Kogenate Bayer 3000 Iu
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
KOGENATE Bayer 250 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 500 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 1000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 2000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 3000 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička nominálně obsahuje 250/500/1000/2000/3000 IU lidského koagulačního faktoru VIII (INN: octocogum alfa).
Lidský koagulační faktor VIII je vyroben rekombinantní DNA technologií (rDNA) v ledvinových buňkách křeččích mláďat obsahujících gen pro lidský faktor VIII.
• Jeden ml přípravku KOGENATE Bayer 250 IU obsahuje po rekonstituci přibližně 100 IU (250 IU / 2,5 ml) lidského koagulačního faktoru VIII (INN: octocogum alfa).
• Jeden ml přípravku KOGENATE Bayer 500 IU obsahuje po rekonstituci přibližně 200 IU (500 IU / 2,5 ml) lidského koagulačního faktoru VIII (INN: octocogum alfa).
• Jeden ml přípravku KOGENATE Bayer 1000 IU obsahuje po rekonstituci přibližně 400 IU (1000 IU / 2,5 ml) lidského koagulačního faktoru VIII (INN: octocogum alfa).
• Jeden ml přípravku KOGENATE Bayer 2000 IU obsahuje po rekonstituci přibližně 400 IU (2000 IU / 5 ml) lidského koagulačního faktoru VIII (INN: octocogum alfa).
• Jeden ml přípravku KOGENATE Bayer 3000 IU obsahuje po rekonstituci přibližně 600 IU (3000 IU / 5 ml) lidského koagulačního faktoru VIII (INN: octocogum alfa).
Síla (IU) je určena jednostupňovou srážecí zkouškou proti standardnímu roztoku FDA Mega, který byl kalibrován proti standardu WHO v mezinárodních jednotkách (IU).
Specifická účinnost přípravku KOGENATE Bayer je přibližně 4000 IU/mg bílkoviny.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok (systém Bio-Set).
Prášek: suchý bílý až nažloutlý prášek nebo koláč. Rozpouštědlo: voda na injekci, čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u nemocných s hemofilií A (vrozený nedostatek faktoru VIII).
Tento přípravek neobsahuje von Willebrandův faktor a není proto indikován pro von Willebrandovu chorobu.
Tento přípravek je indikován k léčbě dospělých, dospívajících a dětí všech věkových kategorií.
4.2 Dávkování a způsob podání
Léčba by měla probíhat pod dozorem lékaře se zkušenostmi s léčbou hemofilie. Dávkování
Počet jednotek podaného faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které jsou odvozeny od současné normy WHO pro přípravky s faktorem VIII. Plazmatická aktivita faktoru VIII je vyjádřena buď jako procento (vztažené k normální lidské plazmě), nebo v mezinárodních jednotkách (odvozených z mezinárodní normy pro faktor VIII v plazmě).
Aktivita jedné mezinárodní jednotky (IU) faktoru VIII odpovídá množství faktoru VIII obsaženému v jednom ml normální lidské plazmy.
Léčba podle potřeby
Výpočet požadované dávky faktoru VIII vychází z empirické zkušenosti, že 1 mezinárodní jednotka (IU) faktoru VIII na jeden kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 1,5 % až 2,5 % normální aktivity. Požadovaná dávka se určí pomocí následujícího vzorce:
I. Požadované jednotky IU = tělesná hmotnost (kg) x požadované zvýšení faktoru VIII
(% normální hladiny) x 0,5
II. Očekávané zvýšení faktoru VIII (% normální hladiny) = 2 x podané IU
tělesnáhmotnost(kg)
Dávkování, frekvence a doba trvání substituční terapie musí být individuálně přizpůsobeny potřebám pacienta (hmotnost, závažnost poruchy hemostatické funkce, místo a míra krvácení, přítomnost inhibitorů a požadovaná hladina faktoru VIII v plazmě).
Následující tabulka obsahuje návod pro minimální hladiny faktoru VIII v krvi. V případě níže uvedených výskytů krvácení by aktivita faktoru VIII neměla v příslušném období klesnout pod danou hladinu (v % normální hladiny):
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Četnost podání dávky (hodiny)/ Délka terapie (dny) |
|
Krvácení Časné hemartrózy, krvácení do svalu nebo krvácení dutiny ústní |
20 - 40 |
Opakujte po 12 až 24 hodinách. Minimálně 1 den, dokud krvácení projevující se bolestí není zastaveno nebo dokud nebylo dosaženo zahojení. |
|
Intenzivnější hemartrózy, krvácení do svalu nebo tvorba hematomů |
30-60 |
Infúzi opakujte po 12 až 24 hodinách po dobu 3 až 4 dnů nebo déle, dokud nevymizí bolest a porucha funkce. |
|
Život ohrožující hemoragie (např. nitrolebeční krvácení, krvácení hrdla, těžké krvácení do břišní dutiny) |
O o 1 o |
Infúzi opakujte po 8 až 24 hodinách, dokud není hrozba odvrácena. |
|
Chirurgické operace | ||
|
Menší včetně extrakce zubů |
30 - 60 |
Každých 24 hodin, minimálně 1 den, dokud není dosaženo zahojení. |
|
Větší |
80- 100 (před- a pooperační) |
a) Bolusovou infúzí Infúzi opakujte po 8 až 24 hodinách, dokud není rána přiměřeně zahojena, pak pokračujte v terapii nejméně dalších 7 dnů, abyste udrželi 30 % až 60 % (IU/dl) aktivity faktoru VIII b) Kontinuální infúzí Zvyšte aktivitu faktoru VIII před operací pomocí počáteční bolusové infúze a okamžitě pokračujte s kontinuální infúzí (v IU/kg/h), kterou upravte dle denní clearance pacienta a požadovaných hladin faktoru VIII, po dobu minimálně 7 dnů. |
U jednotlivých pacientů by dávkování a četnost podání vždy měly být přizpůsobeny klinickému účinku. Za určitých okolností je třeba použít vyšší dávku, nežli byla vypočtena, a to zejména v případě první dávky.
V průběhu léčby se doporučuje určování hladiny faktoru VIII pomocí vhodné metody, aby bylo možné upravit dávku, která má být podávána, a frekvenci opakování infúzí. Zejména v případě větších chirurgických zákroků je přesné sledování substituční terapie koagulační analýzou (aktivita plazmatického faktoru VIII) nezbytné. Jednotliví pacienti mohou reagovat na faktor VIII různě, mohou vykazovat různé poločasy a obnovení.
Kontinuální infúze
Pro účely výpočtu počáteční rychlosti infúze je možné získat clearance vynesením křivky rozpadu před operací, a nebo je možné vycházet z průměrných hodnot populace (3,0 - 3,5 ml/h/kg) a následně ji vhodně upravit.
Rychlost infúze (v IU/kg/h) = clearance (v ml/h/kg) x požadovaná hladina faktoru VIII (v IU/ml)
Pro kontinuální infuze byla prokázána klinická a in vitro stabilita pomocí ambulantních pump s nádrží z PVC. KOGENATE Bayer obsahuje malé množství pomocné látky polysorbát 80, u které je známo, že zvyšuje rychlost extrakce Di (2-ethylhexyl)ftalátu (DEHP) z materiálů PVC. Toto by mělo být zváženo při podávání kontinuální infuze.
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů se závažnou hemofilií A jsou obvykle podávány dávky 20 až 40 IU KOGENATE Bayer na kg tělesné hmotnosti v intervalech 2 až 3 dnů. V některých případech, zejména u mladších pacientů, mohou být nutné kratší intervaly podávání léku nebo vyšší dávky.
Zvláštní _ populace Pediatrická populace
Bezpečnost a účinnost přípravku KOGENATE Bayer byla stanovena u dětí všech věkových skupin. Údaje byly pořízeny z klinických hodnocení, která probíhala u 61 dětí mladších 6 let a neintervenčních studií u dětí všech věkových kategorií.
Pacienti s inhibitory
U pacientů by měl být sledován vznik inhibitorů faktoru VIII. Pokud není očekávaná hladina aktivity plazmatického faktoru VIII dosažena nebo pokud krvácení není zastaveno příslušnou dávkou, měla by být provedena zkouška za účelem určení přítomnosti inhibitoru faktoru VIII. Je-li inhibitor přítomen v hladině nižší než 10 jednotek Bethesda (BU) na ml, může podání dalšího rekombinantního koagulačního faktoru VIII zneutralizovat inhibitor a umožnit pokračování klinicky efektivní terapie s KOGENATE Bayer. Avšak při přítomnosti inhibitoru je požadované dávkování proměnlivé a musí být upravováno podle klinické odpovědi a výsledků sledování aktivity plazmatického faktoru VIII. U pacientů s titry inhibitoru nad 10 BU nebo s vysokou odpovědí v anamnéze musí být zváženo použití (aktivovaného) koncentrátu protrombinového komplexu (PCC) nebo přípravků rekombinantního aktivovaného faktoru VII (rFVIIa). Tyto terapie by měly být vedeny lékaři se zkušenostmi v léčbě pacientů s hemofilií.
Způsob podání
Intravenózní podání.
KOGENATE Bayer by měl být aplikován intravenózně po dobu 2 až 5 minut. Rychlost aplikace by měla být určována podle komfortu pacienta (maximální rychlost infuze: 2 ml/min).
Kontinuální infúze
KOGENATE Bayer může být použit při kontinuální infúzi. Výpočet rychlosti infúze by měl být založen na clearance a požadované hladině FVIII.
Příklad: Pro pacienta s hmotností 75 kg s clearance 3 ml/h/kg bude počáteční rychlost infúze 3 IU/h/kg pro dosažení 100 % hladiny FVIII. Pro výpočet ml/h vynásobte rychlost infúze v IU/h/kg tělesnou hmotností v kg/koncentrace roztoku (IU/ml).
Tabulka 2: Příklad výpočtu rych
osti infúze pro kontinuální infúzi po iniciální bolusové injekci
|
Požadovaná hladina plazmatického FVIII |
Rychlost infúze IU/h/kg |
Rychlost infúze pro 75 kg pacienta ml/h | |||
|
Clearance 3 ml/h/kg |
Koncentrace roztoku rFVIII | ||||
|
100 IU/ml |
200 IU/ml |
400 IU/ml | |||
|
100 % (1 IU/ml) |
3,0 |
2,25 |
1,125 |
0,56 | |
|
60 % (0,6 IU/ml) |
1,8 |
1,35 |
0,68 |
0,34 | |
|
40 % (0,4 IU/ml) |
1,2 |
0,9 |
0,45 |
0,225 | |
V situacích, kdy dochází ke zvýšení clearance v průběhu velkých krvácení, nebo při extenzivním poškození tkání během chirurgických zákroků, může být nutné zvýšit rychlost infúze.
Po iniciální 24 hodinové kontinuální infúzi by měla být clearance každý den přepočtena pomocí rovnice rovnovážného stavu s naměřenou hladinou FVIII a rychlosti infúze pomocí následující rovnice:
clearance = rychlost infúze/aktuální hladina FVIII.
Během kontinuální infúze se musí infúzní vaky měnit každých 24 hodin.
Návod k rekonstituci léčivého přípravku před jeho podáním je uveden v bodě 6.6. a příbalovém informaci.
4.3 Kontraindikace
- Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
- Známé alergické reakce na myší nebo křeččí protein.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Při léčbě přípravkem KOGENATE Bayer může dojít ke vzniku hypersenzitivních reakcí alergického typu. Přípravek obsahuje stopy myších a křeččích proteinů a lidské proteiny jiné než faktor VIII (viz bod 5.1).
Pokud se objeví příznaky hypersenzitivity, pacientům má být doporučeno okamžité přerušení používání tohoto léčivého přípravku a měli by kontaktovat svého lékaře.
Pacienti by měli být informováni, že časné příznaky hypersenzitivních reakcí zahrnují vyrážku, nauzeu, generalizovanou kopřivku, svíravé pocity na hrudi, sípání, hypotenzi a anafylaxi.
V případě šoku musí být provedena standardní léčba šoku.
Inhibitory
Tvorba neutralizačních protilátek (inhibitorů) proti faktoru VIII je známá komplikace v léčbě jednotlivců s hemofilií A. Tyto inhibitory jsou obvykle imunoglobuliny IgG namířené proti prokoagulační aktivitě faktoru VIII, jejichž množství je vyjádřeno v jednotkách Bethesda (BU) na ml plazmy pomocí modifikovaného testu. Riziko vývoje inhibitorů koreluje, mimo jiné, s expozicí faktoru VIII a s genetickými faktory,toto riziko je nejvyšší během prvních 20 dnů expozice. Jen vzácně se mohou inhibitory vyvinout po prvních 100 dnech expozice.
Po expozici delší než 100 dnů byly mezi dříve léčenými pacienty, kteří měli v anamnéze vývoj inhibitorů, pozorovány případy opětného výskytu inhibitorů (nízkého titru) po přechodu z faktoru VIII od jednoho výrobce na faktor VIII od jiného výrobce. Proto se po přechodu na jiný přípravek doporučuje pečlivě sledovat všechny pacienty z důvodu výskytu inhibitorů.
Obecně u pacientů, kteří jsou léčeni přípravky s koagulačním faktorem VIII, musí být pečlivě monitorován vývoj inhibitorů příslušným klinickým pozorováním a laboratorními zkouškami. Pokud nejsou dosaženy očekávané hladiny faktoru VIII nebo pokud není krvácení zvládnuto odpovídající dávkou, měly by být provedeny testy na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitorů nemusí být terapie faktorem VIII účinná a měly by být zváženy jiné možnosti terapie. Léčba takových pacientů by měla být vedena lékaři se zkušenostmi s léčbou hemofilie a s inhibitory faktoru VIII.
Kontinuální infúze
V klinické studii o používání kontinuálních infúzí při operacích byl použit heparin, jako prevence před tromboflebitidou v místě infúze, stejně jako u dalších dlouhodobých intravenózních infúzí.
Obsah sodíku
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) na injekční lahvičku, tzn. v podstatě „neobsahuje sodík“.
