požadavků. U pediatrických pacientů ve věku nad 12 let jsou doporučení pro dávkování stejná jako u dospělých.
Způsob podání
Intravenózní podání.
Přípravek Kovaltry má být aplikován intravenózně po dobu 2 až 5 minut v závislosti na celkovém objemu. Rychlost aplikace má být určována podle komfortu pacienta (maximální rychlost infuze:
2 ml/min). Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6. a příbalové informaci.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Známé alergické reakce na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Při léčbě přípravkem Kovaltry může dojít ke vzniku hypersenzitivních reakcí alergického typu.
Pokud se objeví příznaky hypersenzitivity, pacientům má být doporučeno, aby okamžitě přerušili používání tohoto léčivého přípravku a kontaktovali svého lékaře.
Pacienti mají být informováni o časných příznacích hypersenzitivních reakcí, které zahrnují kopřivku, nauzeu, generalizovanou kopřivku, svíravé pocity na hrudi, sípání, hypotenzi a anafylaxi.
V případě šoku musí být provedena standardní léčba šoku.
Inhibitory
Tvorba neutralizačních protilátek (inhibitorů) proti faktoru VIII je známá komplikace v léčbě jednotlivců s hemofilií A. Tyto inhibitory jsou obvykle imunoglobuliny IgG namířené proti prokoagulační aktivitě faktoru VIII, jejichž množství je vyjádřeno v jednotkách Bethesda (BU) na ml plazmy pomocí modifikovaného testu. Riziko vývoje inhibitorů koreluje, mimo jiné, s expozicí faktoru VIII a s genetickými faktory, toto riziko je nejvyšší během prvních 20 dnů expozice. Jen vzácně se mohou inhibitory vyvinout po prvních 100 dnech expozice.
Po expozici delší než 100 dnů byly mezi dříve léčenými pacienty, kteří měli v anamnéze vývoj inhibitorů, pozorovány případy opětného výskytu inhibitorů (nízkého titru) po přechodu z faktoru VIII od jednoho výrobce na faktor VIII od jiného výrobce. Proto se po přechodu na jiný přípravek doporučuje pečlivě sledovat všechny pacienty z důvodu výskytu inhibitorů.
Obecně u pacientů, kteří jsou léčeni přípravky s koagulačním faktorem VIII, musí být pečlivě monitorován vývoj inhibitorů příslušným klinickým pozorováním a laboratorními zkouškami.
Pokud nejsou dosaženy očekávané hladiny faktoru VIII nebo pokud není krvácení zvládnuto odpovídající dávkou, mají být provedeny testy na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitorů nemusí být terapie faktorem VIII účinná a mají být zváženy jiné možnosti terapie. Léčba takových pacientů má být vedena lékaři se zkušenostmi s léčbou hemofilie a s inhibitory faktoru VIII.
Kardiovaskulární příhody
Hemofiličtí pacienti s kardiovaskulárními rizikovými faktory nebo s kardiovaskulárními chorobami mohou mít to samé riziko rozvoje kardiovaskulárních příhod jako nehemofiličtí pacienti, jestliže byla srážlivost krve normalizována léčbou faktorem VIII. Zvýšení hladiny FVIII po jeho podání, zejména u těch pacientů s přítomnými kardiovaskulárními rizikovými faktory, může pacienta vystavit stejnému riziku uzávěru cév nebo infarktu myokardu jako u nehemofilické populace. Proto mají být pacienti vyšetřeni pro přítomnost kardiovaskulárních rizikových faktorů.
Komplikace související s katetrem
Jestliže je třeba použít centrální žilní vstup (centrál venous access devices, CVAD), musí se uvážit riziko vzniku komplikací souvisejících s CVAD, včetně lokální infekce, bakteriémie a trombózy v místě vstupu katetru. Tyto komplikace nesouvisely se samotným přípravkem.
Dokumentace
Důrazně se doporučuje, aby byl vždy, když je přípravek Kovaltry podán pacientovi, zaznamenán název a číslo šarže přípravku v zájmu zachování spojení mezi pacientem a šarží přípravku.
Pediatrická populace
Uvedená upozornění a opatření platí pro dospělé i děti.
Obsah sodíku
Pro síly 250/500/1 000IU:
Tento léčivý přípravek obsahuje po rekonstituci 0,081 mmol sodíku na injekční lahvičku rekonstituovaného roztoku (což odpovídá 1,86 mg na injekční lahvičku). Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě je „bez sodíku“.
Pro síly 2 000/3 000 IU:
Tento léčivý přípravek obsahuje po rekonstituci 0,156 mmol sodíku na injekční lahvičku rekonstituovaného roztoku (což odpovídá 3,59 mg na injekční lahvičku). Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě je „bez sodíku“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly hlášeny žádné interakce přípravků obsahujících lidský koagulační faktor VIII (rDNA) s jinými léčivými přípravky.
4.6 Fertilita, těhotenství a kojení
Vzhledem k vzácnému výskytu hemofilie A u žen, nejsou k dispozici žádné zkušenosti s používáním faktoru VIII během těhotenství. Reprodukční studie na zvířatech nebyly s faktorem VIII provedeny. Proto má být faktor VIII během těhotenství používán pouze, je-li to jasně indikováno.
Kojení
Není známo, zda se přípravek Kovaltry vylučuje do lidského mateřského mléka. Vylučování u zvířat nebylo hodnoceno. Proto má být faktor VIII během kojení používán pouze, je-li to jasně indikováno.
Fertilita
U přípravku Kovaltry nebyly provedeny žádné studie na zvířatech hodnotící fertilitu a jeho účinky na fertilitu u člověka nebyly stanoveny v kontrolovaných klinických studiích. Protože přípravek Kovaltry je substituční protein endogenního faktoru VIII, nejsou očekávány žádné nežádoucí účinky na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pokud pacienti zaznamenají závrať nebo jiné příznaky ovlivňující jejich schopnost soustředit se a reagovat, doporučuje se, aby neřídili ani neobsluhovali stroje, dokud tyto reakce neustoupí.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Byly pozorovány hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, zimnici, zrudnutí, generalizovanou kopřivku, bolest hlavy, vyrážku, hypotenzi, letargii, nauzeu, neklid, tachykardii, svíravý pocit na hrudi, brnění, zvracení, sípání) a v některých případech mohou vyústit v závažnou anafylaxi (včetně šoku).
V souvislosti s hypersenzitivními reakcemi se mohou vyvinout protilátky proti myšímu a křeččímu proteinu.
U pacientů s hemofilií A se mohou vytvořit neutralizační protilátky (inhibitory) proti faktoru VIII. Pokud se objeví takovéto inhibitory, tento stavse projeví jako nedostatečná klinická odpověď.
V těchto případech se doporučuje kontaktovat specializované centrum pro léčbu hemofilie.
Tabulkový přehled nežádoucích účinků
Tabulka uvedená níže je uspořádána podle standardních tříd orgánových systémů a stanoveného vyjádření frekvence výskytu (MedDRA). Frekvence byly vyhodnoceny podle následující konvence: časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100).
V každé skupině frekvence jsou nežádoucí účinky uvedeny podle klesající závažnosti.
Tabulka 2: Frekvence nežádoucích účinků v klinických studiích
|
Podle MedDRA Třída orgánových systémů |
Nežádoucí účinky |
Frekvence |
|
Poruchy krve a lymfatického systému |
Lymfadenopatie |
Časté |
|
Srdeční poruchy |
Palpitace, sinusová tachykardie |
Časté |
|
Gastrointestinální poruchy |
Bolest břicha, abdominální dyskomfort, dyspepsie |
Časté |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie, hrudní dyskomfort, reakce v místě podání injekce* |
Časté |
|
Poruchy imunitního systému |
Hypersenzitivita |
Méně časté |
|
Poruchy nervového systému |
Bolest hlavy, závrať |
Časté |
|
Dysgeusie |
Méně časté | |
|
Psychiatrické poruchy |
Časté | |
|
Poruchy kůže a podkožní tkáně |
Pruritus, vyrážka**, alergická dermatitida |
Časté |
|
Kopřivka |
Méně časté | |
|
Cévní poruchy |
Zrudnutí |
Méně časté |
* patří sem extravazace v místě podání injekce, hematom, bolest v místě podání injekce, pruritus, otok ** vyrážka, erytematózní vyražka, pruritická vyrážka
Popis vybraných nežádoucích účinků
Imunogenicita
Imunogenicita přípravku Kovaltry byla hodnocena u dříve léčených pacientů. Během klinických studií s přípravkem Kovaltry nebyl zaznamenán žádný výskyt inhibitoru u přibližně 200 pediatrických a dospělých pacientů s diagnózou těžké hemofilie A (FVIII< 1 %) s předchozí expozicí koncentrátům faktoru VIII > 50 ED.