Kardiovaskulární příhody
Hemofiličtí pacienti s kardiovaskulárními rizikovými faktory nebo s kardiovaskulárními chorobami mohou mít to samé riziko rozvoje kardiovaskulárních příhod jako nehemofiličtí pacienti, jestliže byla srážlivost krve normalizována léčbou faktorem VIII. Zvýšení hladiny FVIII po jeho podání, zejména u pacientů s přítomnými kardiovaskulárními rizikovými faktory, může pacienta vystavit minimálně stejnému riziku uzávěru cév nebo infarktu myokardu jako u nehemofilické populace. Proto by pacienti měli být vyšetřeni a sledováni pro přítomnost kardiovaskulárních rizikových faktorů.
Komplikace související s katetrem
Jestliže je třeba použít centrální žilní vstup (centrál venous access devices, CVAD), musí se uvážit riziko vzniku komplikací souvisejících s CVAD, včetně lokální infekce, bakteriemie a trombózy v místě vstupu katetru.
Dokumentace
Důrazně se doporučuje, aby byl vždy, když je KOGENATE Bayer podán pacientovi, zaznamenán název a číslo šarže přípravku v zájmu zachování spojení mezi pacientem a šarží přípravku.
Pediatrická populace
Uvedená upozornění a opatření platí pro dospělé i děti.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly hlášeny žádné interakce přípravku KOGENATE Bayer s jinými léčivými přípravky.
4.6 Fertilita, těhotenství a kojení
S přípravkem KOGENATE Bayer nebyly provedeny zvířecí reprodukční studie.
Těhotenství a kojení
Vzhledem k vzácnému výskytu hemofilie A u žen, nejsou k dispozici žádné zkušenosti s používáním KOGENATE Bayer během těhotenství a kojení. Proto by měl být KOGENATE Bayer během těhotenství a kojení používán pouze, je-li to jasně indikováno.
Fertilita
Údaje týkající se fertility nejsou k dispozici.
4.7 Účinky na schopnost řídit a obsluhovat stroje
KOGENATE Bayer nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, zimnici, zarudnutí, generalizovanou vyrážku, bolest hlavy, kopřivku, hypotenzi, letargii, nauzeu, neklid, tachykardii, svíravý pocit na hrudi, brnění, zvracení, sípání) byly pozorovány v souvislosti s přípravky s rekombinantním faktorem VIII a mohou v některých případech vyústit až v závažnou anafylaxi (včetně šoku). Běžně se mohou vyskytovat zejména nežádoucí účinky projevující se na kůži, zatímco vývoj závažné anafylaxe (včetně šoku) je považován za vzácný.
U pacientů s hemofilií A se mohou vytvořit neutralizační protilátky (inhibitory) proti faktoru VIII. Tento stav se může projevit jako nedostatečná klinická odpověď. V těchto případech se doporučuje kontaktovat specializované centrum pro léčbu hemofilie.
Tabulkový přehled nežádoucích účinků
Tabulka uvedená níže je uspořádána podle standardních tříd orgánových systémů a stanoveného vyjádření frekvence výskytu (MedDRA).
Četnosti výskytu byly definovány podle následující konvence: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
Tabulka 3: Frekvence nežádoucích účinků
|
Podle MedDRA Třída orgánových systémů |
Frekvence | ||||
|
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné/není známo | |
|
Poruchy krve a lymfatického systému |
Vznik inhibitorů FVIII (Hlášeno u předtím neléčených pacientů a minimálně léčených pacientů)* |
Vznik inhibitorů FVIII (Hlášeno u předtím léčených pacientů v klinických studiích a v post-marketingových studiích)* | |||
|
Celkové poruchy a reakce v místě aplikace |
Reakce v místě infúze |
Febrilní stav (pyrexie) související s infúzí | |||
|
Poruchy imunitního systému |
Kožní reakce způsobené hypersenzitivit ou (svědění, kopřivka a vyrážka) |
Systémové hypersenzitivn í reakce (včetně anafylaktické reakce, nauzey, abnormálního krevního tlaku a závratě) | |||
|
Poruchy nervového systému |
Dysgeusie | ||||
* viz níže
Popis vybraných nežádoucích účinků
Vývoj inhibitoru
Byl hlášen vývoj inhibitoru u dříve neléčených a léčených pacientů (viz bod 4.4).
Při klinických studiích byl KOGENATE Bayer použit k léčbě krvácení u 37 pacientů, kteří dosud nebyli léčeni, a u 23 minimálně léčených dětských pacientů, již jsou definováni jako pacienti, kteří měli maximálně 4 dny expozice, se zbytkovým FVIII: C < 2 IU/dl. U 5 z 37 (14 %) pacientů, kteří nebyli dosud léčeni, a u 4 z 23 (17 %) pacientů léčených minimálně, došlo při léčbě přípravkem KOGENATE Bayer k vývoji inhibitorů během 20 denní expozice. Celkem u 9 z 60 (15 %) došlo k vývoji inhibitorů. Jeden pacient byl následně ztracen pro sledování a u jednoho pacienta se vyvinul nízký titr inhibitoru během sledování po ukončení studie.
U jedné observační studie s přípravkem KOGENATE Bayer (sledování až 75 dnů expozice) byla incidence vývoje inhibitoru u pacientů s těžkou hemofilií A, kteří dosud nebyli léčeni, 64/183 (37,7 %).
V klinických studiích s více než 73 pacienty, kteří již byli léčeni, (definováni jako pacienti, kteří měli více než 100 dní expozice), nebyl pozorován nový výskyt inhibitorů v průběhu více než čtyřletého sledování.
V rozsáhlých observačních studiích přípravku KOGENATE Bayer, provedených po registraci, do nichž bylo zařazeno více než 1 000 pacientů, bylo pozorováno následující: U méně než 0,2% pacientů, kteří již byli léčeni, byl pozorován nový výskyt inhibitorů.
Pediatrická populace
Očekává se, že výskyt, typ a závažnost nežádoucích účinků u dětí budou stejné jako u všech skupin v populaci, s výjimkou té, u které se vytvoří inhibitor.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné případy předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: hemostatika: krevní koagulační faktor VIII, ATC kód: B02BD02 Mechanismus účinku
Faktor VIII/von Willebrandův faktor (vWF) se skládá ze dvou molekul (faktor VIII a vWF) s různými fyziologickými funkcemi. Při aplikaci pacientovi s hemofilií se faktor VIII váže na vWF v oběhu pacienta. Aktivovaný faktor VIII působí jako kofaktor pro aktivovaný faktor IX, urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin pak přeměňuje fibrinogen na fibrin a může dojít k vytvoření sraženiny. Hemofilie A je pohlavně vázaná dědičná porucha srážlivosti krve způsobená sníženou hladinou faktoru VIII:C, následkem čehož dochází k profuznímu krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánnímu, nebo jako následek úrazu při nehodě nebo chirurgickém zákroku. Substituční léčbou se hladiny faktoru VIII v plazmě zvýší, čímž je umožněna přechodná úprava nedostatku faktoru VIII a úprava sklonů ke krvácení.
Farmakodynamické účinky
Určování aktivovaného parciálního tromboplastinového času (aPTT) je konvenční zkušební metoda in vitro pro biologickou aktivitu faktoru VIII. aPTT je delší u všech hemofiliků. Stupeň a trvání normalizace aPTT pozorované po podání KOGENATE Bayer jsou podobné těm dosaženým s faktorem VIII získaným z lidské plazmy.
Kontinuální infúze
V klinické studii u dospělých pacientů s hemofilií A, kteří podstupují velký chirurgický zákrok, bylo prokázáno, že přípravek KOGENATE Bayer může být používán ke kontinuální infúzi při operacích (před, během i po operaci). V této klinické studii byl použit heparin, jako prevence tromboflebitidy v místě infúze, stejně jako u dalších dlouhodobých intravenózních infúzí.
Hypersenzitivita
V průběhu studií žádný pacient nevytvořil klinicky významné titry protilátek proti stopovým množstvím myších a křeččích bílkovin přítomných v přípravku. Nicméně stále existuje možnost alergické reakce na složky, např. stopová množství myší a křeččí bílkoviny v přípravku u predisponovaných pacientů (viz body 4.3 a 4.4).
Navození imunotolerance (Immune Tolerance Induction, ITI)
U pacientů s hemofilií A, u kterých se vyvinuly inhibitory proti faktoru FVIII, byly shromážděny údaje týkající se navození imunotolerance. U 40 pacientů byla provedena retrospektivní kontrola a 39 pacientů bylo zařazeno do prospektivní klinické studie iniciované zkoušejícím. Údaje prokázaly, že použití přípravku KOGENATE Bayer vedlo k indukci imunitní tolerance. U pacientů, u kterých bylo dosaženo imunitní tolerance, bylo možné s přípravkem KOGENATE Bayer opět krvácení předcházet nebo krvácení kontrolovat a pacientů mohli pokračovat v profylaktické léčbě jako v léčbě udržovací.
5.2 Farmakokinetické vlastnosti
Absorpce
Analýza všech uváděných in vivo uzdravených dříve léčených pacientů ukázala průměrné zvýšení o 2 % na IU/kg tělesné váhy pro KOGENATE Bayer. Tento výsledek je podobný hodnotám zaznamenaným pro faktor VIII získaný z lidské plazmy.
Distribuce v organismu a eliminace z organismu
Po podání přípravku KOGENATE Bayer klesala nejvyšší aktivita faktoru VIII s dvoufázovým exponenciálním poklesem s průměrným terminálním poločasem kolem 15 hodin. To je podobné jako u faktoru VIII získaného z plazmy, jehož průměrný terminální poločas je přibližně 13 hodin. Další farmakokinetické parametry pro KOGENATE Bayer pro bolusové injekce jsou průměrná doba setrvání v těle [MRT (0-48)], která je přibližně 22 hodin, a clearance, která je asi 160 ml/hod. Průměrná výchozí linka clearance pro 14 dospělých pacientů, kteří podstupují velký chirurgický zákrok s kontinuální infúzí, je 188 ml/h, což odpovídá 3,0 ml/h/kg (rozmezí 1,6 - 4,6 ml/h/kg).
5.3 Předklinické údaje vztahující se k bezpečnosti
Dokonce ani dávky několikrát převyšující doporučenou klinickou dávku (vztaženou na tělesnou hmotnost) neprokázaly jakýkoli akutní nebo subakutní toxický účinek přípravku KOGENATE Bayer na laboratorní zvířata (myš, potkan, králík a pes).
Specifické studie reprodukční toxicity, chronické toxicity a kancerogenity s opakovaným podáváním oktokogu alfa nebyly provedeny kvůli imunitní reakci na heterologní proteiny u všech nelidských druhů savců.
Nebyly provedeny žádné studie o mutagenním potenciálu KOGENATE Bayer, protože u předcházejícího přípravku KOGENATE Bayer nemohl být zjištěn in vitro nebo in vivo žádný mutagenní potenciál.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Glycin
Chlorid sodný Chlorid vápenatý Histidin Polysorbát 80 Sacharosa
Rozpouštědlo
Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
K rozpuštění a injikování mohou být použity pouze dodávané komponenty (injekční lahvička s práškem se systémem Bio-Set, předplněná injekční stříkačka obsahující rozpouštědlo a venepunkční set), protože může dojít k selhání léčby v důsledku adsorpce lidského rekombinantního koagulačního faktoru VIII na vnitřní povrchy některého infúzního zařízení.
6.3 Doba použitelnosti
30 měsíců.
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
Při používání během studií in vitro však byla prokázána chemická a fyzikální stabilita po otevření před použitím ve vacích z PVC pro kontinuální infúzi po dobu 24 hodin při 30 °C.
Během in vitro studií byla po rekonstituci prokázána chemická a fyzikální stabilita po otevření před použitím po dobu 3 hodin.
Po zředění chraňte před chladem.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem. Injekční lahvičku a předplněnou injekční stříkačku uchovávejte ve vnějším obalu, aby byly chráněny před světlem.
V rámci své celkové doby použitelnosti v délce 30 měsíců může být přípravek uchováván ve vnějším obalu při pokojové teplotě (do 25°C) po omezenou dobu 12 měsíců. V takovém případě skončí doba použitelnosti na konci tohoto 12měsíčního období, nebo po uplynutí data použitelnosti uvedeného na injekční lahvičce, podle toho, co nastane dříve. Nová doba použitelnosti musí být uvedena na krabičce.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení a zvláštní vybavení pro použití, podání nebo implantaci
Jedno balení KOGENATE Bayer obsahuje:
• jednu injekční lahvičku plus zařízení Bio-Set obsahující prášek (injekční lahvička typu 1 z čirého skla o objemu 10 ml se zátkou z šedé halogenobutylové pryžové směsi bez obsahu latexu a přepouštěcím adaptérem s ochranným víčkem [Bio-Set])
• jednu předplněnou injekční stříkačku s 2,5 ml (por 250IU, 500 IU a 1000 IU) nebo 5 ml (pro 2000 IU a 3000 IU) rozpouštědla (lahvička cylindrického typu 1 z čirého skla se zátkou z šedé brombutylové pryžové směsi bez obsahu latexu)
• píst pro injekční stříkačku
• jednu venepunkční sadu
• dva tampony napuštěné alkoholem pro jedno použití
• dva suché tampony
• dvě náplasti
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Podrobné pokyny pro přípravu a podání jsou obsaženy v příbalové informaci k přípravku KOGENATE Bayer.
Rekonstituovaný léčivý přípravek je čirý a bezbarvý roztok.
Prášek KOGENATE Bayer by měl být rekonstituován pouze s dodaným rozpouštědlem (2,5 ml (pro 250 IU, 500 IU a 1000 IU) nebo 5 ml (for 2000 IU a 3000 IU) vody na injekci) v předplněné injekční stříkačce a integrovaným přepouštěcím adaptérem (Bio-Set). Pro infuzi musí být přípravek připravován v aseptických podmínkách. Pokud je kterákoli součást balení otevřená nebo poškozená, nepoužívejte ji.
Jemně otáčejte injekční lahvičkou, až se veškerý prášek rozpustí. Po rekomstituci je roztok čirý. Parenterální léčivé přípravky je nutno před podáním vizuálně zkontrolovat, zda se v nich nenalézají částice nebo zda nezměnily barvu. Nepoužívejte KOGENATE Bayer, pokud jsou v roztoku viditelné částečky látky nebo zákal.
Po rekomstituci je roztok natažen zpět do injekční stříkačky. Přípravek KOGENATE Bayer je nutno rekonstituovat a podat s použitím pomůcek, které jsou dodány v jednotlivých balíčcích.
Rekonstituovaný přípravek je nutno před podáním přefiltrovat, aby byly z roztoku odstraněny případné přítomné částice. Filtrace se provádí podle podrobných pokynů pro přípravu a/nebo podání, které jsou obsaženy v příbalové informaci, která je dodávaná s přípravkem KOGENATE Bayer.
K podání je důležité použít venepunkční sadu dodanou spolu s přípravkem, protože její součástí je in-line filtr.
V situacích, kdy nelze použít dodanou venepunkční sadu (např. při infuzi do periferního nebo centrálního katetru), je třeba použít samostatný filtr, kompatibilní s přípravkem KOGENATE Bayer. Tyto kompatibilní filtry mají polyakrylový kryt s adaptérem typu luer s integrovaným filtračním prvkem, který obsahuje polyamidovou síťku s velikostí ok 5 - 20 mikrometrů.
Dodaná venepunkční sada nesmí být použita k odběru krve, protože obsahuje in-line filtr. Jestliže je třeba před infuzí provést odběr krve, použijte aplikační sadu bez filtru a pak podejte infuzi přípravku KOGENATE Bayer přes injekční filtr.
Máte-li jakékoli dotazy, které se týkají přípravku KOGENATE Bayer a kompatibilních samostatných filtrů, obraťte se na společnost Bayer Pharma AG.
Pouze pro jedno použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
8. REGISTRAČNÍ ČÍSLO(A)
|
EU/1/00/143/004 - |
KOGENATE Bayer 250 IU |
|
EU/1/00/143/005 - |
KOGENATE Bayer 500 IU |
|
EU/1/00/143/006 -EU/1/00/143/010 -EU/1/00/143/012 - |
KOGENATE Bayer 1000 IU KOGENATE Bayer 2000 IU KOGENATE Bayer 3000 IU |
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 4. srpna 2000
Datum posledního prodloužení registrace: 6. srpna 2010
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
KOGENATE Bayer 250 IU prášek a rozpouštědlo pro injekční roztok.
KOGENATE Bayer 500 IU prášek a rozpouštědlo pro injekční roztok.
KOGENATE Bayer 1000 IU prášek a rozpouštědlo pro injekční roztok.
KOGENATE Bayer 2000 IU prášek a rozpouštědlo pro injekční roztok.
KOGENATE Bayer 3000 IU prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička nominálně obsahuje 250/500/1000/2000/3000 IU lidského koagulačního faktoru VIII (INN: octocogum alfa).
Lidský koagulační faktor VIII je vyroben rekombinantní DNA technologií (rDNA) v ledvinových buňkách křeččích mláďat obsahujících gen pro lidský faktor VIII.
• Jeden ml přípravku KOGENATE Bayer 250 IU obsahuje po rekonstituci přibližně 100 IU (250 IU / 2,5 ml) lidského koagulačního faktoru VIII (INN: octocogum alfa).
• Jeden ml přípravku KOGENATE Bayer 500 IU obsahuje po rekonstituci přibližně 200 IU (500 IU / 2,5 ml) lidského koagulačního faktoru VIII (INN: octocogum alfa).
• Jeden ml přípravku KOGENATE Bayer1000 IU obsahuje po rekonstituci přibližně 400 IU (1000 IU / 2,5 ml) lidského koagulačního faktoru VIII (INN: octocogum alfa).
• Jeden ml přípravku KOGENATE Bayer 2000 IUobsahuje po rekonstituci přibližně 400 IU (2000 IU / 5 ml) lidského koagulačního faktoru VIII (INN: octocogum alfa).
• Jeden ml přípravku KOGENATE Bayer 3000 obsahuje po rekonstituci přibližně 600 IU (3000 IU / 5 ml) lidského koagulačního faktoru VIII (INN: octocogum alfa).
Síla (IU) je určena jednostupňovou srážecí zkouškou proti standardnímu roztoku FDA Mega, který byl kalibrován proti standardu WHO v mezinárodních jednotkách (IU).
Specifická účinnost přípravku KOGENATE Bayer je přibližně 4000 IU/mg bílkoviny.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok (adaptér na injekční lahvičku).
Prášek: suchý bílý až nažloutlý prášek nebo koláč.
Rozpouštědlo: voda na injekci, čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u nemocných s hemofilií A (vrozený nedostatek faktoru VIII).
Tento přípravek neobsahuje von Willebrandův faktor a není proto indikován pro von Willebrandovu chorobu.
Tento přípravek je indikován k léčbě dospělých, dospívajících a dětí všech věkových kategorií.
4.2 Dávkování a způsob podání
Léčba by měla probíhat pod dozorem lékaře se zkušenostmi s léčbou hemofilie. Dávkování
Počet jednotek podaného faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které jsou odvozeny od současné normy WHO pro přípravky s faktorem VIII. Plazmatická aktivita faktoru VIII je vyjádřena buď jako procento (vztažené k normální lidské plazmě), nebo v mezinárodních jednotkách (odvozených z mezinárodní normy pro faktor VIII v plazmě).
Aktivita jedné mezinárodní jednotky (IU) faktoru VIII odpovídá množství faktoru VIII obsaženému v jednom ml normální lidské plazmy.
Léčba podle potřeby
Výpočet požadované dávky faktoru VIII vychází z empirické zkušenosti, že 1 mezinárodní jednotka (IU) faktoru VIII na jeden kg tělesné hmoptnosti aktivitu plazmatického faktoru VIII o 1,5 % až 2,5 % normální aktivity. Požadovaná dávka se určí pomocí následujícího vzorce:
I. Požadované jednotky IU = tělesná hmotnost (kg) x požadované zvýšení faktoru VIII
(% normální hladiny) x 0,5
II. Očekávané zvýšení faktoru VIII (% normální hladiny) = 2 x podané IU
tělesnáhmotnost(kg)
Dávkování, frekvence a doba trvání substituční terapie musí být individuálně přizpůsobeny potřebám pacienta (hmotnost, závažnost poruchy hemostatické funkce, místo a míra krvácení, přítomnost inhibitorů a požadovaná hladina faktoru VIII v plazmě).
Následující tabulka obsahuje návod pro minimální hladiny faktoru VIII v krvi. V případě níže uvedených výskytů krvácení by aktivita faktoru VIII neměla v příslušném období klesnout pod danou hladinu (v % normální hladiny):
|
Stupeň krvácení / Typ chirurgického zákroku |
Požadovaná hladina faktoru VIII (%) (IU/dl) |
Četnost podání dávky (hodiny)/ Délka terapie (dny) |
|
Krvácení Časné hemartrózy, krvácení do svalu nebo krvácení dutiny ústní |
20 - 40 |
Opakujte po 12 až 24 hodinách. Minimálně 1 den, dokud krvácení projevující se bolestí není zastaveno nebo dokud nebylo dosaženo zahojení. |
|
Intenzivnější hemartrózy, krvácení do svalu nebo tvorba hematomů |
30-60 |
Infúzi opakujte po 12 až 24 hodinách po dobu 3 až 4 dnů nebo déle, dokud nevymizí bolest a porucha funkce. |
|
Život ohrožující hemoragie (např. nitrolebeční krvácení, krvácení hrdla, těžké krvácení do břišní dutiny) |
O o 1 o |
Infúzi opakujte po 8 až 24 hodinách, dokud není hrozba odvrácena. |
|
Chirurgické operace | ||
|
Menší včetně extrakce zubů |
30 - 60 |
Každých 24 hodinách, minimálně 1 den, dokud není dosaženo zahojení. |
|
Větší |
80- 100 (před- a pooperační) |
a) Bolusovou infúzí Infúzi opakujte po 8 až 24 hodinách, dokud není rána přiměřeně zahojena, pak pokračujte v terapii nejméně dalších 7 dnů, abyste udrželi 30 % až 60 % (IU/dl) aktivity faktoru VIII b) Kontinuální infúze Zvyšte aktivitu faktoru VIII před operací pomocí počáteční bolusové infúze a okamžitě pokračujte s kontinuální infúzí (v IU/kg/h), kterou upravte dle denní clearance pacienta a požadovaných hladin faktoru VIII, po dobu minimálně 7 dnů. |
U jednotlivých pacientů by dávkování a četnost podání vždy měly být přizpůsobeny klinickému účinku. Za určitých okolností je třeba použít vyšší dávku, nežli byla vypočtena, a to zejména v případě první dávky.
V průběhu léčby se doporučuje určování hladiny faktoru VIII pomocí vhodné metody, aby bylo možné upravit dávku, která má být podávána, a frekvenci opakování infúzí. Zejména v případě větších chirurgických zákroků je přesné sledování substituční terapie koagulační analýzou (aktivita plazmatického faktoru VIII) nezbytné. Jednotliví pacienti mohou reagovat na faktor VIII různě, mohou vykazovat různé poločasy a obnovení.
Kontinuální infúze
Pro účely výpočtu počáteční rychlosti infúze je možné získat clearance vynesením křivky rozpadu před operací a nebo je možné vycházet z průměrných hodnot populace (3,0 - 3,5 ml/h/kg) a následně ji vhodně upravit.
Rychlost infúze (v IU/kg/h) = clearance (v ml/h/kg) x požadovaná hladina faktoru VIII (v IU/ml)
Pro kontinuální infuze byla prokázána klinická a in vitro stabilita pomocí ambulantních pump s nádrží z PVC. KOGENATE Bayer obsahuje malé množství pomocné látky polysorbát 80, u které je známo, že zvyšuje rychlost extrakce Di (2-ethylhexyl)ftalátu (DEHP) z materiálů PVC. Toto by mělo být zváženo při podávání kontinuální infuze.
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů se závažnou hemofilií A jsou obvykle podávány dávky 20 až 40 IU KOGENATE Bayer na kg tělesné hmotnosti v intervalech 2 až 3 dnů. V některých případech, zejména u mladších pacientů, mohou být nutné kratší intervaly podávání léku nebo vyšší dávky.
Zvláštní _ populace Pediatrická populace
Bezpečnost a účinnost přípravku KOGENATE Bayer byla stanovena u dětí všech věkových skupin. Údaje byly pořízeny z klinických hodnocení, která probíhala u 61 dětí mladších 6 let a neintervenčních studií u dětí všech věkových kategorií.
Pacienti s inhibitory
U pacientů by měl být sledován vznik inhibitorů faktoru VIII. Pokud není očekávaná hladina aktivity plazmatického faktoru VIII dosažena nebo pokud krvácení není zastaveno příslušnou dávkou, měla by být provedena zkouška za účelem určení přítomnosti inhibitoru faktoru VIII. Je-li inhibitor přítomen v hladině nižší než 10 jednotek Bethesda (BU) na ml, může podání dalšího rekombinantního koagulačního faktoru VIII zneutralizovat inhibitor a umožnit pokračování klinicky efektivní terapie s KOGENATE Bayer. Avšak při přítomnosti inhibitoru je požadované dávkování proměnlivé a musí být upravováno podle klinické odpovědi a výsledků sledování aktivity plazmatického faktoru VIII. U pacientů s titry inhibitoru nad 10 BU nebo s vysokou odpovědí v anamnéze musí být zváženo použití (aktivovaného) koncentrátu protrombinového komplexu (PCC) nebo přípravků rekombinantního aktivovaného faktoru VII (rFVIIa). Tyto terapie by měly být vedeny lékaři se zkušenostmi v léčbě pacientů s hemofilií.
Způsob podání
Intravenózní podání.
KOGENATE Bayer by měl být aplikován intravenózně po dobu 2 až 5 minut. Rychlost aplikace by měla být určována podle komfortu pacienta (maximální rychlost infuze: 2 ml/min).
Kontinuální infúze
KOGENATE Bayer může být použit při kontinuální infúzi. Výpočet rychlosti infúze by měl být založen na clearance a požadované hladině FVIII.
Příklad: Pro pacienta s hmotností 75 kg s clearance 3 ml/h/kg bude počáteční rychlost infúze 3 IU/h/kg pro dosažení 100 % hladiny FVIII. Pro výpočet ml/h vynásobte rychlost infúze v IU/h/kg tělesnou hmotností v kg/koncentrace roztoku (IU/ml).
|
Požadovaná hladina plazmatického FVIII |
Rychlost infúze IU/h/kg |
Rychlost infúze pro 75 kg pacienta ml/h | |||
|
Clearance 3 ml/h/kg |
Koncentrace roztoku rFVIII | ||||
|
100 IU/ml |
200 IU/ml |
400 IU/ml | |||
|
100 % (1 IU/ml) |
3,0 |
2,25 |
1,125 |
0,56 | |
|
60 % (0,6 IU/ml) |
1,8 |
1,35 |
0,68 |
0,34 | |
|
40 % (0,4 IU/ml) |
1,2 |
0,9 |
0,45 |
0,225 | |
V situacích, kdy dochází ke zvýšení clearance v průběhu velkých krvácení, nebo při extenzivním poškození tkání během chirurgických zákroků, může být nutné zvýšit rychlost infúze.
Po iniciální 24 hodinové kontinuální infúzi by měla být clearance každý den přepočtena pomocí rovnice rovnovážného stavu s naměřenou hladinou FVIII a rychlosti infúze pomocí následující rovnice:
clearance = rychlost infúze/aktuální hladina FVIII.
Během kontinuální infúze se musí infúzní vaky měnit každých 24 hodin.
Návod k rekonstituci léčivého přípravku před jeho podáním je uveden v bodě 6.6. a příbalovém informaci.
4.3 Kontraindikace
- Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
- Známé alergické reakce na myší nebo křeččí protein.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Při léčbě přípravkem KOGENATE Bayer může dojít ke vzniku hypersenzitivních reakcí alergického typu. Přípravek obsahuje stopy myších a křeččích proteinů a lidské proteiny jiné než faktor VIII (viz bod 5.1).