Pediatrická populace
V dokončených klinických studiích u 71 dříve léčených pediatrických pacientů bylo zjištěno, že frekvence, typ a závažnost nežádoucích účinků u dětí byly podobné jako u dospělých.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování humánním rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: hemostatika: krevní koagulační faktor VIII, ATC kód: B02BD02. Mechanismus účinku
Faktor VIII/von Willebrandův faktor (vWF) se skládá ze dvou molekul (faktor VIII a vWF) s různými fyziologickými funkcemi. Při aplikaci pacientovi s hemofilií se faktor VIII váže na vWF v oběhu pacienta. Aktivovaný faktor VIII působí jako kofaktor pro aktivovaný faktor IX, urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin pak přeměňuje fibrinogen na fibrin a může dojít k vytvoření sraženiny. Hemofilie A je pohlavně vázaná dědičná porucha srážlivosti krve způsobená sníženou hladinou faktoru VIII:C, následkem čehož dochází k profuznímu krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánnímu, nebo jako následek úrazu při nehodě nebo chirurgickém zákroku. Substituční léčbou se hladiny faktoru VIII v plazmě zvýší, čímž je umožněna přechodná úprava nedostatku faktoru VIII a úprava sklonů ke krvácení.
Přípravek Kovaltry neobsahuje von Willebrandův faktor.
Farmakodynamické účinky
Aktivovaný parciální tromboplastinový čas (aPTT) je prodloužený u osob s hemofilií. Stanovení aPTT je konvenční zkušební metoda in vitro pro biologickou aktivitu faktoru VIII. Léčba pomocí rFVIII normalizuje aPTT podobné těm dosaženým s faktorem VIII získaným z lidské plazmy.
Klinická účinnost a bezpečnost
Kontrola a prevence krvácení
Byly provedeny dvě multicentrické, otevřené, zkřížené, nekontrolované, randomizované studie u dříve léčených dospělých/dospívajících s těžkou hemofilií A (< 1 %) a jedna multricentrická, otevřená, nekontrolovaná studie u dříve léčených dětí ve věku < 12 let s těžkou hemofilií A.
Do výzkumného klinického programu bylo zařazeno celkem 204 pacientů, 153 pacientů bylo ve věku > 12 let a 51 pacientů bylo ve věku < 12 let. 140 pacientů bylo léčeno pod dobu minimálně 12 měsíců a 55 z těchto pacientů po střední dobu 24 měsíců.
Tabulka 3: Spotřeba a celková míra úspěšnosti (pacienti léčení pouze profylakticky)
|
Mladší děti (0 < 6 let) |
Starší děti (6 < 12let ) |
Dospívající a dospělí 12-65 let |
Celkem | |||
|
Studie 1 |
Studie 2 Dávková ní 2 x/týdně |
Study 2 Dávková ní 3 x/týdně | ||||
|
Účastníci studie |
25 |
26 |
62 |
28 |
31 |
172 |
|
Injekční dávka/profylaxe, IU/kg BW medián (min, max) |
36 IU/kg (21; 58 IU/kg) |
32 IU/kg (22; 50 IU/kg) |
31 IU/kg (21; 43 IU/kg) |
30 IU/kg (21; 34 IU/kg) |
37 IU/kg (30; 42 IU/kg) |
32 IU/kg (21; 58 IU/kg) |
|
ABR - všechna krvácení (medián, Q1,Q3) |
2,0 (0,0; 6,0) |
0,9 (0,0; 5,8) |
1,0 (0,0; 5,1) |
4,0 (0,0; 8,0) |
2,0 (0,0; 4,9) |
2,0 (0,0; 6,1) |
|
Dávka/injekce pro léčbu krvácení Medián (min; max) |
39 IU/kg (21;72 IU /kg) |
32 IU/kg (22; 50 IU/kg) |
29 IU/kg (13; 54 IU/kg) |
28 IU/kg (19; 39 IU/kg) |
31 IU/kg (21; 49IU/kg) |
31 IU/kg (13; 72 IU/kg) |
|
Četnost úspěšnosti* |
92,4 % |
86,7 % |
86,3 % |
95,0 % |
97,7 % |
91,4 % |
ABR anualizovaná četnost krvácení Q1 první kvartil; Q3 třetí kvartil BW: tělesná hmotnost
*Četnost úspěšnosti je definovaná jako % krvácení léčených úspěšně pomocí =/< 2 infuzí
5.2 Farmakokinetické vlastnosti
Farmakokinetický (FK) profil přípravku Kovaltry byl hodnocen u dříve léčených pacientů s těžkou hemofilií A po 50 IU/kg u 21 pacientů ve věku > 18 let, 5 pacientů ve věku > 12 let a < 18 let a u 19 pacientů ve věku < 12 let.
Populační FK model byl vyvinut na základě všech dostupných měření FVIII (z častých odběrů na FK vyšetření a všech obnovených vzorků) v průběhu 3 klinických studií, což umožnilo výpočet FK parametrů u pacientů v různých studiích. Tabulka 4 níže uvádí FK parametry na základě populačního FK modelu.
Tabulka 4: FK parametry (geometrický průměr (%CV)) na základě chromogenního testu.*, **
|
FK parametr |
> 18 let N=109 |
12-< 18 let N=23 |
6-< 12 let N=27 |
0—< 6 let N=24 |
|
Tl/2 (h) |
14,8 (34) |
13,3 (24) |
14,1 (31) |
13,3 (24) |
|
AUC (IU.h/dl)** |
1 858 (38) |
1 523 (27) |
1 242 (35) |
970 (25) |
|
CL (dl/h/kg) |
0,03 (38) |
0,03 (27) |
0,04 (35) |
0,05 (25) |
|
Vss (dl/kg) |
0,56 (14) |
0,61 (14) |
0,77 (15) |
0,92 (11) |
* Na základě populačních FK odhadů **AUC vypočtená pro dávku 50 IU/kg
Opakovaná FK měření po 6 až 12 měsících profylaktické léčby přípravkem Kovaltry neukázaly žádné významné změny FK charakteristik po dlouhodobé léčbě.
V mezinárodní studii provedené ve 41 klinických laboratořích byla hodnocena účinnost přípravku Kovaltry v testech FVIII:C a porovnána s přípravkem obsahujícím rFVIII plné délky. Byly stanoveny konzistentní výsledky pro oba přípravky. FVIII:C u přípravku Kovalty může být změřen v plazmě pomocí jednofázového koagulačního testu a rovněž pomocí chromogenního testu při použití běžných laboratorních metod.
Analýza všech uváděných inkrementálních uzdravení u dříve léčených pacientů ukázala medián zvýšení o > 2 % (> 2 IU/dl) na IU/kg tělesné váhy pro přípravek Kovaltry. Tento výsledek je podobný hodnotám zaznamenaným pro faktor VIII získaný z lidské plazmy. Během 6-12měsíční léčebné fáze nedošlo k žádné významné změně.
Tabulka 5: Výsledky inkrementálních uzdravení z fáze III
|
Účastníci studie |
N=115 |
|
Výsledky chromogenního testu Medián; (Q1; Q3) (IU/dl / IU/kg) |
2,3 (1,8; 2,6) |
|
Výsledky jednofázového testu Medián; (Q1; Q3) (IU/dl / IU/kg) |
2,2 (1,8; 2,4) |
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě bezpečnostních farmakologických in vitro genotoxických studií a krátkodobých studií toxicity po opakovaném podávání neodhalily žádné zvláštní riziko pro člověka. Studie opakované toxicity delší než 5 dnů, studie reprodukční toxicity a karcinogenity nebyly provedeny. Takové studie nejsou považovány za významné v důsledku tvorby protilátek proti heterogennímu humánnímu proteinu u zvířat. FVIII je také vnitřní protein a není známo, že vyvolává jakékoli reprodukční nebo karcinogenní účinky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Sacharosa
Histidin
Glycin
Chlorid sodný Chlorid vápenatý Polysorbát 80
Rozpouštědlo Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
K rekonstituci a aplikaci mohou být použity pouze dodávané infuzní soupravy, protože může dojít k selhání léčby v důsledku adsorpce lidského rekombinantního koagulačního faktoru VIII na vnitřní povrchy některého infuzního zařízení.
6.3 Doba použitelnosti
30 měsíců
Chemická a fyzikální stabilita po otevření před použitím po rekonstituci byla prokázána po dobu 3 hodin při pokojové teplotě.
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
Po zředění chraňte před chladem.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C-8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku a předplněnou injekční stříkačku v krabičce, aby byl přípravek chráněn před světlem.