Pokud se objeví příznaky hypersenzitivity, pacientům má být doporučeno okamžité přerušení používání tohoto léčivého přípravku a měli by kontaktovat svého lékaře.
Pacienti by měli být informováni, že časné příznaky hypersenzitivních reakcí zahrnují vyrážku, nauzeu, generalizovanou kopřivku, svíravé pocity na hrudi, sípání, hypotenzi a anafylaxi.
V případě šoku musí být provedena standardní léčba šoku.
Inhibitory
Tvorba neutralizačních protilátek (inhibitorů) proti faktoru VIII je známá komplikace v léčbě jednotlivců s hemofilií A. Tyto inhibitory jsou obvykle imunoglobuliny IgG namířené proti prokoagulační aktivitě faktoru VIII, jejichž množství je vyjádřeno v jednotkách Bethesda (BU) na ml plazmy pomocí modifikovaného testu.. Riziko vývoje inhibitorů koreluje, mimo jiné, s expozicí faktoru VIII a s genetickými faktory, toto riziko je nejvyšší během prvních 20 dnů expozice. Jen vzácně se mohou inhibitory vyvinout po prvních 100 dnech expozice.
Po expozici delší než 100 dnů byly mezi dříve léčenými pacienty, kteří měli v anamnéze vývoj inhibitorů, pozorovány případy opětného výskytu inhibitorů (nízkého titru) po přechodu zfaktoru VIII od jednoho výrobce na faktor VIII od jiného výrobce.
Proto se po přechodu na jiný přípravek doporučuje pečlivě sledovat všechny pacienty z důvodu výskytu inhibitorů.
Obecně u pacientů, kteří jsou léčeni přípravky s koagulačním faktorem VIII, musí být pečlivě monitorován vývoj inhibitorů příslušným klinickým pozorováním a laboratorními zkouškami. Pokud
nejsou dosaženy očekávané hladiny faktoru VIII nebo pokud není krvácení zvládnuto odpovídající dávkou, měly by být provedeny testy na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitorů nemusí být terapie faktorem VIII účinná a měly by být zváženy jiné možnosti terapie. Léčba takových pacientů by měla být vedena lékaři se zkušenostmi s léčbou hemofilie a s inhibitory faktoru VIII.
Kontinuální infuze
V klinické studii o používání kontinuálních infúzí při operacích byl použit heparin, jako prevence před tromboflebitidou v místě infuze, stejně jako u dalších dlouhodobých intravenózních infuzí.
Obsah sodíku
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) na injekční lahvičku, tzn. v podstatě „neobsahuje sodík“.
Kardiovaskulární příhody
Hemofiličtí pacienti s kardiovaskulárními rizikovými faktory nebo s kardiovaskulárními chorobami mohou mít to samé riziko rozvoje kardiovaskulárních příhod jako nehemofiličtí pacienti, jestliže byla srážlivost krve normalizována léčbou faktorem VIII. Zvýšení hladiny FVIII po jeho podání, zejména u pacientů s přítomnými kardiovaskulárními rizikovými faktory, může pacienta vystavit minimálně stejnému riziku uzávěru cév nebo infarktu myokardu jako u nehemofilické populace. Proto by pacienti měli být vyšetřeni a sledováni pro přítomnost kardiovaskulárních rizikových faktorů.
Komplikace související s katetrem
Jestliže je třeba použít centrální žilní vstup (centrál venous access devices, CVAD), musí se uvážit riziko vzniku komplikací souvisejících s CVAD, včetně lokální infekce, bakteriemie a trombózy v místě vstupu katetru.
Dokumentace
Důrazně se doporučuje, aby byl vždy, když je KOGENATE Bayer podán pacientovi, zaznamenán název a číslo šarže přípravku v zájmu zachování spojení mezi pacientem a šarží přípravku.
Pediatrická populace
Uvedená upozornění a opatření platí pro dospělé i děti.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly hlášeny žádné interakce přípravku KOGENATE Bayer s jinými léčivými přípravky.
4.6 Fertilita, těhotenství a kojení
S přípravkem KOGENATE Bayer nebyly provedeny zvířecí reprodukční studie.
Těhotenství a kojení
Vzhledem k vzácnému výskytu hemofilie A u žen, nejsou k dispozici žádné zkušenosti s používáním KOGENATE Bayer během těhotenství a kojení. Proto by měl být KOGENATE Bayer během těhotenství a kojení používán pouze, je-li to jasně indikováno.
Fertilita
Údaje týkající se fertility nejsou k dispozici.
4.7 Účinky na schopnost řídit a obsluhovat stroje
KOGENATE Bayer nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, zimnici, zarudnutí, generalizovanou vyrážku, bolest hlavy, kopřivku, hypotenzi, letargii, nauzeu, neklid, tachykardii, svíravý pocit na hrudi, brnění, zvracení, sípání) byly pozorovány v souvislosti s přípravky s rekombinantním faktorem VIII a mohou v některých případech vyústit až v závažnou anafylaxi (včetně šoku). Běžně se mohou vyskytovat zejména nežádoucí účinky projevující se na kůži, zatímco vývoj závažné anafylaxe (včetně šoku) je považován za vzácný.
U pacientů s hemofilií A se mohou vytvořit neutralizační protilátky (inhibitory) proti faktoru VIII. Tento stav se může projevit jako nedostatečná klinická odpověď. V těchto případech se doporučuje kontaktovat specializované centrum pro léčbu hemofilie.
Tabulkový přehled nežádoucích účinků
Tabulka uvedená níže je uspořádána podle standardních tříd orgánových systémů a stanoveného vyjádření frekvence výskytu (MedDRA).
Četnosti výskytu byly definovány podle následující konvence: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
Tabulka 3: Frekvence nežádoucích účinků
|
Podle MedDRA Třída orgánových systémů |
Frekvence | ||||
|
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné/není známo | |
|
Poruchy krve a lymfatického systému |
Vznik inhibitorů FVIII (Hlášeno u předtím neléčených pacientů a minimálně léčených pacientů)* |
Vznik inhibitorů FVIII (Hlášeno u předtím léčených pacientů v klinických studiích a v post-marketingových studiích)* | |||
|
Celkové poruchy a reakce v místě aplikace |
Reakce v místě infúze |
Febrilní stav (pyrexie) související s infúzí | |||
|
Poruchy imunitního systému |
Kožní reakce způsobené hypersenzitivit ou (svědění, kopřivka a vyrážka) |
Systémové hypersenzitivn í reakce (včetně anafylaktické reakce, nauzey, abnormálního krevního tlaku a závratě) | |||
|
Poruchy nervového systému |
Dysgeusie | ||||
* viz níže
Popis vybraných nežádoucích účinků
Vývoj inhibitoru
Byl hlášen vývoj inhibitoru u dříve neléčených a léčených pacientů (viz bod 4.4).
Při klinických studiích byl KOGENATE Bayer použit k léčbě krvácení u 37 pacientů, kteří dosud nebyli léčeni, a u 23 minimálně léčených dětských pacientů, již jsou definováni jako pacienti, kteří měli maximálně 4 dny expozice, se zbytkovým FVIII: C < 2 IU/dl. U 5 z 37 (14 %) pacientů, kteří nebyli dosud léčeni, a u 4 z 23 (17 %) pacientů léčených minimálně, došlo při léčbě přípravkem KOGENATE Bayer k vývoji inhibitorů během 20 denní expozice. Celkem u 9 z 60 (15 %) došlo k vývoji inhibitorů. Jeden pacient byl následně ztracen pro sledování a u jednoho pacienta se vyvinul nízký titr inhibitoru během sledování po ukončení studie.
U jedné observační studie s přípravkem KOGENATE Bayer (sledování až 75 dnů expozice) byla incidence vývoje inhibitoru u pacientů s těžkou hemofilií A, kteří dosud nebyli léčeni, 64/183 (37,7 %).
V klinických studiích s více než 73 pacienty, kteří již byli léčeni, (definováni jako pacienti, kteří měli více než 100 dní expozice), nebyl pozorován nový výskyt inhibitorů v průběhu více než čtyřletého sledování.
V rozsáhlých observačních studiích přípravku KOGENATE Bayer, provedených po registraci, do nichž bylo zařazeno více než 1 000 pacientů, bylo pozorováno následující: U méně než 0,2% pacientů, kteří již byli léčeni, byl pozorován nový výskyt inhibitorů.
Pediatrická populace
Očekává se, že výskyt, typ a závažnost nežádoucích účinků u dětí budou stejné jako u všech skupin v populaci, s výjimkou té, u které se vytvoří inhibitor.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné případy předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: hemostatika: krevní koagulační faktor VIII, ATC kód: B02BD02 Mechanismus účinku
Faktor VIII/von Willebrandův faktor (vWF) se skládá ze dvou molekul (faktor VIII a vWF) s různými fyziologickými funkcemi. Při aplikaci pacientovi s hemofilií se faktor VIII váže na vWF v oběhu pacienta. Aktivovaný faktor VIII působí jako kofaktor pro aktivovaný faktor IX, urychlující konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin pak přeměňuje fibrinogen na fibrin a může dojít k vytvoření sraženiny. Hemofilie A je pohlavně vázaná dědičná porucha srážlivosti krve způsobená sníženou hladinou faktoru VIII:C, následkem čehož dochází k profuznímu krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánnímu, nebo jako následek úrazu při nehodě nebo chirurgickém zákroku. Substituční léčbou se hladiny faktoru VIII v plazmě zvýší, čímž je umožněna přechodná úprava nedostatku faktoru VIII a úprava sklonů ke krvácení.
Farmakodynamické účinky
Určování aktivovaného parciálního tromboplastinového času (aPTT) je konvenční zkušební metoda in vitro pro biologickou aktivitu faktoru VIII. aPTT je delší u všech hemofiliků. Stupeň a trvání normalizace aPTT pozorované po podání KOGENATE Bayer jsou podobné těm dosaženým s faktorem VIII získaným z lidské plazmy.
Kontinuální infíize
V klinické studii u dospělých pacientů s hemofilií A, kteří podstupují velký chirurgický zákrok, bylo prokázáno, že přípravek KOGENATE Bayer může být používán ke kontinuální infúzi při operacích (před, během i po operaci). V této klinické studii byl použit heparin, jako prevence tromboflebitidy v místě infúze, stejně jako u dalších dlouhodobých intravenózních infúzí.
Hypersenzitivita
V průběhu studií žádný pacient nevytvořil klinicky významné titry protilátek proti stopovým množstvím myších a křeččích bílkovin přítomných v přípravku. Nicméně stále existuje možnost alergické reakce na složky, např. stopová množství myší a křeččí bílkoviny v přípravku u predisponovaných pacientů (viz body 4.3 a 4.4).
Navození imunotolerance (Immune Tolerance Induction, ITI)
U pacientů s hemofilií A, u kterých se vyvinuly inhibitory proti faktoru FVIII, byly shromážděny údaje týkající se navození imunotolerance. U 40 pacientů byla provedena retrospektivní kontrola a 39 pacientů bylo zařazeno do prospektivní klinické studie iniciované zkoušejícím. Údaje prokázaly, že použití přípravku KOGENATE Bayer vedlo k indukci imunitní tolerance. U pacientů, u kterých bylo dosaženo imunitní tolerance, bylo možné s přípravkem KOGENATE Bayer opět krvácení předcházet nebo krvácení kontrolovat a pacientů mohli pokračovat v profylaktické léčbě jako v léčbě udržovací.
5.2 Farmakokinetické vlastnosti
Absorpce
Analýza všech uváděných in vivo uzdravených dříve léčených pacientů ukázala průměrné zvýšení o 2 % na IU/kg tělesné váhy pro KOGENATE Bayer. Tento výsledek je podobný hodnotám zaznamenaným pro faktor VIII získaný z lidské plazmy.
Distribuce v organismu a eliminace z organismu
Po podání přípravku KOGENATE Bayer klesala nejvyšší aktivita faktoru VIII s dvoufázovým exponenciálním poklesem s průměrným terminálním poločasem kolem 15 hodin. To je podobné jako u faktoru VIII získaného z plazmy, jehož průměrný terminální poločas je přibližně 13 hodin. Další farmakokinetické parametry pro KOGENATE Bayer pro bolusové injekce jsou průměrná doba setrvání v těle [MRT (0-48)], která je přibližně 22 hodin, a clearance, která je asi 160 ml/hod. Průměrná výchozí clearance pro 14 dospělých pacientů, kteří podstupují velký chirurgický zákrok s kontinuální infúzí, je 188 ml/h, což odpovídá 3,0 ml/h/kg (rozmezí 1,6 - 4,6 ml/h/kg).
5.3 Předklinické údaje vztahující se k bezpečnosti
Dokonce ani dávky několikrát převyšující doporučenou klinickou dávku (vztaženou na tělesnou hmotnost) neprokázaly jakýkoli akutní nebo subakutní toxický účinek přípravku KOGENATE Bayer na laboratorní zvířata (myš, potkan, králík a pes).
Specifické studie reprodukční toxicity, chronické toxicity a kancerogenity s opakovaným podáváním oktokogu alfa nebyly provedeny kvůli imunitní reakci na heterologní proteiny u všech nelidských druhů savců.
Nebyly provedeny žádné studie o mutagenním potenciálu KOGENATE Bayer, protože u předcházejícího přípravku KOGENATE Bayer nemohl být zjištěn in vitro nebo in vivo žádný mutagenní potenciál.
6. FARMACEUTICKÉ ÚDAJE 6.1 Seznam pomocných látek
Prášek
Glycin
Chlorid sodný Chlorid vápenatý Histidin Polysorbát 80 Sacharosa
Rozpouštědlo
Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
K rozpuštění a injikování mohou být použity pouze dodávané komponenty (injekční lahvička s práškem, předplněná injekční stříkačka obsahující rozpouštědlo, nástavec na injekční lahvičku a venepunkční set), protože může dojít k selhání léčby v důsledku adsorpce lidského rekombinantního koagulačního faktoru VIII na vnitřní povrchy některého infúzního zařízení.
6.3 Doba použitelnosti
30 měsíců.
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
Při používání během studií in vitro však byla prokázána chemická a fyzikální stabilita po otevření před použitím ve vacích z PVC pro kontinuální infúzi po dobu 24 hodin při 30 °C.
Během in vitro studií byla po rekonstituci prokázána chemická a fyzikální stabilita po otevření před použitím po dobu 3 hodin.
Po zředění chraňte před chladem.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem. Injekční lahvičku a předplněnou injekční stříkačku uchovávejte ve vnějším obalu, aby byly chráněny před světlem.
V rámci své celkové doby použitelnosti v délce 30 měsíců může být přípravek uchováván ve vnějším obalu při pokojové teplotě (do 25°C) po omezenou dobu 12 měsíců. V takovém případě skončí doba použitelnosti na konci tohoto 12měsíčního období, nebo po uplynutí data použitelnosti uvedeného na injekční lahvičce, podle toho, co nastane dříve. Nová doba použitelnosti musí být uvedena na krabičce.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení a zvláštní vybavení pro použití, podání nebo implantaci
Jedno balení KOGENATE Bayer obsahuje:
• jednu injekční lahvičku s práškem (injekční lahvička typu 1 z čirého skla o objemu 10 ml se zátkou z šedé halogenobutylové pryžové směsi bez obsahu latexu a s hliníkovým víčkem)
• jednu předplněnou injekční stříkačku s 2,5 ml (pro 250 IU, 500 IU a 1000 IU) nebo 5 ml (pro 2000 IU a 30000 IU) rozpouštědla (lahvička cylindrického typu 1 z čirého skla se zátkou z šedé brombutylové pryžové směsi bez obsahu latexu)
• píst pro injekční stříkačku
• nástavec na injekční lahvičku
• jednu venepunkční sadu
• dva tampony napuštěné alkoholem pro jedno použití
• dva suché tampony
• dvě náplasti
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Podrobné pokyny pro přípravu a podání jsou obsaženy v příbalové informaci k přípravku KOGENATE Bayer.
Rekonstituovaný léčivý přípravek je čirý a bezbarvý roztok.
Prášek KOGENATE Bayer by měl být rekonstituován pouze s dodaným rozpouštědlem (2,5 ml (pro 250 IU, 500 IU a 1000 IU) nebo 5 ml (pro 2000 IU a 30000 IU) vody na injekci) v předplněné injekční stříkačce a nástavci na injekční lahvičku. Pro infuzi musí být přípravek připravován v aseptických podmínkách. Pokud je kterákoli součást balení otevřená nebo poškozená, nepoužívejte ji. Jemně otáčejte injekční lahvičkou, až se veškerý prášek rozpustí. Po rekomstituci je roztok čirý. Parenterální léčivé přípravky je nutno před podáním vizuálně zkontrolovat, zda se v nich nenalézají částice nebo zda nezměnily barvu. Nepoužívejte KOGENATE Bayer, pokud jsou v roztoku viditelné částečky látky nebo zákal.
Po rekomstituci je roztok natažen zpět do injekční stříkačky. Přípravek KOGENATE Bayer je nutno rekonstituovat a podat s použitím pomůcek, které jsou dodány v jednotlivých balíčcích.
Rekonstituovaný přípravek je nutno před podáním přefiltrovat, aby byly z roztoku odstraněny případné přítomné částice. Filtrace se provádí pomocí nástavce na injekční lahvičku.
Pouze pro jedno použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
8. REGISTRAČNÍ ČÍSLO(A)
|
EU/1/00/143/007 |
- KOGENATE Bayer 250 IU |
|
EU/1/00/143/008 |
- KOGENATE Bayer 500 IU |
|
EU/1/00/143/009 EU/1/00/143/011 EU/1/00/143/013 |
- KOGENATE Bayer 1000 IU - KOGENATE Bayer 2000 IU - KOGENATE Bayer 3000 IU |
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 4. srpna 2000
Datum posledního prodloužení registrace: 6. srpna 2010 10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE>
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa vvrobce/vvrobců biologické léčivé látky
Bayer Corporation (license holder)
Bayer HealthCare LLC 800 Dwight Way Berkeley, CA 94710 USA
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Bayer HealthCare Manufacturing S.r.l.
Via delle Groane 126
20024 Garbagnate Milanese (MI)
Itálie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2)
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA - PRO SYSTÉM BIO-SET
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
KOGENATE Bayer 250 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 500 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 1000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 2000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 3000 IU prášek a rozpouštědlo pro injekční roztok
Rekombinantní koagulační faktor VIII (octocogum alfa)
2. OBSAH LÉČIVÉ LÁTKY/ LÉČIVÝCH LÁTEK
KOGENATE Bayer 250 IU obsahuje (250 IU / 2,5 ml) = octocogum alfa 100 IU na ml po rekonstituci KOGENATE Bayer 500 IU obsahuje (500 IU / 2,5 ml) = octocogum alfa 200 IU na ml po rekonstituci KOGENATE Bayer 1000 IU obsahuje (1000 IU / 2,5 ml) = octocogum alfa 400 IU na ml po rekonstituci
KOGENATE Bayer 2000 IU obsahuje (2000 IU / 5 ml) = octocogum alfa 400 IU na ml po rekonstituci
KOGENATE Bayer 3000 IU obsahuje (3000 IU / 5 ml) = octocogum alfa 600 IU na ml po rekonstituci
3. SEZNAM POMOCNÝCH LÁTEK
Glycin, chlorid sodný, chlorid vápenatý, histidin, polysorbát 80, sacharosa.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Systém Bio-Set:
1 injekční lahvička se systémem Bio-Set s práškem pro přípravu injekčního roztoku 1 předplněná injekční stříkačka s 2,5 ml nebo 5 ml vody na injekci a samostatným pístem
1 venepunkční sada
2 tampony napuštěné alkoholem pro jednorázové použití 2 suché tampony
2 náplasti
Intravenózní podání, pouze k jednorázové aplikaci. Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST, KÓD DÁRCE A KÓD LÉČIVÉHO PŘÍPRAVKU
EXP:
EXP (Konec 12 měsíčního období, jestliže je uchováván při pokojové teplotě): ...........
Nepoužívejte po tomto datu.
Přípravek může být uchováván při teplotě do 25 °C po dobu 12 měsíců v rámci doby použitelnosti uvedené na obalu. Zapište nové datum použitelnosti na krabičku. Po rekonstituci se přípravek musí použít během 3 hodin. Po rekonstituci roztok chraňte před chladem.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem.
Injekční lahvičku a předplněnou injekční stříkačku uchovávejte ve vnějším obalu, aby byly chráněny před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Všechen nepoužitý roztok musí být zlikvidován.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/00/143/004 - KOGENATE Bayer 250 IU EU/1/00/143/005 - KOGENATE Bayer 500 IU EU/1/00/143/006 - KOGENATE Bayer 1000 IU EU/1/00/143/010 - KOGENATE Bayer 2000 IU EU/1/00/143/012 - KOGENATE Bayer 3000 IU
c.s.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
Před použitím si přečtěte příbalovou informaci.
16. INFORMACE V BRAILLOVĚ PÍSMU
KOGENATE Bayer 250 KOGENATE Bayer 500 KOGENATE Bayer 1000 KOGENATE Bayer 2000 KOGENATE Bayer 3000
KRABIČKA - PRO ADAPTÉR NA INJEKČNÍ LAHVIČKU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
KOGENATE Bayer 250 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 500 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 1000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 2000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 3000 IU prášek a rozpouštědlo pro injekční roztok
Rekombinantní koagulační faktor VIII (octocogum alfa)
2. OBSAH LÉČIVÉ LÁTKY/ LÉČIVÝCH LÁTEK
KOGENATE Bayer 250 IU obsahuje (250 IU / 2,5 ml) = octocogum alfa 100 IU na ml po rekonstituci KOGENATE Bayer 500 IU obsahuje (500 IU / 2,5 ml) = octocogum alfa 200 IU na ml po rekonstituci KOGENATE Bayer 1000 IU obsahuje (1000 IU / 2,5 ml) = octocogum alfa 400 IU na ml po rekonstituci
KOGENATE Bayer 2000 IU obsahuje (2000 IU / 5 ml) = octocogum alfa 400 IU na ml po rekonstituci
KOGENATE Bayer 3000 IU obsahuje (3000 IU / 5 ml) = octocogum alfa 600 IU na ml po rekonstituci
3. SEZNAM POMOCNÝCH LÁTEK
Glycin, chlorid sodný, chlorid vápenatý, histidin, polysorbát 80, sacharosa.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Adaptér na injekční lahvičku:
1 injekční lahvička s práškem pro přípravu injekčního roztoku
1 předplněná injekční stříkačka s 2,5 ml nebo 5 ml vody na injekci a samostatným pístem 1 nástavec na injekční lahvičku
1 venepunkční sada
2 tampony napuštěné alkoholem pro jednorázové použití 2 suché tampony
2 náplasti
Intravenózní podání, pouze k jednorázové aplikaci. Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST, KÓD DÁRCE A KÓD LÉČIVÉHO PŘÍPRAVKU
EXP:
EXP (Konec 12 měsíčního období, jestliže je uchováván při pokojové teplotě): ...........
Nepoužívejte po tomto datu.
Přípravek může být uchováván při teplotě do 25 °C po dobu 12 měsíců v rámci doby použitelnosti uvedené na obalu. Zapište nové datum použitelnosti na krabičku. Po rekonstituci se přípravek musí použít během 3 hodin. Po rekonstituci roztok chraňte před chladem.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem.
Injekční lahvičku a předplněnou injekční stříkačku uchovávejte ve vnějším obalu, aby byly chráněny před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Všechen nepoužitý roztok musí být zlikvidován.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/00/143/007 - KOGENATE Bayer 250 IU EU/1/00/143/008 - KOGENATE Bayer 500 IU EU/1/00/143/009 - KOGENATE Bayer 1000 IU EU/1/00/143/011 - KOGENATE Bayer 2000 IU EU/1/00/143/013 - KOGENATE Bayer 3000 IU
c.s.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
Před použitím si přečtěte příbalovou informaci.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
KOGENATE Bayer 250 KOGENATE Bayer 500 KOGENATE Bayer 1000 KOGENATE Bayer 2000 KOGENATE Bayer 3000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
KOGENATE Bayer 250 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 500 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 1000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 2000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 3000 IU prášek a rozpouštědlo pro injekční roztok
Rekombinantní koagulační faktor VIII (octocogum alfa)
Intravenózní podání.
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
250 IU (octocogum alfa) (100 IU/ml po rekonstituci). 500 IU (octocogum alfa) (200 IU/ml po rekonstituci). 1000 IU (octocogum alfa) (400 IU/ml po rekonstituci). 2000 IU (octocogum alfa) (400 IU/ml po rekonstituci). 3000 IU (octocogum alfa) (600 IU/ml po rekonstituci).
6. JINÉ
Bayer-Logo
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Voda na injekci
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2,5 ml [pro rekonstituci sil 250/500/1000 IU]
5 ml [pro rekonstituci sil 2000/3000 IU]
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
KOGENATE Bayer 250 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 500 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 1000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 2000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 3000 IU prášek a rozpouštědlo pro injekční roztok Rekombinantní koagulační faktor VIII (octocogum alfa)
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek KOGENATE Bayer a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek KOGENATE Bayer používat
3. Jak se přípravek KOGENATE Bayer používá
4. Možné nežádoucí účinky
5. Jak přípravek KOGENATE Bayer uchovávat
6. Obsah balení a další informace
1. Co je přípravek KOGENATE Bayer a k čemu se používá
KOGENATE Bayer obsahuje léčivou látku rekombinantní lidský koagulační faktor VIII (oktokog alfa).
KOGENATE Bayer se používá k léčbě a profylaxi krvácení u dospělých, dospívajících a dětí všech věkových kategorií s hemofilií A (vrozený nedostatek faktoru VIII).
Tento přípravek neobsahuje von Willebrandův faktor, a z tohoto důvodu nemá být používán u von Willebrandovy choroby.
2. Čemu musíte věnovat pozornost, než začnete přípravek KOGENATE Bayer používat
Nepoužívejte přípravek KOGENATE Bayer
• jestliže jste alergický/á na oktokog alfa nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6 a na konci bodu 2).
• jestliže jste alergický/á na myší nebo křeččí bílkovinu.
Jestliže si nejste jistí, zeptejte se svého lékaře.
Upozornění a opatření
Věnujte zvláštní opatrnost při použití přípravku KOGENATE Bayer a poraďte se se svým
lékařem nebo lékárníkem, pokud:
• máte svíravý pocit v hrudní oblasti, točí se vám hlava, je vám nevolno nebo mdlo, nebo máte závratě, jakmile vstanete, může to znamenat závažnou, náhlou alergickou reakci (tak zvanou anafylaktickou reakci) na tento léčivý přípravek. Pokud toto nastane, okamžitě zastavte podávání přípravku a vyhledejte lékařskou pomoc.
• krvácení není kontrolováno obvyklou dávkou tohoto léčivého přípravku, okamžitě se poraďte se svým lékařem. Pravděpodobně u vás došlo k vytvoření inhibitorů faktoru VIII a lékař může chtít provést testy, aby toto potvrdil. Inhibitory faktoru VIII jsou protilátky v krvi, které blokují faktor VIII, který užíváte, a způsobují, že je méně účinný pro prevenci a zastavení krvácení.
• se u vás již objevil inhibitor faktoru VIII a změnil(a) jste produkty s faktorem VIII, existuje tu riziko, že se vám inhibitor vrátí.
• Vám bylo řečeno, že máte onemocnění srdce nebo riziko pro vznik onemocnění srdce.
• je pro podání přípravku KOGENATE Bayer potřeba použít centrální žilní vstup. Můžete mít riziko vzniku komplikací souvisejících s centrálním žilním vstupem, včetně lokální infekce, bakterií v krvi (bakteriemie) a krevních sraženin v cévách (trombóza) v místě vstupu katétru.