V rámci své celkové doby použitelnosti v délce 30 měsíců může být přípravek uchováván ve vnějším obalu při pokojové teplotě (do 25 °C) po omezenou dobu 12 měsíců. V takovém případě skončí doba použitelnosti na konci tohoto 12měsíčního období, nebo po uplynutí data použitelnosti uvedeného na injekční lahvičce, podle toho, co nastane dříve. Nová doba použitelnosti musí být uvedena na krabičce.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení a zvláštní vybavení pro použití, podání nebo implantaci
Jedno balení přípravku Kovaltry obsahuje:
• jednu injekční lahvičku obsahující prášek (injekční lahvička typu 1 z čirého skla o objemu 10 ml se zátkou z šedé halogenobutylové pryžové směsi a hliníkovým těsněním)
• jednu předplněnou injekční stříkačku s 2,5 ml (pro 250 IU, 500 IU a 1 000 IU) nebo 5 ml (pro
2 000 IU a 3 000 IU) rozpouštědla (lahvička cylindrického typu 1 z čirého skla se zátkou z šedé brombutylové pryžové směsi)
• píst pro injekční stříkačku
• adaptér injekční lahvičky
• jednu venepunkční sadu
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Podrobné pokyny pro přípravu a podání jsou obsaženy v příbalové informaci k přípravku Kovaltry. Rekonstituovaný léčivý přípravek je čirý a bezbarvý roztok.
Prášek přípravku Kovaltry musí být rekonstituován pouze s dodaným rozpouštědlem (2,5 ml nebo 5 ml vody na injekci) v předplněné injekční stříkačce a adaptéru injekční lahvičky. Pro infuzi musí být přípravek připravován v aseptických podmínkách. Pokud je kterákoli součást balení otevřená nebo poškozená, nepoužívejte ji.
Po rekonstituci je roztok čirý. Léčivé přípravky pro parenterální podání je nutno před podáním vizuálně zkontrolovat, zda se v nich nenalézají částice nebo zda nezměnily barvu. Nepoužívejte přípravek Kovaltry, pokud jsou v roztoku viditelné částečky látky nebo zákal.
Po rekonstituci je roztok natažen zpět do injekční stříkačky. Přípravek Kovaltry je nutno rekonstituovat a podat s použitím pomůcek (adaptér injekční lahvičky, předplněná injekční stříkačka, venepunkční sada), které jsou dodány v jednotlivých balíčcích.
Rekonstituovaný přípravek je nutno před podáním přefiltrovat, aby byly z roztoku odstraněny případné přítomné částice. Fitrace je dosažena pomocí adaptéru injekční lahvičky.
Dodaná venepunkční sada nesmí být použita k odběru krve, protože obsahuje in-line filtr.
Pouze pro jednorázové použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlín Německo
8. REGISTRAČNÍ ČÍSLA
|
EU/1/15/1076/002 |
- Kovaltry 250 IU |
|
EU/1/15/1076/004 |
- Kovaltry 500 IU |
|
EU/1/15/1076/006 EU/1/15/1076/008 EU/1/15/1076/010 |
- Kovaltry 1000 IU - Kovaltry 2000 IU - Kovaltry 3000 IU |
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA
PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Bayer HealthCare LLC 800 Dwight Way Berkeley, CA 94710 USA
Název a adresa výrobce odpovědného za propouštění šarží
Bayer Pharma AG Kaiser-Wilhelm-Allee 51368 Leverkusen Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Poregistrační studie účinnosti: za účelem zhodnocení bezpečnosti a účinnosti přípravku Kovaltry u dříve neléčených pacientů držitel rozhodnutí o registraci musí předložit výsledky probíhající studie “13400 - Leopold Kids Part B” |
12/2018 |
|
Poregistrační studie účinnosti: za účelem zhodnocení bezpečnosti a účinnosti dlouhodobé léčby přípravkem Kovaltry držitel rozhodnutí o registraci musí předložit výsledky probíhající studie “13400 - Leopold Kids extension” |
12/2020 |
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA - PRO SYSTÉM BIO-SET
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Kovaltry 250 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 500 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 1000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 2000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 3000 IU prášek a rozpouštědlo pro injekční roztok
Rekombinantní lidský koagulační faktor VIII (octocogum alfa)
2. OBSAH LÉČIVÝCH LÁTEK
Kovaltry 250 IU obsahuje (250 IU / 2,5 ml) = octocogum alfa 100 IU na ml po rekonstituci. Kovaltry 500 IU obsahuje (500 IU / 2,5 ml) = octocogum alfa 200 IU na ml po rekonstituci. Kovaltry 1000 IU obsahuje (1 000 IU / 2,5 ml) = octocogum alfa 400 IU na ml po rekonstituci. Kovaltry 2000 IU obsahuje (2 000 IU / 5 ml) = octocogum alfa 400 IU na ml po rekonstituci. Kovaltry 3000 IU obsahuje (3 000 IU / 5 ml) = octocogum alfa 600 IU na ml po rekonstituci.
3. SEZNAM POMOCNÝCH LÁTEK
Sacharosa, histidin, glycin, chlorid sodný, dihydrát chloridu vápenatého, polysorbát 80.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok. Systém Bio Set Systém Bio-Set:
1 injekční lahvička s práškem, 1 předplněná injekční stříkačka s vodou na injekci a 1 venepunkční sada.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Intravenózní podání. Pouze pro jednorázové použití. Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST, KÓD DÁRCE A KÓD LÉČIVÉHO PŘÍPRAVKU
EXP:
EXP (Konec 12 měsíčního období, jestliže je uchováván při teplotě do 25 °C): ...........
Nepoužívejte po tomto datu.
Přípravek může být uchováván při teplotě do 25 °C po dobu 12 měsíců v rámci doby použitelnosti uvedené na obalu. Zapište nové datum použitelnosti na krabičku.
Po rekonstituci se přípravek musí použít během 3 hodin. Po rekonstituci roztok chraňte před chladem.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem.
Injekční lahvičku a předplněnou injekční stříkačku uchovávejte ve vnějším obalu, aby byly chráněny před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý roztok musí být zlikvidován.
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1076/001 - Kovaltry 250 IU EU/1/15/1076/003 - Kovaltry 500 IU EU/1/15/1076/005 - Kovaltry 1000 IU EU/1/15/1076/007 - Kovaltry 2000 IU EU/1/15/1076/009 - Kovaltry 3000 IU
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Kovaltry 250 Kovaltry 500 Kovaltry 1000 Kovaltry 2000 Kovaltry 3000
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
KRABIČKA - PRO ADAPTÉR NA INJEKČNÍ LAHVIČKU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Kovaltry 250 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 500 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 1000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 2000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 3000 IU prášek a rozpouštědlo pro injekční roztok
Rekombinantní lidský koagulační faktor VIII (octocogum alfa)
2. OBSAH LÉČIVÉ LÁTKY/ LÉČIVÝCH LÁTEK
Kovaltry 250 IU obsahuje (250 IU / 2,5 ml) = octocogum alfa 100 IU na ml po rekonstituci. Kovaltry 500 IU obsahuje (500 IU / 2,5 ml) = octocogum alfa 200 IU na ml po rekonstituci. Kovaltry 1000 IU obsahuje (1 000 IU / 2,5 ml) = octocogum alfa 400 IU na ml po rekonstituci. Kovaltry 2000 IU obsahuje (2 000 IU / 5 ml) = octocogum alfa 400 IU na ml po rekonstituci. Kovaltry 3000 IU obsahuje (3 000 IU / 5 ml) = octocogum alfa 600 IU na ml po rekonstituci.
3. SEZNAM POMOCNÝCH LÁTEK
Sacharosa, histidin, glycin, chlorid sodný, dihydrát chloridu vápenatého, polysorbát 80.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok. Adaptér na injekční lahvičku Adaptér na injekční lahvičku:
1 injekční lahvička s práškem, 1 předplněná injekční stříkačka s vodou na injekci, 1 adaptér na injekční lahvičku a 1 venepunkční sada.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Intravenózní podání. Pouze pro jednorázové použití. Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST, KÓD DÁRCE A KÓD LÉČIVÉHO PŘÍPRAVKU
EXP:
EXP (Konec 12 měsíčního období, jestliže je uchováván při teplotě do 25 °C): ...........
Nepoužívejte po tomto datu.
Přípravek může být uchováván při teplotě do 25 °C po dobu 12 měsíců v rámci doby použitelnosti uvedené na obalu. Zapište nové datum použitelnosti na krabičku.
Po rekonstituci se přípravek musí použít během 3 hodin. Po rekonstituci roztok chraňte před chladem.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem.
Injekční lahvičku a předplněnou injekční stříkačku uchovávejte ve vnějším obalu, aby byly chráněny před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Veškerý nepoužitý roztok musí být zlikvidován.
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1076/002 - Kovaltry 250 IU EU/1/15/1076/004 - Kovaltry 500 IU EU/1/15/1076/006 - Kovaltry 1000 IU EU/1/15/1076/008 - Kovaltry 2000 IU EU/1/15/1076/010 - Kovaltry 3000 IU
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Kovaltry 250 Kovaltry 500 Kovaltry 1000 Kovaltry 2000 Kovaltry 3000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Kovaltry 250 IU prášek pro injekční roztok Kovaltry 500 IU prášek pro injekční roztok Kovaltry 1000 IU prášek pro injekční roztok Kovaltry 2000 IU prášek pro injekční roztok Kovaltry 3000 IU prášek pro injekční roztok
Rekombinantní lidský koagulační faktor VIII (octocogum alfa) Intravenózní podání.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
250 IU (octocogum alfa ) (100 IU/ml po rekonstituci). 500 IU (octocogum alfa ) (200 IU/ml po rekonstituci).
1 000 IU (octocogum alfa ) (400 IU/ml po rekonstituci).
2 000 IU (octocogum alfa ) (400 IU/ml po rekonstituci).
3 000 IU (octocogum alfa ) (600 IU/ml po rekonstituci).
6. JINÉ
Bayer-Logo
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Voda na injekci
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2,5 ml [pro rekonstituci sil 250/500/1 000 IU]
5 ml [pro rekonstituci sil 2 000/3 000 IU]
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Kovaltry 250 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 500 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 1000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 2000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 3000 IU prášek a rozpouštědlo pro injekční roztok
Rekombinantní lidský koagulační faktor VIII (octocogum alfa)
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Kovaltry a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Kovaltry používat
3. Jak se přípravek Kovaltry používá
4. Možné nežádoucí účinky
5. Jak přípravek Kovaltry uchovávat
6. Obsah balení a další informace
1. Co je přípravek Kovaltry a k čemu se používá
Přípravek Kovaltry je lék, který obsahuje léčivou látku rekombinantní lidský koagulační faktor VIII, nazývanou také oktokog alfa. Přípravek Kovaltry je připraven rekombinantní technologií bez přidání jakékoliv složky lidského či zvířecího původu ve výrobním procesu. Faktor VIII je bílkovina, která se přirozeně vyskytuje v krvi a pomáhá při jejím srážení.
Přípravek Kovaltry se používá k léčbě a prevenci krvácení u dospělých, dospívajících a dětí všech věkových kategorií s hemofilií A (vrozený nedostatek faktoru VIII).
2. Čemu musíte věnovat pozornost, než začnete přípravek Kovaltry používat
Nepoužívejte přípravek Kovaltry
• jestliže jste alergický(á) na oktokog alfa nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6 a na konci bodu 2).
• jestliže jste alergický(á) na myší nebo křeččí bílkoviny.
Nepoužívejte přípravek Kovaltry, pokud se Vás týká cokoli z výše uvedeného. Pokud si nejste jistý(á), zeptejte se před použitím tohoto přípravku svého lékaře.
Upozornění a opatření
Věnujte zvláštní opatrnost při použití přípravku Kovaltry a poraďte se se svým lékařem nebo lékárníkem, pokud:
• máte svíravý pocit na hrudi, závrať (včetně pokud vstáváte z pozice vsedě nebo vleže), vyrážku, svědivou vyrážku (kopřivku), sípání, je vám nevolno nebo mdlo. Toto mohou být známky vzácné závažné náhlé alergické reakce (anafylaktická reakce) na přípravek Kovaltry. Pokud toto nastane, okamžitě zastavte podávání přípravku a vyhledejte lékařskou pomoc.
• Vaše krvácení není kontrolováno obvyklou dávkou přípravku Kovaltry. Pokud toto nastane, okamžitě se poraďte se svým lékařem. Pravděpodobně u Vás došlo k vytvoření inhibitorů faktoru VIII a Váš lékař může chtít provést testy, aby toto potvrdil. Inhibitory faktoru VIII jsou protilátky v krvi, které blokují faktor VIII, který používáte, a způsobují, že je méně účinný pro prevenci a zastavení krvácení. Tvorba takových protilátek je známá komplikace léčby pacientů s hemofilií A.
• se u Vás již objevil vznik inhibitorů faktoru VIII jiných přípravků. Pokud jste změnil(a) přípravky s faktorem VIII, existuje tu riziko, že se Vám tvorba inhibitoru vrátí.
• Vám bylo řečeno, že máte onemocnění srdce nebo riziko pro vznik onemocnění srdce.
• je pro podání přípravku Kovaltry potřeba použít centrální žilní vstup. Můžete mít riziko vzniku komplikací souvisejících s centrálním žilním vstupem, včetně místní infekce, bakterií v krvi (bakteriemie) a krevních sraženin v cévách (trombóza) v místě vstupu katétru.
Další léčivé přípravky a přípravek Kovaltry
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Děti a dospívající
Uvedená upozornění a opatření se týkají pacientů všech věkových skupin, dospělých a dětí. Těhotenství a kojení
Zkušenosti s použitím přípravků obsahujících faktor VIII během těhotenství a kojení nejsou k dispozici, protože hemofilie A se u žen vyskytuje vzácně. Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek používat.
Přípravek Kovaltry pravděpodobně neovlivní plodnost u mužů nebo žen, protože léčivá látka se normálně vyskytuje v těle.
Řízení dopravních prostředků a obsluha strojů
Pokud máte závratě nebo jiné příznaky ovlivňující Vaši schopnost se koncentrovat a reagovat, neřiďte ani neobsluhujte stroje, dokud reakce neustoupí.
Přípravek Kovaltry obsahuje sodík
Tento přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě je „bez sodíku“.
Dokumentace
Je doporučeno, abyste pokaždé, když použijete přípravek Kovaltry, zapsali název a číslo šarže tohoto přípravku.
3. Jak se přípravek Kovaltry používá
Léčba přípravkem Kovaltry bude zahájena lékařem, který má zkušenosti s léčbou pacientů s hemofilií A. Vždy používejte tento přípravek přesně v souladu s příbalovou informací nebo podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Léčba krvácení
Váš lékař vypočítá dávku tohoto přípravku a stanoví, jak často ho máte používat, aby bylo dosaženo potřebné hladiny úrovně aktivity faktoru VIII v krvi. Lékař má vždy upravit dávku a frekvenci podávání podle Vašich individuálních potřeb. Množství a četnost používání přípravku Kovaltry závisí na mnoha faktorech, jako jsou:
• Vaše hmotnost,
• závažnost hemofilie,
• místo a závažnost krvácení,
• zda máte inhibitory a jak je vysoký jejich titr,
• požadovaná hladina faktoru VIII.
Prevence krvácení
Pokud používáte přípravek Kovaltry za účelem prevence krvácení (profylaxe), Váš lékař vypočte potřebnou dávku. Tato dávka se bude obvykle pohybovat v rozsahu 20 až 40 IU oktokogu alfa na kg tělesné hmotnosti podávaná v injekci dva nebo třikrát týdně. Nicméně v některých případech, zejména u mladších pacientů, může být nutné podávat přípravek v kratších intervalech nebo ve vyšších dávkách.
Laboratorní testy
Velmi se doporučuje, aby byly u Vás ve vhodných intervalech prováděny příslušné laboratorní zkoušky plazmy, aby bylo zajištěno, že byla dosažena a je udržována přiměřená hladina faktoru VIII. Zejména v případě větších chirurgických zákroků je nevyhnutelné přesné monitorování substituční terapie pomocí koagulační analýzy.
Použití u dětí a dospívajících
Přípravek Kovaltry může být používán u dětí všech věkových skupin. U dětí do 12 let věku mohou být zváženy vyšší dávky nebo vyšší četnost dávkování.
Pacienti s inhibitory
Pokud Vás lékař informoval, že se u Vás vytvořily inhibitory faktoru VIII, budete pravděpodobně muset používat k zastavení krvácení větší dávku přípravku Kovaltry. Pokud tato dávka nezastaví krvácení, může Váš lékař zvážit podání jiného přípravku.
Promluvte si se svým lékařem, chcete-li více informací.
Nezvyšujte dávku přípravku Kovaltry, kterou používáte k zastavení krvácení, aniž byste se poradil(a) se svým lékařem.
Doba trvání léčby
Váš lékař Vám sdělí, jak často a v jakých intervalech má být tento léčivý přípravek podáván.