Váš lékař může nechat provést testy, aby se ujistil, že Vaše současná dávka tohoto přípravku zajišťuje dostatečnou hladinu faktoru VIII.
Další léčivé přípravky a přípravek KOGENATE Bayer
Vzájemná působení s jinými léky nejsou známa. Nicméně informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Děti a dospívající
Uvedená upozornění a opatření se týkají pacientů všech věkových skupin, dospělých a dětí. Těhotenství, kojení a fertilita
Nejsou žádné zkušenosti s přípravkem KOGENATE Bayer ohledně fertility nebo s používáním tohoto přípravku během těhotenství a kojení. Z tohoto důvodu, pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
KOGENATE Bayer pravděpodobně neovlivní plodnost u mužů nebo žen, protože léčivá látka se normálně vyskytuje v těle.
Řízení dopravních prostředků a obsluha strojů
Nebyl pozorován žádný účinek na schopnost řídit dopravní prostředek nebo obsluhovat stroje. Přípravek KOGENATE Bayer obsahuje sodík
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku na injekční lahvičku, tzn. v podstatě je „bez sodík“.
Dokumentace
Je doporučeno, abyste pokaždé, když použijete přípravek KOGENATE Bayer, zapsali název a číslo šarže tohoto přípravku.
3. Jak se přípravek KOGENATE Bayer používá
Vždy používejte tento přípravek přesně v souladu s příbalovou informací nebo podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Léčba krvácení
Váš lékař vypočítá dávku tohoto přípravku a stanoví, jak často ho máte používat, aby bylo dosaženo potřebné hladiny úrovně aktivity faktoru VIII v krvi. Lékař má vždy upravit dávku a frekvenci podávání podle Vašich individuálních potřeb. Množství a četnost používání přípravku KOGENATE Bayer závisí na mnoha faktorech, jako jsou:
• Vaše hmotnost,
• závažnost hemofilie,
• místo a závažnost krvácení,
• zda máte inhibitory a jak je vysoký jejich titr,
• požadovaná hladina faktoru VIII.
Prevence krvácení
Pokud užíváte KOGENATE Bayer za účelem prevence krvácení (profylaxe), váš lékař vypočte potřebnou dávku. Tato dávka se bude obvykle pohybovat v rozsahu 20 až 40 IU oktokogu alfa na kg tělesné váhy, podávaná každé 2 až 3 dny. Nicméně v některých případech, zejména u mladších pacientů, může být nutné podávat přípravek v kratších intervalech nebo ve vyšších dávkách.
Laboratorní testy
Velmi se doporučuje, aby byly prováděny příslušné laboratorní zkoušky vaší plazmy ve vhodných intervalech, aby bylo zajištěno, že byla dosažena a je udržována přiměřená hladina faktoru VIII. Zejména v případě větších chirurgických zákroků je nevyhnutelné přesné monitorování substituční terapie pomocí koagulační analýzy.
Použití u dětí a dospívajících
KOGENATE Bayer může být používán u dětí všech věkových skupin.
Pokud není krvácení zastaveno
Pokud faktor VIII ve vaší plazmě nedosáhne očekávané hladiny nebo pokud není krvácení zastaveno ani po zřejmém dostatečném dávkování, je možné, že u vás došlo k rozvoji inhibitorů faktoru VIII. Tato možnost musí být zkontrolována zkušeným lékařem.
Jestliže máte pocit, že účinek tohoto léčivého přípravku je příliš silný nebo příliš slabý, řekněte to svému lékaři.
Pacienti s inhibitory
Pokud vás lékař informoval, že se u vás vytvořily inhibitory faktoru VIII, budete pravděpodobně muset užívat k zastavení krvácení větší množství tohoto léčivého přípravku než dříve. Pokud tato dávka nezastaví krvácení, může váš lékař zvážit podání dalšího přípravku, koncentrátu faktoru VIIa nebo koncentrátu (aktivovaného) protrombinového komplexu.
Tyto terapie by měly být předepsány lékaři, kteří mají zkušenosti v péči o pacienty s hemofilií A. Promluvte si se svým lékařem, chcete-li více informací.
Nezvyšujte dávku tohoto léčivého přípravku, kterou užíváte k zastavení krvácení, aniž byste se poradil(a) se svým lékařem.
Doba trvání léčby
Váš lékař vám sdělí, jak často a v jakých intervalech má být tento léčivý přípravek podáván.
Obvykle je substituční terapie s KOGENATE Bayer celoživotní léčbou.
Jak se přípravek KOGENATE Bayer podává
Tento léčivý přípravek je určen pro injekci do žíly po dobu delší než 2 až 5 minut v závislosti na celkovém objemu a úrovni Vašeho pohodlí a má být použit běhěm tří hodin po rekonstituci.
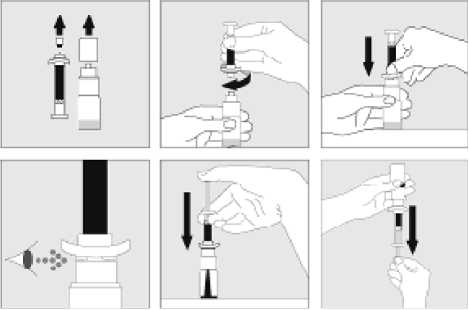
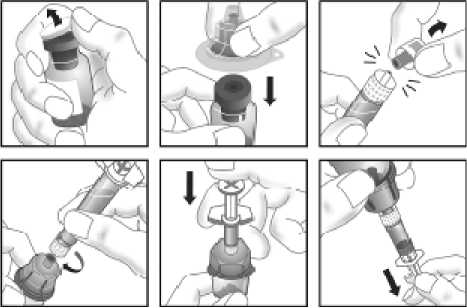
Jak se přípravek KOGENATE Bayer připravuje pro podání
Používejte pouze položky (injekční lahvička s práškem s víčkem Bio-Set, předplněná injekční stříkačka obsahující rozpouštědlo a venepunkční sada), které jsou součástí každého balení tohoto léčivého přípravku. Pokud tyto součásti nelze použít, obraťte se na svého lékaře. Pokud je kterákoli součást balení otevřená nebo poškozená, nepoužívejte ji.
Rozpuštěný přípravek je nutno před podáním přefiltrovat, aby byly z roztoku odstraněny případné přítomné částice. Filtrování provádějte podle pokynů pro rozpuštění a/nebo podání, které jsou popsány níže. Je důležité použít dodanou venepunkční sadu, protože její součástí je in-line filtr. Pokud nelze použít dodanou venepunkční sadu, použijte samostatný filtr podle instrukcí zdravotní sestry nebo lékaře.
Dodanou venepunkční sadu nepoužívejte k odběru krve, protože obsahuje in-line filtr. Jestliže je třeba před infuzí provést odběr krve, použijte aplikační sadu bez filtru a pak podejte infuzi tohoto přípravku přes injekční filtr. S dotazy, které se týkají tohoto přípravku a kompatibilních samostatných filtrů, se obraťte na svého lékaře.
Tento léčivý přípravek nesmí být smíchán s jinými infuzními roztoky. Nepoužívejte roztoky, pokud obsahují viditelné částice nebo jsou zakalené. Dodržujte přesně pokyny svého lékaře a podrobné instrukce pro rozředění a podání uvedené na konci této příbalové informace.
Jestliže jste použil(a) více přípravku KOGENATE Bayer, než jste měl(a)
Nebyly zaznamenány žádné případy předávkování rekombinantním koagulačním faktorem VIII.
Pokud jste použil(a) větší množství KOGENATE Bayer, než jste měl(a), informujte, prosím, svého lékaře.
Jestliže jste zapomněl(a) použít přípravek KOGENATE Bayer
• Pokračujte okamžitě se svou další dávkou a pokračujte v pravidelných intervalech dle pokynů svého lékaře.
• Nezdvojujte následující dávku, abyste doplnil(a) vynechanou dávku.
Jestliže chcete přestat používat přípravek KOGENATE Bayer Nepřestávejte užívat KOGENATE Bayer 250 IU bez porady se svým lékařem.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Mezi nejzávažnější nežádoucí účinky patří alergické reakce nebo anafylaktický šok (vzácný nežádoucí účinek).
Pokud se vyskytne alergická nebo anafylaktická reakce, injekce/infuze musí být okamžitě zastaveny. Prosím, obraťte se okamžitě na svého lékaře.
Celkový seznam možných nežádoucích účinků:
Velmi časté (mohou postihnout více než 1 z 10 uživatelů)
• tvorba neutralizačních protilátek proti faktoru VIII (inhibitory) u dříve neléčených pacientů
Časté (mohou se týkat až 1 z 10 uživatelů)
• vyrážka/svědivá vyrážka,
• lokální reakce v místě vpichu léčiva (např. pálení, přechodné zčervenání).
Méně časté (mohou postihnout až 1 ze 100 uživatelů)
• tvorba neutralizačních protilátek proti faktoru VIII (inhibitory) u dříve léčených pacientů Vzácné (mohou se týkat až 1 z 1 000 uživatelů)
• hypersensitivní reakce, včetně závažné náhlé alergické reakce (která může zahrnovat vyrážku, pocit na zvracení, kopřivku, angioedém, zimnici, návaly horka, bolest hlavy, letargii, dušnost nebo obtížné dýchání, neklid, tachykardii, mravenčení nebo anafylaktický šok, např. svírání
v hrudní oblasti/ celková nevolnost, závratě a nevolnost, mírně snížený krevní tlak, který způsobí, že pocítíte mdloby poté, co vstanete)
• horečka
Není známo (frekvenci nelze z dostupných údajů určit)
• dysgeusie (zvláštní chuť)
Pokud zpozorujete jakýkoli z následujících symptomů během aplikace injekce/infúze:
• svíravý pocit v hrudníku/celkovou nevolnost,
• závratě,
• mírnou hypotenzi (mírně snížený krevní tlak, který způsobí, že pocítíte mdloby poté, jakmile vstanete),
• nevolnost,
může to znamenat časné upozornění na hypersensitivitu a anafylaktické reakce.
Pokud dojde k alergické nebo anafylaktické reakci, musí být injekce/infúze okamžitě zastaveny. Prosím, obraťte se okamžitě na svého lékaře.
Protilátky (inhibitory)
Tvorba neutralizačních protilátek na faktor VIII (inhibitorů) je známá komplikace v léčbě pacientů s hemofilií A. Váš lékař může chtít provést testy sledující rozvoj inhibitorů.
Reakce přecitlivělosti
V průběhu klinických hodnocení se u žádného pacienta nevytvořily klinicky relevantní titry protilátek proti stopovému množství myší a křeččí bílkoviny přítomné v přípravku. Možnost alergické reakce na složky obsažené v tomto léčivu, např. stopová množství myší a křeččí bílkoviny v přípravku, existuje u jistých predisponovaných pacientů.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek KOGENATE Bayer uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem. Injekční lahvičku a předplněnou injekční stříkačku uchovávejte ve vnějším obalu, aby byly chráněny před světlem.
V rámci doby použitelnosti uvedené na obalu může být přípravek uchováván ve vnějším obalu při pokojové teplotě (do 25°C) po omezenou dobu 12 měsíců. V tomto případě skončí doba použitelnosti přípravku po 12 měsících nebo po uplynutí data použitelnosti uvedeného na injekční lahvičce, podle toho, co nastane dříve. Nové datum použitelnosti musíte zapsat na krabičku.
Po rekonstituci roztok chraňte před chladem. Rekonstituovaný roztok musí být použit během 3 hodin. Tento přípravek je určen pouze pro jednorázové použití. Všechen nespotřebovaný roztok musí být zlikvidován.
Nepoužívejte tento léčivý přípravek po uplynutí doby použitelnosti vyznačené na štítcích a krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Nepoužívejte tento léčivý přípravek, jestliže zaznamenáte viditelné částečky nebo zakalení roztoku.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek KOGENATE Bayer obsahuje
Prášek
Léčivou látkou je lidský koagulační faktor VIII (octocogum alfa), který je vyroben rekombinantní DNA technologií. Jedna injekční lahvička s přípravkem KOGENATE Bayer obsahuje nominální množství 250, 500, 1 000, 2 000 nebo 3 000 IU octocogum alfa.
Pomocnými látkami jsou glycin, chlorid sodný, chlorid vápenatý, histidin, polysorbát 80 a sacharóza
(viz poslední část bodu 2).
Rozpouštědlo Voda na injekci.
Jak přípravek KOGENATE Bayer vypadá a co obsahuje toto balení
KOGENATE Bayer se dodává jako prášek a rozpouštědlo pro injekční roztok a je to suchý bílý až nažloutlý prášek nebo tableta. The pre-filled syringe contains water for injections to be used to reconstitute the contents of the vial. Po rozpuštění je roztok čirý. Zdravotnické prostředky pro rozpuštění a podání se dodávají s každým balením tohoto přípravku.
Každé balení přípravku KOGENATE Bayer obsahuje injekční lahvičku s víčkem Bio-Set a předplněnou stříkačku se samostatným pístem, stejně jako venepunkční sadu (pro podání injekce do žíly), dva tampony napuštěné alkoholem, dva suché tampony a dvě náplasti.
Držitel rozhodnutí o registraci
Bayer Pharma AG 13342 Berlin Německo
Výrobce
Bayer HealthCare Manufacturing S.r.l.
Via delle Groane 126
20024 Garbagnate Milanese (MI)
Itálie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Lietuva
UAB Bayer
Tel. +37 05 23 36 868
Luxembourg/Luxemburg
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Magyarország
Bayer Hungária KFT
Tel:+36 14 87-41 00
Malta
Alfred Gera and Sons Ltd. Tel: +35 621 44 62 05 Nederland Bayer B.V.
Tel: +31-(0)297-28 06 66 Norge
Bayer AS
Tlf: +47 23 13 05 00 Osterreich
Bayer Austria Ges.m.b.H. Tel: +43-(0)1-711 46-0 Polska
Bayer Sp. z o.o.
Tel: +48 22 572 35 00 Portugal
Bayer Portugal, Lda..
Tel: +351 21 416 42 00
Románia
SC Bayer SRL
Tel: +40 21 529 59 00
Slovenija
Bayer d. o. o.