Obvykle je terapie hemofilie přípravkem Kovaltry zapotřebí po celý život.
Jak se přípravek Kovaltry podává
Tento léčivý přípravek je určen pro injekci do žíly po dobu delší než 2 až 5 minut v závislosti na celkovém objemu a úrovni Vašeho pohodlí a má být použit běhěm tří hodin po rekonstituci.
Jak se přípravek Kovaltry připravuje pro podání
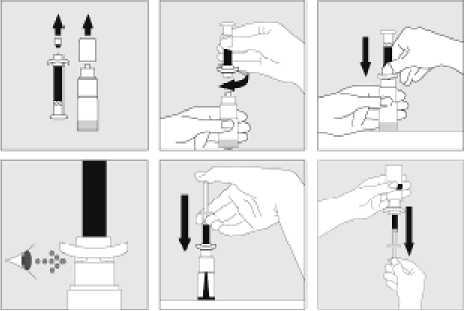
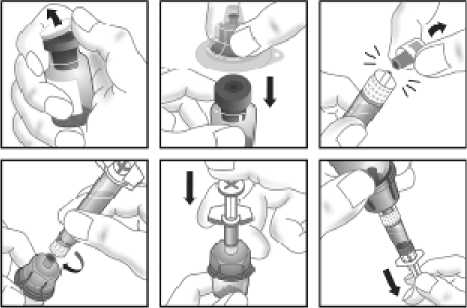
Používejte pouze položky (injekční lahvička s práškem s víčkem Bio-Set, předplněná injekční stříkačka obsahující rozpouštědlo a venepunkční sada), které jsou součástí každého balení tohoto léčivého přípravku. Pokud tyto součásti nelze použít, obraťte se na svého lékaře. Pokud je kterákoli součást balení otevřená nebo poškozená, nepoužívejte ji.
Rozpuštěný přípravek je nutno před podáním přefiltrovat, aby byly z roztoku odstraněny případné přítomné částice. Filtrování provádějte podle pokynů pro rozpuštění a/nebo podání, které jsou popsány níže. Je důležité použít dodanou venepunkční sadu, protože její součástí je in-line filtr. Pokud nelze použít dodanou venepunkční sadu, použijte samostatný filtr podle instrukcí zdravotní sestry nebo lékaře.
Dodanou venepunkční sadu nepoužívejte k odběru krve, protože obsahuje in-line filtr. Jestliže je třeba před infuzí provést odběr krve, použijte aplikační sadu bez filtru a pak podejte infuzi tohoto přípravku přes injekční filtr. S dotazy, které se týkají tohoto přípravku a kompatibilních samostatných filtrů, se obraťte na svého lékaře.
Tento léčivý přípravek nesmí být smíchán s jinými infuzními roztoky. Nepoužívejte roztoky, pokud obsahují viditelné částice nebo jsou zakalené. Dodržujte přesně pokyny svého lékaře a podrobné instrukce pro rozředění a podání uvedené na konci této příbalové informace.
Jestliže jste použil(a) více přípravku Kovaltry, než jste měl(a)
Nebyly zaznamenány žádné případy předávkování rekombinantním koagulačním faktorem VIII.
Pokud jste použil(a) větší množství přípravku Kovaltry, než jste měl(a), řekněte to, prosím, svému lékaři.
Jestliže jste zapomněl(a) použít přípravek Kovaltry
• Aplikajte okamžitě svou další dávku a pokračujte v pravidelných intervalech dle pokynů svého lékaře.
• Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat přípravek Kovaltry Nepřestávejte užívat přípravek Kovaltry bez porady se svým lékařem.
Máte-li jakékoli další otázky týkající setohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Mezi nejzávažnější nežádoucí účinky patří alergické reakce nebo anafylaktický šok (méně častá závažná alergická reakce postihující krevní tlak a dýchání). Pokud se vyskytne alergická nebo anafylaktická reakce, ukončete okamžitě injekce/infuze a řekněte to ihned svému lékaři. Jakýkoliv z následujících příznaků během podávání injekce/infuze může být časným varováním alergických a anafylaktických reakcí:
• pocit tíže na hrudi/celkový pocit nevolnosti,
• závrať,
• mírná hypotenze (mírně snížený krevní tlak, což může způsobit mdloby po vstání),
• pocit na zvracení.
Další možné nežádoucí účinky:
Časté (mohou postihnout až 1 z 10 pacientů):
• zvětšení mízních uzlin (otok pod kůží v oblasti krku, podpažních jamek nebo třísel)
• srdeční palpitace (pocit silného a rychlého nebo nepravidelného bušení srdce)
• zrychlený srdeční tep
• bolest žaludku nebo diskomfort
• porucha trávení
• horečka
• bolest na hrudi nebo diskomfort
• lokální reakce v místě vpichu léčiva (např. krvácení pod kůží, intenzivní svědění, otok, pálení, přechodné začervenání)
• bolest hlavy
• závrať
• problémy s usínáním
• vyrážka/svědivá vyrážka
Méně časté (mohou postihnout až 1 ze 100 pacientů):
• alergické reakce včetně závažné náhlé alergické reakce
• dysgeusie (zvláštní chuť)
• kopřivka (svědivá vyrážka)
• návaly (zrudnutí v obličeji)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Kovaltry uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Uchovávejte v chladničce (2 °C-8 °C). Chraňte před mrazem.
Uchovávejte přípravek v původním obalu, aby byl chráněn před světlem.
Tento přípravek může být uchováván při pokojové teplotě (do 25 °C) po omezenou dobu 12 měsíců, pokud je uchováván ve vnějším obalu. Pokud uchováváte tento přípravek při pokojové teplotě, uplyne doba použitelnosti po 12 měsících nebo po uplynutí data použitelnosti, podle toho, co nastane dříve. Nové datum použitelnosti musíte zapsat na krabičku.
Po rekonstituci roztok chraňte před chladem. Rekonstituovaný roztok musí být použit během 3 hodin. Tento přípravek je určen pouze pro jednorázové použití. Všechen nespotřebovaný roztok musí být zlikvidován.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítcích a krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Nepoužívejte tento přípravek, pokud si všimnete viditelných částic nebo zakalení roztoku.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Kovaltry obsahuje
Prášek
Léčivou látkou je lidský koagulační faktor VIII (octocogum alfa). Jedna injekční lahvička s přípravkem Kovaltry obsahuje nominální množství 250, 500, 1 000, 2 000 nebo 3 000 IU octocogum alfa.
Dalšími složkami jsou sacharosa, histidin, glycin, chlorid sodný, chlorid vápenatý, polysorbát 80 (viz poslední část bodu 2).
Rozpouštědlo Voda na injekci.
Jak přípravek Kovaltry vypadá a co obsahuje toto balení
Přípravek Kovaltry se dodává jako prášek a rozpouštědlo pro injekční roztok a je to suchý bílý až nažloutlý prášek nebo tableta. Po rozpuštění je roztok čirý.
Každé balení přípravku Kovaltry obsahuje injekční lahvičku s víčkem Bio-Set a předplněnou stříkačku se samostatným pístem, stejně jako venepunkční sadu (pro podání injekce do žíly).
Složky pro rozpuštění a podání se dodávají s každým balením tohoto přípravku.
Držitel rozhodnutí o registraci
Bayer Pharma AG 13342 Berlín Německo
Výrobce
Bayer Pharma AG 51368 Leverkusen Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie/Belgique/Belgien
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Bt^rapnn
Eaňep Etnrapna EOOfl Ten.: 359 02 81 401 01 Česká republika Bayer s.r.o.
Tel: +420 266 101 111
Danmark
Bayer A/S
Tlf: +45 45 23 50 00
Deutschland
Bayer Vital GmbH
Tel: +49 (0)214-30 513 48
Eesti
Bayer OU
Tel: +372 655 8565
EXlába
Bayer EMá; ABEE Tr(k: +30-210-61 87 500 Espaňa
Bayer Hispania S.L.
Tel: +34-93-495 65 00 France
Bayer HealthCare
Tél (N° vert): +33-(0)800 87 54 54
Hrvatska
Bayer d.o.o.
Tel: +385-(0)1-6599 900
Ireland
Bayer Limited
Tel: +353 1 2999313
Island
Icepharma hf.
Sími: +354 540 8000 Italia
Bayer S.p.A.
Tel: +39 02 397 81 Kúnpoq
NOVAGEM Limited Tr(k: +357 22 48 38 58 Latvija SIA Bayer
Tel: +371 67 84 55 63
Tato příbalová informace byla ni
Lietuva
UAB Bayer
Tel. +37 05 23 36 868
Luxembourg/Luxemburg
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Magyarország
Bayer Hungária KFT
Tel:+36 14 87-41 00
Malta
Alfred Gera and Sons Ltd. Tel: +35 621 44 62 05 Nederland Bayer B.V.
Tel: +31-(0)297-28 06 66 Norge
Bayer AS
Tlf: +47 23 13 05 00 Osterreich
Bayer Austria Ges.m.b.H. Tel: +43-(0)1-711 46-0 Polska
Bayer Sp. z o.o.
Tel: +48 22 572 35 00 Portugal
Bayer Portugal, Lda.
Tel: +351 21 416 42 00
Románia
SC Bayer SRL
Tel: +40 21 529 59 00
Slovenija
Bayer d. o. o.
Tel: +386 (0)1 58 14 400 Slovenská republika Bayer spol. s r.o.
Tel. +421 2 59 21 31 11 Suomi/Finland
Bayer Oy
Puh/Tel: +358- 20 785 21
Sverige
Bayer AB
Tel: +46 (0) 8 580 223 00 United Kingdom Bayer plc
Tel: +44-(0)1635 563000 revidována {MM/RRRR}
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
Podrobné instrukce pro rekonstituci a podání přípravku Kovaltry pomocí injekční lahvičky s víčkem na rekonstituci (zařízení Bio-Set):
Budete potřebovat tampony napuštěné alkoholem, vatové tampony a náplasti. Tyto položky nejsou součástí balení přípravku Kovaltry.
1. Omyjte si pečlivě ruce mýdlem a teplou vodou. Roztok musí být připravován na čistém a suchém povrchu.
2. Zahřejte neotevřenou injekční lahvičku s práškem a injekční stříkačku s rozpouštědlem ve
svých rukách na tělesnou teplotu. Materiál nemá být zahřátý na vyšší, než je tělesná teplota (nesmí překročit 37 °C).
3.
Odstraňte víčko z injekční lahvičky s práškem tak, že víčkem několikrát pohnete na obě strany a zároveň táhnete víčko směrem nahoru. Vytáhněte zátku připojenou k bílému víčku z injekční stříkačky (A).
4.
Našroubujte šetrně injekční stříkačku na injekční lahvičku s práškem (B).
5.
Umístěte injekční lahvičku na stabilní a neklouzavý povrch a jednou rukou ji pevně držte. Poté silně zatlačte palcem a ukazováčkem na ochranný štítek u konce injekční stříkačky směrem dolů (C), dokud se štítek nenarazí na horní okraj víčka na rekonstituci (Bio-Set).
Toto potvrzuje, že systém je aktivován (D).
6.
Připevněte píst k injekční stříkačce tak, že jej zašroubujete do pryžové zátky (E).
7.
Vstříkněte roztok do injekční lahvičky s práškem tak, že pomalu tlačíte píst injekční stříkačky směrem dolů (F).
8.
Prášek rozpusťte mírným kroužením injekční lahvičkou (G). Injekční lahvičkou netřepejte! Ujistěte se před použitím, že se veškerý prášek rozpustil. Před podáním vizuálně zkontrolujte, zda nejsou přítomny částice nebo zda přípravek nezměnil barvu. Nepoužívejte roztok, který obsahuje viditelné částice nebo je zakalený.
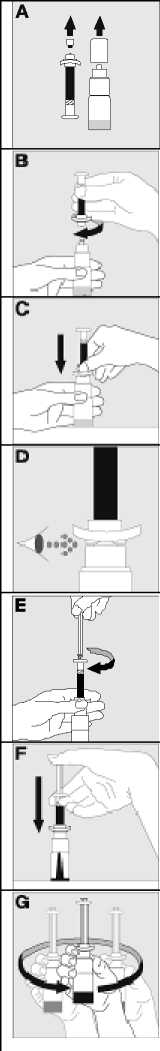
9. Obraťte injekční lahvičku/injekční stříkačku a natáhněte roztok do injekční
stříkačky tak, že pomalu a plynule vytahujete píst ze stříkačky (H). Zkontrolujte, že byl celý obsah injekční lahvičky přemístěn do stříkačky. Držte stříkačku svisle a zatlačte na píst tak, aby ve stříkačce nezbyl žádný vzduch.
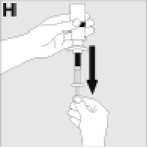
10. Přiložte na paži turniket. Určete místo vpichu a očistěte kůži tamponem napuštěným
alkoholem. Napíchněte žílu a zajistěte venepunkční sadu náplastí.
11. Vyšroubujte injekční stříkačku, abyste oddělili injekční lahvičku (I).
12. Připevněte injekční stříkačku k venepunkční sadě šroubováním ve směru
hodinových ručiček a ujistěte se, že se do injekční stříkačky nedostává krev (J).
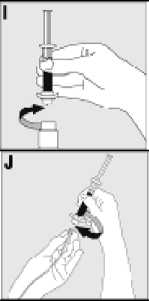
13. Sejměte turniket!
|
14. |
Roztok podávejte injekcí do žíly po dobu 2 až 5 minut, přičemž kontrolujte polohu jehly. Rychlost podání má vycházet z toho, jak je Vám to pohodlné, ale nemá být rychlejší než 2 ml za minutu. |
|
15. |
Je-li nutné podat další dávku, odstraňte prázdnou injekční stříkačku otáčením proti směru hodinových ručiček. Rozřeďte potřebné množství přípravku, zopakujte kroky 2-9, požijte novou injekční stříkačku a připojte ji k venepunkční sadě. |
|
16. |
Pokud není potřebná žádná další dávka, odstraňte venepunkční sadu a injekční stříkačku. Přidržujte tampón pevně k místu vpichu na natažené paži po dobu asi 2 minut. Nakonec přiložte na místo aplikace injekce malý tlakový obvaz a zvažte, je-li nutné použít náplast. |
Příbalová informace: informace pro uživatele
Kovaltry 250 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 500 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 1000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 2000 IU prášek a rozpouštědlo pro injekční roztok Kovaltry 3000 IU prášek a rozpouštědlo pro injekční roztok
Rekombinantní lidský koagulační faktor VIII (octocogum alfa)
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Kovaltry a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Kovaltry používat
3. Jak se přípravek Kovaltry používá
4. Možné nežádoucí účinky
5. Jak přípravek Kovaltry uchovávat
6. Obsah balení a další informace
1. Co je přípravek Kovaltry a k čemu se používá
Přípravek Kovaltry je lék, který obsahuje léčivou látku rekombinantní lidský koagulační faktor VIII, nazývaný také oktokog alfa. Přípravek Kovaltry je připraven rekombinantní technologií bez přidání jakékoliv složky lidského či zvířecího původu ve výrobním procesu Faktor VIII je bílkovina, která se přirozeně vyskytuje v krvi a pomáhá při jejím srážení.
Přípravek Kovaltry se používá k léčbě a prevenci krvácení u dospělých, dospívajících a dětí všech věkových kategorií s hemofilií A (vrozený nedostatek faktoru VIII).
2. Čemu musíte věnovat pozornost, než začnete přípravek Kovaltry používat
Nepoužívejte přípravek Kovaltry
• jestliže jste alergický(á) na oktokog alfa nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6 a na konci bodu 2).
• jestliže jste alergický(á) na myší nebo křeččí bílkoviny.
Nepoužívejte přípravek Kovaltry, pokud se Vás týká cokoli z výše uvedeného. Pokud si nejste jistý(á), zeptejte se před použitím tohoto přípravku svého lékaře.
Upozornění a opatření
Věnujte zvláštní opatrnost při použití přípravku Kovaltry a poraďte se se svým lékařem nebo lékárníkem, pokud:
• máte svíravý pocit na hrudi, závrať (včetně pokud vstáváte z pozice vsedě nebo vleže), vyrážku, svědivou vyrážku (kopřivku), sípání, je vám nevolno nebo mdlo. Toto mohou být známky vzácné závažné náhlé alergické reakce (anafylaktická reakce) na přípravek Kovaltry. Pokud toto nastane, okamžitě zastavte podávání přípravku a vyhledejte lékařskou pomoc.
• Vaše krvácení není kontrolováno obvyklou dávkou přípravku Kovaltry. Pokud toto nastane, okamžitě se poraďte se svým lékařem. Pravděpodobně u Vás došlo k vytvoření inhibitorů faktoru VIII a Váš lékař může chtít provést testy, aby toto potvrdil. Inhibitory faktoru VIII jsou protilátky v krvi, které blokují faktor VIII, který používáte, a způsobují, že je méně účinný pro prevenci a zastavení krvácení. Tvorba takových protilátek je známá komplikace léčby pacientů s hemofilií A.