Tel: +386 (0)1 58 14 400 Slovenská republika Bayer spol. s r.o.
Tel. +421 2 59 21 31 11 Suomi/Finland
Bayer Oy
Puh/Tel: +358- 20 785 21 Sverige
Bayer AB
Tel: +46 (0) 8 580 223 00 United Kingdom Bayer plc
Tel: +44-(0)1635 563000
Belgie/Belgique/Belgien
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Bt^rapnn
Eaňep Etnrapna EOOfl Ten.: 359 02 81 401 01 Česká republika Bayer s.r.o.
Tel: +420 266 101 111
Danmark
Bayer A/S
Tlf: +45 45 23 50 00
Deutschland
Bayer Vital GmbH
Tel: +49 (0)214-30 513 48
Eesti
Bayer OU
Tel: +372 655 8565
EXlába
Bayer EXkáq ABEE Tr(k: +30-210-61 87 500 Espaňa
Bayer Hispania S.L.
Tel: +34-93-495 65 00 France
Bayer HealthCare
Tél (N° vert): +33-(0)800 87 54 54
Hrvatska
Bayer d.o.o.
Tel: +385-(0)1-6599 900
Ireland
Bayer Limited
Tel: +353 1 2999313
Island
Icepharma hf.
Sími: +354 540 8000 Italia
Bayer S.p.A.
Tel: +39 02 397 81 Kúnpoq
NOVAGEM Limited Tr(k: +357 22 48 38 58 Latvija SIA Bayer
Tel: +371 67 84 55 63
Tato příbalová informace byla naposledy revidována {MM/RRRR}.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
Podrobné instrukce pro rekonstituci a podání přípravku KOGENATE Bayer pomocí injekční lahvičky s víčkem na rekonstituci (systém Bio-Set):
1. Omyjte si pečlivě ruce mýdlem a teplou vodou. Roztok musí být připravován na čistém a suchém povrchu.
2. Zahřejte neotevřenou injekční lahvičku s práškem a injekční stříkačku s rozpouštědlem ve
svých rukách na tělesnou teplotu. Materiál nemá být zahřátý na vyšší, než je tělesná teplota (nesmí překročit 37 °C).
3.
Odstraňte víčko z injekční lahvičky s práškem tak, že víčkem několikrát pohnete na obě strany a zároveň táhnete víčko směrem nahoru. Vytáhněte zátku připojenou k bílému víčku z injekční stříkačky (A).
4.
Našroubujte šetrně injekční stříkačku na injekční lahvičku s práškem (B).
5.
Umístěte injekční lahvičku na stabilní a neklouzavý povrch a jednou rukou ji pevně držte. Poté silně zatlačte palcem a ukazováčkem na ochranný štítek u konce injekční stříkačky směrem dolů (C), dokud se štítek nenarazí na horní okraj víčka na rekonstituci (Bio-Set).
Toto potvrzuje, že systém je aktivován (D).
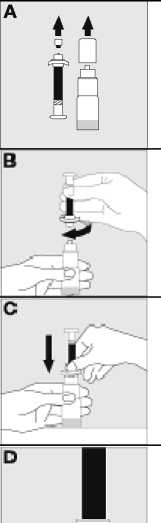

6.
Připevněte píst k injekční stříkačce tak, že jej zašroubujete do pryžové zátky (E).
7.
Vstříkněte roztok do injekční lahvičky s práškem tak, že pomalu tlačíte píst injekční stříkačky směrem dolů (F).
8.
Prášek rozpusťte mírným kroužením injekční lahvičkou (G). Injekční lahvičkou netřepejte! Ujistěte se před použitím, že se veškerý prášek rozpustil. Před podáním vizuálně zkontrolujte, zda nejsou přítomny částice nebo zda přípravek nezměnil barvu. Nepoužívejte roztok, který obsahuje viditelné částice nebo je zakalený.
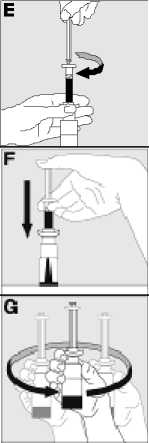
Obraťte injekční lahvičku/injekční stříkačku a natáhněte roztok do injekční stříkačky tak, že pomalu a plynule vytahujete píst ze stříkačky (H). Zkontrolujte, že byl celý obsah injekční lahvičky přemístěn do stříkačky. Držte stříkačku svisle a zatlačte na píst tak, aby ve stříkačce nezbyl žádný vzduch.
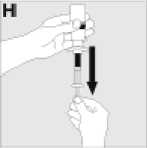
10. Přiložte na paži turniket. Určete místo vpichu, očistěte kůži tamponem napuštěným alkoholem.
a připravte antisepticky místo vpichu tak, jak Vás poučil lékař. Napíchněte žílu a zajistěte venepunkční sadu náplastí.
11. Vyšroubujte injekční stříkačku, abyste oddělili injekční lahvičku (I).
12. Připevněte injekční stříkačku k venepunkční sadě šroubováním ve směru
hodinových ručiček a ujistěte se, že se do injekční stříkačky nedostává krev (J).
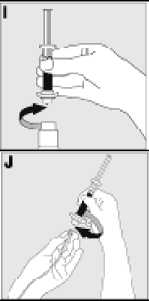
13. Sejměte turniket!
|
14. |
Roztok podávejte injekcí do žíly po dobu 2 až 5 minut, přičemž kontrolujte polohu jehly. Rychlost podání by měla být založena na komfortu pacienta (ale neměla by být rychlejší než maximální rychlost infuze: 2 ml/min). |
|
15. |
Je-li nutné podat další dávku, odstraňte prázdnou injekční stříkačku otáčením proti směru hodinových ručiček. Rozřeďte potřebné množství přípravku, zopakujte kroky 2-9, požijte novou injekční stříkačku a připojte ji k venepunkční sadě. |
|
16. |
Pokud není potřebná žádná další dávka, odstraňte venepunkční sadu a injekční stříkačku. Přidržujte tampón pevně k místu vpichu na natažené paži po dobu asi 2 minut. Nakonec přiložte na místo aplikace injekce malý tlakový obvaz a zvažte, je-li nutné použít náplast. |
Příbalová informace: informace pro uživatele
KOGENATE Bayer 250 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 500 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 1000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 2000 IU prášek a rozpouštědlo pro injekční roztok KOGENATE Bayer 3000 IU prášek a rozpouštědlo pro injekční roztok Rekombinantní koagulační faktor VIII (octocogum alfa)
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek KOGENATE Bayer a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek KOGENATE Bayer používat
3. Jak se přípravek KOGENATE Bayer používá
4. Možné nežádoucí účinky
5. Jak přípravek KOGENATE Bayer uchovávat
6. Obsah balení a další informace
1. Co je přípravek KOGENATE Bayer a k čemu se používá
KOGENATE Bayer obsahuje léčivou látku rekombinantní lidský koagulační faktor VIII (oktokog alfa).
KOGENATE Bayer se používá k léčbě a profylaxi krvácení u dospělých, dospívajících a dětí všech věkových kategorií s hemofilií A (vrozený nedostatek faktoru VIII).
Tento přípravek neobsahuje von Willebrandův faktor, a z tohoto důvodu nemá být používán u von Willebrandovy choroby.
2. Čemu musíte věnovat pozornost, než začnete přípravek KOGENATE Bayer používat
Nepoužívejte přípravek KOGENATE Bayer
• jestliže jste alergický/á na oktokog alfa nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6 a na konci bodui 2).
• jestliže jste alergický/á na myší nebo křeččí bílkovinu.
Jestliže si nejste jistí, zeptejte se svého lékaře.
Upozornění a opatření
Věnujte zvláštní opatrnost při použití přípravku KOGENATE Bayer a poraďte se se svým
lékařem nebo lékárníkem, pokud:
• máte svíravý pocit v hrudní oblasti, točí se vám hlava, je vám nevolno nebo mdlo, nebo máte závratě, jakmile vstanete, může to znamenat závažnou, náhlou alergickou reakci (tak zvanou anafylaktickou reakci) na tento léčivý přípravek. Pokud toto nastane, okamžitě zastavte podávání přípravku a vyhledejte lékařskou pomoc.
• krvácení není kontrolováno obvyklou dávkou tohoto léčivého přípravku, okamžitě se poraďte se svým lékařem. Pravděpodobně u vás došlo k vytvoření inhibitorů faktoru VIII a lékař může chtít provést testy, aby toto potvrdil. Inhibitory faktoru VIII jsou protilátky v krvi, které blokují faktor VIII, který užíváte, a způsobují, že je méně účinný pro prevenci a zastavení krvácení.
• se u vás již objevil inhibitor faktoru VIII a změnil(a) jste produkty s faktorem VIII, existuje tu riziko, že se vám inhibitor vrátí.
• Vám bylo řečeno, že máte onemocnění srdce nebo riziko pro vznik onemocnění srdce.
• je pro podání přípravku KOGENATE Bayer potřeba použít centrální žilní vstup. Můžete mít riziko vzniku komplikací souvisejících s centrálním žilním vstupem, včetně lokální infekce, bakterií v krvi (bakteriemie) a krevních sraženin v cévách (trombóza) v místě vstupu katétru.
Váš lékař může nechat provést testy, aby se ujistil, že Vaše současná dávka tohoto přípravku zajišťuje dostatečnou hladinu faktoru VIII.
Další léčivé přípravky a přípravek KOGENATE Bayer
Vzájemná působení s jinými léky nejsou známa. Nicméně informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Děti a dospívající
Uvedená upozornění a opatření se týkají pacientů všech věkových skupin, dospělých a dětí. Těhotenství, kojení a fertilita
Nejsou žádné zkušenosti s přípravkem KOGENATE Bayer ohledně fertility nebo s používáním tohoto přípravku během těhotenství a kojení. Z tohoto důvodu, pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Přípravek KOGENATE Bayer pravděpodobně neovlivní plodnost u mužů nebo žen, protože léčivá látka se normálně vyskytuje v těle.
Řízení dopravních prostředků a obsluha strojů
Nebyl pozorován žádný účinek na schopnost řídit dopravní prostředek nebo obsluhovat stroje. Přípravek KOGENATE Bayer obsahuje sodík
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku na injekční lahvičku, tzn. v podstatě je „bez sodíku“.
Dokumentace
Je doporučeno, abyste pokaždé, když použijete přípravek KOGENATE Bayer, zapsali název a číslo šarže tohoto přípravku.
3. Jak se přípravek KOGENATE Bayer používá
Vždy používejte tento přípravek přesně v souladu s příbalovou informací nebo podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Léčba krvácení
Váš lékař vypočítá dávku tohoto přípravku a stanoví, jak často ho máte používat, aby bylo dosaženo potřebné hladiny úrovně aktivity faktoru VIII v krvi. Lékař má vždy upravit dávku a frekvenci podávání podle Vašich individuálních potřeb. Množství a četnost používání přípravku KOGENATE Bayer závisí na mnoha faktorech, jako jsou:
• Vaše hmotnost,
• závažnost hemofilie,
• místo a závažnost krvácení,
• zda máte inhibitory a jak je vysoký jejich titr,
• požadovaná hladina faktoru VIII.
Prevence krvácení
Pokud užíváte KOGENATE Bayer za účelem prevence krvácení (profylaxe), váš lékař vypočte potřebnou dávku. Tato dávka se bude obvykle pohybovat v rozsahu 20 až 40 IU oktokogu alfa na kg tělesné váhy, podávaná každé 2 až 3 dny. Nicméně v některých případech, zejména u mladších pacientů, může být nutné podávat přípravek v kratších intervalech nebo ve vyšších dávkách.
Laboratorní testy
Velmi se doporučuje, aby byly prováděny příslušné laboratorní zkoušky vaší plazmy ve vhodných intervalech, aby bylo zajištěno, že byla dosažena a je udržována přiměřená hladina faktoru VIII. Zejména v případě větších chirurgických zákroků je nevyhnutelné přesné monitorování substituční terapie pomocí koagulační analýzy.
Použití u dětí a dospívajících
KOGENATE Bayer může být používán u dětí všech věkových skupin.
Pokud není krvácení zastaveno
Pokud faktor VIII ve vaší plazmě nedosáhne očekávané hladiny nebo pokud není krvácení zastaveno ani po zřejmém dostatečném dávkování, je možné, že u vás došlo k rozvoji inhibitorů faktoru VIII. Tato možnost musí být zkontrolována zkušeným lékařem.
Jestliže máte pocit, že účinek tohoto léčivého přípravku je příliš silný nebo příliš slabý, řekněte to svému lékaři.
Pacienti s inhibitory
Pokud vás lékař informoval, že se u vás vytvořily inhibitory faktoru VIII, budete pravděpodobně muset užívat k zastavení krvácení větší množství tohoto léčivého přípravku než dříve. Pokud tato dávka nezastaví krvácení, může váš lékař zvážit podání dalšího přípravku, koncentrátu faktoru VIIa nebo koncentrátu (aktivovaného) protrombinového komplexu.
Tyto terapie by měly být předepsány lékaři, kteří mají zkušenosti v péči o pacienty s hemofilií A. Promluvte si se svým lékařem, chcete-li více informací.
Nezvyšujte dávku tohoto léčivého přípravku, kterou užíváte k zastavení krvácení, aniž byste se poradil(a) se svým lékařem.
Doba trvání léčby
Váš lékař vám sdělí, jak často a v jakých intervalech má být tento léčivý přípravek podáván.
Obvykle je substituční terapie s KOGENATE Bayer celoživotní léčbou.
Jak se přípravek KOGENATE Bayer podává
Tento léčivý přípravek je určen pro injekci do žíly po dobu delší než 2 až 5 minut v závislosti na celkovém objemu a úrovni Vašeho pohodlí a má být použit běhěm tří hodin po rekonstituci.
Jak se přípravek KOGENATE Bayer připravuje pro podání
Používejte pouze položky (adaptér injekční lahvičky, předplněná injekční stříkačka obsahující rozpouštědlo a venepunkční sada), které jsou součástí každého balení tohoto léčivého přípravku.