• se u Vás již objevil vznik inhibitorů faktoru VIII jiných přípravků. Pokud jste změnil(a) přípravky s faktorem VIII, existuje tu riziko, že se Vám tvorba inhibitoru vrátí.
• Vám bylo řečeno, že máte onemocnění srdce nebo riziko pro vznik onemocnění srdce.
• je pro podání přípravku Kovaltry potřeba použít centrální žilní vstup. Můžete mít riziko vzniku komplikací souvisejících s centrálním žilním vstupem, včetně místní infekce, bakterií v krvi (bakteriemie) a krevních sraženin v cévách (trombóza) v místě vstupu katétru.
Další léčivé přípravky a přípravek Kovaltry
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Děti a dospívající
Uvedená upozornění a opatření se týkají pacientů všech věkových skupin, dospělých a dětí. Těhotenství a kojení
Zkušenosti s použitím přípravků obsahujících faktor VIII během těhotenství a kojení nejsou k dispozici, protože hemofilie A se u žen vyskytuje vzácně. Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek používat.
Přípravek Kovaltry pravděpodobně neovlivní plodnost u mužů nebo žen, protože léčivá látka se normálně vyskytuje v těle.
Řízení dopravních prostředků a obsluha strojů
Pokud máte závratě nebo jiné příznaky ovlivňující Vaši schopnost se koncentrovat a reagovat, neřiďte ani neobsluhujte stroje, dokud reakce neustoupí.
Přípravek Kovaltry obsahuje sodík
Tento přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě je „bez sodíku“.
Dokumentace
Je doporučeno, abyste pokaždé, když použijete přípravek Kovaltry, zapsali název a číslo šarže tohoto přípravku.
3. Jak se přípravek Kovaltry používá
Léčba přípravkem Kovaltry bude zahájena lékařem, který má zkušenosti s léčbou pacientů s hemofilií A. Vždy používejte tento přípravek přesně v souladu s příbalovou informací nebo podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Léčba krvácení
Váš lékař vypočítá dávku tohoto přípravku a stanoví, jak často ho máte používat, aby bylo dosaženo potřebné hladiny úrovně aktivity faktoru VIII v krvi. Lékař má vždy upravit dávku a frekvenci podávání podle Vašich individuálních potřeb. Množství a četnost používání přípravku Kovaltry závisí na mnoha faktorech, jako jsou:
• Vaše hmotnost,
• závažnost hemofilie,
• místo a závažnost krvácení,
• zda máte inhibitory a jak je vysoký jejich titr,
• požadovaná hladina faktoru VIII.
Prevence krvácení
Pokud používáte přípravek Kovaltry za účelem prevence krvácení (profylaxe), váš lékař vypočte potřebnou dávku. Tato dávka se bude obvykle pohybovat v rozsahu 20 až 40 IU oktokogu alfa na kg tělesné hmotnosti podávaná v injekci dva nebo třikrát týdně. Nicméně v některých případech, zejména u mladších pacientů, může být nutné podávat přípravek v kratších intervalech nebo ve vyšších dávkách.
Laboratorní testy
Velmi se doporučuje, aby byly u Vás ve vhodných intervalech prováděny příslušné laboratorní zkoušky plazmy, aby bylo zajištěno, že byla dosažena a je udržována přiměřená hladina faktoru VIII. Zejména v případě větších chirurgických zákroků je nevyhnutelné přesné monitorování substituční terapie pomocí koagulační analýzy.
Použití u dětí a dospívajících
Přípravek Kovaltry může být používán u dětí všech věkových skupin. U dětí do 12 let věku mohou být zváženy vyšší dávky nebo vyšší četnost dávkování.
Pacienti s inhibitory
Pokud Vás lékař informoval, že se u Vás vytvořily inhibitory faktoru VIII, budete pravděpodobně muset používat k zastavení krvácení větší dávku přípravku Kovaltry. Pokud tato dávka nezastaví krvácení, může Váš lékař zvážit podání jiného přípravku.
Promluvte si se svým lékařem, chcete-li více informací.
Nezvyšujte dávku přípravku Kovaltry, kterou používáte k zastavení krvácení, aniž byste se poradil(a) se svým lékařem.
Doba trvání léčby
Váš lékař Vám sdělí, jak často a v jakých intervalech má být tento léčivý přípravek podáván.
Obvykle je terapie hemofilie přípravkem Kovaltry zapotebí po celý život.
Jak se přípravek Kovaltry podává
Tento léčivý přípravek je určen pro injekci do žíly po dobu delší než 2 až 5 minut v závislosti na celkovém objemu a úrovni Vašeho pohodlí a má být použit běhěm tří hodin po rekonstituci.
Jak se přípravek Kovaltry připravuje pro podání
Používejte pouze položky (adaptér injekční lahvičky, předplněná injekční stříkačka obsahující rozpouštědlo a venepunkční sada), které jsou součástí každého balení tohoto léčivého přípravku.
Pokud tyto součásti nelze použít, obraťte se na svého lékaře. Pokud je kterákoli součást balení otevřená nebo poškozená, nepoužívejte ji.
Rozpuštěný přípravek je nutno před podáním přefiltrovat, aby byly z roztoku odstraněny případné přítomné částice. Filtrování provádějte pomocí adaptéru injekční lahvičky.
Dodanou venepunkční sadu nepoužívejte k odběru krve, protože obsahuje in-line filtr.
Tento léčivý přípravek nesmí být smíchán s jinými infuzními roztoky. Nepoužívejte roztoky, pokud obsahují viditelné částice nebo jsou zakalené. Dodržujte přesně pokyny svého lékaře a podrobné instrukce pro rozředění a podání uvedené na konci této příbalové informace.
Jestliže jste použil(a) více přípravku Kovaltry, než jste měl(a)
Nebyly zaznamenány žádné případy předávkování rekombinantním koagulačním faktorem VIII. Pokud jste použil(a) větší množství přípravku Kovaltry, než jste měl(a), řekněte to, prosím, svému lékaři.
Jestliže jste zapomněl(a) použít přípravek Kovaltry
• Aplikujte okamžitě svou další dávku a pokračujte v pravidelných intervalech dle pokynů svého lékaře.
• Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat přípravek Kovaltry Nepřestávejte užívat přípravek Kovaltry bez porady se svým lékařem.
Máte-li jakékoli další otázky týkající se tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Mezi nejzávažnější nežádoucí účinky patří alergické reakce nebo anafylaktický šok (méně častá závažná alergická reakce postihující krevní tlak a dýchání). Pokud se vyskytne alergická nebo anafylaktická reakce, ukončete okamžitě injekce/infuze a řekněte to ihned svému lékaři. Jakýkoliv z následujících příznaků během podávání injekce/infuze může být časným varováním alergických a anafylaktických reakcí:
• pocit tíže na hrudi/celkový pocit nevolnosti,
• závrať,
• mírná hypotenze (mírně snížený krevní tlak, což může způsobit mdloby po vstání),
• pocit na zvracení.
Další možnéh nežádoucí účinky:
Časté (mohou postihnout až 1 z 10 pacientů):
• zvětšení mízních uzlin (otok pod kůží v oblasti krku, podpažních jamek nebo třísel)
• srdeční palpitace (pocit silného a rychlého nebo nepravidelného bušení srdce)
• zrychlený srdeční tep
• bolest žaludku nebo diskomfor,
• porucha trávení
• horečka
• bolest na hrudi nebo diskomfort
• lokální reakce v místě vpichu léčiva (např. krvácení pod kůží, intenzívní svědění, otok, pálení, přechodné začervenání)
• bolest hlavy
• závrať
• problémy s usínáním
• vyrážka/svědivá vyrážka
Méně časté (mohou postihnout až 1 ze 100 pacientů):
• alergické reakce, včetně závažné náhlé alergické reakce,
• dysgeusie (zvláštní chuť)
• kopřivka (svědivá vyrážka)
• návaly (zarudnutí v obličeji)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Kovaltry uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Uchovávejte v chladničce (2 °C-8 °C). Chraňte před mrazem.
Uchovávejte přípravek v původním obalu, aby byl chráněn před světlem.
Tento přípravek může být uchováván při pokojové teplotě (do 25 °C) po omezenou dobu 12 měsíců, pokud je uchováván ve vnějším obalu. Pokud uchováváte tento přípravek při pokojové teplotě, uplyne doba použitelnosti po 12 měsících nebo po uplynutí data použitelnosti, podle toho, co nastane dříve. Nové datum použitelnosti musíte zapsat na krabičku.
Po rekonstituci roztok chraňte před chladem. Rekonstituovaný roztok musí být použit během 3 hodin. Tento přípravek je určen pouze pro jednorázové použití. Všechen nespotřebovaný roztok musí být zlikvidován.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítcích a krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Nepoužívejte tento přípravek, pokud si všimnete viditelných částic nebo zakalení roztoku.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Kovaltry obsahuje
Prášek
Léčivou látkou je lidský koagulační faktor VIII (octocogum alfa). Jedna injekční lahvička s přípravkem Kovaltry obsahuje nominální množství 250, 500, 1 000, 2 000 nebo 3 000 IU octocogum alfa.
Dalšími složkami jsou sacharosa, histidin, glycin, chlorid sodný, chlorid vápenatý, polysorbát 80 (viz poslední část bodu 2).
Rozpouštědlo Voda na injekci.
Jak přípravek Kovaltry vypadá a co obsahuje toto balení
Přípravek Kovaltry se dodává jako prášek a rozpouštědlo pro injekční roztok a je to suchý bílý až nažloutlý prášek nebo tableta. Po rozpuštění je roztok čirý.
Každé balení přípravku Kovaltry obsahuje injekční lahvičku a předplněnou stříkačku se samostatným pístem, stejně jako adaptér injekční lahvičky a venepunkční sadu (pro podání injekce do žíly).
Složky pro rozpuštění a podání se dodávají s každým balením tohoto přípravku.
Držitel rozhodnutí o registraci
Bayer Pharma AG 13342 Berlín Německo
Výrobce
Bayer Pharma AG 51368 Leverkusen Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie/Belgique/Belgien
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Bt^rapnn
Eaňep Etnrapna EOOfl Ten.: 359 02 81 401 01 Česká republika Bayer s.r.o.
Tel: +420 266 101 111
Danmark
Bayer A/S
Tlf: +45 45 23 50 00
Deutschland
Bayer Vital GmbH
Tel: +49 (0)214-30 513 48
Eesti
Bayer OU
Tel: +372 655 8565
EXlába
Bayer EMá; ABEE Tr(k: +30-210-61 87 500 Espaňa
Bayer Hispania S.L.
Tel: +34-93-495 65 00 France
Bayer HealthCare
Tél (N° vert): +33-(0)800 87 54 54
Hrvatska
Bayer d.o.o.
Tel: +385-(0)1-6599 900
Ireland
Bayer Limited
Tel: +353 1 2999313
Island
Icepharma hf.
Sími: +354 540 8000 Italia
Bayer S.p.A.
Tel: +39 02 397 81 Kúnpoq
NOVAGEM Limited Tr(k: +357 22 48 38 58 Latvija SIA Bayer
Tel: +371 67 84 55 63
Tato příbalová informace byla ni
Lietuva
UAB Bayer
Tel. +37 05 23 36 868
Luxembourg/Luxemburg
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Magyarország
Bayer Hungária KFT
Tel:+36 14 87-41 00
Malta
Alfred Gera and Sons Ltd. Tel: +35 621 44 62 05 Nederland Bayer B.V.
Tel: +31-(0)297-28 06 66 Norge
Bayer AS
Tlf: +47 23 13 05 00 Osterreich
Bayer Austria Ges.m.b.H. Tel: +43-(0)1-711 46-0 Polska
Bayer Sp. z o.o.
Tel: +48 22 572 35 00 Portugal
Bayer Portugal, Lda.
Tel: +351 21 416 42 00
Románia
SC Bayer SRL
Tel: +40 21 529 59 00
Slovenija
Bayer d. o. o.
Tel: +386 (0)1 58 14 400 Slovenská republika Bayer spol. s r.o.
Tel. +421 2 59 21 31 11 Suomi/Finland
Bayer Oy
Puh/Tel: +358- 20 785 21
Sverige
Bayer AB
Tel: +46 (0) 8 580 223 00 United Kingdom Bayer plc
Tel: +44-(0)1635 563000 revidována {MM/RRRR}
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
Podrobné instrukce pro rekonstituci a podání přípravku Kovaltry pomocí injekční lahvičky s adaptérem injekční lahvičky:
Budete potřebovat tampony napuštěné alkoholem, vatové tampony a náplasti. Tyto položky nejsou součástí balení přípravku Kovaltry.
1. Omyjte si pečlivě ruce mýdlem a teplou vodou.
2. Zahřejte neotevřenou injekční lahvičku a injekční stříkačku ve svých rukách na příjemnou
teplotu (nesmí překročit 37 °C).
3.
Odstraňte ochranné víčko z injekční lahvičky (A) a očistěte pryžovou zátku injekční lahvičky tamponem namočeným v alkoholu a nechte ji před použitím uschnout.
4.
Umístěte injekční lahvičku s přípravkem na stabilní, neklouzavý povrch. Odloupněte papírový kryt na plastovém obalu adaptéru injekční lahvičky. Nevyndávejte adaptér z plastového obalu. Držte obal adaptéru, nasaďte jej přes injekční lahvičku s přípravkem a pevně jej zatlačte (B). Adaptér zaklapne přes víčko injekční lahvičky. Nesundávejte nyní obal adaptéru.
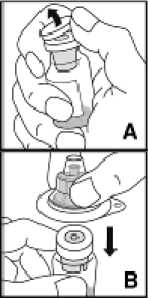
5.
Přidržte injekční stříkačku předplněnou vodou na injekci ve svislé poloze, uchopte píst podle obrázku a připojte jej otočením pevně ve směru hodinových ručiček do zátky se závitem (C).
6.
Držte injekční stříkačku za válec, odlomte víčko injekční stříkačky z jejího konce (D). Nedotýkejte se koncem injekční stříkačky ruky nebo jiného povrchu. Položte injekční stříkačku stranou pro další použití.
7.
Nyní sejměte a vyhoďte obal adaptéru (E).
8.
Připojte předplněnou injekční stříkačku na adaptér injekční lahvičky se závitem otočením ve směru hodinových ručiček (F).
9.
Vstříkněte rozpouštědlo tak, že tlačíte pomalu píst injekční stříkačky směrem dolů (G).
Mírným kroužením injekční lahvičkou rozpusťte veškerý materiál (H). Injekční lahvičkou netřepejte! Ujistěte se před použitím, že se veškerý prášek rozpustil. Před podáním vizuálně zkontrolujte, zda nejsou přítomny částice nebo zda přípravek nezměnil barvu. Nepoužívejte roztoky, které obsahují viditelné částice nebo jsou zakalené.
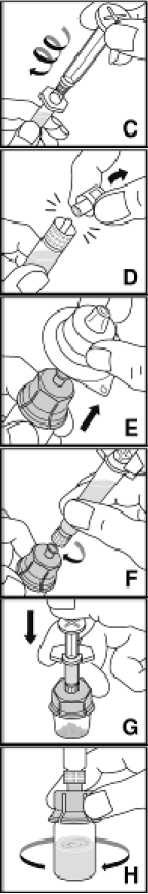
11. Držte injekční lahvičku na jednom konci nad adaptérem injekční
lahvičky a injekční stříkačkou (I). Naplňte injekční stříkačku tak, že pomalu a plynule vytahujete píst z injekční stříkačky. Držte stříkačku svisle a zatlačte na píst tak, aby ve stříkačce nezbyl žádný vzduch.
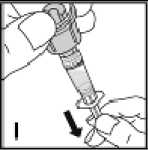
12. Přiložte turniket na paži.
13. Určete místo vpichu a očistěte kůži tamponem napuštěným alkoholem.
14. Napíchněte žílu a zajistěte venepunkční sadu náplastí.
15. Přidržujte adaptér injekční lahvičky na místě, sejměte injekční stříkačku
z adaptéru injekční lahvičky (adaptér by měl zůstat připojený na injekční lahvičku). Připevněte injekční stříkačku k venepunkční sadě a ujistěte se, že se do injekční stříkačky nedostává krev (J).
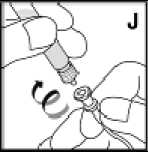
16. Sejměte turniket.
|
17. |
Roztok podávejte injekcí do žíly po dobu 2 až 5 minut, přičemž kontrolujte polohu jehly. Rychlost podání má vycházet z toho, jak je Vám to pohodlné, ale nemá být rychlejší než 2 ml za minutu. |
|
18. |
Je-li nutné podat další dávku, použijte novou injekční stříkačku s rekonstituovaným přípravkem, jak je popsáno výše. |
|
19. |
Pokud není potřebná žádná další dávka, odstraňte venepunkční sadu a injekční stříkačku. Přidržujte tampón pevně k místu vpichu na natažené paži po dobu asi 2 minut. Nakonec přiložte na místo aplikace injekce malý tlakový obvaz a zvažte, je-li nutné použít náplast. |
55