Pokud tyto součásti nelze použít, obraťte se na svého lékaře. Pokud je kterákoli součást balení otevřená nebo poškozená, nepoužívejte ji.
Rozpuštěný přípravek je nutno před podáním přefiltrovat, aby byly z roztoku odstraněny případné přítomné částice. Filtrování provádějte pomocí adaptéru injekční lahvičky.
Dodanou venepunkční sadu nepoužívejte k odběru krve, protože obsahuje in-line filtr.
Tento léčivý přípravek nesmí být smíchán s jinými infuzními roztoky. Nepoužívejte roztoky, pokud obsahují viditelné částice nebo jsou zakalené. Dodržujte přesně pokyny svého lékaře a podrobné instrukce pro rozředění a podání uvedené na konci této příbalové informace.
Jestliže jste použil(a) více přípravku KOGENATE Bayer, než jste měl(a)
Nebyly zaznamenány žádné případy předávkování rekombinantním koagulačním faktorem VIII. Pokud jste použil(a) větší množství KOGENATE Bayer, než jste měl(a), informujte, prosím, svého lékaře.
Jestliže jste zapomněl(a) použít přípravek KOGENATE Bayer
• Pokračujte okamžitě se svou další dávkou a pokračujte v pravidelných intervalech dle pokynů svého lékaře.
• Nezdvojujte následující dávku, abyste doplnil(a) vynechanou dávku.
Jestliže chcete přestat používat přípravek KOGENATE Bayer Nepřestávejte užívat KOGENATE Bayer 250 IU bez porady se svým lékařem.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Mezi nejzávažnější nežádoucí účinky patří alergické reakce nebo anafylaktický šok (vzácný nežádoucí účinek).
Pokud se vyskytne alergická nebo anafylaktická reakce, injekce/infuze musí být okamžitě zastaveny. Prosím, obraťte se okamžitě na svého lékaře.
Celkový seznam možných nežádoucích účinků:
Velmi časté (mohou postihnout více než 1 z 10 uživatelů)
• tvorba neutralizačních protilátek proti faktoru VIII (inhibitory) u dříve neléčených pacientů
Časté (mohou se týkat až 1 z 10 uživatelů)
• vyrážka/svědivá vyrážka,
• lokální reakce v místě vpichu léčiva (např. pálení, přechodné zčervenání).
Méně časté (mohou postihnout až 1 ze 100 uživatelů)
• tvorba neutralizačních protilátek proti faktoru VIII (inhibitory) u dříve léčených pacientů Vzácné (mohou se týkat až 1 z 1 000 uživatelů)
• hypersensitivní reakce včetně závažné náhlé alergické reakce (která může zahrnout vyrážku, pocit na zvracení, kopřivku, angioedém, zimnici, návaly horka, bolest hlavy, letargii, dušnost nebo obtížné dýchání, neklid, tachykardii, mravenčení nebo anafylaktický šok, např. svírání v hrudní oblasti/ celková nevolnost, závratě a nevolnost, mírně snížený krevní tlak, který způsobí, že pocítíte mdloby poté, co vstanete)
• horečka
Není známo (frekvenci nelze z dostupných údajů určit)
• dysgeusie (zvláštní chuť)
Pokud zpozorujete jakýkoli z následujících symptomů během aplikace injekce/infúze:
• svíravý pocit v hrudníku/celkovou nevolnost,
• závratě,
• mírnou hypotenzi (mírně snížený krevní tlak, který způsobí, že pocítíte mdloby poté, jakmile vstanete),
• nevolnost,
může to znamenat časné upozornění na hypersensitivitu a anafylaktické reakce.
Pokud dojde k alergické nebo anafýlaktické reakci, musí být injekce/infúze okamžitě zastaveny. Prosím, obraťte se okamžitě na svého lékaře.
Protilátky (inhibitory)
Tvorba neutralizačních protilátek na faktor VIII (inhibitorů) je známá komplikace v léčbě pacientů s hemofilií A. Váš lékař může chtít provést testy sledující rozvoj inhibitorů.
Reakce přecitlivělosti
V průběhu klinických hodnocení se u žádného pacienta nevytvořily klinicky relevantní titry protilátek proti stopovému množství myší a křeččí bílkoviny přítomné v přípravku. Možnost alergické reakce na složky obsažené v tomto léčivu, např. stopová množství myší a křeččí bílkoviny v přípravku, existuje u jistých predisponovaných pacientů.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek KOGENATE Bayer uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem. Injekční lahvičku a předplněnou injekční stříkačku uchovávejte ve vnějším obalu, aby byly chráněny před světlem.
V rámci doby použitelnosti uvedené na obalu může být přípravek uchováván ve vnějším obalu při pokojové teplotě (do 25°C) po omezenou dobu 12 měsíců. V tomto případě skončí doba použitelnosti přípravku po 12 měsících nebo po uplynutí data použitelnosti uvedeného na injekční lahvičce, podle toho, co nastane dříve. Nové datum použitelnosti musíte zapsat na krabičku.
Po rekonstituci roztok chraňte před chladem. Rekonstituovaný roztok musí být použit během 3 hodin. Tento přípravek je určen pouze pro jednorázové použití. Všechen nespotřebovaný roztok musí být zlikvidován.
Nepoužívejte tento léčivý přípravek po uplynutí doby použitelnosti vyznačené na štítcích a krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Nepoužívejte tento léčivý přípravek, jestliže zaznamenáte viditelné částečky nebo zakalení roztoku.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek KOGENATE Bayer obsahuje
Prášek
Léčivou látkou je lidský koagulační faktor VIII (octocogum alfa), který je vyroben rekombinantní DNA technologií. Jedna injekční lahvička s přípravkem KOGENATE Bayer obsahuje nominální množství 250, 500, 1 000, 2 000 nebo 3 000 IU octocogum alfa.
Pomocnými látkami jsou glycin, chlorid sodný, chlorid vápenatý, histidin, polysorbát 80 a sacharóza
(viz poslední část bodu 2).
Rozpouštědlo Voda na injekci.
Jak přípravek KOGENATE Bayer vypadá a co obsahuje toto balení
KOGENATE Bayer se dodává jako prášek a rozpouštědlo pro injekční roztok a je to suchý bílý až nažloutlý prášek nebo tableta. Předplněná injekční stříkačka obsahuje vodu na injekci a slouží k rozpuštění obsahu injekční lahvičky. Po rozpuštění je roztok čirý. Zdravotnické prostředky pro rozpuštění a podání se dodávají s každým balením tohoto přípravku.
Každé balení přípravku KOGENATE Bayer obsahuje injekční lahvičku a předplněnou stříkačku se samostatným pístem, stejně jako adaptér injekční lahvičky, venepunkční sadu (pro podání injekce do žíly), dva tampony napuštěné alkoholem, dva suché tampony a dvě náplasti.
Držitel rozhodnutí o registraci
Bayer Pharma AG 13342 Berlin Německo
Výrobce
Bayer HealthCare Manufacturing S.r.l.
Via delle Groane 126
20024 Garbagnate Milanese (MI)
Itálie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Lietuva
UAB Bayer
Tel. +37 05 23 36 868
Luxembourg/Luxemburg
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Magyarország
Bayer Hungária KFT
Tel:+36 14 87-41 00
Malta
Alfred Gera and Sons Ltd. Tel: +35 621 44 62 05 Nederland Bayer B.V.
Tel: +31-(0)297-28 06 66 Norge
Bayer AS
Tlf: +47 23 13 05 00 Osterreich
Bayer Austria Ges.m.b.H. Tel: +43-(0)1-711 46-0 Polska
Bayer Sp. z o.o.
Tel: +48 22 572 35 00 Portugal
Bayer Portugal, Lda.
Tel: +351 21 416 42 00
Románia
SC Bayer SRL
Tel: +40 21 529 59 00
Slovenija
Bayer d. o. o.
Tel: +386 (0)1 58 14 400 Slovenská republika Bayer spol. s r.o.
Tel. +421 2 59 21 31 11 Suomi/Finland
Bayer Oy
Puh/Tel: +358- 20 785 21 Sverige
Bayer AB
Tel: +46 (0) 8 580 223 00 United Kingdom Bayer plc
Tel: +44-(0)1635 563000
Belgie/Belgique/Belgien
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Bt^rapnn
Eaňep Etnrapna EOOfl Ten.: 359 02 81 401 01 Česká republika Bayer s.r.o.
Tel: +420 266 101 111
Danmark
Bayer A/S
Tlf: +45 45 23 50 00
Deutschland
Bayer Vital GmbH
Tel: +49 (0)214-30 513 48
Eesti
Bayer OU
Tel: +372 655 8565
EXlába
Bayer EXkáq ABEE Tr(k: +30-210-61 87 500 Espaňa
Bayer Hispania S.L.
Tel: +34-93-495 65 00 France
Bayer HealthCare
Tél (N° vert): +33-(0)800 87 54 54
Hrvatska
Bayer d.o.o.
Tel: +385-(0)1-6599 900
Ireland
Bayer Limited
Tel: +353 1 2999313
Island
Icepharma hf.
Sími: +354 540 8000 Italia
Bayer S.p.A.
Tel: +39 02 397 81 Kúnpoq
NOVAGEM Limited Tr(k: +357 22 48 38 58 Latvija SIA Bayer
Tel: +371 67 84 55 63
Tato příbalová informace byla naposledy revidována {MM/RRRR}.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
Podrobné instrukce pro rekonstituci a podání přípravku KOGENATE Bayer pomocí injekční lahvičky s adaptérem injekční lahvičky:
1. Omyjte si pečlivě ruce mýdlem a teplou vodou.
Zahřejte neotevřenou injekční lahvičku a injekční stříkačku ve svých rukách na příjemnou teplotu (nesmí překročit 37 °C).
3.
Odstraňte ochranné víčko z injekční lahvičky (A) a očistěte pryžovou zátku injekční lahvičky tamponem namočeným v alkoholu a nechte ji před použitím uschnout.
4.
Umístěte injekční lahvičku s přípravkem na stabilní, neklouzavý povrch. Odloupněte papírový kryt na plastovém obalu adaptéru injekční lahvičky. Nevyndávejte adaptér z plastového obalu. Držte obal adaptéru, nasaďte jej přes injekční lahvičku s přípravkem a pevně jej zatlačte (B). Adaptér zaklapne přes víčko injekční lahvičky. Nesundávejte nyní obal adaptéru.
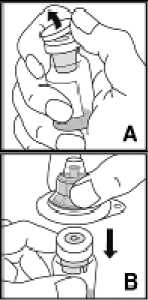
5.
Přidržte injekční stříkačku předplněnou vodou na injekci ve svislé poloze, uchopte píst podle obrázku a připojte jej otočením pevně ve směru hodinových ručiček do zátky se závitem (C).
6.
Držte injekční stříkačku za válec, odlomte víčko injekční stříkačky z jejího konce (D). Nedotýkejte se koncem injekční stříkačky ruky nebo jiného povrchu. Položte injekční stříkačku stranou pro další použití.
7.
Nyní sejměte a vyhoďte obal adaptéru (E).
8.
Připojte předplněnou injekční stříkačku na adaptér injekční lahvičky se závitem otočením ve směru hodinových ručiček (F).
9.
Vstříkněte rozpouštědlo tak, že tlačíte pomalu píst injekční stříkačky směrem dolů (G).
Mírným kroužením injekční lahvičkou rozpusťte veškerý materiál (H). Injekční lahvičkou netřepejte! Ujistěte se před použitím, že se veškerý prášek rozpustil. Před podáním vizuálně zkontrolujte, zda nejsou přítomny částice nebo zda přípravek nezměnil barvu. Nepoužívejte roztoky, které obsahují viditelné částice nebo jsou zakalené.
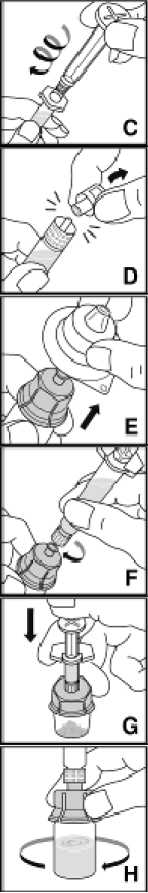
11. Držte injekční lahvičku na jednom konci nad adaptérem injekční
lahvičky a injekční stříkačkou (I). Naplňte injekční stříkačku tak, že pomalu a plynule vytahujete píst z injekční stříkačky. Držte stříkačku svisle a zatlačte na píst tak, aby ve stříkačce nezbyl žádný vzduch.
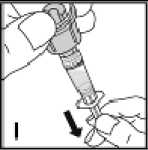
12. Přiložte turniket na paži.
13. Určete místo vpichu, očistěte kůži tamponem napuštěným alkoholem a připravte
antisepticky místo vpichu tak, jak Vás poučil lékař.
14. Napíchněte žílu a zajistěte venepunkční sadu náplastí.
15. Přidržujte adaptér injekční lahvičky na místě, sejměte injekční stříkačku
z adaptéru injekční lahvičky (adaptér by měl zůstat připojený na injekční lahvičku). Připevněte injekční stříkačku k venepunkční sadě a ujistěte se, že se do injekční stříkačky nedostává krev (J).
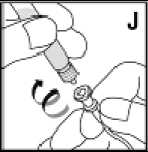
16. Sejměte turniket.
|
17. |
Roztok podávejte injekcí do žíly po dobu 2 až 5 minut, přičemž kontrolujte polohu jehly. Rychlost podání by měla být založena na komfortu pacienta (ale neměla by být rychlejší než maximální rychlost infuze: 2 ml/min). |
|
18. |
Je-li nutné podat další dávku, použijte novou injekční stříkačku s rekonstituovaným přípravkem, jak je popsáno výše. |
|
19. |
Pokud není potřebná žádná další dávka, odstraňte venepunkční sadu a injekční stříkačku. Přidržujte tampón pevně k místu vpichu na natažené paži po dobu asi 2 minut. Nakonec přiložte na místo aplikace injekce malý tlakový obvaz a zvažte, je-li nutné použít náplast. |
57